WO2013161842A1 - ザルトプロフェンおよびその誘導体の製造方法 - Google Patents
ザルトプロフェンおよびその誘導体の製造方法 Download PDFInfo
- Publication number
- WO2013161842A1 WO2013161842A1 PCT/JP2013/061974 JP2013061974W WO2013161842A1 WO 2013161842 A1 WO2013161842 A1 WO 2013161842A1 JP 2013061974 W JP2013061974 W JP 2013061974W WO 2013161842 A1 WO2013161842 A1 WO 2013161842A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- zaltoprofen
- organic solvent
- production method
- formula
- derivative
- Prior art date
Links
Images













Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D337/00—Heterocyclic compounds containing rings of more than six members having one sulfur atom as the only ring hetero atom
- C07D337/02—Seven-membered rings
- C07D337/06—Seven-membered rings condensed with carbocyclic rings or ring systems
- C07D337/10—Seven-membered rings condensed with carbocyclic rings or ring systems condensed with two six-membered rings
- C07D337/14—[b,f]-condensed
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
Definitions
- the present invention relates to a novel method for producing zaltoprofen and its derivatives.
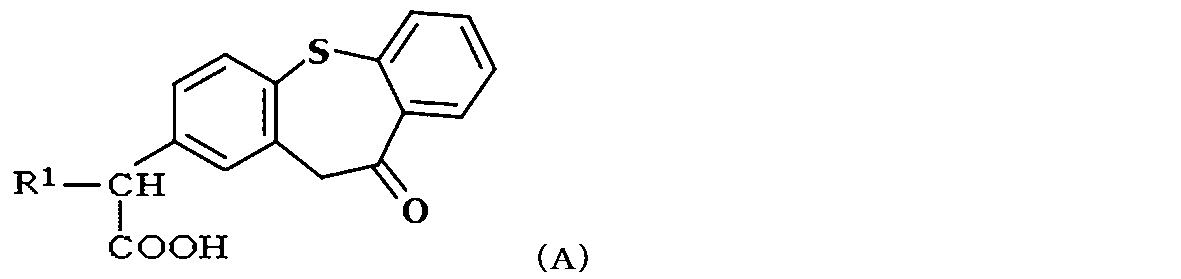
- a compound represented by the following formula (A) (wherein R 1 represents a lower alkyl group) is known to have an excellent anti-inflammatory and analgesic action as a non-steroidal analgesic / anti-inflammatory agent ( JP 61-7199, JP 2005-289949).
- the compound of the above formula (A) is a compound represented by the following formula (B) (wherein R 1 and R 2 each independently represents a lower alkyl group) As a starting material, and can be obtained by subjecting it to a Friedel-Crafts reaction type intramolecular cyclization (Japanese Patent Publication No. 1-292993, Japanese Patent Application Laid-Open No. 2005-289949).
- the temperature during the intramolecular cyclization reaction is reported to be “a temperature higher than 70 ° C. is not advantageous because a side reaction occurs when stirring at a high temperature” (Japanese Patent Laid-Open No. 57-171991). , “Preferably, it can be carried out by heating to a temperature in the vicinity of the boiling point of the organic solvent to be used” (Patent No. 4603276), so that the temperature in the vicinity of the boiling point is suitable or a low temperature. It is still unclear whether is suitable.
- the method of Japanese Patent Publication No. 01-29793 is a method in which the target compound is obtained with an isolation yield as high as 84.8%.
- a large excess of polyphosphoric acid for example, 20 times by weight of the raw material compound
- the reaction is necessary to use for the reaction (Example 2 of JP-B-01-29793).
- the polyphosphoric acid used as the condensing agent cannot be recovered and used, the treatment of the reaction waste is expensive.
- polyphosphoric acid is a very strong acid, and the phosphoric acid waste liquid generated by the decomposition treatment is a cause of marine pollution. Therefore, the influence on the global environment is not preferable in the disposal process. Therefore, the above method has problems in terms of an increase in raw material costs and a complicated waste liquid treatment.
- Patent No. 4603276 a method (Patent No. 4603276) is proposed in which a compound of formula (IV) is heated with phosphoric acid in the presence of an aromatic hydrocarbon solvent or a halogenated hydrocarbon solvent. Has been.
- the organic solvent used for the ring closure reaction is “promoting improvement of the management of grasping of the amount of specific chemical substances released into the environment, etc.” due to concern about environmental burden and human toxicity.
- dichloromethane which is a substance that is monitored for use and disposal by the Law on the Law.
- the purity of the obtained zaltoprofen is only 98.01% (Example 5) to 99.01% (Example 12), which is the purity of zaltoprofen obtained when no inorganic base or inorganic salt is added.
- JP-B-6-51697 has a problem that it is necessary to recrystallize three times and to convert a crystalline salt with an organic amine into a free form or a hydrochloride after the recrystallization. Furthermore, in the example of the method of Japanese Patent Publication No. 6-51697, the spot of impurities was not confirmed in the purified product obtained by recrystallization according to the thin layer chromatography which is a simple measurement method. I can't say I didn't.
- JP-A-2006-290753 10 g of a crude product is dissolved in 100 ml of a ketone solvent, then distilled off to 20 ml, brought to room temperature, added with seed crystals, stirred for 12 hours while cooling to 5 ° C., and then ⁇
- a complicated operation of stirring for 1 hour while cooling to 30 ° C. is required, and a large facility capable of cooling to ⁇ 30 ° C. is required.
- the method disclosed in Japanese Patent Application Laid-Open No. 2006-290753 has a problem that the dimer of 0.01% to 0.12% is still not removed.
- Japanese Patent Laid-Open No. 55-53282 Japanese Patent Laid-Open No. 61-7199
- Japanese Patent Laid-Open No. 57-106678 Japanese Patent Laid-Open No. 01-29793
- Japanese Patent Laid-Open No. 57-171991 Japanese Patent Laid-Open No. 61-251682 Japanese Patent Publication No. 5-41152
- JP 62-108877 A Japanese Patent Publication No. 6-51697
- JP 62-292780 A Patent No. 2612161
- Japanese Patent Laid-Open No. 8-99953 Japanese Patent Laid-Open No. 2549997) JP 2005-247778 A (Patent No. 4603276) JP 2005-289949 A JP 2006-273731 A JP 2006-290753 A
- the compound in the method of ring closure of the compound of formula (II) or formula (IV) by Friedel-Crafts reaction type intramolecular cyclization reaction, the compound is less than 6.5 times the compound of formula (II) or formula (IV)
- the amount of polyphosphoric acid used as a condensing agent is of sufficient purity to be used as a pharmaceutical active ingredient, without producing detectable amounts of impurities such as dimers and industrially usable
- the solvent used in the method must be capable of producing high-purity zaltoprofen or a zaltoprofen derivative at a sufficiently high reaction rate and easily isolated and purified.
- water having a boiling point of 80 ° C. or higher is required.
- JP-B-67-1199 is benzene
- JP-A 57-171991 describes dichloromethane, 1,2-dichloroethane, toluene, xylene and benzene
- JP-B-5-41152 describes dichloromethane, toluene and benzene
- JP-B-6- 51697 describes dichloromethane, 1,2-dichloroethane, chloroform, benzene, toluene and xylene
- Patent No. 4603276 describes benzene, toluene, dichloromethane, dichloroethane, trichloro Tan, chlorobenzene and dichlorobenzene
- JP 2005-289949, JP 2006-273731 and JP 2006-290753 are dichloromethane, chloroform, carbon tetrachloride, toluene, chlorobenzene, nitrobenzene, hexane, heptane, diethyl ether, Diisopropyl ether and dimethyl carbonate are described.
- the organic solvents whose effects have been demonstrated are 1,2-dichloroethane (Japanese Patent Laid-Open No. 57-171991, Japanese Patent No. 4603276), dichloromethane (Japanese Patent Laid-Open No. 57-171991, Japanese Patent Publication No. 5). -41152, Japanese Patent No. 261161, Japanese Patent No. 2549997 and Japanese Patent Application Laid-Open No. 2005-289949), Toluene (Japanese Patent Application Laid-Open No. 57-171991 and Japanese Patent No. 4603276), Xylene (Japanese Patent Application Laid-Open No. 57-171991) and Chlorobenzene (Japanese Patent No. 4603276) only. .
- any of these organic solvents has many problems when used for industrial production of pharmaceuticals.
- dichloromethane is flame retardant, there is a risk of explosion, and in the event of a fire, harmful phosgene, hydrogen chloride and chlorine are generated.
- it is a Class 2 organic solvent that says “the amount of residue should be regulated in order to protect patients from possible harmful effects,” and because of concerns about environmental impact and human toxicity, It is also a substance whose use and disposal is monitored by the Law Concerning Monitoring of Emissions to the Environment and Promotion of Improvements in Management, and also causes dizziness, lethargy, headache, nausea, weakness, and loss of consciousness. It is desirable to avoid use in production.
- Toluene is a class 2 organic solvent, and is volatile at room temperature and flammable. Since the vapor and air mixture is explosive, there is a high risk of fire. Inhalation of toluene has a strong anesthetic or hypnotic effect, and causes cough, sore throat, dizziness, lethargy, headache, nausea and loss of consciousness. Therefore, it is desirable to avoid use in the manufacture of pharmaceuticals.
- toluene is an organic solvent hardly soluble in water, and toluene has a boiling point of 111 ° C.
- Xylene is flammable in all of o-xylene, m-xylene, and p-xylene, and at 32 ° C or higher, an explosive gas mixture of steam and air may be generated, so there is a high risk of fire.
- Xylene is a class 2 organic solvent, and it is a substance whose use and disposal is monitored by the Poisonous and Deleterious Substances Control Law and the “Act on the Promotion of Improvements in Management, etc. of Understanding the Release of Specific Chemical Substances into the Environment”
- inhalation has a strong anesthetic or hypnotic effect, as well as dizziness, lethargy, headache, and nausea.
- o-xylene, m-xylene, and p-xylene are all organic solvents that are hardly soluble in water, and the boiling points of o-xylene, m-xylene, and p-xylene are 144 ° C. and 139 ° C., respectively. 138 ° C.
- 1,2-dichloroethane is highly flammable, and the vapor / air mixture is explosive and contains chlorine, which may cause environmental pollution and harmful hydrogen chloride and phosgene.
- the “Guidelines for Residual Solvents for Pharmaceuticals” notified by the Ministry of Health and Welfare in 1998, “Appropriate by evaluation from the perspective of risk-benefit. Should be avoided in the manufacture of drug substances, pharmaceutical excipients or formulations unless clearly indicated otherwise, and if factory workers inhale May cause abdominal pain, cough, dizziness, lethargy, headache, nausea, sore throat, loss of consciousness, and vomiting.
- 1,2-dichloroethane is an organic solvent hardly soluble in water and has a boiling point of 83.5 ° C.
- Chlorobenzene is a class 2 organic solvent, and is flammable. At 27 ° C or higher, it may produce an explosive vapor / air mixture. Moreover, it contains chlorine and is a concern for environmental pollution and harmful. There is concern that hydrogen chloride and phosgene are generated. Furthermore, inhalation of chlorobenzene causes inhalation somnolence, headache, nausea and loss of consciousness, so it is desirable to avoid use in the manufacture of pharmaceuticals. Chlorobenzene is an organic solvent that is hardly soluble in water, and the boiling point of chlorobenzene is 132 ° C.
- Chloroform is a class 2 organic solvent and contains chlorine, so there are concerns about environmental pollution and the generation of harmful hydrogen chloride and phosgene. Therefore, it is desirable to avoid use in the manufacture of pharmaceuticals. Furthermore, inhalation of chloroform causes inhalation sputum somnolence, headache, nausea and loss of consciousness, and has a strong anesthetic action or hypnotic action. Chloroform is an organic solvent that dissolves in water up to 0.82%, and the boiling point of chloroform is 61 ° C.
- dichloromethane, toluene, xylene, 1,2-dichloroethane, chlorobenzene, and chloroform are organic solvents that should be avoided in pharmaceutical production.
- the inventor of the present application uses a compound of formula (II) or formula (IV) in a method for producing zaltoprofen or a zaltoprofen derivative by cyclization by a Friedel-Crafts reaction type intramolecular cyclization reaction.
- the present invention relates to 1,1,2-trichloroethene, methylcyclohexane, cyclohexane, n-heptane, acetonitrile, 1,2-dimethoxyethane, 1,4-dioxane, tetrahydrofuran, N, N-dimethylformamide, ethyl acetate And cyclization of the compound of formula (II) or formula (IV) by Friedel-Crafts reaction type intramolecular cyclization reaction in the presence of one or more organic solvents selected from the group consisting of methylethylketone Wherein the compound of formula (I) or formula (III) is synthesized.
- the temperature suitable for the heat treatment in carrying out the ring-closing reaction is a temperature in the range of + 25 ° C. to ⁇ 25 ° C. based on the boiling point of the organic solvent used, and preferably the temperature of the organic solvent used. It is a temperature in the range of + 20 ° C. to ⁇ 20 ° C. of the boiling point, more preferably a temperature in the range of + 15 ° C. to ⁇ 15 ° C.
- the temperature is in the range of -10 ° C to -10 ° C, and most preferably the temperature is in the range of + 5 ° C to -5 ° C of the boiling point of the organic solvent used.
- the present invention is specifically as follows.
- R 1 is H or a hydrocarbon group having 1 to 8 carbon atoms
- R 2 is H or a hydrocarbon group having 1 to 8 carbon atoms
- the weight of the condensing agent is preferably 1 to 10 times, more preferably 1 to 6.5 times the weight of the compound represented by the formula (II), according to any one of [1] to [5] Production method.
- zaltoprofen with a purity of 99% or more and a yield of 60% or more Alternatively, a zaltoprofen derivative can be produced.
- the Friedel-Crafts reaction type intramolecular cyclization reaction can be carried out at a high temperature, and therefore the reaction within 8 hours.
- zaltoprofen or zaltoprofen derivatives with a purity of 99% or more can be produced in a yield of 60% or more.
- an organic solvent used for the reaction can be obtained at a sufficiently economical price.
- a zaltoprofen or zaltoprofen derivative having a purity of 99% or more can be produced with a small number of steps, and the formation of a dimer is not detected.
- zaltoprofen or a zaltoprofen derivative can be produced simply and economically on an industrial scale.
- the number of manufacturing steps can be reduced.
- zazartoprofen or a zaltoprofen derivative can be produced using an organic solvent that is less likely to cause health hazards for workers on the production site and that is easy to handle for work.
- the flow rate was 1 mL / min, and the detection was ultraviolet absorption at a wavelength of 240 nm.
- the unit of retention time (RT) on the HPLC chart is minutes.
- FIG. 3 is a high performance liquid chromatography chart of zaltoprofen produced by ring closure in the presence of n-heptane (Example 2).
- the conditions of high performance liquid chromatography and the meaning of the display in the figure are the same as in FIG. As long as it is confirmed on the chart, no peaks other than those of zaltoprofen are observed.
- Example 3 is a high performance liquid chromatography chart of zaltoprofen produced by ring closure in the presence of 1,1,2-trichloroethene (Example 3).
- the conditions of high performance liquid chromatography and the meaning of the display in the figure are the same as in FIG. As long as it is confirmed on the chart, no peaks other than those of zaltoprofen are observed.
- Example 6 which is a sketch of the thin layer chromatography development image of the product by the ring closure reaction in the presence of dimethoxyethane.
- the conditions of thin layer chromatography and the meaning of the display in the figure are the same as in FIG.
- the formation of zaltoprofen was confirmed as the main spot, but at the same time, an unreacted compound of formula (IV), a small amount of impurities (spots near the origin) and unknown impurities (spots near the solvent front) were confirmed.
- Example 8 which is the sketch of the thin-layer-chromatography development image of the product by the ring-closing reaction in tetrahydrofuran presence.
- the conditions of thin layer chromatography and the meaning of the display in the figure are the same as in FIG.
- an unreacted compound of formula (IV) and a small amount of impurities (spots near the origin) were confirmed.
- Example 10 which is a sketch of the development image of the thin layer chromatography of the product by the ring-closing reaction in the presence of ethyl acetate.
- the conditions of thin layer chromatography and the meaning of the display in the figure are the same as in FIG.
- zaltoprofen was confirmed to be the main product, an unreacted compound of the formula (IV) and impurities (spots near the solvent front) considered to be decomposition products were confirmed.
- Example 11 which is the sketch of the thin layer chromatography developed image of the product by the ring-closing reaction in the presence of methyl ethyl ketone.
- the conditions of thin layer chromatography and the meaning of the display in the figure are the same as in FIG.
- an unreacted compound of formula (IV) and four types of impurities spots between the zaltoprofen spot and the solvent front
- FIG. 3 is a high performance liquid chromatography chart of zaltoprofen produced by ring closure in the presence of 1,2-dichloroethane (Comparative Example 2).
- the conditions of high performance liquid chromatography and the meaning of the display in the figure are the same as in FIG. As long as it is confirmed on the chart, no peaks other than those of zaltoprofen are observed.
- Organic Solvent used in the present invention is methylcyclohexane, cyclohexane, n-heptane, 1,1,2-trichloroethene, acetonitrile, dimethoxyethane, 1,4-dioxane, tetrahydrofuran, N, N-dimethylformamide, acetic acid.
- One or more organic solvents selected from the group consisting of ethyl and methyl ethyl ketone.
- a preferable organic solvent used in the present invention is one or more organic solvents selected from the group consisting of methylcyclohexane, cyclohexane, n-heptane and 1,1,2-trichloroethene, and more preferable to be used in the present invention.
- the organic solvent is one or more organic solvents selected from the group consisting of methylcyclohexane, cyclohexane and n-heptane, and more preferable organic solvent is one selected from the group consisting of methylcyclohexane and n-heptane. These are the above organic solvents.
- methylcyclohexane is an organic solvent insoluble in water and has a boiling point of 101 ° C.
- Methylcyclohexane is an organic solvent, so it has a slight anesthetic property, but does not have a strong anesthetic property.
- “Guideline for Residual Solvents of Pharmaceuticals” “To protect patients from possible adverse effects, Although it is classified as a class 2 organic solvent whose residual amount should be regulated, it is not an organic solvent that should be avoided.
- n-heptane is an organic solvent insoluble in water and has a boiling point of 98.4 ° C. Since n-heptane is an organic solvent, it has a slight anesthetic property, but it does not have a strong anesthetic property. Class 3 organic solvents.
- cyclohexane is an organic solvent insoluble in water and has a boiling point of 81.4 ° C. Since cyclohexane is an organic solvent, it has a slight anesthetic property, but does not have a strong anesthetic property.
- the “Guideline for Residual Solvents for Pharmaceuticals” states that “To protect patients from possible harmful effects, Although it is classified as a class 2 organic solvent that should be regulated in terms of residual amount, it is not an organic solvent that should be avoided.
- 1,1,2-trichloroethene is an organic solvent hardly soluble in water and has a boiling point of 87.2 ° C.
- 1,1,2-Trichloroethene has strong anesthetic properties and is an organic solvent containing chlorine. Therefore, there are concerns about environmental pollution and generation of harmful hydrogen chloride and phosgene.
- 1,1,2-Trichloroethene is classified as a class 2 organic solvent in the “Pharmaceutical Residual Solvent Guidelines” that the residual amount should be regulated to protect patients from possible harmful effects It is not an organic solvent that should be avoided.
- acetonitrile is an organic solvent that is extremely soluble in water and has a boiling point of 82 ° C.
- acetonitrile is an organic solvent, it has slight anesthetic properties, but does not have strong anesthetic properties.
- the “Guideline for Residual Solvents for Pharmaceuticals” states that “to protect patients from possible harmful effects, Although it is classified as a class 2 organic solvent that should be regulated in terms of residual amount, it is not an organic solvent that should be avoided.
- 1,2-dimethoxyethane is an organic solvent that is extremely soluble in water and has a boiling point of 82 ° C. Since 1,2-dimethoxyethane is an organic solvent, it has a slight anesthetic property, but it does not have a strong anesthetic property. In order to protect it, it is classified as a class 2 organic solvent whose residual amount should be regulated, but it is not an organic solvent that should be avoided.
- 1,4-dioxane is an organic solvent miscible with water and has a boiling point of 101 ° C. Since 1,4-dioxane is an organic solvent, it has slight anesthetic properties, but it does not have strong anesthetic properties, and the “Pharmaceutical Residual Solvent Guidelines” protects patients from possible adverse effects. Therefore, it is classified as a class 2 organic solvent whose residual amount should be regulated, but it is not an organic solvent that should not be used.
- tetrahydrofuran is an organic solvent miscible with water and has a boiling point of 66 ° C. Tetrahydrofuran is an organic solvent and has slight anesthetic properties, but it does not have strong anesthetic properties.
- the “Guideline for Residual Solvents for Pharmaceuticals” states that “To protect patients from possible adverse effects, Although it is classified as a class 2 organic solvent that should be regulated in terms of residual amount, it is not an organic solvent that should be avoided.
- N, N-dimethylformamide is an organic solvent miscible with water and has a boiling point of 153 ° C. Since N, N-dimethylformamide is an organic solvent, it has a slight anesthetic property, but it does not have a strong anesthetic property. In order to protect it, it is classified as a class 2 organic solvent whose residual amount should be regulated, but it is not an organic solvent that should be avoided.
- ethyl acetate is an organic solvent that dissolves in water up to 8.3 g / 100 mL and has a boiling point of 77.2 ° C. Since ethyl acetate is an organic solvent, it has a slight anesthetic property, but it does not have a strong anesthetic property. Class 3 organic solvents.
- methyl ethyl ketone is an organic solvent that dissolves in water up to 29 g / 100 mL and has a boiling point of 79.6 ° C.
- Methyl ethyl ketone is an organic solvent and has a slight anesthetic property, but does not have a strong anesthetic property. According to the “Guidelines for Residual Solvents for Pharmaceuticals”, it is considered as “a solvent that should be used” and “a low-toxic solvent”. Class 3 organic solvents.
- the condensing agent which can be used in the present invention is polyphosphoric acid, polyphosphoric acid ester, phosphoric acid, sulfuric acid, aluminum chloride, boron trifluoride and tin chloride.
- the condensing agent used in the present invention is preferably polyphosphoric acid.
- the polyphosphoric acid used in the present invention is a linear polymeric phosphoric acid produced by dehydration condensation of orthophosphoric acid. Metaphosphoric acid having the formula (HPO 3 ) n is also included in polyphosphoric acid. Polyphosphoric acid of the general formula H n + 2 P n O 3n + 1 (n is an integer of 2 or more) is indicated by. Examples of polyphosphoric acid include diphosphoric acid, triphosphoric acid, tetraphosphoric acid, and pentaphosphoric acid. The polyphosphoric acid used in the present invention may be a mixture of the above polyphosphoric acids.
- Polyphosphoric acid is, for example, (1) a method of removing water by heating H 3 PO 4 , or (2) a sufficient amount of diphosphorus pentoxide (P 4 O 10 ; phosphoric anhydride) with a sufficient amount of H 3 PO 4 It can manufacture by the method of adding.
- P 4 O 10 diphosphorus pentoxide
- P 4 O 10 diphosphorus pentoxide
- H 3 PO 4 phosphoric anhydride
- the aqueous solution of polyphosphoric acid is easily hydrolyzed, passes through low molecular weight polyphosphoric acid, and finally becomes orthophosphoric acid.
- the degree of polymerization of the polyphosphoric acid used in the present invention is preferably 2 or more, more preferably 5 or more.
- the degree of polymerization of the polyphosphoric acid used in the present invention is preferably 20 or less, more preferably 12 or less.
- the degree of polymerization of polyphosphoric acid depends on (1) the analytical value in terms of P 2 O 5 by acid titration, or (2) polyphosphoric acid condensation rate, ie, orthophosphoric acid generated by hydrolysis of polyphosphoric acid relative to the weight of polyphosphoric acid. Expressed by the weight ratio of the acid, in any case, the larger the value, the higher the degree of polymerization.
- the polyphosphoric acid used in the present invention a commercially available product can be used.
- the analytical value in terms of P 2 O 5 by acid titration is at least 76%, preferably 81% or more, more preferably 83% or more. It is.
- the polyphosphoric acid used in the present invention has a polyphosphoric acid condensation rate of at least 105%, preferably 111% or more, more preferably 115% or more.
- polyphosphoric acid having a condensation rate of 85% to 120% is used.
- the polyphosphoric acid used in the present invention is, for example, a commercially available polyphosphoric acid available from Riedel-de Haen. Heat treatment
- the temperature of the heat treatment in the present invention it is desirable to perform the heat treatment using a temperature at which the ring closure reaction proceeds rapidly and the generation of impurities is minimized. Therefore, the temperature of the heat treatment in the present invention is preferably near the boiling point of the organic solvent to be used.
- the reaction solution for the ring closure reaction is a mixed solution in which polyphosphoric acid and the organic solvent to be used are added to the compound of the formula (II) or the formula (IV) which is the compound before the ring closure.
- the temperature of the treatment can be a temperature not lower than the boiling point of the organic solvent to be used.
- the temperature of the heat treatment suitable in the present invention is a temperature in the range of + 25 ° C. to ⁇ 25 ° C. based on the boiling point of the organic solvent used.
- the boiling points of the organic solvents used in the present invention are 87 ° C for 1,1,2-trichloroethene (trichloroethylene), 101 ° C for methylcyclohexane, 81 ° C for cyclohexane, 98 ° C for n-heptane, acetonitrile.
- the appropriate heat treatment temperatures in the present invention are 62 to 112 ° C. when 1,1,2-trichloroethene (trichloroethylene) is used, 76 to 126 ° C. when methylcyclohexane is used, and cyclohexane is used.
- the temperature of the heat treatment suitable in the present invention is preferably a temperature in the range of + 20 ° C. to ⁇ 20 ° C. of the boiling point of the organic solvent used, more preferably + 15 ° C. to ⁇ 15 of the boiling point of the organic solvent used.
- the preferable heat treatment temperature in each organic solvent is similarly determined according to each preferable temperature range on the basis of the boiling point of each organic solvent.
- the heat treatment time in the present invention is too short, the formation of zaltoprofen or a zaltoprofen derivative is not sufficient, and if it is too long, the amount of impurities increases.
- the heat treatment time in the present invention is desirably a heat treatment time in which a high yield is obtained and the generation of impurities is minimized.
- a preferable heat treatment temperature is 1 to 8 hours, more preferably 3 to 6 hours, and further preferably 3.5 to 4.5 hours.
- the starting material used in the present invention is a compound represented by the following formula (II),
- R 1 and R 2 in the formula each independently represent H or a hydrocarbon group having 1 to 8 carbon atoms, preferably each independently H or a lower alkyl group having 1 to 4 carbon atoms, More preferred is a compound in which R 1 is a methyl group and R 2 is H or a methyl group.
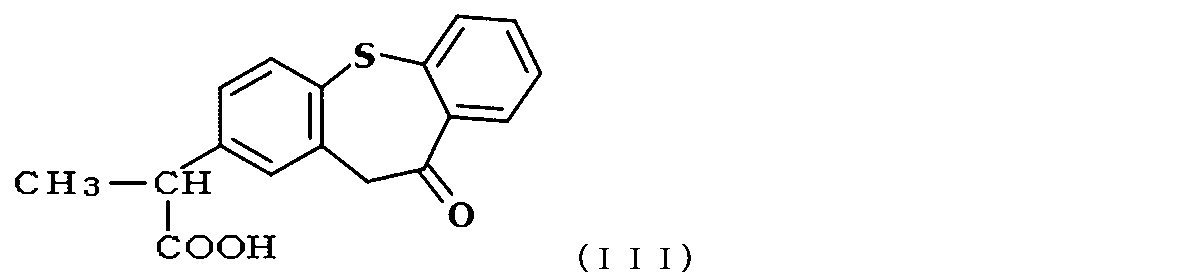
- zaltoprofen is a compound represented by the following formula (III).
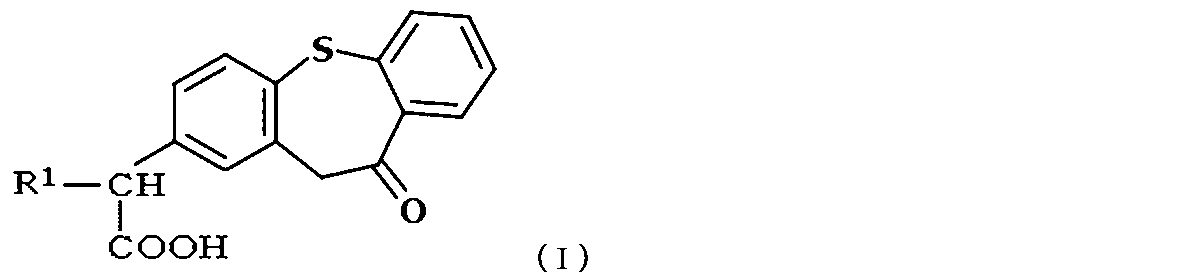
- the zaltoprofen derivative is a compound represented by the following formula (I),
- R 1 is a compound which is H or a hydrocarbon group having 2 to 8 carbon atoms.
- the hydrocarbon group having 2 to 8 carbon atoms is preferably lower alkyl having 2 to 8 carbon atoms, more preferably lower alkyl having 2 to 4 carbon atoms.
- the yield of zaltoprofen or zaltoprofen derivative produced by the production method of the present invention can be increased by extraction with any organic solvent such as ethyl acetate.
- the organic solvent containing the zaltoprofen or the zaltoprofen derivative produced by the production method of the present invention can be dried with anhydrous sodium sulfate or the like. In general, it can be recrystallized for the purpose of isolating and purifying the target product after the reaction for generating the target product or after drying.
- Solvents suitable for extraction and recrystallization are not particularly limited as long as the target product dissolves, but water, methanol, ethanol, acetone, benzene, toluene, which are solvents generally usable in organic chemistry, Optimal ones can be selected from hexane, ethyl acetate, isopropyl ether, and mixed solvents thereof. Isolation and purification of zaltoprofen or zaltoprofen derivatives
- the zaltoprofen or zaltoprofen derivative produced by the production method of the present invention can be used as it is as a pharmaceutical, but an insoluble matter may be filtered off after adding an organic solvent such as acetone and heating to dissolve.
- zaltoprofen or zaltoprofen derivatives produced by the production method of the present invention can be precipitated as crystals by adding an organic solvent such as acetone and lowering the temperature after dissolution by heating.
- the production method of the present invention can be further improved by adding any known technique. For example, in general, since it is an empirically known fact that the production scale and temperature can affect the formation of crystals, the target crystal can be efficiently obtained by controlling the production scale and temperature.
- a single compound has a plurality of crystal forms, and in this case, the crystal form obtained by temperature control such as a set temperature and a cooling rate may be selected.
- temperature control such as a set temperature and a cooling rate
- the aqueous layer was extracted with 150 mL of ethyl acetate, and the separated ethyl acetate and the organic solvent layer were combined. After adding 200 mL of water and 200 mL of brine, the organic solvent layer was separated and washed. After 50 g of anhydrous sodium sulfate was added to the separated organic layer for dehydration, the organic solvent was distilled off under reduced pressure to obtain an orange solid (54.7 g).
- This orange solid (54.7 g) was dissolved by adding 100 mL of acetone, and after acetone was collected and dried up, 240 mL of acetone was added again and heated to 50 ° C. to dissolve. Filtered off. About 1/2 volume of the obtained acetone solution was distilled off, and about 120 mL of the remaining solution was distilled off at room temperature for 22 hours while stirring under reduced pressure. The precipitated crystals were collected by filtration. The crystals collected by filtration were washed with 20 mL of cold acetone and then air-dried to obtain 35.95 g of zaltoprofen as white crystals (yield 76.2%). This white crystal was confirmed to have a purity of 99.9% by high performance liquid chromatography.
- the aqueous layer was extracted with 300 mL of ethyl acetate, and the organic solvent layers were combined and washed by separating the organic solvent layer after adding 500 mL of water and 500 mL of brine. After 50 g of anhydrous sodium sulfate was added to the separated organic layer for dehydration, the organic solvent was distilled off under reduced pressure to obtain a solid (89.9 g).
- the aqueous layer was extracted with 15 mL of ethyl acetate, and the separated ethyl acetate and the organic solvent layer were combined to obtain a zaltoprofen fraction.
- the zaltoprofen fraction was washed by separating the organic solvent layer after adding 30 mL of water and 30 mL of saline.
- the separated organic layer was dehydrated by adding 5 g of anhydrous sodium sulfate, and then the organic solvent was distilled off under reduced pressure to obtain a solid (4.8 g).
- Example 1 was closed in the presence of cyclohexane.
- Zaltoprofen was produced by ring-closing the compound of formula (IV) under the same conditions as in Example 1, and the purity was confirmed by HPLC under the same conditions as in Example 1.
- cyclohexane was used as the solvent for the ring closure
- the heat treatment temperature for the ring closure was 95 to 100 ° C.
- the heat treatment time for the ring closure was 1 hour.
- zaltprofen having a purity of 99.8% was finally obtained as white crystals (yield 63.7%).
- peaks other than the peak of zaltoprofen were not seen. (FIG. 4).
- Example of ring closure in the presence of acetonitrile (CH 3 CN) Sartoprofen was produced by ring closure of the compound of formula (IV) under the same conditions as in Example 1.
- acetonitrile was used as the organic solvent for ring closure
- the heat treatment temperature for ring closure was 100 ° C.
- the heat treatment time for ring closure was 1 hour.
- the compound of IV) and a small amount of impurities were confirmed near the origin (FIG. 5).
- Example of ring closure in the presence of 1,2-dimethoxyethane Sartoprofen was produced by ring closure of the compound of formula (IV) under the same conditions as in Example 1.
- 1,2-dimethoxyethane was used as the organic solvent for the ring closure
- the heat treatment temperature for the ring closure was 95 to 100 ° C.
- a compound of the formula (IV) of the reaction, a small amount of impurities (spots near the origin) and unknown impurities (spots near the solvent front) were confirmed (FIG. 6).
- Example of ring closure in the presence of 1,4-dioxane was produced by ring closure of the compound of formula (IV) under the same conditions as in Example 1.
- 1,4-dioxane was used as the organic solvent for the ring closure
- the heat treatment temperature for the ring closure was 100 to 115 ° C.
- the heat treatment time for the ring closure was 5 hours.
- Example of ring closure in the presence of tetrahydrofuran was produced by ring-closing the compound of formula (IV) under the same conditions as in Example 1.
- tetrahydrofuran was used as the organic solvent for the ring closure
- the heat treatment temperature for the ring closure was 100 ° C.
- the heat treatment time for the ring closure was 5 hours.
- the formation of zaltoprofen was clearly confirmed and unreacted.
- a compound of formula (IV) and a small amount of impurities (spots near the origin) were confirmed (FIG. 7).
- Example of ring closure in the presence of N, N-dimethylformamide Sartoprofen was produced by ring closure of the compound of formula (IV) under the same conditions as in Example 1.
- N, N-dimethylformamide was used as the organic solvent for ring closure
- the heat treatment temperature for ring closure was 115 ° C.
- the heat treatment time for ring closure was 6 hours.
- Example of ring closure in the presence of ethyl acetate was produced by ring-closing the compound of formula (IV) under the same conditions as in Example 1.
- ethyl acetate was used as the organic solvent for ring closure
- the heat treatment temperature for ring closure was 100 ° C.
- the heat treatment time for ring closure was 6 hours.
- zaltoprofen was the main product.
- an unreacted compound of the formula (IV) and impurities (spots near the solvent front end) considered to be decomposition products were confirmed (FIG. 8).
- Example of ring closure in the presence of xylene Zaltoprofen was produced by ring closure of the compound of formula (IV) under the same conditions as in Example 1.
- xylene was used as the organic solvent for the ring closure
- the heat treatment temperature for the ring closure was 97 to 100 ° C.
- the heat treatment time for the ring closure was 2 hours. It was confirmed by high performance liquid chromatography under the same conditions as in Example 1 that zaltprofen having a purity of 99.8% was finally obtained as white crystals (yield 57.8%).
- peaks other than the peak of zaltoprofen were not seen. (FIG. 10).
- Example Zaltoprofen was produced by ring closure of the compound of formula (IV) under the same conditions as in Example 1, and the purity was confirmed by HPLC under the same conditions as in Example 1. did. However, 1,2-dichloroethane was used as the organic solvent for ring closure, the heat treatment temperature for ring closure was 85 to 95 ° C., and the heat treatment time for ring closure was 4.5 hours. It was confirmed by high performance liquid chromatography under the same conditions as in Example 1 that zaltprofen having a purity of 99.9% was finally obtained as white crystals (yield 67.2%). Moreover, as long as it confirmed on the chart of a high performance liquid chromatography, peaks other than the peak of zaltoprofen were not seen. (Fig. 11)
- Example of ring closure in the presence of toluene Under the same conditions as in Example 1, zaltoprofen was produced by ring closure of the compound of formula (IV), and the purity was confirmed by HPLC under the same conditions as in Example 1.
- toluene was used as the organic solvent for ring closure
- the heat treatment temperature for ring closure was 97 ° C.
- the heat treatment time for ring closure was 3 hours. It was confirmed by high performance liquid chromatography under the same conditions as in Example 1 that finally zaltoprofen having a purity of 99.1% was obtained as pale yellow crystals (FIG. 12).
- Example 1 Zaltoprofen was produced by cyclization of the compound of formula (IV) under the same conditions as in Example 1 in which the ring was closed in the presence of chloroform, and the purity was confirmed by HPLC under the same conditions as in Example 1.
- chloroform was used as the organic solvent for ring closure
- the heat treatment temperature for ring closure was 98 ° C.
- the heat treatment time for ring closure was 4 hours. It was confirmed by high performance liquid chromatography under the same conditions as in Example 1 that finally zaltoprofen having a purity of 99.5% was obtained as white crystals (FIG. 13).
- zaltoprofen and a derivative of zaltoprofen are used in a purity of 99% or more by using polyphosphoric acid in an amount of 6.5 times or less of the compound of formula (II) or formula (IV) as a condensing agent. And since zaltoprofen or a zaltoprofen derivative can be manufactured with a yield of 60% or more, it is industrially useful.
- zaltoprofen and a zaltoprofen derivative can be produced using an organic solvent that is less susceptible to health hazards for workers on the production site and that is easy to handle for work. Is suitable.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pain & Pain Management (AREA)
- Pharmacology & Pharmacy (AREA)
- Rheumatology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Compounds Containing Sulfur Atoms (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Description


次式(I):
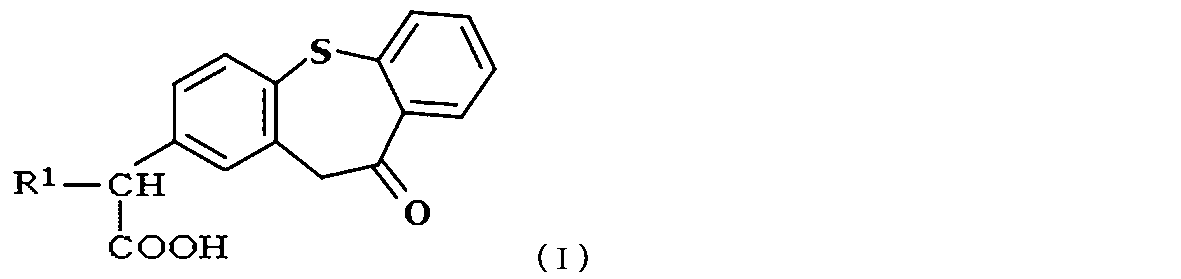
R1は、Hまたは炭素数1~8の炭化水素基である;
で表される化合物の製造方法において、
次式(II):
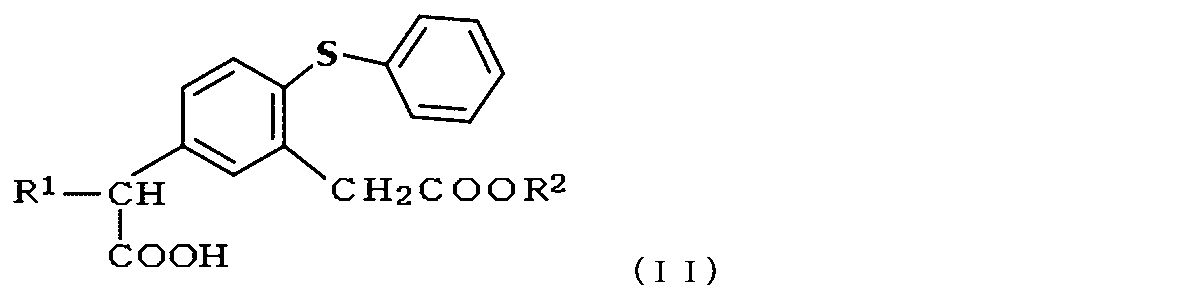
R1は、Hまたは炭素数1~8の炭化水素基であり;
R2は、Hまたは炭素数1~8の炭化水素基である;
で表される化合物を、
1,1,2-トリクロロエテン、メチルシクロヘキサン、シクロヘキサン、n-ヘプタン、アセトニトリル、1,2-ジメトキシエタン、1,4-ジオキサン、テトラヒドロフラン、N,N-ジメチルホルムアミド、酢酸エチル、および、メチルエチルケトンからなる群から選択される少なくとも一つの有機溶媒の存在下に、
ポリリン酸、オルトリン酸および濃硫酸からなる群から選択される少なくとも一つの縮合剤とともに加熱処理することにより閉環することを特徴とする方法。
有機溶媒が、1,1,2-トリクロロエテン、メチルシクロヘキサン、シクロヘキサン、および、n-ヘプタンからなる群から選択される少なくとも一つの有機溶媒である、[1]に記載の製造方法。
[3]
縮合剤が、ポリリン酸である、[1]または[2]に記載の製造方法。
[4]
加熱処理の温度が、使用する有機溶媒の沸点を基準としてその+25℃~-25℃の範囲の温度で行われる、請求項[1]~[3]のいずれかに記載の製造方法。
加熱処理の時間が、1~8時間である、[1]~[4]のいずれかに記載の製造方法。
[6]
縮合剤の重量が、式(II)で表される化合物の好ましくは1~10倍、より好ましくは1~6.5倍の重量である、[1]~[5]のいずれかに記載の製造方法。
[7]
有機溶媒の量が、式(II)で表される化合物の量が50gである場合に、50~100mLの割合である、[1]~[6]のいずれかに記載の製造方法。
R1がメチル基である、[1]~[7]のいずれかに記載の製造方法。
[9]
R2がHまたはメチル基である、[1]~[8]のいずれかに記載の製造方法。
[10]
105%以上の縮合率のポリリン酸を用いる、[1]~[9]のいずれかに記載の製造方法。
ザルトプロフェンまたはザルトプロフェン誘導体を、酢酸エチルを用いて抽出することをさらに含む、[1]~[10]のいずれかに記載の製造方法。
[12]
ザルトプロフェンまたはザルトプロフェン誘導体を、無水硫酸ナトリウムによって乾燥させることをさらに含む、[1]~[11]のいずれかに記載の製造方法。
ザルトプロフェンまたはザルトプロフェン誘導体に、有機溶媒を加えて加温溶解し、不溶物を濾別することをさらに含む、[1]~[12]のいずれかに記載の製造方法。
[14]
ザルトプロフェンまたはザルトプロフェン誘導体を、有機溶媒を加えて加温溶解後に温度を下げて晶析させることをさらに含む、[1]~[13]のいずれかに記載の製造方法。
本発明において使用する有機溶媒は、メチルシクロヘキサン、シクロヘキサン、n-ヘプタン、1,1,2-トリクロロエテン、アセトニトリル、ジメトキシエタン、1,4-ジオキサン、テトラヒドロフラン、N,N-ジメチルホルムアミド、酢酸エチル、および、メチルエチルケトンからなる群から選択される1種以上の有機溶媒である。本発明において使用する好ましい有機溶媒は、メチルシクロヘキサン、シクロヘキサン、n-ヘプタンおよび1,1,2-トリクロロエテンからなる群から選択される1種以上の有機溶媒であり、本発明において使用するより好ましい有機溶媒は、メチルシクロヘキサン、シクロヘキサンおよびn-ヘプタン、からなる群から選択される1種以上の有機溶媒であり、さらに好ましい有機溶媒は、メチルシクロヘキサンおよびn-ヘプタンからなる群から選択される1種以上の有機溶媒である。
本発明において使用可能な縮合剤は、ポリリン酸、ポリリン酸エステル、リン酸、硫酸、塩化アルミニウム、三フツ化ホウ素、塩化スズである。本発明において使用する縮合剤は、好ましくは、ポリリン酸である。
またリン酸と無水リン酸(5酸化二リン)を混合して得られる粘度が700~1000mPa・sでリン酸濃度が110~130%のものも使用することができる。(特開2005-289949)
加熱処理
したがって、本発明における加熱処理の温度は、使用する有機溶媒の沸点付近の温度が好ましい。
ここで、閉環反応の反応液は、閉環前の化合物である式(II)もしくは式(IV)の化合物に対してポリリン酸および使用する有機溶媒を加えた混合液であるので、本発明における加熱処理の温度は、使用する有機溶媒の沸点以上の温度とすることもできる。
本発明において好ましい加熱処理の温度は、1~8時間、より好ましくは3~6時間、さらに好ましくは3.5~4.5時間である。
出発原料

ザルトプロフェンまたはザルトプロフェン誘導体
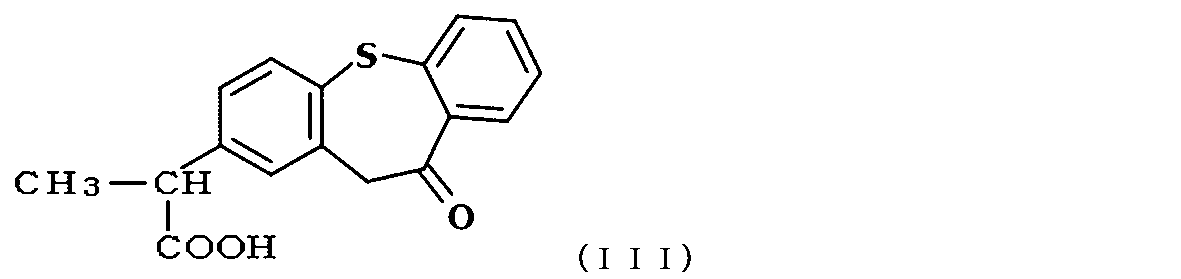

ザルトプロフェンまたはザルトプロフェン誘導体の抽出
ザルトプロフェンまたはザルトプロフェン誘導体の単離・精製
本発明の製造方法には、任意の公知技術を付加してさらに改良することも可能である。例えば、一般に、製造規模および温度が結晶の生成に影響し得ることは経験的に知られた事実であるので、製造規模および温度を制御することで、目的の結晶を効率よく得ることができる。また例えば、一つの化合物に複数の結晶形が存在することも、頻繁に経験することであり、この場合、設定する温度、冷却速度などの温度制御によって得られる結晶形を選別できる場合があることが知られている(PHARM TECH JAPAN vol.17 N0.12(2001)p27-38,特開2006-160766)。また、一般に、種結晶として接種することにより、効率よく目的の結晶を得ることができることも良く知られた事実である。
50g(0.158mol)の下記式(IV)

100g(0.316mol)の下記式(IV)
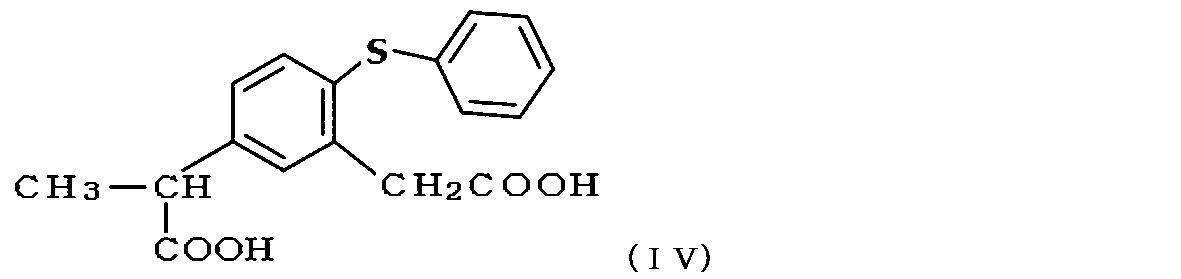
5.20g(16.4mmol)の下記式(IV)

実施例1と同様の条件で、式(IV)の化合物を閉環させることによりザルトプロフェンを製造し、実施例1と同一の条件のHPLCにより純度を確認した。ただし、閉環の際の溶媒はシクロヘキサンを使用し、閉環の際の加熱処理温度は95~100℃、閉環の際の加熱処理時間は1時間であった。最終的に純度99.8%のザルトプロフェンを白色結晶として得たことを、実施例1と同条件の高速液体クロマトグラフィーにより確認した(収率63.7%)。また、高速液体クロマトグラフィーのチャート上で確認する限り、ザルトプロフェンのピーク以外のピークは見られなかった。(図4)。
実施例1と同様の条件で、式(IV)の化合物を閉環させることによりザルトプロフェンを製造した。ただし、閉環の際の有機溶媒はアセトニトリルを使用し、閉環の際の加熱処理温度は100℃、閉環の際の加熱処理時間は1時間であった。薄層クロマトグラフィー(CHCl3:MeOH:AcOH=100:10:1、254nmの波長における紫外吸収で検出)により、純度を確認したところ、ザルトプロフェンの生成が確認されたが、同時に未反応の式(IV)の化合物、および、原点付近に少量の不純物が確認された(図5)。
実施例1と同様の条件で、式(IV)の化合物を閉環させることによりザルトプロフェンを製造した。ただし、閉環の際の有機溶媒は1,2-ジメトキシエタンを使用し、閉環の際の加熱処理温度は95~100℃、閉環の際の加熱処理時間は5時間であった。薄層クロマトグラフィー(CHCl3:MeOH:AcOH=100:10:1、254nmの波長における紫外吸収で検出)により、純度を確認したところ、ザルトプロフェンの生成が主要なスポットとして確認されたが、同時に未反応の式(IV)の化合物、少量の不純物(原点付近のスポット)および不明な不純物(溶媒先端付近のスポット)が確認された(図6)。
実施例1と同様の条件で、式(IV)の化合物を閉環させることによりザルトプロフェンを製造した。ただし、閉環の際の有機溶媒は1,4-ジオキサンを使用し、閉環の際の加熱処理温度は100~115℃、閉環の際の加熱処理時間は5時間であった。薄層クロマトグラフィー(CHCl3:MeOH:AcOH=100:10:1、254nmの波長における紫外吸収で検出)により、少量のザルトプロフェンに生成が確認されたが、未反応の式(IV)の化合物が多量に存在することが確認された。
実施例1と同様の条件で、式(IV)の化合物を閉環させることによりザルトプロフェンを製造した。ただし、閉環の際の有機溶媒はテトラヒドロフランを使用し、閉環の際の加熱処理温度は100℃、閉環の際の加熱処理時間は5時間であった。薄層クロマトグラフィー(CHCl3:MeOH:AcOH=100:10:1、254nmの波長における紫外吸収で検出)により、純度を確認したところ、ザルトプロフェンの生成が明確に確認されると同時に、未反応の式(IV)の化合物、および、少量の不純物(原点付近のスポット)が確認された(図7)。
実施例1と同様の条件で、式(IV)の化合物を閉環させることによりザルトプロフェンを製造した。ただし、閉環の際の有機溶媒はN,N-ジメチルホルムアミドを使用し、閉環の際の加熱処理温度は115℃、閉環の際の加熱処理時間は6時間であった。薄層クロマトグラフィー(CHCl3:MeOH:AcOH=100:10:1、254nmの波長における紫外吸収で検出)により純度を確認したところ、ザルトプロフェンの生成はほとんど確認されなかった。
実施例1と同様の条件で、式(IV)の化合物を閉環させることによりザルトプロフェンを製造した。ただし、閉環の際の有機溶媒は酢酸エチルを使用し、閉環の際の加熱処理温度は100℃、閉環の際の加熱処理時間は6時間であった。薄層クロマトグラフィー(CHCl3:MeOH:AcOH=100:10:1、254nmの波長における紫外吸収で検出)により、純度を確認したところ、ザルトプロフェンが主要な生成物であることが確認されると同時に、未反応の式(IV)の化合物、および、分解産物と思われる不純物(溶媒先端付近のスポット)が確認された(図8)。
実施例1と同様の条件で、式(IV)の化合物を閉環させることによりザルトプロフェンを製造した。ただし、閉環の際の有機溶媒はメチルエチルケトンを使用し、閉環の際の加熱処理温度は95~101℃、閉環の際の加熱処理時間は2時間であった。薄層クロマトグラフィー(CHCl3:MeOH:AcOH=100:10:1、254nmの波長における紫外吸収で検出)により、純度を確認したところ、ザルトプロフェンの生成が確認されると同時に、未反応の式(IV)の化合物、および、分解産物と思われる4種類のの不純物(ザルトプロフェンのスポットと溶媒先端の間のスポット)が確認された(図9)。
実施例1と同様の条件で、式(IV)の化合物を閉環させることによりザルトプロフェンを製造した。ただし、閉環の際の有機溶媒はキシレンを使用し、閉環の際の加熱処理温度は97~100℃、閉環の際の加熱処理時間は2時間であった。最終的に純度99.8%のザルトプロフェンを白色結晶として得たことを、実施例1と同条件の高速液体クロマトグラフィーにより確認した(収率57.8%)。また、高速液体クロマトグラフィーのチャート上で確認する限り、ザルトプロフェンのピーク以外のピークは見られなかった。(図10)。
実施例1と同様の条件で、式(IV)の化合物を閉環させることによりザルトプロフェンを製造し、実施例1と同一の条件のHPLCにより純度を確認した。ただし、閉環の際の有機溶媒は1,2-ジクロロエタンを使用し、閉環の際の加熱処理温度は85~95℃、閉環の際の加熱処理時間は4.5時間であった。最終的に純度99.9%のザルトプロフェンを白色結晶として得たことを、実施例1と同条件の高速液体クロマトグラフィーにより確認した(収率67.2%)。また、高速液体クロマトグラフィーのチャート上で確認する限り、ザルトプロフェンのピーク以外のピークは見られなかった。(図11)
実施例1と同様の条件で、式(IV)の化合物を閉環させることによりザルトプロフェンを製造し、実施例1と同一の条件のHPLCにより純度を確認した。ただし、閉環の際の有機溶媒はトルエンを使用し、閉環の際の加熱処理温度は97℃、閉環の際の加熱処理時間は3時間であった。最終的に純度99.1%のザルトプロフェンを淡黄色結晶として得たことを、実施例1と同条件の高速液体クロマトグラフィーにより確認した(図12)。
実施例1と同様の条件で、式(IV)の化合物を閉環させることによりザルトプロフェンを製造し、実施例1と同一の条件のHPLCにより純度を確認した。ただし、閉環の際の有機溶媒はクロロホルムを使用し、閉環の際の加熱処理温度は98℃、閉環の際の加熱処理時間は4時間であった。最終的に純度99.5%のザルトプロフェンを白色結晶として得たことを、実施例1と同条件の高速液体クロマトグラフィーにより確認した(図13)。
Claims (14)
- 次式(I):
R1は、Hまたは炭素数1~8の炭化水素基である;
で表される化合物の製造方法において、
次式(II):

R1は、Hまたは炭素数1~8の炭化水素基であり;
R2は、Hまたは炭素数1~8の炭化水素基である;
で表される化合物を、
1,1,2-トリクロロエテン、メチルシクロヘキサン、シクロヘキサン、n-ヘプタン、アセトニトリル、1,2-ジメトキシエタン、1,4-ジオキサン、テトラヒドロフラン、N,N-ジメチルホルムアミド、酢酸エチル、および、メチルエチルケトンからなる群から選択される少なくとも一つの有機溶媒の存在下に、
ポリリン酸、オルトリン酸および濃硫酸からなる群から選択される少なくとも一つの縮合剤とともに加熱処理することにより閉環することを特徴とする方法。 - 有機溶媒が、1,1,2-トリクロロエテン、メチルシクロヘキサン、シクロヘキサン、および、n-ヘプタンからなる群から選択される少なくとも一つの有機溶媒である、請求項1に記載の製造方法。
- 縮合剤が、ポリリン酸である、請求項1または2に記載の製造方法。
- 加熱処理の温度が、使用する有機溶媒の沸点を基準としてその+25℃~-25℃の範囲の温度で行われる、請求項1~3のいずれか一項に記載の製造方法。
- 加熱処理の時間が、1~8時間である、請求項1~4のいずれか一項に記載の製造方法。
- 縮合剤の重量が、式(II)で表される化合物の重量の1~10倍の重量である、請求項1~5のいずれか一項に記載の製造方法。
- 有機溶媒の量が、式(II)で表される化合物の量が50gである場合に、50~100mLの割合である、請求項1~6のいずれか一項に記載の製造方法。
- R1がメチル基である、請求項1~7のいずれか一項に記載の製造方法。
- R2がHまたはメチル基である、請求項1~8のいずれか一項に記載の製造方法。
- 105%以上の縮合率のポリリン酸を用いる、請求項1~9のいずれか一項に記載の製造方法。
- ザルトプロフェンまたはザルトプロフェン誘導体を、酢酸エチルを用いて抽出することをさらに含む、請求項1~10のいずれか一項に記載の製造方法。
- ザルトプロフェンまたはザルトプロフェン誘導体を、無水硫酸ナトリウムによって乾燥させることをさらに含む、請求項1~11のいずれか一項に記載の製造方法。
- ザルトプロフェンまたはザルトプロフェン誘導体に、有機溶媒を加えて加温溶解し、不溶物を濾別することをさらに含む、請求項1~12のいずれか一項に記載の製造方法。
- ザルトプロフェンまたはザルトプロフェン誘導体を、有機溶媒を加えて加温溶解後に温度を下げて晶析させることをさらに含む、請求項1~13のいずれか一項に記載の製造方法。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SG11201406873QA SG11201406873QA (en) | 2012-04-24 | 2013-04-24 | Method for preparing zaltoprofen and derivative thereof |
| JP2014512628A JP5937679B2 (ja) | 2012-04-24 | 2013-04-24 | ザルトプロフェンおよびその誘導体の製造方法 |
| CN201380010158.0A CN104185633B (zh) | 2012-04-24 | 2013-04-24 | 扎托布洛芬及其衍生物的制造方法 |
| EP13780982.8A EP2842952A4 (en) | 2012-04-24 | 2013-04-24 | PROCESS FOR PRODUCING ZALTOPROFEN AND ITS DERIVATIVE |
| KR1020147026146A KR101677970B1 (ko) | 2012-04-24 | 2013-04-24 | 잘토프로펜 및 그 유도체의 제조 방법 |
| HK15103875.7A HK1203490A1 (en) | 2012-04-24 | 2015-04-22 | Method for producing zaltoprofen and derivative thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-098736 | 2012-04-24 | ||
| JP2012098736 | 2012-04-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013161842A1 true WO2013161842A1 (ja) | 2013-10-31 |
Family
ID=49483150
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/061974 WO2013161842A1 (ja) | 2012-04-24 | 2013-04-24 | ザルトプロフェンおよびその誘導体の製造方法 |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP2842952A4 (ja) |
| JP (1) | JP5937679B2 (ja) |
| KR (1) | KR101677970B1 (ja) |
| CN (1) | CN104185633B (ja) |
| HK (1) | HK1203490A1 (ja) |
| SG (1) | SG11201406873QA (ja) |
| WO (1) | WO2013161842A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110590737A (zh) * | 2019-09-25 | 2019-12-20 | 华中农业大学 | 一种高比活度氚标记扎托布洛芬及其制备方法 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105198858B (zh) * | 2015-10-30 | 2017-07-21 | 天津药物研究院药业有限责任公司 | 扎托布洛芬的纯化方法 |
| CN105218514A (zh) * | 2015-10-30 | 2016-01-06 | 天津药物研究院药业有限责任公司 | 扎托布洛芬的制备方法 |
| CN110407804A (zh) * | 2019-07-18 | 2019-11-05 | 河南后羿制药有限公司 | 微通道反应器在扎托布洛芬的环合反应中的应用及扎托布洛芬的环合方法 |
| CN113200973B (zh) * | 2021-04-22 | 2022-07-26 | 华中农业大学 | 一种基于扎托布洛芬母体结构的化合物及其抗炎用途 |
Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5553282A (en) | 1978-10-17 | 1980-04-18 | Nippon Chemiphar Co Ltd | Dibenzothiepin derivative and its preparation |
| JPS57106678A (en) | 1980-12-23 | 1982-07-02 | Nippon Chemiphar Co Ltd | Preparation of dibenzothiepinpropionic acid derivative |
| JPS57171991A (en) | 1981-04-14 | 1982-10-22 | Fuji Photo Film Co Ltd | Preparation of 2-(10,11-dihydro-10-oxodibenzo(b,f)thiepin-2- yl)propionic acid |
| JPS61251682A (ja) | 1985-04-30 | 1986-11-08 | Sankyo Kagaku Kk | 2−(10,11−ジヒドロ−10−オキソジベンゾ〔b,f〕チエピン−2−イル)プロピオン酸の製造方法 |
| JPS62108877A (ja) | 1985-11-07 | 1987-05-20 | Nippon Chemiphar Co Ltd | 2−(10,11−ジヒドロ−10−オキソジベンゾ〔b,f〕チエピン−2−イル)プロピオン酸の精製法 |
| JPS62292780A (ja) | 1986-06-10 | 1987-12-19 | Nippon Chemiphar Co Ltd | ジベンゾチエピン誘導体の製造法 |
| JPH0899953A (ja) | 1995-04-24 | 1996-04-16 | Nippon Chemiphar Co Ltd | 新規ハロケタール化合物 |
| JP2005247778A (ja) | 2004-03-05 | 2005-09-15 | Shigeji Maekawa | ジベンゾチエピンプロピオン酸誘導体の製造方法 |
| JP2005289949A (ja) | 2004-04-06 | 2005-10-20 | Tokuyama Corp | ケトン化合物の製造方法 |
| JP2006160766A (ja) | 2005-02-22 | 2006-06-22 | Mitsubishi Pharma Corp | (±)2−(ジメチルアミノ)−1−{〔O−(m−メトキシフェネチル)フェノキシ〕メチル}エチル水素サクシナート塩酸塩の結晶 |
| JP2006273731A (ja) | 2005-03-28 | 2006-10-12 | Tokuyama Corp | 2−(10、11−ジヒドロ−10−オキシジベンゾ〔b、f〕チエピン−2−イル)プロピオン酸の製造方法 |
| JP2006290753A (ja) | 2005-04-06 | 2006-10-26 | Tokuyama Corp | 2−(10、11−ジヒドロ−10−オキシジベンゾ〔b、f〕チエピン−2−イル)プロピオン酸の製造方法 |
-
2013
- 2013-04-24 CN CN201380010158.0A patent/CN104185633B/zh active Active
- 2013-04-24 WO PCT/JP2013/061974 patent/WO2013161842A1/ja active Application Filing
- 2013-04-24 SG SG11201406873QA patent/SG11201406873QA/en unknown
- 2013-04-24 KR KR1020147026146A patent/KR101677970B1/ko active IP Right Grant
- 2013-04-24 JP JP2014512628A patent/JP5937679B2/ja active Active
- 2013-04-24 EP EP13780982.8A patent/EP2842952A4/en not_active Withdrawn
-
2015
- 2015-04-22 HK HK15103875.7A patent/HK1203490A1/xx unknown
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5553282A (en) | 1978-10-17 | 1980-04-18 | Nippon Chemiphar Co Ltd | Dibenzothiepin derivative and its preparation |
| JPS57106678A (en) | 1980-12-23 | 1982-07-02 | Nippon Chemiphar Co Ltd | Preparation of dibenzothiepinpropionic acid derivative |
| JPS57171991A (en) | 1981-04-14 | 1982-10-22 | Fuji Photo Film Co Ltd | Preparation of 2-(10,11-dihydro-10-oxodibenzo(b,f)thiepin-2- yl)propionic acid |
| JPS61251682A (ja) | 1985-04-30 | 1986-11-08 | Sankyo Kagaku Kk | 2−(10,11−ジヒドロ−10−オキソジベンゾ〔b,f〕チエピン−2−イル)プロピオン酸の製造方法 |
| JPS62108877A (ja) | 1985-11-07 | 1987-05-20 | Nippon Chemiphar Co Ltd | 2−(10,11−ジヒドロ−10−オキソジベンゾ〔b,f〕チエピン−2−イル)プロピオン酸の精製法 |
| JPS62292780A (ja) | 1986-06-10 | 1987-12-19 | Nippon Chemiphar Co Ltd | ジベンゾチエピン誘導体の製造法 |
| JPH0899953A (ja) | 1995-04-24 | 1996-04-16 | Nippon Chemiphar Co Ltd | 新規ハロケタール化合物 |
| JP2005247778A (ja) | 2004-03-05 | 2005-09-15 | Shigeji Maekawa | ジベンゾチエピンプロピオン酸誘導体の製造方法 |
| JP2005289949A (ja) | 2004-04-06 | 2005-10-20 | Tokuyama Corp | ケトン化合物の製造方法 |
| JP2006160766A (ja) | 2005-02-22 | 2006-06-22 | Mitsubishi Pharma Corp | (±)2−(ジメチルアミノ)−1−{〔O−(m−メトキシフェネチル)フェノキシ〕メチル}エチル水素サクシナート塩酸塩の結晶 |
| JP2006273731A (ja) | 2005-03-28 | 2006-10-12 | Tokuyama Corp | 2−(10、11−ジヒドロ−10−オキシジベンゾ〔b、f〕チエピン−2−イル)プロピオン酸の製造方法 |
| JP2006290753A (ja) | 2005-04-06 | 2006-10-26 | Tokuyama Corp | 2−(10、11−ジヒドロ−10−オキシジベンゾ〔b、f〕チエピン−2−イル)プロピオン酸の製造方法 |
Non-Patent Citations (2)
| Title |
|---|
| PHARM TECH JAPAN, vol. 17, no. 12, 2001, pages 27 - 38 |
| See also references of EP2842952A4 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110590737A (zh) * | 2019-09-25 | 2019-12-20 | 华中农业大学 | 一种高比活度氚标记扎托布洛芬及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| HK1203490A1 (en) | 2015-10-30 |
| EP2842952A1 (en) | 2015-03-04 |
| EP2842952A4 (en) | 2015-09-09 |
| SG11201406873QA (en) | 2014-12-30 |
| CN104185633A (zh) | 2014-12-03 |
| CN104185633B (zh) | 2018-01-30 |
| KR20140132737A (ko) | 2014-11-18 |
| KR101677970B1 (ko) | 2016-11-21 |
| JP5937679B2 (ja) | 2016-06-22 |
| JPWO2013161842A1 (ja) | 2015-12-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5937679B2 (ja) | ザルトプロフェンおよびその誘導体の製造方法 | |
| AU2006281449A1 (en) | Method for producing betamimetics | |
| FI84913C (fi) | Foerfarande foer framstaellning av 2-alkoxi-n-(1-azabicyclo/2.2.2./oktan-3-yl)aminobensamider. | |
| CN101723929B (zh) | 一种4,5-二甲基-2-氧代-1,3-二氧杂环戊烯的纯化方法 | |
| US20230286901A1 (en) | Process for the synthesis of melphalan | |
| JP4568398B2 (ja) | ヘキサヒドロイソインドリン・酸付加塩およびその使用方法 | |
| CN115093339A (zh) | 一种l-草铵膦中间体的合成方法 | |
| ES2441996T3 (es) | Procedimiento para la preparación de 4a,5,9,10,11,12,-hexahidro-6H-benzofuro[3a,3,2-ef][2]benzazepina altamente pura asi como sus derivados | |
| Linch | CLXXXVII.—3-Aminocoumarin | |
| CN101563309B (zh) | 纯化对苯二甲醛的方法 | |
| FR2593179A1 (fr) | Derives d'imidazo(1,2-a)quinoleines, leur preparation et leur application en therapeutique | |
| IE852338L (en) | 4h-1,2,4 - triazole derivative. | |
| WO2013093928A1 (en) | An improved process for preparing 2-oxindoles of formula i, a key raw material for making pharmaceutical drugs and intermediates thereof | |
| JP2008222593A (ja) | アルキルアミノピリジン類の精製方法 | |
| Hu et al. | Facile synthesis of novel tacrine analogues | |
| JP2007277101A (ja) | 2,3,6,7,10,11−ヘキサヒドロキシトリフェニレンの製造方法 | |
| JP2006290753A (ja) | 2−(10、11−ジヒドロ−10−オキシジベンゾ〔b、f〕チエピン−2−イル)プロピオン酸の製造方法 | |
| JP4303685B2 (ja) | 2−シクロペンテン−1−オンの製造方法 | |
| JP2005082496A (ja) | 2’−(1h−テトラゾール−5−イル)ビフェニル−4−カルボアルデヒド結晶およびその製造方法 | |
| WO2021233884A1 (en) | Continuous process for manufacturing alkyl 7-amino-5-methyl-[1,2,5]oxadiazolo[3,4-b]pyridine-carboxylate | |
| JPH0586000A (ja) | 2−アミノ−4−フルオロ安息香酸の製造方法 | |
| CN115611901A (zh) | 一种氮杂䓬类化合物或其药学上可接受的盐及其制备方法和应用 | |
| JP2011093869A (ja) | 光学活性2−フェノキシブタン酸類の製造方法 | |
| Sánchez et al. | New synthesis of methylfuro [3, 4-b][1, 4] benzoxazine as an intermediate in the preparation of polycyclic compounds | |
| HU227949B1 (en) | Method for producing derivatives of biphenyl-2-carboxylic acid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13780982 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014512628 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20147026146 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013780982 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013780982 Country of ref document: EP |