WO2015194152A1 - 光増感剤および光電変換素子 - Google Patents
光増感剤および光電変換素子 Download PDFInfo
- Publication number
- WO2015194152A1 WO2015194152A1 PCT/JP2015/002978 JP2015002978W WO2015194152A1 WO 2015194152 A1 WO2015194152 A1 WO 2015194152A1 JP 2015002978 W JP2015002978 W JP 2015002978W WO 2015194152 A1 WO2015194152 A1 WO 2015194152A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- dye
- aldehyde
- hydrogen atom
- photosensitizer
- Prior art date
Links
- 238000006243 chemical reaction Methods 0.000 title claims abstract description 35
- 239000003504 photosensitizing agent Substances 0.000 title claims abstract description 27
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 25
- 125000003118 aryl group Chemical group 0.000 claims abstract description 18
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 18
- 150000003839 salts Chemical class 0.000 claims abstract description 10
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims abstract description 7
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims abstract description 4
- 125000001424 substituent group Chemical group 0.000 claims description 13
- 125000003277 amino group Chemical group 0.000 claims description 12
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 11
- 125000003545 alkoxy group Chemical group 0.000 claims description 9
- 125000004122 cyclic group Chemical group 0.000 claims description 8
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 6
- 230000031700 light absorption Effects 0.000 claims description 6
- 230000002378 acidificating effect Effects 0.000 claims description 5
- 125000005843 halogen group Chemical group 0.000 claims description 5
- 125000000623 heterocyclic group Chemical group 0.000 claims description 5
- 125000002723 alicyclic group Chemical group 0.000 claims description 4
- 125000003342 alkenyl group Chemical group 0.000 claims description 4
- 125000004414 alkyl thio group Chemical group 0.000 claims description 4
- 125000002947 alkylene group Chemical group 0.000 claims description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 4
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 3
- 125000001174 sulfone group Chemical group 0.000 claims description 3
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 3
- 229910052717 sulfur Inorganic materials 0.000 claims description 3
- 125000004434 sulfur atom Chemical group 0.000 claims description 3
- 125000001931 aliphatic group Chemical group 0.000 claims description 2
- 239000000126 substance Substances 0.000 abstract description 3
- 150000001299 aldehydes Chemical class 0.000 description 122
- 239000000975 dye Substances 0.000 description 67
- 238000005481 NMR spectroscopy Methods 0.000 description 54
- 239000000543 intermediate Substances 0.000 description 46
- 230000015572 biosynthetic process Effects 0.000 description 44
- 238000003786 synthesis reaction Methods 0.000 description 42
- 229910052739 hydrogen Inorganic materials 0.000 description 41
- 239000010410 layer Substances 0.000 description 19
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 18
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- 238000000034 method Methods 0.000 description 15
- 239000007787 solid Substances 0.000 description 15
- 239000000758 substrate Substances 0.000 description 14
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 13
- -1 alkali metal salts Chemical class 0.000 description 13
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 description 13
- 239000004065 semiconductor Substances 0.000 description 12
- 239000000243 solution Substances 0.000 description 11
- 235000014692 zinc oxide Nutrition 0.000 description 10
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 9
- 239000011787 zinc oxide Substances 0.000 description 9
- 150000001875 compounds Chemical class 0.000 description 8
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 6
- 239000003792 electrolyte Substances 0.000 description 6
- 239000011521 glass Substances 0.000 description 6
- HSZCZNFXUDYRKD-UHFFFAOYSA-M lithium iodide Chemical compound [Li+].[I-] HSZCZNFXUDYRKD-UHFFFAOYSA-M 0.000 description 6
- 229910052751 metal Inorganic materials 0.000 description 6
- 239000002184 metal Substances 0.000 description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 5
- 238000013461 design Methods 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 4
- 230000000052 comparative effect Effects 0.000 description 4
- KXGVEGMKQFWNSR-UHFFFAOYSA-N deoxycholic acid Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 KXGVEGMKQFWNSR-UHFFFAOYSA-N 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 239000007772 electrode material Substances 0.000 description 4
- 239000010408 film Substances 0.000 description 4
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 4
- 238000004519 manufacturing process Methods 0.000 description 4
- 239000010409 thin film Substances 0.000 description 4
- BHQCQFFYRZLCQQ-UHFFFAOYSA-N (3alpha,5alpha,7alpha,12alpha)-3,7,12-trihydroxy-cholan-24-oic acid Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 BHQCQFFYRZLCQQ-UHFFFAOYSA-N 0.000 description 3
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 239000004380 Cholic acid Substances 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 206010070834 Sensitisation Diseases 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 238000006069 Suzuki reaction reaction Methods 0.000 description 3
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- BHQCQFFYRZLCQQ-OELDTZBJSA-N cholic acid Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 BHQCQFFYRZLCQQ-OELDTZBJSA-N 0.000 description 3
- 229960002471 cholic acid Drugs 0.000 description 3
- 235000019416 cholic acid Nutrition 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine group Chemical group N1=CCC2=CC=CC=C12 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 239000000203 mixture Substances 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 230000008313 sensitization Effects 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 3
- 229910052719 titanium Inorganic materials 0.000 description 3
- 239000010936 titanium Substances 0.000 description 3
- ISHFYECQSXFODS-UHFFFAOYSA-M 1,2-dimethyl-3-propylimidazol-1-ium;iodide Chemical compound [I-].CCCN1C=C[N+](C)=C1C ISHFYECQSXFODS-UHFFFAOYSA-M 0.000 description 2
- YSHMQTRICHYLGF-UHFFFAOYSA-N 4-tert-butylpyridine Chemical compound CC(C)(C)C1=CC=NC=C1 YSHMQTRICHYLGF-UHFFFAOYSA-N 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- RAXXELZNTBOGNW-UHFFFAOYSA-O Imidazolium Chemical compound C1=C[NH+]=CN1 RAXXELZNTBOGNW-UHFFFAOYSA-O 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-O Piperidinium(1+) Chemical compound C1CC[NH2+]CC1 NQRYJNQNLNOLGT-UHFFFAOYSA-O 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 2
- 238000005874 Vilsmeier-Haack formylation reaction Methods 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- 239000003463 adsorbent Substances 0.000 description 2
- 125000003368 amide group Chemical group 0.000 description 2
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 2
- 239000001045 blue dye Substances 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 238000005229 chemical vapour deposition Methods 0.000 description 2
- QQVDYSUDFZZPSU-UHFFFAOYSA-M chloromethylidene(dimethyl)azanium;chloride Chemical compound [Cl-].C[N+](C)=CCl QQVDYSUDFZZPSU-UHFFFAOYSA-M 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- MGNZXYYWBUKAII-UHFFFAOYSA-N cyclohexa-1,3-diene Chemical compound C1CC=CC=C1 MGNZXYYWBUKAII-UHFFFAOYSA-N 0.000 description 2
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 2
- BGTOWKSIORTVQH-UHFFFAOYSA-N cyclopentanone Chemical compound O=C1CCCC1 BGTOWKSIORTVQH-UHFFFAOYSA-N 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 239000008151 electrolyte solution Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 239000010419 fine particle Substances 0.000 description 2
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 2
- 238000006170 formylation reaction Methods 0.000 description 2
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 2
- 230000001965 increasing effect Effects 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 125000005647 linker group Chemical group 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- IVSZLXZYQVIEFR-UHFFFAOYSA-N m-xylene Chemical group CC1=CC=CC(C)=C1 IVSZLXZYQVIEFR-UHFFFAOYSA-N 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 229910052758 niobium Inorganic materials 0.000 description 2
- 239000010955 niobium Substances 0.000 description 2
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000006722 reduction reaction Methods 0.000 description 2
- 229910052707 ruthenium Inorganic materials 0.000 description 2
- 238000005245 sintering Methods 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 125000000020 sulfo group Chemical group O=S(=O)([*])O[H] 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- DZLFLBLQUQXARW-UHFFFAOYSA-N tetrabutylammonium Chemical compound CCCC[N+](CCCC)(CCCC)CCCC DZLFLBLQUQXARW-UHFFFAOYSA-N 0.000 description 2
- QEMXHQIAXOOASZ-UHFFFAOYSA-N tetramethylammonium Chemical compound C[N+](C)(C)C QEMXHQIAXOOASZ-UHFFFAOYSA-N 0.000 description 2
- 125000003396 thiol group Chemical group [H]S* 0.000 description 2
- 229910052718 tin Inorganic materials 0.000 description 2
- 239000011135 tin Substances 0.000 description 2
- XOLBLPGZBRYERU-UHFFFAOYSA-N tin dioxide Chemical compound O=[Sn]=O XOLBLPGZBRYERU-UHFFFAOYSA-N 0.000 description 2
- 229910001887 tin oxide Inorganic materials 0.000 description 2
- BWHDROKFUHTORW-UHFFFAOYSA-N tritert-butylphosphane Chemical compound CC(C)(C)P(C(C)(C)C)C(C)(C)C BWHDROKFUHTORW-UHFFFAOYSA-N 0.000 description 2
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 2
- 229910052721 tungsten Inorganic materials 0.000 description 2
- 239000010937 tungsten Substances 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- 125000000027 (C1-C10) alkoxy group Chemical group 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- SFPQDYSOPQHZAQ-UHFFFAOYSA-N 2-methoxypropanenitrile Chemical compound COC(C)C#N SFPQDYSOPQHZAQ-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- OHXPGWPVLFPUSM-KLRNGDHRSA-N 3,7,12-trioxo-5beta-cholanic acid Chemical compound C1CC(=O)C[C@H]2CC(=O)[C@H]3[C@@H]4CC[C@H]([C@@H](CCC(O)=O)C)[C@@]4(C)C(=O)C[C@@H]3[C@]21C OHXPGWPVLFPUSM-KLRNGDHRSA-N 0.000 description 1
- ZVVXXGSRDOMYII-UHFFFAOYSA-N 3-cyanoprop-2-ynoic acid Chemical group OC(=O)C#CC#N ZVVXXGSRDOMYII-UHFFFAOYSA-N 0.000 description 1
- OOWFYDWAMOKVSF-UHFFFAOYSA-N 3-methoxypropanenitrile Chemical compound COCCC#N OOWFYDWAMOKVSF-UHFFFAOYSA-N 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- DLYVTEULDNMQAR-SRNOMOOLSA-N Cholic Acid Methyl Ester Chemical compound C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@H](C)CCC(=O)OC)[C@@]2(C)[C@@H](O)C1 DLYVTEULDNMQAR-SRNOMOOLSA-N 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- KMTRUDSVKNLOMY-UHFFFAOYSA-N Ethylene carbonate Chemical compound O=C1OCCO1 KMTRUDSVKNLOMY-UHFFFAOYSA-N 0.000 description 1
- 238000006783 Fischer indole synthesis reaction Methods 0.000 description 1
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 238000006000 Knoevenagel condensation reaction Methods 0.000 description 1
- WRQNANDWMGAFTP-UHFFFAOYSA-N Methylacetoacetic acid Chemical compound COC(=O)CC(C)=O WRQNANDWMGAFTP-UHFFFAOYSA-N 0.000 description 1
- DLYVTEULDNMQAR-UHFFFAOYSA-N Methylallocholat Natural products OC1CC2CC(O)CCC2(C)C2C1C1CCC(C(C)CCC(=O)OC)C1(C)C(O)C2 DLYVTEULDNMQAR-UHFFFAOYSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 235000005811 Viola adunca Nutrition 0.000 description 1
- 240000009038 Viola odorata Species 0.000 description 1
- 235000013487 Viola odorata Nutrition 0.000 description 1
- 235000002254 Viola papilionacea Nutrition 0.000 description 1
- 238000007239 Wittig reaction Methods 0.000 description 1
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 1
- 239000003377 acid catalyst Substances 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 238000005882 aldol condensation reaction Methods 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000005210 alkyl ammonium group Chemical group 0.000 description 1
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 125000005427 anthranyl group Chemical group 0.000 description 1
- 150000008430 aromatic amides Chemical group 0.000 description 1
- 125000005129 aryl carbonyl group Chemical group 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 125000003828 azulenyl group Chemical group 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000000440 benzylamino group Chemical group [H]N(*)C([H])([H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 230000008033 biological extinction Effects 0.000 description 1
- 150000001639 boron compounds Chemical class 0.000 description 1
- 125000005620 boronic acid group Chemical class 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- YTIVTFGABIZHHX-UHFFFAOYSA-N butynedioic acid Chemical group OC(=O)C#CC(O)=O YTIVTFGABIZHHX-UHFFFAOYSA-N 0.000 description 1
- 238000001354 calcination Methods 0.000 description 1
- VTJUKNSKBAOEHE-UHFFFAOYSA-N calixarene Chemical compound COC(=O)COC1=C(CC=2C(=C(CC=3C(=C(C4)C=C(C=3)C(C)(C)C)OCC(=O)OC)C=C(C=2)C(C)(C)C)OCC(=O)OC)C=C(C(C)(C)C)C=C1CC1=C(OCC(=O)OC)C4=CC(C(C)(C)C)=C1 VTJUKNSKBAOEHE-UHFFFAOYSA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000001728 carbonyl compounds Chemical class 0.000 description 1
- 238000010531 catalytic reduction reaction Methods 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 229920001940 conductive polymer Polymers 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 150000003983 crown ethers Chemical class 0.000 description 1
- 125000004093 cyano group Chemical group *C#N 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960002997 dehydrocholic acid Drugs 0.000 description 1
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 description 1
- 229960003964 deoxycholic acid Drugs 0.000 description 1
- 125000004663 dialkyl amino group Chemical group 0.000 description 1
- 150000001983 dialkylethers Chemical group 0.000 description 1
- 125000004986 diarylamino group Chemical group 0.000 description 1
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- 125000004914 dipropylamino group Chemical group C(CC)N(CCC)* 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000004070 electrodeposition Methods 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 125000004705 ethylthio group Chemical group C(C)S* 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 230000022244 formylation Effects 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229910052733 gallium Inorganic materials 0.000 description 1
- 239000011245 gel electrolyte Substances 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 230000002140 halogenating effect Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 230000005525 hole transport Effects 0.000 description 1
- OAKJQQAXSVQMHS-UHFFFAOYSA-N hydrazine Substances NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 description 1
- 229910003437 indium oxide Inorganic materials 0.000 description 1
- PJXISJQVUVHSOJ-UHFFFAOYSA-N indium(iii) oxide Chemical compound [O-2].[O-2].[O-2].[In+3].[In+3] PJXISJQVUVHSOJ-UHFFFAOYSA-N 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 238000007733 ion plating Methods 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000002462 isocyano group Chemical group *[N+]#[C-] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000001810 isothiocyanato group Chemical group *N=C=S 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 238000010030 laminating Methods 0.000 description 1
- 239000011244 liquid electrolyte Substances 0.000 description 1
- 238000009766 low-temperature sintering Methods 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 1
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000002896 organic halogen compounds Chemical class 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 239000011368 organic material Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- 125000001792 phenanthrenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C=CC12)* 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 125000000286 phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005499 phosphonyl group Chemical group 0.000 description 1
- 230000029553 photosynthesis Effects 0.000 description 1
- 238000010672 photosynthesis Methods 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- UORVCLMRJXCDCP-UHFFFAOYSA-N propynoic acid Chemical group OC(=O)C#C UORVCLMRJXCDCP-UHFFFAOYSA-N 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000005412 pyrazyl group Chemical group 0.000 description 1
- 125000001725 pyrenyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 150000003303 ruthenium Chemical class 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 230000001235 sensitizing effect Effects 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- NRHMKIHPTBHXPF-TUJRSCDTSA-M sodium cholate Chemical compound [Na+].C([C@H]1C[C@H]2O)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC([O-])=O)C)[C@@]2(C)[C@@H](O)C1 NRHMKIHPTBHXPF-TUJRSCDTSA-M 0.000 description 1
- 239000007784 solid electrolyte Substances 0.000 description 1
- 238000004544 sputter deposition Methods 0.000 description 1
- 230000003637 steroidlike Effects 0.000 description 1
- 125000000542 sulfonic acid group Chemical group 0.000 description 1
- 229910052715 tantalum Inorganic materials 0.000 description 1
- GUVRBAGPIYLISA-UHFFFAOYSA-N tantalum atom Chemical compound [Ta] GUVRBAGPIYLISA-UHFFFAOYSA-N 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000000858 thiocyanato group Chemical group *SC#N 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- SRPWOOOHEPICQU-UHFFFAOYSA-N trimellitic anhydride Chemical compound OC(=O)C1=CC=C2C(=O)OC(=O)C2=C1 SRPWOOOHEPICQU-UHFFFAOYSA-N 0.000 description 1
- 238000007738 vacuum evaporation Methods 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- GPPXJZIENCGNKB-UHFFFAOYSA-N vanadium Chemical compound [V]#[V] GPPXJZIENCGNKB-UHFFFAOYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 238000001429 visible spectrum Methods 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 description 1
- RNWHGQJWIACOKP-UHFFFAOYSA-N zinc;oxygen(2-) Chemical class [O-2].[Zn+2] RNWHGQJWIACOKP-UHFFFAOYSA-N 0.000 description 1
- 229910052726 zirconium Inorganic materials 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B23/00—Methine or polymethine dyes, e.g. cyanine dyes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G9/00—Electrolytic capacitors, rectifiers, detectors, switching devices, light-sensitive or temperature-sensitive devices; Processes of their manufacture
- H01G9/20—Light-sensitive devices
- H01G9/2059—Light-sensitive devices comprising an organic dye as the active light absorbing material, e.g. adsorbed on an electrode or dissolved in solution
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B23/00—Methine or polymethine dyes, e.g. cyanine dyes
- C09B23/0066—Methine or polymethine dyes, e.g. cyanine dyes the polymethine chain being part of a carbocyclic ring,(e.g. benzene, naphtalene, cyclohexene, cyclobutenene-quadratic acid)
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B23/00—Methine or polymethine dyes, e.g. cyanine dyes
- C09B23/0075—Methine or polymethine dyes, e.g. cyanine dyes the polymethine chain being part of an heterocyclic ring
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B23/00—Methine or polymethine dyes, e.g. cyanine dyes
- C09B23/02—Methine or polymethine dyes, e.g. cyanine dyes the polymethine chain containing an odd number of >CH- or >C[alkyl]- groups
- C09B23/04—Methine or polymethine dyes, e.g. cyanine dyes the polymethine chain containing an odd number of >CH- or >C[alkyl]- groups one >CH- group, e.g. cyanines, isocyanines, pseudocyanines
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B23/00—Methine or polymethine dyes, e.g. cyanine dyes
- C09B23/02—Methine or polymethine dyes, e.g. cyanine dyes the polymethine chain containing an odd number of >CH- or >C[alkyl]- groups
- C09B23/06—Methine or polymethine dyes, e.g. cyanine dyes the polymethine chain containing an odd number of >CH- or >C[alkyl]- groups three >CH- groups, e.g. carbocyanines
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B23/00—Methine or polymethine dyes, e.g. cyanine dyes
- C09B23/02—Methine or polymethine dyes, e.g. cyanine dyes the polymethine chain containing an odd number of >CH- or >C[alkyl]- groups
- C09B23/08—Methine or polymethine dyes, e.g. cyanine dyes the polymethine chain containing an odd number of >CH- or >C[alkyl]- groups more than three >CH- groups, e.g. polycarbocyanines
- C09B23/083—Methine or polymethine dyes, e.g. cyanine dyes the polymethine chain containing an odd number of >CH- or >C[alkyl]- groups more than three >CH- groups, e.g. polycarbocyanines five >CH- groups
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01G—CAPACITORS; CAPACITORS, RECTIFIERS, DETECTORS, SWITCHING DEVICES, LIGHT-SENSITIVE OR TEMPERATURE-SENSITIVE DEVICES OF THE ELECTROLYTIC TYPE
- H01G9/00—Electrolytic capacitors, rectifiers, detectors, switching devices, light-sensitive or temperature-sensitive devices; Processes of their manufacture
- H01G9/20—Light-sensitive devices
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/615—Polycyclic condensed aromatic hydrocarbons, e.g. anthracene
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/656—Aromatic compounds comprising a hetero atom comprising two or more different heteroatoms per ring
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/657—Polycyclic condensed heteroaromatic hydrocarbons
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/657—Polycyclic condensed heteroaromatic hydrocarbons
- H10K85/6572—Polycyclic condensed heteroaromatic hydrocarbons comprising only nitrogen in the heteroaromatic polycondensed ring system, e.g. phenanthroline or carbazole
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K30/00—Organic devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation
- H10K30/10—Organic devices sensitive to infrared radiation, light, electromagnetic radiation of shorter wavelength or corpuscular radiation comprising heterojunctions between organic semiconductors and inorganic semiconductors
- H10K30/15—Sensitised wide-bandgap semiconductor devices, e.g. dye-sensitised TiO2
- H10K30/151—Sensitised wide-bandgap semiconductor devices, e.g. dye-sensitised TiO2 the wide bandgap semiconductor comprising titanium oxide, e.g. TiO2
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
- Y02E10/542—Dye sensitized solar cells
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E10/00—Energy generation through renewable energy sources
- Y02E10/50—Photovoltaic [PV] energy
- Y02E10/549—Organic PV cells
Definitions
- the present invention relates to a photosensitizer and a photoelectric conversion element using the photosensitizer.
- Organic solar cells that have recently attracted attention as next-generation solar cells are roughly classified into organic thin film types and dye-sensitized types.
- Organic thin-film solar cells use a combination of organic materials to form a pn junction, and the operation mechanism is exactly the same as that of inorganic solar cells represented by silicon.
- dye-sensitized solar cells (Dye Sensitized Solar Cell: DSC) are characterized by the use of modified electrodes that combine organic dyes with inorganic semiconductors such as titanium oxide and zinc oxide as photosensitizers.
- the operating mechanism is also completely different from that of inorganic solar cells, rather it is close to photosynthesis.
- This solar cell that should also be referred to as an inorganic / organic hybrid molecular element is attracting attention as a low-cost solar cell (Non-Patent Document 1).
- the working electrode of the dye-sensitized solar cell is obtained by sintering and laminating an inorganic semiconductor such as titanium oxide or zinc oxide on a conductive substrate and adsorbing the sensitizing dye. It is easy to manufacture because it has a simple structure filled with In addition, special equipment such as a vacuum line is not necessary for manufacturing, and cost reduction is easier than conventional solar cells. In particular, a so-called Gretzell-type DSC has a high possibility of realizing a high cost reduction because a material such as an electrode is inexpensive and a special equipment investment is unnecessary.
- the electrode is made of porous titanium oxide with a high roughness factor made by high-temperature sintering of nanoparticles. By combining this with a ruthenium dye as a photosensitizer, it is now highly converted to over 12%. Efficiency has been achieved, and it is said that commercialization is imminent.
- the present invention has been made in view of the above problems, and is an inexpensive photosensitizer (a novel organic sensitizer) with high conversion efficiency, high durability, and abundant color variations for sensitization of a titanium oxide or zinc oxide electrode. (Pigment). Moreover, this invention aims at providing the photoelectric conversion element which has a light absorption layer containing this photosensitizer.
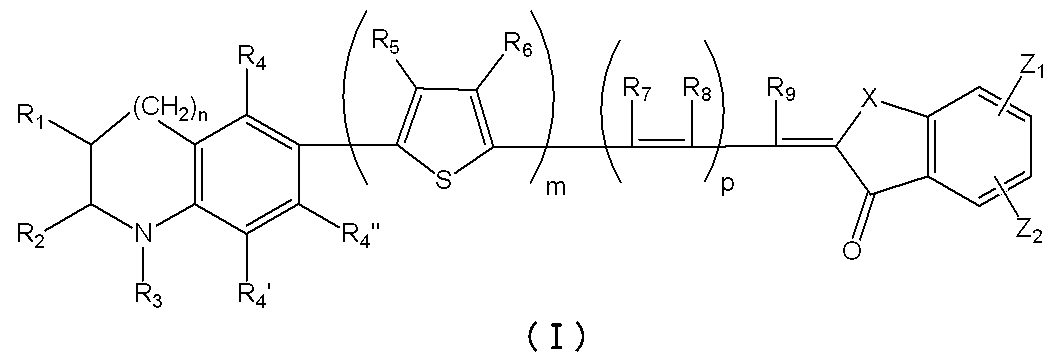
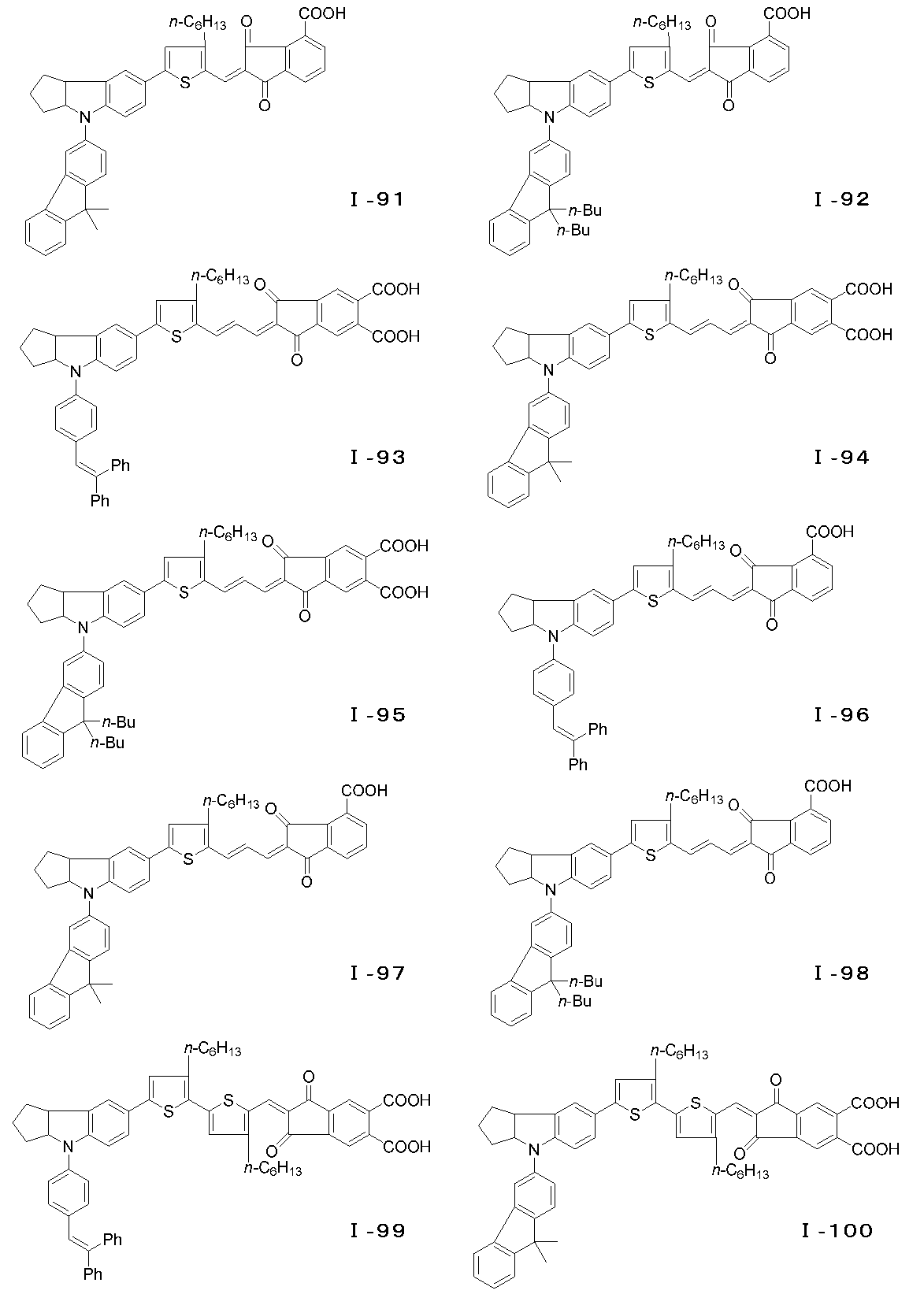
- the photosensitizer of the present invention is characterized by being a dye represented by the following general formula (I) or a salt thereof, and more specifically, having a novel enone structure at the electron acceptor site. It is a feature.
- m represents an integer of 0 to 4.
- n represents an integer of 0 or 1.
- p represents an integer of 0 to 2.
- R 1 and R 2 represent a hydrogen atom, an alkyl group or an aryl.
- R 3 represents an alkyl group, an aryl group, an aralkyl group, or a heterocyclic residue, and may be bonded to form an aromatic ring or an alicyclic ring, and R 4 , R 4 ′, R 4 ′′.
- R 5 and R 6 are A hydrogen atom, an alkyl group, an alkoxy group or an alkylthio group, which may combine with R 5 and R 6 to form a cyclic structure, where R 7 , R 8 and R 9 are a hydrogen atom, an alkyl group or an aralkyl group; , an alkenyl group or an aryl group, linked by R 7 and R 8 or R 8 and R 9 Optionally to form an aliphatic ring .
- X an oxygen atom, a sulfur atom, an alkylene group, a substituted amino group, a carbonyl group, .Z 1, Z 2 showing a sulfonyl group or an aliphatic spiro ring is a hydrogen atom, a hydroxyl group, A carboxyl group, an alkoxycarbonyl group, a sulfone group or a substituted amino group, provided that at least one of the substituents in the molecule has an acidic group.
- the photoelectric conversion element of the present invention is characterized by having a light absorption layer containing a photosensitizer of the dye represented by the above general formula (I) or a salt thereof.
- the dye represented by the above general formula (I) or a salt thereof, which is the photosensitizer of the present invention improves the photoelectric conversion efficiency and improves the adsorption stability of the dye as compared with conventionally known dyes. It is possible. Although its mechanism of action is not always clear, the efficiency of photoelectron transfer by the efficient push-pull action of ⁇ electrons between the donor part of the indoline skeleton or tetrahydroquinoline skeleton and the acceptor part of the novel enone structure is high conversion efficiency It is thought that it has influenced.
- the photosensitizer of the present invention is also advantageous in that the effect of increasing the wavelength by introducing a long linker is extremely high compared to existing dyes, and the hue can be finely adjusted by a substituent.
- the conventional blue dye was a cationic acceptor obtained by quaternizing nitrogen such as indolenine and benzothiazole, so it was strongly influenced by oxygen in the air.
- the dye of the present invention is a nonionic compound. It is considered that the durability was remarkably improved because the oxidation potential of the dye was high and it was difficult to be decomposed by air oxidation. By enhancing the push-pull effect by introducing this conjugated structure, the molar extinction coefficient can be increased, and at the same time, a longer wavelength can be achieved. Therefore, the photosensitizer can be optimal for a photoelectric conversion element.
- the photosensitizer of the present invention when used as a light absorption layer of a photoelectric conversion element, conversion efficiency at a practical level can be obtained.
- the photosensitizer of the present invention can improve the photoelectric conversion efficiency in a wide range of wavelengths, and is particularly effective for a dye-sensitized solar cell in which design is also important.
- the compound represented by the above general formula (I) of the photosensitizer of the present invention may be any of a free acid represented by the above general formula (I) and a salt thereof.
- the salt of the compound represented by the general formula (I) include alkali metal salts or alkaline earth metal salts such as lithium, sodium, potassium, magnesium and calcium of carboxylic acid, or tetramethylammonium, tetrabutylammonium, Examples thereof include alkylammonium salts such as pyridinium, piperidinium and imidazolium.
- R 1 and R 2 in the general formula (I) represent a hydrogen atom, an alkyl group, or an aryl group.
- the alkyl group include linear alkyl groups such as a methyl group, an ethyl group, and a propyl group, an isopropyl group, and an isobutyl group.
- a branched alkyl group such as a cyclopentyl group and a cyclohexyl group. These alkyl groups may be further substituted with a substituent described later.
- aryl group examples include phenyl, naphthyl, anthranyl, phenanthrenyl, pyrenyl, indenyl, azulenyl, and fluorenyl groups, which may further have a substituent.
- R 1 and R 2 may be bonded to each other to form an aromatic ring or an alicyclic ring.
- Examples of the cyclic structure formed at that time include benzene, naphthalene, cyclopentane, cyclopentanone, pyridine, piperidine, piperazine, Examples include pyrazole, pyrrole, imidazole, thiazole, indole, quinoline, carbazole and the like, which may further have a substituent, and may further have a cyclic structure as a substituent.
- substituents examples include cyano group, isocyano group, thiocyanato group, isothiocyanato group, nitro group, nitrosyl group, sulfo group, halogen atom, hydroxyl group, phosphate group, phosphate ester group, substituted or unsubstituted mercapto group, substituted or Examples include unsubstituted amino groups, substituted or unsubstituted amide groups, alkoxy groups, alkoxyalkane groups, carboxyl groups, alkoxycarbonyl groups, alkyl groups, aryl groups, acyl groups, etc. is not.
- the acyl group is preferably, for example, an alkylcarbonyl group having 1 to 10 carbon atoms or an arylcarbonyl group.
- the halogen atom include chlorine, bromine and iodine atoms
- the phosphate ester group include an alkyl phosphate (C1-C4) ester group.
- the substituted mercapto group include alkylthio groups such as methylthio and ethylthio.
- substituted amino groups include mono- or dialkylamino groups, mono- or diarylamino groups, mono- or dimethylamino groups, mono- or diethylamino groups, mono- or dipropylamino groups, monophenylamino groups, or benzylamino groups.
- substituted amide group include an alkylamide group and an aromatic amide group.
- alkoxy group include an alkoxy group having 1 to 10 carbon atoms.
- alkoxyalkyl group include (C1-C10) alkoxy (C1-C4) alkyl groups such as ethoxyethyl group.
- alkoxycarbonyl group examples include an alkoxycarbonyl group having 1 to 10 carbon atoms such as an ethoxycarbonyl group.
- Acid groups such as carboxyl group, sulfo group and phosphate group are metal salts such as lithium, sodium, potassium, magnesium and calcium, and ammonium salts such as tetramethylammonium, tetrabutylammonium, pyridinium, piperidinium and imidazolium.
- An organic salt may be formed.
- R 3 represents an alkyl group, an aryl group, an aralkyl group or a heterocyclic residue, and examples of the alkyl group and the aryl group are the same as those described above.
- the aralkyl group means an alkyl group substituted with an aryl group as described later, and examples thereof include a benzyl group, a phenylethyl group, a methylnaphthyl group, and the like, and these may further have a substituent.
- a heterocyclic residue means a group obtained by removing one hydrogen atom from a heterocyclic compound, such as pyridyl, pyrazyl, piperidyl, pyrazolyl, morpholyl, indolinyl, thiophenyl, furyl, oxazolyl, thiazolyl, indolyl, benzothiazolyl, benzo Examples thereof include oxazolyl, quinolyl, rhodanyl and the like, which may further have a substituent.
- R 4 , R 4 ′ and R 4 ′′ in general formula (I) represent a hydrogen atom, an alkyl group, an aralkyl group, an alkoxy group, a substituted amino group or a halogen atom, and examples thereof are the same as those described above. is there. R 4 ′ and R 4 ′′ may be bonded to each other to form a cyclic structure, and examples thereof are the same as those described above.
- R 5 and R 6 in the general formula (I) represent a hydrogen atom, an alkyl group, an alkoxy group or an alkylthio group, and examples thereof are the same as those described above.
- R 5 and R 6 may be bonded to each other to form a cyclic structure, and examples thereof are the same as those described above.
- R 7 , R 8 and R 9 in formula (I) represent a hydrogen atom, an alkyl group, an aralkyl group, an alkenyl group or an aryl group.
- alkyl group, aralkyl group and aryl group are the same as those described above. is there.
- alkenyl group include a vinyl group and an allyl group, which may further have a substituent. Examples thereof are the same as those described above.
- R 7 and R 8 or R 8 and R 9 may be bonded to each other to form an alicyclic ring, and examples thereof are the same as those described above.
- X in the general formula (I) represents an oxygen atom, a sulfur atom, an alkylene group, a substituted amino group, a carbonyl group, a sulfonyl group or an aliphatic spiro ring.
- alkylene group include a dimethylmethylene group and a dibutylmethylene group.
- aliphatic spiro ring include cyclopentane, cyclohexane, cyclohexene, cyclohexadiene, and the like, which may further have a substituent.
- Z 1 and Z 2 in the general formula (I) represent a hydrogen atom, a hydroxyl group, a carboxyl group, an alkoxycarbonyl group, a sulfone group or a substituted amino group, and examples thereof are the same as those described above.
- the compound represented by the general formula (I) has at least one acidic group as a substituent in the molecule.
- the acidic group is preferably a carboxy group, a sulfonic acid group, a phosphonyl group, a phosphoryl group, or a salt thereof.
- the acidic group may be a group bonded through a linking group.
- a carboxyvinylene group, a dicarboxyvinylene group, a cyanocarboxyvinylene group, a carboxyphenyl group, and the like can be mentioned as preferable examples.
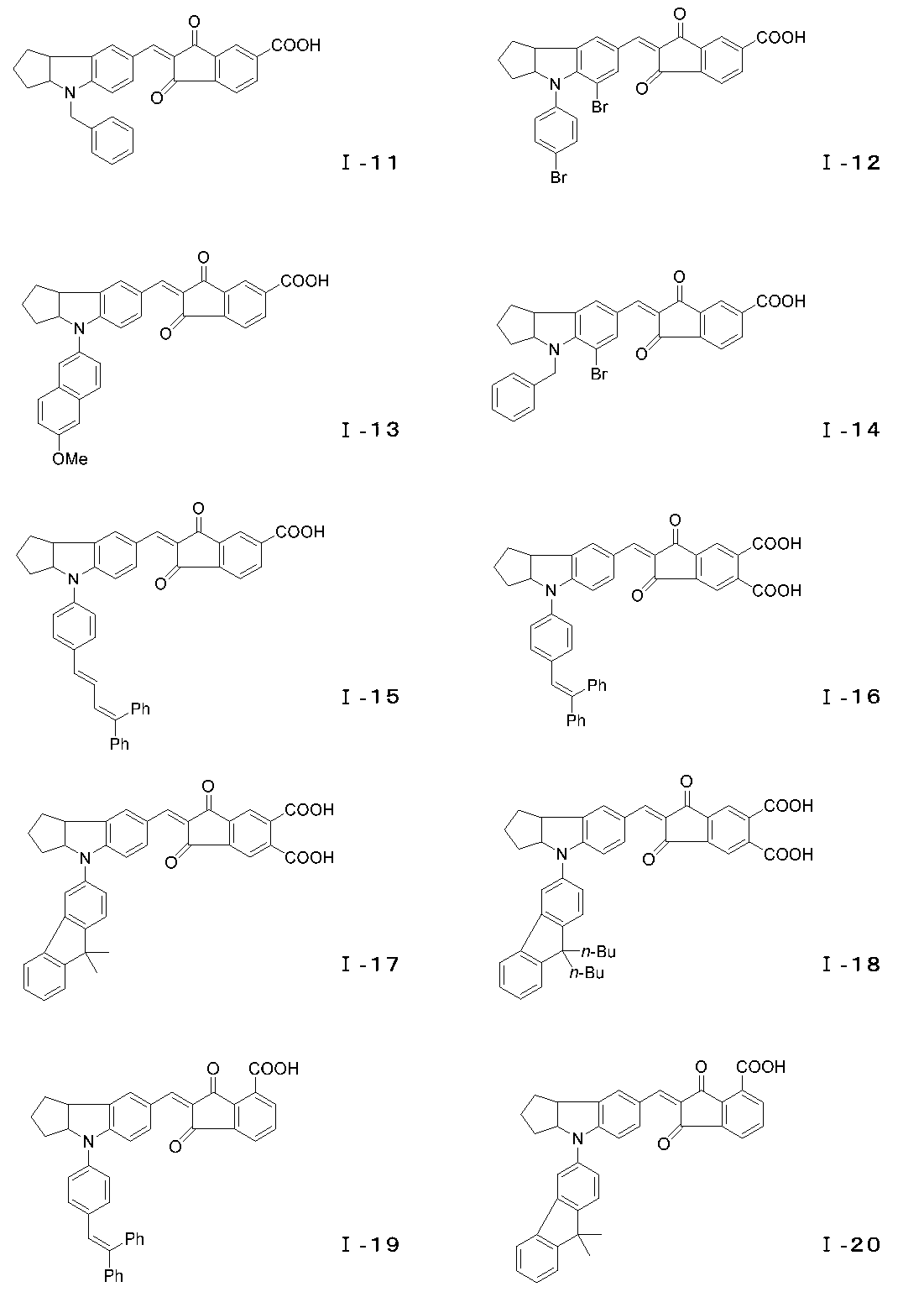
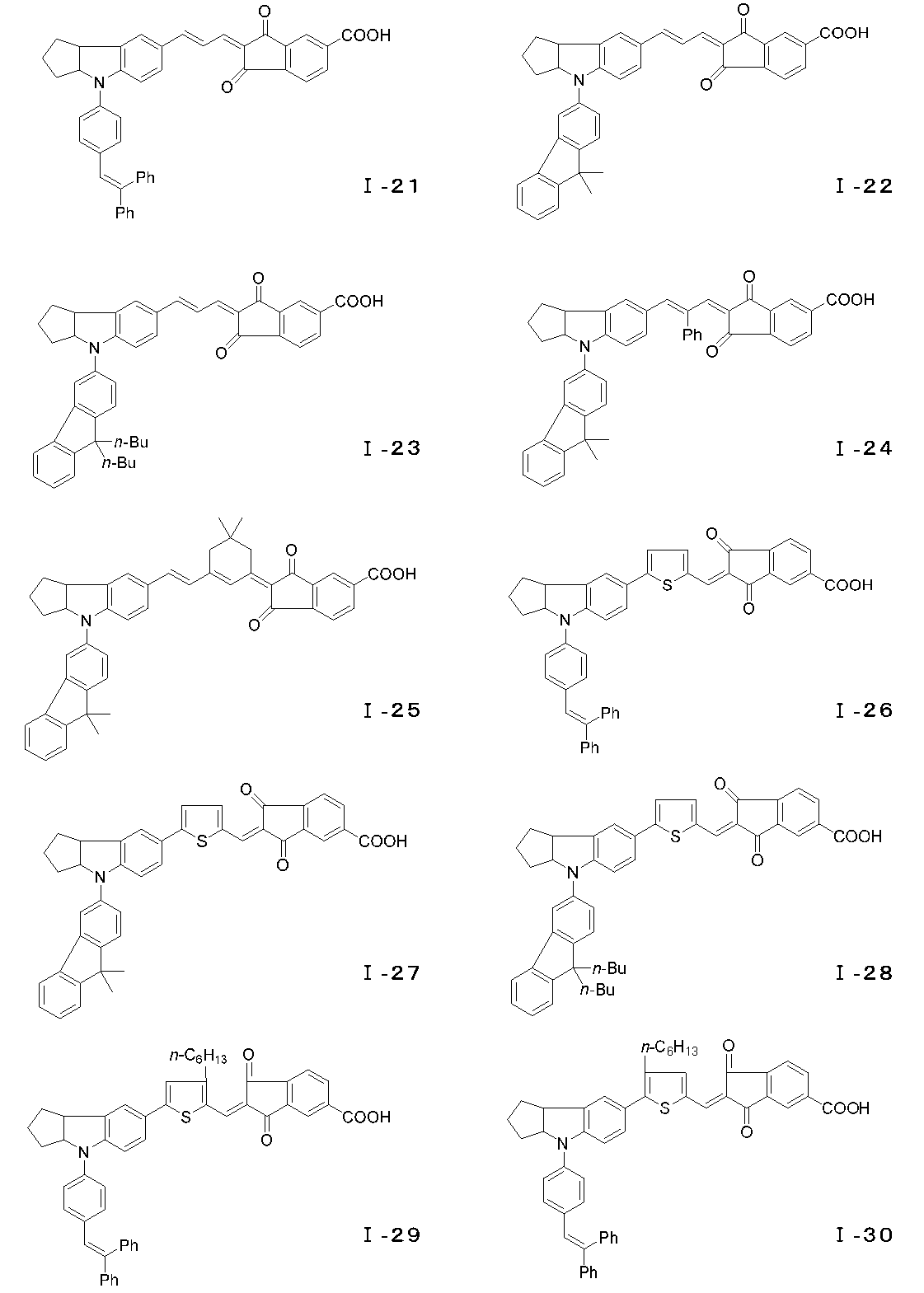
- the compound represented by the general formula (I) can have an E-type or Z-type geometric isomer at the conjugated chain site, and an optical isomer at the 3a position of the indoline skeleton or tetrahydroquinoline skeleton. Is also effective as a photosensitizer and can be used as the dye of the present invention. Examples of the compound of the general formula (I) are specifically shown below, but the present invention is of course not limited to these examples.
- a so-called Fischer indole synthesis method in which an aryl hydrazone formed from a ketone and an aryl hydrazine is heated under an acid catalyst such as sulfuric acid can be used.
- the Scrapup method can be used.
- These heterocycles can be reduced to the intermediate (1) by an appropriate method such as catalytic reduction.
- Intermediate (2) can be easily synthesized by a coupling reaction of intermediate (1) and intermediate (3) using a palladium catalyst.
- the intermediate (2) When introducing a conjugated chain (m ⁇ 0), the intermediate (2) is first made into a bromo-substituted product (5) with a halogenating agent such as NBS (N-bromosuccinimide), and then the conjugated chain is extended by Suzuki coupling or the like. Then, intermediate (4) can be synthesized through formylation by Vilsmeier reaction.
- a borate ester having a formyl group already at the time of Suzuki coupling it is possible to extend the conjugated chain and introduce the formyl group in one step.
- Suzuki coupling is a method in which an organic halogen compound and an organic boron compound are cross-coupled in the presence of a palladium catalyst, and is a useful method that is widely used because of relatively mild conditions and high functional group selectivity.
- Examples of the method of obtaining the dye (6) by condensing the intermediate (4) and the enone compound include a method of reacting a carbonyl compound and active methylene, such as aldol condensation and Knoevenagel condensation, and a method of olefin synthesis by Wittig reaction.
- FIG. 1 is a schematic diagram showing one embodiment of a photoelectric conversion element of the present invention.
- the photoelectric conversion element 1 includes a semiconductor layer 3 and an electrolyte layer 4 each having a light absorption layer formed by adsorbing a dye (photosensitizer of the present invention) to an oxide semiconductor layer on a substrate 2 having conductivity on the surface.
- the counter electrode 5 is laminated
- a support having a conductive property such as metal, or glass or plastic can be used as the support when the surface has conductivity.
- a conductive property such as metal, or glass or plastic
- the material of the conductive layer tin-doped indium oxide (ITO), fluorine-doped tin oxide (FTO), gold, platinum, or a combination of these can be used, and this can be applied to a substrate by vacuum evaporation, By forming the conductive layer directly by a method such as sputter deposition, ion plating, chemical vapor deposition (CVD), or by attaching a film on which these are formed to a substrate, A substrate having conductivity on the surface can be formed.
- oxide semiconductor examples include oxides such as titanium, tin, zinc, tungsten, zirconium, gallium, indium, yttrium, niobium, tantalum, and vanadium. Of these, oxides such as titanium, tin, zinc, niobium, and tungsten are preferable. Among these, (1) low cost, (2) easy formation of a porous body, and (3) conductivity as an electrode. From the viewpoints of properties, durability, stability and safety, and (4) compatibility of energy levels with the photosensitizer synthesized in the present invention, titanium and zinc oxides are preferable. These oxide semiconductors may be used alone or in combination of two or more.
- An oxide semiconductor can be formed porous on a substrate by applying fine particles of the oxide semiconductor on the substrate and then heat-treating or electrodeposition with an electric furnace or microwave.
- a method for adsorbing the dye to the oxide semiconductor layer a method such as immersing the substrate on which the oxide semiconductor layer is formed in a dye solution or a dye dispersion can be used. Can be formed.
- the concentration of the solution can be appropriately determined depending on the dye, and specific examples of the solvent that can be used for dissolving the dye include, for example, methanol, ethanol, acetonitrile, dimethyl sulfoxide, dimethylformamide, acetone, t-butanol and the like. Can be mentioned.
- a co-adsorbent may be added to the dye solution when adsorbing the dye on the thin film of oxide semiconductor fine particles.
- the co-adsorbent include steroidal compounds such as cholic acid, crown ether, cyclodextrin, calixarene, polyethylene oxide, etc., but deoxycholic acid, dehydrocholic acid, cholic acid methyl ester, sodium cholate and the like are more preferable. preferable.
- the electrolyte layer is a mixed liquid of acetonitrile and ethylene carbonate, a liquid electrolyte in which an electrolyte made of iodide such as metal iodine or lithium iodide is added using methoxypropionitrile as a solvent, a polymer gel electrolyte, etc. It can be formed using a solid electrolyte such as a solidified electrolyte, a p-type semiconductor, and a hole transport agent.
- the counter electrode may be prepared in the same manner as the conductive substrate when transparency is required, or it may be prepared using carbon, a conductive polymer, a general metal, etc. when transparency is not required. be able to.
- the photosensitizer of the present invention can also be used as a light absorption layer of an organic thin film solar cell.
- the present invention will be described in more detail with reference to examples.
- intermediates (A-01) to (A-03), intermediate aldehydes (B-01) to (B-13), and intermediate (C-01) in this example are represented by the following chemical formulas. Is.
- Aldehyde (B-02) was also synthesized by the same route as (Synthesis of aldehyde (B-01)) using the corresponding intermediate.
- the structure of the obtained aldehyde (B-02) was identified by NMR analysis.
- Aldehyde (B-03) was also synthesized by the same route as (Synthesis of aldehyde (B-01)) using the corresponding intermediate.
- the structure of the obtained aldehyde (B-03) was identified by NMR analysis.
- Aldehyde (B-05) was also synthesized by the same route as (Synthesis of aldehyde (B-04)) using the corresponding intermediate.
- the structure of the obtained aldehyde (B-05) was identified by NMR analysis.
- Aldehyde (B-06) was also synthesized by the same route as (Synthesis of aldehyde (B-04)) using the corresponding intermediate.
- Aldehyde (B-07) was also synthesized by the same route as (Synthesis of aldehyde (B-04)) using the corresponding intermediate.
- the structure of the obtained aldehyde (B-07) was identified by NMR analysis.
- Aldehyde (B-08) was also synthesized by the same route as (Synthesis of aldehyde (B-04)) using the corresponding intermediate.
- the structure of the obtained aldehyde (B-08) was identified by NMR analysis.
- Aldehyde (B-09) was also synthesized by the same route as (Synthesis of aldehyde (B-04)) using the corresponding intermediate.
- the structure of the obtained aldehyde (B-09) was identified by NMR analysis.
- Aldehyde (B-10) was also synthesized by the same route as (Synthesis of aldehyde (B-04)) using the corresponding intermediate.
- the structure of the obtained aldehyde (B-10) was identified by NMR analysis.
- Aldehyde (B-11) was also synthesized by the same route as (Synthesis of aldehyde (B-04)) using the corresponding intermediate.
- the structure of the obtained aldehyde (B-11) was identified by NMR analysis.
- Aldehyde (B-12) was also synthesized by the same route as (Synthesis of aldehyde (B-04)) using the corresponding intermediate.
- Example 2 ⁇ Synthesis of Example 2 (I-05)>
- the aldehyde intermediate (B-02) was used to obtain the dye (I-05) as a black solid.
- ⁇ max 541nm (CHCl 3).
- Example 3 ⁇ Synthesis of Example 3 (I-06)>
- the aldehyde intermediate (B-03) was used to obtain the dye (I-06) as a black solid.
- ⁇ max 546nm (CHCl 3).
- the structure of the obtained dye (I-06) was identified by NMR analysis.
- Example 4 ⁇ Synthesis of Example 4 (I-26)>
- the aldehyde intermediate (B-04) was used to obtain the dye (I-26) as a black solid.
- ⁇ max 607nm (CHCl 3).
- the structure of the obtained dye (I-26) was identified by NMR analysis.
- Example 5 ⁇ Synthesis of Example 5 (I-27)>
- the aldehyde intermediate (B-05) was used to obtain the dye (I-27) as a black purple solid.
- ⁇ max 616nm (CHCl 3).
- the structure of the obtained dye (I-27) was identified by NMR analysis.
- Example 6 ⁇ Synthesis of Example 6 (I-28)>
- the aldehyde intermediate (B-06) was used to obtain the dye (I-28) as a black solid.
- ⁇ max 619nm (CHCl 3).
- the structure of the obtained dye (I-28) was identified by NMR analysis.
- Example 8 ⁇ Synthesis of Example 8 (I-32)>
- the aldehyde intermediate (B-08) was used to obtain the dye (I-32) as a black solid.
- ⁇ max 621nm (CHCl 3).
- the structure of the obtained dye (I-32) was identified by NMR analysis.
- Example 10 (I-45)>
- the aldehyde intermediate (B-10) was used to obtain the dye (I-45) as a black solid.
- ⁇ max 647nm (CHCl 3).
- the structure of the obtained dye (I-45) was identified by NMR analysis.
- Example 11 ⁇ Synthesis of Example 11 (I-47)>
- the aldehyde intermediate (B-11) was used to obtain the dye (I-47) as a black solid.
- ⁇ max 655nm (CHCl 3).
- Example 12 ⁇ Synthesis of Example 12 (I-48)>
- the aldehyde intermediate (B-12) was used to obtain the dye (I-48) as a black solid.
- ⁇ max 661nm (CHCl 3).
- the structure of the obtained dye (I-48) was identified by NMR analysis.
- Example 13 (I-75)>
- the aldehyde intermediate (B-13) was used to obtain the dye (I-75) as a black purple solid.
- ⁇ max 595nm (CHCl 3).
- the structure of the obtained dye (I-75) was identified by NMR analysis.
- the photoelectric conversion efficiency was improved as compared with the comparative example using the dye of the corresponding hue.
- the photoelectric conversion efficiency could be improved in all the hues from magenta to blue-green.
- the conversion efficiency was greatly improved in the blue-violet to blue-green region of 600 to 700 nm, and a practical level dye capable of covering a wide range of hues was obtained.
- significant improvement in photoelectric conversion efficiency was also observed in zinc oxide.
- the photosensitizer of the present invention can improve the photoelectric conversion efficiency and the adsorption stability of the dye. Moreover, since it can cover a wide range of hues, it can be suitably used for a dye-sensitized solar cell in which design is also important. Furthermore, even with zinc oxide, which is a good electrode material, photoelectric conversion efficiency comparable to that of titanium oxide can be obtained.
Landscapes
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Power Engineering (AREA)
- Organic Chemistry (AREA)
- Materials Engineering (AREA)
- Microelectronics & Electronic Packaging (AREA)
- Physics & Mathematics (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Hybrid Cells (AREA)
- Photovoltaic Devices (AREA)
Abstract
Description
本発明の光増感剤の上記一般式(I)で表される化合物は、上記一般式(I)で示されるフリーの酸及びその塩のいずれでもよい。上記一般式(I)で表される化合物の塩としては、例えばカルボン酸のリチウム、ナトリウム、カリウム、マグネシウム、カルシウムなどのアルカリ金属塩又はアルカリ土類金属塩、又はテトラメチルアンモニウム、テトラブチルアンモニウム、ピリジニウム、ピペリジニウム、イミダゾリウムなどのアルキルアンモニウム塩を挙げることができる。
一般式(I)におけるZ1、Z2は水素原子、ヒドロキシル基、カルボキシル基、アルコキシカルボニル基、スルホン基または置換アミノ基を示し、それらの例としては前述の場合と同様である。
以下に一般式(I)の化合物の例を具体的に示すが、もちろん本発明はこれらの例に限定されるものではない。



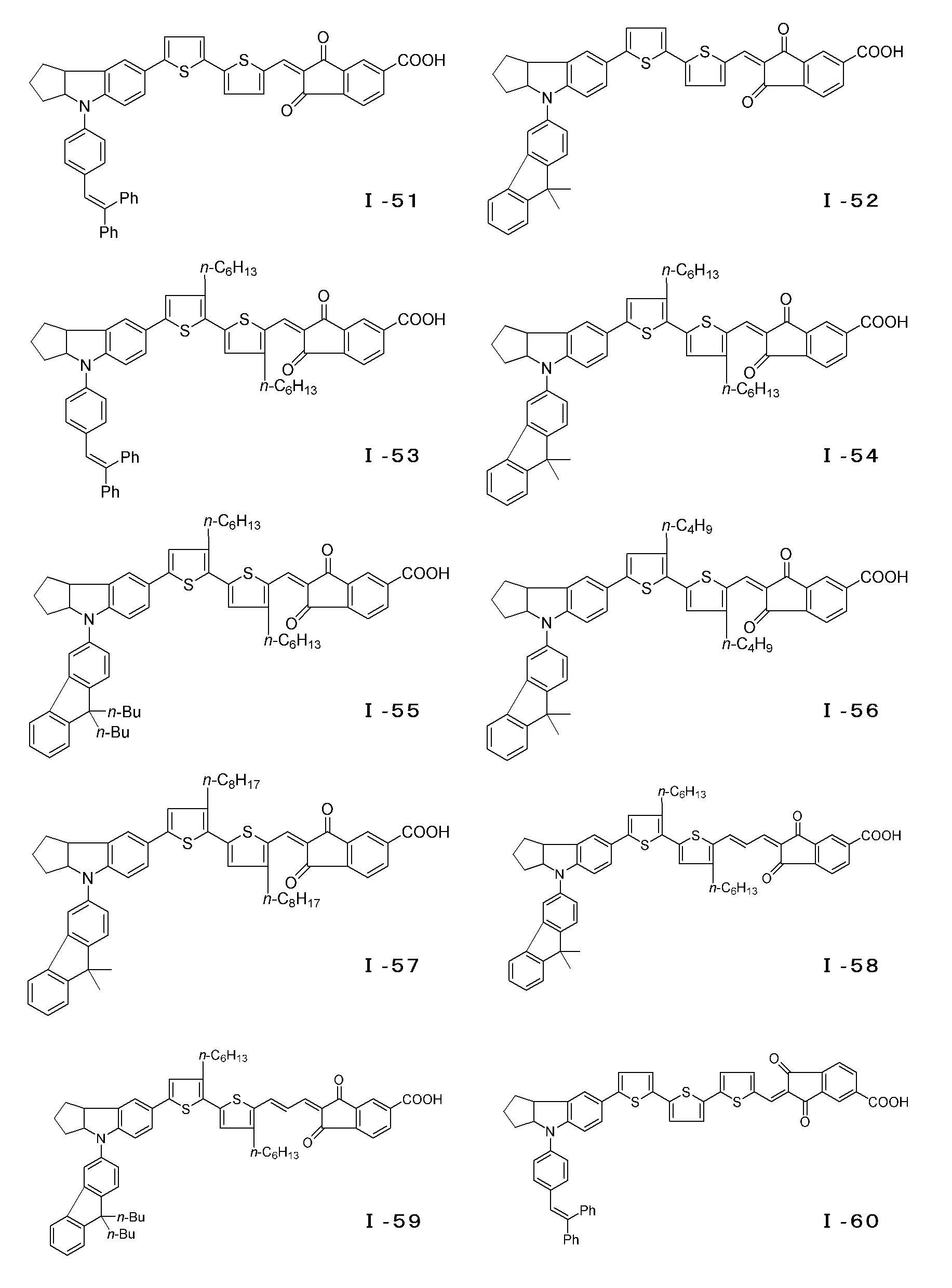


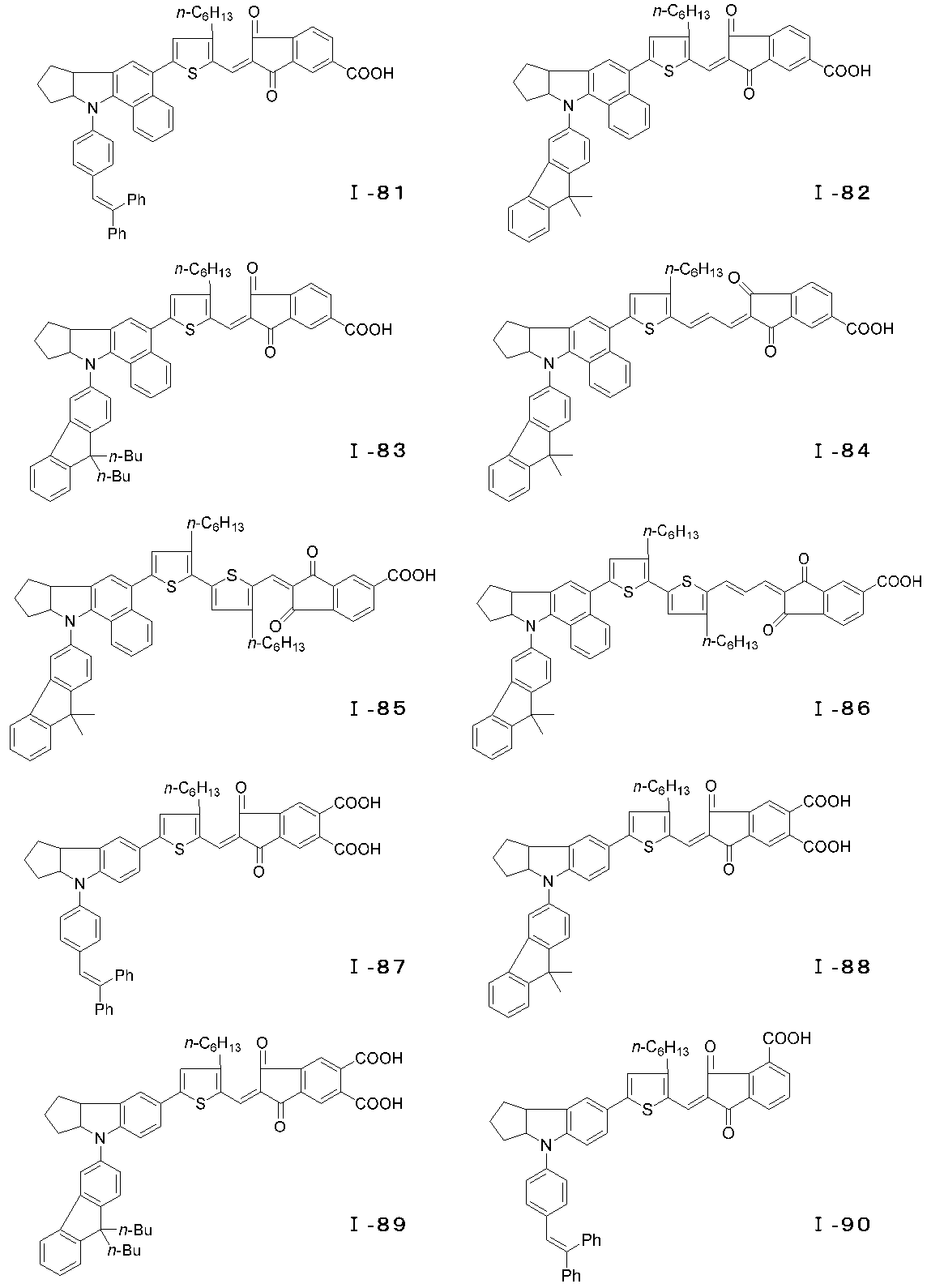




中間体(2)から中間体(4)を合成する方法(m=0)としては、Vilsmeier反応に代表されるホルミル化反応が挙げられる。
酸化物半導体層に色素を吸着させる方法としては、色素溶液中あるいは色素分散液中にこの酸化物半導体層を形成させた基板を浸漬するなどの方法を用いることができ、これによって、半導体層を形成することができる。溶液の濃度は色素によって適宜決めることができ、色素を溶解させるのに使用しうる溶媒の具体例としては、例えば、メタノール、エタノール、アセトニトリル、ジメチルスルホキシド、ジメチルホルムアミド、アセトン、t-ブタノール等が好ましく挙げられる。
以下に本発明を実施例を用いてさらに詳細に説明する。


(アルデヒド(B-01)の合成)
中間体(A-01)(5.2g)、中間体(A-03)(11.7g)、カリウムt-ブトキシド(5.5g)、酢酸パラジウム(74mg)、トリt-ブチルホスフィン(0.3g)をm-キシレン(40mL)に溶解し系内を窒素置換した後、120℃で8時間加熱攪拌した。反応混合物を室温まで冷却後、不溶物をろ過し、ろ液を水洗、無水硫酸ナトリウムで乾燥させた後、減圧濃縮し、褐色オイルを得た(15.0g)。次に氷冷下でDMF(25mL)に塩化ホスホリル(10.0g)を滴下し調整したVilsmeier試薬にこの褐色オイル(15.0g)を滴下し室温で3時間攪拌した。反応液に水(100ml)を加え、ついで25%水酸化ナトリウム水溶液を加えpH11とした。この反応液をクロロホルムで抽出、有機層を無水硫酸ナトリウムで乾燥後、減圧濃縮し、残渣をカラムクロマトグラフィー(シリカゲル、展開溶媒:CHCl3)で分離精製することによりアルデヒド(B-01)の黄土色固体を11.6g得た(収率80%)。
1H NMR(400MHz,CDCl3)
δ(ppm)=9.70(1H,s),7.61(1H,dd,J=1.6,1.2Hz),7.50(1H,dd,J=8.4,1.6Hz),7.24-7.41(10H,m),7.07(2H,d,J=9.2Hz),7.03(2H,d,J=8.8Hz),6.95(1H,s),6.85(1H,d,J=8.4Hz),4.83-4.87(1H,m),3.78-3.83(1H,m),2.00-2.10(1H,m),1.82-1.91(2H,m),1.73-1.80(1H,m),1.62-1.71(1H,m),1.40-1.52(1H,m)
アルデヒド(B-02)も対応する中間体を用いて、(アルデヒド(B-01)の合成)と同様のルートで合成した。
得られたアルデヒド(B-02)についてNMR分析により構造を同定した。
1H NMR(400MHz,CDCl3)
δ(ppm)=9.71(1H,s),7.72(1H,d,J=8.0Hz),7.68(1H, d,J=6.8Hz),7.66(1H,s),7.54(1H,dd,J=8.0,1.2Hz),7.43(1H,d,J=6.8Hz),7.36(1H,d,J=.2.0Hz),7.33(1H,dd,J=7.2,1.2Hz),7.31(1H,dd,J=7.6,2.0Hz),7.26(1H,dd,J=8.4,2.0Hz),6.85(1H,d,J=8.4Hz),5.00(1H,br.t,J=7.2Hz),3.87(1H,dt,J=8.4,2.4Hz),2.04-2.14(1H,m),1.88-1.99(2H,m),1.76-1.85(1H,m),1.67-1.75(1H,m),1.52-1.61(1H,m),1.51(3H,s),1.50(3H,s)
アルデヒド(B-03)も対応する中間体を用いて、(アルデヒド(B-01)の合成)と同様のルートで合成した。
得られたアルデヒド(B-03)についてNMR分析により構造を同定した。
1H NMR(400MHz,CDCl3)
δ(ppm)=9.71(1H,s),7.70(1H,d,J=8.1Hz),7.67(1H, d,J=6.3Hz),7.66(1H,s),7.54(1H,dd,J=8.3,1.4Hz),7.25-7.35(5H,m),6.83(1H,d,J=8.3Hz),5.02(1H,br.t,J=6.8Hz),3.87(1H,br.t,J=7.3Hz),2.05-2.14(1H,m),1.90-1.98(6H,m),1.69-1.81(2H,m),1.50-1.59(1H,m),1.09(4H,m),0.72(3H,t,J=7.3Hz),0.68(3H,t,J=7.3Hz),0.59-0.73(4H,m)
中間体(A-01)と中間体(A-03)を(アルデヒド(B-01)の合成)と同様の方法でカップリングさせた後、NBSでブロモ化し、ボロン酸類と反応させ、これをVilsmeier試薬と反応させることにより合成した。
得られたアルデヒド(B-04)についてNMR分析により構造を同定した。
1H NMR(400MHz,CDCl3)
δ(ppm)=9.82(1H,s),7.67(1H,d,J=4.0Hz),7.24-7.39(13H,m),7.05(2H,d,J=8.8Hz),7.00(2H,d,J=9.2Hz),6.95(1H,d,J=8.0Hz),6.94(1H,s),4.75-4.80(1H,m),3.81-3.86(1H,m),2.02-2.11(1H,m),1.77-1.93(3H,m),1.61-1.71(1H,m),1.43-1.54(1H,m)
アルデヒド(B-05)も対応する中間体を用いて、(アルデヒド(B-04)の合成)と同様のルートで合成した。
得られたアルデヒド(B-05)についてNMR分析により構造を同定した。
1H NMR(400MHz,CDCl3)
δ(ppm)=9.83(1H,s),7.66-7.70(3H,m),7.41-7.44(3H,m),7.23-7.36(5H,m),6.97(1H,d,J=8.0Hz), 4.92-4.95(1H,m),3.87-3.91(1H,m),2.07-2.13(1H,m),1.86-1.98(3H,m),1.58-1.62(1H,m),1.51(3H,s),1.50(3H,s)
アルデヒド(B-06)も対応する中間体を用いて、(アルデヒド(B-04)の合成)と同様のルートで合成した。
得られたアルデヒド(B-06)についてNMR分析により構造を同定した。
1H NMR(400MHz,CDCl3)
δ(ppm)=9.83.(1H,s),7.68(1H,d,J=4.0Hz),7.67(1H, d,J=10.0Hz),7.63(1H,dd,J=8.8,8.8Hz),7.44(1H,br.s),7.41(1H,dd,J=8.8,8.0Hz),7.33(1H,d,J=6.8Hz),7.31(1H,d,J=6.4Hz),7.26-7.28(3H,m),7.25(1H,s),6.94(1H,d,J=8.4Hz),4.94-4.98(1H,m),3.86-3.91(1H,m),2.06-2.16(1H,m),1.90-2.01(6H,m),1.79-1.88(1H,m),1.67-1.75(1H,m),1.52-1.63(1H,m),1.04-1.16(4H,m),0.72(3H,t,J=7.2Hz),0.69(3H,t,J=7.2Hz),0.61-0.71(4H,m)
アルデヒド(B-07)も対応する中間体を用いて、(アルデヒド(B-04)の合成)と同様のルートで合成した。
得られたアルデヒド(B-07)についてNMR分析により構造を同定した。
1H NMR(400MHz,CDCl3)
δ(ppm)=9.96(1H,s),7.69(1H,d,J=8.0Hz),7.67(1H, d,J=7.2Hz),7.43(1H,s),7.40-7.42(2H,m),7.36(1H,d,J=2.0Hz),7.33(1H,dt,J=7.2,1.2Hz),7.27(1H,dt,J=7.2,1.2Hz),7.24(1H,dd,J=8.0,1.6Hz),7.09(1H,s),6.96(1H,d,J=8.0Hz),4.91-4.95(1H,m),3.86-3.91(1H,m),2.93(2H,dd,J=8.0,7.6Hz),2.06-2.16(1H,m),1.82-2.02(3H,m),1.67-1.75(3H,m),1.53-1.63(1H,m),1.51(3H,s),1.50(3H,s),1.30-1.43(6H,m),0.90(3H,t,J=6.8Hz)
アルデヒド(B-08)も対応する中間体を用いて、(アルデヒド(B-04)の合成)と同様のルートで合成した。
得られたアルデヒド(B-08)についてNMR分析により構造を同定した。
1H NMR(400MHz,CDCl3)
δ(ppm)=9.96(1H,s),7.67(1H,d,J=8.0Hz),7.65(1H, d,J=8.8Hz),7.43(1H,s),7.40(1H,dd,J=8.4,1.6Hz),7.32(2H,dd,J=7.2,6.0Hz),7.26(2H,dd,J=7.6,7.6Hz),7.24(1H,s),7.09(1H,s),6.94(1H,d,J=8.4Hz),4.94-4.98(1H,m),3.86-3.90(1H,m),2.93(2H,dd,J=8.0,7.6Hz),2.06-2.15(1H,m),1.91-2.01(6H,m),1.79-1.88(1H,m),1.67-1.75(3H,m),1.52-1.62(1H,m),1.30-1.43(6H,m),1.04-1.16(4H,m),0.90(3H,t,J=6.8Hz),0.71(3H,t,J=7.2Hz),0.69(3H,t,J=7.2Hz),0.60-0.70(4H,m)
アルデヒド(B-09)も対応する中間体を用いて、(アルデヒド(B-04)の合成)と同様のルートで合成した。
得られたアルデヒド(B-09)についてNMR分析により構造を同定した。
1H NMR(400MHz,CDCl3)
δ(ppm)=9.60(1H,d,J=7.6Hz),7.68(2H,dd,J=8.8,8.0Hz),7.53(1H,d,J=15.6Hz),7.42(1H,d,J=7.6Hz),7.39(1H,s),7.37(2H,dd,J=8.4,7.6Hz),7.33(1H,dt,J=7.6,7.2Hz),7.27-7.29(2H,m),7.24(1H,dd,J=8.8,8.4Hz),7.17(1H,d,J=3.6Hz),6.98(1H,d,J=8.4Hz),6.44(1H,dd,J=7.6,7.6Hz),4.90-4.94(1H,m),3.86-3.91(1H,m),2.07-2.16(1H,m),1.83-2.01(3H,m),1.66-1.75(1H,m),1.55-1.64(1H,m),1.51(3H,s),1.50(3H,s)
アルデヒド(B-10)も対応する中間体を用いて、(アルデヒド(B-04)の合成)と同様のルートで合成した。
得られたアルデヒド(B-10)についてNMR分析により構造を同定した。
1H NMR(400MHz,CDCl3)
δ(ppm)=9.60(1H,d,J=8.0Hz),7.66(2H,ddd,J=9.6,9.2,7.2Hz), 7.53(1H,d,J=15.6Hz),7.40(1H,br.s),7.37(1H,dd,J=8.4,2.0Hz),7.33(1H,d,J=7.2Hz),7.32(1H,s),7.28-7.30(2H,m),7.24-7.27(2H,m),7.18(1H, d,J=4.0Hz),6.96(1H,d,J=8.4Hz),6.44(1H,dd,J=15.6,7.6Hz),4.93-4.97(1H,m),3.87-3.90(1H,m),2.06-2.16(1H,m),1.91-2.01(6H,m),1.80-1.89(1H,m),1.67-1.75(1H,m),1.53-1.63(1H,m),1.05-1.14(4H, m),0.72(3H,t,J=7.2Hz),0.69(3H,t,J=7.6Hz),0.61-0.69(4H,m)
アルデヒド(B-11)も対応する中間体を用いて、(アルデヒド(B-04)の合成)と同様のルートで合成した。
得られたアルデヒド(B-11)についてNMR分析により構造を同定した。
1H NMR(400MHz,CDCl3)
δ(ppm)=9.60(1H,d,J=8.0Hz),7.68(1H,d,J=8.0Hz), 7.66(1H,d,J=8.8Hz),7.61(1H,d,J=15.6Hz), 7.41(1H,d,J=7.2Hz),7.39(1H,s),7.35-7.38(2H,m),7.33(1H,dt,J=7.2,1.2Hz),7.27(1H,dt,J=7.2,1.2Hz),7.24(1H, dd,J=8.4,2.0Hz),7.05(1H,s),6.97(1H,d,J=8.0Hz),6.40(1H,dd,J=15.2,7.6Hz),4.90-4.94(1H,m),3.86-3.90(1H,m),2.72(2H,dd,J=8.0,7.6Hz),2.06-2.17(1H,m),1.83-2.03(3H,m),1.62-1.75(3H,m),1.51(3H,s),1.50(3H,s),1.47-1.55(1H,m),1.31-1.42(6H,m),0.90(3H,t,J=6.8Hz)
アルデヒド(B-12)も対応する中間体を用いて、(アルデヒド(B-04)の合成)と同様のルートで合成した。
得られたアルデヒド(B-12)についてNMR分析により構造を同定した。
1H NMR(400MHz,CDCl3)
δ(ppm)=9.61(1H,d,J=7.6Hz),7.67(1H,d,J=8.8Hz),7.63-7.66(1H,m),7.62(1H,d,J=15.2Hz),7.39(1H,s),7.36(1H,dd,J=8.0,1.6Hz),7.33(1H,d,J=7.2Hz),7.31(1H,d,J=6.4Hz),7.25-7.28(2H,m),7.24(1H,s),7.06(1H,s),6.95(1H,d,J=8.4Hz),6.40(1H,dd,J=15.6,8.0Hz),4.93-4.96(1H,m),3.86-3.90(1H,m),2.72(2H,dd,J=8.0,7.6Hz),2.06-2.15(1H,m),1.90-2.01(6H,m),1.80-1.89(1H,m),1.62-1.74(3H,m),1.51-1.61(1H,m),1.28-1.42(6H,m),1.05-1.14(4H,m),0.90(3H,t,J=7.2Hz),0.72(3H,t,J=7.2Hz),0.69(3H,t,J=7.2Hz),0.61-0.70(4H,m)
中間体(A-02)と中間体(A-03)を(アルデヒド(B-01)の合成)と同様のルートで合成した。
得られたアルデヒド(B-13)についてNMR分析により構造を同定した。
1H NMR(400MHz,CDCl3)
δ(ppm)=9.28(1H,d,J=8.0Hz),7.76(1H,s),7.49-7.54(1H,s),7.28-7.38(10H,m),7.23-7.25(2H,m),7.16-7.21(1H,m),6.97-6.99(3H,m),6.89(1H,d,J=7.3Hz),4.58(1H,ddd,J=8.9,6.7,2.5Hz),4.12(1H,dt,J=6.1,2.5Hz),2.03-2.14(2H,m),1.89-1.98(2H,m),1.63-1.72(1H,m),1.41-1.53(1H,m)
トリメリット酸無水物(東京化成工業株式会社製)(10.18g)の無水酢酸溶液(75mL)にアセト酢酸メチル(12.00g)とトリエチルアミン(33.78g)を滴下し100℃で1.5時間加熱撹拌した。反応混合物を室温まで冷却した後溶媒を留去し、カラムクロマトグラフィー(シリカゲル、展開溶媒:CHCl3/MeOH=10/1)で分離精製すると黒色液体を得た(12.65g)。次に、得られた黒色液体(12.65g)のメタノール溶液(150mL)に酢酸アンモニウム(6.49g)を加え75℃で3時間加熱撹拌すると固化した。粗結晶をろ別し、メタノールで洗浄することにより中間体(C-01)を黄色固体として5.76g得た(収率44%)。得られた中間体(C-01)についてNMR分析により構造を同定した。なお、カルボン酸の水素は観測されなかった。
1H NMR(400MHz,DMSO-d6)
δ(ppm)=7.92(1H,d,J=7.6Hz),7.80(1H,s),7.25(1H,d,J=7.6Hz),7.14(1H,br.s),3.49(3H,s)
アルデヒド(B-01)(0.88g)、中間体(C-01)(0.50g)を酢酸(10mL)に溶解し、100℃で4時間加熱攪拌した。反応混合物を室温まで冷却すると固化した。粗結晶をろ別し、カラムクロマトグラフィー(シリカゲル、展開溶媒:CHCl3/MeOH=100/1)で分離精製することにより色素(I-03)を褐色固体として0.85g得た(収率69%)。λmax=545nm(CHCl3)。
得られた色素(I-03)についてNMR分析により構造を同定した。
1H NMR(400MHz,DMSO-d6)
δ(ppm)=13.65(1H,br.s),8.67(1H,br.s),8.33(1H,dd,J=8.0,7.6Hz), 8.24(1H,d,J=11.6Hz), 8.20(1H,br.s),7.94(1H,dd,J=7.6,7.6Hz),7.65(1H,S),7.41-7.49(3H,m),7.30-7.38(5H,m),7.19-7.23(4H,m),7.11(1H,s),7.07(2H,d,J=8.4Hz),6.87(1H,d,J=8.8Hz), 5.07-5.11(1H,m),3.82-3.87(1H,m),2.03-2.12(1H,m),1.69-1.83(2H,m),1.59-1.67(2H,m),1.27-1.38(1H,m)
実施例1と同様の手法を用い、アルデヒド中間体(B-02)を用いることにより色素(I-05)を黒色固体として得た。λmax=541nm(CHCl3)。
得られた色素(I-05)についてNMR分析により構造を同定した。
1H NMR(400MHz,DMSO-d6)
δ(ppm)=13.63(1H,br.s),8.72(1H,br.s),8.35(1H,d,J=8.0Hz), 8.28(1H,br.s),8.25(1H,d,J=11.6Hz),7.95(1H,dd,J=8.0,7.6Hz),7.90(1H,d,J=8.4Hz),7.83(1H,d,J=7.2Hz),7.69(1H,s),7.67(1H,d,J=1.6Hz),7.56(1H,d,J=7.2Hz),7.41(1H,dd,J=8.0,1.2Hz),7.36(1H,dd,J=8.0,7.2Hz),7.32(1H,dd,J=8.4,7.2Hz),6.87(1H,d,J=8.4Hz),5.24-5.27(1H,m),3.90-3.94(1H,m),2.08-2.18(1H,m),1.74-1.88(3H,m),1.64-1.73(1H,m),1.50(3H,s),1.47(3H,s),1.37-1.53(1H,m)
実施例1と同様の手法を用い、アルデヒド中間体(B-03)を用いることにより色素(I-06)を黒色固体として得た。λmax=546nm(CHCl3)。
得られた色素(I-06)についてNMR分析により構造を同定した。
1H NMR(400MHz,DMSO-d6)
δ(ppm)=13.07(1H,br.s),8.17(1H,br.s),7.81(1H,dd,J=7.6, 7.2Hz),7.73(2H,d,J=9.6Hz),7.41(1H,d,J=8.0Hz),7.34(1H,d,J=8.4Hz),7.27(1H,d,J=7.2Hz),7.14(1H,s),6.99(1H,s),6.92(1H,d,J=6.8Hz),6.88(1H,d,J=8.0Hz),6.83(1H,dd,J=7.2,6.0Hz),6.80(1H,dd,J=7.2,6.4Hz),6.30(1H,d,J=8.4Hz),4.72-4.75(1H,m),3.36-3.40(1H,m),1.57-1.65(1H,m),1.43-1.56(4H,m),1.28-1.35(1H,m),1.11-1.26(3H,m),0.84-0.95(1H,m),0.45-0.59(4H,m),0.16(3H,t,J=7.2Hz),0.10(3H,t,J=7.2Hz),0.02-0.04(4H,m)
実施例1と同様の手法を用い、アルデヒド中間体(B-04)を用いることにより色素(I-26)を黒色固体として得た。λmax=607nm(CHCl3)。
得られた色素(I-26)についてNMR分析により構造を同定した。
1H NMR(400MHz,DMSO-d6)
δ(ppm)=13.62(1H,br.s),8.32(1H,dd,J=8.0,7.6Hz),8.23(1H,d,J=10.4Hz),8.17(1H,d,J=4.0Hz),7.95(1H,s),7.92(1H,d,J=8.0Hz),7.59(1H,d,J=4.4Hz),7.57(1H,s),7.45(1H,d,J=6.8Hz),7.42(1H,d,J=7.6Hz),7.39-7.52(2H,m),7.33(1H,d,J=6.8Hz),7.30(1H,s),7.29(1H,d,J=7.2Hz),7.26-7.36(3H,m),7.20(1H,d,J=7.2Hz),7.09(2H,d,J=8.4Hz),7.05(1H,s),7.00(2H,d,J=8.4Hz),6.94(1H,dd,J=8.4,3.2Hz),4.85-4.88(1H,m),3.80-3.84(1H,m),2.00-2.10(1H,m),1.76-1.86(2H,m),1.55-1.70(2H,m),1.27-1.39(1H,m)
実施例1と同様の手法を用い、アルデヒド中間体(B-05)を用いることにより色素(I-27)を黒紫色固体として得た。λmax=616nm(CHCl3)。
得られた色素(I-27)についてNMR分析により構造を同定した。
1H NMR(400MHz,DMSO-d6)
δ(ppm)=13.66(1H,br.s),8.35(1H,d,J=8.0Hz),8.25(1H,br.s),8.24(1H,d,J=8.4Hz),8.20(1H,d,J=4.4Hz),7.98(1H,s),7.94(1H,d,J=8.0Hz),7.81(1H,d,J=8.4Hz),7.76(1H,d,J=7.2Hz),7.62-7.63(1H,m),7.62(1H,s),7.58-7.61(1H,m),7.54(1H,s),7.53(1H,d,J=6.8Hz),7.33(1H,dd,J=8.0,6.8Hz),7.28(1H,dd,J=7.6,6.4Hz),6.98(1H,dd,J=8.4,8.4Hz), 5.04-5.07(1H,m),3.86-3.91(1H,m),2.05-2.16(1H,m),1.73-1.92(3H,m),1.60-1.70(1H,m),1.48(3H,s),1.47(3H,s),1.37-1.52(1H,m)
実施例1と同様の手法を用い、アルデヒド中間体(B-06)を用いることにより色素(I-28)を黒色固体として得た。λmax=619nm(CHCl3)。
得られた色素(I-28)についてNMR分析により構造を同定した。
1H NMR(400MHz,DMSO-d6)
δ(ppm)= 13.67(1H,br.s),8.34(1H,dd,J=9.2,8.4Hz),8.25(1H, d,J=10.4Hz),8.21(1H,d,J=4.0Hz),8.00(1H,s),7.94(1H,d,J=8.0Hz),7.80(1H,d,J=8.4Hz),7.74(1H,d,J=7.2Hz),7.67(1H,d,J=4.4Hz),7.65(1H,s),7.58(1H,d,J=8.4Hz),7.42(1H,d,J=7.2Hz),7.40(1H,s),7.30-7.33(2H,m),7.27(1H,dd,J=7.6,7.2Hz),6.94(1H,dd,J=8.4,2.8Hz),5.08-5.12(1H,m),3.86-3.90(1H,m),1.94-2.15(5H,m),1.72-1.88(3H,m),1.61-1.69(1H,m),1.35-1.46(1H,m),0.99-1.11(4H,m),0.67(3H,t,J=7.6Hz),0.63(3H,t,J=7.2Hz),0.50-0.57(4H,m)
実施例1と同様の手法を用い、アルデヒド中間体(B-07)を用いることにより色素(I-31)を黒色固体として得た。λmax=617nm(CHCl3)。
得られた色素(I-31)についてNMR分析により構造を同定した。
1H NMR(400MHz,DMSO-d6)
δ(ppm)=13.60(1H,br.s),8.28(1H,dd,J=8.0,6.4Hz),8.18(1H,d,J=3.2Hz),7.87(1H,dd,J=7.2,6.8Hz),7.80(1H,d,J=1.2Hz),7.80(1H,d,J=8.0Hz),7.75(1H,d,J=7.6Hz),7.59(1H,s),7.52-7.55(4H,m),6.95(1H,dd,J=8.4,3.6Hz),5.01-5.05(1H,m),3.83-3.87(1H,m),2.82(2H,t,J=7.2Hz),2.06-2.15(1H,m),1.73-1.90(3H,m),1.61-1.68(3H,m),1.48(3H,s),1.47(3H,s),1.40-1.46(1H,m),1.25-1.83(6H,m),0.86(3H,dd,J=6.8,6.4Hz)
実施例1と同様の手法を用い、アルデヒド中間体(B-08)を用いることにより色素(I-32)を黒色固体として得た。λmax=621nm(CHCl3)。
得られた色素(I-32)についてNMR分析により構造を同定した。
1H NMR(400MHz,DMSO-d6)
δ(ppm)=13.61(1H,br.s),8.28(1H,dd,J=7.2,6.4Hz),8.18(1H, d,J=6.0Hz),7.86(1H,dd,J=7.2,6.8Hz),7.79(1H,s),7.78(1H,d,J=9.2Hz),7.73(1H,d,J=7.2Hz),7.59(1H,br.s),7.52(1H,s),7.50-7.55(1H,m),7.41(1H,d,J=6.8Hz),7.38(1H,s),7.31(1H,dd,J=7.6,7.2Hz),7.29(1H,s),7.27(1H,dd,J=8.4,8.4Hz),6.90(1H,dd,J=8.4,8.4Hz),5.04-5.07(1H,m),3.81-3.85(1H,m),2.81(2H,dd,J=7.6,7.2Hz),1.94-2.13(5H,m),1.58-1.86(6H,m),1.19-1.46(7H,m),0.98-1.10(4H,m),0.85(3H,t,J=6.4Hz),0.66(3H,t,J=7.2Hz),0.62(3H,t,J=7.2Hz),0.46-0.48(4H,m)
実施例1と同様の手法を用い、アルデヒド中間体(B-09)を用いることにより色素(I-44)の紫色固体を得た。λmax=642nm(CHCl3)。
得られた色素(I-44)についてNMR分析により構造を同定した。
1H NMR(400MHz,DMSO-d6)
δ(ppm)= 13.61(1H,br.s),8.31-8.34(1H,m),8.23(1H,br.s),7.89-7.97(2H,m),7.85(1H,d,J=14.8Hz),7.79(1H,d,J=8.0Hz),7.74(1H,d,J=7.2Hz),7.61(1H,d,J=11.6Hz),7.48(1H,d,J=14.4Hz),7.45-7.55(5H,m),7.32(1H,t,J=7.2Hz),7.25-7.29(2H,m),6.94(1H,d,J=8.4Hz),4.99-5.02(1H,m),3.82-3.87(1H,m),2.04-2.14(1H,m),1.74-1.94(3H,m),1.60-1.70(1H,m),1.47(6H,m),1.37-1.52(1H,m)
実施例1と同様の手法を用い、アルデヒド中間体(B-10)を用いることにより色素(I-45)を黒色固体として得た。λmax=647nm(CHCl3)。
得られた色素(I-45)についてNMR分析により構造を同定した。
1H NMR(400MHz,DMSO-d6)
δ(ppm)=13.69(1H,br.s),8.34(1H,ddd,J=7.6,3.6,1.2Hz),8.23(1H,d,J=2.4Hz),7.90-7.94(2H,m),7.87(1H,d,J=14.8Hz),7.77(1H,d,J=8.0Hz),7.72(1H,d,J=7.2Hz),7.63(1H,dd,J=11.2,2.0Hz),7.57(1H,s),7.52(1H,d,J=4.0Hz),7.49(1H,d,J=4.0Hz),7.46-7.48(1H,m),7.41(1H,d,J=6.8Hz),7.36(1H,s),7.25-7.33(3H,m),6.91(1H,d,J=8.0Hz),5.03-5.06(1H,m),3.82-3.86(1H,m),1.94-2.11(5H,m),1.70-1.88(3H,m),1.60-1.68(1H,m),1.34-1.44(1H,m),1.01-1.09(4H,m),0.66(3H,t,J=7.6Hz),0.63(3H,t,J=8.0Hz),0.50-0.57(4H,m)
実施例1と同様の手法を用い、アルデヒド中間体(B-11)を用いることにより色素(I-47)を黒色固体として得た。λmax=655nm(CHCl3)。
得られた色素(I-47)についてNMR分析により構造を同定した。
1H NMR(400MHz,DMSO-d6)
δ(ppm)=13.61(1H,br.s),8.30(1H,ddd,J=7.6,1.6,1.6Hz),8.20(1H,s),8.02(1H,d,J=14.4Hz),7.83-7.90(2H,m),7.78(1H,d,J=8.0Hz),7.74(1H,s),7.72(1H,dd,J=6.8,1.6Hz),7.55(1H,s),7.52(1H,d,J=7.6Hz),7.49(1H,d,J=1.2Hz),7.45(1H,ddd,J=8.4,4.0,2.0Hz),7.38(1H,s),7.32(1H,ddd,J=7.2,7.2,1.2Hz),7.25-7.29(2H,m),6.93(1H,dd,J=8.4,1.2Hz),4.98-5.01(1H,m),3.81-3.86(1H,m),2.74(2H,dd,J=8.0,6.8Hz),2.03-2.13(1H,m),1.73-1.91(3H,m),1.57-1.67(3H,m),1.47(3H,s),1.46(3H,s),1.28-1.48(7H,m),0.87(3H,t,J=6.8Hz)
実施例1と同様の手法を用い、アルデヒド中間体(B-12)を用いることにより色素(I-48)を黒色固体として得た。λmax=661nm(CHCl3)。
得られた色素(I-48)についてNMR分析により構造を同定した。
1H NMR(400MHz,DMSO-d6)
δ(ppm)=13.62(1H,br.s),8.28(1H,d,J=8.0Hz),8.19(1H,s),7.99(1H,d,J=14.4Hz),7.81-7.89(2H,m),7.75(1H,d,J=8.0Hz),7.71(1H,d,J=8.4Hz),7.70(1H,dd,J=9.2,3.2Hz),7.52(1H,br.s),7.41-7.44(1H,m),7.40(1H,d,J=7.2Hz),7.36(1H,s),7.34(1H,s),7.30(1H,dd,J=7.2,6.4Hz),7.24-7.28(2H,m),6.87(1H,dd,J=8.8,2.4Hz),4.98-5.01(1H,m),3.78-3.82(1H,m),2.72(2H,dd,J=7.6,7.2Hz),1.93-2.11(5H,m),1.70-1.86(3H,m),1.56-1.65(3H,m),1.25-1.41(7H,m),0.98-1.10(4H,m),0.86(3H,t,J=6.8Hz),0.65(3H,t,J=7.2Hz),0.62(3H,t,J=7.2Hz),0.48-0.57(4H,m)
実施例1と同様の手法を用い、アルデヒド中間体(B-13)を用いることにより色素(I-75)を黒紫色固体として得た。λmax=595nm(CHCl3)。
得られた色素(I-75)についてNMR分析により構造を同定した。
1H NMR(400MHz,DMSO-d6)
δ(ppm)=13.60(1H,br.s),9.59(1H,s),8.54(1H,s),8.35(1H,d,J=7.2Hz),8.32-8.34(1H,m),8.24(1H,d,J=10.0Hz),7.93(1H,dd,J=8.4,7.6Hz),7.63(1H,dd,J=8.0,7.2Hz),7.43-7.47(2H,m),7.32-7.40(6H,m),7.19-7.25(4H,m),7.10-7.16(5H,m),4.87-4.90(1H,m),4.08-4.12(1H,m),2.10-2.20(1H,m),1.93-2.00(1H,m),1.83-1.90(1H,m),1.73-1.82(1H,m),1.63-1.72(1H,m),1.38-1.48(1H,m)
比較用色素一覧を以下に記載した。

(光電極層の作製)
電極基板として片面にFTO電極皮膜が形成されたFTOガラスを用いて、このFTOガラスの電極面に、酸化チタンペースト(日揮触媒化成株式会社製、PST-18NR)をスキージ法にて塗布した。125℃で6分乾燥後、325℃で5分、375℃で5分、450℃で15分、500℃で15分焼成し、膜厚10μmの酸化チタン膜を形成した。この酸化チタン膜が形成されたFTOガラスを、実施例1~13および比較例1~6で得られた各色素を濃度が200μMになるようにアセトニトリル/t-ブチルアルコール=1/1に溶解させ、この溶液に90分間浸漬し光電変換層を作製した。なお、添加剤としてこの色素溶液にコール酸濃度が0.4mMになるようにコール酸を加えた。
3-メトキシプロピオニトリル溶液に、1,2-ジメチル-3-プロピルイミダゾリウムヨージドとヨウ素とヨウ化リチウムと4-t-ブチルピリジンを1,2-ジメチル-3-プロピルイミダゾリウムヨージド(0.60M)、ヨウ素(0.05M)、ヨウ化リチウム(0.10M)、4-t-ブチルピリジン(0.05M)となるように混合し、電解質液とした。この電解質液を上記電極基材と同じFTOガラスを用いた対向基板と先述の光電極層との間に配し電解質層を形成した。
上記で作製した各光電変換素子(受光面積0.20cm2)に分光計器株式会社製「CEP-2000」を用いて100mW/cm2の照射強度で光を当てて、光電変換素子の短絡電流(mA)と開放電圧(V)を測定し、短絡電流と受光面積より短絡電流密度(mA/cm2)を求めた。次いで、光電変換素子の電極間に接続する抵抗値を変化させて最大電力Wmax(mW)を観測し、形状因子FFと光電変換効率(%)を下記計算式により求めた。酸化チタンを用いた場合の評価結果を表1に示す。また、光電極層の作製において酸化チタンを酸化亜鉛に変えた場合の評価結果を表2に示す。


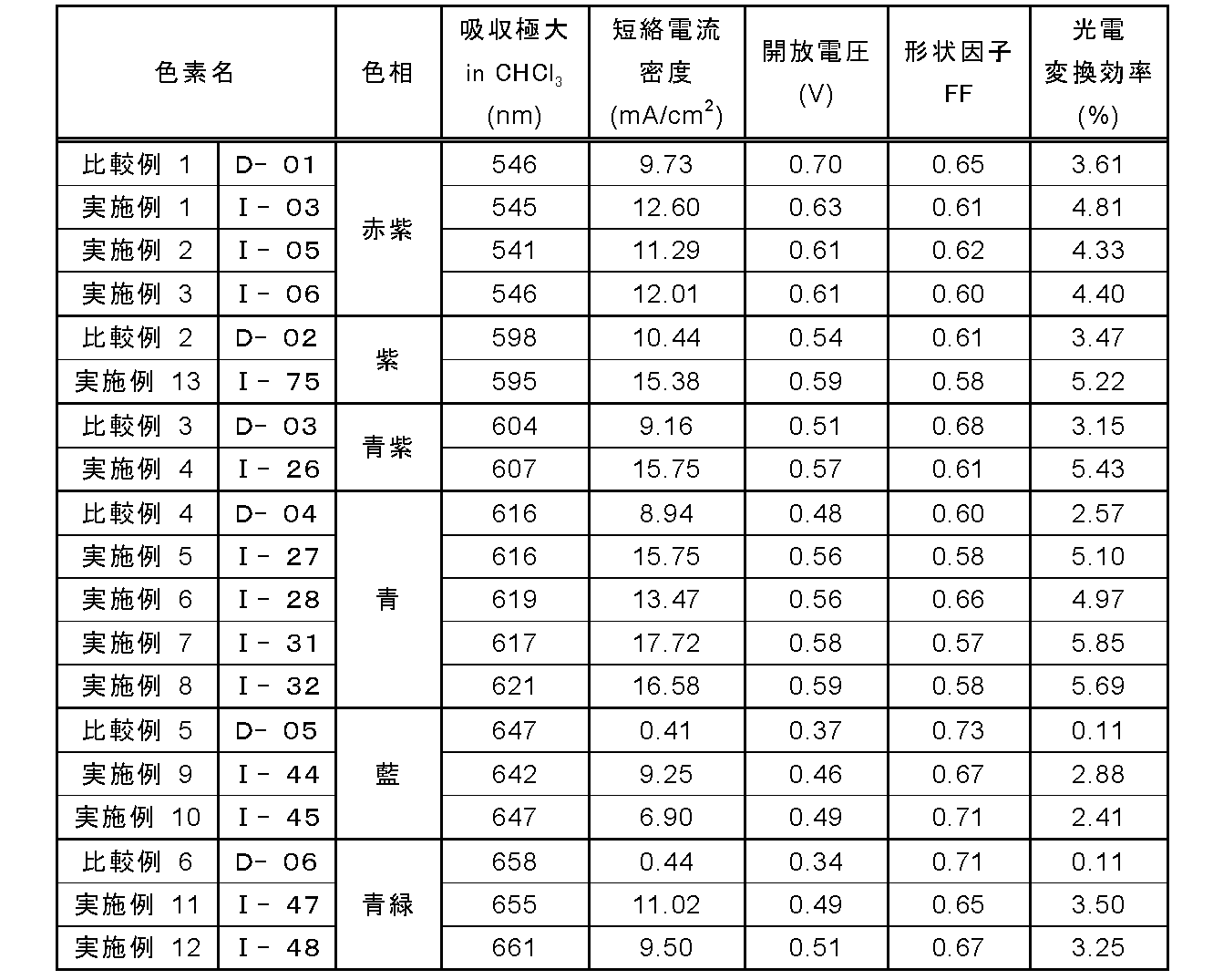

また、表2に示すように、酸化亜鉛においても光電変換効率の大きな改善が認められた。
Claims (2)
- 下記一般式(I)で示される色素またはその塩であることを特徴とする光増感剤。

- 請求項1記載の光増感剤を含む光吸収層を有することを特徴とする光電変換素子。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020167035166A KR101802221B1 (ko) | 2014-06-20 | 2015-06-15 | 광 증감제 및 광전 변환 소자 |
| CN201580031533.9A CN106463272B (zh) | 2014-06-20 | 2015-06-15 | 光敏剂及光电转换元件 |
| EP15809237.9A EP3159905B1 (en) | 2014-06-20 | 2015-06-15 | Photosensitizer and photoelectric conversion element |
| US15/378,875 US9748044B2 (en) | 2014-06-20 | 2016-12-14 | Photosensitizer and photoelectric conversion device |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014126802A JP5898725B2 (ja) | 2014-06-20 | 2014-06-20 | 光増感剤および光電変換素子 |
| JP2014-126802 | 2014-06-20 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/378,875 Continuation US9748044B2 (en) | 2014-06-20 | 2016-12-14 | Photosensitizer and photoelectric conversion device |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015194152A1 true WO2015194152A1 (ja) | 2015-12-23 |
Family
ID=54935158
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2015/002978 WO2015194152A1 (ja) | 2014-06-20 | 2015-06-15 | 光増感剤および光電変換素子 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9748044B2 (ja) |
| EP (1) | EP3159905B1 (ja) |
| JP (1) | JP5898725B2 (ja) |
| KR (1) | KR101802221B1 (ja) |
| CN (1) | CN106463272B (ja) |
| TW (1) | TWI632427B (ja) |
| WO (1) | WO2015194152A1 (ja) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016196318A1 (en) * | 2015-05-29 | 2016-12-08 | Wake Forest University | Thin-film pn junctions and applications thereof |
| WO2017067631A1 (en) | 2015-10-23 | 2017-04-27 | Merck Patent Gmbh | Blue optoelectronic device |
| CN109715736B (zh) | 2016-09-27 | 2021-08-24 | 保土谷化学工业株式会社 | 增感色素、光电转换用增感色素及使用了其的光电转换元件以及色素增感太阳能电池 |
| CN114752227B (zh) * | 2017-03-29 | 2023-09-01 | 保土谷化学工业株式会社 | 增感染料、光电转换用增感染料组合物、使用其的光电转换元件及染料增感太阳能电池 |
| JP2021138935A (ja) | 2020-02-28 | 2021-09-16 | 保土谷化学工業株式会社 | 増感色素、光電変換用増感色素組成物、光電変換素子および色素増感太陽電池 |
| JP2023032132A (ja) | 2021-08-26 | 2023-03-09 | 保土谷化学工業株式会社 | 増感色素、光電変換用増感色素組成物、光電変換素子および色素増感太陽電池 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB818639A (en) * | 1955-06-25 | 1959-08-19 | Agfa Ag | Anti-halation layers and filter layers for photographic materials |
| JPH09216907A (ja) * | 1995-12-05 | 1997-08-19 | Toyobo Co Ltd | 光重合性組成物およびそれを用いた感光性原版およびその露光方法 |
| JP2005019250A (ja) * | 2003-06-26 | 2005-01-20 | Mitsubishi Paper Mills Ltd | 光電変換材料、半導体電極並びにそれを用いた光電変換素子 |
| JP2006039355A (ja) * | 2004-07-29 | 2006-02-09 | Fuji Photo Film Co Ltd | 感光性組成物 |
| JP2007066690A (ja) * | 2005-08-31 | 2007-03-15 | Mitsubishi Paper Mills Ltd | 光電変換材料、半導体電極並びにそれを用いた光電変換素子 |
| JP2010169678A (ja) * | 2008-12-25 | 2010-08-05 | Canon Inc | 生物試料用標識剤並びに該標識剤を用いた標識方法及びスクリーニング方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4162162A (en) * | 1978-05-08 | 1979-07-24 | E. I. Du Pont De Nemours And Company | Derivatives of aryl ketones and p-dialkyl-aminoarylaldehydes as visible sensitizers of photopolymerizable compositions |
| JPS57204550A (en) * | 1981-06-12 | 1982-12-15 | Fuji Photo Film Co Ltd | Electrophotographic receptor |
| GB9217811D0 (en) | 1992-08-21 | 1992-10-07 | Graetzel Michael | Organic compounds |
| JP4080288B2 (ja) | 2002-09-26 | 2008-04-23 | 三菱製紙株式会社 | 太陽電池用メロシアニン色素 |
| JP2013035936A (ja) * | 2011-08-08 | 2013-02-21 | Kemikurea:Kk | 光増感剤および光電変換素子 |
| JP5946652B2 (ja) * | 2012-02-28 | 2016-07-06 | 富士フイルム株式会社 | 光電変換素子、金属錯体色素、色素増感太陽電池用色素吸着液組成物、色素増感太陽電池およびその製造方法 |
-
2014
- 2014-06-20 JP JP2014126802A patent/JP5898725B2/ja active Active
-
2015
- 2015-06-15 CN CN201580031533.9A patent/CN106463272B/zh active Active
- 2015-06-15 KR KR1020167035166A patent/KR101802221B1/ko active IP Right Grant
- 2015-06-15 WO PCT/JP2015/002978 patent/WO2015194152A1/ja active Application Filing
- 2015-06-15 EP EP15809237.9A patent/EP3159905B1/en active Active
- 2015-06-17 TW TW104119487A patent/TWI632427B/zh active
-
2016
- 2016-12-14 US US15/378,875 patent/US9748044B2/en active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB818639A (en) * | 1955-06-25 | 1959-08-19 | Agfa Ag | Anti-halation layers and filter layers for photographic materials |
| JPH09216907A (ja) * | 1995-12-05 | 1997-08-19 | Toyobo Co Ltd | 光重合性組成物およびそれを用いた感光性原版およびその露光方法 |
| JP2005019250A (ja) * | 2003-06-26 | 2005-01-20 | Mitsubishi Paper Mills Ltd | 光電変換材料、半導体電極並びにそれを用いた光電変換素子 |
| JP2006039355A (ja) * | 2004-07-29 | 2006-02-09 | Fuji Photo Film Co Ltd | 感光性組成物 |
| JP2007066690A (ja) * | 2005-08-31 | 2007-03-15 | Mitsubishi Paper Mills Ltd | 光電変換材料、半導体電極並びにそれを用いた光電変換素子 |
| JP2010169678A (ja) * | 2008-12-25 | 2010-08-05 | Canon Inc | 生物試料用標識剤並びに該標識剤を用いた標識方法及びスクリーニング方法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3159905A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CN106463272B (zh) | 2018-06-08 |
| KR20170002651A (ko) | 2017-01-06 |
| CN106463272A (zh) | 2017-02-22 |
| US20170092436A1 (en) | 2017-03-30 |
| EP3159905B1 (en) | 2018-08-15 |
| TW201602725A (zh) | 2016-01-16 |
| TWI632427B (zh) | 2018-08-11 |
| US9748044B2 (en) | 2017-08-29 |
| KR101802221B1 (ko) | 2017-11-28 |
| EP3159905A4 (en) | 2017-11-15 |
| EP3159905A1 (en) | 2017-04-26 |
| JP5898725B2 (ja) | 2016-04-06 |
| JP2016006811A (ja) | 2016-01-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5898725B2 (ja) | 光増感剤および光電変換素子 | |
| JP5940089B2 (ja) | ナフタリンモノイミド誘導体、及び当該誘導体を、太陽電池及び光検出器における光増感剤として用いる使用 | |
| Gupta et al. | Carbazole based A-π-D-π-A dyes with double electron acceptor for dye-sensitized solar cell | |
| WO2007100033A1 (ja) | 色素増感光電変換素子 | |
| JP5086807B2 (ja) | 新規アミノ基含有複素環誘導体および該複素環誘導体を含有する光電変換用増感色素 | |
| WO2006126538A1 (ja) | 色素増感光電変換素子 | |
| Bolisetty et al. | Benzothiadiazole-based organic dyes with pyridine anchors for dye-sensitized solar cells: effect of donor on optical properties | |
| JP2013035936A (ja) | 光増感剤および光電変換素子 | |
| Maglione et al. | Tuning optical absorption in pyran derivatives for DSSC | |
| KR101157743B1 (ko) | 염료 감응 태양전지용 유기염료 및 이를 포함하는 염료 감응 태양전지 | |
| Shivashimpi et al. | Effect of nature of anchoring groups on photosensitization behavior in unsymmetrical squaraine dyes | |
| JP5378725B2 (ja) | 光増感剤および光電変換素子 | |
| JP6005678B2 (ja) | 金属錯体色素、光電変換素子、色素増感太陽電池および金属錯体色素を含有する色素溶液 | |
| TW201604245A (zh) | 光電轉換元件、色素增感太陽電池、金屬錯合物色素、色素溶液以及三聯吡啶化合物或它的酯化物 | |
| JP2016216663A (ja) | 光増感剤および光電変換素子 | |
| JPWO2006041156A1 (ja) | スクアリリウム化合物ならびにこれを用いた光電変換材料、光電変換素子および光電気化学電池 | |
| JP2009173928A (ja) | ベンゾインドール系化合物及びこれを利用した色素増感太陽電池 | |
| JP2012007084A (ja) | 新規光増感剤 | |
| KR20120126498A (ko) | 신규한 유기염료 및 이를 포함하는 염료감응 태양전지 | |
| JP2016157837A (ja) | 光電変換用増感色素、その製造方法および該色素を用いた光電変換素子 | |
| WO2016136164A1 (ja) | 光増感剤および光電変換素子 | |
| KR101690902B1 (ko) | 광전 변환 소자, 색소 증감 태양 전지 및 이에 이용하는 금속 착체 색소 | |
| Arslan | 5, 6, 7-Trimethoxy-2-(Methylthio) Quinoline with Different Anchoring Groups: Synthesis And Dye-Sensitized Solar Cell Applications | |
| JPWO2006041155A1 (ja) | スクアリリウム化合物ならびにこれを用いた光電変換材料、光電変換素子および光電気化学電池 | |
| JP6330553B2 (ja) | 色素増感型太陽電池用の増感色素、及び当該増感色素を備える色素増感型太陽電池。 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15809237 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015809237 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015809237 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20167035166 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |