WO2017171098A1 - カロテノイドの製造方法 - Google Patents
カロテノイドの製造方法 Download PDFInfo
- Publication number
- WO2017171098A1 WO2017171098A1 PCT/JP2017/014162 JP2017014162W WO2017171098A1 WO 2017171098 A1 WO2017171098 A1 WO 2017171098A1 JP 2017014162 W JP2017014162 W JP 2017014162W WO 2017171098 A1 WO2017171098 A1 WO 2017171098A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino acid
- carotenoid
- enzyme
- bacterium
- acid sequence
- Prior art date
Links
Images





























Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/74—Vectors or expression systems specially adapted for prokaryotic hosts other than E. coli, e.g. Lactobacillus, Micromonospora
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N1/00—Microorganisms, e.g. protozoa; Compositions thereof; Processes of propagating, maintaining or preserving microorganisms or compositions thereof; Processes of preparing or isolating a composition containing a microorganism; Culture media therefor
- C12N1/20—Bacteria; Culture media therefor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/52—Genes encoding for enzymes or proenzymes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/1022—Transferases (2.) transferring aldehyde or ketonic groups (2.2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
- C12N9/1085—Transferases (2.) transferring alkyl or aryl groups other than methyl groups (2.5)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P23/00—Preparation of compounds containing a cyclohexene ring having an unsaturated side chain containing at least ten carbon atoms bound by conjugated double bonds, e.g. carotenes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/02—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving viable microorganisms
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/02—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving viable microorganisms
- C12Q1/04—Determining presence or kind of microorganism; Use of selective media for testing antibiotics or bacteriocides; Compositions containing a chemical indicator therefor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y202/00—Transferases transferring aldehyde or ketonic groups (2.2)
- C12Y202/01—Transketolases and transaldolases (2.2.1)
- C12Y202/01007—1-Deoxy-D-xylulose-5-phosphate synthase (2.2.1.7)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y205/00—Transferases transferring alkyl or aryl groups, other than methyl groups (2.5)
- C12Y205/01—Transferases transferring alkyl or aryl groups, other than methyl groups (2.5) transferring alkyl or aryl groups, other than methyl groups (2.5.1)
- C12Y205/01086—Trans,polycis-decaprenyl diphosphate synthase (2.5.1.86)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y205/00—Transferases transferring alkyl or aryl groups, other than methyl groups (2.5)
- C12Y205/01—Transferases transferring alkyl or aryl groups, other than methyl groups (2.5) transferring alkyl or aryl groups, other than methyl groups (2.5.1)
- C12Y205/01091—All-trans-decaprenyl-diphosphate synthase (2.5.1.91)
Definitions
- the present invention relates to a method for producing carotenoids using a mutant of a carotenoid-producing bacterium.
- Carotenoids are useful natural pigments used as feed additives, food additives, pharmaceuticals and the like.
- Carotenoids include astaxanthin, canthaxanthin, zeaxanthin, ⁇ -cryptoxanthin, lycopene, ⁇ -carotene, adonilvin, adonixanthin, echinone, asteroidenone, 3-hydroxyechinenone, and the like.
- astaxanthin is useful as a feed additive for body color improving agents such as salmon, trout and red sea bream that are farmed fish, and egg yolk improving agents for poultry.
- Natural astaxanthin has high industrial value as a safe food additive and health food material. Similar to astaxanthin, adonixanthin and adonilvin are expected to be used as feed additives, food additives, pharmaceuticals and the like.
- ⁇ -carotene is used as a feed additive, food additive, medicine, etc.
- canthaxanthin is used as a feed additive
- zeaxanthin is used as a food additive, feed additive, etc. ing.
- lycopene, echinenone, ⁇ -cryptoxanthin, 3-hydroxyechinenone, asteroidenone and the like are expected to be used as feed additives, food materials, and the like.
- Known methods for producing these carotenoids include chemical synthesis methods, extraction methods from natural products, and production methods by culturing microorganisms.
- astaxanthin As a chemical synthesis method of astaxanthin, a method by conversion of ⁇ -carotene (Pure Appl. Chem., 57, 741, 1985 (Non-patent Document 1)) and a method of synthesizing from C15 phosphonium salt (Helv. Chim. Acta, 64). 2436, 1981 (Non-Patent Document 2)).
- As an extraction method from natural products astaxanthin is present in fish such as salmon and red sea bream and in crustaceans such as shrimp, crab and krill, and can be extracted and collected from these.
- a culture method using the green alga Haematococcus pluvialis Japanese Patent Application Laid-Open No. 2007-97584 (Patent Document 1)
- a fermentation method using red yeast Phaffia rhodozyma Japanese Patent Application Laid-Open No. 11-69969 (Patent Document 2)).
- Fermentation using bacteria belonging to the genus Paracoccus hereinafter also referred to as “paracoccus genus bacteria”
- fermentation using bacteria belonging to the genus Brevundimonas Japanese Patent Laid-Open No.
- Patent Document 6 2001-512030
- Patent Document 7 Paracoccus haeundaensis BC74171 strain (International Journal of Symboletystol 4 System 200 Biostimol 4). 1699-1702 (Non-Patent Document 4)), Paracoccus genus bacteria N-81106 strain (Japanese Patent Laid-Open No. 2007-244205 (Patent Document 7)), Paracoccus zeaxanthinifaciens (International Journal of Systemic and Evolu iffy Microbiology (2003), 53,231-238 (Non-Patent Document 5)), and Paracoccus sp. PC-1 strain (WO2005 / 118812 pamphlet (Patent Document 8)) and the like.
- carotenoids produced by chemical synthesis methods give consumers an unfavorable impression even though they are safe.
- Carotenoids extracted from natural products are much more expensive to manufacture than chemical synthesis methods.
- the production by microorganisms the production of green algae and yeast by cultivation is low in productivity, and these microorganisms have a strong cell wall, which makes it difficult to extract carotenoids from the culture.
- the production of carotenoids by bacteria belonging to the genus Paracoccus has advantages such as a high growth rate of the cells, high productivity of carotenoids, and easy extraction of carotenoids from cultures. Culture methods and production methods have been reported.
- JP 2007-143492 A is a method of adding an iron salt during culture
- WO 2010/044469 pamphlet is a method of adding an amino acid to a culture medium
- JP 2011-188895 A JP 2013-143492 A
- Patent Document 11 discloses a method of adding biotin to the medium
- Patent Document 12 discloses a method of adding a calcium compound to the medium so as to be 3.6 mM or more.
- the details of which gene contributes to the increase in production efficiency in the bacteria producing carotenoids are unknown.
- Patent Document 1 JP 2007-97584
- Patent Document 2 JP 11-69969
- Patent Document 3 JP 2006-340676
- Patent Document 4 JP 2008-259449
- Patent Document 5 Japanese Patent Laid-Open No. 7-7979
- Patent Document 6 Japanese Patent Publication No. 2001-512030
- Patent Document 7 Japanese Patent Application Laid-Open No.
- Patent Document 8 Pamphlet of WO 2005/118812
- Patent Document 9 JP 2007-143492 A
- Patent Document 10 WO 2010/044469 Pamphlet
- Patent Document 11 JP 2011-188895 A
- Patent Document 12 JP 2012-139164 A [Non-patent Document]
- Non-Patent Document 1 Pure Appl. Chem. , 57, 741, 1985
- Non-Patent Document 2 Helv. Chim. Acta, 64, 2436, 1981
- Non Patent Literature 3 International Journal of Systemic Bacteriology (1999), 49, 277-282.
- Non-Patent Document 4 International Journal of Systemic and Evolutionary Microbiology (2004), 54, 1699-1702.
- Non-Patent Document 5 International Journal of Systemic and Evolutionary Microbiology (2003), 53, 231-238.
- An object of the present invention is to provide a mutant carotenoid-producing bacterium and a method for producing carotenoid using the bacterium.
- a mutant carotenoid-producing bacterium comprising any of the following genes (a) to (c): (A) a protein comprising a mutant amino acid sequence in which at least the 225th amino acid residue is substituted with another amino acid residue in the amino acid sequence of 1-deoxy-D-xylulose 5-phosphate synthase in a carotenoid-producing bacterium (B) Gene encoding a protein comprising a mutant amino acid sequence in which at least the 305th amino acid residue is substituted with another amino acid residue in the amino acid sequence of decaprenyl diphosphate synthase in a carotenoid producing bacterium (C) The bacterium according to (1), wherein the amino acid sequences of both genes (2) 1-deoxy-D-xylulose 5-phosphate synthase of (a) and (b) above are those shown in SEQ ID NO: 2.
- the bacterium according to (6) which has acquired the production ability of at least 5 times the amount of carotenoid production of a carotenoid production bacterium that does not have a gene encoding a protein containing a mutant amino acid sequence.
- the bacterium according to (8), wherein the bacterium belonging to the genus Paracoccus is E-396 strain.
- a method for producing a carotenoid comprising culturing the bacterium according to any one of (1) to (10) and collecting carotenoid from the obtained culture. (12) The method according to (11), wherein the amount of carotenoid produced is at least five times the amount of carotenoid produced by a carotenoid producing bacterium that does not have a gene encoding a protein containing a mutant amino acid sequence. . (13) The method according to (11) or (12), wherein the carotenoid is astaxanthin.
- a method for screening a carotenoid-producing bacterium comprising subjecting the carotenoid-producing bacterium to a mutation treatment and selecting a bacterium having any of the following characteristics (a) to (c) from the mutated bacterium: .
- (b) The activity of decaprenyl diphosphate synthase is higher than that in bacteria before mutation treatment
- C Features of both (a) and (b) above (15) Culturing bacteria selected by the method according to (14), and collecting carotenoids from the resulting culture A method for producing carotenoids.
- a gene encoding a protein comprising a mutant amino acid sequence in which at least the 225th amino acid residue is substituted with another amino acid residue in the amino acid sequence of 1-deoxy-D-xylulose 5-phosphate synthase is obtained from the following DNA (a) or (b): (A) DNA containing the base sequence represented by SEQ ID NO: 5 (B) a DNA that hybridizes with a DNA comprising a base sequence complementary to the DNA of (a) above under stringent conditions and encodes a protein having 1-deoxy-D-xylulose 5-phosphate synthase activity (18) A gene encoding a protein comprising a mutant amino acid sequence in which at least the 305th amino acid residue is substituted with another amino acid residue in the amino acid sequence of decaprenyl diphosphate synthase.
- a gene comprising the following DNA (a) or (b): (A) DNA comprising the base sequence represented by SEQ ID NO: 7 (B) a DNA encoding a protein that hybridizes with a DNA comprising a base sequence complementary to the DNA of (a) above under stringent conditions and has a reduced decaprenyl diphosphate synthase activity (20)
- a recombinant vector comprising any of the following genes (a) to (c): (A) the gene according to (16) or (17) (b) the gene according to (18) or (19) (c) the gene according to (a) and (b) above (21) according to (20) A transformant containing a recombinant vector.
- (22) A method for producing a carotenoid, comprising culturing the transformant according to (21) and collecting carotenoid from the obtained culture.
- the present invention provides a high-producing carotenoid bacterium. By using the bacterium of the present invention, carotenoids can be produced efficiently.
- the active site is indicated by a green box, and mutations whose activity was improved in DXS_ECOLI and DXS_VITVI are indicated by ⁇ .
- Rhodobacter capsulatus-derived decaprenyl diphosphate synthase (PDB ID: 3MZV) is shown. It is a figure which shows alignment with the enzyme C and a template structure (3MZV). Inferred active sites are shown in green. It is a figure which shows the template structure and the model structure of the composite_body
- FPP and IPP are indicated by Space-filling. It is a figure which shows the template structure and the model structure of the composite_body
- the structure of the matching amino acid residue is shown in green. The substrate binding region and the surrounding structure all match.
- a chain (light red), B chain (light blue). FPP and IPP are indicated by Space-filling.
- Ala305 is indicated by green, and Val305 is indicated by magenta. It is a figure which shows the comparison of the intramolecular energy of a wild type (blue) and variant A305V (red). It is a figure which shows the structure comparison of a wild type and variant A305V. Due to the mutation of A305V, the structure of amino acid residues around Ala305 (green) and Val305 (magenta) changes (left). This structural change also affects adjacent ⁇ -helices (right). It is a figure which shows the influence of A305V in the enzyme C. It is a figure which shows the influence of A305V in the enzyme C. It is a figure which shows the influence of A305V in the enzyme C. It is a figure which shows the effect of the enzyme C variant guessed by an astaxanthin synthetic pathway.
- the present invention relates to a bacterium that highly produces carotenoids, and is a bacterium containing any one of the following genes (a) and (b), or both of these genes.
- B Gene encoding a protein comprising a mutant amino acid sequence in which at least the 305th amino acid residue is substituted with another amino acid residue in the amino acid sequence of decaprenyl diphosphate synthase in a carotenoid producing bacterium
- the present inventor examined carotenoid-producing ability in the E-396 strain and the strain after the mutation treatment, and in these strains, an enzyme involved in the carotenoid synthesis pathway was selected. Mutations in the encoded gene were analyzed. As a result, a strain (referred to as “ASB-57 strain”) having a higher carotenoid production ability than the E-396 strain used as the parent strain was obtained. As a result of genome analysis of the ASB-57 strain, the amino acid sequence of 1-deoxy-D-xylulose 5-phosphate synthase (DXS) and the amino acid sequence of decaprenyl diphosphate synthase (DPS) are mutated.
- DXS 1-deoxy-D-xylulose 5-phosphate synthase
- DPS decaprenyl diphosphate synthase
- the carotenoid-producing bacterium of the present invention is obtained by performing mutation treatment on the parent strain and using as an indicator that a mutation has occurred in the 225th amino acid residue of DXS and / or the 305th amino acid residue of DPS. It is a bacterium that can produce carotenoids with high efficiency.
- the carotenoid producing bacterium of the present invention is referred to herein as “mutant carotenoid producing bacterium”.
- (1) Parent strain In the present invention, the bacterium used as a parent strain for obtaining a mutant carotenoid-producing bacterium is not limited as long as it is a bacterium that produces carotenoid.
- bacteria belonging to the genus Paracoccus, Brevundimonas, and Erythrobacter can be mentioned.
- a bacterium belonging to the genus Paracoccus, a bacterium belonging to the genus Brevundimonas, or a bacterium belonging to the genus Erythrobacter more preferably a bacterium belonging to the genus Paracoccus is used.
- the Paracoccus genus, the Erythrobacter genus, and the Brevundimonas genus are all classified as Proteobacteria gates and Alphaproteobacteria steels, and since there is a common bacterial taxonomy, bacteria belonging to these genera can be used in the present invention. is there.
- Paracoccus carotifaciens Among the bacteria belonging to the genus Paracoccus, Paracoccus carotifaciens, Paracoccus marcusii, Paracoccus haundaensis, and Paracoccus zeaxanthinifaciens are preferably used, and Paracoccus carotinis is particularly preferable.
- specific strains of bacteria belonging to the genus Paracoccus include Paracoccus carotinifaciens E-396 strain (FERM BP-4283) and Paracoccus genus bacteria A-581-1 strain (FERM BP-4671), and these mutant strains Are also preferably used in the present invention.
- Examples of the carotenoid-producing bacteria belonging to the genus Erythrobacter include the Erythrobacter JPCC M species (Japanese Patent Laid-Open No. 2008-259542), the Erythrobacter JPCC O species (Japanese Patent Laid-Open No. 2008-259449), and the like.
- Examples of the carotenoid-producing bacteria belonging to the genus Brevundimonas include Brevundimonas SD212 strain (JP 2009-27995), Brevundimonas FERM P-20515, 20516 strain (JP 2006-340676), Brevundimonas vesicularis (Gene, Vol. -108,1 Sep 2006).
- a bacterium having a DNA sequence corresponding to 16S ribosomal RNA having high homology with the base sequence of E-396 strain described in SEQ ID NO: 9 is preferably used.
- the homology of the base sequence mentioned here is preferably 95% or more, more preferably 96% or more, still more preferably 97% or more, particularly preferably 98% or more, and most preferably 99% or more.
- the base sequence of DNA corresponding to 16S ribosomal RNA means a base sequence in which U (uracil) in the base sequence of 16S ribosomal RNA is replaced with T (thymine).
- the classification method of microorganisms based on the homology of the base sequence of 16S ribosomal RNA has become mainstream.
- the conventional classification method of microorganisms is based on bacteriological properties such as conventional motility, auxotrophy, and sugar assimilation. Sometimes classified.
- the classification method based on the homology significantly improves the classification reliability compared to the conventional classification method.
- Paracoccus carotinifaciens E-396 16S ribosomal RNA base sequence and other carotenoid producing bacteria Paracoccus marcusii DSM 11574, Paracoccus sp. Paracoccus zeaxanthinifaciens ATCC 21588 strain and Paracoccus sp.
- the homology with the base sequence of 16S ribosomal RNA of the PC-1 strain is 99.7%, 99.7%, 99.6%, 99.4%, 95.7%, and 95.4%, respectively. These show that they are taxonomically closely related strains. Therefore, it can be said that these strains form one group as bacteria producing carotenoids. For this reason, these strains are preferably used in the present invention and can efficiently produce carotenoids.
- known mutant strains with improved carotenoid productivity can also be used.
- Examples of the known mutants include strains with high astaxanthin-producing ability (JP 2001-95500), strains that selectively produce a large amount of canthaxanthin (JP 2003-304875), and zeaxanthin and ⁇ -cryptoxanthin selectively. Strains that produce a large amount (JP-A-2005-87097) and strains that selectively produce lycopene (JP-A-2005-87100).
- the E-396 strain mentioned as an example of a carotenoid-producing bacterium used as a parent strain in the present invention is internationally deposited at the National Institute of Technology and Evaluation (NITE) Patent Organism Depositary (NITE-IPOD) as follows. . International Depositary Authority: National Institute of Technology and Evaluation (NITE) Patent Organism Depositary Center 2-5-8 Kazusa Kamashika, Kisarazu City, Chiba Prefecture 292-0818 Display for identification: E-396 Accession Number: FERM BP-4283 Original deposit date: April 27, 1993
- A-581-1 strain mentioned as another example of the carotenoid-producing bacterium used as the parent strain in the present invention has been deposited internationally with the above institution as follows. Display for identification: A-581-1 Accession Number: FERM BP-4671 Original deposit date: May 20, 1994
- the parent strain is subjected to mutation treatment, and a mutation occurs in the 225th amino acid residue of DXS and / or the 305th amino acid residue of DPS. Can be obtained as an index.
- the method for mutation treatment is not particularly limited as long as it induces mutation.
- chemical methods using mutants such as N-methyl-N′-nitro-N-nitrosoguanidine (NTG) and ethyl methanesulfonate (EMS), physical methods such as ultraviolet irradiation and X-ray irradiation, genetic recombination and Biological methods such as transposon can be used.
- the bacterium to be mutated is not particularly limited, but is preferably a carotenoid-producing bacterium.
- a point mutation can be introduced into a gene (DNA) encoding the protein.
- a mutation introduction kit using a site-directed mutagenesis method such as Kunkel method or Gapped duplex method, for example, QuikChange TM Site-Directed Mutagenesis Kit (Stratagene), GeneTailor TM Site-Directed Detected Mute.
- the screening method of the mutant strain is not particularly limited, but gene analysis is performed using a known genome analysis tool PacBio RS II (manufactured by Pacific Biosciences), MiSeq (manufactured by Illumina), etc., and the 225th amino acid residue of DXS And / or the presence or absence of a mutation in the base sequence corresponding to the 305th amino acid residue of DPS may be confirmed. Furthermore, in parallel with the above genomic analysis, for example, in addition to a method of selecting a target mutant strain by the color of a colony on an agar medium, the mutant strain is cultured in a test tube, a flask, a fermenter, etc.
- Carotenoid production can be selected as an index by carotenoid pigment analysis using chromatography, thin layer chromatography, or the like.
- the mutation and screening steps may be performed once, or, for example, a mutant strain is obtained by mutation treatment and screening, and a mutant strain with improved productivity is obtained by further mutation treatment and screening. Mutation and screening steps may be repeated twice or more.
- the 225th amino acid residue of DXS is mutated to another amino acid and / or the 305th amino acid residue of DPS is changed to another amino acid residue. It has a gene encoding a mutated amino acid sequence.
- a mutation from the 225th amino acid residue of DXS to another amino acid contributes to an increase in the enzyme activity of DXS.
- combination from pyruvic acid to 1-deoxy-D xylulose-5-phosphate is accelerated
- IPP isopentenyl diphosphate
- Mutation from the 305th amino acid residue of DPS to another amino acid residue contributes to a decrease in the enzyme activity of DPS.
- This mutation inhibits the synthesis of farnesyl diphosphate (FPP) to decaprenyl diphosphate (DPP). Since IPP is used for the synthesis from FPP to DPP, the amount of IPP used for DPP synthesis decreases due to the mutation, and the IPP is used as a substrate for the astaxanthin synthesis.
- FPP farnesyl diphosphate
- DPP decaprenyl diphosphate
- the amino acid sequence in which the 225th amino acid residue of DXS is mutated to another amino acid and / or the 305th amino acid residue of DPS is mutated to another amino acid residue.
- the mutant carotenoid-producing bacterium of the present invention can contain the following gene (a), the following gene (b), or both the following genes (a) and (b).
- Examples of such a mutant amino acid sequence include those shown in SEQ ID NO: 6, and examples of the gene include those shown in SEQ ID NO: 5.
- the amino acid sequence shown in SEQ ID NO: 2 is preferably an amino acid sequence in which glycine as the 225th amino acid residue is substituted with aspartic acid.
- the 225th amino acid residue is substituted with another amino acid residue, and one or more (for example, 1) other than the 225th amino acid residue
- a protein having a mutant amino acid sequence in which several amino acid residues are deleted, substituted or added, and having a DXS activity iii) a gene comprising a DNA comprising the base sequence represented by SEQ ID NO: 5
- a gene encoding a protein comprising a mutant amino acid sequence in which at least the 305th amino acid residue is substituted with another amino acid residue is substituted with another amino acid residue.
- DPS amino acid sequence of DPS
- Examples of such a mutant amino acid sequence include those shown in SEQ ID NO: 8, and examples of the gene include those shown in SEQ ID NO: 7.
- the amino acid sequence shown in SEQ ID NO: 4 is preferably an amino acid sequence in which alanine, which is the 305th amino acid residue, is substituted with valine.
- the amino acid sequence of DPS for example, SEQ ID NO: 4
- the 305th amino acid residue is substituted with another amino acid residue, and one or more (for example, 1) other than the 305th amino acid residue
- a protein comprising a mutant amino acid sequence in which several amino acid residues have been deleted, substituted or added, and having a reduced DPS activity
- iv a gene comprising a DNA that hybridizes under stringent conditions with a DNA comprising a base sequence complementary to the DNA comprising the base sequence represented by SEQ ID NO: 7 and which encodes a protein having a reduced DPS activity.
- the base sequence represented by No. 7 is the DNA (SEQ ID No. 3) encoding the amino acid sequence of DPS in carotenoid-producing bacteria.
- the 305th amino acid residue encodes a protein containing an amino acid sequence substituted with another amino acid residue.
- hybridization can be performed according to a known method (for example, Sambrook J. et al., Molecular Cloning, A Laboratory Manual (4th edition) (Cold Spring Harbor Laboratory Press (2012)).
- the conditions refer to conditions in which a so-called specific hybrid is formed and a non-specific hybrid is not formed.
- the sodium concentration is 10 mM to 300 mM, preferably 20 mM to 100 mM
- the temperature is 25 ° C. to 70 ° C., preferably Means conditions at 42 ° C. to 55 ° C.
- mutant carotenoid producing bacteria examples include ASB-57 strain, ASK-8 strain, and ASH-66 strain.
- the ASB-57 strain encodes a protein containing an amino acid sequence in which glycine, which is the 225th amino acid residue of DXS, is mutated to aspartic acid, and alanine, which is the 305th amino acid residue of DPS, is mutated to valine.
- glycine which is the 225th amino acid residue of DXS
- alanine which is the 305th amino acid residue of DPS
- a gene recombinant type is obtained by introducing a gene encoding the mutant DXS and / or a gene encoding the mutant DPS into a host for transformation.
- the mutant carotenoid-producing bacterium can be obtained. Any known method may be adopted for the recombinant vector obtained by introducing the mutant DXS gene and / or the mutant DPS gene into the vector, and the transformant obtained by introducing the recombinant vector into the host.
- Sambrook J. et al. et al. Molecular Cloning, A Laboratory Manual (4th edition) (Cold Spring Harbor Laboratory Press (2012)).
- a DNA encoding the enzyme is designed and synthesized.
- the design and synthesis of DNA can be performed by the PCR method using primers designed to synthesize a desired DNA region using, for example, a vector containing a full-length gene as a template.
- a recombinant vector for protein expression is obtained by ligating the above DNA to an appropriate vector, and a transformant is obtained by introducing this recombinant vector into a host so that the target gene can be expressed (Sambrook). J. et al., Molecular Cloning, A Laboratory Manual (4th edition) (Cold Spring Harbor Laboratory Press (2012)).
- a phage or a plasmid capable of autonomously growing in a host microorganism is used.
- animal virus and insect virus vectors can also be used.
- the purified DNA may be cleaved with an appropriate restriction enzyme, inserted into a restriction enzyme site or the like of an appropriate vector DNA, and ligated to the vector.
- the host used for transformation is not particularly limited as long as it can express the target gene. Examples include bacteria (Bacillus subtilis, Paracoccus bacteria, etc.), yeast, animal cells (COS cells, CHO cells, etc.), plant cells, insect cells or insects. Methods for introducing a recombinant vector into a host are known. Moreover, the method for introducing a mutation into a gene is the same as described above.
- a high concentration of carotenoid can be stably produced by culturing the above carotenoid-producing bacterium or transformant in a predetermined medium.
- Carotenoid produced is not particularly limited, for example, astaxanthin, canthaxanthin, zeaxanthin, ⁇ -cryptoxanthin, lycopene, ⁇ -carotene, adonilvin, adonixanthin, echinenone, asteroidenone or 3-hydroxyechinenone
- it is astaxanthin, canthaxanthin, zeaxanthin or ⁇ -cryptoxanthin, more preferably astaxanthin or zeaxanthin.
- One kind of carotenoid produced from the present invention may be used, or a plurality of kinds may be combined.
- the carotenoid production medium used for the culture of the present invention can be added with any components as long as carotenoid-producing bacteria or transformants grow and produce carotenoids. Any medium containing such additives may be used, but a medium containing a carbon source, a nitrogen source, inorganic salts and, if necessary, vitamins is preferably used.
- Examples of the carbon source include sugars such as glucose, sucrose, lactose, fructose, trehalose, mannose, mannitol and maltose, and organic acids such as acetic acid, fumaric acid, citric acid, propionic acid, malic acid, malonic acid and pyruvic acid. , Alcohols such as ethanol, propanol, butanol, pentanol, hexanol, isobutanol and glycenol, and fats and oils such as soybean oil, nuka oil, olive oil, corn oil, sesame oil and linseed oil, among which glucose or Sucrose is used. Among these carbon sources, one type or two or more types can be used.
- the amount added to the medium before culture varies depending on the type of carbon source and may be adjusted as appropriate, but is usually 1 to 100 g, preferably 2 to 50 g, per liter of the medium.
- the carbon source is preferably added not only to the starting medium, but also to be added sequentially or continuously during the culture.
- the nitrogen source one or more of inorganic salts such as ammonium salts such as ammonium nitrate, ammonium sulfate, ammonium chloride and ammonium phosphate, nitrates such as potassium nitrate, ammonia and urea are used.
- the addition amount varies depending on the type of nitrogen source and may be adjusted as appropriate, but is usually 0.1 to 20 g, preferably 0.2 to 10 g, with respect to 1 L of the medium.
- organic nitrogen sources include corn steep liquor (including filtered products), pharma media, soybean meal, soybean flour, peanut meal, soy peptone, distillers solver, dry yeast, yeast extract, casamino acid, and glutamic acid.
- aspartic acid are used.
- the addition concentration varies depending on the type of nitrogen source and may be adjusted as appropriate, but it is usually 0 to 80 g / L, preferably 1 to 30 g / L.
- the inorganic nitrogen source and the organic nitrogen source are usually added to the starting medium, but it is also preferable to add them sequentially or continuously.
- inorganic salts include phosphates such as potassium dihydrogen phosphate, dipotassium hydrogen phosphate and disodium hydrogen phosphate, magnesium salts such as magnesium sulfate and magnesium chloride, iron salts such as iron sulfate and iron chloride, Calcium salts such as calcium chloride and calcium carbonate, sodium salts such as sodium carbonate and sodium chloride, manganese salts such as manganese sulfate, copper salts such as copper sulfate, zinc salts such as zinc sulfate, molybdenum salts such as sodium molybdate, sulfuric acid 1 or 2 of nickel salts such as nickel, selenium salts such as sodium selenate, tungsten salts such as sodium tungstate, aluminum salts such as aluminum chloride, chromium salts such as chromium chloride, boric acid and potassium
- the amount added varies depending on the type of inorganic salt and may be adjusted as appropriate, but is usually 0.0001 to 15 g with respect to 1 L of the medium.
- the preferred concentration is 0.1 to 15 mg / L.
- Inorganic salts are usually added to the starting medium, but may be additionally supplied sequentially or continuously.
- vitamins for example, cyanocobalamin, riboflavin, pantothenic acid, pyridoxine, thiamine, ascorbic acid, folic acid, niacin, p-aminobenzoic acid, biotin, inositol, choline and the like can be used.
- the addition ratio varies depending on the type of vitamins and may be adjusted as appropriate, but is usually 0.001 to 1000 mg, preferably 0.01 to 100 mg per 1 L of the medium. Vitamins are usually added to the starting medium, but may be added sequentially or continuously.
- an antifoaming agent is preferably used to suppress foaming of the culture solution.
- Any type of antifoaming agent may be used as long as it suppresses the generation of bubbles or eliminates the generated bubbles and has a small inhibitory effect on produced bacteria.
- examples thereof include alcohol-based antifoaming agents, polyether-based antifoaming agents, ester-based antifoaming agents, fatty acid-based antifoaming agents, silicon-based antifoaming agents, and sulfonic acid-based antifoaming agents.
- the amount added varies depending on the type of antifoaming agent and may be adjusted as appropriate, but is usually 0.01 to 10 g per 1 L of the medium.
- Defoamer is usually added to the starting medium before sterilization. Further, it may be added continuously or intermittently during the culture.
- a method of adding an antifoaming agent during the culture a method of automatically adding bubbles by sensing with a sensor, a method of adding at a fixed time with a program timer, a carbon source for feed, a nitrogen source in conjunction with the growth rate Or the method of mixing and adding with a pH adjuster etc. can be illustrated.
- the antifoaming agent added to the initial culture medium and the antifoaming agent added to the culture medium during the culture may be the same, but different types may be used according to the action.
- the initial pH of the medium is adjusted to 2 to 12, preferably 6 to 9, and more preferably 6.5 to 8.0. It is preferable to maintain the pH in the above range during the culture.
- the pH adjuster include an aqueous sodium hydroxide solution, an aqueous potassium hydroxide solution, an aqueous sodium carbonate solution, ammonia water, ammonia gas, an aqueous sulfuric acid solution, or a mixture thereof.
- the medium is sterilized and then used for bacterial culture. Sterilization can be appropriately performed by those skilled in the art.
- the medium in a suitable container may be heat sterilized with an autoclave. Or what is necessary is just to sterilize by filtration with a sterilization filter.
- the mutant carotenoid-producing bacterium or transformant of the present invention is inoculated into the medium prepared as described above and cultured under predetermined conditions. Inoculation is performed by appropriately increasing the number of strains by seed culture using a test tube, flask, or fermenter, and adding the obtained culture solution to the carotenoid production medium.
- the medium used for seed culture is not particularly limited as long as the carotenoid-producing bacteria grow well.
- Culture is performed in a suitable culture vessel.
- the culture vessel can be appropriately selected depending on the culture volume, and examples thereof include a test tube, a flask, and a fermenter.
- the culture temperature is 15 to 40 ° C., preferably 20 to 35 ° C., more preferably 25 to 32 ° C., usually 1 to 18 days, preferably 2 to 12 days, more preferably 3 to 8 days, aerobic. Cultivate under conditions. Examples of aerobic conditions include shaking culture or aeration and agitation culture, and it is preferable to control the dissolved oxygen concentration within a certain range.
- the dissolved oxygen concentration can be controlled, for example, by changing the number of rotations of stirring, the amount of ventilation, the internal pressure, and the like.
- the dissolved oxygen concentration is preferably controlled to 0.3 to 10 ppm, more preferably 0.5 to 7 ppm, and still more preferably 1 to 5 ppm.
- the number of cells or the number of transformants of the carotenoid-producing bacterium after culturing the mutant carotenoid-producing bacterium or transformant of the present invention can be measured by OD.
- quantification of carotenoids in a culture obtained by culturing carotenoid-producing bacteria or transformants, or carotenoids collected from the culture can be performed by high performance liquid chromatography. After carotenoid-producing bacteria or transformants are cultured as described above, carotenoids can be collected from the resulting culture. Examples of the culture include a culture solution, a culture supernatant, a cell concentrate, a wet cell, a dry cell, and a cell lysate.
- the culture supernatant may be prepared by removing the cells from the culture solution by subjecting the culture solution to centrifugation or filtration.
- the bacterial cell concentrate can be obtained by centrifugation or membrane filtration concentration of the culture solution.
- the wet cells can be obtained by centrifuging or filtering the culture solution.
- a dry microbial cell can be obtained by drying a wet microbial cell or a microbial cell concentrate by a general drying method.
- the carotenoid-containing dry cells thus obtained can be used as they are as feed additives.
- the yield during fermentation culture is at least 150 mg / L, and includes, for example, 150 mg / L, 400 mg / L, 2000 mg / L, and 4000 mg / L carotenoids.
- the amount of carotenoid contained in the culture solution varies depending on the microbial cells used, it contains, for example, 400 mg / L to 4000 mg / L, more preferably 500 mg / L to 3500 mg / L carotenoid.
- the bacterium of the present invention produces at least 5 times, preferably 10 times or more the amount of carotenoid produced by a carotenoid producing bacterium that does not have a gene encoding a protein containing a DXS and / or DPS mutant amino acid sequence. Have the ability.
- the method for collecting carotenoids from the culture is not particularly limited, and any method in which carotenoids are stably and efficiently recovered may be used. Those skilled in the art can perform these methods by appropriately selecting from known extraction and purification techniques.
- the culture can also be used as a carotenoid-containing composition. Prior to extraction, the culture is chemically treated with alkaline reagents or surfactants, biochemically treated with lytic enzymes, lipolytic enzymes, proteolytic enzymes, etc., or physically such as ultrasound or grinding. Among the processes, one or more processes may be performed.
- the solvent used for extraction and washing is not particularly limited, but lower alcohols such as methanol, ethanol, isopropanol, acetone, tetrahydrofuran, methyl ethyl ketone, methyl isobutyl ketone, dichloromethane, chloroform, dimethylformamide And dimethyl sulfoxide.
- the treatment may be performed in an inert gas atmosphere such as nitrogen gas.
- you may select the antioxidant currently used by the pharmaceutical and foodstuff, and may add to an extraction solvent. Alternatively, these processes may be combined. Further, in order to prevent the decomposition of carotenoids by light as much as possible, it may be performed under conditions where no light is applied.
- the extract thus obtained can be used as it is as a carotenoid, and can also be used after further purification.
- a method for separating bacteria remaining in the extract after the extraction operation is not particularly limited, and membrane filtration, centrifugation, decantation, and the like are used.
- As a method for obtaining a carotenoid precipitate from the extract generally heating and / or vacuum concentration and crystallization are exemplified.
- the carotenoid pigment may be separated without being concentrated by precipitation of the carotenoid pigment at a low temperature or by precipitation with an acid / alkali agent or various salts. In industrial use, it is desirable to crystallize.
- the obtained carotenoid precipitate may be suspended and stirred using a small amount of a solvent such as a lower alcohol as necessary for washing.
- a solvent such as a lower alcohol
- the method of washing is not particularly limited, for example, a method of filtering after suspension and stirring, a method of passing liquid from above the precipitate, and the like are practically preferable methods.
- the cultures, extracts or purified products obtained as described above can be used alone as carotenoids, or these can be mixed and used in an arbitrary ratio.
- Enzyme A was presumed to be 1-deoxy-D-xylulose 5-phosphate synthase (DXS).
- DXS 1-deoxy-D-xylulose 5-phosphate synthase
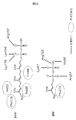
- the identified amino acid mutation G225D is in the disordered region located near the active site. From the three-dimensional structure model, it is inferred that the mutation G225D of enzyme A causes a structural change similar to the mutation of the known disordered region, which is known to improve the activity of DXS, to the enzyme G225D. It was predicted that the enzyme activity was improved. Considering that DXS is controlled by feedback inhibition of IPP, it is suggested that the mutant G225D enzyme loses feedback inhibition and increases the production amount of IPP. An increase in the supply amount of IPP, which is a raw material for astaxanthin, due to the mutation G225D of enzyme A is considered to be a factor that increases the production amount of astaxanthin.
- Enzyme C was assumed to be decaprenyl diphosphate synthase. From the three-dimensional structure model, it was inferred that the identified mutation A305V caused steric hindrance with surrounding amino acid residues and destabilized the three-dimensional structure of enzyme C.
- FPP and IPP which are substrates for enzyme C and raw materials for decaprenyl diphosphate, are also raw materials for astaxanthin synthesis. It is suggested that the amount of FPP and IPP consumed by the enzyme C is reduced by reducing the activity of the enzyme C due to destabilization. As a result, it is considered that the amount of FPP and IPP that can be used for astaxanthin synthesis increases and the production amount of astaxanthin increases.
- Mutation treatment of Paracoccus genus bacteria and genome analysis Mutation treatment method
- the parent strain uses UV, NTG (nitrosoguanidine), etc. as a mutation source, and screens several times using various selection pressures. Carried out. Screening was performed using astaxanthin yield as an index.
- Genome analysis method Genome analysis is performed by reading a genome sequence using a sequencer of PacBio RS II (manufactured by Pacific Biosciences) or MiSeq (Illumina), and then SMART Cell 8 Pac V3 (manufactured by Pacific Biosciences) or MiSeq ControlSwitter. Analysis software such as (MCS) v2.4.1.3, Real Time Analysis (RTA) v1.18.54, bc12fastq v 1.8.4 (Illumina) was used.
- results of genome analysis (identification of mutation sites)
- the identification of the mutation site is highly homologous between the amino acid sequence of the region considered to be a protein having the mutation point of these genome analysis results and the enzyme gene amino acid sequence listed in Kyoto Encyclopedia of Genes and Genomes (KEGG).
- KEGG Kyoto Encyclopedia of Genes and Genomes
- those contained in the genus Paracoccus are identified, and furthermore, by analyzing the three-dimensional structure of the enzyme from these information and searching for a template having a matching sequence, the final amino acid sequence of the protein having the mutation site is determined.
- the enzyme name was determined.
- test tube media for production were prepared in which 7.2 ml of the medium having the following composition was placed in a test tube with a cotton plug having an inner diameter of 18 mm.
- the raw material of the test tube medium for production was a lot that has been confirmed to have insufficient growth of bacterial cells.
- Three-dimensional structure data and method Three-dimensional structure models of enzymes A and C were constructed by homology modeling.
- the software Swiss-Pdb viewer and SWISS-MODEL were used [1,2].
- the mutant model was created with Swiss-Pdb viewer.
- a mutate command was used, for calculation of intramolecular energy, a compute energy command was used, and for energy minimization calculation, an energy minimization command was used.
- Production of a complex model with a substrate, detection of residues near the substrate, measurement of interatomic distances, and display of the three-dimensional structure were performed using software Waals (Altif Labs. Inc.).
- a three-dimensional model of the low molecular weight compound was prepared by Marvin Sketch (ChemAxon Ltd.).
- the coordinate data of the three-dimensional structure of the template structure was obtained from Protein Data Base (PDB) (http://www.rcsb.org/pdb/), which is a protein three-dimensional structure database.
- PDB Protein Data Base
- the template structure the data having the highest amino acid coincidence with enzyme A and enzyme C among the data registered in PDB was used.
- the three-dimensional structure data used as a template for homology modeling is shown in Table 1.
- the mutation G225D identified by genome analysis is in the disordered region near the active site, where multiple cases of DXS enzyme activity improving when mutation occurs have been reported. It was. From the analysis results of the mutant model, it is inferred that the mutation G225D of enzyme A causes the same structural change as the known mutation that improves the activity of DXS, and the enzyme activity of DXS is the same as that of the known mutation. Is expected to improve. As the activity of DXS increases, the amount of astaxanthin produced increases as the supply amount of IPP, the raw material for astaxanthin, increases.
- 3D structure model of enzyme A Construction of 3D structure model of enzyme A by homology modeling 3D structure model of enzyme A is determined by X-ray crystallographic analysis of 3D structure of complex with coenzyme TPP. Based on the three-dimensional structure (PDB ID: 2O1X) [3] of 1-deoxy-D-xyrose-5-phosphate synthase (DXS, template) derived from Deinococcus radiodurans (D. radiodurans) (Fig. 2), constructed by homology modeling did.
- PDB ID: 2O1X 1-deoxy-D-xyrose-5-phosphate synthase
- DXS 1-deoxy-D-xyrose-5-phosphate synthase
- FIG. 4 shows the template structure and the constructed model structure.
- Enzyme A like the template structure, forms a homodimer and has a TPP binding site and a substrate binding site in each subunit.
- the monomer of enzyme A is composed of three domains, domain I (1 to 319 residues), domain II (320 to 495 residues), and domain III (496 to 629 residues).
- FIG. 5 shows the model structure of enzyme A and the substrate complex.
- TPP Binding site of coenzyme TPP
- TPP is located between domain I and domain II, as in the template structure, and the pyrimidine ring of TPP is linked to domain II.
- the acid group is bound to domain I.
- TPP is composed of an aminopyrimidine ring, a thiazoline ring, and pyrophosphate (FIG. 6).
- the aminopyrimidine ring is bound so that it fits snugly inside the binding pocket of enzyme A.
- the side chains of Phe396 and Ile369 are linked by a hydrophobic interaction so that the aminopyrimidine ring is sandwiched from both sides.
- the cyclic phenyl group which is the side chain of Phe396 and the aminopyrimidine ring are presumed to be strongly bonded by ⁇ stacking.
- the main chain oxygen of Gly120 and the side chain oxygen of Glu371 form a hydrogen bond with the nitrogen atom at the 1-position of the aminopyrimidine ring.
- Mg is required for binding of the coenzyme TPP. It was speculated that Mg is coordinated between two phosphate groups of TPP, and the side chain of Asp151 and Asn180 and the main chain of Met182 are bonded to Mg. Regarding DXS, it is known that the sequence of GDGX25-30N is conserved as a TPP-binding motif [3]. The sequence of the amino acid sequence Gly150-Asp151-Gly152-Asn180 containing Asp151 and Asn180 in which Mg binding was estimated by enzyme A matches this motif.
- FIG. 7 shows inferred amino acid residues involved in binding with TPP.
- FIGS. 6, 10 and 11 respectively show the binding mode between enzyme A and coenzyme TPP, the binding mode between enzyme A and pyruvate, and the binding mode between enzyme A and glyceraldehyde triphosphate.
- Enzyme A is a template of D.I. Since it retains an active site that binds the coenzyme TTP, the substrate pyruvate and GAP, which has been clarified in radiodurans-derived DXS, it is presumed to have DXS enzyme activity. These amino acid residues are D.I. radiodurans-derived DXS, DXS such as E. coli and S. It is known that cerevisiae-derived TK also has high conservation [3].
- E. coli DXS has been shown to be inactivated when amino acid residues corresponding to Glu370, Arg399, and Arg479 of enzyme A are substituted with Ala [3].
- Reference [8] shows that the enzyme activity is almost inactivated in experiments in which amino acid residues corresponding to His48, Glu371, Asp428 of enzyme A of E. coli DXS were mutated. Also in enzyme A, these amino acid residues are presumed to be important for DXS activity.
- the enzyme G mutation G225D is present in the disordered region (196 to 238 residues) in which the three-dimensional structure could not be specified. In order to infer the influence of this mutation on the three-dimensional structure of enzyme A, the relationship between the position of the disordered region on the three-dimensional structure and the active site was examined.
- inventions relating to mutations of K284N and R306C in Muscat and K213N and K234C in Escherichia coli are known (Special Tables 2014-500710, US20130276166).
- the present invention relates to a method for increasing the production amount of terpene by improving the activity of DXS. In all four cases, the production amount of terpene is increased by mutation of one residue each.
- FIG. 12 shows the positions of the mutations in which the enzyme activity increased in DXS of enzyme A, E. coli and Muscat. Muscat K284N and R306C and Escherichia coli K213N and K234C are all present in the disordered region (blue).
- the mutation G225D of enzyme A is also present in the disordered region as well. From the amino acid sequence alignment, the active site (green) of DXS is conserved, and it is presumed that enzyme A also has the same reaction pattern as these DXS. An active site exists on the N-terminal side (agenta) of the disordered region.
- FIG. 13 shows the position of the disordered region (residues 196 to 238) where the enzyme G mutation G225D exists.
- This region is located near the binding site of coenzyme TPP, which is essential for DXS activity.
- the side chain of Asn180 and the main chain of Met182 bind Mg.
- the side chain of Ile184 is hydrophobically bound to TPP. In order for TPP and Mg essential for DXS activity to bind, it seems important that this loop has an appropriate structure.
- FIG. 14 shows the result of mapping the electrostatic potential to the surface shape of the produced three-dimensional structure model. Blue indicates a positive (positive charge) region, and red indicates a negative (negative charge) region.
- the mutant G225D enzyme has a weak positive charge and a strong negative charge.
- side chains of Arg227, Arg228, Lys230, and K234 having a positive charge are gathered to form a strong positively charged region. It is considered that the positive charge in the vicinity of Asp225 was weakened by replacing Gly225 having no charge present in this region with Asp having a negative charge.
- This result shows the same tendency as the change in the electrostatic potential of the Muscat mutation K284N shown in the literature [9], that is, the change in the electrostatic potential from the positive charge to the negative charge on the surface of the disordered region.
- the structural change of the disordered region by G225D of enzyme A is predicted to have the same effect as the Muscat mutation on the active site including the TPP binding site, and as a result, enzyme G mutant G225D is also a Muscat mutant K284N. It is suggested that the enzyme activity increases as well.
- enzyme A mutation G225D occurs in the disordered region of enzyme A, and this region exists in the vicinity of the TPP binding site essential for activity. Was confirmed.
- a plurality of mutations that improve the enzyme activity of DXS have been found so far in this region, and the mutation of G225D was predicted to cause the same structural change as a known mutation that improves the activity of DXS.
- IPP isopentenyl diphosphate
- the amount of IPP supplied is controlled by DXS, so simply increasing the enzyme activity Kcat / Km of DXS does not increase the amount of IPP supplied, and the terpene increases significantly. I can't do it.
- the amount of terpene synthesis is increased due to mutation of the disordered region, and it is predicted that the supply amount of IPP is increased due to this mutation, that is, feedback inhibition by DXP IPP is not effective. It is suggested.
- IPP In vitro, IPP has been shown to compete with TPP, bind to DXS and inhibit DXS [10].
- the mutation in the disordered region causes a structural change in the TPP binding region, which not only makes the structure more suitable for TPP binding, but also affects the binding of IPP, so that the inhibition of feedback by IPP may not be effective. It is suggested.
- the mutation G225D of enzyme A is presumed that the production amount of IPP is increased by causing a structural change in the TPP binding region and the feedback inhibition by IPP becoming ineffective. As a result, it is considered that the amount of astaxanthin synthesized is increased by increasing the supply amount of IPP as a raw material for astaxanthin synthesis (FIG. 15).
- Construction of a three-dimensional structure model of enzyme C and analysis of mutant enzyme C is a kind of polyprenyl diphosphate synthase based on amino acid sequence homology and three-dimensional structure comparison analysis, and farnesyl diphosphate (FPP) and seven Decaprenyl diphosphate synthase: Decaprenyl diphosphate synthase that synthesizes decaprenyl diphosphate from isopentenyl diphosphate (IPP).
- FPP farnesyl diphosphate
- IPP isopentenyl diphosphate
- Decaprenyl diphosphate synthase has the activity to condense FPP and IPP, and repeats condensation with IPP to synthesize decaprenyl diphosphate (DPP) from FPP and seven IPPs. It is an enzyme. The enzyme reaction of decaprenyl diphosphate synthase is shown below.
- 3D structure model of enzyme C (1) Construction of 3D structure model of enzyme C by homology modeling In order to investigate the effect of the active site and mutation of enzyme C, the amino acid matching degree is highest in the 3D structure of PDB as a template structure. (Amino acid coincidence is 76.2%), using the three-dimensional structure (PDB ID: 3MZV) of decaprenyl diphosphate synthase derived from Rhodobacter capsulatus (R. capsulatus) whose three-dimensional structure is determined by X-ray crystal structure analysis Then, a three-dimensional structure model was constructed by homology modeling (FIG. 16) [4]. Homology modeling was performed based on the three-dimensional structure alignment of enzyme C and the template structure (FIG. 17).
- Decaprenyl diphosphate synthase of comparative enzyme C based on its three-dimensional structure belongs to the family of polyprenyl diphosphate synthases (Pfam PF00348 Polyprenyl synthetase).
- FIG. 19 shows the substrate binding state of enzyme C.
- the polyprenyl diphosphate synthase condenses IPP from FPP in a head-to-tail direction (herein, the phosphate group side is called head and the isoprenyl group side is called tail according to the literature [4]).
- Various polyprenyl diphosphates are synthesized.
- the decaprenyl diphosphate synthase continues the condensation reaction of FPP and IPPG, so that the prenyl chain is extended toward the back of the substrate binding site (indicated by an arrow on the right of FIG. 19), and C50 decaprenyl diphosphate is Synthesize.
- Enzyme C and template R.I. capsulatus-derived decaprenyl diphosphate synthase has a high amino acid identity of 76.3% and the active site is also conserved (FIG. 17).
- the RMSD of the three-dimensional structure by the superposition of C ⁇ is 0.063 mm, the two enzymes are very similar, and when the amino acid residue matches are displayed in different colors, the regions with different types of amino acid residues are molecules
- the region that binds to the active site and the substrate is limited to the surface, and is composed of all matching amino acid residues (FIG. 20).
- FIG. 22 shows the binding site between FPP and IPP in the A chain (light red).
- FPP is bound to the tunnel-like region of the A chain, and FPP and IPP are bound in a head-to-tail form in which the phosphate group of FPP is directed to the isopentenyl group of IPP.
- the catalytic reaction takes place between the phosphate group of FPP and the isopentenyl group of IPP in the presence of Mg.
- This catalytic reaction requires Mg.
- Mg binding site two known polyprenyl diphosphate synthases have two DDXDD motifs [11].
- the coordinates of Mg are not determined, but also in enzyme C, Asp93, Asp94, Asp97 and Asp220, Asp221, Asp224 corresponding to the DDXXD motif are present in the vicinity of the phosphate group as well as the known Mg binding site. And it is speculated that these amino acid residues bind Mg (FIG. 23 bottom).
- the inferred active sites are shown in Tables 6 to 8 in the attached material. Moreover, the figure about the coupling
- Each amino acid residue whose binding is inferred is R.I. It is consistent with decaprenyl diphosphate synthase from capsulatus (FIG. 17).
- the FPP binding site, IPP binding site, and Mg binding site of enzyme C are the E. coli used for preparing the substrate complex model. Since the binding site of the E. coli-derived octaprenyl diphosphate synthase is retained, it is considered that enzyme C takes the same reaction pattern as octaprenyl diphosphate synthase.
- Arg102, Lys179, Lys244 that bind the phosphate group of FPP, and Asp93, Asp94, Asp97, Asp220, Asp221, and Asp224 that bind Mg are important residues that are directly involved in the catalytic activity of transferring the phosphate group. It appears to be.
- amino acid residues corresponding to the enzyme C phosphate binding sites Arg102 and Arg103 and the Mg binding sites Asp94, Asp97, Asp220, Asp221 and Asp224 It has been reported that these amino acid residues are highly conserved among polyprenyl diphosphate synthases [14, 15].
- the side chains of Phe and Gln are found to be important for substrate binding by producing a mutant of the amino acid residue corresponding to Phe216, Gln217 of enzyme C [16]. Also in the complex model of enzyme C, Phe216 and Gln217 are in the active site region and are predicted to be important for activity.
- FIG. 26 shows a three-dimensional structure model of the wild type and mutant A305V enzyme. Wild-type Ala305 is present in the ⁇ -helix, and the side chain is packed with the amino acid residue of the adjacent ⁇ -helix by hydrophobic interaction.
- FIG. 27 shows the structure of amino acid residues adjacent to Ala305.
- the carbon atom of the methyl group which is the side chain of Ala305 is in contact with the peripheral amino acid residues, Tyr208, Ala211, His301, and Ala302, and the distance between the carbon atoms is less than 4.0 mm.
- the substitution of Ala 305 with Val increases the number of side chain methyl groups by two.
- the interatomic distances between the methyl group carbon of Val and the carbons of Tyr208 and Ala211 were 2.42 mm and 2.47 mm.
- the lower limit of the contact distance due to the non-covalent bond between carbons is 2.9 mm. Since the distance between carbon atoms measured with Val305 is less than this value, it is considered that these carbon atoms collide and steric hindrance with surrounding amino acid residues occurs. In the display by Space-filling, the distance between atoms is shorter than the van der Waals radius, and it can be confirmed that the atoms collide. Based on the above, it is predicted that the mutation of A305V causes steric hindrance of enzyme C due to atom collision, leading to destabilization of the steric structure.
- the mutant A305V enzyme increases the intramolecular energy of -16,295 (KJ / mol) and mutant A305V by 9.02%, and the structure It was confirmed that it became unstable. This increase in intramolecular energy is particularly observed in each amino acid residue of Tyr208, Ala211 and Val305, and it is considered that these amino acid residues are caused to collide due to the increase in intramolecular energy.
- FIG. 28 shows a comparison of the intramolecular energy of enzyme C wild type and mutant A305V.
- the ⁇ helix adjacent to the mutation A305V has active sites (Phe216, Gln217, Asp220) that bind to substrates IPP and Mg. It is considered that the amino acid residue in the active site cannot take its original position due to the structural change in the main chain structure of the ⁇ helix that forms the basis of the active site, and as a result, substrate binding and activity itself may be affected (FIG. 29). right).
- FIG. 29 shows the structure around Ala305 and Val305 of mutant A305V after calculation of wild type and energy minimization
- FIGS. 30, 31, and 32 show the structural changes of mutant A305V.
- Decaprenyl diphosphate synthase is one of the enzymes of the coenzyme C10 (CoQ10) synthesis pathway. It has been revealed that decaprenyl diphosphate synthase derived from Paracoccus zeaxanthinifaciens or Paracoccus denitrificans confirmed to have high homology with enzyme C is an enzyme necessary for the production of CoQ10 [Japanese Patent Laid-Open No. 2005]. 211020, Special Table 2006-517794].
- FPP and IPP which are substrates of decaprenyl diphosphate synthase, are also substrates of CrtE of the geranyl-geranyl pyrophosphate (GGPP) synthase of the astaxanthin synthesis pathway. Therefore, in normal Paracoccus cells, it is considered that decaprenyl diphosphate synthase and CrtE use the substrates FPP and IPP together.
- GGPP geranyl-geranyl pyrophosphate
- enzyme C reduces the activity of enzyme C by destabilizing the three-dimensional structure of the molecule. If the activity of enzyme C decreases, the amount of FPP and IPP used as substrates also decreases, and as a result, the amount of FPP and IPP that can be used in the astaxanthin synthesis pathway increases.
- CrtE synthesizes one molecule of GGPP from one molecule of FPP and one molecule of IPP.
- enzyme C requires one molecule of FPP and seven molecules of IPP to synthesize one molecule of decaprenyl diphosphate.
- enzyme C consumes 7 times as much IPP as CrtE in one reaction. For this reason, reducing the activity of enzyme C is considered to be extremely effective in increasing IPP supplied to the astaxanthin synthesis pathway.
- SWISS-MODEL modeling protein tertiary and quadrature structure using evolutionary information. Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassario TG, Bertoni M, Bordoli L, Schweded T. Nucleic Acids Res. 2014; 42: W252-8. 2. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Guex, N.M. Peitsch, M .; C. , Schwede, T .; Electrophoresis, (2009). 30 (S1), S162-S173. 3.
- Yeast farnesyl-diphosphate synthase site-directed mutations of residues in high constrained predominant domains I and II.
- Song L1 Polter CD. Proc Natl Acad Sci USA. 1994 12; 91 (8): 3044-8. 16.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Molecular Biology (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Cell Biology (AREA)
- Analytical Chemistry (AREA)
- Immunology (AREA)
- Toxicology (AREA)
- Virology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Enzymes And Modification Thereof (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Fodder In General (AREA)
- Coloring Foods And Improving Nutritive Qualities (AREA)
Abstract
Description
しかし、カロテノイドを産生する細菌において、どの遺伝子が生産効率の上昇に寄与すのかについて、その詳細は不明であった。
[特許文献1]特開2007−97584号公報
[特許文献2]特開平11−69969号公報
[特許文献3]特開2006−340676号公報
[特許文献4]特開2008−259449号公報
[特許文献5]特開平7−79796号公報
[特許文献6]特表2001−512030号公報
[特許文献7]特開2007−244205号公報
[特許文献8]WO2005/118812号パンフレット
[特許文献9]特開2007−143492号公報
[特許文献10]WO2010/044469号パンフレット
[特許文献11]特開2011−188795号公報
[特許文献12]特開2012−139164号公報
[非特許文献]
[非特許文献2]Helv.Chim.Acta,64,2436,1981
[非特許文献3]International Journal of Systematic Bacteriology(1999),49,277−282
[非特許文献4]International Journal of Systematic and Evolutionary Microbiology(2004),54,1699−1702
[非特許文献5]International Journal of Systematic and Evolutionary Microbiology(2003),53,231−238
(a)カロテノイド産生細菌における1−デオキシ−D−キシルロース5リン酸合成酵素のアミノ酸配列において、少なくとも第225番目のアミノ酸残基が他のアミノ酸残基に置換された変異型アミノ酸配列を含むタンパク質をコードする遺伝子
(b)カロテノイド産生細菌におけるデカプレニル二リン酸合成酵素のアミノ酸配列において、少なくとも第305番目のアミノ酸残基が他のアミノ酸残基に置換された変異型アミノ酸配列を含むタンパク質をコードする遺伝子
(c)上記(a)及び(b)の両方の遺伝子
(2)1−デオキシ−D−キシルロース5リン酸合成酵素のアミノ酸配列が配列番号2に示されるものである(1)に記載の細菌。
(3)第225番目のアミノ酸残基が、グリシンからアスパラギン酸に置換された、(1)又は(2)に記載の細菌。
(4)デカプレニル二リン酸合成酵素のアミノ酸配列が配列番号4に示されるものである(1)~(3)のいずれか1項に記載の細菌。
(5)第305番目のアミノ酸残基が、アラニンからバリンに置換された、(1)~(4)のいずれか1項に記載の細菌。
(6)変異型アミノ酸配列を含むタンパク質をコードする遺伝子を有さないカロテノイド産生細菌のカロテノイド産生能よりも高い産生能を獲得した、(1)~(5)のいずれか1項に記載の細菌。
(7)変異型アミノ酸配列を含むタンパク質をコードする遺伝子を有さないカロテノイド産生細菌のカロテノイド産生量よりも少なくとも5倍以上の量の産生能を獲得した、(6)に記載の細菌。
(8)カロテノイド産生細菌がパラコッカス属に属するものである(1)~(7)のいずれか1項に記載の細菌。
(9)パラコッカス属に属する細菌がE−396株である(8)に記載の細菌。
(10)カロテノイドがアスタキサンチンである(1)~(9)のいずれか1項に記載の細菌。
(11) (1)~(10)のいずれか1項に記載の細菌を培養し、得られる培養物からカロテノイドを採取することを特徴とするカロテノイドの製造方法。
(12)カロテノイドの産生量が、変異型アミノ酸配列を含むタンパク質をコードする遺伝子を有さないカロテノイド産生細菌のカロテノイド産生量よりも少なくとも5倍以上の産生量である、(11)に記載の方法。
(13)カロテノイドがアスタキサンチンである(11)又は(12)に記載の方法。
(14)カロテノイド産生細菌に変異処理を施し、変異処理された細菌から以下の(a)~(c)のいずれかの特徴を有する細菌を選択することを特徴とする、カロテノイド産生細菌のスクリーニング方法。
(a)1−デオキシ−D−キシルロース5リン酸合成酵素の活性が変異処理前の細菌における活性よりも上昇した特徴
(b)デカプレニル二リン酸合成酵素の活性が変異処理前の細菌における活性よりも低下した特徴
(c)上記(a)及び(b)の両方の特徴
(15) (14)に記載の方法により選択された細菌を培養し、得られる培養物からカロテノイドを採取することを特徴とするカロテノイドの製造方法。
(16)1−デオキシ−D−キシルロース5リン酸合成酵素のアミノ酸配列において、少なくとも第225番目のアミノ酸残基が他のアミノ酸残基に置換された変異型アミノ酸配列を含むタンパク質をコードする遺伝子。
(17)以下の(a)又は(b)のDNAを含む遺伝子。
(a)配列番号5で表される塩基配列を含むDNA
(b)上記(a)のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ1−デオキシ−D−キシルロース5リン酸合成酵素活性を有するタンパク質をコードするDNA
(18)デカプレニル二リン酸合成酵素のアミノ酸配列において、少なくとも第305番目のアミノ酸残基が他のアミノ酸残基に置換された変異型アミノ酸配列を含むタンパク質をコードする遺伝子。
(19)以下の(a)又は(b)のDNAを含む遺伝子。
(a)配列番号7で表される塩基配列を含むDNA
(b)上記(a)のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつデカプレニル二リン酸合成酵素活性が低下したタンパク質をコードするDNA
(20)以下の(a)~(c)のいずれかの遺伝子を含む組換えベクター。
(a) (16)又は(17)に記載の遺伝子
(b) (18)又は(19)に記載の遺伝子
(c) 上記(a)及び(b)の遺伝子
(21) (20)に記載の組換えベクターを含む形質転換体。
(22) (21)に記載の形質転換体を培養し、得られる培養物からカロテノイドを採取することを特徴とするカロテノイドの製造方法。
1.概要
本発明は、カロテノイドを高生産する細菌に関するものであり、以下の(a)及び(b)のいずれかの遺伝子、又はこれらの両遺伝子を含む細菌である。
(a)カロテノイド産生細菌における1−デオキシ−D−キシルロース5リン酸合成酵素のアミノ酸配列において、少なくとも第225番目のアミノ酸残基が他のアミノ酸残基に置換された変異型アミノ酸配列を含むタンパク質をコードする遺伝子
(b)カロテノイド産生細菌におけるデカプレニル二リン酸合成酵素のアミノ酸配列において、少なくとも第305番目のアミノ酸残基が他のアミノ酸残基に置換された変異型アミノ酸配列を含むタンパク質をコードする遺伝子
その結果、親株として使用したE−396株よりも高いカロテノイド産生能を有する株(「ASB−57株」という。)を取得した。ASB−57株のゲノム解析を行った結果、1−デオキシ−D−キシルロース5リン酸合成酵素(DXS)のアミノ酸配列、及びデカプレニル二リン酸合成酵素(DPS)のアミノ酸配列に変異が生じていることが確認された。そこで、アミノ酸立体構造解析により予測される機能解析を行い、少なくとも、DXSの第225番目のアミノ酸残基及び/又はDPSの第305番目のアミノ酸残基に変異が生じていることが、カロテノイドの高生産に寄与するものと考えられた。
本発明は、上記知見に基づいて完成されたものである。
本発明のカロテノイド産生細菌は、親株を変異処理し、DXSの第225番目のアミノ酸残基及び/又はDPSの第305番目のアミノ酸残基に変異が生じたことを指標として得られる変異型細菌であって、カロテノイドを高効率で産生することができる細菌である。本発明のカロテノイド産生細菌を、本明細書において「変異型カロテノイド産生細菌」という。
(1)親株
本発明において、変異型カロテノイド産生細菌を得るための親株として用いる細菌としては、カロテノイドを産生する細菌であれば何ら限定されず、例えばParacoccus属、Brevundimonas属、Erythrobacter属に属する細菌が挙げられる。
好ましくはParacoccus属に属する細菌、Brevundimonas属に属する細菌又はErythrobacter属に属する細菌が用いられ、より好ましくはParacoccus属に属する細菌が用いられる。Paracoccus属、Erythrobacter属及びBrevundimonas属は、いずれもProteobacteria門、Alphaproteobacteria鋼に分類され、細菌分類学上の共通性があるため、本発明においては、これらの属に属する細菌を使用することが可能である。
Erythrobacter属に属するカロテノイド産生細菌としては、例えばErythrobacter JPCC M種(特開2008−259452)、Erythrobacter JPCC O種(特開2008−259449)などが挙げられる。
Brevundimonas属に属するカロテノイド産生細菌としては、例えばBrevundimonas SD212株(特開2009−27995)、Brevundimonas FERM P−20515,20516株(特開2006−340676)、Brevundimonas vesicularis(Gene,Vol.379,p.101−108,1 Sep 2006)などが挙げられる。
16SリボソームRNAに対応するDNAの塩基配列とは、16SリボソームRNAの塩基配列中のU(ウラシル)をT(チミン)に置き換えた塩基配列を意味する。
国際寄託当局:独立行政法人製品評価技術基盤機構(NITE)特許生物寄託センター
〒292−0818 千葉県木更津市かずさ鎌足2−5−8
識別のための表示:E−396
受託番号:FERM BP−4283
原寄託日:平成5年(1993年)4月27日
識別のための表示:A−581−1
受託番号:FERM BP−4671
原寄託日:平成6年(1994年)5月20日
本発明の変異型カロテノイド産生細菌は、前記親株に変異処理を施し、DXSの第225番目のアミノ酸残基及び/又はDPSの第305番目のアミノ酸残基に変異が生じたことを指標として得ることができる。
変異処理する方法は変異を誘発するものであれば特に限定されない。例えば、N−メチル−N’−ニトロ−N−ニトロソグアニジン(NTG)及びエチルメタンスルホネート(EMS)などの変異剤による化学的方法、紫外線照射及びX線照射などの物理的方法、遺伝子組換え及びトランスポゾンなどによる生物学的方法などを用いることができる。変異処理される細菌は特に限定されないが、カロテノイド産生細菌であることが好ましい。
また、本発明においては、上記の変異を有するタンパク質を調製するために、該タンパク質をコードする遺伝子(DNA)に点突然変異を導入することができる。その変異導入方法として、Kunkel法やGapped duplex法等の部位特異的突然変異誘発法を利用した変異導入用キット、例えばQuikChangeTM Site−Directed Mutagenesis Kit(ストラタジーン社製)、GeneTailorTM Site−Directed Mutagenesis System(インビトロジェン社製)、TaKaRa Site−Directed Mutagenesis System(Mutan−K、Mutan−Super Express Km等:タカラバイオ社製)等を用いて行うことができる。また、「Molecular Cloning,A Laboratory Manual(4th edition)」(Cold Spring Harbor Laboratory Press(2012))等に記載された部位特異的変異誘発法等の方法を用いることができる。
さらに、上記ゲノム解析と並行して、例えば、寒天培地上のコロニーの色調で目的の変異株を選択する方法の他、試験管、フラスコ、発酵槽などで変異株を培養し、吸光度、高速液体クロマトグラフィー、薄層クロマトグラフィーなどを利用したカロテノイド色素分析により、カロテノイドの生産量を指標として選択することもできる。
変異及びスクリーニングの工程は1回でもよいし、また、例えば突然変異処理とスクリーニングにより変異株を得て、これをさらに変異処理とスクリーニングにより生産性の改良された変異株を取得するというように、変異及びスクリーニング工程を2回以上繰り返してもよい。
DXSの第225番目のアミノ酸残基から他のアミノ酸への変異は、DXSの酵素活性の上昇に寄与する。これにより、ピルビン酸から1−デオキシ−Dキシルロース−5−リン酸への合成を促進し、ひいてはアスタキサンチン合成の基質となるイソペンテニル二リン酸(IPP)の生産が上昇する。
DPSの第305番目のアミノ酸残基から他のアミノ酸残基への変異は、DPSの酵素活性の低下に寄与する。この変異はファルネシル二リン酸(FPP)からデカプレニル二リン酸(DPP)への合成を抑制する。FPPからDPPへの合成にはIPPが使用されることから、上記変異により、DPP合成に使用されるIPPの量が減少し、当該IPPは前記のアスタキサンチン合成の基質として利用される。
従って、本願発明の変異型カロテノイド産生細菌は、以下の(a)の遺伝子、以下の(b)の遺伝子、又は以下の(a)及び(b)の遺伝子の両者を含むことができる。
上記変異型DXS遺伝子としては、例えば以下のものが挙げられる。
(i)DXSのアミノ酸配列(例えば配列番号2)のうち第225番目のアミノ酸残基が他のアミノ酸残基に置換された変異型アミノ酸配列を含み、かつDXS活性を有するタンパク質をコードする遺伝子
このような変異型アミノ酸配列として、配列番号6に示すものが挙げられ、上記遺伝子として、配列番号5に示されるものが挙げられる。本発明においては、配列番号2に示すアミノ酸配列において、第225番目のアミノ酸残基であるグリシンがアスパラギン酸に置換されたアミノ酸配列であることが好ましい。
(ii)DXSのアミノ酸配列(例えば配列番号2)のうち第225番目のアミノ酸残基が他のアミノ酸残基に置換されるとともに、当該第225番目のアミノ酸残基以外の1若しくは複数(例えば1~数個)のアミノ酸残基が欠失、置換若しくは付加された変異型アミノ酸配列を含み、かつDXS活性を有するタンパク質
(iii)配列番号5で表される塩基配列を含むDNAからなる遺伝子
(iv)配列番号5で表される塩基配列を含むDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつDXS活性を有するタンパク質をコードするDNAからなる遺伝子
上記配列番号5で表される塩基配列は、カロテノイド産生細菌におけるDXSのアミノ酸配列をコードするDNA(配列番号1)において、第225番目のアミノ酸残基が他のアミノ酸残基に置換されたアミノ酸配列を含むタンパク質をコードするものである。
このような遺伝子としては、例えば以下のものが挙げられる。
(i)DPSのアミノ酸配列(例えば配列番号4)のうち第305番目のアミノ酸残基が他のアミノ酸残基に置換された変異型アミノ酸配列を含み、かつDPS活性が低下したタンパク質をコードする遺伝子
このような変異型アミノ酸配列として、配列番号8に示すものが挙げられ、上記遺伝子として、配列番号7に示されるものが挙げられる。本発明においては、配列番号4に示すアミノ酸配列において、第305番目のアミノ酸残基であるアラニンがバリンに置換されたアミノ酸配列であることが好ましい。
(ii)DPSのアミノ酸配列(例えば配列番号4)のうち第305番目のアミノ酸残基が他のアミノ酸残基に置換されるとともに、当該第305番目のアミノ酸残基以外の1若しくは複数(例えば1~数個)のアミノ酸残基が欠失、置換若しくは付加された変異型アミノ酸配列を含み、かつDPS活性が低下したタンパク質
(iii)配列番号7で表される塩基配列を含むDNAからなる遺伝子
(iv)配列番号7で表される塩基配列を含むDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつDPS活性が低下したタンパク質をコードするDNAからなる遺伝子
上記配列番号7で表される塩基配列は、カロテノイド産生細菌におけるDPSのアミノ酸配列をコードするDNA(配列番号3)において、第305番目のアミノ酸残基が他のアミノ酸残基に置換されたアミノ酸配列を含むタンパク質をコードするものである。
ASB−57株は、DXSの第225番目のアミノ酸残基であるグリシンがアスパラギン酸に変異し、DPSの第305番目のアミノ酸残基であるアラニンがバリンに変異したアミノ酸配列を含むタンパク質をコードする遺伝子を有する。ASB−57株においてDXSのアミノ酸配列及び遺伝子の塩基配列をそれぞれ配列番号6、5に示す。また、ASB−57株においてDPSのアミノ酸配列及び遺伝子の塩基配列をそれぞれ配列番号8、7に示す。
本発明においては、上記変異型DXSをコードする遺伝子、及び/又は上記変異型DPSをコードする遺伝子を宿主に導入して形質転換を行うことにより、遺伝子組換え型の変異型カロテノイド産生細菌を得ることができる。
変異型DXS遺伝子及び/又は変異型DPS遺伝子をベクターに導入して得られる組換えベクター、並びに当該組換えベクターを宿主に導入して得られる形質転換体は、任意の公知方法を採用すればよく、例えば、Sambrook J.et al.,Molecular Cloning,A Laboratory Manual(4th edition)(Cold Spring Harbor Laboratory Press(2012)に従って行うことができる。
上記DXS遺伝子及びDPS遺伝子を遺伝子工学的に合成する場合は、まず、当該酵素ををコードするDNAを設計し合成する。DNAの設計及び合成は、例えば、全長の遺伝子を含むベクター等を鋳型とし、所望のDNA領域を合成し得るように設計したプライマーを用いて、PCR法により行うことができる。そして、上記DNAを適当なベクターに連結することによってタンパク質発現用組換えベクターを得て、この組換えベクターを目的遺伝子が発現し得るように宿主中に導入することによって形質転換体を得る(Sambrook J.et al.,Molecular Cloning,A Laboratory Manual(4th edition)(Cold Spring Harbor Laboratory Press(2012))。
ベクターには、宿主微生物で自律的に増殖し得るファージ又はプラスミドが使用される。さらに、動物ウイルス、昆虫ウイルスベクターを用いることもできる。組換えベクターの作製は、精製されたDNAを適当な制限酵素で切断し、適当なベクターDNAの制限酵素部位等に挿入してベクターに連結すればよい。形質転換に使用する宿主としては、目的の遺伝子を発現できるものであれば特に限定されるものではない。例えば、細菌(枯草菌、パラコッカス属細菌等)、酵母、動物細胞(COS細胞、CHO細胞等)、植物細胞、昆虫細胞又は昆虫が挙げられる。宿主への組換えベクターの導入方法は公知である。
また、遺伝子への変異の導入方法は、前記と同様である。
本発明において、上記のカロテノイド産生細菌又は形質転換体を所定の培地で培養することにより、高濃度のカロテノイドを安定的に生産させることができる。
産生されるカロテノイドは特に限定されないが、例えば、アスタキサンチン、カンタキサンチン、ゼアキサンチン、β−クリプトキサンチン、リコペン、β−カロテン、アドニルビン、アドニキサンチン、エキネノン、アステロイデノン又は3−ヒドロキシエキネノンであり、好ましくは、アスタキサンチン、カンタキサンチン、ゼアキサンチン又はβ−クリプトキサンチンであり、より好ましくは、アスタキサンチン又はゼアキサンチンである。本発明より製造されるカロテノイドは一種でもよいし、複数種が組み合わされていてもよい。
また、有機窒素源としては、例えば、コーンスティープリカー(ろ過処理物を含む)、ファーマメディア、大豆粕、大豆粉、ピーナッツミール、ソイペプトン、ディスティラーズソルブル、乾燥酵母、酵母エキス、カザミノ酸、グルタミン酸、アスパラギン酸などの中、1種又は2種以上が用いられる。添加濃度は窒素源の種類により異なり適宜調整すれば足りるが、通常、0~80g/L、好ましくは1~30g/Lである。
無機塩類としては、例えば、リン酸二水素カリウム、リン酸水素二カリウム、リン酸水素二ナトリウムなどのリン酸塩類、硫酸マグネシウム、塩化マグネシウムなどのマグネシウム塩類、硫酸鉄、塩化鉄などの鉄塩類、塩化カルシウム、炭酸カルシウムなどのカルシウム塩類、炭酸ナトリウム、塩化ナトリウムなどのナトリウム塩類、硫酸マンガンなどのマンガン塩類、硫酸銅などの銅塩類、硫酸亜鉛などの亜鉛塩類、モリブデン酸ナトリウムなどのモリブデン塩類、硫酸ニッケルなどのニッケル塩類、セレン酸ナトリウムなどのセレン塩類、タングステン酸ナトリウムなどのタングステン塩類、塩化アルミニウムなどのアルミニウム塩類、塩化クロムなどのクロム塩類、ホウ酸及びヨウ化カリウム等の中、1種又は2種以上が用いられる。添加量は無機塩の種類により異なり適宜調整すれば足りるが、通常、培地1Lに対し0.0001~15gである。リン酸塩類、マグネシウム塩類、カルシウム塩類、ナトリウム塩類及び鉄塩類では、0.02~15g/Lが好ましく、マンガン塩類、銅塩類、亜鉛塩類、モリブデン塩類、ニッケル塩類、セレン塩類、タングステン塩類、アルミニウム塩類、クロム塩類、ホウ酸、ヨウ化カリウムなどを加える場合には、0.1~15mg/Lが好ましい濃度である。無機塩類は通常始発培地に添加するが、逐次的又は連続的に追加供給してもよい。
本発明において、培地は滅菌処理した後、細菌の培養に用いられる。滅菌処理は、当業者であれば、適宜行うことができる。例えば、適切な容器中の培地をオートクレーブで加熱滅菌すればよい。あるいは、滅菌フィルターによりろ過滅菌すればよい。
培養温度は15~40℃、好ましくは20~35℃、より好ましくは25℃~32℃であり、通常1日~18日間、好ましくは2~12日間、より好ましくは3~8日間、好気条件で培養を行う。好気条件としては、例えば、振とう培養又は通気撹拌培養等が挙げられ、溶存酸素濃度を一定の範囲に制御するのが好ましい。溶存酸素濃度の制御は、例えば、攪拌回転数、通気量、内圧などを変化させることにより行うことができる。溶存酸素濃度は好ましくは0.3~10ppm、より好ましくは0.5~7ppm、さらに好ましくは1~5ppmに制御する。
培養物は、例えば、培養液、培養上清、菌体濃縮液、湿菌体、乾燥菌体、菌体溶解物などが挙げられる。培養上清は、培養液を遠心処理又はろ過処理することで、培養液から菌体を除いて調製すればよい。菌体濃縮液は、培養液を遠心分離又は膜ろ過濃縮することにより得ることができる。湿菌体は、培養液を遠心又はろ過することにより得ることができる。乾燥菌体は、湿菌体又は菌体濃縮液を一般的な乾燥方法によって乾燥させることにより得ることができる。このようにして得られたカロテノイド含有乾燥菌体はそのまま飼料添加物として用いることができる。
本発明の細菌は、DXS及び/又はDPSの変異型アミノ酸配列を含むタンパク質をコードする遺伝子を有さないカロテノイド産生細菌のカロテノイド産生量よりも、少なくとも5倍、好ましくは10倍以上の量の産生能を有する。
抽出を行う前に、培養物をアルカリ試薬や界面活性剤などを用いた化学的処理、溶菌酵素、脂質分解酵素及びタンパク分解酵素などを用いた生化学処理、又は超音波若しくは粉砕などの物理的処理の中、1つ又は2つ以上の処理を行ってもよい。
例えば、カロテノイドを培養物から抽出する場合、抽出及び洗浄に用いる溶媒は特に限定されないが、メタノール、エタノール、イソプロパノールなどの低級アルコール類、アセトン、テトラヒドロフラン、メチルエチルケトン、メチルイソブチルケトン、ジクロロメタン、クロロホルム、ジメチルホルムアミド、ジメチルスルホキシドなどが挙げられる。
このように得られた抽出物をカロテノイドとしてそのまま用いることが可能であり、さらに精製して使用することもできる。
抽出液からカロテノイド沈殿物を得る方法としては、一般的には加熱及び/又は減圧濃縮や晶析が挙げられる。この他、低温におけるカロテノイド色素の析出、酸・アルカリ薬剤や各種塩類による析出によってカロテノイド色素を濃縮せずに分離してもよい。工業的に用いる場合には、晶析することが望ましい。
上記のように得られる培養物、抽出物又は精製物は、カロテノイドとしてそれぞれ単独で用いることもできるし、これらを任意の割合で混合して用いることもできる。
前記DXSの第225番目のアミノ酸残基、及びDPSの第305番目のアミノ酸残基の変異がカロテノイド合成に重要な役割を果たすことを示すため、これらの酵素の立体構造解析を行うことができる。
本発明においては、アスタキサンチン合成経路上での酵素2種(酵素A、酵素Cという)について点変異が同定された。アスタキサンチンの産生の増加は、これらの酵素に起こった変異が原因と考えられることから、変異が同定された酵素2種について立体構造モデルを構築し、変異によるアミノ酸置換の影響を推察する。
以下、実施例により本発明をさらに具体的に説明する。但し、本発明の範囲はこれらの実施例により限定されるものではない。
[実施例1]
変異処理方法
親株(E−396株)に変異源にUVやNTG(ニトロソグアニジン)などを使用し、種々選択圧を使用して数度のスクリーニングを実施した。スクリーニングは、アスタキサンチンの収量を指標として行った。
ゲノム解析法
ゲノム解析は、PacBio RS II(Pacific Biosciences社製)やMiSeq(イルミナ社)のシーケンサーを用いてゲノム配列を読んだ後、SMART Cell 8 Pac V3(Pacific Biosciences社製)や、MiSeq Control Software(MCS)v2.4.1.3、Real Time Analysis(RTA)v1.18.54,bc12fastq v 1.8.4(イルミナ社)などの解析ソフトを使用して行った。
変異部位の同定は、これらのゲノム解析結果の変異点を持つタンパクと考えられる領域のアミノ酸配列とKyoto Encyclopedia of Genes and Genomes(KEGG)に収載されている酵素遺伝子アミノ酸配列の間で相同性の高いもののうち、Paracoccus属に含まれるものを洗い出し、さらに、これらの情報から酵素の立体構造解析を行って、配列の一致するテンプレートを探しだすことにより、変異部位を持つタンパク質のアミノ酸配列の最終的な酵素名を決定した。
(i)培養条件
試験管ストローク‐330rpm、28℃、pH7.2、培地量8ml/本
培養時間‐72時間。
培地‐
以下の組成の培地8mlを内径18mmの綿栓付き試験管に入れ121℃で15分間オートクレーブ滅菌し、シード用試験管培地を調製した。シード用試験管培地の原料は、十分に菌体の生育することが確認されているロットのものを使用した。
シュークロース 30g/L
コーンスティープリカー 30g/L
リン酸二水素カリウム 1.5g/L
リン酸水素二ナトリウム12水和物 3.8g/L
塩化カルシウム2水和物 5.0g/L
硫酸マグネシウム7水和物 0.7g/L
硫酸鉄7水和物 1.0g/L
pH7.2
グルコース 30g/L
コーンスティープリカーろ過処理物 30g/L
硫酸アンモニウム 1.5g/L
リン酸二水素カリウム 1.5g/L
リン酸水素二ナトリウム12水和物 3.8g/L
塩化カルシウム2水和物 5.0g/L
硫酸マグネシウム7水和物 0.7g/L
硫酸鉄7水和物 1.0g/L
シリコン系消泡剤 0.2g/L
各株における比生産性を図1に示す。
本実施例により、E−396株と比較して10倍以上のカロテノイド産生能を有するASB−57株を取得した。
[実施例2]
アスタキサンチン合成経路に関与する酵素の立体構造解析
酵素A、酵素Cの立体構造モデルはホモロジーモデリングにより構築した。モデリングには、ソフトウェアSwiss−Pdb viewer及びSWISS−MODELを使用した[1,2]。変異体モデルはSwiss−Pdb viewerで作製した。アミノ酸残基の置換はmutateコマンドを、分子内エネルギーの算出はcompute energyコマンドを、また、エネルギー最小化計算はenergy minimizationコマンドを使用した。基質等との複合体モデルの作製、基質近傍残基の検出、原子間距離の測定、立体構造の表示はソフトウェアWaals(Altif Labs.Inc.)を使用して行った。低分子化合物の立体構造モデルはMarvinSketch(ChemAxon Ltd.)により作製した。

酵素Aは、アスタキサンチン合成の原料となるイソプレニル二リン酸(IPP)を生合成するイソプレノイド生合成経路の1つであるデオキシキシルロース経路において、ピルビン酸とD−グリセルアルデヒド三リン酸から、1−デオキシ−Dキシルロース5リン酸を合成する1−デオキシ−D−キシルロース5リン酸合成酵素:1−deoxy−D−xylurose−5−phosphate synthase(DXS)である。
酵素A、1−デオキシ−D−キシルロース5リン酸合成酵素:1−deoxy−D−xylurose−5−phosphate synthase(DXS)は、ピルビン酸とD−グリセルアルデヒド三リン酸から、マグネシウムイオン(Mg)存在下で、1−デオキシ−D−キシルロース5リン酸を合成する。触媒反応には、補酵素としてチアミンピロリン酸(Thiaminepyrophosphate,TPP)を必要とし、まず、補酵素TPPが基質のピルビン酸に付加し、ヒドロキシエチル−TPP中間体を生じる。この中間体とグリセルアルデヒド三リン酸が反応することにより1−デオキシ−D−キシルロース5リン酸が生成する。以下に酵素Aの酵素反応を示す。

(1)ホモロジーモデリングによる酵素Aの立体構造モデルの構築
酵素Aの立体構造モデルは、補酵素TPPとの複合体の立体構造がX線結晶構造解析により決定されているDeinococcus radiodurans(D.radiodurans)由来の1−deoxy−D−xylurose−5−phosphate synthase(DXS、テンプレート)の立体構造(PDB ID:2O1X)[3]に基づき(図2)、ホモロジーモデリングにより構築した。
酵素AとD.radiodurans由来DXSのアミノ酸一致度は44.1%である。テンプレートのDXSの立体構造では、アミノ酸番号199~242残基の領域(44残基)がdisorderedであり、X線結晶構造解析により原子の位置が特定できていない。そのため、酵素Aのdisordered領域に相当する196~238残基(43残基)を除く、アミノ酸残基番号7~630残基の立体構造モデルを構築した。次に、酵素Aの立体構造モデルとテンプレート構造を重ね合わせることにより、TPP及びMgをはめ込み、酵素AとTPPの複合体モデルを作製した。図4にテンプレート構造と構築したモデル構造を示す。
酵素Aは、テンプレート構造と同様、ホモ二量体を形成しており、それぞれのサブユニットにTPP結合部位及び基質結合部位を持つ。酵素Aの単量体は、ドメインI(1~319残基)、ドメインII(320~495残基)、ドメインIII(496~629残基)の3つのドメインで構成される。
酵素Aの基質との結合に関与するアミノ酸残基を推察するため、酵素Aに基質が結合した複合体モデルを構築した。テンプレート構造の201Xでは、基質であるピルビン酸及びグリセルアルデヒド3リン酸の座標は確定できていないため、先ず、ピルビン酸結合部位を検出するために、補酵素TPPにピルビン酸が付加したヒドロキシエチル−TPP中間体との複合体モデルを、類縁のSaccharomyces cerevisiae(S.cerevisiae)由来のトランスケトラーゼTransketolase(TK)とヒドロキシエチル−TPP中間体が結合した立体構造(PDB ID:1GPU)[6]に基づき、重ね合わせにより、ヒドロキシエチル−TPP中間体をはめ込み、複合体モデルを作製した。同様にS.cerevisiae由来のTKとエリトロース−4−リン酸との立体構造(PDB ID:1NGS)[7]に基づき、エリトロース−4−リン酸をはめ込み、さらにエリトロース−4−リン酸からグリセルアルデヒド3リン酸モデルを作製することにより、酵素Aとグリセルアルデヒド三リン酸との複合体モデルを作製した。
図5に酵素Aと基質複合体のモデル構造を示す。
補酵素及び基質を結合するアミノ酸残基を推定するため、酵素Aと補酵素の複合体モデルにおいて、TPP中間体、GAP及びMgとの相互作用について調べた。
酵素AとTPPとの複合体モデルでは、テンプレート構造と同様、TPPはドメインIとドメインIIの間に位置しており、TPPのピリミジン環はドメインIIに、リン酸基はドメインIに結合している。
基質であるピルビン酸はTPPと反応し、ヒドロキシエチル−TPP中間体となる。酵素Aとヒドロキシエチル−TPP中間体の複合体モデルからは、ピルビン酸由来のヒドロキシエチル基との相互作用として、Val77との疎水性相互作用及びHis432との水素結合が推察された。これらのアミノ酸残基がピルビン酸との結合に関与していると考えられる。図8に、ヒドロキシエチル基との相互作用が推測されるアミノ酸残基を示す。
基質であるグリセルアルデヒド3リン酸(GAP)と相互作用するアミノ酸残基として、His48,Tyr393,Arg421,Asp428,Arg479との水素結合が推察された(図9)。
His48とAsp428はGAPのアルデヒド基と水素結合を形成する。Tyr393,Arg421,Arg479はGAPのリン酸基との水素結合を形成することが推察された。



酵素Aは、テンプレートのD.radiodurans由来DXSで明らかになっている補酵素のTTP、基質のピルビン酸及びGAPを結合する活性部位を保持していることから、DXSの酵素活性を持つと推測される。これらのアミノ酸残基は、D.radiodurans由来DXSの他、大腸菌等のDXSやS.cerevisiae由来TKにおいても保存性が高いことが知られている[3]。
酵素Aの変異G225Dは立体構造が特定できなかったdisordered領域(196~238残基)に存在する。この変異が酵素Aの立体構造に与える影響を推察するため、これまでの知見や立体構造上でのdisordered領域の位置と活性部位との関連について調べた。
これまでに、マスカットと大腸菌のDXSについて、disordered領域で起こった変異、それぞれ2箇所、計4種がいずれも酵素活性を増加させることが報告されている。
マスカット(Vitis vinifera)由来DXSでは、K284Nの変異が野生型に比べVmax及びKcat/Kmで約2倍の活性の増大をもたらすこと、過剰発現によりモノテルペンの産生量が大幅に増加することが報告されている[9]。
マスカットのK284NとR306C、大腸菌のK213NとK234Cは、いずれもdisordered領域(青)に存在する。酵素Aの変異G225Dもこれらと同じくdisordered領域に存在する。アミノ酸配列アライメントから、DXSの活性部位(green)が保存されており、酵素AもこれらのDXSと同様の反応様式を持つと推測される。なお、disordered領域のN末端側(magenta)には、活性部位が存在する。
図13に、酵素Aの変異G225Dが存在するdisordered領域(196~238残基)位置を青の点線で示す。
本領域は、DXSの活性に必須である補酵素TPPの結合部位近傍に位置する。本領域のN末端側のループ(magenta)には活性部位であるAsn180,Met182及びIle184が存在する。Asn180の側鎖とMet182の主鎖はMgを結合する。Ile184の側鎖はTPPと疎水性結合している。DXSの活性に必須なTPPとMgが結合するには、このループが適切な構造をとることが重要であると思われる。文献[9]では、生理的役割は不明ではあるが、本領域は活性部位の近くに存在し、本領域内の変異が合理的に酵素の活性に影響するではないか、と考察されている。図12、13に示すように、本領域は、アミノ酸配列上でも立体構造上でも、TPP結合部位の近傍に存在するため、本領域への変異がこれらの活性部位に影響を与えることは、十分起こりうると考えられる。
次に、酵素Aの変異G225Dがdisordered領域の立体構造に与える影響を調べるため、酵素Aの196~238残基(43残基)のdisordered領域についてモデル構造を作製した。マスカットの変異K284Nについて報告された文献[9]では、マスカットDXSのdisordered領域について、モデルを作製し、変異による静電ポテンシャルの変化を見ている。参考のため、酵素Aについても同様の解析を行った。ホモロジーモデリングにより、酵素Aのdisordered領域と相同性の高いアミノ酸配列のフラグメント構造を基に立体構造モデルを作製した。テンプレート構造として、PDBに登録された立体構造の中で最も高いアミノ酸一致度(34%)を持つ1AL7のフラグメントを使用した。disordered領域はゆらいだ構造をとっていると推測されるが、本領域が形成しやすいフォールディングとしては参考になると思われる。作製したモデルを図14(左)に示す。
以上のように、酵素Aの変異G225Dは酵素Aのdisordered領域で起きていること、本領域は、活性に必須なTPP結合部位の近傍に存在することが確認された。また、本領域にはこれまで複数のDXSの酵素活性を向上させる変異が見つかっており、G225Dの変異は、DXSの活性を向上させる既知の変異と同様の構造変化を起こすことが予測された。
酵素Cは、アミノ酸配列の相同性および立体構造比較解析から、ポリプレニル二リン酸合成酵素の一種であり、ファルネシル二リン酸(FPP)と7個のイソペンテニル二リン酸(IPP)からデカプレニル二リン酸を合成するデカプレニル二リン酸合成酵素:Decaprenyl diphosphate synthaseである。
デカプレニル二リン酸合成酵素は、FPPとIPPを縮合する活性を持ち、IPPとの縮合を繰り返し、FPPと7個のIPPからデカプレニル二リン酸(DPP)を合成する酵素である。デカプレニル二リン酸合成酵素の酵素反応を以下に示す。

(1)ホモロジーモデリングによる酵素Cの立体構造モデルの構築
酵素Cの活性部位及び変異による影響を調べるため、テンプレート構造として、PDBの立体構造でアミノ酸一致度が最も高く(アミノ酸一致度は76.2%)、立体構造がX線結晶構造解析により決定されているRhodobacter capsulatus(R.capsulatus)由来のデカプレニル二リン酸合成酵素の立体構造(PDB ID:3MZV)を使用し、ホモロジーモデリングにより立体構造モデルを構築した(図16)[4]。
ホモロジーモデリングは、酵素Cとテンプレート構造の立体構造アライメントに基づいて行った(図17)。
テンプレート構造の3MZVは基質が結合していないため、Escherichia coli由来のoctaprenyl pyrophosphate synthase(オクタプレニル二リン酸合成酵素)(PDB ID:3WJN,3WJO)の立体構造データ[11]を使用して、酵素Cの立体構造モデルに3WJNを重ね合わせ、基質であるFPPを酵素Cの立体構造モデルにはめ込むことにより、酵素CとFPPの複合体モデルを作製した。次に、同様に、3WJOを重ね合わせて、IPPをはめ込み、酵素CとFPP,IPP複合体モデルを作製した。
図18に、テンプレート構造と構築したモデル構造を示す。酵素Cはテンプレート構造と同様にホモ二量体を形成している。
(1)立体構造による比較
酵素Cのデカプレニル二リン酸合成酵素は、ポリプレニル二リン酸合成酵素のファミリー(Pfam PF00348 Polyprenyl synthetase)に属する。図19に酵素Cの基質結合の様子を示す。
ポリプレニル二リン酸合成酵素は、FPPからIPPをhead−to−tail(ここでは文献[4]に従い、リン酸基側をhead、イソプレニル基側をtailと呼ぶ。)の向きで縮合することにより、各種のポリプレニル二リン酸を合成する。デカプレニル二リン酸合成酵素は、FPPとIPPGの縮合反応を続けることにより、基質結合部位の奥の方へと(図19右に矢印で示す)プレニル鎖が伸長され、C50のデカプレニル二リン酸を合成する。
次に、酵素Cのアミノ酸配列について、Paracoccus由来のデカプレニル二リン酸合成酵素との比較を行った。Paracoccus由来のデカプレニル二リン酸合成酵素として、既に明らかになっているアミノ酸配列として、UniProt(http://www.uniprot.org)に、Paracoccus zeaxnthinifaciens(Q8L1I6)とParacoccus denitrificans(A1B3M9)由来のアミノ酸配列が登録されている。これらと酵素Cのアミノ酸配列を比較したところ、アミノ酸一致度は75.1%(類似度89.2%)であり、高い相同性を示した(表5)。
アミノ酸配列からも酵素Cはデカプレニル二リン酸合成酵素であると推測される。図21にアライメントを示す。

酵素Cと基質の複合体モデルにおいて、基質であるFPP結合部位とIPP結合部位、触媒に必要なMg結合部位を推察した。これらの結果から推察された活性部位と他のポリプレニル二リン酸合成酵素の活性中心や基質結合部位の保存性は高く、酵素Cはポリプレニル二リン酸合成酵素と同様の反応様式を持つと推察される。
酵素Cは、テンプレート構造のデカプレニルニリン酸合成酵素と同様、ホモ二量体を形成すると推測される。図22に、A鎖(light red)のFPPとIPPとの結合部位を示す。FPPは、A鎖のトンネル状の領域に結合し、FPPとIPPは、IPPのイソペンテニル基にFPPのリン酸基を向けたhead−to−tailの形で結合している。触媒反応は、Mg存在下において、FPPのリン酸基とIPPのイソペンテニル基の間で起こる。
IPPについては、リン酸基とLys54,Arg57,His86,Arg103との間に水素結合が、イソペンテニル基とPhe216との間に疎水性相互作用が推察された(図23中央)。



酵素Cで同定されたAla305からValへの変異が立体構造に及ぼす影響を推察するため、酵素Cの変異A305V酵素の立体構造モデルを作製した。
AlaからValへの置換による影響を見るため、まず、他のアミノ酸残基の立体構造を固定して、(rigid bodyと仮定して)、Ala305をValに一残基置換したモデルを作製し、野生型と比較した。
Ala305の側鎖であるメチル基の炭素原子は周辺のアミノ酸残基、Tyr208,Ala211,His301,Ala302と接しており、炭素の原子間の距離はいずれも4.0Å未満である。Ala305からValへの置換により側鎖のメチル基が2つ分増えることになる。Ala305をValに置換した立体構造モデルでは、Valの側鎖のメチル基の炭素とTyr208及びAla211の炭素との原子間距離は2.42Å,2.47Åであった。
A305Vによる構造の不安定化を評価するため、酵素Cの野生型と変異体A305Vの分子内エネルギーを原子間の結合の長さ、結合角、ねじれ、結合エネルギー等の合計により、単位キロジュール/mol(KJ/mol)で算出した。。
次に、変異体モデルに対しエネルギー最小化計算を行い、周辺のアミノ酸残基の立体構造を動かすことにより、A305Vによる衝突を回避できるかどうかを調べた。その結果、Val305の側鎖の衝突を回避して、側鎖を許容するためには、Val305が存在するαヘリックスと隣接するαヘリックスのアミノ酸残基の立体構造を動かさなければならないことがわかった。すなわち、その構造変化は側鎖のみならず、主鎖にも及ぶものであり、変異A305V酵素では、αヘリックス間の本来のパッキングができなくなり、2本のヘリックス周辺の構造が不安定化することが予想される。また、変異A305Vに隣接するαヘリックスには基質であるIPPやMgを結合する活性部位(Phe216,Gln217,Asp220)が存在する。活性部位の土台となるαヘリックスの主鎖構造の構造変化により、活性部位のアミノ酸残基が本来の位置をとれなくなり、その結果、基質結合や活性そのものに影響がでることが考えられる(図29右)。
以上のように、A305Vの立体構造モデルからは、周辺のアミノ酸残基との立体障害、分子内エネルギーの増加および隣接する2本のαヘリックスの構造変化が起きていると推察され、A305Vの変異が酵素Cの立体構造を不安定化することが示唆された。点変異による立体構造の不安定化については、遺伝病等でも数多く報告されている。たとえば、不安定化を引き起こす変異を持ったタンパク質が、溶媒中で本来の立体構造を維持することができずに、通常よりも短かい時間で失活する、また、細胞内では、本来のパッキングができない変異体タンパク質が細胞自身の機能により排除されると考えられている。そのため、本来の活性を有さない、不安定な構造である変異A305V酵素は、細菌内で排除されている可能性があり、結果的に、細菌内での酵素Cの活性が減少するものと推察される。
デカプレニル二リン酸合成酵素は、コエンザイムC10(CoQ10)合成経路の酵素の1つである。今回、酵素Cと高い相同性を持つことが確認されたParacoccus zeaxanthinifaciensあるいはParacoccus denitrificans由来のデカプレニル二リン酸合成酵素は、CoQ10の産生に必要な酵素であることが明らかにされている[特開2005−211020、特表2006−517794]。デカプレニル二リン酸合成酵素の基質であるFPPとIPPは、アスタキサンチン合成経路のゲラニルゲラニル二リン酸(Geranyl−geranyl pyrophosphate;GGPP)合成酵素のCrtEの基質でもある。したがって、通常のParacoccusの細胞内では、デカプレニル二リン酸合成酵素とCrtEが、基質であるFPPとIPPを取り合って使用していると考えられる。
1.SWISS−MODEL:modelling protein tertiary and quaternary structure using evolutionary information.Biasini M,Bienert S,Waterhouse A,Arnold K,Studer G,Schmidt T,Kiefer F,Cassarino TG,Bertoni M,Bordoli L,Schwede T.Nucleic Acids Res.2014;42:W252−8.
2.Automated comparative protein structure modeling with SWISS−MODEL and Swiss−PdbViewer:A historical perspective.Guex,N.,Peitsch,M.C.,Schwede,T.Electrophoresis,(2009).30(S1),S162−S173.
3.Crystal structure of 1−deoxy−D−xylulose 5−phosphate synthase,acrucial enzyme for isoprenoids biosynthesis.Xiang S,Usunow G,Lange G,Busch M,Tong L.J Biol Chem.2007 26;282(4):2676−82.
4.Prediction of function for the polyprenyl transferase subgroup in the isoprenoid synthase superfamily.Wallrapp FH,Pan JJ,Ramamoorthy G,Almonacid DE,Hillerich BS,Seidel R,Patskovsky Y,Babbitt PC,Almo SC,Jacobson MP,Poulter CD.Proc Natl Acad Sci U S A.2013 26;110(13):E1196−202.
14;107(50):21337−42.
6.Snapshot of a key intermediate in enzymatic thiamin catalysis:crystal structure of the alpha−carbanion of(alpha,beta−dihydroxyethyl)−thiamin diphosphate in the active site of transketolase from Saccharomyces cerevisiae.Fiedler E,Thorell S,Sandalova T,Golbik R,K▲o▼nig S,Schneider G.Proc Natl Acad Sci U S A.2002 22;99(2):591−5.
7.Examination of substrate binding in thiamin diphosphate−dependent transketolase by protein crystallography and site−directed mutagenesis.Nilsson U1,Meshalkina L,Lindqvist Y,Schneider G.J Biol Chem.1997 17;272(3):1864−9.
8.Catalytically Important Residues in E.coli 1−Deoxy−D−Xylulose 5−Phosphate Synthase.Jordi Querol−Aud▲i▼,Albert Boronat,Josep J.Centelles,Santiago Imperial Journal of Biosciences and Medicines,2014,2,30−35
9.Functional effect of grapevine 1−deoxy−D−xylulose 5−phosphate synthase substitution K284N on Muscat flavour formation.Battilana J1,Emanuelli F,Gambino G,Gribaudo I,Gasperi F,Boss PK,Grando MS.J Exp Bot.2011;62(15):5497−508.
10.Feedback Inhibition of Deoxy−D−xylulose−5−phosphate Synthase Regulates the Methylerythritol 4−Phosphate Pathway.Banerjee A1,Wu Y,Banerjee R,Li Y,Yan H,Sharkey TD.J Biol Chem.2013 7;288(23):16926−36.
11.Crystal structures of ligand−bound octaprenyl pyrophosphate synthase from Escherichia coli reveal the catalytic and chain−length determining mechanisms.Han X,Chen CC,Kuo CJ,Huang CH,Zheng Y,Ko TP,Zhu Z,Feng X,Wang K,Oldfield E,Wang AH,Liang PH,Guo RT,Ma Y.Proteins.2015;83(1):37−45.
14.Effect of site−directed mutagenesis of conserved aspartate and arginine residues upon farnesyl diphosphate synthase activity.Joly A,Edwards PA.J Biol Chem.1993 25;268(36):26983−9.
15.Yeast farnesyl−diphosphate synthase:site−directed mutagenesis of residues in highly conserved prenyltransferase domains I and II.Song L1,Poulter CD.Proc Natl Acad Sci U S A.1994 12;91(8):3044−8.
16.Significance of Phe−220 and Gln−221 in the catalytic mechanism of farnesyl diphosphate synthase of Bacillus stearothermophilus.Koyama T1,Tajima M,Nishino T,Ogura K.Biochem Biophys Res Commun.1995 7;212(2):681−6.
Claims (22)
- 以下の(a)~(c)のいずれかの遺伝子を含む、変異型カロテノイド産生細菌。
(a)カロテノイド産生細菌における1−デオキシ−D−キシルロース5リン酸合成酵素のアミノ酸配列において、少なくとも第225番目のアミノ酸残基が他のアミノ酸残基に置換された変異型アミノ酸配列を含むタンパク質をコードする遺伝子
(b)カロテノイド産生細菌におけるデカプレニル二リン酸合成酵素のアミノ酸配列において、少なくとも第305番目のアミノ酸残基が他のアミノ酸残基に置換された変異型アミノ酸配列を含むタンパク質をコードする遺伝子
(c)上記(a)及び(b)の両方の遺伝子 - 1−デオキシ−D−キシルロース5リン酸合成酵素のアミノ酸配列が配列番号2に示されるものである請求項1に記載の細菌。
- 第225番目のアミノ酸残基が、グリシンからアスパラギン酸に置換された、請求項1又は2に記載の細菌。
- デカプレニル二リン酸合成酵素のアミノ酸配列が配列番号4に示されるものである請求項1~3のいずれか1項に記載の細菌。
- 第305番目のアミノ酸残基が、アラニンからバリンに置換された、請求項1~4のいずれか1項に記載の細菌。
- 変異型アミノ酸配列を含むタンパク質をコードする遺伝子を有さないカロテノイド産生細菌のカロテノイド産生能よりも高い産生能を獲得した、請求項1~5のいずれか1項に記載の細菌。
- 変異型アミノ酸配列を含むタンパク質をコードする遺伝子を有さないカロテノイド産生細菌のカロテノイド産生量よりも少なくとも5倍以上の量の産生能を獲得した、請求項6に記載の細菌。
- カロテノイド産生細菌がパラコッカス属に属するものである請求項1~7のいずれか1項に記載の細菌。
- パラコッカス属に属する細菌がE−396株である請求項8に記載の細菌。
- カロテノイドがアスタキサンチンである請求項1~9のいずれか1項に記載の細菌。
- 請求項1~10のいずれか1項に記載の細菌を培養し、得られる培養物からカロテノイドを採取することを特徴とするカロテノイドの製造方法。
- カロテノイドの産生量が、変異型アミノ酸配列を含むタンパク質をコードする遺伝子を有さないカロテノイド産生細菌のカロテノイド産生量よりも少なくとも5倍以上の産生量である、請求項11に記載の方法。
- カロテノイドがアスタキサンチンである請求項11又は12に記載の方法。
- カロテノイド産生細菌に変異処理を施し、変異処理された細菌から以下の(a)~(c)のいずれかの特徴を有する細菌を選択することを特徴とする、カロテノイド産生細菌のスクリーニング方法
(a)1−デオキシ−D−キシルロース5リン酸合成酵素の活性が変異処理前の細菌における活性よりも上昇した特徴
(b)デカプレニル二リン酸合成酵素の活性が変異処理前の細菌における活性よりも低下した特徴
(c)上記(a)及び(b)の両方の特徴 - 請求項14に記載の方法により選択された細菌を培養し、得られる培養物からカロテノイドを採取することを特徴とするカロテノイドの製造方法。
- 1−デオキシ−D−キシルロース5リン酸合成酵素のアミノ酸配列において、少なくとも第225番目のアミノ酸残基が他のアミノ酸残基に置換された変異型アミノ酸配列を含むタンパク質をコードする遺伝子。
- 以下の(a)又は(b)のDNAを含む遺伝子。
(a)配列番号5で表される塩基配列を含むDNA
(b)上記(a)のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつ1−デオキシ−D−キシルロース5リン酸合成酵素活性を有するタンパク質をコードするDNA - デカプレニル二リン酸合成酵素のアミノ酸配列において、少なくとも第305番目のアミノ酸残基が他のアミノ酸残基に置換された変異型アミノ酸配列を含むタンパク質をコードする遺伝子。
- 以下の(a)又は(b)のDNAを含む遺伝子。
(a)配列番号7で表される塩基配列を含むDNA
(b)上記(a)のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつデカプレニル二リン酸合成酵素活性が低下したタンパク質をコードするDNA - 以下の(a)~(c)のいずれかの遺伝子を含む組換えベクター。
(a)請求項16又は17に記載の遺伝子
(b)請求項18又は19に記載の遺伝子
(c)上記 (a)及び(b)の遺伝子 - 請求項20に記載の組換えベクターを含む形質転換体。
- 請求項21に記載の形質転換体を培養し、得られる培養物からカロテノイドを採取することを特徴とするカロテノイドの製造方法。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201780020844.4A CN109715796B (zh) | 2016-03-31 | 2017-03-29 | 类胡萝卜素的制造方法 |
| KR1020187026936A KR102316650B1 (ko) | 2016-03-31 | 2017-03-29 | 카로테노이드의 제조 방법 |
| CA3018942A CA3018942A1 (en) | 2016-03-31 | 2017-03-29 | Carotenoid production method |
| US16/088,058 US11268121B2 (en) | 2016-03-31 | 2017-03-29 | Carotenoid production method |
| EP17775629.3A EP3441464A4 (en) | 2016-03-31 | 2017-03-29 | METHOD FOR PRODUCING CAROTINOID |
| AU2017245123A AU2017245123B2 (en) | 2016-03-31 | 2017-03-29 | Carotenoid production method |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016-071303 | 2016-03-31 | ||
| JP2016071303A JP6357186B2 (ja) | 2016-03-31 | 2016-03-31 | カロテノイドの製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017171098A1 true WO2017171098A1 (ja) | 2017-10-05 |
Family
ID=59965870
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/014162 WO2017171098A1 (ja) | 2016-03-31 | 2017-03-29 | カロテノイドの製造方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US11268121B2 (ja) |
| EP (1) | EP3441464A4 (ja) |
| JP (1) | JP6357186B2 (ja) |
| KR (1) | KR102316650B1 (ja) |
| CN (1) | CN109715796B (ja) |
| AU (1) | AU2017245123B2 (ja) |
| CA (1) | CA3018942A1 (ja) |
| TW (1) | TW201738372A (ja) |
| WO (1) | WO2017171098A1 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6357186B2 (ja) * | 2016-03-31 | 2018-07-11 | Jxtgエネルギー株式会社 | カロテノイドの製造方法 |
| US20210346315A1 (en) * | 2018-09-20 | 2021-11-11 | Eneos Corporation | Composition for inhibition or treatment of brain tumors or symptoms attributable thereto |
| KR102323664B1 (ko) * | 2019-04-25 | 2021-11-08 | 부경대학교 산학협력단 | 파라코커스 속 균주를 이용한 사료첨가제 |
| CN110438033B (zh) * | 2019-07-02 | 2021-04-27 | 浙江工业大学 | 一种油脂降解菌、应用及油脂降解方法 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004527265A (ja) * | 2001-06-06 | 2004-09-09 | ロシュ ビタミン アーゲー | 改善されたイソプレノイド産生法 |
| JP2006515174A (ja) * | 2002-12-19 | 2006-05-25 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | 染色体組込みにより細菌において増加するカロテノイド産生 |
| WO2007126639A1 (en) * | 2006-03-31 | 2007-11-08 | E. I. Du Pont De Nemours And Company | Mutant carotenoid ketolase |
| JP2008509689A (ja) * | 2004-08-19 | 2008-04-03 | ディーエスエム アイピー アセッツ ビー.ブイ. | イソプレノイドの生成 |
| JP2009142275A (ja) * | 2007-12-13 | 2009-07-02 | Korea Atom Energ Res Inst | カロチノイドを高濃度で生産する微生物及びそれを使用したカロチノイド生産方法 |
| WO2011115099A1 (ja) * | 2010-03-15 | 2011-09-22 | Jx日鉱日石エネルギー株式会社 | 発酵によるアスタキサンチン製造方法 |
Family Cites Families (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK199887D0 (da) | 1987-04-15 | 1987-04-15 | Danisco Bioteknologi As | Gaerstamme |
| JP3242531B2 (ja) | 1993-07-22 | 2001-12-25 | 日石三菱株式会社 | カロチノイド色素の製造方法 |
| US5607839A (en) | 1993-07-22 | 1997-03-04 | Nippon Oil Company, Ltd. | Bacteria belonging to new genus process for production of carotenoids using same |
| US5935808A (en) | 1997-07-29 | 1999-08-10 | Yissum Research And Development Company Of The Hebrew University Of Jerusalem | Carotenoid-producing bacterial species and process for production of carotenoids using same |
| US7232665B2 (en) | 2002-12-19 | 2007-06-19 | E. I. Du Pont De Nemours And Company | Mutations affecting carotenoid production |
| US7741070B2 (en) | 2003-12-24 | 2010-06-22 | Massachusetts Institute Of Technology | Gene targets for enhanced carotenoid production |
| JP4803739B2 (ja) | 2004-06-04 | 2011-10-26 | キリンホールディングス株式会社 | カロテノイドケトラーゼ及びカロテノイドヒドロキシラーゼ遺伝子を利用したアスタキサンチンまたはその代謝物の製造法 |
| US20060219629A1 (en) | 2005-03-31 | 2006-10-05 | Andrew Noestheden | Oil and water separator |
| JP2006340676A (ja) | 2005-06-10 | 2006-12-21 | Asahi Kasei Corp | アスタキサンチンの製造方法 |
| US20070054351A1 (en) | 2005-09-06 | 2007-03-08 | Yamaha Hatsudoki Kabushiki Kaisha | Green algae having a high astaxanthin content and method for producing the same |
| JP2007097584A (ja) | 2005-09-06 | 2007-04-19 | Yamaha Motor Co Ltd | アスタキサンチン含有量の高い緑藻およびその製造方法 |
| JP2007143492A (ja) | 2005-11-29 | 2007-06-14 | Tosoh Corp | カロテノイド類の生産方法 |
| JP2007244205A (ja) | 2006-03-13 | 2007-09-27 | Tosoh Corp | カロテノイド類の製造方法 |
| JP5066385B2 (ja) | 2007-04-12 | 2012-11-07 | 電源開発株式会社 | アスタキサンチン産生細菌、細菌培養物およびアスタキサンチンの製造方法 |
| KR100936216B1 (ko) | 2008-01-03 | 2010-01-11 | 한국과학기술원 | 코엔자임 q10 생성능을 가지는 재조합 미생물 및 이를이용한 코엔자임 q10의 제조방법 |
| CA2740967C (en) | 2008-10-17 | 2016-02-23 | Kazuaki Hirasawa | Carotenoid fermentation method |
| EP2444415A1 (en) | 2010-10-20 | 2012-04-25 | Genoplante-Valor | 1-Deoxy-D-xylulose 5-phosphate synthase alleles responsible for enhanced terpene biosynthesis |
| JP2012139164A (ja) | 2010-12-28 | 2012-07-26 | Tosoh Corp | 微生物を用いたカロテノイドの製造法 |
| AU2013347891A1 (en) * | 2012-11-21 | 2015-06-11 | Joshua BLAKESLEE | Engineering plants to produce farnesene and other terpenoids |
| KR20170018051A (ko) | 2014-06-13 | 2017-02-15 | 데이노브 | 테르펜 또는 테르페노이드의 생산 방법 |
| JP6357186B2 (ja) * | 2016-03-31 | 2018-07-11 | Jxtgエネルギー株式会社 | カロテノイドの製造方法 |
-
2016
- 2016-03-31 JP JP2016071303A patent/JP6357186B2/ja active Active
-
2017
- 2017-03-29 KR KR1020187026936A patent/KR102316650B1/ko active IP Right Grant
- 2017-03-29 WO PCT/JP2017/014162 patent/WO2017171098A1/ja active Application Filing
- 2017-03-29 AU AU2017245123A patent/AU2017245123B2/en active Active
- 2017-03-29 CN CN201780020844.4A patent/CN109715796B/zh active Active
- 2017-03-29 CA CA3018942A patent/CA3018942A1/en active Pending
- 2017-03-29 TW TW106110500A patent/TW201738372A/zh unknown
- 2017-03-29 US US16/088,058 patent/US11268121B2/en active Active
- 2017-03-29 EP EP17775629.3A patent/EP3441464A4/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004527265A (ja) * | 2001-06-06 | 2004-09-09 | ロシュ ビタミン アーゲー | 改善されたイソプレノイド産生法 |
| JP2006515174A (ja) * | 2002-12-19 | 2006-05-25 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | 染色体組込みにより細菌において増加するカロテノイド産生 |
| JP2008509689A (ja) * | 2004-08-19 | 2008-04-03 | ディーエスエム アイピー アセッツ ビー.ブイ. | イソプレノイドの生成 |
| WO2007126639A1 (en) * | 2006-03-31 | 2007-11-08 | E. I. Du Pont De Nemours And Company | Mutant carotenoid ketolase |
| JP2009142275A (ja) * | 2007-12-13 | 2009-07-02 | Korea Atom Energ Res Inst | カロチノイドを高濃度で生産する微生物及びそれを使用したカロチノイド生産方法 |
| WO2011115099A1 (ja) * | 2010-03-15 | 2011-09-22 | Jx日鉱日石エネルギー株式会社 | 発酵によるアスタキサンチン製造方法 |
Non-Patent Citations (3)
| Title |
|---|
| DATABASE GenBank 23 February 2015 (2015-02-23), XP055429182, Database accession no. JYGY01000003 * |
| IDE, T. ET AL.: "Enhanced production of astaxanthin in Paracoccus sp. strain N-81106 by using random mutagenesis and genetic engineering", BIOCHEM. ENG. J., vol. 65, 2012, pages 37 - 43, XP028423635, ISSN: 1369-703X * |
| See also references of EP3441464A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3441464A4 (en) | 2020-01-08 |
| EP3441464A1 (en) | 2019-02-13 |
| JP2017176099A (ja) | 2017-10-05 |
| AU2017245123A1 (en) | 2018-10-11 |
| US20200299747A1 (en) | 2020-09-24 |
| CA3018942A1 (en) | 2017-10-05 |
| CN109715796B (zh) | 2021-04-09 |
| CN109715796A (zh) | 2019-05-03 |
| JP6357186B2 (ja) | 2018-07-11 |
| KR20180131543A (ko) | 2018-12-10 |
| TW201738372A (zh) | 2017-11-01 |
| KR102316650B1 (ko) | 2021-10-22 |
| US11268121B2 (en) | 2022-03-08 |
| AU2017245123B2 (en) | 2023-05-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10240215B2 (en) | Method for producing astaxanthin by fermentation | |
| WO2017171098A1 (ja) | カロテノイドの製造方法 | |
| CN108431205A (zh) | 牛磺酸在微生物中的异源表达 | |
| Jinendiran et al. | Optimization of submerged fermentation process for improved production of β-carotene by Exiguobacterium acetylicum S01 | |
| US11312981B2 (en) | Carotenoid and amino acid biosynthesis using recombinant corynebacterium glutamicum | |
| AU2011235718B2 (en) | Method of manufacturing zeaxanthin by fermentation | |
| JP6810096B2 (ja) | カロテノイドの製造方法 | |
| KR20220063159A (ko) | 파피아 로도지마의 아스타잔틴 과생산 균주 | |
| CN101675166A (zh) | 参与番茄红素生物合成的基因、含有该基因的重组载体以及带有重组载体的转化的微生物 | |
| JP6291631B2 (ja) | コバルト含有培地によるカロテノイド産生細菌によるカロテノイドの発酵製造方法 | |
| Henke et al. | C50 carotenoids: occurrence, biosynthesis, glycosylation, and metabolic engineering for their overproduction | |
| KR20230129762A (ko) | 바이오라세인 생산능이 증대된 형질전환 메탄올자화균 및 이를 이용한 바이오라세인 생산방법 | |
| Henke et al. | OCCURRENCE, BIOSYNTHESIS, GLYCOSYLATION, AND | |
| Jin et al. | Activated Biosynthetic Genetic Module Chassis for Microbial Zeaxanthin Manufacturing Through Sphingomonasarvum Sp. Nov. Genome Mining |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 20187026936 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3018942 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2017245123 Country of ref document: AU Date of ref document: 20170329 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017775629 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2017775629 Country of ref document: EP Effective date: 20181031 |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17775629 Country of ref document: EP Kind code of ref document: A1 |