WO2012053535A1 - ノルボルネン系モノマー重合用触媒及びノルボルネン系重合体の製造方法 - Google Patents
ノルボルネン系モノマー重合用触媒及びノルボルネン系重合体の製造方法 Download PDFInfo
- Publication number
- WO2012053535A1 WO2012053535A1 PCT/JP2011/074013 JP2011074013W WO2012053535A1 WO 2012053535 A1 WO2012053535 A1 WO 2012053535A1 JP 2011074013 W JP2011074013 W JP 2011074013W WO 2012053535 A1 WO2012053535 A1 WO 2012053535A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- bis
- norbornene
- group
- dipalladium
- general formula
- Prior art date
Links
- 0 CCC[C@@](C)N(CCC(C)(C)N(*)CN(*)CC)C(*)N* Chemical compound CCC[C@@](C)N(CCC(C)(C)N(*)CN(*)CC)C(*)N* 0.000 description 2
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F32/00—Homopolymers and copolymers of cyclic compounds having no unsaturated aliphatic radicals in a side chain, and having one or more carbon-to-carbon double bonds in a carbocyclic ring system
Definitions
- the present invention relates to a norbornene-based monomer polymerization catalyst and a method for producing a norbornene-based monomer copolymer having a polar group using the catalyst.
- cyclic olefin addition polymers represented by norbornene polymers have been industrially used in the fields of optical films and the like as organic materials having excellent heat resistance and transparency. It has been reported variously that such a cyclic olefin addition polymer can be produced by addition polymerization of a cyclic olefin monomer using a catalyst containing a transition metal compound such as Ti, Zr, Cr, Co, Ni, and Pd. .
- Patent Document 1 European Patent Application Publication No. 0445755 (Patent Document 1), a transition metal compound of group 5 to 10 elements is used as a main catalyst, and methylaluminoxane (MAO) is used as a cocatalyst, so that the number average molecular weight is 1,000,000. It has been reported that norbornene homo-addition polymers exceeding 10 can be produced. However, in this catalyst system, polymerization of a norbornene monomer having a polar group, which is more difficult to polymerize, has not been performed, and there has been a concern about catalyst deactivation due to the influence of the polar group.
- MAO methylaluminoxane
- Patent Document 2 using only dichlorobis (benzonitrile) palladium or allylpalladium chloride dimer as a catalyst, a homo-addition polymer of norbornene monomer having a polar group and norbornene A method for producing a copolymer of the above has been reported.
- this patent there is no example of producing a polymer having a number average molecular weight exceeding 10,000, and the polymerization activity of the catalyst is low, which is difficult to say as an industrially useful production method.
- Patent Document 3 Japanese Patent No. 3678754
- Patent Patents Patent Patents
- Document 4 Japanese Patent Application Laid-Open No. 2008-31304
- the polymerization activity and the molecular weight of the polymer are improved by using a combination of allyl palladium chloride dimer and silver tetrafluoroborate or silver hexafluorophosphate as a catalyst.
- the number average molecular weight of the copolymer disclosed in (1) is less than 200,000, and a copolymer having a number average molecular weight of 200,000 or more that has a practical mechanical property value has not been produced.
- Mn number average molecular weight
- Mw weight average molecular weight
- polarities are obtained by combining a group 8-10 transition metal compound having a cyclopentadienyl ligand as a main catalyst and a promoter capable of reacting with the main catalyst to form a cationic transition metal compound. It is disclosed in International Publication No. 2006/064814 (Patent Document 5) (US2009 / 264608 A1) that addition copolymerization of norbornene having a group with norbornene can be efficiently carried out and a high molecular weight copolymer is obtained. ing.
- the norbornene compound having a polar group disclosed in this publication has a structure in which an ester group is directly introduced into the norbornene skeleton, and the distance between the carbon-carbon double bond portion and the polar group is small. Since it was close, it easily coordinated to the transition metal complex as a catalyst, leading to a decrease in catalytic activity. Therefore, norbornene homo-addition polymerization can produce a high activity and high molecular weight polymer. However, when a norbornene monomer having a polar group is used, a high molecular weight copolymer is obtained, but the catalytic activity is low. It was.
- An object of the present invention is to provide a highly active catalyst system capable of producing a high molecular weight addition copolymer of a norbornene monomer having a polar group, and an efficient production method of the copolymer.
- the present inventors have obtained a main catalyst for a binuclear palladium compound having a ⁇ -allyl ( ⁇ 3 -allyl) ligand and a bidentate amidine ligand. And a norbornene compound in which one methylene chain is introduced between the norbornene skeleton and the ester group in order to increase the distance between the polymerizable carbon-carbon double bond and the polar group (ester group).
- a high molecular weight addition copolymer of a norbornene monomer having a polar group can be efficiently produced, and the present invention has been completed.
- the present invention relates to the following catalysts for polymerizing norbornene monomers [1] to [6] and methods for producing norbornene (co) polymers [7] to [12].
- [1] General formula (1) (In the formula, R 1 and R 2 each independently represent a hydrogen atom or a hydrocarbon group having 1 to 20 carbon atoms which may have a substituent, and R 3 , R 4 and R 5 each independently And a transition metal complex (A) represented by a hydrogen atom or a hydrocarbon group having 1 to 6 carbon atoms), a norbornene-based monomer polymerization catalyst.
- R 1 in the general formula (1) is a methyl group or a phenyl group which may have a substituent
- R 2 is a phenyl group which may have a cyclohexyl group or a substituent, 2.
- R 1 in the general formula (1) is a methyl group or a phenyl group
- R 2 is a cyclohexyl group, a phenyl group, a 2-methylphenyl group, a 2-isopropylphenyl group, or a 2,6-diisopropylphenyl group.
- Any one of items 1 to 3 comprising a promoter (B) and a phosphine-based ligand (C) which are ionic compounds capable of reacting with the transition metal complex (A) to form a cationic transition metal compound.
- C phosphine ligand
- a method for producing a norbornene (co) polymer wherein the norbornene monomer is homopolymerized or copolymerized in the presence of the polymerization catalyst according to any one of items 1 to 6.
- a method for producing a norbornene copolymer comprising copolymerizing a norbornene monomer and another vinyl monomer in the presence of the polymerization catalyst according to any one of items 1 to 6.
- a high molecular weight addition copolymer of norbornene and a norbornene monomer having a polar group can be efficiently produced.
- the norbornene-based copolymer obtained by the present invention has excellent transparency, heat resistance, low water absorption, electrical insulation properties, etc. It can be used for various purposes. Specifically, optical molded products such as lenses and polarizing films, films, carrier tapes, film capacitors, electrical insulating materials such as flexible printed boards, press-through packages, infusion bags, medical containers such as liquid vials, wraps, It can be used for food packaging molded products such as trays, casings for electrical appliances, automobile interior parts such as inner panels, and building materials such as carports and glazings.
- 1 is a 1 H-NMR spectrum of a transition metal complex obtained in Examples 1, 2, and 5.
- 2 is a 1 H-NMR spectrum of the copolymer obtained in Example 9. It is a chart of gel permeation chromatography (GPC) of the copolymer obtained in Example 9 and Comparative Example 2.
- the norbornene-based monomer polymerization catalyst of the present invention comprises a transition metal complex (A) as an essential component, and is a promoter (B (Hereinafter abbreviated as “promoter (B)”) and a phosphine-based ligand (C) as optional components.
- A transition metal complex
- B promoter
- C phosphine-based ligand
- Transition metal complex (A) which is a catalyst component for polymerization of the norbornene monomer of the present invention is a binuclear palladium having a structure in which two palladium having a ⁇ -allyl ligand are bridged by two amidine ligands. Rather than a single amidine that is a complex compound, which is a bidentate ligand, bidentates to one palladium to form a four-membered ring, two palladium are bridged by two amidine ligands, respectively. It is characterized by being coordinated to form an eight-membered ring.
- the transition metal complex (A) has the general formula (1) Indicated by
- R 1 and R 2 each independently represents a hydrogen atom or a hydrocarbon group having 1 to 20 carbon atoms which may have a substituent
- R 3 , R 4 and R 5 Each independently represents a hydrogen atom or a hydrocarbon group having 1 to 6 carbon atoms.
- the two CN bonds in the amidine (NCN) which is a ligand are single bonds or double bonds.
- specific examples of the hydrocarbon group having 1 to 20 carbon atoms represented by R 1 and R 2 include a linear or branched alkyl group having 1 to 20 carbon atoms, 3 to And a cycloalkyl group having 20 carbon atoms, an aryl group having 6 to 20 carbon atoms, an alkylaryl group, or an aralkyl group.
- Examples of the substituent for the hydrocarbon group having 1 to 20 carbon atoms include a halogen atom, an alkoxy group, an aryloxy group, a carboxy group, an alkoxycarbonyl group, a cyano group, and a trialkylsilyl group.
- R 1 and R 2 are each a methyl group, an ethyl group, an isopropyl group, a t-butyl group, a cyclohexyl group, a phenyl group, a 2-methylphenyl group, 2, from the viewpoint of the stability of the complex and the ease of synthesis.
- 6-dimethylphenyl group 2-isopropylphenyl group, 2,6-diisopropylphenyl group, 2-t-butylphenyl group, 2,6-di-t-butylphenyl group, benzyl group, naphthyl group, biphenyl group, anthracenyl Group, trimethylsilyl group is preferable, methyl group, isopropyl group, t-butyl group, cyclohexyl group, phenyl group, 2-methylphenyl group, 2,6-dimethylphenyl group, 2-isopropylphenyl group, 2,6-diisopropylphenyl Group, trimethylsilyl group is more preferable, methyl group, isopropyl group, cyclohexyl group, phenyl group Particularly preferred are a dialkyl group, a 2-methylphenyl group, a 2,6-dimethylphenyl group, a 2-isopropylphen
- hydrocarbon group having 1 to 6 carbon atoms represented by R 3 , R 4 and R 5 in the general formula (1) include a linear or branched alkyl group having 1 to 6 carbon atoms, carbon Examples thereof include a cycloalkyl group having a number of 3 to 6 and a phenyl group.
- R 3 , R 4 and R 5 are preferably a hydrogen atom, a methyl group, an ethyl group, an isopropyl group, a t-butyl group or a phenyl group from the viewpoint of the stability of the complex and the ease of synthesis.
- a methyl group and an ethyl group are more preferable, and a hydrogen atom and a methyl group are particularly preferable.
- transition metal compound (A) represented by the general formula (1) examples include di ( ⁇ -allyl) bis [ ⁇ - (N, N′-diphenylbenzamidinato) -N: N ′] dipalladium. , Di ( ⁇ -allyl) bis ⁇ - [N, N′-bis (2-methylphenyl) benzamidinate] -N: N ′ ⁇ dipalladium, di ( ⁇ -allyl) bis ⁇ - [N , N′-bis (2,6-dimethylphenyl) benzamidinate] -N: N ′ ⁇ dipalladium, di ( ⁇ -allyl) bis ⁇ - [N, N′-bis (2-isopropylphenyl) Benzamidinate] -N: N ′ ⁇ dipalladium, di ( ⁇ -allyl) bis ⁇ - [N, N′-bis (2,6-diisopropylphenyl) benzamidinate] —N: N ′ ⁇ Dipalladium, di ( ⁇ -ally
- the transition metal complex (A) according to the present invention includes a precursor ( ⁇ -allyl) palladium (II) compound and an amidine compound (Wherein R1 and R2 represent the same meaning as in general formula (1)), and can be produced by a ligand exchange reaction.
- Specific examples of the production method include the method described in J. Chem. Res., 2005, 702 and the method described in J. Organomet. Chem., 1992, 426, 261.
- the ( ⁇ -allyl) palladium (II) compound is not particularly limited as long as it is a compound having a ligand capable of ligand exchange with an amidine compound.
- a compound having R 3 , R 4 and R 5 corresponding to the target transition metal complex (A) may be selected.
- di ( ⁇ -allyl) di ( ⁇ -chloro) dipalladium (chemical formula (5)) and ( ⁇ -allyl) (acetylacetonato) palladium (chemical formula (6)) are preferable.
- amidine compound which has R ⁇ 1 > and R ⁇ 2 > corresponding to the target transition metal complex (A) as an amidine compound used when manufacturing a transition metal complex (A).
- Specific examples thereof include N, N'-diphenylbenzamidine, N, N'-bis (2-methylphenyl) benzamidine, N, N'-bis (2,6-dimethylphenyl) benzamidine, N, N'- Bis (2-isopropylphenyl) benzamidine, N, N′-bis (2,6-diisopropylphenyl) benzamidine, N, N′-bis (2-tert-butylphenyl) benzamidine, N, N′-bis (2, 6-di-tert-butylphenyl) benzamidine, N, N′-dicyclohexylbenzamidine, N, N′-diisopropylbenzamidine, N, N′-di-tert-butylbenzamidine, N, N′
- a precursor ( ⁇ -allyl) palladium (II) compound dissolved in a solvent is added with an amidine compound or, if necessary, a base added thereto, at a predetermined temperature. It can be carried out by stirring for a predetermined time.
- the solvent used in the ligand exchange reaction is not particularly limited as long as it does not react with each substrate.
- aliphatic hydrocarbons such as pentane, hexane and heptane; alicyclic carbonization such as cyclohexane Hydrogen; aromatic hydrocarbons such as benzene, toluene and xylene; halogenated hydrocarbons such as dichloromethane, chloroform and chlorobenzene; nitrogen-containing hydrocarbons such as nitromethane, nitrobenzene and acetonitrile; ethers such as diethyl ether, dioxane and tetrahydrofuran Can be mentioned. These solvents may be used as a mixture.
- the solvent used is preferably dehydrated and degassed.
- the amount of solvent used is not particularly limited as long as the reaction is not significantly delayed. It can be appropriately determined according to the solubility of the ( ⁇ -allyl) palladium (II) compound as a precursor. Usually, 1 to 100 g of a solvent is used per 1 g of the precursor ( ⁇ -allyl) palladium (II) compound.
- the reaction temperature is not particularly limited, but is generally ⁇ 100 to 150 ° C., preferably ⁇ 50 to 120 ° C. When the temperature is lower than ⁇ 100 ° C., the reaction rate is slow, and when the temperature is higher than 150 ° C., the formed complex may be decomposed.
- the reaction rate can be adjusted by selecting the reaction temperature within the above range.
- the reaction time is not particularly limited, and depends on the reaction temperature, but is, for example, 1 minute to 50 hours, preferably 30 minutes to 3 hours.
- the reaction is desirably performed in an inert gas atmosphere such as nitrogen gas or argon gas.
- the desired transition metal complex (A) can be isolated by carrying out ordinary separation / purification operations. Specifically, when di ( ⁇ -allyl) di ( ⁇ -chloro) dipalladium is used as a raw material, the salt such as LiCl produced in the reaction is removed by centrifugation or filtration, and then recrystallized. The target transition metal complex (A) is isolated. On the other hand, when ( ⁇ -allyl) (acetylacetonato) palladium is used as a raw material, the acetylacetone produced in the reaction is distilled off together with the solvent under reduced pressure, and then the solvent is added again and recrystallized to achieve the desired transition. The metal complex (A) is isolated.
- transition metal complex (A) can be carried out by NMR spectrum, elemental analysis, mass spectrum, X-ray crystallography and the like.
- the transition metal complex (A) obtained as described above is useful as a catalyst component for polymerization of norbornene monomers.
- the norbornene-based monomer polymerization catalyst of the present invention may be any catalyst that contains at least one transition metal complex (A), but can react with the transition metal complex (A) to produce a cationic transition metal compound.
- A transition metal complex
- the promoter (B) which is an ionic compound capable of producing a cationic transition metal compound by reacting with the transition metal complex (A) used in the present invention, is an ionic compound in which a non-coordinating anion and a cation are combined. Is mentioned.
- non-coordinating anions include quaternary anions of Group 13 elements of the 1991 periodic table. Specifically, tetra (phenyl) borate, tetra (fluorophenyl) borate, tetrakis (difluorophenyl) borate, tetrakis (trifluorophenyl) borate, tetrakis (tetrafluorophenyl) borate, tetrakis (pentafluorophenyl) borate, tetrakis (Tetrafluoromethylphenyl) borate, tetrakis [3,5-di (trifluoromethyl) phenyl] borate, tetra (triyl) borate, tetra (xylyl) borate, triphenyl (pentafluorophenyl) borate, [tris (pentafluoro Phenyl) phenyl] borate, tridecahydride-7,8-dic
- Examples of the cation include a carbonium cation, an oxonium cation, an ammonium cation, a phosphonium cation, a cycloheptyltrienyl cation, and a ferrocenium cation having a transition metal.
- the carbonium cation include tri-substituted carbonium cations such as triphenyl carbonium cation and tri-substituted phenyl carbonium cation.
- the tri-substituted phenylcarbonium cation include a tri (methylphenyl) carbonium cation and a tri (dimethylphenyl) carbonium cation.
- oxonium cations include hydroxonium cations, alkyloxonium cations such as methyloxonium cations, dialkyloxonium cations such as dimethyloxonium cations, trialkyloxonium cations such as trimethyloxonium cations and triethyloxonium cations. And cations.
- ammonium cation examples include trialkylammonium cation, triethylammonium cation, tripropylammonium cation, tributylammonium cation, tri (n-butyl) ammonium cation, and the like, N, N-diethylanilinium cation, N N, N-dialkylanilinium cations such as N, N-2,4,6-pentamethylanilinium cation, and dialkylammonium cations such as di (isopropyl) ammonium cation and dicyclohexylammonium cation.
- phosphonium cation examples include triarylphosphonium cations such as triphenylphosphonium cation, tri (methylphenyl) phosphonium cation, and tri (dimethylphenyl) phosphonium cation.
- Preferred examples of the cocatalyst (B) include trityltetrakis (pentafluorophenyl) borate, triphenylcarboniumtetra (fluorophenyl) borate, N, N-dimethylaniliniumtetrakis (pentafluorophenyl) borate, trityltetrakis [3, 5-di (trifluoromethyl) phenyl] borate, N, N-dimethylanilinium tetrakis [3,5-di (trifluoromethyl) phenyl] borate, 1,1′-dimethylferrocenium tetrakis (pentafluorophenyl) Borate and the like.
- Phosphine-based ligand (C) is a trivalent phosphorus compound in which three substituents independently selected from a hydrogen atom, an alkyl group or an aryl group are bonded.
- trialkylphosphines such as trimethylphosphine, triethylphosphine, triisopropylphosphine and tri-t-butylphosphine
- tricycloalkylphosphines such as tricyclopentylphosphine and tricyclohexylphosphine
- triaryl such as triphenylphosphine
- Mention may be made of phosphines.
- tricyclohexylphosphine, tri-t-butylphosphine, and triisopropylphosphine are preferable from the viewpoint of improving catalytic activity.
- R 1 is a methyl group or a phenyl group
- R 2 is a phenyl group or an alkyl-substituted phenyl group
- R 3 , R 4 and R A complex in which all 5 are hydrogen atoms is used, and N, N-dimethylanilinium tetrakis (pentafluorophenyl) borate ⁇ [Ph (Me) 2 NH] [B (C 6 F 5 ) is used as the promoter (B).
- phosphine-based ligand C
- -butylphosphine is a preferred embodiment as a catalyst capable of producing a norbornene-based polymer with high activity.
- R 1 is a phenyl group
- R 2 is a phenyl group or a (2,6-alkyl-substituted) phenyl group
- R 3 , R 4 and A complex in which R 5 is a hydrogen atom is used, and N, N-dimethylanilinium tetrakis (pentafluorophenyl) borate ⁇ [Ph (Me) 2 NH] [B (C 6 F 5 ) 4 ] ⁇ and triisopropylphosphine as the phosphine-based ligand (C) is the most preferable embodiment as a catalyst capable of producing a norbornene-based polymer with high activity.
- the ratio of the transition metal complex (A) and the cocatalyst (B) used in the catalyst of the present invention is not uniquely determined because it varies depending on various conditions, but is usually (A) / (B) (molar ratio). ) In the range of 1 / 0.1 to 1/100, preferably 1 / 0.5 to 1/50, more preferably 1/1 to 1/10.
- the ratio of the transition metal complex (A) and the phosphine-based ligand (C) used in the catalyst of the present invention is not uniquely determined because it varies depending on various conditions, but usually (A) / (C).
- (Molar ratio) is 1 / 0.1 to 1/2, preferably 1 / 0.5 to 1 / 1.8, more preferably 1/1 to 1 / 1.5.
- the temperature at which each catalyst component is brought into contact is not particularly limited, but is generally ⁇ 100 to 150 ° C., preferably ⁇ 50 to 120 ° C. When the temperature is lower than ⁇ 100 ° C., the reaction between the components is delayed, and when the temperature is higher than 150 ° C., the components are decomposed and the activity of the catalyst is lowered.
- the contact temperature within the above range, the polymerization rate, the molecular weight of the produced polymer, and the like can be adjusted when used for polymerization.
- each catalyst component may be performed in the presence of a solvent.
- Solvents that can be used are not particularly limited, but those that are not easily reactive with each catalyst component, are manufactured on an industrial scale, and are easily available are preferred. Specifically, aliphatic hydrocarbons such as pentane, hexane and heptane; alicyclic hydrocarbons such as cyclohexane; aromatic hydrocarbons such as benzene, toluene and xylene; halogenated hydrocarbons such as dichloromethane, chloroform and chlorobenzene; Nitrogen-containing hydrocarbons such as nitromethane, nitrobenzene and acetonitrile; ethers such as diethyl ether, dioxane and tetrahydrofuran can be used. Among these, aliphatic hydrocarbons, aromatic hydrocarbons, and halogenated hydrocarbons are preferable. These solvents may be used as a mixture.
- the method for producing a norbornene-based polymer of the present invention is characterized in that a norbornene-based monomer is addition-polymerized in the presence of the polymerization catalyst of the present invention.
- the production method of the present invention includes (i) a method of obtaining a single addition polymer of a norbornene monomer by addition polymerization of only one kind of norbornene monomer, and (ii) an addition copolymerization of two or more kinds of norbornene monomers.
- a method of obtaining a single addition polymer of a norbornene monomer by addition polymerization of only one kind of norbornene monomer includes (i) a method of obtaining a single addition polymer of a norbornene monomer by addition polymerization of only one kind of norbornene monomer, and (ii) an addition copolymerization of two or more kinds of norbornene monomers.
- To obtain an addition copolymer of a norbornene-based monomer and (iii) addition-copolymerizing one or more norbornene-based monomers with one or more other vinyl monomers copolymerizable with the norbornene-based monomers. It is one of
- Norbornene monomer The norbornene-based monomer used in the present invention is not particularly limited as long as it is a compound having a norbornene ring structure (hereinafter sometimes simply referred to as “norbornenes”). It may have a polar or non-polar substituent and may have a ring structure other than the norbornene ring. As norbornene, what is shown by General formula (4) is preferable.
- R 10 to R 13 each independently represent a hydrogen atom; a halogen atom; a functional group containing a nitrogen atom, an oxygen atom, a sulfur atom, a halogen atom or a silicon atom; a halogen atom or the functional group Represents an optionally substituted hydrocarbon group having 1 to 20 carbon atoms.
- R 10 to R 13 may be bonded to each other to form a ring.
- n is 0 or 1.
- the norbornenes represented by the general formula (4) include bicyclo [2.2.1] hept-2-enes in which n is 0 and tetracyclo [6.2.1.1 3,6 . 0 2,7 ] dodec-4-enes. Any of the production methods of the present invention can be used.
- R 10 to R 13 in the general formula (4) include: hydrogen atom; halogen atom such as chlorine atom, bromine atom and fluorine atom; hydroxyl group, alkoxy group, aryloxy group, carbonyl group, hydroxycarbonyl group, alkoxy A functional group containing an oxygen atom such as a carbonyl group and an aryloxycarbonyl group; a nitrogen atom such as an amino group, an alkylamino group, an arylamino group, an aminocarbonyl group, an alkylaminocarbonyl group, an arylaminocarbonyl group, and a cyano group; Functional groups containing; functional groups containing sulfur atoms such as mercapto groups, alkoxythio groups, and aryloxythio groups; silicon atoms such as silyl groups, alkylsilyl groups, arylsilyl groups, alkoxysilyl groups, and aryloxysilyl groups Mention may be made of functional groups
- hydrocarbon groups such as an alkyl group having 1 to 20 carbon atoms, an alkenyl group, and an aryl group which may have these functional groups are also included.
- R 10 to R 13 may be bonded to each other to form a ring, and examples of such include an acid anhydride structure, a carbonate structure, a dithiocarbonate structure, and the like.
- norbornenes used in the present invention include 2-norbornene, 5-methyl-2-norbornene, 5-ethyl-2-norbornene, 5-n-butyl-2-norbornene, and 5-n-hexyl-2.
- dodec-4-ene 9-ethylidenetetracyclo [6.2.1.1 3,6 . 0 2,7 ] dodec-4-ene, 9-vinyltetracyclo [6.2.1.1 3,6 . 0 2,7 ] dodec-4-ene, 9-phenyltetracyclo [6.2.1.1 3,6 . 0 2,7 ] dodeca-4-ene or other unsubstituted or hydrocarbon-containing tetracyclo [6.2.1.1 3,6 . 0 2,7 ] dodec-4-enes;
- Bicyclo [2.2.1] hept-2-enes having the following alkoxycarbonyl group; tetracyclo [6.2.1.1 3,6 . 0 2,7 ] methyl dodec-9-ene-4-carboxylate, tetracyclo [6.2.1.1 3,6 . 0 2,7 ] ethyl dodeca-9-ene-4-carboxylate, 4-methyltetracyclo [6.2.1.1 3,6 . 0 2,7 ] methyl dodec-9-ene-4-carboxylate, tetracyclo [6.2.1.1 3,6 . 0 2,7 ] methyl dodec-9-ene-4,5-dicarboxylate, tetracyclo [6.2.1.1 3,6 . Tetracyclo [6.2.1.1 3,6 . Having an alkoxycarbonyl group such as 0 2,7 ] dodec-9-ene-4,5-dicarboxylate. 0 2,7 ] dodeca-9-enes;
- acetoxyl group such as 2-acetoxy-5-norbornene, 2-acetoxymethyl-5-norbornene, 2,2-di (acetoxymethyl) -5-norbornene, 2,3-di (acetoxymethyl) -5-norbornene Bicyclo [2.2.1] hept-2-enes; 4-acetoxytetracyclo [6.2.1.1 3,6 . 0 2,7 ] dodec-9-ene, 4-acetoxymethyltetracyclo [6.2.1.1 3,6 . 0 2,7 ] dodec-9-ene, 4,5-di (acetoxymethyl) tetracyclo [6.2.1.1 3,6 . Tetracyclo [6.2.1.1 3,6 . Having an acetoxyl group such as 0 2,7 ] dodec-9-ene. 0 2,7 ] dodeca-9-enes;
- the alkyl group having 1 to 10 carbon atoms represented by R 6 in the general formula (2) may be linear or branched.
- linear alkyl groups include methyl, ethyl, n-propyl, n-butyl, n-pentyl, n-hexyl, n-octyl, n-decyl and the like. It is done.
- branched alkyl group include isopropyl group, isobutyl group, sec-butyl group, neopentyl group, isohexyl group, isooctyl group, and isodecyl group.
- a linear alkyl group having 1 to 3 carbon atoms is preferable from the viewpoint of economy.
- a methyl group is particularly preferred from the viewpoint of monomer production cost.
- R 7 in the general formula (2) and R 8 and R 9 in the general formula (3) each independently represent a hydrogen atom or an alkyl group having 1 to 10 carbon atoms, and the alkyl group having 3 to 10 carbon atoms is branched. You may do it.
- these alkyl groups the same alkyl groups as those described above for R 6 can be mentioned.
- a hydrogen atom is preferable from the viewpoint of monomer production cost.
- the norbornenes that are the basis of the monomer unit represented by the general formula (2) are 2-acetoxymethyl-5-norbornene when R 6 is a C 1 alkyl group, When R 6 is an alkyl group having 2 carbon atoms, 2-[(ethylcarbonyloxy) methyl] -5-norbornene, and when R 6 is a linear alkyl group having 3 carbon atoms, 2-[(propylcarbonyloxy ) Methyl] -5-norbornene.
- R 8 and R 9 are hydrogen atoms
- the norbornenes that are the basis of the monomer unit represented by the general formula (3) are norbornene.
- polymerization of norbornene monomer using transition metal complex (A), promoter (B) and phosphine ligand (C) is bulk polymerization, suspension polymerization, emulsion polymerization, solution polymerization, It can be carried out by precipitation polymerization.
- A transition metal complex
- promoter B
- phosphine ligand C
- Usable solvents include aliphatic hydrocarbons such as pentane, hexane and heptane; alicyclic hydrocarbons such as cyclohexane; aromatic hydrocarbons such as benzene, toluene and xylene; halogenated carbons such as dichloromethane, chloroform and chlorobenzene.
- Nitrogen-containing hydrocarbons such as nitromethane, nitrobenzene and acetonitrile; ethers such as diethyl ether, dioxane and tetrahydrofuran; esters such as ethyl acetate, n-propyl acetate and n-butyl acetate; ⁇ -butyrolactone and ⁇ -valero Examples include lactones such as lactones and water. These solvents may be used as a mixture. When water is used, the reaction solution can be emulsified using an anionic, cationic or nonionic surfactant.
- Precipitation polymerization is a kind of solution polymerization, and a solvent that dissolves the monomer but does not dissolve the polymer is used.
- a polymer is precipitated together with the polymerization, so that a poor solvent (such as methanol) used in a large amount for reprecipitation purification is unnecessary, which is advantageous in terms of production cost.
- a mixed solvent of toluene and ethyl acetate is suitable for precipitation polymerization.
- the main catalyst (A), the cocatalyst (B) and the phosphine-based ligand (C) are mixed, and the mixing order is such that the main catalyst (A) contacts the cocatalyst (B).
- Others are not particularly limited as long as they are mixed with the phosphine-based ligand (C) before the operation.
- a main catalyst (A) component and a phosphine-based ligand (C) are mixed in advance, and a co-catalyst (B) is further mixed to obtain a reaction composition, which is added to a solution containing a monomer to be polymerized. Also good.
- the promoter (B) may be added to the solution containing the monomer to be polymerized, the main catalyst (A) and the phosphine-based ligand (C), and the monomer to be polymerized and the promoter (B).
- a mixture of the main catalyst (A) and the phosphine-based ligand (C) may be added to the mixed solution.
- the main catalyst (A) and the phosphine-based ligand (C) are mixed in advance and contacted for 1 minute or more, preferably about 30 minutes to 1 hour, and then mixed with the promoter (B).
- it is added to the reaction system, or a mixture of the main catalyst (A) and the phosphine-based ligand (C) is added to the reaction system containing the promoter (B).
- the polymerization temperature is not particularly limited, but is generally ⁇ 100 to 150 ° C., preferably ⁇ 50 to 120 ° C. When the temperature is lower than ⁇ 100 ° C., the polymerization rate is slow, and when the temperature is higher than 150 ° C., the activity of the catalyst may be lowered. By selecting the polymerization temperature within the above range, the polymerization rate, molecular weight and the like can be adjusted.
- the polymerization time is not particularly limited, and is, for example, 1 minute to 100 hours.
- the reaction is desirably performed in an inert gas atmosphere such as nitrogen gas.
- the product norbornene-based copolymer is isolated by performing post-treatment by a known operation and treatment method (for example, reprecipitation) as necessary, separation by filtration, and drying. It is.
- the content of the monomer unit represented by the general formula (2) is: It is preferably 10 to 70 mol%.
- the monomer unit represented by the general formula (2) is less than 10 mol%, the hydrophobicity of the copolymer increases and the solubility in an organic solvent decreases, but the water absorption tends to decrease.
- the copolymer becomes hydrophilic and the solubility in an organic solvent is improved, but the water absorption tends to increase. Therefore, it is possible to control the solubility of the copolymer in the solvent and the water absorption by adjusting the content of the monomer unit represented by the general formula (2).
- the content of the monomer unit represented by the general formula (2) is preferably 10 to 80 mol%, more preferably 15 to 70 mol%, and more preferably 20 to 60 mol%. % Is more preferable.
- the content of the monomer unit represented by the general formula (2) is calculated from an integral value obtained by dissolving a powdery or film-like copolymer in an appropriate deuterated solvent, measuring 1 H-NMR. be able to.
- the norbornene copolymer produced by the production method of the present invention is basically composed only of norbornenes. However, even in this case, it does not exclude the presence of a very small amount, for example, 1 mol% or less of the third monomer unit that hardly changes the properties of the norbornene-based copolymer of the present invention. Moreover, the norbornene-type copolymer manufactured with the manufacturing method of this invention may copolymerize a 3rd monomer in the range which does not impair the effect of this invention for a physical property improvement.
- the third monomer is not particularly limited, but a monomer having an ethylenic carbon-carbon double bond is preferable.
- ⁇ -olefins such as ethylene, propylene, 1-butene, 1-pentene and 1-hexene
- Aromatic vinyl compounds such as styrene, ⁇ -methylstyrene and divinylbenzene
- chain conjugated dienes such as 1,3-butadiene and isoprene
- vinyl ethers such as ethyl vinyl ether and propyl vinyl ether; methyl acrylate, ethyl acrylate, 2- Examples thereof include acrylates such as ethylhexyl acrylate; methacrylates such as methyl methacrylate and ethyl methacrylate;
- ⁇ -olefins such as ethylene, propylene and 1-hexene
- aromatic vinyl compounds such as styrene are particularly preferable.
- the copolymerization mode of each monomer unit can be random, block, or alternating depending on the polymerization conditions, but from the viewpoint of improving the physical properties of the copolymer. It is desirable to be random.
- the number average molecular weight (Mn) in terms of polystyrene measured by gel permeation chromatography (GPC) of the norbornene copolymer produced by the production method of the present invention is 50,000 to 2,000,000. Further, 100,000 to 1,500,000 is more preferable. If the number average molecular weight in terms of polystyrene is less than 50,000, the mechanical strength is insufficient. When the number average molecular weight in terms of polystyrene exceeds 2,000,000, not only the solubility in a solvent is lowered when a cast film is formed, but also the solution viscosity is increased and the molding processability is lowered.
- the molecular weight distribution Mw / Mn (weight average molecular weight / number average molecular weight) is preferably 1.00 to 4.00, more preferably 1.30 to 3.50, and even more preferably 1.50 to 3.30.
- Mw / Mn weight average molecular weight / number average molecular weight
- the saturated water absorption at 23 ° C. of the norbornene copolymer produced by the production method of the present invention is usually 0.001 to 1% by mass, preferably 0.005 to 0.7% by mass, and more preferably 0.00. 01 to 0.5% by mass.
- various optical properties such as transparency, retardation, uniformity of retardation, and dimensional accuracy are maintained even under conditions such as high temperature and humidity, and adhesion to other materials.
- compatibility with additives such as an antioxidant is good, so that the degree of freedom of addition is increased.
- the said saturated water absorption is a value calculated
- the glass transition temperature (Tg) of the norbornene-based (co) polymer produced by the production method of the present invention varies depending on the types of constituent monomer units, the composition ratio, the presence or absence of additives, Usually, it is 80 to 350 ° C, preferably 100 to 320 ° C, more preferably 120 to 300 ° C.
- Tg is lower than the above range, the heat distortion temperature is lowered, there is a possibility that a problem occurs in heat resistance, and the change in the optical characteristics depending on the temperature of the obtained optical film may be increased.
- Tg is higher than the said range, when heating to Tg vicinity at the time of an extending
- the norbornene copolymer produced by the production method of the present invention can be formed into a film by a solution casting method (solution casting method) and processed into a film.
- a solution casting method solution casting method
- toluene, tetrahydrofuran (THF), dichloromethane, chloroform or the like can be used as a solvent to be used.
- the catalytic activity is expressed by the following formula. Calculated by The weight average molecular weight (Mw), number average molecular weight (Mn), and molecular weight distribution (Mw / Mn) of the obtained polymer were determined by gel permeation chromatography (GPC) using polystyrene as a standard substance. The composition ratio of norbornene and 5-acetoxymethyl-2-norbornene in the copolymer is the peak [ ⁇ : 3.5-4.5 ppm, 5-acetoxymethyl-2-norbornene obtained by 1 H-NMR.
- Cyclopentadienyl ( ⁇ -allyl) palladium was synthesized according to the method of synthesis by Shaw et al. (Proc. Chem. Soc., 1960, 247).
- Synthesis Example 1 Synthesis of 2-acetoxymethyl-5-norbornene
- dicyclopentadiene Tokyo Chemical Industry, 759.80 g, 5.747 mol
- allyl acetate Tokyo Chemical Industry, 1457. 86 g, 14.561 mol
- hydroquinone Wako Pure Chemical Industries, 2.25 g, 0.0204 mol
- the autoclave was cooled to room temperature, the contents were transferred to a distillation apparatus, and distilled under reduced pressure to obtain 1306.70 g of a colorless transparent liquid as a fraction of 0.07 kPa and 48 ° C.
- Synthesis Example 2 Synthesis of N, N′-diphenylbenzamidine A two-necked flask equipped with a three-way cock was replaced with nitrogen, and 3-methyl-1-phenylphospholene-1-oxide (manufactured by Tokyo Chemical Industry Co., Ltd. 92 g, 4.787 mmol) and phenyl isocyanate (manufactured by Tokyo Chemical Industry Co., Ltd., 75.59 g, 0.635 mol) were added, and the reaction was carried out at 50 ° C. for 3 hours. Thereafter, the reaction solution was transferred to a distillation apparatus and distilled under reduced pressure to obtain 55.13 g of a pale yellow transparent liquid as a 0.03 kPa, 105 ° C. fraction.
- 3-methyl-1-phenylphospholene-1-oxide manufactured by Tokyo Chemical Industry Co., Ltd. 92 g, 4.787 mmol
- phenyl isocyanate manufactured by Tokyo Chemical Industry Co., Ltd., 75.59 g,
- Synthesis Example 3 Synthesis of N, N′-bis (2,6-diisopropylphenyl) benzamidine
- a three-necked flask equipped with a three-way cock and a cooling tube was purged with nitrogen, followed by diphosphorus pentoxide (Wako Pure Chemical Industries, 7 .13 g, 50.23 mmol), hexamethyldisiloxane (Wako Pure Chemical Industries, 17.86 g, 110.00 mmol), dichloromethane (Wako Pure Chemical Industries, 30.0 ml) were added and stirred, The mixture was refluxed at 30 ° C. for 30 minutes. Thereafter, the reaction was carried out at 160 ° C.
- Synthesis Example 4 Synthesis of N, N′-bis (2-isopropylphenyl) benzamidine A three-necked flask equipped with a three-way cock and a condenser tube was purged with nitrogen, followed by diphosphorus pentoxide (Wako Pure Chemical Industries, 8.57 g). , 60.38 mmol), hexamethyldisiloxane (Wako Pure Chemical Industries, 21.52 g, 132.53 mmol), dichloromethane (Wako Pure Chemical Industries, 36.0 ml) are added and stirred at 50 ° C. Reflux was performed for 30 minutes. Thereafter, the reaction was carried out at 160 ° C.
- Synthesis Example 5 Synthesis of N, N′-bis (2-methylphenyl) benzamidine A three-necked flask equipped with a three-way cock and a condenser tube was purged with nitrogen, followed by diphosphorus pentoxide (Wako Pure Chemical Industries, 7.40 g). , 52.13 mmol), hexamethyldisiloxane (Wako Pure Chemical Industries, 18.62 g, 114.67 mmol) and dichloromethane (Wako Pure Chemical Industries, 31.0 ml) were added and stirred at 50 ° C. Reflux was performed for 30 minutes. Thereafter, the reaction was carried out at 160 ° C.
- Synthesis Example 7 Synthesis of N, N'-diphenylacetamidine A three-necked flask equipped with a three-way cock and a condenser tube was purged with nitrogen, followed by diphosphorus pentoxide (Wako Pure Chemical Industries, 7.10 g, 50.00 mmol). , Hexamethyldisiloxane (Wako Pure Chemical Industries, 17.86 g, 110.00 mmol) and dichloromethane (Wako Pure Chemical Industries, 30.0 ml) were added and refluxed at 50 ° C. for 30 minutes with stirring. It was. Thereafter, the reaction was carried out at 160 ° C.
- Example 1 Synthesis of ( ⁇ -allyl) palladium (N, N'-diphenylbenzamidinato) dimer [complex A-1]
- N, N′-diphenylbenzamidine (0.75566 g, 2.778 mmol) prepared in Synthesis Example 2 was added thereto, and dehydrated toluene (manufactured by Wako Pure Chemical Industries, Ltd.). , 20 ml) was added and dissolved.
- Example 2 Synthesis of ( ⁇ -allyl) palladium [N, N′-bis (2,6-diisopropylphenyl) benzamidinate] dimer [complex A-2]
- a two-necked flask equipped with a three-way cock was purged with nitrogen, and N, N′-bis (2,6-diisopropylphenyl) benzamidine (0.8998 g, 2.028 mmol) prepared in Synthesis Example 3 was added thereto, and dehydrated toluene (Wako Pure Chemical Industries, 20 ml) was added and dissolved.
- a two-necked flask equipped with a separately prepared three-way cock was replaced with nitrogen, and allyl palladium chloride dimer (manufactured by Wako Pure Chemical Industries, Ltd., 0.3710 g, 1.014 mmol) was charged into the dehydrated THF (manufactured by Wako Pure Chemical Industries, Ltd. , 20 ml) was added and dissolved.
- This solution was cooled to ⁇ 15 ° C., and a toluene / hexane mixed solution of N, N′-bis (2,6-diisopropylphenyl) benzamidine-lithium complex prepared previously was slowly added dropwise over 10 minutes.
- Example 3 Synthesis of ( ⁇ -allyl) palladium [N, N′-bis (2-isopropylphenyl) benzamidinate] dimer [complex A-3]
- N, N′-bis (2-isopropylphenyl) benzamidine (0.9668 g, 2.712 mmol) prepared in Synthesis Example 4 was added thereto, and dehydrated toluene (Japanese 20 ml) was added and dissolved.
- Example 4 Synthesis of ( ⁇ -allyl) palladium [N, N′-bis (2-methylphenyl) benzamidinate] dimer [complex A-4]
- a two-necked flask equipped with a three-way cock was purged with nitrogen, and N, N′-bis (2-methylphenyl) benzamidine (0.8677 g, 2.888 mmol) prepared in Synthesis Example 5 was added thereto, and dehydrated toluene (Japanese 20 ml) was added and dissolved.
- Example 5 Synthesis of ( ⁇ -allyl) palladium (N, N'-dicyclohexylbenzamidinate) dimer [complex A-5]
- N, N′-dicyclohexylbenzamidine (0.8276 g, 2.910 mmol) prepared in Synthesis Example 6 was charged into the flask, and dehydrated toluene (manufactured by Wako Pure Chemical Industries, Ltd.). , 20 ml) was added and dissolved.
- Example 6 Synthesis of ( ⁇ -allyl) palladium (N, N'-diphenylacetamidinate) dimer [complex A-6]
- a two-necked flask equipped with a three-way cock was purged with nitrogen, and N, N′-diphenylacetamidine (0.5756 g, 2.737 mmol) prepared in Synthesis Example 7 was charged into the flask and dehydrated toluene (manufactured by Wako Pure Chemical Industries, Ltd.). , 20 ml) was added and dissolved.
- Example 7 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene A three-necked flask equipped with a three-way cock and a mechanical stirrer was purged with nitrogen, and norbornene (Tokyo Chemical Industry Co., Ltd., 4.71 g, 0.050 mol) was added.
- norbornene Tokyo Chemical Industry Co., Ltd., 4.71 g, 0.050 mol
- the catalytic activity calculated from the polymer yield and the amount of charged catalyst was 1270 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 27.0 mol%.
- Examples 8 to 9 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene A polymerization reaction and post-treatment were performed to obtain a white powdery polymer. The results are shown in Table 2.
- FIG. 2 shows the 1 H-NMR spectrum of the copolymer obtained in Example 9, and
- FIG. 3 shows the chart of gel permeation chromatography (GPC).
- Example 10 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene A three-necked flask equipped with a three-way cock and a mechanical stirrer was purged with nitrogen, and norbornene (Tokyo Chemical Industry Co., Ltd., 4.71 g, 0.050 mol) was added.
- norbornene Tokyo Chemical Industry Co., Ltd., 4.71 g, 0.050 mol
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 22.1 mol%.
- Examples 11 to 12 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene The same method as in Example 10 except that the polymerization temperature was 80 ° C. in Example 11 and 70 ° C. in Example 12. A polymerization reaction and post-treatment were performed to obtain a white powdery polymer. The results are shown in Table 2.
- Example 13 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene ( ⁇ -allyl) palladium (N, N′-diphenylbenzamidinato) dimer [complex A-1] (4.2 mg, 0. ( ⁇ -allyl) palladium [N, N′-bis (2,6-diisopropylphenyl) benzamidinate] dimer [complex A-2] (5.9 mg, 0.005 mmol) was used instead of (005 mmol). Except for this, the polymerization reaction and post-treatment were carried out in the same manner as in Example 7 to obtain 7.40 g of a white powdery polymer.
- the catalytic activity calculated from the polymer yield and the amount of charged catalyst was 740 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 22.3 mol%.
- Examples 14 to 15 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene The same method as in Example 13 except that the polymerization temperature was 80 ° C. in Example 14 and 70 ° C. in Example 15 respectively. A polymerization reaction and post-treatment were performed to obtain a white powdery polymer. The results are shown in Table 2.
- Example 16 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene ( ⁇ -allyl) palladium (N, N′-diphenylbenzamidinato) dimer [complex A-1] (4.2 mg, 0.2 mg ( ⁇ -allyl) palladium [N, N′-bis (2,6-diisopropylphenyl) benzamidinate] dimer [complex A-2] (5.9 mg, 0.005 mmol) instead of 005 mmol)
- a polymerization reaction and post-treatment were performed in the same manner as in Example 10 except that the polymerization temperature was 70 ° C., to obtain 34.95 g of a white powdery polymer.
- the catalytic activity calculated from the polymer yield and the amount of charged catalyst was 3495 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 19.1 mol%.
- Example 17 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene N, N-dimethylanilinium tetrakis (pentafluorophenyl) borate [(C 6 H 5 ) (CH 3 ) 2 NH] [B (C 6 F 5 ) 4 ] (Strem) was used at 12.0 mg (0.015 mmol), and triisopropylphosphine [P (i-C 3 H 7 ) 3 ] (Strem) was used at 2 Polymerization reaction and post-treatment were performed in the same manner as in Example 16 except that the amount was changed to 0.4 mg (0.015 mmol) to obtain 43.90 g of a white powdery polymer.
- the catalytic activity calculated from the polymer yield and the amount of charged catalyst was 4390 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 20.2 mol%.
- Example 18 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene ( ⁇ -allyl) palladium (N, N′-diphenylbenzamidinato) dimer [complex A-1] (4.2 mg, 0. Except that ( ⁇ -allyl) palladium [N, N′-bis (2-isopropylphenyl) benzamidinate] dimer [complex A-3] (5.0 mg, 0.005 mmol) was used instead of 005 mmol). Were subjected to a polymerization reaction and post-treatment in the same manner as in Example 7 to obtain 18.31 g of a white powdery polymer.
- the catalytic activity calculated from the polymer yield and the amount of charged catalyst was 1831 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 34.1 mol%.
- Example 19 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene A polymerization reaction and post-treatment were performed in the same manner as in Example 18 except that the polymerization temperature was 80 ° C. Obtained. The results are shown in Table 2.
- Example 20 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene A three-necked flask equipped with a three-way cock and a mechanical stirrer was purged with nitrogen, and norbornene (Tokyo Chemical Industry Co., Ltd., 7.07 g, 0.075 mol) was used.
- norbornene Tokyo Chemical Industry Co., Ltd., 7.07 g, 0.075 mol
- the catalyst activity calculated from the polymer yield and the amount of charged catalyst was 2170 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 29.0 mol%.
- Example 21 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene A polymerization reaction and post-treatment were performed in the same manner as in Example 20 except that the polymerization temperature was 80 ° C. .60 g was obtained. The results are shown in Table 2.
- Example 22 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene ( ⁇ -allyl) palladium (N, N′-diphenylbenzamidinato) dimer [complex A-1] (4.2 mg, 0.2 mg Except that ( ⁇ -allyl) palladium [N, N′-bis (2-methylphenyl) benzamidinate] dimer [complex A-4] (4.5 mg, 0.005 mmol) was used instead of 005 mmol) Were subjected to a polymerization reaction and post-treatment in the same manner as in Example 7 to obtain 16.74 g of a white powdery polymer.
- the catalytic activity calculated from the polymer yield and the amount of the charged catalyst was 1,674 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 32.3 mol%.
- Examples 23 to 24 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene The same method as in Example 22 except that the polymerization temperatures were 80 ° C. for Example 23 and 70 ° C. for Example 24, respectively. A polymerization reaction and post-treatment were performed to obtain a white powdery polymer. The results are shown in Table 2.
- Example 25 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene ( ⁇ -allyl) palladium [N, N′-bis (2-isopropylphenyl) benzamidinate] dimer [complex A-3] ( ( ⁇ -allyl) palladium [N, N′-bis (2-methylphenyl) benzamidinate] dimer [complex A-4] (4.5 mg, 0.005 mmol) instead of 5.0 mg, 0.005 mmol)
- the polymerization reaction and post-treatment were performed in the same manner as in Example 20 except that the polymerization temperature was 80 ° C., to obtain 19.95 g of a white powdery polymer.
- the catalytic activity calculated from the polymer yield and the amount of charged catalyst was 1995 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 27.2 mol%.
- Example 26 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene ( ⁇ -allyl) palladium (N, N′-diphenylbenzamidinato) dimer [complex A-1] (4.2 mg, 0. ( ⁇ -allyl) palladium [N, N′-bis (2-methylphenyl) benzamidinate] dimer [complex A-4] (4.5 mg, 0.005 mmol) was used instead of 005 mmol), and the polymerization temperature was The polymerization reaction and post-treatment were performed in the same manner as in Example 10 except that the temperature was changed to 70 ° C., to obtain 28.73 g of a white powdery polymer.
- the catalytic activity calculated from the polymer yield and the amount of charged catalyst was 2873 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 18.8 mol%.
- Example 27 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene N, N-dimethylanilinium tetrakis (pentafluorophenyl) borate [(C 6 H 5 ) (CH 3 ) 2 NH] [B (C 6 F 5 ) 4 ] (Strem) was used at 12.0 mg (0.015 mmol), and triisopropylphosphine [P (i-C 3 H 7 ) 3 ] (Strem) was used at 2 Polymerization reaction and post-treatment were performed under the same conditions as in Example 26 except that the amount was 0.4 mg (0.015 mmol) to obtain 40.70 g of a white powdery polymer.
- the catalytic activity calculated from the polymer yield and the amount of charged catalyst was 4070 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 20.6 mol%.
- Example 28 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene ( ⁇ -allyl) palladium (N, N′-diphenylbenzamidinato) dimer [complex A-1] (4.2 mg, 0. ( ⁇ -allyl) palladium (N, N′-dicyclohexylbenzamidinato) dimer [complex A-5] (4.3 mg, 0.005 mmol) was used instead of (005 mmol).
- the polymerization reaction and post-treatment were carried out by the above method to obtain 13.25 g of a white powdery polymer.
- the catalytic activity calculated from the polymer yield and the amount of charged catalyst was 1325 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 29.8 mol%.
- Examples 29 to 30 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene A polymerization reaction and post-treatment were performed to obtain a white powdery polymer. The results are shown in Table 2.
- Example 31 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene ( ⁇ -allyl) palladium (N, N′-diphenylbenzamidinato) dimer [complex A-1] (4.2 mg, 0.
- Example 7 except that ( ⁇ -allyl) palladium (N, N′-diphenylacetamidinate) dimer [complex A-6] (3.6 mg, 0.005 mmol) was used instead of (005 mmol).
- the polymerization reaction and post-treatment were carried out by the above method to obtain 18.10 g of a white powdery polymer.
- the catalytic activity calculated from the polymer yield and the amount of charged catalyst was 1810 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 28.7 mol%.
- Example 32 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene A polymerization reaction and post-treatment were performed in the same manner as in Example 31 except that the polymerization temperature was 80 ° C. Obtained 0.01 g. The results are shown in Table 2.
- Comparative Example 1 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene (polymerization by the method of Patent Document 4) A three-necked flask equipped with a three-way cock and a mechanical stirrer was purged with nitrogen, and norbornene (4.71 g, 0.050 mol) manufactured by Tokyo Chemical Industry Co., Ltd.
- Allyl palladium chloride dimer [[(C 3 H 5 ) PdCl] 2 ]; metal complex CA-1 (manufactured by Wako Pure Chemical Industries, 1.8 mg, 0.005 mmol) and triisopropylphosphine [P (i-C) 3 H 7 ) 3 ] (Strem, 1.6 mg, 0.010 mmol) dissolved in 3.5 ml of toluene was added, and a polymerization reaction was performed at 90 ° C. for 30 minutes.
- the catalytic activity calculated from the polymer yield and the amount of charged catalyst was 1053 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 24.0 mol%.
- a general solvent such as THF and chloroform
- FIG. 3 shows a gel permeation chromatography (GPC) chart of the obtained copolymer.
- Comparative example 3 addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene (polymerization by the method of Patent Document 4) Instead of ( ⁇ -allyl) palladium (N, N′-diphenylbenzamidinato) dimer [complex A-1] (4.2 mg, 0.005 mmol), allyl palladium chloride dimer [[(C 3 H 5 ) PdCl ] 2 ]; A polymerization reaction and a post-treatment were performed in the same manner as in Example 10 except that the metal complex CA-1 (manufactured by Wako Pure Chemical Industries, Ltd., 1.8 mg, 0.005 mmol) was used. 40.10 g of the following polymer was obtained.
- the metal complex CA-1 manufactured by Wako Pure Chemical Industries, Ltd., 1.8 mg, 0.005 mmol
- the catalytic activity calculated from the polymer yield and the amount of charged catalyst was 4010 g-polymer / mmol-Pd.
- the composition of 2-acetoxymethyl-5-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 23.6 mol%.
- Comparative Example 4 Addition copolymerization of norbornene and 2-acetoxymethyl-5-norbornene A three-necked flask equipped with a three-way cock and a mechanical stirrer was purged with nitrogen, and norbornene (Tokyo Chemical Industry Co., Ltd., 11.80 g, 0.125 mol) 5-acetoxymethyl-2-norbornene (41.50 g, 0.250 mol) and trityltetrakis (pentafluorophenyl) borate [Ph 3 C] [B (C 6 F 5 ) 4 ] prepared in Synthesis Example 1; Catalyst B-2 (manufactured by Tosoh Finechem, 93 mg, 0.100 mmol) was added and dissolved in 60 ml of toluene.
- norbornene Tokyo Chemical Industry Co., Ltd., 11.80 g, 0.125 mol
- 5-acetoxymethyl-2-norbornene 41.50 g
- the composition of the 5-acetoxymethyl-2-norbornene monomer unit in the polymer calculated from the integral value of 1 H-NMR was 26.3 mol%.
- Cocatalyst B-1: N, N-dimethylanilinium tetrakis (pentafluorophenyl) borate [(C 6 H 5 ) (CH 3 ) 2 NH] [B (C 6 F 5 ) 4 ], B-2: Trityltetrakis (pentafluorophenyl) borate [Ph 3 C] [B (C 6 F 5 ) 4 ].
- Phosphine-based ligand C-1: triisopropylphosphine P (iC 3 H 7 ) 3
- C-2 Tricyclohexylphosphine P (C 6 H 11 ) 3 .
- monomer NB: Norbornene
- ANB 2-acetoxymethyl-5-norbornene.
- the polymers obtained in Examples 7 to 32 and Comparative Examples 1 to 4 were all easily dissolved in a general solvent such as THF and chloroform.
- the norbornene-based copolymer obtained by the production method of the present invention has excellent transparency, heat resistance, low water absorption, electrical insulation properties, etc., so that optical molded articles such as lenses and polarizing films, films, and carrier tapes.
- Electrical insulation materials such as film capacitors, flexible printed circuit boards, press-through packages, infusion bags, medical containers such as drug vials, food packaging molded products such as wraps and trays, casings for electrical appliances, automobile interiors such as inner panels It can be used for parts, building materials such as carports and glazings.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
本発明は一般式(1)で示される遷移金属錯体(A)を含有するノルボルネン系モノマー重合用触媒、及び前記重合用触媒の存在下に、一般式(2)及び一般式(3)で示されるモノマーユニットを有するノルボルネン系共重合体の製造方法を提供する。(式中、R1及びR2は水素原子または置換基を有していてもよい炭素数1~20の炭化水素基を表し、R3、R4及びR5は水素原子または炭素数1~6の炭化水素基を表し、R6は炭素数1~10のアルキル基を表し、R7、R8及びR9は水素原子または炭素数1~10のアルキル基を表す。) 本発明によれば、極性基を有するノルボルネン化合物の高分子量付加共重合体を効率よく製造することができる。
Description
本発明は、ノルボルネン系モノマーの重合用触媒及びその触媒を用いた極性基を有するノルボルネン系モノマーの共重合体の製造方法に関する。
従来、ノルボルネン系重合体を代表とする環状オレフィン系付加重合体は耐熱性及び透明性に優れる有機材料として、光学フィルム等の分野で工業的に利用されている。このような環状オレフィン系付加重合体はTi、Zr、Cr、Co、Ni、Pd等の遷移金属化合物を含む触媒を用いて環状オレフィン系モノマーを付加重合することにより製造できることが種々報告されている。
例えば、欧州特許出願公開第0445755号明細書(特許文献1)では、5~10族元素の遷移金属化合物を主触媒とし、メチルアルミノキサン(MAO)を助触媒として用いることにより数平均分子量が100万を超えるノルボルネンの単独付加重合体が製造できることが報告されている。しかし、この触媒系では、重合の難易度がより高い、極性基を有するノルボルネン系モノマーの重合は実施されておらず、極性基の影響による触媒失活が懸念された。
一方、米国特許第3330815号明細書(特許文献2)には、ジクロロビス(ベンゾニトリル)パラジウムやアリルパラジウムクロライドダイマーのみを触媒として用いて、極性基を有するノルボルネン系モノマーの単独付加重合体及びノルボルネンとの共重合体を製造する方法が報告されている。しかし、この特許には、数平均分子量が1万を超えた重合体を製造した例がなく、かつ触媒の重合活性も低く、工業的に有用な製造法とは言い難いものであった。
さらに、極性基を有するノルボルネン系モノマーの単独付加重合及びノルボルネンとの共重合を改善する方法が特許第3678754号明細書(特許文献3)(WO96/37526)や特開2008-31304号公報(特許文献4)に開示されている。これらの方法では、触媒としてアリルパラジウムクロライドダイマーとテトラフルオロホウ酸銀やヘキサフルオロリン酸銀を組み合わせたものを使用することにより重合活性と重合体の分子量がいずれも向上しているものの、実施例で開示されている共重合体の数平均分子量は20万未満であり、機械物性が実用的な値となる数平均分子量が20万以上の共重合体は製造できていなかった。なお、特許文献4の表1では数平均分子量(Mn)と重量平均分子量(Mw)が入れ替わって記載されている。これはMw/Mnが2.5前後であることからも明かであり、表1を正しく解釈すると、数平均分子量が20万以上の共重合体は存在していなかったことが明白である。
これらの方法に対し、シクロペンタジエニル配位子を有する8~10族遷移金属化合物を主触媒とし、これに主触媒と反応してカチオン性遷移金属化合物を生成できる助触媒を組み合わせることにより極性基を有するノルボルネンとノルボルネンとの付加共重合を効率よく実施でき、高分子量の共重合体が得られることが国際公開第2006/064814号パンフレット(特許文献5)(US2009/264608 A1)に開示されている。しかし、この公報に開示されている極性基を有するノルボルネン化合物はノルボルネン骨格に直接エステル基が導入された構造を有しており、その炭素-炭素二重結合部と極性基との間の距離が近いために、触媒である遷移金属錯体に容易に配位し、触媒活性の低下を招いていた。従って、ノルボルネンの単独付加重合では高活性で高分子量の重合体を製造可能であるが、極性基を有するノルボルネン系モノマーを使用した場合には高分子量の共重合体が得られるものの触媒活性は低かった。
これらの先行技術文献の記載から、極性基を有するノルボルネン系モノマーの付加共重合において数平均分子量が20万以上の高分子量の共重合体を得ることができ、高活性で、活性の低下が小さい触媒系は知られていなかったことがわかる。
このように極性基を有するノルボルネン系付加共重合体の製造方法において、高活性で、実用的な機械物性を有する共重合体を得ることのできる触媒の例はなく、そのような触媒の開発が望まれていた。
本発明は、極性基を有するノルボルネン系モノマーの高分子量付加共重合体を製造可能な高活性の触媒系及び当該共重合体の効率的な製造方法を提供することを目的とする。
本発明者らは、上記課題を解決するために鋭意研究を重ねた結果、π-アリル(η3-アリル)配位子と2座のアミジン配位子を有する2核のパラジウム化合物を主触媒とする触媒系と、重合性炭素-炭素二重結合と極性基(エステル基)との間の距離を遠ざけるためにノルボルネン骨格とエステル基の間にメチレン鎖を1つ導入したノルボルネン化合物とを組み合わせることにより、極性基を有するノルボルネン系モノマーの高分子量付加共重合体を効率的に製造できることを見出し、本発明を完成した。
すなわち、本発明は以下の[1]~[6]のノルボルネン系モノマーの重合用触媒、[7]~[12]のノルボルネン系(共)重合体の製造方法に関する。
[1] 一般式(1)

(式中、R1及びR2はそれぞれ独立して、水素原子または置換基を有していてもよい炭素数1~20の炭化水素基を表し、R3、R4及びR5はそれぞれ独立して、水素原子または炭素数1~6の炭化水素基を表す。)で示される遷移金属錯体(A)を含有することを特徴とするノルボルネン系モノマーの重合用触媒。
[2] 一般式(1)中のR1がメチル基または置換基を有していてもよいフェニル基であり、R2がシクロヘキシル基または置換基を有していてもよいフェニル基であり、R3、R4及びR5がいずれも水素原子である前項1に記載のノルボルネン系モノマーの重合用触媒。
[3] 一般式(1)中のR1がメチル基またはフェニル基であり、R2がシクロヘキシル基、フェニル基、2-メチルフェニル基、2-イソプロピルフェニル基、または2,6-ジイソプロピルフェニル基である前項2に記載のノルボルネン系モノマーの重合用触媒。
[4] 遷移金属錯体(A)と反応してカチオン性遷移金属化合物を生成できるイオン性化合物である助触媒(B)及びホスフィン系配位子(C)を含有する前項1~3のいずれかに記載のノルボルネン系モノマーの重合用触媒。
[5] 助触媒(B)が、トリチルテトラキス(ペンタフルオロフェニル)ボレートまたはN,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレートである前項4に記載のノルボルネン系モノマーの重合用触媒。
[6] ホスフィン系配位子(C)がトリシクロヘキシルホスフィン、トリ-t-ブチルホスフィン、またはトリイソプロピルホスフィンである前項4に記載のノルボルネン系モノマーの重合用触媒。
[7] 前項1~6のいずれかに記載の重合用触媒の存在下に、ノルボルネン系モノマーを単独重合または共重合することを特徴とするノルボルネン系(共)重合体の製造方法。
[8] 前項1~6のいずれかに記載の重合用触媒の存在下に、ノルボルネン系モノマーと他のビニルモノマーを共重合することを特徴とするノルボルネン系共重合体の製造方法。
[9] 前項1~6のいずれかに記載の重合用触媒の存在下に、一般式(2)

及び一般式(3)

(式中、R6は炭素数1~10のアルキル基を表し、R7、R8及びR9はそれぞれ独立して水素原子または炭素数1~10のアルキル基を表す。)
で示されるモノマーユニットに対応するノルボルネン系モノマーを重合することを特徴とする、一般式(2)及び一般式(3)で示されるモノマーユニットを含むノルボルネン系共重合体の製造方法。
[10] 一般式(2)及び一般式(3)で示されるモノマーユニットのみからなる前項9に記載のノルボルネン系共重合体の製造方法。
[11] 一般式(2)中のR6がメチル基である前項9または10に記載のノルボルネン系共重合体の製造方法。
[12] 一般式(2)中のR7、及び一般式(3)中のR8及びR9が水素原子である前項9~11のいずれかに記載のノルボルネン系共重合体の製造方法。
[1] 一般式(1)
[2] 一般式(1)中のR1がメチル基または置換基を有していてもよいフェニル基であり、R2がシクロヘキシル基または置換基を有していてもよいフェニル基であり、R3、R4及びR5がいずれも水素原子である前項1に記載のノルボルネン系モノマーの重合用触媒。
[3] 一般式(1)中のR1がメチル基またはフェニル基であり、R2がシクロヘキシル基、フェニル基、2-メチルフェニル基、2-イソプロピルフェニル基、または2,6-ジイソプロピルフェニル基である前項2に記載のノルボルネン系モノマーの重合用触媒。
[4] 遷移金属錯体(A)と反応してカチオン性遷移金属化合物を生成できるイオン性化合物である助触媒(B)及びホスフィン系配位子(C)を含有する前項1~3のいずれかに記載のノルボルネン系モノマーの重合用触媒。
[5] 助触媒(B)が、トリチルテトラキス(ペンタフルオロフェニル)ボレートまたはN,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレートである前項4に記載のノルボルネン系モノマーの重合用触媒。
[6] ホスフィン系配位子(C)がトリシクロヘキシルホスフィン、トリ-t-ブチルホスフィン、またはトリイソプロピルホスフィンである前項4に記載のノルボルネン系モノマーの重合用触媒。
[7] 前項1~6のいずれかに記載の重合用触媒の存在下に、ノルボルネン系モノマーを単独重合または共重合することを特徴とするノルボルネン系(共)重合体の製造方法。
[8] 前項1~6のいずれかに記載の重合用触媒の存在下に、ノルボルネン系モノマーと他のビニルモノマーを共重合することを特徴とするノルボルネン系共重合体の製造方法。
[9] 前項1~6のいずれかに記載の重合用触媒の存在下に、一般式(2)
で示されるモノマーユニットに対応するノルボルネン系モノマーを重合することを特徴とする、一般式(2)及び一般式(3)で示されるモノマーユニットを含むノルボルネン系共重合体の製造方法。
[10] 一般式(2)及び一般式(3)で示されるモノマーユニットのみからなる前項9に記載のノルボルネン系共重合体の製造方法。
[11] 一般式(2)中のR6がメチル基である前項9または10に記載のノルボルネン系共重合体の製造方法。
[12] 一般式(2)中のR7、及び一般式(3)中のR8及びR9が水素原子である前項9~11のいずれかに記載のノルボルネン系共重合体の製造方法。
本発明によればノルボルネンと極性基を有するノルボルネン系モノマーとの高分子量付加共重合体を効率よく製造することができる。本発明により得られるノルボルネン系共重合体は優れた透明性、耐熱性、低吸水性、電気絶縁特性等を有し、光学用途、医療用途、電材用途、包装材料用途、構造材料用途等の多くの用途で利用できる。
具体的には、レンズや偏光フィルム等の光学用成形品、フィルム、キャリアテープ、フィルムコンデンサー、フレキシブルプリント基板等の電気絶縁材料、プレススルーパッケージ、輸液バック、薬液バイアル等の医療用容器、ラップやトレイ等の食品包装成形品、電気器具等のケーシング、インナーパネル等の自動車内装部品、カーポートやグレージング等の建材等に利用可能である。
具体的には、レンズや偏光フィルム等の光学用成形品、フィルム、キャリアテープ、フィルムコンデンサー、フレキシブルプリント基板等の電気絶縁材料、プレススルーパッケージ、輸液バック、薬液バイアル等の医療用容器、ラップやトレイ等の食品包装成形品、電気器具等のケーシング、インナーパネル等の自動車内装部品、カーポートやグレージング等の建材等に利用可能である。
以下、本発明についてより詳細に説明する。
[ノルボルネン系モノマーの重合用触媒]
本発明のノルボルネン系モノマーの重合用触媒は、遷移金属錯体(A)を必須成分とし、遷移金属錯体(A)と反応してカチオン性遷移金属化合物を生成できるイオン性化合物である助触媒(B)(以下、「助触媒(B)」と略すことがある。)及びホスフィン系配位子(C)を任意成分として含有することを特徴とする。
[ノルボルネン系モノマーの重合用触媒]
本発明のノルボルネン系モノマーの重合用触媒は、遷移金属錯体(A)を必須成分とし、遷移金属錯体(A)と反応してカチオン性遷移金属化合物を生成できるイオン性化合物である助触媒(B)(以下、「助触媒(B)」と略すことがある。)及びホスフィン系配位子(C)を任意成分として含有することを特徴とする。
遷移金属錯体(A):
本発明のノルボルネン系モノマーの重合用触媒成分である遷移金属錯体(A)は、π-アリル配位子を有するパラジウム2つが、2つのアミジン配位子により架橋された構造を有する2核のパラジウム錯体化合物であって、2座配位子である1つのアミジンが1つのパラジウムに2座配位して四員環を形成するのではなく、2つのパラジウムを2つのアミジン配位子がそれぞれ架橋するように配位して八員環を形成していることを特徴とする。
本発明のノルボルネン系モノマーの重合用触媒成分である遷移金属錯体(A)は、π-アリル配位子を有するパラジウム2つが、2つのアミジン配位子により架橋された構造を有する2核のパラジウム錯体化合物であって、2座配位子である1つのアミジンが1つのパラジウムに2座配位して四員環を形成するのではなく、2つのパラジウムを2つのアミジン配位子がそれぞれ架橋するように配位して八員環を形成していることを特徴とする。
遷移金属錯体(A)は一般式(1)

で示される。
一般式(1)中、R1及びR2はそれぞれ独立して、水素原子または置換基を有していてもよい炭素数1~20の炭化水素基を表し、R3、R4及びR5はそれぞれ独立して、水素原子または炭素数1~6の炭化水素基を表す。配位子であるアミジン(N-C-N)中の2つのC-N結合は単結合または二重結合である。
一般式(1)中、R1及びR2が表す炭素数1~20の炭化水素基の具体例としては、炭素数1~20の直鎖または分枝鎖を有するアルキル基、炭素数3~20のシクロアルキル基、炭素数6~20のアリール基、アルキルアリール基またはアラルキル基等が挙げられる。
炭素数1~20の炭化水素基の置換基としては、ハロゲン原子、アルコキシ基、アリールオキシ基、カルボキシ基、アルコキシカルボニル基、シアノ基、トリアルキルシリル基等が挙げられる。
R1及びR2としては、錯体の安定性、合成のしやすさの観点から、メチル基、エチル基、イソプロピル基、t-ブチル基、シクロヘキシル基、フェニル基、2-メチルフェニル基、2,6-ジメチルフェニル基、2-イソプロピルフェニル基、2,6-ジイソプロピルフェニル基、2-t-ブチルフェニル基、2,6-ジ-t-ブチルフェニル基、ベンジル基、ナフチル基、ビフェニル基、アントラセニル基、トリメチルシリル基が好ましく、メチル基、イソプロピル基、t-ブチル基、シクロヘキシル基、フェニル基、2-メチルフェニル基、2,6-ジメチルフェニル基、2-イソプロピルフェニル基、2,6-ジイソプロピルフェニル基、トリメチルシリル基がさらに好ましく、メチル基、イソプロピル基、シクロヘキシル基、フェニル基、2-メチルフェニル基、2,6-ジメチルフェニル基、2-イソプロピルフェニル基、2,6-ジイソプロピルフェニル基が特に好ましい。
一般式(1)中、R3、R4及びR5が表す炭素数1~6の炭化水素基の具体例としては、炭素数1~6の直鎖または分枝鎖を有するアルキル基、炭素数3~6のシクロアルキル基、フェニル基が挙げられる。
R3、R4及びR5としては、錯体の安定性、合成のしやすさの観点から、水素原子、メチル基、エチル基、イソプロピル基、t-ブチル基、フェニル基が好ましく、水素原子、メチル基、エチル基がさらに好ましく、水素原子、メチル基が特に好ましい。
一般式(1)で示される遷移金属化合物(A)の具体例としては、ジ(π-アリル)ビス[μ-(N,N’-ジフェニルベンズアミジナート)-N:N’]ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ジシクロヘキシルベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ジイソプロピルベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ジ-t-ブチルベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(トリメチルシリル)ベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス[μ-(N,N’-ジフェニルアセトアミジナート)-N:N’]ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2-メチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ジシクロヘキシルアセトアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ジイソプロピルアセトアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ジ-t-ブチルアセトアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(トリメチルシリル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス[μ-(N,N’-ジフェニルベンズアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ジシクロヘキシルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ジイソプロピルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ジ-t-ブチルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(トリメチルシリル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス[μ-(N,N’-ジフェニルアセトアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ジシクロヘキシルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ジイソプロピルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ジ-t-ブチルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-プロペニル]ビス{μ-[N,N’-ビス(トリメチルシリル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス[μ-(N,N’-ジフェニルベンズアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ジシクロヘキシルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ジイソプロピルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ジ-t-ブチルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(トリメチルシリル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス[μ-(N,N’-ジフェニルアセトアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ジシクロヘキシルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ジイソプロピルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ジ-t-ブチルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-ブテニル]ビス{μ-[N,N’-ビス(トリメチルシリル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス[μ-(N,N’-ジフェニルベンズアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ジシクロヘキシルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ジイソプロピルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ジ-t-ブチルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(トリメチルシリル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチ
ル-2-ブテニル]ビス[μ-(N,N’-ジフェニルアセトアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ジシクロヘキシルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ジイソプロピルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ジ-t-ブチルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(トリメチルシリル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス[μ-(N,N’-ジフェニルベンズアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ジシクロヘキシルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ジイソプロピルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ジ-t-ブチルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(トリメチルシリル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス[μ-(N,N’-ジフェニルアセトアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ジシクロヘキシルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ジイソプロピルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ジ-t-ブチルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(トリメチルシリル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス[μ-(N,N’-ジフェニルベンズアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ジシクロヘキシルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ジイソプロピルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ジ-t-ブチルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(トリメチルシリル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス[μ-(N,N’-ジフェニルアセトアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ジシクロヘキシルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ジイソプロピルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ジ-t-ブチルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(トリメチルシリル)アセトアミジナート]-N:N’}ジパラジウム等が挙げられるが、これらに限定されるものではない。
ル-2-ブテニル]ビス[μ-(N,N’-ジフェニルアセトアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ジシクロヘキシルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ジイソプロピルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ジ-t-ブチルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(トリメチルシリル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス[μ-(N,N’-ジフェニルベンズアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ジシクロヘキシルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ジイソプロピルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ジ-t-ブチルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(トリメチルシリル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス[μ-(N,N’-ジフェニルアセトアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ジシクロヘキシルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ジイソプロピルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ジ-t-ブチルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-2-メチル-2-ブテニル]ビス{μ-[N,N’-ビス(トリメチルシリル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス[μ-(N,N’-ジフェニルベンズアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ジシクロヘキシルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ジイソプロピルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ジ-t-ブチルベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(トリメチルシリル)ベンズアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス[μ-(N,N’-ジフェニルアセトアミジナート)-N:N’]ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-メチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジメチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(2,6-ジ-t-ブチルフェニル)アセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ジシクロヘキシルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ジイソプロピルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ジ-t-ブチルアセトアミジナート]-N:N’}ジパラジウム、ビス[(1,2,3-η)-1,2-ジメチル-2-ブテニル]ビス{μ-[N,N’-ビス(トリメチルシリル)アセトアミジナート]-N:N’}ジパラジウム等が挙げられるが、これらに限定されるものではない。
これらの中でも、ジ(π-アリル)ビス[μ-(N,N’-ジフェニルベンズアミジナート)-N:N’]ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ジシクロヘキシルベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス{μ-[N,N’-ジイソプロピルベンズアミジナート]-N:N’}ジパラジウム、ジ(π-アリル)ビス[μ-(N,N’-ジフェニルアセトアミジナート)-N:N’]ジパラジウムが好ましい。
本発明に係る遷移金属錯体(A)は、前駆体である(π-アリル)パラジウム(II)化合物とアミジン化合物
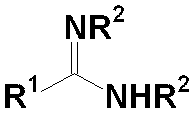
(式中、R1及びR2は一般式(1)と同じ意味を表す。)の配位子交換反応により製造することができる。具体的な製造方法として、例えばJ. Chem. Res., 2005, 702に記載の方法やJ. Organomet. Chem., 1992, 426, 261に記載の方法を挙げることができる。
(π-アリル)パラジウム(II)化合物としては、アミジン化合物と配位子交換可能な配位子を有する化合物であれば特に制限はされない。目的とする遷移金属錯体(A)に対応するR3、R4及びR5を有する化合物を選択すればよい。例えば、ジ(π-アリル)ジ(μ-クロロ)ジパラジウム(化学式(5))や(π-アリル)(アセチルアセトナト)パラジウム(化学式(6))が好ましい。

遷移金属錯体(A)を製造する際に用いるアミジン化合物は目的とする遷移金属錯体(A)に対応するR1及びR2を有するアミジン化合物を選択すればよい。その具体例としては、N,N’-ジフェニルベンズアミジン、N,N’-ビス(2-メチルフェニル)ベンズアミジン、N,N’-ビス(2,6-ジメチルフェニル)ベンズアミジン、N,N’-ビス(2-イソプロピルフェニル)ベンズアミジン、N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジン、N,N’-ビス(2-t-ブチルフェニル)ベンズアミジン、N,N’-ビス(2,6-ジ-t-ブチルフェニル)ベンズアミジン、N,N’-ジシクロヘキシルベンズアミジン、N,N’-ジイソプロピルベンズアミジン、N,N’-ジ-t-ブチルベンズアミジン、N,N’-ビス(トリメチルシリル)ベンズアミジン、N,N’-ジフェニルアセトアミジン、N,N’-ビス(2-メチルフェニル)アセトアミジン、N,N’-ビス(2,6-ジメチルフェニル)アセトアミジン、N,N’-ビス(2-イソプロピルフェニル)アセトアミジン、N,N’-ビス(2,6-ジイソプロピルフェニル)アセトアミジン、N,N’-ビス(2-t-ブチルフェニル)アセトアミジン、N,N’-ビス(2,6-ジ-t-ブチルフェニル)アセトアミジン、N,N’-ジシクロヘキシルアセトアミジン、N,N’-ジイソプロピルアセトアミジン、N,N’-ジ-t-ブチルアセトアミジン、N,N’-ビス(トリメチルシリル)アセトアミジン等が挙げられるが、これらに限定されない。
このようなアミジン化合物は市販されているものをそのまま使用することができる。また、Bull. Chem. Soc. Jpn., 1986, 59, 2171に記載の方法で製造したものを使用することもできる。
前記配位子交換反応は前駆体である(π-アリル)パラジウム(II)化合物を溶媒に溶解したものに、アミジン化合物もしくは必要に応じてそれに塩基を加えたものを添加し、所定の温度で所定の時間撹拌を行うことで実施することができる。
配位子交換反応の際に使用する溶媒としては、各基質と反応しないものであれば特に制限はないが、例えば、ペンタン、ヘキサン、ヘプタン等の脂肪族炭化水素;シクロヘキサン等の脂環族炭化水素;ベンゼン、トルエン、キシレン等の芳香族炭化水素;ジクロロメタン、クロロホルム、クロロベンゼン等のハロゲン化炭化水素;ニトロメタン、ニトロベンゼン、アセトニトリル等の含窒素系炭化水素;ジエチルエーテル、ジオキサン、テトラヒドロフラン等のエーテル類が挙げられる。これらの溶媒は混合して使用してもよい。また、使用する溶媒は脱水処理を施し、脱気処理したものが好ましい。
溶媒の使用量は、反応を著しく遅延しなければ、特に制限はない。前駆体である(π-アリル)パラジウム(II)化合物の溶解性等に応じて適宜定めることができる。通常、前駆体である(π-アリル)パラジウム(II)化合物1gに対して、1~100gの溶媒を用いる。
反応温度は特に制限されないが、一般には、-100~150℃、好ましくは-50~120℃である。温度が-100℃より低いと反応速度が遅くなり、温度が150℃より高いと生成した錯体の分解が起こることがある。上記範囲内で反応温度を選択することにより、反応速度を調整することができる。
反応時間も特に制限はなく、反応温度にもよるが、例えば1分~50時間、好ましくは30分~3時間である。また、反応は窒素ガスやアルゴンガスのような不活性ガス雰囲気下で行うことが望ましい。
反応終了後は、通常の分離・精製操作を行うことにより、目的の遷移金属錯体(A)を単離することができる。具体的には、原料としてジ(π-アリル)ジ(μ-クロロ)ジパラジウムを使用した場合は、反応で生成したLiCl等の塩を遠心分離やろ過で除去した後、再結晶することにより目的の遷移金属錯体(A)を単離する。一方、(π-アリル)(アセチルアセトナト)パラジウムを原料として使用した場合は、反応で生成したアセチルアセトンを溶媒と共に減圧下で留去した後、あらためて溶媒を加え、再結晶することにより目的の遷移金属錯体(A)を単離する。
反応で得られた生成物が目的の遷移金属錯体(A)であることの確認はNMRスペクトル、元素分析、マススペクトル、X線結晶解析等により行うことができる。
以上のようにして得られる遷移金属錯体(A)は、ノルボルネン系モノマーの重合用触媒成分として有用である。
以上のようにして得られる遷移金属錯体(A)は、ノルボルネン系モノマーの重合用触媒成分として有用である。
本発明のノルボルネン系モノマーの重合用触媒は、遷移金属錯体(A)の少なくとも1種を含有するものであればよいが、遷移金属錯体(A)と反応してカチオン性遷移金属化合物を生成できるイオン性化合物である助触媒(B)、及びホスフィン系配位子(C)をさらに含有するものが、より高い触媒活性を発現できる点で好ましい。
助触媒(B):
本発明で用いられる遷移金属錯体(A)と反応してカチオン性遷移金属化合物を生成できるイオン性化合物である助触媒(B)としては、非配位性アニオンとカチオンとを組み合わせたイオン性化合物が挙げられる。
本発明で用いられる遷移金属錯体(A)と反応してカチオン性遷移金属化合物を生成できるイオン性化合物である助触媒(B)としては、非配位性アニオンとカチオンとを組み合わせたイオン性化合物が挙げられる。
非配位性アニオンとしては、1991年版周期表第13族元素の4級アニオンが挙げられる。具体的には、テトラ(フェニル)ボレート、テトラ(フルオロフェニル)ボレート、テトラキス(ジフルオロフェニル)ボレート、テトラキス(トリフルオロフェニル)ボレート、テトラキス(テトラフルオロフェニル)ボレート、テトラキス(ペンタフルオロフェニル)ボレート、テトラキス(テトラフルオロメチルフェニル)ボレート、テトラキス[3,5-ジ(トリフルオルメチル)フェニル]ボレート、テトラ(トリイル)ボレート、テトラ(キシリル)ボレート、トリフェニル(ペンタフルオロフェニル)ボレート、[トリス(ペンタフルオロフェニル)フェニル]ボレート、トリデカハイドライド-7,8-ジカルバウンデカボレート等が挙げられる。
前記カチオンとしては、カルボニウムカチオン、オキソニウムカチオン、アンモニウムカチオン、ホスホニウムカチオン、シクロヘプチルトリエニルカチオン、遷移金属を有するフェロセニウムカチオン等が挙げられる。
カルボニウムカチオンの具体例としては、トリフェニルカルボニウムカチオン、トリ置換フェニルカルボニウムカチオン等のトリ置換カルボニウムカチオンが挙げられる。トリ置換フェニルカルボニウムカチオンの具体例としては、トリ(メチルフェニル)カルボニウムカチオン、トリ(ジメチルフェニル)カルボニウムカチオンが挙げられる。
オキソニウムカチオンの具体例としては、ヒドロキソニウムカチオン、メチルオキソニウムカチオン等のアルキルオキソニウムカチオン、ジメチルオキソニウムカチオン等のジアルキルオキソニウムカチオン、トリメチルオキソニウムカチオン、トリエチルオキソニウムカチオン等のトリアルキルオキソニウムカチオン等が挙げられる。
アンモニウムカチオンの具体例としては、トリメチルアンモニウムカチオン、トリエチルアンモニウムカチオン、トリプロピルアンモニウムカチオン、トリブチルアンモニウムカチオン、トリ(n-ブチル)アンモニウムカチオン等のトリアルキルアンモニウムカチオン、N,N-ジエチルアニリニウムカチオン、N,N-2,4,6-ペンタメチルアニリニウムカチオン等のN,N-ジアルキルアニリニウムカチオン、ジ(イソプロピル)アンモニウムカチオン、ジシクロヘキシルアンモニウムカチオン等のジアルキルアンモニウムカチオンが挙げられる。
ホスホニウムカチオンの具体例としては、トリフェニルホスホニウムカチオン、トリ(メチルフェニル)ホスホニウムカチオン、トリ(ジメチルフェニル)ホスホニウムカチオン等のトリアリールホスホニウムカチオンが挙げられる。
助触媒(B)の好ましい例は、トリチルテトラキス(ペンタフルオロフェニル)ボレート、トリフェニルカルボニウムテトラ(フルオロフェニル)ボレート、N,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート、トリチルテトラキス[3,5-ジ(トリフルオルメチル)フェニル]ボレート、N,N-ジメチルアニリニウムテトラキス[3,5-ジ(トリフルオルメチル)フェニル]ボレート、1,1’-ジメチルフェロセニウムテトラキス(ペンタフルオロフェニル)ボレート等である。
ホスフィン系配位子(C):
本発明で用いられるホスフィン系配位子(C)とは、水素原子、アルキル基もしくはアリール基から独立して選ばれる3つの置換基が結合した3価のリン化合物である。具体的にはトリメチルホスフィン、トリエチルホスフィン、トリイソプロピルホスフィン、トリ-t-ブチルホスフィン等のトリアルキルホスフィン類、トリシクロペンチルホスフィン、トリシクロヘキシルホスフィン等のトリシクロアルキルホスフィン類、ならびにトリフェニルホスフィン等のトリアリールホスフィン類を挙げることができる。これらの中では触媒活性向上の観点から、トリシクロヘキシルホスフィン、トリ-t-ブチルホスフィン、トリイソプロピルホスフィンが好ましい。
本発明で用いられるホスフィン系配位子(C)とは、水素原子、アルキル基もしくはアリール基から独立して選ばれる3つの置換基が結合した3価のリン化合物である。具体的にはトリメチルホスフィン、トリエチルホスフィン、トリイソプロピルホスフィン、トリ-t-ブチルホスフィン等のトリアルキルホスフィン類、トリシクロペンチルホスフィン、トリシクロヘキシルホスフィン等のトリシクロアルキルホスフィン類、ならびにトリフェニルホスフィン等のトリアリールホスフィン類を挙げることができる。これらの中では触媒活性向上の観点から、トリシクロヘキシルホスフィン、トリ-t-ブチルホスフィン、トリイソプロピルホスフィンが好ましい。
本発明では、遷移金属錯体(A)として、一般式(1)において、R1がメチル基またはフェニル基であり、R2がフェニル基またはアルキル置換フェニル基であり、R3、R4及びR5がいずれも水素原子である錯体を用い、助触媒(B)として、N,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート{[Ph(Me)2NH][B(C6F5)4]}またはトリチルテトラキス(ペンタフルオロフェニル)ボレート{[Ph3C][B(C6F5)4]}を用い、さらにホスフィン系配位子(C)として、トリイソプロピルホスフィンまたはトリ-t-ブチルホスフィンを用いる場合が、高活性にノルボルネン系重合体を製造することができる触媒としての好ましい態様である。
また、遷移金属錯体(A)として、一般式(1)において、R1がフェニル基であり、R2がフェニル基または(2,6-アルキル置換)フェニル基であり、R3、R4及びR5がいずれも水素原子である錯体を用い、助触媒(B)として、N,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート{[Ph(Me)2NH][B(C6F5)4]}を用い、さらにホスフィン系配位子(C)として、トリイソプロピルホスフィンを用いる場合が、高活性にノルボルネン系重合体を製造することができる触媒としての最も好ましい態様である。
本発明の触媒における遷移金属錯体(A)と助触媒(B)との使用割合は、各種の条件により異なるため一義的には定められないが、通常は(A)/(B)(モル比)で1/0.1~1/100であり、好ましくは1/0.5~1/50、さらに好ましくは1/1~1/10である。
本発明の触媒における遷移金属錯体(A)とホスフィン系配位子(C)との使用割合は、各種の条件により異なるため一義的には定められないが、通常は(A)/(C)(モル比)で1/0.1~1/2であり、好ましくは1/0.5~1/1.8、さらに好ましくは1/1~1/1.5である。
各触媒成分を接触させる温度も特に制限されないが、一般には、-100~150℃、好ましくは-50~120℃である。温度が-100℃より低いと各成分間の反応が遅くなり、温度が150℃より高いと各成分の分解を招き、触媒の活性が低下する。上記範囲内で接触温度を選択することにより、重合に使用した際に重合速度や生成ポリマーの分子量等を調整することができる。
各触媒成分の混合は溶媒の存在下に行ってもよい。使用可能な溶媒は特に限定はされないが、各触媒成分との反応性がなく、工業的スケールでの製造がされていて、入手が容易なものが好ましい。具体的には、ペンタン、ヘキサン、ヘプタン等の脂肪族炭化水素;シクロヘキサン等の脂環族炭化水素;ベンゼン、トルエン、キシレン等の芳香族炭化水素;ジクロロメタン、クロロホルム、クロロベンゼン等のハロゲン化炭化水素;ニトロメタン、ニトロベンゼン、アセトニトリル等の含窒素系炭化水素;ジエチルエーテル、ジオキサン、テトラヒドロフラン等のエーテル類等を使用することができる。これらの中でも、脂肪族炭化水素、芳香族炭化水素、ハロゲン化炭化水素が好ましい。また、これらの溶媒は混合して使用してもよい。
[ノルボルネン系重合体の製造方法]
本発明のノルボルネン系重合体の製造方法は、本発明の重合用触媒の存在下に、ノルボルネン系モノマーを付加重合することを特徴とする。
本発明のノルボルネン系重合体の製造方法は、本発明の重合用触媒の存在下に、ノルボルネン系モノマーを付加重合することを特徴とする。
本発明の製造方法は、(i)ノルボルネン系モノマー1種類のみを付加重合することにより、ノルボルネン系モノマーの単独付加重合体を得る方法、(ii)ノルボルネン系モノマー2種類以上を付加共重合することにより、ノルボルネン系モノマーの付加共重合体を得る方法、(iii)ノルボルネン系モノマー1種類以上とノルボルネン系モノマーと共重合可能な他のビニルモノマー1種類以上とを付加共重合することにより、ノルボルネン系モノマーの付加共重合体を得る方法のいずれかである。
ノルボルネン系モノマー:
本発明に用いられるノルボルネン系モノマーは、ノルボルネン環構造を有する化合物(以下、単に「ノルボルネン類」ということがある。)であれば、特に制限はされない。極性あるいは非極性の置換基を有していてもよく、ノルボルネン環以外の環構造を有していてもよい。
ノルボルネン類としては、一般式(4)で示されるものが好ましい。
本発明に用いられるノルボルネン系モノマーは、ノルボルネン環構造を有する化合物(以下、単に「ノルボルネン類」ということがある。)であれば、特に制限はされない。極性あるいは非極性の置換基を有していてもよく、ノルボルネン環以外の環構造を有していてもよい。
ノルボルネン類としては、一般式(4)で示されるものが好ましい。

式中、R10~R13は、それぞれ独立して、水素原子;ハロゲン原子;窒素原子、酸素原子、硫黄原子、ハロゲン原子もしくはケイ素原子を含む官能基;ハロゲン原子もしくは前記官能基を有していてもよい炭素数1~20の炭化水素基を表す。また、R10~R13は、互いに結合して環を形成していてもよい。nは0または1である。
一般式(4)で示されるノルボルネン類は、nが0であるビシクロ[2.2.1]ヘプト-2-エン類及びnが1であるテトラシクロ[6.2.1.13,6.02,7]ドデカ-4-エン類に分類することができる。本発明の製造方法ではいずれも使用することができる。
一般式(4)におけるR10~R13の具体例としては、水素原子;塩素原子、臭素原子、フッ素原子等のハロゲン原子;水酸基、アルコキシ基、アリールオキシ基、カルボニル基、ヒドロキシカルボニル基、アルコキシカルボニル基、及びアリールオキシカルボニル基等の酸素原子を含む官能基;アミノ基、アルキルアミノ基、アリールアミノ基、アミノカルボニル基、アルキルアミノカルボニル基、アリールアミノカルボニル基、及びシアノ基等の窒素原子を含む官能基;メルカプト基、アルコキシチオ基、及びアリールオキシチオ基等の硫黄原子を含む官能基;シリル基、アルキルシリル基、アリールシリル基、アルコキシシリル基、及びアリールオキシシリル基等のケイ素原子を含む官能基を挙げることができる。また、これらの官能基を有していてもよい炭素数1~20のアルキル基、アルケニル基、及びアリール基等の炭化水素基も挙げられる。さらに、R10~R13は、互いに結合して環を形成してもよく、このような例としては、酸無水物構造、カーボネート構造、ジチオカーボネート構造等が挙げられる。
本発明に用いられるノルボルネン類の具体例としては、2-ノルボルネン、5-メチル-2-ノルボルネン、5-エチル-2-ノルボルネン、5-n-ブチル-2-ノルボルネン、5-n-ヘキシル-2-ノルボルネン、5-n-デシル-2-ノルボルネン、5-シクロヘキシル-2-ノルボルネン、5-エチリデン-2-ノルボルネン、5-ビニル-2-ノルボルネン、5-フェニル-2-ノルボルネン、5-ベンジル-2-ノルボルネン、ジシクロペンタジエン、ジヒドロジシクロペンタジエン、テトラシクロ[9.2.1.02,10.03,8]テトラデカ-3,5,7,12-テトラエン、テトラシクロ[10.2.1.02,11.04,9]ペンタデカ-4,6,8,13-テトラエン等の無置換または炭化水素基を有するビシクロ[2.2.1]ヘプト-2-エン類;
テトラシクロ[6.2.1.13,6.02,7]ドデカ-4-エン、9-メチルテトラシクロ[6.2.1.13,6.02,7]ドデカ-4-エン、9-エチルテトラシクロ[6.2.1.13,6.02,7]ドデカ-4-エン、9-エチルテトラシクロ[6.2.1.13,6.02,7]ドデカ-4-エン、9-n-ブチルテトラシクロ[6.2.1.13,6.02,7]ドデカ-4-エン、9-シクロヘキシルテトラシクロ[6.2.1.13,6.02,7]ドデカ-4-エン、9-エチリデンテトラシクロ[6.2.1.13,6.02,7]ドデカ-4-エン、9-ビニルテトラシクロ[6.2.1.13,6.02,7]ドデカ-4-エン、9-フェニルテトラシクロ[6.2.1.13,6.02,7]ドデカ-4-エン等の無置換または炭化水素基を有するテトラシクロ[6.2.1.13,6.02,7]ドデカ-4-エン類;
5-ノルボルネン-2-カルボン酸メチル、5-ノルボルネン-2-カルボン酸エチル、5-ノルボルネン-2-カルボン酸n-ブチル、2-メチル-5-ノルボルネン-2-カルボン酸メチル、2-メチル-5-ノルボルネン-2-カルボン酸エチル、2-メチル-5-ノルボルネン-2-カルボン酸n-ブチル、5-ノルボルネン-2,3-ジカルボン酸メチル、5-ノルボルネン-2,3-ジカルボン酸エチル等のアルコキシカルボニル基を有するビシクロ[2.2.1]ヘプト-2-エン類;テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4-カルボン酸メチル、テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4-カルボン酸エチル、4-メチルテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4-カルボン酸メチル、テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4,5-ジカルボン酸メチル、テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4,5-ジカルボン酸エチル等のアルコキシカルボニル基を有するテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン類;
5-ノルボルネン-2-カルボン酸、5-ノルボルネン-2,3-ジカルボン酸等のヒドロキシカルボニル基を有するビシクロ[2.2.1]ヘプト-2-エン類;テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4-カルボン酸、テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4,5-ジカルボン酸等のヒドロキシカルボニル基を有するテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン類;
2-ヒドロキシ-5-ノルボルネン、2-ヒドロキシメチル-5-ノルボルネン、2,2-ジ(ヒドロキシメチル)-5-ノルボルネン、2,3-ジ(ヒドロキシメチル)-5-ノルボルネン等のヒドロキシル基を有するビシクロ[2.2.1]ヘプト-2-エン類;テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4-オール、テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4-メタノール、テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4,5-ジメタノール等のヒドロキシル基を有するテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン類;
2-アセトキシ-5-ノルボルネン、2-アセトキシメチル-5-ノルボルネン、2,2-ジ(アセトキシメチル)-5-ノルボルネン、2,3-ジ(アセトキシメチル)-5-ノルボルネン等のアセトキシル基を有するビシクロ[2.2.1]ヘプト-2-エン類;4-アセトキシテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン、4-アセトキシメチルテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン、4,5-ジ(アセトキシメチル)テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン等のアセトキシル基を有するテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン類;
5-ノルボルネン-2-カルボニトリル、5-ノルボルネン-2-カルボキサミド等の窒素原子を含む官能基を有するビシクロ[2.2.1]ヘプト-2-エン類;テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4-カルボニトリル、テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4-カルボキサミド等の窒素原子を含む官能基を有するテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン類;
2-クロロ-5-ノルボルネン、2-フルオロ-5-ノルボルネン等のハロゲン原子を有するビシクロ[2.2.1]ヘプト-2-エン類;4-クロロテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン、4-フルオロテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン等のハロゲン原子を有するテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン類;
2-トリメチルシロキシ-5-ノルボルネン、2-トリメトキシシリル-5-ノルボルネン、2-トリス(トリメトキシシリロキシ)シリル-5-ノルボルネン等のケイ素原子を含む官能基を有するビシクロ[2.2.1]ヘプト-2-エン類;4-トリメチルシロキシテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン、4-トリメトキシシリルテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン、4-トリス(トリメトキシシリロキシ)シリルテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン等のケイ素原子を含む官能基を有するテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン類;
5-ノルボルネン-2,3-ジカルボン酸無水物、5-ノルボルネン-2,3-カーボネート、5-ノルボルネン-2,3-ジチオカーボネート等の酸無水物構造、カーボネート構造、ジチオカーボネート構造を有するビシクロ[2.2.1]ヘプト-2-エン類;テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4,5-ジカルボン酸無水物、テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4,5-カーボネート、テトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン-4,5-ジチオカーボネート等の酸無水物構造、カーボネート構造、ジチオカーボネート構造を有するテトラシクロ[6.2.1.13,6.02,7]ドデカ-9-エン類;等を挙げることができる。
これらのノルボルネン類はそれぞれ単独で用いることもできるし、2種以上組み合わせて用いることもできる。
これらのノルボルネン類はそれぞれ単独で用いることもできるし、2種以上組み合わせて用いることもできる。
本発明の製造方法においては、これらのノルボルネン類の中でも、下記一般式(2)

及び一般式(3)

(式中、R6は炭素数1~10のアルキル基を表し、R7、R8、及びR9はそれぞれ独立して水素原子または炭素数1~10のアルキル基を表す。)
で示されるモノマーユニットに相当するノルボルネン類を用いることが好ましい。
で示されるモノマーユニットに相当するノルボルネン類を用いることが好ましい。
一般式(2)におけるR6が表す炭素数1~10のアルキル基は直鎖状でも分岐していてもよい。
直鎖状のアルキル基の例としては、メチル基、エチル基、n-プロピル基、n-ブチル基、n-ぺンチル基、n-ヘキシル基、n-オクチル基、n-デシル基等が挙げられる。
分岐を有するアルキル基の例としては、イソプロピル基、イソブチル基、sec-ブチル基、ネオペンチル基、イソヘキシル基、イソオクチル基、イソデシル基等が挙げられる。
分岐を有するアルキル基の例としては、イソプロピル基、イソブチル基、sec-ブチル基、ネオペンチル基、イソヘキシル基、イソオクチル基、イソデシル基等が挙げられる。
これらの中でも、炭素数1~3の直鎖状のアルキル基が経済性の面で好ましい。モノマー製造コストの観点からメチル基が特に好ましい。
一般式(2)におけるR7及び一般式(3)におけるR8及びR9は、それぞれ独立して水素原子または炭素数1~10のアルキル基を表し、炭素数3~10のアルキル基は分岐していてもよい。これらのアルキル基としては前述のR6のアルキル基と同様のものが挙げられる。これらの中でも、モノマー製造コストの観点から水素原子が好ましい。
なお、R7が水素原子である場合、一般式(2)で示されるモノマーユニットの基本になるノルボルネン類は、R6が炭素数1のアルキル基のとき、2-アセトキシメチル-5-ノルボルネン、R6が炭素数2のアルキル基のとき、2-[(エチルカルボニルオキシ)メチル]-5-ノルボルネン、R6が炭素数3の直鎖状のアルキル基のとき、2-[(プロピルカルボニルオキシ)メチル]-5-ノルボルネンとなる。
R8及びR9が水素原子である場合、一般式(3)で示されるモノマーユニットの基本になるノルボルネン類はノルボルネンとなる。
R8及びR9が水素原子である場合、一般式(3)で示されるモノマーユニットの基本になるノルボルネン類はノルボルネンとなる。
本発明の製造方法において、遷移金属錯体(A)、助触媒(B)及びホスフィン系配位子(C)を用いたノルボルネン系モノマーの重合は塊状重合、懸濁重合、乳化重合、溶液重合、沈殿重合で行うことができる。溶媒を用いる重合を行う場合には、触媒活性に悪影響を与えない溶媒を使用する必要がある。使用可能な溶媒としては、ペンタン、ヘキサン、ヘプタン等の脂肪族炭化水素;シクロヘキサン等の脂環族炭化水素;ベンゼン、トルエン、キシレン等の芳香族炭化水素;ジクロロメタン、クロロホルム、クロロベンゼン等のハロゲン化炭化水素;ニトロメタン、ニトロベンゼン、アセトニトリル等の含窒素系炭化水素;ジエチルエーテル、ジオキサン、テトラヒドロフラン等のエーテル類;酢酸エチル、酢酸n-プロピル、酢酸n-ブチル等のエステル類;γ-ブチロラクトン、δ-バレロラクトン等のラクトン類及び水が挙げられる。これらの溶媒は混合して使用してもよい。また、水を使用する際はアニオン型、カチオン形、非イオン型の界面活性剤等を用いて反応液を乳化状態にすることもできる。
沈殿重合は溶液重合の一種であり、溶媒としてモノマーは溶解するが、ポリマーが溶解しないものを使用する。沈殿重合では重合と共にポリマーが析出してくるので、再沈殿精製のために大量に使用する貧溶媒(メタノール等)が不要となり、製造コストの面で有利となる。本発明の(共)重合体ではトルエンと酢酸エチルとの混合溶媒等が沈殿重合に適する。
重合を行う際には、主触媒(A)、助触媒(B)及びホスフィン系配位子(C)を混合するが、その混合順序は、主触媒(A)が助触媒(B)と接触する前にホスフィン系配位子(C)と混合されるようになっていれば、その他は特に限定されない。予め主触媒(A)成分とホスフィン系配位子(C)を混合し、さらに助触媒(B)を混合して反応組成物を得、重合させる単量体を含む溶液にこれを添加してもよい。また、重合させる単量体と主触媒(A)及びホスフィン系配位子(C)を含む溶液に、助触媒(B)を添加してもよく、重合させる単量体と助触媒(B)の混合溶液中に主触媒(A)及びホスフィン系配位子(C)の混合物を添加してもよい。
本発明では、予め主触媒(A)とホスフィン系配位子(C)とを混合し、1分間以上、好ましくは30分~1時間程度接触させた後に、助触媒(B)と混合して反応系に添加するか、もしくは主触媒(A)とホスフィン系配位子(C)との混合物を助触媒(B)を含む反応系に添加することが好ましい。このような操作を行うことにより、より高い重合活性を発現することが可能になる。
重合温度も特に制限されないが、一般には、-100~150℃、好ましくは-50~120℃である。温度が-100℃より低いと重合速度が遅くなり、温度が150℃より高いと触媒の活性が低下することがある。上記範囲内で重合温度を選択することにより、重合速度や分子量等を調整することができる。
重合時間も特に制限はなく、例えば1分~100時間である。また、反応は窒素ガスのような不活性ガス雰囲気下で行うことが望ましい。
重合反応終了後、生成物であるノルボルネン系共重合体は、必要に応じて公知の操作、処理方法(例えば、再沈殿等)により後処理を行い、ろ過分別後、乾燥を行うことにより単離される。
本発明の製造方法で製造される一般式(2)及び一般式(3)で示されるモノマーユニットから構成されるノルボルネン系共重合体において、一般式(2)で示されるモノマーユニットの含有量は10~70モル%であることが好ましい。一般式(2)で示されるモノマーユニットが10モル%未満であると共重合体の疎水性が高くなり、有機溶媒に対する溶解性は低下するが、吸水性が低くなる傾向がある。一方、70モル%を超えると共重合体が親水性となり、有機溶媒に対する溶解性が向上するが、吸水性が高くなる傾向がある。従って、一般式(2)で示されるモノマーユニットの含有量を調整することにより、共重合体の溶媒への溶解性と吸水性を制御することが可能である。
本発明の製造方法で製造される一般式(2)及び一般式(3)で示されるモノマーユニットから構成されるノルボルネン系共重合体をフィルム、シート等へ成形する際に必要となる溶媒への適度な溶解性と低吸水性を両立させる観点からは、一般式(2)で示されるモノマーユニットの含有量は10~80モル%が好ましく、15~70モル%がより好ましく、20~60モル%がさらに好ましい。なお、一般式(2)で示されるモノマーユニットの含有量は粉末状もしくはフィルム状の共重合体を適当な重水素化溶媒に溶解させ、1H-NMRを測定し、その積分値より算出することができる。
本発明の製造方法で製造されるノルボルネン系共重合体は、基本的にはノルボルネン類のみで構成される。ただし、この場合であっても本発明のノルボルネン系共重合体の性質をほとんど変化させないような微少量、例えば1モル%以下の第3のモノマーユニットの存在を除外するものではない。また、本発明の製造方法で製造されるノルボルネン系共重合体は物性改良のため、本発明の効果を損なわない範囲で第3のモノマーを共重合させてもよい。
第3のモノマーには特に制限はないが、エチレン性炭素-炭素二重結合を有するモノマーが好ましく、例えば、エチレン、プロピレン、1-ブテン、1-ペンテン及び1-ヘキセン等のα-オレフィン類;スチレン、α-メチルスチレン、ジビニルベンゼン等の芳香族ビニル化合物類;1,3-ブタジエン、イソプレン等の鎖状共役ジエン類;エチルビニルエーテル、プロピルビニルエーテル等のビニルエーテル類;メチルアクリレート、エチルアクリレート、2-エチルヘキシルアクリレート等のアクリレート類;メチルメタクリレート、エチルメタクリレート等のメタクリレート類;等を挙げることができる。なかでも、エチレン、プロピレン、1-ヘキセンのようなα-オレフィン類やスチレンのような芳香族ビニル化合物類が特に好ましい。
本発明の製造方法で製造されるノルボルネン系共重合体において、各モノマーユニットの共重合様式は重合条件により、ランダム、ブロック、交互のいずれをもとり得るが、共重合体の物性向上の観点からは、ランダムであることが望ましい。
本発明の製造方法で製造されるノルボルネン系共重合体のゲルパーミエイションクロマトグラフィー(GPC)法により測定したポリスチレン換算数平均分子量(Mn)は50,000~2,000,000である。さらには100,000~1,500,000がより好ましい。ポリスチレン換算数平均分子量が50,000未満であると機械強度が不十分である。ポリスチレン換算数平均分子量が2,000,000を超えると、キャストフィルムを成形する際に溶媒への溶解度が低下するばかりでなく、溶液粘度が高くなり、成形加工性が低下する。また、分子量分布Mw/Mn(重量平均分子量/数平均分子量)は、1.00~4.00が好ましく、1.30~3.50がより好ましく、1.50~3.30がさらに好ましい。分子量分布が広いとキャストフィルム成形時の溶液が均一になりにくいため、良好なフィルムが作製しにくくなる。
本発明の製造方法で製造されるノルボルネン系共重合体の23℃における飽和吸水率は、通常、0.001~1質量%、好ましくは0.005~0.7質量%、さらに好ましくは0.01~0.5質量%である。飽和吸水率がこの範囲内であると、各種光学特性、例えば透明性、位相差、位相差の均一性、及び寸法精度が、高温多湿のような条件下でも維持され、他材料との密着性や接着性に優れるため使用途中で剥離等が発生せず、また、酸化防止剤等の添加物との相溶性も良好であるため、添加の自由度が大きくなる。なお、上記飽和吸水率はJIS K7209に準拠し、23℃水中で24時間浸漬して増加質量を測定することにより求められる値である。
本発明の製造方法で製造されるノルボルネン系(共)重合体のガラス転移温度(Tg)は、共重合体の場合、その構成モノマー単位の種類、組成比、添加剤等の有無により異なるが、通常、80~350℃、好ましくは100~320℃、さらに好ましくは120~300℃である。Tgが上記範囲よりも低いと、熱変形温度が低くなり、耐熱性に問題が生じるおそれがあり、また、得られる光学フィルムの温度による光学特性の変化が大きくなることがある。また、Tgが上記範囲よりも高いと、延伸加工時にTg近辺まで加熱する場合に樹脂が熱劣化する可能性が高くなる。
本発明の製造方法で製造されるノルボルネン系共重合体は溶液流延法(溶液キャスト法)により成膜してフィルムに加工することができる。使用する溶媒としてはトルエン、テトラヒドロフラン(THF)、ジクロロメタン、クロロホルム等を用いることができる。
以下、実施例及び比較例を挙げて本発明をさらに詳細に説明するが、本発明はこれらの記載により何らの限定を受けるものではない。
各実施例及び比較例において、触媒活性は以下の式

により算出した。得られたポリマーの重量平均分子量(Mw)、数平均分子量(Mn)、分子量分布(Mw/Mn)は、ポリスチレンを標準物質として用いたゲルパーミエイションクロマトグラフィー(GPC)により求めた。また、共重合体中のノルボルネンと5-アセトキシメチル-2-ノルボルネンの組成比は、1H-NMRにより得られたピーク[δ:3.5-4.5ppm,5-アセトキシメチル-2-ノルボルネン(「ANB」と略す。)の「-COOCH2-」ユニット]と[δ:0.5-3.0ppm,ノルボルネン(「NB」と略す。)及び5-アセトキシメチル-2-ノルボルネンの「CH3COO-」、「-CH2-」及び「-CH=」ユニット)]の積分比から求め、ANB含有率は以下の式

より算出した。
実施例及び比較例で合成した物質の諸物性は、以下の通りに測定した。
1.1H-NMR,13C-NMR
使用機種:JEOL EX-400(400MHz,日本電子社製)、
測定方法:重水素化クロロホルムに溶解し、内部標準物質にテトラメチルシランを使用して測定した。
2.FT-IR
使用機種
システム:Spectrum GX(パーキンエルマー社製)、
ATR:MIRacleTM(Pike Technologies社製)。
測定方法
1回反射ATR法により測定した。
1.1H-NMR,13C-NMR
使用機種:JEOL EX-400(400MHz,日本電子社製)、
測定方法:重水素化クロロホルムに溶解し、内部標準物質にテトラメチルシランを使用して測定した。
2.FT-IR
使用機種
システム:Spectrum GX(パーキンエルマー社製)、
ATR:MIRacleTM(Pike Technologies社製)。
測定方法
1回反射ATR法により測定した。
3.ゲルパーミエイションクロマトグラフィー(GPC)
使用機種
カラム:Shodex GPC K-G+KF-806L×2(昭和電工社製)、
検出器:Shodex SE-61(昭和電工社製)。
測定条件
溶媒:テトラヒドロフラン、
測定温度:40℃、
流速:1.0ml/分、
試料濃度:1.0mg/ml、
注入量:1.0μl、
検量線:Universal Calibration curve、
解析プログラム:SIC 480II(システム インスツルメンツ社製)。
使用機種
カラム:Shodex GPC K-G+KF-806L×2(昭和電工社製)、
検出器:Shodex SE-61(昭和電工社製)。
測定条件
溶媒:テトラヒドロフラン、
測定温度:40℃、
流速:1.0ml/分、
試料濃度:1.0mg/ml、
注入量:1.0μl、
検量線:Universal Calibration curve、
解析プログラム:SIC 480II(システム インスツルメンツ社製)。
また、シクロペンタジエニル(π-アリル)パラジウムは、Shawらの合成法(Proc. Chem. Soc., 1960, 247)に従って合成した。
合成例1:2-アセトキシメチル-5-ノルボルネンの合成
10Lのステンレス製オートクレーブにジシクロペンタジエン(東京化成工業社製,759.80g,5.747mol)、酢酸アリル(東京化成工業社製,1457.86g,14.561mol)及びヒドロキノン(和光純薬工業社製,2.25g,0.0204mol)を加えた。系内を窒素置換した後、500rpmで撹拌しながら、このオートクレーブを190℃まで昇温し、5時間反応させた。反応終了後、オートクレーブを室温まで冷却し、内容物を蒸留装置に移し、減圧下に蒸留を行い、0.07kPa、48℃の留分として、無色透明液状物1306.70gを得た。
得られた液状物の1H-NMRを測定し、目的の2-アセトキシメチル-5-ノルボルネンであることを確認した。また、得られた2-アセトキシメチル-5-ノルボルネンのエキソ異性体とエンド異性体のモル比率はエキソ/エンド=18/82であった。
10Lのステンレス製オートクレーブにジシクロペンタジエン(東京化成工業社製,759.80g,5.747mol)、酢酸アリル(東京化成工業社製,1457.86g,14.561mol)及びヒドロキノン(和光純薬工業社製,2.25g,0.0204mol)を加えた。系内を窒素置換した後、500rpmで撹拌しながら、このオートクレーブを190℃まで昇温し、5時間反応させた。反応終了後、オートクレーブを室温まで冷却し、内容物を蒸留装置に移し、減圧下に蒸留を行い、0.07kPa、48℃の留分として、無色透明液状物1306.70gを得た。
得られた液状物の1H-NMRを測定し、目的の2-アセトキシメチル-5-ノルボルネンであることを確認した。また、得られた2-アセトキシメチル-5-ノルボルネンのエキソ異性体とエンド異性体のモル比率はエキソ/エンド=18/82であった。
合成例2:N,N’-ジフェニルベンズアミジンの合成
三方コックを装備した二口フラスコを窒素置換し、それに3-メチル-1-フェニルホスホレン-1-オキシド(東京化成工業社製,0.92g,4.787mmol)、フェニルイソシアネート(東京化成工業社製,75.59g,0.635mol)を加え、50℃で3時間反応を行った。その後、反応液を蒸留装置に移し、減圧下に蒸留を行い、0.03kPa、105℃の留分として、淡黄色透明液状物55.13gを得た。得られた液状物の1H-NMRを測定し、N,N’-ジフェニルカルボジイミドであることを確認した。
別途、三方コックを装備した二口フラスコを窒素置換し、これに脱水THF(和光純薬工業社製,250ml)と先に合成したN,N’-ジフェニルカルボジイミド(35.06g,0.180mol)を加え、-20℃に冷却後、撹拌を行いながら、フェニルリチウムの1.9mol/lブチルエーテル溶液(東京化成工業社製,95.0ml,0.181mol)を30分かけてゆっくりと滴下し、滴下終了後、室温にもどし、4時間反応を行った。その後、塩化アンモニウムの飽和水溶液100mlを加えて反応を停止し、内容物を分液漏斗に移し有機層を分離後、硫酸ナトリウムで乾燥、ろ過後、溶媒をエバポレーターで留去して、残渣を回収した。この残渣をエタノールに溶解し、再結晶を行って、淡黄色結晶39.1gを得た。得られた結晶の1H-NMRを測定し、N,N’-ジフェニルベンズアミジンであることを確認した。
三方コックを装備した二口フラスコを窒素置換し、それに3-メチル-1-フェニルホスホレン-1-オキシド(東京化成工業社製,0.92g,4.787mmol)、フェニルイソシアネート(東京化成工業社製,75.59g,0.635mol)を加え、50℃で3時間反応を行った。その後、反応液を蒸留装置に移し、減圧下に蒸留を行い、0.03kPa、105℃の留分として、淡黄色透明液状物55.13gを得た。得られた液状物の1H-NMRを測定し、N,N’-ジフェニルカルボジイミドであることを確認した。
別途、三方コックを装備した二口フラスコを窒素置換し、これに脱水THF(和光純薬工業社製,250ml)と先に合成したN,N’-ジフェニルカルボジイミド(35.06g,0.180mol)を加え、-20℃に冷却後、撹拌を行いながら、フェニルリチウムの1.9mol/lブチルエーテル溶液(東京化成工業社製,95.0ml,0.181mol)を30分かけてゆっくりと滴下し、滴下終了後、室温にもどし、4時間反応を行った。その後、塩化アンモニウムの飽和水溶液100mlを加えて反応を停止し、内容物を分液漏斗に移し有機層を分離後、硫酸ナトリウムで乾燥、ろ過後、溶媒をエバポレーターで留去して、残渣を回収した。この残渣をエタノールに溶解し、再結晶を行って、淡黄色結晶39.1gを得た。得られた結晶の1H-NMRを測定し、N,N’-ジフェニルベンズアミジンであることを確認した。
合成例3:N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジンの合成
三方コックと冷却管を装備した三口フラスコを窒素置換し、それに五酸化二りん(和光純薬工業社製,7.13g,50.23mmol)、ヘキサメチルジシロキサン(和光純薬工業社製,17.86g,110.00mmol)、ジクロロメタン(和光純薬工業社製,30.0ml)を加え、撹拌しながら、50℃で30分還流を行った。その後、さらに低沸分を留去しながら、160℃で30分反応を行い、残渣として粘稠な液体を得た。これに2,6-ジイソプロピルアニリン(東京化成工業社製,4.43g,25.00mmol)と安息香酸(東京化成工業社製,1.53g,12.50mmol)を加え、160℃で5時間反応を行った。フラスコを冷却後、内容物を1mol/l水酸化ナトリウム水溶液100mlにゆっくりと加えて中和し、析出した固体部分を回収してエタノールに溶解し、再結晶を行って、淡黄色結晶4.01gを得た。得られた結晶の1H-NMRを測定し、N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジンであることを確認した。
三方コックと冷却管を装備した三口フラスコを窒素置換し、それに五酸化二りん(和光純薬工業社製,7.13g,50.23mmol)、ヘキサメチルジシロキサン(和光純薬工業社製,17.86g,110.00mmol)、ジクロロメタン(和光純薬工業社製,30.0ml)を加え、撹拌しながら、50℃で30分還流を行った。その後、さらに低沸分を留去しながら、160℃で30分反応を行い、残渣として粘稠な液体を得た。これに2,6-ジイソプロピルアニリン(東京化成工業社製,4.43g,25.00mmol)と安息香酸(東京化成工業社製,1.53g,12.50mmol)を加え、160℃で5時間反応を行った。フラスコを冷却後、内容物を1mol/l水酸化ナトリウム水溶液100mlにゆっくりと加えて中和し、析出した固体部分を回収してエタノールに溶解し、再結晶を行って、淡黄色結晶4.01gを得た。得られた結晶の1H-NMRを測定し、N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジンであることを確認した。
合成例4:N,N’-ビス(2-イソプロピルフェニル)ベンズアミジンの合成
三方コックと冷却管を装備した三口フラスコを窒素置換し、それに五酸化二りん(和光純薬工業社製,8.57g,60.38mmol)、ヘキサメチルジシロキサン(和光純薬工業社製,21.52g,132.53mmol)、ジクロロメタン(和光純薬工業社製,36.0ml)を加え、撹拌しながら、50℃で30分還流を行った。その後、さらに低沸分を留去しながら、160℃で30分反応を行い、残渣として粘稠な液体を得た。これに2-イソプロピルアニリン(東京化成工業社製,4.04g,29.88mmol)と安息香酸(東京化成工業社製,1.82g,14.94mmol)を加え、160℃で5時間反応を行った。フラスコを冷却後、内容物を1mol/l水酸化ナトリウム水溶液100mlにゆっくりと加えて中和し、析出した固体部分を回収してエタノールに溶解し、再結晶を行って、淡黄色結晶4.40gを得た。得られた結晶の1H-NMRを測定し、N,N’-ビス(2-イソプロピルフェニル)ベンズアミジンであることを確認した。
三方コックと冷却管を装備した三口フラスコを窒素置換し、それに五酸化二りん(和光純薬工業社製,8.57g,60.38mmol)、ヘキサメチルジシロキサン(和光純薬工業社製,21.52g,132.53mmol)、ジクロロメタン(和光純薬工業社製,36.0ml)を加え、撹拌しながら、50℃で30分還流を行った。その後、さらに低沸分を留去しながら、160℃で30分反応を行い、残渣として粘稠な液体を得た。これに2-イソプロピルアニリン(東京化成工業社製,4.04g,29.88mmol)と安息香酸(東京化成工業社製,1.82g,14.94mmol)を加え、160℃で5時間反応を行った。フラスコを冷却後、内容物を1mol/l水酸化ナトリウム水溶液100mlにゆっくりと加えて中和し、析出した固体部分を回収してエタノールに溶解し、再結晶を行って、淡黄色結晶4.40gを得た。得られた結晶の1H-NMRを測定し、N,N’-ビス(2-イソプロピルフェニル)ベンズアミジンであることを確認した。
合成例5:N,N’-ビス(2-メチルフェニル)ベンズアミジンの合成
三方コックと冷却管を装備した三口フラスコを窒素置換し、それに五酸化二りん(和光純薬工業社製,7.40g,52.13mmol)、ヘキサメチルジシロキサン(和光純薬工業社製,18.62g,114.67mmol)、ジクロロメタン(和光純薬工業社製,31.0ml)を加え、撹拌しながら、50℃で30分還流を行った。その後、さらに低沸分を留去しながら、160℃で30分反応を行い、残渣として粘稠な液体を得た。これにo-トルイジン(東京化成工業社製,2.68g,25.00mmol)と安息香酸(東京化成工業社製,1.53g,12.50mmol)を加え、160℃で5時間反応を行った。フラスコを冷却後、内容物を1mol/l水酸化ナトリウム水溶液100mlにゆっくりと加えて中和し、析出した固体部分を回収してエタノールに溶解し、再結晶を行って、淡黄色結晶3.15gを得た。得られた結晶の1H-NMRを測定し、N,N’-ビス(2-メチルフェニル)ベンズアミジンであることを確認した。
三方コックと冷却管を装備した三口フラスコを窒素置換し、それに五酸化二りん(和光純薬工業社製,7.40g,52.13mmol)、ヘキサメチルジシロキサン(和光純薬工業社製,18.62g,114.67mmol)、ジクロロメタン(和光純薬工業社製,31.0ml)を加え、撹拌しながら、50℃で30分還流を行った。その後、さらに低沸分を留去しながら、160℃で30分反応を行い、残渣として粘稠な液体を得た。これにo-トルイジン(東京化成工業社製,2.68g,25.00mmol)と安息香酸(東京化成工業社製,1.53g,12.50mmol)を加え、160℃で5時間反応を行った。フラスコを冷却後、内容物を1mol/l水酸化ナトリウム水溶液100mlにゆっくりと加えて中和し、析出した固体部分を回収してエタノールに溶解し、再結晶を行って、淡黄色結晶3.15gを得た。得られた結晶の1H-NMRを測定し、N,N’-ビス(2-メチルフェニル)ベンズアミジンであることを確認した。
合成例6:N,N’-ジシクロヘキシルベンズアミジンの合成
三方コックを装備した二口フラスコを窒素置換し、これに脱水THF(和光純薬工業社製,80ml)とN,N’-ジイソプロピルカルボジイミド(東京化成工業社製,4.80g,38.00mmol)を加え、-20℃に冷却後、撹拌を行いながら、フェニルリチウムの1.9mol/lブチルエーテル溶液(東京化成工業社製,20.0ml,38.00mmol)を30分かけてゆっくりと滴下し、滴下終了後、室温にもどし、2時間反応を行った。その後、塩化アンモニウムの飽和水溶液40mlを加えて反応を停止し、塩化メチレン(和光純薬工業社製,100ml)を加え、同時に内容物を分液漏斗に移し有機層を分離後、硫酸ナトリウムで乾燥、ろ過後、溶媒をエバポレーターで留去して、残渣を回収した。この残渣をエタノールに溶解し、再結晶を行って、淡黄色結晶4.20gを得た。得られた結晶の1H-NMRを測定し、N,N’-ジシクロヘキシルベンズアミジンであることを確認した。
三方コックを装備した二口フラスコを窒素置換し、これに脱水THF(和光純薬工業社製,80ml)とN,N’-ジイソプロピルカルボジイミド(東京化成工業社製,4.80g,38.00mmol)を加え、-20℃に冷却後、撹拌を行いながら、フェニルリチウムの1.9mol/lブチルエーテル溶液(東京化成工業社製,20.0ml,38.00mmol)を30分かけてゆっくりと滴下し、滴下終了後、室温にもどし、2時間反応を行った。その後、塩化アンモニウムの飽和水溶液40mlを加えて反応を停止し、塩化メチレン(和光純薬工業社製,100ml)を加え、同時に内容物を分液漏斗に移し有機層を分離後、硫酸ナトリウムで乾燥、ろ過後、溶媒をエバポレーターで留去して、残渣を回収した。この残渣をエタノールに溶解し、再結晶を行って、淡黄色結晶4.20gを得た。得られた結晶の1H-NMRを測定し、N,N’-ジシクロヘキシルベンズアミジンであることを確認した。
合成例7:N,N’-ジフェニルアセトアミジンの合成
三方コックと冷却管を装備した三口フラスコを窒素置換し、それに五酸化二りん(和光純薬工業社製,7.10g,50.00mmol)、ヘキサメチルジシロキサン(和光純薬工業社製,17.86g,110.00mmol)、ジクロロメタン(和光純薬工業社製,30.0ml)を加え、撹拌しながら、50℃で30分還流を行った。その後、さらに低沸分を留去しながら、160℃で30分反応を行い、残渣として粘稠な液体を得た。これにアニリン(東京化成工業社製,2.33g,25.00mmol)と酢酸(和光純薬工業社製,0.75g,12.50mmol)を加え、160℃で5時間反応を行った。フラスコを冷却後、内容物を1mol/l水酸化ナトリウム水溶液100mlにゆっくりと加えて中和し、析出した固体部分を回収してエタノールに溶解し、再結晶を行って、淡黄色結晶2.25gを得た。得られた結晶の1H-NMRを測定し、N,N’-ジフェニルアセトアミジンであることを確認した。
三方コックと冷却管を装備した三口フラスコを窒素置換し、それに五酸化二りん(和光純薬工業社製,7.10g,50.00mmol)、ヘキサメチルジシロキサン(和光純薬工業社製,17.86g,110.00mmol)、ジクロロメタン(和光純薬工業社製,30.0ml)を加え、撹拌しながら、50℃で30分還流を行った。その後、さらに低沸分を留去しながら、160℃で30分反応を行い、残渣として粘稠な液体を得た。これにアニリン(東京化成工業社製,2.33g,25.00mmol)と酢酸(和光純薬工業社製,0.75g,12.50mmol)を加え、160℃で5時間反応を行った。フラスコを冷却後、内容物を1mol/l水酸化ナトリウム水溶液100mlにゆっくりと加えて中和し、析出した固体部分を回収してエタノールに溶解し、再結晶を行って、淡黄色結晶2.25gを得た。得られた結晶の1H-NMRを測定し、N,N’-ジフェニルアセトアミジンであることを確認した。
実施例1:(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1]の合成

三方コックを装備した二口フラスコを窒素置換し、これに合成例2で調製したN,N’-ジフェニルベンズアミジン(0.7566g,2.778mmol)を仕込み、脱水トルエン(和光純薬工業社製,20ml)を加えて溶解した。これを、氷浴に漬けて0℃に冷却した後、n-ブチルリチウムの1.6mol/lヘキサン溶液(和光純薬工業社製,1.74ml,2.784mmol)を5分かけてゆっくりと滴下し、滴下終了後、室温にもどし、2時間反応を行った。
別途用意した三方コックを装備した二口フラスコを窒素置換し、これにアリルパラジウムクロリドダイマー(和光純薬工業社製,0.5083g,1.389mmol)を仕込み、脱水THF(和光純薬工業社製,20ml)を加えて溶解した。
この溶液を-15℃まで冷却し、これに先に調製したN,N’-ジフェニルベンズアミジン-リチウム錯体のトルエン/ヘキサン混合溶液を10分かけてゆっくりと滴下し、滴下終了後、徐々に室温にもどし、室温で2時間反応を行った。その後、減圧下に溶媒を完全に留去し、あらためて脱水トルエン(和光純薬工業社製,50ml)を加えて撹拌した後、窒素下に遠心分離を行って、不溶な塩(LiCl)を取り除き、上澄みのトルエン溶液を回収した。この溶液より減圧下に溶媒留去し、残渣を回収した。この残渣を脱水ヘキサン/脱水ジクロロメタンの混合溶媒に溶解し、再結晶を行って、黄色結晶1.003gを得た。得られた結晶の1H-NMRを測定し、(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1]であることを確認した。1H-NMRスペクトルを図1に示す。
別途用意した三方コックを装備した二口フラスコを窒素置換し、これにアリルパラジウムクロリドダイマー(和光純薬工業社製,0.5083g,1.389mmol)を仕込み、脱水THF(和光純薬工業社製,20ml)を加えて溶解した。
この溶液を-15℃まで冷却し、これに先に調製したN,N’-ジフェニルベンズアミジン-リチウム錯体のトルエン/ヘキサン混合溶液を10分かけてゆっくりと滴下し、滴下終了後、徐々に室温にもどし、室温で2時間反応を行った。その後、減圧下に溶媒を完全に留去し、あらためて脱水トルエン(和光純薬工業社製,50ml)を加えて撹拌した後、窒素下に遠心分離を行って、不溶な塩(LiCl)を取り除き、上澄みのトルエン溶液を回収した。この溶液より減圧下に溶媒留去し、残渣を回収した。この残渣を脱水ヘキサン/脱水ジクロロメタンの混合溶媒に溶解し、再結晶を行って、黄色結晶1.003gを得た。得られた結晶の1H-NMRを測定し、(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1]であることを確認した。1H-NMRスペクトルを図1に示す。
実施例2:(π-アリル)パラジウム[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-2]の合成

三方コックを装備した二口フラスコを窒素置換し、これに合成例3で調製したN,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジン(0.8998g,2.028mmol)を仕込み、脱水トルエン(和光純薬工業社製,20ml)を加えて溶解した。これを、氷浴に漬けて0℃に冷却した後、n-ブチルリチウムの1.6mol/lヘキサン溶液(和光純薬工業社製,1.27ml,2.032mmol)を5分かけてゆっくりと滴下し、滴下終了後、室温にもどし、2時間反応を行った。
別途用意した三方コックを装備した二口フラスコを窒素置換し、これにアリルパラジウムクロリドダイマー(和光純薬工業社製,0.3710g,1.014mmol)を仕込み、脱水THF(和光純薬工業社製,20ml)を加えて溶解した。
この溶液を-15℃まで冷却し、これに先に調製したN,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジン-リチウム錯体のトルエン/ヘキサン混合溶液を10分かけてゆっくりと滴下し、滴下終了後、徐々に室温にもどし、室温で2時間反応を行った。その後、減圧下に溶媒を完全に留去し、あらためて脱水トルエン(和光純薬工業社製,50ml)を加えて撹拌した後、窒素下に遠心分離を行って、不溶な塩(LiCl)を取り除き、上澄みのトルエン溶液を回収した。この溶液より減圧下に溶媒留去し、残渣を回収した。この残渣を脱水ヘキサン/脱水ジクロロメタンの混合溶媒に溶解し、再結晶を行って、黄色結晶0.483gを得た。得られた結晶の1H-NMRを測定し、(π-アリル)パラジウム[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-2]であることを確認した。1H-NMRスペクトルを図1に示す。
別途用意した三方コックを装備した二口フラスコを窒素置換し、これにアリルパラジウムクロリドダイマー(和光純薬工業社製,0.3710g,1.014mmol)を仕込み、脱水THF(和光純薬工業社製,20ml)を加えて溶解した。
この溶液を-15℃まで冷却し、これに先に調製したN,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジン-リチウム錯体のトルエン/ヘキサン混合溶液を10分かけてゆっくりと滴下し、滴下終了後、徐々に室温にもどし、室温で2時間反応を行った。その後、減圧下に溶媒を完全に留去し、あらためて脱水トルエン(和光純薬工業社製,50ml)を加えて撹拌した後、窒素下に遠心分離を行って、不溶な塩(LiCl)を取り除き、上澄みのトルエン溶液を回収した。この溶液より減圧下に溶媒留去し、残渣を回収した。この残渣を脱水ヘキサン/脱水ジクロロメタンの混合溶媒に溶解し、再結晶を行って、黄色結晶0.483gを得た。得られた結晶の1H-NMRを測定し、(π-アリル)パラジウム[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-2]であることを確認した。1H-NMRスペクトルを図1に示す。
実施例3:(π-アリル)パラジウム[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-3]の合成

三方コックを装備した二口フラスコを窒素置換し、これに合成例4で調製したN,N’-ビス(2-イソプロピルフェニル)ベンズアミジン(0.9668g,2.712mmol)を仕込み、脱水トルエン(和光純薬工業社製,20ml)を加えて溶解した。これを、氷浴に漬けて0℃に冷却した後、n-ブチルリチウムの1.6mol/lヘキサン溶液(和光純薬工業社製,1.70ml,2.720mmol)を5分かけてゆっくりと滴下し、滴下終了後、室温にもどし、2時間反応を行った。
別途用意した三方コックを装備した二口フラスコを窒素置換し、これにアリルパラジウムクロリドダイマー(和光純薬工業社製,0.4961g,1.356mmol)を仕込み、脱水THF(和光純薬工業社製,20ml)を加えて溶解した。
この溶液を-15℃まで冷却し、これに先に調製したN,N’-ビス(2-イソプロピルフェニル)ベンズアミジン-リチウム錯体のトルエン/ヘキサン混合溶液を10分かけてゆっくりと滴下し、滴下終了後、徐々に室温にもどし、室温で2時間反応を行った。その後、減圧下に溶媒を完全に留去し、あらためて脱水トルエン(和光純薬工業社製,50ml)を加えて撹拌した後、窒素下に遠心分離を行って、不溶な塩(LiCl)を取り除き、上澄みのトルエン溶液を回収した。この溶液より減圧下に溶媒留去し、残渣を回収した。この残渣を脱水ヘキサン/脱水ジクロロメタンの混合溶媒に溶解し、再結晶を行って、黄色結晶0.885gを得た。得られた結晶の1H-NMRを測定し、(π-アリル)パラジウム[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-3]であることを確認した。
別途用意した三方コックを装備した二口フラスコを窒素置換し、これにアリルパラジウムクロリドダイマー(和光純薬工業社製,0.4961g,1.356mmol)を仕込み、脱水THF(和光純薬工業社製,20ml)を加えて溶解した。
この溶液を-15℃まで冷却し、これに先に調製したN,N’-ビス(2-イソプロピルフェニル)ベンズアミジン-リチウム錯体のトルエン/ヘキサン混合溶液を10分かけてゆっくりと滴下し、滴下終了後、徐々に室温にもどし、室温で2時間反応を行った。その後、減圧下に溶媒を完全に留去し、あらためて脱水トルエン(和光純薬工業社製,50ml)を加えて撹拌した後、窒素下に遠心分離を行って、不溶な塩(LiCl)を取り除き、上澄みのトルエン溶液を回収した。この溶液より減圧下に溶媒留去し、残渣を回収した。この残渣を脱水ヘキサン/脱水ジクロロメタンの混合溶媒に溶解し、再結晶を行って、黄色結晶0.885gを得た。得られた結晶の1H-NMRを測定し、(π-アリル)パラジウム[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-3]であることを確認した。
実施例4:(π-アリル)パラジウム[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]ダイマー[錯体A-4]の合成

三方コックを装備した二口フラスコを窒素置換し、これに合成例5で調製したN,N’-ビス(2-メチルフェニル)ベンズアミジン(0.8677g,2.888mmol)を仕込み、脱水トルエン(和光純薬工業社製,20ml)を加えて溶解した。これを、氷浴に漬けて0℃に冷却した後、n-ブチルリチウムの1.6mol/lヘキサン溶液(和光純薬工業社製,1.80ml,2.880mmol)を5分かけてゆっくりと滴下し、滴下終了後、室温にもどし、2時間反応を行った。
別途用意した三方コックを装備した二口フラスコを窒素置換し、これにアリルパラジウムクロリドダイマー(和光純薬工業社製,0.5283g,1.444mmol)を仕込み、脱水THF(和光純薬工業社製,20ml)を加えて溶解した。
この溶液を-15℃まで冷却し、これに先に調製したN,N’-ビス(2-メチルフェニル)ベンズアミジン-リチウム錯体のトルエン/ヘキサン混合溶液を10分かけてゆっくりと滴下し、滴下終了後、徐々に室温にもどし、室温で2時間反応を行った。その後、減圧下に溶媒を完全に留去し、あらためて脱水トルエン(和光純薬工業社製,50ml)を加えて撹拌した後、窒素下に遠心分離を行って、不溶な塩(LiCl)を取り除き、上澄みのトルエン溶液を回収した。この溶液より減圧下に溶媒留去し、残渣を回収した。この残渣を脱水ヘキサン/脱水ジクロロメタンの混合溶媒に溶解し、再結晶を行って、黄色結晶0.505gを得た。得られた結晶の1H-NMRを測定し、(π-アリル)パラジウム[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]ダイマー[錯体A-4]であることを確認した。
別途用意した三方コックを装備した二口フラスコを窒素置換し、これにアリルパラジウムクロリドダイマー(和光純薬工業社製,0.5283g,1.444mmol)を仕込み、脱水THF(和光純薬工業社製,20ml)を加えて溶解した。
この溶液を-15℃まで冷却し、これに先に調製したN,N’-ビス(2-メチルフェニル)ベンズアミジン-リチウム錯体のトルエン/ヘキサン混合溶液を10分かけてゆっくりと滴下し、滴下終了後、徐々に室温にもどし、室温で2時間反応を行った。その後、減圧下に溶媒を完全に留去し、あらためて脱水トルエン(和光純薬工業社製,50ml)を加えて撹拌した後、窒素下に遠心分離を行って、不溶な塩(LiCl)を取り除き、上澄みのトルエン溶液を回収した。この溶液より減圧下に溶媒留去し、残渣を回収した。この残渣を脱水ヘキサン/脱水ジクロロメタンの混合溶媒に溶解し、再結晶を行って、黄色結晶0.505gを得た。得られた結晶の1H-NMRを測定し、(π-アリル)パラジウム[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]ダイマー[錯体A-4]であることを確認した。
実施例5:(π-アリル)パラジウム(N,N’-ジシクロヘキシルベンズアミジナート)ダイマー[錯体A-5]の合成

三方コックを装備した二口フラスコを窒素置換し、これに合成例6で調製したN,N’-ジシクロヘキシルベンズアミジン(0.8276g,2.910mmol)を仕込み、脱水トルエン(和光純薬工業社製,20ml)を加えて溶解した。これを、氷浴に漬けて0℃に冷却した後、n-ブチルリチウムの1.6mol/lヘキサン溶液(和光純薬工業社製,1.82ml,2.912mmol)を5分かけてゆっくりと滴下し、滴下終了後、室温にもどし、2時間反応を行った。
別途用意した三方コックを装備した二口フラスコを窒素置換し、これにアリルパラジウムクロリドダイマー(和光純薬工業社製,0.5324g,1.455mmol)を仕込み、脱水THF(和光純薬工業社製,20ml)を加えて溶解した。
この溶液を-15℃まで冷却し、これに先に調製したN,N’-ジシクロヘキシルベンズアミジン-リチウム錯体のトルエン/ヘキサン混合溶液を10分かけてゆっくりと滴下し、滴下終了後、徐々に室温にもどし、室温で2時間反応を行った。その後、減圧下に溶媒を完全に留去し、あらためて脱水トルエン(和光純薬工業社製,50ml)を加えて撹拌した後、窒素下に遠心分離を行って、不溶な塩(LiCl)を取り除き、上澄みのトルエン溶液を回収した。この溶液より減圧下に溶媒留去し、残渣を回収した。この残渣を脱水ヘキサン/脱水ジクロロメタンの混合溶媒に溶解し、再結晶を行って、黄色結晶1.100gを得た。得られた結晶の1H-NMRを測定し、(π-アリル)パラジウム(N,N’-ジシクロヘキシルベンズアミジナート)ダイマー[錯体A-5]であることを確認した。1H-NMRスペクトルを図1に示す。
別途用意した三方コックを装備した二口フラスコを窒素置換し、これにアリルパラジウムクロリドダイマー(和光純薬工業社製,0.5324g,1.455mmol)を仕込み、脱水THF(和光純薬工業社製,20ml)を加えて溶解した。
この溶液を-15℃まで冷却し、これに先に調製したN,N’-ジシクロヘキシルベンズアミジン-リチウム錯体のトルエン/ヘキサン混合溶液を10分かけてゆっくりと滴下し、滴下終了後、徐々に室温にもどし、室温で2時間反応を行った。その後、減圧下に溶媒を完全に留去し、あらためて脱水トルエン(和光純薬工業社製,50ml)を加えて撹拌した後、窒素下に遠心分離を行って、不溶な塩(LiCl)を取り除き、上澄みのトルエン溶液を回収した。この溶液より減圧下に溶媒留去し、残渣を回収した。この残渣を脱水ヘキサン/脱水ジクロロメタンの混合溶媒に溶解し、再結晶を行って、黄色結晶1.100gを得た。得られた結晶の1H-NMRを測定し、(π-アリル)パラジウム(N,N’-ジシクロヘキシルベンズアミジナート)ダイマー[錯体A-5]であることを確認した。1H-NMRスペクトルを図1に示す。
実施例6:(π-アリル)パラジウム(N,N’-ジフェニルアセトアミジナート)ダイマー[錯体A-6]の合成

三方コックを装備した二口フラスコを窒素置換し、これに合成例7で調製したN,N’-ジフェニルアセトアミジン(0.5756g,2.737mmol)を仕込み、脱水トルエン(和光純薬工業社製,20ml)を加えて溶解した。これを、氷浴に漬けて0℃に冷却した後、n-ブチルリチウムの1.6mol/lヘキサン溶液(和光純薬工業社製,1.71ml,2.736mmol)を5分かけてゆっくりと滴下し、滴下終了後、室温にもどし、2時間反応を行った。
別途用意した三方コックを装備した二口フラスコを窒素置換し、これにアリルパラジウムクロリドダイマー(和光純薬工業社製,0.5005g,1.368mmol)を仕込み、脱水THF(和光純薬工業社製,20ml)を加えて溶解した。
この溶液を-15℃まで冷却し、これに先に調製したN,N’-ジフェニルアセトアミジン-リチウム錯体のトルエン/ヘキサン混合溶液を10分かけてゆっくりと滴下し、滴下終了後、徐々に室温にもどし、室温で2時間反応を行った。その後、減圧下に溶媒を完全に留去し、あらためて脱水トルエン(和光純薬工業社製,50ml)を加えて撹拌した後、窒素下に遠心分離を行って、不溶な塩(LiCl)を取り除き、上澄みのトルエン溶液を回収した。この溶液より減圧下に溶媒留去し、残渣を回収した。この残渣を脱水ヘキサン/脱水ジクロロメタンの混合溶媒に溶解し、再結晶を行って、黄色結晶0.802gを得た。得られた結晶の1H-NMRを測定し、(π-アリル)パラジウム(N,N’-ジフェニルアセトアミジナート)ダイマー[錯体A-6]であることを確認した。
別途用意した三方コックを装備した二口フラスコを窒素置換し、これにアリルパラジウムクロリドダイマー(和光純薬工業社製,0.5005g,1.368mmol)を仕込み、脱水THF(和光純薬工業社製,20ml)を加えて溶解した。
この溶液を-15℃まで冷却し、これに先に調製したN,N’-ジフェニルアセトアミジン-リチウム錯体のトルエン/ヘキサン混合溶液を10分かけてゆっくりと滴下し、滴下終了後、徐々に室温にもどし、室温で2時間反応を行った。その後、減圧下に溶媒を完全に留去し、あらためて脱水トルエン(和光純薬工業社製,50ml)を加えて撹拌した後、窒素下に遠心分離を行って、不溶な塩(LiCl)を取り除き、上澄みのトルエン溶液を回収した。この溶液より減圧下に溶媒留去し、残渣を回収した。この残渣を脱水ヘキサン/脱水ジクロロメタンの混合溶媒に溶解し、再結晶を行って、黄色結晶0.802gを得た。得られた結晶の1H-NMRを測定し、(π-アリル)パラジウム(N,N’-ジフェニルアセトアミジナート)ダイマー[錯体A-6]であることを確認した。
実施例7:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
三方コックとメカニカルスターラーを装備した三口フラスコを窒素置換し、それにノルボルネン(東京化成工業社製,4.71g,0.050mol)と合成例1で調製した2-アセトキシメチル-5-ノルボルネン(16.62g,0.100mol)を加え、トルエン75mlで溶解し、さらにN,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート[(C6H5)(CH3)2NH][B(C6F5)4](ストレム社製、8.0mg,0.010mmol)をジクロロメタン1mlで溶解した溶液を加えた後、90℃まで昇温した。そこへ実施例1で合成し、別容器中で調製した(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)とトリイソプロピルホスフィン[P(i-C3H7)3](ストレム社製,1.6mg,0.010mmol)をトルエン3.5mlに溶解した触媒溶液を添加し、90℃で30分重合反応を行った。その後、その反応溶液に別途調製したノルボルネン(東京化成工業社製,4.71g,0.050mol)をトルエン5.4mlで溶解した溶液を加え、さらに90℃で30分重合反応を行った。反応終了後、少量の塩酸を添加したメタノール8mlを反応液に加え、反応を停止した後、トルエンで希釈し、さらに多量のメタノール中に注いでポリマーを析出させ、ろ別洗浄後、減圧下に90℃で5時間乾燥して白色粉末状のポリマー12.70gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1270g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=285,400、分子量分布はMw/Mn=2.43であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は27.0mol%であった。
三方コックとメカニカルスターラーを装備した三口フラスコを窒素置換し、それにノルボルネン(東京化成工業社製,4.71g,0.050mol)と合成例1で調製した2-アセトキシメチル-5-ノルボルネン(16.62g,0.100mol)を加え、トルエン75mlで溶解し、さらにN,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート[(C6H5)(CH3)2NH][B(C6F5)4](ストレム社製、8.0mg,0.010mmol)をジクロロメタン1mlで溶解した溶液を加えた後、90℃まで昇温した。そこへ実施例1で合成し、別容器中で調製した(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)とトリイソプロピルホスフィン[P(i-C3H7)3](ストレム社製,1.6mg,0.010mmol)をトルエン3.5mlに溶解した触媒溶液を添加し、90℃で30分重合反応を行った。その後、その反応溶液に別途調製したノルボルネン(東京化成工業社製,4.71g,0.050mol)をトルエン5.4mlで溶解した溶液を加え、さらに90℃で30分重合反応を行った。反応終了後、少量の塩酸を添加したメタノール8mlを反応液に加え、反応を停止した後、トルエンで希釈し、さらに多量のメタノール中に注いでポリマーを析出させ、ろ別洗浄後、減圧下に90℃で5時間乾燥して白色粉末状のポリマー12.70gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1270g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=285,400、分子量分布はMw/Mn=2.43であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は27.0mol%であった。
実施例8~9:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
重合温度をそれぞれ、実施例8は80℃、実施例9は70℃にしたこと以外は実施例7と同様の方法で重合反応と後処理を行い、白色粉末状のポリマーを得た。結果を表2に示す。実施例9で得られた共重合体の1H-NMRスペクトルを図2、ゲルパーミエイションクロマトグラフィー(GPC)のチャートを図3に示す。
重合温度をそれぞれ、実施例8は80℃、実施例9は70℃にしたこと以外は実施例7と同様の方法で重合反応と後処理を行い、白色粉末状のポリマーを得た。結果を表2に示す。実施例9で得られた共重合体の1H-NMRスペクトルを図2、ゲルパーミエイションクロマトグラフィー(GPC)のチャートを図3に示す。
実施例10:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
三方コックとメカニカルスターラーを装備した三口フラスコを窒素置換し、それにノルボルネン(東京化成工業社製,4.71g,0.050mol)と合成例1で調製した2-アセトキシメチル-5-ノルボルネン(16.62g,0.100mol)を加え、トルエン75mlで溶解し、さらにN,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート[(C6H5)(CH3)2NH][B(C6F5)4](ストレム社製、8.0mg,0.010mmol)をジクロロメタン1mlで溶解した溶液を加えた後、90℃まで昇温した。そこへ実施例1で合成し、別容器中で調製した(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)とトリイソプロピルホスフィン[P(i-C3H7)3](ストレム社製,1.6mg,0.010mmol)をトルエン3.5mlに溶解した触媒溶液を添加し、重合を開始した。この後、別途調製したノルボルネン(東京化成工業社製,4.71g,0.050mol)をトルエン5.4mlで溶解した溶液を30分おきに5回、2-アセトキシメチル-5-ノルボルネン(5.00g,0.030mol)とトルエン20mlを1時間おきに2回、反応溶液に加えながら、90℃でトータル3時間重合反応を行った。反応終了後、少量の塩酸を添加したメタノール8mlを反応液に加え、反応を停止した後、トルエンで希釈し、さらに多量のメタノール中に注いでポリマーを析出させ、ろ別洗浄後、減圧下に90℃で5時間乾燥して白色粉末状のポリマー34.25gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は3425g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=281,100、分子量分布はMw/Mn=2.96であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は22.1mol%であった。
三方コックとメカニカルスターラーを装備した三口フラスコを窒素置換し、それにノルボルネン(東京化成工業社製,4.71g,0.050mol)と合成例1で調製した2-アセトキシメチル-5-ノルボルネン(16.62g,0.100mol)を加え、トルエン75mlで溶解し、さらにN,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート[(C6H5)(CH3)2NH][B(C6F5)4](ストレム社製、8.0mg,0.010mmol)をジクロロメタン1mlで溶解した溶液を加えた後、90℃まで昇温した。そこへ実施例1で合成し、別容器中で調製した(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)とトリイソプロピルホスフィン[P(i-C3H7)3](ストレム社製,1.6mg,0.010mmol)をトルエン3.5mlに溶解した触媒溶液を添加し、重合を開始した。この後、別途調製したノルボルネン(東京化成工業社製,4.71g,0.050mol)をトルエン5.4mlで溶解した溶液を30分おきに5回、2-アセトキシメチル-5-ノルボルネン(5.00g,0.030mol)とトルエン20mlを1時間おきに2回、反応溶液に加えながら、90℃でトータル3時間重合反応を行った。反応終了後、少量の塩酸を添加したメタノール8mlを反応液に加え、反応を停止した後、トルエンで希釈し、さらに多量のメタノール中に注いでポリマーを析出させ、ろ別洗浄後、減圧下に90℃で5時間乾燥して白色粉末状のポリマー34.25gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は3425g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=281,100、分子量分布はMw/Mn=2.96であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は22.1mol%であった。
実施例11~12:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
重合温度をそれぞれ、実施例11は80℃、実施例12は70℃にしたこと以外は実施例10と同様の方法で重合反応と後処理を行い、白色粉末状のポリマーを得た。結果を表2に示す。
重合温度をそれぞれ、実施例11は80℃、実施例12は70℃にしたこと以外は実施例10と同様の方法で重合反応と後処理を行い、白色粉末状のポリマーを得た。結果を表2に示す。
実施例13:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-2](5.9mg,0.005mmol)を用いたこと以外は実施例7と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー7.40gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は740g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=421,300、分子量分布はMw/Mn=2.07であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は22.3mol%であった。
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-2](5.9mg,0.005mmol)を用いたこと以外は実施例7と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー7.40gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は740g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=421,300、分子量分布はMw/Mn=2.07であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は22.3mol%であった。
実施例14~15:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
重合温度をそれぞれ、実施例14は80℃、実施例15は70℃にしたこと以外は実施例13と同様の方法で重合反応と後処理を行い、白色粉末状のポリマーを得た。結果を表2に示す。
重合温度をそれぞれ、実施例14は80℃、実施例15は70℃にしたこと以外は実施例13と同様の方法で重合反応と後処理を行い、白色粉末状のポリマーを得た。結果を表2に示す。
実施例16:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-2](5.9mg,0.005mmol)を用い、重合温度を70℃にしたこと以外は実施例10と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー34.95gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は3495g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=928,100、分子量分布はMw/Mn=2.41であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は19.1mol%であった。
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-2](5.9mg,0.005mmol)を用い、重合温度を70℃にしたこと以外は実施例10と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー34.95gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は3495g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=928,100、分子量分布はMw/Mn=2.41であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は19.1mol%であった。
実施例17:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
N,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート[(C6H5)(CH3)2NH][B(C6F5)4](ストレム社製)の使用量を12.0mg(0.015mmol)にし、トリイソプロピルホスフィン[P(i-C3H7)3](ストレム社製)の使用量を2.4mg(0.015mmol)にしたこと以外は実施例16と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー43.90gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は4390g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=612,800、分子量分布はMw/Mn=2.63であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は20.2mol%であった。
N,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート[(C6H5)(CH3)2NH][B(C6F5)4](ストレム社製)の使用量を12.0mg(0.015mmol)にし、トリイソプロピルホスフィン[P(i-C3H7)3](ストレム社製)の使用量を2.4mg(0.015mmol)にしたこと以外は実施例16と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー43.90gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は4390g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=612,800、分子量分布はMw/Mn=2.63であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は20.2mol%であった。
実施例18:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-3](5.0mg,0.005mmol)を用いたこと以外は実施例7と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー18.31gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1831g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=224,800、分子量分布はMw/Mn=3.51であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は34.1mol%であった。
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-3](5.0mg,0.005mmol)を用いたこと以外は実施例7と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー18.31gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1831g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=224,800、分子量分布はMw/Mn=3.51であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は34.1mol%であった。
実施例19:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
重合温度を80℃にしたこと以外は実施例18と同様の方法で重合反応と後処理を行い、白色粉末状のポリマーを得た。結果を表2に示す。
重合温度を80℃にしたこと以外は実施例18と同様の方法で重合反応と後処理を行い、白色粉末状のポリマーを得た。結果を表2に示す。
実施例20:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
三方コックとメカニカルスターラーを装備した三口フラスコを窒素置換し、それにノルボルネン(東京化成工業社製,7.07g,0.075mol)と合成例1で調製した2-アセトキシメチル-5-ノルボルネン(24.93g,0.150mol)を加え、トルエン110mlで溶解し、さらにN,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート[(C6H5)(CH3)2NH][B(C6F5)4](ストレム社製、8.0mg,0.010mmol)をジクロロメタン1mlで溶解した溶液を加えた後、90℃まで昇温した。そこへ実施例2で合成し、別容器中で調製した(π-アリル)パラジウム[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-3](5.0mg,0.005mmol)とトリイソプロピルホスフィン[P(i-C3H7)3](ストレム社製,1.6mg,0.010mmol)をトルエン3.5mlに溶解した触媒溶液を添加し、90℃で30分重合反応を行った。その後、その反応溶液に別途調製したノルボルネン(東京化成工業社製,7.07g,0.075mol)をトルエン8.1mlで溶解した溶液を加え、さらに90℃で30分重合反応を行った。反応終了後、少量の塩酸を添加したメタノール8mlを反応液に加え、反応を停止した後、トルエンで希釈し、さらに多量のメタノール中に注いでポリマーを析出させ、ろ別洗浄後、減圧下に90℃で5時間乾燥して白色粉末状のポリマー21.70gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は2170g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=462,500、分子量分布はMw/Mn=2.70であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は29.0mol%であった。
三方コックとメカニカルスターラーを装備した三口フラスコを窒素置換し、それにノルボルネン(東京化成工業社製,7.07g,0.075mol)と合成例1で調製した2-アセトキシメチル-5-ノルボルネン(24.93g,0.150mol)を加え、トルエン110mlで溶解し、さらにN,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート[(C6H5)(CH3)2NH][B(C6F5)4](ストレム社製、8.0mg,0.010mmol)をジクロロメタン1mlで溶解した溶液を加えた後、90℃まで昇温した。そこへ実施例2で合成し、別容器中で調製した(π-アリル)パラジウム[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-3](5.0mg,0.005mmol)とトリイソプロピルホスフィン[P(i-C3H7)3](ストレム社製,1.6mg,0.010mmol)をトルエン3.5mlに溶解した触媒溶液を添加し、90℃で30分重合反応を行った。その後、その反応溶液に別途調製したノルボルネン(東京化成工業社製,7.07g,0.075mol)をトルエン8.1mlで溶解した溶液を加え、さらに90℃で30分重合反応を行った。反応終了後、少量の塩酸を添加したメタノール8mlを反応液に加え、反応を停止した後、トルエンで希釈し、さらに多量のメタノール中に注いでポリマーを析出させ、ろ別洗浄後、減圧下に90℃で5時間乾燥して白色粉末状のポリマー21.70gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は2170g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=462,500、分子量分布はMw/Mn=2.70であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は29.0mol%であった。
実施例21:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
重合温度を80℃にしたこと以外は実施例20と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー19.60gを得た。結果を表2に示す。
重合温度を80℃にしたこと以外は実施例20と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー19.60gを得た。結果を表2に示す。
実施例22:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]ダイマー[錯体A-4](4.5mg,0.005mmol)を用いたこと以外は実施例7と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー16.74gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1674g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=365,300、分子量分布はMw/Mn=2.61であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は32.3mol%であった。
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]ダイマー[錯体A-4](4.5mg,0.005mmol)を用いたこと以外は実施例7と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー16.74gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1674g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=365,300、分子量分布はMw/Mn=2.61であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は32.3mol%であった。
実施例23~24:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
重合温度をそれぞれ、実施例23は80℃、実施例24は70℃にしたこと以外は実施例22と同様の方法で重合反応と後処理を行い、白色粉末状のポリマーを得た。結果を表2に示す。
重合温度をそれぞれ、実施例23は80℃、実施例24は70℃にしたこと以外は実施例22と同様の方法で重合反応と後処理を行い、白色粉末状のポリマーを得た。結果を表2に示す。
実施例25:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
(π-アリル)パラジウム[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-3](5.0mg,0.005mmol)の代わりに(π-アリル)パラジウム[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]ダイマー[錯体A-4](4.5mg,0.005mmol)を用い、重合温度を80℃にしたこと以外は実施例20と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー19.95gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1995g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=806,400、分子量分布はMw/Mn=2.48であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は27.2mol%であった。
(π-アリル)パラジウム[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]ダイマー[錯体A-3](5.0mg,0.005mmol)の代わりに(π-アリル)パラジウム[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]ダイマー[錯体A-4](4.5mg,0.005mmol)を用い、重合温度を80℃にしたこと以外は実施例20と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー19.95gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1995g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=806,400、分子量分布はMw/Mn=2.48であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は27.2mol%であった。
実施例26:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]ダイマー[錯体A-4](4.5mg,0.005mmol)を用い、重合温度を70℃にしたこと以外は実施例10と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー28.73gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は2873g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=761,100、分子量分布はMw/Mn=2.34であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は18.8mol%であった。
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]ダイマー[錯体A-4](4.5mg,0.005mmol)を用い、重合温度を70℃にしたこと以外は実施例10と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー28.73gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は2873g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=761,100、分子量分布はMw/Mn=2.34であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は18.8mol%であった。
実施例27:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
N,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート[(C6H5)(CH3)2NH][B(C6F5)4](ストレム社製)の使用量を12.0mg(0.015mmol)にし、トリイソプロピルホスフィン[P(i-C3H7)3](ストレム社製)の使用量を2.4mg(0.015mmol)にしたこと以外は実施例26と同様の条件で重合反応と後処理を行い、白色粉末状のポリマー40.70gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は4070g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=573,200、分子量分布はMw/Mn=2.86であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は20.6mol%であった。
N,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート[(C6H5)(CH3)2NH][B(C6F5)4](ストレム社製)の使用量を12.0mg(0.015mmol)にし、トリイソプロピルホスフィン[P(i-C3H7)3](ストレム社製)の使用量を2.4mg(0.015mmol)にしたこと以外は実施例26と同様の条件で重合反応と後処理を行い、白色粉末状のポリマー40.70gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は4070g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=573,200、分子量分布はMw/Mn=2.86であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は20.6mol%であった。
実施例28:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム(N,N’-ジシクロヘキシルベンズアミジナート)ダイマー[錯体A-5](4.3mg,0.005mmol)を用いたこと以外は実施例7と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー13.25gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1325g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=416,000、分子量分布はMw/Mn=2.19であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は29.8mol%であった。
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム(N,N’-ジシクロヘキシルベンズアミジナート)ダイマー[錯体A-5](4.3mg,0.005mmol)を用いたこと以外は実施例7と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー13.25gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1325g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=416,000、分子量分布はMw/Mn=2.19であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は29.8mol%であった。
実施例29~30:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
重合温度をそれぞれ、実施例29は80℃、実施例30は70℃にしたこと以外は実施例28と同様の方法で重合反応と後処理を行い、白色粉末状のポリマーを得た。結果を表2に示す。
重合温度をそれぞれ、実施例29は80℃、実施例30は70℃にしたこと以外は実施例28と同様の方法で重合反応と後処理を行い、白色粉末状のポリマーを得た。結果を表2に示す。
実施例31:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム(N,N’-ジフェニルアセトアミジナート)ダイマー[錯体A-6](3.6mg,0.005mmol)を用いたこと以外は実施例7と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー18.10gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1810g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=579,200、分子量分布はMw/Mn=2.24であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は28.7mol%であった。
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりに(π-アリル)パラジウム(N,N’-ジフェニルアセトアミジナート)ダイマー[錯体A-6](3.6mg,0.005mmol)を用いたこと以外は実施例7と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー18.10gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1810g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=579,200、分子量分布はMw/Mn=2.24であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は28.7mol%であった。
実施例32:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
重合温度を80℃にしたこと以外は実施例31と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー10.01gを得た。結果を表2に示す。
重合温度を80℃にしたこと以外は実施例31と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー10.01gを得た。結果を表2に示す。
比較例1:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合(特許文献4の方法による重合)
三方コックとメカニカルスターラーを装備した三口フラスコを窒素置換し、それにノルボルネン(東京化成工業社製,4.71g,0.050mol)と合成例1で調製した2-アセトキシメチル-5-ノルボルネン(16.62g,0.100mol)を加え、トルエン75mlで溶解し、さらにN,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート[(C6H5)(CH3)2NH][B(C6F5)4](ストレム社製、8.0mg,0.010mmol)をジクロロメタン1mlで溶解した溶液を加えた後、90℃まで昇温した。そこへアリルパラジウムクロライドダイマー[[(C3H5)PdCl]2];金属錯体CA-1(和光純薬工業社製,1.8mg,0.005mmol)とトリイソプロピルホスフィン[P(i-C3H7)3](ストレム社製,1.6mg,0.010mmol)をトルエン3.5mlに溶解した触媒溶液を添加し、90℃で30分重合反応を行った。その後、その反応溶液に別途調製したノルボルネン(東京化成工業社製,4.71g,0.050mol)をトルエン5.4mlで溶解した溶液を加え、さらに90℃で30分重合反応を行った。反応終了後、少量の塩酸を添加したメタノール8mlを反応液に加え、反応を停止した後、トルエンで希釈し、さらに多量のメタノール中に注いでポリマーを析出させ、ろ別洗浄後、減圧下に90℃で5時間乾燥して白色粉末状のポリマー10.53gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1053g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=146,300、分子量分布はMw/Mn=2.20であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は24.0mol%であった。
三方コックとメカニカルスターラーを装備した三口フラスコを窒素置換し、それにノルボルネン(東京化成工業社製,4.71g,0.050mol)と合成例1で調製した2-アセトキシメチル-5-ノルボルネン(16.62g,0.100mol)を加え、トルエン75mlで溶解し、さらにN,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート[(C6H5)(CH3)2NH][B(C6F5)4](ストレム社製、8.0mg,0.010mmol)をジクロロメタン1mlで溶解した溶液を加えた後、90℃まで昇温した。そこへアリルパラジウムクロライドダイマー[[(C3H5)PdCl]2];金属錯体CA-1(和光純薬工業社製,1.8mg,0.005mmol)とトリイソプロピルホスフィン[P(i-C3H7)3](ストレム社製,1.6mg,0.010mmol)をトルエン3.5mlに溶解した触媒溶液を添加し、90℃で30分重合反応を行った。その後、その反応溶液に別途調製したノルボルネン(東京化成工業社製,4.71g,0.050mol)をトルエン5.4mlで溶解した溶液を加え、さらに90℃で30分重合反応を行った。反応終了後、少量の塩酸を添加したメタノール8mlを反応液に加え、反応を停止した後、トルエンで希釈し、さらに多量のメタノール中に注いでポリマーを析出させ、ろ別洗浄後、減圧下に90℃で5時間乾燥して白色粉末状のポリマー10.53gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は1053g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=146,300、分子量分布はMw/Mn=2.20であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は24.0mol%であった。
比較例2:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合(特許文献4の方法による重合)
重合温度を70℃にしたこと以外は比較例1と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー2.56gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は256g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=214,900、分子量分布はMw/Mn=1.93であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は14.1mol%であった。得られた共重合体のゲルパーミエイションクロマトグラフィー(GPC)のチャートを図3に示す。
重合温度を70℃にしたこと以外は比較例1と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー2.56gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は256g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=214,900、分子量分布はMw/Mn=1.93であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は14.1mol%であった。得られた共重合体のゲルパーミエイションクロマトグラフィー(GPC)のチャートを図3に示す。
比較例3:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合(特許文献4の方法による重合)
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりにアリルパラジウムクロライドダイマー[[(C3H5)PdCl]2];金属錯体CA-1(和光純薬工業社製,1.8mg,0.005mmol)を用いたこと以外は実施例10と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー40.10gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は4010g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=121,100、分子量分布はMw/Mn=2.55であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は23.6mol%であった。
(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー[錯体A-1](4.2mg,0.005mmol)の代わりにアリルパラジウムクロライドダイマー[[(C3H5)PdCl]2];金属錯体CA-1(和光純薬工業社製,1.8mg,0.005mmol)を用いたこと以外は実施例10と同様の方法で重合反応と後処理を行い、白色粉末状のポリマー40.10gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は4010g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=121,100、分子量分布はMw/Mn=2.55であった。また、1H-NMRの積分値から算出したポリマー中の2-アセトキシメチル-5-ノルボルネンモノマーユニットの組成は23.6mol%であった。
比較例4:ノルボルネンと2-アセトキシメチル-5-ノルボルネンの付加共重合
三方コックとメカニカルスターラーを装備した三口フラスコを窒素置換し、それにノルボルネン(東京化成工業社製,11.80g,0.125mol)、合成例1で調製した5-アセトキシメチル-2-ノルボルネン(41.50g,0.250mol)及びトリチルテトラキス(ペンタフルオロフェニル)ボレート[Ph3C][B(C6F5)4];助触媒B-2(東ソー・ファインケム社製,93mg,0.100mmol)を加え、トルエン60mlで溶解した。そこへ別途調製したシクロペンタジエニル(π-アリル)パラジウム[(C5H5)Pd(C3H5)];金属錯体CA-2(21mg,0.100mmol)とトリシクロヘキシルホスフィン[P(C6H11)3];ホスフィン系配位子C-2(ストレム社製,28mg,0.100mmol)をトルエン15mlに溶解した触媒溶液を添加し、室温で1.5時間重合反応を行った。その後、その反応溶液に別途調製したノルボルネン(東京化成工業社製,11.80g,0.125mol)をトルエン60mlで溶解した溶液を加え、さらに3時間重合反応を行った。反応終了後、反応液を多量のメタノール中に注いでポリマーを析出させ、ろ別洗浄後、減圧下に90℃で5時間乾燥して白色粉末状のポリマー42.5gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は425g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=435,000、分子量分布はMw/Mn=1.98であった。また、1H-NMRの積分値から算出したポリマー中の5-アセトキシメチル-2-ノルボルネンモノマーユニットの組成は26.3mol%であった。
三方コックとメカニカルスターラーを装備した三口フラスコを窒素置換し、それにノルボルネン(東京化成工業社製,11.80g,0.125mol)、合成例1で調製した5-アセトキシメチル-2-ノルボルネン(41.50g,0.250mol)及びトリチルテトラキス(ペンタフルオロフェニル)ボレート[Ph3C][B(C6F5)4];助触媒B-2(東ソー・ファインケム社製,93mg,0.100mmol)を加え、トルエン60mlで溶解した。そこへ別途調製したシクロペンタジエニル(π-アリル)パラジウム[(C5H5)Pd(C3H5)];金属錯体CA-2(21mg,0.100mmol)とトリシクロヘキシルホスフィン[P(C6H11)3];ホスフィン系配位子C-2(ストレム社製,28mg,0.100mmol)をトルエン15mlに溶解した触媒溶液を添加し、室温で1.5時間重合反応を行った。その後、その反応溶液に別途調製したノルボルネン(東京化成工業社製,11.80g,0.125mol)をトルエン60mlで溶解した溶液を加え、さらに3時間重合反応を行った。反応終了後、反応液を多量のメタノール中に注いでポリマーを析出させ、ろ別洗浄後、減圧下に90℃で5時間乾燥して白色粉末状のポリマー42.5gを得た。ポリマー収量と仕込み触媒量より算出される触媒活性は425g-ポリマー/mmol-Pdであった。
得られたポリマーはTHFやクロロホルム等の一般溶剤に容易に溶解し、数平均分子量はMn=435,000、分子量分布はMw/Mn=1.98であった。また、1H-NMRの積分値から算出したポリマー中の5-アセトキシメチル-2-ノルボルネンモノマーユニットの組成は26.3mol%であった。
実施例7~32及び比較例1~4について、用いた触媒(主触媒、助触媒、配位子)、仕込みノルボルネン(NB)と2-アセトキシメチル-5-ノルボルネン(ANB)のモル比(NB/ANB)、付加共重合で得られた共重合体の数平均分子量(Mn)、重量平均分子量(Mw)及び分子量分布(Mw/Mn)、共重合体中のANB含有率(モル%)をまとめて表1及び2に示す。表1及び2中の各記号の意味は以下の通りである。
金属錯体(A):
A-1:(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー、
A-2:(π-アリル)パラジウム[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]ダイマー、
A-3:(π-アリル)パラジウム[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]ダイマー、
A-4:(π-アリル)パラジウム[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]ダイマー、
A-5:(π-アリル)パラジウム(N,N’-ジシクロヘキシルベンズアミジナート)ダイマー、
A-6:(π-アリル)パラジウム(N,N’-ジフェニルアセトアミジナート)ダイマー、
CA-1:アリルパラジウムクロリドダイマー、
CA-2:シクロペンタジエニル(π-アリル)パラジウム。
助触媒(B):
B-1:N,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート [(C6H5)(CH3)2NH][B(C6F5)4]、
B-2:トリチルテトラキス(ペンタフルオロフェニル)ボレート [Ph3C][B(C6F5)4]。
ホスフィン系配位子(C):
C-1:トリイソプロピルホスフィン P(i-C3H7)3、
C-2:トリシクロヘキシルホスフィン P(C6H11)3。
モノマー:
NB:ノルボルネン、
ANB:2-アセトキシメチル-5-ノルボルネン。
なお、実施例7~32、比較例1~4で得られたポリマーはいずれもTHFやクロロホルム等の一般溶剤に容易に溶解した。
金属錯体(A):
A-1:(π-アリル)パラジウム(N,N’-ジフェニルベンズアミジナート)ダイマー、
A-2:(π-アリル)パラジウム[N,N’-ビス(2,6-ジイソプロピルフェニル)ベンズアミジナート]ダイマー、
A-3:(π-アリル)パラジウム[N,N’-ビス(2-イソプロピルフェニル)ベンズアミジナート]ダイマー、
A-4:(π-アリル)パラジウム[N,N’-ビス(2-メチルフェニル)ベンズアミジナート]ダイマー、
A-5:(π-アリル)パラジウム(N,N’-ジシクロヘキシルベンズアミジナート)ダイマー、
A-6:(π-アリル)パラジウム(N,N’-ジフェニルアセトアミジナート)ダイマー、
CA-1:アリルパラジウムクロリドダイマー、
CA-2:シクロペンタジエニル(π-アリル)パラジウム。
助触媒(B):
B-1:N,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレート [(C6H5)(CH3)2NH][B(C6F5)4]、
B-2:トリチルテトラキス(ペンタフルオロフェニル)ボレート [Ph3C][B(C6F5)4]。
ホスフィン系配位子(C):
C-1:トリイソプロピルホスフィン P(i-C3H7)3、
C-2:トリシクロヘキシルホスフィン P(C6H11)3。
モノマー:
NB:ノルボルネン、
ANB:2-アセトキシメチル-5-ノルボルネン。
なお、実施例7~32、比較例1~4で得られたポリマーはいずれもTHFやクロロホルム等の一般溶剤に容易に溶解した。


既知の方法では分子量Mnが200,000を超える共重合体を工業的に実用化が見込める活性で製造できなかった(比較例1~4)。本発明の製造方法によれば、機械的性質に優れた分子量Mnが200,000を超えるノルボルネン系共重合体が工業的に実用化され得る触媒活性で得られた(実施例7~32)。
本発明の製造方法により得られるノルボルネン系共重合体は優れた透明性、耐熱性、低吸水性、電気絶縁特性等を有することにより、レンズや偏光フィルム等の光学用成形品、フィルム、キャリアテープ、フィルムコンデンサー、フレキシブルプリント基板等の電気絶縁材料、プレススルーパッケージ、輸液バック、薬液バイアル等の医療用容器、ラップやトレイ等の食品包装成形品、電気器具等のケーシング、インナーパネル等の自動車内装部品、カーポートやグレージング等の建材等に利用可能である。
Claims (12)
- 一般式(1)

- 一般式(1)中のR1がメチル基または置換基を有していてもよいフェニル基であり、R2がシクロヘキシル基または置換基を有していてもよいフェニル基であり、R3、R4及びR5がいずれも水素原子である請求項1に記載のノルボルネン系モノマーの重合用触媒。
- 一般式(1)中のR1がメチル基またはフェニル基であり、R2がシクロヘキシル基、フェニル基、2-メチルフェニル基、2-イソプロピルフェニル基、または2,6-ジイソプロピルフェニル基である請求項2に記載のノルボルネン系モノマーの重合用触媒。
- 遷移金属錯体(A)と反応してカチオン性遷移金属化合物を生成できるイオン性化合物である助触媒(B)及びホスフィン系配位子(C)を含有する請求項1~3のいずれかに記載のノルボルネン系モノマーの重合用触媒。
- 助触媒(B)が、トリチルテトラキス(ペンタフルオロフェニル)ボレートまたはN,N-ジメチルアニリニウムテトラキス(ペンタフルオロフェニル)ボレートである請求項4に記載のノルボルネン系モノマーの重合用触媒。
- ホスフィン系配位子(C)がトリシクロヘキシルホスフィン、トリ-t-ブチルホスフィン、またはトリイソプロピルホスフィンである請求項4に記載のノルボルネン系モノマーの重合用触媒。
- 請求項1~6のいずれかに記載の重合用触媒の存在下に、ノルボルネン系モノマーを単独重合または共重合することを特徴とするノルボルネン系(共)重合体の製造方法。
- 請求項1~6のいずれかに記載の重合用触媒の存在下に、ノルボルネン系モノマーと他のビニルモノマーを共重合することを特徴とするノルボルネン系共重合体の製造方法。
- 請求項1~6のいずれかに記載の重合用触媒の存在下に、一般式(2)


で示されるモノマーユニットに対応するノルボルネン系モノマーを重合することを特徴とする、一般式(2)及び一般式(3)で示されるモノマーユニットを含むノルボルネン系共重合体の製造方法。 - 一般式(2)及び一般式(3)で示されるモノマーユニットのみからなる請求項9に記載のノルボルネン系共重合体の製造方法。
- 一般式(2)中のR6がメチル基である請求項9または10に記載のノルボルネン系共重合体の製造方法。
- 一般式(2)中のR7、及び一般式(3)中のR8及びR9が水素原子である請求項9~11のいずれかに記載のノルボルネン系共重合体の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012539743A JPWO2012053535A1 (ja) | 2010-10-20 | 2011-10-19 | ノルボルネン系モノマー重合用触媒及びノルボルネン系重合体の製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-235078 | 2010-10-20 | ||
| JP2010235078 | 2010-10-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012053535A1 true WO2012053535A1 (ja) | 2012-04-26 |
Family
ID=45975246
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/074013 WO2012053535A1 (ja) | 2010-10-20 | 2011-10-19 | ノルボルネン系モノマー重合用触媒及びノルボルネン系重合体の製造方法 |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JPWO2012053535A1 (ja) |
| TW (1) | TW201229073A (ja) |
| WO (1) | WO2012053535A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111362835A (zh) * | 2020-04-20 | 2020-07-03 | 苏州科晟通新材料科技有限公司 | 一种具有阻燃和抗水解功能的碳化二亚胺化合物及其制备方法 |
| CN112679642A (zh) * | 2020-12-24 | 2021-04-20 | 广东新华粤石化集团股份公司 | 一种环烯烃共聚物及其制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006328034A (ja) * | 2005-05-30 | 2006-12-07 | Nippon Zeon Co Ltd | 遷移金属錯体、環状オレフィン重合用触媒、および環状オレフィン重合体の製造方法 |
| JP2008031304A (ja) * | 2006-07-28 | 2008-02-14 | Fujifilm Corp | ノルボルネン系共重合体を用いたフィルム、偏光板、液晶表示装置、ノルボルネン系共重合体 |
| JP2008231361A (ja) * | 2007-03-23 | 2008-10-02 | Jsr Corp | 環状オレフィン系付加共重合体、その製造方法およびその用途 |
-
2011
- 2011-10-19 JP JP2012539743A patent/JPWO2012053535A1/ja active Pending
- 2011-10-19 WO PCT/JP2011/074013 patent/WO2012053535A1/ja active Application Filing
- 2011-10-19 TW TW100138166A patent/TW201229073A/zh unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006328034A (ja) * | 2005-05-30 | 2006-12-07 | Nippon Zeon Co Ltd | 遷移金属錯体、環状オレフィン重合用触媒、および環状オレフィン重合体の製造方法 |
| JP2008031304A (ja) * | 2006-07-28 | 2008-02-14 | Fujifilm Corp | ノルボルネン系共重合体を用いたフィルム、偏光板、液晶表示装置、ノルボルネン系共重合体 |
| JP2008231361A (ja) * | 2007-03-23 | 2008-10-02 | Jsr Corp | 環状オレフィン系付加共重合体、その製造方法およびその用途 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111362835A (zh) * | 2020-04-20 | 2020-07-03 | 苏州科晟通新材料科技有限公司 | 一种具有阻燃和抗水解功能的碳化二亚胺化合物及其制备方法 |
| CN111362835B (zh) * | 2020-04-20 | 2022-08-30 | 苏州科晟通新材料科技有限公司 | 一种具有阻燃和抗水解功能的碳化二亚胺化合物及其制备方法 |
| CN112679642A (zh) * | 2020-12-24 | 2021-04-20 | 广东新华粤石化集团股份公司 | 一种环烯烃共聚物及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW201229073A (en) | 2012-07-16 |
| JPWO2012053535A1 (ja) | 2014-02-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5411407B2 (ja) | ノルボルネン系付加(共)重合体の製造方法 | |
| US8420755B2 (en) | Compounds, catalyst composition comprising the same, and method for preparing of cycloolefin-based polymer using the same | |
| KR101157275B1 (ko) | 환상 올레핀계 부가 중합체의 제조 방법 | |
| JP2010174099A (ja) | 環状オレフィン付加重合体及びその製造方法 | |
| JP5738097B2 (ja) | ノルボルネン系重合体の製造方法 | |
| JP5803035B2 (ja) | ノルボルネン系共重合体及びその製造方法 | |
| JP5834017B2 (ja) | ノルボルネン系モノマー重合用触媒及びノルボルネン系重合体の製造方法 | |
| US8404792B2 (en) | Cyclobutene polymers and methods of making the same | |
| KR101494222B1 (ko) | 촉매 조성물, 촉매 조성물을 사용한 노르보르넨계 공중합체의 제조 방법, 및 노르보르넨계 공중합체, 또한 그 공중합체를 사용한 내열성 필름 | |
| WO2012053535A1 (ja) | ノルボルネン系モノマー重合用触媒及びノルボルネン系重合体の製造方法 | |
| JP5864966B2 (ja) | ノルボルネン系重合体の製造方法 | |
| JP2010254880A (ja) | 架橋重合体 | |
| JP2012153777A (ja) | ノルボルネン系モノマー重合用触媒及びノルボルネン系共重合体の製造方法 | |
| JP2006016606A (ja) | 環状オレフィンの付加重合用触媒、およびこれを用いた環状オレフィン付加重合体の製造方法 | |
| JP6099907B2 (ja) | 極性基含有ノルボルネン系共重合体の製造方法 | |
| JP6467270B2 (ja) | ノルボルネン系付加共重合体の製造方法 | |
| JP2014040532A (ja) | 環状オレフィン付加重合体及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11834374 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012539743 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11834374 Country of ref document: EP Kind code of ref document: A1 |