WO2018042776A1 - Dna結合タンパク質の結合領域の近傍に所望のdna断片を挿入する方法 - Google Patents
Dna結合タンパク質の結合領域の近傍に所望のdna断片を挿入する方法 Download PDFInfo
- Publication number
- WO2018042776A1 WO2018042776A1 PCT/JP2017/019309 JP2017019309W WO2018042776A1 WO 2018042776 A1 WO2018042776 A1 WO 2018042776A1 JP 2017019309 W JP2017019309 W JP 2017019309W WO 2018042776 A1 WO2018042776 A1 WO 2018042776A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dna
- binding
- sequence
- base sequence
- transposase
- Prior art date
Links
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6804—Nucleic acid analysis using immunogens
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6806—Preparing nucleic acids for analysis, e.g. for polymerase chain reaction [PCR] assay
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1093—General methods of preparing gene libraries, not provided for in other subgroups
Definitions
- the present invention relates to a method for inserting a desired DNA fragment in the vicinity of a binding region of a DNA binding protein.
- Chromatin immunoprecipitation (ChIP) analysis ChIP-chip analysis, ChIP-Seq analysis, Chromome Conformation Capture (3C, etc.) as a method for detecting the binding between proteins and specific genomic regions or the interaction between genomic regions.
- Hi-C Chromatin immunoprecipitation
- Non-Patent Document 1 Chromatin immunoprecipitation (ChIP) analysis, ChIP-chip analysis, ChIP-Seq analysis, Chromome Conformation Capture (3C, etc.) as a method for detecting the binding between proteins and specific genomic regions or the interaction between genomic regions.
- Hi-C is widely used (see, for example, Non-Patent Document 1).
- DNA and DNA binding protein are cross-linked by UV irradiation, formaldehyde treatment or the like, and then DNA is fragmented by ultrasonic treatment, restriction enzyme treatment or the like. Subsequently, the DNA fragment to which the DNA binding protein is bound is recovered by immunoprecipitation. Subsequently, the recovered DNA fragment is treated with protease to remove the DNA-binding protein, and the base sequence of the DNA fragment is converted to dot blot hybridization or Southern hybridization using a radiolabeled probe, hybridization with a DNA array (ChIP- analysis by chip analysis), PCR, real-time PCR, base sequence analysis using a next-generation sequencer (ChIP-Seq analysis), and the like.
- an object of this invention is to provide the technique which can analyze the coupling
- the present invention includes the following aspects.
- the method according to (1) further comprising a step of gene amplification of the DNA molecule starting from the desired base sequence to obtain an amplification product, and a step of analyzing the base sequence of the amplification product.
- the desired base sequence includes a promoter sequence, and the gene amplification is performed by contacting the promoter sequence with RNA polymerase to transcribe DNA downstream of the promoter sequence to generate RNA.
- the method according to 2). (4) The method according to (3), wherein the desired base sequence further comprises an identification sequence downstream of the promoter sequence.
- the conjugate according to (6), wherein the desired base sequence further comprises an identification sequence downstream of the promoter sequence.
- the present invention it is possible to provide a technique capable of analyzing the binding between a protein and a specific genomic region without performing immunoprecipitation. Therefore, it is possible to analyze the binding of proteins such as insoluble proteins that are difficult to immunoprecipitate to DNA.
- ChILT Chromatin Integration Labeling Technology
- the present invention relates to a method for inserting a DNA fragment having a desired base sequence in the vicinity of a binding region of a DNA binding protein bound to a DNA molecule, using a specific binding substance for the DNA binding protein.
- the method of the present embodiment may be referred to as a Chromatin Integration Labeling Technology (ChILT) method.
- ChILT Chromatin Integration Labeling Technology
- insoluble proteins cannot be immunoprecipitated. Further, for example, a wide range of proteins can be stained by immunostaining of fixed tissues and cells, whereas antigens that can be recovered by immunoprecipitation are limited, and about 10 of the antigens that can be immunostained. It is said that only about 1% can be immunoprecipitated. In addition, in immunoprecipitation using a small number of cells as a sample, it is necessary to use a carrier, so that contamination of contaminants and loss of target are problematic. On the other hand, according to the ChILT method, it is possible to analyze the binding between a protein and a specific genomic region without performing immunoprecipitation.
- the ChILT method it is possible to analyze the binding of a DNA-binding protein, which has been difficult to analyze by the conventional technique with immunoprecipitation as an essential step, to DNA. In addition, it has been difficult for conventional techniques to analyze DNA-binding proteins at the level of one cell. On the other hand, according to the ChILT method, as described later, it is also possible to analyze a DNA binding protein at a single cell level.
- the DNA binding protein to be analyzed is not particularly limited, and examples thereof include histones, transcription factors, and phosphorylated polymerase.
- histones include histones, transcription factors, and phosphorylated polymerase.
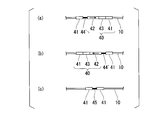
- FIG. 1A to FIG. 5 are schematic diagrams for explaining the ChILT method.
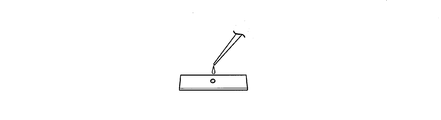
- FIG. 1A is a diagram showing a cell specimen fixed with paraformaldehyde.
- FIG. 1B is an enlarged view of the cell specimen S fixed on the slide glass of FIG. 1A.
- FIG. 1C is a diagram illustrating a reaction that occurs in the nucleus of the cell specimen S when the ChILT method is performed on the cell specimen S.
- FIG. 1C shows a state in which genomic DNA 10 is wound around histone 20 to form a chromatin structure.
- the DNA binding protein is histone 20.
- the specific binding substance 30 is an antibody that recognizes histone 20.
- a DNA fragment 40 is bound to the specific binding substance 30 via a linker 44.
- the DNA fragment 40 has a base sequence including a transposase binding sequence 41 and a desired base sequence.
- the desired base sequence is a base sequence in which an identification sequence 43 is linked downstream of the T7 promoter sequence 42.
- the arrow of the T7 promoter sequence 42 indicates the direction of transcription by T7 polymerase.
- the linker 44 has a role of a tether for bringing the DNA fragment 40 close to the binding region 11 of the histone 20.
- the linker 44 is a DNA fragment, and has a length of, for example, 50 to 70 bases.
- the linker 44 is not particularly limited as long as it functions as a tether.
- the linker 44 may be composed of a polymer such as polyethylene glycol.
- the ChILT method is performed using a cell specimen S fixed with paraformaldehyde as a sample.
- the DNA fragment 40 and the binding region 11 are brought close to each other using the specific binding substance 30 for the histone 20.
- the close proximity using the specific binding substance 30 means that the specific binding substance 30 to which the DNA fragment 40 is bound as shown in the example of FIG.
- the fragment 40 and the binding region 11 may be brought close to each other.
- the specific binding substance 30 is, for example, a mouse IgG antibody against histone 20, this mouse IgG antibody is bound to histone 20 as a primary antibody, and then an anti-mouse IgG antibody to which DNA fragment 40 is bound is secondary.
- the DNA fragment 40 and the binding region 11 may be brought close to each other by binding to the primary antibody as an antibody.
- the step of bringing the DNA fragment 40 and the binding region 11 into close proximity using the specific binding substance 30 for the histone 20 is performed in cells in a tissue section fixed with paraformaldehyde as in the example of FIG. 1C. Can do.
- a plurality of antibodies can be used simultaneously as in immunostaining. Therefore, according to the ChILT method, DNA fragments having different desired base sequences can be inserted in the vicinity of the binding regions of a plurality of DNA binding proteins.
- the identification sequence 43 it is possible to distinguish and detect the binding of a plurality of DNA binding proteins to DNA.
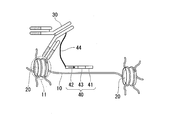
- FIG. 2 shows a state in which the transposase 50 is added to the sample and the transposase 50 is bound to the transposase binding sequence 41.
- the transposase is preferably small in size from the viewpoint of easily entering the nucleus.
- a transposase having a molecular weight of 50 KDa or less is preferable.
- a sequence having a high sequence specificity is preferable.
- a DNA type transposase is preferable.
- a transposase that can control the activity is preferable, and examples thereof include a transposase that is activated when a cation such as a divalent metal ion is added to the buffer.
- Preferred transposases include, for example, TN5 transposase, sleeping beauty transposase (SB10), TN10, and the like.
- the transposase binding sequence 41 may be determined according to the transposase to be used.
- transposase 50 is activated. Activation of the transposase 50 can be carried out, for example, by adding a divalent metal ion to the buffer.
- transposase 50 it may be necessary to form a dimer in order to exhibit the activity of transferring DNA fragments.
- the transposase 50 may need to be bound to the transposase binding sequence 41.
- TN5, SB10, TN10 and the like described above are such transposases.
- the transposase 50 can also be activated by forming a dimer of the transposase 50.
- transposase 50 it may be necessary to form a dimer of transposase 50 and add a divalent metal ion to the buffer in order to activate transposase 50.
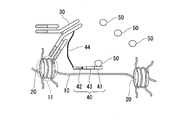
- FIG. 3 shows a state in which a dimer of the transposase 50 is formed.
- a free DNA fragment having the transposase binding sequence 41 is added to the sample, and the transposase 50 is bound to the DNA fragment.
- the transposase 50 bound to the above-described DNA fragment forms a dimer with the transposase 50 bound to the transposase binding sequence 41 constituting the DNA fragment 40.
- a DNA fragment having a desired base sequence in DNA fragment 40 (in the example of FIG. 3, a base sequence in which identification sequence 43 is linked downstream of T7 promoter sequence 42) is inserted in the vicinity of binding region 11.
- the neighborhood may be a spatially close region, and may be, for example, 500 bases or less from the binding region 11, for example, 100 bases or less, for example, 50 bases or less. .
- the neighborhood may be a region that is spatially close, and thus is a region that is very far from the binding region 11 on the base sequence of the genomic DNA 10 and that is close to the interaction between the genomic regions. Also good. That is, it may be a region close to the chromosome structure.
- the DNA fragment insertion reaction by transposase 50 may be completed incompletely. Specifically, for example, when the inserted DNA fragment is partially a single-stranded DNA, the inserted DNA fragment is fragmented, and the like. Therefore, a step of completely terminating the DNA fragment insertion reaction by the transposase 50 may be further performed by a fill in reaction using T4 DNA ligase, T4 DNA polymerase I or the like.
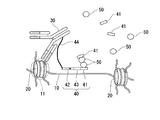
- FIG. 4 is a schematic diagram showing an example of a state in which a DNA fragment having a desired base sequence is inserted in the vicinity of the binding region 11.
- a DNA fragment having a desired base sequence (a base sequence in which an identification sequence 43 is linked downstream of the T7 promoter sequence 42) is inserted in the vicinity of the binding region 11.
- a desired base sequence not only a desired base sequence but also two transposase binding sequences 41 and a base sequence 44 ′ derived from a part of the linker 44 are inserted into the genomic DNA 10.
- FIGS. 5A to 5C are schematic diagrams showing an example in which a DNA fragment is inserted into the genomic DNA 10 as a result of activation of the transposase 50 from the state of FIG. 3 described above.
- FIG. 5A shows the state of FIG. 4 described above.
- FIG. 5B shows a state in which a desired base sequence is inserted in the opposite direction to FIG. 4 described above.
- FIG. 5C shows a base sequence that can be inserted into the genomic DNA 10 when the transposases 50 bound to the free DNA fragment having the transposase binding sequence 41 form a dimer.
- the base sequence inserted into the genomic DNA 10 includes two transposase binding sequences 41, and may include an arbitrary base sequence 45 between them.
- the base sequence 45 is formed by the fill in reaction described above.
- the base sequence shown in FIG. 5 (c) is an unintended by-product and can be inserted not only in the vicinity of the binding region 11 but also in any part on the genomic DNA 10.
- the base sequence shown in FIG. 5C does not have a desired base sequence (in this example, a base sequence in which an identification sequence 43 is linked downstream of the T7 promoter sequence 42). For this reason, the influence in the target analysis can be excluded.
- a DNA fragment having a desired base sequence can be inserted in the vicinity of the binding region of the DNA binding protein bound to the DNA molecule.
- specific binding substances include antibody fragments, aptamers and the like in addition to antibodies.
- An antibody can be produced, for example, by immunizing an animal such as a mouse with a target substance or a fragment thereof as an antigen. Alternatively, for example, it can be prepared by screening a phage library. Examples of antibody fragments include Fv, Fab, scFv and the like.
- the antibody may be a monoclonal antibody or a polyclonal antibody. A commercially available antibody may also be used.
- An aptamer is a substance having a specific binding ability to a target substance.
- examples of aptamers include nucleic acid aptamers and peptide aptamers.
- a nucleic acid aptamer having a specific binding ability to a target substance can be selected by, for example, a systematic evolution of ligand by exponential enrichment (SELEX) method.
- Peptide aptamers having specific binding ability to the target substance can be selected by, for example, the two-hybrid method using yeast.
- the desired base sequence to be inserted into the genomic DNA 10 is a base sequence in which an identification sequence 43 is linked downstream of the T7 promoter sequence 42.
- the desired base sequence is not limited to this.
- another promoter sequence such as an SP6 promoter sequence may be used instead of the T7 promoter sequence 42.
- the desired base sequence may be composed only of a promoter sequence.
- the desired base sequence may be comprised only from the identification sequence.
- the identification sequence is not particularly limited as long as it is a base sequence that has a low probability of occurrence, and an arbitrary base sequence of about 4 to 20 bases can be used, for example.
- the length of the desired base sequence to be inserted into the genomic DNA 10 may be, for example, 20 to 500 bases, for example, 20 to 200 bases, for example, 20 to 100 bases.
- each element (T7 promoter sequence 42, identification sequence 43, and transposase binding sequence 41) constituting the DNA fragment 40 is adjacent to each other, but an arbitrary base is interposed between the elements. Sequence spacers may be present. Further, the order of the elements is not limited to that shown in the examples of FIGS. 1C to 5, and may be appropriately replaced as necessary, or additional elements may be added.
- the DNA fragment 40 may be single-stranded or double-stranded.
- the transposase binding sequence 41 needs to be double-stranded DNA due to the characteristics of the transposase to be used, at least the transposase binding sequence 41 needs to be double-stranded.
- a DNA fragment having a desired base sequence is inserted in the vicinity of the binding region of the DNA-binding protein, and then the cell is excised and collected by laser microdissection or the like.
- the analysis described later can also be performed at the level.
- the ChILT method includes a step of gene amplification of DNA 10 starting from the desired base sequence (in the example of FIGS. 1C to 5, a base sequence in which an identification sequence 43 is linked downstream of the T7 promoter sequence 42) to obtain an amplified product; And a step of analyzing the base sequence of the amplification product. Thereby, the position on the genome where the desired DNA fragment is inserted can be analyzed.
- Gene amplification examples include PCR combining a primer complementary to the inserted base sequence and a random primer, transcription using a polymerase (RNA polymerase, DNA-dependent DNA polymerase, etc.), and the like.
- RNA polymerase RNA polymerase, DNA-dependent DNA polymerase, etc.
- gene amplification using a desired base sequence as a starting point means that the genomic DNA 10 is amplified using part or all of the desired base sequence inserted in the vicinity of the binding region 11. means.
- the base sequence inserted into the genomic DNA 10 includes the T7 promoter sequence 42. Therefore, “amplifying a gene with a desired base sequence as a starting point” means that the genomic DNA 10 downstream of the T7 promoter sequence 42 is transcribed into RNA by causing T7 polymerase to act on the genomic DNA 10 described above. May be.
- “gene amplification using a desired base sequence as a starting point” is a combination of a primer having a base sequence complementary to a part or all of the inserted base sequence and a random primer using the above genomic DNA 10 as a template.
- the base sequence of the genomic DNA 10 in the vicinity of the binding region 11 may be amplified by performing PCR or the like.
- the inserted nucleotide sequence contains a promoter sequence, and gene amplification is performed by bringing RNA polymerase into contact with the promoter sequence to transcribe DNA downstream of the promoter sequence to generate RNA. May be.
- the base sequence shown in FIG. 5C does not have the T7 promoter sequence 42, gene amplification (transcription) is not performed even when T7 polymerase is allowed to act.
- gene amplification may be performed by PCR using the inserted desired base sequence.
- the base sequence shown in FIG. 5C does not have the desired base sequence (the base sequence in which the identification sequence 43 is linked downstream of the T7 promoter sequence 42), the base sequence is used. Gene amplification is not performed even when the PCR reaction is performed.
- FIG. 6A is a diagram showing a state in which T7 polymerase is allowed to act on the DNA 10 shown in FIG. 4 or FIG. 5A and the DNA 10 downstream of the T7 promoter sequence 42 is transcribed into RNA 60.
- FIG. 6B is a diagram showing a state in which T7 polymerase is allowed to act on the DNA 10 shown in FIG. 5B and the DNA 10 downstream of the T7 promoter sequence 42 is transcribed into RNA 60.
- an identification sequence 43 is introduced downstream of the T7 promoter sequence 42. That is, in the ChILT method, the desired base sequence may further include an identification sequence downstream of the promoter sequence.
- the RNA 60 since the identification sequence 43 is introduced downstream of the T7 promoter sequence 42, the RNA 60 has the complementary strand 43 ′ of the identification sequence 43 and the DNA 10 downstream of the identification sequence 43. Complementary strand 10 'is included.
- RNA may be transcribed from a region other than the T7 promoter sequence 42 inserted by the above reaction.
- the presence of the identification sequence 43 makes it possible to distinguish RNA transcribed from the inserted T7 promoter sequence 42 from other RNAs. That is, it can be determined that the RNA 60 having the complementary strand 43 ′ of the identification sequence 43 is RNA transcribed from the inserted T7 promoter sequence 42.
- the ChILT method can simultaneously perform analysis using specific binding substances for a plurality of DNA binding proteins.
- RNAs transcribed from the vicinity of the binding region of any DNA-binding protein are introduced by introducing different identification sequences 43 into the DNA fragments 40 inserted into the DNA 10 using each specific binding substance. It is also possible to identify whether or not.
- RNA 60 by analyzing the base sequence of the transcribed RNA 60, the position on the genome to which the DNA binding protein is bound can be identified. Analysis of the base sequence of RNA 60 may be performed, for example, by a next-generation sequencer or may be performed by hybridization with a DNA array.
- RNA 60 is RNA transcribed from the vicinity of binding region 11. Therefore, by analyzing the base sequence of RNA 60, for example, the position of the binding region of the DNA binding protein (histone 20 in the examples of FIGS. 1 to 6) on the genome can be identified. That is, the direction in which the desired base sequence is inserted does not affect the analysis result.
- the present invention relates to binding of a DNA fragment having a base sequence comprising a transposase binding sequence and a desired base sequence to a specific binding substance for a DNA binding protein or a specific binding substance for the specific binding substance.
- a DNA fragment having a base sequence comprising a transposase binding sequence and a desired base sequence
- a specific binding substance for a DNA binding protein or a specific binding substance for the specific binding substance Provide the body.
- the conjugate of this embodiment is for insertion of a desired base sequence in the vicinity of the binding region of a DNA binding protein bound to a DNA molecule.
- the transposase binding sequence, the desired base sequence, and the specific binding substance are the same as those described above. That is, the desired base sequence may include a promoter sequence. The desired base sequence may further contain an identification sequence downstream of the promoter sequence.
- the DNA fragment is preferably bound to a specific binding substance for a DNA binding protein or a specific binding substance for the specific binding substance via a linker.
- the linker is the same as described above.
- a conjugate of the above DNA fragment and a specific binding substance for a DNA binding protein means, for example, a conjugate in which the above DNA fragment is bound to a primary antibody. That is, it means a conjugate in which the above DNA fragment is directly bound to a specific binding substance for a DNA binding protein.
- the conjugate of the above-mentioned DNA fragment and the specific binding substance for the specific binding substance for DNA binding protein means, for example, that the DNA fragment is bound to the secondary antibody.
- the number of the above DNA fragments per molecule of the specific binding substance is not particularly limited, and may be 1 to 10, for example.
- the method for binding the DNA fragment to the specific binding substance is not particularly limited, and for example, it may be bound using a chemical cross-linking agent having a succinimide group, a maleimide group or the like.
- a chemical cross-linking agent having a succinimide group, a maleimide group or the like.
- an amino group is bonded to the 5 ′ end of the DNA fragment, and the amino group and a functional group such as an amino group or a carboxy group present in the specific binding substance are covalently bonded with an appropriate chemical crosslinking agent. It is done.
- a DNA fragment may be bound to a specific binding substance using a bond between avidin and biotin.
- ChIP buffer (10 mM Tris-HCl (pH 8.0), 200 mM KCl, 1 mM CaCl 2 , 0.5% NP40, PMSF, aprotinin, leupeptin) was added to suspend the cells, and the cells were suspended on ice. Allowed to stand for 10 minutes.
- micrococcal nuclease was added, and enzyme treatment was performed at 37 ° C. for 40 minutes. Subsequently, the reaction was stopped by adding EDTA to a final concentration of 10 mM. Subsequently, the mixture was centrifuged at 15,000 ⁇ g for 10 minutes at 4 ° C., and the supernatant was collected in a 2 mL siliconized tube. Subsequently, ChIP buffer was added and diluted to obtain a soluble chromatin fraction.
- Magnetic particles (Dynabeads M-) in which an antibody complex solution obtained by reacting 2 ⁇ g of rabbit anti-mouse IgG antibody and 2 ⁇ g of mouse anti-H3K4me3 antibody in advance with 20 ⁇ L of goat anti-rabbit IgG antibody is combined with the soluble chromatin fraction. 280) and stirred at 4 ° C. overnight with a rotator.
- the reaction solution was set on a magnetic stand and allowed to stand for 1 minute to collect magnetic particles. Subsequently, the collected magnetic particles were washed three times with ChIP buffer, three times with a washing buffer (10 mM Tris-HCl (pH 8.0), 500 mM KCl, 1 mM CaCl 2 , 0.5% NP40), 1 ⁇ TE (10 mM). Washed 3 times with Tris-HCl (pH 8.0), 1 mM EDTA (pH 8.0)).
- FIG. 7 (a) is a graph showing the result of mapping the binding of H3K4me3 to the Eef1a1 locus using about 1000 cells by ChIP-Seq analysis, which is a conventional method.
- a blocking agent (trade name “Blocking One-P”, Nacalai Tesque) was added to the fixed cell sample, allowed to stand at room temperature for 10 minutes, and then washed with PBS. Subsequently, an antibody complex (2 ⁇ g / mL anti-H3K4me3 antibody and nucleic acid-labeled secondary antibody previously reacted) diluted with 0.1 ⁇ Blocking One-P was added and reacted at 37 ° C. for 2 hours. And washed with PBS. As a result, the DNA fragment labeled with the nucleic acid-labeled secondary antibody was close to the H3K4me3 binding region on the genomic DNA.
- the DNA fragment labeled with the nucleic acid-labeled secondary antibody was obtained by annealing a DNA fragment consisting of the base sequence shown in SEQ ID NO: 1 and a DNA fragment consisting of the base sequence shown in SEQ ID NO: 2. .
- the 49th to 67th nucleotide sequences are TN5 transposase binding sequences
- the 7th to 26th nucleotide sequences are T7 promoter sequences
- the 27th to 34th nucleotide sequences Is an identification sequence
- the first to sixth bases were a partial base sequence of the linker.
- the DNA fragment consisting of the base sequence shown in SEQ ID NO: 2 is a DNA fragment having a base sequence complementary to the 49th to 67th base sequences in the base sequence shown in SEQ ID NO: 1, A binding sequence of TN5 transposase, which is a double-stranded DNA, was formed by annealing with a DNA fragment comprising the described base sequence.
- transposase (Binding of transposase) Subsequently, 85 ⁇ g / mL of TN5 transposase was added to the sample and allowed to stand at room temperature for 10 minutes. Thereby, the transposase was bound to the transposase binding sequence in the DNA fragment labeled with the nucleic acid-labeled secondary antibody.
- oligo3-4 a free DNA fragment having a transposase binding sequence (hereinafter referred to as “oligo3-4”) was added to the sample, reacted at room temperature for 1 hour, and then washed with PBS. Oligo 3-4 was obtained by annealing a DNA fragment consisting of the base sequence shown in SEQ ID NO: 3 and a DNA fragment consisting of the base sequence shown in SEQ ID NO: 4. The double-stranded DNA region of oligo3-4 was a binding sequence for TN5 transposase.
- TN5 dialysis buffer 50 mM HEPES-KOH (pH 7.2), 0.1 M NaCl, 0.1 mM EDTA, 1 mM DTT, 0.1% Triton X-100, 10% glycerol.
- TAPS-DMF buffer (10 mM TAPS-NaOH (pH 8.5), 5 mM MgCl 2 , 10% N, N-dimethylformamide) was added to the sample and allowed to stand at 37 ° C. for 1 hour.
- TAPS-DMF buffer 10 mM TAPS-NaOH (pH 8.5), 5 mM MgCl 2 , 10% N, N-dimethylformamide
- the transposase bound to the DNA fragment labeled with the nucleic acid labeled secondary antibody was activated, and the DNA fragment labeled with the nucleic acid labeled secondary antibody was inserted in the vicinity of the H3K4me3 binding region on the genomic DNA.
- the plate was washed with 1 ⁇ T4 DNA ligase reaction buffer, and a fill in reaction solution (T4 DNA ligase reaction buffer, dNTPmix, T4 DNA ligase I, T4 DNA polymerase I) was added and reacted at room temperature for 30 minutes.
- reaction solution T4 DNA ligase reaction buffer, dNTPmix, T4 DNA ligase I, T4 DNA polymerase I
- T4 DNA ligase and T4 DNA polymerase I were inactivated and removed.
- T7 RNA polymerase transcription by T7 RNA polymerase was performed as gene amplification.
- each excised cell was washed with 1 ⁇ T7 RNA polymerase buffer.
- an in vitro transcription solution (T7 RNA polymerase buffer, ATP, CTP, GTP, UTP, RNase inhibitor, T7 RNA polymerase) was added and reacted at 37 ° C. for 16 hours.
- genomic DNA was transcribed starting from the T7 RNA promoter sequence contained in the inserted DNA fragment.
- RNA transcribed by RNeasy MinElute Cleanup Kit (Qiagen) was purified.
- RNA was reverse transcribed to prepare cDNA, the nucleotide sequence was analyzed using a next-generation sequencer, and mapped onto the genome.
- FIGS. 7B to 7D are graphs showing typical results obtained by analyzing and mapping the binding of H3K4me3 to the Eef1a1 locus by the ChILT method at the level of one cell.
- Example 3 By the ChILT method, the binding of various DNA binding proteins on genomic DNA was analyzed at the level of one cell. Mouse skeletal myoblasts were used as cells. Further, anti-DNA binding protein antibodies shown in Table 1 below were used as primary antibodies. As the secondary antibody, an anti-mouse IgG antibody or an anti-rabbit IgG antibody labeled with the same DNA fragment as used in Experimental Example 2 was used.
- Blocking One-P a blocking agent (trade name “Blocking One-P”, Nacalai Tesque) was added to the fixed cell sample, allowed to stand at room temperature for 10 minutes, and then washed with PBS. Subsequently, an antibody complex diluted with 0.1 ⁇ Blocking One-P (a pre-reacted primary antibody and a nucleic acid-labeled secondary antibody) was added and reacted at 37 ° C. for 2 hours. Washed.
- the antibody complex was prepared by reacting a secondary antibody with a nucleic acid-labeled mouse antibody when the primary antibody was a mouse antibody.
- the primary antibody was a rabbit antibody
- it was prepared by reacting a secondary antibody with a nucleic acid-labeled rabbit antibody.
- a sample in which only a nucleic acid labeled anti-mouse IgG antibody or only a nucleic acid labeled anti-rabbit IgG antibody was reacted was also prepared. That is, a sample not reacted with the primary antibody was prepared as a negative control. Thereafter, binding of transposase, activation of transposase and insertion of a desired base sequence were carried out in the same manner as in Experimental Example 2.
- RNA amplification and nucleotide sequence analysis Each collected cell was transcribed with T7 RNA polymerase in the same manner as in Experimental Example 2, and the transcribed RNA was purified. Subsequently, the purified RNA was reverse transcribed to prepare cDNA, the nucleotide sequence was analyzed using a next-generation sequencer, and mapped onto the genome.
- FIG. 8A is a graph showing the result of analyzing and mapping the binding of a DNA binding protein at the level of one cell by the ChILT method using various primary antibodies shown in Table 1. Two cells were analyzed for each antibody complex.
- the horizontal axis of the graph represents the number of reads obtained by the next-generation sequencer. “Valid” indicates a read that is effectively mapped on the genome, “Duplicate” indicates a read that has two reads mapped at the same position on the genome, and “Multi” indicates the same position on the genome. Represents a lead in which three or more reads mapped to 1 are present, “Unmapped” represents a lead not mapped on the genome, and “Nextera” represents a lead derived from the library used for the base sequence analysis.
- “rH3K27me3” represents the result of reacting a complex of rH3K27me3 antibody and nucleic acid-labeled anti-rabbit IgG antibody, and so on.
- “mH3K4me3-rH3K27me3” represents a result of simultaneously reacting a complex of mH3K4me3 antibody with a nucleic acid-labeled anti-mouse IgG antibody and a complex of rH3K27me3 antibody with a nucleic acid-labeled anti-rabbit IgG antibody. It is the same.
- “# 1” and “# 2” represent the result of the first cell and the result of the second cell of the analyzed two cells, respectively.
- FIG. 8B is a graph in which the horizontal axis of the graph of FIG. 8A is changed to the existence ratio of each lead of “Valid”, “Duplicate”, “Multi”, “Unmapped”, and “Nextera”. As a result, it was revealed that an effective lead of about 50 to 85% can be obtained by the ChILT method using a nucleic acid-labeled anti-mouse IgG antibody or a nucleic acid-labeled anti-rabbit IgG antibody.
- the present invention it is possible to provide a technique capable of analyzing the binding between a protein and a specific genomic region without performing immunoprecipitation. Therefore, it is possible to analyze the binding of proteins such as insoluble proteins that are difficult to immunoprecipitate to DNA. In addition, analysis using a small number of cells as a sample becomes easy.
- SYMBOLS 10 Genomic DNA, 10 '... Complementary strand of genomic DNA 10, 11 ... Binding region, 20 ... Histone, 30 ... Specific binding substance, 40 ... DNA fragment, 41 ... Transposase binding sequence, 41' ... Complement of transposase binding sequence 41 Chain, 42 ... T7 promoter sequence, 43 ... identification sequence, 43 '... complementary strand of identification sequence 43, 44 ... linker, 44' ... base sequence derived from part of linker 44, 45 ... arbitrary sequence, 50 ... transposase, 60 ... RNA, S ... cell specimen.
Abstract
DNA分子に結合したDNA結合タンパク質の結合領域の近傍に所望の塩基配列のDNA断片を挿入する方法であって、前記DNA結合タンパク質に対する特異的結合物質を利用して、トランスポザーゼ結合配列と前記所望の塩基配列とを含む塩基配列を有するDNA断片と、前記結合領域とを近接させる工程と、前記トランスポザーゼ結合配列にトランスポザーゼを結合させる工程と、前記トランスポザーゼを活性化し、その結果、前記所望の塩基配列のDNA断片が前記結合領域の近傍に挿入される工程と、を備える方法。
Description
本発明は、DNA結合タンパク質の結合領域の近傍に所望のDNA断片を挿入する方法に関する。本願は、2016年8月30日に、日本に出願された特願2016-167967号に基づき優先権を主張し、その内容をここに援用する。
タンパク質と特定のゲノム領域との結合、あるいはゲノム領域同士の相互作用を検出する方法として、クロマチン免疫沈降(chromatin immunoprecipitation、ChIP)解析、ChIP-chip解析、ChIP-Seq解析、Chromosome Conformation Capture(3C、Hi-C)等が広く利用されている(例えば、非特許文献1を参照)。
これらの方法は、概ね次のようにして行われる。まず、UV照射、ホルムアルデヒド処理等によりDNAとDNA結合タンパク質とをクロスリンクした後、超音波処理、制限酵素処理等によりDNAを断片化する。続いて、免疫沈降によりDNA結合タンパク質が結合したDNA断片を回収する。続いて、回収したDNA断片をプロテアーゼ処理してDNA結合タンパク質を除去し、DNA断片の塩基配列を、放射線標識プローブを用いたドットブロットハイブリダイゼーション又はサザンハイブリダイゼーション、DNAアレイとのハイブリダイゼーション(ChIP-chip解析)、PCR、リアルタイムPCR、次世代シークエンサーを用いた塩基配列解析(ChIP-Seq解析)等により解析する。
Carey M. F., et al., Chromatin Immunoprecipitation (ChIP), Cold Spring Harbor Protocols, 4 (9), pdb.prot5279, 2009.
しかしながら、例えば、不溶性タンパク質は免疫沈降することができない。また、試料が少数細胞である場合等には、反応時に担体の使用を必要とするため夾雑物の混入や標的の損失が避けられない。このため、免疫沈降を必須の工程とする従来の方法では、不溶性タンパク質のDNAへの結合を解析することや組織等の少数細胞からの解析が困難な場合があった。そこで、本発明は、免疫沈降を行うことなく、タンパク質と特定のゲノム領域との結合を解析することができる技術を提供することを目的とする。
本発明は以下の態様を含む。
(1)DNA分子に結合したDNA結合タンパク質の結合領域の近傍に所望の塩基配列のDNA断片を挿入する方法であって、前記DNA結合タンパク質に対する特異的結合物質を利用して、トランスポザーゼ結合配列と前記所望の塩基配列とを含む塩基配列を有するDNA断片と、前記結合領域とを近接させる工程と、前記トランスポザーゼ結合配列にトランスポザーゼを結合させる工程と、前記トランスポザーゼを活性化し、その結果、前記所望の塩基配列のDNA断片が前記結合領域の近傍に挿入される工程と、を備える方法。
(2)前記所望の塩基配列を起点として前記DNA分子を遺伝子増幅し、増幅産物を得る工程と、前記増幅産物の塩基配列を解析する工程と、を更に備える、(1)に記載の方法。
(3)前記所望の塩基配列がプロモーター配列を含み、前記遺伝子増幅が、前記プロモーター配列にRNAポリメラーゼを接触させて前記プロモーター配列の下流のDNAを転写してRNAを生成することにより行われる、(2)に記載の方法。
(4)前記所望の塩基配列が前記プロモーター配列の下流に識別配列を更に含む、(3)に記載の方法。
(5)トランスポザーゼ結合配列と所望の塩基配列とを含む塩基配列を有するDNA断片と、DNA結合タンパク質に対する特異的結合物質又は前記特異的結合物質に対する特異的結合物質との結合体。
(6)前記所望の塩基配列がプロモーター配列を含む、(5)に記載の結合体。
(7)前記所望の塩基配列が前記プロモーター配列の下流に識別配列を更に含む、(6)に記載の結合体。
(1)DNA分子に結合したDNA結合タンパク質の結合領域の近傍に所望の塩基配列のDNA断片を挿入する方法であって、前記DNA結合タンパク質に対する特異的結合物質を利用して、トランスポザーゼ結合配列と前記所望の塩基配列とを含む塩基配列を有するDNA断片と、前記結合領域とを近接させる工程と、前記トランスポザーゼ結合配列にトランスポザーゼを結合させる工程と、前記トランスポザーゼを活性化し、その結果、前記所望の塩基配列のDNA断片が前記結合領域の近傍に挿入される工程と、を備える方法。
(2)前記所望の塩基配列を起点として前記DNA分子を遺伝子増幅し、増幅産物を得る工程と、前記増幅産物の塩基配列を解析する工程と、を更に備える、(1)に記載の方法。
(3)前記所望の塩基配列がプロモーター配列を含み、前記遺伝子増幅が、前記プロモーター配列にRNAポリメラーゼを接触させて前記プロモーター配列の下流のDNAを転写してRNAを生成することにより行われる、(2)に記載の方法。
(4)前記所望の塩基配列が前記プロモーター配列の下流に識別配列を更に含む、(3)に記載の方法。
(5)トランスポザーゼ結合配列と所望の塩基配列とを含む塩基配列を有するDNA断片と、DNA結合タンパク質に対する特異的結合物質又は前記特異的結合物質に対する特異的結合物質との結合体。
(6)前記所望の塩基配列がプロモーター配列を含む、(5)に記載の結合体。
(7)前記所望の塩基配列が前記プロモーター配列の下流に識別配列を更に含む、(6)に記載の結合体。
本発明によれば、免疫沈降を行うことなく、タンパク質と特定のゲノム領域との結合を解析することができる技術を提供することができる。したがって、例えば不溶性タンパク質等の、免疫沈降を行うことが困難なタンパク質のDNAへの結合を解析することが可能になる。
以下、場合により図面を参照しつつ、本発明の実施形態について詳細に説明する。なお、図面中、同一又は相当部分には同一又は対応する符号を付し、重複する説明は省略する。また、各図における寸法比は、説明のため誇張している部分があり、必ずしも実際の寸法比とは一致しない。
[DNA結合タンパク質の結合領域の近傍に所望のDNA断片を挿入する方法]
1実施形態において、本発明は、DNA分子に結合したDNA結合タンパク質の結合領域の近傍に所望の塩基配列のDNA断片を挿入する方法であって、前記DNA結合タンパク質に対する特異的結合物質を利用して、トランスポザーゼ結合配列と前記所望の塩基配列とを含む塩基配列を有するDNA断片と、前記結合領域とを近接させる工程(a)と、前記トランスポザーゼ結合配列にトランスポザーゼを結合させる工程(b)と、前記トランスポザーゼを活性化し、その結果、前記所望の塩基配列のDNA断片が前記結合領域の近傍に挿入される工程(c)と、を備える方法を提供する。以下、本実施形態の方法をChromatin Integration Labeling Technology(ChILT)法という場合がある。
1実施形態において、本発明は、DNA分子に結合したDNA結合タンパク質の結合領域の近傍に所望の塩基配列のDNA断片を挿入する方法であって、前記DNA結合タンパク質に対する特異的結合物質を利用して、トランスポザーゼ結合配列と前記所望の塩基配列とを含む塩基配列を有するDNA断片と、前記結合領域とを近接させる工程(a)と、前記トランスポザーゼ結合配列にトランスポザーゼを結合させる工程(b)と、前記トランスポザーゼを活性化し、その結果、前記所望の塩基配列のDNA断片が前記結合領域の近傍に挿入される工程(c)と、を備える方法を提供する。以下、本実施形態の方法をChromatin Integration Labeling Technology(ChILT)法という場合がある。
上述したように、不溶性タンパク質は免疫沈降することができない。また、例えば固定した組織や細胞の免疫染色では広範なタンパク質を染色することが可能であるのに対し、免疫沈降で回収することができる抗原は限られており、免疫染色可能な抗原の約10%程度しか免疫沈降できないといわれている。また、少数細胞を試料とした免疫沈降では、担体を使用する必要があるため、夾雑物の混入や標的の損失が問題となる。これに対し、ChILT法によれば、免疫沈降を行うことなく、タンパク質と特定のゲノム領域との結合を解析することが可能になる。
したがって、ChILT法によれば、免疫沈降を必須の工程とする従来技術では解析が困難であったDNA結合タンパク質について、DNAとの結合を解析することが可能になる。また、従来技術では、1細胞レベルでDNA結合タンパク質の解析を行うことは困難であった。これに対し、ChILT法によれば、後述するように、1細胞レベルでDNA結合タンパク質の解析を行うことも可能である。
解析対象となるDNA結合タンパク質は、特に制限されず、例えば、ヒストン、転写因子、リン酸化ポリメラーゼ等が挙げられる。例えば、DNA結合タンパク質として、特定のリン酸化状態のRNAポリメラーゼIIを解析することにより、遺伝子の転写状態を解析することが可能である。
以下、図1A~図5を参照しながらChILT法について説明する。図1A~図5は、ChILT法を説明する模式図である。図1Aは、パラホルムアルデヒド固定された細胞標本を示す図である。図1Bは、図1Aのスライドガラス上に固定された細胞標本Sを拡大した図である。図1Cは、細胞標本Sに対しChILT法を実施した場合に、細胞標本Sの核内で生じる反応を説明する図である。図1Cは、ゲノムDNA10がヒストン20に巻きついてクロマチン構造を形成した状態を示す。図1Cの例では、DNA結合タンパク質はヒストン20である。
図1Cの例では、特異的結合物質30は、ヒストン20を認識する抗体である。特異的結合物質30には、リンカー44を介してDNA断片40が結合されている。DNA断片40は、トランスポザーゼ結合配列41と、所望の塩基配列とを含む塩基配列を有している。図1の例では、所望の塩基配列は、T7プロモーター配列42の下流に識別配列43が連結した塩基配列である。図1C~図6において、T7プロモーター配列42の矢印は、T7ポリメラーゼによる転写の向きを示す。
リンカー44は、DNA断片40をヒストン20の結合領域11に近接させるためのつなぎひも(tether、テザー)の役割を有している。図1Cの例では、リンカー44はDNA断片であり、例えば50~70塩基の長さを有している。しかしながら、リンカー44は、テザーとして機能するものであれば特に制限されず、例えばポリエチレングリコール等のポリマーで構成されていてもよい。
(工程(a))
図1Cの例では、パラホルムアルデヒド固定された細胞標本Sを試料としてChILT法を実施している。まず、ヒストン20に対する特異的結合物質30を利用して、DNA断片40と結合領域11とを近接させる。ここで、特異的結合物質30を利用して近接させるとは、図1Cの例のようにDNA断片40が結合した特異的結合物質30を、その抗原であるヒストン20に結合させることにより、DNA断片40と結合領域11とを近接させることであってもよい。あるいは、特異的結合物質30が、例えばヒストン20に対するマウスIgG抗体であり、このマウスIgG抗体を1次抗体としてヒストン20に結合させ、続いて、DNA断片40が結合した抗マウスIgG抗体を2次抗体として上記の1次抗体に結合させることにより、DNA断片40と結合領域11とを近接させることであってもよい。
図1Cの例では、パラホルムアルデヒド固定された細胞標本Sを試料としてChILT法を実施している。まず、ヒストン20に対する特異的結合物質30を利用して、DNA断片40と結合領域11とを近接させる。ここで、特異的結合物質30を利用して近接させるとは、図1Cの例のようにDNA断片40が結合した特異的結合物質30を、その抗原であるヒストン20に結合させることにより、DNA断片40と結合領域11とを近接させることであってもよい。あるいは、特異的結合物質30が、例えばヒストン20に対するマウスIgG抗体であり、このマウスIgG抗体を1次抗体としてヒストン20に結合させ、続いて、DNA断片40が結合した抗マウスIgG抗体を2次抗体として上記の1次抗体に結合させることにより、DNA断片40と結合領域11とを近接させることであってもよい。
ヒストン20に対する特異的結合物質30を利用して、DNA断片40と結合領域11とを近接させる工程は、図1Cの例のように、パラホルムアルデヒド固定された組織切片中の細胞内で実施することができる。この工程は、免疫染色と同様に、複数の抗体を同時に使用することも可能である。したがって、ChILT法によれば、複数のDNA結合タンパク質の結合領域の近傍にそれぞれ異なる所望の塩基配列のDNA断片を挿入することもできる。ここで、識別配列43として、DNA結合タンパク質ごとに区別可能な配列を使用することにより、複数のDNA結合タンパク質のDNAへの結合を区別して検出することもできる。
(工程(b))
続いて、トランスポザーゼ結合配列41にトランスポザーゼ50を結合させる。図2は、試料にトランスポザーゼ50を添加して、トランスポザーゼ結合配列41にトランスポザーゼ50を結合させた状態をを示す。
続いて、トランスポザーゼ結合配列41にトランスポザーゼ50を結合させる。図2は、試料にトランスポザーゼ50を添加して、トランスポザーゼ結合配列41にトランスポザーゼ50を結合させた状態をを示す。
トランスポザーゼとしては、核内に入りやすい観点から、大きさが小さなものが好ましい。例えば、分子量が50KDa以下のトランスポザーゼが好ましい。また、認識する配列の配列特異性が高いものが好ましい。また、DNA型トランスポザーゼが好ましい。また、活性を制御できるトランスポザーゼが好ましく、例えば、2価の金属イオン等のカチオンをバッファー中に添加すると活性化されるトランスポザーゼが挙げられる。好ましいトランスポザーゼとしては、例えば、TN5トランスポザーゼ、sleeping beauty transposase(SB10)、TN10等が挙げられる。トランスポザーゼ結合配列41は、使用するトランスポザーゼに応じて決定すればよい。
(工程(c))
続いて、上記のトランスポザーゼ50を活性化する。トランスポザーゼ50の活性化は、例えば、2価の金属イオンをバッファー中に添加すること等により実施することができる。
続いて、上記のトランスポザーゼ50を活性化する。トランスポザーゼ50の活性化は、例えば、2価の金属イオンをバッファー中に添加すること等により実施することができる。
また、トランスポザーゼ50の種類によっては、DNA断片を転移させる活性を発揮するためにダイマーを形成する必要がある場合がある。また、トランスポザーゼ50がダイマーを形成するために、トランスポザーゼ50がトランスポザーゼ結合配列41に結合している必要がある場合がある。例えば、上述したTN5、SB10、TN10等はこのようなトランスポザーゼである。このような場合には、トランスポザーゼ50のダイマーを形成させることによってもトランスポザーゼ50を活性化することができる。
また、トランスポザーゼ50の種類によっては、トランスポザーゼ50を活性化するために、トランスポザーゼ50のダイマーを形成させ、且つ2価の金属イオンをバッファー中に添加することが必要な場合がある。
図3は、トランスポザーゼ50のダイマーを形成させた状態を示す。図3では、試料に、トランスポザーゼ結合配列41を有する遊離のDNA断片を添加して、当該DNA断片にトランスポザーゼ50を結合させている。図3に示すように、上記のDNA断片と結合したトランスポザーゼ50は、DNA断片40を構成するトランスポザーゼ結合配列41に結合したトランスポザーゼ50とダイマーを形成する。
トランスポザーゼ50を活性化した結果、DNA断片40中の所望の塩基配列(図3の例ではT7プロモーター配列42の下流に識別配列43が連結した塩基配列)のDNA断片が結合領域11の近傍に挿入される。ここで、近傍とは、空間的に近い領域であればよく、例えば結合領域11から500塩基以下であってもよく、例えば100塩基以下であってもよく、例えば50塩基以下であってもよい。
あるいは、近傍とは、空間的に近い領域であればよいため、ゲノムDNA10の塩基配列上は結合領域11から非常に離れた領域であって、ゲノム領域同士の相互作用により近接した領域であってもよい。すなわち、染色体構造上近接した領域であってもよい。
本工程において、トランスポザーゼ50によるDNA断片の挿入反応が不完全に終了する場合がある。具体的には、例えば、挿入されたDNA断片が部分的に1本鎖DNAとなっている場合、挿入されたDNA断片が断片化している場合等が挙げられる。そこで、T4DNAリガーゼ、T4DNAポリメラーゼI等を用いたfill in反応等により、トランスポザーゼ50によるDNA断片の挿入反応を完全に終了させる工程を更に実施してもよい。
図4は、所望の塩基配列のDNA断片が結合領域11の近傍に挿入された状態の一例を示す模式図である。図4に示すように、この例では、結合領域11の近傍に所望の塩基配列(T7プロモーター配列42の下流に識別配列43が連結した塩基配列)のDNA断片が挿入されている。図4の例では、所望の塩基配列だけでなく、2つのトランスポザーゼ結合配列41、及びリンカー44の一部に由来する塩基配列44’もゲノムDNA10に挿入されている。
図5(a)~(c)は、上述した図3の状態からトランスポザーゼ50が活性化した結果、DNA断片がゲノムDNA10に挿入された場合の例を示す模式図である。図5(a)は、上述した図4の状態を示す。図5(b)は、上述した図4とは逆向きに所望の塩基配列が挿入された状態を示す。図5(c)は、トランスポザーゼ結合配列41を有する遊離のDNA断片に結合したトランスポザーゼ50同士がダイマーを形成した場合に、ゲノムDNA10に挿入されうる塩基配列を示す。
図5(c)においてゲノムDNA10に挿入された塩基配列は、2つのトランスポザーゼ結合配列41を含み、それらの間に、任意の塩基配列45を含む場合がある。塩基配列45は、上述したfill in反応により形成されたものである。
図5(c)に示す塩基配列は、目的外の副産物であり、結合領域11の近傍に限らず、ゲノムDNA10上のあらゆる部分に挿入され得る。しかしながら、図5(c)に示す塩基配列は、所望の塩基配列(この例ではT7プロモーター配列42の下流に識別配列43が連結した塩基配列)を有していない。このため、目的とする解析における影響を排除することができる。
以上の工程により、DNA分子に結合したDNA結合タンパク質の結合領域の近傍に所望の塩基配列のDNA断片を挿入することができる。
(特異的結合物質)
ChILT法において、特異的結合物質としては、抗体の他にも、抗体断片、アプタマー等が挙げられる。抗体は、例えば、マウス等の動物に標的物質又はその断片を抗原として免疫することによって作製することができる。あるいは、例えば、ファージライブラリーのスクリーニングにより作製することができる。抗体断片としては、Fv、Fab、scFv等が挙げられる。抗体は、モノクローナル抗体であってもよく、ポリクローナル抗体であってもよい。また、市販の抗体であってもよい。
ChILT法において、特異的結合物質としては、抗体の他にも、抗体断片、アプタマー等が挙げられる。抗体は、例えば、マウス等の動物に標的物質又はその断片を抗原として免疫することによって作製することができる。あるいは、例えば、ファージライブラリーのスクリーニングにより作製することができる。抗体断片としては、Fv、Fab、scFv等が挙げられる。抗体は、モノクローナル抗体であってもよく、ポリクローナル抗体であってもよい。また、市販の抗体であってもよい。
アプタマーとは、標的物質に対する特異的結合能を有する物質である。アプタマーとしては、核酸アプタマー、ペプチドアプタマー等が挙げられる。標的物質に特異的結合能を有する核酸アプタマーは、例えば、systematic evolution of ligand by exponential enrichment(SELEX)法等により選別することができる。また、標的物質に特異的結合能を有するペプチドアプタマーは、例えば酵母を用いたTwo-hybrid法等により選別することができる。
(所望の塩基配列)
また、図1C~図5の例では、ゲノムDNA10に挿入する所望の塩基配列は、T7プロモーター配列42の下流に識別配列43が連結した塩基配列であったが、所望の塩基配列はこれに限定されない。例えば、所望の塩基配列として、T7プロモーター配列42の代わりにSP6プロモーター配列等の別のプロモーター配列を用いてもよい。あるいは、所望の塩基配列は、プロモーター配列のみから構成されていてもよい。あるいは、所望の塩基配列は、識別配列のみから構成されていてもよい。また、識別配列としては、確率的に出現頻度が低い塩基配列であれば特に制限されず、例えば約4~20塩基程度の任意の塩基配列を用いることができる。
また、図1C~図5の例では、ゲノムDNA10に挿入する所望の塩基配列は、T7プロモーター配列42の下流に識別配列43が連結した塩基配列であったが、所望の塩基配列はこれに限定されない。例えば、所望の塩基配列として、T7プロモーター配列42の代わりにSP6プロモーター配列等の別のプロモーター配列を用いてもよい。あるいは、所望の塩基配列は、プロモーター配列のみから構成されていてもよい。あるいは、所望の塩基配列は、識別配列のみから構成されていてもよい。また、識別配列としては、確率的に出現頻度が低い塩基配列であれば特に制限されず、例えば約4~20塩基程度の任意の塩基配列を用いることができる。
ゲノムDNA10に挿入する所望の塩基配列の長さは、例えば20~500塩基であってもよく、例えば20~200塩基であってもよく、例えば20~100塩基であってもよい。
また、図1C~図5の例では、DNA断片40を構成する各要素(T7プロモーター配列42、識別配列43及びトランスポザーゼ結合配列41)は隣接しているが、各要素の間には任意の塩基配列のスペーサーが存在していてもよい。また、各要素の順序は図1C~図5の例に示したものに限定されず、必要に応じて適宜入れ換えてもよいし、追加の要素を付加してもよい。
また、DNA断片40は、1本鎖であってもよく、2本鎖であってもよい。但し、使用するトランスポザーゼの特性により、トランスポザーゼ結合配列41が2本鎖DNAである必要がある場合には、少なくともトランスポザーゼ結合配列41は2本鎖である必要がある。
上述した反応により、固定された細胞標本を試料として、DNA結合タンパク質の結合領域の近傍に所望の塩基配列のDNA断片を挿入した後、レーザーマイクロダイセクション等により細胞を切り出して回収し、1細胞レベルで後述する解析を行うこともできる。
(ChILT法により挿入したDNA断片を利用した解析)
ChILT法は、前記所望の塩基配列(図1C~図5の例ではT7プロモーター配列42の下流に識別配列43が連結した塩基配列)を起点としてDNA10を遺伝子増幅し、増幅産物を得る工程と、前記増幅産物の塩基配列を解析する工程と、を更に備えていてもよい。これにより、所望のDNA断片が挿入されたゲノム上の位置を解析することができる。
ChILT法は、前記所望の塩基配列(図1C~図5の例ではT7プロモーター配列42の下流に識別配列43が連結した塩基配列)を起点としてDNA10を遺伝子増幅し、増幅産物を得る工程と、前記増幅産物の塩基配列を解析する工程と、を更に備えていてもよい。これにより、所望のDNA断片が挿入されたゲノム上の位置を解析することができる。
(遺伝子増幅)
遺伝子増幅としては、例えば、挿入した塩基配列に相補的なプライマーとランダムプライマーとを組み合わせたPCR、ポリメラーゼ(RNAポリメラーゼ、DNA依存性DNAポリメラーゼ等)を用いた転写等が挙げられる。本明細書において、「所望の塩基配列を起点として遺伝子増幅する」とは、結合領域11の近傍に挿入された所望の塩基配列の一部又は全部を利用してゲノムDNA10を遺伝子増幅することを意味する。
遺伝子増幅としては、例えば、挿入した塩基配列に相補的なプライマーとランダムプライマーとを組み合わせたPCR、ポリメラーゼ(RNAポリメラーゼ、DNA依存性DNAポリメラーゼ等)を用いた転写等が挙げられる。本明細書において、「所望の塩基配列を起点として遺伝子増幅する」とは、結合領域11の近傍に挿入された所望の塩基配列の一部又は全部を利用してゲノムDNA10を遺伝子増幅することを意味する。
例えば、図1C~図5の例では、ゲノムDNA10に挿入した塩基配列にT7プロモーター配列42が含まれている。そこで、「所望の塩基配列を起点として遺伝子増幅する」とは、上記のゲノムDNA10にT7ポリメラーゼを作用させて転写することにより、T7プロモーター配列42の下流のゲノムDNA10をRNAに転写することであってもよい。
あるいは、「所望の塩基配列を起点として遺伝子増幅する」とは、上記のゲノムDNA10を鋳型として、挿入した塩基配列の一部又は全部に相補的な塩基配列を有するプライマーと、ランダムプライマーとを組み合わせたPCRを行うこと等により、結合領域11の近傍のゲノムDNA10の塩基配列を遺伝子増幅することであってもよい。
すなわち、ChILT法では、挿入した塩基配列がプロモーター配列を含んでおり、遺伝子増幅が、プロモーター配列にRNAポリメラーゼを接触させてプロモーター配列の下流のDNAを転写してRNAを生成することにより遺伝子増幅してもよい。なお、上述したように、図5(c)に示す塩基配列は、T7プロモーター配列42を有していないため、T7ポリメラーゼを作用させても遺伝子増幅(転写)されることはない。
あるいは、挿入した所望の塩基配列を利用したPCR等により遺伝子増幅してもよい。なお、上述したように、図5(c)に示す塩基配列は、所望の塩基配列(T7プロモーター配列42の下流に識別配列43が結合した塩基配列)を有していないため、塩基配列を利用したPCR反応を行っても遺伝子増幅されることはない。
(識別配列)
図6(a)は、図4又は図5(a)に示すDNA10にT7ポリメラーゼを作用させて、T7プロモーター配列42の下流のDNA10をRNA60に転写した状態を示す図である。また、図6(b)は、図5(b)に示すDNA10にT7ポリメラーゼを作用させて、T7プロモーター配列42の下流のDNA10をRNA60に転写した状態を示す図である。
図6(a)は、図4又は図5(a)に示すDNA10にT7ポリメラーゼを作用させて、T7プロモーター配列42の下流のDNA10をRNA60に転写した状態を示す図である。また、図6(b)は、図5(b)に示すDNA10にT7ポリメラーゼを作用させて、T7プロモーター配列42の下流のDNA10をRNA60に転写した状態を示す図である。
図6(a)、(b)の例では、T7プロモーター配列42の下流に識別配列43が導入されている。すなわち、ChILT法では、所望の塩基配列は、プロモーター配列の下流に識別配列を更に含んでいてもよい。
図6(a)、(b)の例では、T7プロモーター配列42の下流に識別配列43が導入されているため、RNA60は、識別配列43の相補鎖43’及び識別配列43の下流のDNA10の相補鎖10’を含んでいる。
ゲノムDNAにT7RNAポリメラーゼを作用させると、上記の反応により挿入したT7プロモーター配列42以外の領域からRNAが転写されてしまう場合がある。これに対し、識別配列43が存在することにより、挿入したT7プロモーター配列42を起点として転写されたRNAとそれ以外のRNAとを識別することが可能になる。すなわち、識別配列43の相補鎖43’を有するRNA60は、挿入したT7プロモーター配列42を起点として転写されたRNAであると判断することができる。
また、上述したように、ChILT法では、同時に複数のDNA結合タンパク質に対する特異的結合物質を用いた解析を行うことができる。ここで、各特異的結合物質を利用してDNA10に挿入されるDNA断片40に、それぞれ異なる識別配列43を導入しておくことにより、いずれのDNA結合タンパク質の結合領域の近傍から転写されたRNAであるかを識別することも可能になる。
続いて、転写されたRNA60の塩基配列を解析することにより、DNA結合タンパク質が結合したゲノム上の位置を特定することができる。RNA60の塩基配列の解析は、例えば、次世代シークエンサーにより行ってもよいし、DNAアレイとのハイブリダイゼーションにより行ってもよい。
上述した図6(a)、(b)の例では、RNA60に転写される領域が互いに異なっているが、いずれの例においても、RNA60は結合領域11の近傍から転写されるRNAである。したがって、RNA60の塩基配列を解析することにより、例えば、DNA結合タンパク質(図1~6の例ではヒストン20)の結合領域のゲノム上の位置を特定することができる。すなわち、所望の塩基配列が挿入される向きは、解析結果に影響しない。
[結合体]
1実施形態において、本発明は、トランスポザーゼ結合配列と所望の塩基配列とを含む塩基配列を有するDNA断片と、DNA結合タンパク質に対する特異的結合物質又は前記特異的結合物質に対する特異的結合物質との結合体を提供する。
1実施形態において、本発明は、トランスポザーゼ結合配列と所望の塩基配列とを含む塩基配列を有するDNA断片と、DNA結合タンパク質に対する特異的結合物質又は前記特異的結合物質に対する特異的結合物質との結合体を提供する。
本実施形態の結合体は、DNA分子に結合したDNA結合タンパク質の結合領域の近傍への、所望の塩基配列の挿入用であるということができる。
本実施形態の結合体において、トランスポザーゼ結合配列、所望の塩基配列、特異的結合物質については、上述したものと同様である。すなわち、所望の塩基配列はプロモーター配列を含んでいてもよい。また、所望の塩基配列はプロモーター配列の下流に識別配列を更に含んでいてもよい。
本実施形態の結合体において、DNA断片は、リンカーを介してDNA結合タンパク質に対する特異的結合物質又は前記特異的結合物質に対する特異的結合物質と結合していることが好ましい。リンカーについては上述したものと同様である。
本実施形態の結合体において、「上記のDNA断片と、DNA結合タンパク質に対する特異的結合物質との結合体」とは、例えば、1次抗体に上記のDNA断片が結合した結合体を意味する。すなわち、DNA結合タンパク質に対する特異的結合物質に直接上記のDNA断片が結合した結合体を意味する。
また、本実施形態の結合体において、「上記のDNA断片と、DNA結合タンパク質に対する前記特異的結合物質に対する特異的結合物質との結合体」とは、例えば、2次抗体にDNA断片が結合した結合体を意味する。すなわち、本実施形態では、上記のDNA断片は、DNA結合タンパク質に対する特異的結合物質には結合しておらず、DNA結合タンパク質に対する特異的結合物質に結合する特異的結合物質に結合している。より具体的には、例えば、DNA結合タンパク質に対する特異的結合物質がマウスIgGであった場合、上記のDNA断片と抗マウスIgG抗体との結合体等が挙げられる。
特異的結合物質1分子あたりの上記のDNA断片の数は特に制限されず、例えば1~10個であってもよい。
特異的結合物質にDNA断片を結合する方法は特に制限されず、例えば、スクシンイミド基、マレイミド基等を有する化学架橋剤を用いて結合してもよい。例えば、DNA断片の5’末端にアミノ基を結合させておき、当該アミノ基と特異的結合物質に存在するアミノ基、カルボキシ基等の官能基を適宜の化学架橋剤で共有結合することが挙げられる。あるいは、例えば、アビジンとビオチンの結合等を利用して特異的結合物質にDNA断片を結合させてもよい。
次に実施例を示して本発明を更に詳細に説明するが、本発明は以下の実施例に限定されるものではない。
[実験例1]
従来法であるChIP-Seq解析により、約1000個のマウス骨格筋芽細胞を用いて、DNA結合タンパク質である、リジン4トリメチル化修飾されたヒストンH3(H3K4me3)の、ゲノムDNA上の結合位置を解析した。
従来法であるChIP-Seq解析により、約1000個のマウス骨格筋芽細胞を用いて、DNA結合タンパク質である、リジン4トリメチル化修飾されたヒストンH3(H3K4me3)の、ゲノムDNA上の結合位置を解析した。
(可溶性クロマチン分画の調製)
マウス骨格筋芽細胞1000個を1mLの培地に懸濁し、1.5mLシリコナイズチューブに入れた。続いて、100μLの固定溶液(37%ホルムアルデヒド)を添加し、5分間、室温で静置し、DNAとDNA結合タンパク質とをクロスリンクした。
マウス骨格筋芽細胞1000個を1mLの培地に懸濁し、1.5mLシリコナイズチューブに入れた。続いて、100μLの固定溶液(37%ホルムアルデヒド)を添加し、5分間、室温で静置し、DNAとDNA結合タンパク質とをクロスリンクした。
続いて、100μLの1Mグリシン水溶液を添加し、5分間室温で撹拌した。これにより、グリシンと残存したホルムアルデヒドとを反応させて、クロスリンク反応を停止させた。続いて、3000rpmで5分間、4℃で遠心し、上清を除去し、更にリン酸緩衝液(PBS)で細胞を2回洗浄した。
続いて、2mLのChIPバッファー(10mM Tris-HCl(pH8.0),200mM KCl,1mM CaCl2,0.5%NP40,PMSF,アプロチニン、ロイぺプチン)を添加して細胞を懸濁し、氷上で10分間静置した。
続いて、超音波細胞破砕装置を用いて、氷水で冷却しながら超音波処理を行い、DNAを断片化した。
続いて、マイクロコッカルヌクレアーゼを加え、40分間、37℃で酵素処理を行った。続いて、終濃度10mMとなるようにEDTAを加えて反応を停止させた。続いて、15,000×gで10分間、4℃で遠心し、上清を2mLシリコナイズチューブに回収した。続いて、ChIPバッファーを加えて希釈し、可溶性クロマチン分画を得た。
(免疫沈降)
上記の可溶性クロマチン分画に、2μgのウサギ抗マウスIgG抗体と2μgのマウス抗H3K4me3抗体とを予め反応させた抗体複合体溶液と、20μLのヤギ抗ウサギIgG抗体が結合した磁性粒子(Dynabeads M-280)とを添加し、ローテーターで一晩、4℃で撹拌した。
上記の可溶性クロマチン分画に、2μgのウサギ抗マウスIgG抗体と2μgのマウス抗H3K4me3抗体とを予め反応させた抗体複合体溶液と、20μLのヤギ抗ウサギIgG抗体が結合した磁性粒子(Dynabeads M-280)とを添加し、ローテーターで一晩、4℃で撹拌した。
続いて、マグネティックスタンドに反応溶液をセットして1分間静置し、磁性粒子を回収した。続いて、回収した磁性粒子を、ChIPバッファーで3回、洗浄バッファー(10mM Tris-HCl(pH8.0),500mM KCl,1mM CaCl2,0.5%NP40)で3回、1×TE(10mM Tris-HCl(pH8.0),1mM EDTA(pH8.0))で3回洗浄した。
(DNAの精製)
100μLのChIP溶出バッファー(50mM Tris-HCl(pH8.0),10mM EDTA,1%SDS)を上記の磁性粒子に加え、ボルテックスミキサーで懸濁し、更に5M NaCl 5μLを添加した。続いて、65℃で4時間以上加熱し、DNAとDNA結合タンパク質のクロスリンクをはずした。
100μLのChIP溶出バッファー(50mM Tris-HCl(pH8.0),10mM EDTA,1%SDS)を上記の磁性粒子に加え、ボルテックスミキサーで懸濁し、更に5M NaCl 5μLを添加した。続いて、65℃で4時間以上加熱し、DNAとDNA結合タンパク質のクロスリンクをはずした。
続いて、0.5μLの10mg/mL RNaseAを添加し、ボルテックスミキサーで撹拌し、37℃で30分インキュベートした。続いて、1μLの10mg/mLプロテイナーゼKを添加し、ボルテックスミキサーで撹拌し、50℃で1時間インキュベートした。続いて、PCR purification kit(キアゲン社)を用いてDNAを精製した。
(塩基配列の解析)
続いて、次世代シークエンサーにより回収されたDNAの塩基配列を解析した。
続いて、次世代シークエンサーにより回収されたDNAの塩基配列を解析した。
(結果)
図7(a)は、従来法であるChIP-Seq解析により、約1000個の細胞を用いてEef1a1遺伝子座へのH3K4me3の結合をマッピングした結果を示すグラフである。
図7(a)は、従来法であるChIP-Seq解析により、約1000個の細胞を用いてEef1a1遺伝子座へのH3K4me3の結合をマッピングした結果を示すグラフである。
[実験例2]
マウス骨格筋芽細胞を用いて、ChILT法により、DNA結合タンパク質である、リジン4トリメチル化修飾されたヒストンH3(H3K4me3)の、ゲノムDNA上の結合位置を1細胞レベルで解析した。
マウス骨格筋芽細胞を用いて、ChILT法により、DNA結合タンパク質である、リジン4トリメチル化修飾されたヒストンH3(H3K4me3)の、ゲノムDNA上の結合位置を1細胞レベルで解析した。
(細胞の固定)
まず、スライドガラスにマウス骨格筋芽細胞を播種した。続いて、16時間培養後、培地を除去し、PBSで洗浄した。続いて、1%パラホルムアルデヒド溶液を添加して室温で5分間静置し、細胞を固定後、PBSで洗浄した。
まず、スライドガラスにマウス骨格筋芽細胞を播種した。続いて、16時間培養後、培地を除去し、PBSで洗浄した。続いて、1%パラホルムアルデヒド溶液を添加して室温で5分間静置し、細胞を固定後、PBSで洗浄した。
(DNA断片のH3K4me3結合領域への近接)
続いて、固定した細胞試料にブロッキング剤(商品名「Blocking One-P」、ナカライテスク社)を加え室温で10分間静置後、PBSで洗浄した。続いて、0.1×Blocking One-Pで希釈した抗体複合体(2μg/mL抗H3K4me3抗体及び核酸標識2次抗体を予め反応させたもの)を添加し、37℃で2時間反応させた後、PBSで洗浄した。この結果、核酸標識2次抗体に標識したDNA断片とゲノムDNA上のH3K4me3結合領域とが近接した。
続いて、固定した細胞試料にブロッキング剤(商品名「Blocking One-P」、ナカライテスク社)を加え室温で10分間静置後、PBSで洗浄した。続いて、0.1×Blocking One-Pで希釈した抗体複合体(2μg/mL抗H3K4me3抗体及び核酸標識2次抗体を予め反応させたもの)を添加し、37℃で2時間反応させた後、PBSで洗浄した。この結果、核酸標識2次抗体に標識したDNA断片とゲノムDNA上のH3K4me3結合領域とが近接した。
ここで、核酸標識2次抗体に標識したDNA断片は、配列番号1に記載の塩基配列からなるDNA断片と、配列番号2に記載の塩基配列からなるDNA断片とをアニーリングさせたものであった。
配列番号1に記載の塩基配列において、第49~67番目の塩基配列はTN5トランスポザーゼの結合配列であり、第7~26番目の塩基配列はT7プロモーター配列であり、第27~34番目の塩基配列は識別配列であり、第1~6塩基はリンカーの一部の塩基配列であった。
また、配列番号2に記載の塩基配列からなるDNA断片は、配列番号1に記載の塩基配列における第49~67番目の塩基配列と相補的な塩基配列を有するDNA断片であり、配列番号1に記載の塩基配列からなるDNA断片とアニールすることにより、2本鎖DNAであるTN5トランスポザーゼの結合配列を形成した。
(トランスポザーゼの結合)
続いて、試料にTN5トランスポザーゼを85μg/mLずつ添加し、室温で10分間放置した。これにより、核酸標識2次抗体に標識されたDNA断片中のトランスポザーゼ結合配列にトランスポザーゼが結合した。
続いて、試料にTN5トランスポザーゼを85μg/mLずつ添加し、室温で10分間放置した。これにより、核酸標識2次抗体に標識されたDNA断片中のトランスポザーゼ結合配列にトランスポザーゼが結合した。
(トランスポザーゼの活性化及び所望の塩基配列の挿入)
続いて、試料に、トランスポザーゼ結合配列を有する遊離のDNA断片(以下、「oligo3-4」という。)を添加し、室温で1時間反応させた後、PBSで洗浄した。oligo3-4は、配列番号3に記載の塩基配列からなるDNA断片と、配列番号4に記載の塩基配列からなるDNA断片とをアニーリングさせたものであった。oligo3-4の2本鎖DNA領域は、TN5トランスポザーゼの結合配列であった。
続いて、試料に、トランスポザーゼ結合配列を有する遊離のDNA断片(以下、「oligo3-4」という。)を添加し、室温で1時間反応させた後、PBSで洗浄した。oligo3-4は、配列番号3に記載の塩基配列からなるDNA断片と、配列番号4に記載の塩基配列からなるDNA断片とをアニーリングさせたものであった。oligo3-4の2本鎖DNA領域は、TN5トランスポザーゼの結合配列であった。
続いて、試料を1×TN5透析バッファー(50mM HEPES-KOH(pH7.2),0.1M NaCl,0.1mM EDTA,1mM DTT,0.1%Triton X-100,10%グリセロール)で洗浄した。
続いて、試料に1×TAPS-DMFバッファー(10mM TAPS-NaOH(pH8.5),5mM MgCl2,10%N,N-ジメチルホルムアミド)を添加し、37℃で1時間静置した。これにより、核酸標識2次抗体に標識されたDNA断片に結合したトランスポザーゼが活性化し、核酸標識2次抗体に標識されたDNA断片がゲノムDNA上のH3K4me3結合領域の近傍に挿入された。
続いて、1%ドデシル硫酸ナトリウム溶液を添加し、室温で10分間静置したのち、PBSで洗浄した。これにより、トランスポザーゼを失活させて除去した。
続いて、1×T4DNAリガーゼ反応バッファーで洗浄し、fill in反応溶液(T4DNAリガーゼ反応バッファー、dNTPmix,T4DNAリガーゼI、T4DNAポリメラーゼI)を加え室温で30分間反応させた。これにより、核酸標識2次抗体に標識されたDNA断片をゲノムDNA上のH3K4me3結合領域の近傍に確実に挿入させた。
続いて、1%ドデシル硫酸ナトリウム溶液を添加し、室温で10分間静置したのち、PBSで洗浄した。これにより、T4DNAリガーゼ及びT4DNAポリメラーゼIを失活させて除去した。
(1細胞の切り出し)
続いて、レーザーマイクロダイセクションにより、対象細胞を1個ずつ切り出し、それぞれマイクロチューブに回収した。
続いて、レーザーマイクロダイセクションにより、対象細胞を1個ずつ切り出し、それぞれマイクロチューブに回収した。
(遺伝子増幅)
ここでは、遺伝子増幅として、T7RNAポリメラーゼによる転写を行った。まず、切り出した各細胞を1×T7RNAポリメラーゼバッファーで洗浄した。続いて、インビトロ転写溶液(T7RNAポリメラーゼバッファー,ATP,CTP,GTP,UTP,RNaseインヒビター,T7RNAポリメラーゼ)を加え、37℃で16時間反応させた。これにより、挿入したDNA断片に含まれていたT7RNAプロモーター配列を起点として、ゲノムDNAが転写された。
ここでは、遺伝子増幅として、T7RNAポリメラーゼによる転写を行った。まず、切り出した各細胞を1×T7RNAポリメラーゼバッファーで洗浄した。続いて、インビトロ転写溶液(T7RNAポリメラーゼバッファー,ATP,CTP,GTP,UTP,RNaseインヒビター,T7RNAポリメラーゼ)を加え、37℃で16時間反応させた。これにより、挿入したDNA断片に含まれていたT7RNAプロモーター配列を起点として、ゲノムDNAが転写された。
続いて、DNaseIを加え37℃で30分間反応させた。これにより、回収した細胞中に含まれていたDNAが分解された。続いて、RNeasy MinElute Cleanup Kit(キアゲン社)により転写されたRNAを精製した。
(回収したRNAの塩基配列解析)
続いて、精製したRNAを逆転写してcDNAを調製し、次世代シークエンサーを用いて塩基配列を解析し、ゲノム上にマッピングした。
続いて、精製したRNAを逆転写してcDNAを調製し、次世代シークエンサーを用いて塩基配列を解析し、ゲノム上にマッピングした。
(結果)
図7(b)~(d)は、それぞれ、Eef1a1遺伝子座へのH3K4me3の結合を、ChILT法により1細胞レベルで解析してマッピングした代表的な結果を示すグラフである。
図7(b)~(d)は、それぞれ、Eef1a1遺伝子座へのH3K4me3の結合を、ChILT法により1細胞レベルで解析してマッピングした代表的な結果を示すグラフである。
その結果、図7(b)~(d)に示すように、個々の細胞によって、Eef1a1遺伝子座へのH3K4me3の結合に差があることが明らかとなった。このような結果は、図7(a)の1000個の細胞を用いたChIP-Seq解析では検出することができず、DNAとDNA結合タンパク質との結合を1細胞レベルで解析してはじめて検出することができたものである。
[実験例3]
ChILT法により、様々なDNA結合タンパク質のゲノムDNA上の結合を1細胞レベルで解析した。細胞としてマウス骨格筋芽細胞を用いた。また、1次抗体として、下記表1に示す抗DNA結合タンパク質抗体を使用した。また、2次抗体として、実験例2で用いたものと同様のDNA断片で標識した、抗マウスIgG抗体又は抗ウサギIgG抗体を使用した。
ChILT法により、様々なDNA結合タンパク質のゲノムDNA上の結合を1細胞レベルで解析した。細胞としてマウス骨格筋芽細胞を用いた。また、1次抗体として、下記表1に示す抗DNA結合タンパク質抗体を使用した。また、2次抗体として、実験例2で用いたものと同様のDNA断片で標識した、抗マウスIgG抗体又は抗ウサギIgG抗体を使用した。

(細胞の固定)
実験例2と同様にして、スライドガラス上でマウス骨格筋芽細胞を培養して固定し、PBSで洗浄した。
実験例2と同様にして、スライドガラス上でマウス骨格筋芽細胞を培養して固定し、PBSで洗浄した。
(抗体複合体の反応)
続いて、実験例2と同様にして、固定した細胞試料にブロッキング剤(商品名「Blocking One-P」、ナカライテスク社)を加え室温で10分間静置後、PBSで洗浄した。続いて、0.1×Blocking One-Pで希釈した抗体複合体(1次抗体及び核酸標識2次抗体を予め反応させたもの)を添加し、37℃で2時間反応させた後、PBSで洗浄した。
続いて、実験例2と同様にして、固定した細胞試料にブロッキング剤(商品名「Blocking One-P」、ナカライテスク社)を加え室温で10分間静置後、PBSで洗浄した。続いて、0.1×Blocking One-Pで希釈した抗体複合体(1次抗体及び核酸標識2次抗体を予め反応させたもの)を添加し、37℃で2時間反応させた後、PBSで洗浄した。
抗体複合体は、1次抗体がマウス抗体である場合には2次抗体に核酸標識マウス抗体を反応させて調製した。また、1次抗体がウサギ抗体である場合には2次抗体に核酸標識ウサギ抗体を反応させて調製した。
また、陰性対照として、核酸標識抗マウスIgG抗体のみ、又は核酸標識抗ウサギIgG抗体のみを反応させたサンプルも調製した。すなわち、1次抗体を反応させなかったサンプルを陰性対照として調製した。その後、トランスポザーゼの結合、トランスポザーゼの活性化及び所望の塩基配列の挿入を実験例2と同様にして行った。
(1細胞の切り出し)
続いて、レーザーマイクロダイセクションにより、対象細胞を1個ずつ切り出し、それぞれマイクロチューブに回収した。
続いて、レーザーマイクロダイセクションにより、対象細胞を1個ずつ切り出し、それぞれマイクロチューブに回収した。
(遺伝子増幅及び塩基配列解析)
回収した各細胞について、実験例2と同様にしてT7RNAポリメラーゼによる転写を行い、転写されたRNAを精製した。続いて、精製したRNAを逆転写してcDNAを調製し、次世代シークエンサーを用いて塩基配列を解析し、ゲノム上にマッピングした。
回収した各細胞について、実験例2と同様にしてT7RNAポリメラーゼによる転写を行い、転写されたRNAを精製した。続いて、精製したRNAを逆転写してcDNAを調製し、次世代シークエンサーを用いて塩基配列を解析し、ゲノム上にマッピングした。
(結果)
図8Aは、表1に示す様々な1次抗体を用いたChILT法により、DNA結合タンパク質の結合を1細胞レベルで解析してマッピングした結果を示すグラフである。各抗体複合体につき2細胞ずつ解析を行った。
図8Aは、表1に示す様々な1次抗体を用いたChILT法により、DNA結合タンパク質の結合を1細胞レベルで解析してマッピングした結果を示すグラフである。各抗体複合体につき2細胞ずつ解析を行った。
図8A中、グラフの横軸は次世代シークエンサーにより得られたリード数を表す。また、「Valid」はゲノム上に有効にマッピングされたリードを表し、「Duplicate」はゲノム上の同一位置にマッピングされたリードが2個存在したリードを表し、「Multi」はゲノム上の同一位置にマッピングされたリードが3個以上存在したリードを表し、「Unmapped」はゲノム上にマッピングされなかったリードを表し、「Nextera」は塩基配列解析に使用したライブラリーに由来するリードを表す。
図8A中、例えば「rH3K27me3」はrH3K27me3抗体と核酸標識抗ウサギIgG抗体との複合体を反応させた結果であることを表し、以下同様である。また、例えば「mH3K4me3-rH3K27me3」はmH3K4me3抗体と核酸標識抗マウスIgG抗体との複合体、及びrH3K27me3抗体と核酸標識抗ウサギIgG抗体との複合体を同時に反応させた結果であることを表し、以下同様である。また、「#1」、「#2」は、それぞれ、解析した2細胞のうちの第1の細胞の結果及び第2の細胞の結果であることを表す。
その結果、1次抗体を使用しなかった陰性対照ではほとんどリードが得られておらず、1次抗体の反応依存的にリードが得られたことが確認された。また、細胞内に存在するヒストン修飾や転写因子の量に応じたリードが得られたことが確認された。
図8Bは、図8Aのグラフの横軸を、「Valid」、「Duplicate」、「Multi」、「Unmapped」、「Nextera」の各リードの存在割合に変更したグラフである。その結果、核酸標識抗マウスIgG抗体、又は核酸標識抗ウサギIgG抗体を用いたChILT法により、約50~85%の有効リードを得ることができることが明らかとなった。
本発明によれば、免疫沈降を行うことなく、タンパク質と特定のゲノム領域との結合を解析することができる技術を提供することができる。したがって、例えば不溶性タンパク質等の、免疫沈降を行うことが困難なタンパク質のDNAへの結合を解析することが可能になる。また、少数細胞等を試料とした解析も容易になる。
10…ゲノムDNA、10’…ゲノムDNA10の相補鎖、11…結合領域、20…ヒストン、30…特異的結合物質、40…DNA断片、41…トランスポザーゼ結合配列、41’…トランスポザーゼ結合配列41の相補鎖、42…T7プロモーター配列、43…識別配列、43’…識別配列43の相補鎖、44…リンカー、44’…リンカー44の一部に由来する塩基配列、45…任意配列、50…トランスポザーゼ、60…RNA、S…細胞標本。
Claims (7)
- DNA分子に結合したDNA結合タンパク質の結合領域の近傍に所望の塩基配列のDNA断片を挿入する方法であって、
前記DNA結合タンパク質に対する特異的結合物質を利用して、トランスポザーゼ結合配列と前記所望の塩基配列とを含む塩基配列を有するDNA断片と、前記結合領域とを近接させる工程と、
前記トランスポザーゼ結合配列にトランスポザーゼを結合させる工程と、
前記トランスポザーゼを活性化し、その結果、前記所望の塩基配列のDNA断片が前記結合領域の近傍に挿入される工程と、
を備える方法。 - 前記所望の塩基配列を起点として前記DNA分子を遺伝子増幅し、増幅産物を得る工程と、
前記増幅産物の塩基配列を解析する工程と、
を更に備える、請求項1に記載の方法。 - 前記所望の塩基配列がプロモーター配列を含み、
前記遺伝子増幅が、前記プロモーター配列にRNAポリメラーゼを接触させて前記プロモーター配列の下流のDNAを転写してRNAを生成することにより行われる、
請求項2に記載の方法。 - 前記所望の塩基配列が前記プロモーター配列の下流に識別配列を更に含む、請求項3に記載の方法。
- トランスポザーゼ結合配列と所望の塩基配列とを含む塩基配列を有するDNA断片と、DNA結合タンパク質に対する特異的結合物質又は前記特異的結合物質に対する特異的結合物質との結合体。
- 前記所望の塩基配列がプロモーター配列を含む、請求項5に記載の結合体。
- 前記所望の塩基配列が前記プロモーター配列の下流に識別配列を更に含む、請求項6に記載の結合体。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17845787.5A EP3508574A4 (en) | 2016-08-30 | 2017-05-24 | METHOD FOR INSERTING A DESIRED DNA FRAGMENT INTO A SITE NEXT TO A BINDING DOMAIN OF A DNA-BINDING PROTEIN |
| US16/327,754 US11414680B2 (en) | 2016-08-30 | 2017-05-24 | Method for inserting desired DNA fragment into site located adjacent to binding domain of DNA-binding protein |
| JP2018536938A JP6943376B2 (ja) | 2016-08-30 | 2017-05-24 | Dna結合タンパク質の結合領域の近傍に所望のdna断片を挿入する方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016167967 | 2016-08-30 | ||
| JP2016-167967 | 2016-08-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018042776A1 true WO2018042776A1 (ja) | 2018-03-08 |
Family
ID=61300480
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/019309 WO2018042776A1 (ja) | 2016-08-30 | 2017-05-24 | Dna結合タンパク質の結合領域の近傍に所望のdna断片を挿入する方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US11414680B2 (ja) |
| EP (1) | EP3508574A4 (ja) |
| JP (1) | JP6943376B2 (ja) |
| WO (1) | WO2018042776A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10227574B2 (en) | 2016-12-16 | 2019-03-12 | B-Mogen Biotechnologies, Inc. | Enhanced hAT family transposon-mediated gene transfer and associated compositions, systems, and methods |
| WO2021157589A1 (ja) * | 2020-02-04 | 2021-08-12 | 国立大学法人九州大学 | 対象核酸の塩基配列を1細胞レベルで並列に検出する方法 |
| CN114106196A (zh) * | 2021-10-29 | 2022-03-01 | 陈凯 | 抗体-转座酶融合蛋白及其制备方法和应用 |
| US11278570B2 (en) | 2016-12-16 | 2022-03-22 | B-Mogen Biotechnologies, Inc. | Enhanced hAT family transposon-mediated gene transfer and associated compositions, systems, and methods |
| WO2022176910A1 (ja) | 2021-02-19 | 2022-08-25 | 国立大学法人九州大学 | 核酸断片及びその使用 |
| US11760983B2 (en) | 2018-06-21 | 2023-09-19 | B-Mogen Biotechnologies, Inc. | Enhanced hAT family transposon-mediated gene transfer and associated compositions, systems, and methods |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20060258603A1 (en) * | 2003-02-10 | 2006-11-16 | Max-Delbruck-Centrum Fur Molekulare Medizin (Mdc) | Transposon-based targeting system |
| WO2010048605A1 (en) * | 2008-10-24 | 2010-04-29 | Epicentre Technologies Corporation | Transposon end compositions and methods for modifying nucleic acids |
| WO2014190214A1 (en) * | 2013-05-22 | 2014-11-27 | Active Motif, Inc. | Targeted transposition for use in epigenetic studies |
| WO2014205296A1 (en) * | 2013-06-21 | 2014-12-24 | The Broad Institute, Inc. | Methods for shearing and tagging dna for chromatin immunoprecipitation and sequencing |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011021684A1 (ja) * | 2009-08-21 | 2011-02-24 | 国立大学法人大阪大学 | 特定ゲノム領域の単離方法 |
| WO2013078470A2 (en) | 2011-11-22 | 2013-05-30 | MOTIF, Active | Multiplex isolation of protein-associated nucleic acids |
| EP2783001B1 (en) * | 2011-11-22 | 2018-01-03 | Active Motif | Multiplex isolation of protein-associated nucleic acids |
| EP4321628A3 (en) | 2013-05-23 | 2024-04-24 | The Board of Trustees of the Leland Stanford Junior University | Transposition into native chromatin for personal epigenomics |
| US10914729B2 (en) * | 2017-05-22 | 2021-02-09 | The Trustees Of Princeton University | Methods for detecting protein binding sequences and tagging nucleic acids |
-
2017
- 2017-05-24 JP JP2018536938A patent/JP6943376B2/ja active Active
- 2017-05-24 WO PCT/JP2017/019309 patent/WO2018042776A1/ja active Search and Examination
- 2017-05-24 US US16/327,754 patent/US11414680B2/en active Active
- 2017-05-24 EP EP17845787.5A patent/EP3508574A4/en active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20060258603A1 (en) * | 2003-02-10 | 2006-11-16 | Max-Delbruck-Centrum Fur Molekulare Medizin (Mdc) | Transposon-based targeting system |
| WO2010048605A1 (en) * | 2008-10-24 | 2010-04-29 | Epicentre Technologies Corporation | Transposon end compositions and methods for modifying nucleic acids |
| WO2014190214A1 (en) * | 2013-05-22 | 2014-11-27 | Active Motif, Inc. | Targeted transposition for use in epigenetic studies |
| WO2014205296A1 (en) * | 2013-06-21 | 2014-12-24 | The Broad Institute, Inc. | Methods for shearing and tagging dna for chromatin immunoprecipitation and sequencing |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3508574A4 * |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10227574B2 (en) | 2016-12-16 | 2019-03-12 | B-Mogen Biotechnologies, Inc. | Enhanced hAT family transposon-mediated gene transfer and associated compositions, systems, and methods |
| US11111483B2 (en) | 2016-12-16 | 2021-09-07 | B-Mogen Biotechnologies, Inc. | Enhanced hAT family transposon-mediated gene transfer and associated compositions, systems and methods |
| US11162084B2 (en) | 2016-12-16 | 2021-11-02 | B-Mogen Biotechnologies, Inc. | Enhanced hAT family transposon-mediated gene transfer and associated compositions, systems, and methods |
| US11278570B2 (en) | 2016-12-16 | 2022-03-22 | B-Mogen Biotechnologies, Inc. | Enhanced hAT family transposon-mediated gene transfer and associated compositions, systems, and methods |
| US11760983B2 (en) | 2018-06-21 | 2023-09-19 | B-Mogen Biotechnologies, Inc. | Enhanced hAT family transposon-mediated gene transfer and associated compositions, systems, and methods |
| WO2021157589A1 (ja) * | 2020-02-04 | 2021-08-12 | 国立大学法人九州大学 | 対象核酸の塩基配列を1細胞レベルで並列に検出する方法 |
| WO2022176910A1 (ja) | 2021-02-19 | 2022-08-25 | 国立大学法人九州大学 | 核酸断片及びその使用 |
| CN114106196A (zh) * | 2021-10-29 | 2022-03-01 | 陈凯 | 抗体-转座酶融合蛋白及其制备方法和应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20200010853A1 (en) | 2020-01-09 |
| EP3508574A1 (en) | 2019-07-10 |
| EP3508574A4 (en) | 2020-04-01 |
| US11414680B2 (en) | 2022-08-16 |
| JPWO2018042776A1 (ja) | 2019-08-08 |
| JP6943376B2 (ja) | 2021-09-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2018042776A1 (ja) | Dna結合タンパク質の結合領域の近傍に所望のdna断片を挿入する方法 | |
| JP7234114B2 (ja) | 細胞区画内の生体分子への直交アクセスおよび細胞区画内の生体分子のタグ付けのための分析システム | |
| US8916504B2 (en) | Methods of RNA display | |
| JP7372927B6 (ja) | 遺伝子およびタンパク質の発現を検出する生体分子プローブおよびその検出方法 | |
| JP7026248B2 (ja) | 二本鎖dnaを増幅するための方法およびキット | |
| CN107109698B (zh) | Rna stitch测序:用于直接映射细胞中rna:rna相互作用的测定 | |
| JP2019017281A (ja) | タンパク質−rna相互作用の検出 | |
| JP2023551072A (ja) | Rnaおよびdna修飾の多重プロファイリング | |
| JP2008253176A (ja) | 高親和性分子取得のためのリンカー | |
| US20230416828A1 (en) | Rna and dna analysis using engineered surfaces | |
| US20230332215A1 (en) | Methods for barcoding macromolecules in individual cells | |
| US20210310049A1 (en) | Reagents and methods for blocking non-specific interactions with nucleic acids | |
| JP2004097213A (ja) | 核酸および/またはタンパク質の選択方法 | |
| JP2023508796A (ja) | Dna中のn-4-アセチルデオキシシチジンの検出のための方法およびキット | |
| WO2021157589A1 (ja) | 対象核酸の塩基配列を1細胞レベルで並列に検出する方法 | |
| US8536100B2 (en) | Rapid method for identifying polypeptide-nucleic acid interactions | |
| JPWO2005001086A1 (ja) | 固定化mRNA−ピューロマイシン連結体及びその用途 | |
| WO2021182587A1 (ja) | 光架橋塩基を活用した、新規ペプチドアプタマー探索用標的モジュール、新規ペプチドアプタマー探索用リンカー及びそれらを用いる新規ペプチドアプタマーの探索方法 | |
| WO2022176910A1 (ja) | 核酸断片及びその使用 | |
| Tullius | Fiber-seq reveals the single-molecule architecture of nuclear transcription and the mitochondrial genome | |
| JP6619983B2 (ja) | Head−to−Head架橋を用いるmRNAディスプレイ法 | |
| Ng | Mapping of the Novel mRNA Acetylation, ac4C, in the HEK293 Transcriptome | |
| JPWO2020145405A1 (ja) | 三次元dna構造相互作用分析方法 | |
| Jacobsen | Novel Multiomics Method of RNA and Nuclear Protein Characterization in Single Cells |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17845787 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2018536938 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2017845787 Country of ref document: EP Effective date: 20190401 |