WO2018181425A1 - 癌治療用医薬組成物 - Google Patents
癌治療用医薬組成物 Download PDFInfo
- Publication number
- WO2018181425A1 WO2018181425A1 PCT/JP2018/012644 JP2018012644W WO2018181425A1 WO 2018181425 A1 WO2018181425 A1 WO 2018181425A1 JP 2018012644 W JP2018012644 W JP 2018012644W WO 2018181425 A1 WO2018181425 A1 WO 2018181425A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antibody
- cells
- ccr8
- mouse
- tumor
- Prior art date
Links
- 206010028980 Neoplasm Diseases 0.000 title claims abstract description 272
- 201000011510 cancer Diseases 0.000 title claims abstract description 44
- 239000000203 mixture Substances 0.000 title abstract description 7
- 101000716063 Homo sapiens C-C chemokine receptor type 8 Proteins 0.000 claims abstract description 246
- 102100036305 C-C chemokine receptor type 8 Human genes 0.000 claims abstract description 242
- 210000003289 regulatory T cell Anatomy 0.000 claims description 65
- 208000008839 Kidney Neoplasms Diseases 0.000 claims description 45
- 206010038389 Renal cancer Diseases 0.000 claims description 44
- 201000010982 kidney cancer Diseases 0.000 claims description 44
- 238000000034 method Methods 0.000 claims description 43
- 206010009944 Colon cancer Diseases 0.000 claims description 30
- 208000029742 colonic neoplasm Diseases 0.000 claims description 29
- 230000000694 effects Effects 0.000 claims description 27
- 239000008194 pharmaceutical composition Substances 0.000 claims description 27
- 210000002540 macrophage Anatomy 0.000 claims description 24
- 230000002601 intratumoral effect Effects 0.000 claims description 22
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 claims description 16
- 208000026310 Breast neoplasm Diseases 0.000 claims description 14
- 206010006187 Breast cancer Diseases 0.000 claims description 13
- 230000009471 action Effects 0.000 claims description 13
- 230000003472 neutralizing effect Effects 0.000 claims description 11
- 201000000062 kidney sarcoma Diseases 0.000 claims description 6
- 239000012830 cancer therapeutic Substances 0.000 claims description 2
- 229940126585 therapeutic drug Drugs 0.000 claims description 2
- 210000004027 cell Anatomy 0.000 description 300
- 238000002054 transplantation Methods 0.000 description 76
- 230000014509 gene expression Effects 0.000 description 71
- 241000699666 Mus <mouse, genus> Species 0.000 description 62
- 241000699670 Mus sp. Species 0.000 description 51
- 108090000623 proteins and genes Proteins 0.000 description 47
- 210000001744 T-lymphocyte Anatomy 0.000 description 44
- 238000002513 implantation Methods 0.000 description 31
- 230000000259 anti-tumor effect Effects 0.000 description 30
- 241000700159 Rattus Species 0.000 description 28
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 25
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 25
- 102100027268 Interferon-stimulated gene 20 kDa protein Human genes 0.000 description 25
- 241000282414 Homo sapiens Species 0.000 description 20
- 230000004083 survival effect Effects 0.000 description 19
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 18
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 18
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 18
- 101000738413 Homo sapiens T-cell surface glycoprotein CD3 gamma chain Proteins 0.000 description 17
- 101100005660 Mus musculus Ccr8 gene Proteins 0.000 description 17
- 239000002609 medium Substances 0.000 description 17
- 108091007433 antigens Proteins 0.000 description 16
- 102000036639 antigens Human genes 0.000 description 16
- 239000012981 Hank's balanced salt solution Substances 0.000 description 15
- 102100037911 T-cell surface glycoprotein CD3 gamma chain Human genes 0.000 description 15
- 108020004999 messenger RNA Proteins 0.000 description 15
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 14
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 14
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 14
- 239000007995 HEPES buffer Substances 0.000 description 14
- 241001465754 Metazoa Species 0.000 description 14
- 230000003247 decreasing effect Effects 0.000 description 14
- 239000003550 marker Substances 0.000 description 14
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 13
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 13
- 239000000427 antigen Substances 0.000 description 13
- 238000010186 staining Methods 0.000 description 13
- 238000005406 washing Methods 0.000 description 13
- 239000004698 Polyethylene Substances 0.000 description 12
- 239000003814 drug Substances 0.000 description 12
- 238000005259 measurement Methods 0.000 description 12
- 108020004414 DNA Proteins 0.000 description 11
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 10
- 206010061535 Ovarian neoplasm Diseases 0.000 description 10
- 238000004458 analytical method Methods 0.000 description 10
- 229940079593 drug Drugs 0.000 description 10
- 239000012636 effector Substances 0.000 description 10
- 238000011156 evaluation Methods 0.000 description 10
- 210000004408 hybridoma Anatomy 0.000 description 10
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 9
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 description 9
- 102100022338 Integrin alpha-M Human genes 0.000 description 9
- 206010033128 Ovarian cancer Diseases 0.000 description 9
- 206010060862 Prostate cancer Diseases 0.000 description 9
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 9
- 239000013604 expression vector Substances 0.000 description 9
- 238000000926 separation method Methods 0.000 description 9
- 239000013598 vector Substances 0.000 description 9
- 206010005003 Bladder cancer Diseases 0.000 description 8
- 101150100160 CCR8 gene Proteins 0.000 description 8
- 208000030808 Clear cell renal carcinoma Diseases 0.000 description 8
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 8
- 208000006265 Renal cell carcinoma Diseases 0.000 description 8
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 8
- 206010073251 clear cell renal cell carcinoma Diseases 0.000 description 8
- 239000002299 complementary DNA Substances 0.000 description 8
- 230000002950 deficient Effects 0.000 description 8
- 238000010195 expression analysis Methods 0.000 description 8
- 201000005202 lung cancer Diseases 0.000 description 8
- 208000020816 lung neoplasm Diseases 0.000 description 8
- 201000011330 nonpapillary renal cell carcinoma Diseases 0.000 description 8
- 102000004169 proteins and genes Human genes 0.000 description 8
- 230000001225 therapeutic effect Effects 0.000 description 8
- 201000005112 urinary bladder cancer Diseases 0.000 description 8
- 208000023275 Autoimmune disease Diseases 0.000 description 7
- 206010014733 Endometrial cancer Diseases 0.000 description 7
- 206010014759 Endometrial neoplasm Diseases 0.000 description 7
- 241000282412 Homo Species 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 238000012790 confirmation Methods 0.000 description 7
- 230000017858 demethylation Effects 0.000 description 7
- 238000010520 demethylation reaction Methods 0.000 description 7
- 238000000684 flow cytometry Methods 0.000 description 7
- 230000003053 immunization Effects 0.000 description 7
- 235000018102 proteins Nutrition 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 239000000243 solution Substances 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- 210000001519 tissue Anatomy 0.000 description 7
- 210000004881 tumor cell Anatomy 0.000 description 7
- 241000124008 Mammalia Species 0.000 description 6
- 229930182474 N-glycoside Natural products 0.000 description 6
- 241000283973 Oryctolagus cuniculus Species 0.000 description 6
- 238000001358 Pearson's chi-squared test Methods 0.000 description 6
- 238000003559 RNA-seq method Methods 0.000 description 6
- 239000012228 culture supernatant Substances 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 238000002649 immunization Methods 0.000 description 6
- 230000003834 intracellular effect Effects 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 210000000952 spleen Anatomy 0.000 description 6
- 238000012353 t test Methods 0.000 description 6
- 241000699800 Cricetinae Species 0.000 description 5
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 description 5
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 5
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 5
- 230000027455 binding Effects 0.000 description 5
- 239000011575 calcium Substances 0.000 description 5
- 210000001072 colon Anatomy 0.000 description 5
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 5
- BRZYSWJRSDMWLG-CAXSIQPQSA-N geneticin Natural products O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](C(C)O)O2)N)[C@@H](N)C[C@H]1N BRZYSWJRSDMWLG-CAXSIQPQSA-N 0.000 description 5
- 230000036039 immunity Effects 0.000 description 5
- 206010003445 Ascites Diseases 0.000 description 4
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 4
- 201000009030 Carcinoma Diseases 0.000 description 4
- 241000700199 Cavia porcellus Species 0.000 description 4
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 4
- 238000000692 Student's t-test Methods 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 229910052791 calcium Inorganic materials 0.000 description 4
- 239000012634 fragment Substances 0.000 description 4
- 102000048031 human CCR8 Human genes 0.000 description 4
- 210000001165 lymph node Anatomy 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 210000001616 monocyte Anatomy 0.000 description 4
- 210000000822 natural killer cell Anatomy 0.000 description 4
- 230000000638 stimulation Effects 0.000 description 4
- 230000035899 viability Effects 0.000 description 4
- 108090000672 Annexin A5 Proteins 0.000 description 3
- 102000004121 Annexin A5 Human genes 0.000 description 3
- SHZGCJCMOBCMKK-UHFFFAOYSA-N D-mannomethylose Natural products CC1OC(O)C(O)C(O)C1O SHZGCJCMOBCMKK-UHFFFAOYSA-N 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 3
- PNNNRSAQSRJVSB-SLPGGIOYSA-N Fucose Natural products C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C=O PNNNRSAQSRJVSB-SLPGGIOYSA-N 0.000 description 3
- 206010062016 Immunosuppression Diseases 0.000 description 3
- 108010002350 Interleukin-2 Proteins 0.000 description 3
- SHZGCJCMOBCMKK-DHVFOXMCSA-N L-fucopyranose Chemical compound C[C@@H]1OC(O)[C@@H](O)[C@H](O)[C@@H]1O SHZGCJCMOBCMKK-DHVFOXMCSA-N 0.000 description 3
- 108060001084 Luciferase Proteins 0.000 description 3
- OVRNDRQMDRJTHS-UHFFFAOYSA-N N-acelyl-D-glucosamine Natural products CC(=O)NC1C(O)OC(CO)C(O)C1O OVRNDRQMDRJTHS-UHFFFAOYSA-N 0.000 description 3
- OVRNDRQMDRJTHS-FMDGEEDCSA-N N-acetyl-beta-D-glucosamine Chemical group CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O OVRNDRQMDRJTHS-FMDGEEDCSA-N 0.000 description 3
- MBLBDJOUHNCFQT-LXGUWJNJSA-N N-acetylglucosamine Natural products CC(=O)N[C@@H](C=O)[C@@H](O)[C@H](O)[C@H](O)CO MBLBDJOUHNCFQT-LXGUWJNJSA-N 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 210000000628 antibody-producing cell Anatomy 0.000 description 3
- 230000009460 calcium influx Effects 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 238000012258 culturing Methods 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 239000007850 fluorescent dye Substances 0.000 description 3
- 230000001506 immunosuppresive effect Effects 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 238000002955 isolation Methods 0.000 description 3
- 210000000265 leukocyte Anatomy 0.000 description 3
- 229950006780 n-acetylglucosamine Drugs 0.000 description 3
- 239000013642 negative control Substances 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- DBABZHXKTCFAPX-UHFFFAOYSA-N probenecid Chemical compound CCCN(CCC)S(=O)(=O)C1=CC=C(C(O)=O)C=C1 DBABZHXKTCFAPX-UHFFFAOYSA-N 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- GUAHPAJOXVYFON-ZETCQYMHSA-N (8S)-8-amino-7-oxononanoic acid zwitterion Chemical compound C[C@H](N)C(=O)CCCCCC(O)=O GUAHPAJOXVYFON-ZETCQYMHSA-N 0.000 description 2
- 101150017312 CD3G gene Proteins 0.000 description 2
- 229940045513 CTLA4 antagonist Drugs 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- 230000007067 DNA methylation Effects 0.000 description 2
- 208000008334 Dermatofibrosarcoma Diseases 0.000 description 2
- 206010057070 Dermatofibrosarcoma protuberans Diseases 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- 241000588724 Escherichia coli Species 0.000 description 2
- 102100027581 Forkhead box protein P3 Human genes 0.000 description 2
- 101150112014 Gapdh gene Proteins 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- 101000861452 Homo sapiens Forkhead box protein P3 Proteins 0.000 description 2
- 102000008100 Human Serum Albumin Human genes 0.000 description 2
- 108091006905 Human Serum Albumin Proteins 0.000 description 2
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 2
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 239000005089 Luciferase Substances 0.000 description 2
- 101100005550 Mus musculus Ccl1 gene Proteins 0.000 description 2
- 206010035226 Plasma cell myeloma Diseases 0.000 description 2
- 229910000831 Steel Inorganic materials 0.000 description 2
- 102000004142 Trypsin Human genes 0.000 description 2
- 108090000631 Trypsin Proteins 0.000 description 2
- 229940127174 UCHT1 Drugs 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 210000000601 blood cell Anatomy 0.000 description 2
- 210000000988 bone and bone Anatomy 0.000 description 2
- 230000007910 cell fusion Effects 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 238000010494 dissociation reaction Methods 0.000 description 2
- 230000005593 dissociations Effects 0.000 description 2
- 230000000857 drug effect Effects 0.000 description 2
- 210000003743 erythrocyte Anatomy 0.000 description 2
- 230000005284 excitation Effects 0.000 description 2
- 239000013613 expression plasmid Substances 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 201000005787 hematologic cancer Diseases 0.000 description 2
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 2
- 210000005260 human cell Anatomy 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 238000001325 log-rank test Methods 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 210000004698 lymphocyte Anatomy 0.000 description 2
- 235000013372 meat Nutrition 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 230000011987 methylation Effects 0.000 description 2
- 238000007069 methylation reaction Methods 0.000 description 2
- 201000000050 myeloid neoplasm Diseases 0.000 description 2
- 238000010606 normalization Methods 0.000 description 2
- 235000015097 nutrients Nutrition 0.000 description 2
- 201000008968 osteosarcoma Diseases 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 239000013610 patient sample Substances 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- 229920001992 poloxamer 407 Polymers 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 229960003081 probenecid Drugs 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 239000000941 radioactive substance Substances 0.000 description 2
- 238000005215 recombination Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 230000009870 specific binding Effects 0.000 description 2
- 239000010959 steel Substances 0.000 description 2
- 230000004936 stimulating effect Effects 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 239000012588 trypsin Substances 0.000 description 2
- 238000005199 ultracentrifugation Methods 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 108091093088 Amplicon Proteins 0.000 description 1
- 102000000412 Annexin Human genes 0.000 description 1
- 108050008874 Annexin Proteins 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 206010004593 Bile duct cancer Diseases 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 108091008927 CC chemokine receptors Proteins 0.000 description 1
- 102000005674 CCR Receptors Human genes 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- 108010078791 Carrier Proteins Proteins 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 101150087313 Cd8a gene Proteins 0.000 description 1
- 206010057250 Cell-mediated cytotoxicity Diseases 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 101000837299 Euglena gracilis Trans-2-enoyl-CoA reductase Proteins 0.000 description 1
- 108010087819 Fc receptors Proteins 0.000 description 1
- 102000009109 Fc receptors Human genes 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 102000030782 GTP binding Human genes 0.000 description 1
- 108091000058 GTP-Binding Proteins 0.000 description 1
- 208000022072 Gallbladder Neoplasms Diseases 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 101001043809 Homo sapiens Interleukin-7 receptor subunit alpha Proteins 0.000 description 1
- 101000951234 Homo sapiens Solute carrier family 49 member 4 Proteins 0.000 description 1
- 101000599037 Homo sapiens Zinc finger protein Helios Proteins 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 1
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 102100021593 Interleukin-7 receptor subunit alpha Human genes 0.000 description 1
- 102100020870 La-related protein 6 Human genes 0.000 description 1
- 108050008265 La-related protein 6 Proteins 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 101100179561 Mus musculus Il2ra gene Proteins 0.000 description 1
- 101100519207 Mus musculus Pdcd1 gene Proteins 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 101710160107 Outer membrane protein A Proteins 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- BELBBZDIHDAJOR-UHFFFAOYSA-N Phenolsulfonephthalein Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2S(=O)(=O)O1 BELBBZDIHDAJOR-UHFFFAOYSA-N 0.000 description 1
- 241000276498 Pollachius virens Species 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 108091081062 Repeated sequence (DNA) Proteins 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 102100037945 Solute carrier family 49 member 4 Human genes 0.000 description 1
- 208000000102 Squamous Cell Carcinoma of Head and Neck Diseases 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- 206010057644 Testis cancer Diseases 0.000 description 1
- 208000000728 Thymus Neoplasms Diseases 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 101710120037 Toxin CcdB Proteins 0.000 description 1
- 108090000992 Transferases Proteins 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 208000002495 Uterine Neoplasms Diseases 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 102100037796 Zinc finger protein Helios Human genes 0.000 description 1
- KYIKRXIYLAGAKQ-UHFFFAOYSA-N abcn Chemical compound C1CCCCC1(C#N)N=NC1(C#N)CCCCC1 KYIKRXIYLAGAKQ-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical class N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 230000005809 anti-tumor immunity Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 230000005975 antitumor immune response Effects 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 229960003852 atezolizumab Drugs 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 229950002916 avelumab Drugs 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 208000026900 bile duct neoplasm Diseases 0.000 description 1
- 230000008033 biological extinction Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 238000010804 cDNA synthesis Methods 0.000 description 1
- 238000010805 cDNA synthesis kit Methods 0.000 description 1
- 244000309466 calf Species 0.000 description 1
- 208000035269 cancer or benign tumor Diseases 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000009060 clear cell adenocarcinoma Diseases 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000012136 culture method Methods 0.000 description 1
- 238000013211 curve analysis Methods 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 238000000432 density-gradient centrifugation Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000007847 digital PCR Methods 0.000 description 1
- 229940105990 diglycerin Drugs 0.000 description 1
- GPLRAVKSCUXZTP-UHFFFAOYSA-N diglycerol Chemical compound OCC(O)COCC(O)CO GPLRAVKSCUXZTP-UHFFFAOYSA-N 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 229950009791 durvalumab Drugs 0.000 description 1
- 210000003162 effector t lymphocyte Anatomy 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 239000006274 endogenous ligand Substances 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 201000004101 esophageal cancer Diseases 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000012997 ficoll-paque Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 238000001917 fluorescence detection Methods 0.000 description 1
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 201000010175 gallbladder cancer Diseases 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 210000001156 gastric mucosa Anatomy 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 238000002523 gelfiltration Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 210000004209 hair Anatomy 0.000 description 1
- 201000000459 head and neck squamous cell carcinoma Diseases 0.000 description 1
- 238000007417 hierarchical cluster analysis Methods 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 210000003917 human chromosome Anatomy 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 230000016784 immunoglobulin production Effects 0.000 description 1
- 210000005008 immunosuppressive cell Anatomy 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 229960005386 ipilimumab Drugs 0.000 description 1
- 108010045069 keyhole-limpet hemocyanin Proteins 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 208000037841 lung tumor Diseases 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000002715 modification method Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 210000005087 mononuclear cell Anatomy 0.000 description 1
- 210000000066 myeloid cell Anatomy 0.000 description 1
- 229960003301 nivolumab Drugs 0.000 description 1
- 208000002154 non-small cell lung carcinoma Diseases 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 210000002741 palatine tonsil Anatomy 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229960002621 pembrolizumab Drugs 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229960003531 phenolsulfonphthalein Drugs 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 235000019422 polyvinyl alcohol Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 201000005825 prostate adenocarcinoma Diseases 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000003127 radioimmunoassay Methods 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 230000008844 regulatory mechanism Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 238000005185 salting out Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 238000002336 sorption--desorption measurement Methods 0.000 description 1
- 210000004989 spleen cell Anatomy 0.000 description 1
- 230000010473 stable expression Effects 0.000 description 1
- 238000007447 staining method Methods 0.000 description 1
- 239000012128 staining reagent Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- 229940126622 therapeutic monoclonal antibody Drugs 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 206010044412 transitional cell carcinoma Diseases 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 206010046766 uterine cancer Diseases 0.000 description 1
- 210000005167 vascular cell Anatomy 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
Images






































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2866—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for cytokines, lymphokines, interferons
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/14—Blood; Artificial blood
- A61K35/17—Lymphocytes; B-cells; T-cells; Natural killer cells; Interferon-activated or cytokine-activated lymphocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
- A61K47/6435—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent the peptide or protein in the drug conjugate being a connective tissue peptide, e.g. collagen, fibronectin or gelatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6883—Polymer-drug antibody conjugates, e.g. mitomycin-dextran-Ab; DNA-polylysine-antibody complex or conjugate used for therapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2818—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against CD28 or CD152
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2827—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against B7 molecules, e.g. CD80, CD86
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5011—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics for testing antineoplastic activity
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
- A61K2039/507—Comprising a combination of two or more separate antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/57—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2
- A61K2039/572—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2 cytotoxic response
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
- C07K2317/732—Antibody-dependent cellular cytotoxicity [ADCC]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
Definitions
- the present invention relates to a pharmaceutical composition for cancer treatment containing an antibody against CCR8.
- Non-patent Document 1 A strong negative regulatory mechanism including immunosuppression mediated by regulatory T cells (Treg cells) in the tumor microenvironment is a major obstacle for tumor treatment.
- CD4-positive Treg cells that infiltrate tumors may strongly inhibit the anti-tumor immune response and can be a major obstacle to effective cancer treatment.
- Tumor immunosuppression mediated by CD4-positive FoxP3-positive Treg cells has been well documented in animal tumor models, and anti-tumor effects can be obtained by systemic removal of Treg cells including within the tumor, but on the other hand, about 50% It has been reported that no effect is observed in the removal of Treg cells infiltrating tumors (Non-patent Document 2).
- Non-Patent Documents 3 to 8 Increased ratio of CD4 positive CD25 positive Treg cells (cell population containing Treg cells) in the total CD4 positive T cell population in humans detected in tumors of various cancer patients including lung, breast and ovarian tumors It has been reported that there is a negative correlation between the abundance ratio and the patient survival rate (Non-Patent Documents 3 to 8).
- the antitumor effect has been confirmed by removing CD4-positive CD25-positive Treg cells from the tumor with an anti-CD25 antibody.
- CD25 is also expressed on the cell surface of CD4-positive CD25-positive Treg cells and newly activated effector T cells, so it cannot be said that Treg cells are specifically removed.
- the anti-tumor effect of anti-CD25 antibody administration in mice is limited, and shows therapeutic effect only by antibody administration before tumor transplantation, and there is almost no therapeutic effect when antibody is administered after tumors are engrafted in mice This has been demonstrated in various tumor models.
- Non-patent Document 9 In the case of starting administration of anti-CD25 antibody on the first day after transplantation, the antitumor effect is attenuated, and in the case of starting administration on and after the second day after transplantation, almost no antitumor effect is observed.
- Non-Patent Document 10 Non-Patent Document 10
- CCR8 is a G protein-coupled seven-transmembrane CC chemokine receptor protein expressed in the thymus and spleen, also called CY6, CKR-L1 or TER1, and its gene is 3p21 in human chromosome Gene exists.
- Human CCR8 consists of 355 amino acids (Non-patent Document 11).
- CCL1 is known as an endogenous ligand for CCR8 (Non-patent Document 12).
- Human CCR8 cDNA is composed of the base sequence shown in Genbank ACC No. M_005211.3, and mouse CCR8 cDNA is composed of the base sequence shown in Genbank ACC. No. NM_007720.2.
- the problem to be solved by the present invention is to provide a pharmaceutical composition for cancer treatment through this mechanism by stimulating immunity by inhibiting immunosuppression by Treg cells and the like.
- intratumorally infiltrating Treg cells and intratumorally infiltrating macrophage cells specifically express CCR8, and by administering an antibody against CCR8, intratumorally infiltrating Treg cells and intratumoral tumors.
- the present invention has been completed by finding that the number of infiltrating macrophage cells is decreased and tumor growth is inhibited.
- the present invention (1) A pharmaceutical composition for cancer treatment containing an antibody against CCR8, (2) The pharmaceutical composition according to (1), wherein the antibody against CCR8 is an antibody having ADCC activity, (3) The pharmaceutical composition according to any one of (1) and (2), wherein the antibody against CCR8 is a neutralizing antibody of CCR8, (4) The pharmaceutical composition according to any one of (1) to (3), wherein the antibody against CCR8 has an action of removing intratumoral infiltrating Treg cells, (5) The pharmaceutical composition according to any one of (1) to (4), wherein the antibody against CCR8 has an action of removing intratumoral macrophage cells.
- a cancer therapeutic drug comprising a combination of an antibody against CCR8 and an anti-PD-1 antibody or an anti-PD-L1 antibody
- a method for treating cancer comprising administering the antibody against CCR8 according to any one of (1) to (5), (8-1) A method for treating cancer, comprising administering an antibody against CCR8, (8-2) The treatment method according to (8-1), wherein the antibody against CCR8 is an antibody having ADCC activity, (8-3) The method of treatment according to (8-1) or (8-2), wherein the antibody against CCR8 is a neutralizing antibody of CCR8, (8-4)
- the pharmaceutical composition containing the antibody of the present invention is very useful as a medicament for the treatment of cancer.
- CD25 molecule and FoxP3 molecule were stained with an antibody, and the expression rate was evaluated. It shows that CD25 expressing cells also express FoxP3.
- the flow cytometry analysis result is shown about CD45RA and CD25 expression intensity in the peripheral blood mononuclear cell (henceforth PBMC) of the same patient.
- CD3 + CD4 + T cells were fractionated into 6 (Fr1 to Fr6) by the expression levels of CD45RA and CD25 as shown in the figure, and the cells were collected with a sorter. A numerical value is a cell abundance ratio (%) of each fraction.
- the Treg fractions in this case are Fr1 and Fr2.
- the flow cytometry analysis result is shown about CD45RA and CD25 expression intensity of renal cancer tumor infiltrating cells.
- Intratumoral infiltrating CD3 + 4CD4 + T cells were fractionated into four (Fr2 to Fr5) by the expression levels of CD45RA and CD25 as shown in the figure, and the cells were collected with a sorter.
- a numerical value is a cell abundance ratio (%) of each fraction.
- the vertical axis represents the relative mRNA expression level after normalization.
- anti-mouse CCR8 antibody SA214G2
- ADCC antibody-dependent-cell-mediated-cytotoxicity-
- SA214G2 anti-mouse CCR8 antibody
- FIG. 5 shows that anti-mouse CCR8 antibody (SA214G2) has an activity of inhibiting intracellular calcium influx via CCR8.
- An isotype control antibody was used as a negative subject. It shows that anti-mouse CCR8 antibody (SA214G2) does not recognize CT26 cells. An isotype control antibody was used as a negative subject.
- a control antibody was administered to BALB / c mice transplanted with 3 mouse colon cancer cell lines CT26 cells on the third day (day 3) after transplantation, and the tumor was excised on the fourth or seventh day after administration.
- the result of having analyzed the Treg cell rate which exists in FIG. In the same experiment as FIG. 12, the result of having analyzed the CCR8 + Treg cell rate with the flow cytometer is shown. It is the figure which analyzed the CCR8 positive cell rate in the intratumor CD11b + (gamma) Gr + (+) CD206 + (M) type
- Anti-mouse CCR8 antibody (SA214G2) or isotype control antibody was administered to BALB / c mice transplanted with colorectal cancer cell line CT26 cells on day 3 after transplantation (day 3), and tumors were observed on day 7 or 10 after transplantation.
- the flow of the experiment which extracted T and verified the T lymphocyte and macrophage cell presence rate which exist there is shown.
- the ratio of CD25 + FoxP3 + cells in CD45 + CD4 + cells on day 7 (d7) or day 10 (d10) after transplantation is shown.
- the ratio of CD11b + F4 / 80 + macrophage cells on day 7 (d7) after transplantation is shown.
- the abundance ratio of IA / IE positive (IA / IE +) or IA / IE negative cells (IA / IE ⁇ ) on day 7 (d7) after transplantation is shown.
- a single dose of 400 ⁇ g / mouse of anti-mouse CCR8 antibody (SA214G2) or isotype control antibody (rat anti-KLH) was administered to BALB / c mice transplanted with colon cancer cell line CT26 cells on the third day after transplantation (d3).
- the flow of the experiment in which the tumor diameter was measured from 3 days after transplantation (d7) to 21 days (d21) every 3-4 days is shown. It is the result of measuring the solid tumor diameter of each individual after transplantation and calculating the tumor volume.
- a plot shows the average value (MFI) of the fluorescence intensity of each FACS analysis, a center horizontal line shows the average value of 14 examples of MFI value, and a vertical line shows a standard deviation.
- Significance level *** indicates P ⁇ 0.001.
- Ratio of cells showing CCR8 positive signal above background level with isotype control antibody for CD3 + CD4 + FoxP3 + T cells or CD3 + CD4 + FoxP3- T cells in 14 human renal cancer tumors (positive rate% ) Is plotted for each individual.
- the center horizontal line shows the average value of the positive rates of 14 cases, and the vertical line shows the standard deviation.
- Isotype control antibody (Isotype) was used as a negative subject.
- 3 ⁇ 10 5 osteosarcoma cell lines LM8 cells were transplanted into the back skin of C3H / He mice, and 3 days after transplantation (d3), 400 ⁇ g / mouse of anti-mouse CCR8 antibody (SA214G2) or isotype control antibody (control antibody) Single dose.
- Tumor volumes were measured from 7 days after transplantation until 35 days after every 3-4 days. The average value of the tumor volume at each time point after transplantation in each group is shown. The standard deviation is shown at the same time.
- Significance level *** indicates p ⁇ 0.001, significance level ** indicates p ⁇ 0.01, and significance level * indicates p ⁇ 0.05 (t-test).
- MethA cells 1 ⁇ 10 5 MethA cells were transplanted into the back skin of Balb / c mice, and on the third day after transplantation, anti-mouse CCR8 antibody (SA214G2) or isotype control antibody (control antibody) was administered once at 400 ⁇ g / mouse.
- Tumor volumes were measured from 3 days after transplantation until 21 days after 3 days. The mean value of the tumor volume at each time point after transplantation in each group is shown. Significance level * indicates p ⁇ 0.05 (t-test).
- EMT6 cells were transplanted into the back skin of Balb / c mice, and anti-mouse CCR8 antibody (SA214G2) or isotype control antibody was administered at 100 ⁇ g / mouse on the 3rd and 10th days after transplantation.
- Tumor volume was measured from 4 days after transplantation to every 22 days after 3-4 days. The average value of the tumor volume at each time point after transplantation in each group is shown. Significance level *** indicates p ⁇ 0.001, and significance level ** indicates p ⁇ 0.01 (t-test).
- Colon 26 cells were transplanted into the back skin of BALB / c mice, and on days 3 and 10 after transplantation, anti-isotype control antibody (Isotepe antibody), mouse CCR8 antibody (SA214G2) or anti-PD -1 antibody (RMP1-14) was administered at 400 ⁇ g / mouse. Tumor volume was measured every 3-4 days up to 24 days after 3 days after transplantation. The mean value of the tumor volume at each time point after transplantation in each group is shown.
- Isotepe antibody anti-isotype control antibody
- SA214G2 mouse CCR8 antibody
- RMP1-14 anti-PD -1 antibody
- Colon26 cells were transplanted into the back skin of BALB / c mice, and 3 days and 10 days after transplantation, 400 ⁇ g / ml of anti-mouse CCR8 antibody (SA214G2) or isotype control antibody (control antibody) was used. Administered in mice. 24 days after transplantation, mouse organs were collected and weighed. Average values for 10 cases are shown.
- 1 ⁇ 10 5 mouse breast cancer cell line EMT6 cells were transplanted into the back skin of BALB / c mice, 3 days and 10 days after transplantation for anti-mouse CCR8 antibody, 8 days after transplantation for anti-mouse PD-1 antibody It was administered intravenously on the eyes and on the 13th day.
- isotype control antibody was administered intravenously 3 days and 10 days after transplantation. Tumor volume was measured from 6 days after transplantation to 27 days after every 3-4 days. The mean value of the tumor volume at each time point after transplantation in each group is shown. In the same experiment as FIG. 35, the percentage of individuals whose tumors regressed to 50 mm 3 or less at each time point after transplantation in each group is shown.
- mice 4.5 ⁇ 10 5 mouse kidney cancer-derived cell lines RAG were transplanted into the back skin of BALB / c mice, and saline, anti-mouse CCR8 antibody, anti-mouse PD-1 antibody or anti-mouse 8 days and 15 days after tumor implantation
- Mouse CCR8 antibody and anti-mouse PD-1 antibody were intravenously administered at 100 ⁇ L, and the tumor volume was measured every 3 to 4 days until 33 days after 8 days after transplantation. The median tumor volume at each time point after transplantation for each group is shown.
- Tumor volume was measured every 3-4 days after transplantation. The left figure shows the mean value of the tumor volume at each time point after transplantation of each group for the wild type mouse, and the right figure shows the mean value of the tumor volume at each time point after transplantation of each group for the CCR8 gene homo-deficient mouse. .
- the pharmaceutical composition of the present invention contains an antibody against CCR8,
- the CCR8 of the present invention includes those derived from primate mammals including mice, rats, hamsters, guinea pigs, dogs, pigs, monkeys, and humans. Human CCR8 is preferred.
- the antibody against CCR8 is an antibody that binds to CCR8, it may be any of a human-derived antibody, a mouse-derived antibody, a rat-derived antibody, a rabbit-derived antibody, or a goat-derived antibody, or a polyclonal antibody or a monoclonal antibody thereof.
- a complete antibody, antibody fragment eg, F (ab ′) 2 , Fab ′, Fab or Fv fragment
- chimerized antibody humanized antibody or fully human antibody.
- the antibody of the present invention can be produced according to a known antibody or antiserum production method using the full-length protein or partial protein of CCR8 as an antigen. Since the antibody of the present invention preferably binds to CCR8 expressed on the cell surface, the partial protein is preferably an extracellular region of CCR8. These antigens can be prepared by known protein expression and purification methods.
- examples of antigens suitable for the production of antibodies against CCR8 include cells in which CCR8 is forcibly expressed by an expression vector or the like, CCR8 expression plasmid vectors, CCR8 expression virus vectors and the like (such as adenovirus vectors).
- Polyclonal antibodies can be produced by known methods. For example, it can be produced by immunizing a suitable animal with an antigen protein or a mixture of it and a carrier protein, collecting an antibody-containing substance against the antigen protein from the immunized animal, and performing separation and purification of the antibody. As the animal to be used, mouse, rat, sheep, goat, rabbit and guinea pig are generally mentioned. In order to enhance antibody production ability, complete Freund's adjuvant or incomplete Freund's adjuvant can be administered together with the antigen protein. Administration is generally performed about once every 2 weeks, about 3 to 10 times in total. Polyclonal antibodies can be collected from blood, ascites, etc. of animals immunized by the above method.
- the polyclonal antibody titer in the antiserum can be measured by ELISA.
- Separation and purification of polyclonal antibodies include, for example, a purification method using an antigen-binding solid phase or an active adsorbent such as protein A or protein G, salting-out method, alcohol precipitation method, isoelectric precipitation method, electrophoresis method, ion exchange It can be carried out according to immunoglobulin separation and purification methods such as body adsorption / desorption, ultracentrifugation, and gel filtration.
- Monoclonal antibodies can be prepared by known general production methods. Specifically, the antigen of the present invention is administered to a mammal, preferably a mouse, rat, hamster, guinea pig or rabbit subcutaneously, intramuscularly, intravenously, in a food pad, or intraperitoneally, together with Freund's adjuvant as necessary. Immunization is performed by one to several injections. Usually, immunization is performed 1 to 4 times about every 1 to 21 days from the initial immunization, and antibody-producing cells can be obtained from the immunized mammal about 1 to 10 days after the final immunization. The number of times of immunization and the time interval can be appropriately changed depending on the nature of the immunogen used.
- Hybridomas that secrete monoclonal antibodies can be prepared according to the method of Kohler and Milstein et al. (Nature, 1975, vol. 256, p495-497) and methods analogous thereto. That is, antibody-producing cells contained in the spleen, lymph node, bone marrow, tonsil, etc., preferably from the spleen obtained from the immunized mammal as described above, and preferably mouse, rat, guinea pig, hamster, rabbit or human
- a hybridoma can be prepared by cell fusion of a myeloma cell having no autoantibody-producing ability derived from a mammal such as a mouse, rat or human.
- myeloma cells used for cell fusion cell lines generally obtained from mice such as P3-U1, NS-1, SP-2, 653, X63, AP-1 and the like can be used. .
- Hybridoma clones producing monoclonal antibodies are screened by culturing the hybridomas in, for example, a microtiter plate, and reacting the culture supernatant of the wells in which proliferation has been observed with the antigen of the present invention used in the aforementioned mouse immunization. Sex is measured by a measuring method such as RIA, ELISA, FACS, and the like, and a clone producing a monoclonal antibody exhibiting specific binding to the antigen or hapten is selected. In general, a method is used in which an antibody in a culture supernatant bound to an antigen is immobilized with a secondary antibody labeled with a radioactive substance, a fluorescent substance, an enzyme, or the like.
- the hybridoma culture supernatant is added to the cells, and then a secondary antibody labeled with fluorescence is reacted. Then, the cells are detected using a fluorescence detection device such as a flow cytometer. By measuring the fluorescence intensity, a monoclonal antibody capable of binding to the antigen of the present invention on the cell membrane can be detected.
- Production of the monoclonal antibody from the selected hybridoma can be obtained by culturing the hybridoma in vitro or by culturing it in mouse, rat, guinea pig, hamster or rabbit, preferably mouse or rat, more preferably mouse ascites. It can be carried out by isolation from the culture supernatant or ascites of mammals.
- Examples of the basic medium include a low calcium medium such as Ham'F12 medium, MCDB153 medium or low calcium MEM medium, and a high calcium medium such as MCDB104 medium, MEM medium, D-MEM medium, RPMI1640 medium, ASF104 medium or RD medium.
- the basic medium can contain, for example, serum, hormones, cytokines and / or various inorganic or organic substances depending on the purpose.
- affinity ammonium chromatography such as saturated ammonium sulfate, ion exchange chromatography (DEAE or DE52, etc.), anti-immunoglobulin column or protein A column, etc. Can be performed.
- an antibody gene is cloned from an antibody-producing cell such as a hybridoma, incorporated into an appropriate vector, introduced into a host, and a recombinant antibody produced using gene recombination technology is used.
- an antibody-producing cell such as a hybridoma
- a recombinant antibody produced using gene recombination technology is used.
- variable region (V region) of an antibody is encoded from a hybridoma that produces the target antibody or an immune cell that produces the antibody, for example, a cell in which sensitized lymphocytes have been immortalized by an oncogene or the like.
- Isolate mRNA For isolation of mRNA, total RNA is prepared by a known method such as guanidine ultracentrifugation (Chirgwin, JM, et al., Biochemistry (1979) 18, 5294-5299), mRNA Purification Kit (manufactured by Pharmacia), etc. To prepare mRNA.
- the antibody V region cDNA is synthesized from the obtained mRNA using reverse transcriptase.
- the synthesis of cDNA can be performed using AMV Reverse Transscriptase First-strand cDNA Synthesis Kit or the like.
- 5'-Ampli FINDER RACEKit® manufactured by Clontech
- 5'-RACE method using PCR Frohman, MA, et al., Proc. Natl. Acad. Sci. USA
- the target DNA fragment is purified from the obtained PCR product and ligated with vector DNA.
- a recombinant vector is prepared from this, introduced into Escherichia coli, etc., and colonies are selected to prepare a desired recombinant vector.
- the base sequence of the target DNA is confirmed by a known method such as the deoxy method.
- DNA encoding the desired antibody V region is obtained, it is ligated with DNA encoding the desired antibody constant region (C region) and incorporated into an expression vector.
- DNA encoding the V region of the antibody may be incorporated into an expression vector containing DNA of the antibody C region.
- the antibody gene is incorporated into an expression vector so as to be expressed under the control of an expression control region, eg, an enhancer / promoter.
- host cells can be transformed with this expression vector to express the antibody.
- the expression of the antibody gene may be carried out by co-transforming the host by separately incorporating the heavy chain (H chain) or light chain (L chain) of the antibody into an expression vector, or DNA encoding the H chain and L chain. May be incorporated into a single expression vector to transform the host (see WO94 / 11523).
- phage display technology (Nature Biotechnology® 23, 1105 (2005)) can also be used. Specifically, for example, selection from an antibody gene library prepared by a known method using B lymphocytes of humans or animals (for example, rabbits, mice, rats, hamsters, etc.) or Germ Line sequences of humans or animals. And a modified and fully synthesized antibody gene library is displayed on the surface of cells such as bacteriophage, E. coli, yeast, animal cells, or on ribosomes. In this case, IgG molecules, IgM molecules, Fab fragments, single-chain Fv (scFv) fragments and the like can be mentioned as antibody forms to be presented on the cell surface.
- scFv single-chain Fv
- the antibody fragment gene thus obtained can be combined with a corresponding region of an IgG antibody gene by a known method to obtain an antibody gene. Then, the gene thus obtained can be incorporated into an appropriate vector, introduced into a host, and an antibody can be produced using a gene recombination technique (for example, Carl et al., THERAPEUTIC MONOCLONAL ANTIBODIES, 1990). Issued year).
- a gene recombination technique for example, Carl et al., THERAPEUTIC MONOCLONAL ANTIBODIES, 1990. Issued year).
- the antibodies of the present invention include antibodies artificially modified for the purpose of reducing the heteroantigenicity to humans, such as chimerized antibodies, humanized antibodies, and fully human antibodies.
- the antibody of the present invention may be a conjugated antibody bound to various molecules such as polyethylene glycol (PEG), radioactive substances, toxins, sugar chains.
- PEG polyethylene glycol
- Such a conjugated antibody can be obtained by chemically modifying the obtained antibody.
- Antibody modification methods have already been established in this field. These conjugate antibodies are also included in the antibody of the present invention.
- the antibody of the present invention includes an antibody in which an N-glycoside-linked sugar chain is bound to the Fc region of the antibody and fucose is not bound to N-acetylglucosamine at the reducing end of the N-glycoside-bound sugar chain.
- An antibody in which an N-glycoside-linked sugar chain is bound to the Fc region of the antibody and fucose is not bound to N-acetylglucosamine at the reducing end of the N-glycoside-linked sugar chain includes, for example, ⁇ 1,6-fucose transferase gene And antibodies produced using CHO cells deficient in (WO 2005/035586, WO 02/31140).
- the antibody of the present invention in which an N-glycoside-linked sugar chain is bound to the Fc region of the antibody and fucose is not bound to N-acetylglucosamine at the reducing end of the N-glycoside-linked sugar chain has high ADCC activity.
- the antibody of the present invention may be fused with other proteins at its N-terminus or C-terminus (Clinical Cancer Research, 2004, 10, 1274-1281). Those skilled in the art can appropriately select the protein to be fused.
- the antibody fragment means a fragment that is a part of the antibody of the present invention described above and has a specific binding property to CCR8 like the antibody.
- antibody fragments include Fab, F (ab ′) 2 , Fab ′, single chain antibody (scFv), disulfide stabilized antibody (dsFv), dimerized V region fragment (Diabody), CDR. Peptides and the like can be mentioned (Expert Opinion on Therapeutic Patents, Vol. 6, No. 5, pp. 441-456, 1996).
- the antibody of the present invention may be a bispecific antibody (bispecific antibody) having two different antigen-determining sites and binding to different antigens.
- ADCC Antibody-dependent-cell-mediated-cytotoxicity activity means that an antibody bound to a cell surface antigen such as a tumor cell in vivo is on the Fc region and the effector cell surface of the antibody. It means the activity of activating effector cells through binding to existing Fc receptors and damaging tumor cells and the like. Examples of effector cells include natural killer cells and activated macrophages.
- the antibody of the present invention is preferably an antibody having ADCC activity against cells expressing CCR8 from the viewpoint that Treg cells or macrophage cells can be removed. Whether or not the antibody of the present invention has such an ADCC activity can be measured, for example, by the method described in the Examples below.
- the antibody against CCR8 contained in the pharmaceutical composition of the present invention is preferably a neutralizing antibody of CCR8 from the viewpoint of suppressing accumulation of Treg cells or macrophage cells in the tumor.
- the neutralizing antibody for CCR8 means an antibody having neutralizing activity against CCR8. Whether or not it has a neutralizing activity for CCR8 can be determined by measuring whether or not the physiological action of CCL1 on CCR8 is suppressed. Examples include, but are not limited to, measuring the binding of CCL1 to CCR8, the migration of CCR8-expressing cells by CCL1, or an increase in intracellular Ca ++ or gene expression sensitive to CCL1 stimulation. Can be mentioned. Moreover, it can also measure by the method as described in the below-mentioned Example.
- the antibody against CCR8 of the present invention preferably has an action of removing infiltrating Treg cells in the tumor. Whether or not the antibody of the present invention has an action of removing intratumoral infiltrating Treg cells can be measured, for example, by the method described in Examples described later.
- the antibody against CCR8 of the present invention preferably has an action of removing tumor-infiltrating macrophage cells. Whether or not the antibody of the present invention has an action of removing intratumoral infiltrating macrophage cells can be measured, for example, by the method described in Examples below.
- the antibody of the present invention is useful as a pharmaceutical composition. Therefore, the pharmaceutical composition containing the antibody of the present invention can be administered systemically or locally orally or parenterally.
- parenteral administration for example, intravenous injection such as infusion, intramuscular injection, intraperitoneal injection, subcutaneous injection, intranasal administration, inhalation and the like can be selected.
- the “cancer” in the “pharmaceutical composition for cancer treatment” of the present invention includes all solid cancers and blood cancers. Specifically, breast cancer, endometrial cancer, cervical cancer, ovarian cancer, prostate cancer, lung cancer, stomach (gastric gland) cancer, non-small cell lung cancer, pancreatic cancer, squamous cell carcinoma of the head and neck, esophageal cancer, bladder cancer, melanoma , Colon cancer, renal cancer, non-Hodgkin lymphoma, urothelial cancer, sarcoma, blood cell cancer (leukemia, lymphoma, etc.), bile duct cancer, gallbladder cancer, thyroid cancer, prostate cancer, testicular cancer, thymic cancer, liver cancer, etc.
- breast cancer in the “pharmaceutical composition for cancer treatment” of the present invention is preferably a cancer that expresses a tumor-specific antigen.
- the “cancer” described herein includes not only epithelial malignant tumors such as ovarian cancer and gastric cancer but also non-epithelial malignant tumors including hematopoietic cancers such as chronic lymphocytic leukemia and Hodgkin lymphoma.
- epithelial malignant tumors such as ovarian cancer and gastric cancer
- non-epithelial malignant tumors including hematopoietic cancers such as chronic lymphocytic leukemia and Hodgkin lymphoma.
- the terms “cancer”, “carcinoma”, “tumor”, “neoplasm” and the like are not distinguished from each other and It is exchangeable.
- the antibody against CCR8 of the present invention is: (1) complementation and / or enhancement of the therapeutic effect of the pharmaceutical composition of the present invention, (2) Dynamics, absorption improvement, dose reduction and / or of the pharmaceutical composition of the present invention (3) Reduction of side effects of the pharmaceutical composition of the present invention, Therefore, it may be administered as a concomitant drug in combination with other drugs.
- the combination of the antibody against CCR8 of the present invention and another drug may be administered in the form of a combination preparation in which both components are mixed in one preparation, or may be administered in separate preparations.
- simultaneous administration and administration by time difference are included.
- administration by time difference may be such that the antibody of the present invention is administered first and the other drug may be administered later, or the other drug may be administered first and the compound of the present invention may be administered later.
- the administration method may be the same or different.
- Examples of other drugs that may be used in combination with the antibody against CCR8 of the present invention include anti-PD-1 antibody, anti-PD-L1 antibody, and anti-CTLA-4 antibody.
- Anti-PD-1 antibody or anti-PD-L1 antibody is preferable, and anti-PD-1 antibody is more preferable.
- examples of the anti-PD-1 antibody include nivolumab and pembrolizumab.
- the anti-PD-L1 antibody includes, for example, atezolizumab, avelumab, and durvalumab.
- examples of the anti-CTLA-4 antibody include ipilimumab.
- the target patient of the pharmaceutical composition of the present invention is a cancer patient or suspected.
- the effective dose is selected from the range of 0.01 mg to 100 mg per kg of body weight at a time.
- a dose of 5 to 5000 mg, preferably 10 to 500 mg per patient can be selected.
- the pharmaceutical composition containing the antibody or antibody fragment thereof of the present invention is not limited to these doses.
- the administration period can be appropriately selected depending on the age and symptoms of the patient.
- the pharmaceutical composition of the present invention may contain both pharmaceutically acceptable carriers and additives depending on the route of administration.
- Such carriers and additives include water, pharmaceutically acceptable organic solvents, collagen, polyvinyl alcohol, polyvinyl pyrrolidone, sodium alginate, water-soluble dextran, pectin, methyl cellulose, ethyl cellulose, casein, diglycerin, propylene glycol , Polyethylene glycol, petrolatum, human serum albumin (HSA), mannitol, sorbitol, lactose, surfactants acceptable as pharmaceutical additives, and the like.
- the additive to be used is selected appropriately or in combination from the above depending on the dosage form, but is not limited thereto.
- PBMC Peripheral blood mononuclear cells
- Intratumoral infiltrating cells were reacted for 30 minutes in ice using anti-CD3 antibody (BioLegend, Clone UCHT1), anti-CD4 antibody (BioLegend, Clone OKT4), and anti-CD25 antibody (BioLegend, Clone BC96). Staining was performed. After washing twice with 2% FCS / HEPES / HBSS, the cells were fixed and permeabilized according to the protocol attached to the kit using Foxp3 / TransscriptscFactor Staining Buffer Set (eBioscience, 00-5523-00). Further, FoxP3 was stained with PE-labeled anti-FoxP3 antibody (eBioscience, Clone PCH010).
- the tumor-infiltrating cells and PBMC were stained with anti-CD3 antibody, anti-CD4 antibody, anti-CD45RA antibody (BD Biosciences, Clone HI100) and anti-CD25 antibody, and CD3 + CD4 + T cells were expressed in CD45RA and CD25 expression levels.
- Two-dimensional development. The result of PBMC is FIG. 2, and the result of the tumor infiltrating cells is shown in FIG.
- CD3 + CD4 + CD45RA- ⁇ using a cell sorter (FACSAria II) and CD25 expression intensity as an index as shown in FIG.
- PBMC strongly positive cells
- Fr3 strongly positive cells
- Fr4 and Fr5 weakly positive cells
- PBMC was also developed two-dimensionally in the same manner as the tumor-infiltrating cells, and fractionated into Fr1 to Fr6 as shown in FIG. 2 using the expression intensity of CD45RA and CD25 as an index, and the cells contained in each fraction were collected.
- RNA from fractionated cells were dissolved in RLT buffer (Qiagen), and total RNA was extracted using Agencourt RNAClean XP (Bechman Coulter). The recovered RNA was converted to cDNA using SMART-Seq v4 Ultra Low Input RNA kit for Sequencing (Clontech), and the library was prepared using KAPA Hyper Prep Kit for Illumina (Kapa Biosystems). For cDNA synthesis and library preparation, quality control was always performed using Agilent 2100 Bioanalyzer (Agilet Technologies), and it was confirmed that there was no problem.
- the completed cDNA library was titrated using KAPA library Quantification kit Illumina Platforms (Kapa Biosystems), then subjected to DNA sequencing with paired-end reads using Hiseq 4000 (illumina), and 100 million pairs of sequences per sample. Acquired more than lead (fastq file). The raw data (fastq file) was analyzed by FastQC, and adapter sequences and repeat sequences were removed using CutAdapt. Each paired end lead was aligned using the cmpfastq_pe program. Genome mapping was performed with hg38 as a reference sequence and mapped to the genome with the default setting by the TOPHAT2 program having Bowtie2. The mapped reads were sequence-sorted by the SAMtools program and read-counted by the HTSEQ program. The Deseq2 program was used for normalization of the count data. About each obtained fraction, which fraction contained Treg cell was confirmed with the following method.
- Treg cells constantly express FoxP3 and Ikzf2 genes as marker genes, and hardly secrete IFN ⁇ or IL2 even when activated by stimulation. Whether Treg cells are contained or not can be confirmed to some extent by examining the expression levels of these genes. Based on the RNA-Seq data, the expression levels of these genes were examined for each fraction of tumor infiltrating cells and PBMC. As a result, Ikzf2 and FoxP3 were specifically expressed in Fr2, Fr3, and Fr2 of PBMC in the tumor infiltrating cells. It was found that it was expressed and hardly expressed in other fractions (FIG. 4).
- IFN ⁇ IFNgamma
- IL2 IFNgamma
- Tr cells were contained in Fr2, Fr3, and Fr2 of PBMC in tumor infiltrating cells, but not in other fractions.
- the demethylation rate in the FoxP3 region is an index for accurately determining the proportion of Treg cells
- the depletion of the FoxP3 region in the Fr2 to Fr5 cells of the renal cancer tumor infiltrating cells obtained above is performed.
- the methylation rate was examined.
- the specific CpG region in the first intron of the FoxP3 gene there is a region that is demethylated specifically in Treg cells (chrX, 4918000-491118500, hg19).
- the fraction obtained this time consists only of Treg cells, or other cells are also mixed. Can be verified.
- Genome DNA was treated with Bisulfite using the Methyl Easy Xceed kit (Human Genetic Signatures) and the TREG cell-specific demethylation region FOXP3 intron1 region (chrX, 4918000-491118500, hg19). It was. Detection of DNA methylation was performed using a Quant Studio 3D digital PCR system (Applied Biosystems) with a methylated DNA-specific FAM fluorescent probe and a demethylation-specific VIC fluorescent probe.
- each patient's PBMC-derived CD4 + T-cell fraction is identical to each tumor-derived CD4 + T-cell fraction
- Hierarchical cluster analysis was performed on the expression data, and CCR8 was identified as a gene that was expressed in Fr2 in Treg cells and almost not expressed in tumor-derived Fr5 and Fr4 and Fr5 of PBMC (FIG. 6).
- mouse CCR8 forced expression cells The full length ORF of mouse CCR8 (hereinafter sometimes referred to as mCCR8) was inserted into an expression vector (pcDNA3.4) to construct pcDNA3.4-mCCR8 plasmid.
- the base sequence was changed to a codon that is frequently used in mammals as long as the amino acid was not changed.
- the pcDNA3.4 and pcDNA3.4-mCCR8 expression plasmids were introduced into HEK293 cells using Lipofectamine 3000, respectively, and drug selection was performed at Geneticin (G418) concentration of 1 mg / ml for 2 weeks.
- Cells transformed with pcDNA3.4 were only drug selected and not sorted. In order to confirm the expression, both cells were stained with a commercially available anti-PE-labeled anti-mouse CCR8 antibody (clone SA214G2) and analyzed with a flow cytometer (FACSAria II). The result is shown (FIG. 7). Compared with the cells transformed with pcDNA3.4, cells transformed with pcDNA3.4-mCCR8 showed expression of mCCR8 in 99% or more of the cells.
- shaft shows a light emission amount relative value. From FIG. 8, the maximum activity value was about 6000 Relative Light Unit (R.L.U), and the EC50 value (about 3500 R.L.U) was about 0.1 .mu.g / ml (line in the figure).
- SA214G2 anti-mouse CCR8 antibody
- the mCCR8 stable expression HEK293 cells prepared in Example 5 were used to evaluate the cytotoxic activity of the anti-mCCR8 antibody (SA214G2).
- SA214G2 anti-mCCR8 antibody
- the spleen of C57BL / 6 mice was isolated, and the spleen cells were collected through a cell strainer. After washing the cells, biotinylated anti-CD49b (clone DX5) antibody was reacted at 4 ° C. for 30 minutes, and after washing, NK cells were purified using streptavidin microbeads (Miltenyi) to obtain effector cells.
- Mouse CCR8-expressing HEK293 cells were stained at a final concentration of 2.5 uM using Cell Trace Violet (CTV) (Thermo Fisher, C34557) to obtain target cells (target cells).
- CTV Cell Trace Violet
- Annexin V-PE PE-labeled Annexin V
- MBL MBL
- 4696-100 PE-labeled Annexin V
- the addition of anti-mouse CCR8 antibody significantly increased the Annexin V positive cell rate in the target cells by about 6 times. From the above, it was found that the anti-mouse CCR8 antibody (SA214G2) has ADCC activity.
- HEPES WAKO CAS.NO7365-45-9
- HBSS (+) without Phenol Red
- Pluronic F127 P3000MP; Life Technology
- 10 mM HEPES / HBSS / 0.1% BSA Buffer (added to HBSS to a final concentration of 10 mM HEPES and a final concentration of 0.1% BSA, respectively)
- Fluo3-AM was dissolved in 10 ⁇ M HEPES / HBSS Buffer at a final concentration of 4 ⁇ mol / L and Pluronic F127 at 0.04%.
- the cells were suspended in this solution and incubated at 37 ° C. for 1 hour, so that Fluo3-AM was taken into the cells.
- the cells were then washed 3 times with 10 mM HEPES / HBSS / 0.1% BSA solution and suspended in 10 mM HEPES / HBSS / 0.1% BSA solution containing 1.25 uM probenecid to a cell concentration of 2 ⁇ 10 5 / ml. It became cloudy. Then, it was incubated for 10 minutes at 37 ° C. in a CO 2 incubator.
- anti-mCCR8 antibody SA214G2
- Isotype Control antibody was added at a concentration of 5 ⁇ g / ml.
- the cells were placed in a spectrophotometer HITACHI F7000, in which a 2 mL solution was placed in a quartz glass cuvette and the temperature of the measurement chamber was set to 35 ° C. in advance.
- the measurement conditions are as follows. Excitation wavelength 508.0 nm, fluorescence (measurement) wavelength 527.0 nm, excitation side slit 5 nm, fluorescence side slit 5 nm, photomultiplier voltage 950 V, response 0.5 s
- the mixture was incubated for about 30 seconds with stirring with a stirrer until the fluorescence wavelength stabilized.
- mouse CCL1 was added to a final concentration of 50 nM (4 ⁇ L), and measurement was started.
- anti-mCCR8 antibody in advance, intracellular calcium influx by mCCL1 was almost completely suppressed (FIG. 10). No inhibition was observed when the control antibody was added.
- the gap in the graph is when the lid of the device is opened and closed to administer the agonist to the cells. From the above, it was found that the anti-mCCR8 antibody (SA214G2) antibody has neutralizing activity against mouse CCR8.
- 400 ⁇ g of rat anti-KLH (keyhole limpet hemocyanin, Clone LTF-2) antibody (IgG2b) was intraperitoneally administered.
- the tumor mass of CT26 cells was cut finely with scissors and a commercially available kit (Tumor Dissociation Kit, mouse, Miltenyi and the gentleMACS (TM) Dissociator, Miltenyi Biotech cat. 130-929-929 according to the protocol). Infiltrating cells were prepared. The prepared cells were passed through a 70 um cell strainer and then washed twice with 10 mM HEPES / HBSS / 2% FBS. Thereafter, the cells were treated with an erythrocyte lysate (Miltenyi) for 5 minutes to remove erythrocytes, and further washed twice with 2% FCS (Metal Calf Serum) / 10 mM HEPES / HBSS buffer.
- TM gentleMACS
- Tumor-infiltrating cells were divided into two, one identified Treg cells and the other identified myeloid (macrophage) cells.
- Cells were stained with the following methods and antibodies.
- the used antibodies, staining reagents, and assay buffers are as follows.
- the staining method is as follows. Infiltrating cells were stained on ice for 30 minutes using Zombie NIR Fixable Viability Kit reagent. After washing once with 2% FCS / 10 mM HEPES / HBSS, for Treg and CCR8 positive cells, Bv510-labeled anti-CD45, PerCP. The cells were stained with Cy5.5-labeled anti-mouse CD4, FITC-labeled anti-mouse CD8, Bv421-labeled anti-mouse CD25, and AF647-labeled anti-mouse CCR8 antibody (or AF647-labeled isotype control antibody).
- CD45 + CD4 + T cells were analyzed. A negative cell region was determined by staining with an isotype control antibody in CD45 + CD4 + T cells, and cells positive for both anti-mouse CD25 and anti-mouse FoxP3 antibodies were used as Treg cells 4 days after administration (7 days after transplantation) And the presence frequency 7 days after administration (10 days after transplantation) was calculated. As a result, about 23% (4d) and about 30% (7d) of CD45 + CD4 + T cells in the mouse tumor were CD25 + FoxP3 + cells (FIG. 12).
- CCR8 expression in CD45 + CD4 + CD25 + FoxP3 + T cells was analyzed.
- a negative cell region was determined by staining CD45 + CD4 + CD25 + FoxP3 + T cells with an isotype control antibody, and cells positive with anti-mouse CCR8 antibody were used as CCR8 + Treg cells 4 days after administration (7 days after transplantation) and 7 days after administration.
- the existence frequency of (10 days after transplantation) was calculated (FIG. 13).
- about 50% (4d) and about 67% (7d) of CD45 + CD4 + CD25 + FoxP3 + T cells in the mouse tumor were CCR8 + cells (FIG. 13).
- CCR8 was expressed in at least CD4 + CD25 + FoxP3 + T cells and CD11b + Gr1 + CD206 + macrophages (referred to as M2-type macrophages) among the tumor infiltrating cells.
- Tumors were collected 7 days after tumor implantation (4 days after antibody administration) and 10 days after antibody administration (7 days after antibody administration), and infiltrated cells in the tumor were prepared and analyzed (FIG. 15).
- Intratumoral infiltrating Treg cells were collected by the same method as in Example 10. The antibody used is the same as in Example 10.
- infiltrating cells were stained in ice for 30 minutes using ZombieiNIR Fixable Viability Kit. After washing once with 2% FCS / 10 mM HEPES / HBSS, Bv510-labeled anti-CD45, PerCP. The cells were stained with Cy5.5-labeled anti-mouse CD4, FITC-labeled anti-mouse CD8 antibody, Bv421-labeled anti-mouse CD25, and AF647-labeled anti-mouse CCR8 antibody (or an AF647-labeled isotype control antibody). Staining was performed in ice for 30 minutes.
- the cells were fixed using a commercially available kit (FoxP3 staining kit, eBioscience) according to the attached protocol, and intracellular FoxP3 was stained using a PE-labeled anti-FoxP3 antibody. After washing with the buffer attached to the kit, the cells were analyzed using a flow cytometer.
- CD45 + CD4 + FoxP3 + CD25 + cells were used as mouse Treg cells.
- the negative cell region was determined by staining with an AF647-labeled isotype control antibody in Treg cells, and the frequency of cells that were positive with AF647-labeled anti-mouse CCR8 antibody as compared to the control was determined as CCR8-positive cells. .
- CCR8-positive cells As a result, as shown in FIG.
- Treg cell T cell ratio of the isotype antibody-administered mouse was 100% (10 days later), the same cell (Treg) in the anti-mouse CCR8 (SA214G2) antibody-administered mouse
- the positive rate of cells was about 80% 7 days after tumor implantation (4 days after antibody administration) and about 40% after 10 days (7 days after antibody administration) (FIG. 16).
- the significance level ** was P ⁇ 0.01 (t test). From the above, it was shown that about 60% of the tumor-infiltrating Treg cells were removed by anti-CCR8 antibody 7 days after administration of anti-CCR8 antibody.
- FSC / SSC + Tumor-infiltrating cells were isolated from the tumor on the 7th day after transplantation (d7) in the same manner as described above, and the CD45 + cells were gated to the myeloid population by FSC / SSC (denoted as FSC / SSC +).
- FSC / SSC + a marker for mouse mature macrophages and monocytes.
- F4 / 80 (Ly719) is a marker for mouse mature macrophages and monocytes.
- the graph shows the abundance of F4 / 80 + cells in the CD45 + FSC / SSC + mononuclear cell population.
- FIG. 17 shows the abundance ratio of IA / IE positive or class 2 (IA / IE) negative cells in M4 (tumor histocompatibility antigen) class 2 molecules of F4 / 80 + cells.
- T test significance level *; P ⁇ 0.05
- Tumor volume (mm 3 ) was weighed by major axis (mm) ⁇ minor axis (mm) ⁇ minor axis (mm) / 2 (FIG. 19).
- Tumor volume in the anti-mCCR8 antibody administration group decreased (significance levels were on days 11 and 14, ***; P ⁇ 0.001, days 17 and 21 **; P ⁇ 0.01 ).
- the tumor volume decreased from the 14th day and disappeared almost completely on the 17th day (data by individual is shown in FIG.
- CT26 was used to evaluate the efficacy of anti-PD-1 antibody (clone RMP1-14, Bio X Cell), which is a specific antibody against mouse PD-1, and compared with anti-mCCR8.
- 2 ⁇ 10 5 colon cancer-derived CT26 cells (50 ⁇ L) were transplanted into the back skin of a mouse Balb / c mouse (7 weeks old, female).
- anti-PD-1 antibody 200 ⁇ g / head, ip
- the anti-PD-1 antibody significantly suppressed the increase in tumor volume compared to the control at any time point on days 14, 17, and 20 after transplantation. However, the number of individuals in which the tumor completely disappeared was 1 in 8 during the observation period (up to the 20th day after transplantation).
- Example 12 Confirmation of presence or absence of induction of autoimmune disease in anti-mCCR8 antibody-administered mice
- the condition of mice up to 18 days after administration in Example 12 was evaluated. There was no significant difference in body weight during the period between the control antibody administration group and the anti-CCR8 antibody administration group. Neither group had raised hairs. It was dissected 18 days after administration.
- the anti-CCR8 antibody-administered group compared with the control, it was examined whether the lymph node and intestinal tract were enlarged, but there was no difference between them and no enlargement was observed. From the above findings, it was concluded that no signs of autoimmune disease were observed in the anti-CCR8 antibody-administered mice during the period when the anti-tumor effect was exerted.
- Tumor volume (mm 3 ) was weighed by major axis (mm) x minor axis (mm) x minor axis (mm) / 2. When the tumor reached the endpoint volume (800 mm 3 ), it was taken as the endpoint of each animal. As a result, tumor volume increase was suppressed in the anti-mCCR8 administration group compared to the isotype control antibody administration group on the 14th and 18th days after transplantation.
- Mean tumor volume at day 14 451.3Mm 3 isotype control antibody administered group (standard deviation ⁇ 177.5mm 3), 322.6mm 3 (standard deviation with an anti-CCR8 antibody administered group ⁇ 146.0mm 3
- the number of individuals with a tumor volume on day 14 of 350 mm 3 or more was 9 out of 10 in the isotype control group and 4 out of 10 in the anti-mCCR8 administration group.
- there was a significant difference at P 0.019. Therefore, there was a difference in the number of individuals that reached a tumor volume of 350 mm 3 on day 14.
- the mean tumor volume at day 18 post-transplantation 874.7mm 3 (standard deviation ⁇ 269.2mm 3) with an isotype control antibody administration group, 585.4mm 3 (standard deviation with an anti-CCR8 antibody administration group ⁇ 401.7 mm 3 ) (FIG. 22).
- Individuals with a tumor volume of 600 mm 3 or more on day 18 were 9 out of 10 cases in the isotype control group, whereas 4 out of 10 cases were in the anti-mCCR8 administration group.
- CCR8 expression analysis in 14 human renal cancer intratumoral infiltrating cells was performed.
- the background of 14 patients with renal cancer was 11 males and 3 females, the median age was 68.5 years, the pathological stage was 6 T1A, 2 T1B, 5 T3A, There was one T3b.
- renal tumor primary tumor infiltrating cells in 14 renal cancer (Clear Cell Renal Cell Carcinoma, ccRCC) patients were isolated in the same manner as in FIG.
- Example 1 Anti-CD4 (BioLegend, Clone OKT4), anti-CD3 (BioLegend, Clone UCHT1), anti-CD45RA (BD Biosciences, Clone HI100), anti-CD8 (Biolegend, RPA-T8), anti-CCR8 (BioLegend, Clone L263G8 / E7), stained with an anti-FoxP3 isotype control antibody and analyzed by flow cytometry (BD Biosciences, BD LSRFortessa) .
- CD3 + CD8 + T cells and CD3 + CD4 + T cells were analyzed.
- CD3 + CD4 + T cells were further divided into two groups according to the presence or absence of FoxP3 expression.
- a negative control for FoxP3 expression was obtained by staining with an isotype control antibody.
- an average value (MFI) of FACS analysis values of each patient sample was used.
- Table 1 shows the mean and standard deviation of MFI values of staining with anti-CCR8 antibody and its isotype control antibody.
- FIG. 23 is a graph showing the results of Table 1. Each plot of the graph shows an average value (MFI) of CCR8 expression level of each patient sample by a flow cytometer. The horizontal line of the graph indicates the average value of the MFI values of each sample. Bars indicate standard deviation. Significance level *** indicates P ⁇ 0.001.
- CCR8 protein is specifically expressed on the surface of CD3 + CD4 + FoxP3 + T cells infiltrating in the tumor in human renal cancer (ccRCC). This result is consistent with the mRNA expression analysis by RNA-Seq analysis.
- flow cytometric analysis of FoxP3 and CCR8 was performed on tumor-infiltrating CD4 + T cells. The ratio of CCR8 positive cells in FoxP3 positive cells and the ratio of CCR8 positive cells in FoxP3 negative cells were plotted for each sample. (FIG. 24).
- staining with an isotype control antibody was used as a negative standard, and cells above this threshold were defined as positive cells.
- the CCR8 expression rate of intratumoral CD3 + CD4 + FoxP3 + T cells was about 75%, and the CCR8 expression rate of CD3 + CD4 + FoxP3-T cells was about 10%. From the above results, it was found that CCR8 is expressed in most Treg cells expressing FoxP3 among infiltrating cells in human renal cancer tumors, and CCR8 is expressed in about 10% of CD4 positive T cells other than Treg cells. . As described above, the expression rate of CCR8 in FoxP3-positive Treg cells in human tumors is similar to the expression rate of CCR8 in Treg cells in mouse tumors, and anti-human CCR8-specific antibodies in the tumor infiltrate FoxP3 as in mice. The possibility of removing most of the positive Treg cells was shown.
- FoxP3 gene has been identified as a gene that is specifically expressed in Treg cells and not expressed in tumor cells or most normal human cells.
- marker genes that are expressed only in certain specific cells for example, FoxP3 gene as a marker gene for Treg cells, CD3G gene as a marker gene for T cells and NK cells, CD8A gene as a marker gene for CD8 positive T cells, etc. It has been known.
- FoxP3 gene which is a marker gene of Treg cells, it has also been reported that it can be used as an index of the presence ratio of Treg cells in tumors by measuring the expression level of FoxP3 gene mRNA in each tumor.
- RNA-Seq data is a mixture of mRNAs expressed in both tumor cells and infiltrating cells (lymphocytes, vascular cells, etc.), but they are not expressed in tumor cells.
- the expression level of the marker gene in the tumor mass can be taken as the product of the number of expressed cells of the specific cell corresponding to the marker gene infiltrating there and the expression level of each expressed cell. Assuming that the expression level of the marker gene in each cell is almost constant among individuals, the expression level is directly proportional to the number of infiltrating cells. Therefore, by using this expression level, the number of cells expressed in the tumor can be calculated for each individual, and comparison between individuals becomes possible.
- CCR8 expression analysis at the cell level RNA expression data of 1037 human cell lines is registered in CCLE (Cancer Cell Line Encyclopedia) which is a public database. Using these databases, it was analyzed whether CCR8 and CD3G genes were expressed in cancer cells other than T cells or normal cells. With respect to renal cancer, prostate cancer and bladder cancer-derived cell lines, the mRNA expression of CD3G and CCR8 was analyzed using the CCLE database.
- the cell lines examined were kidney cancer-derived cell lines VMRCRCW, SKRC20, SNU34, SKRC31, UOK10, SLR20, OSRC2, TUHR14TKB, SLR24, HK2, A498, RCC4, KMRC1, RCC10RGB, ACHN, SLR25, SNU1272 40 types of VMRCRCZ, KMRC3, KMRC20, CAKI2, BFTC909, 786O, A704, TUHR10TKB, SLR26, UMRC2, CAL54, FURPNT1, FURPNT2, HEK293, G402
- VMRCRCW kidney cancer-derived cell lines
- SKRC20, SNU34, SKRC31 UOK10
- SLR20 OSRC2
- TUHR14TKB SLR24
- VMRCRCZ VMRC3, KMRC20, CAKI2, BFTC909, 786O
- CCR8 and CD3G were the same level as the background, and mRNA expression was not observed at all (the value showing the maximum expression was 1/500 or less of G3PDH. Except for 1/1000 of the expression level of G3PDH). That is, it was confirmed that CCR8 and CD3G were hardly expressed in solid cancer cells.
- the primary normal cells derived from human tissues were similarly analyzed, and it was found that CCR8 and CD3G are expressed only in a part of blood cells, and are hardly expressed in other primary normal tissue-derived cells. did. From the above, it was shown that CCR8 and CD3G were not expressed in these three types of cancer cells. Therefore, when using TCGA RNA expression data for tumor masses of renal cancer, prostate cancer, and bladder cancer, it is concluded that CCR8 and CD3G reflect mRNA expression in infiltrating normal cells present in the tumor mass other than cancer cells. did.
- CCR8 is expressed in infiltrating cells other than T cells
- a cell population expressing CCR8 is used here. It has already been reported in papers that CD3G is specifically expressed in T cells and NK cells, and since T cells are the main cells infiltrating tumors, the expression level of CD3G is infiltrating T cells. It can be compared with the number. Therefore, the CCR8 / CD3G value can be defined as the number of cells expressing CCR8 per T cell number present in the tumor. The CCR8 / CD3G ratio and patient survival rate of these three types of carcinomas were analyzed using Kaplan-Meier curves.
- Kidney cancer was obtained from Kidney Rene Clear Cell Carcinoma (TCGA, Provisional) data among TCGA data, and 523 cases with RNA expression data and patient survival data were used. Similarly, 490 cases of prostate cancer in which RNA expression data and patient survival rate data were prepared from Prostate Adenocarcinoma (TCGA, Provisional) data among TCGA data were used. For bladder cancer, 392 cases of RNA expression data and patient survival data were used from the TCGA data of the bladder urological carcinoma (TCGA, Provisional).
- CCR8 / CD3G The expression level of CCR8 / CD3G is equally divided into two groups, a high group and a low group (261: 262 because it is an odd number in the case of renal cancer), and Kaplan Meier survival curve analysis is performed using analysis software R (R-Studio). I went. The significant difference test was Log-rank test.
- FIG. 25 shows the results of renal cancer
- FIG. 26 shows the results of prostate cancer
- FIG. 27 shows the results of bladder cancer.
- the vertical line in the graph treats the patient as surviving (corresponding to a so-called sensor) because the patient is alive but the evaluation period is only up to this point.
- the value on the horizontal axis represents the number of months in all graphs.
- the patient survival rate was significantly low in the group having a high CCR8 / CD3G value in all three types of carcinoma. It was found that the survival rate decreased in a group with a high ratio of CCR8-expressing cells infiltrating in human tumors in T cells. This indicates that CCR8-expressing cells also have an inhibitory effect on tumor immunity in humans. It is suggested that From this, in the same way as the anti-tumor effect of anti-mCCR8 antibody administration in mice, tumor immunity can be increased and the survival rate can be increased in humans by specifically removing or killing CCR8-expressing cells in some way. It is suggested that it may increase.
- Tumor volume (mm 3 ) was weighed by major axis (mm) x minor axis (mm) x minor axis (mm) / 2 ( Figure 29).
- the mean value of the tumor volume was significantly decreased in the anti-mCCR8 administration group compared with the isotype control antibody administration group at the time of all measurements after the 18th day of transplantation (significance level was * on the 18th day; P ⁇ 0 .05, 21, 24, 27, 31st day **; P ⁇ 0.01, 35th day ***; P ⁇ 0.001).
- Tumor volume (mm 3 ) was weighed by major axis (mm) x minor axis (mm) x minor axis (mm) / 2 ( Figure 30).
- the mean value of the tumor volume was significantly decreased in the anti-mouse CCR8 antibody-administered group compared with the isotype control antibody-administered group at all measurement points on and after the 11th day after transplantation (significance levels were * at all time points; P ⁇ 0.05).
- Tumor volume (mm 3 ) was weighed by major axis (mm) x minor axis (mm) x minor axis (mm) / 2 (Fig. 31).
- the mean value of the tumor volume was significantly decreased in the anti-mouse CCR8 antibody administration group compared to the isotype control antibody administration group at the time of all measurements after the 10th day after transplantation (the significance level is ** on the 10th day; P ⁇ 0.01, 14, 17, 21 days are ***; P ⁇ 0.001).
- Tumor volume (mm 3 ) was weighed by major axis (mm) x minor axis (mm) x minor axis (mm) / 2 (Fig. 32). As a result, the tumor volume was significantly reduced in the anti-mouse CCR8 administration group on the 17th, 20th, and 24th days compared to the isotype antibody administration group (Steel non-parametric test: significance level was P ⁇ 0.05). ). In the anti-PD-1 antibody administration group, no significant difference was observed at any time point as compared to the isotype antibody administration group.
- the tumor volume was significantly decreased in the anti-mouse CCR8 administration group compared to the anti-mouse PD-1 antibody administration group (Steel-Dbass non-parametric test; the significance level was P ⁇ 0.05).
- the significance level was P ⁇ 0.05.
- kidney cancer-derived cell line RAG kidney cancer-derived cell line
- 4 ⁇ 10 5 renal cancer-derived RAG cells 50 uL were transplanted into the back skin of Balb / c mice (8 weeks old, female).
- RAG cells were transplanted to a Balb / c mouse subcutaneously and engrafted with a tumor again. This operation was repeated twice, and the RAG cells (cell-adapted strains) that had improved the engraftment efficiency into the subcutaneous mouse. )It was used.
- Tumor volume was measured every 3-4 days from 6 days after tumor implantation.
- Tumor volume (mm 3 ) was weighed by major axis (mm) x minor axis (mm) x minor axis (mm) / 2 ( Figure 33).
- the tumor volume decreased significantly on the 14th, 17th, and 21st days after administration in the anti-mouse CCR8 antibody administration group compared to the isotype antibody administration group (Steel non-parametric test: significance level P ⁇ 0 .05). There was no significant difference in the anti-mouse PD-1 antibody administration group compared to the isotype antibody administration group. From the above, an anti-tumor therapeutic effect was observed by administration of anti-mouse CCR8 antibody in a renal cancer cell line.
- the tumor volume was significantly decreased in the anti-mouse CCR8 antibody administration group compared to the anti-mouse PD-1 antibody administration group (Steel-Dbass non-parametric test; the significance level was P ⁇ 0. 05).
- the significance level was P ⁇ 0. 05.
- mice 2 ⁇ 10 5 colon cancer-derived Colon26 cells (50 uL) were transplanted into the back skin of mouse Balb / c mice (7 weeks old, female).
- the body weight on the 24th day of transplantation and the weight of each mouse organ were measured (FIG. 34).
- CCR8 in invasive cells in human renal cancer, ovarian cancer, endometrial cancer, colon cancer, and lung cancer was performed.
- the number of patients with various clinical tumors used for expression analysis is 12 for renal cancer, 14 for ovarian cancer, 21 for endometrial cancer, 10 for colon cancer, and 4 for lung cancer.
- Various clinical tumor infiltrating cells were isolated in the same manner as in FIG. 1 of Example 1, stained with Anti-CD45 (BioLegend, Clone H130) and anti-CCR8 (BioLegend, Clone L263G8) antibodies, and flow cytometry (BD Biosciences, BD LSRFortessa).
- the number of CCR8 positive cells per tumor weight and the ratio of CCR8 positive cells in CD45 positive leukocytes were analyzed.
- Table 2 shows the average number of CCR8 positive cells per tumor weight and its standard deviation.
- Table 3 shows the average value and the standard deviation of the ratio of CCR8 positive cells in CD45 positive leukocytes.
- 15 ⁇ g (100 ⁇ L) of rat anti-mouse CCR8 antibody was intravenously administered on the 3rd and 10th days of tumor implantation, and the 8th and 13th days of tumor implantation.
- was administered intravenously with 200 ⁇ g (100 ⁇ L) of anti-PD-1 antibody (N 10).
- the tumor was small in the combination group compared with each single administration group.
- the complete remission rates of tumors on the 17th and 27th days after transplantation were also compared.
- 0 out of 10 animals in the control group and anti-PD-1 antibody administration group, and 1 out of 10 animals in the anti-mouse CCR8 antibody administration group showed complete tumor remission.
- 6 out of 10 mice were in complete remission.
- Tumor volume (mm 3 ) was weighed by major axis (mm) x minor axis (mm) x minor axis (mm) / 2 ( Figure 37). As a result, it was found that the combination group of anti-PD-1 antibody and anti-mouse CCR8 antibody had a smaller tumor than the group administered with anti-PD-1 antibody or anti-mouse CCR8 antibody alone.
- Wild-type mice were intravenously administered with 100 ⁇ g (100 ⁇ L) of rat anti-mouse CCR8 antibody (clone SA214G2, BioLegend) or isotype control antibody (LTF-2, Bioxcell) 3 and 10 days after tumor implantation. 5).
- a rat anti-mouse CCR8 antibody clone SA214G2, BioLegend
- isotype control antibody LDF-2, Bioxcell
- the antibody against CCR8 of the present invention has an effect of stimulating immunity by reducing tumor infiltrating Treg cells and the like, and is useful as a medicament for cancer treatment.
Abstract
Description
例えば、腫瘍に浸潤するCD4陽性Treg細胞は、抗腫瘍免疫応答を強力に阻害している可能性があり、効果的な癌治療の大きな障害となりうる。
CD4陽性FoxP3陽性Treg細胞が媒介する腫瘍免疫抑制は、動物腫瘍モデルで十分に立証されており、腫瘍内も含めた全身性のTreg細胞除去により抗腫瘍効果が得られるが、一方で50%程度の腫瘍内浸潤Treg細胞除去では効果が認められないことが報告されている(非特許文献2)。
(1)CCR8に対する抗体を含有する、癌治療用医薬組成物、
(2)CCR8に対する抗体がADCC活性を有する抗体である、(1)記載の医薬組成物、
(3)CCR8に対する抗体が、CCR8の中和抗体である、(1)又は(2)のいずれかに記載の医薬組成物、
(4)CCR8に対する抗体が、腫瘍内浸潤Treg細胞除去作用を有する、(1)~(3)のいずれかに記載の医薬組成物、
(5)CCR8に対する抗体が、腫瘍内浸潤マクロファージ細胞除去作用を有する、(1)~(4)のいずれかに記載の医薬組成物、
(6)癌が乳癌、大腸癌、腎癌または肉腫である、(1)~(5)のいずれかに記載の医薬組成物、
(7)CCR8に対する抗体、及び抗PD―1抗体又は抗PD-L1抗体を組み合わせてなる癌治療用の医薬、
(8)(1)~(5)のいずれかに記載のCCR8に対する抗体を投与することを特徴とする、癌の治療方法、
(8-1)CCR8に対する抗体を投与することを特徴とする、癌の治療方法、
(8-2)CCR8に対する抗体がADCC活性を有する抗体である、(8-1)記載の治療方法、
(8-3)CCR8に対する抗体がCCR8の中和抗体である、(8-1)又は(8-2)記載の治療方法、
(8-4)CCR8に対する抗体が、腫瘍内浸潤Treg細胞除去作用を有する、(8-1)~(8-3)記載の治療方法、
(8-5)CCR8に対する抗体が、腫瘍内浸潤マクロファージ細胞除去作用を有する、(8-1)~(8-4)記載の治療方法、
(8-6)癌が乳癌、大腸癌、腎癌または肉腫である、(8-1)~(8-5)記載の治療方法、
(8-7)さらに、抗PD―1抗体又は抗PD-L1抗体を投与することを特徴とする、(8-1)~(8-6)記載の治療方法、
(9)癌を治療するための、(1)~(5)のいずれかに記載のCCR8に対する抗体、
(9-1)癌を治療するための、CCR8に対する抗体、
(9-2)CCR8に対する抗体がADCC活性を有する抗体である、(9-1)記載のCCR8に対する抗体、
(9-3)CCR8に対する抗体がCCR8の中和抗体である、(9-1)~(9-2)記載のCCR8に対する抗体、
(9-4)CCR8に対する抗体が、腫瘍内浸潤Treg細胞除去作用を有する、(9-1)~(9-3)記載のCCR8に対する抗体、
(9-5)CCR8に対する抗体が、腫瘍内浸潤マクロファージ細胞除去作用を有する、(9-1)~(9-4)記載のCCR8に対する抗体、
(9-6)癌が乳癌、大腸癌、腎癌または肉腫である、(9-1)~(9-5)記載のCCR8に対する抗体、
(9-7)癌を治療するため使用される、(9-1)~(9-6)記載のCCR8に対する抗体と抗PD―1抗体又は抗PD-L1抗体との組合せ、
に関する。
細胞融合に用いられるミエローマ細胞としては、一般的にはマウスから得られた株化細胞、例えばP3-U1、NS-1、SP-2、653、X63、AP-1などを使用することができる。
また、本発明の「癌治療用医薬組成物」における「癌」は、腫瘍特異抗原を発現する癌が好ましい。
(1)本発明の医薬組成物の治療効果の補完及び/又は増強、
(2)本発明の医薬組成物の動態、吸収改善、投与量の低減及び/又は、
(3)本発明の医薬組成物の副作用の軽減、
のために他の薬剤と組み合わせて、併用剤として投与してもよい。
術前に抗癌剤や放射線等の治療を行っていない淡明細胞型腎細胞癌(clear cell renal cell carcinoma; ccRCC)の患者(3例)から外科的治療により摘出された原発腫瘍組織の一部を用いて以下の解析を行った.腫瘍重量を測定後、腫瘍塊をハサミで2mm角に切断し、Tumor Dissociation Kit, human (130-095-929, Miltenyi)及び gentleMACS(TM)Dissociator (Miltenyi, 130-093-235)を用いてキットの添付プロトコルにしたがい腫瘍組織のホモジネートを作製した。ホモジネートを70umのセルストレイナーに通し、溶血処理を行った後,30%パーコール/PBS溶液でデブリスおよび死細胞を除去し、腫瘍組織単細胞を得た。
同一患者の末梢血単核球(PBMC)は末梢血からFicoll-paque PLUS (GE Healthcare社)を用いた密度勾配遠心法により分取した. 分離した腫瘍内細胞及びPBMCは細胞数を計測後、Human TruStain FcX(TM)(BioLegend, 422-301)及びZombie NIRTM Fixable Viability kit (BioLegend, 423105)を添付プロトコルにしたがい処理し、氷中で30分間染色した。その後2%FCS/HEPES/HBSSで1回洗浄後、以下の標識抗体、標識抗体添付プロトコルにしたがい染色した。
分離回収された各細胞は、RLT buffer (Qiagen)に溶解し、Agencourt RNAClean XP (Bechman Coulter) を用いてtotal RNAを抽出した。回収したRNAは、SMART-Seq v4 Ultra Low Input RNA kit for Sequencing (Clontech)を用いてcDNA化し、KAPA Hyper Prep Kit for illumina (Kapa Biosystems)を用いてライブラリー調整をおこなった。cDNA合成、ライブラリー調整は、常時Agilent 2100 Bioanalyzer (Agilet Technologies)を用いてクオリティーコントロールをおこない、問題ないことを確認した。完成したcDNAライブラリーはKAPA library Quantification kit Illumina Platforms (Kapa Biosystems)を用いてタイトレーション後、Hiseq4000 (illumina)を用いてペアエンドリードでDNA sequencingをおこない、各サンプルあたり100塩基対の配列データを2000万リード以上取得した(fastqファイル)。
生データ(fastq ファイル)はFastQCにより解析し、アダプター配列及びリピート配列はCutAdaptを用いて除去した。各ペアエンドリードはcmpfastq_peプログラムを使用して各ペアを揃えた。ゲノムマッピングはhg38を参照配列として、Bowtie2を持つTOPHAT2プログラムによりデフォルトセッティングでゲノムにマッピングした。マッピングされたリードはSAMtoolsプログラムにより配列ソートされ、HTSEQプログラムによりリードカウントした。カウントデータの正規化はDeseq2プログラムを使用した。得られた各分画について、どの分画がTreg細胞を含んでいるかを以下の方法で確認した。
FoxP3領域の脱メチル化率はTreg細胞の割合を正確に求める指標であるため、上記で得た腎癌腫瘍浸潤細胞のFr2~Fr5の細胞についてFoxP3領域の脱メチル化率を検討した。FoxP3遺伝子の第一イントロン内の特定のCpG領域にはTreg細胞特異的に脱メチル化されている領域が存在する(chrX, 49118000-49118500, hg19)。腫瘍内浸潤細胞の各分画に含まれる細胞について、この領域の脱メチル化を解析することで、今回得られた分画がTreg細胞のみからなっているのか、それとも他の細胞も混在しているのかを検証可能である。
腫瘍内浸潤CD4+ T細胞の各分画(Fr2,3,4,5)を回収し、フェノール抽出法を用いてgenome DNAを回収した。Genome DNAに対し、MethylEasy Xceed キット (Human Genetic Signatures) を用いてBisulfite処理をおこない、Treg細胞特異的脱メチル化領域であるFOXP3 intron1 領域(chrX, 49118000-49118500, hg19)に対しアンプリコンPCRをおこなった。DNAメチル化の検出は、メチル化DNA特異的FAM蛍光プローブと脱メチル化特異的VIC蛍光プローブを用意し、QuantStudio 3D digital PCRシステム (Applied Biosystems)によりおこなった。アンプリコンPCR後、FAMおよびVIC蛍光プローブの発光数をカウントし、両蛍光数の比率からDNAメチル化率を算出し、各分画(Fr2~Fr5)のメチル化率とした。
その結果腫瘍内浸潤細胞中のFr2及びFr3に含まれる細胞では、FOXP3 intron1 領域(chrX, 49118000-49118500)内のCpG配列の95%以上が脱メチル化されており、Fr4及びFr5の脱メチル化率は50%以下であった。このことから、Fr2及びFr3に含まれる細胞のほぼすべてがTreg細胞であると判明した(図5)。
Treg細胞(腫瘍浸潤細胞中のFr2)に特異的に発現する一群の遺伝子を同定するために腫瘍由来の各CD4+ T細胞分画と同一患者PBMC由来CD4+ T細胞分画の遺伝子発現データについて階層的クラスター解析を行い、Treg細胞中のFr2に発現し、腫瘍由来Fr5及び、PBMCのFr4, Fr5にはほとんど発現していない遺伝子としてCCR8を同定した(図6)。
マウスCCR8(以下、mCCR8と記載する場合がある。)のORF全長を発現ベクター(pcDNA3.4)に挿入し、pcDNA3.4- mCCR8プラスミドを構築した。塩基配列はアミノ酸を変更しない範囲で、哺乳類で使用頻度の高いコドンに変更した。HEK293細胞にリポフェクタミン3000を用いてpcDNA3.4及びpcDNA3.4- mCCR8発現プラスミドをそれぞれ導入し、Geneticin(G418)濃度1mg/mlで2週間薬剤選択を行った。
生き残った細胞をトリプシンで剥離し、DMEM/10%FCS培地で洗浄後、PE標識抗mCCR8抗体(クローンSA214G2)を1/200希釈で添加、30分間氷上で抗体を反応させ、その後DMEM/10%FCSで1回洗浄し、細胞表面に発現するmCCR8を標識した。セルソーター(FACSAriaII)によりmCCR8発現している細胞集団をソーティングにより濃縮した。陽性細胞集団をDMEM/10%FCS(1mg/mlのG418含有培地)存在下で37℃ CO2インキュベーターで2週間培養した。pcDNA3.4 を形質転換した細胞は薬剤選択のみを行い、ソーティングはしなかった。発現確認のため両細胞を市販抗PE標識抗マウスCCR8抗体(クローンSA214G2)で染色し、フローサイトメーター(FACSAriaII)で解析した。その結果を示す(図7)。pcDNA3.4を形質転換した細胞と比較して、pcDNA3.4-mCCR8を形質転換した細胞は、99%以上の細胞でmCCR8の発現を認めた。
抗マウスCCR8抗体(クローンSA214G2, BioLegend社より購入)のADCC活性に必要なFcgR刺激能をmFcγRIV ADCC Reporter Bioassays Core kit (Promega社)を用いて評価した。本キットではエフェクター細胞上のFcγRの活性化が当該細胞のNFATプロモーター下流につないだルシフェラーゼ遺伝子の発現量で示され、これを定量することでFcγRシグナルの活性化を定量可能となる。
以下、簡単に記述する。トリプシンで剥離したmCCR8発現HEK293ターゲット細胞(ターゲット細胞)1x105/wellとキット添付FcγR発現エフェクター細胞を1:1.5の比率で、96wellプレート中で混合した。細胞混合後すぐにmCCR8抗体を添加した。その濃度は図8に示すように33ug/mlから0.033ug/mlでとした(N=2)。エフェクター細胞のみを陰性対象とした。抗体添加後14時間後に細胞を回収し、ルシフェラーゼ活性を測定した(図8)。N=2の平均値を表示する。
結果、陰性対象ではいかなる抗体濃度でもルシフェラーゼ活性は認められなかったのに対し、ターゲット細胞添加群では抗体濃度依存的な活性が認められた。縦軸は発光量相対値を示す。図8より最大活性値は約6000Relative Light Unit (R.L.U)であり、EC50値(約3500R.L.U)は約0.1μg/mlであった(図中のライン)。以上の結果より抗マウスCCR8抗体(SA214G2)がFcγRIVを活性化できることを明らかにした。
実施例5で作製したmCCR8安定発現HEK293細胞を用いて、抗mCCR8抗体(SA214G2)の細胞傷害活性を評価した。
C57BL/6マウスの脾臓を分離し、脾臓細胞をセルストレイナーに通して回収した。細胞を洗浄後、ビオチン化抗CD49b (clone DX5) 抗体を4℃で30分反応させ、洗浄後にストレプトアビジンマイクロビーズ(Miltenyi)を用いてNK細胞を精製し、エフェクター細胞とした。マウスCCR8発現HEK293細胞は、Cell Trace Violet (CTV) (サーモフィッシャー社、C34557)を用いて終濃度2.5uMで染色し、標的細胞(ターゲット細胞)とした。96well プレート中でエフェクター細胞 : 標的細胞 = 5 : 1(エフェクター細胞数は2.5x105)の比率で200μLに混合し、抗マウスCCR8抗体またはアイソタイプコントロール抗体 (ラットIgG2b, clone RTK4530) を終濃度1μg/mlで添加し、37℃のCO2インキュベーター中で一晩培養した。その後, PE標識Annexin V (AnnexinV-PE, MBL社、4696-100)を添付プロトコルに従い1/100希釈で添加し、37℃で30分染色後、1回洗浄した。フローサイトメーターでCTV染色された標的細胞中のAnnexin V陽性アポトーシス細胞率を解析した。トリプリケート(N=3)で実施し、その平均値と標準偏差を示す。2回同様の実験を行った代表例を示す。(図9)。アイソタイプコントロール抗体と比較して、抗マウスCCR8抗体を添加すると、標的細胞中のAnnexin V 陽性細胞率が6倍程度有意に増加した。以上より抗マウスCCR8抗体(SA214G2)はADCC活性を有することが判明した。
マウスCCR8安定発現HEK293細胞を用いてマウスCCR8のリガンドであるマウスCCL1による細胞内カルシウム流入を指標として抗マウスCCR8抗体(SA214G2)のCCR8に対する中和活性を評価した。
カルシウム測定には以下の試薬を使用した。
HEPES (WAKO CAS.NO7365-45-9)
HBSS(+) without Phenol Red (WAKO)
Fluo3-AM (cat F023 同人化学)
プロベネシド (CAS-No:57-66-9, ナカライテスク)
Pluronic F127 (P3000MP; Life Technology社)
10mM HEPES/HBSS/0.1% BSA Buffer (HBSSに終濃度10mM HEPESと終濃度0.1% BSAになるようにそれぞれ添加)
Fluo3-AMを4μmol/L、Pluronic F127を0.04%の終濃度で10mM HEPES/HBSS Bufferに溶解した。この溶液に細胞を懸濁し、37℃で1時間インキュベートすることで、Fluo3-AMを細胞に取り込ませた。その後細胞を10mM HEPES/HBSS/0.1%BSA溶液で3回洗浄し、1.25uM プロベネシドを含む10mM HEPES/HBSS/0.1%BSA 溶液に 2x105/mlの細胞濃度になるように懸濁した。そして、10分間37℃、CO2インキュベーターでインキュベートした。さらに抗mCCR8抗体(SA214G2)あるいはIsotype Control抗体(Clone LTF-2, Bio X Cell)を5μg/mlの濃度で添加した。さらに20分間37℃でインキュベートした。
細胞は2mL溶液を水晶ガラス製キュベットに入れ、予め測定室を35℃に温度設定しておいた分光光度計HITACHI F7000にセットした。測定条件は下記のとおりである。
励起波長 508.0nm、蛍光(測定)波長 527.0nm、励起側スリット5nm、蛍光側スリット 5nm、ホトマル電圧 950V、レスポンス 0.5s
蛍光波長が安定するまで約30秒間スターラーで撹拌しながらインキュベートした。波長が安定したら、マウスCCL1を終濃度50nM(4μL)になるように添加し、測定を開始した。測定の結果、抗mCCR8抗体を予め添加することより、mCCL1による細胞内カルシウム流入がほぼ完全に抑制されることが判明した(図10)。コントロール抗体添加では抑制は認められなかった。なお、グラフ中のギャップはアゴニストを細胞に投与するために機器の蓋の開閉をした際のものである。以上より抗mCCR8抗体(SA214G2)抗体はマウスCCR8に対する中和活性を有することが判明した。
CT26細胞を6ウェルディッシュで培養し、約50%のコンフルエント状態になった時点で培養液を除去し、10mM EDTA/PBSを5ml添加し、37℃で5分間インキュベートした。その結果細胞はほぼすべて剥離され、ピペットでサスペンドすることで、ほぼ単一細胞にまで分離できた。2回D-MEM/10%FCSで洗浄し、D-MEM/10%FCSにサスペンドし、LIVE/DEAD(登録商標) Fixable Near-IR Dead Cell Stain Kit (ThermoFisher Scientific, L34975)とAPC標識抗mCCR8(SA214G2)又はAPC標識Isotype Control抗体で、氷中で細胞を染色した。1時間後に3回D-MEM/10%FCSで洗浄し、フローサイトメーター(FACSCantoII)でmCCR8発現率を解析した。Isotype Control抗体を用いてバックグラウンドを設定し、バックグラウンドレベル以上の陽性細胞率(P6)とAPC蛍光の中央値(Median)を算出した(図11)。その結果APC蛍光強度の中央値に差は認められず、また陽性細胞も0.2%とほとんど認められなかった。以上より、CT26細胞は抗mCCR8抗体には認識されず、CT26細胞はmCCR8を発現しないことが確認された。
マウスBalb/cマウス(7w、メス)の背部皮内に3x105個(50μL)のCT26細胞(N=3)を移植し、移植3日目にラット抗KLH(キーホールリンペットヘモシアニン、Clone LTF-2)抗体(IgG2b)400μgを腹腔内に投与した。投与後4日目(4d)と7日(7d)目に、3例の個体より腫瘍を回収した(N=3)。CT26細胞の腫瘍塊を、ハサミで細かく切断し、市販キット(Tumor Dissociation Kit, mouse, Miltenyi と the gentleMACS(TM) Dissociator, Miltenyi Biotech cat. 130-095-929)を用いて添付キットプロトコルに従い、腫瘍浸潤細胞を調製した。
調製された細胞は70umのセルストレイナーに通した後、2回10mM HEPES/HBSS/2%FBSで洗浄した。その後、赤血球溶解液(Miltenyi社)で5分間処理し、赤血球を除去し、さらに2%FCS(Fetal Calf Serum)/10mM HEPES/HBSSバッファーで2回洗浄した。腫瘍浸潤細胞を2つに分け、一つはTreg細胞の同定、もう一方はミエロイド系(マクロファージ)細胞の同定を行った。以下の方法及び抗体により細胞を染色した。使用した抗体及び染色試薬、アッセイバッファーは下記の通りである。
(Treg細胞確認用抗体セット)
PE anti-mouse/rat FoxP3 (clone FJK-16s) eBiosciences
Anti-mouse CD4 PerCP/Cy5.5 (clone RM4-5)eBiosciences
Anti-mouse CD8a FITC (clone 5H10-1)Biolegend
Bv421 anti-mouse CD25 (clone PC61) BioLegend
Bv510 anti-mouse CD45 (clone 30-F11) Biolegend
AF647 Anti-mouse CCR8 (clone SA214G2) BioLegend
AF647 Isotype Control (clone RTK4530) BioLegend(CCR8の陰性コントロール)
(ミエロイド、マクロファージ細胞確認用抗体セット)
AF647 Anti-mouse CCR8 (clone SA214G2) BioLegend
AF647 Isotype Control (clone RTK4530) BioLegend(CCR8の陰性コントロール)
Bv510 anti-mouse CD45 (clone 30-F11) Biolegend
FITC anti-mouse Gr-1(clone RB6-8C5) Biolegend
Bv421 anti-mouse F4/80 (clone BM8) BioLegend
PECy7 anti-mouse CD11b (clone M1/70) BioLegend
PerCP/Cy5.5 Anti-mouse MHC classII IA/IE (clone M5/114.15.2)BioLegend
PE anti-mouse CD206 (clone C068C2) BioLegend
(その他、使用した試薬)
Zombie NIR Fixable Viability Kit (cat no.423106) BioLegend
BD Pharmingen Transcription Factor buffer Set (cat no.562574)
BD Pharmingen Lysing Buffer (cat no.555899)
HBSS(-) Wako 084-08345
FCS(Hyclone cat no.SH30070.03)
染色は30分間氷中で実施した。2%FCS/HEPES/HBSSで2回洗浄後、細胞を市販キット(FoxP3 staining kit、eBioscience社)を用いて添付プロトコルにしたがい固定し、PE標識抗FoxP3抗体を用いて細胞内FoxP3を染色した。当該キット添付バッファーで洗浄後、フローサイトメーターを用いて細胞を解析した。
マウスBalb/cマウス(7w、メス)の背部皮内に3x105個のCT26細胞(50uL)を移植した。移植3日後にラット抗マウスCD198(CCR8)抗体(クローンSA214G2、BioLegend社)或いはアイソタイプコントロール抗体(Clone LTF-2)を尾静脈内に400μg(液量400μL)投与した(各群N=3)。腫瘍移植7日後(抗体投与後4日)と10日後(抗体投与後7日)に腫瘍を回収し、腫瘍内浸潤細胞を調製し、解析した(図15)。
腫瘍内浸潤Treg細胞は実施例10と同様の方法で回収した。用いた抗体は実施例10と同じである。
その結果、図16のようにアイソタイプ抗体投与マウスの腫瘍内CD45+CD4+CD25+FoxP3+T細胞(Treg細胞)比率を100%とした場合の(10日後)抗マウスCCR8(SA214G2)抗体投与マウスでの同細胞(Treg細胞)の陽性率は、腫瘍移植7日後(抗体投与後4日)で約80%、10日後(抗体投与後7日)で約40%であった(図16)。有意水準**はP<0.01(t検定)であった。以上より抗CCR8抗体投与7日後で腫瘍内浸潤Treg細胞の約60%が抗CCR8抗体により除去されていることが示された。
図17でF4/80+細胞をさらにMHC(腫瘍組織適合抗原)クラス2分子のうち、IA/IE陽性或いはクラス2(IA/IE)陰性細胞の存在比率を示す。図18に示すとおり、アイソタイプコントロール(N=3)と比較して、抗mCCR8抗体投与群(N=3)で、IA/IE陰性群で減少傾向を示し、IA/IE陽性群では有意に減少した(t検定;有意水準*; P<0.05)。以上よりマウスCT26腫瘍内単球/マクロファージ集団或いはその一部の集団の腫瘍内細胞数が減少していることが判明した。
マウスBalb/cマウス(7週齢、メス)の背部皮内に3x105個の大腸癌由来CT26細胞(50uL)を移植した。腫瘍移植3日後にラット抗マウスCD198(CCR8)抗体(クローンSA214G2、BioLegend社)を静脈内に400μg(400μL)投与した(N=10)。コントロールはアイソタイプコントロール抗体を投与した(N=10)。腫瘍移植8日後(抗体投与後5日)より3~4日ごとに腫瘍体積を測定した。腫瘍体積(mm3)は長径(mm)x短径(mm)x短径(mm)/2で計量した(図19)。
その結果移植7日目にはアイソタイプコントロール抗体投与群と比較して、抗mCCR8投与群で有意差は認められなかったが、11日目、14日目、17日目、21日目において有意に抗mCCR8抗体投与群の腫瘍体積が減少した(有意水準は11日目と14日目が、***;P<0.001、17日目と21日目が**; P<0.01)。また抗マウスCCR8抗体投与群では14日目以降腫瘍体積が減少し、17日目にはほぼ完全に消失した(個体別データは図20、平均値データは図21に示す。)。以上の結果より、抗mCCR8抗体投与により、免疫抑制細胞として指摘されているTreg及び単球/マクロファージに発現するmCCR8の機能を抑制し、あるいはそれら発現細胞を抗体のADCC活性により死滅(除去)することで、腫瘍免疫が亢進し、腫瘍の退縮、消滅につながったと結論した。
その結果アイソタイプコントロール抗体を投与した群(N=8)と比較して抗PD-1抗体を投与した群(N=8)で抗腫瘍効果が認められた。移植後14、17、20日目でのアイソタイプコントロールの腫瘍体積の平均と標準偏差は、それぞれ601.7±378.1mm3、956.3±467.7mm3及び1528.4±774.1mm3であり、一方抗PD-1抗体投与群ではそれぞれ175.3±42.6mm3、174.7±55.8mg及び209.6±99.8mm3であった。抗PD-1抗体は移植後14、17、20日目のいずれの時点においてもコントロールと比較して腫瘍体積の増加が有意に抑制されていた。しかし腫瘍が完全に消失した個体は、観察期間(移植後20日目まで)では8匹中1匹であった。一方抗mCCR8抗体投与では同期間中に、10匹全例で、腫瘍の完全消失が認められた。以上より、標準的な投与法での抗PD-1抗体と比較して、抗mCCR8抗体の方が強い薬効を有していると結論した。
次に実施例12での、投与後18日目までのマウスの状態について評価した。コントロール抗体投与群と抗CCR8抗体投与群間で、当該期間中の体重に有意差は無かった。またどちらの群も立毛は認められなかった。投与後18日目に解剖した。コントロールと比較して抗CCR8抗体投与群で、リンパ節及び腸管の肥大があるか検討したが、どちらも違いはなく、肥大は認められなかった。以上の所見より、抗CCR8抗体投与マウスにおいて、抗腫瘍効果を発揮した期間中に、自己免疫疾患の兆候は認められないと結論した。一般的には、抗腫瘍効果が惹起される程度まで、マウスの全身のTregを除去すると、除去後14日程度で重篤な自己免疫疾患が惹起されることが論文で報告されており、Treg抑制療法をふくむ腫瘍免疫療法の懸念材料となっている。今回の結果は、抗CCR8抗体投与により強い抗腫瘍免疫効果が認められたマウスにおいて、抗体投与後18日目においても自己免疫疾患はまったく惹起されていなかった。その一つの説明として、マウスおよびヒトCCR8は腫瘍組織と比較して、PBMC、脾臓、リンパ節での発現が低いことが報告されている。しかしこれら末梢組織でCCR8発現Treg細胞を除去あるいは機能阻害した場合に、自己免疫疾患が惹起されるか否かは今まで報告がなかった。今回初めて、自己免疫疾患を惹起しないことが判明し、従来の知見からは予測できない効果と考えられる。
マウスBalb/cマウス(7週齢、メス)の背部皮内に2x105個の大腸癌由来Colon-26細胞(50μL)を移植した。腫瘍移植3日後にラット抗マウスCCR8抗体(クローンSA214G2、BioLegend社)を静脈内に400μg(400μL)投与した(N=10)。コントロールはアイソタイプコントロール抗体を投与した(N=10)。腫瘍移植3日後(抗体投与後5日)より3~4日ごとに腫瘍体積を測定した。腫瘍体積(mm3)は長径(mm)x短径(mm)x短径(mm)/2で計量した。その腫瘍がエンドポイント体積(800mm3)に達した時点で、各動物のエンドポイントとした。その結果移植後14日目及び18日目にアイソタイプコントロール抗体投与群と比較して、抗mCCR8投与群で腫瘍体積増加が抑制された。14日目での腫瘍体積の平均はアイソタイプコントロール抗体投与群で451.3mm3(標準偏差は±177.5mm3)、抗CCR8抗体投与群で322.6mm3(標準偏差は±146.0mm3)であり、14日目の腫瘍体積が350mm3以上の個体はアイソタイプコントロール群で10例中9例、抗mCCR8投与群で10例中4例であり、この分離態様に関して、ピアソンのカイ2乗検定ではP=0.019で有意差があった。したがって14日目に350mm3の腫瘍体積に達した個体数に差が認められた。また移植後18日目での腫瘍体積の平均は、アイソタイプコントロール抗体投与群で874.7mm3(標準偏差は±269.2mm3)、抗CCR8抗体投与群で585.4mm3(標準偏差は±401.7mm3)であった(図22)。18日目の腫瘍体積が600mm3以上の個体はアイソタイプコントロール群で10例中9例であり、一方抗mCCR8投与群では10例中4例であり、この分離態様に関して、ピアソンのカイ2乗検定ではP=0.019であり有意差があった。したがって18日目で600mm3の腫瘍体積に達した個体数に差が認められた。さらに腫瘍体積が800mm3になった時点をエンドポイントとして予め設定した。腫瘍体積が800mm3を超え、死亡したとみなした個体は、14日まではどちらも認められず、18日目でアイソタイプコントロール群が、10例中7例、抗CCR8抗体群が10例中3例であった。18日目での生存率について、ピアソンのカイ2乗検定を行い、生存率に差があるか検討した結果、P=0.025で生存率に有意差があった。
また同じ細胞株を用いた同様の実験で、アイソタイプコントロール抗体を投与した群と比較して抗PD-1抗体(clone RMP1-14、Bio X Cell 社)の投与群で、抗腫瘍効果は認められなかった。以上より抗mCCR8抗体は抗PD-1抗体耐性のColon26細胞に対してより高い抗腫瘍効果を示した。
14例のヒト腎癌腫瘍内浸潤細胞でのCCR8の発現解析を行った。14例の腎癌患者の背景は、性別は男性11名と女性3名であり、年齢中央値は68.5歳、病理病期はT1Aが6名、T1Bが2名、T3Aが5名、T3bが1名であった。具体的には、腎癌(Clear Cell Renal Cell Carcinoma, ccRCC)患者14名の腎癌原発腫瘍内浸潤細胞を実施例1の図1と同様に単離し、Anti-CD4 (BioLegend, Clone OKT4)、 anti-CD3 (BioLegend, Clone UCHT1), anti-CD45RA (BD Biosciences, Clone HI100), anti-CD8 (Biolegend, RPA-T8), anti-CCR8 (BioLegend, Clone L263G8), anti-FoxP3 (eBioscience, Clone 236A/E7)、anti-FoxP3のアイソタイプコントロール抗体で染色し、フローサイトメトリー (BD Biosciences, BD LSRFortessa) で解析した。CD3+CD8+T細胞及びCD3+CD4+T細胞について解析した。CD3+CD4+T細胞については、さらにFoxP3発現の有無で2群にわけて解析した。アイソタイプコントロール抗体の染色により、FoxP3発現の陰性対照とした。CCR8の発現強度は各患者サンプルのFACS解析値の平均値(MFI)を使用した。表1に抗CCR8抗体及びそのアイソタイプコントロール抗体での染色のMFI値の平均及びその標準偏差を示す。

上記ccRCCの14サンプルについて、腫瘍浸潤CD4+ T細胞について、FoxP3とCCR8のフローサイトメトリー解析を行った。FoxP3陽性細胞中のCCR8陽性細胞の割合、FoxP3陰性細胞でCCR8陽性細胞の割合をサンプルごとにプロットした。(図24)。FoxP3及びCCR8共にアイソタイプコントロール抗体での染色を陰性標準として使用し、この閾値以上の細胞を陽性細胞とした。その結果、腫瘍内CD3+CD4+FoxP3+T細胞のCCR8発現率は約75%であり、CD3+CD4+FoxP3-T細胞のCCR8発現率は、約10%であった。
以上の結果よりヒト腎癌腫瘍内浸潤細胞のうちFoxP3を発現するTreg細胞のほとんどでCCR8が発現し、Treg細胞以外のCD4陽性T細胞では約10%にCCR8が発現していることが判明した。以上よりヒト腫瘍内のFoxP3陽性Treg細胞中のCCR8発現率は、マウス腫瘍内のTreg細胞でのCCR8発現率と類似しており、マウスと同様に抗ヒトCCR8特異的抗体により、腫瘍内浸潤FoxP3陽性Treg細胞のほとんどを除去できる可能性が示された。
Treg細胞に特異的に発現し、腫瘍細胞あるいはヒトのほとんどの正常細胞でも発現していない遺伝子としてFoxP3遺伝子が同定されている。このようにある特異的な細胞でのみ発現するいわゆるマーカー遺伝子として、例えばTreg細胞のマーカー遺伝子としてFoxP3遺伝子、T細胞及びNK細胞のマーカー遺伝子としてCD3G遺伝子、CD8陽性T細胞のマーカー遺伝子としてCD8A遺伝子などが知られている。
Treg細胞のマーカー遺伝子であるFoxP3遺伝子に関しては、各腫瘍内でのFoxP3遺伝子のmRNA発現量を測定することによりTreg細胞の腫瘍内での存在割合の指標とすることが可能であることも報告されている(Cell,2015年、第160巻、p.48-61)。
また同じ論文で報告されているように、TCGAのようなRNA-Seqデータベースを利用してマーカー遺伝子の腫瘍内発現率(Treg存在比率)と患者生存率について、カプランマイヤー生存曲線を描くことで、Treg細胞の腫瘍内での存在率が、生存率と関連するか解析可能である。腫瘍塊のRNA-Seqデータは、腫瘍細胞及びそこに存在する浸潤細胞(リンパ球や血管細胞など)の両方の細胞で発現するmRNAが混在したデータであるが、腫瘍細胞に発現していないことが示された遺伝子であれば、腫瘍内浸潤細胞で発現する遺伝子とみなすことが可能であり、それを利用して上記のような解析、すなわち腫瘍塊のRNA-Seqデータにおけるマーカー遺伝子の発現解析により腫瘍内浸潤細胞の同定が可能である。さらに腫瘍塊でのマーカー遺伝子の発現量は、そこに浸潤するマーカー遺伝子に対応する特定細胞の発現細胞数と各発現細胞の発現量の積と捉えることができる。
ここで各細胞のマーカー遺伝子の発現量は個体間でほぼ一定と仮定すると、その発現量は浸潤細胞数と正比例する。したがってこの発現量を用いることで、腫瘍内の発現細胞数が個体ごとに算定可能となり、個体間比較が可能となる。
公共データベースであるCCLE(Cancer Cell Line Encyclopedia)にヒト各種細胞株1037種類のRNA発現データが登録されている。これらのデータベースを用いてCCR8やCD3G遺伝子がT細胞以外の癌細胞又は正常細胞で発現しているかを解析した。
腎癌、前立腺癌および膀胱癌由来細胞株について、CCLEデータベースを用いてCD3GとCCR8のmRNA発現を解析した。
調べた細胞株は、腎癌由来細胞株では、
VMRCRCW、 SKRC20、 SNU34、 SKRC31、 UOK10、 SLR20、 OSRC2、TUHR14TKB、 SLR24、 HK2、 A498、 RCC4、 KMRC1、 RCC10RGB、 ACHN、 SLR25、 SNU1272、 UMRC6、 SLR23、 769P、 SLR21、 HEKTE、 CAKI1、 TUHR4TKB、 KMRC2、 VMRCRCZ、 KMRC3、 KMRC20、 CAKI2、 BFTC909、 786O、 A704、 TUHR10TKB、 SLR26、 UMRC2、 CAL54、FURPNT1、FURPNT2、HEK293、G402の40種であり、
前立腺癌由来細胞株では、
VCAP、 LNCAPCLONEFGC、 DU145、 PC3、 22RV1、 PRECLH、 MDAPCA2B、 NCIH660 の8種であり、
膀胱癌由来細胞株では、
TCBC14TK、TCBC2TKBの2種であった。これら調べた全ての固形癌細胞株でCCR8及びCD3Gの発現はバックグラウンドと同レベルの値であり、mRNA発現はまったく認められなかった(最大の発現を示す値でもG3PDHの1/500以下。それ以外はすべてG3PDHの発現量の1/1000以下)。 すなわち、CCR8及びCD3Gは固形癌細胞にはほとんど発現していないことが確認できた。ヒト各組織由来のプライマリーな正常細胞についても同様に解析し、CCR8及びCD3Gは血球系細胞の一部にのみ発現し、それ以外のプライマリーな正常組織由来細胞では、ほとんど発現していないことが判明した。
以上よりこれら3種の癌細胞では、CCR8とCD3Gは発現していないことが示された。したがって腎癌、前立腺癌、膀胱癌の腫瘍塊について、TCGAのRNA発現データを用いた場合に、CCR8とCD3Gは癌細胞以外の、腫瘍塊に存在する浸潤正常細胞でのmRNA発現を反映すると結論した。
次にTCGA公共データベースを利用して、腎癌、前立腺癌、膀胱癌の腫瘍中で発現するCD3G遺伝子とCCR8遺伝子の比(CCR8/CD3G)と、患者生存率について解析を行った。これら3種の腫瘍内について、CCR8及びCD3G遺伝子ともっともよく発現が相関(ピアソン相関)する遺伝子は、T細胞で特異的に発現する種々の遺伝子であることが判明した(FoxP3, CD5, IL7R,等で相関係数rは0.7以上)。この結果はCCR8やCD3Gが腫瘍細胞自体には発現せず、腫瘍内に浸潤している発現細胞(特にT細胞)に特異的に発現することを示している。ただしCCR8がT細胞以外の浸潤細胞で発現していることを否定するものではないので、ここではCCR8を発現する細胞集団とする。CD3GについてはT細胞及びNK細胞に特異的に発現することがすでに論文等で報告されており、またT細胞は腫瘍に浸潤する主要な細胞であるので、CD3G発現量は浸潤しているT細胞数と比定できる。したがってCCR8/CD3G値は腫瘍内に存在する、T細胞数あたりのCCR8を発現する細胞数と定義できる。
これら3種の癌腫についてCCR8/CD3G比と患者生存率をカプランマイヤー曲線で解析した。腎癌はTCGAデータのうちKidney Renal Clear Cell Carcinoma (TCGA, Provisional)のデータを使用し、RNA発現データおよび患者生存率データの揃っている523例を使用した。同様に前立腺癌はTCGAデータのうちでProstate Adenocarcinoma (TCGA, Provisional)のデータでRNA発現データおよび患者生存率データの揃っている490例を使用した。
また膀胱癌は TCGAデータのうちでBladder Urothelial Carcinoma (TCGA, Provisional)のデータを使用し、RNA発現データおよび患者生存率データの揃っている392例を使用した。
CCR8/CD3Gの発現値について、高い群と低い群の2群に等分し(腎癌の場合は奇数なので261:262)、カプランマイヤー生存曲線解析を、解析ソフトR (R-Studio)を用いて行った。有意差検定はLog-rank検定を行った。腎癌の結果は図25、前立腺癌の結果は図26、膀胱癌の結果を図27に示す。グラフ中の縦線は、患者は生存しているが、評価期間がこの時点までしか無いために、この時点で脱落したもの(いわゆるセンサーに該当)として扱っている。また横軸の値はすべてのグラフで月数を表す。
骨肉種由来LM8細胞及び皮膚線維肉腫由来MethA細胞を6ウェルディッシュで培養し、約50%のコンフルエント状態になった時点で培養液を除去し、10mM EDTA/PBSを5ml添加し、37℃で5分間インキュベートした。その結果細胞はほぼすべて剥離され、ピペットでサスペンドすることで、ほぼ単一細胞にまで分離できた。2回D-MEM/10%FCSで洗浄し、D-MEM/10%FCSにサスペンドし、LIVE/DEAD(登録商標) Fixable Near-IR Dead Cell Stain Kit (ThermoFisher Scientific, L34975)と抗マウスCCR8抗体(SA214G2)又はIsotype Control抗体で、氷中で細胞を染色した。1時間後に3回D-MEM/10%FCSで洗浄し、フローサイトメーター(FACSCantoII)でマウスCCR8発現率を解析した。Isotype Control抗体を用いてバックグラウンドを設定し、バックグラウンドレベル以上の陽性細胞率と蛍光の中央値(Median)を算出した(図28)。その結果いずれの細胞においてもPEの蛍光強度の中央値に差は認められず、また陽性細胞もまったく認められなかった。以上より、これら細胞は抗マウスCCR8抗体には認識されず、マウスCCR8を発現しないあるいは抗体が反応するエピトープを保持しないことが確認された。
マウスC3H/Heマウス(7週齢、オス)の背部皮内に3x105個のマウス骨肉種由来LM8細胞(50uL)を移植した。腫瘍移植3日後にラット抗マウスCCR8抗体(クローンSA214G2、BioLegend社)を腹腔内に400μg(400μL)投与した(N=11)。コントロールはアイソタイプコントロール抗体を投与した(N=10)。腫瘍移植7日後(抗体投与後4日)より3~4日ごとに腫瘍体積を測定した。腫瘍体積(mm3)は長径(mm)x短径(mm)x短径(mm)/2で計量した(図29)。その結果移植18日目以降全測定時点において、アイソタイプコントロール抗体投与群と比較して、抗mCCR8投与群で有意に腫瘍体積の平均値が減少した(有意水準は18日目が*;P<0.05、21、24、27、31日目が**; P<0.01、35日目が***; P<0.001)。また抗体投与31日目時点で、抗マウスCCR8抗体投与群では11匹中6匹、アイソタイプコントロール抗体投与群では10匹中1匹の腫瘍が消失した。この分離態様に関してピアソンのカイ2乗検定を行ったところ、有意差があった(P=0.031)。
マウスBalb/cマウス(7週齢、メス)の背部皮内に1x105個の皮膚線維肉腫由来MethA(50uL)を移植した。腫瘍移植3日後にラット抗マウスCCR8抗体(クローンSA214G2、BioLegend社)を腹腔内に400μg(400μL)投与した(N=5)。コントロールはアイソタイプコントロール抗体を投与した(N=5)。腫瘍移植11日後(抗体投与後8日)より3~4日ごとに腫瘍体積を測定した。腫瘍体積(mm3)は長径(mm)x短径(mm)x短径(mm)/2で計量した(図30)。
その結果移植11日目以降全測定時点において、アイソタイプコントロール抗体投与群と比較して、抗マウスCCR8抗体投与群で有意に腫瘍体積の平均値が減少した(有意水準はすべての時点で、*;P<0.05)。また抗体投与21日目時点で、抗マウスCCR8抗体投与群では5匹中5匹、アイソタイプコントロール抗体投与群では5匹中0匹の腫瘍が消失した。この分離態様に関してピアソンのカイ2乗検定を行ったところ、有意差があった(P=0.0016)。
マウスBalb/cマウス(7週齢、メス)の背部皮内に1x105個の乳癌由来EMT6(50uL)を移植した。腫瘍移植3日後及び10日後にラット抗マウスCCR8抗体(クローンSA214G2、BioLegend社)を尾静脈内に100μg(100μL)投与した(N=20)。コントロールはアイソタイプコントロール抗体を同量投与した(N=20)。腫瘍移植4日後(抗体投与後1日)より3~4日ごとに腫瘍体積を測定した。腫瘍体積(mm3)は長径(mm)x短径(mm)x短径(mm)/2で計量した(図31)。
その結果移植10日目以降全測定時点において、アイソタイプコントロール抗体投与群と比較して、抗マウスCCR8抗体投与群で有意に腫瘍体積の平均値が減少した(有意水準は10日目が**;P<0.01、14、17、21日目が***; P<0.001)。また抗体投与21日目時点で、抗マウスCCR8抗体投与群では20匹中19匹、アイソタイプコントロール抗体投与群では20匹中2匹の腫瘍が消失した。この分離態様に関してピアソンのカイ2乗検定を行ったところ、有意差があった(P<0.0001)。
マウスBalb/cマウス(7週齢、メス)の背部皮内に2x105個の大腸癌由来Colon26細胞(50uL)を移植した。腫瘍移植3日後及び10日後にアイソタイプコントロール抗体、ラット抗マウスCCR8抗体(クローンSA214G2、BioLegend社)または抗マウスPD-1抗体(RMP1-14,Bioxcell社)を静脈内に400μg(400μL)投与した(N=10)。腫瘍移植3日後より3~4日ごとに腫瘍体積を測定した。腫瘍体積(mm3)は長径(mm)x短径(mm)x短径(mm)/2で計量した(図32)。その結果アイソタイプ抗体投与群と比較して、抗マウスCCR8投与群で、17、20、及び24日目において、有意に腫瘍体積が減少した(Steelのノンパラメトリック検定:有意水準はP<0.05)。抗PD-1抗体投与群ではアイソタイプ抗体投与群と比較して、どの時点においても有意差は認められなかった。
また抗体投与24日目時点で、1000mm3以上の体積の腫瘍を保持するマウス個体はアイソタイプ抗体投与群では10匹中7匹、抗マウスCCR8抗体投与群では10匹中2匹、抗PD-1抗体投与群では10匹中7匹であり、抗CCR8投与群はアイソタイプ抗体投与群及び抗PD-1抗体投与群のどちらに対しても、分離態様において、ピアソンのカイ2乗検定で有意差があった(どちらもP=0.025)。以上より大腸癌細胞株Colon26において、抗マウスCCR8抗体投与により抗腫瘍治療効果が認められた。
さらに移植20日目、24日目において、抗マウスPD-1抗体投与群と比較して、抗マウスCCR8投与群で有意に腫瘍体積が減少した(Steel-Dwassのノンパラメトリック検定;有意水準はP<0.05)。以上よりマウス大腸細胞株において、抗PD-1抗体投与群と比較して、抗マウスCCR8抗体投与群において、より強い抗腫瘍治療効果が認められた。
マウス腎癌由来細胞株RAGを用いて同様の検討をした。Balb/cマウス(8週齢、メス)の背部皮内に4x105個の腎癌由来RAG細胞(50uL)を移植した。なお、RAG細胞は予めBalb/cマウスに皮下移植して生着した腫瘍を再度マウスに移植し、この操作を2回繰り返し、マウス皮下への生着効率を上昇させたRAG細胞(細胞馴化株)を使用した。腫瘍移植6日後にアイソタイプコントロール抗体(N=10、ただし21日目のみN=9)、ラット抗マウスCCR8抗体(N=10)(クローンSA214G2、BioLegend社)または抗マウスPD-1抗体(N=10)(RMP1-14,Bioxcell社)を静脈内に100μg(100μL)投与した。腫瘍移植6日後より3~4日ごとに腫瘍体積を測定した。腫瘍体積(mm3)は長径(mm)x短径(mm)x短径(mm)/2で計量した(図33)。その結果アイソタイプ抗体投与群と比較して、抗マウスCCR8抗体投与群で、投与後14、17、及び21日目において有意に腫瘍体積が減少した(Steelのノンパラメトリック検定:有意水準はP<0.05)。アイソタイプ抗体投与群と比較して、抗マウスPD-1抗体投与群では有意差は認められなかった。以上より腎癌細胞株において抗マウスCCR8抗体投与により抗腫瘍治療効果が認められた。また移植14日目において、抗マウスPD-1抗体投与群と比較して、抗マウスCCR8抗体投与群で有意に腫瘍体積が減少した(Steel-Dwassのノンパラメトリック検定;有意水準はP<0.05)。以上よりマウス腎癌細胞株において、抗マウスPD-1抗体投与群と比較して、抗マウスCCR8抗体投与群において、より強い抗腫瘍治療効果が認められた。
マウスBalb/cマウス(7週齢、メス)の背部皮内に2x105個の大腸癌由来Colon26細胞(50uL)を移植した。腫瘍移植3日後及び10日後にラット抗マウスCD198(CCR8)抗体(クローンSA214G2、BioLegend社)またはアイソタイプコントロール抗体(LTF-2,Bioxcell社)を静脈内に400μg(400μL)投与した(N=10)。移植24日目での体重及びマウス各臓器(肺、肝臓、脾臓、小腸、鼠径リンパ節)の重量を測定した(図34)。その結果、図34に示すように、コントロール投与群(N=10)と抗マウスCCR8抗体投与群(N=10)で体重及び各臓器重量に有意差は認められなかった。以上より抗マウスCCR8抗体投与による炎症反応及び自己免疫疾患は惹起されていないと結論した。
ヒト腎癌、卵巣癌、子宮体癌、大腸癌、及び肺癌の腫瘍内浸潤細胞におけるCCR8の発現解析を行った。発現解析に用いた各種臨床腫瘍の患者数は、腎癌が12名、卵巣癌が14名、子宮体癌が21名、大腸癌が10名、及び肺癌が4名である。各種臨床腫瘍内浸潤細胞を実施例1の図1と同様に単離し、Anti-CD45 (BioLegend, Clone H130)、anti-CCR8 (BioLegend, Clone L263G8) 抗体で染色し、フローサイトメトリー (BD Biosciences, BD LSRFortessa) で測定した。腫瘍重量あたりのCCR8陽性細胞数及びCD45陽性白血球中のCCR8陽性細胞の割合について解析した。
表2に腫瘍重量あたりのCCR8陽性細胞数の平均値及びその標準偏差を示す。表3にCD45陽性白血球中のCCR8陽性細胞の割合の平均値及びその標準偏差を示す。
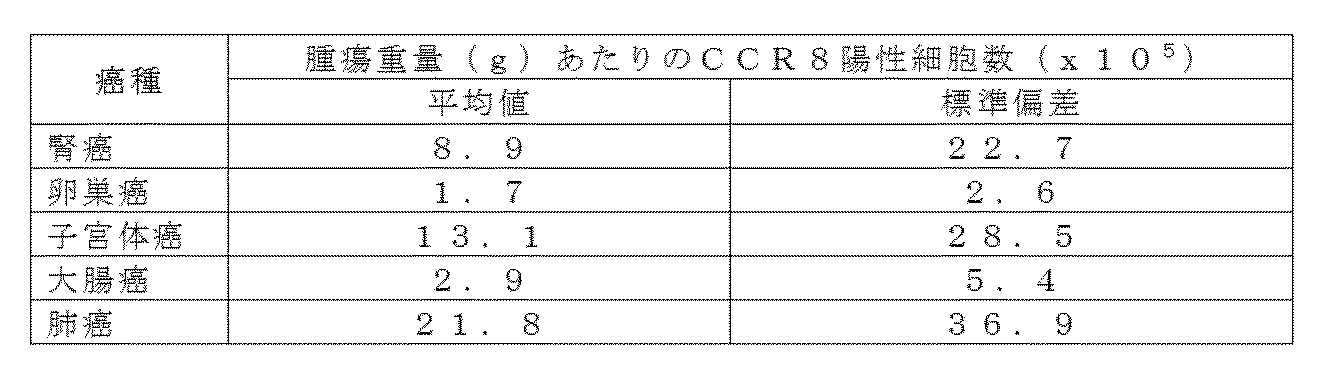
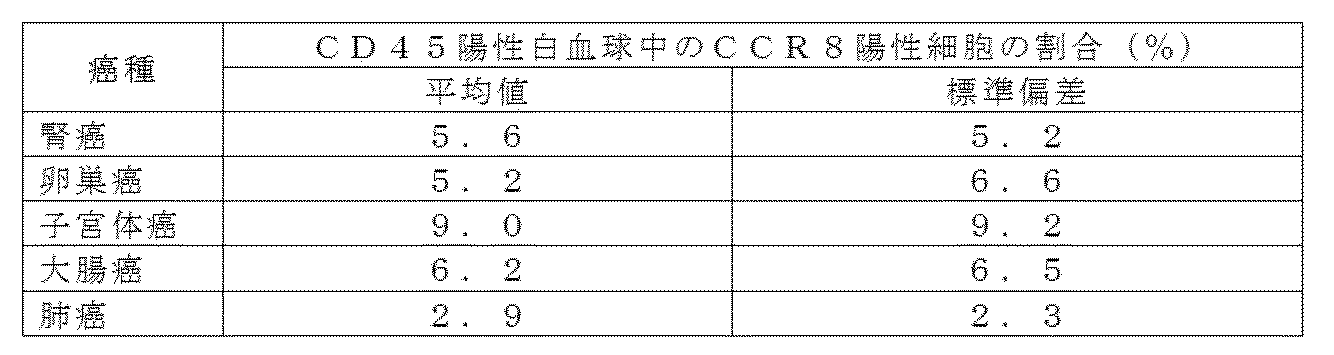
マウスBalb/cマウス(7週齢、メス)の背部皮内に1x105個の乳癌由来EMT6(50uL)を移植した。
抗マウスCCR8抗体単独投与群は、腫瘍移植3日後及び10日後にラット抗マウスCCR8抗体15μg(クローンSA214G2、BioLegend社)を静脈内に投与し(100μL)、腫瘍移植8日目及び13日目にアイソタイプコントロール抗体を200μg(100μL) 投与した(N=10)。抗PD-1抗体単独投与群は、腫瘍移植3日後及び10日後にアイソタイプコントロール抗体15μg(100μL)を、腫瘍移植8日目及び13日目に抗マウスPD-1抗体(RMP1-14、Bioxcell社)200μg(100μL)を静脈内に投与した(N=10)。抗PD-1抗体及び抗マウスCCR8抗体併用投与群は、腫瘍移植3日目及び10日目にラット抗マウスCCR8抗体15μg(100μL)を静脈内に投与し、腫瘍移植8日目及び13日目に抗PD-1抗体200μg(100μL)を静脈内に投与した(N=10)。コントロール群は腫瘍移植3日後及び10日後にアイソタイプコントロール抗体15μg(100μL)を静脈内に投与し、腫瘍移植8日目及び13日目にPBS100μLを静脈内に投与した(N=10)。腫瘍移植3日後(抗体投与後1日)より3~4日ごとに腫瘍体積を測定した。腫瘍体積(mm3)は長径(mm)x短径(mm)x短径(mm)/2で計量した(図35)。
平均腫瘍体積についての単独投与群同士の比較では、抗PD-1抗体投与群と比較して10、14、17、20、23及び27日目において、抗マウスCCR8抗体投与群で平均腫瘍体積が有意に小さかった(Dunnett法による有意水準:P<0.05)。また、各単独投与群と比較して併用群で腫瘍が小さかった。
また、移植後17日及び27日目における腫瘍の完全寛解率についても比較した。移植17日目でコントロール群及び抗PD-1抗体投与群では10匹中0匹、抗マウスCCR8抗体投与群では10匹中1匹が腫瘍の完全寛解を示したが、抗PD-1抗体と抗マウスCCR8抗体併用群では10匹中6匹が完全寛解した。移植27日目では、コントロール群及び抗PD-1抗体投与群ではそれぞれ10匹中2匹と3匹、抗マウスCCR8抗体投与群では10匹中7匹が腫瘍の完全寛解を示したが、抗PD-1抗体と抗マウスCCR8抗体の併用群では10匹中9匹が完全寛解した。
さらに、50mm3以下にまで腫瘍が退縮した個体の割合を算出した(図36)。抗PD-1抗体と抗マウスCCR8抗体の併用群では移植17日目ですべての個体で腫瘍が50mm3以下に退縮し(100%)、その後27日目まで50mm3以下であったが、抗PD-1抗体投与群では17日目で10%、27日目で30%、また抗マウスCCR8抗体投与群では移植後17日目で70%、27日目でも同じ70%であった。
以上の結果より,他の単独投与群と比較して併用群では腫瘍退縮までの時間が早く、また退縮効果が強いことが分かった。
マウスBalb/cマウス(6週齢、メス)の背部皮内に4.5x105個の腎癌由来RAG細胞(50uL)を移植した。なお、RAG細胞は予めBalb/cマウスに皮下移植して生着した腫瘍を再度マウスに移植し、この操作を2回繰り返し、マウス皮下への生着効率を上昇させたRAG細胞(細胞馴化株)を使用した。
抗PD-1抗体単独投与群は腫瘍移植8日後及び15日後に抗PD-1抗体(RMP1-14、Bioxcell社)を静脈内に50μg(100μL)投与した(N=10)。抗マウスCCR8抗体単独投与群は腫瘍移植8日後及び15日後にラット抗マウスCCR8抗体(クローンSA214G2、BioLegend社)を静脈内に25μg(100μL)投与した(N=10)。抗PD-1抗体及び抗マウスCCR8抗体併用投与群は、腫瘍移植8日後及び15日後に抗PD-1抗体(RMP1-14、Bioxcell社)50μgとラット抗マウスCCR8抗体(クローンSA214G2、BioLegend社)25μgを混合し(100μL)、静脈内に投与した(N=10)。コントロール群は腫瘍移植8日後及び15日後に生理食塩水を静脈内に100μL投与した(N=10)。
腫瘍移植8日後より3~4日ごとに腫瘍体積を測定した。腫瘍体積(mm3)は長径(mm)x短径(mm)x短径(mm)/2で計量した(図37)。
その結果抗PD-1抗体と抗マウスCCR8抗体の併用群が、抗PD-1抗体あるいは抗マウスCCR8抗体の単独投与群と比較して、腫瘍が小さくなることが分かった。
Balb/c系統の野生型マウス(N=10)及びCCR8遺伝子ホモ欠損マウス(N=5)の背部皮内に3x105個の大腸癌由来Colon26細胞(50uL)を移植した。野生型マウスには腫瘍移植3日後及び10日後にラット抗マウスCCR8抗体(クローンSA214G2、BioLegend社)またはアイソタイプコントロール抗体(LTF-2,Bioxcell社)を静脈内に100μg(100μL)投与した(N=5)。CCR8遺伝子ホモ欠損マウスについても腫瘍移植3日後及び10日後にラット抗マウスCCR8抗体(クローンSA214G2、BioLegend社)またはアイソタイプコントロール抗体(LTF-2,Bioxcell社)を静脈内に100μg(100μL)投与した(N=5)。投与後7日目より腫瘍の大きさを測定した。
その結果、野生型マウスでは、アイソタイプコントロール抗体投与と比較して、抗マウスCCR8抗体投与により、全例で有意な腫瘍の退縮と、最終的な腫瘍の完全退縮が認められた。一方、CCR8遺伝子ホモ欠損マウスではアイソタイプ抗体と比較して、抗マウスCCR8抗体投与群で腫瘍の体積に変化は認められず、また腫瘍退縮も認められなかった(図38)。
CCR8遺伝子ホモ欠損マウスにおいて抗マウスCCR8抗体の抗腫瘍効果が完全に消失したことから、使用している抗マウスCCR8抗体(SA214G2)はCCR8を介して抗腫瘍効果を発揮していることが証明された。
Claims (9)
- CCR8に対する抗体を含有する癌治療用医薬組成物。
- CCR8に対する抗体がADCC活性を有する抗体である、請求項1記載の医薬組成物。
- CCR8に対する抗体が、CCR8の中和抗体である、請求項1又は請求項2のいずれかに記載の医薬組成物。
- CCR8に対する抗体が、腫瘍内浸潤Treg細胞除去作用を有する、請求項1~請求項3のいずれかに記載の医薬組成物。
- CCR8に対する抗体が、腫瘍内浸潤マクロファージ細胞除去作用を有する、請求項1~請求項4のいずれかに記載の医薬組成物。
- 癌が乳癌、大腸癌、腎癌または肉腫である、請求項1~請求項5のいずれかに記載の医薬組成物。
- CCR8に対する抗体、及び抗PD―1抗体又は抗PD-L1抗体を組み合わせてなる癌治療用の医薬。
- 請求項1~請求項5のいずれかに記載のCCR8に対する抗体を投与することを特徴とする、癌の治療方法。
- 癌を治療するための、請求項1~請求項5のいずれかに記載のCCR8に対する抗体。
Priority Applications (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18775737.2A EP3431105B1 (en) | 2017-03-29 | 2018-03-28 | Medicinal composition for treating cancer |
| CA3057274A CA3057274C (en) | 2017-03-29 | 2018-03-28 | Pharmaceutical composition for cancer treatment |
| ES18775737T ES2776926T3 (es) | 2017-03-29 | 2018-03-28 | Composición medicinal para tratamiento del cáncer |
| PL18775737T PL3431105T3 (pl) | 2017-03-29 | 2018-03-28 | Kompozycja lecznicza do leczenia raka |
| RU2019134417A RU2730984C1 (ru) | 2017-03-29 | 2018-03-28 | Фармацевтическая композиция для лечения злокачественного новообразования |
| CN201880021831.3A CN110573180B (zh) | 2017-03-29 | 2018-03-28 | 癌症治疗用药物组合物 |
| DK18775737.2T DK3431105T3 (da) | 2017-03-29 | 2018-03-28 | Medicinsk sammensætning til behandling af cancer |
| PL19202367T PL3616720T3 (pl) | 2017-03-29 | 2018-03-28 | Kompozycja farmaceutyczna do leczenia nowotworu |
| AU2018243020A AU2018243020B2 (en) | 2017-03-29 | 2018-03-28 | Medicinal composition for treating cancer |
| EP20216033.9A EP3858860A1 (en) | 2017-03-29 | 2018-03-28 | Pharmaceutical composition for cancer treatment |
| EP19202367.9A EP3616720B1 (en) | 2017-03-29 | 2018-03-28 | Pharmaceutical composition for cancer treatment |
| KR1020197029821A KR102144658B1 (ko) | 2017-03-29 | 2018-03-28 | 암 치료용 의약 조성물 |
| JP2018551478A JP6501171B2 (ja) | 2017-03-29 | 2018-03-28 | 癌治療用医薬組成物 |
| US16/183,216 US10550191B2 (en) | 2017-03-29 | 2018-11-07 | Method of treating cancer with an anti-CCR8 having antibody-dependent cell-mediated cytotoxicity (ADCC) activity against cells expressing CCR8 |
| US16/732,764 US20200222463A1 (en) | 2017-03-29 | 2020-01-02 | Pharmaceutical Composition for Cancer Treatment |
| US16/732,850 US20200232973A1 (en) | 2017-03-29 | 2020-01-02 | Pharmaceutical Composition for Cancer Treatment |
| US17/085,040 US20210047421A1 (en) | 2017-03-29 | 2020-10-30 | Pharmaceutical Composition for Cancer Treatment |
| US17/134,775 US11932696B2 (en) | 2017-03-29 | 2020-12-28 | Method of treating cancer with an anti-CCR8 that binds tumor infiltrating cells |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017065603 | 2017-03-29 | ||
| JP2017-065603 | 2017-03-29 | ||
| JP2017185935 | 2017-09-27 | ||
| JP2017-185935 | 2017-09-27 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US16/183,216 Continuation US10550191B2 (en) | 2017-03-29 | 2018-11-07 | Method of treating cancer with an anti-CCR8 having antibody-dependent cell-mediated cytotoxicity (ADCC) activity against cells expressing CCR8 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018181425A1 true WO2018181425A1 (ja) | 2018-10-04 |
Family
ID=63676314
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/012644 WO2018181425A1 (ja) | 2017-03-29 | 2018-03-28 | 癌治療用医薬組成物 |
Country Status (14)
| Country | Link |
|---|---|
| US (5) | US10550191B2 (ja) |
| EP (3) | EP3431105B1 (ja) |
| JP (4) | JP6501171B2 (ja) |
| KR (1) | KR102144658B1 (ja) |
| CN (1) | CN110573180B (ja) |
| AU (1) | AU2018243020B2 (ja) |
| CA (1) | CA3057274C (ja) |
| DK (2) | DK3616720T3 (ja) |
| ES (2) | ES2776926T3 (ja) |
| HU (1) | HUE054316T2 (ja) |
| PL (2) | PL3616720T3 (ja) |
| PT (2) | PT3431105T (ja) |
| RU (1) | RU2730984C1 (ja) |
| WO (1) | WO2018181425A1 (ja) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3431105B1 (en) | 2017-03-29 | 2020-01-01 | Shionogi&Co., Ltd. | Medicinal composition for treating cancer |
| WO2020138489A1 (ja) | 2018-12-27 | 2020-07-02 | 塩野義製薬株式会社 | 新規抗ccr8抗体 |
| WO2021079958A1 (ja) * | 2019-10-25 | 2021-04-29 | 第一三共株式会社 | 抗garp抗体と免疫調節剤の組み合わせ |
| WO2021152186A2 (en) | 2020-06-26 | 2021-08-05 | Bayer Aktiengesellschaft | Ccr8 antibodies for therapeutic applications |
| WO2022003156A1 (en) | 2020-07-02 | 2022-01-06 | Oncurious Nv | Ccr8 non-blocking binders |
| WO2022004760A1 (ja) | 2020-06-30 | 2022-01-06 | 塩野義製薬株式会社 | 抗ccr8抗体と化学療法剤の併用 |
| WO2022117572A2 (en) | 2020-12-02 | 2022-06-09 | Oncurious Nv | An ltbr agonist in combination therapy against cancer |
| WO2022117569A1 (en) | 2020-12-02 | 2022-06-09 | Oncurious Nv | A ccr8 antagonist antibody in combination with a lymphotoxin beta receptor agonist antibody in therapy against cancer |
| WO2022136647A1 (en) | 2020-12-24 | 2022-06-30 | Oncurious Nv | Human ccr8 binders |
| WO2022136649A1 (en) | 2020-12-24 | 2022-06-30 | Oncurious Nv | Non-blocking human ccr8 binders |
| WO2022136650A1 (en) | 2020-12-24 | 2022-06-30 | Oncurious Nv | Murine cross-reactive human ccr8 binders |
| WO2022211046A1 (ja) | 2021-03-31 | 2022-10-06 | 塩野義製薬株式会社 | Ccr8を抗原として認識するキメラ抗原受容体 |
| RU2782462C1 (ru) * | 2018-12-27 | 2022-10-27 | Сионоги Энд Ко., Лтд. | Новое антитело против ccr8 |
| WO2022256563A1 (en) | 2021-06-04 | 2022-12-08 | Amgen Inc. | Anti-ccr8 antibodies and uses thereof |
| US11692038B2 (en) | 2020-02-14 | 2023-07-04 | Gilead Sciences, Inc. | Antibodies that bind chemokine (C-C motif) receptor 8 (CCR8) |
| WO2024062082A1 (en) | 2022-09-21 | 2024-03-28 | Domain Therapeutics | Anti-ccr8 monoclonal antibodies and their therapeutic use |
| WO2024062072A2 (en) | 2022-09-21 | 2024-03-28 | Domain Therapeutics | Anti-ccr8 monoclonal antibodies and their therapeutic use |
| WO2024062076A1 (en) | 2022-09-21 | 2024-03-28 | Domain Therapeutics | Anti-ccr8 monoclonal antibodies and their therapeutic use |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2022008341A (es) * | 2020-01-06 | 2022-08-10 | Vaccinex Inc | Anticuerpos anti-ccr8 y usos de estos. |
| PE20230821A1 (es) | 2020-03-23 | 2023-05-19 | Bristol Myers Squibb Co | Anticuerpos anti-ccr8 para el tratamiento del cancer |
| EP4010378A4 (en) | 2020-10-16 | 2023-07-26 | LaNova Medicines Limited | ANTICCR8 MONOCLONAL ANTIBODIES AND USES THEREOF |
| AU2022310847A1 (en) | 2021-07-14 | 2024-01-25 | Genentech, Inc. | Anti-c-c motif chemokine receptor 8 (ccr8) antibodies and methods of use |
| JP2023018678A (ja) | 2021-07-27 | 2023-02-08 | アッヴィ・インコーポレイテッド | 抗ccr8抗体 |
| CN114565761B (zh) * | 2022-02-25 | 2023-01-17 | 无锡市第二人民医院 | 一种基于深度学习的肾透明细胞癌病理图像肿瘤区域的分割方法 |
| TW202400648A (zh) * | 2022-04-29 | 2024-01-01 | 大陸商江蘇恆瑞醫藥股份有限公司 | 抗ccr8抗體及其用途 |
| WO2024077239A1 (en) | 2022-10-07 | 2024-04-11 | Genentech, Inc. | Methods of treating cancer with anti-c-c motif chemokine receptor 8 (ccr8) antibodies |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994011523A2 (en) | 1992-11-13 | 1994-05-26 | Idec Pharmaceuticals Corporation | Fully impaired consensus kozac sequences for mammalian expression |
| WO2002031140A1 (fr) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cellules produisant des compositions d'anticorps |
| WO2005035586A1 (ja) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | 融合蛋白質組成物 |
| WO2007044756A2 (en) * | 2005-10-11 | 2007-04-19 | Icos Corporation | Monoclonal antibodies recognizing human ccr8 |
| WO2013131010A2 (en) * | 2012-03-02 | 2013-09-06 | Icahn School Of Medicine At Mount Sinai | Function of chemokine receptor ccr8 in melanoma metastasis |
| WO2015179236A1 (en) * | 2014-05-21 | 2015-11-26 | Pfizer Inc. | Combination of an anti-ccr4 antibody and a 4-1bb agonist for treating cancer |
| WO2016092419A1 (en) | 2014-12-09 | 2016-06-16 | Rinat Neuroscience Corp. | Anti-pd-1 antibodies and methods of use thereof |
| JP2017065603A (ja) | 2015-10-01 | 2017-04-06 | 横浜ゴム株式会社 | 空気入りタイヤ |
| JP2017185935A (ja) | 2016-04-07 | 2017-10-12 | 北陸重機工業株式会社 | 鉄道車両と除雪装置との着脱構造 |
| WO2017198631A1 (en) * | 2016-05-16 | 2017-11-23 | Istituto Nazionale Di Genetica Molecolare - Ingm | Markers selectively deregulated in tumor-infiltrating regulatory t cells |
| EP3616720B1 (en) | 2017-03-29 | 2021-02-17 | Shionogi&Co., Ltd. | Pharmaceutical composition for cancer treatment |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6693130B2 (en) | 1999-02-18 | 2004-02-17 | Regents Of The University Of California | Inhibitors of epoxide hydrolases for the treatment of hypertension |
| US6531506B1 (en) | 1996-08-13 | 2003-03-11 | Regents Of The University Of California | Inhibitors of epoxide hydrolases for the treatment of hypertension |
| WO2002062850A2 (en) * | 2001-02-02 | 2002-08-15 | Millennium Pharmaceuticals, Inc. | Hybrid antibodies and uses thereof |
| EP1904050B1 (en) | 2005-06-06 | 2012-11-14 | The Regents of The University of California | Use of cis-epoxyeicosatrienoic acids and inhibitors of soluble epoxide hydrolase to reduce cardiomyopathy |
| WO2008073160A2 (en) * | 2006-08-17 | 2008-06-19 | The Trustees Of Columbia University In The City Of New York | Methods for converting or inducing protective immunity |
| EP2093237B1 (en) * | 2006-10-20 | 2015-12-30 | Chugai Seiyaku Kabushiki Kaisha | Anti-cancer agent comprising anti-hb-egf antibody as active ingredient |
| US8680237B2 (en) | 2007-06-01 | 2014-03-25 | Gliknik Inc. | Immunoglobulin constant region FC receptor binding agents |
| LT2530091T (lt) | 2010-01-29 | 2018-06-11 | Chugai Seiyaku Kabushiki Kaisha | Anti-dll3 antikūnas |
| EP3434695B1 (en) | 2012-08-07 | 2020-12-02 | Roche Glycart AG | Improved immunotherapy |
| GB2519786A (en) | 2013-10-30 | 2015-05-06 | Sergej Michailovic Kiprijanov | Multivalent antigen-binding protein molecules |
| US10087259B1 (en) | 2014-04-28 | 2018-10-02 | Memorial Sloan Kettering Cancer Center | Depleting tumor-specific tregs |
| US11505605B2 (en) * | 2014-05-13 | 2022-11-22 | Chugai Seiyaku Kabushiki Kaisha | T cell-redirected antigen-binding molecule for cells having immunosuppression function |
| EP3835323A1 (en) * | 2014-10-29 | 2021-06-16 | Five Prime Therapeutics, Inc. | Combination therapy for cancer |
| US10596257B2 (en) | 2016-01-08 | 2020-03-24 | Hoffmann-La Roche Inc. | Methods of treating CEA-positive cancers using PD-1 axis binding antagonists and anti-CEA/anti-CD3 bispecific antibodies |
| IL261998B2 (en) * | 2016-04-07 | 2023-03-01 | Chemocentryx Inc | Reducing your burden by giving ccr1 antagonists in combination with pd-1 inhibitors or pd-l1 inhibitors |
| EP3463451A1 (en) | 2016-05-26 | 2019-04-10 | Qilu Puget Sound Biotherapeutics Corporation | Mixtures of antibodies |
| WO2018112033A1 (en) | 2016-12-13 | 2018-06-21 | President And Fellows Of Harvard College | Methods and compositions for targeting tumor-infiltrating tregs |
| WO2018181424A1 (ja) | 2017-03-30 | 2018-10-04 | 日東電工株式会社 | 遮熱断熱基板 |
| WO2019157098A1 (en) | 2018-02-06 | 2019-08-15 | Advaxis, Inc. | Compositions comprising a recombinant listeria strain and an anti-ccr8 antibody and methods of use |
| MX2021007576A (es) | 2018-12-27 | 2021-08-24 | Shionogi & Co | Nuevo anticuerpo anti receptor 8 de motivo c-c de quimiocina (ccr8). |
| WO2021178749A2 (en) | 2020-03-05 | 2021-09-10 | Memorial Sloan Kettering Cancer Center | Anti-ccr8 agents |
| EP4267618A1 (en) | 2020-12-24 | 2023-11-01 | Vib Vzw | Non-blocking human ccr8 binders |
-
2018
- 2018-03-28 DK DK19202367.9T patent/DK3616720T3/da active
- 2018-03-28 PL PL19202367T patent/PL3616720T3/pl unknown
- 2018-03-28 EP EP18775737.2A patent/EP3431105B1/en active Active
- 2018-03-28 ES ES18775737T patent/ES2776926T3/es active Active
- 2018-03-28 AU AU2018243020A patent/AU2018243020B2/en active Active
- 2018-03-28 HU HUE19202367A patent/HUE054316T2/hu unknown
- 2018-03-28 PT PT187757372T patent/PT3431105T/pt unknown
- 2018-03-28 EP EP19202367.9A patent/EP3616720B1/en active Active
- 2018-03-28 WO PCT/JP2018/012644 patent/WO2018181425A1/ja active Application Filing
- 2018-03-28 CN CN201880021831.3A patent/CN110573180B/zh active Active
- 2018-03-28 RU RU2019134417A patent/RU2730984C1/ru active
- 2018-03-28 ES ES19202367T patent/ES2863412T3/es active Active
- 2018-03-28 JP JP2018551478A patent/JP6501171B2/ja active Active
- 2018-03-28 KR KR1020197029821A patent/KR102144658B1/ko active IP Right Grant
- 2018-03-28 CA CA3057274A patent/CA3057274C/en active Active
- 2018-03-28 PL PL18775737T patent/PL3431105T3/pl unknown
- 2018-03-28 DK DK18775737.2T patent/DK3431105T3/da active
- 2018-03-28 PT PT192023679T patent/PT3616720T/pt unknown
- 2018-03-28 EP EP20216033.9A patent/EP3858860A1/en active Pending
- 2018-11-07 US US16/183,216 patent/US10550191B2/en active Active
-
2019
- 2019-01-22 JP JP2019008733A patent/JP6557827B2/ja active Active
- 2019-05-31 JP JP2019102342A patent/JP6656565B2/ja active Active
-
2020
- 2020-01-02 US US16/732,850 patent/US20200232973A1/en not_active Abandoned
- 2020-01-02 US US16/732,764 patent/US20200222463A1/en active Pending
- 2020-01-10 JP JP2020002606A patent/JP6712701B2/ja active Active
- 2020-10-30 US US17/085,040 patent/US20210047421A1/en active Pending
- 2020-12-28 US US17/134,775 patent/US11932696B2/en active Active
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994011523A2 (en) | 1992-11-13 | 1994-05-26 | Idec Pharmaceuticals Corporation | Fully impaired consensus kozac sequences for mammalian expression |
| WO2002031140A1 (fr) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kogyo Co., Ltd. | Cellules produisant des compositions d'anticorps |
| WO2005035586A1 (ja) | 2003-10-08 | 2005-04-21 | Kyowa Hakko Kogyo Co., Ltd. | 融合蛋白質組成物 |
| WO2007044756A2 (en) * | 2005-10-11 | 2007-04-19 | Icos Corporation | Monoclonal antibodies recognizing human ccr8 |
| WO2013131010A2 (en) * | 2012-03-02 | 2013-09-06 | Icahn School Of Medicine At Mount Sinai | Function of chemokine receptor ccr8 in melanoma metastasis |
| WO2015179236A1 (en) * | 2014-05-21 | 2015-11-26 | Pfizer Inc. | Combination of an anti-ccr4 antibody and a 4-1bb agonist for treating cancer |
| WO2016092419A1 (en) | 2014-12-09 | 2016-06-16 | Rinat Neuroscience Corp. | Anti-pd-1 antibodies and methods of use thereof |
| JP2017065603A (ja) | 2015-10-01 | 2017-04-06 | 横浜ゴム株式会社 | 空気入りタイヤ |
| JP2017185935A (ja) | 2016-04-07 | 2017-10-12 | 北陸重機工業株式会社 | 鉄道車両と除雪装置との着脱構造 |
| WO2017198631A1 (en) * | 2016-05-16 | 2017-11-23 | Istituto Nazionale Di Genetica Molecolare - Ingm | Markers selectively deregulated in tumor-infiltrating regulatory t cells |
| EP3616720B1 (en) | 2017-03-29 | 2021-02-17 | Shionogi&Co., Ltd. | Pharmaceutical composition for cancer treatment |
Non-Patent Citations (53)
| Title |
|---|
| "Data Highlighting Advaxis’ Antigen Delivery Platform Accepted for Multiple Presentations at 2018 Keystone Symposia Conference on Cancer Immunotherapies | Business Wire", 29 January 2018 (2018-01-29), XP093001652, Retrieved from the Internet <URL:https://www.businesswire.com/news/home/20180129005112/en/Data-Highlighting-Advaxis’-Antigen-Delivery-Platform-Accepted-for-Multiple-Presentations-at-2018-Keystone-Symposia-Conference-on-Cancer-Immunotherapies> |
| "GenBank", Database accession no. M_005201.3 |
| "GenBank", Database accession no. NM_007720.2 |
| "Methods described in Molecular Cloning: A Laboratory Manual", COLD SPRING HARBOR LABORATORY |
| ALI, S. O'BOYLE, G. MELLOR, P. KIRBY, J.A.: "An apparent paradox: Chemokine receptor agonists can be used for anti-inflammatory therapy", MOLECULAR IMMUNOLOGY, PERGAMON, GB, vol. 44, no. 7, 26 November 2006 (2006-11-26), GB , pages 1477 - 1482, XP005780827, ISSN: 0161-5890, DOI: 10.1016/j.molimm.2006.08.011 |
| ANONYMOUS: "Antibody Isotype Families Clinically relevant antibodies in various Ig isotypes", INTERNET ARCHIVE WAYBACK MACHINE, 29 October 2016 (2016-10-29), pages 1 - 2, XP055887479, Retrieved from the Internet <URL:https://web.archive.org/web/20161029145322/www.invivogen.com/antibody-isotypes> [retrieved on 20220204] |
| ANONYMOUS: "OMIM Entry * 604836 CHEMOKINE, CC MOTIF, RECEPTOR 4; CCR4", INTERNET ARCHIVE WAYBACK MACHINE, 13 March 2017 (2017-03-13), pages 1 - 4, XP055887564, Retrieved from the Internet <URL:https://web.archive.org/web/20170313100720/www.omim.org/entry/604836> [retrieved on 20220204] |
| ARIAN D. J. LAURENCE: "Location, movement and survival: the role of chemokines in haematopoiesis and malignancy", BRITISH JOURNAL OF HAEMATOLOGY, JOHN WILEY, HOBOKEN, USA, vol. 132, no. 3, 5 December 2005 (2005-12-05), Hoboken, USA, pages 255 - 267, XP071121207, ISSN: 0007-1048, DOI: 10.1111/j.1365-2141.2005.05841.x |
| CANCER RES., vol. 59, no. 13, 1 July 1999 (1999-07-01), pages 3128 - 33 |
| CANCER RES., vol. 70, no. 7, 2010, pages 2665 - 74 |
| CANCER, vol. 107, 2006, pages 2866 - 2872 |
| CARL ET AL., THERAPEUTIC MONOCLONAL ANTIBODIES, 1990 |
| CELL, vol. 160, 2015, pages 48 - 61 |
| CELL. MOL. IMMUNOL., vol. 8, 2011, pages 59 - 66 |
| CHEMICAL ABSTRACTS, Columbus, Ohio, US; abstract no. 57-66-9 |
| CHEMICAL ABSTRACTS, Columbus, Ohio, US; abstract no. 7365-45-9 |
| CHIRGWIN, J.M. ET AL., BIOCHEMISTRY, vol. 18, 1979, pages 5294 - 5299 |
| CLINICAL CANCER RESEARCH, vol. 10, 2004, pages 1274 - 1281 |
| DANIEL O. VILLARREAL, ANDREW L'HUILLIER, SUSAN ARMINGTON, CRISTINA MOTTERSHEAD, ELENA V. FILIPPOVA, BRANDON D. CODER, ROBERT G. PE: "Targeting CCR8 Induces Protective Antitumor Immunity and Enhances Vaccine-Induced Responses in Colon Cancer", CANCER RESEARCH, vol. 78, no. 18, 15 September 2018 (2018-09-15), pages 5340 - 5348, XP055631142, ISSN: 0008-5472, DOI: 10.1158/0008-5472.CAN-18-1119 |
| E. J. KIM: "Immunopathogenesis and therapy of cutaneous T cell lymphoma", THE JOURNAL OF CLINICAL INVESTIGATION, B M J GROUP, GB, vol. 115, no. 4, GB , pages 798 - 812, XP055271996, ISSN: 0021-9738, DOI: 10.1172/JCI200524826 |
| EUR. J. CANCER, vol. 44, 2008, pages 1875 - 1882 |
| EUR. J. IMMUNOL., vol. 40, 2010, pages 3325 - 3335 |
| EXPERT OPINION ON THERAPEUTIC PATENTS, vol. 6, no. 5, 1996, pages 441 - 456 |
| FROHMAN, M.A. ET AL., PROC. NATL. ACAD. SCI. USA, vol. 85, 1988, pages 8998 |
| GANGULY, ET AL.: "The role of dendritic cells in autoimmunity", NAT REV IMMUNOL, vol. 13, no. 8, 1 January 2013 (2013-01-01), pages 566 - 577, XP055868977 |
| GROSSOJURE-KUNKEL: "CTLA-4 blockade in tumor models: an overview of preclinical and translational research", CANCER IMMUN. REV., vol. 13, no. 5, 22 January 2013 (2013-01-22), XP002739455 |
| GUPTA, ET AL.: "Antibodies against G-protein coupled receptors: novel uses in screening and drug development", COMB CHEM HIGH THROUGHPUT SCREEN., vol. 11, no. 6, 1 January 2008 (2008-01-01), pages 463 - 467, XP055868956 |
| HOELZINGER, D. B. ET AL.: "Blockade of CCL1 Inhibits T Regulatory Cell Suppressive Function Enhancing Tumor Immunity without Affecting T Effector Responses", JOURNAL OF IMMUNOLOGY, vol. 184, no. 12, 2010, pages 6833 - 6842, XP055306968 * |
| INVITROGEN: "CCR8 Recombinant Rabbit Monoclonal Antibody (ARC0956)", PRODUCT DATA SHEET, pages 1 - 2 |
| J. BIOL. CHEM., vol. 272, no. 28, 1997, pages 17251 - 4 |
| J. CLIN. ONCOL., vol. 24, 2006, pages 5373 - 5380 |
| J. CLIN. ONCOL., vol. 25, 2007, pages 2586 - 2593 |
| J. IMMUNOL., vol. 157, no. 7, 1996, pages 2759 - 63 |
| JIAWEN HAN: "Monoclonal Antibodies as Cancer Therapeutics", NORTH AMERICAN JOURNAL OF MEDICINE AND SCIENCE, vol. 3, no. 3, 1 July 2010 (2010-07-01), pages 146 - 151, XP055887475 |
| JIM STALLARD: "FDA Approves 'Game Changer' Immunotherapy Drug for Bladder Cancer", MSKCC, 18 May 2016 (2016-05-18), pages 1 - 11, XP055869007, Retrieved from the Internet <URL:https://www.mskcc.org/news/fda- approves-game-changer-immunotherapy-drug-bladder> [retrieved on 20211203] |
| KOHLER; MILSTEIN, NATURE, vol. 256, 1975, pages 495 - 497 |
| LIEPING CHEN, XUE HAN: "Anti–PD-1/PD-L1 therapy of human cancer: past, present, and future", THE JOURNAL OF CLINICAL INVESTIGATION, B M J GROUP, GB, vol. 125, no. 9, 1 September 2015 (2015-09-01), GB , pages 3384 - 3391, XP055432081, ISSN: 0021-9738, DOI: 10.1172/JCI80011 |
| MATTHEW TONTONOZ: "Combination Immunotherapy Shows New Promise for Lung Cancer", MSKCC, 1 June 2016 (2016-06-01), pages 1 - 12, XP055869000, Retrieved from the Internet <URL:https://www.mskcc.org/news/new-promise-combination-immunotherapy-lung-cancer#:~:text=Combination%20Immunotherapy%20Shows%20New%20Promise%20for%20Lung%20Cancer,-Share&text=Immunotherapy%20has%20emerged%20as%20a,from%20immunotherapies%2C%20at%20earlier%20stages.> [retrieved on 20211203] |
| MATTHEW TONTONOZ: "Immunotherapy Pioneered at MSK Receives FDA Approval for Advanced Kidney Cancer", MSKCC, 4 December 2015 (2015-12-04), pages 1 - 9, XP055869004, [retrieved on 20211203] |
| NAT. MED., vol. 10, 2004, pages 942 - 949 |
| NAT. REV. IMMUNOL., vol. 6, no. 4, 2006, pages 295 - 307 |
| NATURE BIOTECHNOLOGY, vol. 23, 2005, pages 1105 |
| PERNAMBUCO-HOLSTEN C.: "Immunotherapy Combination Nivolumab Plus Ipilimumab Receives FDA Approval for Metastatic Melanoma", MSKCC, 1 October 2015 (2015-10-01), pages 1 - 15, XP055868997, Retrieved from the Internet <URL:https://www.mskcc.org/news/immunotherapy-combination-nivolumab-plus-ipilimumab-receives-fda-approval-metastatic-melanoma> [retrieved on 20211203] |
| PLITAS, G. ET AL.: "Preferential expression of the chemokine receptor 8 (CCR8) on regulatory T cells (Treg) infiltrating human breast cancers represents a novel immunotherapeutic target", CANCER RESEARCH, vol. 76, no. 4, February 2016 (2016-02-01), XP055396219, Retrieved from the Internet <URL:http://cancerres.aacrjournals.org/content/76/ 4_Supplement/P4-04-ll.short> [retrieved on 20180618] * |
| R&D SYSTEMS: "Mouse CCR8 Antibody", PRODUCT DATA SHEET, 5 December 2018 (2018-12-05), pages 1, XP093001637 |
| ROSENBERG HELENE F.; DYER KIMBERLY D.; FOSTER PAUL S.: "Eosinophils: changing perspectives in health and disease", NATURE REVIEWS IMMUNOLOGY, NATURE PUBLISHING GROUP UK, LONDON, vol. 13, no. 1, 16 November 2012 (2012-11-16), London, pages 9 - 22, XP037065594, ISSN: 1474-1733, DOI: 10.1038/nri3341 |
| SLAUENWHITE, ET AL.: "Regulation of NKT cell localization in homeostasis and infection", FRONTIIERS IMMUNOLOGY, vol. 6, 255, 1 January 2015 (2015-01-01), pages 1 - 19, XP055868973 |
| SUGIYAMA, D. ET AL.: "Anti-CCR4 mAb selectively depletes effector-type FoxP3+ CD 4+ regulatory T cells, evoking antitumor immune responses in humans", PNAS, vol. 110, no. 44, 29 October 2013 (2013-10-29), pages 17945 - 17950, XP055284822 * |
| SUZUKI SUSUMU, ISHIDA TAKASHI, YOSHIKAWA KAZUHIRO, UEDA RYUZO: "Current status of immunotherapy", JAPANESE JOURNAL OF CLINICAL ONCOLOGY, TOKYO., JP, vol. 46, no. 3, 1 March 2016 (2016-03-01), JP , pages 191 - 203, XP055866688, ISSN: 0368-2811, DOI: 10.1093/jjco/hyv201 |
| VARGAS FREDERICK ARCE, FURNESS ANDREW J S, SOLOMON ISABELLE, JOSHI KROOPA, MEKKAOUI LEILA, LESKO MARTA H, ROTA ENRIQUE MIRANDA, DA: "Supplemental Information to Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors", IMMUNITY, 1 January 2017 (2017-01-01), pages 14pp, XP093001623, Retrieved from the Internet <URL:https://ars.els-cdn.com/content/image/1-s2.0-S1074761317301231-mmc1.pdf> |
| VILLARREAL DANIEL O, ARMINGTON SUSAN R, HUILLIER ANDREW L, MOTTERSHEAD CRISTINA, RAMOS KIMBERLY, FILIPPOVA ELENA V, KELKAR DIPTI M: "Targeting of CCR8 induces antitumor activity as a monotherapy that is further enhanced in com- bination with a Listeria-based immunotherapy", POSTER KEYSTONE SYMPOSIA, 1 January 2018 (2018-01-01), XP093001728, Retrieved from the Internet <URL:https://www.advaxis.com/static-files/a17de4c6-e593-443a-8503-8f70c4870a16> |
| WILCOX RYAN A: "Mogamulizumab: 2 birds, 1 stone", BLOOD, vol. 125, no. 12, 1 January 2015 (2015-01-01), pages 1847 - 1848, XP055866757 |
| XINGRUI LI; EFTERPI KOSTARELI; JANINE SUFFNER; NATALIO GARBI; GÜNTER J HÄMMERLING: "Efficient Treg depletion induces T-cell infiltration and rejection of large tumors", EUROPEAN JOURNAL OF IMMUNOLOGY, WILEY-VCH, HOBOKEN, USA, vol. 40, no. 12, 1 December 2010 (2010-12-01), Hoboken, USA, pages 3325 - 3335, XP055844150, ISSN: 0014-2980, DOI: 10.1002/eji.201041093 |
Cited By (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3616720B1 (en) | 2017-03-29 | 2021-02-17 | Shionogi&Co., Ltd. | Pharmaceutical composition for cancer treatment |
| EP3431105B1 (en) | 2017-03-29 | 2020-01-01 | Shionogi&Co., Ltd. | Medicinal composition for treating cancer |
| EP3903817A4 (en) * | 2018-12-27 | 2022-08-17 | Shionogi & Co., Ltd. | NEW ANTI-CCR8 ANTIBODY |
| RU2782462C1 (ru) * | 2018-12-27 | 2022-10-27 | Сионоги Энд Ко., Лтд. | Новое антитело против ccr8 |
| JP7452818B2 (ja) | 2018-12-27 | 2024-03-19 | 塩野義製薬株式会社 | 新規抗ccr8抗体 |
| JPWO2020138489A1 (ja) * | 2018-12-27 | 2021-02-18 | 塩野義製薬株式会社 | 新規抗ccr8抗体 |
| WO2020138489A1 (ja) | 2018-12-27 | 2020-07-02 | 塩野義製薬株式会社 | 新規抗ccr8抗体 |
| KR20210108996A (ko) | 2018-12-27 | 2021-09-03 | 시오노기세이야쿠가부시키가이샤 | 신규 항 ccr8 항체 |
| CN113260381A (zh) * | 2018-12-27 | 2021-08-13 | 盐野义制药株式会社 | 新型抗ccr8抗体 |
| WO2021079958A1 (ja) * | 2019-10-25 | 2021-04-29 | 第一三共株式会社 | 抗garp抗体と免疫調節剤の組み合わせ |
| CN114630679A (zh) * | 2019-10-25 | 2022-06-14 | 第一三共株式会社 | 抗garp抗体和免疫调节剂的组合 |
| US11692038B2 (en) | 2020-02-14 | 2023-07-04 | Gilead Sciences, Inc. | Antibodies that bind chemokine (C-C motif) receptor 8 (CCR8) |
| WO2021152186A2 (en) | 2020-06-26 | 2021-08-05 | Bayer Aktiengesellschaft | Ccr8 antibodies for therapeutic applications |
| WO2021260206A2 (en) | 2020-06-26 | 2021-12-30 | Bayer Aktiengesellschaft | Chemokine receptor antibodies binding sulfated trd motifs |
| WO2021260209A3 (en) * | 2020-06-26 | 2022-03-10 | Bayer Aktiengesellschaft | Ccr8 antibodies and uses thereof |
| WO2021260208A2 (en) | 2020-06-26 | 2021-12-30 | Bayer Aktiengesellschaft | Sulfated peptides for chemokine receptor antibody generation |
| WO2021260210A2 (en) | 2020-06-26 | 2021-12-30 | Bayer Aktiengesellschaft | Anti ccr8 antibody therapy: biomarkers & combination therapies |
| WO2021260209A2 (en) | 2020-06-26 | 2021-12-30 | Bayer Aktiengesellschaft | Ccr8 antibodies and uses thereof |
| US11427640B1 (en) | 2020-06-26 | 2022-08-30 | Bayer Aktiengesellschaft | CCR8 antibodies for therapeutic applications |
| WO2022004760A1 (ja) | 2020-06-30 | 2022-01-06 | 塩野義製薬株式会社 | 抗ccr8抗体と化学療法剤の併用 |
| WO2022003156A1 (en) | 2020-07-02 | 2022-01-06 | Oncurious Nv | Ccr8 non-blocking binders |
| WO2022117572A2 (en) | 2020-12-02 | 2022-06-09 | Oncurious Nv | An ltbr agonist in combination therapy against cancer |
| WO2022117569A1 (en) | 2020-12-02 | 2022-06-09 | Oncurious Nv | A ccr8 antagonist antibody in combination with a lymphotoxin beta receptor agonist antibody in therapy against cancer |
| WO2022136650A1 (en) | 2020-12-24 | 2022-06-30 | Oncurious Nv | Murine cross-reactive human ccr8 binders |
| WO2022136647A1 (en) | 2020-12-24 | 2022-06-30 | Oncurious Nv | Human ccr8 binders |
| WO2022136649A1 (en) | 2020-12-24 | 2022-06-30 | Oncurious Nv | Non-blocking human ccr8 binders |
| WO2022211046A1 (ja) | 2021-03-31 | 2022-10-06 | 塩野義製薬株式会社 | Ccr8を抗原として認識するキメラ抗原受容体 |
| WO2022256563A1 (en) | 2021-06-04 | 2022-12-08 | Amgen Inc. | Anti-ccr8 antibodies and uses thereof |
| WO2024062076A1 (en) | 2022-09-21 | 2024-03-28 | Domain Therapeutics | Anti-ccr8 monoclonal antibodies and their therapeutic use |
| WO2024062082A1 (en) | 2022-09-21 | 2024-03-28 | Domain Therapeutics | Anti-ccr8 monoclonal antibodies and their therapeutic use |
| WO2024062072A2 (en) | 2022-09-21 | 2024-03-28 | Domain Therapeutics | Anti-ccr8 monoclonal antibodies and their therapeutic use |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6557827B2 (ja) | 癌治療用医薬組成物 | |
| JP2022066304A (ja) | Carの抗腫瘍活性のための毒性管理 | |
| CN113573719A (zh) | 治疗和t细胞调节的方法和组合 | |
| US10611839B2 (en) | Anti CD84 antibodies, compositions comprising same and uses thereof | |
| JP6890546B2 (ja) | Tnfrsf14/hvemタンパク質およびその使用の方法 | |
| CN116209461A (zh) | Cd-30阳性癌症的治疗 | |
| JP2021503952A5 (ja) | ||
| WO2022049867A1 (ja) | がん免疫微小環境の調節物質およびそれによる予防・診断・治療的利用 | |
| JP2021503952A (ja) | Il−1rapを標的とするcar−t細胞及びその使用 | |
| WO2012133914A1 (ja) | Ranklアンタゴニストを含む癌免疫増強剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2018551478 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018775737 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2018775737 Country of ref document: EP Effective date: 20181019 |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18775737 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2018243020 Country of ref document: AU Date of ref document: 20180328 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3057274 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20197029821 Country of ref document: KR Kind code of ref document: A |