WO2014166324A1 - 替格瑞洛的中间体及其制备方法和替格瑞洛的制备方法 - Google Patents
替格瑞洛的中间体及其制备方法和替格瑞洛的制备方法 Download PDFInfo
- Publication number
- WO2014166324A1 WO2014166324A1 PCT/CN2014/073388 CN2014073388W WO2014166324A1 WO 2014166324 A1 WO2014166324 A1 WO 2014166324A1 CN 2014073388 W CN2014073388 W CN 2014073388W WO 2014166324 A1 WO2014166324 A1 WO 2014166324A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- group
- ticagrelor
- preparation
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CCCSc(nc1Cl)nc(*[C@@](CC2)C[C@@]2OCCO)c1N Chemical compound CCCSc(nc1Cl)nc(*[C@@](CC2)C[C@@]2OCCO)c1N 0.000 description 1
- DAHCXXNZOJUUTO-YKDSUIRESA-N CCCSc(nc1Cl)nc(N[C@H](C[C@@H]([C@H]2O)OCCO)[C@@H]2O)c1NC(C(F)(F)F)=O Chemical compound CCCSc(nc1Cl)nc(N[C@H](C[C@@H]([C@H]2O)OCCO)[C@@H]2O)c1NC(C(F)(F)F)=O DAHCXXNZOJUUTO-YKDSUIRESA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/47—One nitrogen atom and one oxygen or sulfur atom, e.g. cytosine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the invention relates to a novel synthetic method for the selective anticoagulant ticagrelor and its key intermediates, and belongs to the technical field of medicine manufacturing. Background technique
- the drug belongs to the cyclopentyl triazolopyrimidine compound, a novel, selective anticoagulant developed by AstraZeneca, UK. It is the first reversible binding type P2Y12 adenosine diphosphate. Receptor (ADP) antagonist.
- ADP adenosine diphosphate
- the drug can reversibly act on the ⁇ 2 receptor subtype P2Y12 on vascular smooth muscle cells (VCMC), which has a significant inhibitory effect on platelet aggregation induced by ADP, and can effectively improve the symptoms of patients with acute coronary heart disease. Since the antiplatelet effect of this drug is reversible, it is especially suitable for patients who require prior anticoagulation therapy before surgery. Compared with its competitor clopidogrel, ticagrelor has a promising anticoagulant with a significant reduction in myocardial infarction, stroke or cardiovascular death.
- the patent CN102149716A, WOO 192263 shows that the route is the production process of the original industrial preparation of ticagrelor bulk drug in the original research company, and the synthesis of key intermediates (1-2) in the process is the key of the process.
- Control point the reaction requires high temperature conditions, long-term reaction in an oxygen-free environment and certain pressure conditions, so this condition has a certain equipment bottleneck, the reaction is under high temperature and pressure for a long time, there is a potential danger in industrial production. Sex.
- Chinese patent application CN102675321A discloses 2-[[(3 AR,4S,6R,6AS)-6-aminotetrahydro-2,2-dimethyl-) protected on the hydroxyl group by an alkoxy or silicon-based reagent. 4H-cyclopenteno-1,3-dioxolan-4-yl]oxy]ethanol (II-1) and 4,6-dichloro-2-(propylthio)-5-amino
- the pyrimidine is used as a starting material to obtain a key intermediate (11-2).
- the intermediate is subjected to a ring-closing reaction under the action of an appropriate alkali metal nitrite to obtain an intermediate (11-3), an intermediate ( ⁇ -3).
- Reaction with a suitable salt of (lR,2S)-REL-2-(3,4-difluorophenyl)cyclopropylamine followed by removal of the protecting group affords the synthetic route of ticagrelor (I).
- the method has the same characteristics as the synthetic technology of CN102149716A and WO0192263.
- the disadvantage is that there is still no way to avoid the need to react under high temperature conditions for a long time in an oxygen-free environment and under certain pressure conditions. Low, darker color, making the final product more difficult to purify, the pigment is more difficult to remove, and the overall yield of the process is relatively low.
- Patent US6525060 discloses 4,6-dichloro-2-(;propylthio 5-nitropyrimidine with (3AR,4S,6R,6AS)-6-aminotetrahydro-2,2-dimethyl-4H- Cyclopentene-1,3-dioxol-4-ol (III-1) is reacted to obtain an intermediate (111-2) which is reduced in an iron powder acetic acid system and then subjected to nitrite. Under the cyclization, a series of reactions such as ammoniation, bromination, and substitution reaction are carried out to obtain a synthetic route of ticagrelor (I).
- Patent CN102875537A discloses the reaction of 4,6-dichloro-2-propylthio-5-nitropyrimidine with a protected amino alcohol of formula IV-2 to give an intermediate of formula IV-3, followed by palladium on carbonation. Reduction of the nitro group provides the intermediate of formula IV-4, which is closed under the action of nitrite to give the intermediate of formula IV-5. The intermediate of formula IV-5 is reacted with a chiral cyclopropylamine to give the intermediate of formula IV-6.
- the intermediate of formula IV-6 is reduced by sodium borohydride catalyzed by sodium borohydride to give the intermediate of formula IV-7, and finally subjected to deprotection of the propylidene protecting group under acidic conditions to give the ticagrelor product of formula I.
- This route has the advantage of a relatively simple route.
- the use of formyl protection can activate the chlorine atom in the aminopyrimidine molecule, making the substitution reaction conditions milder, but the subsequent process uses an acid system to remove the formyl group and inevitably the side chain.
- the removal of the propylidene protecting group makes the polarity of the subsequent intermediates very large, which is not conducive to the purification of the intermediate.
- the stability of the intermediate V-5 itself is lowered, and the intermolecular phase is prone to occur. Self-reaction makes it difficult to purify the final product. Summary of the invention
- the ortho-halogen atom is very easily substituted by the nucleophilic reagent, resulting in harsh reaction conditions, such as low temperature, no oxygen, no water, etc., usually many by-products, the intermediate is difficult to purify; conversely, when the 5-position is a free amino group
- the halogen atom leaving ability on the pyrimidine ring is passivated, and the substitution reaction requires relatively strong conditions such as high temperature, pressure, and long-term reaction. Therefore, selecting the appropriate amino protecting group, activating the leaving ability of the ortho-halogen atom, achieving a mild substitution reaction condition, and the intermediate is easy to purify. It is necessary to use a fine amino protecting group.
- the acetylating agent protects the amino group on the pyrimidine ring
- the protective intermediate can be quantitatively obtained at room temperature in less than one hour
- the trifluoroacetyl group protects the amino group
- the halogen atom in the ortho position is effectively activated, so that the substitution reaction can be carried out under very mild conditions, such as a reaction temperature of 30-80 ° C and a reaction time of 1-5 hours.
- the system does not require inert gas protection and the like.
- the method of protecting the amino group with a formyl group shows that the amino group selective reaction of the formyl group on the pyrimidine ring requires a large excess of anhydrous formic acid and acetic anhydride as a solvent system, and the reaction time is longer, and the obtained amino group is obtained.
- the protected intermediate usually contains an acetyl-protected by-product, which makes purification of the intermediate difficult, and the yield is usually maintained at about 70%.
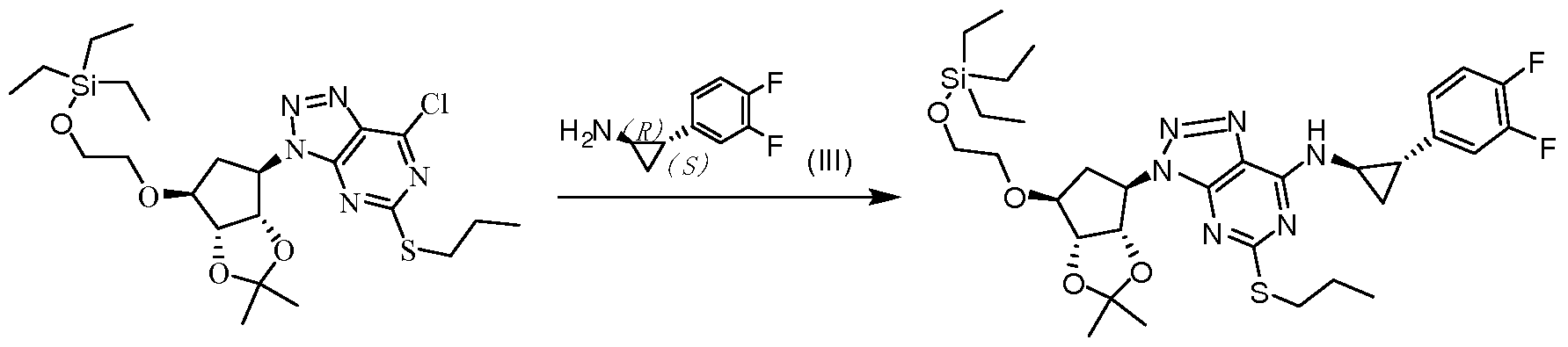
- One aspect of the invention provides a compound of (VI),
- R is hydrogen or a hydroxy protecting group
- the hydroxy protecting group is preferably selected from the group consisting of triethylsilyl, triisopropylsilyl, tert-butyldimethylsilyl, tert-butyldiphenylsilyl, methoxy Methyl, tetrahydropyranyl or tetrahydrofuranyl.
- the compound of the formula (VI) can be conveniently used for the preparation of ticagrelor.
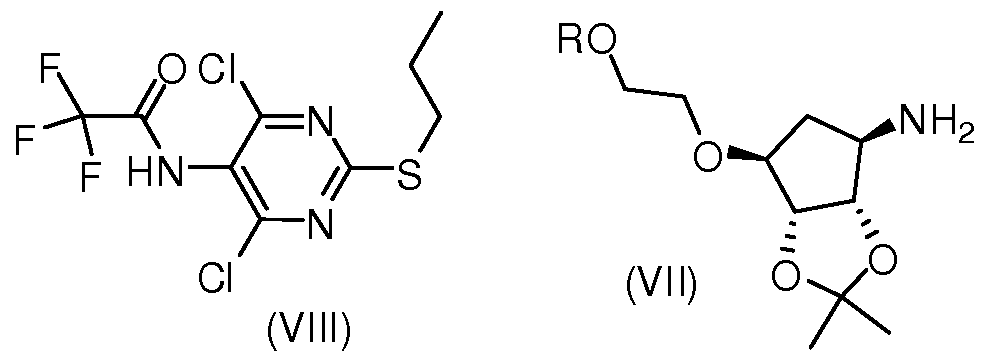
- the present invention provides a process for the preparation of a compound of formula (VI), which comprises reacting a compound of formula (VIII) with formula (VII),
- R is as defined in the compound represented by formula (VI).
- the compound (VIII) can be produced by reacting the compound (VIII) with a suitable base in a solvent at 0 to 100 ° C with a suitable salt of the compound (VII) or the compound (VII).
- the solvent is selected from the group consisting of toluene, tetrahydrofuran, 2-methyltetrahydrofuran, dioxane, N,N-dimethylformamide, N,N-dimethylacetamide, acetonitrile, dichloromethane, chloroform, One or more of dichloroethane, xylene, trimethylbenzene, methyl tert-butyl ether, methylcyclopentyl ether, preferably dioxane;
- the base is triethylamine, diisopropyl Ethylamine, pyridine or a 2,3,4-position monoalkyl-substituted pyridine, preferably diisopropylethylamine; suitable salts of the
- the trifluoroacetylating agent may be selected from the group consisting of ethyl trifluoroacetate, trifluoroacetic anhydride, trifluoroacetic acid-succinimide, trifluoroacetylbenzotriazole, 5-fluorophenolic acid trifluoroacetate, 2-trifluoroacetoxypyridine, preferably trifluoroacetic anhydride.
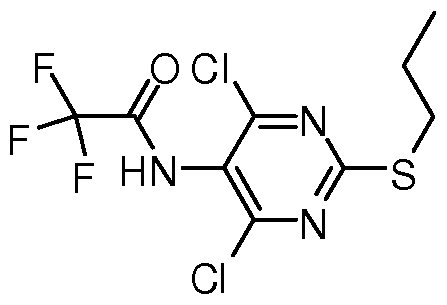
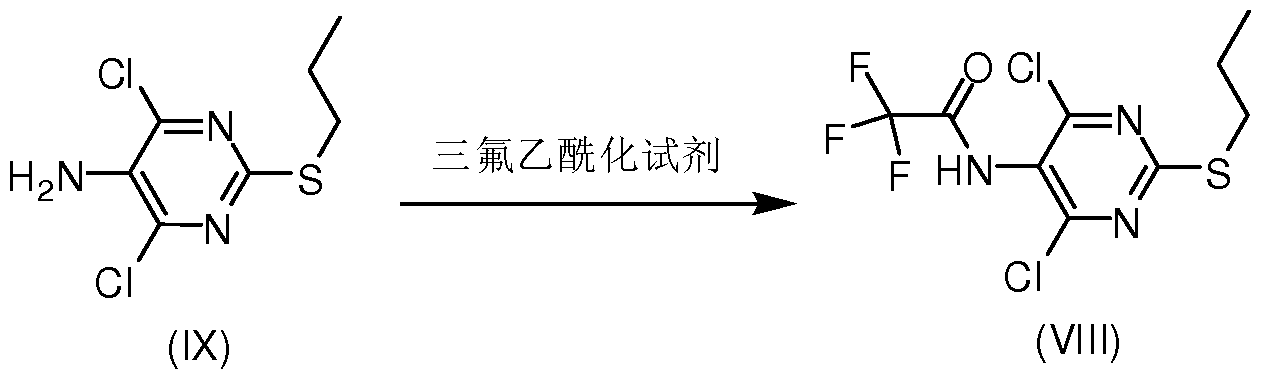
- the present invention provides a compound represented by VIII,
- a process for the preparation of ticagrelor which comprises the steps of preparing the compound of the formula (VI) and the step of further preparing ticagrelor by the compound of the formula (VI).
- the method for further preparing ticagrelor by the compound represented by the formula (VI) comprises the steps of first reacting the compound (VI) with a base in water or a suitable solvent, and reacting at 0-150 ° C to prepare a compound ( V), and then the compound (V) is prepared according to a known method to obtain ticagrelor, for example
- compound (VI) is selected from the group consisting of water, dC 8 lower aliphatic monohydric alcohol, dC 8 lower aliphatic diol, toluene, tetrahydrofuran, 2-methyltetrahydrofuran, dioxane, N, N-dimethylformamide, N, N-dimethylacetamide, acetonitrile, dichloromethane, chloroform, dichloroethane, xylene, three One or more of toluene, methyl tert-butyl ether, methylcyclopentyl ether, preferably a mixed solution of ethanol and water;
- the base is potassium carbonate, sodium carbonate, cesium carbonate, sodium hydrogencarbonate, sodium phosphate, Potassium phosphate, sodium hydroxide, potassium hydroxide, lithium hydroxide, calcium hydroxide or barium hydroxide, preferably potassium carbonate.
- the complete route of the process for the preparation of ticagrelor of the invention is as follows:
- the compound (V) is reacted with an alkali metal nitrite at -10 to 30 ° C in an appropriate acid water to obtain a compound (IV).
- the acid is one or more of formic acid, acetic acid, dilute hydrochloric acid, dilute sulfuric acid, dilute phosphoric acid, preferably acetic acid;
- the alkali metal nitrite is sodium nitrite, potassium nitrite, preferably sodium nitrite.
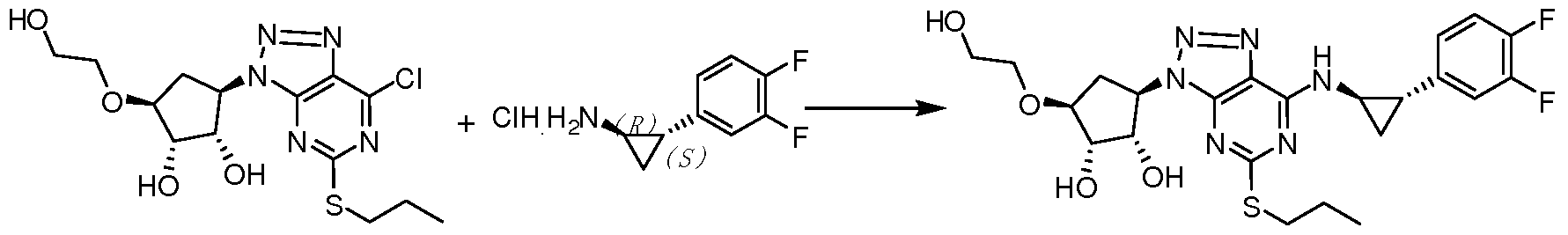
- the compound (11) can be produced by reacting (1R,2S)-2-(3,4-difluorophenyl)cyclopropylamine (III) with a suitable base in a solvent.
- the solvent is selected from the group consisting of dC 8 lower aliphatic monohydric alcohol, dC 8 lower aliphatic diol, toluene, tetrahydrofuran, 2-methyltetrahydrofuran, dioxane, N, N-dimethylformamide, N, N - dimethylacetamide, acetonitrile, dichloromethane, chloroform, dichloroethane, xylene, trimethylbenzene, methyl tert-butyl ether, methyl One or more of cyclopentyl ether, preferably toluene; the base is triethylamine, diisopropylethylamine, pyridine, 2,3,4-position monoalkyl-substituted pyridine, potassium carbonate, sodium carbonate , cesium carbonate, sodium hydrogencarbonate, sodium phosphate, potassium phosphate, preferably triethylamine.
- the compound ( ⁇ ) is prepared by adding an appropriate acidic deprotecting reagent to water or a solvent, and removing the protecting group to prepare ticagrelor (I);
- the solvent is selected from the group consisting of methanol, ethanol, isopropanol, ethylene glycol, ethylene glycol monomethyl ether, ethylene glycol monobutyl ether, tetrahydrofuran, 2-methyltetrahydrofuran, dioxane, acetonitrile.
- the deprotecting reagent acid is a certain concentration of hydrochloric acid or a certain concentration of trifluoroacetic acid, preferably hydrochloric acid.
- R is a hydroxy protecting group, preferably selected from the group consisting of triethylsilyl, triisopropyl, tert-butyldimethylsilyl, tert-butyldiphenylsilyl, methoxymethyl, tetrahydropyranyl , tetrahydrofuranyl.
- the hydroxy protecting group R can also be removed in advance, and the hydroxy protecting group in the obtained compound (VI) is removed to obtain a compound of the formula (VI'), and then ticagrelor can be obtained by a similar method; (In the compound VII, R directly takes hydrogen, that is, directly from the (VII') compound, to obtain a compound of the formula (VI'),
- the compound of the formula (v) is prepared by reacting a compound of the formula (IV'), the acid is selected from the group consisting of hydrochloric acid, methanesulfonic acid, trifluoromethanesulfonic acid, trifluoroacetic acid, sulfuric acid; or the base is selected from the group consisting of triethylamine, Diisopropylethylamine, pyridine, 2,3,4-position monoalkyl substituted pyridine, N-methylmorpholine, anhydrous potassium carbonate, anhydrous sodium carbonate, anhydrous sodium hydrogencarbonate, anhydrous sodium phosphate, Anhydrous potassium phosphate, sodium hydroxide or lithium hydroxide.
- the acid is selected from the group consisting of hydrochloric acid, methanesulfonic acid, trifluoromethanesulfonic acid, trifluoroacetic acid, sulfuric acid
- the base is selected from the group consisting of triethylamine, Diisopropylethylamine
- the compound of the formula (IV) is reacted with a base in a solvent selected from the group consisting of toluene, tetrahydrofuran, 2-methyltetrahydrofuran, dioxane, hydrazine, hydrazine-dimethylformamide, hydrazine, hydrazine. - dimethyl acetamide, One or more of dichloromethane, chloroform, dichloroethane, ethyl acetate, xylene, trimethylbenzene, diethyl ether, diisopropyl ether, methyl tert-butyl ether, methylcyclopentyl ether.
- a solvent selected from the group consisting of toluene, tetrahydrofuran, 2-methyltetrahydrofuran, dioxane, hydrazine, hydrazine-dimethylformamide, hydrazine, hydrazine. - dimethyl
- the compound represented by the formula ⁇ ') is reacted with an acid in a solvent selected from the group consisting of a fatty alcohol having a carbon number of less than 6, preferably methanol or ethanol.
- the synthesis method of the intermediate (VI) provided by the invention is simple, and the quantitative selective protection of the aminopyrimidine by the trifluoroacetyl group greatly enhances the reactivity of the chlorine atom on the aminopyrimidine, so the compound (VIII) can be very mild. Under the condition, the hydroxyl group-protected chiral amino alcohol (VII) or its suitable salt is reacted, thereby avoiding the disadvantages of the prior method requiring high temperature and long-term reaction, suitable for industrial scale-up production, reducing energy consumption, and reducing environmental impact. Pollution, effectively reduce emissions of three wastes.
- Hydrophilic protecting group is a suitable group for hydroxyl protection known in the art, see literature
- the hydroxy protecting group in "Protective Groups in Organic Synthesis", 5 Th Ed. TW Greene & PGM Wuts).
- the hydroxy protecting group may be a (Cwo alkyl or aryl) 3 silane group, for example: triethylsilyl, triisopropylsilyl, tert-butyldimethylsilyl, Tert-butyldiphenylsilyl or the like; may be Cw.
- alkyl or substituted alkyl group for example: methyl, tert-butyl, allyl, benzyl, methoxymethyl, ethoxyethyl, 2-tetrahydropyranyl (THP), etc.; Cwo alkyl or aryl) acyl groups such as: formyl, acetyl, benzoyl and the like; may be (d_ 6 alkyl or C 6 _ 1Q aryl) sulfonyl; may be (d_ 6 alkoxy or Aryloxy)carbonyl.
- Method A Compound IX (476.3 g, 2.0 mole) (IX purchased from Shanghai Qianyuan Chemical Technology Co., Ltd.) was added to 4.0 L of dichloromethane, triethylamine (404 g, 4.0 mole) was added, and the system was cooled to ice water. After the internal temperature is below 10 °C, trifluoroacetic anhydride (630 g, 3.0 mole) is slowly added dropwise. After the dropwise addition is completed, the temperature is naturally raised to room temperature, stirred for 1 hour, and the reaction is traced by HPLC until the conversion of the starting material is complete. 2.0 is added to the reaction solution.
- the compound ⁇ - ⁇ (100 g, 0.15 mole) was dissolved in 3.0 L of methanol, 1.0 L of 3M hydrochloric acid solution was added, and the temperature was raised to 30 degrees for 24 hours. The reaction was completely monitored by HPLC, and most of the solvent was removed by concentration. After adding 2.0 L of ethyl acetate, the aqueous phase was separated, and the aqueous phase was extracted with ethyl acetate (1 mL), and the organic phase was combined, and the organic phase was washed with brine, dried and concentrated to give 70.0 g of crude compound I.
- the crude product was dissolved in 300 ml of ethyl acetate, heated to 50 °C, dissolved completely, 500 ml of n-heptane was added, and the temperature was slowly lowered to 20 °C, and filtered to obtain a pure white solid product of 1 60 g, the content was 99%.
- the yield was 85%.
- the compound VI-A (100 g, 0.16 mole) was dissolved in 1.0 L of anhydrous methanol, and the trifluoroacetic acid was added dropwise at a temperature not exceeding 50 °C. After the addition was completed, the mixture was stirred at room temperature for 24 hours, and the reaction was completely monitored by HPLC. Most of the solvent was removed, and lkg of deionized water was added to the residue to precipitate a large amount of a yellow solid, which was filtered and dried in vacuo to give compound V 73 g, yield 96%.
- the compound III (40 g, 0.10 mole) and the hydrochloride salt of the compound II (30.8 g, 0.15 mole) were dissolved in 1.0 L of dry tetrahydrofuran, and potassium carbonate (42 g, 0.3 mole) was added, and the reaction was stirred at room temperature for 24 hours. The reaction was monitored completely, 500 ml of deionized water was added to the system, the organic phase was washed with brine, dried and concentrated to give compound I 46.5 g (yield: 89%).
- the crude product was dissolved in 150 ml of ethyl acetate. In the middle, the temperature was raised to 50 degrees, and the solution was completely dissolved. 300 ml of n-heptane was added, and the temperature was slowly lowered to 20 °C, and filtered to obtain 1 44 g of tigrelor as a white solid, the content was 99%, and the yield was 95%.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
























Claims









Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016506761A JP6332818B2 (ja) | 2013-04-10 | 2014-03-13 | チカグレロルの中間体およびその製造法、およびチカグレロルの製造法 |
| EP14782391.8A EP2985286B1 (en) | 2013-04-10 | 2014-03-13 | Midbody of ticagrelor and preparation method therefor, and preparation method for ticagrelor |
| US14/781,803 US9359366B2 (en) | 2013-04-10 | 2014-03-13 | Intermediate of Ticagrelor and preparation method therefor, and preparation method for Ticagrelor |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201310122613 | 2013-04-10 | ||
| CN201310122613.8 | 2013-04-10 | ||
| CN201310383474.4 | 2013-08-28 | ||
| CN201310383474.4A CN104098553B (zh) | 2013-04-10 | 2013-08-28 | 替格瑞洛的中间体及其制备方法和替格瑞洛的制备方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014166324A1 true WO2014166324A1 (zh) | 2014-10-16 |
Family
ID=51667139
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2014/073388 Ceased WO2014166324A1 (zh) | 2013-04-10 | 2014-03-13 | 替格瑞洛的中间体及其制备方法和替格瑞洛的制备方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9359366B2 (zh) |
| EP (1) | EP2985286B1 (zh) |
| JP (1) | JP6332818B2 (zh) |
| CN (2) | CN104098553B (zh) |
| HK (1) | HK1202533A1 (zh) |
| TW (1) | TWI634116B (zh) |
| WO (1) | WO2014166324A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015067230A1 (en) * | 2013-11-08 | 2015-05-14 | Zentiva, K.S. | A production method and a new crystalline form of an intermediate of synthesis of ticagrelor |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI229674B (en) | 1998-12-04 | 2005-03-21 | Astra Pharma Prod | Novel triazolo[4,5-d]pyrimidine compounds, pharmaceutical composition containing the same, their process for preparation and uses |
| CN105801583A (zh) * | 2014-12-31 | 2016-07-27 | 徐州万邦金桥制药有限公司 | 一种替格瑞洛的纯化方法 |
| CN105859720A (zh) * | 2015-01-20 | 2016-08-17 | 上海方楠生物科技有限公司 | 一种替格瑞洛溶液的制备方法 |
| CN105669681A (zh) * | 2016-04-11 | 2016-06-15 | 成都华宇制药有限公司 | 一种替格瑞洛的合成方法 |
| CN111848632A (zh) * | 2020-09-07 | 2020-10-30 | 河南师范大学 | 一种血小板聚集抑制剂替格瑞洛的制备方法 |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001092263A1 (en) | 2000-06-02 | 2001-12-06 | Astrazeneca Ab | Novel triazolo pyrimidine compounds |
| US6525060B1 (en) | 1998-12-04 | 2003-02-25 | Astrazeneca Uk Limited | Triazolo(4,5-d)pyrimidine compounds |
| WO2010030224A1 (en) * | 2008-09-09 | 2010-03-18 | Astrazeneca Ab | A process for preparing [1s- [1-alpha, 2-alpha, 3-beta (1s*, 2r*) 5-beta] ] -3- [7- [2- (3, 4-dif luorophenyl) -cyclopropylamino] - 5- (propylthio) -3h-1, 2, 3-triazolo [4, 5-d] pyrimidin-3-yl] -5- (2- hydroxyethoxy) cyclopentane-1, 2-diol and to its intermediates |
| WO2011017108A2 (en) | 2009-07-27 | 2011-02-10 | Auspex Pharmaceuticals, Inc. | Cyclopropyl modulators of p2y12 receptor |
| WO2012085665A2 (en) | 2010-12-20 | 2012-06-28 | Actavis Group Patc Ehf | Novel processes for preparing triazolo[4,5-d]pyrimidine derivatives and intermediates thereof |
| CN102659815A (zh) | 2012-05-04 | 2012-09-12 | 开原亨泰制药股份有限公司 | 一种制备选择性抗凝血药替卡格雷及其中间体的方法 |
| CN102675321A (zh) | 2012-05-11 | 2012-09-19 | 上海皓元化学科技有限公司 | 一种替卡格雷的制备方法 |
| WO2012138981A2 (en) | 2011-04-06 | 2012-10-11 | Teva Pharmaceutical Industries Ltd. | New intermediates and processes for preparing ticagrelor |
| CN102875537A (zh) | 2012-09-10 | 2013-01-16 | 常州制药厂有限公司 | 一种新的抗血栓药物的制备方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0577558A2 (de) * | 1992-07-01 | 1994-01-05 | Ciba-Geigy Ag | Carbocyclische Nukleoside mit bicyclischen Ringen, Oligonukleotide daraus, Verfahren zu deren Herstellung, deren Verwendung und Zwischenproduckte |
| WO1995010506A1 (en) * | 1993-10-12 | 1995-04-20 | The Du Pont Merck Pharmaceutical Company | 1n-alkyl-n-arylpyrimidinamines and derivatives thereof |
| AR017014A1 (es) * | 1997-07-22 | 2001-08-22 | Astrazeneca Ab | Compuestos de triazolo [4,5-d]pirimidina, composiciones farmaceuticas, uso de los mismos para preparar medicamentos y procesos para la preparacionde dichos compuestos |
| GB0013407D0 (en) * | 2000-06-02 | 2000-07-26 | Astrazeneca Ab | Forms of a chemical compound |
| CN101148450A (zh) * | 2007-11-02 | 2008-03-26 | 严红芳 | 一种核苷化合物的制备方法 |
| US9469614B2 (en) * | 2011-10-27 | 2016-10-18 | Lek Pharmaceuticals D.D. | Synthesis of triazolopyrimidine compounds |
| IN2014KN01491A (zh) * | 2011-12-23 | 2015-10-23 | Lek Pharmaceuticals | |
| US9233966B2 (en) * | 2012-04-05 | 2016-01-12 | Dr. Reddy's Laboratories Limited | Preparation of ticagrelor |
-
2013
- 2013-08-28 CN CN201310383474.4A patent/CN104098553B/zh not_active Withdrawn - After Issue
- 2013-08-28 CN CN201710739212.5A patent/CN107573333B/zh not_active Withdrawn - After Issue
-
2014
- 2014-03-13 US US14/781,803 patent/US9359366B2/en not_active Expired - Fee Related
- 2014-03-13 EP EP14782391.8A patent/EP2985286B1/en not_active Not-in-force
- 2014-03-13 JP JP2016506761A patent/JP6332818B2/ja not_active Expired - Fee Related
- 2014-03-13 WO PCT/CN2014/073388 patent/WO2014166324A1/zh not_active Ceased
- 2014-03-24 TW TW103110842A patent/TWI634116B/zh not_active IP Right Cessation
-
2015
- 2015-03-24 HK HK15102985.6A patent/HK1202533A1/zh unknown
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6525060B1 (en) | 1998-12-04 | 2003-02-25 | Astrazeneca Uk Limited | Triazolo(4,5-d)pyrimidine compounds |
| WO2001092263A1 (en) | 2000-06-02 | 2001-12-06 | Astrazeneca Ab | Novel triazolo pyrimidine compounds |
| US20030148888A1 (en) | 2000-06-02 | 2003-08-07 | Ulf Larsson | Novel triazolo pyrimidine compounds |
| WO2010030224A1 (en) * | 2008-09-09 | 2010-03-18 | Astrazeneca Ab | A process for preparing [1s- [1-alpha, 2-alpha, 3-beta (1s*, 2r*) 5-beta] ] -3- [7- [2- (3, 4-dif luorophenyl) -cyclopropylamino] - 5- (propylthio) -3h-1, 2, 3-triazolo [4, 5-d] pyrimidin-3-yl] -5- (2- hydroxyethoxy) cyclopentane-1, 2-diol and to its intermediates |
| CN102149716A (zh) | 2008-09-09 | 2011-08-10 | 阿斯利康(瑞典)有限公司 | 制备[1S-[1α,2α,3β(1S*,2R*),5β]]-3-[7-[2-(3,4-二氟苯基)-环丙氨基]-5-(丙硫基)-3H-1,2,3-三唑并[4,5-d]嘧啶-3-基]-5-(2-羟乙氧基)环戊烷-1,2-二醇的方法及其中间体 |
| WO2011017108A2 (en) | 2009-07-27 | 2011-02-10 | Auspex Pharmaceuticals, Inc. | Cyclopropyl modulators of p2y12 receptor |
| WO2012085665A2 (en) | 2010-12-20 | 2012-06-28 | Actavis Group Patc Ehf | Novel processes for preparing triazolo[4,5-d]pyrimidine derivatives and intermediates thereof |
| WO2012138981A2 (en) | 2011-04-06 | 2012-10-11 | Teva Pharmaceutical Industries Ltd. | New intermediates and processes for preparing ticagrelor |
| CN102659815A (zh) | 2012-05-04 | 2012-09-12 | 开原亨泰制药股份有限公司 | 一种制备选择性抗凝血药替卡格雷及其中间体的方法 |
| CN102675321A (zh) | 2012-05-11 | 2012-09-19 | 上海皓元化学科技有限公司 | 一种替卡格雷的制备方法 |
| CN102875537A (zh) | 2012-09-10 | 2013-01-16 | 常州制药厂有限公司 | 一种新的抗血栓药物的制备方法 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015067230A1 (en) * | 2013-11-08 | 2015-05-14 | Zentiva, K.S. | A production method and a new crystalline form of an intermediate of synthesis of ticagrelor |
Also Published As
| Publication number | Publication date |
|---|---|
| CN107573333A (zh) | 2018-01-12 |
| EP2985286A1 (en) | 2016-02-17 |
| HK1202533A1 (zh) | 2015-10-02 |
| EP2985286A4 (en) | 2016-09-14 |
| JP6332818B2 (ja) | 2018-05-30 |
| US20160052928A1 (en) | 2016-02-25 |
| CN104098553B (zh) | 2017-11-28 |
| TWI634116B (zh) | 2018-09-01 |
| US9359366B2 (en) | 2016-06-07 |
| JP2016515635A (ja) | 2016-05-30 |
| EP2985286B1 (en) | 2017-10-11 |
| TW201439087A (zh) | 2014-10-16 |
| CN104098553A (zh) | 2014-10-15 |
| CN107573333B (zh) | 2019-10-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10647728B2 (en) | Process for preparing (3S,11aR)-6-methoxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-Hexahydrooxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxylic acid | |
| EP3527573B9 (en) | Synthesis of polycyclic-carbamoylpyridone compounds | |
| CN108055842B (zh) | 亚磺酰基苯基或磺亚胺酰基苯基苯并氮杂䓬 | |
| WO2014166324A1 (zh) | 替格瑞洛的中间体及其制备方法和替格瑞洛的制备方法 | |
| CN101203519A (zh) | 用于治疗癌症和病毒感染如丙型肝炎的用作Toll样受体调节剂的2-酰氨基-6-氨基-8-氧代嘌呤衍生物 | |
| CN116987112A (zh) | 制备氨基嘧啶衍生物的改善方法 | |
| CN106068271B (zh) | 2′-取代-2,2′-脱水尿苷或2′-取代-2,2′-脱水胞苷化合物及其制备方法和用途 | |
| US6087497A (en) | Process for producing purine derivatives | |
| US20120330013A1 (en) | Preparation method of mln4924 as an e1 activating inhibitor | |
| EP2714691B1 (en) | Process for the preparation of 2-amino-9-((2-phenyl-1,3-dioxan-5-yloxy)methyl)-1h-purin-6(9h)-one compound useful in the preparation of valganciclovir | |
| CN102216274A (zh) | (R)-3-(2,3-二羟基丙基)-6-氟-5-(2-氟-4-碘苯基氨基)-8-甲基吡啶并[2,3-d]嘧啶-4,7(3H,8H)-二酮及其中间体的制备方法 | |
| WO2012022217A1 (zh) | N-[4-甲基-3-(4-吡啶-3-基-嘧啶-2-基氨基)苯基]苯甲酰胺衍生物及制备方法与在伊马替尼合成中的应用 | |
| CN104592222A (zh) | 抗血小板药物azd6482的制备方法 | |
| TWI389912B (zh) | 用於製備n-〔5-(3-二甲胺基-丙烯醯基)-2-氟苯基〕-n-甲基-乙醯胺之方法 | |
| CN102143938B (zh) | α-三氟甲基-β-取代-β-氨基酸类的制造方法 | |
| CN117088818A (zh) | 一种4,6-二氯-2-丙硫基-5-氨基嘧啶的合成方法 | |
| JPH10120682A (ja) | プリン誘導体の製造方法 | |
| CN106854208A (zh) | 肿瘤抑制剂mln4924的合成方法 | |
| HK40011503A (zh) | 多環氨基甲醯基吡啶酮化合物的合成 | |
| HK40011503B (zh) | 多環氨基甲醯基吡啶酮化合物的合成 | |
| JPH069462A (ja) | アフィディコラン誘導体およびその製造法 | |
| JPH04283571A (ja) | 新規なシクロプロパン誘導体 | |
| HK1236512B (zh) | 多環氨基甲醯基吡啶酮化合物的合成 | |
| HK1236512A1 (zh) | 多環氨基甲醯基吡啶酮化合物的合成 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14782391 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016506761 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14781803 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014782391 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014782391 Country of ref document: EP |