WO2019065774A1 - 放射性薬剤 - Google Patents
放射性薬剤 Download PDFInfo
- Publication number
- WO2019065774A1 WO2019065774A1 PCT/JP2018/035786 JP2018035786W WO2019065774A1 WO 2019065774 A1 WO2019065774 A1 WO 2019065774A1 JP 2018035786 W JP2018035786 W JP 2018035786W WO 2019065774 A1 WO2019065774 A1 WO 2019065774A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- fab
- fgk
- acceptable salt
- Prior art date
Links
- 0 *C(C(NC(CCCCN(*)*)C(O)O)=O)NC(C(Cc1ccccc1)NC(c1ccc(CI)cc1)=O)=O Chemical compound *C(C(NC(CCCCN(*)*)C(O)O)=O)NC(C(Cc1ccccc1)NC(c1ccc(CI)cc1)=O)=O 0.000 description 5
- ZMEGUEVQYVCTHE-UHFFFAOYSA-N CC(C)CN1CCN(CC(O)=O)C(Cc(cc2)ccc2C(NC(Cc2ccccc2)C(NCC(NC(CCCCN(C(C=C2)=O)C2=O)C(O)=O)=O)=O)=O)CN(C/C(/O)=[O]/C)CCN(CC(O)=O)CC1 Chemical compound CC(C)CN1CCN(CC(O)=O)C(Cc(cc2)ccc2C(NC(Cc2ccccc2)C(NCC(NC(CCCCN(C(C=C2)=O)C2=O)C(O)=O)=O)=O)=O)CN(C/C(/O)=[O]/C)CCN(CC(O)=O)CC1 ZMEGUEVQYVCTHE-UHFFFAOYSA-N 0.000 description 1
- DDXFZRZHQOCGAW-UHFFFAOYSA-N CC(C)CN1CCN(CC(O)=O)C(Cc(cc2)ccc2C(NC(Cc2ccccc2)C(NCC(NC(CCCCN)C(O)=O)=O)=O)=O)CN(CC(O)=O)CCN(CC(O)=O)CC1 Chemical compound CC(C)CN1CCN(CC(O)=O)C(Cc(cc2)ccc2C(NC(Cc2ccccc2)C(NCC(NC(CCCCN)C(O)=O)=O)=O)=O)CN(CC(O)=O)CCN(CC(O)=O)CC1 DDXFZRZHQOCGAW-UHFFFAOYSA-N 0.000 description 1
- HFYWWSMTUZUNAY-UHFFFAOYSA-N CC(C)CN1CCN(CC(O)=O)CC(C)N(CC(O)=O)CCN(CC(O)=O)CC1 Chemical compound CC(C)CN1CCN(CC(O)=O)CC(C)N(CC(O)=O)CCN(CC(O)=O)CC1 HFYWWSMTUZUNAY-UHFFFAOYSA-N 0.000 description 1
- FZKXNQPVALOUMN-UHFFFAOYSA-N CC(C)CN1CCN(CC(O)=O)CCN(CC(O)=O)CCN(CC(O)=O)CC1 Chemical compound CC(C)CN1CCN(CC(O)=O)CCN(CC(O)=O)CCN(CC(O)=O)CC1 FZKXNQPVALOUMN-UHFFFAOYSA-N 0.000 description 1
Images








Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
- A61K51/088—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins conjugates with carriers being peptides, polyamino acids or proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
- A61K51/10—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody
- A61K51/1093—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody conjugates with carriers being antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/0474—Organic compounds complexes or complex-forming compounds, i.e. wherein a radioactive metal (e.g. 111In3+) is complexed or chelated by, e.g. a N2S2, N3S, NS3, N4 chelating group
- A61K51/0482—Organic compounds complexes or complex-forming compounds, i.e. wherein a radioactive metal (e.g. 111In3+) is complexed or chelated by, e.g. a N2S2, N3S, NS3, N4 chelating group chelates from cyclic ligands, e.g. DOTA
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0812—Tripeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
- A61K51/10—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody
Definitions
- the present invention relates to a novel compound, a radioactive drug containing the same, a drug for preparing the radioactive drug, and the like.
- Radioactive agents such as radioisotope (RI) labeled antibodies are capable of tumor selective accumulation of RI utilizing the high specificity and affinity of the antibodies. Therefore, it is used for radiotherapy such as isotope therapy and image diagnosis (Non-patent Document 1).
- RI radioisotope
- Non-patent Document 1 Radioactive agents
- a radioactive drug is administered to a living body, nonspecific accumulation in the kidney is observed in addition to specific accumulation in the target tissue. Accumulation of radioactivity in the kidney (hereinafter also referred to as "renal accumulation") is that the RI labeled low molecular weight peptide is taken up into the kidney and is then transported to the lysosome where it is metabolized and the radioactive metabolite produced remains in the kidney Based on that.
- NOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid is used as a radiolabeled drug capable of reducing accumulation in the kidney from the early stage of administration.
- a radiolabeled drug capable of reducing accumulation in the kidney from the early stage of administration.
- compounds having a polypeptide site bound to a chelating agent such as X) and radiopharmaceuticals using the same have been reported.
- gallium-67 and technetium-99m radioactive isotopes can be used.
- various atoms including those with relatively large atomic radius such as lutetium-177 and yttrium-90, which are widely used as therapeutic radioisotopes, and their companion agents, such as indium-111, are now widely used as therapeutic radioisotopes. Labeling agents that can be applied to have not been developed. Therefore, the present invention relates to a compound or the like which can be labeled with various atoms including atoms having a relatively large atomic radius and which can reduce accumulation in the kidney.
- a compound represented by the following formula (1), or a pharmacologically acceptable salt thereof [In the formula, A 1 and A 2 are each independently an amino acid residue, m is an integer of 0 to 3, A 3 is an amino acid residue having an amino group or a carboxy group in the side chain, A 4 is an amino acid residue, n is an integer of 0 to 3, R 1 is bound to the amino group or carboxy group of the side chain of A 3, group having a binding functional group as a target molecule recognition element or a linking group, or, an amino group or carboxy group of the side chain of A 3 And R 1 may form a heterocyclic group having 3 to 10 carbon atoms, including the nitrogen atom of the amino group of the side chain of A 3 as one of the ring-constituting atoms, L is the formula (L1): (Wherein, R 3 , R 4 , R 5 , and R 6 are each independently a hydrogen atom, a —CH 2 COOR
- a pharmaceutical preparation for a radiopharmaceutical which comprises the compound according to the above-mentioned [1] or [2], or a pharmacologically acceptable salt thereof.
- a radiopharmaceutical comprising the metal complex compound according to the above [3], or a pharmacologically acceptable salt thereof.
- a radioactive diagnostic imaging agent of the metal complex compound of the above-mentioned [3], or a pharmacologically acceptable salt thereof [9] The compound according to [1] or [2] above, or a pharmacologically acceptable salt thereof, wherein the compound or the pharmacologically acceptable salt thereof is for the preparation of a radiopharmaceutical. [10] Use of the metal complex compound according to [3] above, or a pharmacologically acceptable salt thereof for the preparation of a radiopharmaceutical. [11] Use of the metal complex compound according to the above [3], or a pharmaceutically acceptable salt thereof for radiotherapy. [12] Use of the metal complex compound according to the above [3], or a pharmaceutically acceptable salt thereof for radiological imaging diagnosis.
- a method for diagnosing a radiographic image which comprises administering the metal complex compound according to [3] or a pharmacologically acceptable salt thereof.
- a target molecule recognition element comprising the compound of the above-mentioned [1] or a pharmacologically acceptable salt thereof, or the compound of the above-mentioned [1], or a pharmacologically acceptable salt thereof
- a kit comprising, as separate packaging units, a compound to be bound, or a pharmaceutically acceptable salt thereof, and an agent comprising one metal selected from the group consisting of radioactive metals and radioactive atom-labeled metals.
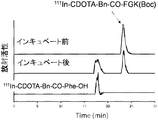
- FIG. 1 shows the experimental results of the incubation of 111 In-CDO3AEt-FGK (Boc) with BBMVs.
- FIG. 2 shows the experimental results of the incubation of 111 In-CDOTA-Bn-CO-FGK (Boc) solution with BBMVs.
- FIG. 3 shows the experimental results of incubation of 111 In-DO3A-Bn-SCN-MVK (Bzo) with BBMVs.
- FIG. 4 shows the experimental results of incubation of 111 In-DO3A-Bn-CO-FGK (Boc) with BBMVs.
- FIG. 5 shows the experimental results of the incubation of 111 In-CDO3AiBu-FGK (Boc) with BBMVs.
- FIG. 6 shows a comparison of the results of 111 In-CDO3AEt-FGK-Fab (derived from Rabbit serum IgG) and 111 In-DO3A-EDA-Fab (derived from Rabbit serum IgG).
- FIG. 7 shows 111 In-CDO3AiBu-FGK-Fab (derived from anti-c-kit IgG), 111 In-DOTA-Bn-SCN-Fab (derived from anti-c-kit IgG), and 111 In-CDO3AEt-FGK-Fab (derived from Figure 2 shows a comparison of the results of anti-c-kit IgG).
- FIG. 7 shows 111 In-CDO3AiBu-FGK-Fab (derived from anti-c-kit IgG), 111 In-DOTA-Bn-SCN-Fab (derived from anti-c-kit IgG), and 111 In-CDO3AEt-FGK-Fab (derived from Figure 2 shows a comparison of the results of anti-c-kit IgG).
- FIG. 8 shows the results of analysis of the chemical form of urinary radioactivity excreted up to 24 hours after administration of 111 In-CDO3AEt-FGK-Fab (derived from Rabbit serum IgG) to mice.
- FIG. 9 shows the results of analysis of the chemical form of urinary radioactivity excreted up to 6 hours after administration of 111 In-CDOTA-Bn-CO-FGK-Fab (derived from anti-c-kit IgG) to mice.
- FIG. 10 shows the results of analysis of chemical forms of urinary radioactivity excreted by 24 hours after administration of 111 In-CDO3AEt-FGK-Fab (derived from Rabbit serum IgG) to mice.
- FIG. 11 shows the results of analysis of chemical forms of urinary radioactivity excreted by 24 hours after administration of 111 In-CDO3AiBu-FGK-Fab (derived from Rabbit serum IgG) to mice.
- FIG. 12 shows that 111 In-DO3A-Bn-SCN-MVK-Fab It is a SPECT / CT image 2.5 hours after administering a solution (anti-c-kit IgG origin) solution or 111 In-CDO3AEt-FGK-Fab (anti-c-kit IgG origin) solution to SY subcutaneous tumor model mice.
- a 1 and A 2 are each independently an amino acid residue
- m is an integer of 0 to 3
- a 3 is an amino acid residue having an amino group or a carboxy group in the side chain
- a 4 is an amino acid residue
- n is an integer of 0 to 3
- R 1 is bound to the amino group or carboxy group of the side chain of A 3, group having a binding functional group as a target molecule recognition element or a linking group, or, an amino group or carboxy group of the side chain of A 3
- R 1 may form a heterocyclic group having 3 to 10 carbon atoms, including the nitrogen atom of the amino group of the side chain of A 3 as one of the ring-constituting atoms
- L is the formula (L1): (Wherein, R 3 , R 4 , R
- the radioactive agent of the present invention has a target molecule recognition element, it can specifically bind to a target site, and thus efficiently accumulate at the target site. Due to such properties, the radiopharmaceutical of the present invention can be specifically accumulated at a tumor site in radiation therapy, and its sensitivity and accuracy can be improved in radiological image diagnosis.
- the reason why the effects of the present invention can be obtained is not clear, but is considered as follows. It is thought that accumulation of radioactivity in the kidney can be reduced if the urinary radioactive metabolite can be efficiently released when the drug is taken into renal cells after administration of the radioactive drug. Therefore, when the radiopharmaceutical is incorporated into renal cells, the renal brush border membrane enzyme is located between the polypeptide and the chelating ligand site so that the chelating ligand site that incorporates the radioactive labeling element can be efficiently released. Introduce the substrate sequence.
- a 1 is preferably a residue of phenylalanine, methionine, valine, leucine, isoleucine, proline, tyrosine, glycine, alanine or tryptophan from the viewpoint of reducing accumulation in the kidney from the early stage of administration, more preferably phenylalanine It is a residue of glycine, alanine or methionine, more preferably a residue of phenylalanine from the viewpoint of making the effect of reducing the accumulation in the kidney from the early stage of administration more pronounced.
- a 2 is preferably a residue of glycine, phenylalanine, methionine, valine, leucine, isoleucine, proline, tyrosine, alanine or tryptophan from the viewpoint of reducing accumulation in the kidney from the early stage of administration, more preferably glycine Or a phenylalanine, alanine, valine or isoleucine residue, more preferably a glycine residue from the viewpoint of making the effect of reducing accumulation in the kidney early from administration more pronounced.
- m is an integer of 0 to 3, preferably 1.
- a 3 is an amino acid residue having an amino group or a carboxy group in the side chain, from the viewpoint of introducing a functional group capable of binding to the polypeptide or its linking group to the side chain of the amino acid sequence, preferably lysine, It is a residue of ornithine, arginine, aspartic acid or glutamic acid, more preferably a residue of lysine, ornithine or arginine, still more preferably a residue of lysine. As A 4, it may also have other amino acid residues. The A 4, any amino acids are used. n is an integer of 0 to 3, preferably 0.
- R 1 is a group having a functional group capable of binding to a target molecule recognition element or its linking group, or a hydrogen atom of an amino group or carboxy group of the side chain of A 2 , and an amino group of the side chain of A 3 or It bonds to a carboxy group.
- R 1 may form a heterocyclic group having a carbon number of 3 to 10, which contains the nitrogen atom of the amino group of the side chain of A 3 as one of the ring-constituting atoms.
- R 1 functions as a spacer and can bind a target molecule recognition element such as a polypeptide to the compound of the present invention via a functional group.
- R 1 can be linked to the amino group or carboxy group of the side chain of A 3 so that the compound of the present invention and the polypeptide can be linked without chemically modifying the end of the amino acid sequence.
- R 1 may be bonded to the nitrogen atom of the side chain amino group or may form an ester bond with the side chain carboxy group.
- the active ester of carboxy group examples include chloroacetyl group, bromoacetyl group, iodoacetyl group and the like.
- the total carbon number of R 1 is not particularly limited, but is preferably 1 or more, more preferably 2 or more, still more preferably 3 or more, and preferably 20 or less, more preferably 10 or less, still more preferably It is 8 or less.
- R 1 for example, an acyl group having 2 to 20 carbon atoms having a functional group a, an alkyl group having 2 to 20 carbon atoms having a functional group a, and an alkyl having 2 to 20 carbon atoms having a functional group a Examples thereof include a carbamoyl group and an alkylthiocarbamoyl group having 2 to 20 carbon atoms in total and having a functional group a.
- the heterocyclic group is preferably a maleimide group.
- the carbon number of the heterocyclic group is preferably 3 to 10, more preferably 3 to 5 and still more preferably 4 or 5.
- R 1 may be a hydrogen atom of an amino group or a carboxy group in the side chain of A 3 . That is, the amino group or carboxy group of A 3 may not be modified.
- R 1 heterocyclic group having 3 to 10 carbon atoms containing a nitrogen atom of the amino group of the side chain of A 3 as a ring-constituting atoms are preferred, the side chain of A 3 as a ring-constituting atom A maleimide group containing a nitrogen atom of an amino group is more preferred.
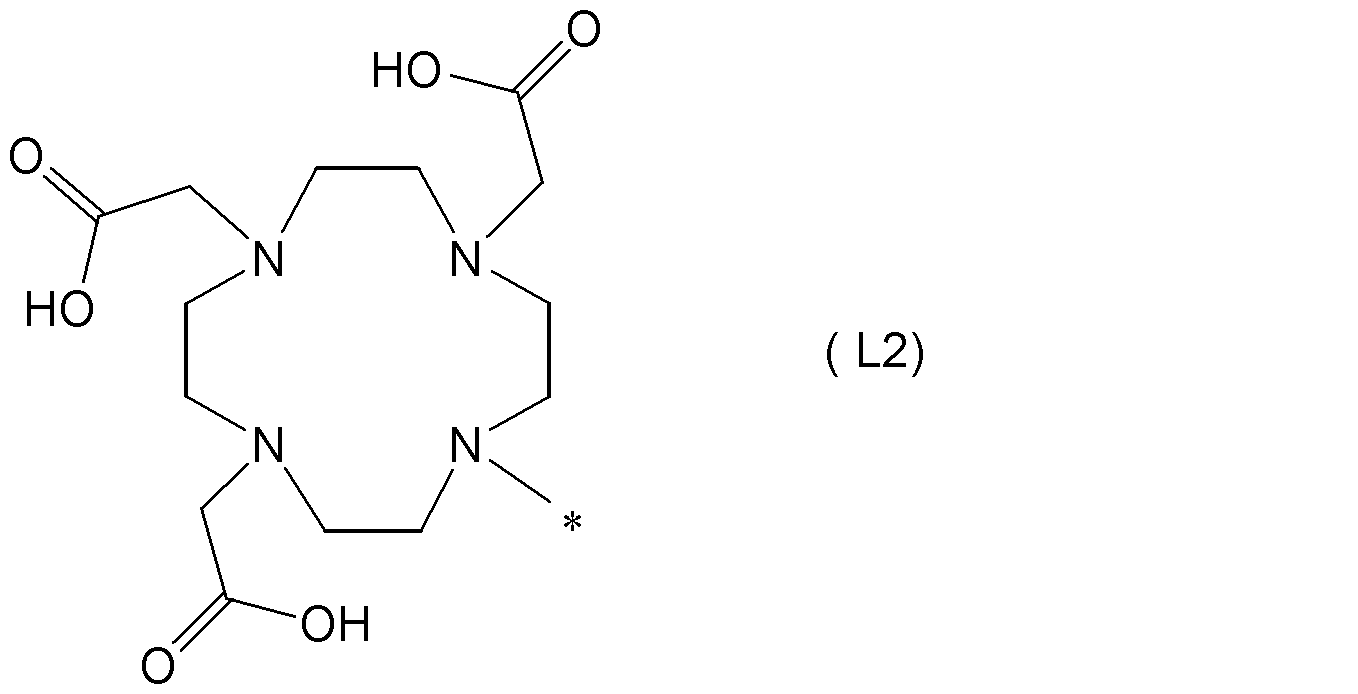
- L is the formula (L1): Or a group represented by the formula (L2): Is a group represented by Among the groups represented by formula (L1), among R 3 , R 4 , R 5 and R 6 , preferably 3 or more and 4 or less are —CH 2 COOH groups, more preferably 3 or more —CH 2 2 COOH group.
- R 10 is a hydrogen atom or a hydrocarbon group having 1 to 8 carbon atoms.
- groups other than —CH 2 COOH group are preferably hydrocarbon groups having 1 to 8 carbon atoms, and more preferably hydrocarbon groups having 1 to 4 carbon atoms It is.
- At least one group of R 3 , R 4 , R 5 and R 6 is preferably a hydrocarbon group having 3 to 8 carbon atoms, more preferably 4 carbon atoms. It is a hydrocarbon group of -6.
- the hydrocarbon group is preferably an aliphatic hydrocarbon group, more preferably a branched aliphatic hydrocarbon group. Examples of the hydrocarbon group include methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, sec-butyl group and isobutyl group.
- L is preferably represented by the formula (L1): (Wherein, R 3 , R 4 , R 5 and R 6 each represent a —CH 2 COOH group or a hydrocarbon group having 1 to 8 carbon atoms, and * represents a bonding site.
- R 3 and R 4 , R 5 and R 6 three of which are a —CH 2 COOH group.
- L a formula (L1-1), a formula (L1-2), a formula (L1-3), a formula (L1-4), a formula (L1-5), a formula (L1-6), a formula (L1-) 7) At least one selected from the group consisting of Formula (L1-8), Formula (L1-9) and Formula (L2) is preferable. (Hereinafter, * is a binding site.)
- L is the formula (L1): (Wherein, R 3 , R 4 , R 5 , and R 6 are each independently a hydrogen atom, a —CH 2 COOR 10 group, or a hydrocarbon group having 1 to 8 carbon atoms, and R 10 is a hydrogen atom Or a hydrocarbon group having 1 to 8 carbon atoms, and * represents a binding site, provided that at least three or more of R 3 , R 4 , R 5 , and R 6 are —CH 2 COOH groups.
- R 8 and R 9 may form a heterocyclic ring containing an adjacent nitrogen atom, in which case the formula:
- the group represented by is of the formula: Is a group represented by ]
- R 8 and R 9 are preferably an acyl group having 2 to 20 carbon atoms in total having a functional group a, and more preferably an acyl group having 3 to 6 carbon atoms in total having a carbamoyl group.
- L is a group of the formula (L1): (Wherein, R 3 , R 4 , R 5 and R 6 are each independently a —CH 2 COOH group or a hydrocarbon group having 1 to 8 carbon atoms, and * is a bonding site. However, R is And R 3 , R 4 , R 5 and R 6 each represent three groups of —CH 2 COOH group, and one group is a hydrocarbon group having 1 to 8 carbon atoms.
- L is a group of formula (L1): (Wherein, R 3 , R 4 , R 5 , and R 6 are each independently a —CH 2 COOH group, an ethyl group, or a butyl group, and * is a bonding site. However, R 3 , R 4 And R 5 and R 6 each represent three groups of —CH 2 COOH and one of ethyl and butyl. More preferably, in the formula, L is a group of formula (L1): (Wherein, R 3 , R 4 , R 5 and R 6 are each independently a —CH 2 COOH group and an isobutyl group, and * is a binding site. However, R 3 , R 4 , R 5 , Among R 6 , three are a —CH 2 COOH group, and one is an isobutyl group.
- Preferred specific examples of the compound (1) of the present invention include the following compound 1-1 to compound 1-6.
- the compound (1) etc. of the present invention may be a pharmacologically acceptable salt of the above-mentioned compound.
- pharmacologically acceptable salt for example, an acid addition salt, a base addition salt and the like can be mentioned.
- the acid addition salt may be either an inorganic acid salt or an organic acid salt.
- the inorganic acid salts include, for example, hydrochlorides, hydrobromides, sulfates, hydroiodides, nitrates and phosphates.
- the base addition salt may be either an inorganic base salt or an organic base salt.
- Inorganic base salts include, for example, sodium salts, potassium salts, calcium salts, magnesium salts and ammonium salts.
- Examples of the organic base salt include triethyl ammonium salt, triethanol ammonium salt, pyridinium salt and diisopropyl ammonium salt.
- the compound (2) etc. of the present invention is a compound obtained by binding a target molecule recognition element to the compound (1) or a pharmacologically acceptable salt thereof, or a pharmacologically acceptable salt thereof .
- the target molecule recognition element may be bound to Compound (1), or a pharmacologically acceptable salt thereof via a linking group, or may be directly bound.
- the linking group includes iminothiol derived from 2-iminothiolane.
- target molecule recognition element is a molecule, a substituent, a functional group or an atomic group capable of recognizing a target molecule such as binding to a target molecule in vivo.
- Target molecule recognition elements include, for example, polypeptides and other ligands that bind to target molecules.
- the polypeptide is usually a polypeptide that binds to a target molecule, preferably a polypeptide that specifically binds to a target molecule. Specific binding means binding to a target molecule but not binding to a molecule other than the target molecule or weak binding.
- the target molecule refers to a target site to be diagnosed by a radioactive drug, for example, a molecule present in a tissue or a cell, preferably a molecule specifically expressed. "Specifically expressed” refers to expression at a target site but not at a site other than the target site or low expression.
- Target molecule recognition elements include, for example, proteins that show high expression in tissue construction associated with inflammation, tumor cell infiltration, etc., ligands that bind to proteins specifically expressed in tumor cells, and antibodies and antibody antigen binding regions Fragments can be mentioned.
- Examples of the antibody include monoclonal antibodies such as anti-CD25 antibody and anti-CD20 antibody.
- Examples of antigen binding region fragments of antibodies include Fab fragments (hereinafter also referred to simply as “Fab”), F (ab ′) 2 fragments, F (ab) 2 fragments, and variable region fragments (hereinafter also referred to as “Fv fragments”).
- Fab fragment means a product having N-terminal side generated by papain degradation of antibody and a fragment having a domain structure similar to this.
- the F (ab ') 2 fragment means a fragment obtained by reducing the disulfide bond of the hinge region of F (ab') 2 of the antibody and a fragment having a domain structure similar to this.
- the F (ab) 2 fragment means a dimer in which two Fab fragments are linked to each other by a disulfide bond.
- Fv fragment is meant a fragment of an antibody that is the smallest fragment that has antigen binding activity. More specifically, the antigen-binding region fragment of the antibody includes an antibody against a protein specifically expressed in a specific cancer cell, and its Fab fragment or Fv fragment.
- cyclic pentapeptides having affinity to integrins that are highly expressed in cancer neovascularization such as cyclo-Arg-Gly-Asp-D-Phe-Lys (hereinafter, “c (c Also referred to as “RGD f K)”.
- fMLP fMet-Leu-Phe
- folic acid which binds to the folate receptor which is recognized in cancer cells, and derivatives thereof.
- the target molecule recognition element is not limited to these exemplified polypeptides, and any polypeptide that binds to a target molecule can be used.
- the target molecule recognition element may be bound using, for example, a thiolating reagent such as 2-iminothiolane to introduce a linking group that reacts with a functional group of the compound.
- a thiolating reagent such as 2-iminothiolane
- the introduction of the linking group to the Fab fragment can add a sulfhydryl group to an amino group of the Fab cross section by reacting the above-mentioned thiolating reagent under conditions of pH 7-9.
- a ligand having an Asn-urea-Lys site or a Glu-urea-Lys site may be used. According to the ligand, it selectively binds to a receptor of prostate specific membrane antigen whose expression is significantly elevated in prostate cancer.
- the Asn-urea-Lys site has the formula: [Wherein, * is a binding site. It is a part represented by].
- the Glu-urea-Lys site has the formula: [Wherein, * is a binding site. It is a part represented by].
- the above-mentioned polypeptide into which a specific functional group f 1 has been introduced, and other ligands or ligands that bind to target molecules are proteins or tumor cells that show high expression in tissue construction associated with inflammation, tumor cell infiltration, etc.
- Target compound such as a protein that is specifically expressed in the compound, and administering a compound (2) having a functional group f 2 that reacts with the functional group f 1 to form a bond as a target molecule recognition element
- Methods to recognize include [Chemical Society Reviews 45: 6409-6658, 2016, Chemical Society Reviews 42: 5131-5142, 2013].
- Examples of the functional group f 1 include groups represented by the following formula (f 1 -1), formula (f 1 -2), or formula (f 1 -3). [Wherein, * is a binding site. ]
- the compound (2) and the like of the present invention can be used to provide a preparation for preparing a radiopharmaceutical comprising the compound.
- the drug for preparation of the radioactive drug may contain, in addition to the compound, a pH regulator such as an aqueous buffer, a stabilizer such as ascorbic acid or p-aminobenzoic acid, and the like.
- Examples of the compound (2) and the like of the present invention include a compound represented by the following formula (2), or a pharmacologically acceptable salt thereof.
- L 1 is a linking group linking R 1 and P 1 , p is 0 or 1, P 1 is a target molecule recognition element.
- L 1 forms a bond with a functional group that can be linked to the linking group of R 1 and also forms a bond with the target molecule recognition element.
- L 1 is preferably iminothiol or the like derived from 2-iminothiolane.
- p is preferably 1.
- P 1 is, for example, the target molecule recognition element described above, preferably a polypeptide, a ligand that binds to another target molecule, or a group represented by the formula (f 2 -1), a formula (f 2 -2), a formula (f 2 ) A functional group f 2 represented by the formula -3), the formula (f 2 -4), or the formula (f 2 -5);
- Preferred specific examples of the compound (2) of the present invention include the following compound 2-1 to compound 2-4.
- Fab means a Fab fragment site.
- Metal complex compound (3) etc. The metal complex compound of the present invention, or a pharmacologically acceptable salt thereof (hereinafter also referred to as “metal complex compound (3) etc.”) is a metal selected from the group consisting of radioactive metals and radioactive atom-labeled metals. And a compound of the present invention or a pharmaceutically acceptable salt thereof coordinated to the metal.
- the radioactive agent containing the metal complex compound (3) etc. of the present invention may contain unreacted substances or impurities in addition to the metal complex compound (3) etc. After production, high performance liquid chromatography (HPLC) method etc. The metal complex compound (3) etc. which were refine
- HPLC high performance liquid chromatography
- complex means a substance in which a ligand is coordinated with an atom or ion of metal and metal-like element at the center, and is also referred to as a coordination compound.
- the coordination means that the ligand forms a coordinate bond with the central metal and aligns around the central metal.
- a complex is formed by the coordination bond of a ligand and a metal. The formation of a complex with a ligand and a metal is sometimes referred to as complex formation.
- a coordinate bond means a bond in which two valence electrons involved in one bond are provided from only one atom.
- ⁇ metal ⁇ examples of the metal include 111 In, 223 Ra, 67 Ga, 68 Ga, 44 Sc, 90 Y, 177 Lu, 225 Ac, 212 Bi, 213 Bi, 212 Pb, 227 Th, 64 Cu, and 67 Cu. .
- the metal is preferably at least one selected from the group consisting of 111 In, 223 Ra, 67 Ga, 68 Ga, 90 Y, 177 Lu, 225 Ac, 212 Bi, 213 Bi, 212 Pb, and 227 Th, More preferably, it is at least one selected from the group consisting of 111 In, 90 Y, 177 Lu, 225 Ac, 212 Bi, 213 Bi and 212 Pb, still more preferably 111 In, 90 Y, 177 Lu and 225 Ac is there.
- the metal is not limited to these specific examples, and any metal can be used as long as it has appropriate radiation, radiation dose and half life for the purpose of diagnosis using a radioactive drug.
- Short half-life metal radioactive isotopes are preferably used from the viewpoint of reducing the influence on normal tissues and cells in radiological imaging.
- the production of the metal complex compound (3) and the like can be carried out by in vitro complexation with a metal radioactive isotope using the above-mentioned compound bound to the target molecule recognition element as a ligand.
- the complex formation can be carried out by a simple operation utilizing a conventionally known complexation reaction.
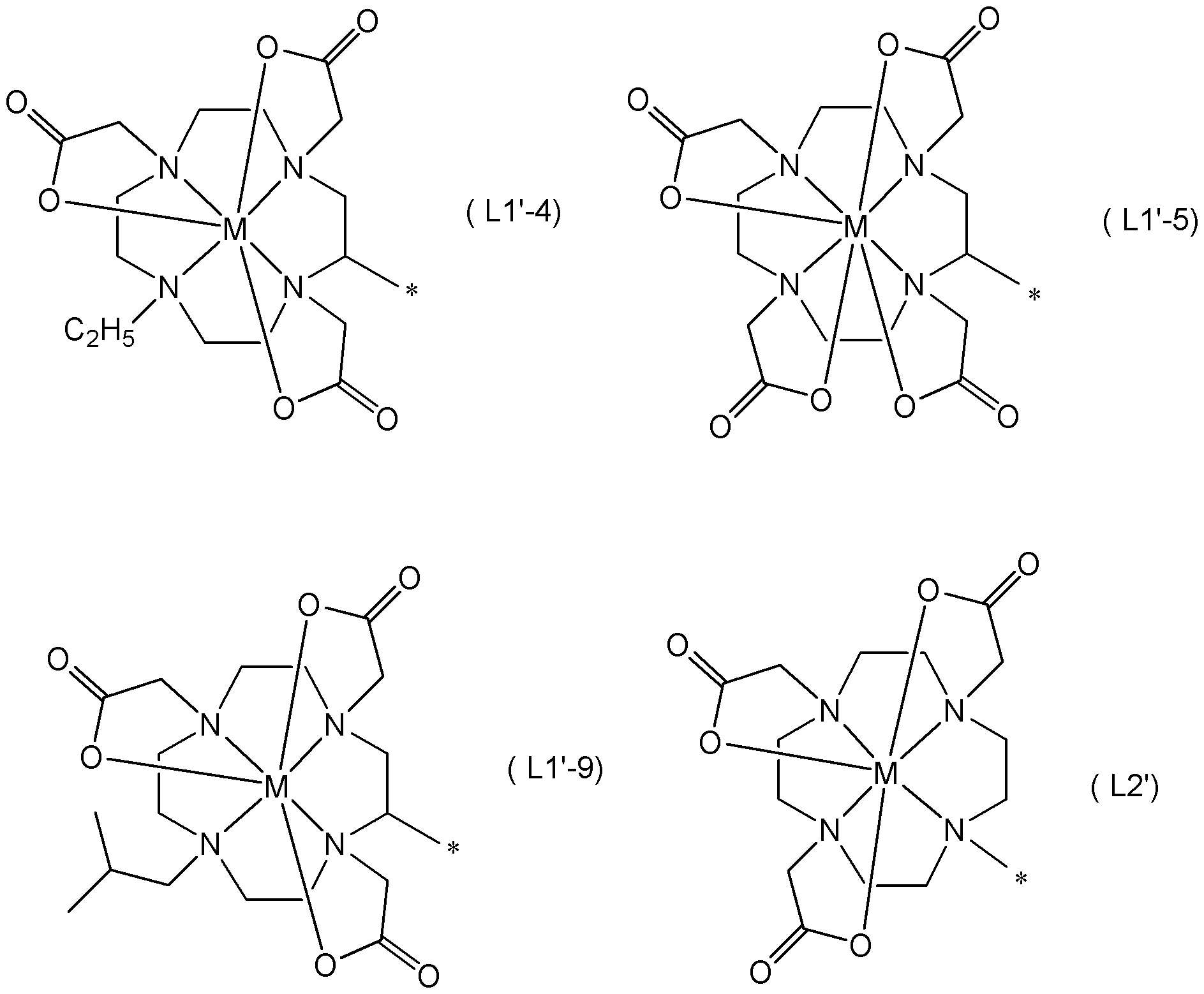
- L of the compound (1) is represented by the formula (L1-4), the formula (L1-5), the formula (L1-9) or the formula (L2)
- a metal complex compound represented by the following formula (3-1) may be mentioned.
- L ′ is represented by formula (L1′-4), formula (L1′-5), formula (L1′-9), or formula (L2 ′):
- M is 111 In, 223 Ra, 67 Ga, 68 Ga, 44 Sc, 90 Y, 177 Lu, 225 Ac, 212 Bi, 213 Bi, 212 Pb, 227 Th, 64 Cu, or 67 Cu
- L of the compound (2) is represented by the formula (L1-4), the formula (L1-5), the formula (L1-9), or the formula (L2)
- metal complex compounds represented by the following formula (3-2) may be mentioned.
- L ′ is a group represented by formula (L1′-4), formula (L1′-5), formula (L1′-9) or formula (L2 ′):
- M is 111 In, 223 Ra, 67 Ga, 68 Ga, 44 Sc, 90 Y, 177 Lu, 225 Ac, 212 Bi, 213 Bi, 212 Pb, 227 Th, 64 Cu, or 67 Cu
- It is a group represented by ]
- the radiopharmaceutical of the present invention is prepared as a pharmaceutical composition containing the above-mentioned radiolabeled polypeptide as an active ingredient and optionally containing one or more types of pharmaceutically acceptable carriers (pharmaceutical carriers).
- pharmaceutically acceptable carriers pharmaceutical carriers, aqueous buffers, pH adjusters such as acids and bases, stabilizers such as ascorbic acid and p-aminobenzoic acid, excipients such as D-mannitol, isotonic agents, preservatives Etc. can be illustrated.
- compounds such as citric acid, tartaric acid, malonic acid, sodium gluconate, sodium glucoheptonate may be added to help improve radiochemical purity.
- the radioactive agent of the present invention can be provided either in the form of an aqueous solution, in the form of a frozen solution, and in a lyophilizate.
- the kit of the present invention comprises the above-mentioned compound and an agent containing the above-mentioned metal as separate packaging units.
- the kit of the present invention separately packages, for example, compound (1) and the like, a drug containing a target molecule recognition element, and a drug containing one metal selected from the group consisting of radioactive metals and radioactive atom-labeled metals.
- the compounds and agents contained in the kit can optionally include one or more types of pharmaceutically acceptable carriers (pharmaceutical carriers) as described above.
- the compound (1) or the like of the present invention, and the compound (2) or the like obtained by binding a target molecule recognition element to the compound (1) or the like can be synthesized using a known method. It can be produced by the method described in the examples of the document.
- the metal complex compound (3) and the like of the present invention can be produced by in vitro complex formation with a metal radioactive metal or a radioactive atom labeled metal using the compound (2) or the like as a ligand. Complexation can be carried out using known methods.
- the metal complex compound etc. of the present invention is used, for example, as a radiopharmaceutical used for radiotherapy or radiography diagnosis.
- the metal complex compound and the like of the present invention can be used for radiation therapy for suppressing cancer by administering an effective amount thereof to a mammal including human.
- an anticancer agent for example, the preventive action of preventing or preventing the occurrence of cancer or metastasis / implantation, relapse, and suppressing the growth of cancer cells or preventing the progression of cancer by reducing cancer It has the broadest meaning, including both therapeutic action to ameliorate the symptoms, and is not to be construed as limiting in any case.
- Examples of one metal selected from the group consisting of radioactive metals and radioactive atom labeled metals used as radiotherapeutic agents include alpha ray emitting nuclides, beta ray emitting nuclides, gamma ray emitting nuclides, and positron emitting nuclides.
- alpha ray emitting nuclides i.e., nuclides that emit beta radiation
- beta radiation emitting nuclides i.e., nuclides that emit beta radiation
- radiographic image diagnosis examples include single photon emission computed tomography (hereinafter referred to simply as "SPECT”), positron emission tomography (hereinafter referred to simply as “PET”) and the like.
- SPECT single photon emission computed tomography
- PET positron emission tomography
- Be The diagnosis is not particularly limited, and it is used for various diseases such as tumor, inflammation, infectious disease, cardiovascular disease, brain and central nervous system diseases, and radiation imaging diagnosis of organs and tissues, preferably cancer radiation Used for diagnostic imaging.
- SPECT single photon emission computed tomography
- PET positron emission tomography
- the administration route of the radioactive drug of the present invention includes, for example, parenteral administration such as intravenous administration or intraarterial administration, oral administration, and preferably intravenous administration.
- parenteral administration such as intravenous administration or intraarterial administration
- oral administration and preferably intravenous administration.
- the administration route is not limited to these routes, and any route that can effectively exert its action after administration of the radioactive drug can be used.
- the radioactive intensity of the radioactive agent of the present invention is an intensity which can achieve the purpose by administering the agent, and is arbitrary as long as the radiation exposure of the subject is as low as possible.
- the radioactive intensity can be determined with reference to the radioactive intensity used in general diagnostic and therapeutic methods using a radioactive drug.
- the dose is determined in consideration of conditions such as the patient's age, body weight, appropriate radiation imaging device, and condition of target disease, and the radioactivity and dose considered to be capable of imaging.
- the amount of radioactivity in the radioactive drug is as follows.
- the dose of the diagnostic agent is not particularly limited, but is, for example, 1.0 MBq / kg to 3.0 MBq / kg as the radioactive amount of radioactive metal (eg, 111 In) .
- TLC Thin-layer chromatography
- a silica plate TLC aluminum sheets 60 RP-18 F 254 s, Merck & Co., Inc.
- the expanded sample was cut by 5 mm, and the radioactivity of each fraction was measured by the Autowell gamma system (WIZARD3, Perkin Elmer).
- CAE Cellulose acetate membrane electrophoresis
- RP-HPLC reverse-phase high-performance liquid chromatography
- SE-HPLC molecular sieve high-performance liquid chromatography
- analysis Analysis by reverse phase high performance liquid chromatography (hereinafter also referred to as “RP-HPLC”) is L-7405 (Hitachi, Ltd.) as a UV detector, L-7100 (Hitachi, Ltd.) as a liquid transfer pump, as an analytical column A Cosmosil 5C 18 -AR-300 column (4.6 x 150 mm, Nacalai Tesque, Inc.) was used.
- the mobile phase is 0.1% (v / v) TFA / H 2 O (phase A) and 0.1% (v / v) TFA / MeCN (phase B), and the phase A 95% (v / v) for 0-20 min.
- SE-HPLC Analysis by molecular sieve HPLC
- the eluate measures absorbance at 254 nm for RP-HPLC and 280 nm for SE-HPLC, and a ⁇ -ray detector Gabi star (Raytest) is connected online for analysis of 67 Ga labeled compounds, or the eluate is fractionated. After aliquoting with a collector (Frac-920, GE Healthcare Japan Co., Ltd.) at intervals of 0.5 minutes, it was analyzed by measuring the radioactivity with the Autowell gamma system.
- ⁇ reagent ⁇ “DO3A-EDA (Mal)” in the following synthesis example is a compound represented by the following formula, and a trade name “B-272” manufactured by Macrocyclics Co., Ltd. was used.
- mice Male ddY-based SPF mice 6 weeks old and male BALB / c-nu / nu mice (Nippon SLC Co., Ltd.) were used.
- Synthesis Example L1 (a): Synthesis of Compound (a3) Compound (a1) (7.97 g, 20.4 mmol) was dissolved in 40 mL of THF and cooled to ⁇ 15 ° C. under an argon atmosphere, N-methylmorpholine (NMM) , 3.36 mL, 30.5 mmol) and isobutyl chloroformate (4.01 mL, 30.5 mmol) were sequentially added. After stirring for 5 minutes, 24 mL of a DMF solution of a compound (a2) (5.87 g, 30.5 mmol) and NMM (3.36 mL, 30.5 mmol) was added dropwise, and stirred under cooling for 30 minutes and at room temperature for 1 hour.
- NMM N-methylmorpholine
- reaction solution is stirred at room temperature for 1 hour, and the reaction solution is filtered to remove dicyclohexylurea (hereinafter also referred to as "DC-urea"), and the filtrate is used as an anhydride solution of compound (a5) to perform the next reaction Used for Compound (a4) (8.08 g, 17.4 mmol) is suspended in 80 mL of ethyl acetate, Et 3 N (3.63 mL, 26.0 mmol) is added, cooled, stirred for 10 minutes, and then the compound previously prepared under argon atmosphere Anhydride solution of (a5) was added dropwise.
- DC-urea dicyclohexylurea
- Synthesis Example L2 (III): Compound (a17) The compound (a16) was dissolved in 100 ⁇ L of saturated aqueous sodium hydrogen carbonate solution, 100 ⁇ L of dioxane in which 1.5 equivalents of (Boc) 2 O was dissolved was added, and the mixture was vigorously stirred at room temperature for 2 hours. After removing dioxane by evaporation under reduced pressure, the aqueous layer was washed with chloroform.
- the aqueous phase is further analyzed by HPLC using Imtakt Cadenza 5CD-C18 150 ⁇ 20 mm as mobile phase, 0.1% TFA / MilliQ for phase A, 0.1% TFA / MeCN for phase B, 100% phase A up to 2 minutes
- the phase is changed from 100% phase A, 0% phase B to 95% phase A, 5% phase B in 2 to 5 minutes, 95% phase A, 5% phase B to 5 phase in 5-40 minutes.
- Synthesis Example L3 (a): Synthesis of Compound (b1) Cyclen (523.6 mg, 3.04 mmol) was dissolved in acetonitrile (25 mL), NaHCO 3 (893.6 mg, 10.6 mmol) was added, and ice cooled under Ar atmosphere. After that, tert-butyl bromoacetate (1.39 mL, 3.34 mmol) was added dropwise. After stirring at room temperature for 48 hours, the reaction solution was filtered and the filtrate was evaporated under reduced pressure. The residue was purified by recrystallization using toluene to give compound (b1) (672.4 mg, yield 43.1%) as white crystals.
- Synthesis Example L3 (g): Synthesis of Compound (b7) Compound (b6) (106.1 mg, 160.0 ⁇ mol) was dissolved in methanol (1 mL), 1 N NaOH (1 mL) was added, and the mixture was stirred at room temperature for 2 hours did. After evaporating methanol under reduced pressure from the solution, extraction was performed with chloroform (5 mL ⁇ 3). The organic layer was added with Na 2 SO 4 and dried, and the solvent was evaporated under reduced pressure to obtain compound (b7) (50.5 mg, yield 48.6%) as a yellow oil.
- the amount of Fmoc-Lys (Bzo) -OH introduced into the resin is determined by measuring the absorbance at A301 of N- (9-fluorenylmethyl) piperidine formed during piperidine treatment. (0.96 mmol / g).
- Compound (b10) was produced by adding 20% piperidine / DMF (mL) and stirring at room temperature for 20 minutes. A portion of the resin was collected and subjected to kaiser test to confirm the deprotection of the N ⁇ -Fmoc group.
- Synthesis Example L4 (g): Synthesis of Compound (b14) Compound (b5) (5.5 mg, 5.78 ⁇ mol) and methionine (1.29 mg, 8.67 ⁇ mol) are used as starting materials to conduct the same operation as in Synthesis Example L4 (f) Thus, a TFA salt (2.3 mg, 36.2% yield) of a compound (b14) (hereinafter, also referred to as “DO3A-Bn-SCN-Met-OH”) was obtained as white crystals.
- ESI-MS (M + H) + : m / z 643.2, found: .643.2
- Synthesis Example L5 (a): Synthesis of Compound (b15) Starting from Cl-Trt (2-Cl) Resin (104.4 mg, 114.8 ⁇ mol, Watanabe Chemical Industries, Ltd.) as a starting material, Fmoc-Lys (Dde) -OH (61.2) mg, 114.8 ⁇ mol) and DIPEA (81 ⁇ L, 459.2 ⁇ mol) were reacted in dichloromethane (2 mL) for 1.5 hours. The reaction was quenched by adding methanol and DIPEA. The resin was washed with DMF then dichloromethane.
- Synthesis Example L6 (a): Synthesis of Compound (b22) Starting from Cl-Trt (2-Cl) Resin (22.1 mg, 24.3 ⁇ mol, Watanabe Chemical Industry Co., Ltd.) as a starting material, Fmoc-Lys (Boc) -OH (17.1) mg, 36.5 ⁇ mol) and DIPEA (16.5 ⁇ L, 97.2 ⁇ mol) were reacted in dichloromethane (1 mL) for 1.5 hours. The reaction was quenched by adding methanol and DIPEA. The resin was washed with DMF then dichloromethane.
- Synthesis Example L6 (b) Synthesis of Compound (b23) Compound (b22) (12.1 ⁇ mol) was used as a starting material, and protected amino acids were changed sequentially to Fmoc-Gly-OH and Fmoc-Phe-OH to obtain Synthesis Example L4 (d Compound (b23) was obtained by performing the same operation as in 2.).
- the aqueous layer is purified by preparative RP-HPLC, whereby the TFA salt (0.8 mg, yield 26.9%) of compound (b25) (hereinafter, also referred to as “DO3A-Bn-CO-FGK (Boc)”) is white Obtained as a crystal.
- Synthesis Example L6 (f): Synthesis of Compound (b27) Fmoc-Phe-OH (6.01 ⁇ mol) and Cl-Trt (2-Cl) Resin (5 mg, 5.50 ⁇ mol, Watanabe Chemical Industry Co., Ltd.) as a starting material DIPEA (3.73 ⁇ L, 21.9 ⁇ mol) was reacted in dichloromethane (0.5 mL) for 1.5 hours. The reaction was quenched by adding methanol and DIPEA. The resin was washed with DMF then dichloromethane. The amount of Fmoc-AA-OH introduced into the resin was quantified (0.917 mmol / g) in the same manner as in Synthesis Example L4 (c).
- Compound (b27) was produced by adding 20% piperidine / DMF (2 mL) and stirring at room temperature for 20 minutes. A portion of the resin was collected and subjected to kaiser test to confirm the deprotection of the N ⁇ -Fmoc group.
- Synthesis Example L6 (g): Synthesis of Compound (b28) Compound (b28) (hereinafter referred to as “DO3A-”) by using Compound (b27) (5.5 ⁇ mol) as a starting material and operating in the same manner as Synthesis Example L6 (c).
- the TFA salt also referred to as “Bn—CO—Phe—OH”
- ESI-MS (M + H) + : m / z 628.3, found: 628.3.
- Synthesis Example L7 (a): Synthesis of Compound (b29)
- Compound (b29) was prepared by the same procedure as in Synthesis Example L6 (c), using p-COOH-Bn-DOTA (tBu) 4 (52.4 ⁇ mol) as a starting material.
- the TFA salt (2.4 mg, 1.3% yield) (hereinafter also referred to as “CDOTA-Bn-CO-FGK”) was obtained as white crystals.
- ESI-MS [M + K] -H) - : m / z 907.36, found: 907.31.
- Synthesis Example L7 (b): Synthesis of Compound (b30) Compound (b30) (hereinafter referred to as “CDOTA-”) by using Compound (b29) (5.75 ⁇ mol) as a starting material and operating in the same manner as in Synthesis Example L6 (d).
- the TFA salt (0.4 mg, 71.8% yield) of Bn-CO-FGK (Boc) was obtained as white crystals.
- ESI-MS [M + K] -H) - : m / z 1007.41, found: 1007.31.
- Synthesis Example L7 (c) Synthesis of Compound (b31) Compound (b31) (hereinafter referred to as “CDOTA-”) was synthesized by the same procedure as Synthesis Example L6 (e), using Compound (b29) (0.694 ⁇ mol) as a starting material.
- the TFA salt (0.6 mg, 61.4% yield) of Bn-CO-FGK (mal) was obtained as white crystals.
- ESI-MS (MH) - m / z 949.39, found: 949.43.
- Synthesis Example L8 (a): Synthesis of Compound (a19) Compound (a8) (913 mg, 1.96 mmol) was dissolved in THF (20 mL), isopropylaldehyde (357 ⁇ L, 3.91 mmol), sodium triacetoxyborohidride (498 mg, 2.35) mmol) was added and stirred overnight at room temperature under Ar atmosphere. After adding isobutylaldehyde (179 ⁇ L, 1.96 mmol) and sodium triacetoxyborohidride (249 mg, 1.18 mmol), the mixture was further stirred at room temperature for 2 hours.
- reaction solution was ice-cooled, water was added, and then THF was evaporated under reduced pressure to extract an aqueous solution obtained three times with chloroform. After adding sodium sulfate to the organic layer and drying it, the solvent is distilled off under reduced pressure, and the residue obtained is purified by a flash chromatography system using chloroform and methanol to produce compound (a19) (403 mg, 829 ⁇ mol, 42.4%) were obtained as white crystals.
- Synthesis Example L8 (b): Synthesis of Compound (a20) Compound (a19) (403 mg, 829 ⁇ mol) was suspended in 2 mL of THF, the solution was ice-cooled, and then 0.95 M borane-THF complex under argon atmosphere. The reaction solution was slowly added with 13 mL of THF solution, stirred for 1 hour, and refluxed for 22 hours. The solution was ice-cooled, 13 mL of methanol was added, and the mixture was stirred for 1 hour, and the solvent was evaporated under reduced pressure. Thereafter, 13 mL of methanol was again added to the residue, and the solvent was evaporated under reduced pressure.
- the resin was removed by filtration, the filtrate was evaporated under reduced pressure, and azeotropic distillation with toluene was performed three times.
- the residue is dissolved in water, and the aqueous solution obtained by washing three times with diethyl ether is lyophilized to obtain white crystals obtained by HPLC using Imtakt Cadenza 5CD-C18 150 ⁇ 20 mm as the mobile phase A phase 0.1% TFA / MilliQ, 0.1% TFA / MeCN for phase B, and changing from 95% phase A, 5% phase B to 50% phase A, 50% phase B in 0-35 minutes, 35-45
- the target compound (a27) is purified at a flow rate of 5 mL / min (retention time 24.8 minutes) by a linear gradient method in which A phase 50%, B phase 50% to A phase 0%, B phase 100% in minutes.
- Synthesis Example L9 Synthesis Example of Compound (a28)
- Compound (a27) (1.8 mg, 2.6 ⁇ mol) was dissolved in 150 ⁇ L of saturated aqueous sodium hydrogen carbonate solution, N-methoxycarbonylmaleimide (1.0 mg, 6.5 ⁇ mol) was added under ice-cooling, and the mixture was stirred for 2 hours under ice-cooling. After completion of the reaction, it was adjusted to acidity with a 5% by mass aqueous citric acid solution.
- phase A is 100%, phase B 0% to phase A 40%, phase B 60% in 5-35 minutes, phase A 40%, phase B 60% to phase A 0%, in 35-50 minutes.
- the target compound (a28) (hereinafter, also referred to as “CDO3AiBu-FGK (Mal)”) (0.8) is purified by purification at a flow rate of 5 mL / min (retention time 30.7 min) by a linear gradient method in which B phase is changed to 100%. mg, 0.57 ⁇ mol, yield 45.5%).
- reaction solution is prepared by spin column method [Analytical Biochemistry, 1984, 142, 68-78] using Sephadex G-50 Fine equilibrated with 0.1 M phosphate buffer (pH 6.0) containing 2 mM EDTA sufficiently degassed. The excess 2-IT in it was removed to obtain an IT-Fab (from Rabbit serum IgG derived) solution. The number of thiol groups introduced per Fab molecule was measured using 2,2'-dipyridyl disulfide [Archives of Biochemistry and Biophysics, 1967, 119, 41-49].
- Synthesis Example F2 Preparation of Fab (derived from anti-c-kit IgG) and IT-Fab (derived from anti-c-kit IgG) Rabbit serum IgG (9 mg) was treated as anti-c-kit IgG (9 mg), except that In the same manner as in Synthesis Example F1, Fab (derived from anti-c-kit IgG) and IT-Fab (derived from anti-c-kit IgG) were produced.
- CDO3AEt-FGK-Fab from Rabbit serum IgG
- DTPA a final concentration of 10 mM
- CDO3AEt-FGK-Fab (derived from Rabbit serum IgG) solution, CDO3AEt-FGK-Fab (derived from anti-c-kit IgG) solution, DO3A-EDA-Fab (derived from Rabbit serum IgG) solution, CDOTA-Bn-CO-FGK- Fab (anti-c-kit IgG derived) solution, DO3A-Bn-SCN-MVK-Fab solution (derived from Rabbit serum IgG), DO3A-Bn-CO-FGK-Fab (derived from Rabbit serum IgG) solution, CDO3AiBu-FGK-Fab (From Rabbit serum IgG) solution, DOTA-Bn-SCN-Fab (from anti-c-kit IgG) solution, CDO3AEt-FGK (Boc) solution, CDOTA-Bn-CO-FGK (Boc) solution, DO3A-Bn-SCN In the same manner as above, 111 In-CDO3 AEt-FGK-Fab (derived from
- 111 In-DO3A-EDA-Fab from Rabbit serum IgG
- 111 In-CDOTA-Bn-CO-FGK-Fab from anti-c-kit IgG
- 111 In-DO3A-Bn-SCN-MVK-Fab Rabbit serum IgG
- 111 In-DO3A-Bn-CO-FGK-Fab derived from Rabbit serum IgG
- 111 In-CDO3AiBu-FGK-Fab derived from Rabbit serum IgG
- 111 In-DOTA-Bn-SCN-Fab derived from serum IgG
- Anti-c-kit IgG derived 111 In-CDO3AEt-FGK (Boc)
- 111 In-CDOTA-Bn-CO-FGK Boc
- 111 In-DO3A-Bn-SCN-MVK Bz) o
- 111 In-DO3A-Bn-CO-FGK Boc
- BBMVs Renal brush border membrane vesicles
- BBMVs Renal brush border membrane vesicles
- 1.0 M MgCl 2 aqueous solution was added to a final concentration of 10 mM, the suspension was allowed to stand for 15 minutes, and then the homogenate was centrifuged at 1,900 g and the supernatant was further centrifuged at 24,000 g for 30 minutes.
- the resulting precipitate was suspended in 0.1 M phosphate buffer (pH 7.0) equivalent to 10 times the cortex weight, and again homogenized using a Teflon (registered trademark) homogenizer (1,000 rpm, 10 strokes). Then. The homogenate was centrifuged at 24,000 g for 30 minutes to obtain BBMVs as a precipitate.
- BBMVs pellet was then resuspended in 0.1 M phosphate buffer (pH 7.0) and vesicles sized by passing 10 times through a 0.4 x 19 mm needle. In the incubation experiment, it was diluted to a protein concentration of 10 mg / mL and used.
- lysosomal enzyme contamination was evaluated by measuring the activity of the lysosomal marker enzyme ⁇ -galactosidase using p-nitrophenyl- ⁇ -D-galacto-pyranoside [Plant Physiology 55: 94-98, 1975].
- BBMVs (Incubation test) Incubation experiments of BBMVs and 111 In-labeled small molecule model substrates were performed by the following method.
- BBMVs (10 ⁇ L) which was prepared to have a protein concentration of 10 mg / mL, were preincubated at 37 ° C. for 10 minutes. Thereafter, the excess ligand was removed by reverse phase HPLC, and a solution of 111 In-CDO3AEt-FGK (Boc) (10 ⁇ L) dissolved in PBS was added, and incubated at 37 ° C. for 2 hours. Ethanol was added to the BBMVs solution so that the ethanol concentration was 60%, and centrifuged at 5000 rpm for 10 minutes.
- Boc 111 In-CDO3AEt-FGK
- FIG. 1 shows the experimental results of the incubation of 111 In-CDO3AEt-FGK (Boc) with BBMVs.
- FIG. 2 shows the experimental results of the incubation of 111 In-CDOTA-Bn-CO-FGK (Boc) solution with BBMVs.
- FIG. 3 shows the experimental results of incubation of 111 In-DO3A-Bn-SCN-MVK (Bzo) with BBMVs.
- FIG. 4 shows the experimental results of incubation of 111 In-DO3A-Bn-CO-FGK (Boc) with BBMVs.
- FIG. 5 shows the experimental results of the incubation of 111 In-CDO3AiBu-FGK (Boc) with BBMVs.
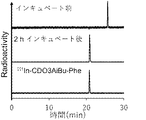
- the 111 In-CDO3AEt-FGK-Fab (derived by Rabbit serum IgG), then changed to 111 In-CDO3AEt-FGK-Fab ( derived anti c-kit IgG), others in the same manner as described above in mouse plasma stability The test was done. After 2 hours of incubation, the percentage of radioactivity of the unchanged form ( 111 In-CDO3AEt-FGK-Fab) was 95.2 ⁇ 0.3%.
- 111 In-CDO3AEt-FGK-Fab (derived from Rabbit serum IgG) was changed to 111 In-CDOTA-Bn-CO-FGK-Fab (derived from anti-c-kit IgG), and the other procedures were the same as above. Plasma stability studies were performed. After 2 hours of incubation, the percentage of radioactivity of the unchanged form ( 111 In-CDOTA-Bn-CO-FGK-Fab) was 95.2 ⁇ 0.3%.
- the 111 In-CDO3AEt-FGK-Fab (derived from Rabbit serum IgG) was changed to 111 In-CDO3AiBu-FGK-Fab (derived from Rabbit serum IgG), and in the same manner as above, the mouse plasma stability test was performed went. Table 2 shows the experimental results.
- feces and urine for up to 6 and 24 hours were collected, and the radioactivity was measured.
- 111 In-CDO3AEt-FGK-Fab derived from anti-c-kit IgG
- radioactivity was measured for the organ of interest and feces by intravenous injection of 6 week old ddY male mice.
- 111 In-DO3A-EDA-Fab derived from Rabbit serum IgG
- Table 3 shows the results of measurement of radioactivity in the mouse by 111 In-CDO3AEt-FGK-Fab (derived from Rabbit serum IgG).
- Table 4 shows the results of measurement of radioactivity in mice by 111 In-CDO3AEt-FGK-Fab (derived from anti-c-kit IgG).
- Table 5 shows the results of measurement of radioactivity in mice with 111 In-DO3A-EDA-Fab (derived from Rabbit serum IgG).
- 111 In-CDOTA-Bn-CO-FGK-Fab (derived from anti-c-kit IgG) solution
- 111 In-DO3A-Bn-SCN-MVK-Fab (derived from Rabbit serum IgG) solution
- 111 in the same manner as described above In-DO3A-Bn-CO-FGK-Fab (derived from Rabbit serum IgG), 111 In-CDO3AiBu-FGK-Fab (derived from Rabbit serum IgG), 111 In-CDO3AiBu-FGK-Fab (derived from anti-c-kit IgG)
- the radioactivity was measured for the organ of interest and feces by intravenous injection of 6-week-old ddY male mice using a solution, 111 In-DOTA-Bn-SCN-Fab (derived from anti-c-kit IgG) solution .
- Table 6 shows the results of measurement of radioactivity in mice with 111 In-CDOTA-Bn-CO-FGK-Fab (derived from anti-c-kit IgG).
- Table 7 shows the results of measurement of radioactivity in mice by 111 In-DO3A-Bn-SCN-MVK-Fab (derived from Rabbit serum IgG).
- Table 8 shows the results of measurement of radioactivity in the mouse by 111 In-DO3A-Bn-CO-FGK-Fab (derived from Rabbit serum IgG).
- Table 9 shows the results of measurement of radioactivity in the mouse by 111 In-CDO3AiBu-FGK-Fab (derived from Rabbit serum IgG).
- Table 10 shows the measurement results of radioactivity in the mouse body by 111 In-CDO3AiBu-FGK-Fab (derived from anti-c-kit IgG).
- Table 11 shows the measurement results of radioactivity in the mouse by 111 In-DOTA-Bn-SCN-Fab (derived from anti-c-kit IgG).
- 6A-C show the comparison of the results of 111 In-CDO3AEt-FGK-Fab and 111 In-DO3A-EDA-Fab.
- 111 In-CDO3AEt-FGK-Fab which is the compound of the present invention and 111 In-DO3A-EDA-Fab which is the comparison compound, 111 In-CDO3AEt-FGK-Fab is 111 In-DO3A-EDA- It is understood that accumulation in the kidney is suppressed and a low kidney-blood ratio can be obtained while showing blood concentration similar to that of Fab.
- compound 111 In-CDO3AEt-FGK-Fab (derived from Rabbit serum IgG) of the present invention having a benzylamide structure as a linking group, 111 In-CDO3AEt-FGK-Fab (derived from anti-c-kit IgG) , 111 In-CDOTA-Bn-CO-FGK-Fab (derived from anti-c-kit IgG), 111 In-DO3A-Bn-CO-FGK-Fab (derived from Rabbit serum IgG), the radioactivity in the kidney is remarkable Comparative compound 111 In-DO3A-Bn-SCN-MVK-Fab having a thiourea structure as a linking group while being suppressed, and 111 In-DO3A-EDA-Fab having an ethylene structure (derived from Rabbit serum IgG) In, the radioactivity in the kidney shows a high value.
- 111 In-CDO3AEt-FGK-Fab (derived from Rabbit serum IgG) was diluted with D-PBS (-).
- Administer 111 In-CDO3AEt-FGK-Fab (from Rabbit serum IgG derived) solution (4 ⁇ Ci / 100 ⁇ L / animal) whose Fab concentration has been adjusted to 5 ⁇ g / 100 ⁇ L from the mouse tail vein, and collect up to 24 hours later
- the filtered urine was filtered through a 0.45 ⁇ m filter, and the chemical form was analyzed by SE-HPLC.
- protein was precipitated by adding double volume of EtOH to the collected urine, and the supernatant was recovered after centrifugation at 15,000 g for 5 minutes.
- FIG. 8 shows the results of analysis of the chemical form of urinary radioactivity excreted up to 24 hours after administration of 111 In-CDO3AEt-FGK-Fab (derived from Rabbit serum IgG) to mice. As described above, most of the radioactivity is excreted in the low molecular weight fraction by analysis by SE-HPLC shown in FIG.
- 111 In-CDO3AEt-FGK-Fab (derived from Rabbit serum IgG) was changed to 111 In-CDOTA-Bn-CO-FGK-Fab (derived from anti-c-kit IgG) and was excreted by 6 hours after administration
- Urinary radioactivity was analyzed in the same manner as described above except that urine was collected and analyzed.
- FIG. 9 shows the results of analysis of the chemical form of urinary radioactivity excreted up to 6 hours after administration of 111 In-CDOTA-Bn-CO-FGK-Fab (derived from anti-c-kit IgG) to mice.
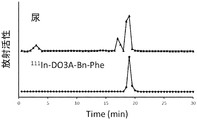
- FIG. 11 shows the results of analysis of chemical forms of urinary radioactivity excreted by 24 hours after administration of 111 In-CDO3AiBu-FGK-Fab (derived from Rabbit serum IgG) to mice. As described above, most of the radioactivity is excreted as a low molecular weight fraction in the analysis by SE-HPLC shown in FIG. 11A by the analysis of radioactivity in urine, and from the result of RP-HPLC shown in FIG.
- 111 In-CDO3AiBu In -FGK-Fab derived from Rabbit serum IgG
- the main radioactivity in the low molecular weight fraction is 111 In-CDO3AiBu-Phe (a compound in which 111 In-CDO3AiBu-FGK-Fab is cleaved between phenylalanine and glycine) I understand.
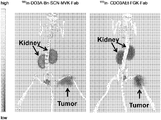
- SPECT 4CT Trifoil Imaging
- FIG. 12 shows a SY subcutaneous tumor model with 111 In-DO3A-Bn-SCN-MVK-Fab (anti-c-kit IgG derived) solution or 111 In-CDO3AEt-FGK-Fab (anti-c-kit IgG derived) solution It is a SPECT / CT image 2.5 hours after administration to a mouse. At 2.5 hours after administration, 111 In-CDO3AEt-FGK-Fab had low accumulation in the kidney, and the tumor was clearly imaged. On the other hand, in 111 In-DO3A-Bn-SCN-MVK-Fab, although the tumor was imaged, high radioactivity was also observed in the kidney. As described above, radiolabeled drugs have low accumulation in the kidney and can improve the sensitivity and accuracy of radiological image diagnosis.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Optics & Photonics (AREA)
- Epidemiology (AREA)
- Physics & Mathematics (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Immunology (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
Abstract
Description
これに対して、特許文献1では、腎臓への集積を投与早期から低減できる放射性標識薬剤として、NOTA(1,4,7,10-テトラアザシクロドデカン-1,4,7,10-テトラ酢酸)等のキレート試薬と結合したポリペプチド部位を有する化合物及びそれを用いた放射性薬剤が報告されている。
そこで本発明は、比較的大きな原子半径を有する原子を含め多様な原子による標識が可能であり、腎臓への集積を低減できる放射性薬剤が得られる化合物等に関する。
〔1〕下記式(1)で表される化合物、又はその薬理学的に許容可能な塩。
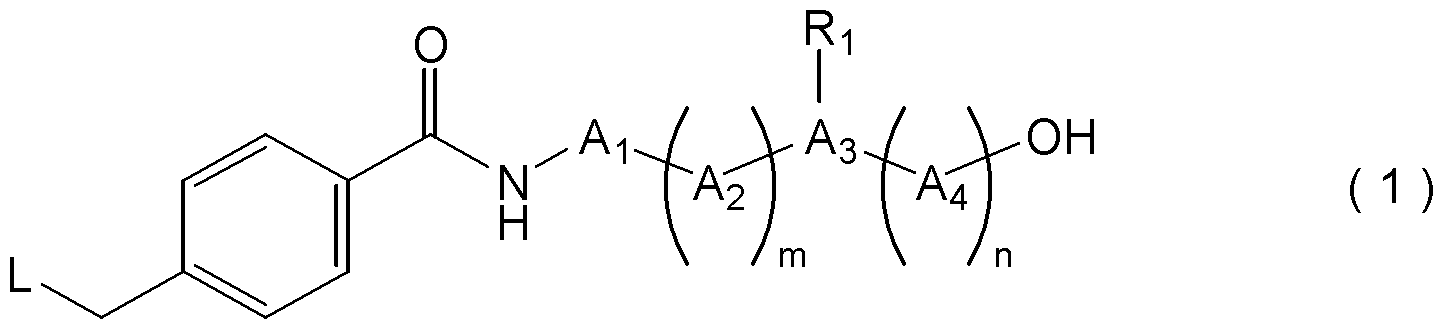
〔式中、
A1、A2は、それぞれ独立にアミノ酸残基であり、
mは、0~3の整数であり、
A3は、側鎖にアミノ基又はカルボキシ基を有するアミノ酸残基であり、
A4は、アミノ酸残基であり、
nは、0~3の整数であり、
R1は、A3の側鎖のアミノ基又はカルボキシ基と結合し、標的分子認識素子又はその連結基と結合可能な官能基を有する基、又は、A3の側鎖のアミノ基若しくはカルボキシ基の水素原子であり、ただし、R1は、環構成原子の一つとしてA3の側鎖のアミノ基の窒素原子を含む炭素数3~10の複素環基を形成していてもよく、
Lは、式(L1):

(式中、R3,R4,R5,R6は、それぞれ独立に、水素原子、-CH2COOR10基、又は炭素数1~8の炭化水素基であり、R10は、水素原子又は炭素数1~8の炭化水素基であり、*は結合部位である。ただし、R3,R4,R5,R6のうち、少なくとも3以上は-CH2COOH基である。)で表される基、又は、式(L2):

(式中、*は結合部位である。)で表される基である。〕
〔2〕上記〔1〕に記載の化合物、又はその薬理学的に許容可能な塩に、標的分子認識素子を結合させてなる化合物、又はその薬理学的に許容可能な塩。
〔3〕放射性金属及び放射性原子標識金属からなる群から選ばれる1種の金属と、前記金属に配位した〔1〕又は〔2〕に記載の化合物又はその薬理的に許容可能な塩と、を有する金属錯体化合物、又はその薬理学的に許容可能な塩。
〔4〕上記〔1〕又は〔2〕に記載の化合物、又はその薬理学的に許容可能な塩を含む、放射性薬剤の調製用薬剤。
〔5〕上記〔1〕又は〔2〕に記載の化合物、又はその薬理学的に許容可能な塩の放射性薬剤の製造のための使用。
〔6〕上記〔3〕に記載の金属錯体化合物、又はその薬理学的に許容可能な塩を含む、放射性薬剤。
〔7〕上記〔3〕に記載の金属錯体化合物、又はその薬理学的に許容可能な塩の放射線治療薬。
〔8〕上記〔3〕に記載の金属錯体化合物、又はその薬理学的に許容可能な塩の放射性画像診断薬。
〔9〕前記化合物、又はその薬理学的に許容可能な塩が、放射性薬剤の調製用である、上記〔1〕又は〔2〕に記載の化合物、又はその薬理学的に許容可能な塩。
〔10〕上記〔3〕に記載の金属錯体化合物、又はその薬理学的に許容可能な塩の放射性薬剤の製造のための使用。
〔11〕上記〔3〕に記載の金属錯体化合物、又はその薬理学的に許容可能な塩の放射線治療のための使用。
〔12〕上記〔3〕に記載の金属錯体化合物、又はその薬理学的に許容可能な塩の放射線画像診断のための使用。
〔13〕上記〔3〕に記載の金属錯体化合物、又はその薬理学的に許容可能な塩を投与する放射線治療方法。
〔14〕上記〔3〕に記載の金属錯体化合物、又はその薬理学的に許容可能な塩を投与する放射線画像診断方法。
〔15〕上記〔1〕に記載の化合物若しくはその薬理学的に許容可能な塩、又は、上記〔1〕に記載の化合物、又はその薬理学的に許容可能な塩に、標的分子認識素子を結合させてなる化合物、又はその薬理学的に許容可能な塩と、放射性金属及び放射性原子標識金属からなる群から選ばれる1種の金属を含む薬剤とを、別々の包装単位として含む、キット。
<化合物(1)等>
本発明の化合物、又はその薬理学的に許容可能な塩(以下、単に「化合物(1)等」ともいう)は、下記式(1)で表される。
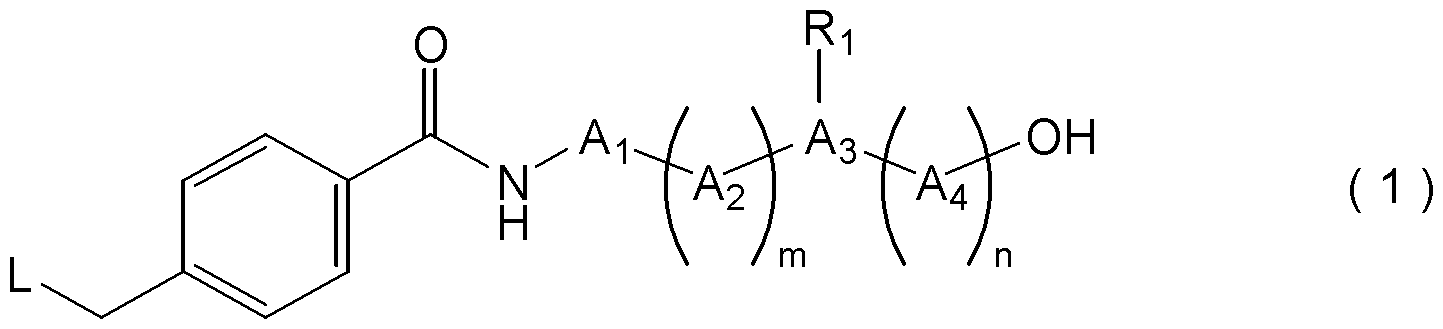
〔式中、
A1、A2は、それぞれ独立にアミノ酸残基であり、
mは、0~3の整数であり、
A3は、側鎖にアミノ基又はカルボキシ基を有するアミノ酸残基であり、
A4は、アミノ酸残基であり、
nは、0~3の整数であり、
R1は、A3の側鎖のアミノ基又はカルボキシ基と結合し、標的分子認識素子又はその連結基と結合可能な官能基を有する基、又は、A3の側鎖のアミノ基若しくはカルボキシ基の水素原子であり、ただし、R1は、環構成原子の一つとしてA3の側鎖のアミノ基の窒素原子を含む炭素数3~10の複素環基を形成していてもよく、
Lは、式(L1):

(式中、R3,R4,R5,R6は、それぞれ独立に、水素原子、-CH2COOR10基、又は炭素数1~8の炭化水素基であり、R10は、水素原子又は炭素数1~8の炭化水素基であり、*は結合部位である。ただし、R3,R4,R5,R6のうち、少なくとも3以上は-CH2COOH基である。)で表される基、又は、式(L2):
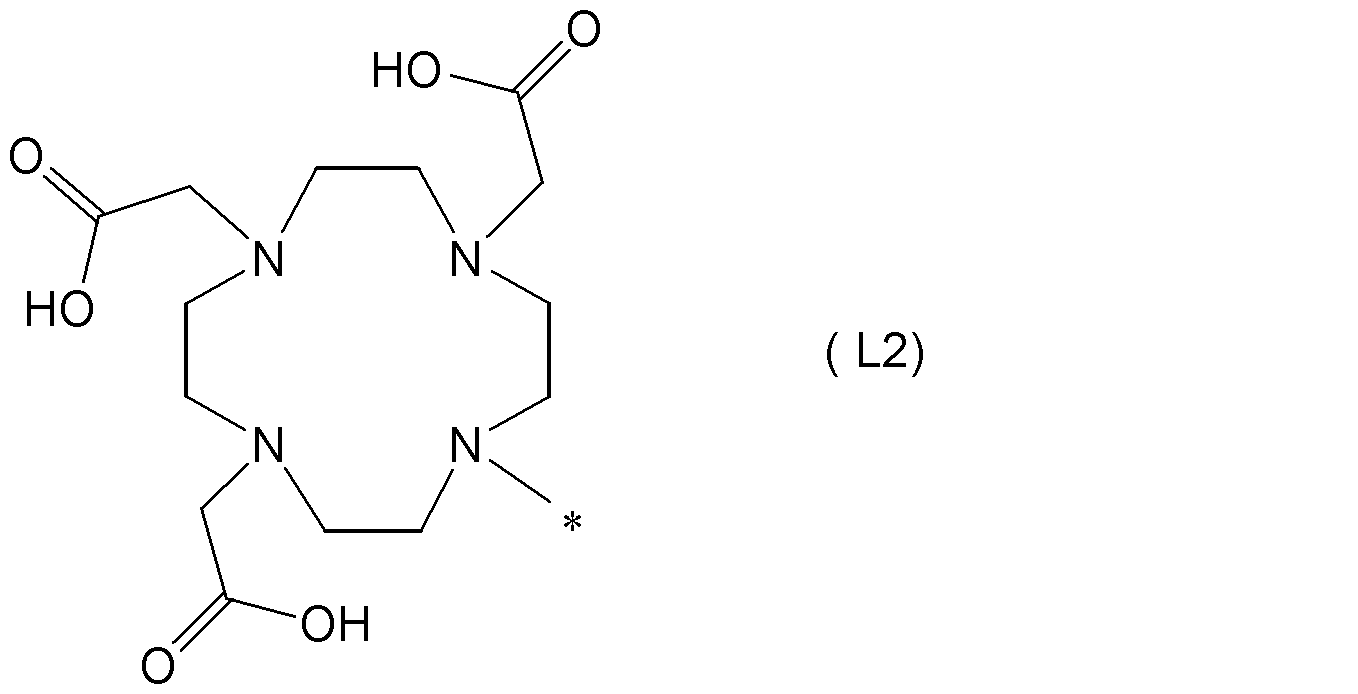
(式中、*は結合部位である。)で表される基である。〕
本発明によれば、インジウム-111による標識が可能であり、腎臓への集積を低減できる放射性薬剤が得られる化合物等が提供できる。更に、本発明の放射性薬剤は、標的分子認識素子を有するため、標的部位に特異的に結合することができ、そのため標的部位に効率的に集積する。このような性質のため、本発明の放射性薬剤は、放射線治療において腫瘍部位に特異的に集積し、放射線画像診断においては、その感度、精度を向上させることができる。
放射性薬剤の投与後、当該薬剤が腎細胞に取り込まれる際に、効率よく尿排泄性の放射性代謝物を遊離することができれば、腎臓への放射活性の集積を低減することができると考えられる。そのため、放射性薬剤が腎細胞に取り込まれる際に、効率よく放射性標識元素を包摂するキレート配位子部位を遊離できるように、ポリペプチドとキレート配位子部位との間に腎刷子縁膜酵素の基質配列を導入する。そうすることで、腎細胞に取り込まれる前に、ポリペプチドとキレート配位子部位がポリペプチドから遊離され、腎臓への放射性物質の取り込みを抑制し投与早期から腎臓への集積を低減することができるものと推測される。
インジウム-111による標識を可能とするため、式(L1)又は式(L2)で表される基を、キレート配位子部位として、導入することを検討した。この場合、例えば、特許文献1に記載された化合物のように、キレート薬剤部位と、ポリペプチド部位とを連結する構造として、チオウレア構造を導入することも考えられた。しかしながら、キレート薬剤として、式(L1)又は式(L2)で示される基を有する化合物を用いた場合には、当該チオウレア構造を有する連結基を導入すると、腎刷子縁膜酵素による分解が進行しないことが、発明者らの実験から明らかになった。これに対して、式(1)で示される化合物のように特定構造の連結基を導入することで、腎刷子縁膜酵素による分解が進行し、腎臓への放射活性の集積を低減することができることが明らかになった。
A1は、腎臓への集積を投与早期から低減する観点から、好ましくは、フェニルアラニン、メチオニン、バリン、ロイシン、イソロイシン、プロリン、チロシン、グリシン、アラニン又はトリプトファンの残基であり、より好ましくは、フェニルアラニン、グリシン、アラニン又はメチオニンの残基であり、腎臓への集積を投与早期から低減する効果をより顕著にする観点から、更に好ましくはフェニルアラニンの残基である。
なお、mは、0~3の整数であり、好ましくは1である。
なお、A4として、他のアミノ酸残基を有していてもよい。A4としては、任意のアミノ酸が用いられる。
nは、0~3の整数であり、好ましくは0である。
R1は、スペーサーとして機能し、官能基を介してポリペプチド等の標的分子認識素子を本発明の化合物に結合することができる。R1は、A3の側鎖のアミノ基又はカルボキシ基と結合することで、アミノ酸配列末端を化学修飾せず、本発明の化合物とポリペプチドとを結合することができる。
R1は、側鎖のアミノ基の窒素原子と結合していてもよいし、側鎖のカルボキシ基とエステル結合を形成していてもよい。
R1の標的分子認識素子又はその連結基と結合可能な官能基は、特に限定されないが、例えば、カルボキシ基又はその活性エステル;マレイミド基、アクリロイル基等のC=C結合を有する基;カルバモイル基、イソチオシアナート基、及びアミノ基からなる群から選ばれる少なくとも1種の官能基(以下「官能基a」ともいう)が挙げられる。カルボキシ基の活性エステルとしては、クロロアセチル基、ブロモアセチル基、ヨードアセチル基等が挙げられる。これらの中でも、官能基aは、C=C結合を有する基、カルバモイル基が好ましい。
R1の総炭素数は、特に限定されないが、例えば、好ましくは1以上、より好ましくは2以上、更に好ましくは3以上であり、そして、好ましくは20以下、より好ましくは10以下、更に好ましくは8以下である。
R1としては、例えば、官能基aを有する総炭素数2~20のアシル基、官能基aを有する総炭素数2~20のアルキル基、官能基aを有する総炭素数2~20のアルキルカルバモイル基、官能基aを有する総炭素数2~20のアルキルチオカルバモイル基が挙げられる。
R1が複素環基を形成する場合、複素環基はマレイミド基が好ましい。
R1が複素環基を形成する場合、複素環基の炭素数は、好ましくは3~10、より好ましくは3~5、更に好ましくは4又は5である。
R1は、A3の側鎖のアミノ基若しくはカルボキシ基の水素原子であってもよい。つまり、A3のアミノ基若しくはカルボキシ基は、修飾されていなくてもよい。
R1の中でも、環構成原子の一つとしてA3の側鎖のアミノ基の窒素原子を含む炭素数3~10の複素環基が好ましく、環構成原子の一つとしてA3の側鎖のアミノ基の窒素原子を含むマレイミド基がより好ましい。

で表される基、又は、式(L2):
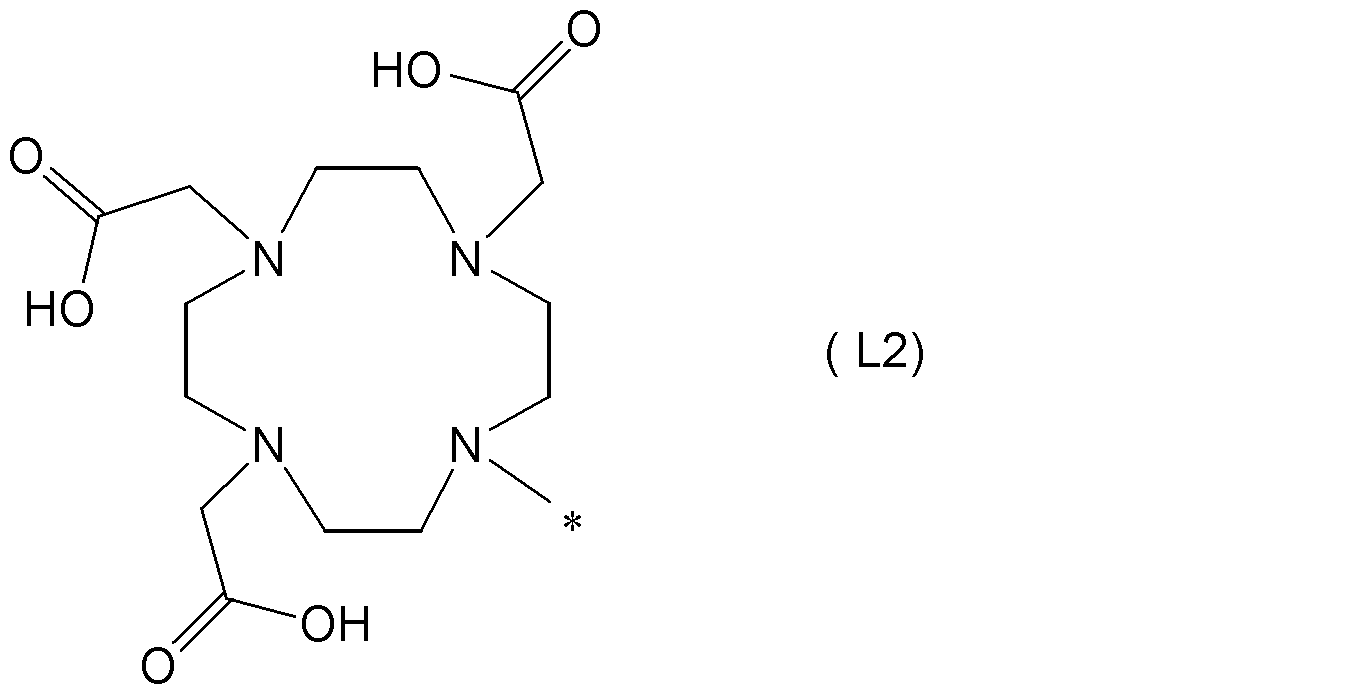
で表される基である。
式(L1)で表される基に関して、R3,R4,R5,R6のうち、好ましくは3個以上4個以下が-CH2COOH基であり、より好ましくは3個が-CH2COOH基である。
-CH2COOR10基として、R10は、水素原子又は炭素数1~8の炭化水素基である。
R3,R4,R5,R6のうち、-CH2COOH基以外の基は、好ましくは炭素数1~8の炭化水素基であり、より好ましくは炭素数1~4の炭化水素基である。
R3,R4,R5,R6のうち、少なくとも1つの基は、腎臓への集積を低減する観点から、好ましくは炭素数3~8の炭化水素基であり、より好ましくは炭素数4~6の炭化水素基である。
当該炭化水素基は、好ましくは、脂肪族炭化水素基であり、より好ましくは分岐鎖脂肪族炭化水素基である。
当該炭化水素基としては、例えば、メチル基、エチル基、n-プロピル基、イソプロピル基、n-ブチル基、sec-ブチル基、イソブチル基が挙げられる。
これらの中でも、腎臓への集積を低減する観点から、エチル基、n-ブチル基、sec-ブチル基、イソブチル基が好ましく、n-ブチル基、sec-ブチル基、イソブチル基がより好ましく、イソブチル基が更に好ましい。
これらの中でも、Lは、好ましくは、式(L1):

(式中、R3,R4,R5,R6は、-CH2COOH基、又は炭素数1~8の炭化水素基であり、*は結合部位である。ただし、R3,R4,R5,R6のうち、3個は-CH2COOH基である。)で表される基である。
Lとしては、式(L1-1),式(L1-2),式(L1-3),式(L1-4),式(L1-5),式(L1-6),式(L1-7),式(L1-8),式(L1-9)及び式(L2)からなる群から選ばれる少なくとも1種が好ましい。


(以上、式中、*は結合部位である。)

〔式中、
Lは、式(L1):

(式中、R3,R4,R5,R6は、それぞれ独立に、水素原子、-CH2COOR10基、又は炭素数1~8の炭化水素基であり、R10は、水素原子又は炭素数1~8の炭化水素基であり、*は結合部位である。ただし、R3,R4,R5,R6のうち、少なくとも3個以上は-CH2COOH基である。)で表される基、又は、式(L2):
(式中、*は結合部位である。)で表される基であり、
R7は、水素原子又はメチル基であり、
R8,R9は、それぞれ独立に、水素原子、又は、官能基aを有する総炭素数2~20のアシル基、官能基aを有する総炭素数2~20のアルキル基、官能基aを有する総炭素数2~20のアルキルカルバモイル基、若しくは官能基aを有する総炭素数2~20のアルキルチオカルバモイル基である。ただし、R8及びR9は、隣接する窒素原子を含む複素環を形成していてもよく、その場合、式:

で表される基は、式:

で表される基である。〕
R8,R9は、好ましくは官能基aを有する総炭素数2~20のアシル基であり、より好ましくはカルバモイル基を有する総炭素数3~6のアシル基である。カルバモイル基を有する総炭素数3~6のアシル基としては、例えば、式:―C(=O)(CH2)a(=O)NH2〔式中、aは1~4の整数である。〕で表される基が挙げられる。

(式中、R3,R4,R5,R6は、それぞれ独立に、-CH2COOH基、又は炭素数1~8の炭化水素基であり、*は結合部位である。ただし、R3,R4,R5,R6のうち、3個が-CH2COOH基であり、1個が炭素数1~8の炭化水素基である。)で表される基であり、
より好ましくは、式中、Lが、式(L1):

(式中、R3,R4,R5,R6は、それぞれ独立に、-CH2COOH基、エチル基、又はブチル基であり、*は結合部位である。ただし、R3,R4,R5,R6のうち、3個が-CH2COOH基であり、1個がエチル基、又はブチル基である。)で表される基であり、
更に好ましくは、式中、Lが、式(L1):
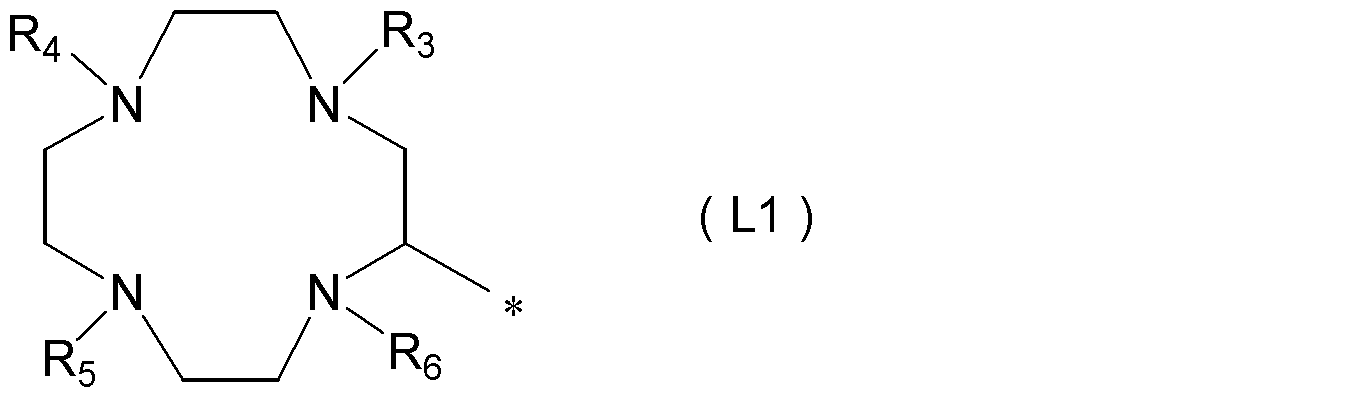
(式中、R3,R4,R5,R6は、それぞれ独立に、-CH2COOH基、イソブチル基であり、*は結合部位である。ただし、R3,R4,R5,R6のうち、3個が-CH2COOH基であり、1個がイソブチル基である。)で表される基である。



薬理学的に許容可能な塩としては、例えば、酸付加塩、塩基付加塩が挙げられる。
酸付加塩としては、無機酸塩、有機酸塩のいずれであってもよい。
無機酸塩としては、例えば、塩酸塩、臭化水素酸塩、硫酸塩、ヨウ化水素酸塩、硝酸塩、リン酸塩が挙げられる。
有機酸塩としては、例えば、クエン酸塩、シュウ酸塩、酢酸塩、ギ酸塩、プロピオン酸塩、安息香酸塩、トリフルオロ酢酸塩、マレイン酸塩、酒石酸塩、メタンスルホン酸塩、ベンゼンスルホン酸塩、パラトルエンスルホン酸塩が挙げられる。
塩基付加塩としては、無機塩基塩、有機塩基塩のいずれであってもよい。
無機塩基塩としては、例えば、ナトリウム塩、カリウム塩、カルシウム塩、マグネシウム塩、アンモニウム塩が挙げられる。
有機塩基塩としては、例えば、トリエチルアンモニウム塩、トリエタノールアンモニウム塩、ピリジニウム塩、ジイソプロピルアンモニウム塩が挙げられる。
本発明の化合物(2)等は、化合物(1)、又はその薬理学的に許容可能な塩に、標的分子認識素子を結合させてなる化合物、又はその薬理学的に許容可能な塩である。標的分子認識素子は、化合物(1)、又はその薬理学的に許容可能な塩に、連結基を介して結合していてもよいし、直接結合していてもよい。連結基としては、2-イミノチオランから誘導されるイミノチオールが挙げられる。
「標的分子認識素子」とは、生体内において、標的分子に結合する等の標的分子を認識可能な分子、置換基、官能基又は原子団である。
標的分子認識素子としては、例えば、ポリペプチド、その他、標的分子に結合するリガンドが挙げられる。
ポリペプチドは、通常、標的分子に結合するポリペプチドであり、好ましくは標的分子に特異的に結合するポリペプチドである。特異的に結合するとは、標的分子に結合するが、標的分子以外の分子には結合しないか、弱い結合であることをいう。
標的分子とは、放射性薬剤による診断の対象となる標的部位、例えば、組織や細胞に存在する分子、好ましくは特異的に発現する分子をいう。「特異的に発現する」とは、標的部位に発現するが、標的部位以外の部位には発現しないか、低い発現であることをいう。
抗体の抗原結合領域断片としては、例えば、Fab断片(以下単に「Fab」ともいう)、F(ab')2断片、F(ab)2断片、可変領域断片(以下、「Fv断片」ともいう)が挙げられる。
Fab断片とは、抗体のパパイン分解により生ずるN末端側の産物及びこれと同様のドメイン構造を有する断片を意味する。
F(ab')2断片とは、抗体のF(ab')2のヒンジ領域のジスルフィド結合を還元することにより得られる断片及びこれと同様のドメイン構造を有する断片を意味する。
F(ab)2断片とは、2分子のFab断片が互いにジスルフィド結合で結合した二量体を意味する。
Fv断片とは、抗体の断片であって抗原との結合活性を有する最小の断片を意味する。
抗体の抗原結合領域断片としては、より具体的には、特定のがん細胞に特異的に発現するタンパク質に対する抗体、及び、そのFab断片若しくはFv断片が挙げられる。
なお、標的分子認識素子は、これら例示されたポリペプチドに限定されず、標的分子に結合するポリペプチドであればいずれを使用することもできる。
Asn-urea-Lys 部位とは、式:

〔式中、*は結合部位である。〕で表される部位である。
Glu-urea-Lys 部位とは、式:

〔式中、*は結合部位である。〕で表される部位である。
〔式中、*は結合部位である。〕

〔式中、*は結合部位である。〕
放射性薬剤の調製用薬剤は、当該化合物の他に、水性緩衝液などのpH調節剤、アスコルビン酸、p-アミノ安息香酸等の安定化剤等を含んでいてもよい。

〔式中、A1,A2,m,A3,A4,n,R1,Lは式(1)と同様であり、
L1は、R1とP1を連結する連結基であり、
pは0又は1であり、
P1は、標的分子認識素子である。〕
L1は、R1の連結基と連結可能な官能基により結合を形成し、標的分子認識素子とも結合を形成する。L1は、好ましくは、2-イミノチオランから誘導されるイミノチオール等である。
pは、好ましくは1である。
P1は、例えば、上述の標的分子認識素子であり、好ましくはポリペプチド、その他標的分子に結合するリガンド、又は、式(f2-1),式(f2-2),式(f2-3),式(f2-4),又は式(f2-5)で表される官能基f2である。




本発明の金属錯体化合物、又はその薬理学的に許容可能な塩(以下「金属錯体化合物(3)等」ともいう)は、放射性金属及び放射性原子標識金属からなる群から選ばれる1種の金属と、前記金属に配位した本発明の化合物又はその薬理的に許容可能な塩と、を有する。
金属としては、例えば、111In、223Ra、67Ga、68Ga、44Sc、90Y、177Lu、225Ac、212Bi、213Bi、212Pb、227Th、64Cu、67Cuが挙げられる。
金属は、好ましくは111In、223Ra、67Ga、68Ga、90Y、177Lu、225Ac、212Bi、213Bi、212Pb、及び227Thからなる群から選ばれる少なくとも1種であり、より好ましくは111In、90Y、177Lu、225Ac、212Bi、213Bi及び212Pbからなる群から選ばれる少なくとも1種であり、更に好ましくは111In、90Y、177Lu及び225Acである。

〔式中、A1,A2,m,A3,A4,n,R1は、式(1)と同様であり、
L’は、式(L1’-4)、式(L1’-5)、式(L1’-9)、又は式(L2’):

〔式中、A1,A2,m,A3,A4,n,R1は、式(1)と同様であり、
L1,p,P1は、式(2)と同様であり、
L’は、式(L1’-4)、式(L1’-5)、式(L1’-9)又は式(L2’):

本発明のキットは、例えば、化合物(1)等と、標的分子認識素子を含む薬剤と、放射性金属及び放射性原子標識金属からなる群から選ばれる1種の金属を含む薬剤とを、別々の包装単位として含むキット;化合物(1)等に標的分子認識素子を結合させてなる化合物(2)等と、放射性金属及び放射性原子標識金属からなる群から選ばれる1種の金属を含む薬剤とを、別々の包装単位として含むキットが挙げられる。
キットに含まれる化合物及び薬剤はいずれも、必要に応じて、上記のような1種類又は2種類以上の医薬的に許容される担体(医薬用担体)を含むことができる。
本発明の化合物(1)等、及び、当該化合物(1)等に標的分子認識素子を結合させてなる化合物(2)等は、公知の方法を用いて合成することができ、例えば、本明細書の実施例に記載された方法により製造することができる。
本発明の金属錯体化合物(3)等は、化合物(2)等を配位子として用い、金属放射性金属又は放射性原子標識金属とインビトロで錯形成させることにより、製造することができる。錯形成は、公知の方法を用いて行うことができる。
本発明の金属錯体化合物等は、例えば、放射線治療又は放射線画像診断に用いられる放射性薬剤として使用される。
放射線治療薬として用いられる、放射性金属及び放射性原子標識金属からなる群から選ばれる1種の金属は、例えば、アルファ線放出核種、ベータ線放出核種、ガンマ線放出核種、ポジトロン放出核種が挙げられる。これらの中でも、放射線治療の用途では、ベータ線放出核種(即ち、β線を放出する核種)が好ましい。
診断としては、特に限定されず、腫瘍、炎症、感染症、心循環器疾患、脳・中枢系疾患等の各種疾患及び臓器・組織の放射線画像診断等に用いられ、好ましくは、がんの放射線画像診断に使用される。
診断の対象となる標的の特性にしたがって、標的分子認識素子を選択することにより、多種類多様な標的の診断や治療が可能であり、本発明の放射性薬剤は診断の分野で放射性画像診断薬として広く使用できる。
投与経路はこれら経路に限定されず、放射性薬剤の投与後に、その作用が有効に発現し得る経路であればいずれも利用できる。
放射性強度は、放射性薬剤を使用する一般的な診断方法や治療方法で使用されている放射活性強度を参考にして決定できる。その投与量は患者の年齢、体重、適当な放射線イメージング装置、及び対象疾患の状態等の諸条件を考慮し、イメージングが可能と考えられる放射能及び投与量が決定される。
通常、放射線治療に使用されることが想定され、その診断薬剤の投与量は、特に限定されないが、例えば、放射性金属(例えば111In)の放射能量として1.0MBq/kg~3.0MBq/kgである。
Fmoc:フルオレニルメトキシカルボニル基
Boc:tert-ブトキシカルボニル基
THF:テトラヒドロフラン
NMM:N-メチルモルフォリン
DMF:ジメチルホルムアミド
Tfa:トリフルオロアセテイト基
DCC: N,N'-ジシクロヘキシルカルボジイミド
Trt(2-Cl):2-クロロトリチル基
Cl-Trt(2-Cl)Resin:2‐クロロトリチルクロライドレジン(2-Chlorotrityl chloride resin)
Fmoc-Lys(Dde)-OH:N-α-(9-フルオレニルメトキシカルボニル)-N-ε-[1-(4,4-ジメチル-2,6-ジオキソシクロヘキシリデン)エチル]-L-リシン
TFA:トリフルオロ酢酸
MeCN:アセトニトリル
DIPEA:N,N-ジイソプロピルエチルアミン
DIC:N,N'-ジイソプロピルカルボジイミド
HOBt:1-ヒドロキシベンゾトリアゾール
NMCM:N-メトキシカルボニルマレイミド
EDTA:エチレンジアミン四酢酸
DPS:2,2'-ジピリジルジスルフィド
EtOH:エタノール
FBS:ウシ胎児血清
D-PBS:ダルベッコリン酸緩衝生理食塩液
EGTA:グリコールエーテルジアミン四酢酸
下記実施例及び比較例において、各種物性等の測定方法は下記の方法で行った。
1H-NMRによる分析はJEOL ECS - 400 spectrometer (日本電子株式会社)を使用した。
〔ESI-MS(エレクトロスプレーイオン化質量分析)〕
ESI-MSによる分析はHPLC1200 series-6130 quadrupole LC/MS mass spectrometer (アジレント・テクノロジー株式会社)を使用した。
薄層クロマトグラフィ(TLC)による分析にはシリカプレート (TLC aluminium sheets Silica gel 60 RP-18 F254s, メルク株式会社)を使用し、0.1 M 酢酸アンモニウム水溶液:メタノール= 1 : 1の展開溶媒で10 cm展開したものを5 mmずつ切断し、オートウェルガンマシステム(WIZARD3, パーキンエルマー社)によりそれぞれの画分の放射活性を測定した。
セルロースアセテート膜電気泳動(以下「CAE」ともいう)では泳動膜に11 cm x 1 cmの大きさに切ったセルロースアセテート膜(ADVANTEC SELECA-V, 東洋濾紙株式会社), 緩衝液に(veronal buffer, pH 8.6, I=0.06)又はsolvent 2 (20 mM P.B. (pH 6.0))を用いて、一定電流(1 mA/cm)にて泳動した。泳動後のセルロースアセテート膜を5 mmずつ切断し、オートウェルガンマシステムによりそれぞれの画分の放射活性を測定した。
(分析)
逆相高速液体クロマトグラフィ(以下「RP-HPLC」ともいう)による分析はUV検出器としてL-7405 (株式会社日立製作所)、送液ポンプとしてL-7100(株式会社日立製作所)、分析用カラムとしてCosmosil 5C18-AR-300 column (4.6 x 150 mm,ナカライテスク株式会社)を使用した。
移動相には0.1% (v/v) TFA/H2O (A相)と0.1% (v/v) TFA/MeCN (B相)を用い、0-20 minまでA相95% (v/v)、B相5%(v/v)からA相70%(v/v)、B相30%(v/v)まで変化させ、20-40 minでA相70%(v/v)、B相30%(v/v)からA相0%(v/v)、B相100%(v/v)まで変化させる直線グラジエント法により流速1.0 mL/minで溶出した。
RP-HPLCによる分取にはガードカラムCadenza 5CD-C18 guard column (10 x 8 mm,インタクト株式会社)を連結したCadenza 5CD-C18 column (20 x 150 mm,インタクト株式会社)を使用した。
移動相には0.1%(v/v) TFA/H2O (A相)と0.1%(v/v) TFA/MeCN (B相)を用い、0-30 minまでA相90%(v/v)、B相10%(v/v)からA相20%(v/v)、B相80%(v/v)まで変化させ、30-40 minでA相20%(v/v)、B相80%(v/v)からA相0%(v/v)、B相100%(v/v)まで変化させる直線グラジエント法により流速5.0 mL/minで溶出した。
下記合成例における、「DO3A-EDA (Mal)」は、下記の式で表される化合物であり、Macrocyclics社製の商品名「B-272」を使用した。

動物実験は、雄性のddY系SPFマウス6週齢 及び雄性のBALB/c-nu/nuマウス(日本エスエルシー株式会社)を使用した。
合成例L1:化合物(a13)の合成

化合物(a1)(7.97 g、20.4 mmol)をTHF 40 mLに溶解し-15°Cに冷却後、アルゴン雰囲気下、 N-メチルモルフォリン(NMM, 3.36 mL、30.5 mmol)及びクロロギ酸イソブチル(4.01 mL、30.5 mmol)を順に加えた。5分間攪拌した後、化合物(a2)(5.87 g、30.5 mmol)とNMM (3.36 mL、30.5 mmol)のDMF溶液24 mLを滴下し、冷却下で30分、室温で1 時間攪拌した。溶媒を減圧留去後、残渣を酢酸エチル100 mL及び5質量%炭酸水素ナトリウム水溶液100 mLにて溶解し、5質量%炭酸水素ナトリム水溶液(50 mL × 3)、5質量%クエン酸水溶液(50 mL × 3)で洗浄した。有機層に無水硫酸マグネシウムを加えて乾燥した後、溶媒を除去して得られた結晶を減圧乾燥することで化合物(a3)(10.6 g、20.0 mmol、収率98.0%)を淡黄色結晶として得た。
1H NMR (CDCl3) : δ 1.42 [9H, s, Boc], 2.96-3.03 [2H, m, CHCH 2 ], 3.39-3.51 [4H, CH2CH 2 ], 4.22-4.24 [1H, dd, NHCH], 4.85, 6.42, 7.74 [3H, t, NH], 6.93-6.95 [2H, d, CCH], 7.62-7.66 [2H, d, ICCH].
ESI-MS (M+Na)+: m/z 552.07. found 552.09.
化合物(a3)(10.6 g、20.0 mmol)を4 N塩酸/酢酸エチル50 mLに溶解し、室温で3 時間攪拌した。溶媒を減圧留去し、減圧乾燥することで化合物(a4)(8.08 g、20.0 mmol、収率99.8%)を淡黄色結晶として得た。
1H NMR (CDCl3) : δ 3.42-3.55 [6H, overlapped, CH 2 ], 3.86-3.88 [1H, dd, CH2CH], 7.04-7.06 [2H, d, CCH], 7.62-7.64 [2H, d, ICCH].
ESI-MS (M+Na)+: m/z 452.02. found 452.03.
化合物(a5)(4.45 g、19.1 mmol)をTHF 70 mLに溶解し、溶液を氷冷した後、アルゴン雰囲気下でDCC(4.30 g、20.8 mmol)のTHF 溶液20 mLを滴下した。滴下完了後、室温で1時間攪拌し、反応液をろ過することでジシクロヘキシル尿素(以下、「DC-urea」ともいう)を除去し、ろ液を化合物(a5)の無水物溶液として次の反応に用いた。化合物(a4)(8.08 g、17.4 mmol)を酢酸エチル80 mLに懸濁し、Et3N(3.63 mL、26.0 mmol)を加えて冷却、10分間攪拌した後、アルゴン雰囲気下、先に調製した化合物(a5)の無水物溶液を滴下した。滴下完了後、室温にて1 時間攪拌し、反応液を5質量%クエン酸水溶液(50 mL × 3)で洗浄した。有機層に無水硫酸マグネシウムを加えて乾燥した後、溶媒を減圧留去して得られた残渣を、酢酸エチルを溶出溶媒とするシリカゲルカラムクロマトグラフィーにより精製を行うことで化合物(a6)(10.2 g、15.8 mmol、収率91.2%)を淡黄色結晶として得た。
1H NMR (CDCl3) : δ 1.37 [9H, s, Boc], 3.00-3.19 [2H, m, CHCH 2 ], 3.42-3.52 [5H, overlapped, NCH 2 , NHCH 2 ], 3.77-4.03 [2H, m, COCH 2 ], 4.11-4.16 [1H, dd, NCH 2 ], 4.60-4.62 [1H, t, CH2CH], 6.96-7.01 [2H, d, CCH], 7.58-7.60 [2H, d, ICCH].
ESI-MS (M+Na)+: m/z 667.09. found 667.02.
化合物(a6)(3.52 g、5.46 mmol)を25質量%アンモニア水50 mLに溶解し、室温で3 時間攪拌した。溶媒を減圧留去後、減圧乾燥して得られた油状物をDMF 90 mLに溶解したものをA液とした。O-(7-アザ-1H-ベンゾトリアゾール-1-イル)-N,N,N',N'-テトラメチルウロニウムヘキサフルオロホスフェート(HATU, 3.12 g、8.21 mmol)をDMF 90 mLに溶解したものをB液とした。DMF 1600 mLにDIEA(3.81 mL、21.9 mmol)、1-ヒドロキシ-7-アザベンゾトリアゾール(1.12 g、8.22 mmol)を溶解し、A液及びB液を、シリンジポンプを用いて1.2 mL/hにて低速同時滴下し、滴下終了後24 時間攪拌した。溶媒を減圧留去して得られた残渣を酢酸エチル及びヘキサンで洗浄することで化合物(a7)(1.57 g、2.96 mmol、収率54.2%)を白色結晶として得た。
1H NMR (CD3OD) : δ 1.46 [9H, s, Boc], 2.82-2.89 [1H, m, CH 2 ], 2.91-2.96 [2H, m, CH 2 ], 3.03-3.14 [1H, m, CH 2 ], 3.56-3.64 [2H, m, CH 2 ], 3.82-4.02 [3H, m, CH 2 ], 4.10-4.18 [1H, m, CH 2 ], 4.46-4.50 [1H, dd, CH], 7.02-7.04 [2H, d, CH], 7.60-7.62 [2H, d, CH].
ESI-MS (M+Na)+: m/z 553.09. found 553.10.
化合物(a7)(1.57 g、2.96 mmol)を4 N塩酸/酢酸エチル40 mLに懸濁し、室温にて4 時間攪拌した。溶媒を減圧留去して得られた結晶をヘキサンで洗浄し、減圧乾燥することで化合物(a8)(1.36 g、2.92 mmol、収率98.4%)を白色結晶として得た。
1H NMR (CDCl3) : δ 2.83-2.89 [1H, m, CH 2 ], 2.96 [1H, s, CH 2 ], 3.12-3.19 [1H, m, CH 2 ], 3.40-3.52 [2H, m, CH 2 ], 3.62-3.59 [2H, m, CH 2 ], 4.07-4.13 [1H, m, CH 2 ], 4.19-4.42 [1H, m, CH 2 ], 4.60-4.62 [1H, dd, CH], 6.11 [1H, s, NH], 6.54 [1H, s, NH], 6.96-7.01 [2H, d, CH], 7.58-7.60 [2H, d, CH].
ESI-MS (M+H)+: m/z 431.06. found 431.03.
化合物(a8)(880 mg、2.04 mmol)をDMF13.5mLに懸濁し、さらに炭酸カリウム(424 mg、 3.06 mmol)を加えて懸濁させた。溶液を氷冷し、アルゴン雰囲気下ヨードエタン(327 μL、 4.08 mmol)を滴下した。滴下完了後、80℃で四日間攪拌した。溶媒を減圧留去して得られた残渣を酢酸エチルに懸濁しろ過を行った。ろ液を5質量%炭酸水素ナトリウム水溶液で洗浄した。有機層に硫酸ナトリウムを加えて乾燥した後、溶媒を減圧留去して得られた残渣をクロロホルムとメタノールを用いたフラッシュクロマトシステムにより生成を行うことで化合物(a9)(340 mg、 0.742 mmol、収率36.3%)を白色結晶として得た。
1H NMR (CDCl3) : δ 1.10 [3H, t, CH 3 ], 2.76-2.84 [2H, q, CH 2 ], 2.85-2.89 [1H, m, CH 2 ], 3.08-3.25 [6H, m, CH 2 ], 3.37-3.43 [1H, m, CH 2 ], 3.53-3.57 [1H, m, CH 2 ], 3.69-3.75 [1H, m, CH 2 ], 4.61-4.66 [1H, dd, CH], 6.59 [1H, s, NH], 6.84 [1H, s, NH], 6.97-6.99 [2H, d, CH], 7.16 [1H, s, NH], 7.58-7.60 [2H, d, CH].
ESI-MS (M+H)+: m/z 459.09. found 459.17.
化合物(a9)(340 mg、0.742 mmol)をTHF 1.4 mLに懸濁し、溶液を氷冷した後、アルゴン雰囲気下、0.95 Mのボラン-THF錯体/THF溶液13.5 mLを緩徐に加え、1時間攪拌した後、24時間還流した。溶液を氷冷し、メタノール13.5 mLを加えた後、1時間攪拌し、溶媒を減圧留去した。その後、残渣に再度メタノール13.5 mLを加え、溶媒を減圧留去した。残渣に濃塩酸13.5 mLを加え、24 時間室温で攪拌した後、1 時間還流した。溶液を氷冷し、12.5 N水酸化ナトリウム水溶液を加えて塩基性とした後、クロロホルムで抽出した。有機層に硫酸ナトリウムを加えて乾燥した後、溶媒を減圧留去して得られた残渣をクロロホルム:メタノール:25質量%アンモニア水(10:1:0.1)を溶出溶媒とするフラッシュクロマトシステムにより精製を行うことで化合物(a10)(163 mg、0.391 mmol、収率52.7%)を黄色油状物として得た。
化合物(a10)(197 mg、0.401 mmol)をアセトニトリル2.75 mLとDMF 0.55 mLの混液に溶解し、さらに炭酸カリウム(229 mg、1.65 mmol)を加えて懸濁させた。溶液を氷冷し、アルゴン雰囲気下、ブロモ酢酸tert-ブチル(229 μL、1.40 mmol)を滴下した。滴下完了後、室温で48時間攪拌し、懸濁液をろ過した後、ろ液から溶媒を減圧留去した。残渣を少量のクロロホルムに溶解し、厚さ1 mmの分取用TLCにアプライし、クロロホルム:メタノール=8:1を展開溶媒とすることで精製し、化合物(a11)(306 mg、 0.403 mmol、100%)を赤褐色油状物として得た。
1H NMR (CDCl3) : δ 0.98-1.03 [3H, t, CH 3 ], 1.41-1.48 [27H, m, t Bu], 1.90-4.90 [27H, m, CH 2 , DOTA], 6.89-7.07 [2H, m, CH], 7.50-7.61 [2H, m, CH].
ESI-MS (M+H)+: m/z 759.36. found 759.24.
化合物(a11)(306 mg、0.403 mmol)をDMF 2.8 mLに懸濁し、Pd(OAc)2(9.1 mg、0.0403 mmol)、1,2-ビス(ジフェニルホスフィノ)エタン(24.12 mg、0.0604 mmol)、Et3N(168 μL、1.21 mmol)、ベンジルアルコール(840 μL、8.06 mmol)を加え、一酸化炭素雰囲気下、24時間還流した。反応後、溶媒を減圧留去し、酢酸エチルに溶解後、ろ過を行い、ろ液を5質量%炭酸水素ナトリウム水溶液で洗浄した。有機層に硫酸ナトリウムを加え乾燥した後、溶媒を減圧留去して得られた残渣をクロロホルムとメタノールを用いたフラッシュクロマトシステムにより精製することで化合物(a12)(113 mg、 0.166 mmol、収率41.4%)を黄色油状物として得た。
ESI-MS (M+H)+: m/z 767.50. found 767.60.
化合物(a12)(46 mg、0.0600 mmol)をメタノール1 mLに溶解した後、10質量% Pd/Cを100mg加えて水素雰囲気下、室温で2時間攪拌した。反応溶液をろ過後、溶媒を減圧留去することで化合物(a13)(25.5mg、0.0377 mmol、収率62.8%)を黄色油状物として得た。
ESI-MS (M+H)+: m/z 677.45. found 677.62.
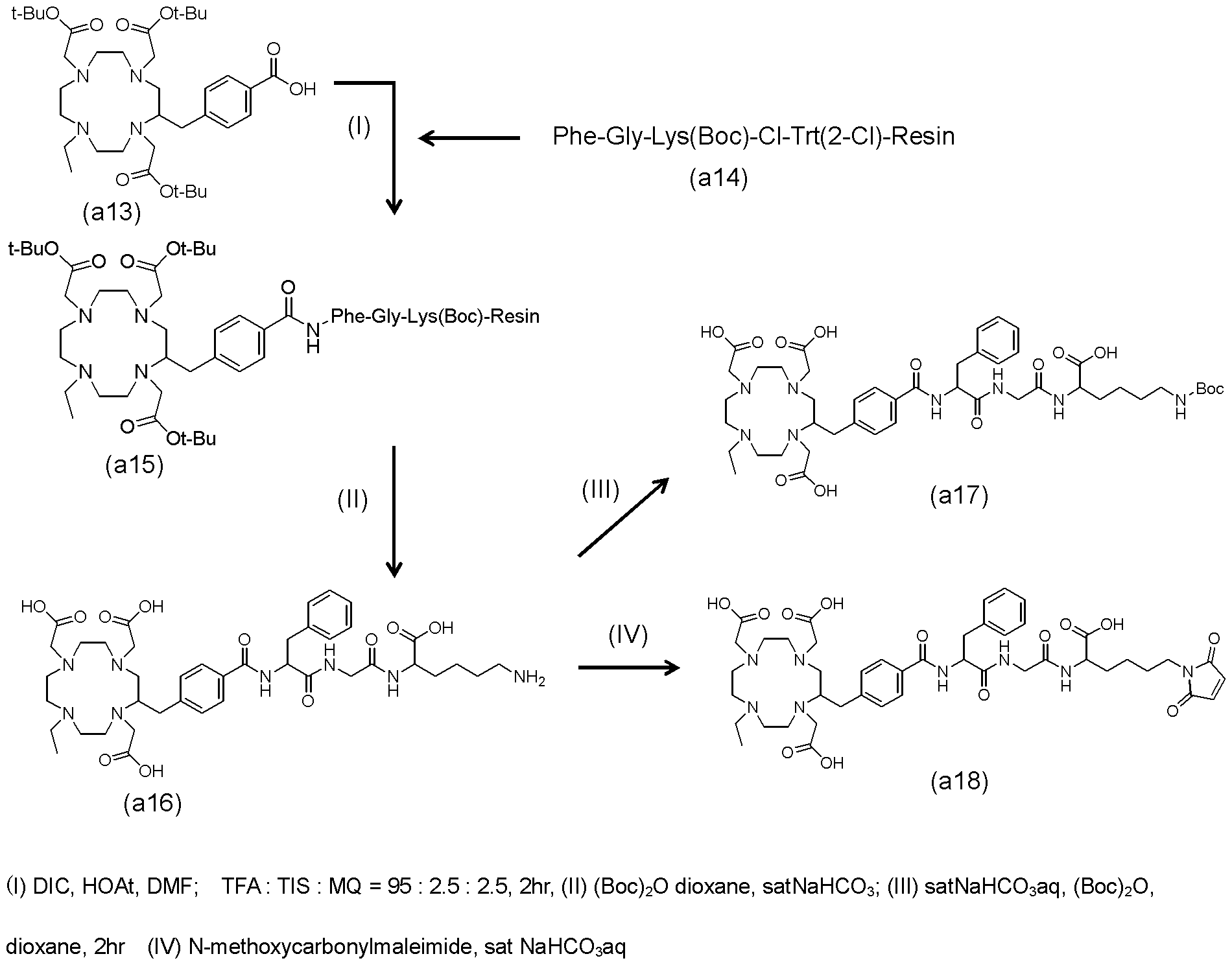
Fmoc固相合成法を用いてペプチドを伸長した樹脂(a14) (22 μmol)、1-ヒドロキシ-7-アザベンゾトリアゾール(15.1 mg、110 μmol)、化合物(a13) (15 mg、22 μmol)をDMF溶解し、N,N'-ジイソプロピルカルボジイミド(17.2 μL、110 μmol)を加えて室温で16時間緩やかに攪拌した。反応終了後DMF及びCH2Cl2を用いて樹脂を洗浄した。
得られた樹脂(a15)をトリフルオロ酢酸: トリイソプロピルシラン: 水= 95 : 2.5 : 2.5を組成とする溶液に懸濁し2時間緩やかに攪拌した。反応終了後、ろ過にて樹脂を除去し、ろ液を減圧留去することで白色結晶を得た。さらにHPLCにてImtakt Cadenza 5CD-C18 150×20 mmを用い移動相にはA相に0.1%TFA/MilliQ、B相に0.1% TFA/MeCNを使用し、0-35分でA相95%、B相5%からA相70%、B相30%まで変化させ、35-40分でA相70%、B相30%からA相0%、B相100%まで変化させる直線gradient法により流速5 mL/minで精製することで目的とする化合物(a16)(以下、「CDO3AEt-FGK」ともいう)(5.7 mg、6.78 μmol、収率30.8%)を得た。
ESI-MS (M-H)-: m/z 839.4, found: 839.3
化合物(a16)を飽和炭酸水素ナトリウム水溶液100 μLに溶解し、1.5当量の(Boc)2Oを溶解したジオキサン100 μLを加え、室温で二時間激しく攪拌した。減圧留去によりジオキサンを除去後、水層をクロロホルムにより洗浄を行った。さらに水層をHPLCにてImtakt Cadenza 5CD-C18 150×20 mmを用い移動相にはA相に0.1%TFA/MilliQ、B相に0.1% TFA/MeCNを使用し、2分までA相 100%に保った後に、2-5分でA相 100%、B相 0%からA相 95%、B相 5%まで変化させ、5-40分でA相 95%、B相 5%からA相 70%、B相 30%まで変化させ、40-45分でA相 70%、B相 30%からA相 0%、B相 100%まで変化させる直線gradient法により流速5 mL/minで精製することで目的とする化合物(a17)(以下、「CDO3AEt-FGK-Boc」ともいう)を得た。
ESI-MS (M-H)-: m/z 939.5, found: 940.49
化合物(a16)(2.2 mg、 2.6 μmol)を飽和炭酸水素ナトリウム水溶液200 μLに溶解し、氷冷下N-メトキシカルボニルマレイミド(0.8 mg、 5.2 μmol)を加え、氷冷下で二時間攪拌した。反応終了後、10質量%クエン酸水溶液で酸性に調整した。さらにHPLCにてImtakt Cadenza 5CD-C18 150×20 mmを用い移動相にはA相に0.1%TFA/MilliQ、B相に0.1% TFA/MeCNを使用し、5分までA相 100%に保った後に、5-35分でA相 100%、B相 0%からA相 55%、B相 45%まで変化させ、35-50分でA相 55%、B相 45%からA相 0%、B相 100%まで変化させる直線gradient法により流速5 mL/minで精製することで目的とする化合物(a18) (以下、「CDO3AEt-FGK(Mal)」ともいう)(0.8 mg、 0.870 μmol、収率33.4%)を得た。
ESI-MS (M-H)-: m/z 919.42, found: 919.45.
合成例L3:化合物(b5)及び化合物(b7)の合成

サイクレン(523.6 mg, 3.04 mmol)をアセトニトリル(25 mL)に溶解し、NaHCO3 (893.6 mg, 10.6 mmol)を加え、Ar雰囲気下、氷冷した後、ブロモ酢酸tert-ブチル(1.39 mL, 3.34 mmol)を滴下した。室温で48時間攪拌した後、反応液をろ過し、ろ液を減圧留去した。残渣をトルエンを用いた再結晶により精製することで化合物(b1) (672.4 mg, 収率43.1%)を白色結晶として得た。
1H NMR (CDCl3): δ 1.44 (27H, s, t Bu), 2.86-3.35 (22H, overlapped, CH 2 ).
ESI-MS (M+H)+: m/z 515.3, found: 515.3.
化合物(b1) (212.9 mg, 413.6 μmol)をアセトニトリル(4.0 mL)に溶解し、Na2CO3 (87.7 mg, 827.4 μmol)を加え、Ar雰囲気下、氷冷した後、アセトニトリル(1.0 mL)に溶解した4-ニトロベンジルブロミド(134.1 mg, 620.7 μmol)を滴下した。60 ℃で18時間攪拌した後、反応液をろ過し、溶媒を減圧留去した。残渣をクロロホルム:メタノール=10:1を溶出溶媒とするシリカゲルクロマトグラフィーにより精製することで化合物(b2) (263.4 mg, 収率98.0%)を黄色油状物質として得た。
1H NMR (CDCl3): δ 1.36 (27H, s, t Bu), 2.06-3.58 (24H, overlapped, CH 2 ), 7.61-7.63 (2H, d, aromatic), 8.07-8.09 (2H, d, aromatic).
ESI-MS (M+Na)+: m/z 672.4, found: 672.3.
化合物(b2) (150 mg, 231 μmol)をメタノール(3.5 mL)に溶解した後、1 N NaOH (0.5 mL)、10%Pd/C (15.4 mg)を加えた。水素雰囲気下、室温で1.5時間攪拌した。溶液をろ過した後、ろ液からメタノールを減圧留去し、クロロホルム(5 mL×3)により抽出した。有機層にNa2SO4を加えて乾燥した後、溶媒を減圧留去することで化合物(b3) (124.6 mg, 収率87.2%)を黄色油状物質として得た。
1H NMR (CDCl3): δ 1.43 (27H, s, t Bu), 2.85-3.35 (24H, overlapped, CH 2 ), 6.59-6.61 (2H, d, aromatic), 6.93-6.95 (2H, d, aromatic).
ESI-MS (M+H)+: m/z 620.4, found: 620.5.
化合物(b3) (124.6 mg, 201 μmol)を10% アニソール/トリフルオロ酢酸(TFA) (2 mL)に溶解し、室温で4時間攪拌した。溶媒を減圧留去し、ジエチルエーテル(5 mL)を加えることで結晶化させた。結晶をろ取し、ジエチルエーテルで洗浄した後、減圧乾燥することで化合物(b4)のTFA塩(118.4 mg, 収率64.9%)を赤褐色結晶として得た。
1H NMR (D2O): δ 2.50-3.50 (24H, overlapped, CH 2 ), 6.78-6.81 (2H, d, aromatic), 7.36-7.38 (2H, d, aromatic).
化合物(b4) (82.5 mg, 80.8 μmol)をmilliQ水(1 mL)に溶解し、1 M チオホスゲン/クロロホルム (1 mL)を加えた。室温で2時間攪拌した後、クロロホルム(5 mL×4)で洗浄した。水層を凍結乾燥することで化合物(b5) (57.4 mg, 収率74.9%)を淡黄色結晶として得た。
1H NMR (D2O): δ 2.50-3.65 (24H, overlapped, CH 2 ), 7.18-7.22 (2H, d, aromatic), 7.38-7.41 (2H, d, aromatic).
化合物(b1) (100.0 mg, 194.4 μmol)をアセトニトリル(2.0 mL)に溶解し、Na2CO3 (26.5 mg, 252.9 μmol)を加え、Ar雰囲気下、氷冷した後、アセトニトリル(0.5 mL)に溶解したメチル-4-(ブロモメチル)ベンゾエート(58.0 mg, 253.0 μmol)を滴下した。60 ℃で18時間攪拌した後、ろ過し、溶媒を減圧留去した。残渣をクロロホルム:メタノール=10:1を溶出溶媒とするシリカゲルクロマトグラフィーにより精製することで化合物(b6) (106.1 mg, 収率82.6%)を黄色油状物質として得た。
1H NMR (CDCl3): δ 1.46 (27H, s, t Bu), 2.01-3.58 (24H, overlapped, CH 2 ), 3.88 (3H, s, OCH 3 ), 7.53-7.55 (2H, d, aromatic), 7.95-7.97 (2H, d, aromatic).
ESI-MS (M+H)+: m/z 663.4, found: 663.4
化合物(b6) (106.1 mg, 160.0 μmol)をメタノール(1 mL)に溶解した後、1 N NaOH (1 mL)を加えて室温で2時間攪拌した。溶液からメタノールを減圧留去した後、クロロホルム(5 mL×3)により抽出した。有機層にNa2SO4を加えて乾燥した後、溶媒を減圧留去することで化合物(b7) (50.5 mg, 収率48.6%)を黄色油状物質として得た。
1H NMR (CDCl3): δ 1.44 (27H, s, t Bu), 2.13-3.58 (24H, overlapped, CH 2 ), 7.31-7.33 (2H, d, aromatic), 8.09-8.11 (2H, d, aromatic). ESI-MS (M+Na)+: m/z 671.4, found: 671.5

安息香酸 (560 mg, 4.59 mmol)とN-ヒドロキシスクシンイミド(NHS, 581 mg, 5.05 mmol)をCH2Cl2 (15 mL)に溶解し、氷冷した後、CH2Cl2 (8 mL)に溶解したN,N’-ジシクロヘキシルカルボジイミド (DCC, 1.05 g, 5.05 mmol)を滴下した。室温で3時間攪拌した後、ろ過し、溶媒を減圧留去した。残渣を酢酸エチル(10 mL)に溶解し、sat. NaHCO3 (10 mL×3)で洗浄した。有機層にMgSO4を加えて乾燥した後、溶媒を減圧留去することで化合物(b8) (749.7 mg, 収率74.7%)を白色結晶として得た。
1H NMR (CDCl3): δ 2.89 (4H, s, succinimide), 7.47-7.51 (2H, m, aromatic), 7.64-7.68 (1H, m, aromatic), 8.11-8.13 (2H, m, aromatic)
Fmoc-Lys-OH (86.7 mg, 100 μmol)をDMF (1.5 mL)に溶解し、N,N-ジイソプロピルエチルアミン(DIPEA, 40 μL, 246 μmol)を加え、Ar雰囲気下、氷冷したのち、DMF (0.5 mL)に溶解した化合物(b8)を滴下した。室温で3時間攪拌した後、溶媒を減圧留去した。残渣を酢酸エチル(5 mL)に溶解し、10質量% クエン酸(5 mL×3)で洗浄した。有機層にNa2SO4を加え乾燥した後、溶媒を減圧留去し、クロロホルム:メタノール=5:1を展開溶媒とするTLC分取により精製することで、化合物(b9) (82.0 mg, 収率82.0%)を白色結晶として得た。
ESI-MS (M+Na)+: m/z 473.2, found: 473.2
Cl-Trt(2-Cl)Resin (62.5 mg, 100 μmol、渡辺化学工業株式会社)を出発物質として、Fmoc-Lys(Bzo)-OH (47.2 mg, 100 μmol)及びDIPEA (65 μL, 400 μmol)をジクロロメタン(1.5 mL)中1.5時間反応させた。メタノール(1.5 mL)及びDIPEA (65 μL)を加えて反応を停止させた。樹脂をDMF次いでジクロロメタンで洗浄した。樹脂を乾燥させた後、ピペリジン処理の際に生成するN-(9-フルオレニルメチル)ピペリジンのA301における吸光度を測定することによりFmoc-Lys(Bzo)-OHの樹脂への導入量を定量(0.96 mmol/g)した。20% ピペリジン/DMF (mL)を加え、室温で20分間攪拌することで、化合物(b10)を作製した。樹脂の一部を採取してkaiser testを行い、Nα-Fmoc基の脱保護を確認した。
化合物(b10) (22.9 mg, 22.0 μmol)を出発物質として、Fmoc固相合成法により2.5等量のFmoc-Met-OH (55.0 μmol)、N,N’-ジイソプロピルカルボジイミド(DIC, 8.5 μL, 55.0 μmol)、1-ヒドロキシベンゾトリアゾール一水和物(HOBt, 8.43 mg, 55.0 μmol)をDMF中、室温で2時間攪拌した。樹脂の一部を採取してKaiser testを行うことで、縮合反応の終了を確認した後、20% ピペリジン/DMF (2 mL)を加え、20分間室温で攪拌した。樹脂の一部を採取してKaiser testを行いNα-Fmoc基の脱保護を確認した。さらに、保護アミノ酸Boc-Met-OHを用いて同様の操作を行うことで、化合物(b11)を作製した。
化合物(b11)をTFA:トリイソプロピルシラン(TIS):H2O=95:2.5:2.5 (1 mL)の混液中室温で2時間攪拌した。樹脂をろ去した後、ろ液を減圧留去して得られた残渣にジエチルエーテルを加えることで結晶化させた。結晶をろ取し、ジエチルエーテルで洗浄した後、減圧乾燥することで化合物(b12)のTFA塩(9.7 mg, 収率69.0%)を白色結晶として得た。
ESI-MS (M+H)+: m/z 481.2, found: 481.2.
化合物(b5) (1.0 mg, 1.05 μmol)と化合物(b12) (1.58 μmol)を0.16 M ホウ酸緩衝液 pH 11.0 (100 μL)に溶解した後、1 N NaOHでpH 9.0に調整した。室温で2時間攪拌した後、分取用RP-HPLCにより精製し、化合物(b13)(以下、「DO3A-Bn-SCN-MVK(Bzo)」ともいう)のTFA塩(0.8 mg, 収率53.3%)を白色結晶として得た。
ESI-MS (M+H)+: m/z 974.4, found: 974.4.
化合物(b5) (5.5 mg, 5.78 μmol)とメチオニン(1.29 mg, 8.67 μmol)を出発物質として、合成例L4(f)と同様の操作を行うことで、化合物(b14)(以下、「DO3A-Bn-SCN-Met-OH」ともいう)のTFA塩(2.3 mg, 収率36.2%)を白色結晶として得た。
ESI-MS (M+H)+: m/z 643.2, found: .643.2

Cl-Trt(2-Cl)Resin (104.4 mg, 114.8 μmol、渡辺化学工業株式会社)を出発物質として、Fmoc-Lys(Dde)-OH (61.2 mg, 114.8 μmol)及びDIPEA (81 μL, 459.2 μmol)をジクロロメタン(2 mL)中1.5時間反応させた。メタノール及びDIPEAを加えて反応を停止させた。樹脂をDMF次いでジクロロメタンで洗浄した。合成例L4(c)と同様の方法でFmoc-Lys(Bzo)-OHの樹脂への導入量を定量(0.769 mmol/g)した。20% ピペリジン/DMF (2 mL)を加え、室温で20分間攪拌することで、化合物(b15)を作製した。樹脂の一部を採取してkaiser testを行い、Nα-Fmoc基の脱保護を確認した。
化合物(b15) (153.4 mg, 114.8 μmol)を出発物質として、保護アミノ酸を順次Fmoc-Val-OH、Boc-Met-OHと変えて、合成例L4(d)と同様の操作を行うことで、化合物(b16)を得た。
化合物(b16) (117. 9 μmol)を2% ヒドラジン/DMF (2 mL)中室温で1時間攪拌した後、樹脂の一部を採取しKaiser testを行い、反応の終了を確認した。樹脂をDMF次いでジクロロメタンで洗浄した後、減圧乾燥することで化合物(b17)を得た。
化合物(b17)を酢酸:2,2,2-トリフルオロエタノール(TFE):ジクロロメタン=3:1:6 (2 mL)の混液中室温で2時間攪拌した。樹脂をろ去した後、ろ液を減圧留去して得られた残渣にジエチルエーテルを加えることで結晶化させた。結晶をろ取し、ジエチルエーテルで洗浄した後、減圧乾燥することで化合物(b18)の酢酸塩(51.4 mg, 収率93.9 %)を白色結晶として得た。
ESI-MS (M+H)+: m/z 477.2, found: 477.2.
化合物(b18) (6.60 mg, 13.9 μmol)を氷冷化でsat.NaHCO3 (100 μL)に溶解し、N-メトキシカルボニルマレイミド(NMCM, 3.22 mg, 20.8 μmol)を加えた。氷冷下2時間攪拌した後、5質量% クエン酸を加え中和し、クロロホルム (5 mL×3)により抽出した。Na2SO4を加えて乾燥した後、溶媒を減圧留去することで、化合物(b19) (7.26 mg, 収率94.1 %)を白色結晶として得た。
ESI-MS (M+H)+: m/z 557.2, found: 557.2.
化合物(b19) (7.26 mg, 13.1μmol)を4 M HCl/酢酸エチル(1 mL)に溶解し、室温で1時間攪拌した。溶媒を減圧留去し、ヘキサンで共沸することにより、化合物(b20)の塩酸塩(5.92 mg, 収率92.4%)を白色結晶として得た。
ESI-MS (M+H)+: m/z 457.2, found: 457.2.
化合物(b20)の塩酸塩(3.38 mg, 4.74 μmol)をDMF(100 μL)に溶解し、トリエチルアミン (TEA, 6 μL, 43.3 μmol)を加えた。化合物(b5) (3.0 mg, 3.16 μmol)を加え、室温で2時間攪拌した後、H2Oで10倍に希釈し、分取用RP-HPLCにより精製することで化合物(b21)(以下「DO3A-Bn-SCN-MVK(Mal)」ともいう)のTFA塩(1.5 mg, 収率33.2%)を白色結晶として得た。
ESI-MS (M+H)+: m/z 972.5, found: 972.5.
合成例L6:DO3A-Bn-CO-FGK(化合物(b24)(上述の化合物1-3と同じ))、DO3A-Bn-CO-FGK(Boc)(化合物(b25))、DO3A-Bn-CO-FGK(Mal)(化合物(b26) (上述の化合物1-4と同じ))、DO3A-Bn-CO-Phe-OH(化合物(b28))の合成

Cl-Trt(2-Cl)Resin (22.1 mg, 24.3 μmol、渡辺化学工業株式会社)を出発物質として、Fmoc-Lys(Boc)-OH (17.1 mg, 36.5 μmol)及びDIPEA (16.5 μL, 97.2 μmol)をジクロロメタン (1 mL)中1.5時間反応させた。メタノール及びDIPEAを加えて反応を停止させた。樹脂をDMF次いでジクロロメタンで洗浄した。合成例L4(c)と同様の方法でFmoc-Lys(Boc)-OHの樹脂への導入量を定量(0.884 mmol/g)した。20% ピペリジン/DMF (2 mL)を加え、室温で20分間攪拌することで、化合物(b22)を作製した。樹脂の一部を採取してkaiser testを行い、Nα-Fmoc基の脱保護を確認した。
化合物(b22) (12.1 μmol)を出発物質として、保護アミノ酸を順次Fmoc-Gly-OH、Fmoc-Phe-OHと変えて、合成例L4(d)と同様の操作を行うことで、化合物(b23)を得た。
化合物(b23) (12.1 μmol)に対して、化合物(b7) (15.7 mg, 24.2 μmol)、DIC (3.7 μL, 24.2 μmol)、HOAt (3.29 mg, 24.2 μmol)をDMF中、室温で一晩攪拌した。樹脂の一部を採取しKaiser testを行うことで、縮合反応の終了を確認した後、TFA:TIS:H2O=95:2.5:2.5 (1 mL)の混液中室温で2時間攪拌した。樹脂をろ去した後、ろ液を減圧留去してえられた残渣にジエチルエーテルを加えることで結晶化した。結晶をろ取し、ジエチルエーテルで洗浄した後、減圧乾燥することで化合物(b24) (以下、「DO3A-Bn-CO-FGK」ともいう)のTFA塩(9.0 mg, 収率53.7%)を白色結晶として得た。
ESI-MS (M+H)+: m/z 813.4, found: 813.4.
化合物(b24) (2.17 μmol)をsat.NaHCO3 (100 μL)に溶解した後、ジオキサン(100 μL)に溶解した(Boc)2O (7.1 mg, 3.26 μmol)を加え室温で2時間激しく攪拌した。ジオキサンを減圧留去した後、クロロホルム(3 mL×3)で洗浄した。水層を分取用RP-HPLCにより精製することで化合物(b25)(以下、「DO3A-Bn-CO-FGK(Boc)」ともいう)のTFA塩(0.8 mg, 収率26.9%)を白色結晶として得た。
ESI-MS (M+H)+: m/z 913.5, found: 913.5.
化合物(b24) (2.17 μmol)をsat.NaHCO3 (100 μL)に溶解した後、氷冷しNMCM (0.5 mg, 3.26 μmol)を加えた。氷冷下2時間攪拌した後、分取用RP-HPLCにより精製することで化合物(b26)(以下、「DO3A-Bn-CO-FGK(mal)」ともいう)のTFA塩(1.3 mg, 収率44.4%)を白色結晶として得た。
ESI-MS (M+H)+: m/z 893.4, found: 893.4.
Cl-Trt(2-Cl)Resin (5 mg, 5.50 μmol、渡辺化学工業株式会社)を出発物質として、Fmoc-Phe-OH (6.01 μmol)及びDIPEA (3.73 μL, 21.9 μmol)をジクロロメタン (0.5 mL)中1.5時間反応させた。メタノール及びDIPEAを加えて反応を停止させた。樹脂をDMF次いでジクロロメタンで洗浄した。合成例L4(c)と同様の方法でFmoc-A.A.-OHの樹脂への導入量を定量した(0.917 mmol/g)。20% ピペリジン/DMF (2 mL)を加え、室温で20分間攪拌することで、化合物(b27)を作製した。樹脂の一部を採取してkaiser testを行い、Nα-Fmoc基の脱保護を確認した。
化合物(b27) (5.5 μmol)を出発物質として、合成例L6(c)と同様の操作をすることで化合物(b28) (以下、「DO3A-Bn-CO-Phe-OH」ともいう)のTFA塩(4.5 mg, 収率59.1%)を白色結晶として得た。
ESI-MS (M+H)+: m/z 628.3, found: 628.3.
合成例L7:CDOTA-Bn-CO-FGK(化合物(b29))、CDOTA-Bn-CO-FGK(Boc)(化合物(b30))、CDOTA-Bn-CO-FGK(Mal)(化合物(b31))の合成

p-COOH-Bn-DOTA(tBu)4(52.4 μmol)を出発物質として、合成例L6(c)と同様の操作をすることで化合物(b29)(以下、「CDOTA-Bn-CO-FGK」ともいう)のTFA塩(2.4 mg, 収率1.3%)を白色結晶として得た。
ESI-MS ([M+K]-H)-: m/z 907.36, found: 907.31.
化合物(b29) (5.75 μmol)を出発物質として、合成例L6(d)と同様の操作をすることで化合物(b30) (以下、「CDOTA-Bn-CO-FGK(Boc)」ともいう)のTFA塩(0.4 mg, 収率71.8%)を白色結晶として得た。
ESI-MS ([M+K]-H)-: m/z 1007.41, found: 1007.31.
化合物(b29) (0.694 μmol)を出発物質として、合成例L6(e)と同様の操作をすることで化合物(b31) (以下、「CDOTA-Bn-CO-FGK(mal)」ともいう)のTFA塩(0.6 mg, 収率61.4%)を白色結晶として得た。
ESI-MS (M-H)-: m/z 949.39, found: 949.43.
合成例L8:化合物(a23)の合成

化合物(a8) (913 mg、1.96 mmol)をTHF (20 mL)に溶解し、isobutylaldehyde (357 μL、3.91 mmol) 、sodium triacetoxyborohidride (498 mg、2.35 mmol)を加え、Ar雰囲気下、室温で一晩拌した。Isobutylaldehyde (179 μL、1.96 mmol) 、sodium triacetoxyborohidride (249 mg、1.18 mmol)を加えた後、さらに室温で2時間攪拌した。反応液を氷冷し、水を加えた後、THFを減圧留去して得られた水溶液からクロロホルムで三回抽出した。有機層に硫酸ナトリウムを加えて乾燥した後、溶媒を減圧留去して得られた残渣をクロロホルムとメタノールを用いたフラッシュクロマトシステムにより生成を行うことで化合物(a19) (403 mg、829 μmol, 42.4%)を白色結晶として得た。
1H NMR (CDCl3): δ 0.92-0.95 (6H, m, CH 3 ), 1.68-1.71 (1H, m, CH), 2.40-2.44 (2H, m, CH 2 ), 2.82-3.30 (8H, overlapped, CH 2 ), 3.61-3.74 (2H, m, CH 2 ), 4.58-4.62 (1H, m, CH), 6.48 (1H, s, NH), 6.66 (1H, s, NH), 6.96-6.98 (2H, d, CH 2 ), 7.56-7.58 (2H, d, CH 2 ), 8.00 (1H, s, NH). ESI-MS (M+H)+: m/z 487.12, found: 487.18.
化合物(a19) (403 mg、829 μmol)をTHF 2 mLに懸濁し、溶液を氷冷した後、アルゴン雰囲気下、0.95 Mのボラン-THF錯体/THF溶液13 mLを緩徐に加え、1時間攪拌した後、22時間還流した。溶液を氷冷し、メタノール13 mLを加えた後、1時間攪拌し、溶媒を減圧留去した。その後、残渣に再度メタノール13 mLを加え、溶媒を減圧留去した。残渣に濃塩酸13 mLを加え、24 時間室温で攪拌した後、1 時間還流した。溶液を氷冷し、12.5 N水酸化ナトリウム水溶液を加えて塩基性とした後、クロロホルムで抽出した。有機層に硫酸ナトリウムを加えて乾燥した後、溶媒を減圧留去して得られた残渣をクロロホルム:メタノール:25質量%アンモニア水(10:1:0.1)を溶出溶媒とするフラッシュクロマトシステムにより精製を行うことで化合物(a20)(275 mg、619 μmol、収率74.7%)を黄色油状物として得た。
1H NMR (CDCl3): δ 0.89-0.92 (6H, m, CH 3 ), 1.80-1.84 (1H, m, CH), 2.06-2.89 (19H, overlapped, CH, CH 2 ), 6.92-6.94 (2H, d, CH 2 ), 7.58-7.60 (2H, d, CH 2 ).
化合物(a20) (275 mg、619 μmol)をアセトニトリル 2 mLに懸濁し、炭酸ナトリウム(428 mg、3.10 mmol)を加えた。溶液を氷冷し、アルゴン雰囲気下、ブロモ酢酸tert-ブチル(272 μL、1.86 mmol)を滴下した。滴下完了後、室温で24時間攪拌し、懸濁液をろ過した後、ろ液から溶媒を減圧留去した。残渣をクロロホルムとメタノールを用いたフラッシュクロマトシステムにより生成を行うことで化合物(a21) (286 mg、364 μmol、58.8%)を黄色油状物として得た。
1H NMR (CDCl3): δ 0.86-0.89 (6H, m, CH 3 ), 1.41-1.50 (27H, m, tBu), 1.86-3.90 (26H, overlapped, CH, CH 2 ), 6.99-7.01 (2H, d, CH 2 ), 7.59-7.61 (2H, d, CH 2 ). ESI-MS (M+H)+: m/z 787.39, found: 787.45.
化合物(a21) (286 mg、364 μmol)をDMF 3.0 mLに懸濁し、Pd(OAc)2(16.3 mg、0.0728 mmol)、1,2-ビス(ジフェニルホスフィノ)エタン(58.0 mg、0.146 mmol)、Et3N(156 μL、1.12 mmol)、ベンジルアルコール(753 μL、7.28 mmol)を加え、一酸化炭素雰囲気下、一晩還流した。反応後、溶媒を減圧留去して得られた残渣をクロロホルムとメタノールを用いたフラッシュクロマトシステムにより精製することで化合物(a22)(73.8 mg、92.8 μmol、収率25.5%)を黄色油状物として得た。
ESI-MS (M+H)+: m/z 795.53, found: 795.40.
化合物(a22) (73.8 mg、92.8 μmol) をメタノール1.5 mLに溶解した後、10質量% Pd/Cを150 mg加えて水素雰囲気下、室温で5時間攪拌した。反応溶液をろ過後、溶媒を減圧留去することで化合物(a23)(31.9 mg、45.3 μmol、収率48.8%)を白色結晶として得た。
ESI-MS (M+H)+: m/z 705.48, found: 705.40.

化合物(a23) (23.7 mg、33.6 μmol) をDMF 0.9 mLに溶解した後、2,3,5,6-テトラフルオロフェノール (8.4 mg、50.6 μmol)、トリエチルアミン (9.3 μL、66.9 μmol)を加えた。溶液を氷冷し、EDC (9.6 mg、50.6 μmol)を加えた後、室温に戻し、4時間攪拌した。溶媒を減圧留去した後、残渣を酢酸エチルに溶解し、飽和塩化アンモニウム水溶液で三回洗浄した。有機層に硫酸ナトリウムを加えて乾燥した後、溶媒を減圧留去することで化合物(a24)(18.3 mg、23.2 μmol、収率68.9%)を黄色油状物として得た。
Fmoc固相合成法を用いてペプチドを伸長した樹脂(64.0 mg、57.6 μmol)、N,N-ジイソプロピルエチルアミン(23.6 μL、135 μmol)、化合物(a24) (18.3 mg、23.2 μmol)をDMFに溶解し、室温で16時間緩やかに攪拌した。反応終了後DMF及びCH2Cl2を用いて樹脂を洗浄した。
得られた樹脂(a25)を酢酸: 2,2,2-トリフルオロエタノール: ジクロロメタン= 3 :1 : 6を組成とする溶液に懸濁し2時間緩やかに攪拌した。反応終了後、ろ過にて樹脂を除去し、ろ液を減圧留去し、さらにトルエンで三回共沸した。残渣をHPLCにてImtakt Cadenza 5CD-C18 150×20 mmを用い移動相にはA相に0.1%TFA/MilliQ、B相に0.1% TFA/MeCNを使用し、0-35分でA相95%、B相5%からA相50%、B相50%まで変化させ、35-45分でA相50%、B相50%からA相0%、B相100%まで変化させる直線gradient法により流速5 mL/minで精製(保持時間 34.7分)することで目的とする化合物(a26)(以下、「CDO3AiBu-FGK(Boc)」ともいう) (1.0 mg、0.70 μmol、収率2.1%)を得た。
ESI-MS (M+H)+: m/z 969.53, found: 969.51.
Fmoc固相合成法を用いてペプチドを伸長した樹脂(48.3 mg、43.4 μmol)、N,N-ジイソプロピルエチルアミン(17.8 μL、102 μmol)、化合物(a24) (18.3 mg、17.5 μmol)を用いて作製した上記と同様の方法で作製した樹脂(a25)をトリフルオロ酢酸: トリイソプロピルシラン: 水= 95 : 2.5 : 2.5を組成とする溶液に懸濁し2時間緩やかに攪拌した。反応終了後、ろ過にて樹脂を除去し、ろ液を減圧留去し、さらにトルエンで三回共沸した。残渣を水に溶解し、ジエチルエーテルで三回洗浄して得られた水溶液を凍結乾燥させて得られた白色結晶をHPLCにてImtakt Cadenza 5CD-C18 150×20 mmを用い移動相にはA相に0.1%TFA/MilliQ、B相に0.1% TFA/MeCNを使用し、0-35分でA相95%、B相5%からA相50%、B相50%まで変化させ、35-45分でA相50%、B相50%からA相0%、B相100%まで変化させる直線gradient法により流速5 mL/minで精製(保持時間 24.8分)することで目的とする化合物(a27)(以下、「CDO3AiBu-FGK」ともいう)(1.8 mg、1.25 μmol、収率6.4%)を得た。ESI-MS (M+H)+: m/z 869.48, found: 869.38.
化合物 (a27)(1.8 mg、2.6 μmol)を飽和炭酸水素ナトリウム水溶液150 μLに溶解し、氷冷下N-メトキシカルボニルマレイミド(1.0 mg、 6.5 μmol)を加え、氷冷下で二時間攪拌した。反応終了後、5質量%クエン酸水溶液で酸性に調整した。さらにHPLCにてImtakt Cadenza 5CD-C18 150×20 mmを用い移動相にはA相に0.1%TFA/MilliQ、B相に0.1% TFA/MeCNを使用し、5分までA相 100%に保った後に、5-35分でA相 100%、B相 0%からA相 40%、B相 60%まで変化させ、35-50分でA相 40%、B相 60%からA相 0%、B相 100%まで変化させる直線gradient法により流速5 mL/minで精製(保持時間 30.7分)することで目的とする化合物(a28) (以下、「CDO3AiBu-FGK(Mal)」ともいう)(0.8 mg、0.57 μmol、収率45.5%)を得た。
ESI-MS (M+H)+: m/z 949.47, found: 949.36.
化合物 (a23)(2.7 mg、3.83 μmol)をDMF(0.3 mL)に溶解し、H-Phe-OtBu・HCl(1.5 mg、5.75 μmol)、N,N-ジイソプロピルエチルアミン(2.9 μL、17.2 μmol)、(1-シアノ-2-エトキシ-2-オキソエチリデンアミノオキシ)ジメチルアミノ-モルホリノ-カルベニウムヘキサフルオロリン酸塩(4.9 mg、11.5 μmol)を添加し、室温で一晩攪拌した。溶媒を減圧留去した後、残渣を酢酸エチルに再溶解し、5質量%炭酸水素ナトリウム水溶液で三回、5質量%クエン酸水溶液で三回洗浄した。有機層に硫酸ナトリウムを加えて乾燥した後、溶媒を減圧留去することで得られた残渣に10質量%アニソール/TFA溶液(2.0 mL)を加えて、室温で2時間攪拌した。溶媒を減圧留去した後、トルエンで三回共沸した。残渣を水に溶解し、ジエチルエーテルで三回洗浄して得られた水溶液から溶媒を減圧留去して得られた白色結晶をHPLCにてImtakt Cadenza 5CD-C18 150×20 mmを用い移動相にはA相に0.1%TFA/MilliQ、B相に0.1% TFA/MeCNを使用し、0-30分でA相100%、B相0%からA相40%、B相60%まで変化させ、30-35分でA相40%、B相60%からA相0%、B相100%まで変化させる直線gradient法により流速5 mL/minで精製(保持時間 25.2分)することで目的とする化合物(a29)(以下、「CDO3AiBu-Phe」ともいう) (0.1 mg、8.8 μmol、収率2.3%)を得た。
ESI-MS (M+H)+: m/z 684.36ound: 684.27
合成例F1: Fab(Rabbit serum IgG由来)及びIT-Fab(Rabbit serum IgG由来)の作製
(Fab(Rabbit serum IgG由来)の作製)
Rabbit serum IgG (9 mg)を10 mM Na2EDTA及び20 mM cysteineを含む20 mMリン酸緩衝液(pH 7.0) 1.5 mLに溶解し、immobilized papain 50% slurry (Thermo Scientific, Yokohama, Japan) 500 μLを加え、37°Cで42時間インキュベートした。反応終了後、10 mMトリス塩酸緩衝液(pH 7.5)を2 mL加え、0.45 μmのフィルターでろ過し、ろ液を回収した。ろ液を10 kDaの限外ろ過膜を用いて20 mMリン酸緩衝液(pH 7.0)に置換し、1 mLに濃縮した。その後、protein Aカラムを用いて精製を行い、Fabを得た。得られたFabは、0.1 Mリン酸緩衝液(pH 6.8)を溶出溶媒とした流速1.0 mL/minで溶出するSE-HPLCにより生成を確認し、A280を測定することで濃度を算出した。
十分に脱気した2 mM EDTA含有0.16 Mホウ酸緩衝液(pH 8.0)を用いてFab溶液(100 μL, 5 mg/mL)を調製し、同じ緩衝液に溶解した2-イミノチオラン(2-IT) (5 μL, 2.88 mg/mL)を1 μLずつ攪拌しながら加え、37 ℃で静かに30分間攪拌した。反応後、十分脱気した2 mM EDTA含有0.1 Mリン酸緩衝液(pH 6.0)で平衡化したSephadex G-50 Fineを用いるスピンカラム法[Analytical Biochemistry, 1984, 142, 68-78]により反応溶液中の過剰の2-ITを除去し、IT-Fab(Rabbit serum IgG由来)溶液を得た。Fab一分子あたりに導入されたチオール基の数は2,2’-ジピリジルジスルフィドを用いて測定した[Archives of Biochemistry and Biophysics, 1967, 119, 41-49]。
Rabbit serum IgG (9 mg)を、抗c-kit IgG (9 mg)とした以外は、合成例F1と同様の方法で、Fab(抗c-kit IgG由来)及びIT-Fab(抗c-kit IgG由来)を作製した。
合成例:CDO3AEt-FGK-Fab(Rabbit serum IgG由来)、CDO3AEt-FGK-Fab(抗c-kit IgG由来)、DO3A-EDA-Fab(Rabbit serum IgG由来)、CDOTA-Bn-CO-FGK-Fab(抗c-kit IgG由来)、DO3A-Bn-SCN-MVK-Fab(Rabbit serum IgG由来)、DO3A-Bn-CO-FGK-Fab(Rabbit serum IgG由来)、CDO3AiBu-FGK-Fab(Rabbit serum IgG由来) 及びCDO3AiBu-FGK-Fab(抗c-kit IgG由来)の作製
IT-Fab(Rabbit serum IgG由来)溶液(100 μL)にH2Oに溶解したCDO3AEt-FGK(Mal) (5 μL, チオール基に対して20等量)を1 μLずつ加え、37 ℃で2時間反応した。次いで、0.1 Mリン酸緩衝液(pH 6.0)を用いてヨードアセトアミド溶液を調製し、これを残存チオール基に対して500等量加えた後、37 ℃で1時間反応を行い、未反応のチオール基をアルキル化した。その後、0.25 M酢酸緩衝液(pH 5.5)で平衡化したSephadex G-50 Fineを用いるスピンカラム法で精製し、CDO3AEt-FGK-Fab(Rabbit serum IgG由来)溶液を得た。Fab一分子あたりに導入されたCDO3AEt-FGK(Mal)由来の単位数は、ヨードアセトアミドを加える前にDPSを用いて測定した[Archives of Biochemistry and Biophysics, 1967, 119, 41-49]チオール数を先に求めたチオール数を差し引くことにより求めた。
CDO3AEt-FGK(Mal)を、DO3A-EDA (Mal)、DO3A-Bn-SCN-MVK(Mal)、DO3A-Bn-CO-FGK(Mal)、及びCDO3AiBu-FGK(Mal)にそれぞれ変更し、その他は上記と同様の方法で、DO3A-EDA-Fab(Rabbit serum IgG由来)溶液、DO3A-Bn-SCN-MVK-Fab(Rabbit serum IgG由来)溶液、DO3A-Bn-CO-FGK-Fab(Rabbit serum IgG由来)溶液、及びCDO3AiBu-FGK-Fab(Rabbit serum IgG由来)溶液を得た。
IT-Fab(Rabbit serum IgG由来)溶液を、IT-Fab(抗c-kit IgG由来)溶液に変更し、その他は上記と同様の方法で、CDO3AEt-FGK-Fab(抗c-kit IgG由来)溶液を得た。
IT-Fab(Rabbit serum IgG由来)溶液を、IT-Fab(抗c-kit IgG由来)溶液に変更し、CDO3AEt-FGK(Mal)を、CDOTA-Bn-CO-FGK(Mal)に変更し、その他は上記と同様の方法で、CDOTA-Bn-CO-FGK-Fab(抗c-kit IgG由来)溶液を得た。
IT-Fab(Rabbit serum IgG由来)溶液を、IT-Fab(抗c-kit IgG由来)溶液に変更し、CDO3AEt-FGK(Mal)を、CDO3AiBu-FGK(Mal)に変更し、その他は上記と同様の方法で、CDO3AiBu-FGK-Fab(抗c-kit IgG由来)溶液を得た。
0.16 Mホウ酸緩衝液(pH 8.5)を用いてFab溶液(100 μL, 5.0 mg/mL)を調整し、同じ緩衝液に溶解したDOTA-Bn-SCN (Macrocyclics, USA) (14 μL, 10mg/mL)を加え、4℃で一晩静置した。反応後、十分に脱気した0.25 M酢酸緩衝液 (pH 5.5)で平衡化したSephadex G-50 Fineを用いるスピンカラム法により過剰のDOTA-Bn-SCNを除去し、DOTA-Bn-SCN-Fab(抗c-kit IgG由来)を得た。
なお、上述のCDO3AEt-FGK-Fab、DO3A-Bn-CO-FGK-Fab、CDOTA-Bn-CO-FGK-Fab、CDO3AiBu-FGK-Fabの化学構造は上述のとおりである。また、DO3A-EDA-Fab、DO3A-Bn-SCN-MVK-Fab、及びDOTA-Bn-SCN-Fabの化学構造は以下のとおりである。

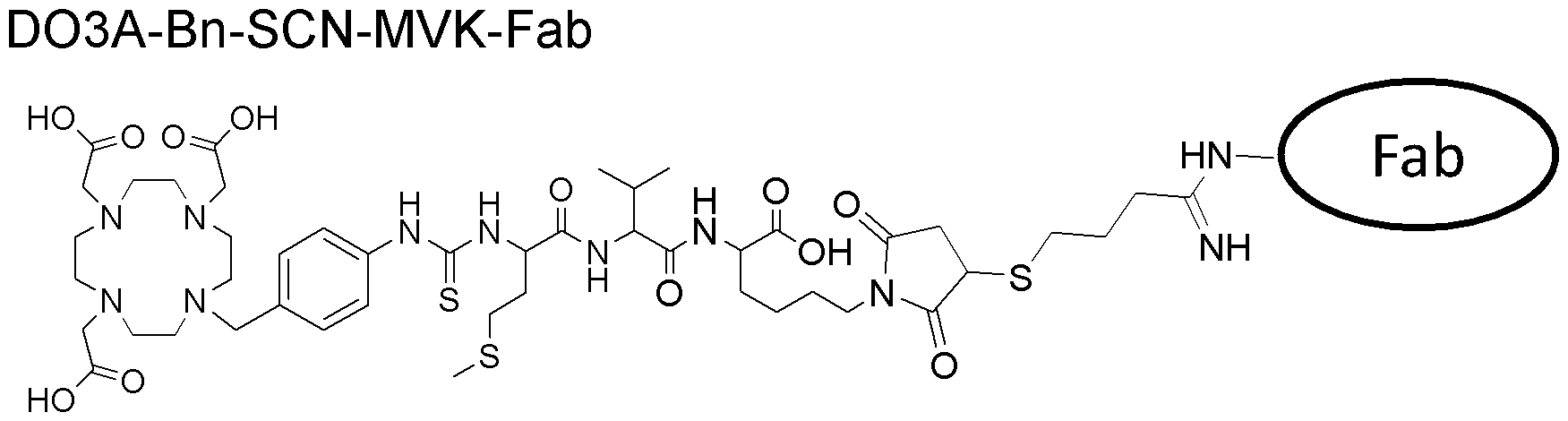

合成例:111In-CDO3AEt-FGK-Fab(Rabbit serum IgG由来)、111In- CDO3AEt-FGK-Fab(抗c-kit IgG由来)、111In-DO3A-EDA-Fab(Rabbit serum IgG由来)、111In-CDOTA-Bn-CO-FGK-Fab(抗c-kit IgG由来)、111In-DO3A-Bn-SCN-MVK-Fab(Rabbit serum IgG由来)、111In-DO3A-Bn-CO-FGK-Fab(Rabbit serum IgG由来)、111In-CDO3AiBu-FGK-Fab(Rabbit serum IgG由来)、111In-CDO3AiBu-FGK-Fab(抗c-kit IgG由来)、111In-DOTA-Bn-SCN-Fab(抗c-kit IgG由来)、111In-CDO3AEt-FGK(Boc)、111In-CDOTA-Bn-CO-FGK(Boc)、111In-DO3A-Bn-SCN-MVK(Bzo)、及び111In-DO3A-Bn-CO-FGK(Boc)の作製
111InCl3(45 μL)を1 M酢酸緩衝液(pH 5.5, 5 μL)に混和し、室温で5分間静置した。CDO3AEt-FGK-Fab(Rabbit serum IgG由来)溶液(30 μL)を混合した後、40 oCで90分間インキュベートした。最終濃度が10 mMとなるようにDTPAを加えた後、室温で18時間静置した。0.1 M D-PBS (pH 7.4)で平衡化したSephadex G-50 Fineを用いるスピンカラム法で精製することで、111In-CDO3AEt-FGK-Fabを作製した。CDO3AEt-FGK-Fab(Rabbit serum IgG由来)溶液を、CDO3AEt-FGK-Fab(抗c-kit IgG由来)溶液、DO3A- EDA -Fab(Rabbit serum IgG由来)溶液、CDOTA-Bn-CO-FGK-Fab(抗c-kit IgG由来)溶液、DO3A-Bn-SCN-MVK-Fab溶液(Rabbit serum IgG由来)、DO3A-Bn-CO-FGK-Fab(Rabbit serum IgG由来)溶液、CDO3AiBu-FGK-Fab(Rabbit serum IgG由来)溶液、DOTA-Bn-SCN-Fab(抗c-kit IgG由来)溶液、CDO3AEt-FGK(Boc)溶液、CDOTA-Bn-CO-FGK(Boc)溶液、DO3A-Bn-SCN-MVK(Bzo)溶液、及びDO3A-Bn-CO-FGK(Boc)溶液にそれぞれ変更し、その他は上記と同様の方法で、111In-CDO3AEt-FGK-Fab(抗c-kit IgG由来)、111In-DO3A-EDA-Fab(Rabbit serum IgG由来)、111In-CDOTA-Bn-CO-FGK-Fab(抗c-kit IgG由来)、111In-DO3A-Bn-SCN-MVK-Fab(Rabbit serum IgG由来)、111In-DO3A-Bn-CO-FGK-Fab(Rabbit serum IgG由来)、111In-CDO3AiBu-FGK-Fab(Rabbit serum IgG由来)、111In-DOTA-Bn-SCN-Fab(抗c-kit IgG由来)、111In-CDO3AEt-FGK(Boc)、111In-CDOTA-Bn-CO-FGK(Boc)、111In-DO3A-Bn-SCN-MVK(Bzo)、及び111In-DO3A-Bn-CO-FGK(Boc)、を作製した。
〔BBMVsとのインキュベート試験〕
(腎刷子縁膜小胞)
腎刷子縁膜小胞(BBMVs)はWistar系雄性ラット (200-250 g) の腎臓からHoriらの方法に従って作製した[Biochemical Pharmacology 45: 1763-1768, 1993]。全ての操作は氷上で行った。皮質に重量で4-5倍の300 mM mannitol、及び5 mM EGTA を含む12 mM トリス-塩酸緩衝液 (pH 7.1) を加え、ポリトロンホモジナイザー(PT-3100 Kinematica GmgH Littau, Switzerland)で2分間ホモジナイズし,同じ緩衝液で希釈することで10%ホモジネートとした。次いで、蒸留水で2倍に希釈した後,最終濃度が10 mM となるよう、1.0 Mに調整したMgCl2水溶液を加え、15分間放置した。その後、ホモジネートを1,900 gで遠心し、上清をさらに30分間24,000 gで遠心した。沈殿を皮質重量の20倍に相当する150 mM mannitol、及び2.5 mM EGTA を含む6 mM トリス-塩酸緩衝液 (pH 7.1) に再懸濁し、テフロン(登録商標)ホモジナイザー (1,000 rpm, 10 strokes) によりホモジナイズした。次いで、最終濃度が10 mMとなるように1.0 M MgCl2水溶液を加え、懸濁液を15分間放置し、その後、ホモジネートを1,900 gで遠心し、上清をさらに30分間24,000 gで遠心した。得られた沈殿を皮質重量の10倍に相当する0.1 M リン酸緩衝液 (pH 7.0) に懸濁し、再度テフロン(登録商標)ホモジナイザー (1,000 rpm, 10 strokes) によりホモジナイズした。次いで。ホモジネートを30分間24,000 gで遠心し、BBMVsを沈殿として得た。次いで、BBMVsの沈殿を0.1 Mリン酸緩衝液 (pH 7.0) に再懸濁し、0.4 x 19 mmの針に10回通すことで小胞の大きさを一定にした。インキュベート実験にはタンパク質濃度が10 mg/mLとなるように希釈して用いた。調製したBBMVsについて、リソソームマーカー酵素であるβ-galactosidase活性をp-nitrophenyl-β-D-galacto- pyranosideを用いて測定することにより、リソソーム酵素の混入を評価した[Plant Physiology 55: 94-98, 1975]。また、γ-glutamyl transferase、aminopeptidaseの活性を、Glossmannら[FEBS Letters 19: 340-344, 1972]、Kramersら[European Journal of Biochemistry 99: 345-351, 1979]の方法に従い、L-γ-glutamyl- p-nitroanilide、L-leucine-p-nitroanilideを用いて測定した。
BBMVsと111In標識低分子モデル基質のインキュベート実験は以下の方法で行った。タンパク質濃度を10 mg/mLとなるように調製したBBMVs (10 μL)を、37 ℃で10分間プレインキュベートした。その後、逆相HPLCによって過剰の配位子を除去してPBSに溶解した111In-CDO3AEt-FGK(Boc)溶液(10 μL)を加え、37 ℃で二時間インキュベートした。BBMVs溶液に対してエタノール濃度が60%となるようにエタノールを加え、5000rpmで10分間遠心した。上清を回収後、沈殿に60%エタノールを加え再度同様の方法で遠心した後に、上清を回収した。得られた上清を、HPLCにてImtakt Unison US-C18 150×4.6 mmを用い移動相にはA相に0.1%TFA/MilliQ、B相に0.1% TFA/MeCNを使用し、0-30分でA相 100%、B相 0%からA相 55%、B相 45%まで変化させる直線gradient法により流速1 mL/minで分析を行った。
図1は、111In-CDO3AEt-FGK(Boc)とBBMVsのインキュベートの実験結果を示す。
111In-CDO3AEt-FGK(Boc)溶液を、111In-CDOTA-Bn-CO-FGK(Boc)溶液、111In-DO3A-Bn-CO-FGK(Boc)溶液、111In-DO3A-Bn-SCN-MVK(Bzo)溶液、111In-CDO3AiBu-FGK(Boc)溶液に代えて同様のインキュベート実験及びBBMVsを使用せずコントロール実験を行った。
図2は、111In-CDOTA-Bn-CO-FGK(Boc)溶液とBBMVsのインキュベートの実験結果を示す。
図3は、111In-DO3A-Bn-SCN-MVK(Bzo)とBBMVsのインキュベートの実験結果を示す。
図4は、111In-DO3A-Bn-CO-FGK(Boc)とBBMVsのインキュベートの実験結果を示す。
図5は、111In-CDO3AiBu-FGK(Boc)とBBMVsのインキュベートの実験結果を示す。
上記の実験結果から、連結基として、本願発明の化合物と同様に、ベンジルアミド構造を有する、111In-CDO3AEt-FGK(Boc)、111In-CDOTA-Bn-CO-FGK(Boc)、111In-DO3A-Bn-CO-FGK(Boc)、111In-CDO3AiBu-FGK(Boc)では、BBMVsのインキュベートにより、キレート配位子部位が遊離されることが理解できる。これに対して、連結基としてチオウレア構造を有する化合物111In-DO3A-Bn-SCN-MVK(Bzo)では、遊離物が観測されない。
PBSに溶解した111In-CDO3AEt-FGK-Fab(Rabbit serum IgG由来) (10 μL)をマウス血漿(90 μL)に加え、37 ℃でインキュベートした。1、3、6、24時間後に溶液の一部を採取しメタノール:10質量% 酢酸アンモニウム水溶液=3:2を展開溶媒とするRP-TLCにより分析することで、未変化体(111In-CDO3AEt-FGK-Fab)の放射活性の割合を算出し、表1に示した。

111In-CDO3AEt-FGK-Fab(Rabbit serum IgG由来)を、111In-CDOTA-Bn-CO-FGK-Fab(抗c-kit IgG由来)に変更し、その他は上記と同様の方法で、マウス血漿中安定性試験を行った。インキュベート2時間後、未変化体(111In-CDOTA- Bn-CO-FGK-Fab)の放射活性の割合は、95.2±0.3%であった。

実施例及び比較例で作製した金属錯体化合物をD-PBS(-) (pH 7.4)を用いて希釈した。6週齢のddY系雄性マウス尾静注により、未修飾Fab濃度を5 μg/100 μLに調整した111In-CDO3AEt-FGK-Fab(Rabbit serum IgG由来)溶液(0.3 μCi/100 μL/匹)を投与した。投与後10、30分、1、3、6、24時間後に各群3匹のマウスを屠殺し、関心臓器を採取、重量を測定後、オートウェルガンマシステムにより放射活性を測定した。また、6、24時間後までの糞尿をそれぞれ採取し、放射活性を測定した。同様の方法にて111In-CDO3AEt-FGK-Fab(抗c-kit IgG由来)溶液を用いて、6週齢のddY系雄性マウス尾静注により、関心臓器、糞尿について、放射活性を測定した。対照化合物として同様の方法にて調製した111In-DO3A-EDA-Fab(Rabbit serum IgG由来)を用いた。
表3は、111In-CDO3AEt-FGK-Fab(Rabbit serum IgG由来)によるマウス体内の放射活性の測定結果を示す。
表4は、111In-CDO3AEt-FGK-Fab(抗c-kit IgG由来)によるマウス体内の放射活性の測定結果を示す。
表5は、111In-DO3A-EDA-Fab(Rabbit serum IgG由来)によるマウス体内の放射活性の測定結果を示す。
上記と同様の方法にて111In-CDOTA-Bn-CO-FGK-Fab(抗c-kit IgG由来)溶液、111In-DO3A-Bn-SCN-MVK-Fab(Rabbit serum IgG由来)溶液、111In-DO3A-Bn-CO-FGK-Fab(Rabbit serum IgG由来)溶液、111In-CDO3AiBu-FGK-Fab(Rabbit serum IgG由来)溶液、111In-CDO3AiBu-FGK-Fab(抗c-kit IgG由来)溶液、111In-DOTA-Bn-SCN-Fab(抗c-kit IgG由来)溶液を用いて、6週齢のddY系雄性マウス尾静注により、関心臓器、糞尿について、放射活性を測定した。
表6は、111In-CDOTA-Bn-CO-FGK-Fab(抗c-kit IgG由来)によるマウス体内の放射活性の測定結果を示す。
表7は、111In-DO3A-Bn-SCN-MVK-Fab(Rabbit serum IgG由来)によるマウス体内の放射活性の測定結果を示す。
表8は、111In-DO3A-Bn-CO-FGK-Fab(Rabbit serum IgG由来)によるマウス体内の放射活性の測定結果を示す。
表9は、111In-CDO3AiBu-FGK-Fab(Rabbit serum IgG由来)によるマウス体内の放射活性の測定結果を示す。
表10は、111In-CDO3AiBu-FGK-Fab(抗c-kit IgG由来)によるマウス体内の放射活性の測定結果を示す。
表11は、111In-DOTA-Bn-SCN-Fab(抗c-kit IgG由来)によるマウス体内の放射活性の測定結果を示す。
図6A~Cに、111In-CDO3AEt-FGK-Fabと111In-DO3A-EDA-Fabとの結果の比較を示した。
これら本発明の化合物である111In-CDO3AEt-FGK-Fabと比較化合物である111In-DO3A-EDA-Fabとの結果から、111In-CDO3AEt-FGK-Fabは、111In-DO3A-EDA-Fabと同程度の血中濃度を示しながら、腎臓への蓄積が抑制され、低い腎臓-血液比が得られることがわかる。
上記の実験結果から、連結基として、ベンジルアミド構造を有する本願発明の化合物111In-CDO3AEt-FGK-Fab(Rabbit serum IgG由来)、111In-CDO3AEt-FGK-Fab(抗c-kit IgG由来)、111In-CDOTA-Bn-CO-FGK-Fab(抗c-kit IgG由来)、111In-DO3A-Bn-CO-FGK-Fab(Rabbit serum IgG由来)では、腎臓での放射活性が顕著に抑えられているのに対して、連結基としてチオウレア構造を有する比較化合物111In-DO3A-Bn-SCN-MVK-Fab、及びエチレン構造を有する111In-DO3A-EDA-Fab(Rabbit serum IgG由来)では、腎臓での放射活性が高い値を示す。上記結果及びBBMVsとのインキュベート試験の試験結果から、ベンジルアミド構造を有する本願発明の化合物では、酵素による代謝が行われ、腎臓に取り込まれる前に、放射性金属部位が遊離されたものと考えられ、一方で、チオウレア構造を有する比較化合物では、酵素認識されず、代謝物の遊離がなかったと考えられる。
図7A~Cに、111In-CDO3AiBu-FGK-Fab(抗c-kit IgG由来)、111In-DOTA-Bn-SCN-Fab(抗c-kit IgG由来)、及び111In-CDO3AEt-FGK-Fab(抗c-kit IgG由来)の結果の比較を示した。
これらの結果から、111In-CDO3AiBu-FGK-Fabは、111In-CDO3AEt-FGK-Fabよりも、更に腎臓への蓄積が抑制され、より低い腎臓-血液比が得られることがわかる。









111In-CDO3AEt-FGK-Fab(Rabbit serum IgG由来)をD-PBS (-)で希釈した。マウス尾静脈より、Fab濃度を5 μg/100 μLに調整した111In-CDO3AEt-FGK-Fab(Rabbit serum IgG由来)溶液(4 μCi/100 μL/匹)を投与し、24時間後までに集積した尿を0.45 μmのフィルターで濾過した後、SE-HPLCにて化学形を分析した。また、回収した尿に2倍量のEtOHを加えることでタンパクを沈殿させ、15,000 gで5分間遠心した後に上清を回収した。上清を回収した後に66% EtOH溶液100 μLを用いてペレットを洗浄し、再度遠心して上清を回収する作業を2回行い、放射活性の上清への回収率を算出した。その後、EtOH濃度が15%以下になるよう上清をD-PBS (-)で希釈し、RP-HPLCによる分析を行った。
図8は、111In-CDO3AEt-FGK-Fab(Rabbit serum IgG由来)をマウスに投与後24時間までに排泄された尿中放射活性の化学形の分析結果を示す。以上、尿中の放射活性の分析により図8Aに示すSE-HPLCによる分析で、大部分の放射活性が低分子画分で排泄され、図8Bに示すRP-HPLCの結果から、111In-CDO3AEt-FGK-Fabでは、低分子画分における主たる放射活性111In-CDO3AEt-Phe(111In-CDO3AEt-FGK-Fabがフェニルアラニン-グリシン間で切断された化合物)であることがわかる。
111In-CDO3AEt-FGK-Fab(Rabbit serum IgG由来)を、111In-CDOTA-Bn-CO-FGK-Fab(抗c-kit IgG由来)に変更し、投与後6時間後までに排泄された尿を集積し分析した以外は、上記と同様の方法で、尿中放射活性の分析を行った。
図9は、111In-CDOTA-Bn-CO-FGK-Fab(抗c-kit IgG由来)をマウスに投与後6時間までに排泄された尿中放射活性の化学形の分析結果を示す。RP-HPLCの結果から、111In-CDOTA-Bn-CO-FGK-Fabでは、低分子画分における主たる放射活性111In-CDOTA-Phe (111In-CDOTA-Bn-CO-FGK-Fabがフェニルアラニン-グリシン間で切断された化合物)であることがわかる。
111In-CDO3AEt-FGK-Fab(Rabbit serum IgG由来)を、111In-DO3A-Bn-CO-FGK-Fab(Rabbit serum IgG由来)に変更した以外は、上記と同様の方法で、尿中放射活性の分析を行った。
図10は、111In-DO3A-Bn-CO-FGK-Fab(Rabbit serum IgG由来)をマウスに投与後24時間までに排泄された尿中放射活性の化学形の分析結果を示す。RP-HPLCの結果から、111In-DO3A-Bn-CO-FGK-Fabでは、低分子画分における主たる放射活性111In-DO3A-Phe (111In-DO3A-Bn-CO-FGK-Fabがフェニルアラニン-グリシン間で切断された化合物)であることがわかる。
111In-CDO3AEt-FGK-Fab(Rabbit serum IgG由来)を、111In-CDO3AiBu-FGK-Fab(Rabbit serum IgG由来)に変更した以外は、上記と同様の方法で、尿中放射活性の分析を行った。
図11は、111In-CDO3AiBu-FGK-Fab(Rabbit serum IgG由来)をマウスに投与後24時間までに排泄された尿中放射活性の化学形の分析結果を示す。以上、尿中の放射活性の分析により図11Aに示すSE-HPLCによる分析で、大部分の放射活性が低分子画分として排泄され、図11Bに示すRP-HPLCの結果から、111In-CDO3AiBu-FGK-Fab(Rabbit serum IgG由来)では、低分子画分における主たる放射活性が111In-CDO3AiBu-Phe (111In-CDO3AiBu-FGK-Fabがフェニルアラニン-グリシン間で切断された化合物)であることがわかる。
上述の方法により調製した111In-CDO3AEt-FGK-Fab(抗c-kit IgG由来)をD-PBS (-)で希釈した。上記SY皮下腫瘍モデルマウスの尾静脈より、Fab濃度を25 μg/100 μLに調整した111In- CDO3AEt-FGK-Fab溶液(45 μCi/100 μL/匹)を投与した。各群2匹のマウスをSPECT/CT装置(SPECT4CT, Trifoil Imaging, CA)を用いて、開口径1 mm、5穴ピンホールコリメータを360度収集、16プロジョエクション、14分/プロジェクションの条件にて、投与後2.5時間から撮像した。
111In-CDO3AEt-FGK-Fab(抗c-kit IgG由来)溶液(45 μCi/100 μL/匹)を111In-DO3A-Bn-SCN-MVK-Fab(抗c-kit IgG由来)溶液(14 μCi/100 μL/匹)に変更した以外は、同様にして、SY皮下腫瘍モデルマウスの尾静脈より投与して、投与後2.5時間から撮像した。
図12は、111In-DO3A-Bn-SCN-MVK-Fab(抗c-kit IgG由来)溶液を、又は111In-CDO3AEt-FGK-Fab(抗c-kit IgG由来)溶液をSY皮下腫瘍モデルマウスに投与後2.5時間のSPECT/CT画像である。
投与2.5時間後において、111In-CDO3AEt-FGK-Fabは腎臓への集積が低く、腫瘍を明瞭に画像化した。一方で、111In-DO3A-Bn-SCN-MVK-Fabでは、腫瘍を画像化したが、腎臓にも高い放射活性が観察された。
以上、放射性標識薬剤は、腎臓への集積が低く、放射線画像診断の感度、精度を向上させることができる。
Claims (16)
- 下記式(1)で表される化合物、又はその薬理学的に許容可能な塩。
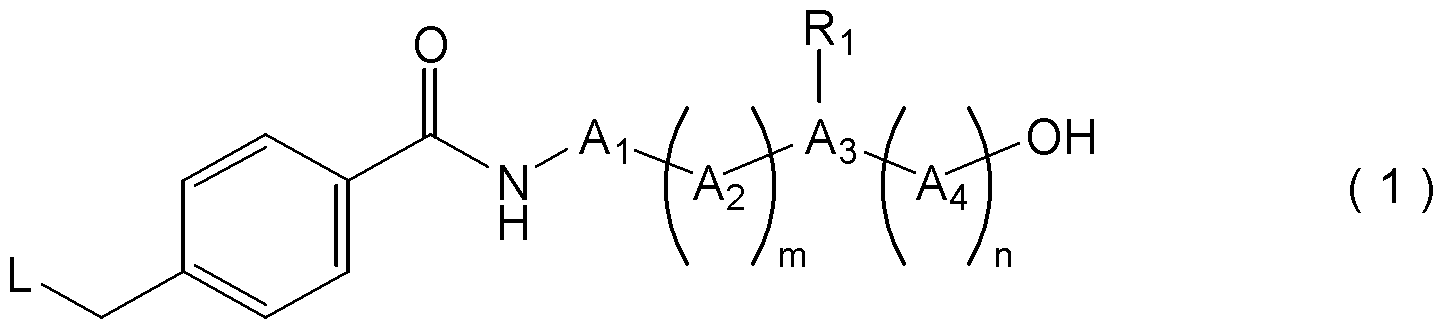
〔式中、
A1、A2は、それぞれ独立にアミノ酸残基であり、
mは、0~3の整数であり、
A3は、側鎖にアミノ基又はカルボキシ基を有するアミノ酸残基であり、
A4は、アミノ酸残基であり、
nは、0~3の整数であり、
R1は、A3の側鎖のアミノ基又はカルボキシ基と結合し、標的分子認識素子又はその連結基と結合可能な官能基を有する基、又は、A3の側鎖のアミノ基若しくはカルボキシ基の水素原子であり、ただし、R1は、環構成原子の一つとしてA3の側鎖のアミノ基の窒素原子を含む炭素数3~10の複素環基を形成していてもよく、
Lは、式(L1):

(式中、R3,R4,R5,R6は、それぞれ独立に、水素原子、-CH2COOR10基、又は炭素数1~8の炭化水素基であり、R10は、水素原子又は炭素数1~8の炭化水素基であり、*は結合部位である。ただし、R3,R4,R5,R6のうち、少なくとも3個以上は-CH2COOH基である。)で表される基、又は、式(L2):

(式中、*は結合部位である。)で表される基である。〕 - A1からA4のアミノ酸配列(ただし、n=0の場合、A1からA3のアミノ酸配列)が、腎刷子縁膜酵素の基質配列の一部と同一の配列である、請求項1に記載の化合物、又はその薬理学的に許容可能な塩。
- R3,R4,R5,R6のうち、3個が-CH2COOH基である、請求項1又は2に記載の化合物、又はその薬理学的に許容可能な塩。
- A3が、リシン、オルニチン又はアルギニンの残基である、請求項1~3のいずれか1項に記載の化合物、又はその薬理学的に許容可能な塩。
- mが、1である、請求項1~4のいずれか1項に記載の化合物、又はその薬理学的に許容可能な塩。
- nが、0である、請求項1~5のいずれか1項に記載の化合物、又はその薬理学的に許容可能な塩。
- 前記化合物が、下記式(1a)で表される、請求項1~6のいずれか1項に記載の化合物、又はその薬理学的に許容可能な塩。
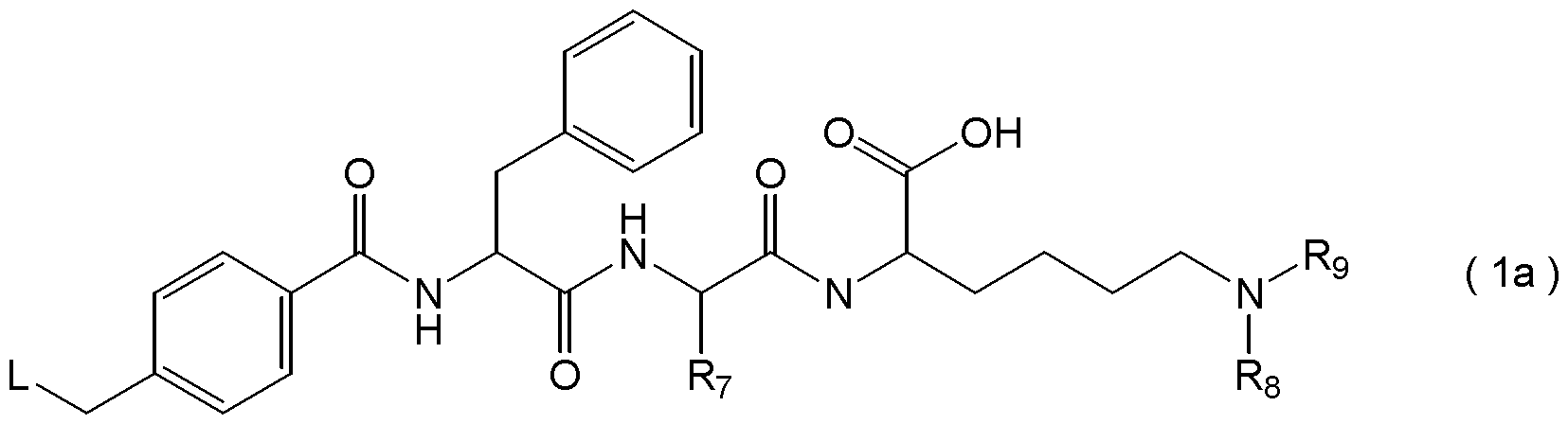
〔式中、
Lは、式(L1):
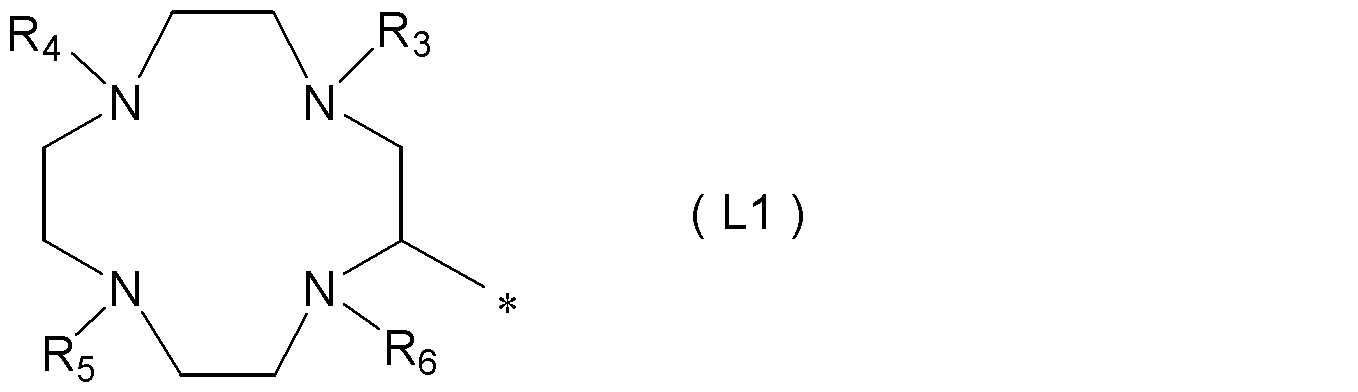
(式中、R3,R4,R5,R6は、それぞれ独立に、水素原子、-CH2COOR10基、又は炭素数1~8の炭化水素基であり、R10は、水素原子又は炭素数1~8の炭化水素基であり、*は結合部位である。ただし、R3,R4,R5,R6のうち、少なくとも3以上は-CH2COOH基である。)で表される基、又は、式(L2):

(式中、*は結合部位である。)で表される基であり、
R7は、水素原子又はメチル基であり、
R8,R9は、それぞれ独立に、水素原子、又は、官能基を有する総炭素数2~20のアシル基、官能基を有する総炭素数2~20のアルキル基、官能基を有する総炭素数2~20のアルキルカルバモイル基、若しくは官能基を有する総炭素数2~20のアルキルチオカルバモイル基である。ただし、R8及びR9は、隣接する窒素原子を含む複素環を形成していてもよく、その場合、式:

で表される基は、式:

で表される基である。〕 - Lが、式(L1):

(式中、R3,R4,R5,R6は、それぞれ独立に、-CH2COOH基、イソブチル基であり、*は結合部位である。ただし、R3,R4,R5,R6のうち、3個が-CH2COOH基であり、1個がイソブチル基である。)で表される基である、請求項1~7のいずれか1項に記載の化合物、又はその薬理学的に許容可能な塩。 - 請求項1~8のいずれか1項に記載の化合物、又はその薬理学的に許容可能な塩に、標的分子認識素子を結合させてなる化合物、又はその薬理学的に許容可能な塩。
- 放射性金属及び放射性原子標識金属からなる群から選ばれる1種の金属と、前記金属に配位した請求項1~9のいずれか1項に記載の化合物又はその薬理的に許容可能な塩と、を有する金属錯体化合物、又はその薬理学的に許容可能な塩。
- 前記金属が、111In、223Ra、67Ga、68Ga、44Sc、90Y、177Lu、225Ac、212Bi、213Bi、212Pb、227Th、64Cu、又は67Cuである、請求項10に記載の金属錯体化合物、又はその薬理学的に許容可能な塩。
- 請求項1~11のいずれか1項に記載の化合物、又はその薬理学的に許容可能な塩を含む、放射性薬剤の調製用薬剤。
- 請求項1~11のいずれか1項に記載の化合物、又はその薬理学的に許容可能な塩の放射性薬剤の製造のための使用。
- 請求項10又は11に記載の金属錯体化合物、又はその薬理学的に許容可能な塩を含む、放射性薬剤。
- 請求項10又は11に記載の金属錯体化合物、又はその薬理学的に許容可能な塩を含む、放射線治療薬。
- 請求項10又は11に記載の金属錯体化合物、又はその薬理学的に許容可能な塩を含む、放射性画像診断薬。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201880055323.7A CN111065646B (zh) | 2017-09-26 | 2018-09-26 | 放射性药物 |
| US16/648,703 US11167050B2 (en) | 2017-09-26 | 2018-09-26 | Metal complex forming compound, metal complex compound formed thereof, radioactive drug containing the metal complex compound, and method of using and preparing the metal complex compound |
| SG11202002528SA SG11202002528SA (en) | 2017-09-26 | 2018-09-26 | Radioactive drug |
| CA3086454A CA3086454A1 (en) | 2017-09-26 | 2018-09-26 | Radioactive drug |
| EP18861877.1A EP3689892B1 (en) | 2017-09-26 | 2018-09-26 | Radioactive drug |
| KR1020207003700A KR20200056979A (ko) | 2017-09-26 | 2018-09-26 | 방사성 약제 |
| JP2019545585A JP7207740B2 (ja) | 2017-09-26 | 2018-09-26 | 放射性薬剤 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017185484 | 2017-09-26 | ||
| JP2017-185484 | 2017-09-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019065774A1 true WO2019065774A1 (ja) | 2019-04-04 |
Family
ID=65903027
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/035786 WO2019065774A1 (ja) | 2017-09-26 | 2018-09-26 | 放射性薬剤 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US11167050B2 (ja) |
| EP (1) | EP3689892B1 (ja) |
| JP (1) | JP7207740B2 (ja) |
| KR (1) | KR20200056979A (ja) |
| CN (1) | CN111065646B (ja) |
| CA (1) | CA3086454A1 (ja) |
| SG (1) | SG11202002528SA (ja) |
| WO (1) | WO2019065774A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019203191A1 (ja) * | 2018-04-16 | 2019-10-24 | 日本メジフィジックス株式会社 | 修飾抗体、および、放射性金属標識抗体 |
| WO2020145227A1 (ja) | 2019-01-07 | 2020-07-16 | アステラス製薬株式会社 | 配位子、スペーサー、ペプチドリンカーおよび生物分子からなる複合体 |
| WO2021177390A1 (ja) | 2020-03-04 | 2021-09-10 | 日本メジフィジックス株式会社 | 化合物及び放射性標識化合物 |
| WO2022186311A1 (ja) | 2021-03-04 | 2022-09-09 | 日本メジフィジックス株式会社 | 化合物及び放射性標識化合物 |
| US11667724B2 (en) | 2017-07-07 | 2023-06-06 | Astellas Pharma Inc. | Anti-human CEACAM5 antibody Fab fragment |
| WO2023100852A1 (ja) | 2021-11-30 | 2023-06-08 | 日本メジフィジックス株式会社 | 安定化放射性医薬組成物 |
| US11679166B2 (en) | 2016-11-18 | 2023-06-20 | Astellas Pharma Inc. | Anti-human MUC1 antibody Fab fragment |
| US12065503B2 (en) | 2018-10-10 | 2024-08-20 | Astellas Pharma Inc. | Pharmaceutical composition containing tagged site-antihuman antibody fab fragment complex |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11919972B2 (en) | 2018-11-02 | 2024-03-05 | Regeneron Pharmaceuticals, Inc. | Peptide libraries with non-canonical amino acids |
| WO2022098745A1 (en) * | 2020-11-03 | 2022-05-12 | Indi Molecular, Inc. | Compositions, delivery systems, and methods useful in tumor therapy |
| CN114685389B (zh) * | 2022-03-04 | 2023-10-27 | 复旦大学附属中山医院 | 一种基于迈克尔加成的pet显像剂、制备方法与用途 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015086213A (ja) * | 2013-03-25 | 2015-05-07 | 国立大学法人 千葉大学 | 放射性核種標識オクトレオチド誘導体 |
| WO2017150549A1 (ja) | 2016-03-01 | 2017-09-08 | 国立大学法人 千葉大学 | 放射性標識薬剤 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005047821A (ja) * | 2003-07-30 | 2005-02-24 | Nihon Medi Physics Co Ltd | シクロペンタジエニルカルボニル基を含む放射性化合物を用いた診断及び治療用薬剤 |
| JP6164556B2 (ja) * | 2011-12-01 | 2017-07-19 | 国立大学法人 千葉大学 | 非特異的腎集積が低減された放射性標識ポリペプチド作製用薬剤 |
-
2018
- 2018-09-26 EP EP18861877.1A patent/EP3689892B1/en active Active
- 2018-09-26 US US16/648,703 patent/US11167050B2/en active Active
- 2018-09-26 CA CA3086454A patent/CA3086454A1/en active Pending
- 2018-09-26 JP JP2019545585A patent/JP7207740B2/ja active Active
- 2018-09-26 CN CN201880055323.7A patent/CN111065646B/zh active Active
- 2018-09-26 KR KR1020207003700A patent/KR20200056979A/ko not_active Application Discontinuation
- 2018-09-26 SG SG11202002528SA patent/SG11202002528SA/en unknown
- 2018-09-26 WO PCT/JP2018/035786 patent/WO2019065774A1/ja unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015086213A (ja) * | 2013-03-25 | 2015-05-07 | 国立大学法人 千葉大学 | 放射性核種標識オクトレオチド誘導体 |
| WO2017150549A1 (ja) | 2016-03-01 | 2017-09-08 | 国立大学法人 千葉大学 | 放射性標識薬剤 |
Non-Patent Citations (11)
| Title |
|---|
| ANALYTICAL BIOCHEMISTRY, vol. 142, 1984, pages 68 - 78 |
| ARCHIVES OF BIOCHEMISTRY AND BIOPHYSICS, vol. 119, 1967, pages 41 - 49 |
| CHEMICAL SOCIETY REVIEWS, vol. 42, 2013, pages 5131 - 5142 |
| CHEMICAL SOCIETYREVIEWS, vol. 45, 2016, pages 6409 - 6658 |
| GLOSSMANN ET AL., FEBS LETTERS, vol. 19, 1972, pages 340 - 344 |
| HORI ET AL., BIOCHEMICAL PHARMACOLOGY, vol. 45, 1993, pages 1763 - 1768 |
| KRAMERS ET AL., EUROPEAN JOURNAL OF BIOCHEMISTRY, vol. 99, 1979, pages 345 - 351 |
| MOLECULAR ONCOLOGY, vol. 8, 2014, pages 799 - 812 |
| PLANT PHYSIOLOGY, vol. 55, 1975, pages 94 - 98 |
| See also references of EP3689892A4 |
| ZIMMERMANN, KURT ET AL.: "A Triglycine Linker Improves Tumor Uptake and Biodistributions of 67-Cu-Labeled Anti- Neuroblastoma MAb chCE7 F (ab')2 Fragments", NUCLEAR MEDICINE & BIOLOGY, vol. 26, 1999, pages 943 - 950, XP055585908, ISSN: 0969-8051 * |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11679166B2 (en) | 2016-11-18 | 2023-06-20 | Astellas Pharma Inc. | Anti-human MUC1 antibody Fab fragment |
| US11667724B2 (en) | 2017-07-07 | 2023-06-06 | Astellas Pharma Inc. | Anti-human CEACAM5 antibody Fab fragment |
| JPWO2019203191A1 (ja) * | 2018-04-16 | 2021-04-22 | 日本メジフィジックス株式会社 | 修飾抗体、および、放射性金属標識抗体 |
| US11701440B2 (en) | 2018-04-16 | 2023-07-18 | Nihon Medi-Physics Co., Ltd. | Modified antibody and radioactive metal-labelled antibody |
| WO2019203191A1 (ja) * | 2018-04-16 | 2019-10-24 | 日本メジフィジックス株式会社 | 修飾抗体、および、放射性金属標識抗体 |
| EP3783016A4 (en) * | 2018-04-16 | 2021-06-16 | Nihon Medi-Physics Co., Ltd | MODIFIED ANTIBODY AND RADIOACTIVE METAL ANTIBODY |
| CN112041337A (zh) * | 2018-04-16 | 2020-12-04 | 日本医事物理股份有限公司 | 修饰抗体、及放射性金属标记抗体 |
| JP7327762B2 (ja) | 2018-04-16 | 2023-08-16 | 日本メジフィジックス株式会社 | 修飾抗体、および、放射性金属標識抗体 |
| US12065503B2 (en) | 2018-10-10 | 2024-08-20 | Astellas Pharma Inc. | Pharmaceutical composition containing tagged site-antihuman antibody fab fragment complex |
| KR20210113201A (ko) | 2019-01-07 | 2021-09-15 | 아스텔라스세이야쿠 가부시키가이샤 | 배위자 및 CEACAM5 항체 Fab 프래그먼트를 포함하는 복합체 |
| KR20210113202A (ko) | 2019-01-07 | 2021-09-15 | 아스텔라스세이야쿠 가부시키가이샤 | 배위자, 스페이서, 펩티드 링커 및 생물 분자를 포함하는 복합체 |
| WO2020145228A1 (ja) | 2019-01-07 | 2020-07-16 | アステラス製薬株式会社 | 配位子及びCEACAM5抗体Fabフラグメントからなる複合体 |
| WO2020145227A1 (ja) | 2019-01-07 | 2020-07-16 | アステラス製薬株式会社 | 配位子、スペーサー、ペプチドリンカーおよび生物分子からなる複合体 |
| WO2021177390A1 (ja) | 2020-03-04 | 2021-09-10 | 日本メジフィジックス株式会社 | 化合物及び放射性標識化合物 |
| WO2022186311A1 (ja) | 2021-03-04 | 2022-09-09 | 日本メジフィジックス株式会社 | 化合物及び放射性標識化合物 |
| WO2023100852A1 (ja) | 2021-11-30 | 2023-06-08 | 日本メジフィジックス株式会社 | 安定化放射性医薬組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20200268913A1 (en) | 2020-08-27 |
| CN111065646B (zh) | 2023-05-30 |
| EP3689892A4 (en) | 2021-05-12 |
| SG11202002528SA (en) | 2020-04-29 |
| JPWO2019065774A1 (ja) | 2021-01-28 |
| CN111065646A (zh) | 2020-04-24 |
| US11167050B2 (en) | 2021-11-09 |
| EP3689892B1 (en) | 2024-07-17 |
| EP3689892A1 (en) | 2020-08-05 |
| JP7207740B2 (ja) | 2023-01-18 |
| CA3086454A1 (en) | 2019-04-04 |
| KR20200056979A (ko) | 2020-05-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7207740B2 (ja) | 放射性薬剤 | |
| JP6966741B2 (ja) | 放射性標識薬剤 | |
| RU2592685C2 (ru) | Радиоактивно меченые пептиды, связывающиеся с her2 | |
| JP2022513256A (ja) | デュアルモード18f標識セラノスティック化合物及びその使用 | |
| JP6164556B2 (ja) | 非特異的腎集積が低減された放射性標識ポリペプチド作製用薬剤 | |
| JP7370520B2 (ja) | 放射性医薬 | |
| Lee et al. | Synthesis and biological evaluation of RGD peptides with the 99m Tc/188 Re chelated iminodiacetate core: Highly enhanced uptake and excretion kinetics of theranostics against tumor angiogenesis | |
| TWI381852B (zh) | 生物素二胺基衍生物類及其與大環螯合劑之共軛物 | |
| JP5481673B2 (ja) | 放射性標識薬剤 | |
| JP2022507296A (ja) | ガストリン放出ペプチド受容体(grpr)のインビボイメージングおよびgrpr関連障害の治療のための放射性標識ボンベシン由来化合物 | |
| Pruszynski et al. | D-amino acid peptide residualizing agents bearing N-hydroxysuccinimido-and maleimido-functional groups and their application for trastuzumab radioiodination | |
| JP5604680B2 (ja) | 放射性標識薬剤 | |
| JP2015086213A (ja) | 放射性核種標識オクトレオチド誘導体 | |
| WO2022080481A1 (ja) | 抗her2抗体の放射性複合体、及び、放射性医薬 | |
| WO2022211051A1 (ja) | 抗egfr抗体の放射性複合体、及び、放射性医薬 | |
| WO2024143314A1 (ja) | 放射性標識医薬 | |
| JP2024517879A (ja) | 放射性金属用キレート剤ならびにその製造方法および使用方法 | |
| JP2005068046A (ja) | ソマトスタチンアナログ誘導体およびその利用 | |
| CN103491984B (zh) | 放射性标记的her2结合肽 | |
| JP2015083546A (ja) | 癌の原発巣・骨転移の検査・治療用放射性標識薬剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18861877 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019545585 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2018861877 Country of ref document: EP Effective date: 20200428 |
|
| ENP | Entry into the national phase |
Ref document number: 3086454 Country of ref document: CA |