WO2013104597A1 - Substituierte triazine derivate und ihre verwendung als stimulatoren der löslichen guanylatcyclase - Google Patents
Substituierte triazine derivate und ihre verwendung als stimulatoren der löslichen guanylatcyclase Download PDFInfo
- Publication number
- WO2013104597A1 WO2013104597A1 PCT/EP2013/050179 EP2013050179W WO2013104597A1 WO 2013104597 A1 WO2013104597 A1 WO 2013104597A1 EP 2013050179 W EP2013050179 W EP 2013050179W WO 2013104597 A1 WO2013104597 A1 WO 2013104597A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- alkyl
- substituted
- trifluoromethyl
- Prior art date
Links
- 0 CC(*)(CCC1)CCc2c1[n](*)nc2C Chemical compound CC(*)(CCC1)CCc2c1[n](*)nc2C 0.000 description 4
- UXVZTMLFTJWIHY-UHFFFAOYSA-N CS(Nc1c(N)nc(-c2n[n](Cc(cccc3)c3F)c3ncccc23)nn1)(=O)=O Chemical compound CS(Nc1c(N)nc(-c2n[n](Cc(cccc3)c3F)c3ncccc23)nn1)(=O)=O UXVZTMLFTJWIHY-UHFFFAOYSA-N 0.000 description 1
- GFJWFOUAJQUZOK-UHFFFAOYSA-N Nc1c(-c2cccnc2)nnc(-c2n[n](Cc(cccc3)c3F)c3ncccc23)n1 Chemical compound Nc1c(-c2cccnc2)nnc(-c2n[n](Cc(cccc3)c3F)c3ncccc23)n1 GFJWFOUAJQUZOK-UHFFFAOYSA-N 0.000 description 1
- ZXRWOWYQXVQTGH-UHFFFAOYSA-N Nc1c(C2CCCC2)nnc(-c2n[n](Cc(cccc3)c3F)c3ncccc23)n1 Chemical compound Nc1c(C2CCCC2)nnc(-c2n[n](Cc(cccc3)c3F)c3ncccc23)n1 ZXRWOWYQXVQTGH-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Definitions
- the present application relates to novel substituted triazines, processes for their preparation, their use alone or in combinations for the treatment and / or prophylaxis of diseases and their use for the preparation of medicaments for the treatment and / or prophylaxis of diseases, in particular for the treatment and / or prophylaxis of cardiovascular diseases.
- cyclic guanosine monophosphate cGMP
- NO nitric oxide
- the guanylate cyclases catalyze the biosynthesis of cGMP from guanosine triphosphate (GTP).
- GTP guanosine triphosphate
- the previously known members of this family can be divided into two groups according to both structural features and the nature of the ligands: the particulate guanylate cyclases stimulable by natriuretic peptides and the soluble guanylate cyclases stimulable by NO.
- the soluble guanylate cyclases consist of two subunits and most likely contain one heme per heterodimer that is part of the regulatory center. This is central to the activation mechanism. NO can bind to the iron atom of the heme and thus significantly increase the activity of the enzyme. On the other hand, heme-free preparations can not be stimulated by NO. Also, carbon monoxide (CO) is able to bind to the central iron atom of the heme, with stimulation by CO being significantly less than by NO.
- CO carbon monoxide
- guanylate cyclase plays a crucial role in various physiological processes, in particular in the relaxation and proliferation of smooth muscle cells, platelet aggregation and adhesion, neuronal signaling and diseases based on a disturbance of the above operations.
- the NO / cGMP system may be suppressed, which may, for example, lead to hypertension, platelet activation, increased cell proliferation, endothelial dysfunction, arteriosclerosis, angina pectoris, heart failure, myocardial infarction, thrombosis, stroke and sexual dysfunction.
- a NO-independent treatment option for such diseases which is aimed at influencing the cGMP pathway in organisms, is a promising approach on account of the expected high efficiency and low side effects.
- WO 2004/009590 describes pyrazolopyridines with substituted 4-aminopyrimidines for the treatment of CNS diseases.
- WO 2010/065275 discloses substituted pyrrolo and dihydropyridopyrimidines as sGC activators.
- the object of the present invention was to provide new substances which act as stimulators of soluble guanylate cyclase and have a similar or improved therapeutic profile over the compounds known from the prior art, for example with respect to their in vivo properties, such as their pharmacokinetic and pharmacodynamic Behavior and / or its metabolism profile and / or its dose-response relationship.
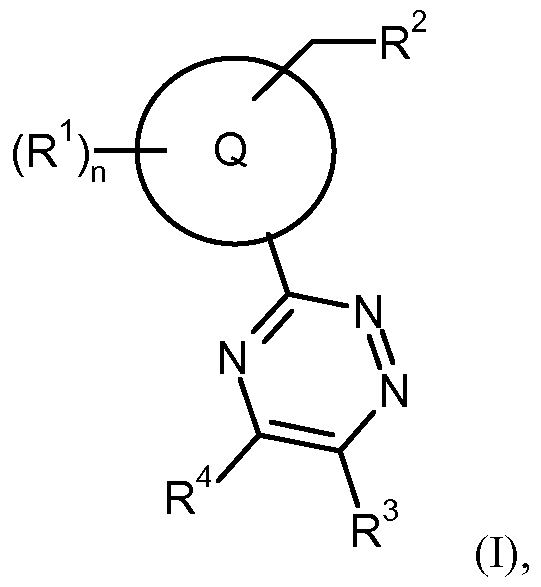
- the present invention relates to compounds of the general formula (I)
- ring Q is 8- or 9-membered heteroaryl
- R 1 is halogen, cyano, difluoromethyl, trifluoromethyl, (C 1 -C 4) -alkyl, hydroxy, oxo or (C 1 -C 4) -alkoxy, n is a number 0, 1 or 2,
- R 2 is trifluoromethyl, (Ci -C 6 ) alkyl, (C 3 -C 8 ) cycloalkyl, phenyl or 5- or 6-membered heteroaryl, wherein (Ci-C6) alkyl is substituted with a substituent selected from the group consisting of difluoromethyl and trifluoromethyl, wherein (C 1 -C 6) -alkyl may be substituted by 1 to 3 substituents of fluorine, wherein (C 3 -C 9) -cycloalkyl having 1 or 2 substituents independently selected from the group of fluorine, methyl and methoxy may be substituted, wherein phenyl with 1 to 3 substituents fluorine
- R 3 is difluoromethyl, trifluoromethyl, (C 1 -C 6 ) -alkyl, (C 3 -C 7 ) -cycloalkyl, (C 1 -C 6 ) -alkylsulfonylamino, (C 1 -C 6 ) -alkoxycarbonylamino, phenyl or 5 or 6 is a heteroaryl-containing heterocycle, where (C 1 -C 6) -alkyl, phenyl and 5 or 6-membered heteroaryl having 1 to 3 substituents independently of one another are selected from the group consisting of halogen, trifluoromethyl, (C 1 -C 4) -alkyl, (C 3 -C 4) -Cycloalkyl, difluoromethoxy, trifluoromethoxy and (Ci-C6) alkoxy, R is hydroxy or amino, and their N-oxides, salts, solvates, salts of N-oxides and
- Compounds according to the invention are the compounds of the formula (I) and their salts, solvates and solvates of the salts comprising the compounds of the formulas below and their salts, solvates and solvates of the salts and of the formula (I) encompassed by formula (I), hereinafter referred to as exemplary compounds and their salts, solvates and solvates of the salts, as far as the compounds of formula (I), the compounds mentioned below are not already salts, solvates and solvates of the salts.
- Salts used in the context of the present invention are physiologically acceptable salts of the compounds according to the invention. Also included are salts which are themselves unsuitable for pharmaceutical applications but can be used, for example, for the isolation or purification of the compounds of the invention.
- Physiologically acceptable salts of the compounds of the invention include acid addition salts of mineral acids, carboxylic acids and sulfonic acids, e.g. Salts of hydrochloric, hydrobromic, sulfuric, phosphoric, methanesulfonic, ethanesulfonic, toluenesulfonic, benzenesulfonic, naphthalenedisulfonic, formic, acetic, trifluoroacetic, propionic, lactic, tartaric, malic, citric, fumaric, maleic and benzoic acids.
- Salts of hydrochloric, hydrobromic, sulfuric, phosphoric, methanesulfonic, ethanesulfonic, toluenesulfonic, benzenesulfonic, naphthalenedisulfonic formic, acetic, trifluoroacetic, propionic, lactic, tartaric, malic, citric, fumaric, maleic and benzoic
- Physiologically acceptable salts of the compounds according to the invention also include salts of customary bases, such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts) and ammonium salts derived from ammonia or organic amines having 1 to 16 carbon atoms, such as, by way of example and by way of illustration, ethylamine, diethylamine, triethylamine, ethyldiisopropylamine, monoethanolamine, diethanolamine, triethanolamine, dicyclohexylamine, dimethylaminoethanol, procaine, dibenzylamine, N-methylmorpholine, arginine, lysine, ethylenediamine and N-methylpiperidine.
- customary bases such as, by way of example and by way of preference, alkali metal salts (for example sodium and potassium salts), alkaline earth salts (for example calcium and magnesium salts
- Solvates in the context of the invention are those forms of the compounds according to the invention which form a complex in the solid or liquid state by coordination with solvent molecules. Hydrates are a special form of solvates that coordinate with water. As solvates, hydrates are preferred in the context of the present invention.
- the compounds according to the invention may exist in different stereoisomeric forms, ie in the form of configurational isomers or optionally also as conformational isomers (enantiomers and / or diastereomers, including those in the case of atropisomers).
- the present invention therefore includes the enantiomers and diastereomers and their respective mixtures. From such mixtures of enantiomers and / or Diastereomers can be the stereoisomerically uniform components in a known manner to isolate; Preferably, chromatographic methods are used for this, in particular HPLC chromatography on achiral or chiral phase.
- the present invention encompasses all tautomeric forms.
- the present invention also includes all suitable isotopic variants of the compounds of the invention.
- An isotopic variant of a compound according to the invention is understood to mean a compound in which at least one atom within the compound according to the invention is exchanged for another atom of the same atomic number but with a different atomic mass than the atomic mass that usually or predominantly occurs in nature.
- isotopes which can be incorporated into a compound of the invention are those of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, chlorine, bromine and iodine, such as 2 H (deuterium), 3 H (tritium), 13 C, 14 C, 15 N, 17 0, 18 0, 32 P, 33 P, 33 S, 34 S, 35 S, 36 S, 18 F, 36 Cl, 82 Br, 123 I, 124 I, 129 I and 131 I.
- isotopic variants of a compound of the invention such as, in particular, those in which one or more radioactive isotopes are incorporated, may be useful, for example, for the study of the mechanism of action or drug distribution in the body; Due to the comparatively easy production and detectability, compounds labeled with 3 H or 14 C isotopes in particular are suitable for this purpose.
- isotopes such as deuterium may result in certain therapeutic benefits as a result of greater metabolic stability of the compound, such as prolonging the body's half-life or reducing the required effective dose;
- Such modifications of the compounds of the invention may therefore optionally also constitute a preferred embodiment of the present invention.
- Isotopic variants of the compounds according to the invention can be prepared by the processes known to the person skilled in the art, for example by the methods described below and the rules given in the exemplary embodiments, by using appropriate isotopic modifications of the respective reagents and / or starting compounds.
- the present invention also includes prodrugs of the compounds of the invention.
- prodrugs refers to compounds which themselves may be biologically active or inactive, but are converted during their residence time in the body to compounds of the invention (for example metabolically or hydrolytically). Unless otherwise specified, in the context of the present invention, the substituents have the following meaning:
- alkyl is a linear or branched alkyl radical having in each case the number of carbon atoms specified.
- alkyl is a linear or branched alkyl radical having in each case the number of carbon atoms specified.
- a 5- or 7-membered saturated or partially unsaturated carbocycle is a saturated or partially unsaturated cyclic alkyl radical having in each case the number of carbon atoms specified.
- Examples which may be mentioned by way of example include cyclopentyl, cyclohexyl, cycloheptyl, cyclopentenyl, cyclohexenyl and cycloheptenyl.
- Cycloalkyl or carbocycle in the context of the invention is a monocyclic, saturated alkyl radical having in each case the number of carbon atoms specified. Examples which may be mentioned by way of example include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl.
- Alkoxy in the context of the invention is a linear or branched alkoxy radical having 1 to 6 or 1 to 4 carbon atoms.
- Examples include: methoxy, ethoxy, n-propoxy, isopropoxy, 1-methylpropoxy, n-butoxy, iso-butoxy, tert-butoxy, n-pentoxy, iso-pentoxy, 1-ethylpropoxy, 1-methylbutoxy, 2-methylbutoxy , 3-methylbutoxy and n-hexoxy. Preference is given to a linear or branched alkoxy radical having 1 to 4 carbon atoms. Examples which may be mentioned are: methoxy, ethoxy, n-propoxy, isopropoxy, 1-methylpropoxy, n-butoxy, isobutoxy, tert-butoxy.
- Alkoxycarbonylamino in the context of the invention represents an amino group having a linear or branched alkoxycarbonyl substituent which has 1 to 4 carbon atoms in the alkyl chain and is linked via the carbonyl group to the nitrogen atom.
- alkoxycarbonylamino represents an amino group having a linear or branched alkoxycarbonyl substituent which has 1 to 4 carbon atoms in the alkyl chain and is linked via the carbonyl group to the nitrogen atom.
- Alkylsulfonylamino in the context of the invention is an amino group having a linear or branched alkylsulfonyl substituent which has 1 to 6 carbon atoms and is linked via the sulfonyl group to the N-atom.
- alkylsulfonylamino is an amino group having a linear or branched alkylsulfonyl substituent which has 1 to 6 carbon atoms and is linked via the sulfonyl group to the N-atom.
- 5- to 7-membered saturated or partially unsaturated heterocycle in the context of the invention represents a saturated or partially unsaturated heterocycle having a total of 5 to 7 ring atoms, which is a ring heteroatom from the series N, O, S, SO and / or SO 2 contains.
- Examples include: pyrrolidinyl, tetrahydrofuranyl, piperidinyl, tetrahydropyranyl, dihydropyrrolyl, dihydropyridyl.
- 5- or 6-membered heteroaryl is in the context of the invention for a monocyclic aromatic heterocycle (heteroaromatic) with a total of 5 or 6 ring atoms, which contains up to three identical or different ring heteroatoms from the series N, O and / or S. a ring carbon atom or optionally linked via a ring nitrogen atom.
- Examples which may be mentioned are: furyl, pyrrolyl, thienyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isoxazolyl, isothiazolyl, triazolyl, oxadiazolyl, thiadiazolyl, pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl and triazinyl. Preference is given to pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl and triazinyl.
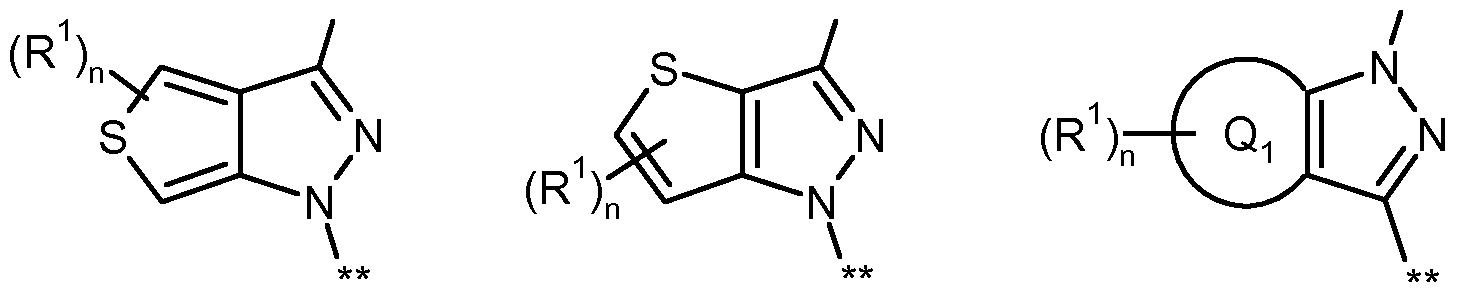
- 8- or 9-membered heteroaryl is in the context of the invention for a bicyclic aromatic or partially unsaturated heterocycle having a total of 8 or 9 ring atoms, the at least two nitrogen atoms and up to two further, identical or different ring heteroatoms from the series N, O and / or S contains.
- Examples which may be mentioned are: dihydrothienopyrazolyl, thienopyrazolyl, pyrazolopyrazolyl, imidazothiazolyl, tetrahydrocyclopentapyrazolyl, dihydrocyclopentapyrazolyl, tetrahydroindazolyl, dihydroindazolyl, indazolyl, pyrazolopyridinyl, tetrahydropyrazolopyridinyl, pyrazolopyrimidinyl, imidazopyridinyl and imidazopyridazinyl.
- Halogen is in the context of the invention for fluorine, chlorine, bromine and iodine. Preference is given to bromine and iodine.
- An oxo group in the context of the invention is an oxygen atom which is bonded via a double bond to a carbon atom.
- the end point of the line on which the symbol * and ** stands does not stand for a carbon atom or a CH 2 group but is part of the bond to the respectively designated atom, is bound to the Q.
- radicals are substituted in the compounds according to the invention, the radicals can, unless otherwise specified, be monosubstituted or polysubstituted. In the context of the present invention, the meaning is independent of each other for all radicals which occur repeatedly. Substitution with one, two or three identical or different substituents is preferred.
- the term “treatment” or “treating” includes inhibiting, delaying, arresting, alleviating, attenuating, restraining, reducing, suppressing, restraining or curing a disease, a disease, a disease, an injury or a medical condition , the unfolding, the course or progression of such conditions and / or the symptoms of such conditions.
- the term “therapy” is understood to be synonymous with the term “treatment”.
- prevention means the avoidance or reduction of the risk, a disease, a disease, a disease, an injury or a health disorder, a development or a Progression of such conditions and / or to get, experience, suffer or have the symptoms of such conditions.
- the treatment or the prevention of a disease, a disease, a disease, an injury or a health disorder can be partial or complete.
- ** represents the point of attachment to the triazine ring, the ring Qi together with the atoms to which it is attached forms a 5- to 7-membered saturated or partially unsaturated carbocycle or a 5- to 7-membered saturated or partially unsaturated heterocycle , R 1 is fluorine, chlorine or methyl, n is a number 0, 1 or 2,
- a 1 , A 2 , A 3 and A 4 are each independently N, CH or CR 1 , with the proviso that at most two of the groups A 1 , A 2 , A 3 and A 4 are N, R is trifluoromethyl, 2,2,2-trifluoroethyl, 3,3,3-trifluoroprop-1-yl, 2,2,3,3,3-pentafluoroprop-1-yl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, phenyl, Pyridyl, pyrimidinyl, pyrazinyl or pyridazinyl, where phenyl is substituted by 1 to 3 fluorine substituents, and where cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, pyridyl, pyrimidinyl, pyrazinyl and pyridazinyl may be substituted by 1 or 2
- R 3 is difluoromethyl, trifluoromethyl, (Ci-C6) alkyl, cyclopropyl, cyclobutyl, cyclopentyl, methylsulfonylamino, methoxycarbonylamino, phenyl, pyrazolyl, oxazolyl or pyridyl, wherein (Ci-C6) alkyl having 1 to 3 substituents selected independently from the group of fluorine, trifluoromethyl, cyclopropyl, cyclobutyl, cyclopentyl, difluoromethoxy, trifluoromethoxy, methoxy and ethoxy, and where phenyl, pyrazolyl, oxazolyl and pyridyl having 1 or 2 substituents independently of one another selected from the group fluorine, chlorine, difluoromethyl, Trifluoromethyl, methyl, ethyl, cyclopropyl, cyclobutyl, cyclopen
- R 4 is hydroxy or amino, and their salts, solvates and solvates of the salts.
- R lc is hydrogen or chlorine, N is N or CH, A is N, CH or CF,
- R 2 is 3,3,3-trifluoroprop-1-yl, 2,2,3,3-tetrafluoroprop-1-yl, 2,2,3,3,3-pentafluoroprop-1-yl, phenyl or pyridyl, wherein phenyl is substituted with 1 to 3 substituents fluorine, and wherein pyridyl may be substituted with 1 substituent fluorine, R is difluoromethyl, trifluoromethyl, (Ci-C6) alkyl, cyclopropyl, cyclobutyl, cyclopentyl, methylsulfonylamino, methoxycarbonylamino, phenyl, pyrazolyl, oxazolyl or pyridyl, wherein (Ci-C6) alkyl having 1 to 3 substituents independently selected from the group fluorine, trifluoromethyl, cyclopropyl, cyclobutyl, cyclopentyl, difluorometh
- R 4 is hydroxy or amino, and their salts, solvates and solvates of the salts.
- R la represents hydrogen or fluorine
- R lb represents hydrogen or methyl
- R 1b represents hydrogen or methyl
- R 2 is 2-fluorophenyl, 2,3-difluorophenyl or 2,3,6-trifluorophenyl, and their salts, solvates and solvates of the salts.
- Another object of the invention is a process for the preparation of compounds of the formula (I) according to the invention characterized in that
- n, Q, R 1 and R 2 each have the abovementioned meanings, in an inert solvent in the presence of a suitable transition metal catalyst with a compound of the formula (III)
- T 1 is hydrogen or (Ci-C4) -alkyl, or the two radicals R 11 together are a -C (CH 3) 2-C (CH 3) 2 bridge form,
- n, Q, R 1 , R 2 and R 3A each have the meanings given above, or
- R 3B is difluoromethyl, trifluoromethyl, (C 1 -C 6 ) -alkyl or (C 3 -C 7 ) -cycloalkyl, where (C 1 -C 6 ) -alkyl having 1 to 3 substituents independently of one another selected from the group consisting of halogen, trifluoromethyl, (C 1 -C 4) -alkyl, (C 3 -C 7) -cycloalkyl, difluoromethoxy, trifluoromethoxy and (C 1 -C 6) -alkoxy may be substituted, and
- T is (C 1 -C 4 ) -alkyl, to give a compound of the formula (IB)
- n, Q, R 1 , R 2 and R 3B each have the meanings given above, implements, or
- n, Q, R 1 , R 2 and R 3B are each as defined above, and these are reacted directly with ammonia to give a compound of the formula (IC)
- n, Q, R 1 , R 2 and R 3B each have the meanings given above, and optionally the resulting compounds of the formula (IA), (IB) and (IC) optionally with the corresponding (i) solvents and / or (ii) converting acids or bases into their solvates, salts and / or solvates of the salts.
- Suitable solvents are, for example, ethers, such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, or other solvents, such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone (NMP), pyridine, acetonitrile or even water. It is likewise possible to use mixtures of the solvents mentioned. Preference is given to acetonitrile.
- ethers such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether
- other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone (NMP), pyridine, ace
- reaction (II) + (III) ⁇ (I-A) can be carried out in the presence of a suitable palladium and / or copper catalyst.
- a suitable palladium catalyst for example, palladium on activated carbon, palladium (II) acetate, tetrakis (triphenylphosphine) palladium (0), bis-
- Suitable copper catalysts are, for example, copper bronze, copper (I) oxide, copper (I) iodide or copper (I) bromide.
- Suitable bases for this reaction are the usual inorganic or organic bases. These include preferably alkali metal hydroxides such as lithium, sodium or potassium hydroxide, alkali metal or alkaline earth metal carbonates such as lithium, sodium, potassium, calcium or cesium carbonate, alkali metal alcoholates such as sodium or potassium, sodium or potassium or sodium or potassium tert-butoxide, alkali metal hydrides such as sodium or potassium hydride, amides such as sodium amide, lithium, sodium or potassium bis (trimethylsilyl) amide or lithium diisopropylamide, or organic amines such as triethylamine, N-methylmorpholine, N-methylpiperidine, N, N-diisopropylethylamine, pyridine, 1,5-diazabicyclo [4.3.0] non-5-ene (DBN), 1,8-diazabicyclo [5.4.0] undec-7
- reaction (II) + (III) -> (IA) is generally carried out in a temperature range of 0 ° C to + 200 ° C, preferably at + 10 ° C to + 150 ° C.
- the reaction can be carried out at normal, elevated or at reduced pressure (for example from 0.5 to 5 bar). Generally, one works at normal pressure.
- radical R 3A is unsaturated, this can then be completely or partially saturated.
- the reduction is carried out with hydrogen in conjunction with transition metal catalysts such as palladium (10% on activated carbon), Raney nickel or palladium hydroxide.
- the reduction is generally carried out in a temperature range from + 20 ° C to + 50 ° C.
- the Reaction can be carried out at normal or elevated pressure (eg in the range from 0.5 to 5 bar). Generally, one works at normal pressure.
- Inert solvents for process step (IV) + (V) - > (IB) are, for example, alcohols, such as methanol, ethanol, n-propanol, isopropanol, n-butanol or tert-butanol, ethers, such as diethyl ether, dioxane, dimethoxyethane, tetrahydrofuran , Glycol dimethyl ether or diethylene glycol dimethyl ether, hydrocarbons such as benzene, xylene, toluene, hexane, cyclohexane or petroleum fractions, or other solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone (NMP), Pyridine or acetonitrile.
- alcohols such as methanol, ethanol, n-propanol, isopropan
- reaction (IV) + (V) -> (IB) is generally carried out in a temperature range from + 50 ° C to + 120 ° C, preferably from + 50 ° C to + 100 ° C, optionally in a microwave.
- the reaction can be carried out at normal or elevated pressure (for example in the range from 0.5 to 5 bar). Generally, one works at normal pressure.
- the reaction (I-B) -> (VI) can be carried out in a solvent which is inert under the reaction conditions or without a solvent.
- Preferred solvent is sulfolane.
- the reaction (I-B) -> (VI) is generally carried out in a temperature range from + 70 ° C to + 150 ° C, preferably from + 80 ° C to + 130 ° C, optionally in a microwave.
- the reaction may be carried out at normal or elevated pressure (e.g., in the range of 0.5 to 5 bar). Generally, one works at normal pressure.
- the reaction is preferably carried out (I-B) without solvent in a temperature range from 0 ° C. to + 50 ° C. under atmospheric pressure.
- Process step (VI) -> (I-C) takes place in a solvent which is inert under the reaction conditions.
- Suitable solvents are, for example, ethers, such as diethyl ether, dioxane, tetrahydrofuran, glycol dimethyl ether or diethylene glycol dimethyl ether, or other solvents, such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), N, N'-dimethylpropyleneurea (DMPU), N-methylpyrrolidone ( ⁇ ), pyridine, acetonitrile or else Water. It is likewise possible to use mixtures of the solvents mentioned. Preference is given to acetonitrile.
- the reaction (VI) -> (IC) is generally carried out in a temperature range from + 20 ° C to + 100 ° C, preferably from + 40 ° C to + 70 ° C, optionally in a microwave.
- the reaction can be carried out at normal or elevated pressure (for example in the range from 0.5 to 5 bar). Generally, one works at normal pressure. Preference is given to the reactions (IB) - » ⁇ (VI) -» ⁇ (IC) without isolation of the intermediate (VI)
- Other compounds of the invention may optionally also be prepared by conversions of functional groups of individual substituents, in particular those listed under L and R 3 , starting from the compounds of the formula (I) obtained by the above methods.
- transformations are carried out by conventional methods known to those skilled in the art and include, for example, reactions such as nucleophilic and electrophilic substitutions, oxidations, reductions, hydrogenations, transition metal-catalyzed coupling reactions, elimination, alkylation, amination, esterification, ester cleavage, etherification, ether cleavage, formation of carbonamides, and introduction and removal of temporary protection groups.
- the compounds of the formula (II) can be prepared by reacting a compound of the formula (VI)
- the compounds of the formula (VI) are known from the literature (see, for example, WO 2010/065275, WO 2011/115804 and WO 2011/149921) or can be prepared in analogy to processes known from the literature.
- the compounds of the invention are potent stimulators of soluble guanylate cyclase, have valuable pharmacological properties, and have an improved therapeutic profile, such as in their in vivo properties and / or their pharmacokinetic behavior. They are therefore suitable for the treatment and / or prophylaxis of diseases in humans and animals.
- the compounds of the invention cause vasorelaxation and inhibition of platelet aggregation and lead to a reduction in blood pressure and to an increase in coronary blood flow. These effects are mediated by a direct stimulation of soluble guanylate cyclase and an intracellular cGMP increase.
- the compounds according to the invention enhance the effect of substances which increase cGMP levels, such as, for example, endothelium-derived relaxing factor (EDRF), NO donors, protoporphyrin IX, arachidonic acid or phenylhydrazine derivatives.
- EDRF endothelium-derived relaxing factor
- NO donors NO donors
- protoporphyrin IX arachidonic acid or phenylhydrazine derivatives.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of cardiovascular, pulmonary, thromboembolic and fibrotic disorders.
- the compounds according to the invention can therefore be used in medicaments for the treatment and / or prophylaxis of cardiovascular diseases such as hypertension, acute and chronic heart failure, coronary heart disease, stable and unstable angina pectoris, peripheral and cardiac vascular diseases, arrhythmias, arrhythmia of the atria and the chambers and conduction disorders such for example atrio-ventricular blockades grade I-III (AB block I-III), supraventricular tachyarrhythmia, atrial fibrillation, atrial flutter, ventricular fibrillation, ventricular tachyarrhythmia, torsade de pointes tachycardia, atrial and ventricular extrasystoles, atrioventricular extrasystoles, Sick sinus syndrome, syncope, AV nodal reentrant tachycardia, Wolff-
- cardiac insufficiency also encompasses more specific or related forms of disease such as acute decompensated heart failure, right heart failure, left heart failure, global insufficiency, ischemic cardiomyopathy, dilated cardiomyopathy, hypertrophic cardiomyopathy, idiopathic cardiomyopathy, congenital heart defects, heart valve defects, heart failure in cardiac valve defects, mitral stenosis, Mitral valve insufficiency, aortic valve stenosis, aortic valve insufficiency, tricuspid stenosis, tricuspid insufficiency, pulmonary valve stenosis, pulmonary valvular insufficiency, combined valvular heart failure, myocarditis, chronic myocarditis, acute myocarditis, viral myocarditis, diabetic heart failure, alcoholic cardiomyopathy, cardiac memory disease, diastolic heart failure and systolic heart failure.
- ischemic cardiomyopathy dilated cardiomyopathy
- the compounds according to the invention may also be used for the treatment and / or prophylaxis of arteriosclerosis, lipid metabolism disorders, hypolipoproteinemias, dyslipidemias, hypertriglyceridemias, hyperlipidemias, hypercholesterolemias, abetelipoproteinaemia, sitosterolemia, xanthomatosis, Tangier's disease, obesity (obesity) and combined hyperlipidemias and the metabolic syndrome.
- the compounds of the invention may be used for the treatment and / or prophylaxis of primary and secondary Raynaud's phenomenon, microcirculatory disorders, claudication, peripheral and autonomic neuropathies, diabetic microangiopathies, diabetic retinopathy, diabetic ulcers on the extremities, gangrenous, CREST syndrome, erythematosis, onychomycosis , rheumatic diseases and to promote wound healing.
- the compounds according to the invention are suitable for the treatment of urological diseases such as benign prostatic syndrome (BPS), benign prostatic hyperplasia (BPH), benign prostate enlargement (BPE), bladder emptying disorder (BOO), lower urinary tract syndromes (LUTS, including Feiine's urological syndrome ( FUS)), diseases of the urogenital system including neurogenic overactive bladder (OAB) and (IC), incontinence (UI) such as mixed, urge, stress, or overflow incontinence (MUI, UUI, SUI, OUI), Pelvic pain, benign and malignant diseases of the organs of the male and female urogenital system.
- BPS benign prostatic syndrome
- BPH benign prostatic hyperplasia
- BPE benign prostate enlargement
- BOO bladder emptying disorder
- LUTS lower urinary tract syndromes
- FUS lower urinary tract syndromes
- UI incontinence
- MUI mixed, urge, stress, or overflow incontinence
- UUI UUI
- SUI S
- kidney diseases in particular of acute and chronic renal insufficiency, as well as of acute and chronic renal failure.
- renal insufficiency includes both acute and chronic manifestations of renal insufficiency, as well as underlying or related renal diseases such as renal hypoperfusion, intradialytic hypotension, obstructive uropathy, glomerulopathies, glomerulonephritis, acute glomerulonephritis, glomerulosclerosis, tubulo-interstitial disorders, nephropathic disorders such as primary and congenital kidney disease, nephritis, immunological kidney diseases such as renal transplant rejection, immune complex-induced kidney disease, nephropathy induced by toxic substances, contrast agent-induced nephropathy, diabetic and non-diabetic nephropathy, pyelonephritis, renal cysts, nephrosclerosis, hyperten
- the present invention also encompasses the use of the compounds of the invention for the treatment and / or prophylaxis of sequelae of renal insufficiency, such as pulmonary edema, cardiac insufficiency, uremia, anemia, electrolyte imbalances (e.g., hyperkalemia, hyponatremia) and disorders in bone and carbohydrate metabolism.
- sequelae of renal insufficiency such as pulmonary edema, cardiac insufficiency, uremia, anemia, electrolyte imbalances (e.g., hyperkalemia, hyponatremia) and disorders in bone and carbohydrate metabolism.
- the compounds according to the invention are also suitable for the treatment and / or prophylaxis of asthmatic diseases, pulmonary arterial hypertension (PAH) and other forms of pulmonary hypertension (PH), including left heart disease, HIV, sickle cell anemia, thromboembolism (CTEPH), sarcoidosis, COPD or Pulmonary fibrosis-associated pulmonary hypertension, chronic obstructive pulmonary disease (COPD), acute respiratory tract syndrome (ARDS), acute lung injury (ALI), alpha-1-antitrypsin deficiency (AATD), pulmonary fibrosis, pulmonary emphysema (eg, cigarette smoke-induced Pulmonary emphysema) and cystic fibrosis (CF).
- PAH pulmonary arterial hypertension
- PH pulmonary hypertension
- COPD chronic obstructive pulmonary disease
- ARDS acute respiratory tract syndrome
- ALI acute lung injury
- AATD alpha-1-antitrypsin deficiency
- CF
- the compounds described in the present invention are also agents for combating diseases in the central nervous system caused by disorders of the NO / cGMP- Systems are marked.
- they are suitable for improving the perception, concentration performance, learning performance or memory performance after cognitive disorders such as occur in situations / diseases / syndromes such as mild cognitive impairment, age-associated learning and memory disorders, age-associated memory loss, vascular dementia, cranial brain -Trauma, stroke, post-stroke dementia, post-traumatic traumatic brain injury, generalized concentration disorder, difficulty concentrating in children with learning and memory problems, Alzheimer's disease, dementia with Lewy bodies , Dementia with degeneration of the frontal lobes including Pick's syndrome, Parkinson's disease, progressive nuclear palsy, dementia with corticobasal degeneration, amyolateral sclerosis (ALS), Huntington's disease, demyelinization, multiple sclerosis, thalamic degeneration, Creutzfeld-Jacob dementia, HIV dementia, schizophrenia with dementia or Korsakoff's psychosis. They are also
- the compounds according to the invention are also suitable for regulating cerebral blood flow and are effective agents for combating migraine. They are also suitable for the prophylaxis and control of the consequences of cerebral infarct events (Apoplexia cerebri) such as stroke, cerebral ischaemias and craniocerebral trauma , Likewise, the compounds according to the invention can be used to combat pain and tinnitus.
- cerebral infarct events Apoplexia cerebri
- cerebral infarct events such as stroke, cerebral ischaemias and craniocerebral trauma
- the compounds according to the invention can be used to combat pain and tinnitus.
- the compounds of the invention have anti-inflammatory action and can therefore be used as anti-inflammatory agents for the treatment and / or prophylaxis of sepsis (SIRS), multiple organ failure (MODS, MOF), inflammatory diseases of the kidney, chronic inflammatory bowel disease (IBD, Crohn's Disease, UC), pancreatitis , Peritonitis, rheumatoid diseases, inflammatory skin diseases as well as inflammatory eye diseases.
- SIRS sepsis
- MODS multiple organ failure
- IBD chronic inflammatory bowel disease
- UC chronic inflammatory bowel disease
- pancreatitis atitis
- Peritonitis rheumatoid diseases
- inflammatory skin diseases as well as inflammatory eye diseases.
- the compounds of the invention can also be used for the treatment and / or prophylaxis of autoimmune diseases.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of fibrotic disorders of the internal organs such as, for example, the lung, the heart, the kidney, the bone marrow and in particular the liver, as well as dermatological fibroses and fibrotic disorders of the eye.
- fibrotic disorders includes in particular the following terms liver fibrosis, liver cirrhosis, Pulmonary fibrosis, endomyocardial fibrosis, nephropathy, glomerulonephritis, interstitial renal fibrosis, fibrotic damage resulting from diabetes, bone marrow fibrosis and related fibrotic diseases, scleroderma, morphea, keloids, hypertrophic scarring (also after surgery), nevi, diabetic retinopathy, proliferative vitroretinopathy and connective tissue disorders (eg sarcoidosis).
- the compounds of the invention are useful for controlling postoperative scarring, e.g. as a result of glaucoma surgery.
- the compounds according to the invention can likewise be used cosmetically for aging and keratinizing skin.
- the compounds according to the invention are suitable for the treatment and / or prophylaxis of hepatitis, neoplasm, osteoporosis, glaucoma and gastroparesis.
- Another object of the present invention is the use of the compounds of the invention for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases.
- Another object of the present invention is the use of the compounds of the invention for the treatment and / or prophylaxis of heart failure, angina pectoris, hypertension, pulmonary hypertension, ischaemia, vascular disease, renal insufficiency, thromboembolic disorders, fibrotic diseases and arteriosclerosis.
- the present invention furthermore relates to the compounds according to the invention for use in a method for the treatment and / or prophylaxis of cardiac insufficiency, angina pectoris, hypertension, pulmonary hypertension, ischaemias, vascular disorders, renal insufficiency, thromboembolic disorders, fibrotic disorders and atherosclerosis.
- Another object of the present invention is the use of the compounds of the invention for the manufacture of a medicament for the treatment and / or prophylaxis of Erkrankun- gene, in particular the aforementioned diseases.
- Another object of the present invention is the use of the compounds of the invention for the manufacture of a medicament for the treatment and / or prophylaxis of heart failure, angina pectoris, hypertension, pulmonary hypertension, ischemia, vascular diseases, renal insufficiency, thromboembolic disorders, fibrotic diseases and arteriosclerosis.
- Another object of the present invention is a method for the treatment and / or prophylaxis of diseases, in particular the aforementioned diseases, using an effective amount of at least one of the compounds of the invention.
- the present invention further provides a method for the treatment and / or prophylaxis of cardiac insufficiency, angina pectoris, hypertension, pulmonary hypertension, ischaemias, vascular diseases, renal insufficiency, thromboembolic disorders, fibrotic diseases and atherosclerosis, using an effective amount of at least one of the compounds according to the invention ,
- the compounds of the invention may be used alone or as needed in combination with other agents.
- Another object of the present invention are pharmaceutical compositions containing at least one of the compounds of the invention and one or more other active ingredients, in particular for the treatment and / or prophylaxis of the aforementioned diseases.
- suitable combination active ingredients may be mentioned by way of example and preferably:
- organic nitrates and NO donors such as sodium nitroprusside, nitroglycerin, isosorbide mononitrate, isosorbide dinitrate, molsidomine or SIN-1, and inhaled NO;
- cGMP cyclic guanosine monophosphate
- PDE phosphodiesterases
- Antithrombotic agents by way of example and preferably from the group of thrombocyte aggregation inhibitors, anticoagulants or profibrinolytic substances;
- Antihypertensive agents by way of example and preferably from the group of calcium antagonists, angiotensin AII antagonists, ACE inhibitors, endothelin antagonists, renin inhibitors, alpha-receptor blockers, beta-receptor blockers, mineralocorticoid receptor Antagonists and diuretics; and / or ⁇ fat metabolism-altering agents, by way of example and preferably from the group of thyroid receptor agonists, cholesterol synthesis inhibitors such as by way of example and preferably HMG-CoA reductase or squalene synthesis inhibitors, ACAT inhibitors, CETP inhibitors, MTP inhibitors , PPAR alpha, PPAR gamma and / or PPAR delta agonists, cholesterol absorption inhibitors, lipase inhibitors, polymeric bile acid adsorbers, bile acid reabsorption inhibitors, and lipoprotein (a) antagonists.
- Antithrombotic agents are preferably understood as meaning compounds from the group of plate
- the compounds according to the invention are administered in combination with a platelet aggregation inhibitor, such as, by way of example and by way of preference, aspirin, clopidogrel, ticlopidine or dipyridamole.
- a platelet aggregation inhibitor such as, by way of example and by way of preference, aspirin, clopidogrel, ticlopidine or dipyridamole.
- the compounds according to the invention are administered in combination with a thrombin inhibitor such as, by way of example and by way of preference, ximelagatran, dabigatran, melagatran, bivalirudin or Clexane.
- a thrombin inhibitor such as, by way of example and by way of preference, ximelagatran, dabigatran, melagatran, bivalirudin or Clexane.
- the compounds according to the invention are administered in combination with a GPIIb / IIIa antagonist, such as, by way of example and by way of preference, tirofiban or abciximab.
- the compounds according to the invention are used in combination with a factor Xa inhibitor, such as by way of example and preferably rivaroxaban (BAY 59-7939), DU-176b, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux, PMD No. 3112, YM-150, KFA-1982, EMD-503982, MCM-17, MLN-1021, DX 9065a, DPC 906, JTV 803, SSR-126512 or SSR-128428.
- a factor Xa inhibitor such as by way of example and preferably rivaroxaban (BAY 59-7939), DU-176b, apixaban, otamixaban, fidexaban, razaxaban, fondaparinux, idraparinux, PMD No. 3112, YM-150, KFA-1982, EMD-503982, MCM
- the compounds according to the invention are administered in combination with heparin or a low molecular weight (LMW) heparin derivative.
- LMW low molecular weight
- the compounds according to the invention are administered in combination with a vitamin K antagonist, such as by way of example and preferably coumarin.
- antihypertensive agents are preferably compounds from the group of calcium antagonists, angiotensin AII antagonists, ACE inhibitors, endothelin antagonists, renin inhibitors, alpha-receptor blocker, beta-receptor blocker, mineralocorticoid receptor - understood antagonists and diuretics.
- the compounds according to the invention are administered in combination with a calcium antagonist, such as, by way of example and by way of preference, nifedipine, amlodipine, verapamil or diltiazem.
- a calcium antagonist such as, by way of example and by way of preference, nifedipine, amlodipine, verapamil or diltiazem.
- the compounds according to the invention are administered in combination with an alpha-1-receptor blocker, such as by way of example and preferably prazosin.
- the compounds according to the invention are used in combination with a beta-receptor blocker, such as by way of example and preferably propranolol, atenolol, timolol, pindolol, alprenolol, oxprenolol, penbutolol, bupranolol, metipranolol, nadolol, mepindolol, carazalol, sotalol, Metoprolol, betaxolol, celiprolol, bisoprolol, carteolol, esmolol, labetalol, carvedilol, adaprolol, landiolol, nebivolol, epanolol or bucindolol.
- a beta-receptor blocker such as by way of example and preferably propranolol, atenolol, timolol
- the compounds according to the invention are administered in combination with an angiotensin AII antagonist, such as by way of example and preferably losartan, candesartan, valsartan, telmisartan or embursatan.
- an angiotensin AII antagonist such as by way of example and preferably losartan, candesartan, valsartan, telmisartan or embursatan.
- the compounds according to the invention are administered in combination with an ACE inhibitor, such as by way of example and preferably enalapril, captopril, lisinopril, ramipril, delapril, fosinopril, quinopril, perindopril or trandopril.
- an ACE inhibitor such as by way of example and preferably enalapril, captopril, lisinopril, ramipril, delapril, fosinopril, quinopril, perindopril or trandopril.
- an endothelin antagonist such as, by way of example and by way of preference, bosentan, darusentan, ambrisentan or sitaxsentan.
- the compounds according to the invention are administered in combination with a renin inhibitor, such as by way of example and preferably aliskiren, SPP-600 or SPP-800.
- a renin inhibitor such as by way of example and preferably aliskiren, SPP-600 or SPP-800.
- the compounds according to the invention are administered in combination with a mineralocorticoid receptor antagonist, such as, by way of example and by way of preference, spironolactone or eplerenone.
- a mineralocorticoid receptor antagonist such as, by way of example and by way of preference, spironolactone or eplerenone.
- the compounds of the present invention are used in combination with a loop diuretic such as furosemide, torasemide, bumetanide and piretanide with potassium sparing diuretics such as amiloride and triamterene with aldosterone antagonists such as spironolactone, potassium canrenoate and eplerenone and thiazide diuretics such as Hydrochlorothiazide, chlorthalidone, xipamide, and indapamide.
- a loop diuretic such as furosemide, torasemide, bumetanide and piretanide
- potassium sparing diuretics such as amiloride and triamterene with aldosterone antagonists such as spironolactone, potassium canrenoate and eplerenone and thiazide diuretics
- Hydrochlorothiazide chlorthalidone
- xipamide xipamide
- indapamide indapamide
- lipid metabolizing agents are preferably compounds from the group of CETP inhibitors, thyroid receptor agonists, cholesterol synthesis inhibitors such as HMG-CoA reductase or squalene synthesis inhibitors, ACAT inhibitors, MTP inhibitors, PPAR-alpha, PPAR-gamma and / or PPAR-delta agonists, cholesterol absorption inhibitors, polymeric bile acid adsorbers, bile acid reabsorption inhibitors, lipase Inhibitors as well as the lipoprotein (a) antagonists understood.
- the compounds according to the invention are administered in combination with a CETP inhibitor, such as, for example and preferably, dalcetrapib, BAY 60-5521, anacetrapib or CETP vaccine (CETi-1).
- a CETP inhibitor such as, for example and preferably, dalcetrapib, BAY 60-5521, anacetrapib or CETP vaccine (CETi-1).
- the compounds of the invention are administered in combination with a thyroid receptor agonist such as, by way of example and by way of preference, D-thyroxine, 3,5,3'-triiodothyronine (T3), CGS 23425 or axitirome (CGS 26214).
- a thyroid receptor agonist such as, by way of example and by way of preference, D-thyroxine, 3,5,3'-triiodothyronine (T3), CGS 23425 or axitirome (CGS 26214).
- the compounds according to the invention are administered in combination with an HMG-CoA reductase inhibitor from the class of statins, such as by way of example and preferably lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastatin or pitavastatin.
- statins such as by way of example and preferably lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rosuvastatin or pitavastatin.
- the compounds according to the invention are administered in combination with a squalene synthesis inhibitor, such as by way of example and preferably BMS-188494 or TAK-475.
- the compounds according to the invention are administered in combination with an ACAT inhibitor, such as by way of example and preferably avasimibe, melinamide, pactimibe, eflucimibe or SMP-797.
- an ACAT inhibitor such as by way of example and preferably avasimibe, melinamide, pactimibe, eflucimibe or SMP-797.
- the compounds according to the invention are administered in combination with an MTP inhibitor such as, for example and preferably, implitapide, BMS-201038, R-103757 or JTT-130.
- an MTP inhibitor such as, for example and preferably, implitapide, BMS-201038, R-103757 or JTT-130.
- the compounds of the invention are administered in combination with a PPAR gamma agonist such as, by way of example and by way of preference, pioglitazone or rosiglitazone.
- a PPAR gamma agonist such as, by way of example and by way of preference, pioglitazone or rosiglitazone.
- the compounds according to the invention are administered in combination with a PPAR delta agonist, such as by way of example and preferably GW 501516 or BAY 68-5042.
- a PPAR delta agonist such as by way of example and preferably GW 501516 or BAY 68-5042.
- the compounds according to the invention are administered in combination with a cholesterol absorption inhibitor, such as by way of example and preferably ezetimibe, tiqueside or pamaqueside.
- the compounds according to the invention are administered in combination with a lipase inhibitor, such as, for example and preferably, orlistat.
- a lipase inhibitor such as, for example and preferably, orlistat.
- the compounds of the invention are administered in combination with a polymeric bile acid adsorbent such as, by way of example and by way of preference, cholestyramine, colestipol, colesolvam, cholesta gel or colestimide.
- a polymeric bile acid adsorbent such as, by way of example and by way of preference, cholestyramine, colestipol, colesolvam, cholesta gel or colestimide.
- ASBT IBAT
- AZD-7806 S-8921
- AK-105 AK-105
- BARI-1741 AK-105
- SC-435 SC-635.
- the compounds according to the invention are administered in combination with a lipoprotein (a) antagonist, such as, by way of example and by way of preference, gemcabene calcium (CI-1027) or nicotinic acid.
- a lipoprotein (a) antagonist such as, by way of example and by way of preference, gemcabene calcium (CI-1027) or nicotinic acid.
- compositions containing at least one compound of the invention are pharmaceutical compositions containing at least one compound of the invention, usually together with one or more inert, non-toxic, pharmaceutically suitable excipients, and their use for the purposes mentioned above.
- the compounds according to the invention can act systemically and / or locally. For this purpose, they may be applied in a suitable manner, e.g. oral, parenteral, pulmonary, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctival, otic or as an implant or stent.
- the compounds according to the invention can be administered in suitable administration forms.
- the compounds of the invention rapidly and / or modified donating application forms containing the compounds of the invention in crystalline and / or amorphized and / or dissolved form, such as tablets (uncoated or coated Tablets, for example with enteric or delayed-dissolving or insoluble coatings which control the release of the compound of the invention), rapidly disintegrating in the oral cavity Tablets or films / wafers, films / lyophilisates, capsules (for example hard or soft gelatin capsules), dragees, granules, pellets, powders, emulsions, suspensions, aerosols or solutions.
- tablets uncoated or coated Tablets, for example with enteric or delayed-dissolving or insoluble coatings which control the release of the compound of the invention
- Tablets or films / wafers, films / lyophilisates capsules (for example hard or soft gelatin capsules), dragees, granules, pellets, powders, emulsions, suspensions, aerosols or solutions
- Parenteral administration may be by circumvention of a resorption step (e.g., intravenous, intraarterial, intracardiac, intraspinal, or intralumbar) or by absorption (e.g., intramuscular, subcutaneous, intracutaneous, percutaneous or intraperitoneal).
- a resorption step e.g., intravenous, intraarterial, intracardiac, intraspinal, or intralumbar
- absorption e.g., intramuscular, subcutaneous, intracutaneous, percutaneous or intraperitoneal.
- parenteral administration are suitable as application forms u.a. Injection and infusion preparations in the form of solutions, suspensions, emulsions, lyophilisates or sterile powders.
- Inhalation medicines including powder inhalers, nebulizers
- nasal drops solutions or sprays
- lingual, sublingual or buccal tablets films / wafers or capsules
- suppositories ear or eye preparations
- vaginal capsules aqueous suspensions (lotions, shake mixtures ), lipophilic suspensions, ointments, creams, transdermal therapeutic systems (eg patches), milk, pastes, foams, powdered powders, implants or stents.
- Preference is given to oral or parenteral administration, in particular oral administration.
- the compounds according to the invention can be converted into the stated administration forms. This can be done in a conventional manner by mixing with inert, non-toxic, pharmaceutically suitable excipients.
- excipients for example microcrystalline cellulose, lactose, mannitol
- solvents for example liquid polyethylene glycols
- emulsifiers and dispersants or wetting agents for example sodium dodecyl sulfate, polyoxysorbitol oleate
- binders for example polyvinylpyrrolidone
- synthetic and natural polymers for example albumin
- stabilizers for example, antioxidants such as ascorbic acid
- dyes eg, inorganic pigments such as iron oxides
- flavor and / or odoriferous for example, antioxidants such ascorbic acid
- dyes eg, inorganic pigments such as iron oxides
- the dosage is about 0.001 to 2 mg / kg, preferably about 0.001 to 1 mg / kg of body weight.
- Example 1 Exemplary embodiments: Example 1
- the force of contraction is detected with Statham UC2 cells, amplified and digitized via A / D converter (DAS-1802 HC, Keithley Instruments Munich) and registered in parallel on a chart recorder.
- a / D converter DAS-1802 HC, Keithley Instruments Munich
- phenylephrine is added cumulatively to the bath in increasing concentration.
- the substance to be examined is added in each subsequent course in increasing dosages and the height of the contraction is compared with the height of the contraction achieved in the last predistortion. This is used to calculate the concentration required to reduce the level of the control value by 50% (IC50 value).
- the standard application volume is 5 ⁇ , the DMSO content in the bath solution corresponds to 0.1%.
- the cellular activity of the compounds of the invention is measured on a recombinant guanylate cyclase reporter cell line as described in F. Wunder et al., Anal. Biochem. 339, 104-112 (2005).
- a commercially available telemetry system from DATA SCIENCES INTERNATIONAL DSI, USA is used for the blood pressure measurement on awake rats described below.
- the system consists of 3 main components: - Implantable transmitters (Physiotel® telemetry transmitters)
- Data acquisition computer are connected.
- the telemetry system allows a continuous recording of blood pressure heart rate and body movement on awake animals in their habitual habitat.
- the experimental animals are kept individually in macroion cages type 3 after transmitter implantation. You have free access to standard food and water.
- the day - night rhythm in the experimental laboratory is changed by room lighting at 6:00 in the morning and at 19:00 in the evening.
- the TAH PA - C40 telemetry transmitters are surgically implanted into the experimental animals under aseptic conditions at least 14 days before the first trial.
- the animals so instrumented are repeatedly used after healing of the wound and ingrowth of the implant.
- the fasting animals are anaesthetized with pentobabital (Nembutal, Sanofi: 50 mg / kg ip) and shaved and disinfected on the ventral side.
- pentobabital Nembutal, Sanofi: 50 mg / kg ip
- the system's liquid-filled measuring catheter above the bifurcation is inserted cranially into the descending aorta and secured with tissue adhesive (VetBonD TM, 3M).
- the transmitter housing is fixed intraperitoneally to the abdominal wall musculature and the wound is closed in layers.
- an antibiotic is administered for infection prophylaxis (Tardomyocel COMP Bayer 1 ml / kg sc)
- a solvent-treated group of animals is used as a control.
- Experimental procedure The existing telemetry measuring device is configured for 24 animals. Each trial is registered under a trial number (VYear month day).
- the instrumented rats living in the plant each have their own receiving antenna (1010 receivers, DSI).
- the implanted transmitters can be activated externally via a built-in magnetic switch. They will be put on the air during the trial run.
- the emitted signals can be recorded online by a data acquisition system (Dataquest TM A.R.T. for Windows, DSI) and processed accordingly. The storage of the data takes place in each case in a folder opened for this purpose which carries the test number.
- SBP Systolic blood pressure
- ACT Activity - Activity
- the measured value acquisition is repeated computer-controlled in 5-minute intervals.
- the absolute value of the source data is corrected in the diagram with the currently measured barometric pressure (Ambient Pressure Reference Monitor, APR-1) and in individual data stored. Further technical details can be found in the extensive documentation of the manufacturer (DSI).
- test substances will take place at 9 o'clock on the day of the experiment. Following the application, the parameters described above are measured for 24 hours.
- the collected individual data are sorted with the analysis software (DATAQUEST TM A.RT. TM ANALYSIS).
- the blank value is assumed here 2 hours before application, so that the selected data record covers the period from 7:00 am on the day of the experiment to 9:00 am on the following day.
- the data is smoothed over a presettable time by averaging (15 minutes average) and transferred as a text file to a disk.
- the presorted and compressed measured values are transferred to Excel templates and displayed in tabular form.
- the filing of the collected data takes place per experiment day in a separate folder that bears the test number. Results and test reports are sorted in folders and sorted by paper.
- mice Male CD-1 mice, male Wister rats and female beagle dogs.
- Intravenous administration is in mice and rats using a species-specific plasma / DMSO formulation and in dogs using a water / PEG400 / ethanol formulation.
- Oral administration of the solute by gavage is performed in all species based on a water / PEG400 / ethanol formulation. Rats are placed in the right external jugular vein for ease of blood sampling prior to drug administration.
- the operation is at least one day before the experiment under isoflurane anesthesia and with administration of an analgesic (atropine / rimadyl (3/1) 0.1 mL sc).
- an analgesic atropine / rimadyl (3/1) 0.1 mL sc.
- the blood collection (usually more than 10 times) takes place in a time window, which includes terminal times of at least 24 to a maximum of 72 hours after substance administration.
- the blood is transferred to heparinized tubes at collection. So then the blood plasma is recovered by centrifugation and optionally stored at -20 ° C until further processing.
- the pharmacokinetic parameters such as AUC, Cmax , ti 2 (terminal half-life), MRI (Mean Residence Time) and CL (clearance) are calculated from the plasma concentration-time profiles determined by means of a validated pharmacokinetic calculation program.
- the blood / plasma distribution of the substance must be determined in order to adjust the pharmacokinetic parameters accordingly.
- a defined amount of substance is incubated in heparinized whole blood of the corresponding species for 20 min in a tumble roll mixer. After centrifugation at 1000 g, the concentration in the plasma is measured (by means of LC-MS / MS, see above) and determined by quotient formation of the CBiut / Cpksma value.
- CYP cytochrome P450
- the compounds of the invention were incubated at a concentration of about 0.1-10 ⁇ .
- stock solutions of the compounds according to the invention with a concentration of 0.01-1 mM in acetonitrile were prepared, and then pipetted with a 1: 100 dilution into the incubation mixture.
- Liver microsomes and recombinant enzymes were detected in 50 mM potassium phosphate buffer pH 7.4 with and without NADPH-generating system, consisting of 1 mM NADP + , 10 mM glucose-6-phosphate and 1 unit of glucose-6-phosphate dehydrogenase, incubated at 37 ° C.
- Primary hepatocytes were also incubated in suspension in Williams E medium also at 37 ° C.
- the incubation mixtures were stopped with acetonitrile (final concentration about 30%) and the protein was centrifuged off at about 15,000 ⁇ g. The samples thus stopped were either analyzed directly or stored at -20 ° C until analysis.
- the analysis is carried out by high performance liquid chromatography with ultraviolet and mass spectrometric detection (HPLC-UV-MS / MS).
- HPLC-UV-MS / MS ultraviolet and mass spectrometric detection
- the supernatants of the incubation samples are chromatographed with suitable C18-reversed-phase columns and variable eluent mixtures of acetonitrile and 10 mM aqueous ammonium formate solution or 0.05% formic acid.
- the UV chromatograms in combination with mass spectrometry data serve to identify, structure elucidate and quantitatively estimate the metabolites, and quantitative metabolic decrease of the compound of the invention in the incubation approaches.
- the compounds according to the invention can be converted into pharmaceutical preparations as follows:
- composition
- the mixture of compound of the invention, lactose and starch is granulated with a 5% solution (m / m) of the PVP in water.
- the granules are mixed after drying with the magnesium stearate for 5 minutes.
- This mixture is compressed with a conventional tablet press (for the tablet format see above).
- a pressing force of 15 kN is used as a guideline for the compression.
- the rhodigel is suspended in ethanol, the compound according to the invention is added to the suspension. While stirring, the addition of water. Until the completion of the swelling of Rhodigels is stirred for about 6 h.
- the compound of the invention is suspended in the mixture of polyethylene glycol and polysorbate with stirring. The stirring is continued until complete dissolution of the compound according to the invention. iv -Solution:
- the compound of the invention is dissolved at a concentration below saturation solubility in a physiologically acceptable solvent (e.g., isotonic saline, 5% glucose solution, and / or 30% PEG 400 solution).
- a physiologically acceptable solvent e.g., isotonic saline, 5% glucose solution, and / or 30% PEG 400 solution.
- the solution is sterile filtered and filled into sterile and pyrogen-free injection containers.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Epidemiology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Hospice & Palliative Care (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description


















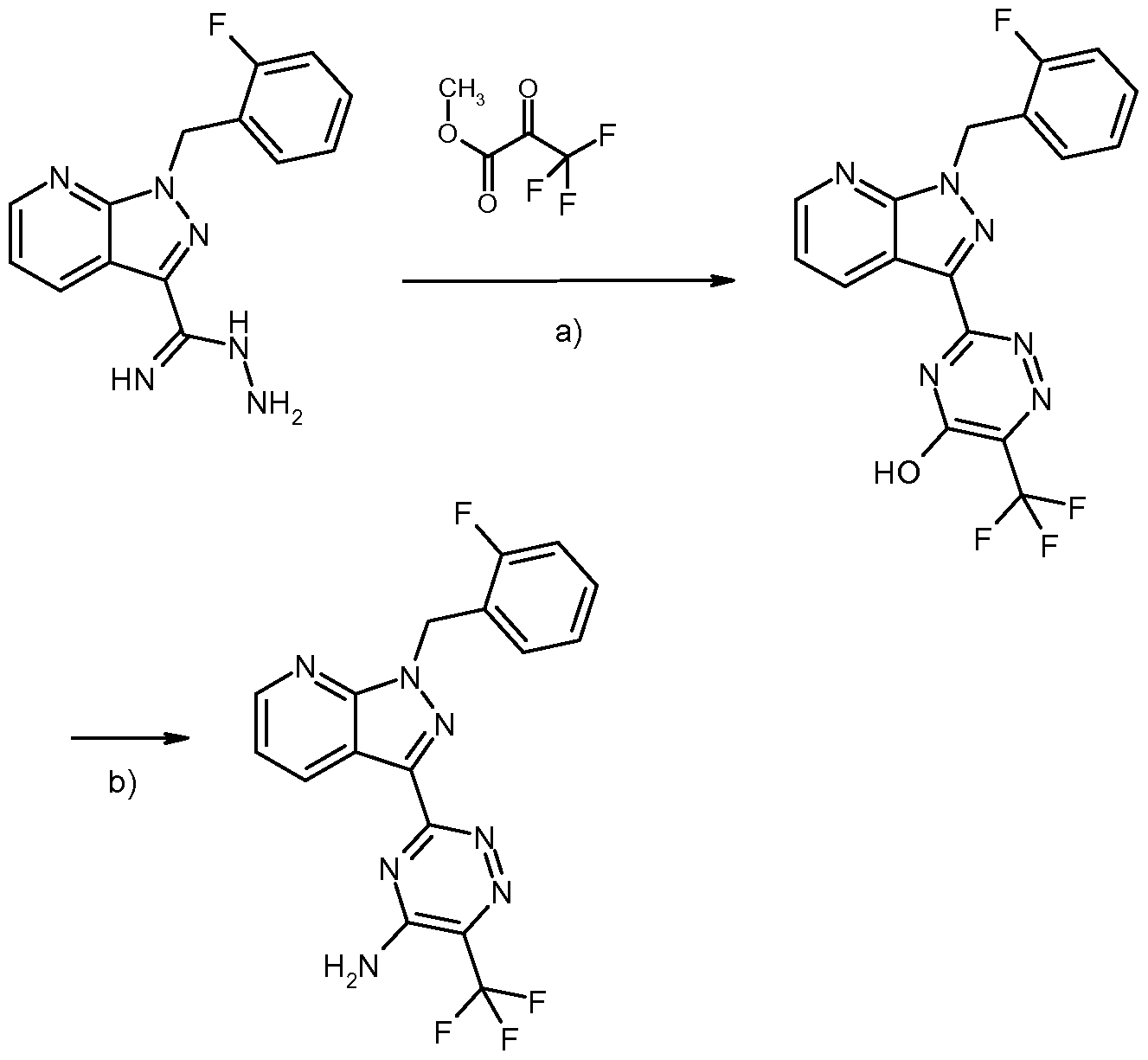















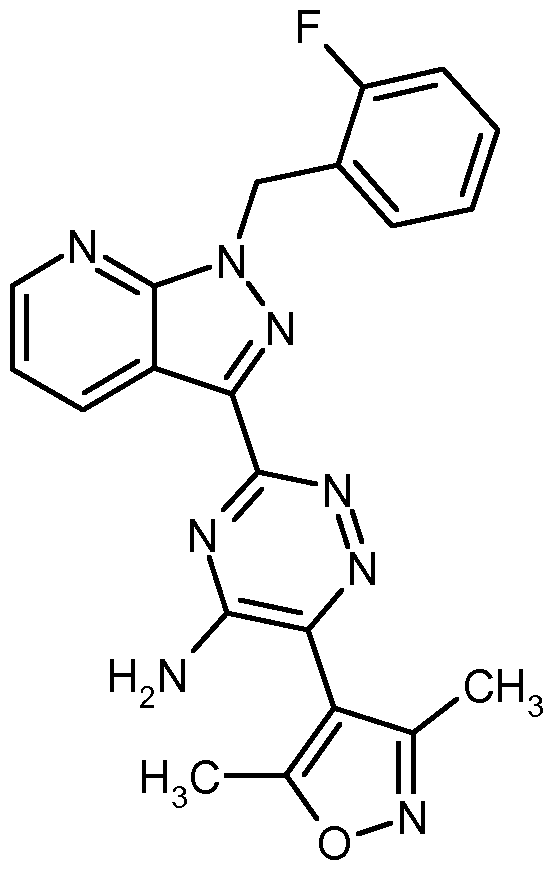




















Claims














Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES13700278.8T ES2597863T3 (es) | 2012-01-11 | 2013-01-08 | Derivados de triazina sustituida y su uso como estimuladores de la guanilato ciclasa soluble |
| CN201380013508.9A CN104159899B (zh) | 2012-01-11 | 2013-01-08 | 取代的三嗪衍生物及其作为可溶性鸟苷酸环化酶激发物的用途 |
| CA2860826A CA2860826A1 (en) | 2012-01-11 | 2013-01-08 | Substituted triazine derivatives and use thereof as stimulators of soluble guanylate cyclase |
| JP2014551586A JP6234938B2 (ja) | 2012-01-11 | 2013-01-08 | 置換トリアジン誘導体および可溶性グアニル酸シクラーゼの刺激剤としてのその使用 |
| EP13700278.8A EP2802580B1 (de) | 2012-01-11 | 2013-01-08 | Substituierte triazine derivate und ihre verwendung als stimulatoren der löslichen guanylatcyclase |
| US14/371,054 US9133191B2 (en) | 2012-01-11 | 2013-01-08 | Substituted triazine derivatives and use thereof as stimulators of soluble guanylate cyclase |
| HK15100007.4A HK1199641A1 (en) | 2012-01-11 | 2015-01-02 | Substituted triazine derivatives and use thereof as stimulators of soluble guanylate cyclase |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102012200360A DE102012200360A1 (de) | 2012-01-11 | 2012-01-11 | Substituierte Triazine und ihre Verwendung |
| DE102012200360.6 | 2012-01-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013104597A1 true WO2013104597A1 (de) | 2013-07-18 |
Family
ID=47559470
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2013/050179 WO2013104597A1 (de) | 2012-01-11 | 2013-01-08 | Substituierte triazine derivate und ihre verwendung als stimulatoren der löslichen guanylatcyclase |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US9133191B2 (de) |
| EP (1) | EP2802580B1 (de) |
| JP (1) | JP6234938B2 (de) |
| CN (1) | CN104159899B (de) |
| CA (1) | CA2860826A1 (de) |
| DE (1) | DE102012200360A1 (de) |
| ES (1) | ES2597863T3 (de) |
| HK (1) | HK1199641A1 (de) |
| WO (1) | WO2013104597A1 (de) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014084312A1 (ja) | 2012-11-30 | 2014-06-05 | アステラス製薬株式会社 | イミダゾピリジン化合物 |
| WO2015106268A1 (en) | 2014-01-13 | 2015-07-16 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF NEUROMUSCULAR DISORDERS |
| US9266871B2 (en) | 2013-03-01 | 2016-02-23 | Bayer Pharma Aktiengesellschaft | Trifluoromethyl-substituted fused pyrimidines and their use |
| WO2016044441A1 (en) * | 2014-09-17 | 2016-03-24 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| WO2016177660A1 (en) | 2015-05-06 | 2016-11-10 | Bayer Pharma Aktiengesellschaft | The use of sgc stimulators, sgc activators, alone and combinations with pde5 inhibitors for the treatment of digital ulcers (du) concomitant to systemic sclerosis (ssc) |
| US9505786B2 (en) | 2012-01-11 | 2016-11-29 | Bayer Pharma Aktiengesellschaft | Substituted annulated triazines and use thereof |
| WO2017013010A1 (de) | 2015-07-23 | 2017-01-26 | Bayer Pharma Aktiengesellschaft | Stimulatoren und/oder aktivatoren der löslichen guanylatzyklase (sgc) in kombination mit einem inhibitor der neutralen endopeptidase (nep inhibitor) und/oder einem angiotensin aii-antagonisten und ihre verwendung |
| US9605008B2 (en) | 2013-07-10 | 2017-03-28 | Bayer Pharma Aktiengesellschaft | Benzyl-1H-pyrazolo[3,4-b]pyridines and use thereof |
| WO2017106175A2 (en) | 2015-12-14 | 2017-06-22 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF GASTROINTESTINAL SPHINCTER DYSFUNCTION |
| EP3135668A4 (de) * | 2014-04-24 | 2018-03-21 | Mitsubishi Tanabe Pharma Corporation | Neuartige disubstituierte 1,2,4-triazin-verbindung |
| WO2018069126A1 (de) | 2016-10-11 | 2018-04-19 | Bayer Pharma Aktiengesellschaft | Kombination enthaltend sgc stimulatoren und mineralocorticoid-rezeptor-antagonisten |
| WO2018111795A2 (en) | 2016-12-13 | 2018-06-21 | Ironwood Pharmaceuticals, Inc. | Use of sgc stimulators for the treatment of esophageal motility disorders |
| WO2019219672A1 (en) | 2018-05-15 | 2019-11-21 | Bayer Aktiengesellschaft | 1,3-thiazol-2-yl substituted benzamides for the treatment of diseases associated with nerve fiber sensitization |
| WO2020014504A1 (en) | 2018-07-11 | 2020-01-16 | Cyclerion Therapeutics, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF MITOCHONRIAL DISORDERS |
| WO2020165010A1 (en) | 2019-02-13 | 2020-08-20 | Bayer Aktiengesellschaft | Process for the preparation of porous microparticles |
| US11331308B2 (en) | 2016-10-11 | 2022-05-17 | Bayer Pharma Aktiengesellschaft | Combination containing sGC activators and mineralocorticoid receptor antagonists |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TR201816146T4 (tr) | 2009-11-27 | 2018-11-21 | Adverio Pharma Gmbh | Meti̇l-{4,6-di̇ami̇no-2-[1-(2-florobenzi̇l)-1h-pi̇razolo[3,4-b]pi̇ri̇di̇n-3-i̇l]pi̇ri̇mi̇di̇n-5-i̇lmeti̇l}karbamatin farmasöti̇k etken madde olarak kullanima yöneli̇k olarak üreti̇lmesi̇ne yöneli̇k yöntem. |
| EP3609883B1 (de) | 2017-04-11 | 2022-06-29 | Sunshine Lake Pharma Co., Ltd. | Fluorsubstituierte indazolverbindungen und verwendungen davon |
| WO2022093714A1 (en) * | 2020-10-26 | 2022-05-05 | Minerals Therapeutics, Inc. | Cyp11b2 beta hydroxylase inhibitors for hypertension |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998016223A1 (de) | 1996-10-14 | 1998-04-23 | Bayer Aktiengesellschaft | Verwendung von 1-benzyl-3-(substituiertes-hetaryl)-kondensierten pyrazol-derivaten zur belandlung von speziellen erkrankungen des herz-kreislaufsystems und des zentralnervensystems |
| WO2000006568A1 (de) | 1998-07-29 | 2000-02-10 | Bayer Aktiengesellschaft | Substituierte pyrazolderivate |
| WO2000006569A1 (de) | 1998-07-29 | 2000-02-10 | Bayer Aktiengesellschaft | Mit sechsgliedrigen heterocyclischen ringen kondensierte substituierte pyrazolderivate |
| WO2003095451A1 (de) | 2002-05-08 | 2003-11-20 | Bayer Healthcare Ag | Carbamat-substituierte pyrazolopyridine |
| WO2004009590A1 (de) | 2002-07-18 | 2004-01-29 | Bayer Healthcare Ag | 4-aminosubstituierte pyrimidinderivate |
| WO2006081230A2 (en) * | 2005-01-26 | 2006-08-03 | Schering Corporation | 3-(indazol-5-yl)-(1,2, 4) triazine derivatives and related compounds as protein kinase inhibitors for the treatment of cancer |
| WO2010065275A1 (en) | 2008-11-25 | 2010-06-10 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2011115804A1 (en) | 2010-03-17 | 2011-09-22 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| WO2011149921A1 (en) | 2010-05-27 | 2011-12-01 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
Family Cites Families (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9314412D0 (en) | 1993-07-13 | 1993-08-25 | Rhone Poulenc Agriculture | New compositions of matter |
| EP0743066A3 (de) | 1995-05-16 | 1998-09-30 | Mitsui Pharmaceuticals, Inc. | Wundheilmittel |
| US6166027A (en) | 1996-10-14 | 2000-12-26 | Bayer Aktiengesellschaft | Heterocyclylmethyl-substituted pyrazole derivatives and their use for treating cardiovascular diseases |
| US6451805B1 (en) | 1997-11-14 | 2002-09-17 | Bayer Aktiengesellschaft | Substituted pyrazole derivatives for the treatment of cardiocirculatory diseases |
| DE10021069A1 (de) | 2000-04-28 | 2001-10-31 | Bayer Ag | Substituiertes Pyrazolderivat |
| DE10031584A1 (de) * | 2000-06-29 | 2002-01-10 | Merck Patent Gmbh | 5-Aminoalkyl-pyrazolo[4,3-d]pyrimidine |
| EP1333833B1 (de) | 2000-10-23 | 2011-08-24 | GlaxoSmithKline LLC | Neues trisubstitutiertes 8H-Pyrido[2,3-d]pyrimidin-7-onderivat zur Behandlung von durch CSBP/p38kinase vermittelten Krankheiten |
| AU2002221827A1 (en) | 2000-11-22 | 2002-06-03 | Bayer Aktiengesellschaft | Novel lactame-substituted pyrazolopyridine derivatives |
| DE10132416A1 (de) | 2001-07-04 | 2003-01-16 | Bayer Ag | Neue Morpholin-überbrückte Pyrazolopyridinderivate |
| CA2521695C (en) | 2003-05-09 | 2011-08-16 | Asahi Glass Company, Limited | Method for producing 3-substituted 2-chloro-5-fluoro-pyridine or its salt |
| CN100355732C (zh) | 2003-11-03 | 2007-12-19 | 上海药明康德新药开发有限公司 | 2-氯-5-氟-烟酸酯及酸的制备方法 |
| ATE433452T1 (de) | 2004-01-31 | 2009-06-15 | Actimis Pharmaceuticals Inc | Imidazoä1,2-cüpyrimidinylessigsäurederivate |
| WO2006130673A1 (en) | 2005-05-31 | 2006-12-07 | Janssen Pharmaceutica, N.V. | 3-benzoimidazolyl-pyrazolopyridines useful in treating kinase disorders |
| DE102006043443A1 (de) * | 2006-09-15 | 2008-03-27 | Bayer Healthcare Ag | Neue aza-bicyclische Verbindungen und ihre Verwendung |
| AU2008282156B2 (en) | 2007-07-31 | 2014-07-17 | Vertex Pharmaceuticals Incorporated | Process for preparing 5-fluoro-1H-pyrazolo [3, 4-b] pyridin-3-amine and derivatives thereof |
| JP2011513483A (ja) | 2008-03-10 | 2011-04-28 | バーテックス ファーマシューティカルズ インコーポレイテッド | タンパク質キナーゼの阻害剤として有用なピリミジンおよびピリジン |
| DE102008063992A1 (de) * | 2008-12-19 | 2010-09-02 | Lerner, Zinoviy, Dipl.-Ing. | Neue aliphatisch substituierte Pyrazolopyridine und ihre Verwendung |
| DE102009004245A1 (de) | 2009-01-09 | 2010-07-15 | Bayer Schering Pharma Aktiengesellschaft | Neue anellierte, Heteroatom-verbrückte Pyrazol- und Imidazol-Derivate und ihre Verwendung |
| CA2762680C (en) | 2009-05-21 | 2018-04-17 | Chlorion Pharma, Inc. | Methyl sulfanyl pyrmidmes useful as antiinflammatories, analgesics, and antiepileptics |
| DE102010021637A1 (de) | 2010-05-26 | 2011-12-01 | Bayer Schering Pharma Aktiengesellschaft | Substituierte 5-Fluor-1H-Pyrazolopyridine und ihre Verwendung |
| CA2803688A1 (en) | 2010-06-25 | 2011-12-29 | Bayer Intellectual Property Gmbh | Use of stimulators and activators of soluble guanylate cyclase for treating sickle-cell anemia and conserving blood substitutes |
| EP2590987B1 (de) | 2010-07-09 | 2016-03-09 | Bayer Intellectual Property GmbH | Annellierte 4-aminopyrimidine und ihre verwendung als stimulatoren der löslichen guanylatcyclase |
| PE20130779A1 (es) * | 2010-07-09 | 2013-06-21 | Bayer Ip Gmbh | Pirimidinas y triazinas condensadas y su uso |
| DE102010040233A1 (de) | 2010-09-03 | 2012-03-08 | Bayer Schering Pharma Aktiengesellschaft | Bicyclische Aza-Heterocyclen und ihre Verwendung |
| CA2833698A1 (en) | 2011-04-21 | 2012-10-26 | Bayer Intellectual Property Gmbh | Fluoroalkyl-substituted pyrazolopyridines and use thereof |
| EP2705037B1 (de) | 2011-05-06 | 2016-06-22 | Bayer Intellectual Property GmbH | Substituierte imidazopyridine und imidazopyridazine und ihre verwendung |
| MX2014000029A (es) | 2011-07-06 | 2014-02-17 | Bayer Ip Gmbh | Pirazolopiridinas sustituidas con heteroarilo y uso de las mismas como estimuladores de guanilato ciclasas solubles. |
| JP6096778B2 (ja) | 2011-09-01 | 2017-03-15 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | ピロロピラジンキナーゼ阻害剤 |
| CN104039784B (zh) | 2011-09-02 | 2017-08-22 | 拜耳知识产权有限责任公司 | 取代的增环嘧啶及其用途 |
| DE102012200349A1 (de) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituierte annellierte Pyrimidine und Triazine und ihre Verwendung |
| DE102012200352A1 (de) | 2012-01-11 | 2013-07-11 | Bayer Intellectual Property Gmbh | Substituierte, annellierte Imidazole und Pyrazole und ihre Verwendung |
| EP2822951B1 (de) | 2012-03-06 | 2017-07-26 | Bayer Intellectual Property GmbH | Substituierte azabicyclen und ihre verwendung |
| KR20150121007A (ko) | 2013-03-01 | 2015-10-28 | 바이엘 파마 악티엔게젤샤프트 | 트리플루오르메틸-치환된 고리-융합된 피리미딘 및 그의 용도 |
-
2012
- 2012-01-11 DE DE102012200360A patent/DE102012200360A1/de not_active Withdrawn
-
2013
- 2013-01-08 WO PCT/EP2013/050179 patent/WO2013104597A1/de active Application Filing
- 2013-01-08 CA CA2860826A patent/CA2860826A1/en not_active Abandoned
- 2013-01-08 EP EP13700278.8A patent/EP2802580B1/de not_active Not-in-force
- 2013-01-08 JP JP2014551586A patent/JP6234938B2/ja not_active Expired - Fee Related
- 2013-01-08 CN CN201380013508.9A patent/CN104159899B/zh not_active Expired - Fee Related
- 2013-01-08 US US14/371,054 patent/US9133191B2/en not_active Expired - Fee Related
- 2013-01-08 ES ES13700278.8T patent/ES2597863T3/es active Active
-
2015
- 2015-01-02 HK HK15100007.4A patent/HK1199641A1/xx not_active IP Right Cessation
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998016223A1 (de) | 1996-10-14 | 1998-04-23 | Bayer Aktiengesellschaft | Verwendung von 1-benzyl-3-(substituiertes-hetaryl)-kondensierten pyrazol-derivaten zur belandlung von speziellen erkrankungen des herz-kreislaufsystems und des zentralnervensystems |
| WO2000006568A1 (de) | 1998-07-29 | 2000-02-10 | Bayer Aktiengesellschaft | Substituierte pyrazolderivate |
| WO2000006569A1 (de) | 1998-07-29 | 2000-02-10 | Bayer Aktiengesellschaft | Mit sechsgliedrigen heterocyclischen ringen kondensierte substituierte pyrazolderivate |
| WO2003095451A1 (de) | 2002-05-08 | 2003-11-20 | Bayer Healthcare Ag | Carbamat-substituierte pyrazolopyridine |
| WO2004009590A1 (de) | 2002-07-18 | 2004-01-29 | Bayer Healthcare Ag | 4-aminosubstituierte pyrimidinderivate |
| WO2006081230A2 (en) * | 2005-01-26 | 2006-08-03 | Schering Corporation | 3-(indazol-5-yl)-(1,2, 4) triazine derivatives and related compounds as protein kinase inhibitors for the treatment of cancer |
| WO2010065275A1 (en) | 2008-11-25 | 2010-06-10 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
| WO2011115804A1 (en) | 2010-03-17 | 2011-09-22 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| WO2011149921A1 (en) | 2010-05-27 | 2011-12-01 | Merck Sharp & Dohme Corp. | Soluble guanylate cyclase activators |
Non-Patent Citations (12)
| Title |
|---|
| E. M. BECKER ET AL., BMC PHARMACOLOGY, vol. 1, no. 13, 2001 |
| F. WUNDER ET AL., ANAL. BIOCHEM., vol. 339, 2005, pages 104 - 112 |
| GOLDBERG ET AL., J. BIOL. CHEM., vol. 252, 1977, pages 1279 |
| HASSAN J. ET AL., CHEM. REV., vol. 102, 2002, pages 1359 - 1469 |
| J. MED. CHEM., vol. 9, 1966, pages 149 - 151 |
| KLAUS WITTE; KAI HU; JOHANNA SWIATEK; CLAUDIA MÜSSIG; GEORG ERTL; BJÖRN LEMMER: "Experimental heart failure in rats: effects on cardiovascular circadian rhythms and on myocardial ?-adrenergic signaling", CARDIOVASC RES, vol. 47, no. 2, 2000, pages 203 - 405 |
| KOZO OKAMOTO: "Spontaneous hypertension in rats", INT REV EXP PATHOL, vol. 7, 1969, pages 227 - 270 |
| MAARTEN VAN DEN BUUSE: "Circadian Rhythms of Blood Pressure, Heart Rate, and Locomotor Activity in Spontaneously Hypertensive Rats as Measured With Radio-Telemetry", PHYSIOLOGY & BEHAVIOR, vol. 55, no. 4, 1994, pages 783 - 787 |
| MÜLSCH ET AL., BRIT. J. PHARMACOL, vol. 120, 1997, pages 681 |
| PETTIBONE ET AL., EUR. J. PHARMACOL., vol. 116, 1985, pages 307 |
| WU ET AL., BLOOD, vol. 84, 1994, pages 4226 |
| YU ET AL., BRIT. J. PHARMACOL., vol. 114, 1995, pages 1587 |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9505786B2 (en) | 2012-01-11 | 2016-11-29 | Bayer Pharma Aktiengesellschaft | Substituted annulated triazines and use thereof |
| WO2014084312A1 (ja) | 2012-11-30 | 2014-06-05 | アステラス製薬株式会社 | イミダゾピリジン化合物 |
| US9266871B2 (en) | 2013-03-01 | 2016-02-23 | Bayer Pharma Aktiengesellschaft | Trifluoromethyl-substituted fused pyrimidines and their use |
| US9605008B2 (en) | 2013-07-10 | 2017-03-28 | Bayer Pharma Aktiengesellschaft | Benzyl-1H-pyrazolo[3,4-b]pyridines and use thereof |
| WO2015106268A1 (en) | 2014-01-13 | 2015-07-16 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF NEUROMUSCULAR DISORDERS |
| EP3135668A4 (de) * | 2014-04-24 | 2018-03-21 | Mitsubishi Tanabe Pharma Corporation | Neuartige disubstituierte 1,2,4-triazin-verbindung |
| US10029993B2 (en) | 2014-04-24 | 2018-07-24 | Mitsubishi Tanabe Pharma Corporation | Disubstituted 1, 2, 4-triazine compound |
| US10329263B2 (en) | 2014-04-24 | 2019-06-25 | Mitsubishi Tanabe Pharma Corporation | Disubstituted 1, 2, 4-triazine compound |
| WO2016044441A1 (en) * | 2014-09-17 | 2016-03-24 | Ironwood Pharmaceuticals, Inc. | Sgc stimulators |
| JP2017527605A (ja) * | 2014-09-17 | 2017-09-21 | アイアンウッド ファーマシューティカルズ インコーポレイテッド | sGC刺激剤 |
| CN107223125A (zh) * | 2014-09-17 | 2017-09-29 | 铁木医药有限公司 | sGC刺激剂 |
| US10047095B2 (en) | 2014-09-17 | 2018-08-14 | Ironwood Pharmaceuticals, Inc. | sGC stimulators |
| CN107223125B (zh) * | 2014-09-17 | 2019-09-27 | 赛科理音医疗有限公司 | sGC刺激剂 |
| WO2016177660A1 (en) | 2015-05-06 | 2016-11-10 | Bayer Pharma Aktiengesellschaft | The use of sgc stimulators, sgc activators, alone and combinations with pde5 inhibitors for the treatment of digital ulcers (du) concomitant to systemic sclerosis (ssc) |
| US11166932B2 (en) | 2015-07-23 | 2021-11-09 | Bayer Pharma Aktiengesellschaft | Stimulators and/or activators of soluble guanylate cyclase (sGC) in combination with an inhibitor of neutral endopeptidase (NEP inhibitor) and/or an angiotensin AII antagonist and the use thereof |
| WO2017013010A1 (de) | 2015-07-23 | 2017-01-26 | Bayer Pharma Aktiengesellschaft | Stimulatoren und/oder aktivatoren der löslichen guanylatzyklase (sgc) in kombination mit einem inhibitor der neutralen endopeptidase (nep inhibitor) und/oder einem angiotensin aii-antagonisten und ihre verwendung |
| WO2017106175A2 (en) | 2015-12-14 | 2017-06-22 | Ironwood Pharmaceuticals, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF GASTROINTESTINAL SPHINCTER DYSFUNCTION |
| WO2018069126A1 (de) | 2016-10-11 | 2018-04-19 | Bayer Pharma Aktiengesellschaft | Kombination enthaltend sgc stimulatoren und mineralocorticoid-rezeptor-antagonisten |
| US10918639B2 (en) | 2016-10-11 | 2021-02-16 | Bayer Pharma Aktiengesellschaft | Combination containing SGC stimulators and mineralocorticoid receptor antagonists |
| US11331308B2 (en) | 2016-10-11 | 2022-05-17 | Bayer Pharma Aktiengesellschaft | Combination containing sGC activators and mineralocorticoid receptor antagonists |
| US11684621B2 (en) | 2016-10-11 | 2023-06-27 | Bayer Pharma Aktiengesellschaft | Combination containing sGC stimulators and mineralocorticoid receptor antagonists |
| WO2018111795A2 (en) | 2016-12-13 | 2018-06-21 | Ironwood Pharmaceuticals, Inc. | Use of sgc stimulators for the treatment of esophageal motility disorders |
| WO2019219672A1 (en) | 2018-05-15 | 2019-11-21 | Bayer Aktiengesellschaft | 1,3-thiazol-2-yl substituted benzamides for the treatment of diseases associated with nerve fiber sensitization |
| WO2020014504A1 (en) | 2018-07-11 | 2020-01-16 | Cyclerion Therapeutics, Inc. | USE OF sGC STIMULATORS FOR THE TREATMENT OF MITOCHONRIAL DISORDERS |
| WO2020165010A1 (en) | 2019-02-13 | 2020-08-20 | Bayer Aktiengesellschaft | Process for the preparation of porous microparticles |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2802580B1 (de) | 2016-07-13 |
| ES2597863T3 (es) | 2017-01-23 |
| US20150094308A1 (en) | 2015-04-02 |
| DE102012200360A1 (de) | 2013-07-11 |
| JP6234938B2 (ja) | 2017-11-22 |
| HK1199641A1 (en) | 2015-07-10 |
| EP2802580A1 (de) | 2014-11-19 |
| CA2860826A1 (en) | 2013-07-18 |
| US9133191B2 (en) | 2015-09-15 |
| JP2015506938A (ja) | 2015-03-05 |
| CN104159899A (zh) | 2014-11-19 |
| CN104159899B (zh) | 2017-02-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2802580B1 (de) | Substituierte triazine derivate und ihre verwendung als stimulatoren der löslichen guanylatcyclase | |
| EP2822951B1 (de) | Substituierte azabicyclen und ihre verwendung | |
| EP2705037B1 (de) | Substituierte imidazopyridine und imidazopyridazine und ihre verwendung | |
| EP2635577B1 (de) | Substituierte 6-fluor-1h-pyrazolo[4,3-b]pyridine und ihre verwendung | |
| EP2961755B1 (de) | Trifluormethyl-substituierte annellierte pyrimidine und ihre verwendung | |
| EP2699578B1 (de) | Fluoralkyl-substituierte pyrazolopyridine und ihre verwendung | |
| EP2635576B1 (de) | Benzyl-substituierte carbamate und ihre verwendung | |
| DE102010021637A1 (de) | Substituierte 5-Fluor-1H-Pyrazolopyridine und ihre Verwendung | |
| DE102012200349A1 (de) | Substituierte annellierte Pyrimidine und Triazine und ihre Verwendung | |
| EP2729476A1 (de) | Heteroaryl-substituierte pyrazolopyridine und ihre verwendung als stimulatoren der löslichen guanylatcyclase | |
| EP2961754A1 (de) | Benzyl-substituierte pyrazolopyridine und ihre verwendung | |
| EP2705038A1 (de) | Substituierte imidazopyridazine und ihre verwendung | |
| WO2012010578A1 (de) | Substituierte methyl-pyrimidin-5-ylcarbamate und ihre verwendung | |
| WO2012010576A1 (de) | Carbamat-substituierte diaminopyrimidine und ihre verwendung | |
| DE102011075399A1 (de) | Substituierte Imidazopyridine und Imidazopyridazine und ihre Verwendung | |
| WO2017121692A1 (de) | Substituierte sulfamide und ihre verwendung | |
| DE102012200356A1 (de) | Substituierte Imidazopyridine und Imidazopyridazine und ihre Verwendung | |
| DE102011007891A1 (de) | Annellierte 4-Aminopyrimidine und ihre Verwendung | |
| DE102011007890A1 (de) | Fluoralkyl-substituierte Pyrazolopyridine und ihre Verwendung | |
| DE102011078715A1 (de) | Heteroaryl-substituierte Pyrazolopyridine und ihre Verwendung | |
| DE102012200351A1 (de) | Substituierte annellierte Pyrimidine und ihre Verwendung | |
| DE102011082041A1 (de) | Substituierte annellierte Pyrimidine und ihre Verwendung | |
| DE102010031148A1 (de) | Annellierte 4-Aminopyrimidine und ihre Verwendung | |
| DE102012200354A1 (de) | Heteroaryl-substituierte Pyrazolopyridine und ihre Verwendung | |
| DE102012200357A1 (de) | Fluoralkyl-substituierte Pyrazolopyridine und ihre Verwendung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13700278 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2860826 Country of ref document: CA |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013700278 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14371054 Country of ref document: US Ref document number: 2013700278 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2014551586 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |