WO2017170637A1 - ペプチド誘導体及びその用途 - Google Patents
ペプチド誘導体及びその用途 Download PDFInfo
- Publication number
- WO2017170637A1 WO2017170637A1 PCT/JP2017/012797 JP2017012797W WO2017170637A1 WO 2017170637 A1 WO2017170637 A1 WO 2017170637A1 JP 2017012797 W JP2017012797 W JP 2017012797W WO 2017170637 A1 WO2017170637 A1 WO 2017170637A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- reaction
- group
- derivative
- peptide derivative
- compound
- Prior art date
Links
- BTCMFEAGCIBXHX-VIFPVBQESA-N CC(C)[C@@H](C(NCC(NC)=O)=O)N(C)C Chemical compound CC(C)[C@@H](C(NCC(NC)=O)=O)N(C)C BTCMFEAGCIBXHX-VIFPVBQESA-N 0.000 description 1
- 0 CC[C@](C)[C@](C)[C@@](C1)*=C1N1C=CC[C@]1[C@](C)[C@@](C)C(N[C@@](Cc1ccccc1)c1nc(C(N(C)C/C=S/SCCN(C)C(*=*)=O)=O)c[s]1)=* Chemical compound CC[C@](C)[C@](C)[C@@](C1)*=C1N1C=CC[C@]1[C@](C)[C@@](C)C(N[C@@](Cc1ccccc1)c1nc(C(N(C)C/C=S/SCCN(C)C(*=*)=O)=O)c[s]1)=* 0.000 description 1
- SEEYREPSKCQBBF-UHFFFAOYSA-N CN(C(C=C1)=O)C1=O Chemical compound CN(C(C=C1)=O)C1=O SEEYREPSKCQBBF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/42—Proteins; Polypeptides; Degradation products thereof; Derivatives thereof, e.g. albumin, gelatin or zein
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/07—Tetrapeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/34—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyesters, polyamino acids, polysiloxanes, polyphosphazines, copolymers of polyalkylene glycol or poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/43504—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from invertebrates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
Definitions
- the present invention relates to peptide derivatives and uses thereof.
- a plurality of types of peptide derivatives having cytotoxicity have been isolated from sea snails present in the ocean.
- Dolastatin 10 is known as one of these peptide derivatives and a compound having extremely strong cytotoxicity (Patent Document 1 and Non-Patent Document 1).
- Patent Documents 2 and 3 and Non-Patent Document 2 Another peptide derivative having a similar tubulin polymerization inhibitory action is monomethyl auristatin (Patent Documents 4 and 5), and this compound binds to a specific amino acid of an antibody via a specific structure called a linker. It is known to be used as an antibody drug complex.
- the drug that can be used in the antibody drug complex needs to have a specific substituent (for example, an amino group, a sulfhydryl group, or a hydroxy group) that can be linked to a linker or an antibody.
- a specific substituent for example, an amino group, a sulfhydryl group, or a hydroxy group
- the use of a plurality of types of substituents has been reported so far because the binding method greatly affects the drug efficacy of the complex (Non-Patent Documents 3 to 5).
- a derivative having a functional group that can be used in a conjugate with a tubulin inhibitor the above-mentioned monomethyl auristatin or PF-063801010 (non-patent document 6) having an amino group at the N-terminus or at the C-terminus can be used.
- Auristatin F derivatives having an amino group, hydroxy group, carboxylic acid, hydroxylamine or alkyne (Patent Document 6 and Non-Patent Documents 7 to 9) have been reported.
- Non-patent Document 10 a derivative having ethyl ester, ethylamide or thiazoleamide has been reported.
- an object of the present invention is to provide a dolastatin 10 derivative having a functional group that can be used for forming a complex at the C-terminus.
- the present inventors have found that a novel peptide derivative having a specific functional group at the C-terminus or a pharmacologically acceptable salt thereof can be used against a plurality of cancer cells.
- the inventors have found that it exhibits high cytotoxicity, and have completed the present invention.
- the present invention provides a peptide derivative represented by the following general formula (I) or a pharmacologically acceptable salt thereof.
- X represents an oxygen atom or NR
- Y represents NH 2 , N (Me) H, SH, OH or a phenyl group in which any one hydrogen atom is substituted with NH 2 or OH
- R represents a hydrogen atom or an alkyl group having 1 to 3 carbon atoms.
- a derivative in which X is NH and Y is NH 2 and a derivative in which X is NH and Y is N (Me) H are excluded.
- X is preferably an oxygen atom.
- X is an oxygen atom
- Y is NH 2 , N (Me) H, SH or OH. Is more preferable.
- X is preferably NR.
- X is preferably NR, and R is more preferably an alkyl group having 1 to 3 carbon atoms.
- X is more preferably NR and R is more preferably a methyl group.
- the present invention also provides a complex comprising the peptide derivative represented by the above general formula (I) and a targeting ligand or polymer, or a pharmacologically acceptable salt thereof.
- the present invention provides the peptide derivative represented by the above general formula (I) or a pharmacologically acceptable salt thereof, or the above complex or a pharmacologically acceptable salt thereof as an active ingredient.
- a cytotoxic agent is provided.
- the peptide derivative of the present invention or a pharmacologically acceptable salt thereof has high cytotoxicity, it can be used as a cytotoxic agent.
- the peptide derivative of the present invention has various functional groups at the C-terminus, the peptide derivative or a prodrug thereof and a targeting ligand or polymer can be complexed. Acceptable salts can be used as cytotoxic agents.
- the peptide derivative of the present invention is characterized by being represented by the following general formula (I).
- X represents an oxygen atom or NR
- Y represents NH 2 , N (Me) H, SH, OH or a phenyl group in which any one hydrogen atom is substituted with NH 2 or OH
- R represents a hydrogen atom or an alkyl group having 1 to 3 carbon atoms.
- a derivative in which X is NH and Y is NH 2 and a derivative in which X is NH and Y is N (Me) H are excluded.
- a phenyl group in which any one hydrogen atom is substituted with NH 2 or OH means 2-aminophenyl group, 3-aminophenyl group, 4-aminophenyl group, 2-hydroxyphenyl group, 3-hydroxy A phenyl group or a 4-hydroxyphenyl group is meant.
- Alkyl group having 1 to 3 carbon atoms means a methyl group, an ethyl group, a propyl group, or an isopropyl group.
- the compounds described in Table 1 also include pharmacologically acceptable salts thereof.
- the peptide derivative represented by the above general formula (I) contains a conformer, a rotational isomer, a tautomer, an optical isomer, a diastereomer, an epimer, or the like. In some cases, either isomer or mixture is included in the peptide derivative (I). Furthermore, when the peptide derivative (I) has an optical isomer, the optical isomer resolved from the racemate is also included in the peptide derivative (I).
- the configuration of the peptide derivative (I) is preferably the following formula (II).
- the present invention also includes a prodrug of the peptide derivative (I) or a pharmacologically acceptable salt thereof.
- the prodrug of the peptide derivative (I) is a compound that is enzymatically or chemically converted into the peptide derivative (I) in vivo.
- the active substance of the prodrug of peptide derivative (I) is peptide derivative (I), but the prodrug itself of peptide derivative (I) may have activity.
- Examples of the prodrug of the peptide derivative (I) include compounds in which the hydroxy group of the peptide derivative (I) is esterified, carbonated, carbamated, alkylated, phosphorylated or borated. These compounds can be synthesized from the peptide derivative (I) or a synthetic intermediate thereof according to a known method.
- Examples of the prodrug of the peptide derivative (I) include compounds in which the amino group of the peptide derivative (I) is carbamate- ed or amidated. These compounds can be synthesized from the peptide derivative (I) or a synthetic intermediate thereof according to a known method.
- Examples of the prodrug of the peptide derivative (I) include a compound in which the sulfhydryl group of the peptide derivative (I) forms a disulfide bond. These compounds can be synthesized from the peptide derivative (I) or a synthetic intermediate thereof according to a known method.
- prodrug of the above peptide derivative (I) are shown in Table 2, but the present invention is not limited thereto.
- the compounds described in Table 2 also include their salts.
- Q is a known method (for example, Ellen M. Sletten et al., Agewante Chimie International Edition, 2009, Vol. 48, p. 6974-6998, Greg T. Hermanson., “Bioconjugate Tech”, “Bioconjugate Tech,” , Xi Chem et al., Organic & Biomolecular Chemistry, 2016, Vol. 14, p.
- the “spacer” represents a peptide derivative (I) or a structure connecting a prodrug of the peptide derivative (I) and Q.
- “Straight or branched alkyl group having 1 to 12 carbon atoms” means, for example, — (CH 2 ) n —, —CH (Me) —, —C (Me) 2 —, — (CH 2 ) o CH (Me)-, -CH (Me) (CH 2 ) o -,-(CH 2 ) p C (Me) 2 -or -C (Me) 2 (CH 2 ) p- .
- n represents an integer of 1 to 12
- o represents an integer of 1 to 10
- p represents an integer of 1 to 9.
- PEG represents a linear polyethylene glycol having a number average molecular weight of 200 to 2,000 represented by a repeating structure of — (CH 2 CH 2 O) m —, and m represents an integer of 5 to 45.
- amino acids and the like are indicated by abbreviations, they are based on abbreviations by IUPAC-IUB Commission on Biochemical Nomenclature or conventional abbreviations in the field, and examples thereof are described below.
- L forms are indicated (for example, “Lys” is L form Lys).
- D- indicates D form (eg, “D-Lys” indicates D form Lys)
- DL- indicates D form and L form racemates.
- DL-Lys is a racemic form of D-form Lys and L-form Lys).
- Amino acid means DL-Ala, DL-Arg, DL-Asn, DL-Asp, DL-Cit, DL-Cys, DL-Gln, DL-Glu, DL-Gly, DL-His, DL-Ile, DL -One selected from Leu, DL-Lys, DL-Met, DL-Phe, DL-Pro, DL-Ser, DL-Thr, DL-Trp, DL-Tyr or DL-Val, but A 1 Lys, A 2 and A 3 are absent, A 1 is Cit or Lys, A 2 is Val or Phe, A 3 is absent, or A 1 is Asp Yes, A 2 and A 3 are preferably Ala.
- a 1 described in Table 2 is bonded to NH of the phenyl group at the carbonyl terminal, and A 1 , A 2 and A 3 are each bonded to the main chain via an amide bond.
- Table 3 shows preferred structures of the prodrug of the peptide derivative (I) described in No. 6 in Table 2, but the present invention is not limited thereto.
- the compounds listed in Table 3 also include their salts.
- Alk 1 and Alk 2 each independently represent a linear or branched alkyl group having 1 to 12 carbon atoms, and E is absent or —C ( ⁇ O) N (Z) — or —N (Z) C ( ⁇ O) — is represented, and other symbols are the same as defined above.
- Alk 1 is more preferably — (CH 2 ) 2 —, X is NR, and Alk 1 is — (CH 2 ) 2. More preferably, E is —N (Z) C ( ⁇ O) —, PEG is — (CH 2 CH 2 O) 12 —, and Q is a maleimide group, The present invention is not limited to these.
- J is -Alk 1 -E-Alk 2 -or -Alk 1 -E-PEG- (CH 2 ) 2 -E-Alk 2 It is preferable that J is — (CH 2 ) 2 —N (Z) C ( ⁇ O) —Alk 2 — or — (CH 2 ) 2 —N (Z) C ( ⁇ O) — (CH 2 CH 2 O) 12 — (CH 2 ) 2 —N (Z) C ( ⁇ O) —Alk 2 — and Q is more preferably a maleimide group, but the present invention is not limited thereto. is not.
- prodrugs of the peptide derivative (I) are known in the literature (“Development of Pharmaceuticals”, Hirokawa Shoten, 1990, Vol. 7, p.163-198, and Progress in Medicine, Vol. 5, 1985, p. 2157 to 2161) may be converted to the peptide derivative (I) under the physiological conditions described in 2157-2161).
- the peptide derivative (I) may be labeled with an isotope.
- the labeled isotope include 2 H, 3 H, 13 C, 14 C, 15 N, 15 O, 17 O, and 18 O. And / or 125 I.
- Examples of the “pharmacologically acceptable salt” of the peptide derivative (I) include inorganic acid salts such as hydrochloride, sulfate, nitrate, hydrobromide, hydroiodide or phosphate, or Oxalate, malonate, citrate, fumarate, lactate, malate, succinate, tartrate, acetate, trifluoroacetate, maleate, gluconate, benzoate, Ascorbate, glutarate, mandelate, phthalate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, camphorsulfonate, aspartate, glutamate or Organic acid salts such as cinnamate are exemplified, and hydrochloride, sulfate, hydrobromide, maleate, benzoate or methanesulfonate is preferable.
- inorganic acid salts such as hydrochlor
- the peptide derivative (I) or a pharmacologically acceptable salt thereof may be an anhydride, or may form a solvate such as a hydrate.
- the solvate is preferably a pharmacologically acceptable solvate.
- the pharmacologically acceptable solvate may be either a hydrate or a non-hydrate, but a hydrate is preferable.
- the solvent constituting the solvate include alcohol solvents such as methanol, ethanol, and n-propanol, N, N-dimethylformamide, dimethyl sulfoxide, and water.
- the peptide derivative (I) can be produced by an appropriate method based on characteristics derived from the basic skeleton and the type of substituent.
- the starting materials and reagents used in the production of these compounds can be generally purchased, or can be produced by a known method or a method analogous thereto.
- the peptide derivative (I) and intermediates and starting materials used for the production thereof can be isolated and purified by known means.
- Known means for isolation and purification include, for example, solvent extraction, recrystallization or chromatography.
- each isomer can be obtained as a single compound by a known method.
- Known methods include, for example, crystallization, enzyme resolution, or chiral chromatography.
- a protective group may be introduced into these groups, and protection is performed as necessary after the reaction.
- the target compound can be obtained by deprotecting the group.
- Examples of the protective group for the hydroxy group include a trityl group, a tetrahydropyranyl group, an aralkyl group having 7 to 10 carbon atoms (for example, a benzyl group) or a substituted silyl group (for example, a trimethylsilyl group, a triethylsilyl group, or tert-butyldimethyl). Silyl group).
- amino-protecting group examples include an alkylcarbonyl group having 2 to 6 carbon atoms (for example, acetyl group), a benzoyl group, an alkyloxycarbonyl group having 2 to 8 carbon atoms (for example, tert-butoxycarbonyl group or benzyloxy group) Carbonyl group), an aralkyl group having 7 to 10 carbon atoms (for example, benzyl group) or a phthaloyl group.
- alkylcarbonyl group having 2 to 6 carbon atoms for example, acetyl group
- benzoyl group an alkyloxycarbonyl group having 2 to 8 carbon atoms (for example, tert-butoxycarbonyl group or benzyloxy group) Carbonyl group)
- an aralkyl group having 7 to 10 carbon atoms for example, benzyl group
- a phthaloyl group examples include an alkylcarbonyl group having 2 to 6 carbon atoms (for example
- Examples of the protecting group for the sulfhydryl group include a trityl group, a 2-mercaptopyridyl group, and a 2-mercapto-5-nitropyridyl group.
- Examples of the protecting group for the carboxyl group include an alkyl group having 1 to 6 carbon atoms (for example, a methyl group, an ethyl group, or a tert-butyl group) or an aralkyl group having 7 to 10 carbon atoms (for example, a benzyl group).
- the deprotection of the protecting group varies depending on the type of the protecting group, but is performed according to a known method (for example, Greene, TW, “Green's Protective Groups in Organic Synthesis”, Wiley-Interscience) or a method equivalent thereto. be able to.
- the peptide derivative (I) can be obtained, for example, by deprotecting the protected peptide derivative (III) as shown in Scheme 1.
- PG represents a protecting group
- X is NR
- Y represents NH 2 or SH
- Y ′ represents NH or S.
- X is an oxygen atom
- Y represents NH 2 , SH, OH, or a phenyl group in which any one hydrogen atom is substituted with NH 2 or OH
- Y ′ represents NH, S, O, or any one of It represents a phenyl group in which a hydrogen atom is substituted with NH or O.
- the deprotection of the protecting group varies depending on the type of the protecting group, but is performed according to a known method (for example, Greene, TW, “Green's Protective Groups in Organic Synthesis”, Wiley-Interscience) or a method equivalent thereto. be able to.
- the protected peptide derivative (III) can be obtained, for example, by a condensation reaction of a carboxylic acid derivative (IV) and a nucleophile (V) as shown in Scheme 2. [Wherein each symbol is the same as defined above. ]
- the amount of the nucleophile (V) used in the condensation reaction is preferably 0.5 to 10 equivalents, more preferably 1 to 3 equivalents, relative to the carboxylic acid derivative (IV).
- Examples of the condensing agent used in the condensation reaction include ethyl chloroformate, oxalyl chloride, 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, hexafluorophosphoric acid 2- (7-aza-1H-benzotriazole- 1-yl) -1,1,3,3-tetramethyluronium, O- (benzotriazol-1-yl) -N, N, N′N′-tetramethyluronium hexafluorophosphate, 1H-benzo Examples include triazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate or 2-methyl-6-nitrobenzoic anhydride, ethyl chloroformate, hexafluorophosphate 2- (7-aza-1H-benzotriazole -1-yl) -1,1,3,3-tetramethyluronium or 2-methyl-6- Toro benzoic anhydr
- the amount of the condensing agent used in the condensation reaction is preferably 0.1 to 100 equivalents, more preferably 0.3 to 30 equivalents, relative to the carboxylic acid derivative (IV).
- the reaction solvent used for the condensation reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethylacetamide Aprotic polar solvents such as N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate, dichloromethane , Chlorinated solvents such as chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, or mixed solvents thereof, including chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, or N , - dimethylformamide, N, N- dimethylacetamide, apro
- a base may be used if desired.
- the base to be used include inorganic bases such as sodium hydride, sodium bicarbonate or potassium carbonate, organic bases such as triethylamine, diisopropylethylamine, 4-dimethylaminopyridine or pyridine, or a mixture thereof.
- organic bases such as ethylamine, 4-dimethylaminopyridine or pyridine or mixtures thereof are preferred.
- the amount of base used in the condensation reaction is preferably 0.5 to 20 equivalents, more preferably 1 to 5 equivalents, relative to the carboxylic acid derivative (IV).
- the reaction temperature of the condensation reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the condensation reaction is appropriately selected according to the reaction temperature and other conditions, but is preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (IV) used for the condensation reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- the nucleophilic agent (V) can be purchased or synthesized by a known method or a method analogous thereto.
- the peptide derivative (I) can also be obtained, for example, by a condensation reaction between a carboxylic acid derivative (IV) and a nucleophile (VI) as shown in Scheme 3.
- X represents NR
- Y represents OH or a phenyl group in which any one hydrogen atom is substituted with NH 2 or OH, and other symbols are the same as defined above].
- the amount of the nucleophile (VI) used in the condensation reaction is preferably 0.5 to 10 equivalents, more preferably 1 to 3 equivalents, relative to the carboxylic acid derivative (IV).
- Examples of the condensing agent used in the condensation reaction include ethyl chloroformate, oxalyl chloride, 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, hexafluorophosphoric acid 2- (7-aza-1H-benzotriazole- 1-yl) -1,1,3,3-tetramethyluronium, O- (benzotriazol-1-yl) -N, N, N′N′-tetramethyluronium hexafluorophosphate or 1H-benzo Triazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate, such as ethyl chloroformate or hexafluorophosphate 2- (7-aza-1H-benzotriazol-1-yl) -1,1,3 3-tetramethyluronium is preferred.
- the amount of the condensing agent used in the condensation reaction is preferably 0.1 to 100 equivalents, more preferably 0.3 to 30 equivalents, relative to the carboxylic acid derivative (IV).
- the reaction solvent used for the condensation reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethylacetamide Aprotic polar solvents such as N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate, dichloromethane , Chlorinated solvents such as chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, or mixed solvents thereof, including chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, or N , - dimethylformamide, N, N- dimethylacetamide, apro
- a base may be used if desired.
- the base to be used include inorganic bases such as sodium hydride, sodium bicarbonate or potassium carbonate, organic bases such as triethylamine, diisopropylethylamine, 4-dimethylaminopyridine or pyridine, or a mixture thereof.
- organic bases such as ethylamine, 4-dimethylaminopyridine or pyridine or mixtures thereof are preferred.
- the amount of base used in the condensation reaction is preferably 0.5 to 20 equivalents, more preferably 1 to 5 equivalents, relative to the carboxylic acid derivative (IV).
- the reaction temperature of the condensation reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the condensation reaction is appropriately selected according to the reaction temperature and other conditions, but is preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (IV) used for the condensation reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- the nucleophilic agent (VI) can be purchased or synthesized by a known method or a method analogous thereto.
- the carboxylic acid derivative (IV) can be obtained, for example, by deprotecting the ester derivative (VII) as shown in Scheme 4. [Wherein each symbol is the same as defined above. ]
- the deprotection of the protecting group varies depending on the type of the protecting group, but is performed according to a known method (for example, Greene, TW, “Green's Protective Groups in Organic Synthesis”, Wiley-Interscience) or a method equivalent thereto. be able to.
- the ester derivative (VII) can be synthesized by a known method or a method analogous thereto.
- the protected phenoxycarbamate derivative (IX) can be obtained, for example, by a condensation reaction of a phenol derivative (Ia) and an electrophile (VIII-a) or (VIII-b) as shown in Scheme 5. it can. [Wherein each symbol is the same as defined above. ]
- the amount of the electrophile (VIII-a) or (VIII-b) used in the condensation reaction is preferably 0.5 to 10 equivalents, more preferably 1 to 3 equivalents, relative to the phenol derivative (Ia).
- the reaction solvent used for the condensation reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethylacetamide Aprotic polar solvents such as N-methyl-2-pyrrolidone, pyridine or dimethylsulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate
- a chlorinated solvent such as dichloromethane, chloroform or 1,2-dichloroethane, a nitrile solvent such as acetonitrile or propionitrile, or a mixed solvent thereof, but a chlorinated solvent such as dichloromethane, chloroform or 1,2-dichloroethane.
- a base may be used if desired.
- the base to be used include inorganic bases such as sodium hydride, sodium hydrogen carbonate or potassium carbonate, organic bases such as triethylamine, diisopropylethylamine or pyridine, or mixtures thereof, and organic bases such as triethylamine, diisopropylethylamine or pyridine.
- a base is preferred.
- the amount of the base used in the condensation reaction is preferably 0.5 to 20 equivalents, more preferably 1 to 5 equivalents, relative to the phenol derivative (Ia).
- the reaction temperature of the condensation reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the condensation reaction is appropriately selected according to the reaction temperature and other conditions, but is preferably 30 minutes to 30 hours.
- the concentration of the phenol derivative (Ia) used for the condensation reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- the electrophile (VIII-a) or (VIII-b) can be generated from a known amine derivative or carboxylic acid derivative by a known method or a method analogous thereto or in a reaction system.
- the protected polyethylene glycol derivative (XI) can be used, for example, as shown in Scheme 6 from basic to medium deprotection of the protected phenoxycarbamate derivative (IX) followed by carboxylic acid derivative (X). Coupling reaction (when G is a succinimidyl group or p-nitrophenyl group) or a condensation reaction (when G is a hydrogen atom). [Wherein G represents a hydrogen atom, a succinimidyl group or a p-nitrophenyl group, and other symbols are the same as defined above. ]
- the deprotection of the protecting group varies depending on the type of the protecting group, but is performed according to a known method (for example, Greene, TW, “Green's Protective Groups in Organic Synthesis”, Wiley-Interscience) or a method equivalent thereto. be able to.
- the amount of the carboxylic acid derivative (X) used in the coupling reaction or condensation reaction is preferably 0.5 to 10 equivalents, more preferably 0.5 to 4 equivalents, relative to the protected phenoxycarbamate derivative (IX).
- the pH of the coupling reaction can be adjusted with a base.
- the base to be used include organic bases such as triethylamine or diisopropylethylamine, inorganic bases such as sodium hydrogencarbonate or potassium carbonate, metal hydride compounds such as sodium hydride, potassium hydride or calcium hydride, methyllithium or butyllithium.
- lithium amides such as lithium hexamethyldisilazide or lithium diisopropylamide or a mixture thereof
- an inorganic base such as sodium hydrogen carbonate or potassium carbonate or an organic base such as triethylamine or diisopropylethylamine is preferred.
- the amount of the base used for the coupling reaction is preferably 0.001 to 10 equivalents, more preferably 0.001 to 4 equivalents, relative to the protected phenoxycarbamate derivative (IX).
- the pH of the coupling reaction can also be adjusted with a buffer solution.
- the buffer used include a phosphate buffer, a citrate buffer, a citrate phosphate buffer, a borate buffer, a tartrate buffer, or a Tris buffer, and have a pH of 7.0 to 8.0.
- a buffer is preferred.
- the concentration of the buffer used for the coupling reaction is preferably 10 mmol / L to 1 mol / L.
- the reaction solvent used in the coupling reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethyl Aprotic polar solvents such as acetamide, N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate
- Examples include chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, water or mixed solvents thereof, but chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane.
- nitrile solvents such as acetonitrile or propion
- the reaction temperature of the coupling reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the coupling reaction is appropriately selected according to the reaction temperature and other conditions, but preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (X) used for the coupling reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- Examples of the condensing agent used in the condensation reaction include carbodiimides such as dicyclohexylcarbodiimide, diisopropylcarbodiimide, or 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, ethyl chloroformate, oxalyl chloride, hexafluorophosphate 2- ( 7-aza-1H-benzotriazol-1-yl) -1,1,3,3-tetramethyluronium, O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyl
- Examples include uronium hexafluorophosphate, 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate, or 2-methyl-6-nitrobenzoic anhydride, but dicyclohexylcarbodiimide, diisopropylcarbodiimi
- the amount of the condensing agent used in the condensation reaction is preferably 0.1 to 100 equivalents, more preferably 0.3 to 30 equivalents, relative to the carboxylic acid derivative (X).
- the reaction solvent used for the condensation reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethylacetamide Aprotic polar solvents such as N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate, dichloromethane , Chlorinated solvents such as chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, or mixed solvents thereof, including chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, or N , - dimethylformamide, N, N- dimethylacetamide, apro
- a base may be used if desired.
- the base to be used include inorganic bases such as sodium hydride, sodium bicarbonate or potassium carbonate, organic bases such as triethylamine, diisopropylethylamine, 4-dimethylaminopyridine or pyridine, or a mixture thereof.
- organic bases such as ethylamine, 4-dimethylaminopyridine or pyridine or mixtures thereof are preferred.
- the amount of base used in the condensation reaction is preferably 0.5 to 20 equivalents, more preferably 1 to 5 equivalents, relative to the carboxylic acid derivative (X).
- the reaction temperature of the condensation reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the condensation reaction is appropriately selected according to the reaction temperature and other conditions, but is preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (X) used for the condensation reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- the carboxylic acid derivative (X) can be purchased or synthesized by a known method or a method analogous thereto.
- the prodrug (XIII) of the peptide derivative (I) is prepared by, for example, deprotection of the protected phenoxycarbamate derivative (IX) followed by the basic reaction with the carboxylic acid derivative (XII) as shown in Scheme 7.
- a coupling reaction under neutral conditions when G is a succinimidyl group or p-nitrophenyl group
- a condensation reaction when G is a hydrogen atom.
- the deprotection of the protecting group varies depending on the type of the protecting group, but is performed according to a known method (for example, Greene, TW, “Green's Protective Groups in Organic Synthesis”, Wiley-Interscience) or a method equivalent thereto. be able to.
- the amount of the carboxylic acid derivative (XII) used in the coupling reaction or condensation reaction is preferably 0.5 to 10 equivalents, more preferably 0.5 to 4 equivalents, relative to the protected phenoxycarbamate derivative (IX).
- the pH of the coupling reaction can be adjusted with a base.
- the base to be used include organic bases such as triethylamine or diisopropylethylamine, inorganic bases such as sodium hydrogencarbonate or potassium carbonate, metal hydride compounds such as sodium hydride, potassium hydride or calcium hydride, methyllithium or butyllithium.
- lithium amides such as lithium hexamethyldisilazide or lithium diisopropylamide or a mixture thereof
- an inorganic base such as sodium hydrogen carbonate or potassium carbonate or an organic base such as triethylamine or diisopropylethylamine is preferred.
- the amount of the base used for the coupling reaction is preferably 0.001 to 10 equivalents, more preferably 0.001 to 4 equivalents, relative to the protected phenoxycarbamate derivative (IX).
- the pH of the coupling reaction can also be adjusted with a buffer solution.
- the buffer used include a phosphate buffer, a citrate buffer, a citrate phosphate buffer, a borate buffer, a tartrate buffer, or a Tris buffer, and have a pH of 7.0 to 8.0.
- a buffer is preferred.
- the concentration of the buffer used for the coupling reaction is preferably 10 mmol / L to 1 mol / L.
- the reaction solvent used in the coupling reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethyl Aprotic polar solvents such as acetamide, N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate
- Examples include chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, water or mixed solvents thereof, but chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane.
- nitrile solvents such as acetonitrile or propion
- the reaction temperature of the coupling reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the coupling reaction is appropriately selected according to the reaction temperature and other conditions, but preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (XII) used for the coupling reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- Examples of the condensing agent used in the condensation reaction include carbodiimides such as dicyclohexylcarbodiimide, diisopropylcarbodiimide, or 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, ethyl chloroformate, oxalyl chloride, hexafluorophosphate 2- ( 7-aza-1H-benzotriazol-1-yl) -1,1,3,3-tetramethyluronium, O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyl
- Examples include uronium hexafluorophosphate, 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate, or 2-methyl-6-nitrobenzoic anhydride, but dicyclohexylcarbodiimide, diisopropylcarbodiimi
- the amount of the condensing agent used in the condensation reaction is preferably 0.1 to 100 equivalents, more preferably 0.3 to 30 equivalents, relative to the carboxylic acid derivative (XII).
- the reaction solvent used for the condensation reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethylacetamide Aprotic polar solvents such as N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate, dichloromethane , Chlorinated solvents such as chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, or mixed solvents thereof, including chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, or N , - dimethylformamide, N, N- dimethylacetamide, apro
- a base may be used if desired.
- the base to be used include inorganic bases such as sodium hydride, sodium bicarbonate or potassium carbonate, organic bases such as triethylamine, diisopropylethylamine, 4-dimethylaminopyridine or pyridine, or a mixture thereof.
- organic bases such as ethylamine, 4-dimethylaminopyridine or pyridine or mixtures thereof are preferred.
- the amount of base used in the condensation reaction is preferably 0.5 to 20 equivalents, more preferably 1 to 5 equivalents, relative to the carboxylic acid derivative (XII).
- the reaction temperature of the condensation reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the condensation reaction is appropriately selected according to the reaction temperature and other conditions, but is preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (XII) used for the condensation reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- the carboxylic acid derivative (XII) can be purchased, or can be synthesized by a known method or a method analogous thereto.
- the prodrug (XIII) of the peptide derivative (I) is a basic compound of the protected polyethylene glycol derivative (XI) followed by the deprotection reaction with the carboxylic acid derivative (XIV) as shown in Scheme 8, for example.
- a coupling reaction under neutral conditions when G is a succinimidyl group or p-nitrophenyl group
- a condensation reaction when G is a hydrogen atom.
- the deprotection of the protecting group varies depending on the type of the protecting group, but is performed according to a known method (for example, Greene, TW, “Green's Protective Groups in Organic Synthesis”, Wiley-Interscience) or a method equivalent thereto. be able to.
- the amount of the carboxylic acid derivative (XIV) used in the coupling reaction or condensation reaction is preferably 0.5 to 10 equivalents, more preferably 0.5 to 4 equivalents, relative to the protected polyethylene glycol derivative (XI).
- the pH of the coupling reaction can be adjusted with a base.
- the base to be used include organic bases such as triethylamine or diisopropylethylamine, inorganic bases such as sodium hydrogencarbonate or potassium carbonate, metal hydride compounds such as sodium hydride, potassium hydride or calcium hydride, methyllithium or butyllithium.
- lithium amides such as lithium hexamethyldisilazide or lithium diisopropylamide or a mixture thereof
- an inorganic base such as sodium hydrogen carbonate or potassium carbonate or an organic base such as triethylamine or diisopropylethylamine is preferred.
- the amount of base used in the coupling reaction is preferably 0.001 to 10 equivalents, more preferably 0.001 to 4 equivalents, relative to the protected polyethylene glycol derivative (XI).
- the pH of the coupling reaction can also be adjusted with a buffer solution.
- the buffer used include a phosphate buffer, a citrate buffer, a citrate phosphate buffer, a borate buffer, a tartrate buffer, or a Tris buffer, and have a pH of 7.0 to 8.0.
- a buffer is preferred.
- the concentration of the buffer used for the coupling reaction is preferably 10 mmol / L to 1 mol / L.
- the reaction solvent used in the coupling reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethyl Aprotic polar solvents such as acetamide, N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate
- Examples include chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, water or mixed solvents thereof, but chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane.
- nitrile solvents such as acetonitrile or propion
- the reaction temperature of the coupling reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the coupling reaction is appropriately selected according to the reaction temperature and other conditions, but preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (XIV) used for the coupling reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- Examples of the condensing agent used in the condensation reaction include carbodiimides such as dicyclohexylcarbodiimide, diisopropylcarbodiimide, or 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, ethyl chloroformate, oxalyl chloride, hexafluorophosphate 2- ( 7-aza-1H-benzotriazol-1-yl) -1,1,3,3-tetramethyluronium, O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyl
- Examples include uronium hexafluorophosphate, 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate, or 2-methyl-6-nitrobenzoic anhydride, but dicyclohexylcarbodiimide, diisopropylcarbodiimi
- the amount of the condensing agent used in the condensation reaction is preferably from 0.1 to 100 equivalents, more preferably from 0.3 to 30 equivalents, based on the carboxylic acid derivative (XIV).
- the reaction solvent used for the condensation reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethylacetamide Aprotic polar solvents such as N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate, dichloromethane , Chlorinated solvents such as chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, or mixed solvents thereof, including chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, or N , - dimethylformamide, N, N- dimethylacetamide, apro
- a base may be used if desired.
- the base to be used include inorganic bases such as sodium hydride, sodium bicarbonate or potassium carbonate, organic bases such as triethylamine, diisopropylethylamine, 4-dimethylaminopyridine or pyridine, or a mixture thereof.
- organic bases such as ethylamine, 4-dimethylaminopyridine or pyridine or mixtures thereof are preferred.
- the amount of the base used in the condensation reaction is preferably 0.5 to 20 equivalents, more preferably 1 to 5 equivalents, relative to the carboxylic acid derivative (XIV).
- the reaction temperature of the condensation reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the condensation reaction is appropriately selected according to the reaction temperature and other conditions, but is preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (XIV) used for the condensation reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- the carboxylic acid derivative (XIV) can be purchased or synthesized by a known method or a method analogous thereto.
- the prodrug (XV) of the peptide derivative (I) is a basic compound of the protected phenoxycarbamate derivative (IX) followed by the deprotection reaction with the carboxylic acid derivative (XIV), for example, as shown in Scheme 9.
- a coupling reaction under neutral conditions when G is a succinimidyl group or p-nitrophenyl group
- a condensation reaction when G is a hydrogen atom.
- the deprotection of the protecting group varies depending on the type of the protecting group, but is performed according to a known method (for example, Greene, TW, “Green's Protective Groups in Organic Synthesis”, Wiley-Interscience) or a method equivalent thereto. be able to.
- the amount of the carboxylic acid derivative (XIV) used in the coupling reaction or condensation reaction is preferably 0.5 to 10 equivalents, more preferably 0.5 to 4 equivalents, relative to the protected phenoxycarbamate derivative (IX).
- the pH of the coupling reaction can be adjusted with a base.
- the base to be used include organic bases such as triethylamine or diisopropylethylamine, inorganic bases such as sodium hydrogencarbonate or potassium carbonate, metal hydride compounds such as sodium hydride, potassium hydride or calcium hydride, methyllithium or butyllithium.
- lithium amides such as lithium hexamethyldisilazide or lithium diisopropylamide or a mixture thereof
- an inorganic base such as sodium hydrogen carbonate or potassium carbonate or an organic base such as triethylamine or diisopropylethylamine is preferred.
- the amount of the base used for the coupling reaction is preferably 0.001 to 10 equivalents, more preferably 0.001 to 4 equivalents, relative to the protected phenoxycarbamate derivative (IX).
- the pH of the coupling reaction can also be adjusted with a buffer solution.
- the buffer used include a phosphate buffer, a citrate buffer, a citrate phosphate buffer, a borate buffer, a tartrate buffer, or a Tris buffer, and have a pH of 7.0 to 8.0.
- a buffer is preferred.
- the concentration of the buffer used for the coupling reaction is preferably 10 mmol / L to 1 mol / L.
- the reaction solvent used in the coupling reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethyl Aprotic polar solvents such as acetamide, N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate
- Examples include chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, water or mixed solvents thereof, but chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane.
- nitrile solvents such as acetonitrile or propion
- the reaction temperature of the coupling reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the coupling reaction is appropriately selected according to the reaction temperature and other conditions, but preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (XIV) used for the coupling reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- Examples of the condensing agent used in the condensation reaction include carbodiimides such as dicyclohexylcarbodiimide, diisopropylcarbodiimide, or 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, ethyl chloroformate, oxalyl chloride, hexafluorophosphate 2- ( 7-aza-1H-benzotriazol-1-yl) -1,1,3,3-tetramethyluronium, O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyl
- Examples include uronium hexafluorophosphate, 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate, or 2-methyl-6-nitrobenzoic anhydride, but dicyclohexylcarbodiimide, diisopropylcarbodiimi
- the amount of the condensing agent used in the condensation reaction is preferably from 0.1 to 100 equivalents, more preferably from 0.3 to 30 equivalents, based on the carboxylic acid derivative (XIV).
- the reaction solvent used for the condensation reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethylacetamide Aprotic polar solvents such as N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate, dichloromethane , Chlorinated solvents such as chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, or mixed solvents thereof, including chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, or N , - dimethylformamide, N, N- dimethylacetamide, apro
- a base may be used if desired.
- the base to be used include inorganic bases such as sodium hydride, sodium bicarbonate or potassium carbonate, organic bases such as triethylamine, diisopropylethylamine, 4-dimethylaminopyridine or pyridine, or a mixture thereof.
- organic bases such as ethylamine, 4-dimethylaminopyridine or pyridine or mixtures thereof are preferred.
- the amount of the base used in the condensation reaction is preferably 0.5 to 20 equivalents, more preferably 1 to 5 equivalents, relative to the carboxylic acid derivative (XIV).
- the reaction temperature of the condensation reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the condensation reaction is appropriately selected according to the reaction temperature and other conditions, but is preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (XIV) used for the condensation reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- the disulfide derivative (XVII) can be obtained, for example, by a condensation reaction between a carboxylic acid derivative (IV) and a nucleophile (XVI) as shown in Scheme 10.
- W represents a 2-mercaptopyridyl group, a 2-mercapto-5-nitropyridyl group or -Alk 1 -N (Z) (PG), and other symbols are the same as defined above.
- the amount of the nucleophile (XVI) used for the condensation reaction is preferably 0.5 to 10 equivalents, more preferably 1 to 3 equivalents, relative to the carboxylic acid derivative (IV).
- Examples of the condensing agent used in the condensation reaction include ethyl chloroformate, oxalyl chloride, 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, hexafluorophosphoric acid 2- (7-aza-1H-benzotriazole- 1-yl) -1,1,3,3-tetramethyluronium, O- (benzotriazol-1-yl) -N, N, N′N′-tetramethyluronium hexafluorophosphate, 1H-benzo Examples include triazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate or 2-methyl-6-nitrobenzoic acid anhydride, such as ethyl chloroformate, hexafluorophosphate 2- (7-aza-1H-benzotriazole -1-yl) -1,1,3,3-tetramethyluronium, 1H-benzotria Lum
- the amount of the condensing agent used in the condensation reaction is preferably 0.1 to 100 equivalents, more preferably 0.3 to 30 equivalents, relative to the carboxylic acid derivative (IV).
- the reaction solvent used for the condensation reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethylacetamide Aprotic polar solvents such as N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate, dichloromethane , Chlorinated solvents such as chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, or mixed solvents thereof, including chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, or N , - dimethylformamide, N, N- dimethylacetamide, apro
- a base may be used if desired.
- the base to be used include inorganic bases such as sodium hydride, sodium bicarbonate or potassium carbonate, organic bases such as triethylamine, diisopropylethylamine, 4-dimethylaminopyridine or pyridine, or a mixture thereof.
- organic bases such as ethylamine, 4-dimethylaminopyridine or pyridine or mixtures thereof are preferred.
- the amount of base used in the condensation reaction is preferably 0.5 to 20 equivalents, more preferably 1 to 5 equivalents, relative to the carboxylic acid derivative (IV).
- the reaction temperature of the condensation reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the condensation reaction is appropriately selected according to the reaction temperature and other conditions, but is preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (IV) used for the condensation reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- the nucleophile (XVI) can be purchased or synthesized by a known method or a method analogous thereto.
- the protected polyethylene glycol derivative (XVIII) can be obtained, for example, from basic to neutral conditions by deprotection of the disulfide derivative (XVII-a) and subsequent carboxylic acid derivative (X) as shown in Scheme 11. It can be obtained by a coupling reaction (when G is a succinimidyl group or a p-nitrophenyl group) or a condensation reaction (when G is a hydrogen atom). [Wherein each symbol is the same as defined above. ]
- the deprotection of the protecting group varies depending on the type of the protecting group, but is performed according to a known method (for example, Greene, TW, “Green's Protective Groups in Organic Synthesis”, Wiley-Interscience) or a method equivalent thereto. be able to.
- the amount of the carboxylic acid derivative (X) used in the coupling reaction or condensation reaction is preferably 0.5 to 10 equivalents, more preferably 0.5 to 4 equivalents, relative to the disulfide derivative (XVII-a).
- the pH of the coupling reaction can be adjusted with a base.
- the base to be used include organic bases such as triethylamine or diisopropylethylamine, inorganic bases such as sodium hydrogencarbonate or potassium carbonate, metal hydride compounds such as sodium hydride, potassium hydride or calcium hydride, methyllithium or butyllithium.
- lithium amides such as lithium hexamethyldisilazide or lithium diisopropylamide or a mixture thereof
- an inorganic base such as sodium hydrogen carbonate or potassium carbonate or an organic base such as triethylamine or diisopropylethylamine is preferred.
- the amount of the base used for the coupling reaction is preferably 0.001 to 10 equivalents, more preferably 0.001 to 4 equivalents, relative to the disulfide derivative (XVII-a).
- the pH of the coupling reaction can also be adjusted with a buffer solution.
- the buffer used include a phosphate buffer, a citrate buffer, a citrate phosphate buffer, a borate buffer, a tartrate buffer, or a Tris buffer, and have a pH of 7.0 to 8.0.
- a buffer is preferred.
- the concentration of the buffer used for the coupling reaction is preferably 10 mmol / L to 1 mol / L.
- the reaction solvent used in the coupling reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethyl Aprotic polar solvents such as acetamide, N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate
- Examples include chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, water or mixed solvents thereof, but chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane.
- nitrile solvents such as acetonitrile or propion
- the reaction temperature of the coupling reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the coupling reaction is appropriately selected according to the reaction temperature and other conditions, but preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (X) used for the coupling reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- Examples of the condensing agent used in the condensation reaction include carbodiimides such as dicyclohexylcarbodiimide, diisopropylcarbodiimide, or 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, ethyl chloroformate, oxalyl chloride, hexafluorophosphate 2- ( 7-aza-1H-benzotriazol-1-yl) -1,1,3,3-tetramethyluronium, O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyl
- Examples include uronium hexafluorophosphate, 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate, or 2-methyl-6-nitrobenzoic anhydride, but dicyclohexylcarbodiimide, diisopropylcarbodiimi
- the amount of the condensing agent used in the condensation reaction is preferably 0.1 to 100 equivalents, more preferably 0.3 to 30 equivalents, relative to the carboxylic acid derivative (X).
- the reaction solvent used for the condensation reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethylacetamide Aprotic polar solvents such as N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate, dichloromethane , Chlorinated solvents such as chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, or mixed solvents thereof, including chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, or N , - dimethylformamide, N, N- dimethylacetamide, apro
- a base may be used if desired.
- the base to be used include inorganic bases such as sodium hydride, sodium bicarbonate or potassium carbonate, organic bases such as triethylamine, diisopropylethylamine, 4-dimethylaminopyridine or pyridine, or a mixture thereof.
- organic bases such as ethylamine, 4-dimethylaminopyridine or pyridine or mixtures thereof are preferred.
- the amount of base used in the condensation reaction is preferably 0.5 to 20 equivalents, more preferably 1 to 5 equivalents, relative to the carboxylic acid derivative (X).
- the reaction temperature of the condensation reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the condensation reaction is appropriately selected according to the reaction temperature and other conditions, but is preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (X) used for the condensation reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- the prodrug (XIX) of the peptide derivative (I) can be obtained from a basic to medium deprotection reaction of the disulfide derivative (XVII-a) followed by the carboxylic acid derivative (XII). Coupling reaction (when G is a succinimidyl group or p-nitrophenyl group) or a condensation reaction (when G is a hydrogen atom). [Wherein each symbol is the same as defined above. ]
- the deprotection of the protecting group varies depending on the type of the protecting group, but is performed according to a known method (for example, Greene, TW, “Green's Protective Groups in Organic Synthesis”, Wiley-Interscience) or a method equivalent thereto. be able to.
- the amount of the carboxylic acid derivative (XII) used in the coupling reaction or condensation reaction is preferably 0.5 to 10 equivalents, more preferably 0.5 to 4 equivalents, relative to the disulfide derivative (XVII-a).
- the pH of the coupling reaction can be adjusted with a base.
- the base to be used include organic bases such as triethylamine or diisopropylethylamine, inorganic bases such as sodium hydrogencarbonate or potassium carbonate, metal hydride compounds such as sodium hydride, potassium hydride or calcium hydride, methyllithium or butyllithium.
- lithium amides such as lithium hexamethyldisilazide or lithium diisopropylamide or a mixture thereof
- an inorganic base such as sodium hydrogen carbonate or potassium carbonate or an organic base such as triethylamine or diisopropylethylamine is preferred.
- the amount of the base used for the coupling reaction is preferably 0.001 to 10 equivalents, more preferably 0.001 to 4 equivalents, relative to the disulfide derivative (XVII-a).
- the pH of the coupling reaction can also be adjusted with a buffer solution.
- the buffer used include a phosphate buffer, a citrate buffer, a citrate phosphate buffer, a borate buffer, a tartrate buffer, or a Tris buffer, and have a pH of 7.0 to 8.0.
- a buffer is preferred.
- the concentration of the buffer used for the coupling reaction is preferably 10 mmol / L to 1 mol / L.
- the reaction solvent used in the coupling reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethyl Aprotic polar solvents such as acetamide, N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate
- Examples include chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, water or mixed solvents thereof, but chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane.
- nitrile solvents such as acetonitrile or propion
- the reaction temperature of the coupling reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the coupling reaction is appropriately selected according to the reaction temperature and other conditions, but preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (XII) used for the coupling reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- Examples of the condensing agent used in the condensation reaction include carbodiimides such as dicyclohexylcarbodiimide, diisopropylcarbodiimide, or 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, ethyl chloroformate, oxalyl chloride, hexafluorophosphate 2- ( 7-aza-1H-benzotriazol-1-yl) -1,1,3,3-tetramethyluronium, O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyl
- Examples include uronium hexafluorophosphate, 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate, or 2-methyl-6-nitrobenzoic anhydride, but dicyclohexylcarbodiimide, diisopropylcarbodiimi
- the amount of the condensing agent used in the condensation reaction is preferably 0.1 to 100 equivalents, more preferably 0.3 to 30 equivalents, relative to the carboxylic acid derivative (XII).
- the reaction solvent used for the condensation reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethylacetamide Aprotic polar solvents such as N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate, dichloromethane , Chlorinated solvents such as chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, or mixed solvents thereof, including chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, or N , - dimethylformamide, N, N- dimethylacetamide, apro
- a base may be used if desired.
- the base to be used include inorganic bases such as sodium hydride, sodium bicarbonate or potassium carbonate, organic bases such as triethylamine, diisopropylethylamine, 4-dimethylaminopyridine or pyridine, or a mixture thereof.
- organic bases such as ethylamine, 4-dimethylaminopyridine or pyridine or mixtures thereof are preferred.
- the amount of base used in the condensation reaction is preferably 0.5 to 20 equivalents, more preferably 1 to 5 equivalents, relative to the carboxylic acid derivative (XII).
- the reaction temperature of the condensation reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the condensation reaction is appropriately selected according to the reaction temperature and other conditions, but is preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (XII) used for the condensation reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- the prodrug (XIX) of the peptide derivative (I) is prepared by, for example, deprotection of a protected polyethylene glycol derivative (XVIII) followed by a basic reaction with a carboxylic acid derivative (XIV) as shown in Scheme 13.
- a coupling reaction under neutral conditions when G is a succinimidyl group or p-nitrophenyl group
- a condensation reaction when G is a hydrogen atom.
- the deprotection of the protecting group varies depending on the type of the protecting group, but is performed according to a known method (for example, Greene, TW, “Green's Protective Groups in Organic Synthesis”, Wiley-Interscience) or a method equivalent thereto. be able to.
- the amount of the carboxylic acid derivative (XIV) used for the coupling reaction or the condensation reaction is preferably 0.5 to 10 equivalents, more preferably 0.5 to 4 equivalents, relative to the protected polyethylene glycol derivative (XVIII).
- the pH of the coupling reaction can be adjusted with a base.
- the base to be used include organic bases such as triethylamine or diisopropylethylamine, inorganic bases such as sodium hydrogencarbonate or potassium carbonate, metal hydride compounds such as sodium hydride, potassium hydride or calcium hydride, methyllithium or butyllithium.
- lithium amides such as lithium hexamethyldisilazide or lithium diisopropylamide or a mixture thereof
- an inorganic base such as sodium hydrogen carbonate or potassium carbonate or an organic base such as triethylamine or diisopropylethylamine is preferred.
- the amount of the base used for the coupling reaction is preferably 0.001 to 10 equivalents, more preferably 0.001 to 4 equivalents with respect to the protected polyethylene glycol derivative (XVII).
- the pH of the coupling reaction can also be adjusted with a buffer solution.
- the buffer used include a phosphate buffer, a citrate buffer, a citrate phosphate buffer, a borate buffer, a tartrate buffer, or a Tris buffer, and have a pH of 7.0 to 8.0.
- a buffer is preferred.
- the concentration of the buffer used for the coupling reaction is preferably 10 mmol / L to 1 mol / L.
- the reaction solvent used in the coupling reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethyl Aprotic polar solvents such as acetamide, N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate
- Examples include chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, water or mixed solvents thereof, but chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane.
- nitrile solvents such as acetonitrile or propion
- the reaction temperature of the coupling reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the coupling reaction is appropriately selected according to the reaction temperature and other conditions, but preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (XIV) used for the coupling reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- Examples of the condensing agent used in the condensation reaction include carbodiimides such as dicyclohexylcarbodiimide, diisopropylcarbodiimide, or 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, ethyl chloroformate, oxalyl chloride, hexafluorophosphate 2- ( 7-aza-1H-benzotriazol-1-yl) -1,1,3,3-tetramethyluronium, O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyl
- Examples include uronium hexafluorophosphate, 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate, or 2-methyl-6-nitrobenzoic anhydride, but dicyclohexylcarbodiimide, diisopropylcarbodiimi
- the amount of the condensing agent used in the condensation reaction is preferably from 0.1 to 100 equivalents, more preferably from 0.3 to 30 equivalents, based on the carboxylic acid derivative (XIV).
- the reaction solvent used for the condensation reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethylacetamide Aprotic polar solvents such as N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate, dichloromethane , Chlorinated solvents such as chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, or mixed solvents thereof, including chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, or N , - dimethylformamide, N, N- dimethylacetamide, apro
- a base may be used if desired.
- the base to be used include inorganic bases such as sodium hydride, sodium bicarbonate or potassium carbonate, organic bases such as triethylamine, diisopropylethylamine, 4-dimethylaminopyridine or pyridine, or a mixture thereof.
- organic bases such as ethylamine, 4-dimethylaminopyridine or pyridine or mixtures thereof are preferred.
- the amount of the base used in the condensation reaction is preferably 0.5 to 20 equivalents, more preferably 1 to 5 equivalents, relative to the carboxylic acid derivative (XIV).
- the reaction temperature of the condensation reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the condensation reaction is appropriately selected according to the reaction temperature and other conditions, but is preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (XIV) used for the condensation reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- the prodrug (XX) of the peptide derivative (I) can be obtained from a basic to medium deprotection reaction of the disulfide derivative (XVII-a) followed by the carboxylic acid derivative (XIV).
- Coupling reaction when G is a succinimidyl group or p-nitrophenyl group
- a condensation reaction when G is a hydrogen atom.
- the deprotection of the protecting group varies depending on the type of the protecting group, but is performed according to a known method (for example, Greene, TW, “Green's Protective Groups in Organic Synthesis”, Wiley-Interscience) or a method equivalent thereto. be able to.
- the amount of the carboxylic acid derivative (XIV) used for the coupling reaction or condensation reaction is preferably 0.5 to 10 equivalents, more preferably 0.5 to 4 equivalents, relative to the disulfide derivative (XVII-a).
- the pH of the coupling reaction can be adjusted with a base.
- the base to be used include organic bases such as triethylamine or diisopropylethylamine, inorganic bases such as sodium hydrogencarbonate or potassium carbonate, metal hydride compounds such as sodium hydride, potassium hydride or calcium hydride, methyllithium or butyllithium.
- lithium amides such as lithium hexamethyldisilazide or lithium diisopropylamide or a mixture thereof
- an inorganic base such as sodium hydrogen carbonate or potassium carbonate or an organic base such as triethylamine or diisopropylethylamine is preferred.
- the amount of the base used for the coupling reaction is preferably 0.001 to 10 equivalents, more preferably 0.001 to 4 equivalents, relative to the disulfide derivative (XVII-a).
- the pH of the coupling reaction can also be adjusted with a buffer solution.
- the buffer used include a phosphate buffer, a citrate buffer, a citrate phosphate buffer, a borate buffer, a tartrate buffer, or a Tris buffer, and have a pH of 7.0 to 8.0.
- a buffer is preferred.
- the concentration of the buffer used for the coupling reaction is preferably 10 mmol / L to 1 mol / L.
- the reaction solvent used in the coupling reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethyl Aprotic polar solvents such as acetamide, N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate
- Examples include chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, water or mixed solvents thereof, but chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane.
- nitrile solvents such as acetonitrile or propion
- the reaction temperature of the coupling reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the coupling reaction is appropriately selected according to the reaction temperature and other conditions, but preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (XIV) used for the coupling reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- Examples of the condensing agent used in the condensation reaction include carbodiimides such as dicyclohexylcarbodiimide, diisopropylcarbodiimide, or 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, ethyl chloroformate, oxalyl chloride, hexafluorophosphate 2- ( 7-aza-1H-benzotriazol-1-yl) -1,1,3,3-tetramethyluronium, O- (benzotriazol-1-yl) -N, N, N ′, N′-tetramethyl
- Examples include uronium hexafluorophosphate, 1H-benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate, or 2-methyl-6-nitrobenzoic anhydride, but dicyclohexylcarbodiimide, diisopropylcarbodiimi
- the amount of the condensing agent used in the condensation reaction is preferably from 0.1 to 100 equivalents, more preferably from 0.3 to 30 equivalents, based on the carboxylic acid derivative (XIV).
- the reaction solvent used for the condensation reaction is appropriately selected depending on the type of reagent used, but is not particularly limited as long as it does not inhibit the reaction.
- N, N-dimethylformamide, N, N-dimethylacetamide Aprotic polar solvents such as N-methyl-2-pyrrolidone or dimethyl sulfoxide, ether solvents such as diethyl ether, tetrahydrofuran, dimethoxyethane or 1,4-dioxane, ester solvents such as ethyl acetate or propyl acetate, dichloromethane , Chlorinated solvents such as chloroform or 1,2-dichloroethane, nitrile solvents such as acetonitrile or propionitrile, or mixed solvents thereof, including chlorinated solvents such as dichloromethane, chloroform or 1,2-dichloroethane, or N , - dimethylformamide, N, N- dimethylacetamide, apro
- a base may be used if desired.
- the base to be used include inorganic bases such as sodium hydride, sodium bicarbonate or potassium carbonate, organic bases such as triethylamine, diisopropylethylamine, 4-dimethylaminopyridine or pyridine, or a mixture thereof.
- organic bases such as ethylamine, 4-dimethylaminopyridine or pyridine or mixtures thereof are preferred.
- the amount of the base used in the condensation reaction is preferably 0.5 to 20 equivalents, more preferably 1 to 5 equivalents, relative to the carboxylic acid derivative (XIV).
- the reaction temperature of the condensation reaction is preferably ⁇ 40 ° C. to 200 ° C., more preferably ⁇ 20 ° C. to 150 ° C.
- the reaction time of the condensation reaction is appropriately selected according to the reaction temperature and other conditions, but is preferably 30 minutes to 30 hours.
- the concentration of the carboxylic acid derivative (XIV) used for the condensation reaction at the start of the reaction is preferably 1 mmol / L to 1 mol / L.
- the complex of the present invention is characterized by containing the peptide derivative (I) and a targeting ligand or polymer.
- the “targeting ligand” is a substance for transporting a physiologically active compound including the peptide derivative (I) to a specific cell, and examples thereof include an antigen-binding protein, a cytokine, or a low molecular weight ligand.
- an “antigen-binding protein” is a protein having the property of binding to a specific protein present on the cell surface, such as an immunoglobulin molecule, a single chain antibody, a scFv, a Fab fragment, or an F (ab ′) fragment.
- an immunoglobulin molecule such as an immunoglobulin molecule, a single chain antibody, a scFv, a Fab fragment, or an F (ab ′) fragment.
- Cytokine refers to interleukin, chemokine, interferon, cell growth factor, cytotoxic factor, or modified proteins thereof, such as interleukin-2, interleukin-2 fusion protein, interleukin-3, Interleukin-4, Interleukin-6, Interleukin-8, Interleukin-12, Interleukin-17, CCL1 to 28, CXCL1 to 17, XCL1, XCL2, CX3CL1 Interferon ⁇ , Interferon ⁇ , Interferon ⁇ , Interferon ⁇ , Interferon ⁇ , consensus interferon, granule colony-stimulating factor (GCSF), granulocyte-macrophage colony-stimulating factor (GMCSF), CD-40 ligand, luteinizing hormone release Hormone (LHRH), insulin-like growth factor (IGF), macrophage colony stimulating factor (M-CSF), nerve growth factor (NGF), platelet-derived growth factor, tissue growth factor, transforming growth factor-1, vascular endo
- “Small molecule ligand” refers to an organic compound or peptide derivative having a property of binding to a specific protein present on the cell surface and having a molecular weight of less than 1000, for example, biotin, folic acid, integrin inhibitor, cRGD, RGD , PSMA inhibitors or VEGF inhibitors, but are not limited to these.
- Polymer means a high molecular weight organic compound having a number average molecular weight of 1,000 to 1,000,000 composed of a constant repeating structure, and examples thereof include polyethylene glycol, polyglutamic acid, polyamino acid or polysaccharide. Alternatively, polyglutamic acid is preferable, and polyethylene glycol having a number average molecular weight of 20000 to 100,000 or polyglutamic acid having a number average molecular weight of 20000 to 100,000 is more preferable.
- the antigen binding proteins, cytokines and peptide derivatives described above are, for example, their chemically synthesized, their recombinant, their natural products, their glycosylated or non-glycosylated forms, non-natural to them.
- the form in which an amino acid is introduced the form in which a functional group (for example, an aldehyde group or a sulfhydryl group) is introduced by chemical reaction or enzymatic reaction such as oxidation or reduction, and biologically active fragments thereof are also included.
- an “unnatural amino acid” is an amino acid that exists in nature and is not included in the amino acids that make up a protein (natural amino acids), and is naturally occurring or produced by chemical synthesis, ie, naturally occurring It means an amino acid that exists and does not constitute a protein, an amino acid obtained by chemically modifying a natural amino acid, or an amino acid that is not included in a natural amino acid and that is produced by chemical synthesis.
- D-amino acid, citrulline, ornithine (S) -2-amino-3- (3-methyl-3H-diazilin-3-yl) propanoic acid ((S) -2-amino-3- ( 3-methyl-3H-diazirin-3-yl) propanoic acid, also referred to as photo-L-leucine), (S) -2-amino-4- (3-methyl-3H-diazilin-3-yl) butanoic acid ( (S) -2-amino-4- (3-methyl-3H-diazirin-3-yl) butanoic acid, also referred to as photo-L-methionine), (S) -3- (4-acetylphenyl) -2- Aminopropanoic acid ((S) -3- (4-acetylphenyl) -2-aminopropanoic acid, 4-acetyl-L-fu Nylalanine) or
- the above-mentioned low-molecular-weight ligands appropriately include analogs thereof, derivatives thereof, agonists thereof, antagonists thereof, inhibitors thereof, isomers thereof, and the like.
- “Complex” means a substance containing peptide derivative (I) or a prodrug thereof and the above targeting ligand, a substance containing peptide derivative (I) or a prodrug thereof and the above polymer, or a peptide derivative. (I) or a prodrug thereof, a substance containing the above targeting ligand and the above polymer (hereinafter collectively referred to as a complex containing the peptide derivative (I)), each of which is directly bound or other The substance is indirectly bonded via a substance (hereinafter referred to as a linker).
- Examples of the complex containing the peptide derivative (I) include an antibody drug complex in which the peptide derivative (I) or a prodrug thereof and an immunoglobulin molecule are directly bound or indirectly bound via a linker. Is mentioned.
- Examples of the complex containing the peptide derivative (I) include a low molecule in which the peptide derivative (I) or a prodrug thereof and a low molecular ligand are directly bonded or indirectly bonded via a linker.
- a pharmaceutical complex is mentioned.
- a complex containing the peptide derivative (I) for example, a polymer pharmaceutical complex in which the peptide derivative (I) or a prodrug thereof and the polymer are directly bonded or indirectly bonded via a linker.
- the body is mentioned.
- the complex containing the peptide derivative (I) includes, for example, the peptide derivative (I) or a prodrug thereof, each of which is independently bonded to the polymer and the targeting ligand or via a linker. And indirectly bound substances.
- the complex containing the peptide derivative (I) is, for example, that the peptide derivative (I) or a prodrug thereof and the targeting ligand are directly bonded to the polymer or via a linker. And indirectly bound substances.
- the complex containing the peptide derivative (I) includes, for example, the above-described targeting ligand, wherein the peptide derivative (I) or a prodrug thereof and the polymer are directly bonded to each other or via a linker. And indirectly bound substances.
- “Containing peptide derivative (I)” means that peptide derivative (I) or a prodrug thereof is a known method (for example, Ellen M. Sletten et al., Angelwante Chimie International Edition, 2009, Vol. 48, p. 6974). -6998, Greg T. Hermanson., "Bioconjugate Technique", Elsevier, Xi Chem et al., Organic & Biomolecular Chemistry, 2016, Vol. 14, p. 5417-5439, or the corresponding method. Or it is directly bonded to the polymer by a covalent bond or indirectly linked via a linker. .
- covalent bond examples include, for example, a bond by addition of a sulfhydryl group to a maleimide group, a disulfide bond, a bond by condensation of an amino group or a hydroxy group and a carboxyl group, a bond by a click reaction of an alkyne group and an azide group, Examples include a bond by condensation of an aminooxy group and a carbonyl group or a bond by an ene-type reaction of a diazo group and a phenol group, a bond by addition of a sulfhydryl group to a maleimide group, a disulfide bond, an amino group or a hydroxy group and a carboxyl group.
- a bond by condensation or a bond by condensation of an aminooxy group and a carbonyl group is preferred, but is not limited thereto.
- Linker means peptide derivative (I) or a prodrug thereof, the above targeting ligand and the above polymer and a functional group (eg, maleimide group, carboxyl group, activated carboxyl group, carbonyl group, aminooxy group, hydrazide group) , A diazo group, an alkyne group, or a hydroxy group).
- a functional group eg, maleimide group, carboxyl group, activated carboxyl group, carbonyl group, aminooxy group, hydrazide group
- Examples of compounds having these functional groups include N-succinimidyl 4- (maleimidomethyl) cyclohexanecarboxylate (SMCC), sulfosuccinimidyl-4- (N-maleimidomethyl) -cyclohexane-1-carboxylate ( Sulfo-SMCC), N-succinimidyl-4- (N-maleimidomethyl) -cyclohexane-1-carboxy- (6-amidocaproate) (LC-SMCC), ⁇ -maleimidoundecanoic acid N-succinimidyl ester (KMUA) ), ⁇ -maleimidobutyric acid N-succinimidyl ester (GMBS), ⁇ -maleimidocaproic acid N-hydroxysuccinimide ester (EMCS), m-maleimidobenzoyl-N-hydroxysuccinimide ester (MBS), N- ( ⁇ -male Imidoacetoxy)
- the “specific protein present on the cell surface” is a protein whose expression on the cell surface increases in cancer, autoimmune disease or infection, for example, CD19, CD22, CD30, CD33, CD37, CD56, CD66e, CD70, CD74, CD79b, DLL-3, PSMA, GPNMB, Her2, CA6, CA9, Mesothelin, Nectin-4, SLC44A4, Cripto, Folate receptor, STEAP-1, MUC16, NaPi2b, GCC, EGFRViii, 5T4, TROP-2, LIV-1, SLITRK6, Tissue Factor, Guanylyl Cycle C, CEACAM5, integrin receptor, interleukin receptor, PSMA, chemokine receptor, inter Ferron receptor, although cell growth factor receptors or cytotoxic factor receptors include, but are not limited thereto.
- Examples of the pharmacologically acceptable salt of the complex containing the peptide derivative (I) include the same salts as the pharmacologically acceptable salts of the peptide derivative (I).
- the complex containing the peptide derivative (I) or a pharmacologically acceptable salt thereof may be an anhydride, or may form a solvate such as a hydrate.
- the solvate is preferably a pharmacologically acceptable solvate.
- the pharmacologically acceptable solvate may be either a hydrate or a non-hydrate, but a hydrate is preferable.
- the solvent constituting the solvate include alcohol solvents such as methanol, ethanol, and n-propanol, N, N-dimethylformamide, dimethyl sulfoxide, and water.
- the compounds described in Table 4 also include pharmacologically acceptable salts thereof.
- r represents an integer of 1 to 50
- L represents a targeting ligand, a polymer, or a polymer and a targeting ligand, and other symbols are the same as defined above.
- a 4 are left (the side bonded to the amino group) is carbonyl end, the right (the side bound to a carbonyl group) is an amino terminus. Also, when A 4 is Lys, Glu and Asp can be attached from the amino group or carboxyl group of the side chain.
- X is NR
- Alk 1 is — (CH 2 ) 2 —
- PEG is — (CH 2 CH 2 O) 12 —.
- a 4 is more preferably Glu bonded with a main chain
- X is NR
- R and Z are methyl groups
- Alk 1 and Alk 2 are — (CH 2 ) 2 —.
- PEG is — (CH 2 CH 2 O) 12 —
- a 4 is Glu bonded with a main chain.
- X is N (Me)
- Alk 1 is — (CH 2 ) 2 —
- PEG is — (CH 2 CH 2 O) 12. More preferably, A 4 is Glu bonded by a main chain
- X is N (Me)
- Z is a methyl group
- Alk 1 and Alk 2 are — (CH 2 ) 2 -
- PEG is - (CH 2 CH 2 O) 12 -
- a 4 is more preferably a Glu bound in the main chain.
- the complex containing the peptide derivative (I) can be produced by an appropriate method based on characteristics derived from the basic skeleton and the type of substituent.
- the starting materials and reagents used in the production of these compounds can be generally purchased, or can be produced by a known method or a method analogous thereto.
- the complex containing the peptide derivative (I) and the intermediates and starting materials used for the production thereof can be isolated and purified by known means.
- Known means for isolation and purification include, for example, solvent extraction, recrystallization or chromatography.
- each isomer can be obtained as a single compound by a known method.
- Known methods include, for example, crystallization, enzyme resolution, or chiral chromatography.
- a protective group may be introduced into these groups, and the protective group may be removed as necessary after the reaction. By protecting, the target compound can be obtained.
- Examples of the protective group for the hydroxy group include a trityl group, a tetrahydropyranyl group, an aralkyl group having 7 to 10 carbon atoms (for example, a benzyl group) or a substituted silyl group (for example, a trimethylsilyl group, a triethylsilyl group, or tert-butyldimethyl). Silyl group).
- amino-protecting group examples include an alkylcarbonyl group having 2 to 6 carbon atoms (for example, acetyl group), a benzoyl group, an alkyloxycarbonyl group having 2 to 8 carbon atoms (for example, tert-butoxycarbonyl group or benzyloxy group) Carbonyl group), an aralkyl group having 7 to 10 carbon atoms (for example, benzyl group) or a phthaloyl group.
- alkylcarbonyl group having 2 to 6 carbon atoms for example, acetyl group
- benzoyl group an alkyloxycarbonyl group having 2 to 8 carbon atoms (for example, tert-butoxycarbonyl group or benzyloxy group) Carbonyl group)
- an aralkyl group having 7 to 10 carbon atoms for example, benzyl group
- a phthaloyl group examples include an alkylcarbonyl group having 2 to 6 carbon atoms (for example
- Examples of the protecting group for the sulfhydryl group include a trityl group, a 2-mercaptopyridyl group, and a 2-mercapto-5-nitropyridyl group.
- Examples of the protecting group for the carboxyl group include an alkyl group having 1 to 6 carbon atoms (for example, a methyl group, an ethyl group, or a tert-butyl group) or an aralkyl group having 7 to 10 carbon atoms (for example, a benzyl group).
- the deprotection of the protecting group varies depending on the type of the protecting group, but is performed according to a known method (for example, Greene, TW, “Green's Protective Groups in Organic Synthesis”, Wiley-Interscience) or a method equivalent thereto. be able to.
- the complex containing the peptide derivative (I) is obtained by subjecting the peptide derivative (I) or a prodrug thereof or a salt thereof to the above targeting ligand and / or the above polymer or a salt thereof by a known method (for example, Ellen).
- a known method for example, Ellen.
- the complex containing the peptide derivative (I) may be labeled with an isotope, and examples of the labeled isotope include 2 H, 3 H, 13 C, 14 C, 15 N, 15 O, 17 O, 18 O and / or 125 I.
- Examples of the peptide derivative (I) or a prodrug thereof, the targeting ligand, or the salt of the polymer used for the above-mentioned conjugation include, for example, hydrochloride, sulfate, nitrate, hydrobromide, hydroiodide, Inorganic acid salts such as phosphate, oxalate, malonate, citrate, fumarate, lactate, malate, succinate, tartrate, acetate, trifluoroacetate, maleate , Gluconate, benzoate, ascorbate, glutarate, mandelate, phthalate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-toluenesulfonate, camphorsulfonate Salt, organic acid salt such as aspartate, glutamate or cinnamate, triethylamine salt, diisopropylethylamine salt, ethanol A salt with an organic
- the peptide derivative (I) or a prodrug thereof, the targeting ligand or the polymer salt may be an anhydride or may form a solvate such as a hydrate.
- the solvent constituting the solvate include alcohol solvents such as methanol, ethanol or n-propanol, N, N-dimethylformamide, dimethyl sulfoxide or water.
- the cytotoxic agent of the present invention is effective for peptide derivative (I) or a pharmacologically acceptable salt thereof, or a complex containing peptide derivative (I) or a pharmacologically acceptable salt thereof. It is characterized by containing as a component.
- “Cytotoxic agent” means a composition containing, as an active ingredient, a compound that causes cell death by impairing cell function or inhibiting cell proliferation or a pharmacologically acceptable salt thereof.
- the “cytotoxic agent” includes a complex containing a compound that causes cell death by impairing cell function or inhibiting cell proliferation, or a prodrug thereof, and the above targeting ligand or the above polymer, or the like
- a composition containing a pharmacologically acceptable salt as an active ingredient is also included.
- the peptide derivative (I) or a pharmacologically acceptable salt thereof, or a complex containing the peptide derivative (I) or a pharmacologically acceptable salt thereof can be used in mammals (eg, mouse, rat, hamster, Rabbits, cats, dogs, monkeys, cows, sheep or humans), particularly when administered to humans, can be used as useful cytotoxic agents.
- mammals eg, mouse, rat, hamster, Rabbits, cats, dogs, monkeys, cows, sheep or humans
- the peptide derivative (I) or a pharmacologically acceptable salt thereof, or a complex containing the peptide derivative (I) or a pharmacologically acceptable salt thereof is used as it is or a pharmacologically acceptable carrier. It can be formulated and administered as a cytotoxic agent to the above mammals orally or parenterally.
- parenteral administration examples include injection administration, nasal administration, pulmonary administration, transdermal administration, sublingual administration, and rectal administration, and injection administration is preferable.
- injection administration refers to administration of a cytotoxic agent systemically or locally by injection or infusion, and examples of the administration site include intravenous, intramuscular, intraperitoneal, and subcutaneous.
- a cytotoxic agent containing the peptide derivative (I) or a pharmacologically acceptable salt thereof, or a complex containing the peptide derivative (I) or a pharmacologically acceptable salt thereof as an active ingredient is a pharmaceutical technique. It can be produced by a known method generally used in the field (for example, Remington's Pharmaceutical Science, latest edition, Mark Publishing Company, Easton, USA). In addition, the cytotoxic agent of the present invention can be prepared by using excipients, binders, disintegrants, lubricants, sweeteners, surfactants, suspending agents, emulsifiers, which are usually used in the pharmaceutical field, if necessary.
- additives such as colorants, preservatives, fragrances, flavoring agents, stabilizers, thickeners, buffering agents, tonicity agents, fluidity promoters, and the like can be appropriately contained.
- pharmacologically acceptable carriers include these additives.
- a cytotoxic agent comprising the peptide derivative (I) or a pharmacologically acceptable salt thereof, or a complex containing the peptide derivative (I) or a pharmacologically acceptable salt thereof as an active ingredient is Based on the mechanism, it can be used as a therapeutic agent for diseases that can be expected to improve pathological conditions or relieve symptoms, such as cancer, autoimmune diseases, or infectious diseases.
- Cancer is a disease caused by an uncontrolled and abnormally growing cell population, such as pharyngeal cancer, laryngeal cancer, tongue cancer, non-small cell lung cancer, breast cancer, esophageal cancer, stomach cancer, colon cancer, uterus.
- Cancer endometrial cancer, ovarian cancer, liver cancer, pancreatic cancer, gallbladder cancer, bile duct cancer, kidney cancer, renal pelvic and ureteral cancer, bladder cancer, prostate cancer, malignant melanoma, thyroid cancer, neuroosteosarcoma, chondrosarcoma, Rhabdomyosarcoma, hemangiosarcoma, fibrosarcoma, glioma, leukemia, malignant lymphoma, neuroblastoma, myeloma or brain tumor.
- autoimmune disease is a disease caused by an abnormal immune response to a substance or cells in a living body, such as active chronic hepatitis, Addison's disease, ankylosing spondylitis, antiphospholipid syndrome, arthritis, atopic Allergy, Behcet's disease, cardiomyopathy, celiac disease, Kogan syndrome, cold coagulopathy, Crohn's disease, Cushing's syndrome, dermatomyositis, discoid lupus erythematosus, erythema, fibromyalgia, glomerulonephritis, Goodpasture's syndrome, graft pair Host disease, Graves' disease, Guillain-Barre syndrome, Hashimoto's disease, idiopathic adrenal atrophy, idiopathic pulmonary fibrosis, IgA nephropathy, inflammatory bowel disease, insulin-dependent diabetes, juvenile arthritis, Lambert-Eaton myasthenia syndrome, Lichen planus,
- infectious disease is a disease caused by infection with bacteria, fungi, pathogenic protozoa, yeast or virus, and the causes thereof include, for example, staphylococci, streptococci, enterococci, corynebacterium, bacillus, Listeria, Peptococcus, Peptostreptococcus, Clostridium, Eubacterium, Propionibacterium, Lactobacillus, Neisseria, Blanchamella, Hemophilus, Bordetella, Escherichia coli, Citrobacter, Salmonella, Shigella, Klebsiella, Enterobacter, Serratia, Hafnia, Proteus, Morganella, Providencia, Yersinia, Campylobacter, Vibrio, Aeromonas, Pseudomonas, Xanthomonas, Acinetobacter, Flavobacterium, Brucella, Legionella, Beironella, Ba Teloides, Fusobacterium, Mycoplasm
- the peptide derivative (I) or a pharmacologically acceptable salt thereof, or a complex containing the peptide derivative (I) or a pharmacologically acceptable salt thereof is cytotoxic using an in vitro test. Can be evaluated. Examples of in vitro tests include the trypan blue dye exclusion method, lactate dehydrogenase (LDH) activity method, [ 3 H] thymidine incorporation method, propidium iodide, which measures the number of dead cells or viable cells after compound treatment.
- LDH lactate dehydrogenase
- ATP using a nuclear staining method a method using a dye such as MTT, MTS and WST (Current Protocol in Toxicology, 2000, 2.6.1-2.26) or CellTiter-Glow (product of Promega) Examples include a method of measuring the amount, and the method of measuring the number of viable cells after treatment with the compound using the MTS method and determining the cytotoxicity value at that time by the 50% working concentration (EC 50 ) is preferable.
- the cytotoxicity of the complex containing the peptide derivative (I) or a pharmacologically acceptable salt thereof the cells are treated with a medium containing the compound for a certain period of time, and then the medium is removed.
- a method of measuring the number of viable cells after changing and further culturing for a certain time is preferable (Bioconjugate Chemistry. 2014, Vol. 25, p. 560-568).
- human ovarian cancer cell SKOV-3 cell human alveolar basal epithelial adenocarcinoma cell A549 cell, mouse lymphocytic leukocyte L1210 cell and the like are used.
- the permeability of the peptide derivative (I) or a pharmacologically acceptable salt thereof through the cell membrane can be measured using an in vitro cell membrane permeability evaluation method.
- an in vitro cell membrane permeability evaluation method for example, a method using a single-layer cell membrane system such as Caco-2 or Madin-Darby canine kidney (MDCK), or a Parallel Artificial Membrane Permeation using an artificial lipid membrane holding a lipid or the like in a filter.
- Assay PAMPA (Japanese Patent Laid-Open No. 2007-11803) and the like.
- LC / MS liquid chromatograph mass spectrometer
- combined by the commercially available or well-known method or a method according to it was used.
- the solvent name shown in the NMR data indicates the solvent used for the measurement.
- the 400 MHz NMR spectrum was measured using a JNM-AL400 type nuclear magnetic resonance apparatus (manufactured by JEOL Ltd.) or a JNM-ECS400 type nuclear magnetic resonance apparatus (manufactured by JEOL Ltd.).
- the chemical shift is represented by ⁇ (unit: ppm) based on tetramethylsilane, and the signals are s (single line), d (double line), t (triple line), q (quadruplex line), m, respectively.
- ⁇ unit: ppm
- the signals are s (single line), d (double line), t (triple line), q (quadruplex line), m, respectively.
- the proton NMR spectrum which cannot be confirmed by broad such as OH or NH proton is not described in the data.
- the molecular weight is Agilent Technologies 1200 Series, 6130A (manufactured by Agilent Technologies) (hereinafter LC / MS-1) or Agilent Technologies 1260 Infinity II Series, 6130B (manufactured by Agilent Technologies) (hereinafter LC / MS-2).
- ESI electrospray ionization
- HRMS Accurate mass spectrometry
- t R The retention time (hereinafter referred to as t R ) was measured by high performance liquid chromatography (hereinafter referred to as HPLC).
- HATU -1,1,3,3-tetramethyluronium
- Example 1 2-((S) -1-(((2S, 3R) -3-((S) -1-((3R, 4S, 5S) -4-((S) -2- ( (S) -2- (Dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl) -3-methoxy-2- Synthesis of methylpropanamide) -2-phenylethyl) -N- (2-mercaptoethyl) thiazole-4-carboxamide: To the compound of Reference Example 2 (25 mg, 0.022 mmol) was added 0.02M triethylsilane-containing trifluoroacetic acid (hereinafter TFA) / dichloromethane (volume ratio 1: 1) solution (4.5 mL), and the mixture was stirred at room temperature for 1 hour.
- TFA triethylsilane
- Example 2 2-((S) -1-((2R, 3R) -3-((S) -1-((3R, 4S, 5S) -4-((S) -2-(( S) -2- (Dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl) -3-methoxy-2-methyl
- the compound of Reference Example 3 was used instead of the compound of Reference Example 2, and the rest was synthesized in the same manner as in Example 1, and the title compound (hereinafter referred to as the compound of Example 2) was synthesized as a white solid (32 mg, Yield 83%).
- Reference Example 5 Synthesis of N-methyl-2- (tritylthio) ethan-1-amine: To a dichloromethane solution (4.0 ml) of Reference Example 4 (150 mg, 0.35 mmol) was added 4N hydrogen chloride-dioxane (3.0 ml), and the mixture was stirred at room temperature for 4 hours. To the reaction solution, an excessive amount of 5% by volume of methanol-containing dichloromethane was added, washed with saturated sodium bicarbonate, dehydrated with anhydrous sodium sulfate, and concentrated under reduced pressure. The title compound (hereinafter referred to as the compound of Reference Example 5) was obtained as an oily substance (15 mg, yield 40%). The obtained oily substance was used in the next reaction without purification.
- Example 3 2-((S) -1-((2R, 3R) -3-((S) -1-((3R, 4S, 5S) -4-((S) -2-(( S) -2- (Dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl) -3-methoxy-2-methyl
- propanamide) -2-phenylethyl) -N- (2-mercaptoethyl) -N-methylthiazole-4-carboxamide The compound of Reference Example 6 was used in place of the compound of Reference Example 2, and the others were synthesized in the same manner as in Example 1, and the title compound (hereinafter referred to as the compound of Example 3) was converted into a white amorphous (27 mg, Yield 77%).
- the solvent of the obtained reaction solution was distilled off under reduced pressure, ethyl acetate was added, and the mixture was washed with a saturated aqueous sodium hydrogen carbonate solution and saturated brine.
- the organic layer was dehydrated with anhydrous sodium sulfate and concentrated under reduced pressure.
- Reference Example 8 (2- (2-((S) -1-((2R, 3R) -3-((S) -1-((3R, 4S, 5S) -4-((S)- 2-((S) -2- (dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl) -3-methoxy -2-Methylpropanamide) -2-phenylethyl) -N-methylthiazole-4-carboxamido) ethyl) Synthesis of tert-butyl carbamate: The title compound (hereinafter referred to as Reference Example) was synthesized in the same manner as in Reference Example 2 except that tert-butyl (2- (methylamino) ethyl) carbamate was used instead of 2- (tritylthio) ethanamine.
- Example 5 N- (2-aminoethyl) -2-((S) -1-((2R, 3R) -3-((S) -1-((3R, 4S, 5S) -4- ((S) -2-((S) -2- (dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl ) -3-Methoxy-2-methylpropanamide) -2-phenylethyl) -N-methylthiazole-4-carboxamide dihydrochloride: A 4N hydrogen chloride-dioxane solution (126 ⁇ L, 0.50 mmol) was added to a methanol solution (1.0 mL) of the compound of Reference Example 8 (33 mg, 0.033 mmol), and the mixture was stirred at room temperature for 15 hours.
- Example 6 2-((S) -1-((2R, 3R) -3-((S) -1-((3R, 4S, 5S) -4-((S) -2-(( S) -2- (Dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl) -3-methoxy-2-methyl Propanamido) -2-phenylethyl) -N-methyl-N- (2- (methylamino) ethyl) thiazole-4-carboxamide dihydrochloride Synthesis: The compound of Reference Example 9 was used in place of the compound of Reference Example 8, and the others were synthesized in the same manner as in Example 5.
- Example 7 2-((S) -1-((2R, 3R) -3-((S) -1-((3R, 4S, 5S) -4-((S) -2-(( S) -2- (Dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl) -3-methoxy-2-methyl
- propanamide) -2-phenylethyl) -N- (2-hydroxyethyl) thiazole-4-carboxamide Synthesis was performed in the same manner as in Reference Example 2 except that 2-aminoethanol was used instead of 2- (tritylthio) ethanamine, and the title compound (hereinafter referred to as the compound of Example 7) was synthesized as a white solid (26 mg, Yield, quantitative).
- Example 8 2-((S) -1-((2R, 3R) -3-((S) -1-((3R, 4S, 5S) -4-((S) -2-(( S) -2- (Dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl) -3-methoxy-2-methyl Propanamido) -2-phenylethyl) thiazole-4-carboxylic acid Synthesis of 2-hydroxyethyl: P-Toluenesulfonic acid monohydrate (7.2 mg) was added to an ethanol solution (1.0 mL) of the compound of Reference Example 10 (36 mg, 0.038 mmol), and the mixture was stirred at room temperature for 18 hours.
- the solvent of the obtained reaction solution was distilled off under reduced pressure, diluted with dichloromethane, saturated aqueous potassium carbonate solution was added, and the mixture was extracted 3 times with chloroform.
- the organic layer was dehydrated with anhydrous sodium sulfate and concentrated under reduced pressure.
- Example 9 2-((S) -1-((2R, 3R) -3-((S) -1-((3R, 4S, 5S) -4-((S) -2-(( S) -2- (Dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl) -3-methoxy-2-methyl
- propanamide) -2-phenylethyl) -N- (2-hydroxyethyl) -N-methylthiazole-4-carboxamide The title compound (hereinafter referred to as the compound of Example 9) was synthesized in the same manner as in Reference Example 2, except that 2- (methylamino) ethan-1-ol was used instead of 2- (tritylthio) ethanamine.
- Example 10 N- (4-aminophenethyl) -2-((S) -1-(((2S, 3R) -3-((S) -1-((3R, 4S, 5S) -4 -((S) -2-((S) -2- (dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidine-2- Yl) -3-methoxy-2-methylpropanamide) -2-phenylethyl) thiazole-4-carboxamide: Synthesis was performed in the same manner as in Reference Example 2 except that 4- (2-aminoethyl) aniline was used instead of 2- (tritylthio) ethanamine, and the title compound (hereinafter referred to as the compound of Example 10) was obtained.
- Example 11 2-((S) -1-((2R, 3R) -3-((S) -1-((3R, 4S, 5S) -4-((S) -2-(( S) -2- (Dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl) -3-methoxy-2-methyl
- propanamide) -2-phenylethyl) -N- (4-hydroxyphenethyl) thiazole-4-carboxamide Synthesis was performed in the same manner as in Reference Example 2 except that 4- (2-aminoethyl) phenol was used instead of 2- (tritylthio) ethanamine, and the title compound (hereinafter referred to as the compound of Example 11) was prepared.
- Reference Example 13 (2- (2-((S) -1-(((2S, 3R) -3-((S) -1-((3R, 4S, 5S) -4-((S)) -2-((S) -2- (dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl) -3- Synthesis of tert-butyl methoxy-2-methylpropanamide) -2-phenylethyl) thiazole-4-carboxamido) ethyl) (methyl) carbamate: The title compound (hereinafter referred to as Reference Example) was synthesized in the same manner as in Reference Example 2 except that tert-butyl (2-aminoethyl) (methyl) carbamate was used instead of 2- (tritylthio) ethanamine.
- Reference Example 14 2-((S) -1-(((2S, 3R) -3-((S) -1-((3R, 4S, 5S) -4-((S) -2- ( (S) -2- (Dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl) -3-methoxy-2- Synthesis of methylpropanamide) -2-phenylethyl) -N- (2- (methylamino) ethyl) thiazole-4-carboxamide dihydrochloride: The compound of Reference Example 13 was used in place of the compound of Reference Example 8, and the others were synthesized in the same manner as in Example 5.
- Reference Example 16 2-((S) -1-(((2S, 3R) -3-((S) -1-((3R, 4S, 5S) -4-((S) -2- ( (S) -2- (Dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl) -3-methoxy-2- Synthesis of methylpropanamide) -2-phenylethyl) -N- (2- (3-mercaptopropanamido) ethyl) thiazole-4-carboxamide: The compound of Reference Example 15 was used in place of the compound of Reference Example 2, and the others were synthesized in the same manner as in Example 1, and the title compound (hereinafter referred to as the compound of Reference Example 16) was synthesized as a white solid (7.
- THF diethylamine-tetrahydrofuran
- Reference Example 18 tert-butyl N5-((S) -5- (tert-butoxy) -1,5-dioxo-1-((2- (tritylthio) ethyl) amino) pentan-2-yl) -L -Glutamate synthesis: Reference Example 17 (480 mg, 0.53 mmol) was dissolved in 20% by volume diethylamine-THF solution (5.0 ml) and stirred overnight at room temperature, and then the reaction solution was concentrated under reduced pressure.
- the obtained residue (7.5 mg) was dissolved in dehydrated pyridine (1.0 ml) and ice-cooled, and then a dehydrated pyridine solution (3.0 ml) of the compound of Example 11 (25 mg, 0.03 mmol) was added, The mixture was stirred overnight while returning to room temperature. After adding an excess amount of ethyl acetate, the mixture was washed three times with water and once with a saturated aqueous ammonium chloride solution and saturated brine, and the organic layer was dehydrated over anhydrous sodium sulfate and concentrated under reduced pressure.
- Example 12 N2- (4-(((2-Amino-4-oxo-4,8-dihydropteridin-6-yl) methyl) amino) benzoyl) -N5-((2S) -4-carboxy- 1-((2-((1- (1- (4- (2-((S) -1-((2R, 3R) -3-((S) -1-((3R, 4R , 5S) -4-((S) -2-((S) -2- (dimethylamino) -3-methylbutanamide) -N, 3-dimethylbutanamide) -3-methoxy-5-methylheptanoyl ) Pyrrolidin-2-yl) -3-methoxy-2-methylpropanamide) -2-phenylethyl) thiazole-4-carboxamide) ethyl) phenoxy) -2,9-dimethyl-1,10,50-trioxo-13 16, 19, 22, 25, 28, 31, 34,
- LC / MS-1 The analysis conditions of LC / MS-1 are as follows.
- LC / MS-1) Liquid chromatograph system: LC1200 (manufactured by Agilent Technologies) Mass spectrometer: 6130A (manufactured by Agilent Technologies)
- Mobile phase A 0.1% by volume formic acid-distilled water
- HPLC mobile phase B 0.1% by volume formic acid-acetonitrile
- HPLC gradient condition mobile bed B% by volume 0-1.5 min: 20% -95% 1.5-3.0 min: 95%
- Flow rate 0.5 mL / min
- Injection volume 2.0 ⁇ L
- Column temperature 40 ° C
- LC / MS-2 The analysis conditions of LC / MS-2 are as follows.
- LC / MS-2 Liquid chromatograph system: LC1260 Infinity II (manufactured by Agilent Technologies) Mass spectrometer: 6130B (manufactured by Agilent Technologies)
- Mobile phase A 0.1% by volume formic acid-distilled water
- HPLC mobile phase B 0.1% by volume formic acid-acetonitrile
- HPLC gradient condition mobile bed B% by volume 0-1.5 min: 20% -95% 1.5-3.0 min: 95%
- Flow rate 0.5 mL / min
- Injection volume 2.0 ⁇ L
- Column temperature 40 ° C
- ODS column purification condition column Yamazen high flash column ODS size M Flow rate: 10 ml / min Column temperature: Room temperature Detection wavelength: 254 nm Moving bed A: 0.05% by volume TFA in H 2 O Moving layer B: 0.05% by volume TFA in CH 3 CN Gradient: moving bed B volume% 0-5 min: 5.0% 5-20 min: Increase from 5% to 95% 20-25 min: 95%
- HPLC HPLC
- Example 14 In vitro cytotoxicity evaluation of peptide derivative (I): The compounds of Examples 1 to 11, the compound of Reference Example 1, the compound of Reference Example 12, the compound of Reference Example 14, the compound of Reference Example 16 and 2-((S) -1-(((2S, 3R) -3 -((S) -1-((3R, 4S, 5S) -4-((S) -2-((S) -2- (dimethylamino) -3-methylbutanamide) -N, 3-dimethyl Butanamide) -3-methoxy-5-methylheptanoyl) pyrrolidin-2-yl) -3-methoxy-2-methylpropanamide) -2-phenylethyl) -N-ethylthiazole-4-carboxamide (hereinafter, comparison) The cytotoxicity of Example Compound) against SKOV-3 cells, A549 cells and L1210 cells was measured using the MTS method.
- Table 5 shows the cytotoxicity of each test substance.
- EC 50 means 50% effective concentration, which represents 50% of the maximum response from the lowest value of the pharmacological action exhibited by the drug
- SKOV-3 represents SKOV-3 cells
- L1210 represents L1210 cells.
- test substance was dissolved in DMSO to 10 mmol / L and then diluted to 200 ⁇ mol / L with PBS containing 20% methanol as a standard solution.
- the pre-coated PAMPA Plate System that was stored frozen was left at room temperature for 30 minutes or more.
- 320 ⁇ L of the standard solution was added to the Donor side plate of the pre-coated PAMPA Plate System, 200 ⁇ L of PBS containing 20% methanol was added to the Acceptor side plate, both plates were set, and allowed to stand at room temperature for 5 hours.
- the reaction solution in each well of the Donor side and Acceptor side plates was subjected to LC / MS analysis (hereinafter, LC / MS-3), and the compound concentration in each well was calculated.
- LC / MS analysis hereinafter, LC / MS-3
- the membrane permeation coefficient (Pe) (cm / s) was calculated from the following equation using the compound concentration of the reaction solution obtained in each well.
- C equilibrium equilibrium concentration of Donor side and Acceptor side plate wells
- CD concentration after 5 hours of Donor side plate wells (mmol / L)
- VD Volume of standard solution added to Donor side plate well (0.32 mL)
- CA concentration (mmol / L) after 5 hours in the acceptor side plate well
- VA volume of PBS containing 20% methanol added to the acceptor side plate well (0.2 mL)
- Table 6 shows the artificial membrane permeability of each test substance.
- Example 16 In Vitro Cytotoxicity Evaluation of Complex Containing Peptide Derivative (I): The cytotoxicity of the compound of Example 12 and the compound of Example 13 against SKOV-3 cells, A549 cells and L1210 cells was measured using the MTS method.
- the compound of Example 12 is a complex obtained by conjugating the prodrug of the compound of Example 11 with folic acid as a targeting ligand by an appropriate method.
- the evaluation method of the compound of Example 12 is as follows. Each cell cultured in a medium not containing folic acid was treated with the compound of Example 12 for 2 hours, then changed to a new medium containing no compound, and continued to culture for 48 hours. The number of viable cells after 48 hours was measured using the MTS method.
- the compound of Example 13 is a complex obtained by complexing the prodrug of the compound of Example 3 with folic acid as a targeting ligand by an appropriate method.
- the method for evaluating the compound of Example 13 is as follows. Each cell cultured in a medium containing no folic acid was treated with the compound of Example 13 for 6 hours, then changed to a new medium containing no compound, and continued to culture for 48 hours. The number of viable cells after 48 hours was measured using the MTS method.
- SKOV-3 cells and L1210 cells have been reported to express medium to high folate receptors on the cell surface.
- A549 cells have been reported to be expressed to the extent that folate receptors are lowly expressed or not detected on the cell surface (Analytical Biochemistry, 2005, 338, 284-293, Molecular Cancer Therapeutics, 2015. , Vol. 14, pp. 1605-13).
- Table 7 shows the cytotoxicity of each test substance.
- EC 50 means 50% effective concentration, which represents 50% of the maximum response from the lowest pharmacological action exhibited by the drug
- SKOV-3 represents SKOV-3 cells
- L1210 represents L1210 cells.
- the complex containing the peptide derivative (I) or a pharmacologically acceptable salt thereof is selectively used against cancer cells in which the folate receptor is expressed on the cell surface. It was shown to have high cytotoxicity.
- the peptide derivative (I) Since the peptide derivative (I) has various functional groups at the C-terminal, it is converted into a complex by conjugating the peptide derivative (I) or a prodrug thereof with a targeting ligand or polymer by an appropriate method. It was shown that it can be done.
- the complex obtained by conjugating the peptide derivative (I) or its prodrug and the targeting ligand by an appropriate method is a cell that is selectively high for specific cells by the action of the targeting ligand. It was shown to have toxicity.
- the peptide derivative (I) of the present invention or a pharmacologically acceptable salt thereof has high cytotoxicity, it can be used as a cytotoxic agent. Moreover, since the peptide derivative (I) of the present invention has various functional groups at the C-terminus, the peptide derivative (I) or a prodrug thereof can be complexed with a targeting ligand or polymer. The body or a pharmacologically acceptable salt thereof can be used as a cytotoxic agent.
Abstract
Description





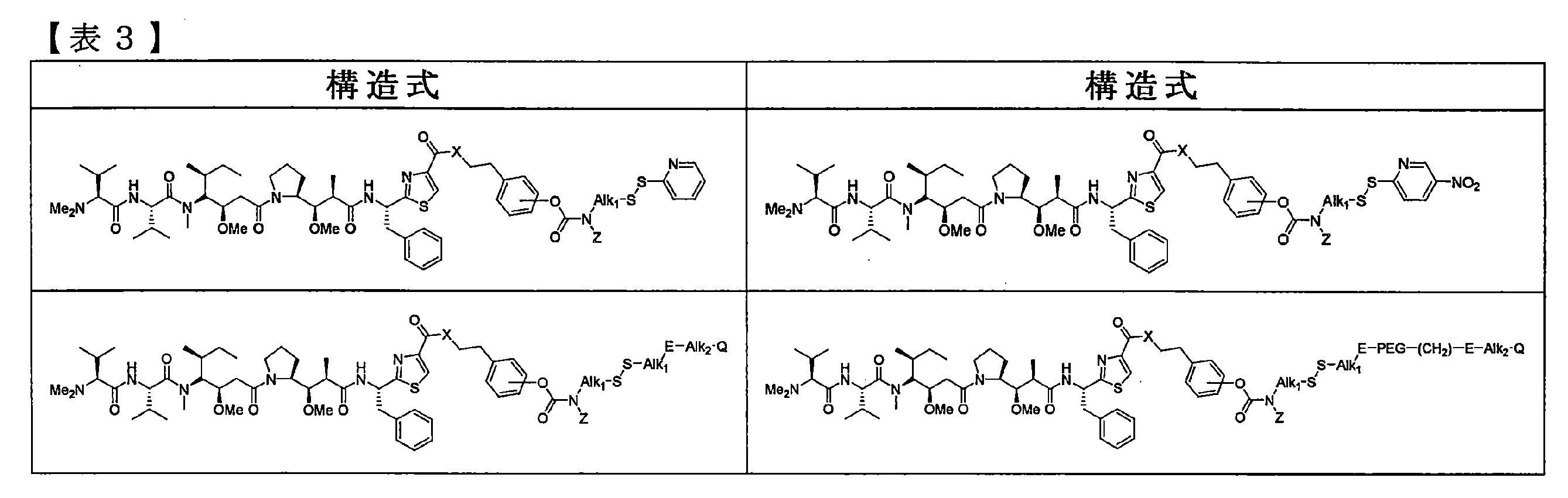



















Trt:トリフェニルメチル基; Boc:tert-ブトキシカルボニル基; THP:テトラヒドロピラニル基。

1H-NMR(400MHz,CDCl3)δ:7.97(1H,s),7.70-7.42(1H,m),7.30-7.15(5H,m),7.13-7.09(1H,m),5.62-5.55(1H,m),4.87-4.67(2H,m),4.11-3.10(7H,m),3.32(3H×2,s),3.04(3H,s),2.70-2.28(4H,m),2.53(6H,s),2.19-2.06(2H,m),1.98-1.55(4H,m),1.40-1.20(1H,m),1.18-0.69(23H,m).
MS m/z (ESI)[M+H]+:829.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.92(1H,s),7.45-7.13(20H,m),6.89(1H,d,J=9.1Hz),5.54-5.47(1H,m),4.87-4.73(2H,m),4.16-4.09(1H,m),4.08-4.03(1H,m),3.87(1H,d,J=8.6Hz),3.46-3.18(6H,m),3.33(3H,s),3.31(3H,s),3.03(3H,s),2.51(2H,t,J=6.6Hz),2.45-2.30(4H,m),2.25(6H,s),2.10-1.90(2H,m),1.81-1.55(4H,m),1.27-1.19(1H,m),1.10(3H,d,J=6.8Hz),1.07-0.89(17H,m),0.81(3H,t,J=7.5Hz).
MS m/z (ESI)[M+H]+:1130.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.97(1H,s),7.70(1H,t,J=6.1Hz),7.49(1H,brs),7.29-7.12(5H,m),6.90(1H,d,J=8.2Hz),5.54-5.48(1H,m),4.82-4.74(2H,m),4.14-4.03(2H,m),3.86(1H,d,J=7.7Hz),3.63(2H,q,J=6.5Hz),3.51-3.16(4H,m),3.33(3H,s),3.32(3H,s),3.04(3H,s),2.81-2.74(2H,m),2.48-2.33(4H,m),2.25(6H,s),2.11-1.92(2H,m),1.85-1.60(4H,m),1.46(1H,t,J=8.6Hz),1.40-1.22(1H,m),1.10(3H,d,J=7.2Hz),1.05-0.90(17H,m),0.81(3H,t,J=7.5Hz).
MS m/z (ESI)[M+H]+:888.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:8.00(1H,s),7.44-7.40(6H,m),7.37(1H,d,J=7.8Hz),7.30-7.10(14H,m),6.90(1H,d,J=9.6Hz),5.55-5.46(1H,m),4.79(2H,t,J=48.0Hz),4.15-4.07(1H,m),4.11(2H,t,J=6.9Hz),4.05-4.00(1H,m),3.87(1H,d,J=7.8Hz),3.80-3.15(4H,m),3.32(3H,s),3.31(3H,s),3.02(3H,s),2.61(2H,t,J=6.9Hz),2.45-2.28(4H,m),2.24(6H,s),2.12-1.96(2H,m),1.90-1.50(4H,m),1.40-1.30(1H,m),1.09(3H,d,J=6.9Hz),1.03-0.89(17H,m),0.81(3H,t,J=7.3Hz).
MS m/z (ESI)[M+H]+:1131.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:8.07(1H,s),7.41(1H,brs),7.30-7.11(5H,m),6.90(1H,d,J=9.1Hz),5.55-5.49(1H,m),4.83-4.74(2H,m),4.47(2H,t,J=6.9Hz),4.13-3.11(7H,m),3.32(6H,s),3.03(3H,s),2.88(2H,t,J=6.9Hz),2.47-2.28(4H,m),2.24(6H,s),2.11-1.98(2H,m),1.98-1.55(4H,m),1.41-1.30(1H,m),1.10(3H,d,J=6.9Hz),1.05-0.91(17H,m),0.81(3H,t,J=7.3Hz).
MS m/z (ESI)[M+H]+:889.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.42(d,J=8.2Hz,6H),7.29(t,J=7.5Hz,6H),7.21(t,J=7.3Hz,3H),3.03(t,J=7.5Hz,2H),2.60(brs,3H),2.33(brs,2H),1.40(s,9H).
MS m/z (ESI)[M+Na]+:456.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.44(dd,J=10.7,4.8Hz,6H),7.32-7.26(m,6H),7.24-7.19(m,3H),3.71(t,J=1.8Hz,1H),3.07(t,J=2.3Hz,1H),2.51(t,J=6.6Hz,1H),2.36(dd,J=11.2,4.8Hz,1H),2.29(t,J=1.8Hz,3H),1.50(s,2H).
MS m/z (ESI)[Trt]+:243.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.84(1H,s),7.82-7.76(1H,m),7.30-7.00(5H,m),6.92(1H,d,J=9.1Hz),5.56-5.48(1H,m),5.07(1H,brs),4.87-4.70(2H,m),4.18-3.20(17H,m),3.10(3H,s),3.02(3H,s),2.45-2.20(4H,m),2.23(6H,s),2.11-1.89(2H,m),1.87-1.47(4H,m),1.40-1.30(1H,m),1.09(3H,d,J=6.9Hz),1.05-0.86(17H,m),0.80(3H,t,J=7.3Hz).
MS m/z (ESI)[M+H]+:1144.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.90(1H,s),7.60-7.45(1H,m),7.28-7.02(5H,m),6.90(1H,d,J=9.1Hz),5.58-5.51(1H,m),4.82-4.74(2H,m),4.20-3.20(9H,m),3.33(3H,s),3.32(3H,s),3.11(3H,s),3.03(3H,s),2.91-2.77(2H,m),2.45-2.31(4H,m),2.24(6H,s),2.10-1.89(2H,m),1.86-1.60(4H,m),1.43-1.20(1H,m),1.11(3H,d,J=6.9Hz),1.05-0.90(17H,m),0.81(3H,t,J=7.3Hz).
MS m/z (ESI)[M+H]+:902.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:8.07(1H,s),7.44(1H,brs),7.30-7.12(5H,m),6.90(1H,d,J=9.1Hz),5.55-5.49(1H,m),5.02(1H,brs),4.82-4.70(2H,m),4.41(2H,t,J=4.6Hz),4.18-4.08(1H,m),4.05-4.00(1H,m),3.87(1H,d,J=7.3Hz),3.70-3.14(6H,m),3.32(3H,s),3.31(3H,s),3.03(3H,s),2.48-2.33(4H,m),2.24(6H,s),2.13-1.87(2H,m),1.80-1.60(4H,m),1.43(9H,s),1.35-1.16(1H,m),1.10(3H,d,J=7.3Hz),1.05-0.87(17H,m),0.81(3H,t,J=7.3Hz).
MS m/z (ESI)[M+H]+:972.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.98(1H,s),7.78(2H,brs),7.54(1H,brs),7.32-7.00(5H,m),6.90(1H,d,J=9.1Hz),5.51-5.45(1H,m),4.80-4.70(2H,m),4.18-4.00(2H,m),3.82(2H,t,J=4.8Hz),3.80-3.10(7H,m),3.33(3H,s),3.32(3H,s),3.05(3H,s),2.52-2.30(4H,m),2.24(6H,s),2.14-1.92(2H,m),1.86-1.60(4H,m),1.40-1.19(1H,m),1.15-0.91(20H,m),0.82(3H,t,J=6.4Hz).
MS m/z (ESI)[M+H]+:872.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.88-7.75(1H,m),7.70-7.60(1H,m),7.34-7.00(5H,m),6.90(1H,d,J=8.7Hz),6.42-6.35(1H,m),5.63-5.48(1H,m),4.86-4.70(2H,m),4.18-4.00(2H,m),3.90-3.17(9H,m),3.32(3H,s),3.31(3H,s),3.09(3H,s),3.02(3H,s),2.45-2.30(4H,m),2.23(6H,s),2.17-1.85(2H,m),1.80-1.60(4H,m),1.35(9H,s),1.30-1.15(1H,m),1.10-0.86(20H,m),0.80(3H,t,J=7.5Hz).
MS m/z (ESI)[M+H]+:985.(LC/MS-1)

1H-NMR(400MHz,CD3OD)δ:8.98-8.60(1H,m),8.14-7.92(1H,m),7.37-7.18(5H,m),5.66-5.57(1H,m),4.85-4.63(2H,m),4.20-3.07(20H,m),3.16(3H,s),2.92(3H,s),2.91(3H,s),2.48-1.39(10H,m),1.30-0.98(21H,m),0.87(3H,t,J=7.1Hz).
MS m/z (ESI)[M+Na]+:907.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.78(1H,s),7.70-7.40(1H,brm),7.30-7.11(5H,m),6.90(1H,d,J=8.7Hz),5.57-5.48(1H,m),4.85-4.73(2H,m),4.18-4.00(2H,m),3.90-2.62(12H,m),3.33(3H,s),3.32(3H,s),3.13(3H,s),3.03(3H,s),2.45-2.30(4H,m),2.24(6H,s),2.13-1.90(2H,m),1.80-1.60(4H,m),1.45(9H,s),1.38-1.30(1H,m),1.16-0.91(20H,m),0.81(3H,t,J=7.3Hz).
MS m/z (ESI)[M+H]+:999.(LC/MS-1)

1H-NMR(400MHz,CD3OD)δ:8.15-8.02(1H,m),7.37-7.18(5H,m),5.66-5.57(1H,m),4.85-4.63(2H,m),4.20-3.07(20H,m),3.27(3H,s),3.15(3H,s),2.91(3H,s),2.89(3H,s),2.51-1.39(10H,m),1.30-0.87(21H,m),0.86(3H,t,J=7.3Hz).
MS m/z (ESI)[M+H]+:899.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.96(1H,s),7.85-7.75(1H,m),7.60(1H,brs),7.30-7.07(5H,m),6.91(1H,d,J=8.7Hz),5.50-5.43(1H,m),4.83-4.72(2H,m),4.16-4.09(1H,m),4.05-4.01(1H,m),3.84-3.78(3H,m),3.65-3.20(6H,m),3.32(3H,s),3.31(3H,s),3.04(3H,s),2.48-2.32(4H,m),2.23(6H,s),2.16-1.63(6H,m),1.40-1.25(1H,m),1.08(3H,d,J=6.9Hz),1.04-0.88(17H,m),0.80(3H,t,J=6.4Hz).
MS m/z (ESI)[M+H]+:872.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:8.05(1H,s),7.36(1H,d,J=6.9Hz),7.30-7.11(5H,m),6.89(1H,d,J=9.6Hz),5.55-5.48(1H,m),4.83-4.75(2H,m),4.69(1H,t,J=3.4Hz),4.60-4.46(2H,m),4.18-4.08(1H,m),4.07-4.00(2H,m),3.91-3.84(2H,m),3.82-3.75(1H,m),3.55-3.25(5H,m),3.33(3H,s),3.32(3H,s),3.02(3H,s),2.46-2.30(4H,m),2.24(6H,s),2.13-1.97(2H,m),1.93-1.51(10H,m),1.42-1.30(1H,m),1.10(3H,d,J=6.9Hz),1.05-0.87(17H,m),0.81(3H,t,J=7.5Hz).
MS m/z (ESI)[M+H]+:957.(LC/MS-1)
1H-NMR(400MHz,CDCl3)δ:8.10(1H,s),7.49-7.44(1H,m),7.30-7.13(5H,m),6.90(1H,d,J=9.1Hz),5.55-5.48(1H,m),4.82-4.74(2H,m),4.50(2H,t,J=4.6Hz),4.20-3.26(9H,m),3.33(3H,s),3.32(3H,s),3.02(3H,s),2.46-2.34(4H,m),2.24(6H,s),2.11-1.90(2H,m),1.84-1.60(4H,m),1.43-1.30(1H,m),1.09(3H,d,J=7.3Hz),1.03-0.89(17H,m),0.81(3H,t,J=7.5Hz).
MS m/z (ESI)[M+H]+:873.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.84(1H,s),7.82-7.76(1H,m),7.30-7.00(5H,m),6.92(1H,d,J=9.1Hz),5.56-5.48(1H,m),5.07(1H,brs),4.87-4.70(2H,m),4.18-3.20(17H,m),3.10(3H,s),3.02(3H,s),2.45-2.20(4H,m),2.23(6H,s),2.11-1.89(2H,m),1.87-1.47(4H,m),1.40-1.30(1H,m),1.09(3H,d,J=6.9Hz),1.05-0.86(17H,m),0.80(3H,t,J=7.3Hz).
MS m/z (ESI)[M+H]+:886.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.95(1H,s),7.43-7.36(2H,m),7.30-7.17(5H,m),7.04(2H,d,J=8.2Hz),6.89(1H,d,J=9.1Hz),6.65(2H,d,J=8.2Hz),5.53-5.46(1H,m),4.83-4.72(2H,m),4.15-4.09(1H,m),4.08-4.03(1H,m),3.87(1H,d,J=8.2Hz),3.70-3.18(6H,m),3.33(3H,s),3.31(3H,s),3.03(3H,s),2.82(2H,t,J=7.1Hz),2.45-2.35(4H,m),2.24(6H,s),2.18-1.89(2H,m),1.84-1.64(4H,m),1.40-1.20(1H,m),1.10(3H,d,J=7.3Hz),1.03-0.90(17H,m),0.81(3H,t,J=7.3Hz).
MS m/z (ESI)[M+H]+:947.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.93(1H,s),7.41-7.30(2H,m),7.27-7.10(5H,m),7.04(2H,d,J=8.2Hz),6.95-6.85(1H,m),6.82(2H,d,J=8.2Hz),5.49-5.44(1H,m),4.85-4.72(2H,m),4.14-3.15(9H,m),3.33(3H,s),3.26(3H,s),3.03(3H,s),2.82(2H,t,J=6.9Hz),2.51-2.30(4H,m),2.23(6H,s),2.17-1.94(2H,m),1.90-1.60(4H,m),1.40-1.20(1H,m),1.15-0.88(20H,m),0.80(3H,t,J=7.3Hz).
MS m/z (ESI)[M+H]+:949.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ(ppm):7.96(1H,s),7.69(1H,brs),7.50(1H,brs),7.34-7.09(5H,m),6.90(1H,d,J=9.1Hz),5.55-5.44(1H,m),5.03(1H,brs),4.89-4.67(2H,m),4.20-3.20(11H,m),3.33(3H,s),3.32(3H,s),3.04(3H,s),2.50-2.33(4H,m),2.25(6H,s),2.10-1.92(2H,m),1.85-1.66(4H,m),1.42(9H,s),1.40-1.20(1H,m),1.15-0.79(23H,m).
MS m/z (ESI)[M+H]+:971.(LC/MS-1)

1H-NMR(400MHz,CD3OD)δ:8.65(1H,brs),[8.19(0.5H,s),8.16(0.5H,s)],7.32-7.16(5H,m),5.69-5.56(1H,m),4.80-4.63(2H,m),4.15-3.05(14H,m),3.33(6H,s),2.91(6H,s),2.51-1.38(10H,m),1.30-0.91(21H,m),0.85(3H,t,J=7.1Hz).
MS m/z (ESI)[M+H]+:871.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.94(1H,s),7.79(1H,brs),7.47(1H,brs),7.26-7.09(5H,m),6.89(1H,d,J=9.1Hz),5.53-5.46(1H,m),4.82-4.74(2H,m),4.15-3.11(11H,m),3.33(3H,s),3.31(3H,s),3.03(3H,s),2.92(3H,s),2.47-2.34(4H,m),2.24(6H,s),2.12-1.94(2H,m),1.77-1.65(4H,m),1.43(9H,s),1.40-1.20(1H,m),1.12-0.78(23H,m).
MS m/z (ESI)[M+H]+:985.(LC/MS-1)

1H-NMR(400MHz,CD3OD)δ:8.63(1H,brs),8.22-8.14(1H,m),7.35-7.15(5H,m),5.70-5.52(1H,m),4.83-4.63(2H,m),4.17-3.04(20H,m),2.91(3H,s),2.90(3H,s),2.74(3H,s),2.51-1.98(6H,m),1.91-1.50(4H,m),1.42-0.80(24H,m).
MS m/z (ESI)[M+H]+:885.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.91(1H,s),7.69(1H,t,J=6.1Hz),7.46(1H,brs),7.42-7.10(20H,m),6.92(1H,d,J=9.5Hz),6.16(1H,brs),5.51-5.45(1H,m),4.88-4.74(2H,m),4.13-4.09(1H,m),4.07-4.02(1H,m),3.87(1H,d,J=7.7Hz),3.60-3.18(8H,m),3.33(3H,s),3.32(3H,s),3.04(3H,s),2.50-2.32(6H,m),2.25(6H,s),2.09-1.96(4H,m),1.90-1.63(4H,m),1.40-1.20(1H,m),1.10(3H,d,J=7.2Hz),1.05-0.88(17H,m),0.81(3H,t,J=7.2Hz).
MS m/z (ESI)[M+H]+:1201.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.97(1H,s),7.78(1H,brs),7.51(1H,brs),7.32-7.10(5H,m),7.00-6.85(1H,m),6.58(1H,brs),5.53-5.45(1H,m),4.82-4.74(2H,m),4.15-4.08(1H,m),4.06-4.00(1H,m),3.86(1H,d,J=7.7Hz),3.64-3.18(8H,m),3.33(3H,s),3.32(3H,s),3.04(3H,s),2.82-2.75(2H,m),2.50(2H,t,J=6.8Hz),2.48-2.37(4H,m),2.26(6H,s),2.12-1.92(2H,m),1.83-1.57(4H,m),1.27-1.19(1H,m),1.11-0.86(20H,m),0.81(3H,t,J=7.2Hz).
MS m/z (ESI)[M+H]+:959.(LC/MS-1)

1H-NMR(400MHz,CDCl3)δ:7.76(d,J=7.3Hz,2H),7.61(d,J=7.8Hz,2H),7.40(td,J=4.9,2.9Hz,7H),7.30(ddt,J=19.7,8.5,3.3Hz,9H),7.22-7.18(m,2H),6.65(d,J=6.4Hz,1H),6.41(s,1H),5.59(d,J=8.2Hz,1H),4.36(dt,J=16.6,6.1Hz,2H),4.21(t,J=6.9Hz,2H),3.04(dd,J=9.8,6.2Hz,2H),2.39(dt,J=18.8,6.6Hz,3H),2.25(dt,J=23.0,6.6Hz,3H),2.01(t,J=6.9Hz,1H),1.87(t,J=6.6Hz,1H),1.46(s,9H),1.42(s,9H),1.13(dt,J=13.3,5.7Hz,2H).
MS m/z (ESI)[M+Na]+:934.(LC/MS-2)

1H-NMR(400MHz,CDCl3)δ:7.41(t,J=4.5Hz,6H),7.29(dd,J=10.4,5.0Hz,7H),7.21(dd,J=8.4,6.1Hz,3H),6.91(s,1H),6.85(d,J=7.2Hz,1H),4.36-4.31(m,1H),3.31-3.26(m,1H),3.12-3.06(m,1H),2.95(dd,J=13.6,6.3Hz,1H),2.45-2.22(m,7H),2.10-2.01(m,3H),1.95-1.87(m,7H),1.46(s,9H),1.43(d,J=3.6Hz,12H),1.30-1.24(m,2H),1.16(t,J=7.0Hz,1H),1.11(t,J=7.2 Hz,1H).
MS m/z (ESI)[M+H]+:690.(LC/MS-1)
1H-NMR(400MHz,CD3OD)δ:8.67(s,1H),7.89(d,J=8.6Hz,3H),7.55(d,J=8.6Hz,2H),7.35(dd,J=5.2,3.4Hz,6H),7.27-7.23(m,6H),7.18(td,J=5.7,2.9Hz,3H),4.40(dd,J=9.5,4.5Hz,1H),4.28(dd,J=8.4,5.7Hz,1H),3.08(dd,J=13.4,6.6Hz,1H),2.99(q,J=6.9Hz,1H),2.40(dd,J=10.9,4.5Hz,2H),2.28(dt,J=17.2,5.0Hz,4H),2.05-1.98(m,2H),1.82(dd,J=13.8,8.4Hz,1H),1.46(s,9H),1.39(s,9H),1.28(s,1H).
MS m/z (ESI)[M-H]-:1079.(LC/MS-1)

1H-NMR(DMSO-d6)δ:8.66(s,1H),8.23(d,J=7.7Hz,1H),8.05(q,J=6.2Hz,2H),7.66(d,J=8.6Hz,2H),6.64(d,J=8.6Hz,2H),4.50(s,2H),4.28(t,J=10.9Hz,1H),4.18(dd,J=13.1,8.2Hz,1H),3.23-3.13(m,2H),2.35(t,J=8.2Hz,1H),2.22(dd,J=16.5,7.5Hz,5H),2.04(d,J=6.8Hz,2H),1.90(dd,J=24.7,10.2Hz,3H),1.70(t,J=7.2Hz,1H),1.25(s,1H).
MS m/z (ESI)[M+H]+:630.(LC/MS-2)
tR=12.21 min.(HPLC)

1H-NMR(400MHz,CDCl3)δ:7.96(1H,s),7.41-7.30(2H,m),7.27-7.10(5H,m),7.06(2H,d,J=8.2Hz),5.49-5.44(1H,m),4.85-4.77(2H,m),4.13-3.64(9H,m),3.60-3.29(15H,m),3.13(3H,s),3.03(3H,s),2.51-2.30(4H,m),2.25(6H,s),2.17-1.98(2H,m),1.78(12H,s),1.11-0.88(24H,m),0.82(5H,m).
MS m/z (ESI)[M-Boc]2+:577.(LC/MS-1)

MS m/z (ESI)[M+H]3+:635.(LC/MS-1)
tR=17.11 min.(HPLC)

MS m/z (ESI)[M-H]2-:1266.(LC/MS-1)
HRMS m/z (ESI)[M-H]-:2532.1754.
C118H180N20O33S4の計算精密質量:2532.1832.
tR=16.40 min.(HPLC)

1H-NMR(400MHz,CDCl3)δ:8.01(1H,s),7.87(1H,brs),7.46(1H,brs),7.26-7.15(5H,m),6.90(1H,d,J=8.2Hz),5.56(1H,brs),4.81-4.77(2H,m),4.12(1H,brs),4.05(1H,brs),3.97(1H,brs),3.88(1H,d,J=7.7Hz),3.80(2H,m),3.51-3.16(4H,m),3.33(3H,s),3.32(3H,s),3.13(3H,s),3.04(3H,s),2.96(3H,s),2.81-2.74(2H,m),2.89(3H,s),2.46-2.34(3H,m),2.25(6H,s),2.12-1.97(2H,m),1.66(15H,d,J=8.8Hz),1.45(9H,t,J=8.6Hz),1.10(2H,d,J=7.2Hz),1.05-0.90(12H,m),0.82(3H,t,J=7.5Hz).
MS m/z (ESI)[M+H]+:1092.(LC/MS-1)

MS m/z (ESI)[M+H]2+:871.(LC/MS-1)
tR=16.66 min.(HPLC)

MS m/z (ESI)[M-H]2-:1184.(LC/MS-1)
HRMS m/z (ESI)[M-H]-:2369.1162.
C109H171N19O31S4の計算精密質量:2369.1198.
tR=15.85 min.(HPLC)
(LC/MS-1)
液体クロマトグラフシステム:LC1200(アジレント・テクノロジー社製)
質量分析計:6130A(アジレント・テクノロジー社製)
分析条件
カラム:ラピッドレゾリューションHTカートリッジ
移動相A:0.1体積%ギ酸-蒸留水、HPLC用
移動相B:0.1体積%ギ酸-アセトニトリル、HPLC用
グラジエント条件:移動層B体積%
0-1.5 min:20%-95%
1.5-3.0 min:95%
流速:0.5 mL/min
注入量:2.0μL
カラム温度:40℃
(LC/MS-2)
液体クロマトグラフシステム:LC1260 Infinity II(アジレント・テクノロジー社製)
質量分析計:6130B(アジレント・テクノロジー社製)
分析条件
カラム:ラピッドレゾリューションHTカートリッジ
移動相A:0.1体積%ギ酸-蒸留水、HPLC用
移動相B:0.1体積%ギ酸-アセトニトリル、HPLC用
グラジエント条件:移動層B体積%
0-1.5 min:20%-95%
1.5-3.0 min:95%
流速:0.5 mL/min
注入量:2.0μL
カラム温度:40℃
液体クロマトグラフシステム:NexeraX2(島津製作所製)
質量分析計:LCMS-IT-TOF質量分析計(島津製作所製)
分析条件
移動相:メタノール
流速:0.1 mL/min
注入量:2.0μL
ODSカラム精製条件
カラム:山善ハイフラッシュカラム ODS サイズM
流速:10 ml/min
カラム温度:室温
検出波長:254 nm
移動層A:0.05体積%TFA in H2O
移動層B:0.05体積%TFA in CH3CN
グラジエント:移動層B体積%
0-5 min:5.0%
5-20 min:5%から95%まで上昇
20-25 min:95%
(HPLC)
カラム:COSMOSIL 5C18-AR-IIl, 4.6mmI.D. x 150mm
流速:1.0 ml/min
カラム温度:40℃
検出波長:254 nm
移動層A:0.05体積%TFA in H2O
移動層B:0.05体積%TFA in CH3CN
グラジエント:移動層B体積%
0-5min:5.0%
5-20min:5%から95%まで上昇
20-25min:95%
実施例1~11の化合物、参考例1の化合物、参考例12の化合物、参考例14の化合物、参考例16の化合物及び2-((S)-1-(((2S,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-(ジメチルアミノ)-3-メチルブタンアミド)-N,3-ジメチルブタンアミド)-3-メトキシ-5-メチルヘプタノイル)ピロリジン-2-イル)-3-メトキシ-2-メチルプロパンアミド)-2-フェニルエチル)-N-エチルチアゾール-4-カルボキサミド(以下、比較例化合物)のSKOV-3細胞、A549細胞及びL1210細胞に対する細胞毒性をMTS法を用いて測定した。
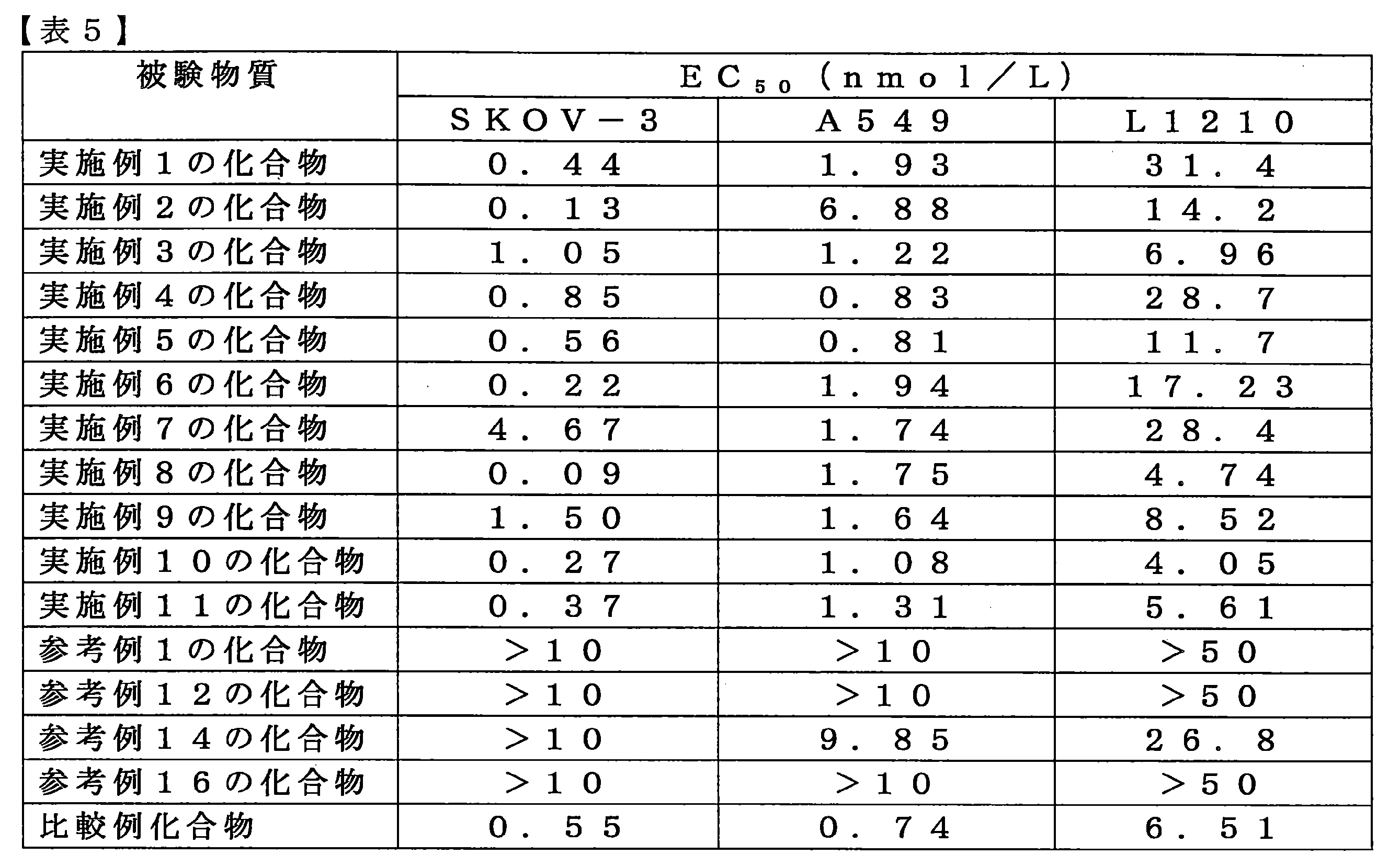
実施例1~11の化合物、参考例1の化合物、参考例12の化合物、参考例14の化合物、参考例16の化合物及び比較例化合物の人工膜透過性をPre-coated PAMPA PlateSystem(Corning)を使用し、評価した。
液体クロマトグラフシステム:Waters Acquity UPLC(Waters社製)
質量分析計:Waters SQD Detector(Waters社製)
カラム:Ascentis Express C18,2.7 μm,2.1 mm ID×20mm(Sigma-Aldrich社)
移動相A:0.1体積% ギ酸水溶液
移動相B:アセトニトリル
流速:0.6 mL/min
グラジエント:移動層B体積%
0.0~2.0 min:3.0→100%
2.0~2.4 min:100%
2.4~2.5 min:100→3.0%
Cequilibrium=(CD×VD-CA×VA)/(VD-VA)
Pe=[-ln(1-CA/Cequilibrium)]/S×(1/VD+1/VA)×t
Cequilibrium:Donor側及びAcceptor側プレートウェルの平衡濃度
CD:Donor側プレートウェルの5時間後の濃度(mmol/L)
VD:Donor側プレートウェルに添加した標準溶液の容積(0.32 mL)
CA:Acceptor側プレートウェルの5時間後の濃度(mmol/L)
VA:Acceptor側プレートウェルに添加した20%メタノール含むPBSの容積(0.2 mL)
S:膜表面積(0.3 cm2)
t:静置時間18,000 s(=5 hr)

実施例12の化合物及び実施例13の化合物のSKOV-3細胞、A549細胞及びL1210細胞に対する細胞毒性をMTS法を用いて測定した。

Claims (8)
- 以下の一般式(I)で示されるペプチド誘導体又はその薬理学的に許容される塩。

- Xは、酸素原子である、請求項1記載のペプチド誘導体又はその薬理学的に許容される塩。
- Yは、NH2、N(Me)H、SH又はOHである、請求項2記載のペプチド誘導体又はその薬理学的に許容される塩。
- Xは、NRである、請求項1記載のペプチド誘導体又はその薬理学的に許容される塩。
- Rは、炭素数1~3のアルキル基である、請求項4記載のペプチド誘導体又はその薬理学的に許容される塩。
- Rは、メチル基である、請求項5記載のペプチド誘導体又はその薬理学的に許容される塩。
- 請求項1~6のいずれか一項記載のペプチド誘導体と、ターゲッティングリガンド又はポリマーと、を含む複合体又はその薬理学的に許容される塩。
- 請求項1~6のいずれか一項記載のペプチド誘導体若しくはその薬理学的に許容される塩、又は、請求項7記載の複合体若しくはその薬理学的に許容される塩、を有効成分として含有する、細胞毒性剤。
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17775174.0A EP3438118B1 (en) | 2016-03-29 | 2017-03-29 | Peptide derivative and use thereof |
| BR112018016008-3A BR112018016008A2 (ja) | 2016-03-29 | 2017-03-29 | A peptide derivative and its use |
| US16/084,994 US10792365B2 (en) | 2016-03-29 | 2017-03-29 | Peptide derivative and use thereof |
| CN201780017518.8A CN108779146B (zh) | 2016-03-29 | 2017-03-29 | 肽衍生物及其用途 |
| CA3014421A CA3014421C (en) | 2016-03-29 | 2017-03-29 | Peptide derivative and use thereof |
| FIEP17775174.0T FI3438118T3 (fi) | 2016-03-29 | 2017-03-29 | Peptidijohdannainen ja sen käyttö |
| JP2017517819A JP6858348B2 (ja) | 2016-03-29 | 2017-03-29 | ペプチド誘導体及びその用途 |
| PL17775174.0T PL3438118T3 (pl) | 2016-03-29 | 2017-03-29 | Pochodna peptydu i jej zastosowanie |
| ES17775174T ES2948846T3 (es) | 2016-03-29 | 2017-03-29 | Derivado peptídico y uso del mismo |
| DK17775174.0T DK3438118T3 (da) | 2016-03-29 | 2017-03-29 | Peptidderivat og anvendelse deraf |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016-065737 | 2016-03-29 | ||
| JP2016065737 | 2016-03-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017170637A1 true WO2017170637A1 (ja) | 2017-10-05 |
Family
ID=59964715
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/012797 WO2017170637A1 (ja) | 2016-03-29 | 2017-03-29 | ペプチド誘導体及びその用途 |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US10792365B2 (ja) |
| EP (1) | EP3438118B1 (ja) |
| JP (1) | JP6858348B2 (ja) |
| CN (1) | CN108779146B (ja) |
| BR (1) | BR112018016008A2 (ja) |
| CA (1) | CA3014421C (ja) |
| DK (1) | DK3438118T3 (ja) |
| ES (1) | ES2948846T3 (ja) |
| FI (1) | FI3438118T3 (ja) |
| HU (1) | HUE062120T2 (ja) |
| PL (1) | PL3438118T3 (ja) |
| PT (1) | PT3438118T (ja) |
| WO (1) | WO2017170637A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20210125484A (ko) * | 2019-03-08 | 2021-10-18 | 레베나 바이오파마 컴퍼니 리미티드 | 항체 약물 결합체에 사용되는 약물-링커 mc-mmaf의 제조 방법 및 이의 중간체 |
| WO2023033129A1 (ja) | 2021-09-03 | 2023-03-09 | 東レ株式会社 | 癌の治療及び/又は予防用医薬組成物 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993003054A1 (en) * | 1991-08-09 | 1993-02-18 | Teikoku Hormone Mfg. Co., Ltd. | Novel tetrapeptide derivative |
| WO2013173393A1 (en) * | 2012-05-15 | 2013-11-21 | Concortis Biosystems, Corp | Drug-conjugates, conjugation methods, and uses thereof |
| WO2014086952A1 (en) * | 2012-12-05 | 2014-06-12 | Institut Curie | Conjugates of the b-subunit of shiga toxin for anticancer therapies |
| WO2015113760A1 (en) * | 2014-01-28 | 2015-08-06 | Tube Pharmaceuticals Gmbh | Cytotoxic tubulysin compounds for conjugation |
| WO2015118497A1 (fr) * | 2014-02-07 | 2015-08-13 | Centre National De La Recherche Scientifique | Conjugués et pro-drogues pour le traitement du cancer et de maladies inflammatoires |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4816444A (en) | 1987-07-10 | 1989-03-28 | Arizona Board Of Regents, Arizona State University | Cell growth inhibitory substance |
| EP0731106B1 (en) | 1993-10-01 | 2004-11-17 | Teikoku Hormone Mfg. Co., Ltd. | Dolastatin derivatives |
| US6884869B2 (en) | 2001-04-30 | 2005-04-26 | Seattle Genetics, Inc. | Pentapeptide compounds and uses related thereto |
| US6737409B2 (en) * | 2001-07-19 | 2004-05-18 | Hoffmann-La Roche Inc. | Dolastatin 10 derivatives |
| EP2357006B1 (en) | 2002-07-31 | 2015-09-16 | Seattle Genetics, Inc. | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| WO2012171020A1 (en) | 2011-06-10 | 2012-12-13 | Mersana Therapeutics, Inc. | Protein-polymer-drug conjugates |
| FR3005051A1 (fr) * | 2013-04-25 | 2014-10-31 | Pf Medicament | Derives de la dolastatine 10 et d'auristatines |
-
2017
- 2017-03-29 ES ES17775174T patent/ES2948846T3/es active Active
- 2017-03-29 US US16/084,994 patent/US10792365B2/en active Active
- 2017-03-29 PT PT177751740T patent/PT3438118T/pt unknown
- 2017-03-29 CN CN201780017518.8A patent/CN108779146B/zh active Active
- 2017-03-29 PL PL17775174.0T patent/PL3438118T3/pl unknown
- 2017-03-29 HU HUE17775174A patent/HUE062120T2/hu unknown
- 2017-03-29 EP EP17775174.0A patent/EP3438118B1/en active Active
- 2017-03-29 WO PCT/JP2017/012797 patent/WO2017170637A1/ja active Application Filing
- 2017-03-29 FI FIEP17775174.0T patent/FI3438118T3/fi active
- 2017-03-29 DK DK17775174.0T patent/DK3438118T3/da active
- 2017-03-29 JP JP2017517819A patent/JP6858348B2/ja active Active
- 2017-03-29 BR BR112018016008-3A patent/BR112018016008A2/ja unknown
- 2017-03-29 CA CA3014421A patent/CA3014421C/en active Active
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993003054A1 (en) * | 1991-08-09 | 1993-02-18 | Teikoku Hormone Mfg. Co., Ltd. | Novel tetrapeptide derivative |
| WO2013173393A1 (en) * | 2012-05-15 | 2013-11-21 | Concortis Biosystems, Corp | Drug-conjugates, conjugation methods, and uses thereof |
| WO2014086952A1 (en) * | 2012-12-05 | 2014-06-12 | Institut Curie | Conjugates of the b-subunit of shiga toxin for anticancer therapies |
| WO2015113760A1 (en) * | 2014-01-28 | 2015-08-06 | Tube Pharmaceuticals Gmbh | Cytotoxic tubulysin compounds for conjugation |
| WO2015118497A1 (fr) * | 2014-02-07 | 2015-08-13 | Centre National De La Recherche Scientifique | Conjugués et pro-drogues pour le traitement du cancer et de maladies inflammatoires |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20210125484A (ko) * | 2019-03-08 | 2021-10-18 | 레베나 바이오파마 컴퍼니 리미티드 | 항체 약물 결합체에 사용되는 약물-링커 mc-mmaf의 제조 방법 및 이의 중간체 |
| JP2022518601A (ja) * | 2019-03-08 | 2022-03-15 | 聯寧(蘇州)生物製薬有限公司 | 抗体薬物複合体用薬物リンカーmc-mmafの調製方法及びその中間体 |
| JP7292751B2 (ja) | 2019-03-08 | 2023-06-19 | 聯寧(蘇州)生物製薬有限公司 | 抗体薬物複合体用薬物リンカーmc-mmafの調製方法及びその中間体 |
| KR102590042B1 (ko) * | 2019-03-08 | 2023-10-17 | 레베나 (쑤저우) 바이오파마 컴퍼니 리미티드 | 항체 약물 결합체에 사용되는 약물-링커 mc-mmaf의 제조 방법 및 이의 중간체 |
| WO2023033129A1 (ja) | 2021-09-03 | 2023-03-09 | 東レ株式会社 | 癌の治療及び/又は予防用医薬組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| PT3438118T (pt) | 2023-06-07 |
| JPWO2017170637A1 (ja) | 2019-02-07 |
| EP3438118A1 (en) | 2019-02-06 |
| HUE062120T2 (hu) | 2023-09-28 |
| CA3014421C (en) | 2022-07-19 |
| US10792365B2 (en) | 2020-10-06 |
| CA3014421A1 (en) | 2017-10-05 |
| EP3438118B1 (en) | 2023-03-29 |
| PL3438118T3 (pl) | 2023-07-10 |
| US20190038757A1 (en) | 2019-02-07 |
| ES2948846T3 (es) | 2023-09-20 |
| FI3438118T3 (fi) | 2023-06-15 |
| CN108779146A (zh) | 2018-11-09 |
| CN108779146B (zh) | 2022-03-18 |
| EP3438118A4 (en) | 2019-12-18 |
| BR112018016008A2 (ja) | 2018-12-18 |
| JP6858348B2 (ja) | 2021-04-14 |
| DK3438118T3 (da) | 2023-05-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN108863963B (zh) | 作为pd-l1抑制剂的杂环类化合物 | |
| CN108794422B (zh) | 作为pd-l1抑制剂的杂环类化合物 | |
| ES2647317T3 (es) | Análogos de CC-1065 y sus conjugados | |
| RU2618523C2 (ru) | Аналоги сплицеостатина | |
| EP2026775B1 (en) | Use of syk tyrosine kinase inhibitors for the treatment of cell proliferative disorders | |
| CN111065623A (zh) | 用于抗体药物缀合物的接头 | |
| DK1876169T3 (en) | PHENYL-CONTAINING N-ACYL-AMINE AND AMINO ACID DERIVATIVES, PROCEDURES FOR PRODUCING THEREOF, PHARMACEUTICAL COMPOSITION AND APPLICATION OF THEREOF | |
| KR20170106472A (ko) | 접합 및 요법을 위한 헤미아스텔린 유도체 | |
| TW202309042A (zh) | 依沙替康(exatecan)衍生物及其抗體藥物結合物 | |
| EA022288B1 (ru) | Антипролиферативные соединения, их конъюгаты, способы их получения и их применение | |
| WO2020145374A1 (ja) | ピリミジン化合物又はその塩 | |
| JP5685440B2 (ja) | 3位において二置換されているインドール−2−オン誘導体、これらの調製およびこれらの治療上の使用 | |
| US20240067616A1 (en) | Peptide conjugates of microtubule-targeting agents as therapeutics | |
| JP2002509930A (ja) | ベンゾ複素環ジスタマイシン誘導体、その製造方法および抗腫瘍薬としてのそれの使用 | |
| JP6858348B2 (ja) | ペプチド誘導体及びその用途 | |
| US10472395B2 (en) | Cyclic peptide analogs and conjugates thereof | |
| EA014874B1 (ru) | Соединения для ингибирования апоптоза | |
| WO2023022231A1 (ja) | ウイルス感染症を処置または予防するための可逆的共有結合阻害物質 | |
| US20110275689A1 (en) | Preparation of 3-Pyrrole Substituted 2-Indolinone Derivatives | |
| WO2019074824A1 (en) | INDOLEAMINE 2,3-DIOXYGENASE INHIBITORS AND METHODS OF USE | |
| US8993515B2 (en) | Peptidomimetics comprising N-amino cyclic urea residues and uses thereof | |
| US7018981B2 (en) | Cyclic motilin receptor antagonists | |
| JP2019522674A (ja) | 抗腫瘍活性を有するベンゾ−n−ヒドロキシアミド化合物 | |
| TW200918065A (en) | Imidazopyridazine derivatives | |
| EP4294802A1 (en) | Cryptophycin compounds and conjugates thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2017517819 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3014421 Country of ref document: CA |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112018016008 Country of ref document: BR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017775174 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2017775174 Country of ref document: EP Effective date: 20181029 |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17775174 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 112018016008 Country of ref document: BR Kind code of ref document: A2 Effective date: 20180806 |