WO2012011591A1 - 縮合複素環化合物およびその用途 - Google Patents
縮合複素環化合物およびその用途 Download PDFInfo
- Publication number
- WO2012011591A1 WO2012011591A1 PCT/JP2011/066766 JP2011066766W WO2012011591A1 WO 2012011591 A1 WO2012011591 A1 WO 2012011591A1 JP 2011066766 W JP2011066766 W JP 2011066766W WO 2012011591 A1 WO2012011591 A1 WO 2012011591A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- group
- reaction
- optionally substituted
- alkyl
- Prior art date
Links
- 0 *c1c(C(N(C(*=C)=N2)[Al])=*)c2nc(*)n1 Chemical compound *c1c(C(N(C(*=C)=N2)[Al])=*)c2nc(*)n1 0.000 description 2
- LYKFDYITYYFUCR-UHFFFAOYSA-N CCC(N1c(cc2)ccc2[AlH2])=Nc2ncccc2C1=O Chemical compound CCC(N1c(cc2)ccc2[AlH2])=Nc2ncccc2C1=O LYKFDYITYYFUCR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/04—Drugs for disorders of the respiratory system for throat disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D475/00—Heterocyclic compounds containing pteridine ring systems
- C07D475/02—Heterocyclic compounds containing pteridine ring systems with an oxygen atom directly attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the present invention relates to a novel condensed heterocyclic compound having excellent properties as a pharmaceutical and its use.
- the present invention has a delta-5-desaturase inhibitory action, exhibits various pharmacological actions based on suppressing the production of eicosanoids, has excellent physical properties such as good crystallinity and stability, Specific structures useful as prophylactic and therapeutic agents for eicosanoid-related diseases such as atherosclerosis, atherothrombosis, diabetes, obesity, asthma, fever, pain, cancer, rheumatism, osteoarthritis, and atopic dermatitis
- the present invention relates to a condensed heterocyclic compound having a salt thereof, a salt thereof, a prodrug thereof, a use thereof, and the like.
- Eicosanoids such as prostaglandins, leukotrienes and thromboxanes are considered to play an important role in various diseases.
- inflammatory diseases such as arteriosclerosis, diabetes, obesity, asthma, rheumatism, osteoarthritis, and inflammatory pain
- the production pathway of inflammatory eicosanoids is increased, and is involved in the onset and exacerbation of the disease. It is believed that.
- drugs such as cyclooxygenase inhibitors and thromboxane A2 receptor antagonists that suppress the production of prostanoids have been clinically applied.
- the need for prophylactic / therapeutic drugs for inflammatory diseases is still high, and there is a strong demand for the development of powerful and few side effects.
- Non-Patent Document 1 Bioorganic & Medicinal Chemistry Letters (2009), 19 (17), 5200-5204 (Non-Patent Document 1)
- CXCR3 antagonists for therapeutic use in diseases such as inflammatory diseases, multiple sclerosis, psoriasis, rheumatism, allograft rejection, inflammatory bowel disease and the like.
- Non-patent Document 2 reports that the progress of heterocycle synthesis is important for the foundation of many pharmaceutically active substances.
- Patent Document 1 includes the following formula
- Z is a member selected from the group consisting of a bond, —N ⁇ , —O—, —S—, —C (R 7 ) ⁇ and N (R 14 ) —.
- L is a bond, C (O)-(C 1 -C 8 ) alkylene, (C 1 -C 8 ) alkylene
- Q is a member selected from the group consisting of (C 2 -C 8 ) heteroalkylene
- Q is (C 1 -C 8 ) alkylene, —C (O) —, —OC (O) —, —N ( R 8) C (O) - , - CH 2 CO -, - CH 2 SO-, and CH 2 SO 2 - member selected from the group consisting of There; or, optionally, L and Q may form a 5 or 6-membered heterocyclic group having 1-3 heteroatoms linked together.
- R 1 and R 2 are independently selected from the group consisting of H, (C 1 -C 8 ) alkyl, (C 2 -C 8 ) heteroalkyl, aryl and heteroaryl, or optionally linked. Forming a 3- to 8-membered ring having 0 to 2 heteroatoms as ring vertices; optionally, R 2 is linked with L to form a 5- or 6-membered heterocyclic group having 1 to 4 heteroatoms Can be formed.
- R 3 is absent or is hydroxy, (C 1 -C 8 ) alkoxy, amino, (C 1 -C 8 ) alkylamino, di (C 1 -C 8 ) alkylamino, (C 1 -C 20 ) Alkyl, (C 2 -C 8 ) heteroalkyl, cyclo (C 3 -C 9 ) heteroalkyl, (C 1 -C 8 ) acylamino, amidino, guanidino, ureido, cyano, heteroaryl, —CONR 9 R 10 and CO 2 is a member selected from the group consisting of R 11 or, optionally, R 3 is bound to R 2 to have 1 to 3 heteroatoms selected from the group consisting of N, O and S
- a 4-membered ring, a 5-membered ring, a 6-membered ring, a 7-membered ring or an 8-membered ring may be formed.
- R 4 is (C 2 -C 20 ) alkyl, (C 2 -C 20 ) heteroalkyl, heteroaryl, aryl, heteroaryl (C 1 -C 6 ) alkyl, heteroaryl (C 2 -C 6 ) heteroalkyl And a member selected from the group consisting of aryl (C 1 -C 6 ) alkyl and aryl (C 2 -C 6 ) heteroalkyl.
- R 5 and R 6 are each independently a member selected from the group consisting of H, (C 1 -C 8 ) alkyl, (C 2 -C 8 ) heteroalkyl, heteroaryl and aryl, or any And R 5 and R 6 combine to form a 3- to 7-membered ring.
- R 7 and R 8 are independently selected from the group consisting of H, (C 1 -C 8 ) alkyl, (C 2 -C 8 ) heteroalkyl, heteroaryl and aryl.
- R 9 , R 10 and R 11 are each independently H, (C 1 -C 8 ) alkyl, (C 2 -C 8 ) heteroalkyl, heteroaryl, aryl, heteroaryl (C 1 -C 6 ) alkyl. , Heteroaryl (C 2 -C 8 ) heteroalkyl, aryl (C 1 -C 8 ) alkyl and aryl (C 2 -C 8 ) heteroalkyl.
- R x , R y, and R z are each independently H, F, or cyano, wherein at least one of R x , R Y, and R z is cyano.
- Y 3 is N or C, and when Y 3 is C, Y 3 shares a double bond with Y 2 , Y 4 or Z.
- Y 4 is N or C, and when Y 4 is C, Y 4 shares a double bond with X, Y 1 or Y 3 .
- Each R 12 is H, halogen, hydroxy, amino, alkylamino, dialkylamino, (C 1 -C 8 ) alkyl, cyclo (C 3 -C 6 ) alkyl, (C 2 -C 8 ) heteroalkyl, hetero A member selected from the group consisting of aryl and aryl, or optionally two R 1 when Y 1 and Y 2 are each one of —C (R 12 ) ⁇ or CH (R 12 ) —
- the 12 groups can be joined to form a substituted or unsubstituted 5- to 6-membered cycloalkyl ring, cycloheteroalkyl ring, aryl ring or heteroaryl ring.
- R 12 and R 5 can be joined to form a substituted or unsubstituted 5- to 6-membered cycloalkyl ring, cycloheteroalkyl ring, aryl ring or heteroaryl ring.
- Each R 13 is H, (C 1 -C 8 ) alkyl, (C 2 -C 8 ) heteroalkyl, heteroaryl, aryl, heteroaryl (C 1 -C 6 ) alkyl, cyclo (C 3 -C 6) A member selected from the group consisting of alkyl, heteroaryl (C 2 -C 8 ) heteroalkyl, aryl (C 1 -C 8 ) alkyl and aryl (C 2 -C 8 ) heteroalkyl, optionally Y When 1 and Y 2 are —C (R 12 ) ⁇ or CH (R 12 ) — and the others are —N (R 13 ) —, R 12 and R 13 are bonded to be substituted or non-substituted 5-6 membered cycloalkyl ring substituted cycloheteroalkyl ring, they can form an aryl or heteroaryl ring, or optionally, -N Y 1 and Y 2 are both (
- R 14 is H, (C 1 -C 8 ) alkyl, (C 2 -C 8 ) heteroalkyl, cyclo (C 3 -C 6 ) alkyl, heteroaryl, aryl, heteroaryl (C 1 -C 6 ) alkyl Or a member selected from the group consisting of heteroaryl (C 2 -C 8 ) heteroalkyl, aryl (C 1 -C 8 ) alkyl and aryl (C 2 -C 8 ) heteroalkyl, or optionally Y 2 Is —C (R 12 ) ⁇ , —CH (R 12 ) — or N (R 13 ) —, R 14 or R 7 is bonded to R 12 or R 13 to form a substituted or unsubstituted A 5- to 6-membered cycloalkyl ring, cycloheteroalkyl ring, aryl ring or heteroaryl ring can be formed.
- the ring comprising X, Y 1 , Y 2 , Y 3 , Y 4 and Z can be aromatic.
- Specific inflammation such as asthma, psoriasis, inflammatory bowel disease, allergic disease, rheumatoid arthritis, multiple sclerosis, etc. It has been disclosed to have therapeutic or prophylactic use for sex and immunoregulatory disorders or diseases.
- Patent Document 2 includes the following formula:
- X represents O or S
- Ar 1 and Ar 2 are the same or different and each independently substituted or unsubstituted aryl, 5- to 7-membered heteroaryl or heterocycle group
- R 1 and R 2 are the same or different and are each independently a hydrogen atom, hydroxy, thiol, nitro, formyl, azide, cyano, halo or optionally substituted or unsubstituted alkyl, haloalkyl, alkoxy, aryl, Aryloxy, aralkyl, aralkoxy, acyl, acyloxy, amino, hydrazine, monoalkylamino, dialkylamino, acylamino, alkylsulfonyl, arylsulfonyl, alkylsulfinyl, arylsulfinyl, alkylthio, arylthio, alkoxycarbo Nyl, aryloxycarbonyl, alkoxyalkyl,
- Patent Document 3 includes the following formula
- A is C 6 -C 14 aryl or 5 to 7 membered heteroaryl;
- R 1 , R 2 and R 3 are the same or different and are hydrogen, hydroxy, nitro, cyano, amino, halogen, carboxy, carbamoyl, mercapto , C 1 -C 6 alkyl, C l -C 6 alkyl substituted by 1 to 7 halogens, C 2 -C 7 alkylcarbonyloxy, C 1 -C 6 alkoxy, C 1 -C 6 alkylthio, C 1 -C 6 alkylsulfinyl, C 1 -C 6 alkylsulfonyl, C 1 -C 6 alkylamino, di (C 1 -C 6 alkyl) amino (alkyl may be the same or different), C 2 -C 7 alkyl Carbonylamino, N- (C 2 -C 7 alkylcarbonyl) -N- (C 1 -C 6 alkyl)
- Non-Patent Document 4 Indian of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry ⁇ (2000), 39B (3), 210-214 (Non-Patent Document 4)
- Patent Document 4 has the following formula
- R 1 represents hydrogen or 1-7C-alkyl
- R 2 represents 1-7C-alkyl, phenyl, phenyl-1-4C-alkyl, Ar-1-4C-alkyl, Ar or N
- R 7 Represents 1-7C-alkylene substituted by R 8
- Ar represents phenyl substituted by R 9 , R 10 and R 11 and R 3 substituted by phenyl or R 31 and R 32
- R 31 is hydrogen, hydroxy, halogen atom, nitro, cyano, carboxy, trifluoromethyl, 1-4C-alkyl, 1-4C-alkoxy, 1 fully or partially substituted by fluorine -4C-alkoxy, 1-4C-alkoxycarbonyl, 1-4C-alkylcarbonyl, amino or mono-1-4C-alkyl
- R 32 represents hydrogen, hydroxy, halogen atom, nitro, trifluoromethyl, 1 to 4C-alkyl or 1 to 4C-alkyl
- the substituted 1,2,3,4-tetrahydroisoquinoline group is 1 to 4 C-alkyl at the 1-position, 3-position and / or 4-position , Carboxy, phenyl, phenyl, phenyl 1-4C-alkyl and phenyl groups substituted by R 9 , R 10 and R 11
- R 9 , R 10 and R 11 Optionally substituted with 1 or 2 identical or different substituents selected from the group consisting of phenyl-1 to 4C-alkyl substituted by R 9 , R 10 and R 11 and in the benzo moiety,
- Non-Patent Document 5 Journal of the Institution of Chemists (India) (1985), 57 (4), 156-158 (Non-Patent Document 5)
- any compound is distinguished from the present compound and there is no description that it has a delta-5-desaturase inhibitory action.
- the object of the present invention is useful for the prevention and treatment of eicosanoid-related diseases such as atherosclerosis, atherothrombosis, diabetes, obesity, asthma, fever, pain, cancer, rheumatism, osteoarthritis, atopic dermatitis, etc. And providing a compound having excellent pharmacological action, physicochemical properties and the like.
- the condensed heterocyclic compound represented by the following formula (I) has a delta-5-desaturase inhibitory action and exhibits various pharmacological actions based on suppressing the production of eicosanoids.
- Eicosanoids with excellent physical properties such as crystallinity and stability, such as atherosclerosis, atherothrombosis, diabetes, obesity, asthma, fever, pain, cancer, rheumatism, osteoarthritis, atopic dermatitis It was found for the first time that it is useful for the prevention and treatment of related diseases. Based on this finding, the present inventors have conducted intensive studies and have completed the present invention.
- Ring A has the formula:
- Each ring may be further substituted with a substituent selected from a halogen atom, a C 1-6 alkyl group and a C 1-6 alkoxy group
- Ring B represents a 6-membered aromatic ring which may be further substituted with a substituent selected from a halogen atom, a C 1-6 alkyl group and a C 1-6 alkoxy group
- X 1 represents a bond, S, SO 2 , O or NR 5
- X 2 represents CR 6 R 7 or O
- Y 1 represents CR 8 R 9 , S, SO, SO 2 or O
- R 1 represents an optionally substituted C 1-6 alkoxy group, an optionally substituted amino group, an optionally substituted carbamoyl group or an optionally substituted C 1-6 alkylthio group
- R 2 represents a hydrogen atom or an optionally substituted C 1-6 alkyl group
- R 3 represents an optionally substituted C 1-6 alkyl group or an optionally substituted 3- to 11-member
- a ring represented by Ring B is benzene;
- X 1 is a bond, S, O or NH;
- X 2 is O;
- Y 1 is CH 2 or S;
- R 1 is (1) a C 1-6 alkoxy group which may be substituted with 1 to 3 C 1-6 alkoxy groups; (2) amino; or (3) carbamoyl;
- Is is;
- R 2 is a hydrogen atom or a C 1-6 alkyl group;
- R 3 is (1) (A) a C 1-6 alkylsulfonyl group, (B) a hydroxy group, (C) C 1-6 alkoxy group, and (D) 1 selected from a halogen atom to 3 substituents optionally substituted by a C 1-6 alkyl group or (2) 1 to 3 halogen,
- the above [1] description which is a C 3-10 cycloalkyl group optionally substituted with an atom; and
- R 4 is a C 1-6 alkyl group optionally
- a ring represented by Ring B is benzene; X 1 is a bond, S or O; X 2 is O; Y 1 is CH 2 ; R 1 is a C 1-6 alkoxy group; R 2 is a hydrogen atom; R 3 is (1) a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms, or (2) a C 3-10 cyclo optionally substituted with 1 to 3 halogen atoms.
- Ring A is represented by the formula:
- a ring represented by Ring B is benzene; X 1 is a bond, S or O; X 2 is O; Y 1 is CH 2 ; R 2 is a hydrogen atom; R 3 is (1) a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms, or (2) a C 3-10 cyclo optionally substituted with 1 to 3 halogen atoms.
- Compound (I) has a delta-5-desaturase inhibitory action, and is atherosclerosis, atherothrombosis, diabetes, obesity, asthma, fever, pain, cancer, rheumatism, osteoarthritis, atopic dermatitis It is useful for prevention and treatment of eicosanoid-related diseases such as, and has excellent medicinal effects. (Detailed description of the invention)
- halogen atom in the present specification include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom.
- C 1-6 alkyl (group) examples include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, tert-pentyl, Examples include hexyl, 2,2-dimethylbutyl, 3,3-dimethylbutyl, 2-ethylbutyl and the like.
- Examples of the “C 1-6 alkoxy (group)” in the present specification include methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, sec-butoxy, tert-butoxy, pentyloxy, isopentyloxy, neopentyl And oxy, tert-pentyloxy, hexyloxy, 2-ethylbutoxy and the like.
- Examples of the “C 3-10 cycloalkyl (group)” in the present specification include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl.
- Examples of the “C 6-14 aryl (group)” in the present specification include phenyl, naphthyl (eg, 1-naphthyl, 2-naphthyl), anthryl, phenanthryl and the like.
- Examples of the “C 7-13 aralkyl (group)” in the present specification include benzyl, phenethyl, naphthylmethyl, biphenylylmethyl and the like.
- C 2-6 alkenyl (group) examples include ethenyl, 1-propenyl, 2-propenyl, 2-methyl-1-propenyl, 1-butenyl, 2-butenyl, 3-butenyl, Examples include 3-methyl-2-butenyl, 1-pentenyl, 2-pentenyl, 3-pentenyl, 4-pentenyl, 4-methyl-3-pentenyl, 1-hexenyl, 3-hexenyl, 5-hexenyl and the like.
- Ring A is the formula:
- each ring may be further substituted with a substituent selected from a halogen atom, a C 1-6 alkyl group and a C 1-6 alkoxy group.
- Ring A is preferably of the formula:
- Ring A is more preferably of the formula:
- Ring A is more preferably of the formula:
- Ring B represents a 6-membered aromatic ring which may be further substituted with a substituent selected from a halogen atom, a C 1-6 alkyl group and a C 1-6 alkoxy group.
- “6-membered aromatic ring” in the “6-membered aromatic ring optionally substituted with a substituent selected from a halogen atom, a C 1-6 alkyl group and a C 1-6 alkoxy group” represented by ring B is as follows: , Benzene, pyridine, pyridazine, pyrimidine, pyrazine, triazine and the like. Preferred are benzene, pyridine, pyridazine, pyrimidine, pyrazine and the like, and more preferred is benzene.
- the “6-membered aromatic ring” in the “6-membered aromatic ring optionally substituted with a substituent selected from a halogen atom, a C 1-6 alkyl group and a C 1-6 alkoxy group” represented by ring B is: It may have 1 to 3 substituents selected from a halogen atom, a C 1-6 alkyl group and a C 1-6 alkoxy group at the substitutable position. When there are two or more substituents, each substituent may be the same or different.
- Ring B is preferably benzene.
- X 1 represents a bond, S, SO 2 , O, or NR 5 . In one preferred embodiment, X 1 is S, O or NR 5 . In another preferred embodiment, X 1 is a bond, S, O or NR 5 .
- R 5 represents a hydrogen atom or an optionally substituted C 1-6 alkyl group.
- the “C 1-6 alkyl group” in the “ optionally substituted C 1-6 alkyl group” for R 5 may have 1 to 3 substituents at substitutable positions.
- substituents for example, similar to the "C 1-6 alkoxy group” substituents which may be possessed by the "optionally substituted C 1-6 alkoxy group” represented by R 1 described later Things.
- each substituent may be the same or different.
- R 5 is preferably a hydrogen atom.
- X 1 is preferably a bond, S, O, or NH. X 1 is more preferably a bond, S or O.
- X 2 represents CR 6 R 7 or O.
- R 6 represents a hydrogen atom, a halogen atom, a C 1-6 alkyl group or a C 1-6 alkoxy group.
- R 6 is preferably a hydrogen atom.
- R 7 represents a hydrogen atom, a halogen atom, a C 1-6 alkyl group or a C 1-6 alkoxy group.
- R 7 is preferably a hydrogen atom.
- X 2 is preferably O.
- Y 1 represents CR 8 R 9 , S, SO, SO 2 or O. In one preferred embodiment, Y 1 is CR 8 R 9 or S.
- R 8 represents a hydrogen atom, a halogen atom, a C 1-6 alkyl group or a C 1-6 alkoxy group.
- R 8 is preferably a hydrogen atom.
- R 9 represents a hydrogen atom, a halogen atom, a C 1-6 alkyl group or a C 1-6 alkoxy group.
- R 9 is preferably a hydrogen atom.
- Y 1 is preferably CH 2 or S. Y 1 is more preferably CH 2 .
- R 1 represents an optionally substituted C 1-6 alkoxy group, an optionally substituted amino group, an optionally substituted carbamoyl group, or an optionally substituted C 1-6 alkylthio group.
- C 1-6 alkoxy group in the "optionally substituted C 1-6 alkoxy groups” represented by R 1 may have 1 to 3 substituents at a substitutable position.
- substituents examples include: (1) a C 3-10 cycloalkyl group (eg, cyclopropyl, cyclohexyl); (2) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group optionally substituted with 1 to 3 halogen atoms, and (d) a C 6- optionally substituted with 1 to 3 substituents selected from halogen atoms.
- a C 3-10 cycloalkyl group eg, cyclopropyl, cyclohexyl
- substituents include: (1) a C 3-10 cycloalkyl group (eg, cyclopropyl, cyclohexyl); (2) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group optionally
- aryl groups eg, phenyl, naphthyl
- (3) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms, and (d) 4 to 7 which may be substituted with 1 to 3 substituents selected from halogen atoms.
- aromatic heterocyclic groups eg, thienyl, furyl, pyridyl, pyrazolyl, imidazolyl, tetrazolyl, oxazolyl, thiazolyl, oxadiazolyl, thiadiazolyl; (4) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms, (d) a halogen atom, and (e) a 4- to 7-membered non-aromatic heterocyclic group (eg, tetrahydrofuryl, morpholinyl, thiomorpholinyl, piperidyl) optionally substituted with 1 to 3 substituents selected from oxo groups , Pyrrolidinyl, piperazinyl); (5) (a) a C 1-6 alkyl group
- a C 1-6 alkoxy-carbonyl group (eg, methoxycarbonyl) optionally substituted by 1 to 3 substituents selected from: (8) a C 1-6 alkylsulfonyl group which may be substituted with 1 to 3 halogen atoms (eg, methylsulfonyl, ethylsulfonyl, isopropylsulfonyl); (9) a carbamoyl group which may be mono- or di-substituted with a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms; (10) a thiocarbamoyl group optionally mono- or disubstituted with a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms; (11) a sulfamoyl group optionally mono- or di-substituted with a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atom
- the “optionally substituted C 1-6 alkoxy group” represented by R 1 is preferably C 1 -C 1 optionally substituted by 1 to 3 C 1-6 alkoxy groups (eg, methoxy). 6 alkoxy groups (eg, methoxy, ethoxy) and the like.
- amino group in the “optionally substituted amino group” represented by R 1 may have 1 or 2 substituents at substitutable positions.
- substituents for example, those similar to the "optionally substituted C 1-6 alkoxy group” substituents which may "C 1-6 alkoxy group” have in represented by R 1 Can be mentioned.
- each substituent may be the same or different.
- the “optionally substituted amino group” represented by R 1 is preferably amino or the like.
- the “carbamoyl group” in the “optionally substituted carbamoyl group” represented by R 1 may have 1 or 2 substituents at substitutable positions.
- substituents for example, those similar to the "optionally substituted C 1-6 alkoxy group” substituents which may "C 1-6 alkoxy group” have in represented by R 1 Can be mentioned.
- each substituent may be the same or different.
- the “optionally substituted carbamoyl group” represented by R 1 is preferably carbamoyl or the like.
- C 1-6 alkylthio group” in the “optionally substituted C 1-6 alkylthio group” represented by R 1 include, for example, methylthio, ethylthio, propylthio, isopropylthio, butylthio, sec-butylthio, tert- Examples include butylthio.
- C 1-6 alkylthio group" of the “optionally substituted C 1-6 alkylthio group” is not 1 at substitutable positions represented by R 1 may have three substituents.
- substituents for example, those similar to the "optionally substituted C 1-6 alkoxy group” substituents which may "C 1-6 alkoxy group” have in represented by R 1 Can be mentioned.
- each substituent may be the same or different.
- R 1 is preferably (1) 1 to 3 C 1-6 alkoxy groups (e.g., methoxy) optionally substituted by C 1-6 alkoxy group (e.g., methoxy, ethoxy); (2) amino; (3) carbamoyl; Etc.
- R 1 is more preferably a C 1-6 alkoxy group (eg, methoxy).
- R 2 represents a hydrogen atom or an optionally substituted C 1-6 alkyl group.
- the “C 1-6 alkyl group” in the “ optionally substituted C 1-6 alkyl group” for R 2 may have 1 to 3 substituents at substitutable positions.
- substituents for example, those similar to the "optionally substituted C 1-6 alkoxy group” substituents which may "C 1-6 alkoxy group” have in represented by R 1 Can be mentioned.
- each substituent may be the same or different.
- the “optionally substituted C 1-6 alkyl group” for R 2 is preferably methyl or the like.
- R 2 is preferably a hydrogen atom or a C 1-6 alkyl group (eg, methyl). R 2 is more preferably a hydrogen atom.
- R 3 represents an optionally substituted C 1-6 alkyl group or an optionally substituted 3- to 11-membered cyclic group.
- the “C 1-6 alkyl group” in the “ optionally substituted C 1-6 alkyl group” represented by R 3 may have 1 to 3 substituents at substitutable positions.
- substituents for example, those similar to the "optionally substituted C 1-6 alkoxy group” substituents which may "C 1-6 alkoxy group” have in represented by R 1 Can be mentioned.
- each substituent may be the same or different.
- the “optionally substituted C 1-6 alkyl group” represented by R 3 is preferably (1) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl), (2) hydroxy group, (3) C 1-6 alkoxy group (eg, methoxy), and (4) halogen atom (eg, fluorine atom, chlorine atom, bromine atom)
- a C 1-6 alkyl group eg, methyl, ethyl, propyl, isopropyl which may be substituted with 1 to 3 substituents selected from
- the “3- to 11-membered cyclic group” in the “optionally substituted 3- to 11-membered cyclic group” represented by R 3 is, for example, a 6- to 10-membered aromatic hydrocarbon group, a 5- to 11-membered aromatic complex. Ring group (eg, 5- to 7-membered monocyclic aromatic heterocyclic group, 8- to 11-membered condensed aromatic heterocyclic group), 3- to 10-membered non-aromatic cyclic hydrocarbon group, and 3- to 8-membered non-aromatic heterocycle A cyclic group etc. are mentioned.
- 6- to 10-membered aromatic hydrocarbon group examples include a C 6-10 aryl group. Specific examples include phenyl, 1-naphthyl, 2-naphthyl and the like.
- Examples of the 5- to 7-membered monocyclic aromatic heterocyclic group include hetero atoms selected from oxygen atoms, sulfur atoms (the sulfur atoms may be oxidized) and nitrogen atoms in addition to carbon atoms as ring-constituting atoms. And 5- to 7-membered monocyclic aromatic heterocyclic group containing 1 to 4 atoms.
- 5- to 7-membered monocyclic aromatic heterocyclic group examples include furyl (eg, 2-furyl, 3-furyl), thienyl (eg, 2-thienyl, 3-thienyl), pyridyl (eg, 2-pyridyl, 3-pyridyl, 4-pyridyl), pyrimidinyl (eg, 2-pyrimidinyl, 4-pyrimidinyl, 5-pyrimidinyl, 6-pyrimidinyl), pyridazinyl (eg, 3-pyridazinyl, 4-pyridazinyl), pyrazinyl ( Eg, 2-pyrazinyl), pyrrolyl (eg, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl), imidazolyl (eg, 1-imidazolyl, 2-imidazolyl, 4-imidazolyl, 5-imidazolyl), pyrazolyl (eg, 1 -Pyrazolyl,
- the 8- to 11-membered condensed aromatic heterocyclic group includes, for example, a hetero atom selected from an oxygen atom, a sulfur atom (the sulfur atom may be oxidized) and a nitrogen atom in addition to a carbon atom as a ring constituent atom.
- a group derived from a condensed ring containing 1 to 4 5- to 7-membered monocyclic aromatic heterocycle and the like and C 6-10 aromatic hydrocarbon or the like; the above 5- to 7-membered monocyclic aroma A group derived from a condensed ring obtained by condensing group heterocycles.
- Examples of the 5- to 7-membered monocyclic aromatic heterocyclic ring include rings corresponding to the aforementioned 5- to 7-membered monocyclic aromatic heterocyclic group.
- Examples of the C 6-10 aromatic hydrocarbon include a ring corresponding to the above C 6-10 aryl group.
- 8- to 11-membered condensed aromatic heterocyclic group examples include quinolyl (eg, 2-quinolyl, 3-quinolyl, 4-quinolyl), isoquinolyl (eg, 1-isoquinolyl, 3-isoquinolyl, 4- Isoquinolyl), quinazolyl (eg, 2-quinazolyl, 4-quinazolyl), quinoxalyl (eg, 2-quinoxalyl), benzofuryl (eg, 2-benzofuryl, 3-benzofuryl), benzothienyl (eg, 2-benzothienyl, 3-quinolyl) Benzothienyl), benzoxazolyl (eg, 2-benzoxazolyl), benzothiazolyl (eg, 2-benzothiazolyl, 5-benzothiazolyl, 6-benzothiazolyl), benzimidazolyl (eg, benzimidazol-1-yl, benzimidazole) -2-yl, benzimid
- Examples of the 3- to 10-membered non-aromatic cyclic hydrocarbon group include a C 3-10 cycloalkyl group, a C 3-10 cycloalkenyl group, and a C 4-10 cycloalkali group, each of which may be condensed with a benzene ring.
- a dienyl group etc. are mentioned.
- a C 3-10 cycloalkyl group eg, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl
- C 3-10 Cycloalkenyl groups eg, cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, cycloheptenyl, cyclooctenyl
- C 4-10 cycloalkadienyl groups eg, cyclobutadienyl, cyclopentadienyl, cyclohexadienyl, cyclohepta
- condensed ring groups obtained by condensing these groups with
- Examples of the 3- to 8-membered non-aromatic heterocyclic group include 3- to 8-membered (preferably 5- or 6-membered) saturated or unsaturated (preferably saturated) non-aromatic heterocyclic group.
- 3- to 8-membered non-aromatic heterocyclic group examples include oxiranyl (eg, 2-oxiranyl), azetidinyl (eg, 2-azetidinyl), oxetanyl (eg, 2-oxetanyl), thietanyl (eg, 2-thietanyl), pyrrolidinyl (eg, 1-pyrrolidinyl, 2-pyrrolidinyl, 3-pyrrolidinyl), tetrahydrofuryl (eg, 2-tetrahydrofuryl, 3-tetrahydrofuryl), thiolanyl (eg, 2-thiolanyl), piperidyl (eg, 1-piperidyl, 2-piperidyl, 3-piperidyl, 4-piperidyl), tetrahydropyranyl (eg, 2-tetrahydropyranyl, 3-tetrahydropyranyl, 4-tetrahydropyranyl), thianyl (
- 6- to 10-membered aromatic hydrocarbon group exemplified as “3- to 11-membered cyclic group” in the “optionally substituted 3- to 11-membered cyclic group” represented by R 3 , 5- to 7-membered monocyclic aromatic Heterocyclic group, 8- to 11-membered condensed aromatic heterocyclic group, 3- to 10-membered non-aromatic cyclic hydrocarbon group, and 3- to 8-membered non-aromatic heterocyclic group have 1 to 3 substituents at substitutable positions You may have.
- substituents examples include: (1) a C 3-10 cycloalkyl group (eg, cyclopropyl, cyclohexyl); (2) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group optionally substituted with 1 to 3 halogen atoms, and (d) a C 6- optionally substituted with 1 to 3 substituents selected from halogen atoms.
- a C 3-10 cycloalkyl group eg, cyclopropyl, cyclohexyl
- substituents include: (1) a C 3-10 cycloalkyl group (eg, cyclopropyl, cyclohexyl); (2) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group optionally
- aryl groups eg, phenyl, naphthyl
- (3) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms, and (d) 4 to 7 which may be substituted with 1 to 3 substituents selected from halogen atoms.
- aromatic heterocyclic groups eg, thienyl, furyl, pyridyl, pyrazolyl, imidazolyl, tetrazolyl, oxazolyl, thiazolyl, oxadiazolyl, thiadiazolyl; (4) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms, (d) a halogen atom, and (e) a 4- to 7-membered non-aromatic heterocyclic group (eg, tetrahydrofuryl, morpholinyl, thiomorpholinyl, piperidyl) optionally substituted with 1 to 3 substituents selected from oxo groups , Pyrrolidinyl, piperazinyl); (5) (a) a C 1-6 alkyl group
- a C 1-6 alkoxy-carbonyl group (eg, methoxycarbonyl) optionally substituted by 1 to 3 substituents selected from: (8) a C 1-6 alkylsulfonyl group which may be substituted with 1 to 3 halogen atoms (eg, methylsulfonyl, ethylsulfonyl, isopropylsulfonyl); (9) a carbamoyl group which may be mono- or di-substituted with a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms; (10) a thiocarbamoyl group optionally mono- or disubstituted with a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms; (11) a sulfamoyl group optionally mono- or di-substituted with a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atom
- a 1-6 alkyl group (33) (a) a halogen atom, (b) a carboxy group, (c) a hydroxy group, (d) a C 1-6 alkoxy-carbonyl group, (e) a C 1-6 alkoxy group, and (f) a C 1-6 alkyl group optionally substituted with 1 to 3 substituents selected from amino groups optionally mono- or di-substituted.
- 2-6 alkenyl groups (eg, ethenyl, 1-propenyl); (34) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group, and (d) a C 7-13 aralkyl group (eg, benzyl) optionally substituted with 1 to 3 substituents selected from halogen atoms; Etc. When there are two or more substituents, each substituent may be the same or different.
- the “optionally substituted 3- to 11-membered cyclic group” represented by R 3 is preferably a C 3-10 cycloalkyl optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom). Group (eg, cyclohexyl) and the like.
- R 3 is preferably (1) (A) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl), (B) a hydroxy group, (C) C 1-6 alkoxy group (eg, methoxy), and (D) halogen atom (eg, fluorine atom, chlorine atom, bromine atom)
- a C 1-6 alkyl group eg, methyl, ethyl, propyl, isopropyl
- R 3 is more preferably (1) a C 1-6 alkyl group (eg, ethyl, propyl) optionally substituted by 1 to 3 halogen atoms (eg, fluorine atom); (2) a C 3-10 cycloalkyl group (eg, cyclohexyl) optionally substituted by 1 to 3 halogen atoms (eg, fluorine atom); Etc.
- a C 1-6 alkyl group eg, ethyl, propyl
- 1 to 3 halogen atoms eg, fluorine atom
- a C 3-10 cycloalkyl group eg, cyclohexyl
- R 4 represents an optionally substituted C 1-6 alkyl group or an optionally substituted 3- to 11-membered cyclic group.
- the “C 1-6 alkyl group” in the “ optionally substituted C 1-6 alkyl group” represented by R 4 may have 1 to 3 substituents at substitutable positions.
- substituents for example, those similar to the "optionally substituted C 1-6 alkoxy group” substituents which may "C 1-6 alkoxy group” have in represented by R 1 Can be mentioned.
- each substituent may be the same or different.
- Optionally substituted C 1-6 alkyl group represented by R 4 is preferably, one to three C 1-6 alkyl group optionally substituted with a halogen atom.
- R 4 As “optionally substituted 3 to be to 11-membered cyclic group" represented by R 4 include those similar to the "optionally substituted 3- or to 11-membered cyclic group" represented by R 3.
- R 4 is preferably a C 1-6 alkyl group (eg, ethyl) optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom).
- Preferred examples of compound (I) include the following compounds.
- Ring A has the formula:
- a ring represented by Ring B is benzene; X 1 is S, O or NR 5 ; X 2 is O; Y 1 is CR 8 R 9 or S; R 1 is (1) 1 to 3 C 1-6 alkoxy groups (e.g., methoxy) optionally substituted by C 1-6 alkoxy group (e.g., methoxy, ethoxy); (2) amino; or (3) carbamoyl; Is; R 2 is a hydrogen atom or a C 1-6 alkyl group (eg, methyl); R 3 is (1) (A) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl), (B) a hydroxy group, (C) C 1-6 alkoxy group (eg, methoxy), and (D) halogen atom (eg, fluorine atom, chlorine atom, bromine atom) A C 1-6 alkyl group (eg, methyl, ethyl, propyl, isopropyl
- a ring represented by Ring B is benzene;
- X 1 is a bond, S, O or NR 5 ;
- X 2 is O;
- Y 1 is CR 8 R 9 or S;
- R 1 is (1) 1 to 3 C 1-6 alkoxy groups (e.g., methoxy) optionally substituted by C 1-6 alkoxy group (e.g., methoxy, ethoxy); (2) amino; or (3) carbamoyl;
- R 2 is a hydrogen atom or a C 1-6 alkyl group (eg, methyl);
- R 3 is (1) (A) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl), (B) a hydroxy group, (C) C 1-6 alkoxy group (eg, methoxy), and (D) halogen atom (eg, fluorine atom, chlorine atom, bromine atom)
- a C 1-6 alkyl group eg
- a ring represented by Ring B is benzene; X 1 is a bond, S or O; X 2 is O; Y 1 is CH 2 ; R 1 is a C 1-6 alkoxy group (eg, methoxy); R 2 is a hydrogen atom; R 3 is (1) a C 1-6 alkyl group (eg, ethyl, propyl) optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom), or (2) 1 to 3 halogen atoms A C 3-10 cycloalkyl group (eg, cyclohexyl) optionally substituted with an atom (eg, fluorine atom); and R 4 is substituted with 1 to 3 halogen atoms (eg, fluorine atom) Compound (I) which may be a C 1-6 alkyl group (eg, ethyl).
- a ring represented by Ring B is benzene;
- X 1 is a bond, S or O;
- X 2 is O;
- Y 1 is CH 2 ;
- R 2 is a hydrogen atom;
- R 3 is (1) a C 1-6 alkyl group (eg, ethyl, propyl) optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom), or (2) 1 to 3 halogen atoms
- a C 3-10 cycloalkyl group eg, cyclohexyl
- R 4 is substituted with 1 to 3 halogen atoms (eg, fluorine atom)
- the salt of the compound represented by formula (I) is preferably a pharmacologically acceptable salt.
- a salt with an inorganic base examples include a salt with an inorganic base, a salt with an organic base, and a salt with an inorganic acid.
- the salt with an inorganic base include alkali metal salts such as sodium salt and potassium salt; alkaline earth metal salts such as calcium salt and magnesium salt; aluminum salt; ammonium salt and the like.
- the salt with an organic base include trimethylamine, triethylamine, pyridine, picoline, ethanolamine, diethanolamine, triethanolamine, tromethamine [tris (hydroxymethyl) methylamine], tert-butylamine, cyclohexylamine, benzylamine, And salts with dicyclohexylamine, N, N-dibenzylethylenediamine and the like.
- salt with inorganic acid examples include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like.
- salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, phthalic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, and benzenesulfonic acid And salts with p-toluenesulfonic acid and the like.
- salts with basic amino acids include salts with arginine, lysine, ornithine and the like.
- salt with acidic amino acid include salts with aspartic acid, glutamic acid and the like.
- Compound (I) may be labeled with an isotope (eg, 3 H, 11 C, 14 C, 18 F, 35 S, 125 I) or the like.
- Compound (I) may be a solvate (for example, an anhydride) or a solvate (for example, a hydrate).
- a deuterium converter obtained by converting 1 H into 2 H (D) is also encompassed in compound (I).
- Compound (I) may be crystalline or amorphous.
- compound (I) is a crystal, it is included in compound (I) whether it is a single crystal form or a crystal form mixture.
- the crystal can be produced by crystallization by applying a crystallization method known per se.
- the melting point is measured using, for example, a trace melting point measuring device (Yanako, MP-500D type or Buchi, B-545 type) or a DSC (differential scanning calorimetry) apparatus (SEIKO, EXSTAR6000). Mean melting point.
- the melting point may vary depending on measurement equipment, measurement conditions, and the like.
- the crystal in the present specification may be a crystal having a value different from the melting point described in the present specification as long as it is within a normal error range.
- Compound (I) may be a pharmaceutically acceptable cocrystal or cocrystal salt.
- co-crystals or co-crystal salts are two or more unique at room temperature, each having different physical properties (eg structure, melting point, heat of fusion, hygroscopicity, solubility and stability). It means a crystalline substance composed of a simple solid.
- the cocrystal or cocrystal salt can be produced according to a cocrystallization method known per se.
- the crystals of the present invention are excellent in physicochemical properties (eg, melting point, solubility, stability) and biological properties (eg, pharmacokinetics (absorbability, distribution, metabolism, excretion), expression of medicinal properties), and are extremely useful as pharmaceuticals. Useful.
- Compound (I) may also be a prodrug.
- the prodrug of compound (I) is a compound that is converted to compound (I) by a reaction with an enzyme or gastric acid under physiological conditions in vivo, that is, compound (I) that undergoes oxidation, reduction, hydrolysis, etc. enzymatically. ), A compound that undergoes hydrolysis or the like due to gastric acid or the like and changes to compound (I).
- the prodrug of compound (I) is a compound that changes to compound (I) under physiological conditions as described in Hirokawa Shoten 1990, “Drug Development”, Volume 7, Molecular Design, pages 163 to 198. It may be.
- each raw material compound may form a salt as long as it does not inhibit the reaction.
- examples of such salts include those exemplified as the salts of the compound represented by the aforementioned formula (I). Used.
- the raw material compound can be easily obtained commercially, or can be produced according to a method known per se or a method analogous thereto, unless a specific production method is described.
- the solvent used in the reaction in each of the following schemes is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material to some extent.
- aromatic hydrocarbons such as benzene, toluene and xylene; hexane Aliphatic hydrocarbons such as heptane; ethers such as diethyl ether, diisopropyl ether, tert-butyl methyl ether, tetrahydrofuran, dioxane and 1,2-dimethoxyethane; ketones such as acetone and 2-butanone; acetonitrile, pro Nitriles such as pionitrile; esters such as ethyl acetate, isopropyl acetate and tert-butyl acetate; amides such as N, N-dimethylformamide, N, N-dimethylacetamide and 1-methyl-2-pyrrolidinone; Imides such as 3-dimethyl-2-imidazolidinone; for
- the reaction temperature is usually carried out below the boiling point of the solvent at -100 to 250 ° C. However, depending on the case, the reaction may be carried out at a temperature above the boiling point of the solvent using pressure resistant reaction conditions.
- the reaction time is usually 0.5 to 100 hours.
- Ra represents a C 1-6 alkyl group
- Xa represents S, SO or SO 2 .
- Compound (I) can be produced by the route shown in Scheme 1. That is, it can be produced by subjecting compound (1) as a starting material to aromatic nucleophilic substitution reaction for compound (4) or (5) via compound (3).
- Compound (1) can be easily obtained as a commercially available product, or can be produced according to a method known per se or a method analogous thereto.
- Step A (cyclization reaction)
- Compound (3) can be produced by a ring-closing reaction performed under the basic conditions of compound (1) and compound (2).
- the compound (2) is used in an amount of about 1.0 to 5.0 mol, preferably about 1.0 to 2.0 mol, per 1 mol of the compound (1).
- Bases include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogenated Examples thereof include metal hydrides such as potassium, organic bases such as triethylamine and N, N-diisopropylethylamine, and about 1.0 to 10.0 mol, preferably about 1.0 to 5.0 mol, per 1.0 mol of compound (1).
- This reaction is preferably carried out using a solvent inert to the reaction.
- the solvent is not particularly limited as long as the reaction proceeds.
- aromatic hydrocarbons such as benzene and toluene
- ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane
- N, N-dimethylformamide N Amides such as N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide or a mixed solvent thereof are preferable.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 100 ° C.
- Compound (3) can be used in the next reaction as the reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, and can be isolated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Step B Compound (4) is produced by S-alkylation reaction of compound (3) using a base and an alkylating agent corresponding to R 3 .
- the base is used in an amount of 1.0 to 10.0 mol, preferably 1.0 to 5.0 mol, and 1.0 to 20.0 mol, preferably 1.0 to 10.0 mol, of the alkylating agent corresponding to R 3 with respect to 1 mol of compound (3).
- Bases include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium bicarbonate, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, hydrogenation Examples thereof include metal hydrides such as sodium and potassium hydride, and organic bases such as triethylamine and N, N-diisopropylethylamine.
- alkylating agent corresponding to R 3 examples include various alkyl halides such as alkyl chloride, alkyl bromide and alkyl iodide and derivatives thereof, sulfonate esters such as p-toluenesulfonate and methylsulfonate, dimethyl Examples thereof include sulfuric acid esters such as sulfuric acid.
- This reaction is preferably carried out using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, Aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, acetonitrile, propionitrile, etc.
- Nitriles, sulfoxides such as dimethyl sulfoxide, water or a mixed solvent thereof are preferred.
- the reaction time is usually 15 minutes to 60 hours, preferably 15 minutes to 24 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- Compound (4) can be used as it is in the reaction solution or as a crude product for the next reaction, but can be isolated from the reaction mixture according to a conventional method, and can be isolated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Process C Compound (5) is produced by an oxidation reaction of compound (4).
- the oxidizing agent is used in an amount of 1.0 to 30.0 mol, preferably 1.0 to 3.0 mol, per 1 mol of compound (4).
- oxidizing agents include hydrogen peroxide, oxone (registered trademark), peracids such as peracetic acid, perbenzoic acid, and metachloroperbenzoic acid, oxoacids such as hypochlorous acid and periodic acid, and salts thereof, chromic acid, and the like.
- examples thereof include metal oxo acids and salts thereof or other oxidizing agents.
- This reaction is preferably performed using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- a solvent inert for example, alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, formic acid, etc. Of these, carboxylic acids, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 60 hours, preferably 30 minutes to 5 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- Compound (5) can be used as it is in the reaction solution or as a crude product for the next reaction, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Process D aromatic nucleophilic substitution reaction
- Compound (I) can be produced by a substitution reaction using a base of compound (4) or (5) and a nucleophile corresponding to X 1 -R 3 .
- the compound (4) or (5) relative to 1 mol base 1.0-20.0 moles, preferably 1.0-10.0 moles used, 1.0 to 100.0 moles nucleophiles corresponding to X 1 -R 3, preferably 1.0 to 10.0 moles are used.
- Bases include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogenated Examples thereof include metal hydrides such as potassium.
- Nucleophiles corresponding to X 1 -R 3 include methanol, ethanol, propanol, 1,1-dimethylethanol, 2,2,2-trifluoroethanol, 2,2,3,3,3-pentafluoropropanol, etc.
- Alcohols various phenol derivatives having an aromatic hydroxyl group, organic thiols such as ethanethiol and thioglycolic acid amide, various aromatic thiol derivatives such as thiophenol, various organic bases such as methylamine and ethylamine, various types such as aniline And organometallic reagents such as aromatic amines, organic Grignard reagents (n-propylmagnesium bromide, n-butylmagnesium bromide), and organic lithium reagents (n-propyllithium, n-butyllithium).
- the base can be used as a nucleophile. This reaction is preferably carried out without solvent or using a solvent inert to the reaction.
- Such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol and propanol
- aromatic hydrocarbons such as benzene and toluene
- saturated hydrocarbons such as cyclohexane and hexane
- Ethers such as dioxane and 1,2-dimethoxyethane
- amides such as N, N-dimethylformamide and N, N-dimethylacetamide
- nitriles such as acetonitrile and propionitrile
- ketones such as acetone and methyl ethyl ketone
- a solvent such as water or a mixed solvent thereof is preferred.
- the reaction time is usually 10 minutes to 24 hours, preferably 15 minutes to 12 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 100 ° C.
- Compound (I) can be used in the next reaction as a reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, and can be isolated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- compound (6a) can be produced by a route according to steps A to D in scheme 1. That is, starting from compound (1a), compound (4a) is produced via compound (3a) by cyclization reaction, and compound (6a) is produced by aromatic substitution reaction of compound (4a) or (5a). Can do.
- Compound (1a) can be produced according to the method of Scheme 4 described below.
- compound (3a) can alternatively be produced by an aromatic nucleophilic substitution reaction (step E) on compound (3b) as shown in Scheme 3.
- Compound (3b) is produced from compound (1b) by a method according to Step A (Scheme 1).
- Compound (1b) can be produced according to a method according to Scheme 4 described later.
- Process E aromatic nucleophilic substitution reaction
- the reaction is carried out using 1.0 to 10 mol, preferably 1.0 to 5.0 mol, of the nucleophile corresponding to R 1 with respect to 1 mol of compound (3b).
- Nucleophiles corresponding to R 1 include alcohols such as methanol, ethanol, propanol and ethylene glycol, metal alkoxides such as sodium methoxide and sodium ethoxide, methylamine, ethylamine, benzylamine, 4-methoxybenzylamine and the like Organic amines and the like.
- a base can be used as necessary. The base is used in an amount of 1.0 to 10 mol, preferably 1.0 to 5.0 mol, per 1 mol of compound (3b).
- bases include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate and potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide, sodium hydride And metal hydrides such as potassium hydride.
- inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate and potassium carbonate
- metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide
- sodium hydride And metal hydrides such as potassium hydride.
- copper iodide, copper bromide, palladium acetate, palladium chloride and the like can be used as the metal catalyst, and 0.01 to 1.0 mol, preferably 0.1 to 0.5 mol can be used with respect to 1 mol of compound (3b).
- Compound (3a) can be used in the next reaction as a reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, and can be isolated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Rb represents a 4-methoxybenzyl group or a 2,4-dimethoxybenzyl group.
- Compound (1a) can be produced by the route shown in Scheme 4. That is, the compound (7) can be produced by an aromatic nucleophilic substitution reaction (Step F) and subsequent deprotection (Step G). Compound (7) can be easily obtained as a commercially available product, or can be produced according to a method known per se or a method analogous thereto.
- Process F aromatic nucleophilic substitution reaction
- Compound (8) can be produced by subjecting compound (7) to a nucleophilic substitution reaction of an amine derivative corresponding to Rb in the presence of a base.
- the amine derivative corresponding to Rb is used in an amount of 1.0 to 5.0 mol, preferably 1.0 to 3.0 mol, per 1 mol of compound (7).
- Examples of the amine derivative corresponding to Rb include 4-methoxybenzylamine or 2,4-dimethoxybenzylamine.
- This reaction is preferably carried out without solvent or using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent, but N, N-dimethylformamide, N, N-dimethylacetamide, N -Amides such as methylpyrrolidone, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 60 hours, preferably 15 minutes to 24 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 180 ° C.
- Compound (8) can be used in the next reaction as a reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, and can be isolated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Process G (deprotection reaction)
- Compound (1a) can be produced by deprotection reaction of compound (8) under acidic conditions.
- the acid is used in an amount of 1.0 to 50 mol, preferably 1.0 to 10 mol, per 1 mol of compound (8).
- the acid used include organic acids such as p-toluenesulfonic acid, pyridine p-toluenesulfonate, trifluoroacetic acid, formic acid and acetic acid, and inorganic acids such as hydrochloric acid and hydrobromic acid.
- This reaction is preferably carried out using no solvent or a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent, but alcohols such as methanol, ethanol, isopropyl alcohol, N, N— Preference is given to amides such as dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone, organic acids such as acetic acid, formic acid and trifluoroacetic acid, sulfoxides such as dimethyl sulfoxide, acetone, water or a mixed solvent thereof.
- the reaction time is usually 10 minutes to 60 hours, preferably 15 minutes to 24 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 180 ° C.
- Anisole can be added to this reaction as necessary, and 0.5 to 20 mol, preferably 1.0 to 10 mol, can be used per 1 mol of compound (8).
- Compound (1a) can be used as it is in the reaction solution or as a crude product for the next reaction, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Compound (6d) can be produced by the route shown in Scheme 5.
- Compound (6d) can be produced from compound (4d) or (5d) according to Step D in Scheme 1.
- Compound (4d) can be produced via compound H from compound (4c) obtained from compound (3c) by a method according to scheme B, step B.
- Compound (3c) can be produced by a method according to step E in scheme 3.
- Process H (demethylation reaction)
- Compound (4d) can be produced by demethylation reaction of compound (4c).
- pyridine hydrochloride is used in an amount of 1.0 to 100 mol, preferably 1.0 to 20.0 mol, per 1 mol of compound (4c).
- This reaction is preferably carried out without solvent or using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent, but N, N-dimethylformamide, N, N-dimethylacetamide, N Preferred are amides such as methylpyrrolidone, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, carboxylic acids such as acetic acid and formic acid, water, or a mixed solvent thereof.
- the reaction time is usually 15 minutes to 60 hours, preferably 30 minutes to 24 hours.
- the reaction temperature is usually 30 to 250 ° C, preferably 50 to 180 ° C.
- the compound (4d) can be used as it is in the reaction solution or as a crude product for the next reaction, but can be isolated from the reaction mixture according to a conventional method, and can be isolated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- a demethylation reaction using a Lewis acid can also be performed. Specifically, it is carried out using 1.0 to 10 moles, preferably 1.0 to 5.0 moles of Lewis acid per 1 mole of compound (4c).
- the Lewis acid include boron such as boron tribromide, boron trichloride, and boron trifluoride diethyl ether complex, and aluminum salts such as aluminum bromide and aluminum chloride.
- This reaction is preferably performed using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent, but halogenated hydrocarbons such as dichloromethane and 1,2-dichloroethane, toluene, And aromatic hydrocarbons such as xylene.
- Additives can be used for this reaction as needed, and sulfur compounds such as dimethyl sulfide and ethanethiol can be added as such additives.
- the reaction time is usually 10 minutes to 60 hours, preferably 30 minutes to 24 hours.
- the reaction temperature is usually -30 to 150 ° C, preferably 0 to 100 ° C.
- the compound (4d) can be used as it is in the reaction solution or as a crude product for the next reaction, but can be isolated from the reaction mixture according to a conventional method, and can be isolated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Rc represents a 4-methoxybenzyl group or a 2,4-dimethoxybenzyl group.
- Compound (6f) can be produced by the route shown in Scheme 6. That is, compound (4e) can be produced from compound (3e) by a method according to Step B (Scheme 1), and can be produced by a deprotection reaction according to Step G (Scheme 4). Compound (3e) can be produced by reacting compound (3b) according to Step E (Scheme 3) and an amine corresponding to Rc.
- Compound (2) can be produced by the route shown in Scheme 7, that is, the isothiocyanate reaction of compound (2h). Specifically, the reaction is carried out using about 1.0 to 5.0 moles, preferably about 1.0 to 2.0 moles of an isothiocyanating agent per mole of compound (2h).
- the isothiocyanating agent include thiophosgene, 1,1′-thiocarbonyldi-2 (1H) -pyridone, di-2-pyridylthionocarbonate, 1,1′-thiocarbonyldiimidazole and the like.
- the reaction can be performed in the presence of a base for the purpose of removing the released hydrogen halide from the reaction system.
- bases include inorganic bases such as sodium carbonate, potassium carbonate, and sodium bicarbonate, aromatic amines such as pyridine and lutidine, triethylamine, tripropylamine, tributylamine, cyclohexyldimethylamine, and 4-dimethylamino.
- aromatic amines such as pyridine, N, N-dimethylaniline, N-methylpiperidine, N-methylpyrrolidine and N-methylmorpholine are desirable.
- This reaction is preferably carried out using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, methanol, ethanol, propanol, 1,1-dimethylethanol.
- Alcohols such as benzene, toluene, saturated hydrocarbons such as cyclohexane and hexane, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, N, N-dimethylformamide, N, Amides such as N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, ketones such as acetone and methyl ethyl ketone, sulfoxides such as dimethyl sulfoxide, solvents such as water or a mixed solvent thereof are preferable.
- the reaction time is usually 10 minutes to 60 hours, preferably 15 minutes to 12 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 120 ° C.
- Compound (2) can be used as it is in the reaction solution or as a crude product for the next reaction, but can be isolated from the reaction mixture according to a conventional method, and can be easily separated by means of separation such as recrystallization, distillation, chromatography, etc. Can be purified.
- Compound (2h) can be easily obtained as a commercially available product, or can be produced according to a method known per se or a method analogous thereto.
- Compound (9) can be produced from compound (10) or compound (11) via compound (12) by the route shown in Scheme 8.
- Compound (12) can be produced by a substitution reaction using the base of compound (10) and an alcohol corresponding to R 4 .
- the base is used in an amount of about 1.0 to 10.0 mol, preferably about 1.0 to 5.0 mol
- the alcohol corresponding to R 4 is about 1.0 to 100.0 mol, preferably 1.0 to 2.0 mol, per 1 mol of compound (10).
- the base include inorganic bases such as sodium carbonate and potassium carbonate, and metal hydrides such as sodium hydride and potassium hydride.
- the alcohol include ethanol, 2,2,2-trifluoroethanol, cyclopropylmethanol, 2-propanol, 2-methylpropanol, 2,2,3,3,3-pentafluoropropanol and the like.
- This reaction is preferably carried out using no solvent or a solvent inert to the reaction.
- a solvent is not particularly limited as long as the reaction proceeds.
- aromatic hydrocarbons such as benzene and toluene
- ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, N, N-dimethylformamide
- solvents such as amides such as N, N-dimethylacetamide
- sulfoxides such as dimethyl sulfoxide, or mixed solvents thereof.
- the reaction time is usually 1 hour to 60 hours, preferably 5 hours to 12 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- (12) can also be produced by O-alkylation of compound (11) using a base and an alkylating agent corresponding to R 4 .
- the base is used in an amount of about 1.0 to 5.0 mol, preferably about 1.0 to 2.0 mol
- the alkylating agent corresponding to R 4 is about 1.0 to 10.0 mol, preferably about 1.0 to 3.0 mol, per 1 mol of compound (11).
- Bases include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogenated Examples thereof include metal hydrides such as potassium.
- Alkylating agents include various alkyl halides such as alkyl chloride, alkyl bromide and alkyl iodide and their derivatives, sulfonic acid esters such as p-toluenesulfonic acid ester and methylsulfonic acid ester, and sulfuric acid ester such as dimethylsulfuric acid. And the like.
- This reaction is preferably carried out using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol, propanol and 1,1-dimethylethanol
- aromatic hydrocarbons such as benzene and toluene, tetrahydrofuran, dioxane
- solvents such as ethers such as 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, sulfoxides such as dimethyl sulfoxide, and mixed solvents thereof.
- the reaction time is usually 1 hour to 60 hours, preferably 5 hours to 24 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- Compound (12) can be used in the next reaction as the reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Compound (9) can be synthesized by a reduction reaction of compound (12). Specifically, it is produced by reducing in a hydrogen atmosphere using about 0.01 to 5.0 mol, preferably about 0.01 to 2.0 mol, of a metal catalyst with respect to 1 mol of compound (12).
- a metal catalyst include palladium-carbon, palladium hydroxide-carbon, platinum oxide, platinum and the like. This reaction is preferably carried out using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol and propanol
- aromatic hydrocarbons such as benzene and toluene
- saturated hydrocarbons such as cyclohexane and hexane
- ethers such as dioxane and 1,2-dimethoxyethane
- amides such as N, N-dimethylformamide and N, N-dimethylacetamide
- sulfoxides such as dimethyl sulfoxide
- solvents such as water, or a mixed solvent thereof.
- the reaction time is usually 1 hour to 60 hours, preferably 5 hours to 36 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- the pressure is about 1 to 10 atmospheres, preferably about 1 to 5 atmospheres.
- reduction metal can be used.
- the reducing metal is used in an amount of about 5.0 to 20.0 mol, preferably about 5.0 to 10.0 mol, per 1 mol of compound (12).
- the reduced metal include reduced iron, tin, and zinc.
- hydrochloric acid or hydrochloride such as ammonium chloride or calcium chloride can be added. This reaction is preferably carried out using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol and propanol
- aromatic hydrocarbons such as benzene and toluene
- saturated hydrocarbons such as cyclohexane and hexane
- Ethers such as dioxane and 1,2-dimethoxyethane
- amides such as N, N-dimethylformamide and N, N-dimethylacetamide
- ketones such as acetone and methyl ethyl ketone
- sulfoxides such as dimethyl sulfoxide, aqueous ammonia, water Or a mixed solvent thereof.
- the reaction time is usually 1 hour to 60 hours, preferably 5 hours to 36 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- Compound (9) can be used as it is in the reaction solution or as a crude product for the next reaction, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- compound (10) and compound (11) commercially available ones can be easily obtained, or they can be produced according to a method known per se or a method analogous thereto.
- Compound (4j) can be produced by the route shown in Scheme 9. That is, it can be produced by a method according to Step A and Step B (Scheme 1), and further according to a method according to Step G (Scheme 4).
- Compound (4k) can be produced from compound (1k) by the route shown in Scheme 10. That is, it can be produced by a method according to Step A (Scheme 1), Step B (Scheme 1) and Step G (Scheme 4).
- Compound (6k) is produced from compound (4k) or compound (5k) produced by Step C (Scheme 1) using a method according to Step D (Scheme 1).
- Compound (1k) is produced by an oxidation reaction of compound (1j) (Step I).
- Oxidizing agent is used in an amount of 0.1 to 10.0 mol, preferably 0.5 to 5.0 mol, per 1 mol of compound (1j).
- the oxidizing agent include 2,3-dichloro-5,6-dicyano-1,4-benzoquinone, 2,3,5,6-tetrachloro-1,4-benzoquinone, palladium-carbon, and the like.
- This reaction is preferably carried out using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol and propanol
- aromatic hydrocarbons such as benzene, toluene and xylene
- saturated hydrocarbons such as cyclohexane and hexane
- amides such as N, N-dimethylformamide and N, N-dimethylacetamide
- sulfoxides such as dimethyl sulfoxide
- solvents such as water or mixed solvents thereof.
- the reaction time is usually 5 minutes to 60 hours, preferably 10 minutes to 12 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- Compound (1k) can be used in the next reaction as the reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Rd represents an optionally substituted C 1-6 alkyl group.
- Compound (14) is produced by O-alkylation of compound (6k) as shown in Scheme 11. That is, the alkylating agent corresponding to Rd is used in an amount of 1.0 to 5.0 mol, preferably 1.5 to 3.0 mol, per 1 mol of compound (6k).
- alkylating agents corresponding to Rd include borate complexes such as trimethyloxonium tetrafluoroborate and triethyloxonium tetrafluoroborate, various alkyl halides such as alkyl chloride, alkyl bromide, alkyl iodide and their derivatives, Examples thereof include sulfonic acid esters such as p-toluenesulfonic acid ester and methylsulfonic acid ester, and sulfuric acid esters such as dimethylsulfuric acid.
- a base can be used, and the base is used in an amount of 1.0 to 5.0 mol, preferably 1.1 to 3.0 mol, per 1 mol of compound (6k).
- Bases include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium bicarbonate, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, hydrogenation Examples thereof include metal hydrides such as sodium and potassium hydride, and organic bases such as triethylamine and N, N-diisopropylethylamine.
- This reaction is preferably carried out using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol and propanol
- aromatic hydrocarbons such as benzene and toluene
- halogenated compounds such as dichloromethane and 1,2-dichloroethane.
- a mixed solvent or the like is preferred.
- the reaction time is usually 1 hour to 60 hours, preferably 5 hours to 36 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- Compound (14) can be used as it is in the reaction solution or as a crude product for the next reaction, but can also be isolated from the reaction mixture according to a conventional method, and separated by means such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- compound (14) can be produced from compound (13) by a method according to step E (Scheme 3).
- Compound (13) can be produced by chlorination reaction of compound (6k). That is, the chlorinating agent is used in an amount of 1.0 to 50 mol, preferably 1.0 to 10 mol, per 1 mol of compound (6k).
- the chlorinating agent include acid chlorides such as phosphorus oxychloride, thionyl chloride, and oxalyl chloride.
- This reaction is preferably carried out using a chlorinating agent as a solvent or using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- aromatic hydrocarbons such as benzene and toluene
- halogenated hydrocarbons such as dichloromethane and 1,2-dichloroethane
- tetrahydrofuran dioxane
- a solvent such as an ether such as 2-dimethoxyethane or a mixed solvent thereof is preferable.
- the reaction time is usually 1 hour to 60 hours, preferably 5 hours to 36 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- Compound (13) can be used as it is in the reaction solution or as a crude product for the next reaction, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Compound (1j) can be produced by the route shown in Scheme 12. That is, the compound (16) can be produced by a reductive amination reaction, the compound (17) produced by the subsequent acylation reaction is cyclized, and the compound (18) can be produced by an imination reaction.
- Process J (reductive amination reaction)
- Compound (16) can be produced from 2,4-dimethoxybenzaldehyde and compound (15) by a reductive amination reaction using a reducing agent. More specifically, 2,4-dimethoxybenzaldehyde is used in an amount of 1.0 to 3.0 mol, preferably 1.0 to 1.5 mol, and the reducing agent is used in an amount of 1.0 to 5.0 mol, preferably 1,0 to 2.0 mol, per 1 mol of compound (15).
- the reducing agent include sodium borohydride, lithium borohydride, sodium triacetoxyborohydride and the like.
- This reaction is preferably carried out using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol and propanol, halogenated hydrocarbons such as dichloromethane and 1,2-dichloroethane, tetrahydrofuran, dioxane, 1,2 -Ethers such as dimethoxyethane, amides such as N, N-dimethylformamide, N, N-dimethylacetamide, sulfoxides such as dimethyl sulfoxide, nitriles such as acetonitrile and propionitrile, solvents such as acetic acid and water, or Those mixed solvents are preferred.
- the reaction time is usually 30 minutes to 60 hours, preferably 1 hour to 36 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- Compound (16) can be used as it is in the reaction solution or as a crude product for the next reaction, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Compound (17) can be produced by acylation reaction of compound (16). Specifically, C 1-6 alkyl chloroglyoxylate is used in an amount of 1.0 to 3.0 mol, preferably 1.0 to 1.5 mol, per 1 mol of compound (16).
- the reaction can be performed in the presence of a base for the purpose of removing hydrogen halide released in this reaction from the reaction system.
- the base is used in an amount of 1.0 to 5.0, preferably 1.0 to 3.0 mol based on compound (16).
- bases include inorganic bases such as sodium carbonate, potassium carbonate, and sodium bicarbonate, aromatic amines such as pyridine and lutidine, triethylamine, tripropylamine, tributylamine, cyclohexyldimethylamine, and 4-dimethylamino. It is desirable to add tertiary amines such as pyridine, N, N-dimethylaniline, N-methylpiperidine, N-methylpyrrolidine and N-methylmorpholine. This reaction is preferably carried out using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- halogenated hydrocarbons such as dichloromethane and 1,2-dichloroethane
- ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane
- N Solvents
- amides such as N-dimethylformamide and N, N-dimethylacetamide
- sulfoxides such as dimethyl sulfoxide
- nitriles such as acetonitrile and propionitrile, or a mixed solvent thereof are preferable.
- the reaction time is usually 30 minutes to 60 hours, preferably 1 hour to 36 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- Compound (17) can be used in the next reaction as the reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Compound (18) can be produced by Dieckmann-condensation of compound (17) under basic conditions. Specifically, the reaction is carried out using 1.0 to 10 mol, preferably 2.0 to 5.0 mol, of base with respect to 1 mol of compound (17).
- bases include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate and potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide, sodium hydride And metal hydrides such as potassium hydride.
- This reaction is preferably carried out using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol and propanol
- ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane
- N N-dimethylformamide
- Amides such as N-dimethylacetamide
- sulfoxides such as dimethyl sulfoxide
- nitriles such as acetonitrile and propionitrile, or a mixed solvent thereof is preferable.
- the reaction time is usually 30 minutes to 60 hours, preferably 1 hour to 36 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- Compound (18) can be used as it is in the reaction solution or as a crude product for the next reaction, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Compound (1j) can be produced by an imination reaction of compound (18). Specifically, it is carried out using 1.0 to 30 moles, preferably 1.0 to 20 moles of an ammonium salt per mole of the compound (18). Examples of ammonium salts include ammonium chloride, ammonium acetate, and ammonium formate. This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol and propanol, tetrahydrofuran, dioxane, 1, Ethers such as 2-dimethoxyethane, amides such as N, N-dimethylformamide, N, N-dimethylacetamide, sulfoxides such as dimethyl sulfoxide, solvents such as nitriles such as acetonitrile and propionitrile, acetic acid, formic acid Organic acids such as these or mixed solvents thereof are preferred.
- the reaction time is usually 30 minutes to 60 hours, preferably 1 hour to 36 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- Compound (1j) can be used as it is in the reaction solution or as a crude product for the next reaction, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. It can be easily purified.
- Compound (6m) can be produced by the route shown in Scheme 13. That is, the compound (4m) is produced from the compound (3m) by a method according to Step B (Scheme 1), and from the compound (5m) or the compound (5m) that can be produced by a method according to Step C (Scheme 1), Step D ( Compound (6m) can be produced by a method according to Scheme 1).
- Rf represents a C 1-6 alkyl group
- Rg represents a C 1-6 alkyl group
- Compound (3m) can be produced by the route shown in Scheme 14. That is, the compound (3m) can be produced by the cyclization reaction under the basic condition using the compound (2) via the compound (20) obtained by Michael addition of the compound (19) to the acrylate ester. it can.
- Compound (20) can be produced by a “Michael” addition reaction to an acrylate ester corresponding to Rg of Compound (19) in the presence of a base.
- the acrylate ester corresponding to Rg is used in an amount of 1.0 to 5.0 mol, preferably 1.0 to 2.0 mol, and the base is 2.0 to 5.0 mol, preferably 2.0 to 3.0 mol, per 1 mol of the compound (19).
- the base examples include inorganic bases such as sodium bicarbonate, sodium carbonate and potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide, metal hydrides such as sodium hydride and potassium hydride, And organic bases such as triethylamine and N, N-diisopropylethylamine.
- This reaction is preferably performed using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- halogenation such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons alcohols such as methanol, ethanol, propanol and 1,1-dimethylethanol, aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, N, N Preferred are amides such as dimethylformamide and N, N-dimethylacetamide, sulfoxides such as dimethyl sulfoxide, solvents such as water, or mixed solvents thereof.
- the reaction time is usually 30 minutes to 30 hours, preferably 45 minutes to 24 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 40 ° C.
- Compound (20) can be used as it is in the reaction solution or as a crude product for the next reaction, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. Can be purified.
- Compound (19) can be produced by a method known per se, for example, the method described in Chemical and Pharmaceutical Bulletin (Chem. Pharm. Bull.), Vol.43, Page 788 (1995) or a method analogous thereto. it can.
- Compound (3m) can be produced by addition reaction of compound (20) to compound (2) and cyclization reaction in the presence of a base.
- compound (2) is used in an amount of 1.0 to 3.0 mol, preferably 1.0 to 1.5 mol
- the base is used in an amount of 1.0 to 10 mol, preferably 2.0 to 5.0 mol, per 1 mol of compound (20).
- the base include inorganic bases such as sodium carbonate and potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide, and metal hydrides such as sodium hydride and potassium hydride.
- This reaction is preferably performed using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, Aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, and sulfoxides such as dimethyl sulfoxide Nitriles such as acetonitrile and propionitrile, solvents such as water, or mixed solvents thereof are preferable.
- the reaction time is usually 30 minutes to 30 hours, preferably 45 minutes to 24 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 100 ° C.
- the compound (3m) can be used as it is in the reaction solution or as a crude product for the next reaction, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. Can be purified.
- Compound (6n) can be produced by the route shown in Scheme 15. That is, it can be produced by cyclization reaction between compound (23) and mercaptoacetic acid, which can be produced by bromination reaction of compound (22).
- Compound (23) can be produced by bromination reaction of compound (22).
- the brominating agent is used in an amount of 1.0 mol to 3.0 mol, preferably 1.0 to 1.5 mol, relative to 1 mol of the compound (22).
- brominating agents include bromine, N-bromosuccinimide, and pyridinium bromide perbromide.
- sodium acetate etc. can be added as an additive for the purpose of adjusting the reactivity.
- This reaction is preferably carried out using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- a solvent inert for example, halogenation such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Solvents such as hydrocarbons, methanol and acetic acid, or mixed solvents thereof are preferred.
- the reaction time is usually 30 minutes to 30 hours, preferably 45 minutes to 5 hours.
- the reaction temperature is usually ⁇ 10 to 100 ° C., preferably 0 to 40 ° C.
- Compound (23) can be used in the next reaction as the reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. Can be purified.
- Compound (6n) can be produced by a cyclization reaction using compound (23) and mercaptoacetic acid. Specifically, the reaction is carried out using 1.0 to 10 mol, preferably 1.0 to 2.0 mol of mercaptoacetic acid per 1 mol of compound (23).
- This reaction is preferably performed using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- halogenated hydrocarbons such as carbon tetrachloride and 1,2-dichloroethane
- Aromatic hydrocarbons such as benzene and toluene
- solvents such as methanol and acetic acid, or mixed solvents thereof are preferred.
- the reaction time is usually 30 minutes to 30 hours, preferably 45 minutes to 5 hours.
- the reaction temperature is usually 0 to 300 ° C, preferably 10 to 190 ° C. Microwave irradiation can also be performed during heating.
- Compound (6n) can be used as it is in the reaction solution or as a crude product for the next reaction, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. Can be purified.
- Compound (22) can be produced by the route shown in Scheme 16. That is, compound (25) was produced from compound (24) by a cyclization reaction according to Step A (Scheme 1), S-alkylation according to Step B (Scheme 1), and Step G (Scheme 4). Compound (22) can be produced by a similar deprotection.
- Compound (24) can be produced by nucleophilic substitution reaction of compound (26) to compound (27). Specifically, compound (27) is used in an amount of 1.0 to 5.0 mol, preferably 1.0 to 3.0 mol, per 1 mol of compound (26). For the purpose of accelerating the reaction, acetic acid can be added in an amount of 1.0 to 5.0 mol, preferably 1.0 to 2.0 mol.
- This reaction is preferably carried out using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent, for example, alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, Aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, and sulfoxides such as dimethyl sulfoxide A solvent such as nitriles such as acetonitrile and propionitrile, or a mixed solvent thereof is preferable.
- a solvent for example, alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, Aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and
- the reaction time is usually 30 minutes to 30 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 100 ° C.
- Compound (24) can be used in the next reaction as the reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, and separated by means of separation such as washing, recrystallization, distillation, chromatography, etc. Can be purified.
- Compound (26) can be produced by a reductive amination reaction according to Step J (Scheme 12).
- M is a metal (for example, potassium, sodium, lithium, magnesium, calcium, copper, mercury, zinc, etc., which may be complexed)
- Compounds (6p) and (6r) can be produced by base treatment of compounds (6d) and (6m), respectively (Scheme 18).
- the base is used in an amount of 1.0 to 10 mol, preferably 2.0 to 5.0 mol, per 1 mol of the compounds (6d) and (6m).
- the base include inorganic bases such as sodium carbonate and potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide, and metal hydrides such as sodium hydride and potassium hydride.
- This reaction is preferably performed using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, Aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, and sulfoxides such as dimethyl sulfoxide Nitriles such as acetonitrile and propionitrile, solvents such as water, or mixed solvents thereof are preferable.
- the reaction time is usually 1 minute to 30 hours, preferably 3 minutes to 30 minutes.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 60 ° C.
- Compounds (6q) and (6s) can also be produced by treating compounds (6p) and (6r) with an alkylating agent corresponding to R 2 (Scheme 18).
- the alkylating agent corresponding to R 2 is used in an amount of 1.0 to 10 mol, preferably 1.0 to 3.0 mol, per 1 mol of the compounds (6p) and (6r).
- Alkylating agents corresponding to R 2 include various alkyl halides such as alkyl chloride, alkyl bromide and alkyl iodide and derivatives thereof, sulfonate esters such as p-toluenesulfonic acid ester and methylsulfonic acid ester, dimethyl Examples thereof include sulfuric acid esters such as sulfuric acid.
- This reaction is preferably carried out using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- a solvent inert for example, alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, Aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, acetonitrile, propionitrile, etc. Nitriles, sulfoxides such as dimethyl sulfoxide, water or a mixed solvent thereof are preferred.
- the reaction time is usually 15 minutes to 60 hours, preferably 15 minutes to 24 hours.
- the reaction temperature is usually ⁇ 10 to 200 ° C., preferably 0 to 150 ° C.
- Compounds (6q) and (6s) can be used as they are in the reaction solution or as a crude product for the next reaction, but can also be isolated from the reaction mixture according to conventional methods, washing, recrystallization, distillation, chromatography, etc. It can be easily purified by this separation means.
- Rh represents a halogen atom or a trifluoromethanesulfonyloxy group
- Ri represents a benzyl group which may be substituted with an alkyl group or an alkoxy group
- Rj represents an alkyl group or A benzophenone imino group which may be substituted with an alkoxy group is shown.
- compound (28) obtained in scheme 20 described later is converted into compound (29) or compound (31), then deprotected to give compound (30), and then subjected to a cyclization reaction to give compound (6t ).
- Compound (29) or compound (31) is produced by a coupling reaction between compound (28) and the corresponding nitrogen-containing reagent.
- the amine reagent or imine reagent is 1.0 to 10.0 mol, preferably 1.0 to 3.0 mol
- the organometallic reagent is 0.01 to 1 mol, preferably 0.1 mol, per 1 mol of the compound (28).
- the phosphine ligand is used in an amount of 0.01 to 1 mol, preferably 0.1 to 0.5 mol, the base 1.0 to 10.0 mol, preferably 2.0 to 6.0 mol. To do. In some cases, a phosphine ligand may not be used.
- benzylamine dibenzylamine, 2-methoxybenzylamine, 3-methoxybenzylamine, 4-methoxybenzylamine, 2,3-dimethoxybenzylamine, 2,4-dimethoxybenzylamine, 3,4-dimethoxy Benzylamine, 2,4,6-trimethoxybenzylamine, 3,4,5-trimethoxybenzylamine, 2,3,4-trimethoxybenzylamine, 2,4,5-trimethoxybenzylamine, 2-methyl Benzylamine, 3-methylbenzylamine, 4-methylbenzylamine, 2,3-dimethylbenzylamine, 2,4-dimethylbenzylamine, 3,4-dimethylbenzylamine, 2,4,6-trimethylbenzylamine, 2 , 4,5-trimethylbenzylamine and the like.
- organometallic reagents include tris (dibenzylideneacetone) dipalladium (0), tetrakis (triphenylphosphine) palladium, [1,1′-bis (diphenylphosphino) ferrocene] palladium (II) dichloride dichloromethane complex, palladium acetate, etc. Is mentioned.
- Bases include sodium hydroxide, potassium hydroxide, barium hydroxide, sodium bicarbonate, sodium carbonate, potassium carbonate, sodium acetate, potassium acetate, cesium carbonate and other basic salts, sodium methoxide, sodium ethoxide, potassium Examples thereof include metal alkoxides such as butoxide, and metal hydrides such as sodium hydride and potassium hydride.
- This reaction is preferably carried out using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane) Etc.
- alcohols eg, methanol, ethanol, etc.
- esters eg, ethyl acetate, etc.
- aromatic hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- Amides eg, N, N-dimethylformamide, N, N-dimethylacetamide, etc.
- halogenated hydrocarbons eg, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, Acetonitrile, propionitrile, etc.
- sulfoxides eg, dimethyl sulfoxide, etc.
- organic acids eg, acetic acid, etc.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 0 ° C. to 300 ° C., preferably 20 ° C. to 200 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (30) is produced by acid-treating or hydrogenating compound (29).
- the acid reagent is used in an amount of 1.0 to 200 mol, preferably 3.0 to 20.0 mol, per 1 mol of compound (29).
- the acid reagent include Lewis acids such as aluminum chloride, organic acids such as acetic acid and trifluoroacetic acid, and mineral acids such as hydrochloric acid.
- the amount of the metal reagent used is 5% to 1000% by weight, preferably 10% to 300% by weight, based on the compound (29).
- the metal reagent include palladium carbon, palladium hydroxide, platinum oxide, Raney nickel, Raney cobalt and the like.
- the hydrogen pressure is usually 1 to 100 atm.
- This reaction is preferably performed in the absence of a solvent or in the presence of a suitable solvent.
- the solvent is not particularly limited as long as the reaction proceeds.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane etc.
- alcohols eg, methanol, ethanol etc.
- esters eg, , Ethyl acetate, etc.
- aromatic hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- amides eg, N, N-dimethylformamide, N, N-dimethylacetamide
- halogenated hydrocarbons eg, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, acet
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 25 ° C to 300 ° C, preferably 50 ° C to 200 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (30) is produced by acid treatment of compound (31).
- the acid reagent is used in an amount of 1.0 to 200 mol, preferably 3.0 to 20.0 mol, per 1 mol of compound (31).
- the acid reagent include Lewis acids such as aluminum chloride, organic acids such as acetic acid and trifluoroacetic acid, and mineral acids such as hydrochloric acid.
- This reaction is preferably performed in the absence of a solvent or in the presence of a suitable solvent.
- the solvent is not particularly limited as long as the reaction proceeds.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane etc.
- alcohols eg, methanol, ethanol etc.
- esters eg, , Ethyl acetate, etc.
- aromatic hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- amides eg, N, N-dimethylformamide, N, N-dimethylacetamide) Etc.
- halogenated hydrocarbons eg, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, acetonitrile, propionitrile, etc.
- sulfoxides eg, dimethyl sulfoxide, etc.
- Organic acids eg, acetic acid, etc.
- the reaction time is usually about 10 minutes to about 50 hours, preferably about 30 minutes to about 12 hours.
- the reaction temperature is usually 25 ° C to 300 ° C, preferably 50 ° C to 200 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (6t) can be produced by a substitution reaction using an electrophile corresponding to compound (30) followed by a cyclization reaction. Specifically, the electrophile is used in an amount of 1.0 to 20.0 mol, preferably 1.0 to 5.0 mol, per 1 mol of compound (30).
- electrophiles prop-2-enoyl chloride, 2-methylprop-2-enoyl chloride, (2E) -but-2-enoyl chloride, (2Z) -but-2-enoyl chloride, (2E)- Examples include 2-methylbut-2-enoyl chloride, (2Z) -2-methylbut-2-enoyl chloride, and the like.
- This reaction is preferably performed in the absence of a solvent or in the presence of a solvent inert to the reaction.
- the solvent is not particularly limited as long as the reaction proceeds.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane etc.
- alcohols eg, methanol, ethanol etc.
- esters eg, , Ethyl acetate, etc.
- aromatic hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- amides eg, N, N-dimethylformamide, N, N-dimethylacetamide
- Etc. halogenated hydrocarbons
- dichloromethane chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, acetonitrile, propionitrile
- the reaction time is usually about 1 minute to about 100 hours, preferably about 5 minutes to about 4 hours.
- the reaction temperature is generally about 0 ° C to about 200 ° C, preferably about 0 ° C to about 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- compound (28), which is the starting compound of scheme 19, is obtained from compound (32) obtained in scheme 21 described later via compound (33).
- Compound (33) is produced by a cyclization reaction performed under basic conditions of compound (32) and dialkyl malonate. Specifically, the dialkyl malonate is used in an amount of 1.0 to 10.0 mol, preferably 1.0 to 3.0 mol, and the base 1.0 to 100.0 mol, preferably 2. 0 to 10.0 moles are used.
- Bases include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogenated Examples thereof include metal hydrides such as potassium, organic amines such as triethylamine, pyridine, diisopropylethylamine, and 1,8-diazabicyclo [5.4.0] undec-7-ene.
- This reaction is preferably carried out using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane) Etc.
- alcohols eg, methanol, ethanol, etc.
- esters eg, ethyl acetate, etc.
- aromatic hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- Amides eg, N, N-dimethylformamide, N, N-dimethylacetamide, etc.
- halogenated hydrocarbons eg, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, Acetonitrile, propionitrile, etc.
- sulfoxides eg, dimethyl sulfoxide, etc.
- organic acids eg, acetic acid, etc.
- the reaction time is usually 10 minutes to 72 hours, preferably 15 minutes to 24 hours.
- the reaction temperature is usually 0 ° C. to 150 ° C., preferably 0 ° C. to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (28) is produced by the substitution reaction of compound (33). Specifically, the reaction is performed using 1.0 to 100.0 mol, preferably 3.0 to 10.0 mol, of phosphorus oxychloride or phosphorus oxybromide with respect to 1 mol of compound (33). This reaction is advantageously performed in the absence of a solvent or in the presence of a solvent inert to the reaction.
- the solvent is not particularly limited as long as the reaction proceeds.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane etc.
- esters eg, ethyl acetate etc.
- aromatic hydrocarbons Eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- amides eg, N, N-dimethylformamide, N, N-dimethylacetamide, etc.
- halogenated hydrocarbons eg, Dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, acetonitrile, propionitrile, etc.
- sulfoxides eg, dimethyl sulfoxide, etc.
- the reaction time is usually 10 minutes to 72 hours, preferably 30 minutes to 3 hours.
- the reaction temperature is usually 0 ° C. to 150 ° C., preferably 0 ° C. to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- a trifluoromethylsulfonylating reagent As another method of compound (28), 1.0 to 10.0 mol, preferably 1.0 to 3.0 mol of a trifluoromethylsulfonylating reagent is used per 1 mol of compound (33), and 1 base is added. The reaction is carried out using 0.0 to 20.0 mol, preferably 1.0 to 10.0 mol.
- the trifluoromethyl sulfonation reagent include 1,1,1-trifluoro-N-phenyl-N-[(trifluoromethyl) sulfonyl] methanesulfonamide, 2- [N, N-bis (trifluoromethylsulfonyl).
- Bases include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogenated Examples thereof include metal hydrides such as potassium, organic amines such as triethylamine, pyridine, diisopropylethylamine, and 1,8-diazabicyclo [5.4.0] undec-7-ene.
- inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogenated
- metal hydrides such as potassium, organic amines such as triethylamine, pyridine, diisopropylethylamine, and 1,8-diazabicyclo [5.4.0] undec-7
- This reaction is preferably carried out using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane) Etc.
- esters eg, ethyl acetate, etc.
- aromatic hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- amides eg, N, N-dimethylformamide) N, N-dimethylacetamide, etc.
- halogenated hydrocarbons eg, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, acetonitrile, propionitrile, etc.
- sulfoxides Example, dimethyl sulfoxide,
- the reaction time is usually 10 minutes to 72 hours, preferably 15 minutes to 24 hours.
- the reaction temperature is usually -78 ° C to 100 ° C, preferably 0 ° C to 50 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Rk represents an optionally substituted C 1-6 alkyl group.
- compound (32), which is the starting compound of scheme 20, is obtained from compound (34).
- Compound (34) which is commercially available can be used as it is, and can be produced according to a method known per se or a method analogous thereto.
- Compound (35) is obtained by substitution reaction of compound (34). Specifically, 1.0 to 10.0 mol, preferably 1.0 to 3.0 mol of a chlorinating agent is used with respect to 1 mol of the compound (34), and 28% aqueous ammonia solution is added to 1.0 to 20.0 mol. Preferably, 1.0 to 3.0 mol is used.
- 0.001 to 10.0 mol, preferably 0.001 to 3.0 mol of pyridine, dicyclohexylamine, N, N-dimethylformamide, phase transfer catalyst, etc. may be used as necessary.
- the chlorinating agent include oxalyl chloride, thionyl chloride, phosphorus oxychloride and the like. This reaction is preferably carried out using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane) Etc.
- esters eg, ethyl acetate, etc.
- aromatic hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- amides eg, N, N-dimethylformamide
- N-dimethylacetamide eg.
- halogenated hydrocarbons eg, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, acetonitrile, propionitrile, etc.
- sulfoxides Example, dimethyl sulfoxide, etc.
- sulfoxides Example, dimethyl sulfoxide, etc.
- the reaction time is usually 10 minutes to 72 hours, preferably 30 minutes to 24 hours.
- the reaction temperature is usually ⁇ 78 ° C. to 100 ° C., preferably ⁇ 10 ° C. to 25 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (36) which is commercially available can be used as it is, and can be produced according to the method described in the Examples, a method known per se or a method analogous thereto. For example, it is produced by O-alkylation reaction of compound (35). Specifically, the corresponding alkylating agent is used in an amount of 1.0 to 50.0 mol, preferably 1.0 to 10.0 mol, based on 1 mol of compound (35), and the base is 1.0 to 100.0 mol, preferably It is carried out using 3.0 to 10.0 mol.
- alkylating agent examples include trimethyloxonium tetrafluoroborate, dimethyl sulfate, methyl trifluoromethanesulfonate, methylfluorosulfonate, and the like.
- Bases include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen phosphate, sodium phosphate, sodium methoxide, sodium ethoxide, potassium tert-butoxide Metal alkoxides and the like. This reaction is preferably carried out using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane) Etc.
- esters eg, ethyl acetate, etc.
- aromatic hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- amides eg, N, N-dimethylformamide
- N-dimethylacetamide eg.
- halogenated hydrocarbons eg, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, acetonitrile, propionitrile, etc.
- sulfoxides Example, dimethyl sulfoxide, etc.
- sulfoxides Example, dimethyl sulfoxide, etc.
- the reaction time is usually 10 minutes to 72 hours, preferably 30 minutes to 24 hours.
- the reaction temperature is usually ⁇ 78 ° C. to 100 ° C., preferably ⁇ 10 ° C. to 25 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (37) is produced by a substitution reaction from compound (35).
- the nucleophilic agent is used in an amount of 1.0 to 3.0 mol, preferably 1.0 to 1.30 mol, relative to 1 mol of the compound (35).
- the nucleophilic agent include Lawson's reagent, phosphorus pentasulfide, and phosphorus pentasulfide. This reaction is preferably carried out using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane) Etc.
- alcohols eg, methanol, ethanol, etc.
- esters eg, ethyl acetate, etc.
- aromatic hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- Amides eg, N, N-dimethylformamide, N, N-dimethylacetamide, etc.
- halogenated hydrocarbons eg, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, Acetonitrile, propionitrile, etc.
- sulfoxides eg, dimethyl sulfoxide, etc.
- organic acids eg, acetic acid, etc.
- the reaction time is usually 10 minutes to 72 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually 0 ° C. to 150 ° C., preferably 25 ° C. to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (38) is produced by an S-alkylation reaction using compound (37) and a corresponding alkylating agent.
- the alkylating agent is used in an amount of 1.0 to 10.0 mol, preferably 1.0 to 5.0 mol, per 1 mol of compound (37).
- Alkylating agents include various alkyl halides such as alkyl chloride, alkyl bromide and alkyl iodide and derivatives thereof, sulfonic acid esters such as p-toluenesulfonic acid ester and methylsulfonic acid ester, and sulfuric acid ester such as dimethylsulfuric acid. And the like.
- This reaction is preferably carried out using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane) Etc.
- alcohols eg, methanol, ethanol, etc.
- esters eg, ethyl acetate, etc.
- aromatic hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- Amides eg, N, N-dimethylformamide, N, N-dimethylacetamide, etc.
- halogenated hydrocarbons eg, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, Acetonitrile, propionitrile, etc.
- the reaction time is usually 15 minutes to 60 hours, preferably 30 minutes to 24 hours.
- the reaction temperature is usually 0 ° C. to 150 ° C., preferably 25 ° C. to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (32) can be obtained by substitution reaction from compound (36) or compound (38). Specifically, the compound (2h) is used in an amount of 1.0 to 20.0 mol, preferably 1.0 to 2.0 mol, and the base is used in an amount of 1.0 to 20 with respect to 1 mol of the compound (36) or the compound (38). The reaction is carried out using 0.0 mol, preferably 1.0 to 10.0 mol.
- Bases include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogenated Examples thereof include metal hydrides such as potassium, organic amines such as triethylamine, pyridine, diisopropylethylamine, and 1,8-diazabicyclo [5.4.0] undec-7-ene.
- a base is not required for the reaction, it can be produced without using a base. This reaction is preferably carried out using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane) Etc.
- alcohols eg, methanol, ethanol, etc.
- esters eg, ethyl acetate, etc.
- aromatic hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- Amides eg, N, N-dimethylformamide, N, N-dimethylacetamide, etc.
- halogenated hydrocarbons eg, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, Acetonitrile, propionitrile, etc.
- sulfoxides eg, dimethyl sulfoxide, etc.
- organic acids eg, acetic acid, etc.
- the reaction time is usually 30 minutes to 100 hours, preferably 1 hour to 72 hours.
- the reaction temperature is usually 0 ° C. to 150 ° C., preferably 25 ° C. to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Rm represents an optionally substituted C 1-6 alkyl group.
- compound (6v) is obtained from compound (6u).
- Compound (6v) is produced by an alkylation reaction of compound (6u). Specifically, the corresponding alkylating agent is used in an amount of 1.0 to 50.0 mol, preferably 1.0 to 3.0 mol, based on 1 mol of the compound (6u), and the base is 1.0 to 100.0 mol, preferably 2 0.0 to 20.0 mol is used.
- Alkylating agents include various alkyl halides such as alkyl chloride, alkyl bromide and alkyl iodide and derivatives thereof, sulfonic acid esters such as p-toluenesulfonic acid ester and methylsulfonic acid ester, and sulfuric acid ester such as dimethylsulfuric acid. And the like.
- the base include inorganic bases such as sodium carbonate and potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide, and metal hydrides such as sodium hydride and potassium hydride. This reaction uses a solvent inert to the reaction.
- the reaction is not particularly limited as long as the reaction proceeds, but this reaction is preferably performed using a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- a solvent inert to the reaction, and is not particularly limited as long as the reaction proceeds as such a solvent.
- diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane, etc. alcohols (eg, methanol, ethanol, etc.), esters (eg, ethyl acetate, etc.), aromatic hydrocarbons (eg, benzene, toluene) Etc.), aliphatic hydrocarbons (eg, hexane, etc.), amides (eg, N, N-dimethylformamide, N, N-dimethylacetamide, etc.), halogenated hydrocarbons (eg, dichloromethane, chloroform, tetrachloride) Carbon, 1,2-dichloroethan
- the reaction time is usually 15 minutes to 100 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually ⁇ 10 ° C. to 200 ° C., preferably 0 ° C. to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- amino protecting groups include formyl group, C 1-6 alkyl-carbonyl group, C 1-6 alkoxy-carbonyl group, benzoyl group, C 7-10 aralkyl-carbonyl group (eg, benzylcarbonyl), C 7 -14 Aralkyloxy-carbonyl groups (eg, benzyloxycarbonyl, 9-fluorenylmethoxycarbonyl), trityl groups, phthaloyl groups, N, N-dimethylaminomethylene groups, substituted silyl groups (eg, trimethylsilyl, triethylsilyl, dimethyl) Phenylsilyl, tert-butyldimethylsilyl, tert-butyldiethylsilyl), C 2-6 alkenyl group (eg, 1-allyl), substituted C 7-10 aralkyl group (eg, 2,4-dimethoxybenzyl) and the like. It is done. These groups may be substituted with
- Examples of the carboxy protecting group include a C 1-6 alkyl group, a C 7-11 aralkyl group (eg, benzyl), a phenyl group, a trityl group, a substituted silyl group (eg, trimethylsilyl, triethylsilyl, dimethylphenylsilyl, tert -Butyldimethylsilyl, tert-butyldiethylsilyl), C 2-6 alkenyl group (eg, 1-allyl) and the like. These groups may be substituted with 1 to 3 substituents selected from a halogen atom, a C 1-6 alkoxy group and a nitro group.
- hydroxy protecting group examples include a C 1-6 alkyl group, a phenyl group, a trityl group, a C 7-10 aralkyl group (eg, benzyl), a formyl group, a C 1-6 alkyl-carbonyl group, a benzoyl group, C 7-10 aralkyl-carbonyl group (eg, benzylcarbonyl), 2-tetrahydropyranyl group, 2-tetrahydrofuranyl group, substituted silyl group (eg, trimethylsilyl, triethylsilyl, dimethylphenylsilyl, tert-butyldimethylsilyl, tert- Butyldiethylsilyl), C 2-6 alkenyl groups (eg, 1-allyl) and the like. These groups may be substituted with 1 to 3 substituents selected from a halogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy
- Examples of the protected carbonyl include cyclic acetals (eg, 1,3-dioxane), acyclic acetals (eg, di-C 1-6 alkylacetal) and the like.
- Examples of mercapto-protecting groups include C 1-6 alkyl groups, phenyl groups, trityl groups, C 7-10 aralkyl groups (eg, benzyl), C 1-6 alkyl-carbonyl groups, benzoyl groups, C 7-10.
- Aralkyl-carbonyl group eg, benzylcarbonyl
- C 1-6 alkoxy-carbonyl group C 6-14 aryloxy-carbonyl group (eg, phenyloxycarbonyl), C 7-14 aralkyloxy-carbonyl group (eg, benzyl) Oxycarbonyl, 9-fluorenylmethoxycarbonyl), 2-tetrahydropyranyl group, C 1-6 alkylamino-carbonyl group (eg, methylaminocarbonyl, ethylaminocarbonyl) and the like.
- These groups may be substituted with 1 to 3 substituents selected from a halogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group or a nitro group.
- the method for removing the protecting group described above can be carried out according to a method known per se, for example, the method described in Protective Groups in Organic Synthesis, published by John Wiley and Sons (1980). . Specifically, acid, base, ultraviolet light, hydrazine, phenylhydrazine, sodium N-methyldithiocarbamate, tetrabutylammonium fluoride, palladium acetate, trialkylsilyl halide (eg, trimethylsilyl iodide, trimethylsilyl bromide), etc. are used. And a reduction method.
- a method known per se for example, the method described in Protective Groups in Organic Synthesis, published by John Wiley and Sons (1980). . Specifically, acid, base, ultraviolet light, hydrazine, phenylhydrazine, sodium N-methyldithiocarbamate, tetrabutylammonium fluoride, palladium acetate, trialkylsily
- the compound (I) obtained by each of the above production methods can be isolated and purified by known means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, transfer dissolution, chromatography and the like. Moreover, each raw material compound used in each of the above production methods can be isolated and purified by the same known means as described above. On the other hand, you may use these raw material compounds as a reaction mixture as it is as a raw material for the next step without isolation.
- compound (I) has an isomer such as an optical isomer, a stereoisomer, a positional isomer, or a rotational isomer
- any one isomer or a mixture of isomers is included in compound (I). Is done.
- compound (I) has an optical isomer
- the optical isomer resolved from the racemate is also encompassed in compound (I).
- Each of these isomers can be obtained as a single product by a known synthesis method or separation method (concentration, solvent extraction, column chromatography, recrystallization, etc.).
- Compound (I) or a prodrug thereof (hereinafter, sometimes simply abbreviated as the compound of the present invention) has low toxicity and should be used as it is or mixed with a pharmacologically acceptable carrier to form a pharmaceutical composition.
- a pharmaceutical composition e.g, a preventive or therapeutic agent for various diseases described below for mammals (eg, humans, mice, rats, rabbits, dogs, cats, cows, horses, pigs, monkeys).
- the pharmacologically acceptable carrier various organic or inorganic carrier substances commonly used as pharmaceutical materials are used, and excipients, lubricants, binders, disintegrants in solid preparations; solvents in liquid preparations , Solubilizing agents, suspending agents, isotonic agents, buffers, soothing agents and the like. If necessary, preparation additives such as preservatives, antioxidants, colorants, sweeteners and the like can also be used.
- excipients include lactose, sucrose, D-mannitol, D-sorbitol, starch, pregelatinized starch, dextrin, crystalline cellulose, low-substituted hydroxypropylcellulose, sodium carboxymethylcellulose, gum arabic, pullulan, light
- excipients include anhydrous silicic acid, synthetic aluminum silicate, and magnesium aluminate metasilicate.
- lubricant examples include magnesium stearate, calcium stearate, talc and colloidal silica.
- Preferred examples of the binder include pregelatinized starch, sucrose, gelatin, gum arabic, methylcellulose, carboxymethylcellulose, sodium carboxymethylcellulose, crystalline cellulose, sucrose, D-mannitol, trehalose, dextrin, pullulan, hydroxypropylcellulose, hydroxy Examples include propylmethylcellulose and polyvinylpyrrolidone.
- disintegrant examples include lactose, sucrose, starch, carboxymethyl cellulose, carboxymethyl cellulose calcium, croscarmellose sodium, carboxymethyl starch sodium, light anhydrous silicic acid, and low-substituted hydroxypropyl cellulose.
- Suitable examples of the solvent include water for injection, physiological saline, Ringer's solution, alcohol, propylene glycol, polyethylene glycol, sesame oil, corn oil, olive oil, and cottonseed oil.
- solubilizer examples include polyethylene glycol, propylene glycol, D-mannitol, trehalose, benzyl benzoate, ethanol, trisaminomethane, cholesterol, triethanolamine, sodium carbonate, sodium citrate, sodium salicylate, sodium acetate. Is mentioned.
- suspending agent examples include surfactants such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalkonium chloride, benzethonium chloride, glyceryl monostearate; polyvinyl alcohol, polyvinylpyrrolidone , Hydrophilic polymers such as sodium carboxymethylcellulose, methylcellulose, hydroxymethylcellulose, hydroxyethylcellulose, hydroxypropylcellulose; polysorbates, and polyoxyethylene hydrogenated castor oil.
- surfactants such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalkonium chloride, benzethonium chloride, glyceryl monostearate
- polyvinyl alcohol, polyvinylpyrrolidone Hydrophilic polymers such as sodium carboxymethylcellulose, methylcellulose, hydroxymethylcellulose, hydroxyethylcellulose,
- Preferable examples of the isotonic agent include sodium chloride, glycerin, D-mannitol, D-sorbitol and glucose.
- buffers such as phosphate, acetate, carbonate and citrate.
- a preferred example of the soothing agent is benzyl alcohol.
- Preferable examples of the preservative include p-hydroxybenzoates, chlorobutanol, benzyl alcohol, phenethyl alcohol, dehydroacetic acid and sorbic acid.
- Preferable examples of the antioxidant include sulfite and ascorbate.
- the colorant examples include water-soluble edible tar dyes (eg, edible dyes such as edible red Nos. 2 and 3, edible yellows Nos. 4 and 5, edible blue Nos. 1 and 2, etc.), water-insoluble lake dyes (Eg, the aluminum salt of the water-soluble edible tar dye) and natural dyes (eg, ⁇ -carotene, chlorophyll, bengara).
- water-soluble edible tar dyes eg, edible dyes such as edible red Nos. 2 and 3, edible yellows Nos. 4 and 5, edible blue Nos. 1 and 2, etc.
- water-insoluble lake dyes Eg, the aluminum salt of the water-soluble edible tar dye
- natural dyes eg, ⁇ -carotene, chlorophyll, bengara
- Suitable examples of sweeteners include saccharin sodium, dipotassium glycyrrhizinate, aspartame, and stevia.
- the medicament containing the compound of the present invention can be used alone or mixed with a pharmacologically acceptable carrier according to a method known per se as a method for producing a pharmaceutical preparation (eg, a method described in the Japanese Pharmacopoeia).
- tablets including sugar-coated tablets, film-coated tablets, sublingual tablets, orally disintegrating tablets, buccal tablets, etc.
- pills powders, granules, capsules (including soft capsules and microcapsules), troches Agent, syrup, solution, emulsion, suspension, controlled release formulation (eg, immediate release formulation, sustained release formulation, sustained release microcapsule), aerosol, film agent (eg, orally disintegrating film, Oral mucosa adhesive film), injection (eg, subcutaneous injection, intravenous injection, intramuscular injection, intraperitoneal injection), drip, transdermal preparation, ointment, lotion, patch, sitting Suppositories (eg, rectal suppositories) Vaginal suppositories), pellets,
- the pharmaceutical composition can be produced by a method commonly used in the field of pharmaceutical technology, for example, a method described in the Japanese Pharmacopoeia.
- the content of the compound of the present invention in the pharmaceutical composition varies depending on the dosage form, the dose of the compound of the present invention, etc., but is, for example, about 0.1 to 100% by weight.
- coating may be performed for the purpose of taste masking, enteric properties or sustainability.
- coating base used for coating examples include sugar coating base, water-soluble film coating base, enteric film coating base and sustained-release film coating base.
- sucrose is used, and one or more selected from talc, precipitated calcium carbonate, gelatin, gum arabic, pullulan, carnauba wax and the like may be used in combination.
- water-soluble film coating base examples include cellulose polymers such as hydroxypropylcellulose, hydroxypropylmethylcellulose, hydroxyethylcellulose, and methylhydroxyethylcellulose; polyvinyl acetal diethylaminoacetate, aminoalkyl methacrylate copolymer E [Eudragit E (trade name) ], Synthetic polymers such as polyvinylpyrrolidone; polysaccharides such as pullulan.
- enteric film coating bases include cellulose polymers such as hydroxypropylmethylcellulose phthalate, hydroxypropylmethylcellulose acetate succinate, carboxymethylethylcellulose, and cellulose acetate phthalate; methacrylic acid copolymer L [Eudragit L (trade name) ] Acrylic acid polymers such as methacrylic acid copolymer LD [Eudragit L-30D55 (trade name)], methacrylic acid copolymer S [Eudragit S (trade name)]; natural products such as shellac.
- cellulose polymers such as hydroxypropylmethylcellulose phthalate, hydroxypropylmethylcellulose acetate succinate, carboxymethylethylcellulose, and cellulose acetate phthalate
- methacrylic acid copolymer L (Eudragit L (trade name) ]
- Acrylic acid polymers such as methacrylic acid copolymer LD [Eudragit L-30D55 (trade name)], methacrylic acid copolymer
- sustained-release film coating base examples include cellulose polymers such as ethyl cellulose; aminoalkyl methacrylate copolymer RS [Eudragit RS (trade name)], ethyl acrylate-methyl methacrylate copolymer suspension [Eudragit Acrylic polymer such as NE (trade name)].
- cellulose polymers such as ethyl cellulose; aminoalkyl methacrylate copolymer RS [Eudragit RS (trade name)], ethyl acrylate-methyl methacrylate copolymer suspension [Eudragit Acrylic polymer such as NE (trade name)].
- the above-mentioned coating bases may be used by mixing two or more of them in an appropriate ratio. Moreover, you may use light-shielding agents, such as a titanium oxide, ferric oxide, etc. in the case of coating.
- the compound of the present invention has low toxicity (eg, acute toxicity, chronic toxicity, genotoxicity, reproductive toxicity, cardiotoxicity, carcinogenicity), few side effects, and mammals (eg, humans, cows, horses, dogs, cats, Monkeys, mice, rats) can be used as preventive or therapeutic agents for various diseases described later, or as diagnostic agents.
- toxicity eg, acute toxicity, chronic toxicity, genotoxicity, reproductive toxicity, cardiotoxicity, carcinogenicity
- mammals eg, humans, cows, horses, dogs, cats, Monkeys, mice, rats
- the compound of the present invention has a potent delta-5-saturase inhibitory action, the prevention or prevention of a disease (or a disease whose onset is promoted) associated with an eicosanoid produced via delta-5-desaturase Useful as a therapeutic agent.
- diseases include heart diseases (cardiac hypertrophy, chronic heart failure including acute heart failure and congestion, cardiomyopathy, angina pectoris, myocarditis, arrhythmia, tachycardia, myocardial infarction, etc.), myocardial ischemia, vein Dysfunction, heart failure transition after myocardial infarction, hypertension, pulmonary heart, atherosclerosis including atherosclerosis (aneurysm, coronary sclerosis, cerebral arteriosclerosis, peripheral arteriosclerosis, etc.), vascular thickening, intervention ( Percutaneous coronary angioplasty, stenting, coronary endoscopy, intravascular ultrasound, coronary thrombolysis, etc.) and vascular thickening or occlusion and organ damage after heart transplantation, vascular re-occlusion / restenosis after bypass surgery , Respiratory disease (cold syndrome, pneumonia, asthma, pulmonary hypertension, pulmonary thrombus / pulmonary embolism, etc.), bone disease (fracture, re
- the compound of the present invention is particularly preferably used for the prevention or treatment of arteriosclerosis, diabetes and obesity.
- arteriosclerosis includes ischemic heart disease (unstable angina pectoris, acute myocardial infarction, acute heart failure, heart death) and stroke fistula (transient brain failure) Prevention of so-called atherothrombosis such as blood, etc., delay of their severity, risk of high incidence of cardiovascular events based on the effect of inhibiting the progression of arteriosclerosis (Acute coronary artery disease patient, stroke patient, metabolic disease patient, hypertension / obesity) -Prevention of the onset of cardiovascular events, prevention of recurrence of ischemic heart disease, prevention of primary onset of cardiovascular events, prevention or treatment of peripheral arterial angiopathy, etc.
- arteriosclerosis acute coronary artery disease patient, stroke patient, metabolic disease patient, hypertension / obesity
- diabetes is a fasting blood glucose level (glucose concentration in venous plasma) of 126 mg / dl or higher, and a 75 g oral glucose tolerance test (75 gOGTT) 2-hour value (glucose concentration in venous plasma) of 200 mg / dl or higher.
- 75 gOGTT 75 g oral glucose tolerance test
- a fasting blood glucose level (glucose concentration in venous plasma) is less than 110 mg / dl or a 75 g oral glucose tolerance test (75 g OGTT) 2 hour value (glucose concentration in venous plasma) is 140 mg / dl.
- a state that is not “a state indicating less than dl” (normal type) is referred to as a “boundary type”.
- diabetes is a fasting blood glucose level (glucose concentration in venous plasma) of 126 mg / dl or more, and a 75 g oral glucose tolerance test 2 hour value (glucose concentration in venous plasma) is 200 mg / dl. This is a state showing dl or more.
- glucose intolerance is a fasting blood glucose level (glucose concentration in venous plasma) of less than 126 mg / dl, and a 75-g oral glucose tolerance test 2 hour value (glucose concentration in venous plasma). Is a state showing 140 mg / dl or more and less than 200 mg / dl. Furthermore, according to the report of ADA, the state where the fasting blood glucose level (glucose concentration in venous plasma) is 110 mg / dl or more and less than 126 mg / dl is called IFG (Impaired Fasting Glucose).
- the IFG is a state where the 75 g oral glucose tolerance test 2 hour value (glucose concentration in venous plasma) is less than 140 mg / dl as IFG (Impaired Fasting Glycemia). Call.
- the compound of the present invention is also used as a prophylactic / therapeutic agent for diabetes, borderline type, impaired glucose tolerance, IFG (Impaired Fasting Glucose) and IFG (Impaired Fasting Glycemia) determined by the above criteria. Furthermore, the compound of the present invention can also prevent progression from borderline type, impaired glucose tolerance, IFG (Impaired Fasting Glucose) or IFG (Impaired Fasting Glycemia) to diabetes.
- the compound of the present invention is also used for secondary prevention and progression suppression of the various diseases described above (eg, cardiovascular events such as myocardial infarction).
- cardiovascular events such as myocardial infarction
- the compound of the present invention continuously suppresses the production of eicosanoid over a long period of time, thereby causing an inflammatory disease, for example, asthma, allergic airway hypersensitivity, which has been pointed out to be associated with inflammatory eicosanoids, Fever, pain, thrombosis, cerebral infarction, myocardial infarction, cancer, autoimmune encephalomyelitis, pain, renal failure, rheumatism, osteoarthritis, pruritus, atopic dermatitis, rhinitis, inflammatory bowel disease, clone It can be used for prevention or treatment of diseases and the like.
- an inflammatory disease for example, asthma, allergic airway hypersensitivity, which has been pointed out to be associated with inflammatory eicosanoids, Fever, pain, thrombosis, cerebral infarction, myocardial infarction, cancer, autoimmune encephalomyelitis, pain, renal failure, rheumatism, osteoarthritis, pruritus,
- disorders or abnormalities of biological functions and physiological actions include facial flushing and skin pain (including those associated with administration of nicotinic acid derivative preparations, prostacyclin preparations, etc.), overactive bladder, cerebral circulation / kidney Impaired or abnormal circulatory regulation, circulatory disorders (eg, peripheral, brain, microcirculation, etc.), cerebral blood barrier disorders, salt sensitivity, abnormal coagulation / fibrinolytic system, abnormal blood / blood cell components (eg, sickle cell red blood cells) Disease, increased platelet aggregation, abnormal red blood cell deformability, increased leukocyte adhesion, increased blood viscosity, etc., growth factors and cytokines (eg, PDGF, VEGF, FGF, interleukin, TNF- ⁇ , MCP-1, etc.) ) Production and action,
- the compound of the present invention can also be used as an analgesic agent as a prophylactic / therapeutic agent for pain.
- pain disorders include acute pain due to inflammation, pain associated with chronic inflammation, pain associated with acute inflammation, postoperative pain (incisional pain, deep pain, visceral pain, postoperative chronic pain, etc.), muscle pain (chronic) Muscle pain associated with pain, stiff shoulder, etc.), joint pain, toothache, temporomandibular joint pain, headache (migraine, tension headache, fever headache, high blood pressure headache), visceral pain (heart pain, angina pain, abdominal pain) , Kidney pain, ureter pain, bladder pain), gynecological pain (intermediate pain, dysmenorrhea, labor pain), neuralgia (disc herniation, nerve root pain, postherpetic neuralgia, trigeminal neuralgia), cancer Pain, reflex sympathetic atrophy, complex local pain syndrome and the like.
- the compound of the present invention is effective for directly and immediately ameliorating various pains such as neuropathic pain, cancer pain, inflammatory pain, etc., and patients and pathological conditions in which the pain threshold is lowered (eg, hypertension, etc.) , And their complications).
- the content of the compound of the present invention in the pharmaceutical composition is usually about 0.01 to about 99.9% by weight, preferably about 0.1 to about 50% by weight, based on the whole preparation.
- the dose of the compound of the present invention depends on age, body weight, general health condition, sex, meal, administration time, administration method, excretion rate, combination of drugs, and the degree of the medical condition being treated at that time of the patient. It is decided in consideration of these and other factors.
- the dose varies depending on the target disease, symptom, administration subject, administration method, etc.
- the compound of the present invention when it is orally administered to an adult as a therapeutic agent for arteriosclerosis, it is usually about 0.01-100 mg / dose as a single dose.
- kg body weight preferably 0.05 to 30 mg / kg body weight, more preferably 0.5 to 10 mg / kg body weight, and this amount is preferably administered once to 3 times a day.
- the compound of the present invention is excellent in safety, it can be administered over a long period of time.
- the compounds of the present invention include, for example, anti-arteriosclerotic agents, antithrombotic agents, heart failure treatment agents, arrhythmia treatment agents, antihypertensive agents, diabetes treatment agents, diabetic complication treatment agents, HDL increase agents, antihyperlipidemic agents, anti-hyperlipidemic agents, Drugs such as obesity agents, diuretics, anti-inflammatory agents, anti-gout agents, chemotherapeutic agents, immunotherapeutic agents, osteoporosis treatment agents, anti-dementia agents, erectile dysfunction agents, urinary incontinence and dysuria treatment agents (hereinafter, (Abbreviated as a concomitant drug).
- concomitant drugs may be low molecular weight compounds, or may be high molecular weight proteins, polypeptides, antibodies, vaccines, or the like.
- anti-arteriosclerotic agent examples include Lp-PLA2 inhibitor (eg, dalapradiv, rilapradib etc.), FLAP inhibitor (eg, AM-103, AM-803, DG-031 etc.), sPLA2 inhibitor ( E.g., Valespradiv), 5-lipoxygenase inhibitor (e.g., VIA-2291), acylcoenzyme A cholesterol acyltransferase (ACAT) inhibitor (e.g., melinamide, abashimive, eflusimib, etc.), lipid rich plaque regression Drugs (eg, compounds described in WO02 / 06264, WO03 / 059900, etc.), HDL preparations (eg, CSL-111, etc.), CTEP inhibitors (eg, torcetrapib, anacetrapib, darcetrapib etc.), MMP inhibitors, chymase inhibitors , SPT inhibitors, ApoA-1 and related molecules (e
- antithrombotic agent examples include blood coagulation inhibitors (eg, heparin sodium, heparin calcium, warfarin calcium (warfarin), antithrombin drugs (eg, argatroban, dabigatran), activated blood, and the like.
- blood coagulation inhibitors eg, heparin sodium, heparin calcium, warfarin calcium (warfarin)
- antithrombin drugs eg, argatroban, dabigatran
- Coagulation factor Xa inhibitor eg, rivaroxaban, apixaban, edoxaban, YM-150, WO02 / 06234, WO2004 / 048363, WO2005 / 030740, WO2005 / 058823, WO2005 / 113504 or WO2004 / Etc.
- thrombolytic drugs eg, tPA, urokinase, tisokinase,reteplase, nateplase, monteplase, pamitepase (pamite) plase
- antiplatelet drugs eg, aspirin, sulfinpyrazone (antulan), dipyridamole (persantin), ticlopidine (panaldin), cilostazol (pretal), GPIIb / IIIa antagonists (eg, leopro, etc.), clopidogrel, prasugrel ( prasugrel),
- cardiotonic drugs eg, digitoxin, digoxin, methyldigoxin, lanatoside C, prossilaridin, etc.
- ⁇ , ⁇ stimulants eg, epinephrine, norepinephrine, isoproterenol, dopamine, Docarpamine, dobutamine, denopamine, etc.
- phosphodiesterase inhibitors eg, amrinone, milrinone, olprinone hydrochloride, etc.
- calcium channel sensitivity enhancers eg, pimobentan, etc.
- nitrate drugs eg, nitroglycerin, isosorbide nitrate, etc.
- angiotensin conversion Enzyme inhibitors eg, angiotensin converting enzyme inhibitors described below
- angiotensin II antagonists eg, angiotensin II antagonists described below
- ⁇ -blockers eg, ⁇ blockers described below
- arrhythmic therapeutic agent examples include sodium channel blockers (eg, quinidine, procainamide, disopyramide, ajmarin, cibenzoline, lidocaine, diphenylhydantoin, mexiletine, propafenone, flecainide, pildicinide, phenytoin), ⁇ -blockers (Eg, propranolol, alprenolol, bufetrol, oxprenolol, atenolol, acebutolol, metoprolol, bisoprolol, pindolol, carteolol, arotilolol, etc.), potassium channel blockers (eg, amiodarone, etc.), calcium channel blockers ( Examples, verapamil, diltiazem, etc.).
- sodium channel blockers eg, quinidine, procainamide, disopyramide, ajmarin, cibenzoline, lid
- angiotensin converting enzyme inhibitors eg, captopril, enalapril, delapril, etc.
- angiotensin II antagonists eg, candesartan cilexetil, candesartan, losartan, losartan potassium, eprosartan, valsartan, telmisartan, Irbesartan, tasosartan, olmesartan, olmesartan medoxomil, azilsartan, azilsartan medoxomil, etc.
- calcium antagonists eg, manidipine, nifedipine, amlodipine, efonidipine, nicardipine, amlodipine, sinildipine, etc.
- ⁇ -blockers eg, metoprolol aproteol, teprolol tetrol
- diuretic examples include xanthine derivatives (eg, sodium salicylate theobromine, calcium salicylate theobromine, etc.), thiazide preparations (eg, etiazide, cyclopenthiazide, trichloromethiazide, hydrochlorothiazide, hydroflumethiazide, benchylhydrochlorothiazide, penflux.
- xanthine derivatives eg, sodium salicylate theobromine, calcium salicylate theobromine, etc.
- thiazide preparations eg, etiazide, cyclopenthiazide, trichloromethiazide, hydrochlorothiazide, hydroflumethiazide, benchylhydrochlorothiazide, penflux.
- anti-aldosterone preparations eg, spironolactone, triamterene, etc.
- carbonic anhydrase inhibitors eg, acetazolamide, etc.
- chlorobenzenesulfonamide preparations eg, chlorthalidone, mefluside, indapamide, etc.
- Azosemide is
- insulin preparations eg, animal insulin preparations extracted from bovine and porcine pancreas; human insulin preparations synthesized by genetic engineering using Escherichia coli and yeast; insulin zinc; protamine insulin zinc; insulin Fragment or derivative (eg, INS-1), oral insulin preparation
- insulin resistance improving agent eg, pioglitazone or a salt thereof (preferably hydrochloride), rosiglitazone or a salt thereof (preferably maleate)
- Metaglidasen AMG-131, Balaglitazone, MBX-2044, Riboglitazone, Aleglitazar, Chiglitazar, Lobeglitazone, PLX-204, PN-2034 , GFT-505, THR-0921, WO2007 / 013694, WO2007 / 018314, WO2008 / 093639 or WO2008 / 0 Compounds described in 99794)
- ⁇ -glucosidase inhibitors e.g, ⁇ -glucosidas
- diabetic complication therapeutic agents include aldose reductase inhibitors (eg, tolrestat, epalrestat, zopolrestat, fidarestat, CT-112, ranirestat (AS-3201), ridressat), neurotrophic factor and its increase drug (Eg, NGF, NT-3, BDNF, neurotrophin production / secretion promoter described in WO01 / 14372 (eg, 4- (4-chlorophenyl) -2- (2-methyl-1-imidazolyl) -5- [3- (2-methylphenoxy) propyl] oxazole), compounds described in WO2004 / 039365), PKC inhibitors (eg, ruboxistaurin mesylate), AGE inhibitors (eg, ALT946, N-phenol) Nasyl thiazolium bromide (ALT766), EXO-226, pyridoline (Pyridorin), pyridoxamine), GABA receptor .
- HDL increasing agent examples include squalene synthase inhibitors, CETP inhibitors (eg, torcetrapib, anacetrapib, darcetrapib, etc.), LPL activators, nicotinic acid drugs (eg, nicomol, niceritrol) (niceritrol)), endothelial lipase inhibitors and the like.
- antihyperlipidemic agent examples include, for example, statins that are cholesterol synthesis inhibitors (eg, cerivastatin, pravastatin, simvastatin, lovastatin, rosuvastatin, atorvastatin, fluvastatin, pitavastatin, or salts thereof (eg, sodium) Salt, etc.), squalene synthase inhibitors or triglyceride-lowering fibrate compounds (eg, bezafibrate, clofibrate, synfibrate, clinofibrate, etc.), cholesterol absorption inhibitors (eg, zetia), anion exchange resins (Eg, cholestyramine), probucol, nicotinic acid drugs (eg, nicomol, niceritrol), plant sterols (eg, soysterol, gamma-oryzanol), fish oil Drug acupuncture (EPA, DHA, omacol etc.), PPAR
- anti-obesity agents include monoamine uptake inhibitors (eg, phentermine, sibutramine, mazindol, floxetine, tesofensin), serotonin 2C receptor agonists (eg, lorcaserin), serotonin 6 receptor antagonist, histamine H3 receptor Body, GABA modulator (eg, topiramate), neuropeptide Y antagonist (eg, Berneperit), cannabinoid receptor antagonist (eg, rimonabant, taranaban), ghrelin antagonist, ghrelin receptor antagonist, ghrelin acylating enzyme Inhibitors, opioid receptor antagonists (eg, GSK-1521498), orexin receptor antagonists, melanocortin 4 receptor agonists, 11 ⁇ -hydroxysteroid dehydrogenase inhibitors (eg, AZD-4017), pancreatic lipase inhibitors (eg, , Orlistat, cetilistat), ⁇ 3
- monoamine uptake inhibitors
- FGF21 preparations eg, extracted from bovine, porcine pancreas
- Animal FGF21 preparations Animal FGF21 preparations
- human FGF21 preparations synthesized by genetic engineering using Escherichia coli and yeast
- fragments or derivatives of FGF21 antifeedants (eg, P-57) and the like.
- diuretic agent examples include xanthine derivatives (eg, sodium salicylate theobromine, calcium salicylate theobromine), thiazide preparations (eg, etiazide, cyclopenthiazide, trichloromethiazide, hydrochlorothiazide, hydroflumethiazide, benzylhydrochlorothiazide).
- xanthine derivatives eg, sodium salicylate theobromine, calcium salicylate theobromine
- thiazide preparations eg, etiazide, cyclopenthiazide, trichloromethiazide, hydrochlorothiazide, hydroflumethiazide, benzylhydrochlorothiazide.
- Penfluthiazide poly-5thiazide, methiclotiazide, etc.
- anti-aldosterone preparations eg, spironolactone, eplerenone, triamterene, etc.
- carbonic anhydrase inhibitors eg, acetazolamide, etc.
- chlorobenzenesulfonamide preparations eg, chlorthalidone
- azosemide isosorbide, ethacrynic acid, piretanide, bumetanide, furosemide and the like.
- anti-inflammatory agent examples include acetaminophen, phenacetin, etenzamide, sulpyrine, antipyrine, miglenin, aspirin, mefenamic acid, flufenamic acid, diclofenac sodium, loxoprofen sodium, phenylbutazone, indomethacin, ibuprofen, ketoprofen, naproxen.
- anti-gout agent examples include febuxostat, allopurinol, probenecid, colchicine, benzbromarone, febuxostat, citrate and the like.
- chemotherapeutic agent examples include alkylating agents (eg, cyclophosphamide, ifosfamide, etc.), antimetabolites (eg, methotrexate, 5-fluorouracil, etc.), anticancer antibiotics (eg, mitomycin, Adriamycin, etc.), plant-derived anticancer agents (eg, vincristine, vindesine, taxol, etc.), cisplatin, carboplatin, etoposide and the like. Of these, 5-fluorouracil derivatives such as furtulon or neofluturon are preferred.
- alkylating agents eg, cyclophosphamide, ifosfamide, etc.
- antimetabolites eg, methotrexate, 5-fluorouracil, etc.
- anticancer antibiotics eg, mitomycin, Adriamycin, etc.
- plant-derived anticancer agents eg, vincristine, vindesine, taxol, etc
- immunotherapy agent examples include microorganisms or bacterial components (eg, muramyl dipeptide derivatives, picibanil, etc.), polysaccharides having immunopotentiating activity (eg, lentinan, schizophyllan, krestin, etc.), genetic engineering techniques.
- cytokines obtained eg, interferon, interleukin (IL), etc.
- colony stimulating factors eg, granulocyte colony stimulating factor, erythropoietin, etc.
- IL-1 interferon, interleukin (IL), etc.
- IL-12 e.g. preferable.
- osteoporosis therapeutic agent '' examples include, for example, alfacalcidol, calcitriol, elcaltonin, salmon calcitonin (calcitoninolsalmon), estriol, ipriflavone, and pamidronate.
- examples include sodium (pamidronate disodium), alendronate sodium hydrate, minderonate disodium ⁇ , and the like.
- anti-dementia agent examples include tacrine, donepezil, rivastigmine, galantamine, and the like.
- erectile dysfunction ameliorating agent examples include apomorphine, PDE5 (phosphodiesterase 5) inhibitor (eg, sildenafil citrate), and the like.
- urine incontinence therapeutic agent examples include flavoxate hydrochloride, oxybutynin hydrochloride, propiverine hydrochloride and the like.
- examples of the above “difficulty to urinate” include acetylcholinesterase inhibitors (eg, distigmine) and the like.
- concomitant drugs include prostacyclin preparations / derivatives (eg, beraprost, epoprostenol, iloprost, treprostinil, etc.), prostaglandin preparations / derivatives (eg, emprostil, alprostadil, limaprost, misoprostol, orno Prostil, etc.), anti-asthma drugs (eg, salmeterol, fluticasone, montelukast), rheumatoid arthritis drugs (eg, etanercept, infliximab, adalimumab), nerve regeneration-promoting drugs (eg, Y-128, VX-853, prosaptide), Antidepressants (eg, desipramine, amitriptyline, imipramine), antiepileptic drugs (eg, lamotrigine), antiarrhythmic drugs (eg, mexiletine), acetylcholine receptor ligands (e
- the administration time of the aforementioned concomitant drug is not limited, and the compound of the present invention and the concomitant drug may be administered simultaneously to the administration subject or may be administered with a time difference.
- the dose of the concomitant drug may be determined according to the dose used clinically, and can be appropriately selected depending on the administration subject, administration route, disease, combination and the like.
- these concomitant drugs may be combined in an appropriate ratio of two or more.
- the administration time of the compound of the present invention and the concomitant drug is not limited, and the compound of the present invention and the concomitant drug may be combined at the time of administration.
- Examples of such administration forms include (1) administration of a single preparation obtained by simultaneously formulating the compound of the present invention and a concomitant drug, and (2) formulating the compound of the present invention and the concomitant drug separately. Simultaneous administration of the two obtained preparations by the same administration route, (3) administration of the two preparations obtained by separately formulating the compound of the present invention and the concomitant drug with a time difference in the same administration route, (4) Simultaneous administration by different administration routes of two types of preparations obtained by separately formulating the compound of the present invention and a concomitant drug, (5) 2 obtained by formulating the compound of the present invention and the concomitant drug separately Administration of different types of preparations at different time intervals (for example, administration in the order of the compound of the present invention ⁇ concomitant drug or administration in the reverse order) and the like.
- the dose of the concomitant drug can be appropriately selected based on the clinically used dose.
- the compounding ratio of the compound of the present invention and the concomitant drug can be appropriately selected depending on the administration subject, administration route, target disease, symptom, combination and the like.
- the concomitant drug may be used in an amount of 0.01 to 100 parts by weight per 1 part by weight of the compound of the present invention.
- the compound of the present invention By combining the compound of the present invention and a concomitant drug, (1) The dose can be reduced compared to the case where the compound of the present invention or the concomitant drug is administered alone. (2) By selecting a concomitant drug having a different mechanism of action from the compound of the present invention, the treatment period can be set longer. (3) By selecting a concomitant drug having a different mechanism of action from the compound of the present invention, the therapeutic effect can be sustained. (4) By using the compound of the present invention and a concomitant drug in combination, excellent effects such as a synergistic effect can be obtained.
- Methyl 6-chloro-2-[(4-methoxybenzyl) amino] pyridine-3-carboxylate Methyl 2,6-dichloropyridine-3-carboxylate (2.06 g), sodium bicarbonate (1.26 g), 4 A mixture of -methoxybenzylamine (1.50 ml), copper (I) iodide (190 mg) and N, N-dimethylformamide (20 ml) was heated to 80 ° C. and stirred for 1 hour 30 minutes. Thereafter, the reaction mixture was concentrated under reduced pressure, and the resulting residue was diluted with ethyl acetate (150 ml).
- methyl 2-amino-6-chloropyridine-3-carboxylate (80 mg) was dissolved in N, N-dimethylformamide (5 ml), and the resulting mixture was added dropwise to the reaction mixture under ice cooling, followed by further stirring for 20 minutes.
- 0.2M hydrochloric acid 35 ml was poured into the reaction mixture, and the precipitated white precipitate was collected by filtration.
- This precipitate was dissolved in tetrahydrofuran (50 ml), diluted with ethyl acetate (100 ml), washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give a white residue (1.2 g). .
- the obtained residue was dissolved in methanol (20 ml), sodium methoxide (28% methanol solution, 3.0 g) was added, and the mixture was heated to reflux for 30 min. After allowing to cool, the reaction mixture was poured into 0.2 M hydrochloric acid solution (100 ml) under ice cooling, and the precipitated white precipitate was collected by filtration. The obtained precipitate was dissolved in tetrahydrofuran (100 ml), diluted with ethyl acetate (200 ml), washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure to obtain a residue. Washing with diethyl ether gave the title compound (861 mg) as a white solid.
- Example 2 7-Methoxy-2- (methylsulfanyl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidin-4 (3H) -one 7-methoxy-2 -Thioxo-3- [4- (2,2,2-trifluoroethoxy) phenyl] -2,3-dihydropyrido [2,3-d] pyrimidin-4 (1H) -one (1.2 g), 1M bicarbonate A mixture of aqueous sodium (3.1 ml), iodomethane (0.98 ml) and N, N-dimethylformamide (10 ml) was stirred at 60 ° C.
- Example 3 7-methoxy-2- (propylsulfanyl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidin-4 (3H) -one 7-methoxy-2 -Thioxo-3- [4- (2,2,2-trifluoroethoxy) phenyl] -2,3-dihydropyrido [2,3-d] pyrimidin-4 (1H) -one (153 mg), 1M bicarbonate A mixture of aqueous sodium (439 ⁇ l), 1-iodopropane (117 ⁇ l) and N, N-dimethylformamide (3 ml) was stirred at 80 ° C.
- the reaction mixture was poured into 0.2M hydrochloric acid (15 ml), and the deposited precipitate was collected by filtration.
- the precipitate collected by filtration was dissolved in tetrahydrofuran (15 ml), diluted with ethyl acetate (30 ml), and dried over anhydrous magnesium sulfate.
- the solvent was distilled off under reduced pressure and the resulting residue was purified by silica gel column chromatography (ethyl acetate / hexane) to give the title compound (95 mg).
- the reaction mixture was poured into 0.2M hydrochloric acid (10 ml), and the precipitated white solid was collected by filtration and washed with water.
- the obtained solid was dissolved in tetrahydrofuran (5 ml), diluted with ethyl acetate (70 ml), washed with saturated brine, and dried over anhydrous magnesium sulfate.
- the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography (ethyl acetate / hexane) to give the title compound (137 mg) as a white solid.
- Example 8 2- (Ethylsulfanyl) -8-methyl-3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidine-4,7 (3H, 8H) -dione 2 -(Ethylsulfanyl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidine-4,7 (3H, 8H) -dione (120 mg) and iodomethane (37.8 ⁇ l) was dissolved in N, N-dimethylformamide (2 ml), sodium hydride (60% oily, 86 mg) was added under ice-cooling, and the mixture was stirred for 45 min.
- Example 12 2- (2,2,2-trifluoroethoxy) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidine-4,7 (3H, 8H) -Dione sodium salt 2- (2,2,2-trifluoroethoxy) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidine-4,7 ( 3H, 8H) -dione (93 mg) was suspended in methanol (3 ml), sodium methoxide (28% methanol solution, 41.2 mg) was added at room temperature, and the mixture was heated to 30-40 ° C to obtain a transparent solution It was.
- Example 17 2- (2,2-difluoro-3-hydroxypropoxy) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidine-4,7 (3H, 8H ) -Dione 2- (propylsulfanyl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidine-4,7 (3H, 8H) -dione and 2
- the title compound was obtained from 1,2-difluoropropane-1,3-diol by a method according to Example 13B).
- Example 18 2-Methoxy-3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidine-4,7 (3H, 8H) -dione 2- (methylsulfinyl) -3 -[4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidine-4,7 (3H, 8H) -dione (60 mg) was suspended in tetrahydrofuran (2 ml). Under ice-cooling, sodium methoxide (28% methanol solution, 200 mg) was added and stirred for 10 minutes.
- Example 19 2- (ethylamino) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidine-4,7 (3H, 8H) -dione in tetrahydrofuran solution of ethylamine ( 2M, 2 ml) diluted with tetrahydrofuran (2 ml) to give 2- (methylsulfinyl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidine- 4,7 (3H, 8H) -dione (100 mg) was added at room temperature.
- Example 20 2- (1-Methylethoxy) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidine-4,7 (3H, 8H) -dione 2- ( Methylsulfinyl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [2,3-d] pyrimidine-4,7 (3H, 8H) -dione (100 mg) was added to tetrahydrofuran (4 2-propanol (77 ⁇ l) was added, sodium hydride (25.0 mg) was added under ice-cooling, and the mixture was stirred for 10 minutes and further stirred at room temperature for 15 minutes.
- reaction mixture was stirred for 5 minutes under ice-cooling, diethyl 2-carbamimidoylpentane-1,5-dioate (2.5 g) was added, and the mixture was stirred for 30 minutes under ice-cooling.
- 1M Hydrochloric acid was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was dissolved in ethanol (25 mL), sodium ethoxide (20% ethanol solution, 10.6 mL) was added, and the mixture was stirred for 30 min.
- methyl 2-amino-6-chloropyridine-3-carboxylate (2.95 g) was dissolved in N, N-dimethylformamide (25 ml), added dropwise to the reaction mixture under ice-cooling, stirred for 20 minutes, and stirred at room temperature. Stir for 1 hour 30 minutes.
- the reaction mixture was poured into 0.5M hydrochloric acid (80 ml), and the precipitated white precipitate was collected by filtration. This precipitate was dissolved in tetrahydrofuran (100 ml), diluted with ethyl acetate (200 ml), washed with saturated brine and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give a white residue.
- the extract was washed with water, saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give a residue.
- the product was purified by silica gel column chromatography (ethyl acetate / hexane) and (NH, ethyl acetate / hexane), and recrystallized from a mixed solvent of ethyl acetate / hexane to give the title compound (28 mg) as white.
- reaction mixture was poured into 0.5M hydrochloric acid (4 ml) and extracted with ethyl acetate. The extract was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure to give the colorless title compound (72 mg).
- Example 30 2- (propylsulfanyl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] -3,7-dihydropyrido [3,4-d] pyrimidine-4,8-dione 7- (2, 4-Dimethoxybenzyl) -2-thioxo-3- [4- (2,2,2-trifluoroethoxy) phenyl] -1,2,3,7-tetrahydropyrido [3,4-d] pyrimidine-4
- the title compound was obtained from 1,8-dione and 1-iodopropane by a method according to C) and D) of Example 29.
- Example 31 2- (2,2,2-trifluoroethoxy) -3- [4- (2,2,2-trifluoroethoxy) phenyl] -3,7-dihydropyrido [3,4-d] pyrimidine-4,8 -To a solution of dione 2,2,2-trifluoroethanol (0.052 ml) in N, N-dimethylformamide (3 ml), sodium hydride (60% oily, 24 mg) was added at room temperature and stirred for 5 minutes.
- the mixture was further stirred at 60 ° C. for 5 hours. After cooling to room temperature, water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate / hexane), the obtained residue was suspended in a mixed solvent of ethyl acetate / hexane, and the resulting solid was collected by filtration to give the title compound (21 mg).
- Example 35 2,8-bis (2-methoxyethoxy) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrido [3,4-d] pyrimidin-4 (3H) -one of Example 34
- the title compound (6 mg) was obtained as a byproduct of C).
- Example 36 2- (Ethylsulfanyl) -7-methyl-3- [4- (2,2,2-trifluoroethoxy) phenyl] -3,7-dihydropyrido [3,4-d] pyrimidine-4,8-dione 2 -(Ethylsulfanyl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] -3,7-dihydropyrido [3,4-d] pyrimidine-4,8-dione (40 mg) N , N-dimethylformamide (5 ml) solution was added sodium hydride (60% oily, 193 mg) at room temperature and stirred for 10 minutes.
- Example 38 2- (Ethylsulfanyl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pteridine-4,7 (3H, 8H) -dione 2- (Ethylsulfanyl) -7-methoxy-3- A mixture of [4- (2,2,2-trifluoroethoxy) phenyl] pteridin-4 (3H) -one (92 mg), pyridine hydrochloride (640 mg) and N, N-dimethylformamide (3 ml) The mixture was stirred at 120 ° C. for 10 hours, and the title compound (50 mg) was obtained as a white powder by the method according to Example 6.
- reaction mixture was concentrated under reduced pressure. THF was added to the residue, the insoluble matter was removed by filtration, and the filtrate was concentrated under reduced pressure. 1N Hydrochloric acid (100 mL) was added to a THF (100 mL) solution of the obtained residue at 0 ° C., and the mixture was stirred for 2 hr. The reaction mixture was extracted with ethyl acetate, and the extract was washed with saturated brine, dried over magnesium sulfate, and concentrated under reduced pressure.
- N- [4- (2,2,2-trifluoroethoxy) phenyl] butanimidoethyl ethyl butyrimidate hydrochloride (10.0 g), 4- (2,2,2-trifluoroethoxy) aniline (12.6 g), a mixture consisting of triethylamine (6.67 g) and THF (250 mL) was stirred at 70 ° C. for 2 days.
- Ethyl acetate and water were added to the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, dried over magnesium sulfate, and concentrated under reduced pressure.
- Tables 1 to 4 show the chemical names, structural formulas, and measured values of MS of the example compounds.
- the reaction mixture was concentrated under reduced pressure, diluted with ethyl acetate, washed successively with saturated aqueous citric acid solution, water, and saturated aqueous sodium hydrogen carbonate solution, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- the resulting pale yellow solid was purified by silica gel column chromatography (ethyl acetate / hexane) to give a colorless solid. This solid was recrystallized from ethyl acetate / hexane to give the title compound (0.22 g) as a colorless solid.
- Test Example 1 (Delta-5-desaturase inhibitory activity using rat liver microsomes) The inhibitory activity of the test compound against delta-5-desaturase was measured by the method described below. Buffer (300 mM NaH 2 PO 4 [pH 7.4], 450 mM KCl, 30 mM NaF, 9 mM MgCl 2 , 4.5 mM glutathione [reduced form], 0.3% BSA [fatty] against a DMSO solution of a test compound prepared in advance. acid free, SIGMA]) was used to make an assay buffer.
- Buffer 300 mM NaH 2 PO 4 [pH 7.4], 450 mM KCl, 30 mM NaF, 9 mM MgCl 2 , 4.5 mM glutathione [reduced form], 0.3% BSA [fatty] against a DMSO solution of a test compound prepared in advance. acid free, SIGMA]
- rat liver microsomes diluted to 3 mg / ml with microsome buffer (10 mM Tris-HCl [pH 7.4], 1 mM EDTA, 250 mM sucrose). 10 ⁇ l fraction was added. The enzymatic reaction, 9mM NADH, 9mM ATP, 0.9mM CoA, 10 ⁇ Ci / ml (8E, 11E, 14E) - (1- 14 C) eicosa-8,11,14-trienoic acid (PerkinElmer Inc.) and 10 ⁇ l added To start.
- the enzyme reaction was carried out for 120 minutes at room temperature, and the enzyme reaction was stopped by adding 10 ⁇ l of 2.5 M NaOH. After the reaction was stopped, plate sealing was performed, and saponification was performed by incubation overnight in a dry heat set at 55 ° C. Fatty acid solvent extraction is based on the Bligh & Dyer method described in Canadian Journal of Biochemistry and Physiology (Vol. 37, 911 (1959)), formic acid: methanol: chloroform (1 : 6: 3) 200 ⁇ l was added to maintain one layer, and after sufficient stirring, 120 ⁇ l of pure water was added to separate into two layers.
- Test Example 2 (N- [2-( ⁇ 4-oxo-3- [4- (2,2,2-trifluoroethoxy) [2,6- 3 H 2 ] phenyl]-to delta-5-desaturase)- 3,4-Dihydrothieno [3,4-d] pyrimidin-2-yl ⁇ sulfanyl) ethyl] acetamide (Reference Example 1) binding inhibitory activity) All tests were performed at room temperature and 200 ⁇ L / well.
- Assay buffer (10 mM Tris-HCl (pH7.5 ), 100 mM KCl, 10 mM NaF, 3mM MgCl 2, 0.005% tween20 and, 1 mM GSH) in the rat liver microsomal fraction of 30 [mu] g / well Preincubation with test compound at a final concentration of 10 ⁇ M for 15 min.
- N- [2-( ⁇ 4-oxo-3- [4- (2,2,2-trifluoroethoxy) [2,6- 3 H 2 ] phenyl] -3,4-dihydrothieno [3,4-d ] Pyrimidin-2-yl ⁇ sulfanyl) ethyl] acetamide was added to a final concentration of 3 nM and incubated for 150 minutes.
- the membrane fraction was captured on a GF / C glass filter using a cell harvester.
- the ligand bound to delta-5-desaturase was separated by washing the GF / C glass filter 5 times with 300 ⁇ l of ice-cold assay buffer (excluding 0.005% tween).
- the compound of the present invention can be prepared by N- [2-( ⁇ 4-oxo-3- [4- (2,2,2-trifluoroethoxy) [2, 6- 3 H 2] exhibited excellent inhibitory action on phenyl] -3,4-dihydrothieno [3,4-d] pyrimidin-2-yl ⁇ sulfanyl) ethyl] binding acetamide.
- Test Example 3 (Delta-5-desaturase inhibitory activity using HepG2 cells) HepG2 was seeded in a 96-well plate at 1 ⁇ 10 5 cells / well and cultured overnight in DMEM (Dulbecco's modified Eagle medium) containing 10% FBS (fetal bovine serum). After washing with PBS (phosphate buffered saline) (200 ⁇ L x 2), add 40 ⁇ L of reaction medium (DMEM, penicillin / streptomycin, 0.3% BSA) containing the test compound, and in a CO 2 incubator at 37 ° C Pre-incubation was performed for 30 minutes at 30 ° C.
- DMEM Dulbecco's modified Eagle medium
- FBS fetal bovine serum
- the reaction was initiated by the addition of 20 ⁇ L of reaction medium containing 0.1 ⁇ Ci of [ 14 C] Eicosatrienoic acid and incubated in a CO 2 incubator set at 37 ° C. After washing for 3 hours, the cells were washed twice with 200 ⁇ L of PBS, and the cells detached with 50 ⁇ L of trypsin-EDTA were transferred to a 96-deep well block, and 20 ⁇ L of 2.5N NaOH was added to the plate. After sealing, saponification was carried out by incubation overnight in a dry heat oven set at 55 ° C. Fatty acid extraction was acidified by adding 10 ⁇ L formic acid, followed by chloroform: methanol (1: 4) Add and stir, and add 200 ⁇ L of pure water. The procedure after TLC development was in accordance with the detection system method using TLC in the rat liver microsome system.Inhibition of delta-5-desaturase at 1 ⁇ M of the test compound The rate (%) was determined and the results are shown in Table 5.
- the compound of the present invention showed an excellent delta-5-desaturase inhibitory action.
- Test Example 4 (In vivo delta-5-desaturase inhibitory action) The in vivo delta-5-desaturase inhibitory effect of the test compound was evaluated by the following method. 7-9 week-old male normal mice (C57BL / 6J, Nippon Claire) conditioned on general breeding feed (CE-2, Nippon Claire) using individual cages, 0.5 mg of test compound 3 mg / kg It was suspended in a methylcellulose solution and orally administered by gavage every morning for 4 days at 10 mL / kg. On the morning of the day after the last administration, the mouse was anesthetized and the liver was removed.
- YMC ester type Labeling was performed using a fatty acid labeling reagent (YMC).
- YMC fatty acid labeling reagent
- the compound of the present invention showed an excellent in vivo delta-5-desaturase inhibitory action.
- Example 1 (1) Compound of Example 1 10.0 g (2) Lactose 70.0g (3) Corn starch 50.0g (4) 7.0g of soluble starch (5) Magnesium stearate 3.0 g 10.0 g of the compound of Example 1 and 3.0 g of magnesium stearate are granulated with 70 ml of an aqueous solution of soluble starch (7.0 g as soluble starch), then dried and mixed with 70.0 g of lactose and 50.0 g of corn starch. (Lactose, corn starch, soluble starch and magnesium stearate are all conforming to the 14th revised Japanese Pharmacopoeia). The mixture is compressed to obtain tablets.
- the compound of the present invention has a delta-5-desaturase inhibitory action and prevents arteriosclerosis, atherothrombosis, diabetes, obesity, asthma, fever, pain, cancer, rheumatism, osteoarthritis, atopic dermatitis, etc. ⁇ Useful for treatment.
Abstract
Description
(発明の背景)
エイコサノイド関連疾患の治療薬としては、プロスタノイド類の産生を抑制するシクロオキシゲナーゼ阻害剤やトロンボキサンA2受容体拮抗剤等の薬剤が臨床応用されている。しかし炎症性疾患等の予防・治療薬のニーズは依然として高く、強力かつ副作用の少ない治療薬の開発が切望されている。


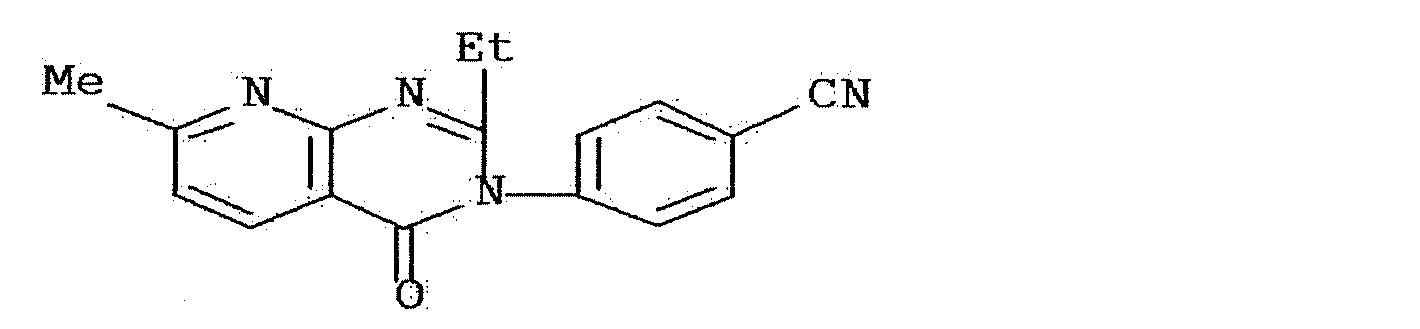

等の化合物が開示されている。
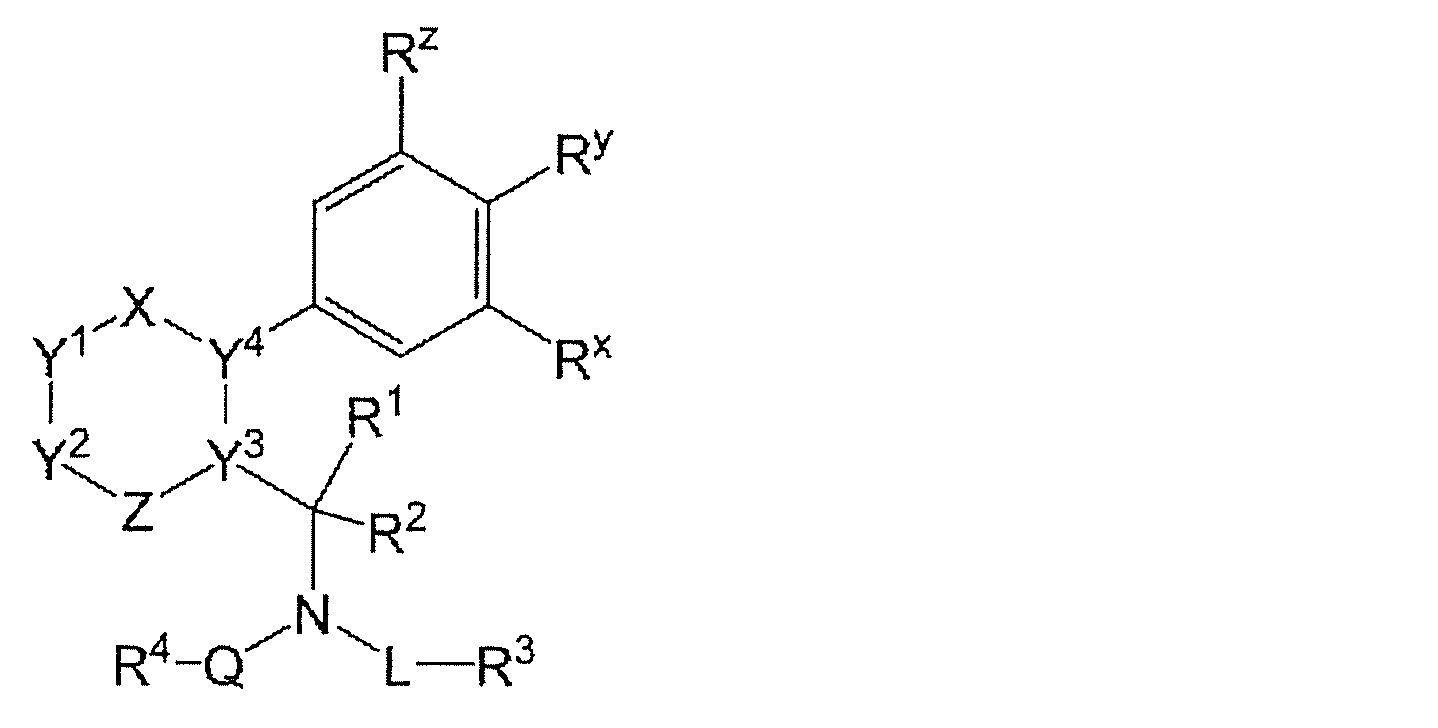
R1およびR2は、独立してH、(C1-C8)アルキル、(C2-C8)ヘテロアルキル、アリールおよびヘテロアリールからなる群から選択されるか、または任意に結合して環頂点として0~2個のヘテロ原子を有する3~8員環を形成し;任意に、R2は、Lと共に連結して1~4個のヘテロ原子を有する5員または6員複素環基を形成することができる。
R3は、存在しないか、またはヒドロキシ、(C1-C8)アルコキシ、アミノ、(C1-C8)アルキルアミノ、ジ(C1-C8)アルキルアミノ、(C1-C20)アルキル、(C2-C8)ヘテロアルキル、シクロ(C3-C9)ヘテロアルキル、(C1-C8)アシルアミノ、アミジノ、グアニジノ、ウレイド、シアノ、ヘテロアリール、-CONR9R10およびCO2R11からなる群から選択されるメンバーであり、または、任意に、R3は、R2と結合して、N、OおよびSからなる群から選択される1~3個のヘテロ原子を含む4員環、5員環、6員環、7員環または8員環を形成してもよい。
R4は、(C2-C20)アルキル、(C2-C20)ヘテロアルキル、ヘテロアリール、アリール、ヘテロアリール(C1-C6)アルキル、ヘテロアリール(C2-C6)ヘテロアルキル、アリール(C1-C6)アルキルおよびアリール(C2-C6)ヘテロアルキルからなる群から選択されるメンバーである。
R5およびR6は、それぞれ独立してH、(C1-C8)アルキル、(C2-C8)ヘテロアルキル、ヘテロアリールおよびアリールからなる群から選択されるメンバーであるか、または任意に、R5およびR6は、結合して3~7員環を形成する。
R7およびR8は、独立してH、(C1-C8)アルキル、(C2-C8)ヘテロアルキル、ヘテロアリールおよびアリールからなる群から選択される。それぞれR9、R10およびR11は、独立してH、(C1-C8)アルキル、(C2-C8)ヘテロアルキル、ヘテロアリール、アリール、ヘテロアリール(C1-C6)アルキル、ヘテロアリール(C2-C8)ヘテロアルキル、アリール(C1-C8)アルキルおよびアリール(C2-C8)ヘテロアルキルからなる群から選択される。
Rx、RyおよびRzは、それぞれ独立してH、F、またはシアノであり、式中Rx、RYおよびRzの少なくとも1つは、シアノである。
次に環頂点Y1、Y2、Y3およびY4に移って:Y1およびY2は、独立して-C(R12)=、-CH(R12)-、-N=、-O-、-S-、およびN(R13)-からなる群から選択されるメンバーである。Y3は、NまたはCであり、式中Y3がCであるときに、Y3は、Y2、Y4またはZと二重結合を共有する。Y4は、NまたはCであり、式中Y4がCであるときに、Y4は、X、Y1またはY3と二重結合を共有する。それぞれのR12は、H、ハロゲン、ヒドロキシ、アミノ、アルキルアミノ、ジアルキルアミノ、(C1-C8)アルキル、シクロ(C3-C6)アルキル、(C2-C8)ヘテロアルキル、ヘテロアリールおよびアリールからなる群から選択されるメンバーであり、または任意に、Y1およびY2がそれぞれ-C(R12)=またはCH(R12)-の1つであるときに、2つのR12基は結合して、置換または非置換の5~6員シクロアルキル環、シクロヘテロアルキル環、アリール環またはヘテロアリール環を形成することができる。任意に、Y1が-C(R12)=またはCH(R12)-であり、かつXが-C(R5)=またはC(R5)(R6)-であるときに、R12およびR5は結合して、置換または非置換の5~6員シクロアルキル環、シクロヘテロアルキル環、アリール環またはヘテロアリール環を形成することができる。それぞれのR13は、H、(C1-C8)アルキル、(C2-C8)ヘテロアルキル、ヘテロアリール、アリール、ヘテロアリール(C1-C6)アルキル、シクロ(C3-C6)アルキル、ヘテロアリール(C2-C8)ヘテロアルキル、アリール(C1-C8)アルキルおよびアリール(C2-C8)ヘテロアルキルからなる群から選択されるメンバーであり、任意に、Y1およびY2が-C(R12)=またはCH(R12)-であり、かつその他が-N(R13)-であるときに、R12およびR13は結合して、置換または非置換の5~6員シクロアルキル環、シクロヘテロアルキル環、アリール環またはヘテロアリール環を形成することができ、あるいは任意に、Y1およびY2が両方とも-N(R13)-であるときに、2つのR13基は結合して、置換または非置換の5~6員シクロアルキル環、シクロヘテロアルキル環、アリール環またはヘテロアリール環を形成することができる。R14は、H、(C1-C8)アルキル、(C2-C8)ヘテロアルキル、シクロ(C3-C6)アルキル、ヘテロアリール、アリール、ヘテロアリール(C1-C6)アルキル、ヘテロアリール(C2-C8)ヘテロアルキル、アリール(C1-C8)アルキルおよびアリール(C2-C8)ヘテロアルキルからなる群から選択されるメンバーであり、あるいは任意に、Y2が-C(R12)=、-CH(R12)-またはN(R13)-であるときに、R14またはR7は、R12またはR13と結合して、置換または非置換の5~6員シクロアルキル環、シクロヘテロアルキル環、アリール環またはヘテロアリール環を形成することができる。当業者には明らかであるように、X、Y1、Y2、Y3、Y4およびZを含む環は、芳香族であることができる。)で表される化合物、塩、溶媒和物、プロドラッグまたは異性体がCXCR3ケモカインレセプターによって介される喘息、乾癬、炎症性腸疾患、アレルギー疾患、リウマチ様関節炎、多発性硬化症等の特定の炎症性および免疫調節性の障害または疾患の治療または予防用途を有することが開示されている。
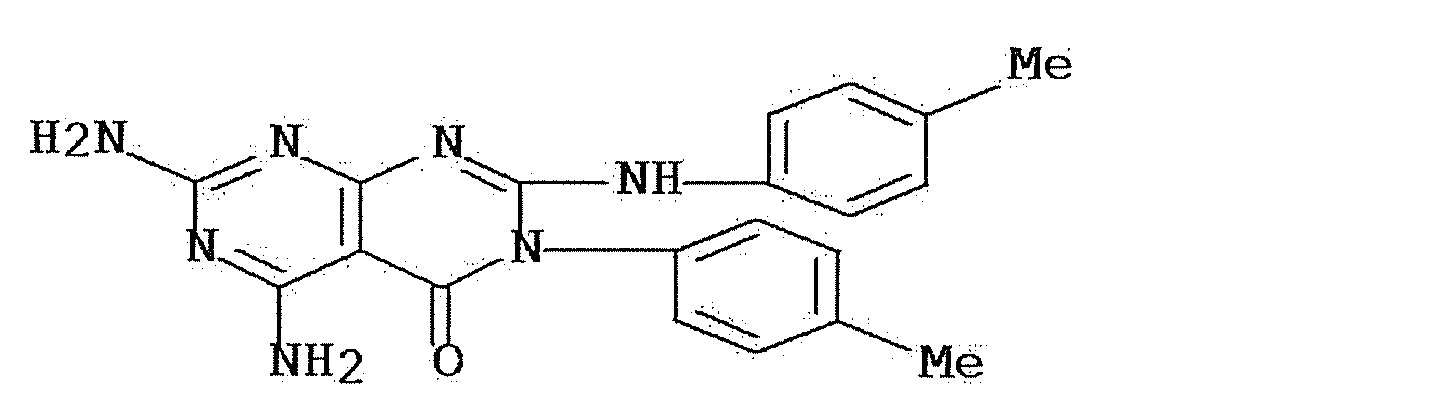


R4およびR5は同一でも異なっていてもよく、それぞれ水素、ヒドロキシ、アミノ、ハロゲン、メルカプト、C1-C6アルキル、1から7個のハロゲンで置換されたC1-C6アルキル、C1-C6アルコキシ、C2-C7アルコキシカルボニル、またはC1-C6アルキルチオを示し;
Xは水素、ヒドロキシ、ハロゲン、C1-C6アルコキシ、または1から7個のハロゲンで置換されたC1-C6アルコキシを示し;
YはC1-C6アルキル、1から7個の置換基で置換されたC1-C6アルキル(該置換基は同一でも異なっていてもよく、置換基グループαから選ばれる)、C3-C10シクロアルキル、1から7個の置換基で置換されたC3-C10シクロアルキル(該置換基は同一でも異なっていてもよく、置換基グループαから選ばれる)、5から9員ヘテロサイクル、1から7個の置換基で置換された5から9員ヘテロサイクル(該置換基は同一でも異なっていてもよく、置換基グループαから選ばれる)、C6-C10アリール、1から4個の置換基で置換されたC6-C10アリール(該置換基は同一でも異なっていてもよく、置換基グループβから選ばれる)、C4-C14シクロアルキルアルキル、1から7個の置換基で置換されたC4-C14シクロアルキルアルキル(該置換基は同一でも異なっていてもよく、置換基グループαから選ばれる)、(5から9員ヘテロサイクル)-(C1-C4アルキル)、1から7個の置換基で置換された(5から9員ヘテロサイクル)-(C1-C4アルキル)(該置換基は同一でも異なっていてもよく、置換基グループαから選ばれる)、C7-C14アラルキル、または1から5個の置換基で置換されたC7-C14アラルキル(該置換基は同一でも異なっていてもよく、置換基グループβから選ばれる)を示し;
置換基グループαは、ハロゲン、ヒドロキシ、シアノ、アミノ、C2-C7アルキルカルボニルオキシ、C1-C6アルキル、C1-C6アルコキシ、C1-C6アルキルチオ、C1-C6アルキルスルフィニル、C1-C6アルキルスルホニル、フェニル、C1-C6アルキルアミノ、ジ(C1-C6アルキル)アミノ(アルキルは同一でも異なっていてもよい)、C2-C7アルキルカルボニルアミノ、C1-C6アルキルスルホニルアミノ、およびC1-C6ハロアルキルスルホニルアミノ(該C1-C6ハロアルキルスルホニルアミノは1から7個のハロゲンで置換されたC1-C6アルキルスルホニルアミノ)からなるグループを示し;
置換基グループβはハロゲン、ヒドロキシ、ニトロ、シアノ、アミノ、C1-C6アルキル、1から7個のハロゲンで置換されたC1-C6アルキル、C2-C7アルキルカルボニルオキシ、C1-C6アルコキシ、C1-C6アルキルチオ、C1-C6アルキルスルフィニル、C1-C6アルキルスルホニル、C1-C6アルキルアミノ、ジ(C1-C6アルキル)アミノ(アルキルは同一でも異なっていてもよい)、C2-C7アルキルカルボニルアミノ、N-(C2-C7アルキルカルボニル)-N-(Cl-C6アルキル)アミノ、C2-C7アルコキシカルボニルアミノ、N-(C2-C7アルコキシカルボニル)-N-(Cl-C6アルキル)アミノ、C1-C6アルキルスルホニルアミノ、N-(C1-C6アルキルスルホニル)-N-(C1-C6アルキル)アミノ、C1-C6ハロアルキルスルホニルアミノ(該C1-C6ハロアルキルスルホニルアミノは1から7個のハロゲンで置換されたC1-C6ハロアルキルスルホニルアミノ)、N-(C1-C6ハロアルキルスルホニル)-N-(C1-C6アルキル)アミノ(該C1-C6ハロアルキルスルホニルは1から7個のハロゲンで置換されたC1-C6アルキルスルホニル)、C6-C10アリール、C7-C14アラルキルオキシ、C1-C4アルキレンジオキシ、C2-C7アルキルカルボニル、C2-C7アルコキシカルボニル、C2-C7アルキルアミノカルボニル、およびジ(C1-C6アルキル)アミノカルボニル(アルキルは同一でも異なっていてもよい)からなるグループを示し;但し、Yが次の(i)から(vii)の一つであり、Aがフェニルの場合、R4およびR5はともに水素を示し、-C(CF3)2(X)はフェニルグループの3または4位に結合する-C(CF3)2(OH)を示す:
(i)アミノ、アルキルアミノ、ジアルキルアミノ、アルキルカルボニルアミノ、アルキルスルホニルアミノまたはハロアルキルスルホニルアミノがその1位に置換されるアルキルであり、さらにアルキルまたはフェニルがその1位に置換されていてもよい;
(ii)アミノ、アルキルアミノ、ジアルキルアミノ、アルキルカルボニルアミノ、アルキルスルホニルアミノまたはハロアルキルスルホニルアミノで置換されるシクロアルキルであり、さらに置換基グループαから選ばれる1から6個の置換基で置換されていてもよい;
(iii)アルキル、アルキルスルフィニル、アルキルスルホニルおよびフェニルの中から選ばれる1または2個の置換基で置換されていてもよい、少なくとも1個の窒素原子を有するヘテロサイクル;
(iv)シクロアルキルアルキルであり、該アルキルに、アミノ、アルキルアミノ、ジアルキルアミノ、アルキルカルボニルアミノ、アルキルスルホニルアミノまたはハロアルキルスルホニルアミノがその1位に置換されており、該シクロアルキルアルキルは置換基グループαから選ばれる1から6個の置換基でさらに置換されていてもよい;
(v)ヘテロサイクルアルキルであり、該アルキルに、アミノ、アルキルアミノ、ジアルキルアミノ、アルキルカルボニルアミノ、アルキルスルホニルアミノまたはハロアルキルスルホニルアミノがその1位に置換されており、該ヘテロサイクルアルキルは置換基グループαから選ばれる1から6個の置換基でさらに置換されていてもよい;
(vi)ヘテロサイクルメチルであり、該ヘテロサイクルは少なくとも1個は窒素原子を有し、置換基グループαから選ばれる1から7個の置換基でさらに置換されていてもよく、該メチルはアルキルまたはフェニルで置換されていてもよい;および
(vii)アラルキルであり、該アルキルに、アミノ、アルキルアミノ、ジアルキルアミノ、アルキルカルボニルアミノ、アルコキシカルボニルアミノ、アルキルスルホニルアミノ、ハロアルキルスルホニルアミノ、N-(アルキルカルボニル)-N-(アルキル)アミノ、N-(アルコキシカルボニル)-N-(アルキル)アミノ、N-(アルキルスルホニル)-N-(アルキル)アミノまたはN-(ハロアルキルスルホニル)-N-(アルキル)アミノがその1位に置換されており、該アラルキルは置換基グループβから選ばれる1から6個の置換基でさらに置換されていてもよい。)で表される化合物またはその塩がLXRモジュレーターとして抗動脈硬化作用および抗炎症作用を有することが開示されている。

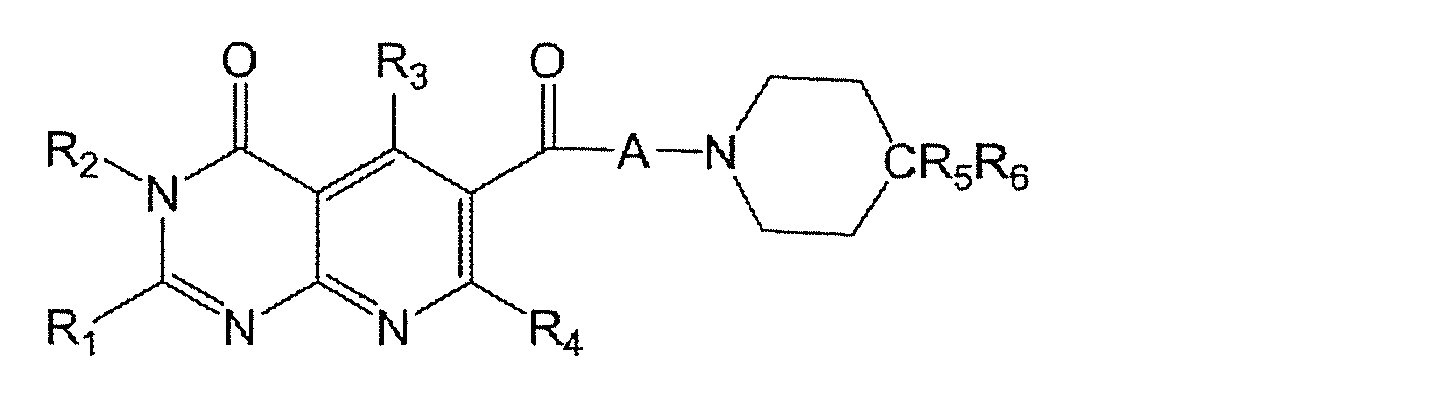
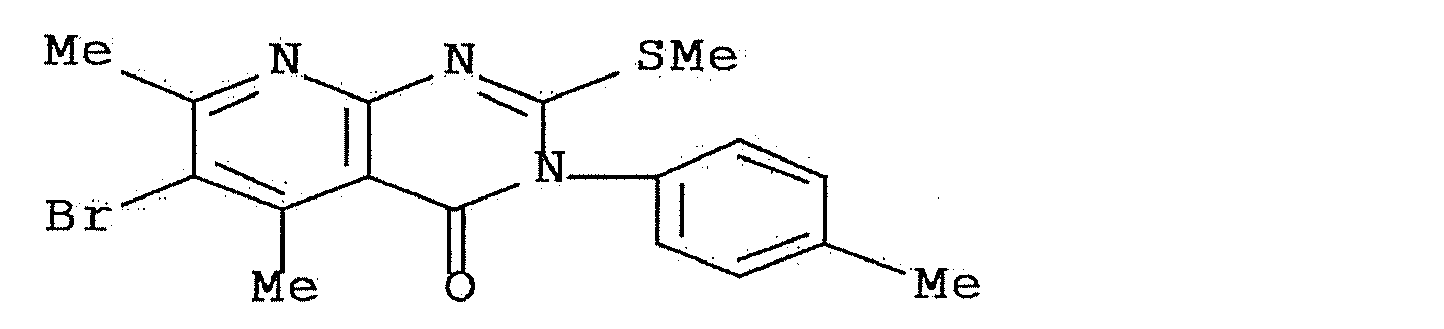
[1]式(I):

(I)
環Aは式:
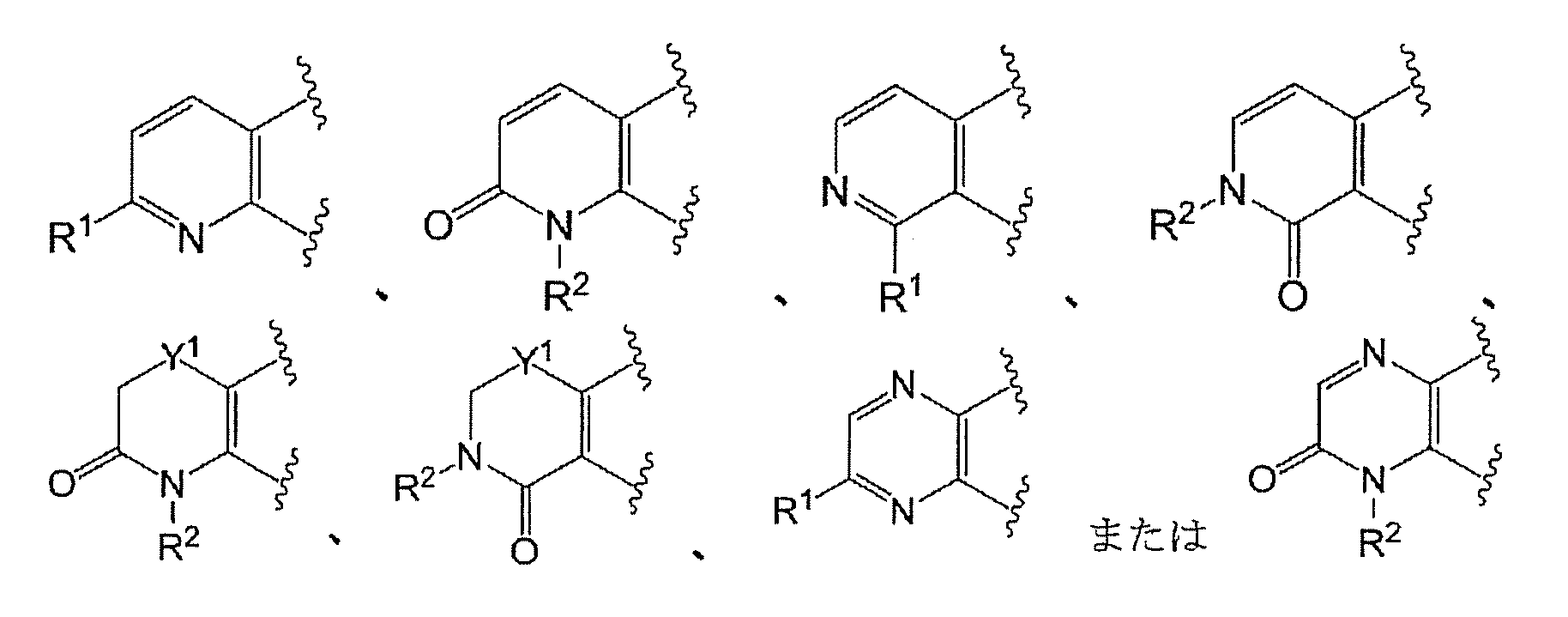
環Bはさらにハロゲン原子、C1-6アルキル基およびC1-6アルコキシ基から選択される置換基で置換されていてもよい6員芳香環を示し、
X1は結合手、S、SO2、OまたはNR5を示し、
X2はCR6R7またはOを示し、
Y1はCR8R9、S、SO、SO2またはOを示し、
R1は置換されていてもよいC1-6アルコキシ基、置換されていてもよいアミノ基、置換されていてもよいカルバモイル基または置換されていてもよいC1-6アルキルチオ基を示し、
R2は水素原子または置換されていてもよいC1-6アルキル基を示し、
R3は置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示し、
R4は置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示し、
R5は水素原子または置換されていてもよいC1-6アルキル基を示し、および
R6、R7、R8およびR9はそれぞれ水素原子、ハロゲン原子、C1-6アルキル基またはC1-6アルコキシ基を示す。〕で表される化合物またはその塩(以下、化合物(I)ともいう);
[2]X2がOであり;
R4が1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、上記[1]記載の化合物またはその塩;
[3]環Aが、式:
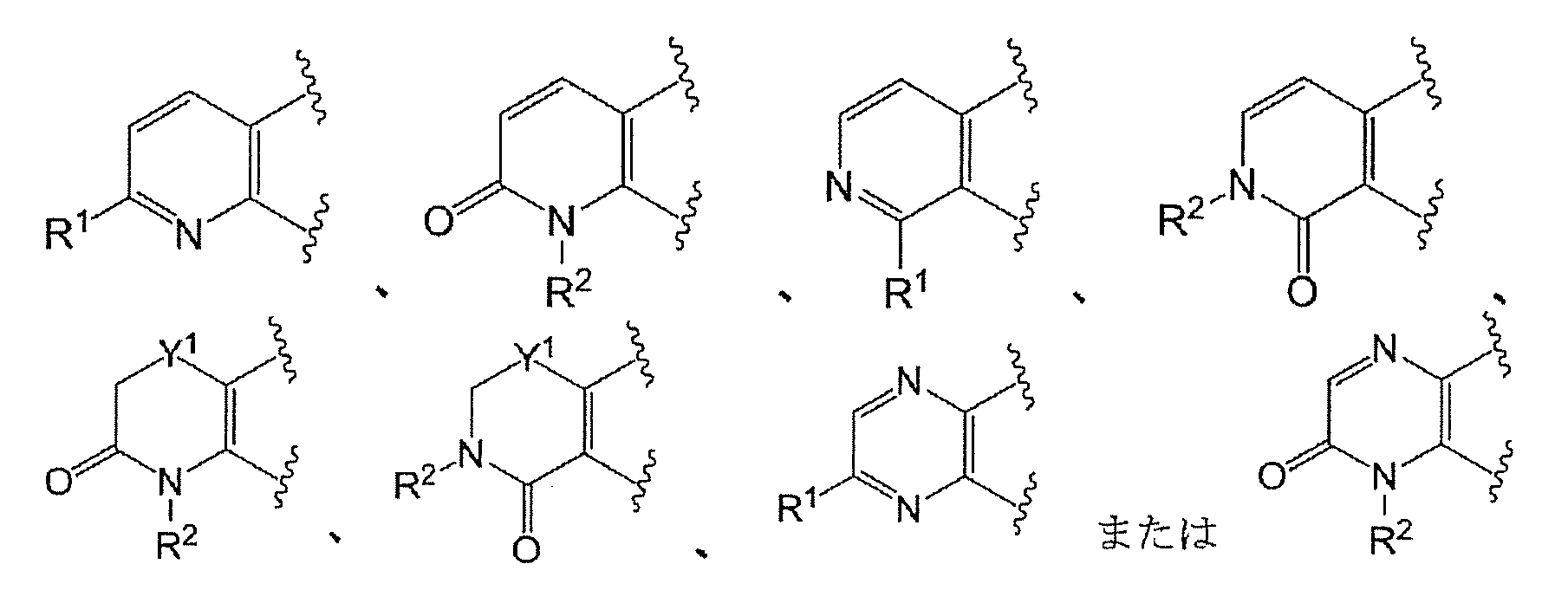
環Bがベンゼンであり;
X1が結合手、S、OまたはNHであり;
X2がOであり;
Y1がCH2またはSであり;
R1が、
(1)1ないし3個のC1-6アルコキシ基で置換されていてもよいC1-6アルコキシ基;
(2)アミノ;または
(3)カルバモイル;
であり;
R2が水素原子またはC1-6アルキル基であり;
R3が
(1) (A)C1-6アルキルスルホニル基、
(B)ヒドロキシ基、
(C)C1-6アルコキシ基、および
(D)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基、または
(2)1ないし3個のハロゲン原子で置換されていてもよいC3-10シクロアルキル基であり;および
R4が1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、上記[1]記載の化合物またはその塩;
[4]環Aが、式:

環Bがベンゼンであり;
X1が結合手、SまたはOであり;
X2がOであり;
Y1がCH2であり;
R1がC1-6アルコキシ基であり;
R2が水素原子であり;
R3が
(1)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、または
(2)1ないし3個のハロゲン原子で置換されていてもよいC3-10シクロアルキル基であり;および
R4が1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、上記[1]記載の化合物またはその塩;
[5]環Aが、式:
環Bがベンゼンであり;
X1が結合手、SまたはOであり;
X2がOであり;
Y1がCH2であり;
R2が水素原子であり;
R3が
(1)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、または
(2)1ないし3個のハロゲン原子で置換されていてもよいC3-10シクロアルキル基であり;および
R4が1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、上記[1]記載の化合物またはその塩;
[6]2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオンまたはその塩;
[7]2-エトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオンまたはその塩;
[8]3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオンまたはその塩;
[9]上記[1]~[8]のいずれかに記載の化合物またはその塩を含有してなる医薬;
[10]デルタ-5-デサチュラーゼ阻害薬である上記[9]記載の医薬;
[11]エイコサノイドが媒介する疾患の予防または治療剤である上記[9]記載の医薬;
[12]動脈硬化症の予防または治療剤である上記[9]記載の医薬;
[13]糖尿病または肥満症の予防または治療剤である上記[9]記載の医薬;
[14]上記[1]~[8]のいずれかに記載の化合物またはその塩を哺乳動物に有効量投与することを特徴とする、該哺乳動物における動脈硬化症の予防または治療方法;
[15]上記[1]~[8]のいずれかに記載の化合物またはその塩を哺乳動物に有効量投与することを特徴とする、該哺乳動物における糖尿病または肥満症の予防または治療方法;
[16]動脈硬化症の予防または治療剤を製造するための、上記[1]~[8]のいずれかに記載の化合物またはその塩の使用;
[17]糖尿病または肥満症の予防または治療剤を製造するための、上記[1]~[8]のいずれかに記載の化合物またはその塩の使用;
[18]動脈硬化症の予防または治療のために使用する上記[1]~[8]のいずれかに記載の化合物またはその塩;
[19]糖尿病または肥満症の予防または治療のために使用する上記[1]~[8]のいずれかに記載の化合物またはその塩;
等に関する。
(発明の詳細な説明)
本明細書中の「ハロゲン原子」としては、例えば、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられる。
本明細書中の「C3-10シクロアルキル(基)」としては、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチルが挙げられる。
本明細書中の「C6-14アリール(基)」としては、例えば、フェニル、ナフチル(例、1-ナフチル、2-ナフチル)、アントリル、フェナントリル等が挙げられる。
本明細書中の「C7-13アラルキル(基)」としては、例えば、ベンジル、フェネチル、ナフチルメチル、ビフェニリルメチル等が挙げられる。
本明細書中の「C2-6アルケニル(基)」としては、例えば、エテニル、1-プロペニル、2-プロペニル、2-メチル-1-プロペニル、1-ブテニル、2-ブテニル、3-ブテニル、3-メチル-2-ブテニル、1-ペンテニル、2-ペンテニル、3-ペンテニル、4-ペンテニル、4-メチル-3-ペンテニル、1-ヘキセニル、3-ヘキセニル、5-ヘキセニル等が挙げられる。
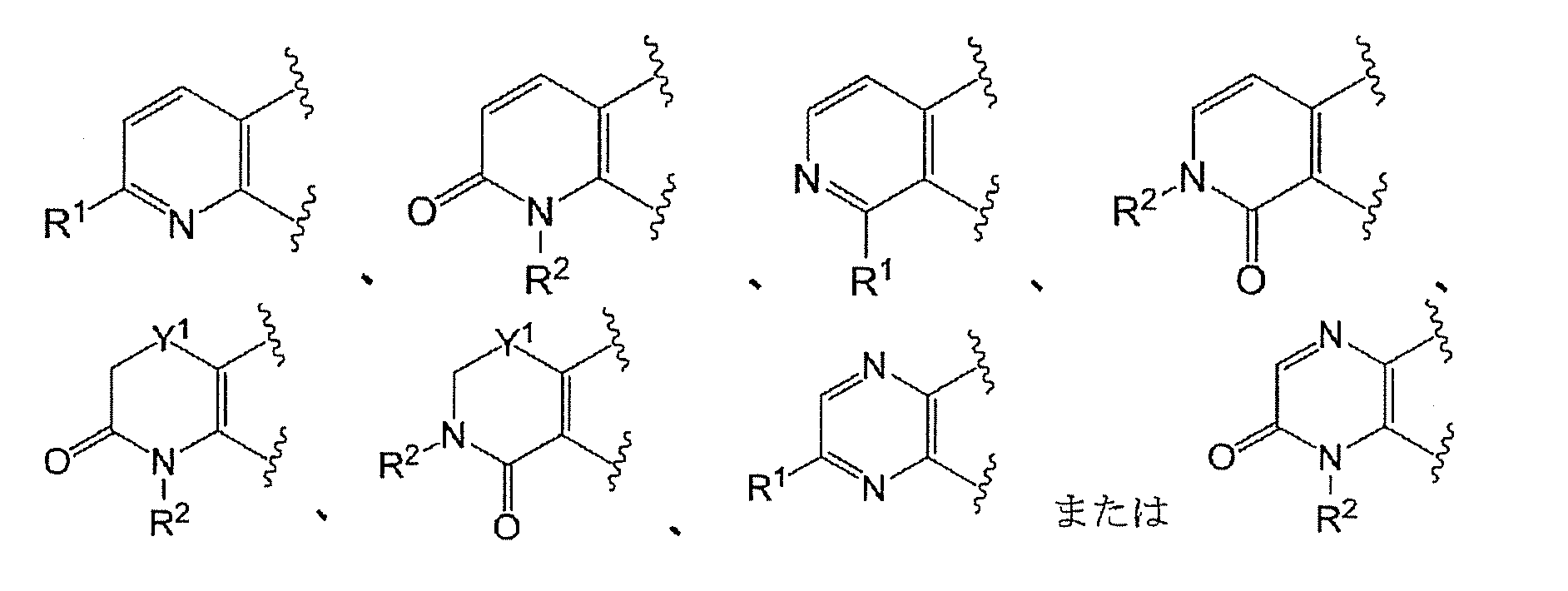
環Aは、より好ましくは、式:
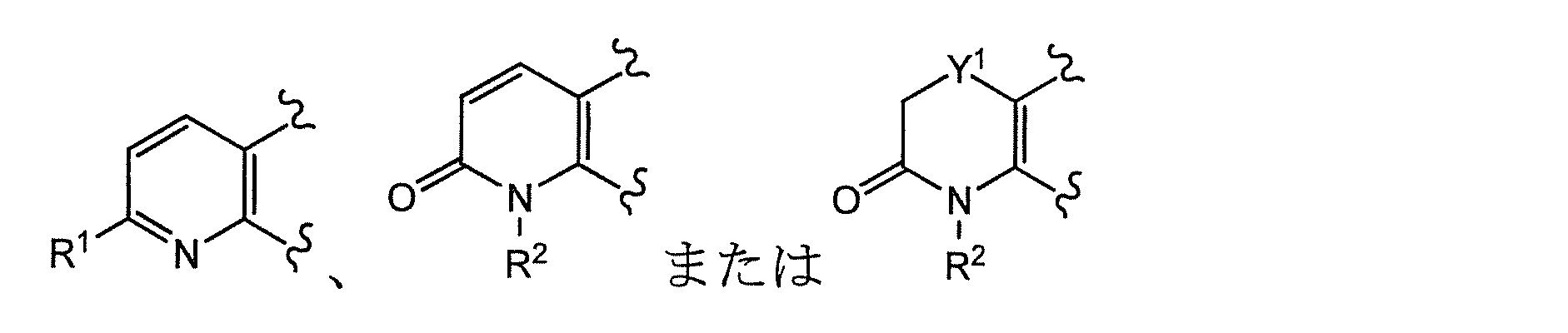

1つの好ましい実施形態では、X1は、S、OまたはNR5である。
別の好ましい実施形態では、X1は、結合手、S、OまたはNR5である。
X1は、より好ましくは、結合手、SまたはOである。
1つの好適な実施形態では、Y1はCR8R9またはSである。
Y1は、より好ましくは、CH2である。
(1)C3-10シクロアルキル基(例、シクロプロピル、シクロヘキシル);
(2)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール基(例、フェニル、ナフチル);
(3)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル);
(4)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、
(d)ハロゲン原子、および
(e)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員非芳香族複素環基(例、テトラヒドロフリル、モルホリニル、チオモルホリニル、ピペリジル、ピロリジニル、ピペラジニル);
(5)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ-カルボニル基、
(d)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル)、
(e)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基、および
(f)4ないし7員芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル)
から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基;
(6)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基;
(7)(a)ハロゲン原子、
(b)C1-6アルコキシ基、
(c)C6-14アリール基(例、フェニル)、および
(d)4ないし7員複素環基(例、テトラヒドロフリル)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ-カルボニル基(例、メトキシカルボニル);
(8)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル、エチルスルホニル、イソプロピルスルホニル);
(9)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基;
(10)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいチオカルバモイル基;
(11)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいスルファモイル基;
(12)カルボキシ基;
(13)ヒドロキシ基;
(14)(a)ハロゲン原子、
(b)カルボキシ基、
(c)C1-6アルコキシ基、
(d)1ないし3個のC6-14アリール基(例、フェニル)で置換されていてもよいC1-6アルコキシ-カルボニル基、
(e)C1-6アルキル基およびC1-6アルコキシ-カルボニル基から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基、
(f)4ないし7員複素環基(例、テトラヒドロフリル)、および
(g)C3-10シクロアルキル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ基;
(15)1ないし3個のハロゲン原子で置換されていてもよいC2-6アルケニルオキシ基(例、エテニルオキシ);
(16)C7-13アラルキルオキシ基(例、ベンジルオキシ);
(17)C6-14アリールオキシ基(例、フェニルオキシ、ナフチルオキシ);
(18)C1-6アルキル-カルボニルオキシ基(例、アセチルオキシ、tert-ブチルカルボニルオキシ);
(19)(a)ハロゲン原子、および
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール-カルボニル基(例、ベンゾイル);
(20)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員非芳香族複素環カルボニル基(例、ピロリジニルカルボニル、モルホリニルカルボニル);
(21)メルカプト基;
(22)(a)ハロゲン原子、および
(b)C1-6アルコキシ-カルボニル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキルチオ基(例、メチルチオ、エチルチオ);
(23)C7-13アラルキルチオ基(例、ベンジルチオ);
(24)C6-14アリールチオ基(例、フェニルチオ、ナフチルチオ);
(25)シアノ基;
(26)ニトロ基;
(27)ハロゲン原子(例、フッ素原子、塩素原子、臭素原子);
(28)C1-3アルキレンジオキシ基;
(29)C1-3アルキレンオキシ基(例、メチレンオキシ、エチレンオキシ);
(30)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員芳香族複素環カルボニル基(例、ピラゾリルカルボニル、ピラジニルカルボニル、イソキサゾリルカルボニル、ピリジルカルボニル、チアゾリルカルボニル);
等が挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
(1)1ないし3個のC1-6アルコキシ基(例、メトキシ)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ);
(2)アミノ;
(3)カルバモイル;
等である。
R2は、より好ましくは、水素原子である。
(1)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(2)ヒドロキシ基、
(3)C1-6アルコキシ基(例、メトキシ)、および
(4)ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル)等である。
該5ないし7員単環式芳香族複素環としては、上述の5ないし7員単環式芳香族複素環基に対応する環が挙げられる。
該C6-10芳香族炭化水素としては、上述のC6-10アリール基に対応する環が挙げられる。
(1)C3-10シクロアルキル基(例、シクロプロピル、シクロヘキシル);
(2)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール基(例、フェニル、ナフチル);
(3)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル);
(4)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、
(d)ハロゲン原子、および
(e)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員非芳香族複素環基(例、テトラヒドロフリル、モルホリニル、チオモルホリニル、ピペリジル、ピロリジニル、ピペラジニル);
(5)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ-カルボニル基、
(d)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル)、
(e)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基、および
(f)4ないし7員芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル)
から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基;
(6)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基;
(7)(a)ハロゲン原子、
(b)C1-6アルコキシ基、
(c)C6-14アリール基(例、フェニル)、および
(d)4ないし7員複素環基(例、テトラヒドロフリル)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ-カルボニル基(例、メトキシカルボニル);
(8)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル、エチルスルホニル、イソプロピルスルホニル);
(9)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基;
(10)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいチオカルバモイル基;
(11)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいスルファモイル基;
(12)カルボキシ基;
(13)ヒドロキシ基;
(14)(a)ハロゲン原子、
(b)カルボキシ基、
(c)C1-6アルコキシ基、
(d)1ないし3個のC6-14アリール基(例、フェニル)で置換されていてもよいC1-6アルコキシ-カルボニル基、
(e)C1-6アルキル基およびC1-6アルコキシ-カルボニル基から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基、
(f)4ないし7員複素環基(例、テトラヒドロフリル)、および
(g)C3-10シクロアルキル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ基;
(15)1ないし3個のハロゲン原子で置換されていてもよいC2-6アルケニルオキシ基(例、エテニルオキシ);
(16)C7-13アラルキルオキシ基(例、ベンジルオキシ);
(17)C6-14アリールオキシ基(例、フェニルオキシ、ナフチルオキシ);
(18)C1-6アルキル-カルボニルオキシ基(例、アセチルオキシ、tert-ブチルカルボニルオキシ);
(19)(a)ハロゲン原子、および
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール-カルボニル基(例、ベンゾイル);
(20)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員非芳香族複素環カルボニル基(例、ピロリジニルカルボニル、モルホリニルカルボニル);
(21)メルカプト基;
(22)(a)ハロゲン原子、および
(b)C1-6アルコキシ-カルボニル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキルチオ基(例、メチルチオ、エチルチオ);
(23)C7-13アラルキルチオ基(例、ベンジルチオ);
(24)C6-14アリールチオ基(例、フェニルチオ、ナフチルチオ);
(25)シアノ基;
(26)ニトロ基;
(27)ハロゲン原子(例、フッ素原子、塩素原子、臭素原子);
(28)C1-3アルキレンジオキシ基;
(29)C1-3アルキレンオキシ基(例、メチレンオキシ、エチレンオキシ);
(30)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員芳香族複素環カルボニル基(例、ピラゾリルカルボニル、ピラジニルカルボニル、イソキサゾリルカルボニル、ピリジルカルボニル、チアゾリルカルボニル);
(31)(a)ハロゲン原子(例、フッ素原子)、および
(b)C1-6アルコキシ基(例、メトキシ)
から選ばれる1ないし3個の置換基で置換されていてもよいC3-10シクロアルコキシ基(例、シクロプロポキシ、シクロペンチルオキシ);
(32)(a)ハロゲン原子、
(b)カルボキシ基、
(c)ヒドロキシ基、
(d)C1-6アルコキシ-カルボニル基、
(e)C1-6アルコキシ基、および
(f)C1-6アルキル基でモノまたはジ置換されていてもよいアミノ基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基;
(33)(a)ハロゲン原子、
(b)カルボキシ基、
(c)ヒドロキシ基、
(d)C1-6アルコキシ-カルボニル基、
(e)C1-6アルコキシ基、および
(f)C1-6アルキル基でモノまたはジ置換されていてもよいアミノ基
から選ばれる1ないし3個の置換基で置換されていてもよいC2-6アルケニル基(例、エテニル、1-プロペニル);
(34)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)C1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC7-13アラルキル基(例、ベンジル);
等が挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
(1) (A)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(B)ヒドロキシ基、
(C)C1-6アルコキシ基(例、メトキシ)、および
(D)ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル);
(2)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-10シクロアルキル基(例、シクロヘキシル);
等である。
(1)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル、プロピル);
(2)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-10シクロアルキル基(例、シクロヘキシル);
等である。
環Aが、式:
環Bがベンゼンであり;
X1がS、OまたはNR5であり;
X2がOであり;
Y1がCR8R9またはSであり;
R1が、
(1)1ないし3個のC1-6アルコキシ基(例、メトキシ)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ);
(2)アミノ;または
(3)カルバモイル;
であり;
R2が水素原子またはC1-6アルキル基(例、メチル)であり;
R3が
(1) (A)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(B)ヒドロキシ基、
(C)C1-6アルコキシ基(例、メトキシ)、および
(D)ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル)、
(2)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-10シクロアルキル基(例、シクロヘキシル)であり;
R4が1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル)であり;
R5が水素原子であり;
R8が水素原子であり;および
R9が水素原子である、化合物(I)。
環Aが、式:
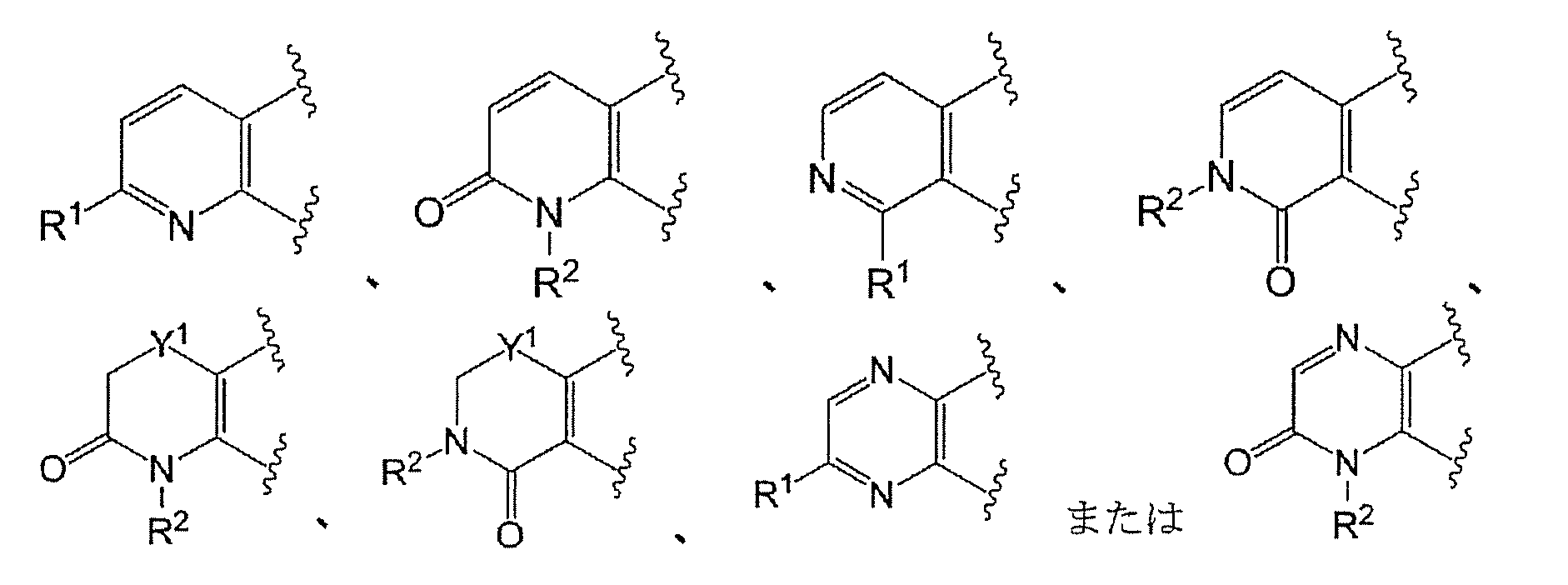
環Bがベンゼンであり;
X1が結合手、S、OまたはNR5であり;
X2がOであり;
Y1がCR8R9またはSであり;
R1が、
(1)1ないし3個のC1-6アルコキシ基(例、メトキシ)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ);
(2)アミノ;または
(3)カルバモイル;
であり;
R2が水素原子またはC1-6アルキル基(例、メチル)であり;
R3が
(1) (A)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(B)ヒドロキシ基、
(C)C1-6アルコキシ基(例、メトキシ)、および
(D)ハロゲン原子(例、フッ素原子、塩素原子、臭素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル)、
(2)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-10シクロアルキル基(例、シクロヘキシル)であり;
R4が1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル)であり;
R5が水素原子であり;
R8が水素原子であり;および
R9が水素原子である、化合物(I)。
環Aが、式:

環Bがベンゼンであり;
X1が結合手、SまたはOであり;
X2がOであり;
Y1がCH2であり;
R1がC1-6アルコキシ基(例、メトキシ)であり;
R2が水素原子であり;
R3が
(1)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル、プロピル)、または
(2)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-10シクロアルキル基(例、シクロヘキシル)であり;および
R4が1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基である(例、エチル)、化合物(I)。
環Aが、式:

環Bがベンゼンであり;
X1が結合手、SまたはOであり;
X2がOであり;
Y1がCH2であり;
R2が水素原子であり;
R3が
(1)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル、プロピル)、または
(2)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-10シクロアルキル基(例、シクロヘキシル)であり;および
R4が1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル)である、請求項1記載の化合物またはその塩。
2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン;
2-エトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン;または
3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン;
あるいはその塩。
また、化合物(I)は、無溶媒和物(例えば、無水物)であっても、溶媒和物(例えば、水和物)であってもよい。
さらに、1Hを2H(D)に変換した重水素変換体も、化合物(I)に包含される。
本明細書中、融点は、例えば、微量融点測定器(ヤナコ、MP-500D型またはBuchi、B-545型)またはDSC(示差走査熱量分析)装置(SEIKO、EXSTAR6000)等を用いて測定される融点を意味する。
一般に、融点は、測定機器、測定条件等によって変動する場合がある。本明細書中の結晶は、通常の誤差範囲内であれば、本明細書に記載の融点と異なる値を示す結晶であってもよい。
化合物(I)のアミノ基がアシル化、アルキル化またはリン酸化された化合物(例、化合物(I)のアミノ基がエイコサノイル化、アラニル化、ペンチルアミノカルボニル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メトキシカルボニル化、テトラヒドロフラニル化、ピロリジルメチル化、ピバロイルオキシメチル化またはtert-ブチル化された化合物);
化合物(I)のヒドロキシ基がアシル化、アルキル化、リン酸化またはホウ酸化された化合物(例、化合物(I)のヒドロキシ基がアセチル化、パルミトイル化、プロパノイル化、ピバロイル化、サクシニル化、フマリル化、アラニル化またはジメチルアミノメチルカルボニル化された化合物);
化合物(I)のカルボキシ基がエステル化またはアミド化された化合物(例、化合物(I)のカルボキシ基がエチルエステル化、フェニルエステル化、カルボキシメチルエステル化、ジメチルアミノメチルエステル化、ピバロイルオキシメチルエステル化、エトキシカルボニルオキシエチルエステル化、フタリジルエステル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メチルエステル化、シクロヘキシルオキシカルボニルエチルエステル化またはメチルアミド化された化合物)等が挙げられる。これらの化合物は自体公知の方法によって化合物(I)から製造することができる。
化合物(I)は、例えば、以下に示す方法またはそれに準ずる方法で製造することができる。
以下の各スキームにおいて、各原料化合物は、反応を阻害しないのであれば、塩を形成していてもよく、かかる塩としては、前述の式(I)で示される化合物の塩として例示したものが用いられる。
また、以下の各スキームにおいて、原料化合物は、具体的製法を述べない場合、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。

化合物(1)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。
化合物(3)は化合物(1)と化合物(2)の塩基性条件下で行う閉環反応によって製造することができる。詳しくは、化合物(1)1モルに対して化合物(2)を約1.0~5.0モル、好ましくは約1.0~2.0モル用いて行う。塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、N,N-ジイソプロピルエチルアミン等の有機塩基類等が挙げられ、化合物(1)1.0モルに対して約1.0~10.0モル、好ましくは約1.0~5.0モル用いる。本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒として反応が進行する限り特に限定されないが、例えばベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類またはそれらの混合溶媒等が好ましい。反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-10~200℃、好ましくは0~100℃である。化合物(3)は反応液のままか粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(4)は化合物(3)の、塩基とR3に対応するアルキル化剤を用いたS-アルキル化反応によって製造される。詳しくは化合物(3)1モルに対して塩基1.0~10.0モル、好ましくは1.0~5.0モル、R3に対応するアルキル化剤は1.0~20.0モル、好ましくは1.0~10.0モル用いて行う。塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、N,N-ジイソプロピルエチルアミン等の有機塩基類等が挙げられる。R3に対応するアルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等の各種ハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、水またはそれらの混合溶媒等が好ましい。反応時間は通常15分~60時間、好ましくは15分~24時間である。反応温度は通常-10~200℃、好ましくは0~150℃である。化合物(4)は反応液のままか粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(5)は化合物(4)の酸化反応によって製造される。詳しくは化合物(4)1モルに対して酸化剤1.0~30.0モル、好ましくは1.0~3.0モル用いる。酸化剤としては過酸化水素、オキソン(登録商標)、過酢酸、過安息香酸、メタクロロ過安息香酸等の過酸類、次亜塩素酸、過ヨウ素酸等のオキソ酸類とその塩、クロム酸等の金属オキソ酸とその塩またはその他の酸化剤等が挙げられる。本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、ギ酸等のカルボン酸類、水またはそれらの混合溶媒等が好ましい。反応時間は通常10分~60時間、好ましくは30分~5時間である。反応温度は通常-10~200℃、好ましくは0~150℃である。化合物(5)は反応液のままか粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(I)は、化合物(4)または(5)の塩基とX1-R3に対応する求核剤を用いた置換反応によって製造することができる。詳しくは、化合物(4)または(5)1モルに対し、塩基を1.0~20.0モル、好ましくは1.0~10.0モル用い、X1-R3に対応する求核剤を1.0~100.0モル、好ましくは1.0~10.0モル用いて行う。塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。X1-R3に対応する求核剤としてはメタノール、エタノール、プロパノール、1,1-ジメチルエタノール、2,2,2-トリフルオロエタノール、2,2,3,3,3-ペンタフルオロプロパノール等のアルコール類、芳香族水酸基を有する各種フェノール誘導体、エタンチオール、チオグリコール酸アミド等の有機チオール類、チオフェノール等の各種芳香族チオール誘導体、メチルアミン、エチルアミン等の有機塩基類、アニリン等の各種芳香族アミン類、有機グリニャール試薬(n-プロピルマグネシウムブロミド、n-ブチルマグネシウムブロミド)、有機リチウム試薬(n-プロピルリチウム、n-ブチルリチウム)等の有機金属試薬類等が挙げられる。なお必要に応じて塩基は求核剤として用いることができる。本反応は無溶媒で行うか、反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、シクロヘキサン、ヘキサン等の飽和炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、アセトン、メチルエチルケトン等のケトン類、ジメチルスルホキシド等のスルホキシド類、水等の溶媒またはそれらの混合溶媒等が好ましい。反応時間は通常10分~24時間、好ましくは15分~12時間である。反応温度は通常-10~200℃、好ましくは0~100℃である。化合物(I)は反応液のままか粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

化合物(1a)は、後述のスキーム4の方法に従って製造することができる。
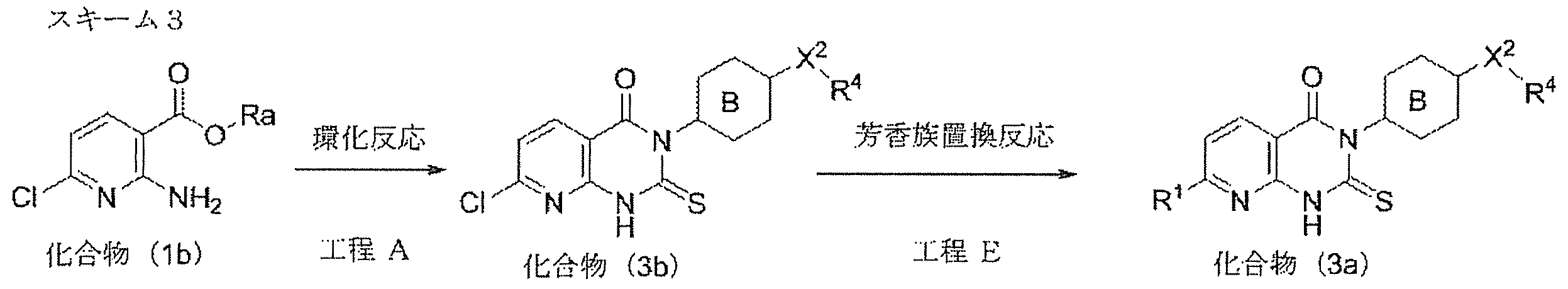
化合物(1b)は、後述のスキーム4に準ずる方法に従って製造することができる。
化合物(3b)1モルに対して、R1に対応する求核剤を1.0~10モル、好ましくは1.0~5.0モル用いて行う。R1に対応する求核剤としてはメタノール、エタノール、プロパノール、エチレングリコール等のアルコール類、ナトリウムメトキシド、ナトリウムエトキシド等の金属アルコキシド類、メチルアミン、エチルアミン、ベンジルアミン、4-メトキシベンジルアミン等の有機アミン類等が挙げられる。必要に応じて塩基を用いることができる。化合物(3b)1モルに対して塩基を1.0~10モル、好ましくは1.0~5.0モル用いる。このような塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。また金属触媒としてヨウ化銅、臭化銅、酢酸パラジウム、塩化パラジウム等を用いることができ、化合物(3b)1モルに対して、0.01~1.0モル、好ましくは0.1~0.5モル用いることができる。本反応は無溶媒または反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、メタノール、エタノール、プロパノール、エチレングリコール等のアルコール類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチルピロリドン等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、水またはそれらの混合溶媒等が好ましい。反応時間は通常10分~60時間、好ましくは15分~24時間である。反応温度は通常-10~200℃、好ましくは0~180℃である。化合物(3a)は反応液のままか粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
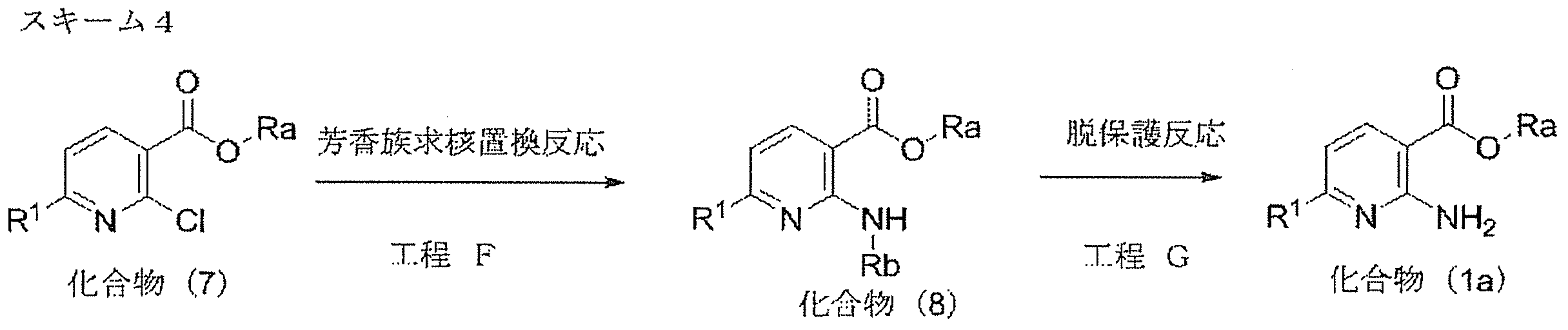
化合物(7)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。
化合物(8)は化合物(7)に塩基存在下、Rbに対応するアミン誘導体の求核置換反応を行い製造することができる。詳しくは化合物(7)1モルに対して、Rbに対応するアミン誘導体を1.0~5.0モル、好ましくは1.0~3.0モル用いて行う。Rbに対応するアミン誘導体としては、4-メトキシベンジルアミンまたは2,4-ジメトキシベンジルアミンが挙げられる。塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。また金属触媒としてヨウ化銅、臭化銅、酢酸パラジウム、塩化パラジウム等を用いることができ、化合物(7)1モルに対して、0.01~1.0モル、好ましくは0.1~0.5モル用いることができる。本反応は無溶媒または反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチルピロリドン等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、水またはそれらの混合溶媒等が好ましい。反応時間は通常10分~60時間、好ましくは15分~24時間である。反応温度は通常-10~200℃、好ましくは0~180℃である。化合物(8)は反応液のままか粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(1a)は化合物(8)の酸性条件下での脱保護反応により製造することができる。詳しくは化合物(8)1モルに対して酸1.0~50モル、好ましくは1.0~10モル用いる。用いる酸として、p-トルエンスルホン酸、ピリジンp-トルエンスルホン酸塩、トリフルオロ酢酸、ギ酸、酢酸等の有機酸、塩酸、臭化水素酸等の無機酸が挙げられる。本反応は無溶媒または反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、メタノール、エタノール、イソプロピルアルコール等のアルコール類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチルピロリドン等のアミド類、酢酸、ギ酸、トリフルオロ酢酸等の有機酸類、ジメチルスルホキシド等のスルホキシド類、アセトン、水またはそれらの混合溶媒等が好ましい。反応時間は通常10分~60時間、好ましくは15分~24時間である。反応温度は通常-10~200℃、好ましくは0~180℃である。本反応には必要に応じアニソールを添加することができ、化合物(8)1モルに対して0.5~20モル好ましくは1.0~10モル用いることができる。化合物(1a)は反応液のままか粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
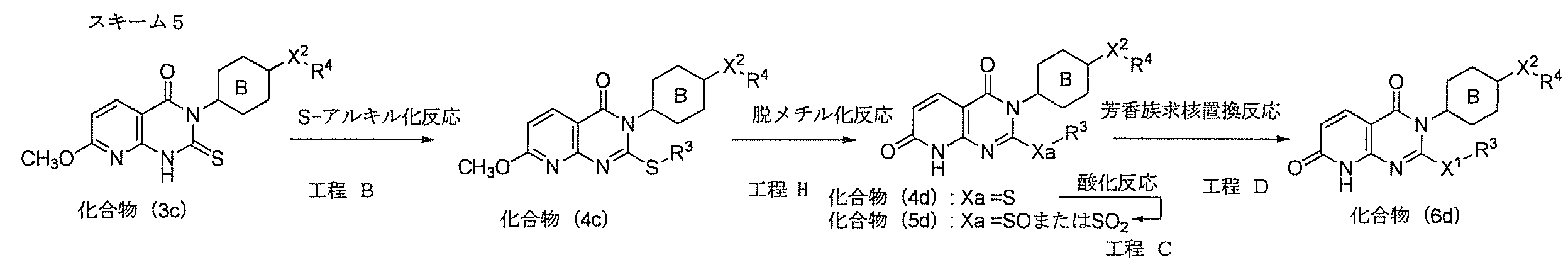
化合物(4d)は化合物(4c)の脱メチル化反応により製造することができる。詳しくは化合物(4c)1モルに対してピリジン塩酸塩を1.0~100モル、好ましくは1.0~20.0モル用いて行う。本反応は無溶媒または反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチルピロリドン等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、ギ酸等のカルボン酸類、水またはそれらの混合溶媒等が好ましい。反応時間は通常15分~60時間、好ましくは30分~24時間である。反応温度は通常30~250℃、好ましくは50~180℃である。化合物(4d)は反応液のままか粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

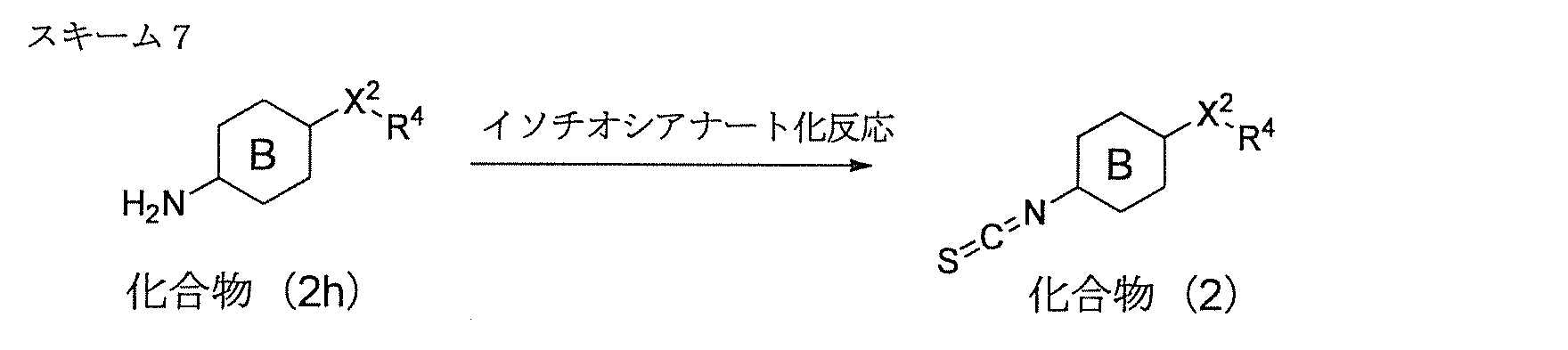
化合物(2h)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。

化合物(10)および化合物(11)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。
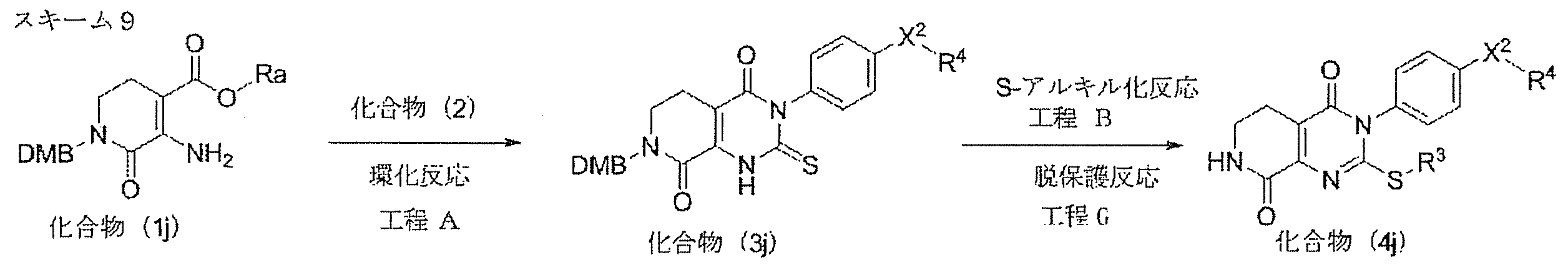

なお、化合物(1k)は化合物(1j)の酸化反応により製造される(工程I)。
化合物(1j)1モルに対し酸化剤0.1~10.0モル、好ましくは0.5~5.0モル用いて行う。酸化剤としては、2,3-ジクロロ-5,6-ジシアノ-1,4-ベンゾキノン、2,3,5,6-テトラクロロ-1,4-ベンゾキノンまたはパラジウム-炭素等が挙げられる。本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール等のアルコール類、ベンゼン、トルエン、キシレン等の芳香族炭化水素類、シクロヘキサン、ヘキサン等の飽和炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、ジメチルスルホキシド等のスルホキシド類、水等の溶媒またはそれらの混合溶媒等が好ましい。反応時間は通常5分~60時間、好ましくは10分~12時間である。反応温度は通常-10~200℃、好ましくは0~150℃である。化合物(1k)は反応液のままか粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

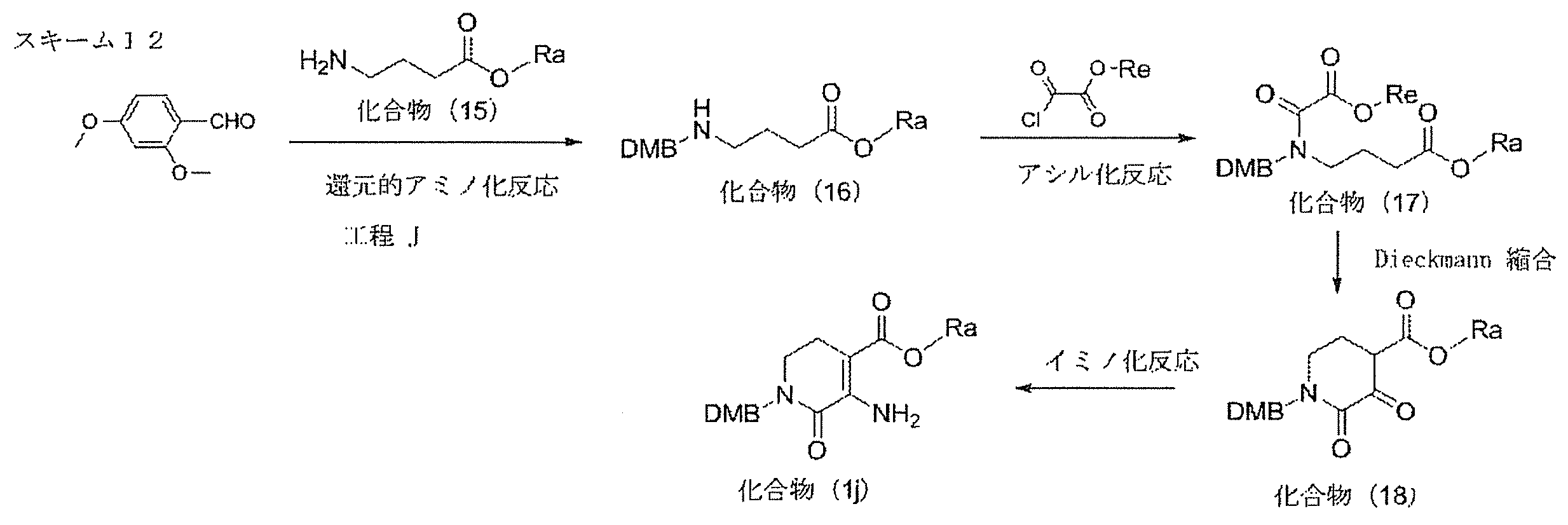
化合物(16)は2,4-ジメトキシベンズアルデヒドと化合物(15)から、還元剤を用いた還元的アミノ化反応により製造することができる。詳しくは化合物(15)1モルに対し2,4-ジメトキシベンズアルデヒドを1.0~3.0モル、好ましくは1.0~1.5モル用い、還元剤は1.0~5.0モル、好ましくは1,0~2.0モル用いて行う。還元剤としては水素化ホウ素ナトリウム、水素化ホウ素リチウム、水素化トリアセトキシホウ素ナトリウム等が挙げられる。本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール等のアルコール類、ジクロロメタン、1,2-ジクロロエタンのようなハロゲン化炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、ジメチルスルホキシド等のスルホキシド類、アセトニトリル、プロピオニトリル等のニトリル類、酢酸、水等の溶媒またはそれらの混合溶媒等が好ましい。反応時間は通常30分~60時間、好ましくは1時間~36時間である。反応温度は通常-10~200℃、好ましくは0~150℃である。化合物(16)は反応液のままか粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。


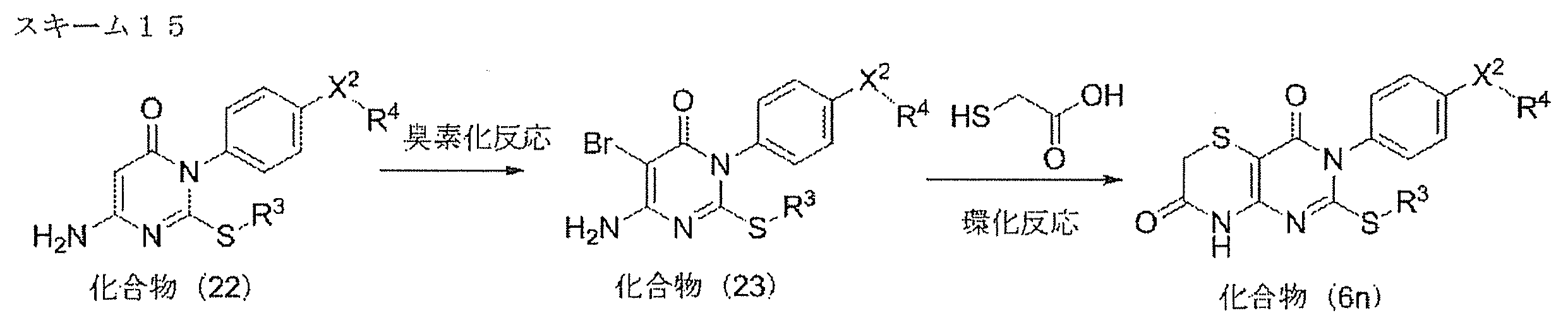
化合物(23)は化合物(22)の臭素化反応により製造することができる。詳しくは化合物(22)1モルに対し臭素化剤1.0モル~3.0モル、好ましくは1.0~1.5モル用いて行う。臭素化剤としては、臭素、N-ブロモスクシンイミド、臭化過臭化ピリジニウム等が挙げられる。また反応性を調節する目的で酢酸ナトリウム等を添加物として加えることができる。本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、酢酸等の溶媒またはそれらの混合溶媒等が好ましい。反応時間は通常30分~30時間、好ましくは45分~5時間である。反応温度は通常-10~100℃、好ましくは0~40℃である。化合物(23)は反応液のままか粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により精製することができる。

化合物(22)はスキーム16に示した経路で製造することができる。すなわち、化合物(24)から工程A(スキーム1)に準じた環化反応により化合物(25)が製造され、工程B(スキーム1)に準じたS-アルキル化、および工程G(スキーム4)に準じた脱保護により化合物(22)は製造することができる。
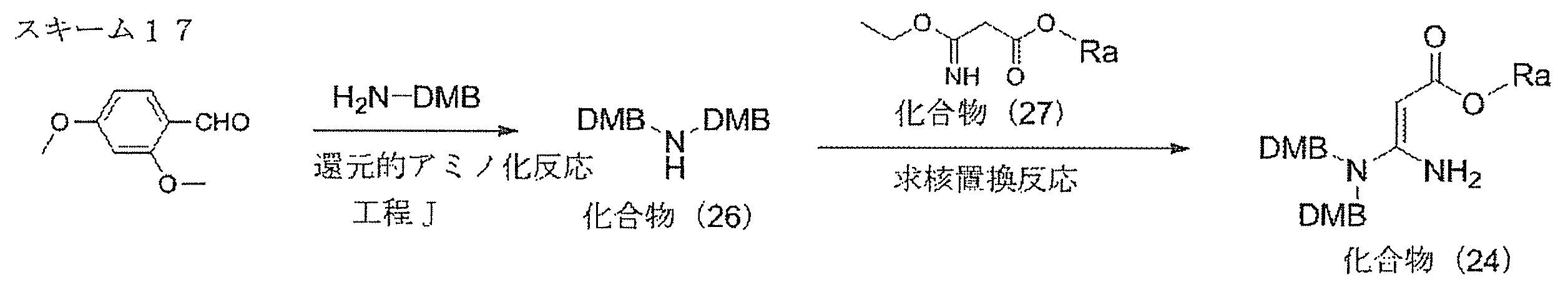
化合物(26)は工程J(スキーム12)に準ずる還元的アミノ化反応により製造することができる。

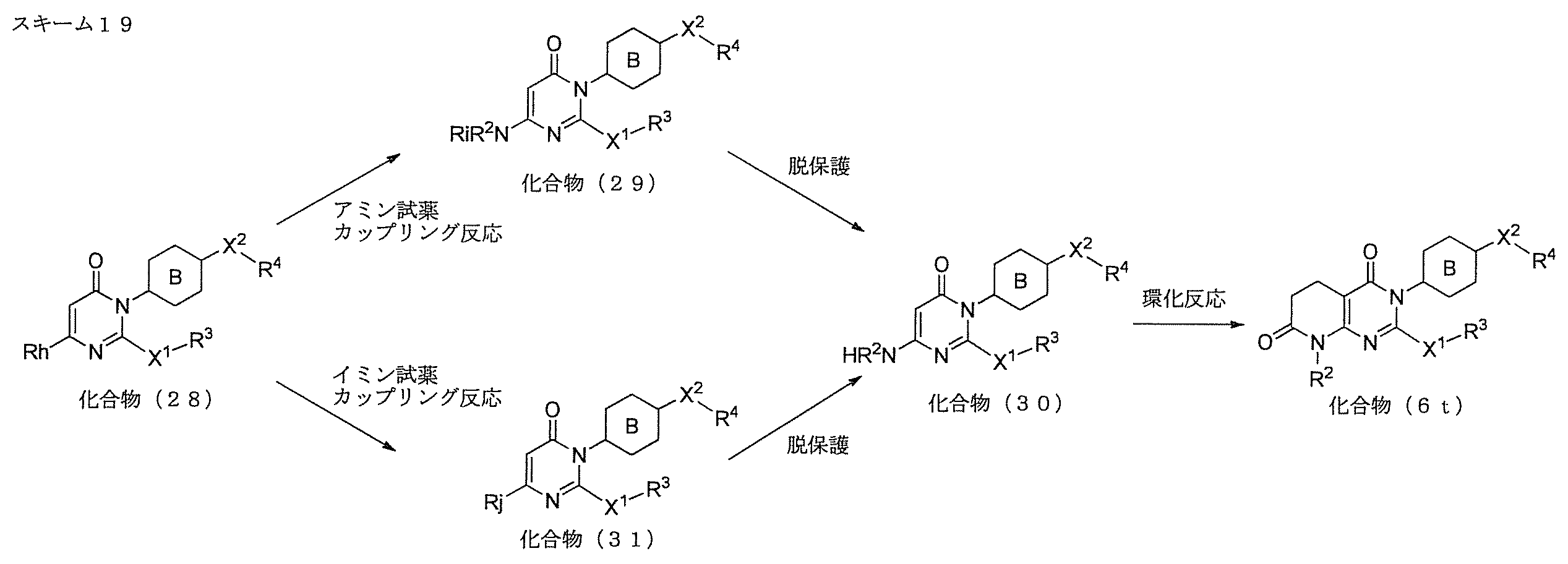
化合物(29)あるいは化合物(31)は、化合物(28)と対応する含窒素試薬とのカップリング反応により製造される。詳しくは化合物(28)1モルに対してアミン試薬あるいはイミン試薬1.0~10.0モル、好ましくは1.0~3.0モル、有機金属試薬0.01~1モル、好ましくは0.05~0.2モル用い、ホスフィン配位子0.01~1モル、好ましくは0.1~0.5モル、塩基1.0~10.0モル、好ましくは2.0~6.0モルを用いて行う。場合により、ホスフィン配位子を用いなくとも良い。
アミン試薬としてはベンジルアミン、ジベンジルアミン、2-メトキシベンジルアミン、3-メトキシベンジルアミン、4-メトキシベンジルアミン、2,3-ジメトキシベンジルアミン、2,4-ジメトキシベンジルアミン、3,4-ジメトキシベンジルアミン、2,4,6-トリメトキシベンジルアミン、3,4,5-トリメトキシベンジルアミン、2,3,4-トリメトキシベンジルアミン、2,4,5-トリメトキシベンジルアミン、2-メチルベンジルアミン、3-メチルベンジルアミン、4-メチルベンジルアミン、2,3-ジメチルベンジルアミン、2,4-ジメチルベンジルアミン、3,4-ジメチルベンジルアミン、2,4,6-トリメチルベンジルアミン、2,4,5-トリメチルベンジルアミン等が挙げられ、該イミン試薬としてはベンゾフェノンイミン、1,1-ビス(4-メトキシフェニル)メタンイミン、9-イミノフルオレン等が挙げられる。
有機金属試薬としてはトリス(ジベンジリデンアセトン)ジパラジウム(0)、テトラキス(トリフェニルホスフィン)パラジウム、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリドジクロロメタン錯体、酢酸パラジウム等が挙げられる。
ホスフィン配位子としては2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル、(9,9-ジメチル-9H-キサンテン-4,5-ジイル)ビス(ジフェニルホスファン)、2,2’-ビス(ジフェニルホスフィノ)-1,1’-ビナフチル等が挙げられる。
塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム、酢酸カリウム、炭酸セシウム等の塩基性塩類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、アルコール類(例、メタノール、エタノール等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、有機酸類(例、酢酸等)、水またはこれら二種以上の混合物等が用いられる。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0℃~300℃、好ましくは20℃~200℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
酸処理の場合、化合物(29)1モルに対して酸試薬1.0~200モル、好ましくは3.0~20.0モルを用いて行う。酸試薬としては、塩化アルミニウム等のルイス酸や酢酸、トリフルオロ酢酸等の有機酸、塩酸等の鉱酸が挙げられる。
水素添加の場合、使用する金属試薬の使用量は、化合物(29)に対して5重量%~1000重量%、好ましくは10重量%~300重量%である。金属試薬としては、パラジウム炭素、水酸化パラジウム、酸化白金、ラネーニッケル、ラネーコバルト等が挙げられる。また水素の圧力は通常1気圧~100気圧である。
本反応は、無溶媒中または適切な溶媒存在下で行うのが好ましい。該溶媒は、反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、アルコール類(例、メタノール、エタノール等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、有機酸類(例、酢酸等)、水またはこれら二種以上の混合物等が用いられる。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常25℃~300℃、好ましくは50℃~200℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
酸試薬としては、塩化アルミニウム等のルイス酸や酢酸、トリフルオロ酢酸等の有機酸、塩酸等の鉱酸が挙げられる。
本反応は、無溶媒中または適切な溶媒存在下で行うのが好ましい。該溶媒は、反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、アルコール類(例、メタノール、エタノール等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、有機酸類(例、酢酸等)、水またはこれら二種以上の混合物等が用いられる。
反応時間は、通常約10分~約50時間、好ましくは約30分~約12時間である。反応温度は、通常25℃~300℃、好ましくは50℃~200℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
求電子剤としてプロパ-2-エノイルクロリド、2-メチルプロパ-2-エノイルクロリド、(2E)-ブタ-2-エノイルクロリド、(2Z)-ブタ-2-エノイルクロリド、(2E)-2-メチルブタ-2-エノイルクロリド、(2Z)-2-メチルブタ-2-エノイルクロリド等が挙げられる。
本反応は、無溶媒中または反応に不活性な溶媒存在下で行うのが好ましい。該溶媒は、反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、アルコール類(例、メタノール、エタノール等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、有機酸類(例、酢酸等)、水またはこれら二種以上の混合物等が用いられる。
反応時間は、通常約1分~約100時間、好ましくは約5分~約4時間である。反応温度は、通常約0℃~約200℃、好ましくは約0℃~約100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
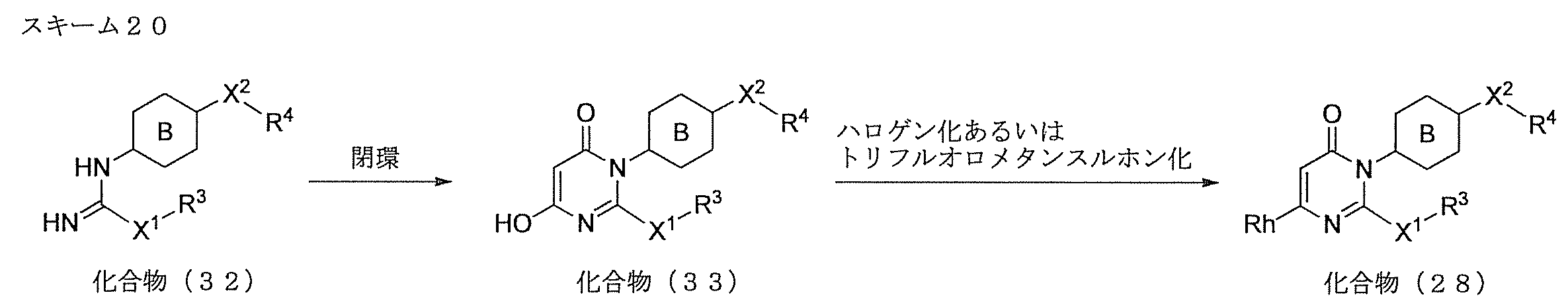
化合物(33)は、化合物(32)とジアルキルマロネートとの塩基性条件化で行う環化反応により製造される。詳しくは化合物(32)1モルに対してジアルキルマロネート1.0~10.0モル、好ましくは1.0~3.0モルを用い、塩基1.0~100.0モル、好ましくは2.0~10.0モルを用いて行う。
塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、アルコール類(例、メタノール、エタノール等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、有機酸類(例、酢酸等)、水またはこれら二種以上の混合物等が用いられる。
反応時間は通常10分~72時間、好ましくは15分~24時間である。反応温度は通常0℃~150℃、好ましくは0℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
本反応は、無溶媒中または反応に不活性な溶媒存在下で行うのが有利である。該溶媒は、反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、またはこれら二種以上の混合物等が用いられる。
反応時間は通常10分~72時間、好ましくは30分~3時間である。反応温度は通常0℃~150℃、好ましくは0℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
トリフルオロメチルスルホン化試薬としては、1,1,1-トリフルオロ-N-フェニル-N-[(トリフルオロメチル)スルホニル]メタンスルホンアミド、2-[N,N-ビス(トリフルオロメチルスルホニル)アミン]-5-クロロピリジン、トリフルオロメタンスルホン酸 無水物等が挙げられる。
塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、またはこれら二種以上の混合物等が用いられる。
反応時間は通常10分~72時間、好ましくは15分~24時間である。反応温度は通常-78℃~100℃、好ましくは0℃~50℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
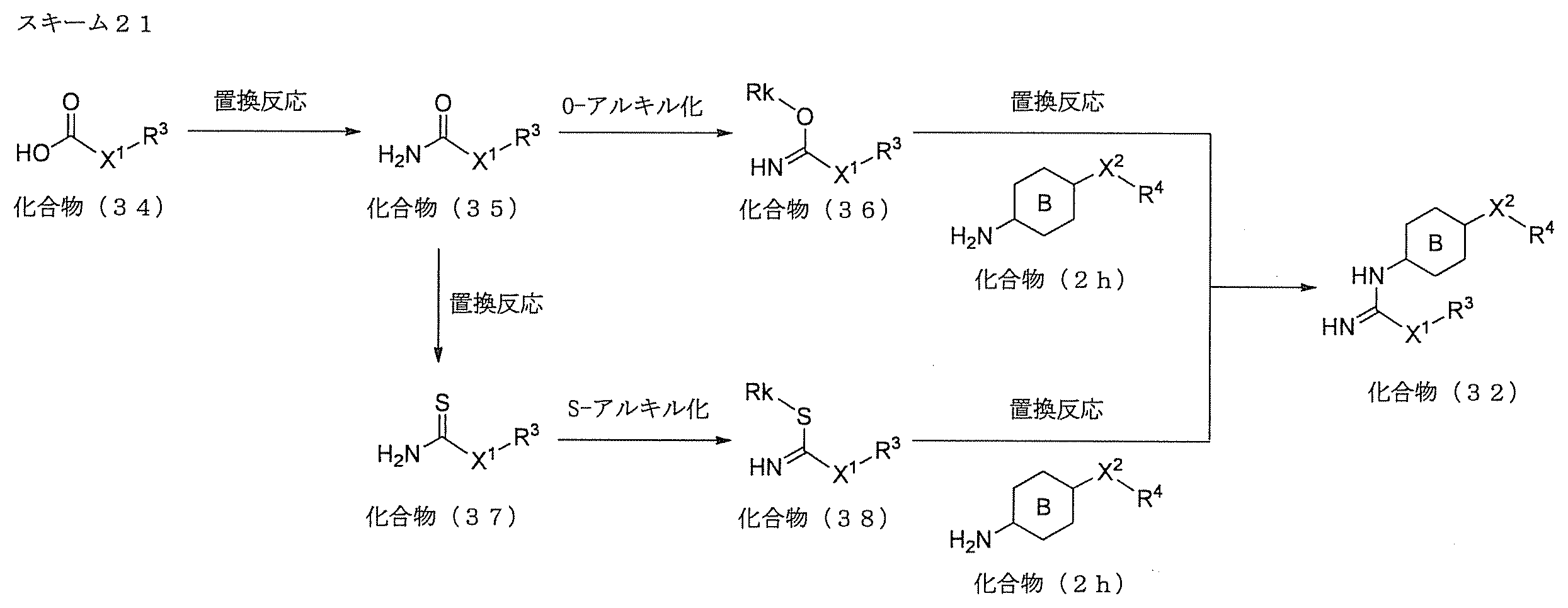
化合物(34)は市販されている場合には市販品をそのまま用いることができ、自体公知の方法あるいはこれらに準じた方法に従って製造することができる。
化合物(35)は化合物(34)の置換反応により得られる。詳しくは、化合物(34)1モルに対してクロル化剤1.0~10.0モル、好ましくは1.0~3.0モルを用い、28%アンモニア水溶液を1.0~20.0モル、好ましくは1.0~3.0モルを用いる。クロル化の際に必要に応じてピリジン、ジシクロヘキシルアミン、N,N-ジメチルホルムアミドや相間移動触媒等を0.001~10.0モル、好ましくは0.001~3.0モルを用いても良い。
クロル化剤としてはオキサリルクロリド、チオニルクロリド、オキシ塩化リン等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、またはこれら二種以上の混合物等が用いられる。
反応時間は通常10分~72時間、好ましくは30分~24時間である。反応温度は通常-78℃~100℃、好ましくは-10℃~25℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
アルキル化剤としてはトリメチルオキソニウムテトラフルオロボラート、ジメチルスルファート、メチルトリフルオロメタンスルホナート、メチルフルオロスルホナート等が挙げられる。
塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、リン酸水素ナトリウム、リン酸ナトリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、またはこれら二種以上の混合物等が用いられる。
反応時間は通常10分~72時間、好ましくは30分~24時間である。反応温度は通常-78℃~100℃、好ましくは-10℃~25℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
求核剤としてはローソン試薬、五硫化リン、五硫化二リン等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、アルコール類(例、メタノール、エタノール等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、有機酸類(例、酢酸等)、水またはこれら二種以上の混合物等が用いられる。
反応時間は通常10分~72時間、好ましくは1時間~24時間である。反応温度は通常0℃~150℃、好ましくは25℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等の各種ハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、アルコール類(例、メタノール、エタノール等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、有機酸類(例、酢酸等)、水またはこれら二種以上の混合物等が用いられる。
反応時間は通常15分~60時間、好ましくは30分~24時間である。反応温度は通常0℃~150℃、好ましくは25℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。反応に塩基が必要でない場合には、塩基を使用せずに製造することができる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、アルコール類(例、メタノール、エタノール等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、有機酸類(例、酢酸等)、水またはこれら二種以上の混合物等が用いられる。
反応時間は通常30分~100時間、好ましくは1時間~72時間である。反応温度は通常0℃~150℃、好ましくは25℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

化合物(6v)は、化合物(6u)のアルキル化反応によって製造される。詳しくは化合物(6u)1モルに対し、対応するアルキル化剤1.0~50.0モル、好ましくは1.0~3.0モル用い、塩基1.0~100.0モル、好ましくは2.0~20.0モル用いる。
アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等の各種ハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
塩基としては炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いる。
このような溶媒として反応が進行する限り特に限定されないが、本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒として反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、アルコール類(例、メタノール、エタノール等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、有機酸類(例、酢酸等)、水またはこれら二種以上の混合物等が用いられる。
反応時間は通常15分~100時間、好ましくは30分~12時間である。反応温度は通常-10℃~200℃、好ましくは0℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
リルヨージド、トリメチルシリルブロミド)等を使用する方法、還元法等が挙げられる。
無痛化剤の好適な例としては、ベンジルアルコールが挙げられる。
抗酸化剤の好適な例としては、亜硫酸塩、アスコルビン酸塩等が挙げられる。
また、本発明化合物は安全性に優れていることから、長期に亘って投与することが可能である。
上記「排尿困難治療剤」としては、例えばアセチルコリンエステラーゼ阻害薬(例、ジスチグミン)等が挙げられる。
(1)本発明化合物または併用薬剤を単独で投与する場合に比べて、その投与量を軽減することができる、
(2)本発明化合物と作用機序が異なる併用薬剤を選択することにより、治療期間を長く設定することができる、
(3)本発明化合物と作用機序が異なる併用薬剤を選択することにより、治療効果の持続を図ることができる、
(4)本発明化合物と併用薬剤とを併用することにより、相乗効果が得られる、等の優れた効果を得ることができる。
以下の実施例中の「室温」は通常約 10 ℃ないし約 35 ℃を示す。混合溶媒において示した比は、特に断らない限り容量比を示す。%は、特に断らない限り重量%を示す。
シリカゲルカラムクロマトグラフィーにおいて、NHと記載した場合は、アミノプロピルシラン結合シリカゲルを用いた。HPLC (高速液体クロマトグラフィー) において、C18と記載した場合は、オクタデシル結合シリカゲルを用いた。溶出溶媒の比は、特に断らない限り容量比を示す。
1H NMR (プロトン核磁気共鳴スペクトル) はフーリエ変換型NMRで測定した。解析にはACD/SpecManager (商品名) 等を用いた。水酸基およびアミノ基等のプロトンが非常に緩やかなピークについては記載していない。
MS (マススペクトル) は、LC/MS (液体クロマトグラフ質量分析計) により測定した。
イオン化法としては、ESI (ElectroSpray Ionization、エレクトロスプレーイオン化) 法、または、APCI (Atomospheric Pressure Cheimcal Ionization、大気圧化学イオン化) 法を用いた。データは実測値 (found) を記載した。塩の場合は、通常、フリー体の分子イオンピークまたはフラグメントイオンピークが観測される。
s :シングレット
d :ダブレット
t :トリプレット
q :カルテット
dd :ダブルダブレット
ddd :ダブルダブルダブレット
dt :ダブルトリプレット
td :トリプルダブレット
tt :トリプルトリプレット
tq :トリプルカルテット
spt :セプテット
sxt :セクステット
br. s. :ブロード(幅広い)シングレット
m :マルチプレット
J :カップリング定数
Hz :ヘルツ
CHLOROFORM-d:重クロロホルム
DMSO-d6:重ジメチルスルホキシド
1H NMR:プロトン核磁気共鳴
下記の実施例においてHPLC-マススペクトル(LC-MS)は以下の条件により測定した。
測定機器:ウォーターズ社 Micromass ZQ-Alliance HT
カラム:CAPCELL PAK C18UG120, S-3 μm, 1.5 X 35 mm
溶媒:A液;0.05%トリフルオロ酢酸含有水、B液;0.04%トリフルオロ酢酸含有アセトニトリル
グラジエントサイクル:0.00分(A液/B液=90/10), 2.00分(A液/B液=5/95), 2.75分(A液/B液=5/95), 2.76分(A液/B液=90/10), 3.45分(A液/B液=90/10)
注入量:2 μl、流速:0.5 ml/min、検出法:UV 220 nm
イオン化法:電子衝撃イオン化法 (Electron Spray Ionization: ESI)
2-(エチルスルファニル)-7-メトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン
メチル 2,6-ジクロロピリジン-3-カルボキシラート (2.06 g) 、炭酸水素ナトリウム(1.26 g)、4-メトキシベンジルアミン(1.50 ml)、ヨウ化銅(I)(190 mg)およびN,N-ジメチルホルムアミド(20 ml)の混合物を80℃に加熱し、1時間30分撹拌した。その後反応混合物を減圧濃縮し、得られた残渣を酢酸エチル(150 ml)で希釈し、不溶固体をろ別した後、ろ液に28%アンモニア水(5 ml)を加え、水および飽和食塩水で順に洗浄し、無水硫酸マグネシウムで乾燥後減圧濃縮した。得られた黄色の油状残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(1.58 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ3.73 (3H, s), 3.81 (3H, s), 4.55 (2H, d, J=5.7 Hz), 6.68 (1H, d, J=8.3 Hz), 6.89 (2H, d, J=8.7 Hz), 7.29 (2H, d, J=8.7 Hz), 8.08 (1H, d, J=8.3 Hz), 8.43 (1H, t, J=5.7 Hz).
メチル 6-クロロ-2-[(4-メトキシベンジル)アミノ]ピリジン-3-カルボキシラート(1.0 g)のトリフルオロ酢酸(5 ml)溶液にアニソール(0.5 ml)を加え、室温で2時間撹拌した。その後、反応混合物を減圧濃縮後、トルエンで共沸して得られた油状残渣を酢酸エチル(80 ml)で希釈し、飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(596 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ3.81 (3H, s), 6.66 (1H, d, J=8.3 Hz), 7.54 (2H, brs), 8.05 (1H, d, J=8.3 Hz).
1-フルオロ-4-ニトロベンゼン(10.6 g)および2,2,2-トリフルオロエタノール(12.0 g)をN,N-ジメチルホルムアミド(80 ml)に溶かし、そこへ炭酸カリウム(15.5 g)を加えて80℃で2時間攪拌した。放冷後、反応混合物に酢酸エチル(100 ml)を加えて、白色沈殿をろ別し、ろ液を減圧濃縮して橙色の残渣を得た。得られた残渣を再び酢酸エチル(400 ml)に溶かし、水、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して橙色の粗製固体を得た。得られた固体を10%ジエチルエーテル/ヘキサン混合溶媒で洗浄し、黄白色、針状結晶の標題化合物(15.8 g)を得た。
1H NMR (400 MHz, DMSO-d6) δ4.99 (2H, q, J=8.8 Hz), 7.30 (2H, d, J=9.3 Hz), 8.26 (2H, d, J=9.3 Hz).
1-ニトロ-4-(2,2,2-トリフルオロエトキシ)ベンゼン(5.5 g)をメタノール(100 ml)に溶かし、そこへ10%パラジウム/活性炭素(50% 含水,2.5 g)を加え、水素雰囲気下室温で24時間攪拌した。その後パラジウム/活性炭をろ別し、ろ液を減圧濃縮して濃橙色油状の残渣を得た。得られた残渣を酢酸エチル(200 ml)に溶かし、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して得られた濃橙色油状の残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して橙色油状物の標題化合物(4.5 g)を得た。
1H NMR (400 MHz, DMSO-d6) δ4.53 (2H, q, J=9.0 Hz), 4.76 (2H, s), 6.52 (2H, d, J=8.9 Hz), 6.75 (2H, d, J=8.9 Hz).
4-(2,2,2-トリフルオロエトキシ)アニリン(10 g)をテトラヒドロフラン(100 ml)に溶かし、6M塩酸(9 ml)を加えた後、-5℃に冷却した。そこへチオホスゲン(4.01 ml)のテトラヒドロフラン(20ml)溶液を5分間かけて滴下した。-5℃で10分間撹拌後、飽和炭酸水素ナトリウム水溶液(125 ml)を注ぎ、酢酸エチル(200 ml)で2回抽出した。抽出層を合わせ、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた褐色の粗製物をシリカゲルカラムクロマトグラフィーで精製し、得られた薄黄色固体をヘキサンで洗浄して標題化合物(10.7 g)を白色結晶として得た。
1H NMR (300 MHz, DMSO-d6) δ4.81 (2H, q, J=8.8 Hz), 7.13 (2H, d, J=9.1 Hz), 7.45 (2H, d, J=9.1 Hz).
1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(725 mg)をN,N-ジメチルホルムアミド(10 ml)に溶かし、氷冷下水素化ナトリウム(60%油性、 274 mg)を加えた。その後メチル 2-アミノ-6-クロロピリジン-3-カルボキシラート(580 mg)をN,N-ジメチルホルムアミド(5 ml)に溶かし、反応混合物中に氷冷下滴下し、さらに20分間撹拌した。反応混合液に0.2M 塩酸(35ml)を注ぎ、析出した白色沈殿をろ取した。この沈殿をテトラヒドロフラン(50 ml)に溶かし、酢酸エチル(100ml)で希釈後、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去して白色残渣 (1.2 g)を得た。得られた残渣をメタノール(20 ml)に溶かし、ナトリウムメトキシド(28%メタノール溶液, 3.0 g)を加えた後30分間加熱還流した。放冷後、反応混合物を氷冷下0.2M 塩酸溶液(100 ml)中に注ぎ、析出した白色沈殿をろ取した。得られた沈殿物をテトラヒドロフラン(100 ml)に溶かし、酢酸エチル(200 ml)で希釈後、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去して得られた残渣をジエチルエーテルで洗浄して白色固体の標題化合物(861 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ3.99 (3H, s), 4.82 (2H, q, J=8.9 Hz), 6.77 (1H, d, J =8.7 Hz), 7.12 (2H, d, J=9.1 Hz), 7.21 (2H, d, J=9.1 Hz), 8.16 (1H d, J=8.7 Hz), 13.34 (1H, s).
7-メトキシ-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリド[2,3-d]ピリミジン-4(1H)-オン(383 mg)、1M炭酸水素ナトリウム水溶液(1.0 ml)、ヨードエタン(0.4 ml)およびアセトニトリル(10 ml)の混合物を100℃で1時間撹拌した。反応混合物を放冷後、酢酸エチル(80 ml)で希釈し、水、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去して得られた残渣をシリカゲルカラムクロマトグラフィー (NH、酢酸エチル/ヘキサン) で精製して標題化合物 (375 mg) を得た。
1H NMR (300 MHz, DMSO-d6) δ1.27 (3H, t), 3.14 (2H, q, J=7.4 Hz), 4.00 (3H, s), 4.88 (2H, q, J=8.9 Hz), 6.88 (1H, d, J=8.7 Hz), 7.22 (2H, d, J=9.0 Hz), 7.42 (2H, d, J=9.0 Hz), 8.27 (1H, d, J=8.7 Hz).
7-メトキシ-2-(メチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン
7-メトキシ-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリド[2,3-d]ピリミジン-4(1H)-オン(1.2 g)、1M炭酸水素ナトリウム水溶液(3.1 ml)、ヨードメタン(0.98 ml)およびN,N-ジメチルホルムアミド(10 ml)の混合物を60℃で1時間撹拌した。反応混合物を放冷後、酢酸エチル(100 ml)で希釈し、水、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去して得られた残渣をシリカゲルカラムクロマトグラフィー (NH、酢酸エチル/ヘキサン) で精製して標題化合物 (1.24 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ2.51 (3H, s), 4.00 (3H, s), 4.88 (2H, q, J=8.9 Hz), 6.89 (1H, d, J=8.5 Hz), 7.22 (2H, d, J=9.0 Hz), 7.43 (2H, d, J=9.0 Hz), 8.28 (1H, d, J=8.7 Hz).
7-メトキシ-2-(プロピルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン
7-メトキシ-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリド[2,3-d]ピリミジン-4(1H)-オン(153 mg)、1M炭酸水素ナトリウム水溶液(439 μl)、1-ヨードプロパン(117 μl)およびN,N-ジメチルホルムアミド(3 ml)の混合物を80℃で10分間撹拌した。反応混合物を室温に戻したのち水(20 ml)を加えて析出した白色沈殿をろ取し、水で洗浄後テトラヒドロフラン(10 ml)に溶かし、酢酸エチル(20 ml)で希釈し、水、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去して得られた残渣を酢酸エチル/ヘプタン混合溶媒から再結晶し綿状針状晶の標題化合物(114 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ0.94 (3H, t, J=7.4 Hz), 1.65 (2H, qt, J=7.4, 7.2 Hz), 3.14 (2H, t, J=7.2 Hz), 4.00 (3H, s), 4.88 (2H, q, J=8.9 Hz), 6.89 (1H, d, J=8.7 Hz), 7.22 (2H, d, J=9.1 Hz), 7.42 (2H, d, J=9.1 Hz), 8.28 (1H, d, J=8.7 Hz).
2-エトキシ-7-メトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン
7-メトキシ-2-(プロピルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン(200 mg)をエタノール(15 ml)に溶かし、ナトリウムエトキシド(20%エタノール溶液、850 mg)を加え60℃で1時間撹拌した。反応混合物を0.2M塩酸(15 ml)に注いで析出した沈殿をろ取した。ろ取した沈殿物をテトラヒドロフラン(15 ml)に溶かした後、酢酸エチル(30 ml)で希釈し、無水硫酸マグネシウムで乾燥した。溶媒を減圧留去して得られた残渣シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(95 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.18 (3H, t, J=7.1 Hz), 3.98 (3H, s), 4.43 (2H, q, J=7.1 Hz), 4.85 (2H, q, J=8.9 Hz), 6.81 (1H, d, J=8.7 Hz), 7.17 (2H, d, J=9.0 Hz), 7.35 (2H, d, J=9.0 Hz), 8.24 (1H, d, J=8.7 Hz).
7-メトキシ-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン
7-メトキシ-2-(プロピルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン(350 mg)および2,2,2-トリフルオロエタノール(592μl)をテトラヒドロフラン(3 ml)に溶かし、水素化ナトリウム(60%油性、165 mg)を室温で加えた後、反応混合物を3日間加熱還流した。室温に戻した後、反応混合物を0.5M塩酸(10 ml)に注ぎ、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して得られた白色固体をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)および(NH、酢酸エチル/ヘキサン)で精製し、白色の標題化合物(63mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ3.99 (3 H, s), 4.85 (2 H, q, J=8.9 Hz), 5.10 (2 H, q, J=8.8 Hz), 6.89 (1 H, d, J=8.7 Hz), 7.19 (2 H, d, J=9.0 Hz), 7.38 (2 H, d, J=9.0 Hz), 8.28 (1 H, d, J=8.7 Hz).
2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
2-(エチルスルファニル)-7-メトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン(150 mg)、ピリジン塩酸塩(1156 mg)およびN,N-ジメチルホルムアミド(1 ml)の混合物を120℃で15分間撹拌した。反応混合物を0.2M塩酸(10 ml)に注ぎ、析出した白色の固体をろ取した後水で洗浄した。得られた固体をテトラヒドロフラン(5 ml)に溶かした後酢酸エチル(70 ml)で希釈し、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧留去して得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し白色固体の標題化合物(137 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.26 (3 H, t, J=7.4 Hz), 3.10 (2 H, q, J=7.3 Hz), 4.86 (2 H, q, J=8.9 Hz), 6.28 (1 H, d, J=9.5 Hz), 7.20 (2 H, d, J=9.1 Hz), 7.38 (2H, d, J=9.1 Hz), 7.83 (1 H, d, J=9.5 Hz), 12.29 (1 H, brs).
2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオンナトリウム塩
2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(100 mg)をメタノール(2 ml)に懸濁し、ナトリウムメトキシド (28%メタノール溶液、53.4 mg)を室温で加え30-40℃に加温し透明な溶液を得た。溶媒を減圧留去して得られた白色固体をエタノール/ジエチルエーテルから再結晶して白色綿状針状結晶の標題化合物(79 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.22 (3H, t, J=7.3 Hz), 3.02 (2H, q, J=7.3 Hz), 4.85(2H, q, J=8.9 Hz), 5.88 (1H, d, J=8.8 Hz), 7.14 (2H, d, J=9.0 Hz), 7.25 (2H, d,J=9.0 Hz), 7.52 (1H, d, J=8.9 Hz).
2-(エチルスルファニル)-8-メチル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(120 mg)およびヨードメタン(37.8 μl)をN,N-ジメチルホルムアミド(2 ml)に溶解し、氷冷下水素化ナトリウム(60%油性、86 mg)を加えて45分間撹拌した。氷冷下0.5M塩酸(6 ml)を加えたのち酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後減圧濃縮して得られた白色固体の残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して白色固体の標題化合物(123 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.31 (3H, t, J=7.4 Hz), 3.16 (2H, q, J=7.4 Hz), 3.64(3H, s), 4.87 (2H, q, J=8.9 Hz), 6.43 (1H, d, J=9.4 Hz), 7.22 (2H, d, J=9.1 Hz), 7.40 (2H, d, J=9.1 Hz), 7.88 (1H, d, J=9.4 Hz).
2-エトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
7-メトキシ-2-(メチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン(1.55 g)およびピリジン塩酸塩(3.1 g)をN,N-ジメチルホルムアミド(20 ml)に入れ120℃で2時間撹拌した。放冷後、反応混合物を0.5M塩酸(22ml)に注ぎ、析出した白色沈殿をろ取した。この沈殿物を水で洗浄後テトラヒドロフラン(30 ml)に溶かし、酢酸エチル(60 ml)で希釈した後無水硫酸マグネシウムで乾燥し、減圧濃縮して得られた残渣を酢酸エチルで洗浄して白色固体の標題化合物(1.33 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ2.47 (3H, s), 4.87 (2H, q, J=8.9 Hz), 6.29 (1H, d, J=9.4 Hz), 7.21 (2H, d, J=9.1 Hz), 7.40 (2H, d, J=9.1 Hz), 7.84 (1H, d, J=9.4 Hz), 12.31 (1H, s).
2-(メチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(300 mg)をエタノール(9 ml)に懸濁し、ナトリウムエトキシド(20%エタノール溶液、1.33 g)を室温で加えた後、3時間加熱還流した。放冷後、反応混合物を氷冷下0.5M塩酸(9 ml)に注ぎ、析出した褐色沈殿をろ取した。この沈殿物を水で洗浄後、テトラヒドロフラン(100 ml)に溶かし、酢酸エチル(150 ml)で希釈した後無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去して得られた褐色残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製後、再びシリカゲルカラムクロマトグラフィー(NH、メタノール/酢酸エチル)で精製し、白色固体の標題化合物(251 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.17 (3H, t, J=7.1 Hz), 4.40 (2H, q, J=7.1 Hz), 4.84(2H, q, J=8.9 Hz), 6.21 (1H, d, J=9.4 Hz), 7.15 (2H, d, J=9.1 Hz), 7.32 (2H, d,J=9.1 Hz), 7.80 (1H, d, J=9.4 Hz), 12.23 (1H, s).
2-エトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオンナトリウム塩
2-エトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(100 mg)をメタノール(3 ml)に懸濁し、ナトリウムメトキシド (28%メタノール溶液、55.7 mg)を室温で加え30-40℃に加温し透明な溶液を得た。溶媒を減圧留去して得られた白色固体をエタノール/ジエチルエーテルから再結晶して白色粉末の標題化合物(100 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.16 (3H, t, J=7.1 Hz), 4.35 (2H, q, J=7.1 Hz), 4.83(2H, q, J=8.9 Hz), 6.05 (1H, d, J=9.2 Hz), 7.13 (2H, d, J=9.1 Hz), 7.26 (2H, d,J=9.1 Hz), 7.68 (1H, d, J=9.2 Hz).
2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
2-(メチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(505 mg)を酢酸(12 ml)に溶かし、オキソン(登録商標)(1.05 g)を水(3 ml)に溶かして加え、室温で1時間撹拌した。反応混合物に水(50 ml)を加えて析出した白色沈殿をろ取し、ジイソプロピルエーテルで洗浄した。得られた固体を減圧下乾燥し、アセトニトリルで共沸して白色固体の標題化合物(528 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ2.72 (3H, s), 4.87 (2H, q, J=8.9 Hz), 6.48 (1H, d, J=9.4 Hz), 7.18 - 7.29 (2H, m), 7.44 - 7.53 (1H, m), 7.58 - 7.67 (1H, m), 7.93 (1H, d, J=9.4 Hz), 12.83 (1H, brs).
2-(メチルスルフィニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(150 mg)をテトラヒドロフラン(5 ml)に懸濁し、2,2,2-トリフルオロエタノール(108 μl)を加えた後、氷冷下水素化ナトリウム(60%油性、37.6 mg)を加えて10分間撹拌した。反応混合物に0.5M塩酸(4 ml)を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し減圧濃縮して得られた白色残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して白色固体の標題化合物(128 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ4.85 (2H, q, J=8.9 Hz), 5.03 (2H, q, J=8.7 Hz), 6.28(1H, d, J=9.4 Hz), 7.18 (2H, d, J=9.1 Hz), 7.35 (2H, d, J=9.1 Hz), 7.84 (1H, d,J=9.4 Hz), 12.39 (1H, s).
2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオンナトリウム塩
2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(93 mg)をメタノール(3 ml)に懸濁し、ナトリウムメトキシド (28%メタノール溶液、41.2 mg)を室温で加え、30-40℃まで加温し透明の溶液を得た。溶媒を減圧留去して得られた白色固体をメタノール/ジエチルエーテルから再結晶して白色粉末の標題化合物(84 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ4.83 (2H, q, J=8.9 Hz), 4.97 (2H, q, J=8.9 Hz), 5.89(1H, d, J=8.9 Hz), 7.13 (2H, d, J=9.1 Hz), 7.23 (2H, d, J=9.1 Hz), 7.53 (1H, d,J=8.9 Hz).
2-[3-(メチルスルホニル)プロポキシ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
7-メトキシ-2-(プロピルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン(213 mg)、ピリジン塩酸塩(1156 mg)およびN,N-ジメチルホルムアミド(10 ml)の混合物を120℃で3時間撹拌した。室温まで冷却後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を炭酸水素ナトリウム水溶液および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(183 mg)を得た。
MS (ESI+): [M+H]+ 412.3.
水素化ナトリウム(60%油性, 48 mg)のテトラヒドロフラン(5 ml)の懸濁液に室温で3-(メチルスルファニル)プロパン-1-オール(0.206 ml)加え、室温で10分間撹拌した。反応混合物に2-(プロピルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(165 mg)を加え、室温で5時間撹拌した。反応混合物を水に注ぎ、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸
エチル/ヘキサン)で精製して標題化合物(96 mg)を得た。
MS (ESI+): [M+H]+ 442.0.
2-[3-(メチルスルファニル)プロポキシ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(90 mg)の酢酸エチル溶液に3-クロロ過安息香酸(135 mg)を0℃で加え、室温で2時間撹拌した。反応混合物に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/メタノール)で精製して標題化合物(70 mg)を得た。
2-(2,2-ジフルオロ-3-メトキシプロポキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
2,2-ジフルオロプロパン-1,3-ジオール(1121 mg)のテトラヒドロフラン(20 ml)溶液に室温で水素化ナトリウム(60%油性, 320 mg)を加え、室温で10分撹拌した。反応混合物にヨードメタン(0.934 ml)を加え、室温で終夜撹拌した。反応混合物に飽和食塩水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(191 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ3.34 (3H, s), 3.53-3.68 (4H, m), 5.50 (1H, t, J=6.2 Hz).
水素化ナトリウム(60%油性, 29 mg)のテトラヒドロフラン(5 ml)の懸濁液に室温で2,2-ジフルオロ-3-メトキシプロパン-1-オール(151 mg)加え、室温で10分撹拌した。反応混合物に2-(プロピルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(165 mg)を加え、室温で5時間撹拌した。反応混合物を水に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、ヘキサン/酢酸エチルの混合溶媒で再結晶して標題化合物(8 mg)を得た。
1H NMR (300 MHz, CDCl3) δ3.26-3.43 (5H, m), 4.42 (2H, q, J = 8.0 Hz), 4.66 (2H,t, J = 12.1 Hz), 6.47 (1H, d, J = 9.4 Hz), 7.02-7.14 (2H, m), 7.16-7.24(2H, m), 8.02 (1H, d, J = 9.4 Hz), 10.32 (1H, brs).
2-[(4,4-ジフルオロシクロヘキシル)オキシ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
水素化ナトリウム(60%油性, 19 mg)のテトラヒドロフラン(5 ml)の懸濁液に室温で4,4-ジフルオロシクロヘキサノール(109 mg)加え、室温で10分撹拌した。反応混合物に2-(プロピルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(165 mg)を加え、室温で5時間撹拌した。反応混合物を水に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン) で精製し、ヘキサン/酢酸エチルの混合溶媒で再結晶して標題化合物(75 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.43 (2H, brs), 1.78 (6H, brs), 4.84 (2H, q, J = 8.7Hz), 5.19-5.34 (1H, m), 6.21 (1H, d, J = 9.4 Hz), 7.10-7.25 (2H, m),7.30-7.43 (2H, m), 7.82 (1H, d, J = 9.4 Hz), 12.24 (1H, s).
2-(2,2-ジフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
水素化ナトリウム(60%油性, 36 mg)のテトラヒドロフラン(5 ml)の懸濁液に室温で2,2-ジフルオロエタノール(0.095 ml)加え、室温で10分撹拌した。反応混合物に2-(プロピルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(123 mg)を加え、室温で5時間撹拌した。反応混合物を水に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン) で精製し、ヘキサン/酢酸エチルの混合溶媒で再結晶して標題化合物(73 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ4.63 (2H, td, J = 14.5, 3.4 Hz), 4.84 (2H, q, J = 9.0 Hz), 6.08-6.52 (2H, m), 7.12-7.22 (2H, m), 7.27-7.40 (2H, m), 7.83 (1H,d, J = 9.4 Hz), 12.33 (1H, s).
2-(2,2-ジフルオロ-3-ヒドロキシプロポキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
2-(プロピルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオンと2,2-ジフルオロプロパン-1,3-ジオールから実施例13のB)に準じた方法により、標題化合物を得た。
2-メトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
2-(メチルスルフィニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(60 mg)をテトラヒドロフラン(2 ml)に懸濁し、氷冷下ナトリウムメトキシド (28%メタノール溶液、200 mg)を加えて10分間撹拌した。反応混合物に1M塩酸(3 ml)を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し減圧濃縮して得られた白色残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して白色固体の標題化合物(39 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ3.89 (3H, s), 4.83 (2H, q, J=8.9 Hz), 6.22 (1H, d, J=9.4 Hz), 7.16 (2H, d, J=9.1 Hz), 7.32 (2H, d, J=9.1 Hz), 7.81 (1H, d, J=9.4 Hz), 12.28 (1H, s).
2-(エチルアミノ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
エチルアミンのテトラヒドロフラン溶液(2M、2 ml)をテトラヒドロフラン(2 ml)で希釈し、2-(メチルスルフィニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(100 mg)を室温で加えた。室温で10分間撹拌した後、反応混合物を0.5M塩酸(約4ml)でpH7付近に調整し、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄し無水硫酸マグネシウムで乾燥後減圧濃縮して得られた白色残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し白色固体の標題化合物(52 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.04 (3H, t, J=7.1 Hz), 3.25 - 3.35 (2H, m), 4.84 (2H, q, J=8.9 Hz), 5.95 (1H, d, J=9.4 Hz), 6.52 (1H, t, J=5.8 Hz), 7.22 (2H, d, J=9.0 Hz), 7.29 (2H, d, J=9.0 Hz), 7.68 (1H, d, J=9.4 Hz), 11.63 (1H, s).
2-(1-メチルエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
2-(メチルスルフィニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン(100 mg)をテトラヒドロフラン(4 ml)に懸濁し、2-プロパノール(77μl)を加えた後、氷冷下水素化ナトリウム(25.0 mg)を加えて10分間撹拌し、室温でさらに15分撹拌した。反応混合物に0.5M塩酸(3 ml)を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し減圧濃縮して得られた白色残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して白色固体の標題化合物(35.4 mg)を得た。
MS (ESI+): [M+H]+ 396.4.
2-(メチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
3-アミノ-3-イミノプロピオン酸エチル塩酸塩 (10 g)とトリエチルアミン (18.4 mL)のエタノール (100 mL) 溶液にアクリル酸エチル (7.15 mL) を加え室温で終夜撹拌した。反応混合物に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出し、抽出液を飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー (酢酸エチル/ヘキサン) で精製して標題化合物 (2.5g) を得た。
1H NMR (300 MHz, DMSO-d6) δ1.11 (3H, t, J=7.2 Hz), 1.17 (3H, t, J=7.2 Hz), 2.15-2.40 (3H, m), 3.91 (2H, q, J=7.2 Hz), 4.02 (2H, q, J=7.2 Hz), 5.66 (2H, s).
1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン (1.77 g) のN,N-ジメチルホルムアミド (25 mL) 溶液に水素化ナトリウム (60%油性、0.478 g) を氷冷下加えた。反応混合物を氷冷下で5分間撹拌後、ジエチル 2-カルバムイミドイルペンタン-1,5-ジオアート (2.5 g)を加え、氷冷下で30分間撹拌した。反応混合物に1M 塩酸を加え、酢酸エチルで抽出し、抽出液を飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をエタノール (25 mL) に溶解し、ナトリウムエトキシド(20%エタノール溶液、10.6 mL)を加え、30分間撹拌した。反応混合物に1M 塩酸を加え、酢酸エチルで抽出し、抽出液を飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー (酢酸エチル/ヘキサン) で精製して標題化合物 (0.75 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ2.54-2.62 (4H, m), 4.81 (2H, q, J=9.1 Hz), 7.00-7.20(4H, m), 9.16 (1H, s), 12.00 (1H, brs).
2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3,5,6-テトラヒドロピリド[2,3-d]ピリミジン-4,7(1H,8H)-ジオン (2.05 g)のアセトニトリル (40 mL) 溶液に1M炭酸水素ナトリウム水溶液 (5.52 mL)、ヨードメタン (1.73 mL)を加えて50℃で30分間撹拌した。反応混合物に水を加え、酢酸エチルで抽出し、抽出液を飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー (酢酸エチル/ヘキサン) で精製して標題化合物 (2.1 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ2.40 (3H, s), 2.45-2.67 (4H, m), 4.86 (2H, q, J=9.1 Hz), 7.13-7.38 (4H, m), 10.45 (1H, s).
2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
2-(メチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン (2.1 g) の酢酸 (40 mL) 溶液にオキソン(登録商標)(4.02 g) の水 (8 mL)溶液を加えた。反応混合物を室温で2時間撹拌後、水を加え、析出した沈殿をろ取して標題化合物 (1.68 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ2.41-2.80 (7H, m), 4.86 (2H, q, J=9.0 Hz), 7.08-7.59(4H, m), 10.87 (1H, s).
2,2,2-トリフルオロエタノール (0.472 mL) のテトラヒドロフラン (20 mL)溶液に水素化ナトリウム(60%油性、0.184 g) を加えた。反応混合物を5分間撹拌後、2-(メチルスルフィニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン (0.88 g)を加え、室温で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出し、抽出液を飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー (酢酸エチル/ヘキサン) で精製した後、酢酸エチル/ヘキサンで再結晶して標題化合物 (0.80 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ2.44-2.68 (4H, m), 4.84 (2H, q, J=8.7 Hz), 4.96 (2H,q, J=8.8 Hz), 7.08-7.36 (4H, m), 10.49 (1H, s).
2-エトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
2-(メチルスルフィニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン (0.64 g) のテトラヒドロフラン (10 mL)溶液にナトリウムエトキシド(20%エタノール溶液、1.56 mL)を加え、室温で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出し、抽出液を飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー (酢酸エチル/ヘキサン) で精製した後、酢酸エチル/ヘキサン混合溶媒から再結晶して標題化合物 (0.40 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ1.14 (3H, t, J=7.2 Hz), 2.41-2.64 (4H, m), 4.31 (2H,q, J=7.1 Hz), 4.83 (2H, q, J=8.7 Hz), 7.03-7.34 (4H, m), 10.37 (1H, s).
2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3,5,6-テトラヒドロピリド[2,3-d]ピリミジン-4,7(1H,8H)-ジオン (0.1 g)のアセトニトリル (2 mL) 溶液に1M炭酸水素ナトリウム水溶液 (0.269 mL)、ヨードエタン (0.109 mL)を加えて70℃で2時間撹拌した。溶媒を減圧下留去した後、残渣をシリカゲルカラムクロマトグラフィー (酢酸エチル/ヘキサン) で精製して標題化合物 (0.08 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ1.22 (3H, t, J=7.4 Hz) 2.44 - 2.67 (4H, m) 3.02 (2H,q, J=7.3 Hz) 4.86 (2H, q, J=8.7 Hz) 7.12 - 7.37 (4H, m) 10.46 (1H, s).
2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3H-ピリミド[5,4-b][1,4]チアジン-4,7(6H,8H)-ジオン
2,4-ジメトキシベンジルアミン (8.98 mL)と2,4-ジメトキシベンズアルデヒド (9.94 g) のエタノール (200 mL) 溶液を室温で2時間撹拌した。反応混合物に水素化トリアセトキシホウ素ナトリウム(20.3 g) を加え室温で1時間撹拌した。反応混合物に水を加え、溶媒を減圧下留去した後、残渣をシリカゲルカラムクロマトグラフィー (酢酸エチル/ヘキサン) で精製して標題化合物 (8 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ2.01 (1H, brs) 3.55 (4H, s) 3.74 (6H, s) 3.75 (6H, s) 6.46 (2H, dd, J=8.1, 2.3 Hz) 6.51 (2H, d, J=2.3 Hz) 7.18 (2H, d, J=8.1 Hz).
ビス(2,4-ジメトキシベンジル)アミン (8 g)、エチル 3-エトキシ-3-イミノプロパノアート (8.02 g)、酢酸 (1.44 mL)、エタノール (420 mL) の混合液を終夜還流した。溶媒を減圧下留去した後、残渣をシリカゲルカラムクロマトグラフィー (酢酸エチル/ヘキサン) で精製して標題化合物 (17 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ1.07 (3H, t, J=7.0 Hz) 3.75 (12H, s) 3.85 (2H, q, J=7.2 Hz) 4.26 (4H, s) 6.51 (2H, dd, J=8.3, 2.3 Hz) 6.56 (2H, d, J=2.3 Hz) 6.90 (2H, d, J=8.3 Hz) 7.32 (2H, brs).
1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン (3.96 g) のN,N-ジメチルホルムアミド (120 mL) 溶液に水素化ナトリウム (60%油性、0.68 g) を氷冷下加えた。反応混合物を氷冷下で5分間撹拌後、エチル (2E)-3-アミノ-3-[ビス(2,4-ジメトキシベンジル)アミノ]プロパ-2-エノアート (7.3 g)を加え、氷冷下で30分間撹拌した。反応混合物に1M塩酸を加え、酢酸エチルで抽出し、抽出液を飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をエタノール (120 mL) に溶解し、ナトリウムエトキシド(20%エタノール溶液、14.5 mL)を加え、1時間撹拌した。反応混合物に1M 塩酸を加え、析出した固体をろ取して標題化合物 (2.8 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ3.77 (6H, s) 3.86 (6H, s) 4.51 (4H, s) 4.70 - 4.91 (3H, m) 6.53 (2H, dd, J=8.3, 2.3 Hz) 6.62 (2H, d, J=2.3 Hz) 7.03 - 7.15 (6H, m) 11.23 (1H, s).
6-[ビス(2,4-ジメトキシベンジル)アミノ]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン (0.65 g)のアセトニトリル (7 mL)溶液に1M炭酸水素ナトリウム水溶液 (1.05 mL)、ヨードエタン (0.425 mL)を加えて70℃で2時間撹拌した。溶媒を減圧下留去した後、残渣をシリカゲルカラムクロマトグラフィー (酢酸エチル/ヘキサン) で精製して標題化合物(0.68 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ1.02 (3H, t, J=7.4 Hz) 2.82 (2H, q, J=7.3 Hz) 3.75 (6H, s) 3.77 (6H, s) 4.83 (2H, q, J=8.7 Hz) 4.96 (1H, s) 6.46 - 6.64 (4H, m) 6.99 (2H, d, J=8.3 Hz) 7.06 - 7.28 (4H, m).
6-[ビス(2,4-ジメトキシベンジル)アミノ]-2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(0.68 g)、アニソール (0.459 mL)、トリフルオロ酢酸 (7 mL) の混合物を室温で1時間撹拌した。溶媒を減圧下留去した後、残渣をシリカゲルカラムクロマトグラフィー (酢酸エチル/ヘキサン) で精製して標題化合物 (0.19 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ1.20 (3H, t, J=7.4 Hz) 2.97 (2H, q, J=7.2 Hz) 4.83 (2H, q, J=8.7 Hz) 4.95 (1H, s) 6.57 (2H, brs) 7.09 - 7.24 (4H, m).
6-アミノ-2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン (0.5 g)、酢酸ナトリウム (0.143 g)の酢酸 (10 mL) 溶液に臭素 (0.09 mL) を滴下し、室温で2時間撹拌した。反応混合物に水を加え、析出した結晶をろ取して標題化合物 (0.1 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ1.21 (3H, t, J=7.3 Hz) 3.00 (2H, q, J=7.3 Hz) 4.85 (2H, q, J=9.0 Hz) 6.95 (2H, brs) 7.10 - 7.34 (4H, m).
6-アミノ-5-ブロモ-2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン (0.1 g)、メルカプト酢酸 (0.168 mL)のトルエン (2 mL)溶液をマイクロ波照射下170℃で2時間撹拌した。反応混合物に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出後、抽出液を飽和食塩水で洗浄し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー (酢酸エチル/ヘキサン) で精製して標題化合物 (0.04 g) を得た。
1H NMR (300 MHz, DMSO-d6) δ1.23 (3H, t, J=7.3 Hz) 3.03 (2H, q, J=7.3 Hz) 3.50 (2H, s) 4.86 (2H, q, J=9.0 Hz) 7.19 (2H, d, J=9.0 Hz) 7.34 (2H, d, J=9.0 Hz) 11.06 (1H, s).
7-アミノ-2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン
1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(4.06 g)をN,N-ジメチルホルムアミド(50 ml)に溶かし、氷冷下水素化ナトリウム(60%油性, 1.39 g)を加えた。その後メチル 2-アミノ-6-クロロピリジン-3-カルボキシラート(2.95 g)をN,N-ジメチルホルムアミド(25 ml)に溶かし、反応混合物中に氷冷下滴下し、20分間撹拌し、室温で1時間30分撹拌した。反応混合液を0.5M 塩酸(80 ml)に注ぎ、析出した白色沈殿をろ取した。この沈殿をテトラヒドロフラン(100 ml)に溶かし、酢酸エチル(200ml)で希釈後、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去して白色残渣を得た。この残渣をジエチルエーテル(100 ml)で懸濁し、ろ液を減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、ジエチルエーテルで洗浄して白色の標題化合物(1.1 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ4.83 (2H, q), 7.14 (2H, d, J=9.1 Hz), 7.23 (2H, d, J=9.1 Hz), 7.44 (1H, d, J=8.3 Hz), 8.32 (1H, d, J=8.3 Hz), 13.65 (1H, s).
7-クロロ-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン(200 mg)、4-メトキシベンジルアミン(181 μl)、ヨウ化銅(I)(57 mg)、炭酸カリウム(166 mg)およびN,N-ジメチルホルムアミド(3 ml)の混合物を130℃で2時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液および水を加えて、析出した沈殿をろ取し、テトラヒドロフランに溶かし、セライトろ過したのち減圧濃縮した。残渣を酢酸エチル/テトラヒドロフラン混合溶媒に溶かし、アンモニア水溶液、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して白色の標題化合物(108 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ3.73 (3H, s), 4.52 (2H, d, J=5.3 Hz), 4.82 (2H, q, J=9.0 Hz), 6.45 (1H, d, J=8.7 Hz), 6.90 (2H, d, J=8.7 Hz), 7.10 (2H, d, J=9.1 Hz), 7.17 (2H, d, J=9.1 Hz), 7.37 (2H, d, J=8.3 Hz), 7.77 (1H, d, J=8.7 Hz), 8.35 (1H, brs), 12.89 (1H, s).
7-[(4-メトキシベンジル)アミノ]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリド[2,3-d]ピリミジン-4(1H)-オン(108 mg)、ヨードエタン(53 μl)および1M水酸化ナトリウム水溶液(220 μl)をN,N-ジメチルホルムアミド(2 ml)に入れ50℃で1時間撹拌した。反応混合物に1M塩酸および水を加え、酢酸エチルで抽出した。抽出液を水、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した後、減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(NH、酢酸エチル/ヘキサン)精製して白色の標題化合物(63 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.24 (3H, t, J=7.3 Hz), 3.09 (2H, q, J=7.5 Hz), 3.73(3H, s), 4.46 - 4.66 (2H, m), 4.87 (2H, q, J=8.7 Hz), 6.60 (1H, d, J=8.7 Hz), 6.91 (2H, d, J=8.7 Hz), 7.18 (2H, d, J=9.0 Hz), 7.30 (2H, d, J=8.7 Hz), 7.35 (2H,d, J=9.0 Hz), 7.93 (1H, d, J=8.7 Hz), 8.07 (1H, brs).
2-(エチルスルファニル)-7-[(4-メトキシベンジル)アミノ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン(63 mg)およびアニソール(26 μl)をトリフルオロ酢酸(1 ml)に入れ、1時間30分加熱還流した。反応混合液に飽和炭酸水素ナトリウム水溶液を加えてアルカリ性とし、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して白色固体の標題化合物(29 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.25 (3H, t, J=7.4 Hz), 3.05 (2H, q, J=7.2 Hz), 4.86(2H, q, J=8.7 Hz), 6.50 (1H, d, J=8.7 Hz), 7.12 (2H, s), 7.18 (2H, d, J=9.1 Hz), 7.35 (2H, d, J=9.1 Hz), 7.94 (1H, d, J=8.7 Hz).
2-(エチルスルファニル)-4-オキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,4-ジヒドロピリド[2,3-d]ピリミジン-7-カルボキサミド
実施例1のA)に準じた方法で、ジメチル 6-クロロピリジン-2,5-ジカルボキシラート(1.61 g)、炭酸水素ナトリウム(882 mg)、4-メトキシベンジルアミン(1.37 ml)、ヨウ化銅(I)(133 mg)およびN,N-ジメチルホルムアミド(20 ml)を用いて、80℃で3時間撹拌して行い、薄黄色油状の標題化合物(943 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ3.72 (3H, s), 3.84 (3H, s), 3.87 (3H, s), 4.62 (2H, s), 6.88 (2H, d, J=8.7 Hz), 7.26 (1H, d, J=7.9 Hz), 7.37 (2H, d, J=8.7), 8.25 (1H, d, J=7.9 Hz), 8.26-8.34 (1H, m).
実施例1のB)に準じた方法で、ジメチル 6-[(4-メトキシベンジル)アミノ]ピリジン-2,5-ジカルボキシラート(943 mg)、アニソール(311 μl)、トリフルオロ酢酸(3 ml)を用いて、1時間加熱還流して行い、薄黄色固体の標題化合物(491 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ3.84 (6H, s), 7.25 (1H, d, J=7.9 Hz), 7.44 (2H, brs), 8.23 (1H, d, J=7.9 Hz).
ジメチル 6-アミノピリジン-2,5-ジカルボキシラート(395 mg)をテトラヒドロフラン/エタノール(1/1)混合溶媒(16 ml)に懸濁し、氷冷下塩化カルシウム(834 mg)および水素化ホウ素ナトリウム(85.5 mg)を加え、2時間撹拌した。反応混合物に1M塩酸を加えて酸性溶液とした後、飽和炭酸カリウム水溶液で塩基性として酢酸エチルで抽出した。抽出液を水、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し減圧濃縮して白色固体の標題化合物(296 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ3.80 (3H, s), 4.38 (2H, d, J=6.1 Hz), 5.36 (1H, t, J=5.9 Hz), 6.75 (1H, d, J=8.0 Hz), 7.12 (2H, brs), 8.07 (1H, d, J=8.0 Hz).
メチル 2-アミノ-6-(ヒドロキシメチル)ピリジン-3-カルボキシラート(296 mg)、tert-ブチル(クロロ)ジメチルシラン(298 mg)、イミダゾール(218 mg)およびN,N-ジメチルホルムアミド(8 ml)の混合物を室温で15分間撹拌した。反応溶液に水を加え、酢酸エチルで抽出した。抽出液を水と飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧留去して白色固体を得た。この白色固体を、1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(670 mg)、水素化ナトリウム(60%油性、132 mg)およびN,N-ジメチルホルムアミド(15 ml)の混合物に氷冷下で加え、30分間撹拌した。反応混合物を1M塩酸を加えて酸性とし、酢酸エチルで抽出した。抽出液を水、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して白色固体の標題化合物(547 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ-0.14 (6H, s), 0.80 (9H, s), 4.57-4.84 (4H, m), 6.99(2H, d, J=9.1 Hz), 7.10 (2H, d, J=9.1 Hz), 7.30 (1H, d, J=8.0 Hz), 8.23 (1H, d,J=8.0 Hz), 13.27 (1H, s).
7-({[tert-ブチル(ジメチル)シリル]オキシ}メチル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリド[2,3-d]ピリミジン-4(1H)-オン(498 mg)をテトラヒドフラン(15 ml)に懸濁し、テトラブチルアンモニウムフルオリドのテトラヒドロフラン溶液(1M、3 ml)、を加えて室温で1時間撹拌した。反応混合物に水を加えた後、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して得られた残渣をN,N-ジメチルホルムアミド(15 ml)に溶かし、ヨードエタン(0.24 ml)および1M炭酸水素ナトリウム水溶液(1 ml)を加えて室温で1時間撹拌した。反応混合物に1M塩酸および水を加え、析出した沈殿をろ取し、水で洗浄した後テトラヒドロフランに溶かし、無水硫酸マグネシウムで乾燥後減圧濃縮して得れらた残渣を酢酸エチル/ヘキサン混合溶媒から再結晶して白色固体の標題化合物(373 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.28 (3H, t, J=7.4 Hz), 3.13 (2H, q, J=7.2 Hz), 4.69(2H, d, J=5.7 Hz), 4.88 (2H, q, J=9.1 Hz), 5.70 (1H, t, J=5.9 Hz), 7.22 (2H, d,J=8.7 Hz), 7.44 (2H, d, J=8.7 Hz), 7.59 (1H, d, J=7.9 Hz), 8.46 (1H, d, J=8.3 Hz).
2-(エチルスルファニル)-7-(ヒドロキシメチル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[2,3-d]ピリミジン-4(3H)-オン(373 mg)、トリエチルアミン(6 ml)をジメチルスルホキシド(18 ml)に溶かし、三酸化硫黄ピリジン錯体(1.16 g)を数回に分けて加えて室温で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水、1M塩酸、および飽和食塩水で洗浄し無水硫酸マグネシウムで乾燥後減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して橙色固体の標題化合物(299 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.31 (3H, t, J=7.2 Hz), 3.18 (2H, q, J=7.4 Hz), 4.88(2H, q, J=8.7 Hz), 7.24 (2H, d, J=9.1 Hz), 7.47 (2H, d, J=9.1 Hz), 7.91 (1H, d,J=7.9 Hz), 8.66 (1H, d, J=7.9 Hz), 10.11 (1H, s).
2-(エチルスルファニル)-4-オキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,4-ジヒドロピリド[2,3-d]ピリミジン-7-カルバルデヒド(299 mg)、2-メチルブタ-2-エン(387 mg)、リン酸二水素カリウム(88 mg)および2-メチル-2-プロパノール/水(4/1、70 ml)の混合物に亜塩素酸ナトリウム(232 mg)を少しずつ加え、室温で1時間撹拌した。反応混合物に氷冷下、1M塩酸、および水を加えテトラヒドロフランで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して薄黄色の残渣を得た。この残渣(117 mg)、1-ヒドロキシベンゾトリアゾールアンモニウム塩(40 mg)をN,N-ジメチルホルムアミド(2 ml)に溶かし、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩(56 mg)を加えて室温で16時間撹拌した。反応混合物に水を加えて、酢酸エチルで抽出後、抽出液を水、飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去して得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)および(NH、酢酸エチル/ヘキサン)で精製し、酢酸エチル/ヘキサン混合溶媒から再結晶して白色の標題化合物(28 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.29 (3H, t, J=7.3 Hz), 3.17 (2H, q, J=7.2 Hz), 4.88(2H, q, J=8.5 Hz), 7.23 (2H, d, J=9.0 Hz), 7.46 ((2H, d, J=9.0 Hz), 7.87 (1H, s), 8.06 (1H, d, J=7.9 Hz), 8.19 (1H, s), 8.62 (1H, d, J=8.3 Hz).
2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,5,6,7-テトラヒドロピリド[3,4-d]ピリミジン-4,8-ジオン
エチル 4-アミノブタノアート塩酸塩(1.77 g)および2,4-ジメトキシベンズアルデヒド(1.75 g)をエタノール(50 ml)に溶かし1時間加熱還流を行った。室温に戻した後、水素化トリアセトキシホウ素ナトリウム(2.2 g)を加えて1時間撹拌した。反応混合物を減圧濃縮し、残渣に飽和炭酸水素ナトリウム水溶液(40 ml)を加えた後、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、溶媒を減圧留去して黄色油状の標題化合物(2.77 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.16 (3H, t, J=7.2 Hz), 1.66 (2H, tt, J=7.4, 7.0 Hz), 2.31 (2H, t, J=7.4 Hz), 2.47 (2H, t, J=7.0 Hz), 3.57 (2H, s), 3.74 (3H, s), 3.76 (3H, s), 4.03 (2H, q, J=7.2 Hz), 6.46 (1H, dd, J=8.3, 2.5 Hz), 6.52 (1H, d, J=2.5 Hz), 7.16 (1H, d, J=8.3 Hz).
エチル 4-[(2,4-ジメトキシベンジル)アミノ]ブタノアート(2.77 g)のテトラヒドロフラン(40 ml)溶液にトリエチルアミン(1.78 ml)を加えた後、水浴下、メチルオキサリルクロリド(998 μl)のテトラヒドロフラン(5 ml)溶液を5分かけて滴下し、室温で10分間撹拌した。反応混合物に酢酸エチル(50 ml)を加え、白色沈殿をろ別し、酢酸エチル(50 ml)で洗浄した。ろ液を飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧濃縮して得られた黄色の油状物をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して黄色油状物(2.53 g)を得た。この黄色油状物(2.14 g)をエタノール(15 ml)に溶かし、ナトリウムエトキシド(20%エタノール溶液、4.95g)を室温で加えた後、30分間加熱還流した。放冷後、反応混合物を減圧濃縮し、得られた固体残渣を水(5 ml)に溶かし、1M塩酸(16 ml)を加えて酸性にした後、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧濃縮して橙色油状物を得た。この油状物(1.95 g)およびギ酸アンモン(1.8 g)をエタノール(20 ml)に入れ、30分間加熱還流した。放冷後、反応混合物を減圧濃縮して得られた残渣に水(10 ml)および飽和炭酸水素ナトリウム水溶液(25 ml)を加えてアルカリ性とし、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧濃縮して得られた濃橙色油状物をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し無色油状の標題化合物(650 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.21 (3H, t, J=7.2 Hz), 2.41 (2H, t, J=6.9 Hz), 3.28(2H, t, J=6.9 Hz), 3.75 (3H, s), 3.78 (3H, s), 4.11 (2H, q, J=7.1 Hz), 4.47 (2H, s), 6.49 (1H, dd, J=8.3, 2.5 Hz), 6.57 (1H, d, J=2.5 Hz), 6.93 (2H, brs), 7.06 (1H, d, J=8.3 Hz).
1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(115 mg)のN,N-ジメチルホルムアミド(2 ml)溶液に水素化ナトリウム(60%油性、45 mg)を加えた後、エチル 5-アミノ-1-(2,4-ジメトキシベンジル)-6-オキソ-1,2,3,6-テトラヒドロピリジン-4-カルボキシラート(150 mg)のN,N-ジメチルホルムアミド(2 ml)溶液を氷冷下5分間かけて滴下した。10分間撹拌後、室温に上げ30分間撹拌した。反応混合物を氷冷下0.5M塩酸(4 ml)に注いだ後、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して得られた黄色の固体をジエチルエーテルで洗浄してベージュ色の細針状晶の標題化合物(70 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ2.57 (2H, t), 3.49 (2H, t, J=7.0 Hz), 3.76 (3H, s), 3.81 (3H, s), 4.55 (2H, s), 4.82 (2H, q, J=8.9 Hz), 6.51 (1H, dd, J=8.3, 2.3 Hz), 6.60 (1H, d, J=2.3 Hz), 7.15 (2H, d, J=8.3 Hz), 11.74 (1H, s).
7-(2,4-ジメトキシベンジル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,2,3,5,6,7-ヘキサヒドロピリド[3,4-d]ピリミジン-4,8-ジオン(68 mg)のN,N-ジメチルホルムアミド(2 ml)溶液に1M水酸化ナトリウム水溶液(140 μl)およびヨードエタン(11 μl)を加え室温で30分間撹拌した。反応混合物を0.5M塩酸(4 ml)に注ぎ、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去して無色の標題化合物(72 mg)が得られた。
1H NMR (300 MHz, DMSO-d6) δ1.24 (3H, t, J=7.3 Hz), 2.66 (2H, t, J=6.9 Hz), 3.08(2H, q, J=7.3 Hz), 3.49 (2H, t, J=6.9 Hz), 3.76 (3H, s), 3.81 (3H, s), 4.55 (2H, s), 4.86 (2H, q, J=8.9 Hz), 6.51 (1H, dd, J=8.4, 2.4 Hz), 6.59 (1H, d, J=2.4 Hz), 7.13 (1H, d, J=8.4 Hz), 7.20 (2H, d, J=9.1 Hz), 7.34 (2H, d, J=9.1 Hz).
7-(2,4-ジメトキシベンジル)-2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,5,6,7-テトラヒドロピリド[3,4-d]ピリミジン-4,8-ジオン(71 mg)をギ酸(3 ml)に溶かし、100℃で1時間撹拌した。室温に放冷後、反応混合物を減圧濃縮し、トルエンで共沸して得られた白色固体をシリカゲルカラムクロマトグラフィー(メタノール/酢酸エチル)で精製して白色固体の標題化合物(38 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.23 (3H, t, J=7.3 Hz), 2.64 (2H, t, J=6.8 Hz), 3.07(2H, q, J=7.3 Hz), 3.36 (2H, td, J=6.8, 2.7 Hz), 4.87 (2H, q, J=8.9 Hz), 7.20 (2H, d, J=9.1 Hz), 7.35 (2H, d, J=9.1 Hz), 8.21 (1H, t, J=2.7 Hz).
2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,7-ジヒドロピリド[3,4-d]ピリミジン-4,8-ジオン
エチル 5-アミノ-1-(2,4-ジメトキシベンジル)-6-オキソ-1,2,3,6-テトラヒドロピリジン-4-カルボキシラート(890 mg)のトルエン(20 ml)溶液に2,3-ジクロロ-5,6-ジシアノ-1,4-ベンゾキノン(785 mg)を室温で加え、30分間加熱還流した。室温まで冷却した後、反応混合物を酢酸エチルで希釈し、不溶物をろ過した。ろ液を減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(535 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.28 (3H, t, J=7.2 Hz), 3.74 (3H, s), 3.80 (3H, s), 4.24 (2H, q, J=7.2 Hz), 4.95 (2H, s), 6.41 (1H, d, J=7.6 Hz), 6.47 (1H, dd, J=8.3, 2.6 Hz), 6.58 (1H, d, J=2.3 Hz), 6.75 (1H, d, J=7.6 Hz), 6.88 (2H, brs), 6.98 (1H, d, J=8.3 Hz).
1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(375 mg)のN,N-ジメチルホルムアミド(5 ml)溶液に、氷冷下水素化ナトリウム(60%油性, 193 mg)を加え、0℃に冷却した。エチル 3-アミノ-1-(2,4-ジメトキシベンジル)-2-オキソ-1,2-ジヒドロピリジン-4-カルボキシラート(535 mg)のN,N-ジメチルホルムアミド(5 ml)溶液を滴下し、さらに0℃で15分間撹拌した。反応混合液を0.3M 塩酸(15ml)に注ぎ、析出物をろ取した。得られた固体をトルエンに懸濁し、減圧濃縮した。残渣をジイソプロピルエーテルで洗浄して標題化合物(527 mg)を得た。
MS (ESI+): [M+H]+ 520.3.
7-(2,4-ジメトキシベンジル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,2,3,7-テトラヒドロピリド[3,4-d]ピリミジン-4,8-ジオンから、実施例1のG)に準じた方法により標題化合物を得た。
MS (ESI+): [M+H]+ 548.1.
7-(2,4-ジメトキシベンジル)-2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,7-ジヒドロピリド[3,4-d]ピリミジン-4,8-ジオン(219 mg)をギ酸(5 ml)に溶解し、100℃で3時間撹拌した。室温まで冷却した後、反応混合物を減圧濃縮した。残渣にトルエンを加え、再度減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(NH、メタノール/酢酸エチル)で精製し、酢酸エチル/ヘキサン/メタノールの混合溶媒で再結晶して標題化合物 (132 mg) を得た。
2-(プロピルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,7-ジヒドロピリド[3,4-d]ピリミジン-4,8-ジオン
7-(2,4-ジメトキシベンジル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,2,3,7-テトラヒドロピリド[3,4-d]ピリミジン-4,8-ジオンおよび1-ヨードプロパンから、実施例29のC)およびD)に準じた方法により標題化合物を得た。
2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,7-ジヒドロピリド[3,4-d]ピリミジン-4,8-ジオン
2,2,2-トリフルオロエタノール(0.052 ml)のN,N-ジメチルホルムアミド(3 ml)溶液に、水素化ナトリウム(60%油性, 24 mg)を室温で加え、5分間撹拌した。その後 2-(プロピルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,7-ジヒドロピリド[3,4-d]ピリミジン-4,8-ジオン(535 mg)のN,N-ジメチルホルムアミド(3 ml)溶液を反応混合物に加え、60℃で4時間撹拌した。反応混合物を室温まで冷却した後、飽和食塩水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(NH、メタノール/酢酸エチル) で精製し、酢酸エチル/ヘキサンの混合溶媒で再結晶して標題化合物(8 mg)を得た。
2-エトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,7-ジヒドロピリド[3,4-d]ピリミジン-4,8-ジオン
2-(プロピルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,7-ジヒドロピリド[3,4-d]ピリミジン-4,8-ジオン(535 mg)のN,N-ジメチルホルムアミド(5 ml)溶液に、ナトリウムエトキシド(20%エタノール溶液、0.863 ml)を反応混合物に加え、60℃で4時間撹拌した。反応混合物を室温まで冷却した後、飽和食塩水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(NH、メタノール/酢酸エチル) で精製して標題化合物(140 mg)を得た。
2-(エチルスルファニル)-8-メトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[3,4-d]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,7-ジヒドロピリド[3,4-d]ピリミジン-4,8-ジオン(40 mg)および(トリフルオロメチル)ベンゼン(10 ml)の混合物にトリメチルオキソニウム テトラフルオロボラート(22 mg)を室温で加え、反応混合物を3時間加熱還流した。室温まで冷却し、反応混合物を酢酸エチルで希釈した。水および飽和食塩水にて反応混合物を順次洗浄した後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、酢酸エチル/ヘキサンの混合溶媒で再結晶して標題化合物(16 mg)を得た。
2-エトキシ-8-(2-メトキシエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[3,4-d]ピリミジン-4(3H)-オン
2-エトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,7-ジヒドロピリド[3,4-d]ピリミジン-4,8-ジオン(763 mg)およびオキシ塩化リン(10 ml)の混合物を100℃で5時間撹拌した。室温まで冷却後、反応混合物を減圧濃縮した。残渣にトルエンを加え、再度減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル) で精製して標題化合物(437 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ4.75-4.90 (2H, m), 7.17 (2H, d, J=9.0 Hz), 7.30 (2H,d, J=9.0 Hz), 7.85 (1H, d, J=5.3 Hz), 8.24 (1H, d, J=4.9 Hz), 11.43 (1H, s).
8-クロロ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[3,4-d]ピリミジン-2,4(1H,3H)-ジオン(437 mg)、ヨードエタン(0.19 ml)およびN,N-ジメチルホルムアミド(10 ml)の混合物に炭酸カリウム(326 mg)を室温で加え、80℃で15時間撹拌した。室温まで冷却後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(メタノール/酢酸エチル)で精製して標題化合物(116 mg)を得た。
MS (ESI+): [M+H]+ 400.0.
2-メトキシエタノール(0.019 ml)のN,N-ジメチルホルムアミド(3 ml)溶液に、水素化ナトリウム(60%油性, 9 mg)を室温で加え、30分間撹拌した。8-クロロ-2-エトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[3,4-d]ピリミジン-4(3H)-オン(80 mg)を反応混合物に加え、60℃で5時間撹拌した。室温まで冷却した後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン) で精製し、得られた残渣を酢酸エチル/ヘキサンの混合溶媒に懸濁し、生成した固体をろ取して標題化合物(21 mg)を得た。
2,8-ビス(2-メトキシエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリド[3,4-d]ピリミジン-4(3H)-オン
実施例34のC)の副生成物として標題化合物(6 mg)を得た。
2-(エチルスルファニル)-7-メチル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,7-ジヒドロピリド[3,4-d]ピリミジン-4,8-ジオン
2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,7-ジヒドロピリド[3,4-d]ピリミジン-4,8-ジオン(40 mg)のN,N-ジメチルホルムアミド(5 ml)溶液に、水素化ナトリウム(60%油性, 193 mg)を室温で加え、10分間撹拌した。反応混合物にヨードメタン(0.009 ml)を加え、室温で3時間撹拌した。反応混合物に水を加え、析出物をろ取した。得られた固体をトルエンに懸濁し、減圧濃縮した。残渣をヘキサン/酢酸エチル/メタノールの混合溶媒で再結晶し、標題化合物 (33 mg) を得た。
2-(エチルスルファニル)-7-メトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]プテリジン-4(3H)-オン
1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(88 mg)、水素化ナトリウム(60%油性, 37.7 mg)およびメチル 3-アミノ-5-メトキシピラジン-2-カルボキシラート(68 mg)を用いて、実施例1のF)と同様の反応を行い、ベージュ固体の標題化合物(122 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ4.05 (3H, s), 4.83 (2H, q, J=8.9 Hz), 7.14 (2H, d, J=9.0 Hz), 7.22 (2H, d, J=9.0 Hz), 8.26 (1H, s), 13.56 (1H, s).
7-メトキシ-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロプテリジン-4(1H)-オン(135 mg)、1M炭酸水素ナトリウム水溶液(351 μl)、ヨードエタン(142 μl)およびアセトニトリル(6 ml)を用いて実施例1のG)と同様の反応を行い、白色固体の標題化合物(110 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.27 (3H, t, J=7.4 Hz), 3.15 (2H, q, J=7.4 Hz), 4.06(3H, s), 4.88 (2H, q, J=8.9 Hz), 7.23 (2H, d, J=9.0 Hz), 7.44 (2H, d, J=9.0 Hz), 8.38 (1H, s).
2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]プテリジン-4,7(3H,8H)-ジオン
2-(エチルスルファニル)-7-メトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]プテリジン-4(3H)-オン(92 mg)、ピリジン塩酸塩(640 mg)およびN,N-ジメチルホルムアミド(3 ml)の混合物を120℃で10時間撹拌し、実施例6に準じた方法で白色粉体の標題化合物(50 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.26 (3H, t, J=7.4 Hz), 3.10 (2H, q, J=7.4 Hz), 4.87(2H, q, J=8.8 Hz), 7.22 (2H, d, J=9.0 Hz), 7.39 (2H, d, J=9.0 Hz), 7.89 (1H, s), 13.04 (1H, s).
3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
4,4,4-トリフルオロブタン酸(19.9 g)のTHF(200 mL)溶液に塩化オキサリル(30.5 mL)、DMF(0.54 mL)を0℃で加え、4時間撹拌した。得られた反応混合物を28%アンモニア水(150 mL)に0℃で加え、1時間撹拌した。反応混合物をろ過した後、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、硫酸マグネシウムで乾燥し、減圧濃縮した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿をろ取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(17.9 g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.27-2.35 (2H, m), 2.37-2.55 (2H, m), 6.96 (1H, brs), 7.44 (1H, brs).
4,4,4-トリフルオロブタンアミド(17.9 g)、2,4-ビス(4-メトキシフェニル)-1,3,2,4-ジチアジホスフェタン 2,4-ジスルフィド(51.3 g)、トルエン(400 mL)からなる混合物を80℃で3時間撹拌した。反応混合物をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル)に付した後、再度アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)にて精製し、標題化合物(14.6 g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.59-2.79 (4H, m), 9.37 (1H, brs), 9.60 (1H, brs).
4,4,4-トリフルオロブタンチオアミド(8.27 g)のアセトン(160 mL)溶液にヨードメタン(16.4 mL)を室温で加え、40℃で3時間撹拌した。反応混合物を減圧濃縮することにより標題化合物(15.4 g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.68 (3H, s), 2.72-2.87 (2H, m), 3.02-3.10 (2H, m), 11.64 (2H, brs).
メチル 4,4,4-トリフルオロブタンイミドチオアート ヨウ素酸塩(15.3 g)、4-(2,2,2-トリフルオロエトキシ)アニリン(8.90 g)、エタノール(150 mL)からなる混合物を室温で2時間撹拌した。反応混合物に酢酸エチル、飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(10.9 g)を固体として得た。
MS (ESI+): [M+H]+ 315.1.
4,4,4-トリフルオロ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]ブタンイミドアミド(20.9 g)、マロン酸ジエチル(20.2 mL)、ナトリウムメトキシド(10.8 g)および2-メトキシエタノール(400 mL)からなる混合物を終夜加熱還流し、反応混合物を減圧濃縮した。同様に、4,4,4-トリフルオロ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]ブタンイミドアミド(22.6 g)、マロン酸ジエチル(21.8 mL)、ナトリウムメトキシド(11.7 g)および2-メトキシエタノール(400 mL)からなる混合物を終夜加熱還流し、反応混合物を減圧濃縮した。各々から得られた残渣を混合し、ジエチルエーテルと水で分配した。水層を分離後、有機層に1規定塩酸を加え酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、硫酸マグネシウムで乾燥し、減圧濃縮した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿をろ取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(41.6 g)を固体として得た。
MS (ESI+): [M+H]+ 383.1.
6-ヒドロキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン(2.63 g)、トリエチルアミン(1.44 mL)のTHF(50 mL)溶液に1,1,1-トリフルオロ-N-フェニル-N-[(トリフルオロメチル)スルホニル]メタンスルホンアミド(2.46 g)を0℃で加え、室温で終夜撹拌した。反応混合物に氷冷した飽和食塩水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(3.21 g)を固体として得た。
MS (ESI+): [M+H]+ 515.2.
トリス(ジベンジリデンアセトン)ジパラジウム(0)(537 mg)、4,5-ビス(ジフェニルホスフィノ)-9,9-ジメチルキサンテン(679 mg)のトルエン(50 mL)溶液に6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル トリフルオロメタンスルホナート(5.03 g)、1,1-ジフェニルメタンイミン(1.97 mL)および炭酸セシウム(7.97 g)を室温で加え、100℃で終夜撹拌した。反応混合物を減圧濃縮した。残渣にTHFを加え、ろ過して不溶物を取り除き、ろ液を減圧濃縮した。得られた残留物のTHF(100 mL)溶液に1規定塩酸(100 mL)を0℃で加え、2時間撹拌した。反応混合物を酢酸エチルで抽出し、抽出液を飽和食塩水で洗浄後、硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)およびシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(2.64 g)を固体として得た。
MS (ESI+): [M+H]+ 382.1.
6-アミノ-3-(4-(2,2,2-トリフルオロエトキシ)フェニル)-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン(1.0 g)のN,N-ジメチルアセトアミド(30 mL)溶液を60℃に加熱し、塩化アクリロイル(0.85 mL)を加え、10分間撹拌した。氷冷下反応混合物に水を加え、酢酸エチルで抽出した。抽出液を硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)、ジオールプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)、更にもう一度シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)にて精製し、得られた固体を酢酸エチル-ヘキサンから再結晶して標題化合物(0.72 g)を固体として得た。この固体(487 mg)を酢酸エチルから再結晶して標題化合物(300 mg)を固体として得た。
1H NMR (300 MHz, CDCl3) δ 2.45-2.62 (4H, m), 2.62-2.73 (2H, m), 2.79-2.93 (2H, m), 4.42 (2H, q, J = 7.9 Hz), 7.03-7.20 (4H, m), 7.42 (1H, s).
2-プロピル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオン
エチル ブチルイミダート 塩酸塩(10.0 g)、4-(2,2,2-トリフルオロエトキシ)アニリン(12.6 g)、トリエチルアミン(6.67 g)、THF(250 mL)からなる混合物を70℃で2日間撹拌した。反応混合物に酢酸エチルおよび水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(4.80 g)を固体として得た。
MS (ESI+): [M+H]+ 261.4.
実施例39の工程Eと同様の方法により標題化合物を得た。
MS (ESI+): [M+H]+ 329.3.
実施例39の工程Fと同様の方法により標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.80 (3H, t, J = 7.3 Hz), 1.40-1.70 (2H, m), 2.31 (2H, t, J = 7.3 Hz), 4.86 (2H, q, J = 9.0 Hz), 6.62 (1H, s), 7.23 (2H, d, J = 9.0 Hz), 7.43 (2H, d, J = 9.0 Hz).
酢酸パラジウム(II)(49.0 mg)、4,5-ビス(ジフェニルホスフィノ)-9,9-ジメチルキサンテン(189 mg)のトルエン(20 mL)溶液に6-オキソ-2-プロピル-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,6-ジヒドロピリミジン-4-イル トリフルオロメタンスルホナート(1.0 g)、4-メトキシベンジルアミン(0.43 mL)および炭酸セシウム(1.77 g)を室温で加え、100℃で3時間撹拌した。反応混合物に酢酸エチルおよび水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(462 mg)を固体として得た。
MS (ESI+): [M+H]+ 448.3.
6-[(4-メトキシベンジル)アミノ]-2-プロピル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(400 mg)のトリフルオロ酢酸溶液(3 mL)をマイクロウェーブ照射下100℃で30分間撹拌した。反応混合物を減圧下濃縮し、残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(270 mg)を固体として得た。
MS (ESI+): [M+H]+ 328.1.
6-アミノ-2-プロピル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(270 mg)のN,N-ジメチルアセトアミド(15 mL)溶液に塩化アクリロイル(0.20 mL)を0℃で加え、そのまま30分間撹拌後、室温で終夜撹拌した。反応混合物に炭酸カリウム(570 mg)を室温で加えて1時間攪拌した。氷冷下反応混合物に飽和塩化アンモニウム水溶液を加え、酢酸エチルとTHFの混合溶媒で抽出した。抽出液を硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(59.6 mg)を固体として得た。



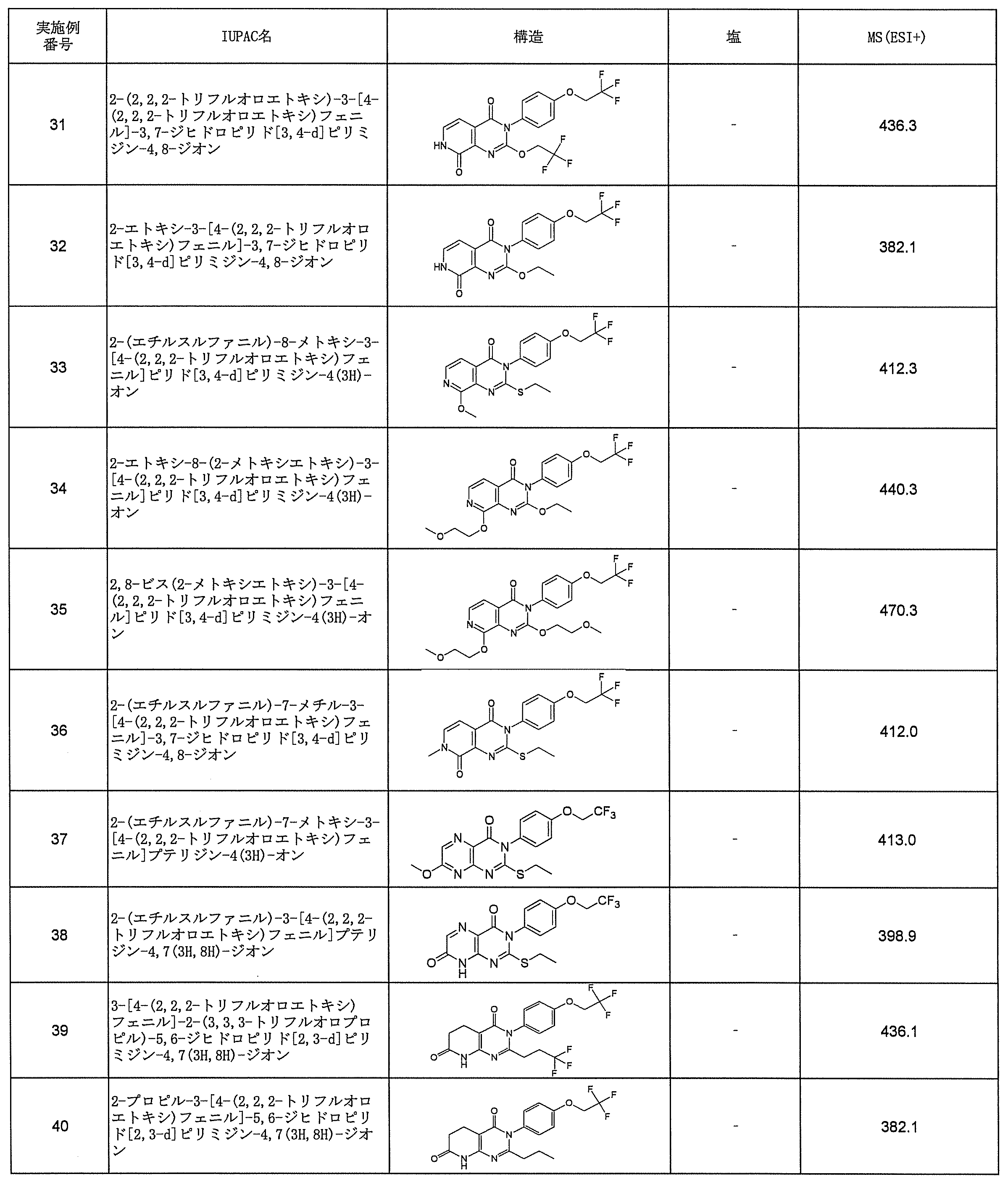
N-[2-({4-オキソ-3-[4-(2,2,2-トリフルオロエトキシ)[2,6-3H2]フェニル]-3,4-ジヒドロチエノ[3,4-d]ピリミジン-2-イル}スルファニル)エチル]アセトアミド
A) エチル 4-イソチオシアナトチオフェン-3-カルボキシラート
4-アミノチオフェン-3-カルボン酸エチル塩酸塩(69g)を1M炭酸水素ナトリウム水溶液(400ml)に加え、混合物を酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、溶媒を減圧留去して褐色油状物(56g)を得た。この褐色油状物をクロロホルム(150ml)に溶かし、チオホスゲン(12.5ml)のクロロホルム(300ml)溶液に加えた。この混合溶液を炭酸水素ナトリウム(18.5g)の水溶液(115ml)に室温で滴下し、4時間撹拌した。有機層と水層を分液し、水層をクロロホルムで抽出した。得られた有機層を合わせて飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥後、減圧濃縮して得られた粗製物をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)にて精製し標題化合物(15.6g)を薄黄色固体として得た。
1H NMR (400 MHz, CDCl3) δ1.39 (3H, t, J=7.2 Hz), 4.36 (2H, q, J=7.2 Hz), 7.14 (1H, d, J=3.6 Hz), 8.06 (1H, d, J=3.6 Hz).
エチル 4-イソチオシアナトチオフェン-3-カルボキシラート(1.1g)および4-(2,2,2-トリフルオロエトキシ)アニリン(0.99g)をアセトニトリル(40ml)に溶かし、1時間加熱還流した。放冷後、カリウムtert-ブトキシド(1.27g)をエタノール(15ml)に溶かし、室温下反応混合物に注いだ。反応混合物を10分間加熱還流した後放冷し、減圧濃縮した。得られた褐色残渣をエタノール(10ml)に溶かし、氷冷下0.3M塩酸(35ml)に注いだ。生じた沈殿物をろ取し、水で洗浄後、テトラヒドロフラン(40ml)に溶かし、酢酸エチル(80ml)で希釈した。この有機層を無水硫酸マグネシウムで乾燥後、溶媒を減圧留去し固体を得た。この固体をテトラヒドロフラン/イソプロピルエーテル(1/10)混合溶媒で洗浄し、標題化合物(1.59g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ4.82 (2 H, q, J=8.9 Hz), 7.10 (2 H, d, J=9.0 Hz), 7.12 (1 H, d, J=3.3 Hz), 7.20 (2 H, d, J=9.0 Hz), 8.51 (1 H, d, J=3.3 Hz), 12.92 (1 H, s).
2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロチエノ[3,4-d]ピリミジン-4(1H)-オン(1.59g)をN,N-ジメチルホルムアミド(20ml)に溶かし、氷冷下、水素化ナトリウム(60%油性、0.25g)およびtert-ブチル(2-ブロモエチル)カルバマート(1.49g)を順に加えた後、室温で1時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液(30ml)を注ぎ、混合物を酢酸エチルで抽出した。有機層を水、飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥後、減圧濃縮した。残留物をシリカゲルカラムクロマトグラフィー(NH、酢酸エチル/ヘキサン)にて精製し、標題化合物(2.12g)を白色固体として得た。
1H NMR (300 MHz, DMSO-d6) δ1.35 (9 H, s), 3.10 (2 H, t, J=6.3 Hz), 3.23 (2 H, td, J=6.3, 5.3 Hz), 4.86 (2 H, q, J=8.9 Hz), 6.97 (1 H, t, J=5.3 Hz), 7.18 (2 H, d, J=9.0 Hz), 7.37 (2 H, d, J=9.0 Hz), 7.66 (1 H, d, J=3.3 Hz), 8.50 (1 H, d, J=3.3 Hz).
4規定塩化水素酢酸エチル溶液(20ml)とメタノール(10ml)の混合溶液にtert-ブチル [2-({4-オキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,4-ジヒドロチエノ[3,4-d]ピリミジン-2-イル}スルファニル)エチル]カルバマート(1.7g)を加え、室温で14時間撹拌した。反応液を減圧濃縮した後、残留物を酢酸エチル/エーテル(1/1)の混合液で洗浄し、標題化合物(1.48g)を白色固体として得た。
1H NMR (300 MHz, DMSO-d6)δ3.03 - 3.21 (2 H, m), 3.29 (2 H, t, J=6.8 Hz), 4.87 (2 H, q, J=8.8 Hz), 7.20 (2 H, d, J=9.1 Hz), 7.43 (2 H, d, J=9.1 Hz), 7.70 (1 H, d, J=3.3 Hz), 8.17 (3 H, br. s.), 8.53 (1 H, d, J=3.3 Hz).
2-[(2-アミノエチル)スルファニル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]チエノ[3,4-d]ピリミジン-4(3H)-オン塩酸塩(0.33g)をテトラヒドロフラン(30ml)に懸濁し、トリエチルアミン(0.31ml)、無水酢酸(0.28ml)および4-ジメチルアミノピリジン(37mg)を順に加え、室温で18時間撹拌した。反応混合液を減圧濃縮した後酢酸エチルで希釈し、飽和クエン酸水溶液、水、飽和炭酸水素ナトリウム水溶液で順次洗浄し、無水硫酸マグネシウムで乾燥後、減圧濃縮した。得られた薄黄色固体をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、無色固体を得た。この固体を酢酸エチル/ヘキサンで再結晶し、標題化合物(0.22g)を無色固体として得た。
1H NMR (300 MHz, DMSO-d6)δ1.77 (3H, s), 3.01-3.20 (2H, m), 3.21-3.38 (2H, m), 4.86 (2H, q, J = 8.7 Hz), 7.10-7.27 (2H, m), 7.31-7.45 (2H, m), 7.66 (1H, d, J = 3.4 Hz), 8.01 (1H, t, J = 5.3 Hz), 8.50 (1H, d, J = 3.4 Hz).
MS (ESI+): [M+H]+ 444.0.
N-[2-({4-オキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,4-ジヒドロチエノ[3,4-d]ピリミジン-2-イル}スルファニル)エチル]アセトアミド(3mg)とクラブトリー触媒:(1,5-シクロオクタジエン)(ピリジン)(トリシクロヘキシルホスフィン)イリジウム(I) ヘキサフルオロリン酸塩(6mg)をジクロロメタン(1.5ml)に溶解し、トリチウムガス(2Ci)雰囲気下4時間撹拌した。反応液を減圧濃縮した後、残渣にエタノールを加え再度減圧濃縮した。残留物を逆層高速液体クロマトグラフィー(C18 column、溶出液:水/アセトニトリル/トリフルオロ酢酸)で精製し、標題化合物(44 Ci/mmol)を得た。高速液体クロマトグラフィーで測定した標題化合物の放射化学的純度は99.9%であった(カラム:Spherisorb ODS2 5μ 150 x 4.6 mm、溶媒:A液;0.1%トリフルオロ酢酸含有水、B液;0.1%トリフルオロ酢酸含有アセトニトリル、グラジエントサイクル:0分(A液/B液=80/20)、15分(A液/B液=0/100)、注入量:5 μl、流速:1 ml/min)。
MS (ESI+): [M+H]+ 448.1.
デルタ-5-デサチュラーゼに対する試験化合物の阻害活性を、以下に記述する方法で測定した。あらかじめ調製した試験化合物のDMSO溶液に対してバッファー(300mM NaH2PO4[pH7.4]、450mM KCl、30mM NaF、9mM MgCl2、4.5mM glutathione[reduced form]、0.3%BSA[fatty acid free、SIGMA])を用いて二次希釈を行い、アッセイバッファーとした。このアッセイバッファー10μlをポリプロピレン製の96穴ディープウェルブロックに分注した後、ミクロソームバッファー(10mM Tris-HCl[pH7.4]、1mM EDTA、250mM sucrose)を用いて3mg/mlに希釈したラット肝ミクロソーム画分10μlを添加した。酵素反応は、9mM NADH、9mM ATP、0.9mM CoA、10μCi/ml(8E,11E,14E)-(1-14C)eicosa-8,11,14-trienoic acid(PerkinElmer Inc.)を10μl添加することで開始した。120分間、室温で酵素反応を行い、10μlの2.5M NaOHの添加によって酵素反応を停止した。反応停止後プレートシールを施したうえで55℃に設定した乾熱器で一晩インキュベーションしてケン化を行った。脂肪酸の溶媒抽出はカナディアン ジャーナル オブ バイオケミストリー アンド フィジオロジー(Can.J.Biochem.Physiol.)、第37巻、911頁(1959)に記載のBligh & Dyer法に基づき、ギ酸:メタノール:クロロホルム(1:6:3)200μlを添加して一層状態を保ち、十分な撹拌を行った後に純水120μlを添加して、二層への分離を行った。下層のクロロホルム層10μlを逆相TLCプレート(RP-18、1154230001、Merck Japan, Ltd.)にスポットして、アセトニトリル:純水:酢酸(95:4.5:0.5)で展開し、乾燥後のTLCプレートをImaging Plate(Fuji Photo Film Co.,Ltd.)に5時間以上転写させた。検出はBAS-5000(Fuji Photo Film Co.,Ltd)を用いて行い、得られたスポット画像をMulti Gauge Ver2.3(Fuji Photo Film Co.,Ltd)を用いて数値化し、試験化合物の10μMでのデルタ-5-デサチュラーゼ阻害率(%)を求めた。結果を表5に示す。
表5から明らかなように、本発明化合物は優れたデルタ-5-デサチュラーゼ阻害作用を示した。
試験はすべて室温で、200 μL/ウェルで実施した。アッセイ緩衝液 (10 mM Tris-HCl (pH7.5)、100 mM KCl、10 mM NaF、3mM MgCl2、0.005% tween20 および、1 mM GSH) 中で、30 μg/ウェルのラット肝ミクロソーム画分を終濃度 10 μM の試験化合物と 15 分プレインキュベーションした。N-[2-({4-オキソ-3-[4-(2,2,2-トリフルオロエトキシ)[2,6-3H2]フェニル]-3,4-ジヒドロチエノ[3,4-d]ピリミジン-2-イル}スルファニル)エチル]アセトアミド を終濃度 3 nM になるよう添加し 150 分インキュベーションした。Bound/Free 分離はセルハーベスターを用いて膜画分を GF/C glass filter 上に補足した。GF/C glass filter を 300 μl の氷冷したアッセイ緩衝液(除 0.005% tween)で 5 回洗浄することによりデルタ-5-デサチュラーゼと結合したリガンドを分離した.50 μl のマイクロシンチ 0 を添加し、トップカウントにてフィルター上の放射活性を測定した。非特異的結合の判定には 10 μM N-[2-({4-オキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-3,4-ジヒドロチエノ[3,4-d]ピリミジン-2-イル}スルファニル)エチル]アセトアミドを用いた。デルタ-5-デサチュラーゼへのN-[2-({4-オキソ-3-[4-(2,2,2-トリフルオロエトキシ)[2,6-3H2]フェニル]-3,4-ジヒドロチエノ[3,4-d]ピリミジン-2-イル}スルファニル)エチル]アセトアミドの結合に対する、試験化合物 10 μM での阻害率 (%) を求めた。結果を表5に示す。
表5から明らかなように、本発明化合物は、デルタ-5-デサチュラーゼへのN-[2-({4-オキソ-3-[4-(2,2,2-トリフルオロエトキシ)[2,6-3H2]フェニル]-3,4-ジヒドロチエノ[3,4-d]ピリミジン-2-イル}スルファニル)エチル]アセトアミドの結合に対する優れた阻害作用を示した。
96-ウェルプレートに1×105 cells/ウェルとなるようにHepG2を播種し, 10% FBS(ウシ胎児血清)を含むDMEM(ダルベッコ改変イーグル培地)で一晩培養を行った。PBS(リン酸緩衝生理食塩水)を用いて洗浄後(200 μL×2), 試験化合物を含む反応培地(DMEM, penicillin/streptomycin, 0.3% BSA を40 μL添加し, CO2インキュベータにて37℃で 30 分のプレインキュベーションを行った。反応は, 0.1 μCiの[14C]Eicosatrienoic acid を含む反応培地20 μLの添加によって開始し, 37℃に設定したCO2インキュベータにてインキュベーションを行った。インキュベーションを3時間行った後, 200 μLのPBSで2回洗浄し, 50 μLのトリプシン-EDTAで剥離した細胞を96-deepウェルブロックに移した。さらに, 20 μLの2.5N NaOHを添加し, プレートシールを施した上で, 55℃ に設定した乾熱器で一晩インキュベーションし, ケン化を行った。脂肪酸抽出は, 10 μLの蟻酸添加で酸性化を行った後, クロロホルム:メタノール(1:4)を添加して攪拌し, さらに200 μLの純水を添加して二層化を行った。TLC展開以降の操作は, ラット肝ミクロソームを用いた系におけるTLCを用いた検出系の手法に従った。試験化合物の1μMでのデルタ-5-デサチュラーゼ阻害率(%)を求めた。結果を表5に示す。

試験化合物の in vivoにおけるデルタ-5-デサチュラーゼ阻害作用の評価は以下の方法で行った。個別ケージを用いて一般飼育飼料 (CE-2、日本クレア社)にて馴化した7-9週齢の雄性正常マウス (C57BL/6J, 日本クレア社)に、試験化合物 3mg/kgを 0.5%のメチルセルロース溶液に懸濁し、10mL/kgで 4日間、毎朝強制経口投与した。最終投与翌日の朝に、マウスを麻酔し、肝臓を摘出した。約 40mgの肝臓サンプルから、ヘキサン-プロパノール溶液を用いて総脂質を抽出し、固相抽出カラム (Sep-Pak Vac NH2, Waters社)を用いてリン脂質画分を抽出した後、YMC社エステル型脂肪酸ラベル化試薬 (YMC社)を用いてラベル化した。ラベル化後のサンプル中のアラキドン酸およびジホモ-γ-リノレン酸含有量は高速液体クロマトグラフィー (Agilent 1200、アジレント・テクノロジー社)を用いて測定した。アラキドン酸 (AA)含有量をジホモ-γ-リノレン酸(DGLA)含有量で除することで、AA/DGLA比を算出し、溶媒投与群の AA/DGLA比を 100%としたときの試験化合物投与群の低下率を算出することで、in vivoにおける肝臓デルタ-5-デサチュラーゼ阻害作用の指標とした。結果を表6に示す。
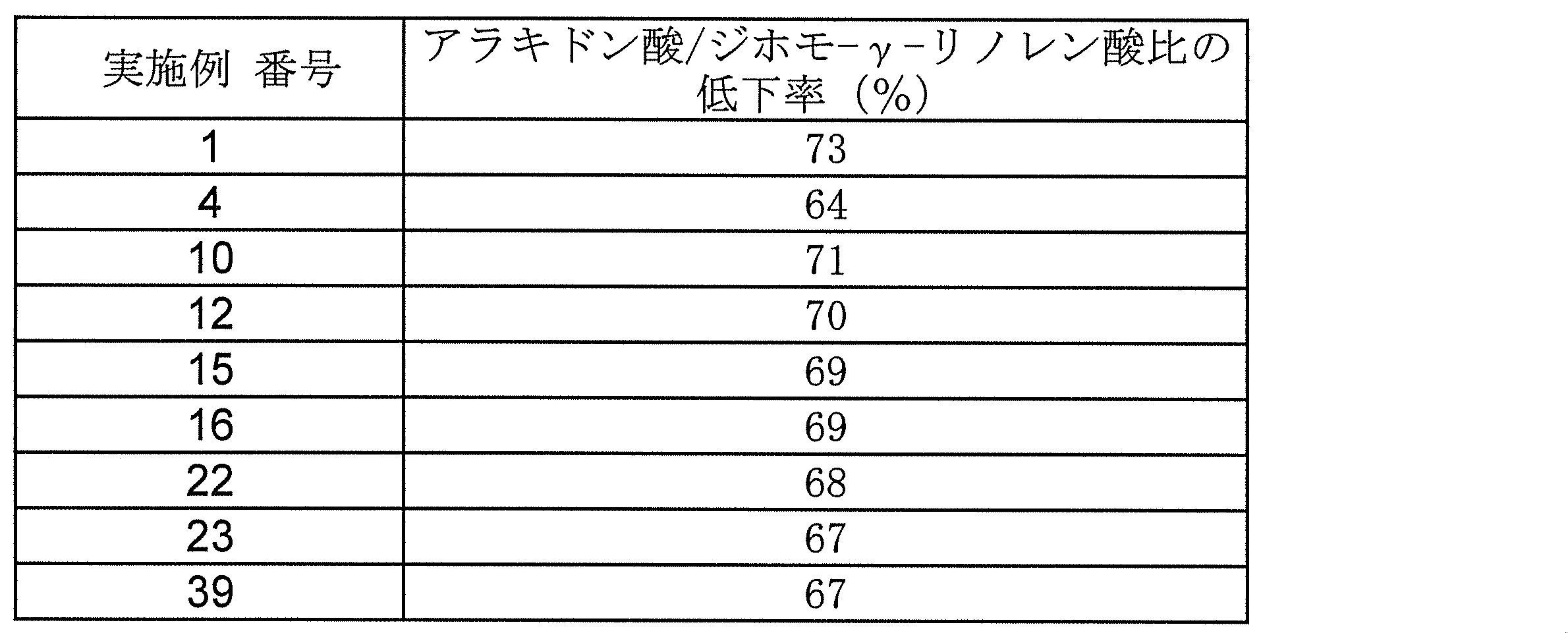
(1)実施例1の化合物 10.0g
(2)乳糖 70.0g
(3)コーンスターチ 50.0g
(4)可溶性デンプン 7.0g
(5)ステアリン酸マグネシウム 3.0g
実施例1の化合物10.0gとステアリン酸マグネシウム3.0gを可溶性デンプンの水溶液70ml(可溶性デンプンとして7.0g)で顆粒化した後、乾燥し、乳糖70.0gおよびコーンスターチ50.0gと混合する(乳糖、コーンスターチ、可溶性デンプンおよびステアリン酸マグネシウムはいずれも第十四改正日本薬局方適合品)。混合物を圧縮して錠剤を得る。
Claims (19)
- 式(I):
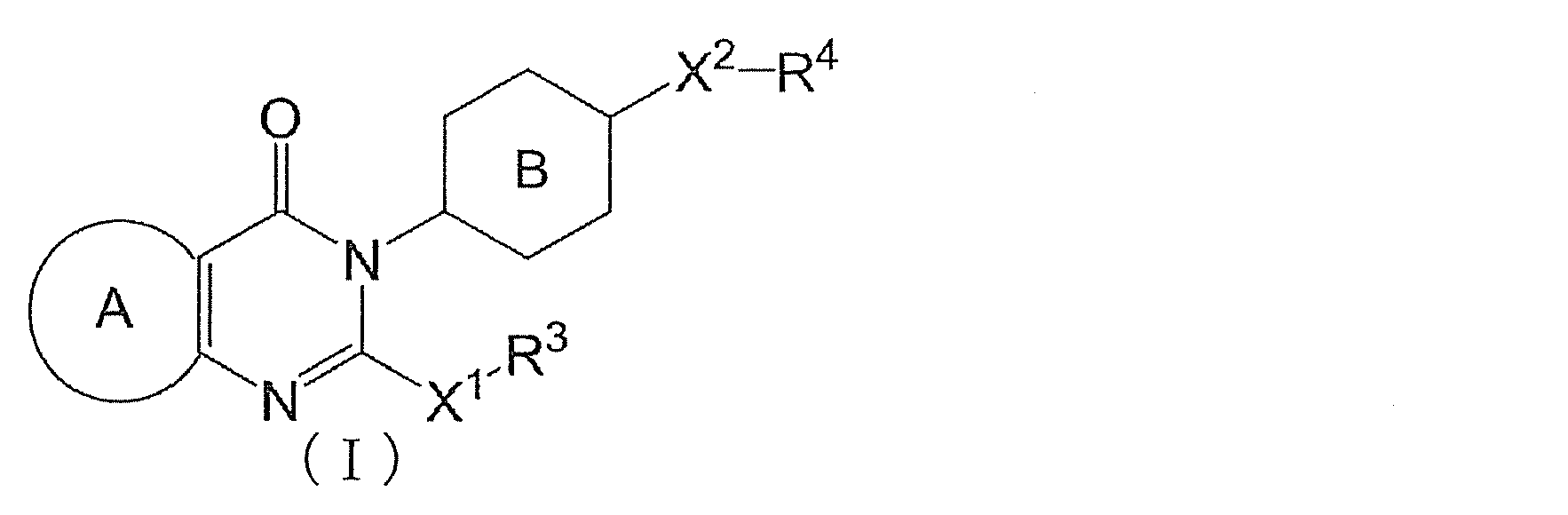
(I)
〔式中、
環Aは式:
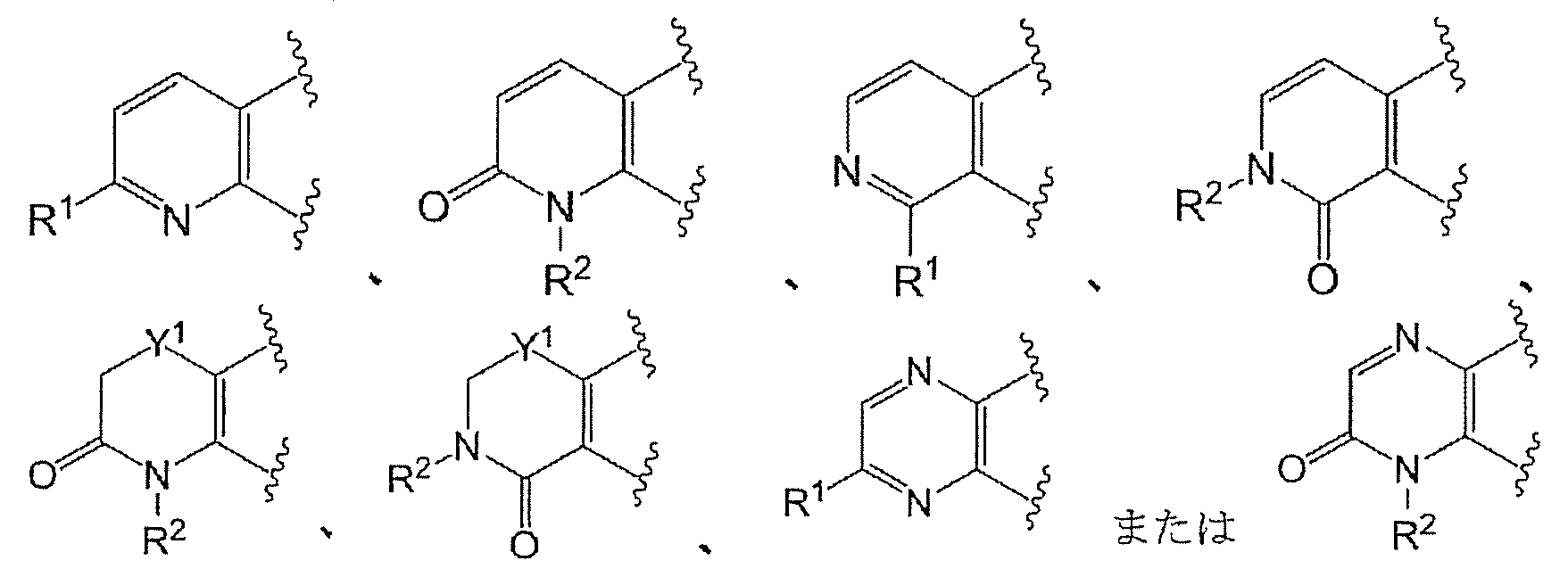
で表される環であり、それぞれの環はさらにハロゲン原子、C1-6アルキル基およびC1-6アルコキシ基から選択される置換基で置換されていてもよく、
環Bはさらにハロゲン原子、C1-6アルキル基およびC1-6アルコキシ基から選択される置換基で置換されていてもよい6員芳香環を示し、
X1は結合手、S、SO2、OまたはNR5を示し、
X2はCR6R7またはOを示し、
Y1はCR8R9、S、SO、SO2またはOを示し、
R1は置換されていてもよいC1-6アルコキシ基、置換されていてもよいアミノ基、置換されていてもよいカルバモイル基または置換されていてもよいC1-6アルキルチオ基を示し、
R2は水素原子または置換されていてもよいC1-6アルキル基を示し、
R3は置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示し、
R4は置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示し、
R5は水素原子または置換されていてもよいC1-6アルキル基を示し、および
R6、R7、R8およびR9はそれぞれ水素原子、ハロゲン原子、C1-6アルキル基またはC1-6アルコキシ基を示す。〕で表される化合物またはその塩。 - X2がOであり;
R4が1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、請求項1記載の化合物またはその塩。 - 環Aが、式:
で表される環であり;
環Bがベンゼンであり;
X1が結合手、S、OまたはNHであり;
X2がOであり;
Y1がCH2またはSであり;
R1が、
(1)1ないし3個のC1-6アルコキシ基で置換されていてもよいC1-6アルコキシ基;
(2)アミノ;または
(3)カルバモイル;
であり;
R2が水素原子またはC1-6アルキル基であり;
R3が
(1) (A)C1-6アルキルスルホニル基、
(B)ヒドロキシ基、
(C)C1-6アルコキシ基、および
(D)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基、または
(2)1ないし3個のハロゲン原子で置換されていてもよいC3-10シクロアルキル基であり;および
R4が1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、請求項1記載の化合物またはその塩。 - 環Aが、式:
環Bがベンゼンであり;
X1が結合手、SまたはOであり;
X2がOであり;
Y1がCH2であり;
R1がC1-6アルコキシ基であり;
R2が水素原子であり;
R3が
(1)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、または
(2)1ないし3個のハロゲン原子で置換されていてもよいC3-10シクロアルキル基であり;および
R4が1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、請求項1記載の化合物またはその塩。 - 環Aが、式:

で表される環であり;
環Bがベンゼンであり;
X1が結合手、SまたはOであり;
X2がOであり;
Y1がCH2であり;
R2が水素原子であり;
R3が
(1)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、または
(2)1ないし3個のハロゲン原子で置換されていてもよいC3-10シクロアルキル基であり;および
R4が1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、請求項1記載の化合物またはその塩。 - 2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオンまたはその塩。
- 2-エトキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオンまたはその塩。
- 3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-5,6-ジヒドロピリド[2,3-d]ピリミジン-4,7(3H,8H)-ジオンまたはその塩。
- 請求項1記載の化合物またはその塩を含有してなる医薬。
- デルタ-5-デサチュラーゼ阻害薬である請求項9記載の医薬。
- エイコサノイドが媒介する疾患の予防または治療剤である請求項9記載の医薬。
- 動脈硬化症の予防または治療剤である請求項9記載の医薬。
- 糖尿病または肥満症の予防または治療剤である請求項9記載の医薬。
- 請求項1記載の化合物またはその塩を哺乳動物に有効量投与することを特徴とする、該哺乳動物における動脈硬化症の予防または治療方法。
- 請求項1記載の化合物またはその塩を哺乳動物に有効量投与することを特徴とする、該哺乳動物における糖尿病または肥満症の予防または治療方法。
- 動脈硬化症の予防または治療剤を製造するための、請求項1記載の化合物またはその塩の使用。
- 糖尿病または肥満症の予防または治療剤を製造するための、請求項1記載の化合物またはその塩の使用。
- 動脈硬化症の予防または治療のために使用する請求項1記載の化合物またはその塩。
- 糖尿病または肥満症の予防または治療のために使用する請求項1記載の化合物またはその塩。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2806258A CA2806258A1 (en) | 2010-07-23 | 2011-07-22 | Fused heterocyclic compound and application thereof |
| JP2012525456A JPWO2012011591A1 (ja) | 2010-07-23 | 2011-07-22 | 縮合複素環化合物およびその用途 |
| EP11809761.7A EP2597095A4 (en) | 2010-07-23 | 2011-07-22 | FUSED HETEROCYCLIC COMPOUND AND APPLICATION THEREOF |
| EA201390158A EA026142B1 (ru) | 2010-07-23 | 2011-07-22 | Конденсированное гетероциклическое соединение и его применение |
| US13/811,755 US9023858B2 (en) | 2010-07-23 | 2011-07-22 | Substituted pyrido[2,3-d]pyrimidines as delta-5-desaturase inhibitors |
| CN2011800453754A CN103119043A (zh) | 2010-07-23 | 2011-07-22 | 稠合的杂环化合物及其用途 |
| BR112013001634A BR112013001634A2 (pt) | 2010-07-23 | 2011-07-22 | composto ou sal, medicamento, métodos para a profilaxia ou tratamento da arteriosclerose, da diabete ou obesidade em um mamífero, e, uso do composto ou sal |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010-166475 | 2010-07-23 | ||
| JP2010166475 | 2010-07-23 | ||
| JP2011-122796 | 2011-05-31 | ||
| JP2011122796 | 2011-05-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012011591A1 true WO2012011591A1 (ja) | 2012-01-26 |
Family
ID=45497008
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/066766 WO2012011591A1 (ja) | 2010-07-23 | 2011-07-22 | 縮合複素環化合物およびその用途 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US9023858B2 (ja) |
| EP (1) | EP2597095A4 (ja) |
| JP (1) | JPWO2012011591A1 (ja) |
| CN (1) | CN103119043A (ja) |
| BR (1) | BR112013001634A2 (ja) |
| CA (1) | CA2806258A1 (ja) |
| EA (1) | EA026142B1 (ja) |
| WO (1) | WO2012011591A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017003723A1 (en) * | 2015-07-01 | 2017-01-05 | Crinetics Pharmaceuticals, Inc. | Somatostatin modulators and uses thereof |
| US11028068B2 (en) | 2017-07-25 | 2021-06-08 | Crinetics Pharmaceuticals, Inc. | Somatostatin modulators and uses thereof |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015195928A1 (en) * | 2014-06-19 | 2015-12-23 | Attillaps Holdings | Acetylcholinesterase inhibitors for treatment of dermatological conditions |
| US11446241B2 (en) | 2013-07-29 | 2022-09-20 | Attillaps Holdings Inc. | Treatment of ophthalmological conditions with acetylcholinesterase inhibitors |
| JPWO2017082132A1 (ja) * | 2015-11-11 | 2018-08-30 | 住友化学株式会社 | 縮合複素環化合物 |
| US10639313B2 (en) | 2017-09-01 | 2020-05-05 | Ndsu Research Foundation | Compound for inhibition of delta-5-desaturase (D5D) and treatment of cancer and inflammation |
Citations (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998045285A1 (en) | 1997-04-04 | 1998-10-15 | Merck & Co., Inc. | Somatostatin agonists |
| WO1998044921A1 (en) | 1997-04-04 | 1998-10-15 | Merck & Co., Inc. | Somatostatin agonists |
| WO1999022735A1 (en) | 1997-10-30 | 1999-05-14 | Merck & Co., Inc. | Somatostatin agonists |
| US5925642A (en) | 1995-11-24 | 1999-07-20 | Byk Gulden Lomberg Chemische Fabrik Gmbh | Pyridopyrimidines |
| WO2001014372A2 (en) | 1999-08-25 | 2001-03-01 | Takeda Chemical Industries, Ltd. | Oxazole and thiazole derivatives as neurotrophin production/secretion promoting agent |
| WO2001025228A1 (fr) | 1999-10-07 | 2001-04-12 | Tadeka Chemical Industries, Ltd. | Derives d'amines |
| WO2001038325A1 (en) | 1999-11-10 | 2001-05-31 | Takeda Chemical Industries, Ltd. | 5-membered n-heterocyclic compounds with hypoglycemic and hypolipidemic activity |
| WO2002006234A1 (fr) | 2000-07-17 | 2002-01-24 | Takeda Chemical Industries, Ltd. | Derives de sulfonate, procede de production et utilisation de ces derives |
| WO2002006264A1 (fr) | 2000-07-13 | 2002-01-24 | Takeda Chemical Industries, Ltd. | Inhibiteurs de plaques riches en lipide |
| WO2003042204A1 (fr) | 2001-10-19 | 2003-05-22 | Takeda Chemical Industries, Ltd. | Derive d'amine |
| WO2003059900A1 (fr) | 2002-01-11 | 2003-07-24 | Takeda Chemical Industries, Ltd. | Derives de coumarine, leur procede de production et d'utilisation |
| WO2003106435A1 (en) | 2002-06-18 | 2003-12-24 | Sankyo Company, Limited | Fused-ring pyrimidin-4(3h)-one derivatives, processes for the preparation and uses thereof |
| WO2004048363A1 (ja) | 2002-11-22 | 2004-06-10 | Takeda Pharmaceutical Company Limited | イミダゾール誘導体、その製造法および用途 |
| WO2005030740A1 (ja) | 2003-09-30 | 2005-04-07 | Takeda Pharmaceutical Company Limited | チアゾリン誘導体およびその用途 |
| WO2005058823A1 (ja) | 2003-12-17 | 2005-06-30 | Takeda Pharmaceutical Company Limited | ウレア誘導体、その製造法及び用途 |
| WO2005077905A1 (ja) * | 2004-02-13 | 2005-08-25 | Banyu Pharmaceutical Co., Ltd. | 縮環4-オキソ-ピリミジン誘導体 |
| WO2005091711A2 (en) | 2004-03-24 | 2005-10-06 | Orchid Chemicals & Pharmaceuticals Ltd | Novel condensed pyrimidones |
| WO2005113504A1 (en) | 2004-05-21 | 2005-12-01 | Takeda Pharmaceutical Company Limited | Cyclic amide derivatives, and their production and use as antithrombotic agents |
| WO2007002701A2 (en) | 2005-06-27 | 2007-01-04 | Amgen Inc. | Anti-inflammatory aryl nitrile compounds |
| JP2008504301A (ja) * | 2004-06-28 | 2008-02-14 | アムジェン エスエフ エルエルシー | 炎症性及び免疫調節性の障害及び疾患の予防及び治療に有用である、cxcr3受容体モジュレーターとしての縮合ピリミジン誘導体及びその組成物 |
| WO2008089307A2 (en) * | 2007-01-18 | 2008-07-24 | Lexicon Pharmaceuticals, Inc. | Delta 5 desaturase inhibitors for the treatment of pain, inflammation and cancer |
| JP2010166475A (ja) | 2009-01-19 | 2010-07-29 | Nec Corp | 移動通信ネットワークにおける通信設定方法及び移動通信端末 |
| WO2010087467A1 (en) * | 2009-01-27 | 2010-08-05 | Takeda Pharmaceutical Company Limited | Delta-5-desaturase inhibitors |
| JP2011122796A (ja) | 2009-12-14 | 2011-06-23 | Sanyo Electric Co Ltd | 断熱箱体及びその製造方法 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8281542B2 (en) | 2006-09-11 | 2012-10-09 | Dana Innovations | Mechanisms for locking and removing flush mounted inserts |
| US7963076B2 (en) | 2006-09-11 | 2011-06-21 | Dana Innovations | Devices and methods for flangeless installations |
| US20090107083A1 (en) | 2006-09-11 | 2009-04-30 | Ray Call | Multicategory Collection of Ceiling/Wall Devices With Familial Appearances |
| US20090249705A1 (en) | 2006-12-04 | 2009-10-08 | Trufig | Mounting Receivers with Spackling Rim Gradient |
| US8209921B2 (en) | 2006-09-11 | 2012-07-03 | Dana Innovations | Flush mount panels with multiple aligned receiving brackets |
| US8061094B2 (en) | 2006-09-11 | 2011-11-22 | Dana Innovations | Sanding shield for in-wall components |
| US7461483B2 (en) | 2006-09-11 | 2008-12-09 | Dana Innovations | Devices and methods for flangeless installations |
| US20100050538A1 (en) | 2006-12-04 | 2010-03-04 | Dana Innovations | Mounting system with component and cover plates |
| US20110067320A1 (en) | 2006-09-11 | 2011-03-24 | Raymond Lee Call | Wall-Mount Adjustment Systems And Methods |
| US8245453B2 (en) | 2006-09-11 | 2012-08-21 | Dana Innovatoions | Flush mounting apparatus and methods using component cover |
| EP2079739A2 (en) * | 2006-10-04 | 2009-07-22 | Pfizer Products Inc. | Pyrido[4,3-d]pyrimidin-4(3h)-one derivatives as calcium receptor antagonists |
| US20090064627A1 (en) | 2006-12-04 | 2009-03-12 | Scott Struthers | Flush Mount Panels With Interconnects |
| US8922518B2 (en) | 2007-12-20 | 2014-12-30 | Samsung Electronics Co., Ltd. | Mobile terminal having touch screen and function controlling method of the same |
-
2011
- 2011-07-22 EA EA201390158A patent/EA026142B1/ru not_active IP Right Cessation
- 2011-07-22 EP EP11809761.7A patent/EP2597095A4/en not_active Withdrawn
- 2011-07-22 BR BR112013001634A patent/BR112013001634A2/pt not_active IP Right Cessation
- 2011-07-22 CA CA2806258A patent/CA2806258A1/en not_active Abandoned
- 2011-07-22 US US13/811,755 patent/US9023858B2/en not_active Expired - Fee Related
- 2011-07-22 WO PCT/JP2011/066766 patent/WO2012011591A1/ja active Application Filing
- 2011-07-22 JP JP2012525456A patent/JPWO2012011591A1/ja active Pending
- 2011-07-22 CN CN2011800453754A patent/CN103119043A/zh active Pending
Patent Citations (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5925642A (en) | 1995-11-24 | 1999-07-20 | Byk Gulden Lomberg Chemische Fabrik Gmbh | Pyridopyrimidines |
| WO1998045285A1 (en) | 1997-04-04 | 1998-10-15 | Merck & Co., Inc. | Somatostatin agonists |
| WO1998044921A1 (en) | 1997-04-04 | 1998-10-15 | Merck & Co., Inc. | Somatostatin agonists |
| WO1999022735A1 (en) | 1997-10-30 | 1999-05-14 | Merck & Co., Inc. | Somatostatin agonists |
| WO2001014372A2 (en) | 1999-08-25 | 2001-03-01 | Takeda Chemical Industries, Ltd. | Oxazole and thiazole derivatives as neurotrophin production/secretion promoting agent |
| WO2001025228A1 (fr) | 1999-10-07 | 2001-04-12 | Tadeka Chemical Industries, Ltd. | Derives d'amines |
| WO2001038325A1 (en) | 1999-11-10 | 2001-05-31 | Takeda Chemical Industries, Ltd. | 5-membered n-heterocyclic compounds with hypoglycemic and hypolipidemic activity |
| WO2002006264A1 (fr) | 2000-07-13 | 2002-01-24 | Takeda Chemical Industries, Ltd. | Inhibiteurs de plaques riches en lipide |
| WO2002006234A1 (fr) | 2000-07-17 | 2002-01-24 | Takeda Chemical Industries, Ltd. | Derives de sulfonate, procede de production et utilisation de ces derives |
| WO2003042204A1 (fr) | 2001-10-19 | 2003-05-22 | Takeda Chemical Industries, Ltd. | Derive d'amine |
| WO2003059900A1 (fr) | 2002-01-11 | 2003-07-24 | Takeda Chemical Industries, Ltd. | Derives de coumarine, leur procede de production et d'utilisation |
| WO2003106435A1 (en) | 2002-06-18 | 2003-12-24 | Sankyo Company, Limited | Fused-ring pyrimidin-4(3h)-one derivatives, processes for the preparation and uses thereof |
| WO2004048363A1 (ja) | 2002-11-22 | 2004-06-10 | Takeda Pharmaceutical Company Limited | イミダゾール誘導体、その製造法および用途 |
| WO2005030740A1 (ja) | 2003-09-30 | 2005-04-07 | Takeda Pharmaceutical Company Limited | チアゾリン誘導体およびその用途 |
| WO2005058823A1 (ja) | 2003-12-17 | 2005-06-30 | Takeda Pharmaceutical Company Limited | ウレア誘導体、その製造法及び用途 |
| WO2005077905A1 (ja) * | 2004-02-13 | 2005-08-25 | Banyu Pharmaceutical Co., Ltd. | 縮環4-オキソ-ピリミジン誘導体 |
| WO2005091711A2 (en) | 2004-03-24 | 2005-10-06 | Orchid Chemicals & Pharmaceuticals Ltd | Novel condensed pyrimidones |
| WO2005113504A1 (en) | 2004-05-21 | 2005-12-01 | Takeda Pharmaceutical Company Limited | Cyclic amide derivatives, and their production and use as antithrombotic agents |
| JP2008504301A (ja) * | 2004-06-28 | 2008-02-14 | アムジェン エスエフ エルエルシー | 炎症性及び免疫調節性の障害及び疾患の予防及び治療に有用である、cxcr3受容体モジュレーターとしての縮合ピリミジン誘導体及びその組成物 |
| WO2007002701A2 (en) | 2005-06-27 | 2007-01-04 | Amgen Inc. | Anti-inflammatory aryl nitrile compounds |
| WO2008089307A2 (en) * | 2007-01-18 | 2008-07-24 | Lexicon Pharmaceuticals, Inc. | Delta 5 desaturase inhibitors for the treatment of pain, inflammation and cancer |
| JP2010166475A (ja) | 2009-01-19 | 2010-07-29 | Nec Corp | 移動通信ネットワークにおける通信設定方法及び移動通信端末 |
| WO2010087467A1 (en) * | 2009-01-27 | 2010-08-05 | Takeda Pharmaceutical Company Limited | Delta-5-desaturase inhibitors |
| JP2011122796A (ja) | 2009-12-14 | 2011-06-23 | Sanyo Electric Co Ltd | 断熱箱体及びその製造方法 |
Non-Patent Citations (10)
| Title |
|---|
| "Design of Molecules", vol. 7, 1990, HIROKAWA SHOTEN, article "IYAKUHIN NO KAIHATSU", pages: 163 - 198 |
| "Protective Groups in Organic Synthesis", 1980, JOHN WILEY AND SONS |
| ARZNEIMITTEL FORSCHUNG, vol. 56, no. 6, 2006, pages 377 - 381 |
| BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 19, no. 17, 2009, pages 5200 - 5204 |
| BLIGH; DYER, CAN. J. BIOCHEM. PHYSIOL., vol. 37, 1959, pages 911 |
| CHEMICAL AND PHARMACEUTICAL BULLETIN, vol. 43, 1995, pages 788 |
| INDIAN JOURNAL OF CHEMISTRY, SECTION B: ORGANIC CHEMISTRY INCLUDING MEDICINAL CHEMISTRY, vol. 39B, no. 3, 2000, pages 210 - 214 |
| JOURNAL OF THE INSTITUTION OF CHEMISTS, vol. 57, no. 4, 1985, pages 156 - 158 |
| See also references of EP2597095A4 * |
| SYNTHESIS, no. 23, 2007, pages 3678 - 3682 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017003723A1 (en) * | 2015-07-01 | 2017-01-05 | Crinetics Pharmaceuticals, Inc. | Somatostatin modulators and uses thereof |
| US9957267B2 (en) | 2015-07-01 | 2018-05-01 | Crinetics Pharmaceuticals, Inc. | Somatostatin modulators and uses thereof |
| US11028068B2 (en) | 2017-07-25 | 2021-06-08 | Crinetics Pharmaceuticals, Inc. | Somatostatin modulators and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2012011591A1 (ja) | 2013-09-09 |
| CA2806258A1 (en) | 2012-01-26 |
| US9023858B2 (en) | 2015-05-05 |
| BR112013001634A2 (pt) | 2016-05-24 |
| EA026142B1 (ru) | 2017-03-31 |
| US20130131050A1 (en) | 2013-05-23 |
| EA201390158A1 (ru) | 2013-08-30 |
| EP2597095A1 (en) | 2013-05-29 |
| EP2597095A4 (en) | 2014-10-01 |
| CN103119043A (zh) | 2013-05-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5657518B2 (ja) | 縮合複素環化合物 | |
| US9957251B2 (en) | Heterocyclic compound | |
| TW201249845A (en) | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors | |
| JP5722781B2 (ja) | 縮合複素環誘導体およびその用途 | |
| US9751878B2 (en) | Tetrahydronaphthyridine somatostatin receptor 5 antagonists | |
| US9023858B2 (en) | Substituted pyrido[2,3-d]pyrimidines as delta-5-desaturase inhibitors | |
| WO2010021381A1 (ja) | 縮合複素環誘導体およびその用途 | |
| WO2012011592A1 (ja) | 複素環化合物およびその用途 | |
| JP5361733B2 (ja) | ピリドオキサゼピン誘導体およびその用途 | |
| JP5722892B2 (ja) | 複素環化合物 | |
| WO2011006066A1 (en) | Cb receptor agonists | |
| US10519110B2 (en) | Heterocyclic compound | |
| WO2016121782A1 (ja) | スルホンアミド化合物 | |
| WO2010076884A1 (ja) | 新規縮合環化合物およびその用途 | |
| WO2011055770A1 (ja) | 縮合複素環化合物 | |
| JP5659159B2 (ja) | 縮合複素環誘導体およびその用途 | |
| JP5599802B2 (ja) | 縮合複素環誘導体およびその用途 | |
| WO2013147026A1 (ja) | 芳香環化合物 | |
| EP4046688A1 (en) | Heterocyclic compound | |
| TWI454473B (zh) | 稠環化合物及其用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180045375.4 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11809761 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012525456 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 224290 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2806258 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011809761 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13811755 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201390158 Country of ref document: EA |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112013001634 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112013001634 Country of ref document: BR Kind code of ref document: A2 Effective date: 20130122 |