WO2014097799A1 - 植物性バイオマスの加水分解方法 - Google Patents
植物性バイオマスの加水分解方法 Download PDFInfo
- Publication number
- WO2014097799A1 WO2014097799A1 PCT/JP2013/081181 JP2013081181W WO2014097799A1 WO 2014097799 A1 WO2014097799 A1 WO 2014097799A1 JP 2013081181 W JP2013081181 W JP 2013081181W WO 2014097799 A1 WO2014097799 A1 WO 2014097799A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- plant biomass
- reaction solution
- reaction
- equivalent concentration
- Prior art date
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C13—SUGAR INDUSTRY
- C13K—SACCHARIDES OBTAINED FROM NATURAL SOURCES OR BY HYDROLYSIS OF NATURALLY OCCURRING DISACCHARIDES, OLIGOSACCHARIDES OR POLYSACCHARIDES
- C13K1/00—Glucose; Glucose-containing syrups
- C13K1/02—Glucose; Glucose-containing syrups obtained by saccharification of cellulosic materials
Definitions
- the present invention relates to a method for hydrolyzing plant biomass. More specifically, the present invention relates to a hydrolysis method capable of obtaining a high glucose yield in which a reaction inhibition factor of hydrolysis by hydrothermal treatment of plant biomass is eliminated.
- Patent Document 1 describes a method of hydrolyzing cellulose powder by contacting it with pressurized hot water heated to 200 to 300 ° C. (hydrolysis method by hydrothermal treatment).
- Patent Document 2 describes a method in which an activated carbon solid acid catalyst treated with sulfuric acid is used as a solid catalyst for hydrothermal reaction.
- Patent Document 3 discloses a method for obtaining a glucose yield of 60% or more by bringing a raw material containing cellulose and an aqueous solution containing an inorganic acid into contact with each other and subjecting the mixture to heat and pressure treatment.
- JP 2011-206044 A discloses a method for obtaining a glucose yield of 60% or more by bringing a raw material containing cellulose and an aqueous solution containing an inorganic acid into contact with each other and subjecting the mixture to heat and pressure treatment.
- An object of the present invention is to provide a method for obtaining a high glucose yield by eliminating a reaction inhibition factor in a method for hydrolyzing plant biomass.
- the present inventors have intensively studied to solve the above problems. As a result, in the hydrolysis of plant biomass by hydrothermal treatment, by adding an acid according to the equivalent concentration of hydroxide ions and cations in the reaction solution, the reaction inhibition factor is released and a high glucose yield can be obtained. As a result, the present invention has been completed.
- the present invention provides the following plant biomass hydrolysis method [1] to [7] and glucose production method [8].
- Hydrothermal treatment is performed by adding an acid having an equivalent concentration of 30 to 1000% of the equivalent concentration of cations in the hydrolysis reaction solution of plant biomass and the equivalent concentration of hydroxide ions to the reaction solution.
- a method for hydrolyzing plant biomass characterized by the above.
- [2] The method for hydrolyzing plant biomass according to item 1 above, wherein a solid catalyst is used for the hydrothermal treatment.
- the method for hydrolyzing plant biomass according to item 1 or 2 wherein the acid is at least one selected from inorganic mineral acids, organic carboxylic acids, and organic sulfonic acids
- the cation in the reaction solution is 4.
- hydrothermal treatment is performed by adding an acid to an equivalent concentration of 30 to 1000% of the equivalent concentration of the cation contained in the reaction solution.
- a method for producing glucose wherein the method for hydrolyzing plant biomass according to any one of items 1 to 7 above is used.
- reaction inhibition factors such as hydroxide ions and cations in the hydrolysis reaction solution are released, and a high glucose yield can be obtained.
- Example 3 shows a comparison of the product yield when sulfuric acid is added before the reaction using dehydrated dry bagasse (Example 2).
- the method for hydrolyzing plant biomass of the present invention is characterized in that the reaction inhibition of hydroxide ions and cations is canceled by allowing a specific amount of acid to coexist in the reaction solution.
- Plant biomass generally refers to “renewable biological organic resources excluding fossil resources”.
- plant biomass means mainly cellulose such as rice straw, straw, sugar cane leaf, rice husk, bagasse, hardwood, bamboo, conifer, kenaf, furniture waste wood, building waste wood, waste paper, food residue, etc.
- plant biomass is used as a solid substrate for the hydrolysis reaction.
- Plant biomass can be used as it is as a solid substrate, but pretreatments such as alkali cooking, alkaline sulfite cooking, neutral sulfite cooking, alkaline sodium sulfide cooking, ammonia cooking, sulfuric acid cooking, hydrothermal cooking, etc. Later, a residue that has been subjected to operations such as neutralization, water washing, dehydration, and drying to reduce the content of lignin and hemicellulose, and containing two or more of cellulose, hemicellulose, and lignin, Industrially prepared cellulose, xylan, cellooligosaccharide, xylooligosaccharide, and the like can be used.
- plant biomass may be dry or wet and may be crystalline or non-crystalline.
- the particle size of the plant biomass is not limited as long as it can be pulverized, but it is preferably 20 ⁇ m or more and several thousand ⁇ m or less from the viewpoint of pulverization efficiency.
- Solid catalyst In the hydrolysis method by hydrothermal treatment of the present invention, a solid catalyst can also be used.
- the solid catalyst is not particularly limited as long as it is a catalyst capable of hydrolyzing plant biomass polysaccharides. For example, it is represented by ⁇ -1,4 glycosidic bond between glucose forming cellulose as a main component. It preferably has an activity of hydrolyzing glycosidic bonds.
- the solid catalyst for example, carbon materials and transition metals can be used alone or in combination of two or more.
- the carbon material for example, activated carbon, carbon black, graphite and the like can be used alone or in combination of two or more.
- the shape of the carbon material is preferably porous and / or fine particles in terms of improving reactivity by expanding the contact area with the substrate, and in terms of promoting acid hydrolysis by expressing acid sites. It preferably has a surface functional group such as a phenolic hydroxyl group, a carboxyl group, a sulfonyl group, or a phosphate group.
- Porous carbon materials possessing surface functional groups include woody materials such as palm, bamboo, pine, walnut, and bagasse, coke, and phenol at high temperatures using gases such as water vapor, carbon dioxide, and air.
- activated carbon prepared by a chemical method such as a chemical method of treating at a high temperature using a chemical such as alkali or zinc chloride. Specifically, an alkali activated porous carbon material or the like can be used.
- transition metal for example, one selected from the group consisting of ruthenium, platinum, rhodium, palladium, iridium, nickel, cobalt, iron, copper, silver and gold may be used alone or in combination of two or more. Also good. From the viewpoint of high catalytic activity, those selected from the platinum group metals of ruthenium, platinum, rhodium, palladium and iridium are preferred, and from the viewpoint of high cellulose conversion and glucose selectivity, they are selected from ruthenium, platinum, palladium and rhodium. Those are particularly preferred.
- Cellulose which is the main component of polysaccharides contained in plant biomass, exhibits crystallinity by binding two or more cellulose molecules by hydrogen bonding.
- cellulose having such crystallinity can be used as a raw material, but cellulose having crystallinity lowered by a treatment for lowering crystallinity can also be used.
- Cellulose with reduced crystallinity can be partially reduced in crystallinity, or completely or almost completely lost.
- the type of the crystallinity reduction treatment is not particularly limited, but is preferably a crystallinity reduction treatment that can break the hydrogen bond and at least partially generate a single-chain cellulose molecule.
- Examples of a method for breaking hydrogen bonds between cellulose molecules include pulverization.
- the pulverizing means is not particularly limited as long as it has a function capable of being pulverized.
- the system of the apparatus may be either dry or wet, and the grinding system of the apparatus may be either batch or continuous.
- pulverization force such as an impact, compression, shear, friction, can be used.
- Specific devices include rolling ball mills such as pot mills, tube mills, and conical mills, vibration ball mills such as circular vibration type vibration mills, swivel type vibration mills, and centrifugal mills, stirring tank mills, annular mills, flow type mills, and tower type grinding.
- rolling ball mills such as pot mills, tube mills, and conical mills
- vibration ball mills such as circular vibration type vibration mills, swivel type vibration mills, and centrifugal mills
- stirring tank mills such as annular mills, flow type mills, and tower type grinding.
- Agitator mills swirl type jet mills, impingement type jet mills, fluidized bed type jet mills, wet type jet mills, etc., jet mills, roughing machines (crushers), shear mills such as ong mills, mortars, Impact type crushers such as colloid mills such as stone mills, hammer mills, cage mills, pin mills, disintegrators, screen mills, turbo type mills, centrifugal classification mills, and planetary pulverizers that employ rotation and revolving motions. Examples include a ball mill.
- the pulverizer is preferably a rolling ball mill, a vibrating ball mill, a stirring mill, or a planetary ball mill, which is used for pretreatment for reducing the crystallinity of the substrate, and is classified as a pot mill or stirring mill classified as a rolling ball mill.
- the ratio of the solid catalyst and the solid substrate to be simultaneously pulverized is not particularly limited, but from the viewpoint of hydrolysis efficiency during the reaction, reduction of the substrate residue after the reaction, and recovery rate of the produced sugar,
- the mass ratio is preferably 1: 100 to 1: 1, and more preferably 1:10 to 1: 1.
- the raw material obtained by individually pulverizing the substrate and the raw material obtained by simultaneously pulverizing the substrate and the catalyst were both determined as the average particle size after pulverization (cumulative median diameter (median diameter): 100% of the total volume of the powder group. From the viewpoint of improving the reactivity, the particle diameter at the point where the cumulative curve becomes 50% is preferably 1 to 100 ⁇ m, more preferably 1 to 30 ⁇ m.
- a rougher such as a shredder, jaw crusher, gyratory crusher, cone crusher, hammer crusher, roll crusher, roll mill, etc.
- Preliminary pulverization can be performed using a pulverizer and a medium pulverizer such as a stamp mill, an edge runner, a cutting / shearing mill, a rod mill, an autogenous pulverizer, and a roller mill.
- a medium pulverizer such as a stamp mill, an edge runner, a cutting / shearing mill, a rod mill, an autogenous pulverizer, and a roller mill.
- the processing time of a raw material will not be limited if the raw material after a process is pulverized uniformly.
- the hydroxide ions in the reaction solution are generally derived from the alkali chemicals used for the pretreatment of the hydrolysis reaction of the plant biomass that is the raw material.
- the equivalent concentration of hydroxide ions in the reaction solution can be determined from the measured pH by the following formula.
- the cation in the reaction solution refers to alkali metal ions and alkaline earths derived from plant biomass and solid catalyst as raw materials and / or derived from alkali chemicals used for the pretreatment of the hydrolysis reaction.
- the equivalent concentration of cations in the reaction solution is determined by ion chromatography analysis, indophenol blue absorptiometry, ICP (inductively coupled plasma), EPMA (electron beam microanalyzer), ESCA (X-ray photoelectron spectrometer), SIMS (secondary ion). Mass spectrometry), atomic absorption method and the like can be obtained by summing up the results. It is preferable to use ion chromatographic analysis from the viewpoint that the main cations in the reaction solution can be directly measured with high sensitivity.
- Acids include inorganic mineral acids such as hydrochloric acid, sulfuric acid, nitric acid and phosphoric acid, organic carboxylic acids such as acetic acid, formic acid, phthalic acid, lactic acid, malic acid, fumaric acid, citric acid and succinic acid, methanesulfonic acid and ethanesulfone Acid, organic sulfonic acid such as benzenesulfonic acid, toluenesulfonic acid and the like can be used alone or in combination of two or more.
- inorganic mineral acids such as hydrochloric acid, sulfuric acid, nitric acid and phosphoric acid
- organic carboxylic acids such as acetic acid, formic acid, phthalic acid, lactic acid, malic acid, fumaric acid, citric acid and succinic acid, methanesulfonic acid and ethanesulfone Acid
- organic sulfonic acid such as benzenesulfonic acid, toluen
- inorganic mineral acids are preferable, and sulfuric acid, hydrochloric acid, and nitric acid are more preferable from the viewpoint that the acid itself is not easily decomposed and denatured during hydrothermal treatment, and the inhibitory property when using the target product sugar is low. .
- the lower limit value of the acid concentration can be set from the viewpoint of recovering the glucose saccharification rate to a higher level, and the upper limit value can be set from the viewpoint of suppressing the excessive decomposition of glucose and suppressing the corrosiveness by the acid.
- the acid is preferably present in the reaction solution at an equivalent concentration in the range of 30 to 1000% of the equivalent concentration of the cation in the reaction solution, more preferably in the range of 50 to 500%. More preferably, an equivalent concentration in the range of ⁇ 300% is present. Therefore, when hydroxide ions are present in the reaction solution before the acid is added, it is necessary to add an acid having a total equivalent concentration obtained by adding the equivalent concentration of hydroxide ions to the equivalent concentration in the above range. This is because the presence of hydroxide ions consumes the acid by neutralization.
- Hydrolysis using plant biomass as a substrate is performed by hydrothermal treatment.
- the hydrothermal treatment is carried out by heating the substrate in the presence of water, preferably adding a solid catalyst, and heating at a temperature at which the substrate is pressurized.
- the temperature for heating to be in a pressurized state is suitably in the range of 110 to 380 ° C., from the viewpoint of rapidly hydrolyzing cellulose and suppressing the conversion of the product glucose to other sugars.
- a relatively high temperature is preferable, for example, a temperature in the range of 170 to 320 ° C., more preferably 180 to 300 ° C. is appropriate.
- the hydrothermal treatment in the hydrolysis method of the present invention is usually carried out in a closed container such as an autoclave, even if the reaction is started at normal pressure, if the reaction system is heated at the above temperature, Become. Further, the reaction can be carried out by pressurizing the inside of the sealed container before or during the reaction.
- the pressure to be applied is, for example, 0.1 to 30 MPa, preferably 1 to 20 MPa, and more preferably 2 to 10 MPa.
- the reaction can also be carried out by heating and pressurizing the reaction solution with a high-pressure pump.
- the amount of water for hydrolysis is an amount capable of hydrolyzing at least cellulose and hemicellulose in the plant biomass, and considering the fluidity and agitation of the reaction mixture,
- the mass ratio is preferably in the range of 1 to 500, more preferably in the range of 2 to 200.
- the atmosphere for the hydrolysis is not particularly limited. Industrially, it is preferably performed in an air atmosphere, but may be performed in an atmosphere of a gas other than air, for example, oxygen, nitrogen, hydrogen, or a mixture thereof.
- the heating of the hydrothermal treatment is terminated when the conversion rate by hydrolysis of cellulose is between 10 and 100% and the selectivity of glucose is between 20 and 80%. Is preferable.
- the heating time is in the range of 5 to 60 minutes, preferably 5 to 30 minutes from the start of heating for the hydrolysis reaction under normal conditions, but is not limited to this range.
- the heating for hydrolysis is such that the conversion by hydrolysis of cellulose is preferably in the range of 30 to 100%, more preferably in the range of 40 to 100%, still more preferably in the range of 50 to 100%, most preferably. Is in the range of 55-100% and ends when the glucose selectivity is preferably in the range of 25-80%, more preferably in the range of 30-80%, most preferably in the range of 40-80%. Is appropriate.
- the form of the hydrolysis reaction may be either a batch type or a continuous type.
- the reaction is preferably carried out while stirring the reaction mixture.
- it is possible to produce a sugar-containing liquid containing glucose as a main component and containing a small amount of a hyperdegradation product such as 5-hydroxymethylfurfural by a hydrolysis reaction at a relatively high temperature for a relatively short time.
- the reaction solution After completion of heating, it is preferable to cool the reaction solution from the viewpoint of suppressing the conversion of glucose to other sugars and increasing the glucose yield. From the viewpoint of increasing the glucose yield, the reaction solution is preferably cooled under the condition that the selectivity of glucose is maintained in the range of 20 to 80%, more preferably in the range of 25 to 80%, and 30 to 80%. Is more preferable, and the range of 40 to 80% is most preferable.
- the reaction solution is preferably cooled as quickly as possible to a temperature at which the conversion of glucose into other sugars does not occur, for example, at a rate in the range of 1 to 200 ° C./min.
- the rate is preferably in the range of 5 to 150 ° C./min.
- the temperature at which the conversion of glucose into other sugars does not occur is, for example, 150 ° C. or lower, preferably 110 ° C. or lower. That is, the reaction solution is suitably cooled to a temperature of 150 ° C. or lower at a rate of 1 to 200 ° C./min, preferably 5 to 150 ° C./min.
- the obtained reaction liquid can be separated and recovered into a liquid phase containing glucose and a solid phase containing a solid catalyst and an unreacted substrate by solid-liquid separation treatment.
- a centrifuge, a centrifugal filter, a pressure filter, a Nutsche filter, or a filter press can be used for solid-liquid separation, but it is limited as long as the liquid phase and the solid phase can be separated. It is not something.
- Coke (coal coke, manufactured by Showa Denko KK) was heat-treated at 700 ° C and finely pulverized with a jet mill, and then potassium hydroxide was added and heat-treated again at 700 ° C to activate.
- the obtained activated coke is washed with water, neutralized with hydrochloric acid, boiled with hot water, dried, and sieved to obtain an alkali-activated porous carbon material having a particle size of 1 ⁇ m to 30 ⁇ m (median diameter 13 ⁇ m). (Hereinafter referred to as a carbon catalyst).
- centrifugal filter manufactured by Kokusan Co., Ltd. 1000 g of water-containing solid content (water content 70%, dry product equivalent 300 g) from which the supernatant was removed by H-110A) was recovered.
- Washing pretreatment bagasse 10.00 g as a solid substrate and carbon catalyst 1.54 g (substrate to catalyst mass ratio 6.5: 1.0) were placed in an alumina sphere having a diameter of 1.5 cm in a ceramic pot mill with a capacity of 3600 mL. Added with 2000 g.
- This ceramic pot mill was set on a desktop pot mill rotary table (manufactured by Nissho Scientific Co., Ltd., desktop pot mill model ANZ-51S), and ball milled at 60 rpm for 48 hours to mix and grind simultaneously.
- the obtained raw material is referred to as a cleaning mixed pulverized raw material.
- the reaction solution After cooling, the reaction solution is separated into a liquid and a solid by a centrifugal separator, and the product in the liquid phase is a high performance liquid chromatograph (device: Shodex high performance liquid chromatography manufactured by Showa Denko KK, column: Shodex (registered trademark)).
- KS801 mobile phase: water 0.6 mL / min, 75 ° C., detection: differential refractive index).
- the cellulose conversion rate was calculated
- PH measurement The pH was measured at 25 ° C. in a glass bottle using a pH meter D-51 (manufactured by Horiba, Ltd.) calibrated at three points using Horiba Seisakusho pH STANDARD 100-4, 100-7, and 100-9. After immersing the glass electrode of the device in the solution, the mixture was lightly stirred and then allowed to stand and measured until it became stable (about 1 minute).
- Reference Example 1, Example 1 and Comparative Examples 1 to 2 Solid catalyst addition desensitized individually pulverized starting material 0.324g by reaction inhibition and acid by the hydroxide ions and cations in the reaction (C 6 H 10 O 5 units 2. 00 mmol) and 0.050 g of a solid catalyst, 40 mL of an aqueous dispersion prepared by adjusting the inhibitor (NaOH) and acid (H 2 SO 4 ) shown in Table 1 so as to have the equivalent concentration shown in Table 1. The mixture was placed in a high-pressure reactor (internal volume: 100 mL, autoclave manufactured by OM Labtech Co., Ltd., Hastelloy (registered trademark) C22), and heated from room temperature to a reaction temperature of 200 ° C.
- a high-pressure reactor internal volume: 100 mL, autoclave manufactured by OM Labtech Co., Ltd., Hastelloy (registered trademark) C22
- Example 1 in which the amount of sulfuric acid minus the neutralization content is 90% of the relative ratio to the equivalent concentration of the cation exhibits the effect of releasing the inhibition and recovers to the same level of saccharification results as in Reference Example 1. (Fig. 1).
- the reaction inhibition factor is eliminated by a simple method in which an acid is allowed to coexist depending on the equivalent concentration of hydroxide ions and cations in the reaction solution. Yields can be obtained.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Emergency Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Organic Chemistry (AREA)
- Processing Of Solid Wastes (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description
しかしながら、これらの特許文献では、原料に純品のセルロースを用いた実施例しか記載されておらず、実バイオマスを処理する場合の不純物による反応阻害の影響やその解除法については言及していない。
実バイオマス原料を水熱処理により糖化する場合、実バイオマスに含有されているヘミセルロース、リグニン、灰分などの非セルロース成分のために引き起こされる糖化効率の低減や得られる糖液純度の低下を改善するために、脱リグニン剤などの薬剤を使用した前処理が行われ、前処理物は固液分離され、その不溶残渣が水熱処理に供される。
以上のように、植物性バイオマスの水熱反応による加水分解反応においては、共存する不純物による反応阻害を解除した、高いグルコース収率が得られるセルロースの糖化方法の確立が求められている。
[1]植物性バイオマスの加水分解反応液中のカチオンの当量濃度の30~1000%の当量濃度と水酸化物イオンの当量濃度の合計値の当量濃度の酸を反応液に加えて水熱処理することを特徴とする植物性バイオマスの加水分解方法。
[2]前記水熱処理に固体触媒を用いる前項1に記載の植物性バイオマスの加水分解方法。
[3]酸が、無機鉱酸、有機カルボン酸、及び有機スルホン酸から選択される少なくとも1種である前項1または2に記載の植物性バイオマスの加水分解方法
[4]反応液中のカチオンが、アルカリ金属イオン、アルカリ土類金属イオン、アンモニウムイオンのうちの少なくとも1種である前項1~3のいずれかに記載の植物性バイオマスの加水分解方法。
[5]固体触媒が炭素材料である前項2~4のいずれかに記載の植物性バイオマスの加水分解方法。
[6]植物性バイオマスがセルロースである前項1~5のいずれかに記載の植物性バイオマスの加水分解方法。
[7]酸を添加して反応液を中和した後、反応液に含有するカチオンの当量濃度の30~1000%の当量濃度になるように酸を添加して水熱処理する前項1~6のいずれかに記載の植物性バイオマスの加水分解方法。
[8]前項1~7のいずれかに記載の植物性バイオマスの加水分解方法を用いることを特徴とするグルコースの製造方法。
本発明の植物性バイオマスの加水分解の方法は、反応液中に特定量の酸を共存させることにより水酸化物イオン及びカチオンの反応阻害を解除することを特徴とする。
「バイオマス」とは一般的には「再生可能な生物由来の有機性資源で化石資源を除いたもの」を指す。本発明において、「植物性バイオマス」とは、例えば稲わら、麦わら、サトウキビ葉、籾殻、バガス、広葉樹、竹、針葉樹、ケナフ、家具廃木材、建築廃木材、古紙、食品残渣等の主にセルロースやヘミセルロースを含むバイオマスを云い、本発明では植物性バイオマスを加水分解反応の固体基質として用いる。
植物性バイオマスの形態は、乾体でも湿体でもかまわず、結晶性でも非結晶性でもかまわない。植物性バイオマスの粒径は、粉砕処理ができる大きさであれば限定されないが、粉砕効率の観点から、20μm以上数1000μm以下であることが好ましい。
本発明の水熱処理による加水分解方法においては、固体触媒を用いることもできる。固体触媒は、植物系バイオマス多糖類を加水分解できる触媒であれば特に限定されるものではなく、例えば、主成分であるセルロースを形成しているグルコース間のβ-1,4グリコシド結合に代表されるような、グリコシド結合を加水分解する活性を有することが好ましい。
固体触媒としては、例えば炭素材料、遷移金属などを、単独でまたは2種類以上を併用して用いることができる。
植物系バイオマスに含まれる多糖類の主成分であるセルロースは、2本またはそれ以上のセルロース分子が水素結合により結合して結晶性を示す。本発明では、そのような結晶性を有するセルロースを原料として使用することもできるが、結晶性低下のための処理を施して結晶性を低下させたセルロースも用いることができる。結晶性を低下させたセルロースは、結晶性を部分的に低下させたものでも、完全に、またほぼ完全に消失したものであることもできる。結晶性低下処理の種類には特に制限はないが、上記水素結合を切断して、1本鎖のセルロース分子を少なくとも部分的に生成できる結晶性低下処理であることが好ましい。少なくとも部分的に1本鎖のセルロース分子を含むセルロースを原料とすることにより加水分解の効率を大幅に向上することができる。
同時粉砕処理は、混合に加え、基質の結晶性を低下させる前処理を兼ねることができる。その観点から、粉砕装置は、基質の結晶性を低下させる前処理に用いられる、転動ボールミル、振動ボールミル、撹拌ミル、遊星ボールミルが好ましく、転動ボールミルに分類されるポットミル、撹拌ミルに分類される撹拌槽ミル、遊星ボールミルがより好ましい。さらに、固体触媒と固体基質との同時粉砕処理された原料の嵩密度が大きい方が反応性が高い傾向が認められることから、固体触媒の粉砕物と固体基質の粉砕物とが食い込むような圧縮力が強く加わる転動ボールミル、撹拌ミル、遊星ボールミルを用いることがより好ましい。
処理する原料の粒径が大きい場合などは、粉砕を効率的に行うために、粉砕の前に、例えば、シュレッダー、ジョークラッシャー、ジャイレトリクラッシャー、コーンクラッシャー、ハンマークラッシャー、ロールクラッシャー、ロールミルなどの粗粉砕機、並びにスタンプミル、エッジランナ、切断・せん断ミル、ロッドミル、自生粉砕機、ローラミルなどの中粉砕機を用いて、予備的な粉砕処理を実施することができる。原料の処理時間は、処理後原料が均一に微粉化されるのであれば限定されるものではない。
本発明は、反応液中に水酸化物イオンとカチオンが共存すると、植物性バイオマスの加水分解が阻害され、転化率、グルコース糖化率が低下するという発見、及びその阻害が水酸化物イオンとカチオンの含有当量濃度に応じて一定量の酸を添加することにより解除できるという発見に基づくものである。
本発明における、反応液中の水酸化物イオンは、一般的には、原料である植物性バイオマスの加水分解反応の前処理に用いたアルカリ薬剤などに由来するものである。
反応液中の水酸化物イオンの当量濃度は、測定したpHより、以下の式で求められる。
酸としては、塩酸、硫酸、硝酸、リン酸などの無機鉱酸、酢酸、蟻酸、フタル酸、乳酸、リンゴ酸、フマル酸、クエン酸、コハク酸などの有機カルボン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、トルエンスルホン酸などの有機スルホン酸等を、単独または2種類以上併用して用いることができる。これらの中でも、水熱処理時に酸そのものが分解変質されにくい点及び目的生成物である糖を利用する際の阻害性が低いという点から、無機鉱酸が好ましく、硫酸、塩酸、及び硝酸がより好ましい。
植物性バイオマスを基質とする加水分解は水熱処理により行う。水熱処理は、基質を水の存在下、好ましくは固体触媒を添加し、加圧状態となる温度で加熱して行う。加圧状態となる加熱の温度は、110~380℃の範囲が適当であり、セルロースの加水分解を迅速に行い、かつ生成物であるグルコースの他の糖への転化を抑制するという観点から、比較的高い温度が好ましく、例えば、170~320℃、より好ましくは180~300℃の範囲とすることが適当である。
本発明においては、比較的高温で比較的短時間の加水分解反応により、グルコースを主成分とし、5-ヒドロキシメチルフルフラールなどの過分解物が少ない糖含有液を製造することができる。
得られた反応液は、固液分離処理によりグルコースを含む液相と固体触媒と未反応基質を含む固相に分離し回収することができる。固液分離には、例えば、遠心分離機、遠心ろ過機、加圧ろ過機、ヌッチェろ過機、フィルタープレスなどの装置を用いることができるが、液相と固相を分離できるのであれば限定されるものではない。
コークス(石炭コークス、昭和電工株式会社製)を700℃で加熱処理し、ジェットミルにて微粉砕した後、水酸化カリウムを添加し再度700℃で加熱処理して賦活化した。得られた賦活化コークスを、水洗後、塩酸で中和し、さらに熱水で煮沸した後、乾燥したものを篩分し、粒径1μm以上30μm以下のアルカリ賦活多孔質炭素材料(メジアン径13μm)(以下、炭素触媒という。)を得た。
各実施例及び比較例では、試薬グレードの固体基質としてアビセル(Avicel,Merck社製結晶性微粉セルロース)を個別粉砕したものを用い、また実バイオマスグレードの固体基質として後述の方法で前処理したバガスを固体触媒と混合粉砕したものを用いた。
固体基質としてのアビセル3.00gを、容量500mLのセラミックポットミルの中に直径1.5cmのジルコニア球300gと共に入れた。このセラミックポットミルを卓上ポットミル回転台((株)入江商会製,卓上ポットミル型式V-1M)にセットし、60rpmで48時間ボールミル処理して粉砕した。得られた原料を、以下、個別粉砕原料という。
高圧反応器(内容積10L,オーエムラボテック社デスクトップリアクターOML-10,SUS316製、ヘリカル翼撹拌付き)に、ロータリースピードミル(フリッチュ・ジャパン製、篩リング0.12mm)で粗粉砕した乾燥バガス(セルロース含有率51%、ヘミセルロース含有率23%)430g、及び水5Lを加え、600rpmで撹拌しながら、温度200℃、9分間の加熱処理を行い、冷却後、遠心ろ過機((株)コクサン製、H-110A)により上澄み液を除去した含水固形分(含水率70%、乾燥品換算300g)1000gを回収した。
精製バガス中のセルロース含有率はNREL(米国・国立再生可能エネルギー研究所)の分析方法(Technical Report NREL/TP-510-42618)により求めた結果、64%であった。
固体基質としての未洗浄前処理バガス10.00gと、炭素触媒1.54g(基質と触媒の質量比6.5:1.0)を、容量3600mLのセラミックポットミルの中に直径1.5cmのアルミナ球2000gと共に入れた。このセラミックポットミルを卓上ポットミル回転台(日陶科学(株)製,卓上ポットミル型式ANZ-51S)にセットし、60rpmで48時間ボールミル処理して混合と同時に粉砕した。得られた原料を、以下、未洗浄混合粉砕原料という。
セルロースの加水分解反応は、各実施例または比較例に記載した通り調整した原料を、高圧反応器(内容積100mL,日東高圧社製オートクレーブ,SUS316製)にセットした後、600rpmで撹拌しながら室温から検討する反応温度(200℃~240℃)まで約20分で加熱して行った。反応温度に到達すると同時に加熱を止め、反応器を水槽に入れ冷却した。冷却後、反応液を遠心分離装置により液体と固体に分離し、液相の生成物は、高速液体クロマトグラフ(装置:昭和電工(株)製Shodex高速液体クロマトグラフィー,カラム:Shodex(登録商標)KS801,移動相:水0.6mL/min,75℃,検出:示差屈折率)により定量分析した。また、水洗した固体残渣を110℃で24時間乾燥した後、未反応セルロースの質量からセルロース転化率を求めた。

pHは、堀場製作所pH STANDARD100-4、100-7、及び100-9を用いて3点校正したpH計D-51((株)堀場製作所製)を用いて、ガラス瓶に入れた25℃の試料溶液に、機器のガラス電極を浸した後、軽く撹拌してから静置し安定するまで(1分程度)待ち計測した。
反応液に含有されているカチオンの当量濃度は、反応液を遠心分離装置により固液分離した上清をイオンクロマトグラフ(装置:昭和電工(株)製Shodex高速液体クロマトグラフィー,カラム:Shodex(登録商標)IC IK-421、移動相:酒石酸0.75g/L、ホウ酸1.5g/L、2-6ピリジンカルボン酸0.267g/L、1mL/min,40℃,検出:電気伝導度)によりNa+、K+、Mg2+、Ca2+、NH4 +を定量分析して求めた。
個別粉砕原料0.324g(C6H10O5単位で2.00mmol)と固体触媒0.050gを用いて、表1に記載の阻害剤(NaOH)及び酸(H2SO4)を、表1に記載の当量濃度になるように調整した水分散液40mLを、高圧反応器(内容積100mL,オーエムラボテック(株)製オートクレーブ,ハステロイ(登録商標)C22製)に入れた後、600rpmで撹拌しながら室温から反応温度200℃まで約15分で加熱した。反応温度に到達すると同時に加熱を止め、反応器を風冷した。冷却開始から150℃に到達するまでの時間は3分であった。冷却後、反応液を遠心分離装置により液体と固体に分離し、液相の生成物は、高速液体クロマトグラフ(装置:昭和電工(株)製Shodex高速液体クロマトグラフィー,カラム:Shodex(登録商標)KS801,移動相:水0.6mL/min,75℃,検出:示差屈折率)によりグルコース、その他糖類及び過分解物を定量分析した。また、固体残渣を110℃で24時間乾燥したものを未反応セルロースと炭素触媒とし、その質量からセルロース転化率を求めた。
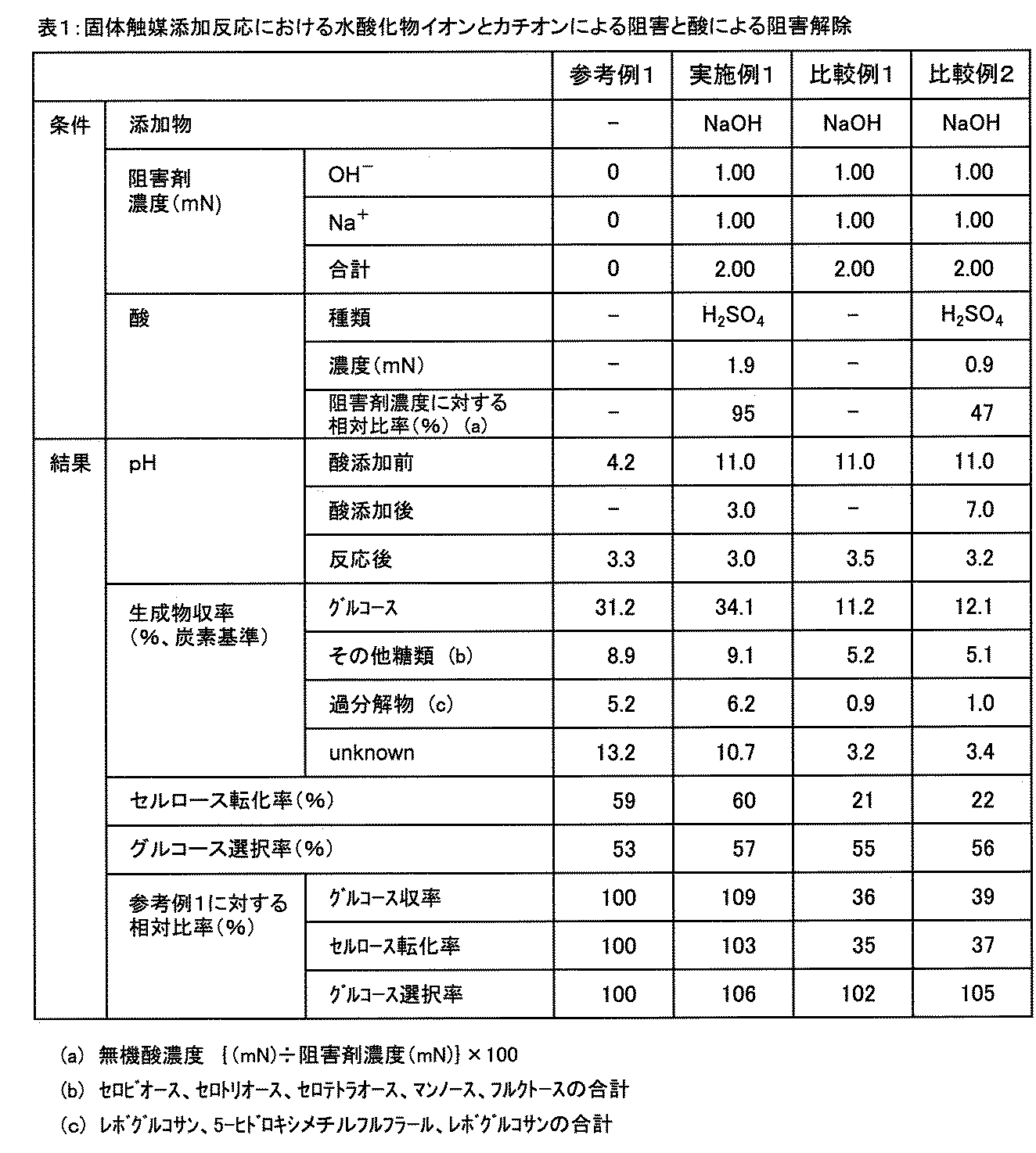
表2に記載の混合粉砕原料0.374g(C6H10O5単位で2.00mmol)を用いて、硫酸を表2に記載の当量濃度になるように調整した水分散液40mLを、高圧反応器(内容積100mL,オーエムラボテック(株)製オートクレーブ,ハステロイC22製)に入れた後、600rpmで撹拌しながら室温から反応温度200℃まで約15分で加熱した。反応温度に到達すると同時に加熱を止め、反応器を風冷した。冷却開始から150℃に到達するまでの時間は3分であった。冷却後、反応液を遠心分離装置により液体と固体に分離し、液相の生成物は、高速液体クロマトグラフ(装置:昭和電工(株)製Shodex高速液体クロマトグラフィー,カラム:Shodex(登録商標)KS801,移動相:水0.6mL/min,75℃,検出:示差屈折率)によりグルコース、その他糖類及び過分解物を定量分析した。反応液のカチオン当量濃度と酸添加前pHは、事前に粉砕原料を反応液と同じ組成に水分散した液を作製して、イオンクロマト及びpH計により分析した。
未洗浄混合粉砕原料を用い、反応液の水酸化物イオン濃度及びカチオンの合計当量濃度に対し115%(対カチオン分としては119%)である33.0mNになるように硫酸を添加した実施例2は、グルコース収率は33.4%で、参考例2に対する相対比で102%となり、阻害は完全に解除された。これは実施例1と同様、水酸化物イオンとカチオン両方の阻害が解除された結果と言える(図2)。

Claims (8)
- 植物性バイオマスの加水分解反応液中のカチオンの当量濃度の30~1000%の当量濃度と水酸化物イオンの当量濃度の合計値の当量濃度の酸を反応液に加えて水熱処理することを特徴とする植物性バイオマスの加水分解方法。
- 前記水熱処理に固体触媒を用いる請求項1に記載の植物性バイオマスの加水分解方法。
- 酸が、無機鉱酸、有機カルボン酸、及び有機スルホン酸から選択される少なくとも1種である請求項1または2に記載の植物性バイオマスの加水分解方法。
- 反応液中のカチオンが、アルカリ金属イオン、アルカリ土類金属イオン、アンモニウムイオンのうちの少なくとも1種である請求項1~3のいずれかに記載の植物性バイオマスの加水分解方法。
- 固体触媒が炭素材料である請求項2~4のいずれかに記載の植物性バイオマスの加水分解方法。
- 植物性バイオマスがセルロースである請求項1~5のいずれかに記載の植物性バイオマスの加水分解方法。
- 酸を添加して反応液を中和した後、反応液に含有されているカチオンの当量濃度の30~1000%の当量濃度になるように酸を添加して水熱処理する請求項1~6のいずれかに記載の植物性バイオマスの加水分解方法。
- 請求項1~7のいずれかに記載の植物性バイオマスの加水分解方法を用いることを特徴とするグルコースの製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/652,980 US20150337401A1 (en) | 2012-12-18 | 2013-11-19 | Plant-biomass hydrolysis method |
| BR112015014153A BR112015014153A2 (pt) | 2012-12-18 | 2013-11-19 | método de hidrólise de biomassa de planta |
| JP2014553028A JPWO2014097799A1 (ja) | 2012-12-18 | 2013-11-19 | 植物性バイオマスの加水分解方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-275514 | 2012-12-18 | ||
| JP2012275514 | 2012-12-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014097799A1 true WO2014097799A1 (ja) | 2014-06-26 |
Family
ID=50978142
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/081181 WO2014097799A1 (ja) | 2012-12-18 | 2013-11-19 | 植物性バイオマスの加水分解方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20150337401A1 (ja) |
| JP (1) | JPWO2014097799A1 (ja) |
| BR (1) | BR112015014153A2 (ja) |
| WO (1) | WO2014097799A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111747410A (zh) * | 2019-03-26 | 2020-10-09 | 天津科技大学 | 一种水热法制备汉麻秆芯基炭材料的方法 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10327900A (ja) * | 1997-06-02 | 1998-12-15 | Agency Of Ind Science & Technol | 水溶性オリゴ糖類及び単糖類の製造方法 |
| JP2009201405A (ja) * | 2008-02-27 | 2009-09-10 | Kochi Univ | グルコースの製造方法およびスルホン化活性炭の製造方法 |
| WO2011036955A1 (ja) * | 2009-09-25 | 2011-03-31 | 国立大学法人北海道大学 | セルロースまたはヘミセルロースの加水分解用触媒、並びにこの触媒を用いる糖含有液の製造方法 |
| JP2011142894A (ja) * | 2010-01-18 | 2011-07-28 | Ihi Corp | バイオマス処理装置 |
| JP2011206044A (ja) * | 2009-09-30 | 2011-10-20 | Sekisui Chem Co Ltd | セルロース糖化方法 |
| JP2011217634A (ja) * | 2010-04-06 | 2011-11-04 | Toyota Motor Corp | 植物バイオマスの処理方法、植物バイオマスからの糖の製造方法、植物バイオマスからのアルコール及び/又は有機酸の製造方法 |
| JP2012000022A (ja) * | 2010-06-15 | 2012-01-05 | Tsukishima Kikai Co Ltd | バイオマスの処理装置及び処理方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7503981B2 (en) * | 2004-12-02 | 2009-03-17 | The Trustees Of Dartmouth College | Removal of minerals from cellulosic biomass |
| WO2007100052A1 (ja) * | 2006-03-01 | 2007-09-07 | National University Corporation Hokkaido University | セルロースの加水分解および/または加水分解物の還元用触媒およびセルロースから糖アルコールの製造方法 |
| JP4765073B2 (ja) * | 2006-07-05 | 2011-09-07 | 国立大学法人広島大学 | リグノセルロースの水熱加水分解方法 |
| JP2011103874A (ja) * | 2009-10-22 | 2011-06-02 | Idemitsu Kosan Co Ltd | バイオマスの処理方法 |
| JP2011101608A (ja) * | 2009-11-10 | 2011-05-26 | Ihi Corp | バイオマス処理システム及び方法 |
| CN102947421B (zh) * | 2010-04-07 | 2015-02-25 | 莱斯拉有限公司 | 生物燃料的生产方法 |
| US9109049B2 (en) * | 2012-06-21 | 2015-08-18 | Iowa State University Research Foundation, Inc. | Method for pretreating lignocellulosic biomass |
-
2013
- 2013-11-19 US US14/652,980 patent/US20150337401A1/en not_active Abandoned
- 2013-11-19 WO PCT/JP2013/081181 patent/WO2014097799A1/ja active Application Filing
- 2013-11-19 JP JP2014553028A patent/JPWO2014097799A1/ja active Pending
- 2013-11-19 BR BR112015014153A patent/BR112015014153A2/pt not_active IP Right Cessation
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH10327900A (ja) * | 1997-06-02 | 1998-12-15 | Agency Of Ind Science & Technol | 水溶性オリゴ糖類及び単糖類の製造方法 |
| JP2009201405A (ja) * | 2008-02-27 | 2009-09-10 | Kochi Univ | グルコースの製造方法およびスルホン化活性炭の製造方法 |
| WO2011036955A1 (ja) * | 2009-09-25 | 2011-03-31 | 国立大学法人北海道大学 | セルロースまたはヘミセルロースの加水分解用触媒、並びにこの触媒を用いる糖含有液の製造方法 |
| JP2011206044A (ja) * | 2009-09-30 | 2011-10-20 | Sekisui Chem Co Ltd | セルロース糖化方法 |
| JP2011142894A (ja) * | 2010-01-18 | 2011-07-28 | Ihi Corp | バイオマス処理装置 |
| JP2011217634A (ja) * | 2010-04-06 | 2011-11-04 | Toyota Motor Corp | 植物バイオマスの処理方法、植物バイオマスからの糖の製造方法、植物バイオマスからのアルコール及び/又は有機酸の製造方法 |
| JP2012000022A (ja) * | 2010-06-15 | 2012-01-05 | Tsukishima Kikai Co Ltd | バイオマスの処理装置及び処理方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112015014153A2 (pt) | 2017-07-11 |
| JPWO2014097799A1 (ja) | 2017-01-12 |
| US20150337401A1 (en) | 2015-11-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6779505B2 (ja) | セロオリゴ糖の製造方法 | |
| Yu et al. | Characterization of mechanical pulverization/phosphoric acid pretreatment of corn stover for enzymatic hydrolysis | |
| JP5943489B2 (ja) | 植物性バイオマスの加水分解反応用原料の前処理方法及び植物性バイオマスの糖化方法 | |
| WO2009084492A1 (en) | Process for producing saccharide | |
| Zhao et al. | Extracting xylooligosaccharides in wheat bran by screening and cellulase assisted enzymatic hydrolysis | |
| Sambusiti et al. | One-pot dry chemo-mechanical deconstruction for bioethanol production from sugarcane bagasse | |
| WO2014097801A1 (ja) | 植物性バイオマスの加水分解方法 | |
| WO2014097800A1 (ja) | 植物性バイオマスの加水分解方法 | |
| JP2015157792A (ja) | リグニン分解物の製造方法 | |
| WO2013136940A1 (ja) | 糖の製造方法 | |
| WO2014007295A1 (ja) | 植物性バイオマスの分解方法及びグルコースの製造方法 | |
| JP2012029567A (ja) | 木質系バイオマスの糖化方法 | |
| WO2014097799A1 (ja) | 植物性バイオマスの加水分解方法 | |
| JPWO2009004950A1 (ja) | セルロースを含む材料の加水分解による単糖類および/または水溶性多糖類の製造方法 | |
| WO2022210558A1 (ja) | ペレットおよびペレットの製造方法 | |
| CN115109102A (zh) | 含纤维寡糖的组合物的制造方法和含纤维寡糖的组合物 | |
| WO2013114962A1 (ja) | バイオエタノールの製造方法及び製造システム | |
| JP6431756B2 (ja) | バイオマスの成分分離方法 | |
| JP7191313B2 (ja) | セロオリゴ糖の製造方法 | |
| JPS63137693A (ja) | 木材の粉砕方法 | |
| CN116376997A (zh) | 利用纤维素制备葡萄糖的方法 | |
| US20160177357A1 (en) | Method for pretreating cellulose-containing biomass, method for producing biomass composition for saccharification use, and method for producing sugar | |
| WO2014109345A1 (ja) | 糖化用バイオマス組成物、糖化用バイオマス組成物の選定方法、及び糖の製造方法 | |
| JP2012005359A (ja) | 単糖類、二糖類、及び/又はオリゴ糖の製造方法 | |
| JP2013192472A (ja) | 糖の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13865352 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014553028 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14652980 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112015014153 Country of ref document: BR |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13865352 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 112015014153 Country of ref document: BR Kind code of ref document: A2 Effective date: 20150616 |