WO2014021329A1 - カワハギ由来の細胞株 - Google Patents
カワハギ由来の細胞株 Download PDFInfo
- Publication number
- WO2014021329A1 WO2014021329A1 PCT/JP2013/070627 JP2013070627W WO2014021329A1 WO 2014021329 A1 WO2014021329 A1 WO 2014021329A1 JP 2013070627 W JP2013070627 W JP 2013070627W WO 2014021329 A1 WO2014021329 A1 WO 2014021329A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cells
- cell
- cell line
- fish
- present
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images























Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2510/00—Genetically modified cells
- C12N2510/04—Immortalised cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2517/00—Cells related to new breeds of animals
- C12N2517/02—Cells from transgenic animals
Definitions
- the present invention relates to a cell line derived from a living part of Monacanthidae fish, which exhibits a fibroblast-like morphology, or a substrain thereof.
- Fish cells derived from fish have many properties similar to animal cells derived from mammals.
- fish cells can generally be cultured at a relatively low temperature, and the temperature range in which they can grow is wide. Therefore, fish cells are regarded as promising as cultured cells in industrial applications and research applications such as production of biological substances as an alternative to animal cells.
- Known cultured cells of fish include cell lines derived from sturgeon eyeball epithelial cells and cell lines derived from scorpion tailfish (see, for example, Patent Documents 1 and 2 below, which are described here) Incorporated herein by reference). These established cell lines are expected not only to replace animal cells but also to elucidate the ecology and culture of sturgeons and scorpions, respectively.
- the fishes of Monacanthidae including frogfish (Stephanolepis cirrhifer), are covered with strong skin throughout the body, and the entire skin can be peeled off at the time of cooking.
- frogfish Stephanolepis cirrhifer
- the riverfish is widely preferred as a raw or cooking ingredient.
- fish cell lines may contribute to diagnosis of viral infections, production of vaccines effective against viral infections, cytotoxicity tests, and the like. Therefore, the present inventors thought that if there was an immortalized cell line of eelfish, it would be possible to find a clue to solve the problem of moribundity of cultivated cultivated fish, and that significant progress could be made in the development of cultivated eelfish.
- fish cell lines can be cultured at low temperatures, but have a low growth rate compared to mammalian cell lines. Therefore, an immortalized cellfish cell line that can be cultured at a low temperature and has a growth rate comparable to that of a mammalian cell line can be cultured easily and quickly. We can expect rapid progress in development.
- pluripotent stem cells of fish not only kingfishes, tissue cells that were previously difficult to collect can be obtained by inducing differentiation into specific cells, and the pathogenesis of fish diseases And study the onset mechanism.
- cells that have developed a specific disease after differentiation induction are used, it is possible to evaluate the effect and toxicity of the drug against the disease. Therefore, fish pluripotent stem cells can also be expected to contribute to the development of fish culture techniques.
- an immortalized cell line or a pluripotent stem cell may significantly advance the research and development of the technique for cultivating the river moth.
- established cell lines and pluripotent stem cells are not known.
- a first object of the present invention is to provide an immortalized cell line derived from a river snail.
- the second object of the present invention is to provide a pluripotent stem cell derived from a river moth.
- the third object of the present invention is to provide a method for producing and utilizing a cell line derived from brookfish.
- the inventors examined the establishment of fish cells, focusing on the fact that fish cells can be cultured at low temperatures. Therefore, we tried to grow various tissue cells using fish from various fish such as Thailand, horse mackerel and shark. However, in addition to obtaining cells with a high growth rate, most of them could not be subcultured. Therefore, the present inventors changed the type of fish and the subculture method as appropriate, and as a result of further earnest research, they have produced an immortalized cell line that can be subcultured from the cormorant's dorsal fin. Successful. Moreover, surprisingly, the obtained cell line can be cultured at a growth rate comparable to or higher than that of a mammalian cell line even when cultured at a temperature of 25 ° C.
- the cell line derived from the river moth obtained by the present inventors expresses a part of cell markers exhibiting pluripotency, and further changes in various forms by changing the culture conditions. It changed to cells.
- the present invention has been completed based on these successful examples and findings.
- the present invention preferably, it further has the following property (4).
- the maximum value is 66, the minimum value is 32, and the mode has 33 chromosomes according to the frequency distribution
- the frequency of the 33 chromosomes as a mode is about 90% of the total frequency in the frequency distribution.
- the present invention preferably, it further has the following property (5).
- the initial cell number is about 1.0 ⁇ 10 6 cells / ml, and Leibovitz's L-15 medium containing 10% FBS is used in a culture vessel having a bottom area of 75 cm 2 , and CO 2 is absent.
- the doubling time when cultured at 25 ° C. is about 14 to 28 hours
- a fish of the river coral fish that is positive for at least one cell marker selected from the group consisting of TRA-1-60, OCT4 and SSEA-3.
- a cell line derived from a biological site or a passage thereof is provided.
- the cell line or its passage is a cell line exhibiting a fibroblast-like morphology or its passage.
- the cell line or its subculture line is a myocyte and a myocyte-like cell, an epithelial cell and an epithelial cell-like cell, a nerve cell and a neuron-like cell, an adipocyte and an adipocyte-like cell, an immunity It has the ability to differentiate into at least one cell selected from the group consisting of cells and immune cell-like cells and liver cells and liver cell-like cells.
- the fishes of the genus Sphaerophyceae are Stephanolepis, Thamnaconus, Cantherhines, Aluterus and Rudus rus. It is a fish selected from a group.
- the fishes of the family Saccharidae are smeltfish (Stephanolepis cirrhifer).
- the biological part is a heel located on the back, chest, abdomen, buttocks or tail.
- a method for producing the cell line of the present invention or a subculture thereof comprising a step of subjecting a cell isolated from a living part of a river fish species to subculture for 70 times or more. Provided.
- a transformant obtained by introducing a foreign gene into the cell line of the present invention or its passage.
- a method for producing a transformant comprising a step of obtaining a transformant by introducing a foreign gene into the cell line of the present invention or its passage.
- an expression product of a foreign gene comprising a step of obtaining an expression product of the foreign gene from a transformant obtained by introducing the foreign gene into the cell line of the present invention or its passage.
- a method of manufacturing is provided.
- a kit for producing a transformant comprising the cell line of the present invention or its passage, a vector, and equipment for transfection.
- a kit for producing differentiated cells comprising the cell line of the present invention or its passage, a culture medium, and a culture vessel.
- serum is further included.
- the serum is at least one serum selected from the group consisting of mammalian serum, fish serum and serum replacement.
- the medium is at least one medium selected from the group consisting of a medium for mammalian cells, a medium for insect cells, and a medium for fish cells.
- the culture vessel is at least one type of culture vessel selected from the group consisting of a culture vessel whose bottom surface is coated with cell adhesion molecules and a culture vessel whose bottom surface is not coated with cell adhesion molecules. .
- the cell line of the present invention or a passage thereof is cultured in a culture vessel whose bottom surface is coated with a cell adhesion molecule or a culture vessel whose bottom surface is not coated with a cell adhesion molecule.
- a method for producing differentiated cells comprising a step of culturing using a medium for mammalian cells, a medium for insect cells, or a medium for fish cells that does not contain serum or contains mammalian serum, fish serum or serum substitutes Is provided.
- a cultured cell sheet comprising the cell line of the present invention or its passage.
- cells that are distinguished from the cell line of the present invention or its passage by staining for alkaline phosphatase activity and are positive for NANOG, OCT4, TRA-1-60 and SSEA-3 Alternatively, a method for producing a giant rod-shaped colony comprising a step of producing a giant rod-shaped colony formed by the cells is provided.
- the cell line of the present invention or its subculture includes a cell line that can be cultured at a growth rate comparable to that of a mammalian cell line. Since the cell line of the present invention or its passages can be cultured at room temperature, it can be used for research use, industrial production use, etc. in place of mammalian cell lines. Can be expected.
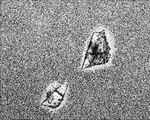
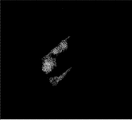
- FIG. 1A is a diagram showing cells migrating from the dorsal fin piece of a river snail.
- FIG. 1B is a diagram showing cells migrating from the tail of a hawkfish.
- FIG. 1C is a diagram showing cells migrating from the buttock piece of a river snail.
- FIG. 1D is a view showing cells migrating from the flank of a riverfish.
- FIG. 2 shows the primary cell (at the time of primary culture), the first passage cell (passage 1), the tenth passage cell (passage 10), the 20th passage cell (passage 20) and the 30th passage cell. It is the figure which showed the cell viability when each cell of (passage 30) was frozen and thawed.
- FIG. 1A is a diagram showing cells migrating from the dorsal fin piece of a river snail.
- FIG. 1B is a diagram showing cells migrating from the tail of a hawkfish.
- FIG. 1C is a diagram showing cells migrating
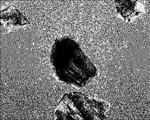
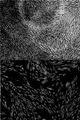
- FIG. 3A is a view of the morphology of KSC cells observed using an optical microscope at a magnification of x460.
- FIG. 3B is a view of the morphology of KSC cells forming two layers, observed using an optical microscope at a magnification of x460.
- FIG. 3C is a view obtained by observing the morphology of KSC cells in which multiple layers were formed using an optical microscope at a magnification of x460.
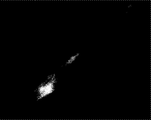
- FIG. 4 shows cells expressing human-AP.
- FIG. 5 shows the activity of human-AP expression products.
- FIG. 6 is a diagram showing expression by a fusion protein of PON and Halotag.
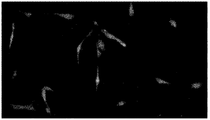
- FIG. 7A is a view observing cells expressing the fluorescent fusion proteins cyan fluorescent protein-yellow fluorescent protein and calmodulin.
- FIG. 7B is a view observing cells expressing a fluorescent fusion protein, red fluorescein protein and vesicular stomatitis virus G-glycoprotein.
- FIG. 7C is a diagram observing cells in which green fluorescent protein, which is a fluorescent protein, is expressed.
- FIG. 8 is a diagram in which the cytotoxicity after each cell was transfected by the lipofectamine method was verified.
- FIG. 9A is a diagram showing that KSC cells are TRA-1-60 positive cells, which are cell markers unique to pluripotent stem cells. The upper side shows the optical microscope observation result, and the lower side shows the fluorescence microscope observation result.
- FIG. 9A is a diagram showing that KSC cells are TRA-1-60 positive cells, which are cell markers unique to pluripotent stem cells. The upper side shows the optical microscope observation result, and the lower side shows the fluorescence microscope observation result.
- FIG. 9B is a diagram showing that KSC cells are OCT4-positive cells, which are cell markers unique to pluripotent stem cells.
- the upper side shows the optical microscope observation result, and the lower side shows the fluorescence microscope observation result.
- FIG. 9C is a diagram showing that KSC cells are SSEA-3 positive cells, which are cell markers unique to pluripotent stem cells.
- the upper side shows the optical microscope observation result, and the lower side shows the fluorescence microscope observation result.
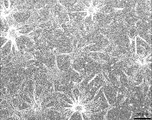
- FIG. 10A shows that KSC cells are fibroblast-like.
- FIG. 10B shows that KSC cells differentiated into myocyte-like cells.
- FIG. 10C shows that KSC cells have differentiated into epithelial cell-like cells.
- FIG. 10D shows that KSC cells have differentiated into neuron-like cells.
- FIG. 10A shows that KSC cells are fibroblast-like.
- FIG. 10B shows that KSC cells differentiated into myocyte-like cells.
- FIG. 10C shows that KSC cells have differentiated
- FIG. 10E shows that KSC cells have differentiated into liver-like cells.
- FIG. 10F shows that KSC cells differentiated into fibrocyte-like cells.
- FIG. 10G shows that KSC cells have differentiated into adipose-like cells.
- FIG. 10H is a diagram showing that KSC cells have differentiated into rose branch cells that may be immune cells.
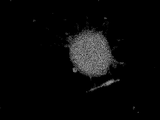
- FIG. 10I shows that KSC cells have differentiated into giant rod-shaped cells.
- FIG. 11A is a diagram showing that the giant cocoon-shaped fused cells derived from KSC cells are positive for the activity of alkaline phosphatase, a cell marker unique to undifferentiated cells.
- FIG. 11A is a diagram showing that the giant cocoon-shaped fused cells derived from KSC cells are positive for the activity of alkaline phosphatase, a cell marker unique to undifferentiated cells.
- FIG. 11B is a diagram showing that the giant rod-shaped fusion cells derived from KSC cells are NANOG positive cells, which are cell markers unique to undifferentiated cells.
- FIG. 11C shows that the giant rod-shaped fusion cells derived from KSC cells are TRA-1-60 positive cells, which are cell markers unique to undifferentiated cells.
- FIG. 11D shows that the giant rod-shaped fused cells derived from KSC cells are OCT4-positive cells, which are cell markers unique to undifferentiated cells.
- FIG. 11E shows that the giant rod-shaped fused cells derived from KSC cells are SSEA-3-positive cells, which are cell markers peculiar to undifferentiated cells.
- FIG. 12 is a view showing a cell sheet obtained by culturing KSC cells.
- the first cell line of the present invention or its passage strain is derived from a living part of a fish of the family Monacanthidae.
- the term “cell line” is understood in the broadest sense by those skilled in the art, and is also referred to as a cell line, an established cell line or the like, and is particularly a cell line that can be continuously subcultured. It is not limited.
- the “passage strain” means all cells obtained by subculturing the cell line.
- a cell line for convenience, a cell that can be evaluated as a cell line is called a cell line, and a cell line obtained after subculture is used. It is called a passage stock.
- the cell line and the passage line are collectively referred to simply as “cell line”.
- the first cell line of the present invention can be produced on the basis of cells isolated from a biological site such as a tissue or an organ excised from a fish of the family Saccharidae.
- the fish species are fishes that have the feature that the skin (outer skin) covering the entire body can be easily peeled off. For example, if a part of the skin is cut with a knife or the like, the skin can be easily peeled off by hand without using a special instrument.
- the organism from which the first cell line of the present invention is derived is not particularly limited as long as it is a riverfish having the characteristics described above. Examples include, ) Fish, Aluterus fish and Rudarius fish.
- suitable specific examples of the otterfish can include kingfish (Stephanolepis cirrhifer), horse mackerel (Thamnoconus modestus), and euph olgi, and more preferred is terfoli. is there.
- the living body part of the fish species which is used for producing the first cell line of the present invention, does not indicate only a specific part of the fish body, but includes two or more parts of tissues or organs in the fish body. Including the whole fish body.
- a suitable living body part in the smeltfish is a part corresponding to a heel, for example, a heel on the back, chest, abdomen, buttocks or tail is preferable, and a ridge on the back is more preferable .
- the method for producing the first cell line of the present invention from the living part of the fish of the family Saccharidae is not particularly limited.
- a specific example of the step of obtaining primary cultured cells comprises, for example, the following procedure. That is, the buttocks of the snapper are cut out as square pieces of an appropriate size without peeling off the skin. The excised square pieces are washed in an appropriate manner and then treated with various concentrations of antibiotics in one to several times. The treated section is aseptically minced into small pieces in the presence of a proteolytic enzyme solution such as trypsin. The operation so far is carried out at a low temperature, preferably under ice cooling. Subsequently, the micro piece immersed in the trypsin solution is left still at room temperature.
- a proteolytic enzyme solution such as trypsin
- the fine pieces after standing are collected by centrifugation or the like, and the collected fine pieces are repeatedly washed several times using a medium suitable for cell growth in a non-CO 2 environment such as Leibovitz's L-15 medium.
- a medium suitable for cell growth in a non-CO 2 environment such as Leibovitz's L-15 medium.
- a standard mammalian cell culture medium such as RPMI 1640 medium or IMDM medium (both Life Technologies) can be used.
- the cells then cover about 80% or more of the bottom surface of the culture container under appropriate culture conditions such as seeding the collagen-coated culture container containing the serum-containing medium and placing it on the bottom of the culture container. Incubate until The cells collected in the minute pieces at this time are used as primary cultured cells.
- the culture medium in the culture vessel is removed, and the cultured cells adhering to the bottom surface of the vessel are washed by an appropriate method.
- the cells seeded in a new culture container are used as the first subculture cells.
- a specific example of the step of subculturing primary cultured cells to obtain the first cell line of the present invention comprises, for example, the following procedure. That is, the subculture for obtaining the first cell line of the present invention from the primary cultured cells is achieved under appropriate culture conditions using a serum-containing medium, for example, in the same manner as the step of obtaining the primary cultured cells.
- the number of cells at the start of the culture is 4 ⁇ 10 5 cells / ml
- the standard at the end of the culture is when the confluency is 90% or more
- the medium is replaced when the culture medium becomes dirty with dead cells
- these are not particularly limited.
- the first cell line of the present invention can be stored and grown by a conventional method known by those skilled in the art, and the method is not particularly limited.
- the method for cryopreserving the first cell line of the present invention includes, for example, adding a commercially available medium to the cell line, collecting the cell by centrifugation, and then collecting the cell into a commercially available serum-free cultured cell. It can be achieved by adding a cell cryopreservation solution for use and cryopreserving.
- the frozen cell line of the present invention can be thawed by placing it in a half-thawed state at room temperature and replacing it with a fresh serum-free medium an appropriate number of times.
- the above subculture can be performed by adding a serum-containing medium to the thawed cells.
- the method for determining that the first cell line of the present invention has been produced is not particularly limited, but primary cultured cells are used 30 times or more, preferably 50 times or more, more preferably 70 times or more, and further preferably. Can be determined to be the first cell line of the present invention by subjecting to subculture for 100 times or more.
- the first cell line of the present invention is not particularly limited as long as it is a cell line that can be continuously subcultured, but is preferably one that can be subcultured substantially without limitation. That is, one aspect of the first cell line of the present invention is one that can be repeatedly cultured for many generations if cultured according to the above-described subculture method with appropriate attention and technical skills possessed by those skilled in the art. It is.
- the first cell line of the present invention may have various properties, but preferably has a fibroblast-like morphology. Fibroblasts have a distinctive morphology such as a flat and long profile, often irregular protrusions, and an elliptical nucleus. When the first cell line of the present invention exhibits a fibroblast-like morphology, the first cell line of the present invention is particularly limited as long as it has any form characteristic to fibroblasts. However, it is preferably at least a flat and long outer shape. The fact that the first cell line of the present invention exhibits a fibroblast-like morphology can be confirmed by various methods for observing the morphology of cells known by those skilled in the art. This can be confirmed by observing the first cell line of the present invention under a microscope.
- the number of divisions is 30 times or more, preferably 50 times or more, more preferably 70 times or more, More preferably, it can be 100 times or more.
- the first cell line of the present invention can be regarded as an immortalized cell. Whether or not the first cell line of the present invention can be subcultured substantially without limitation is determined by, for example, subjecting the cell line of the present invention to the subculture described above, It can be determined by confirming that the number of times is reached.
- the doubling time refers to the time for the cell number to double, as is known by those skilled in the art.
- the doubling time is in principle consistent with the cell cycle for cells in the growth phase.
- the method for measuring the doubling time of the first cell line of the present invention is not particularly limited. For example, when subjected to the above-mentioned subculture, that is, using Leibovitz's L-15 medium containing 10% FBS. When cultured at 25 ° C., it can be determined from the number of cells measured by selecting 2 to several points, preferably 3 points or more, for cells in the growth phase.
- the doubling time of the first cell line of the present invention is not particularly limited.
- the initial cell number is about 1.0 ⁇ 10 6 cells / ml, preferably 0.1 to 1.5 ⁇ 10 6 cells / ml, More preferably 0.4 to 1.0 ⁇ 10 6 cells / ml using Leibovitz's L-15 medium containing 10% FBS in a culture vessel having a bottom area of 75 cm 2 in the absence of CO 2
- When cultured at 25 ° C. at a temperature shorter than the primary cultured cells preferably 10 to 48 hours, more preferably 12 to 36 hours, more preferably 14 to 28 hours, still more preferably 16 to 24 hours, particularly preferably.
- the method for calculating the doubling time from the number of cells is not particularly limited.
- the cell is measured by, for example, attaching an index (marker) to the bottom surface of the culture container in advance, and subjecting the first cell line of the present invention to culturing, and then measuring the number of cells in the vicinity of the marker by an inverted routine microscope. It can be measured by photographing and observing.
- an average of the number of cells in a plurality of squares (for example, 1 mm ⁇ 1 mm) attached to the eyepiece may be used as the number of cells.
- a cell growth curve is obtained. From the growth curve, it is possible to know the growth phase of the delayed phase, logarithmic growth phase, stationary phase and death phase in the cell growth process.
- the time required for doubling the number of cells in the logarithmic growth phase (doubling time) can be calculated from the following formula.
- Double time (t ⁇ t 0 ) log 2 / (log N ⁇ log N 0 ) t: time [h], N: number of cells at time t 0 : initial time [h], N 0 : number of cells at time t 0
- the cultured cells When the cultured cells are cultured in a state where they are adhered to the bottom surface of the culture vessel, the cultured cells usually stop growing when they reach a confluent state covering the entire bottom surface. That is, the cultured cells can grow until a single cell layer is formed on the bottom surface of the culture container.
- another cell layer can be formed on the monolayer cell layer by continuing to grow after the monolayer cell layer is formed. Yes, and preferably a culture in which a multilayer structure is formed.
- the first cell line of the present invention includes those capable of culturing in a three-dimensional structure in which cell layers overlap.
- the fact that the first cell line of the present invention can form a multilayer structure indicates that the culture is further performed after the first cell line of the present invention becomes confluent and cultured by a method known by those skilled in the art. This can be confirmed by observing the cell structure.
- the multilayer structure is not particularly limited as long as another cell layer is formed on a single cell layer, and is a layer structure of two layers or three or more layers.
- the number of chromosomes of the first cell line of the present invention is not particularly limited because it varies depending on the species and male and female of the fish species that are origin organisms. For example, when the number is obtained from the female dorsal fin, The number of chromosomes can be 66, 33 and 32.
- the first cell line of the present invention may be regarded not only as one independent cell but also as a group of cell populations. In that case, when the first cell line of the present invention is obtained from the female dorsal fin of a river moth, it may be a population of cell lines having 66, 33 or 32 chromosomes.
- the first cell line of the present invention preferably has a frequency distribution in which the maximum value is 66 cells, the minimum value is 32 cells, and the mode (mode) is 33 cells. More preferably, it is a population of cell lines having the number of chromosomes, and in the frequency distribution, it is a population of cell lines in which the frequency of 33 chromosomes which are modes is about 90% of the total frequency.
- a method for analyzing the number of chromosomes of the first cell line of the present invention a method for analyzing the number of chromosomes of cells known by those skilled in the art can be adopted without any particular limitation. Can be analyzed by the method described in.
- a specific example of the first cell line of the present invention is a cell line derived from a living part of a cormorant fish, and has one or more of the following properties (1) to (5):
- it has the following property (1) and has at least one of the following properties (2) to (5), more preferably the following properties (1) and (2):
- the second cell line of the present invention is at least one selected from the group consisting of TRA-1-60, OCT4 and SSEA-3. It is a cell line derived from a living part of a cormorant fish that has a positive cell marker or its passage.
- the second cell line of the present invention can be produced from a living part of a fish species of the fish species, by the same method as the first cell line of the present invention.
- the second cell line of the present invention preferably exhibits a fibroblast-like morphology.
- a cell line that is positive for TRA-1-60, OCT4, or SSEA-3 refers to a cell line that expresses an epitope recognized by an antibody against human TRA-1-60, human OCT4, or human SSEA-3, respectively.
- Antibodies to TRA-1-60 react with proteins (epitope) expressed in undifferentiated human embryonic stem (ES) cells, embryonic cancer (EC) cells and embryonic germ (EG) cells, the epitopes being cells It is known to be lost by differentiation.
- OCT4 is a transcription factor highly expressed in ES cells, and is said to be involved in maintaining the undifferentiation of ES cells and maintaining pluripotency.
- SSEA-3 is known as a cell marker for pluripotent stem cells such as ES cells.
- the gene sequences encoding TRA-1-60, OCT4, and SSEA-3 are, for example, ACC_SION No.
- NM_001018111 or NM_001018111 or TRA_ 1-60 as ACCESSION numbers, respectively, by GenBank, a public database of National Center for Biotechnology Information (NCBI).
- GenBank a public database of National Center for Biotechnology Information
- NM_005397, OCT4 are registered as NM_002701
- SSEA-3 is registered as NM_033149 (ACCESSION number of the amino acid sequence is Q9JI67).
- TRA-1-60, OCT4 and SSEA-3 are cell markers exhibiting pluripotency, and are common in that they are not found on the cell surface after differentiation. From these, TRA-1-60, OCT4, SSEA-3, or cell lines that are positive for two or three of these cell markers have pluripotency, the ability to differentiate into various cells. Can be inferred. Therefore, the second cell line of the present invention is highly likely to be a pluripotent cell line.
- pluripotency is not limitedly interpreted, and in addition to the meaning of pluripotency and multipotency as an academic term, totipotency It may be interpreted as having a meaning, having pluripotency or totipotency, or having a probability, and it is interpreted in the broadest sense regardless of this.
- a primary antibody reaction is carried out by adding a primary antibody against TRA-1-60, OCT4 or SSEA-3.
- a secondary antibody against the fluorescently labeled primary antibody is added to carry out a secondary antibody reaction.
- the cells labeled with TRA-1-60 positive, OCT4 positive or SSEA-3 positive can be confirmed by observing the fluorescently labeled cells.
- the primary antibody structure globulin type, fragmented structure, etc.
- the primary antibody recognition specificity of the secondary antibody the wavelength of the fluorescent label, etc. -1-60, OCT4 and SSEA-3 can be confirmed to be positive, but the cells are divided into three populations and TRA-1-60, OCT4 or SSEA-3 are positive for each population It is preferable to confirm.
- the second cell line of the present invention may be capable of expressing a cell marker that is specifically expressed in pluripotent stem cells other than TRA-1-60, OCT4, and SSEA-3.
- the second cell line of the present invention has a low affinity for human anti-NANOG antibodies, and it can be said that NANOG is weakly positive or substantially negative depending on background conditions and image processing methods.
- one aspect of the second cell line of the present invention is, for example, myocytes and myocyte-like cells, epithelial cells and epithelial cell-like cells, nerve cells and neuron-like cells, adipocytes and adipocyte-like cells, liver A cell line that can differentiate into cells such as cells and liver cell-like cells.
- cell differentiation means that expression of at least one cell marker selected from the group consisting of TRA-1-60, OCT4 and SSEA-3 disappears, and a cell marker peculiar to differentiated cells is expressed.
- cell marker selected from the group consisting of TRA-1-60, OCT4 and SSEA-3
- myocyte-like cells, epithelial cell-like cells, neuronal cell-like cells, adipocyte-like cells, and liver cell-like cells are not completely differentiated into muscle cells, but are in the middle of differentiation or differentiation. It may include cells that exhibit a morphology similar to cells.
- a preferred embodiment of the second cell line of the present invention is a myocyte and myocyte-like cell, epithelial cell and epithelial cell-like cell, nerve cell and neuron-like cell, fat cell and adipocyte-like cell, immune cell and immunity
- a cell line having the ability to differentiate into one, preferably two or more, more preferably three or more, and even more preferably four or more cells selected from the group consisting of cell-like cells and liver cells and liver cell-like cells. is there.
- these differentiated cells are exemplary, and those having the ability to differentiate into other cells are also included in the second cell line of the present invention.
- Differentiation into each cell in the second cell line of the present invention can be achieved by a technique for differentiating pluripotent stem cells known to those skilled in the art. This can also be achieved by variously setting the type and presence, the type of medium, and the like.
- a myocyte or myocyte-like cell, epithelial cell or epithelial cell-like cell, nerve cell or neuron-like cell, liver cell or liver from the second cell line of the invention.
- Cell-like cells, fiber cells or fiber cell-like cells, adipocytes or adipocyte-like cells, immune cells or immune cell-like cells can be obtained.
- the cell shown in FIG. 10B is a myocyte-like cell having an elongated spindle-like or fibrous form and capable of proliferating.
- the cells shown in FIG. 10C are epithelial cell-like cells in which individual cells have a flat shape with an indefinite periphery, and the cells grow densely.
- the cell shown in FIG. 10D is a neuron-like cell that exhibits a cell form having an extensible axon or dendrite and has no proliferative ability.
- the cell shown in FIG. 10E is a liver-cell-like cell that has a form that looks like a lipid droplet in the cytoplasm and that can finitely divide.
- the cell shown in FIG. 10B is a myocyte-like cell having an elongated spindle-like or fibrous form and capable of proliferating.
- the cells shown in FIG. 10C are epithelial cell-like cells in which individual cells have a flat shape with an indefinite periphery, and
- FIG. 10F is a fibrous cell-like cell having a fibrous form comparable to or higher than that of muscle cells and having a proliferation ability.
- the cell shown in FIG. 10G is an adipocyte-like cell having a number of cavities, a form having extensible axons, and no proliferation ability.
- the cell shown in FIG. 10H is a dendritic immune cell-like cell that exhibits a branch-like form of a rose having an extensible tip with a sharp tip, has no proliferation ability, and is linked to a neighboring cell. is there. Further, from the second cell line of the present invention, there is a fused cell that stretches like rubber and fuses cells having proliferative ability to form a giant cocoon-like structure as shown in FIG. 10I. can get.
- This fused cell is positive for alkaline phosphatase activity as shown in FIG. 11A and positive for NANOG as shown in FIG. 11B. Furthermore, the fusion cells are also positive for TRA-1-60, OCT4 and SSEA-3 as shown in FIGS. 11C-E, respectively.
- NANOG is registered as ACCESS — NM_024865, for example, by GenBank, a public database of NCBI.
- Differentiation of the cell line can be confirmed by a change in morphology by comparing with a fibroblast-like morphology at the time of subculture, for example, a morphology as shown in FIG. 10A. It can also be confirmed by a cell determination method. For example, it may be confirmed that a cell line has differentiated using a cell marker of each differentiated cell or a substance produced by each differentiated cell as an index.
- a method for producing each differentiated cell from the second cell line of the present invention is provided as another aspect of the present invention. That is, the method for producing differentiated cells of the present invention comprises the second cell line of the present invention in a culture container whose bottom surface is coated with a cell adhesion molecule or a culture container whose bottom surface is not coated with a cell adhesion molecule. Culturing using a mammalian cell culture medium, insect cell culture medium or fish cell culture medium that does not contain serum or contains mammalian serum, fish serum or serum replacement.
- kits for producing differentiated cells comprising the second cell line of the present invention.
- a kit for producing differentiated cells preferably contains serum, a culture medium, a culture vessel, and the like, and more preferably contains a buffer (buffer), a pH adjuster, and the like. Since the second cell line of the present invention has the possibility of differentiating into cells other than the differentiated cells described above, the serum, medium, culture vessel, buffer and pH adjuster are not particularly limited. Can be used.
- mammalian serum, fish serum and serum substitute can be used, fetal bovine serum (Gibco; No. 26140-087 etc.), horse Serum (Gibco; No. 16050-130, etc.), goat serum (Gibco; No. 16210-064, etc.), rabbit serum (Gibco; No. 16120-099, etc.), mouse (KAC; No. AS3054, etc.), chicken serum (Gibco; No. 16110-082, etc.), Lamb serum (Gibco; No. 16070-096, etc.), swine serum (Gibco; No. 26250-084, etc.), dogs (KAC; No.
- the serum contained in the kit for producing the differentiated cell of the present invention may be one type or two or more types.
- the method for obtaining serum is not particularly limited, and commercially available products may be used.
- a serum substitute can be used as the serum contained in the kit for producing the differentiated cell of the present invention.
- Serum substitutes include, for example, Knockout (registered trademark) Serum Replacement (KSR), lactalbumin hydrolyzate, fish-derived cell culture medium additive Hy-Fish (Maruhachi Muramatsu), Nu-Serum (BD; No. BSE 355100) ), SERUM PLUS (Sigma; No. 14008C-500ML), L-Glutamine solution (Sigma; No. 59202C-100ML), etc., and one or more of these serum substitutes are used. be able to.
- KSR Knockout (registered trademark) Serum Replacement
- lactalbumin hydrolyzate lactalbumin hydrolyzate
- fish-derived cell culture medium additive Hy-Fish Maruhachi Muramatsu
- Nu-Serum BD; No. BSE 355100
- SERUM PLUS Sigma; No. 14008C-500ML
- L-Glutamine solution Sigma; No.
- kits for producing differentiated cells of the present invention examples include L-15 (Gibco; No. 11415-064), AIM V (Life Technologies; No. 087-0112DK), IMDM (Gibco; No. 12440-053), RPMI (Gibco; No. 11835030), EX-CELL 420 with L-glutamine (Nichirei; No. 14420C, for insects), D-MEM (Gibco), mTeSR1 (stem cell; No. ST- 05850, for stem cells), Ham's F-12 (Gibco; No. 11765-054), primate ES / iPS cell culture medium (Reprocell; No. RCHEMD001), one of these or Two or more types of media can be used.
- the kit for producing the differentiated cell of the present invention can contain a growth factor or a medium additive as a constituent component.
- growth factors and medium additives include epithelial cell growth factor, fibroblast growth factor, nerve growth factor, insulin-like growth factor, platelet-derived growth factor, vascular endothelial growth factor, transforming growth factor, cytokine, stem cell Factors, T-STIM culture supplements, IL-3 culture supplements, vascular endothelial cell growth supplements, MITO + Sealam extender, ITS culture additives, and one or more of these media can be used be able to.
- Methods for obtaining growth factors and medium additives are not particularly limited, and commercially available products may be used.
- the bottom surface of the culture container included in the kit for producing differentiated cells of the present invention is coated with cell adhesion molecules such as collagen, fibronectin, laminin, poly-D-lysine, poly-L-lysine, gelatin, and streptavidin.
- the culture vessel is a culture vessel whose bottom surface is not coated with a cell adhesion molecule, or two or more of these culture vessels.
- a method for obtaining a culture vessel whose bottom surface is coated with a cell adhesion molecule is not particularly limited, but a commercially available coated culture vessel may be used, or a non-coated culture vessel and a cell adhesion molecule may be separately prepared and used by those skilled in the art.
- the method for obtaining the cell adhesion molecule is not particularly limited.
- collagen such as collagen I (thermo: 132706), IV (cosmobio: 354534); poly-D-lysine (thermo: 132703); gelatin (CR: 354654) Fibronectin (BD: 356242); poly-L-lysine (thermo); poly-L-ornithine (biocoat); laminin (BD); matrigel biocoat (BD); three-dimensional culture scaffold AteloCell (Koken; CSM-50) or the like can be used.
- the shape and material of the culture vessel are not particularly limited, but a polystyrene flask, a petri dish and a multiwell plate are preferable.
- kits for producing differentiated cells of the present invention includes commercially available media (liquid and solid), serum (liquid and solid), serum substitutes, growth factors, media additives (various amino acids)
- a culture vessel is used, but is not particularly limited.
- a medium prepared with reference to the composition disclosed by the manufacturer may be used.
- a more preferable embodiment of the first cell line and the second cell line of the present invention is a cell line having the characteristics of the first cell line and the second cell line of the present invention.
- a cultured cell sheet comprising these cell lines obtained by culturing the first cell line and / or the second cell line of the present invention.
- the cultured cell sheet of the present invention can be expected to be applied to the treatment of high-grade fish.
- the cultured cell sheet of the present invention can contribute to the reduction of injury lethality of cultured fish.
- the cultured cell sheet of the present invention is expected to be used as a biologically-derived selective membrane to overcome this problem because artificial substances such as synthetic polymers and cellophane limit the selective permeability of substances. it can.
- the cultured cell sheet of the present invention is used in vivo from the viewpoint of use as a scaffold when forming a human cultured cell sheet because individual cells produce collagen and fibronectin, and immunity and antibacterial properties derived from the cells. It can be expected to be used for medical use of material transport capsule materials, adhesive bandages, patches, etc., for use in cancer cell trend tests, for use in immune tests, and as a beauty pack for cosmetics that secrete fish components. .
- the cultured cell sheet of the present invention is peeled off from the bottom during sheet formation and can be cultured in a floating state.
- the cultured cell sheet of the present invention has an advantage that it can be cultured in a floating state.
- the production method of the cell line of the present invention is a method of producing the first cell line of the present invention and the second cell line of the present invention.
- the method includes a step of subjecting the cells isolated from the above to subculture for 30 times or more, preferably 50 times or more, more preferably 70 times or more (hereinafter also referred to as subculture step).
- the subculture process is not particularly limited as long as the cell line of the present invention is finally obtained.
- the above-mentioned “1. First cell line of the present invention” and “2. Based on the matter described in the “second cell line of the invention”, determination of materials, methods, conditions, products to be used, and the like can be carried out.
- the cell line production method of the present invention is capable of subculture, for example, by storing and restoring cells during the subculture process as long as the object of producing the cell line of the present invention can be achieved. Appropriate steps can be provided before, during and after the steps.
- the transformant of the present invention is obtained by introducing a foreign gene into the cell line of the present invention.
- the method for producing a transformant of the present invention is a method including a step of obtaining a transformant by introducing a foreign gene into the cell line of the present invention.
- the method for producing a foreign gene product of the present invention is a method including a step of obtaining an expression product of a foreign gene from the transformant of the present invention.
- the method for introducing a foreign gene into the cell line of the present invention is not particularly limited.
- a transfer known by a person skilled in the art such as a lipofection method, an electroporation method, a microinjection method, a calcium phosphate method, a method using a baculovirus, or the like.
- the foreign gene can be introduced into the cell line by introducing the foreign gene itself into the nucleus of the cell line of the present invention, or by introducing a foreign gene, for example, using a gene introduction substance such as a vector.
- the recombinant vector may be introduced into the cell line of the present invention.
- the vector is not particularly limited as long as it is an autonomously replicable vector.
- vectors for mammalian cells more specifically pcDNA3.1, Flexi HaloTag, pcDNA3.2, pcDNA4.
- Plasmid vectors such as pcDNA5, pcDNA6, and pCMV; viral vectors such as ⁇ phage and RSV; amphibian-derived vectors having the EF1 ⁇ promoter derived from Xenopus.
- the foreign gene to be introduced is not particularly limited as long as it is a gene encoding a desired protein such as a functional protein, edible protein, enzyme, marker protein, or labeling protein. Since the cell line of the present invention is a fish-derived cell line, a gene encoding a fish-specific protein can be used as a foreign gene.
- the foreign gene can be prepared based on information on the amino acid sequence of a desired protein, and a commercially available gene can be appropriately modified and used.
- the protein encoded by the foreign gene may be one type or two or more types. For example, in the case of a foreign gene encoding a desired protein and an antibiotic resistance marker, it is possible to efficiently obtain only a transformant into which the foreign gene has been introduced due to antibiotic selectivity after gene introduction.
- a transformant has been obtained by the method for producing a transformant of the present invention can be confirmed using the presence or absence of an expression product of the introduced foreign gene as an indicator. Similarly, whether or not a foreign gene product was obtained by the method for producing a foreign gene product of the present invention can be confirmed using the presence or absence of an expression product of the foreign gene as an indicator.
- a foreign gene product may be obtained by expressing a foreign gene by culturing the transformant of the present invention, or the transformant of the present invention is cultured and proliferated. Thereafter, foreign gene expression may be induced to obtain a foreign gene product.
- the foreign gene product can be obtained as an expression product accumulated in the transformant of the present invention or an expression product accumulated in the culture solution, depending on the kind of the foreign gene.
- the confirmation of the foreign gene product is not particularly limited as long as it is a technique known by a person skilled in the art to know the presence of a specific protein. For example, Western blotting using a molecular weight of the foreign gene product or an antibody against the foreign gene product, an immunoassay Method, chromatography method and the like can be employed.
- the kit for producing the transformant of the present invention is not particularly limited as long as the cell line of the present invention is included, but in addition to the cell line of the present invention, a vector for introducing a foreign gene and a transfection vector It is preferable to include equipment such as instruments, reagents, and devices.
- the equipment for transfection is not particularly limited as long as it is used when performing each transfection technique.
- the transfection technique is a lipofection technique
- liposomes liposomes, culture media, buffers, etc.
- Instruments such as reagents and culture vessels are preferred.
- the uses of the cell line of the present invention are not particularly limited.
- fish cell physiology, fish genes and their expression systems, fish disease viruses, water environment pollution assessment In addition to appropriate research such as experimental systems, it can also be used for evaluation of physiologically active substances, screening for antiviral agents in combination with fish viruses, and the like.
- the cell line of the present invention comprises a step of introducing a test substance into the culture system of the cell line of the present invention and then evaluating the influence of the test substance on living cells;
- a virus propagation method comprising inoculating a culture system with a virus and growing the virus in a nutrient medium suitable for the growth of the virus; introducing the test substance into the virus-growing medium in the virus growth method; It can be used in a method for screening a virus therapeutic agent, including a step of evaluating the action of a test substance on a virus.
- the cell line of the present invention can be used in methods for diagnosing viral infections, methods for producing vaccines effective against viral infections, methods for evaluating cytotoxicity, and the like.
- the cytotoxicity of the test substance is evaluated by comparing the survival number of the cell line of the present invention with the survival number of the cell line after contacting the test substance.
- the step of bringing the test substance into contact with the cell line of the present invention and the step of determining the test substance as a differentiation-inducing substance by detecting muscle cells, skin cells, nerve cells or fat cells a differentiation-inducing substance Can be expected to be screened.
- the piece after standing was replaced three times with PBS containing 1% penicillin / streptomycin under ice-cooling.
- trypsin EDTA MP Biomedicals
- the tissue pieces are minced into 1 mm squares under ice cooling, and then at room temperature. Allowed to stand for 20 minutes.
- the tissue piece after standing was subjected to a centrifuge for several seconds, and then the supernatant was removed.
- the precipitated tissue pieces were treated several times with Leibovitz's L-15 medium (Life Technologies). That is, the tissue piece in the tube was spun down and the supernatant was removed, and then a fresh medium was added and stirred.
- FIGS. 1A to 1D show the appearance of cells that have migrated throughout the flask centering on each piece. Below, the subculture procedure of the cell extracted from the dorsal fin piece is described.
- the medium in the subculture culture flask was removed, and the cells adhering to the bottom of the flask were washed 3 times with PBS. Cells were detached and collected by treatment with Tryp Express (Gibco) for 10 minutes at 38 ° C. Leibovitz's L-15 medium containing 10% FBS was added to the collected cells and cultured at 25 ° C.
- the mixture was centrifuged at 1,100 rpm for 4 minutes at room temperature, then the supernatant was removed, and then the cell bunker 2 (Juji Field) was used at ⁇ 80 ° C. saved.
- the cell bunker 2 Juji Field
- the liquid portion was taken from a semi-thawed tube, transferred to a 15 ml centrifuge tube, 4 ml of L-15 medium was added, and the mixture was stirred and centrifuged.
- KSC cell As a result of performing the above 1 and 2, the present inventors have succeeded in obtaining a cell having a passage number of 70 or more and a division number of 276 or more. This cell was named KSC cell. KSC cells were almost the same as the first passage cells in terms of doubling time. Therefore, KSC cells were evaluated as established. This is in accordance with the definition of the JRCB cell bank (http://cellbank.nibio.go.jp/visitcenter/whatsculture/cellculture03.html, the description of which is incorporated herein by reference).
- CHO cell data is the second largest cell bank in the world, and the only DSMZ (http://www.dsmz.de) in Germany that seems to be highly reliable with complete cell data in the world. (The description of this document is incorporated herein by reference).
- CHO cells reached confluence (cells proliferated until they expanded to the whole flask bottom) in 72 to 96 hours when cells of 10 6 cells / ml were seeded at about 80 cm 2 , and the doubling time Is calculated as 24 hours.
- KSC cells reached confluence in 48 to 72 hours when 10 6 cells / ml cells were seeded at about 80 cm 2 , and the doubling time was calculated as 18.34 hours. Therefore, it was found that KSC cells are cells having the same or better proliferative ability as CHO cells.
- KSC cells about 0.4 ⁇ 10 6 cells / ml of cells were seeded in an area of 75 cm 2 and Leibovitz's L-15 medium containing 10% FBS was used at 25 ° C. in the absence of CO 2.
- the total number of cells when cultured until 48 hours after reaching confluence was measured using a TC10 Automated Cell Counter (BioRad).
- the number of cells after 24 hours of culture was about 1.0 ⁇ 10 6 cells / cell.
- the number of cells after 48 hours of culture was about 2.5 ⁇ 10 6 cells / ml.
- the doubling time is calculated from the number of cells immediately after seeding and the number of cells after 48 hours of culture. Was calculated to be 18.16 hours.
- the doubling time calculated from the number of cells immediately after seeding and the number of cells after 24 hours of culture is 18.16 hours, and is calculated from the number of cells after 24 hours of culture and the number of cells after 48 hours of culture. The doubling time performed was 18.16 hours.
- FIG. 2 shows that the KSC cells after thawing had a survival rate of 75% or more regardless of the number of passages.
- pdf # search % 27 FemtoJetMicroinjector% 27; http: // probes. invitrogen. com / media / pis / mp10582.
- pdf # search % 27celllight% 20pdf% 27, the description of this document is incorporated herein by reference.
- Cytotoxicity (resistance) in gene transfer by the Lipofectamine method is KSC cells, rat mesenchymal stem cells (MSC), human acute T-cell leukemia cell line (Jurkat), and mice. Comparative verification was performed using embryonic tumor cells (P19). As a result, KSC cells have a survival rate of 90% or more after gene introduction (transfected) compared to before introduction of foreign genes (control), and show superior cell stability compared to other cells. ( Figure 8).
- the cell detachment solution was put into a 15 ml tube, centrifuged at 1,100 rpm for 4 minutes, the supernatant was removed, and the cells were collected. This operation was performed quickly because the cells affected the colcemid treatment time.
- hypotonic solution was added to the 15 ml tube containing the collected cells with a pipette and gently pipetted to disperse the cells.
- a hypotonic solution was added to a 15 ml tube containing the dispersed cells until the final volume was 1.5 ml, and then the cells were dispersed again by pipetting. The cells dispersed in the hypotonic solution were allowed to stand at room temperature for 20 minutes for hypotonic treatment.
- the fixative was slowly added so that the total amount was about 10 ml, and gently agitated to immobilize the cells. Further, the supernatant was removed by centrifugation at 1,100 rpm for 4 minutes, and a few drops of the new fixative solution were added to the centrifuged cells, and the cells were dispersed by pipetting. Further, about 10 ml of fixative was added, and the whole was stirred. This operation was repeated two more times to completely fix the cells. The fixed cells were stained with Hoechst quinacrine (Wako Pure Chemical Industries) and observed with an optical microscope.
- the cell population of KSC cells includes three types of cell lines having 32, 33 and 66 chromosomes.
- a normal riverfish has 33 female and 34 male chromosomes.
- Ninety percent of the KSC cell population had 33 chromosomes, similar to the kawaha female. However, the remaining 10% of the cell population were cells with 32 and 66 chromosomes.
- Multipotency analysis using stem cell markers (1) Method According to the following procedure, KSC cells were immunochemically stained. As a blocking buffer, a PBS solution containing 10% FBS and 0.1% Triton X-100 was used. As an antibody dilution buffer, a PBS solution containing 3% goat serum and 0.1% Triton X-100 was used.
- KSC cells were cultured to confluence in a culture flask, the medium was removed, and the cells were washed twice with PBS at room temperature. 4% paraformaldehyde was added to the cells, and the cells were fixed by allowing to stand at room temperature for 20 minutes. Paraformaldehyde was removed and the immobilized cells were washed 3 times with PBS at 10 minute intervals. Blocking buffer was added and allowed to stand at room temperature for 1 hour. The blocking buffer was removed, and the cells after the blocking treatment were washed once with PBS. A primary antibody (ES / iPS Cell Charactarization Kit; Funakoshi) diluted 100 times using an antibody dilution buffer was added, and a primary antibody reaction was performed overnight at 4 ° C.
- ES / iPS Cell Charactarization Kit Funakoshi
- the cells after the reaction were washed with PBS 5 times at 10-minute intervals.
- a secondary antibody diluted with an antibody dilution buffer was added, and left at room temperature for 1 hour to carry out a secondary antibody reaction.
- the plate on which the cells were immobilized was protected from light.
- the secondary antibody was removed, and the reacted cells were washed 4 times with PBS at 10 minute intervals. Again, the plate was protected from light. The washed cells were observed using a FLoid cell imaging station (Life Technologies).
- KSC cells were presumed to be a pluripotent cell line
- differentiation into various cells was attempted by changing the culture medium and culture conditions.
- the cell morphology was observed with a light microscope at a magnification of 460 times.
- KSC cell morphology (before differentiation induction) KSC cells were cultured in a collagen-coated flask using Leibovitz's L-15 medium containing 10% (v / v) FBS. The observation result of the cells after the culture is shown in FIG. 10A.
- the fibroblast-like morphology shown in FIG. 10A is presumed to be a morphology before the KSC cells differentiate into other cells.
- FIGS. 10E-I when KSC cells were cultured using various culture containers, combinations of serum and medium, or differentiation-induced for KSC cells, adipocyte-like cells, fiber cell-like cells, liver cell-like cells, rose cells, Branched immune cell-like cells and giant rod-like fusion cells could be obtained (see FIGS. 10E-I).
- the fused cells shown in FIG. 10I were positive for alkaline phosphatase activity (see FIG. 11A) and positive for NANOG (see FIG. 11B).
- this fusion cell was also positive for TRA-1-60, OCT4 and SSEA-3 (see FIGS. 11C-E).
Landscapes
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Biotechnology (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Cell Biology (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13825193.9A EP2881460B1 (en) | 2012-07-31 | 2013-07-30 | Cell strain derived from monacanthidae |
| US14/418,781 US9777254B2 (en) | 2012-07-31 | 2013-07-30 | Cell line derived from thread-sail filefish (Stephanolepis cirrhifer) |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-169378 | 2012-07-31 | ||
| JP2012169378A JP5846384B2 (ja) | 2012-07-31 | 2012-07-31 | カワハギ由来の細胞株 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014021329A1 true WO2014021329A1 (ja) | 2014-02-06 |
Family
ID=50028002
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/070627 Ceased WO2014021329A1 (ja) | 2012-07-31 | 2013-07-30 | カワハギ由来の細胞株 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US9777254B2 (enExample) |
| EP (1) | EP2881460B1 (enExample) |
| JP (1) | JP5846384B2 (enExample) |
| WO (1) | WO2014021329A1 (enExample) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017176005A (ja) * | 2016-03-29 | 2017-10-05 | テルモ株式会社 | 細胞培養物およびその製造方法 |
| CN108893474A (zh) * | 2018-02-07 | 2018-11-27 | 电子科技大学 | 草鱼细胞间粘附分子基因的分离和鉴定 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113637632A (zh) * | 2021-07-20 | 2021-11-12 | 宁波大学 | 一种银鲳肌肉组织细胞系 |
| CN115595297B (zh) * | 2022-09-22 | 2023-10-13 | 中国水产科学研究院南海水产研究所 | 一种卵形鲳鲹肌肉细胞系及构建方法和应用 |
| CN119979723B (zh) * | 2025-03-13 | 2025-10-03 | 中国水产科学研究院北戴河中心实验站 | 鉴定绿鳍马面鲀性别的特异性dna片段、引物及其应用和鉴定绿鳍马面鲀性别的方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004024192A (ja) | 2002-06-28 | 2004-01-29 | Fujikin Inc | チョウザメ由来の新規な株化細胞 |
| JP2008220198A (ja) | 2007-03-09 | 2008-09-25 | Japan Agengy For Marine-Earth Science & Technology | カサゴ細胞株 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7132230B2 (en) | 2002-06-28 | 2006-11-07 | Fujikin Incorporated | Cytotoxic assay and new established cell line of sturgeon origin |
-
2012
- 2012-07-31 JP JP2012169378A patent/JP5846384B2/ja not_active Expired - Fee Related
-
2013
- 2013-07-30 EP EP13825193.9A patent/EP2881460B1/en not_active Not-in-force
- 2013-07-30 US US14/418,781 patent/US9777254B2/en not_active Expired - Fee Related
- 2013-07-30 WO PCT/JP2013/070627 patent/WO2014021329A1/ja not_active Ceased
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004024192A (ja) | 2002-06-28 | 2004-01-29 | Fujikin Inc | チョウザメ由来の新規な株化細胞 |
| JP2008220198A (ja) | 2007-03-09 | 2008-09-25 | Japan Agengy For Marine-Earth Science & Technology | カサゴ細胞株 |
Non-Patent Citations (11)
| Title |
|---|
| "Current Protocols in Molecular Biology", 1987, JOHN WILEY & SONS |
| "Hayflick L. Moorhead POSITION", EXPOSURE CELL RES., vol. 25, 1961, pages 585 - 621 |
| "Molecular Cloning: A laboratory Manual", 1989, COLD SPRING HARBOR LABORATORY |
| EBITANI N ET AL.: "An established marine fish cell line with high plating efficiency.", ZOOL SCI, vol. 5, no. 1, 1988, pages 183 - 186, XP008176430 * |
| FAN TING-JUN ET AL.: "Establishment of a turbot fin cell line and its susceptibility to turbot reddish body iridovirus.", CYTOTECHNOLOGY, vol. 62, no. 3, 2010, pages 217 - 223, XP019816403 * |
| HASEGAWA S ET AL.: "A Cell Line (CFK) from Fin of Isogeneic Ginbuna Crusian Carp.", FISH PATHOLOGY, vol. 32, no. 2, 1997, pages 127 - 128, XP003034322 * |
| MAKIKO KIKUCHI ET AL.: "Kasago Saibokabu no Sangyo Riyo o Mezashita Baiyo Joken no Kento", KANTO GAKUIN UNIVERSITY KOGAKUBU KENKYU HAPPYO KOEN RONBUNSHU, vol. 2009, 2009, pages 170 - 171, XP008176398 * |
| MEGURO Y ET AL.: "A Cell Line Derived from the Fin of Japanese Flounder, Paralichthys olivaceus.", FISH PATHOLOGY, vol. 26, no. 2, 1991, pages 69 - 75, XP003034323 * |
| NOBUO EGAMI: "Gyorui no Bunshi Seibutsugaku Sakana no Saibo Baiyo", PROTEIN, NUCLEIC ACID AND ENZYME, vol. 34, no. 3, 1989, pages 186 - 192, XP008176429 * |
| SHOHEI WAKAO ET AL., PNAS, vol. 108, no. 24, 14 June 2011 (2011-06-14), pages 9875 - 9880 |
| YUKI NAKATANI: "Shu no Kotonaru Gyoruikan de Harabire no Tayosei o Motarasu Mechanism no Kaimei", THE ZOOLOGICAL SOCIETY OF JAPAN DAI 81 KAI TAIKAI YOKOSHU PROGRAM, 2010, pages 24, XP055191256, Retrieved from the Internet <URL:http://www.zoology.or.jp/annual-meeting/2/img/f_users/r_1005407img20100813185906.pdf> [retrieved on 20131024] * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017176005A (ja) * | 2016-03-29 | 2017-10-05 | テルモ株式会社 | 細胞培養物およびその製造方法 |
| CN108893474A (zh) * | 2018-02-07 | 2018-11-27 | 电子科技大学 | 草鱼细胞间粘附分子基因的分离和鉴定 |
| CN108893474B (zh) * | 2018-02-07 | 2021-11-16 | 电子科技大学 | 草鱼细胞间粘附分子基因的分离和鉴定 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2881460A1 (en) | 2015-06-10 |
| US20150175958A1 (en) | 2015-06-25 |
| JP5846384B2 (ja) | 2016-01-20 |
| EP2881460A4 (en) | 2015-12-30 |
| EP2881460B1 (en) | 2019-01-02 |
| JP2014027888A (ja) | 2014-02-13 |
| US9777254B2 (en) | 2017-10-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101582483B1 (ko) | 심근 세포의 세포괴의 제조 방법 및 상기 심근 세포괴의 용도 | |
| Jin et al. | Integration-free induced pluripotent stem cells derived from retinitis pigmentosa patient for disease modeling | |
| KR100812856B1 (ko) | 영장류의 배아 줄기 세포로부터 배상체를 제조하는 방법 | |
| JP5846384B2 (ja) | カワハギ由来の細胞株 | |
| US11801267B2 (en) | Cardiac cell culture material | |
| WO2016127908A1 (zh) | 一种三维复合细胞聚集体模型及其制备方法与应用 | |
| KR20190112090A (ko) | 다능성줄기세포의 분화 제어 방법 | |
| CN111263807A (zh) | 人工脂肪组织及其制造方法、人工皮肤的制造方法以及脂肪细胞的培养试剂 | |
| US20190185816A1 (en) | Cardiac microtissue and uses thereof | |
| Weinstein et al. | Isolation and purification of primary Schwann cells | |
| Morrison et al. | Expression patterns of Oct4, Cdx2, Tead4, and Yap1 proteins during blastocyst formation in embryos of the marsupial, Monodelphis domestica Wagner | |
| KR102241349B1 (ko) | 각막상피세포 집단의 제조 방법 | |
| WO2017150294A1 (ja) | 多能性幹細胞様スフェロイドの製造方法および多能性幹細胞様スフェロイド | |
| Heinonen et al. | The development and application of key technologies and tools | |
| WO2025063278A1 (ja) | コアシェル構造の三次元構造物 | |
| WO2017073761A1 (ja) | 多能性幹細胞の胚様体形成方法および多能性幹細胞の胚様体形成用組成物 | |
| WO2024086370A1 (en) | Compositions and methods relating to adipocyte production | |
| JP2015198645A (ja) | 細胞構造体の作製方法 | |
| Abdullah et al. | Differentiation of Mouse Bone-Marrow Mesenchymal Stem Cells into Motor Neuron Cells in vitro. | |
| Elbaghdady et al. | Isolation and Molecular Characterization of Spermatogonial Stem Cells in Nile Tilapia |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13825193 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14418781 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013825193 Country of ref document: EP |