WO2012011592A1 - 複素環化合物およびその用途 - Google Patents
複素環化合物およびその用途 Download PDFInfo
- Publication number
- WO2012011592A1 WO2012011592A1 PCT/JP2011/066767 JP2011066767W WO2012011592A1 WO 2012011592 A1 WO2012011592 A1 WO 2012011592A1 JP 2011066767 W JP2011066767 W JP 2011066767W WO 2012011592 A1 WO2012011592 A1 WO 2012011592A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- reaction
- optionally substituted
- substituted
- Prior art date
Links
- 0 **c(cc1)ccc1N(C(*)=NC(*)=C1*)C1=O Chemical compound **c(cc1)ccc1N(C(*)=NC(*)=C1*)C1=O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/47—One nitrogen atom and one oxygen or sulfur atom, e.g. cytosine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to a novel heterocyclic compound having excellent properties as a pharmaceutical and its use.
- the present invention has a delta-5-desaturase inhibitory action, exhibits various pharmacological actions based on suppressing the production of eicosanoids, has excellent physical properties such as good crystallinity and stability, Specific structures useful as prophylactic and therapeutic agents for eicosanoid-related diseases such as atherosclerosis, atherothrombosis, diabetes, obesity, asthma, fever, pain, cancer, rheumatism, osteoarthritis, and atopic dermatitis
- the present invention relates to a heterocyclic compound having a salt thereof, a salt thereof, a prodrug thereof, a use thereof, and the like.
- Eicosanoids such as prostaglandins, leukotrienes and thromboxanes are considered to play an important role in various diseases.
- inflammatory diseases such as arteriosclerosis, diabetes, obesity, asthma, rheumatism, osteoarthritis, and inflammatory pain
- the production pathway of inflammatory eicosanoids is increased, and is involved in the onset and exacerbation of the disease. It is believed that.
- drugs such as cyclooxygenase inhibitors and thromboxane A2 receptor antagonists that suppress the production of prostanoids have been clinically applied.
- the need for prophylactic / therapeutic drugs for inflammatory diseases is still high, and there is a strong demand for the development of powerful and few side effects.
- Patent Document 1 includes the following formula:
- A is CR 1 or N;
- Ar 1 and Ar 2 are each selected from phenyl, naphthyl and 5- to 10-membered heteroaryl, each of which may be substituted, each preferably from R A Optionally substituted with 0 to 6 independently selected substituents;
- R 1 is: (i) hydrogen, halogen, hydroxy, cyano, amino, nitro, aminocarbonyl, aminosulfonyl or —COOH; (ii) C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, (C 3 -C 8 cycloalkyl) C 0 -C 4 alkyl, C 2 -C 6 alkyl ester, C 1 -C 6 alkoxy, C 1 -C 6 alkylthio, C 3 -C 6 alkanone, C 1 -C 6 alkoxycarbonyl, mono- or di- (C 1 -C 6 alkyl) amino C 0
- R 3 is: (i) hydrogen; or (ii) C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 1 -C 6 alkyl sulfonyl, (C 3 -C 8 carbocycle) ) C 0 -C 4 alkyl or (4- to 8-membered heterocycle) C 0 -C 4 alkyl, each optionally substituted, each preferably selected independently from R B Optionally substituted with 1 substituent; R 4a and R 4b are independently: (i) hydrogen, halogen, cyano
- Rw is hydrogen or C 1 -C 6 alkyl; (iii) phenyl, naphthyl or 5- to 10-membered heteroaryl, each of which may be substituted, each of which is preferably substituted with 0 to 6 substituents independently selected from R A Good; or (iv) forming a fused 4- to 7-membered cycloalkyl or heterocycle optionally linked and substituted, preferably substituted with 0 to 4 substituents independently selected from R B May be; R 5 and R 6 are independently (i) hydrogen; or (ii) C 1 -C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, C 1 -C 6 alkanoyl, C 1 -C 6 alkylcarbonyl, C 1 -C 6 alkylsulfonyl, (C 3 -C 8 cycloalkyl) C 0 -C 4 alkyl, (4 to 8 membered heterocycle) C 0
- Non-Patent Document 1 Journal of Chemical Society, Perkin Transactions, 1 (2002), (6), 774-784 (Non-Patent Document 1) has the following formula
- Non-Patent Document 2 Heterocycles (1995), 40 (2), 787-800
- any compound is distinguished from the present compound and there is no description that it has a delta-5-desaturase inhibitory action.
- the object of the present invention is useful for the prevention and treatment of eicosanoid-related diseases such as atherosclerosis, atherothrombosis, diabetes, obesity, asthma, fever, pain, cancer, rheumatism, osteoarthritis, atopic dermatitis, etc. And providing a compound having excellent pharmacological action, physicochemical properties and the like.
- the heterocyclic compound represented by the following formula (I) has a delta-5-desaturase inhibitory action, exhibits various pharmacological actions based on suppressing the production of eicosanoids, Eicosanoid-related, such as atherosclerosis, atherothrombosis, diabetes, obesity, asthma, fever, pain, cancer, rheumatism, osteoarthritis, atopic dermatitis It was found for the first time that it is useful for the prevention and treatment of diseases. Based on this finding, the present inventors have conducted intensive research and have completed the present invention. That is, the present invention [1]
- the compound is represented by the formula (I):
- Ring A represents a 6-membered aromatic ring which may be further substituted;
- X 1 represents a bond or O,
- R 1 represents a C 1-6 alkyl group substituted with a 3- to 11-membered cyclic group, an optionally substituted 3- to 11-membered heterocyclic group, or an optionally substituted C 3-11 cycloalkyl group.
- R 2 represents a C 1-6 alkyl group which may be substituted with a hydrogen atom or a halogen atom
- R 3 is (1) Formula: —X 2 —R 5 (wherein X 2 represents O, S, SO 2 or NR 6 , R 5 represents an optionally substituted C 1-6 alkyl group or a substituted group) A 3 to 11-membered cyclic group which may be (2) an optionally substituted C 1-6 alkyl group; (3) an optionally substituted C 3-11 cycloalkyl group; or (4) an optionally substituted 3- to 11-membered non-aromatic heterocyclic group; R 4 represents an optionally substituted C 1-6 alkyl group or an optionally substituted 3- to 11-membered cyclic group, and R 6 represents a hydrogen atom or an optionally substituted C 1-6 alkyl group Indicates.
- X 1 is O
- R 4 is a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms
- Ring A is substituted with 1 to 3 halogen atoms, benzene which may be further substituted, 1 to 3 halogen atoms which may be further substituted with pyridine, or 1 or 2 halogen atoms.
- X 1 is O;
- R 1 is (1) a C 1-6 alkyl group substituted with a 5- to 7-membered monocyclic aromatic heterocyclic group, or (2) (A) a C 1-6 alkoxy group, (B) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms, (C) a halogen atom, and (D) a 5- or 6-membered heterocyclic group optionally substituted with 1 to 3 substituents selected from an oxo group;
- R 2 is a hydrogen atom or a methyl group;
- R 3 is (1) Formula: -X 2 -R 5 [Where: X 2 is O, S or NH, and R 5 is (A) a 3- to 8-membered non-aromatic heterocyclic group, (B) (i) a C 1-6 alkylsulfonyl group, (Ii) a hydroxy group, (Iii
- Compound (I) has a delta-5-desaturase inhibitory action, and is atherosclerosis, atherothrombosis, diabetes, obesity, asthma, fever, pain, cancer, rheumatism, osteoarthritis, atopic dermatitis It is useful for prevention and treatment of eicosanoid-related diseases such as, and has excellent medicinal effects. (Detailed description of the invention)
- halogen atom examples include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom.
- C 1-6 alkyl (group) examples include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, tert-pentyl, Examples include hexyl, 2,2-dimethylbutyl, 3,3-dimethylbutyl, 2-ethylbutyl and the like.
- C 1-6 alkoxy (group) examples include methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, sec-butoxy, tert-butoxy, pentyloxy, isopentyloxy, neopentyl And oxy, tert-pentyloxy, hexyloxy, 2-ethylbutoxy and the like.
- Examples of the “C 3-11 cycloalkyl (group)” in the present specification include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, cycloundecyl and the like.
- Examples of the “C 3-10 cycloalkyl (group)” in the present specification include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl and the like.
- Examples of the “C 6-14 aryl (group)” in the present specification include phenyl, naphthyl (eg, 1-naphthyl, 2-naphthyl), anthryl, phenanthryl and the like.
- Examples of the “C 7-13 aralkyl (group)” in the present specification include benzyl, phenethyl, naphthylmethyl, biphenylylmethyl and the like.
- C 2-6 alkenyl (group) examples include ethenyl, 1-propenyl, 2-propenyl, 2-methyl-1-propenyl, 1-butenyl, 2-butenyl, 3-butenyl, Examples include 3-methyl-2-butenyl, 1-pentenyl, 1-hexenyl, 3-hexenyl, 5-hexenyl and the like.
- Examples of the “6- to 10-membered aromatic hydrocarbon group” in the present specification include phenyl, 1-naphthyl, 2-naphthyl and the like.
- Examples of the “5- to 7-membered monocyclic aromatic heterocyclic group” in the present specification include, for example, an oxygen atom, a sulfur atom (the sulfur atom may be oxidized) in addition to a carbon atom as a ring-constituting atom, and And a 5- to 7-membered monocyclic aromatic heterocyclic group containing 1 to 4 heteroatoms selected from nitrogen atoms, specifically, furyl (eg, 2-furyl, 3-furyl), Thienyl (eg, 2-thienyl, 3-thienyl), pyridyl (eg, 2-pyridyl, 3-pyridyl, 4-pyridyl), pyrimidinyl (eg, 2-pyrimidinyl, 4-pyrimidinyl, 5-pyrimidinyl, 6-pyrimidinyl) , Pyridazinyl (eg, 3-pyridazinyl, 4-pyridazinyl), pyrazinyl (eg,
- Examples of the “8- to 11-membered condensed aromatic heterocyclic group” in the present specification include, for example, an oxygen atom, a sulfur atom (the sulfur atom may be oxidized) and a nitrogen atom as a ring constituent atom in addition to a carbon atom.
- Examples of the 5- to 7-membered monocyclic aromatic heterocyclic ring include rings corresponding to the aforementioned 5- to 7-membered monocyclic aromatic heterocyclic group.
- Examples of the C 6-10 aromatic hydrocarbon include a ring corresponding to the above C 6-10 aryl group.
- the “8 to 11-membered fused aromatic heterocyclic group” include quinolyl (eg, 2-quinolyl, 3-quinolyl, 4-quinolyl), isoquinolyl (eg, 1-isoquinolyl, 3-isoquinolyl, 4-isoquinolyl), quinazolyl (eg, 2-quinazolyl, 4-quinazolyl), quinoxalyl (eg, 2-quinoxalyl), benzofuryl (eg, 2-benzofuryl, 3-benzofuryl), benzothienyl (eg, 2-benzothienyl, 3-benzothienyl), benzoxazolyl (eg, 2-benzoxazolyl), benzothiazolyl (eg, 2-benzothiazolyl, 5-benzothiazolyl, 6-benzothiazolyl), benzimidazolyl (eg, benzimidazol-1-yl, Benzimidazol-2-yl, benzimidazol-5-yl
- Examples of the “3- to 11-membered non-aromatic cyclic hydrocarbon group” in the present specification include a C 3-11 cycloalkyl group, a C 3-11 cycloalkenyl group, a C 4-11 cycloalkadienyl group, and the like.
- a C 3-11 cycloalkyl group eg, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, cycloundecyl
- a C 3-11 cycloalkenyl group Examples, cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, cycloheptenyl, cyclooctenyl, cyclononenyl, cyclodecenyl, cycloundecenyl
- C 4-11 cycloalkadienyl groups eg, cyclobutadienyl, cyclopentadienyl, cyclohexenyl) Sadienyl, cyclo Phthaldienyl, cyclooctadienyl
- Examples of the “3- to 10-membered non-aromatic cyclic hydrocarbon group” in the present specification include a C 3-10 cycloalkyl group, a C 3-10 cycloalkenyl group, a C 4-10 cycloalkadienyl group, and the like.
- a C 3-10 cycloalkyl group eg, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl
- a C 3-10 cycloalkenyl group eg, cyclopropenyl, cyclobutenyl, cyclo Pentenyl, cyclohexenyl, cycloheptenyl, cyclooctenyl
- C 4-10 cycloalkadienyl groups eg, cyclobutadienyl, cyclopentadienyl, cyclohexadienyl, cycloheptadienyl, cyclooctadienyl, cyclononadienyl
- Cyclodecadienyl these groups and benze Fused Hajime Tamaki the ring and is fused (e.g., indanyl
- the “3- to 8-membered non-aromatic heterocyclic group” in the present specification includes, for example, a 3- to 8-membered (preferably 5- or 6-membered) saturated or unsaturated (preferably saturated) non-aromatic heterocyclic group.
- oxiranyl eg, 2-oxiranyl
- azetidinyl eg, 2-azetidinyl
- oxetanyl eg, 2-oxetanyl, 3-oxetanyl
- thietanyl eg, 2-thietanyl
- Pyrrolidinyl eg, 1-pyrrolidinyl, 2-pyrrolidinyl, 3-pyrrolidinyl
- tetrahydrofuryl eg, 2-tetrahydrofuryl, 3-tetrahydrofuryl
- thiolanyl eg, 2-thiolanyl
- piperidyl eg, 1-piperidyl, 2-piperidyl, 3-piperidyl, 4-piperidyl
- tetrahydropyranyl eg, 2-tetrahydropyranyl, 3 Tetrahydropyranyl, 4-tetrahydropyranyl
- thianyl eg, 2-
- Ring A represents a 6-membered aromatic ring which may be further substituted.
- Examples of the “6-membered aromatic ring” in the “optionally substituted 6-membered aromatic ring” represented by ring A include benzene, pyridine, pyridazine, pyrimidine, pyrazine, triazine and the like. Preferred are benzene, pyridine, pyridazine, pyrimidine, pyrazine and the like, more preferred are benzene, pyridine, pyrimidine and the like, and further preferred are benzene, pyridine and the like.
- the “6-membered aromatic ring” in the “optionally substituted 6-membered aromatic ring” represented by ring A may have 1 to 3 substituents at substitutable positions.
- substituents examples include: (1) a C 3-10 cycloalkyl group (eg, cyclopropyl, cyclohexyl); (2) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group optionally substituted with 1 to 3 halogen atoms, and (d) a C 6- optionally substituted with 1 to 3 substituents selected from halogen atoms.
- a C 3-10 cycloalkyl group eg, cyclopropyl, cyclohexyl
- substituents include: (1) a C 3-10 cycloalkyl group (eg, cyclopropyl, cyclohexyl); (2) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group optionally
- aryl groups eg, phenyl, naphthyl
- (3) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms, and (d) 4 to 7 which may be substituted with 1 to 3 substituents selected from halogen atoms.
- aromatic heterocyclic groups eg, thienyl, furyl, pyridyl, pyrazolyl, imidazolyl, tetrazolyl, oxazolyl, thiazolyl, oxadiazolyl, thiadiazolyl; (4) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms, (d) a halogen atom, and (e) a 4- to 7-membered non-aromatic heterocyclic group (eg, tetrahydrofuryl, morpholinyl, thiomorpholinyl, piperidyl) optionally substituted with 1 to 3 substituents selected from oxo groups , Pyrrolidinyl, piperazinyl); (5) (a) a C 1-6 alkyl group
- a 1-6 alkyl group (33) (a) a halogen atom, (b) a carboxy group, (c) a hydroxy group, (d) a C 1-6 alkoxy-carbonyl group, (e) a C 1-6 alkoxy group, and (f) a C 1-6 alkyl group optionally substituted with 1 to 3 substituents selected from amino groups optionally mono- or di-substituted.
- 2-6 alkenyl groups (eg, ethenyl, 1-propenyl); (34) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group, and (d) a C 7-13 aralkyl group (eg, benzyl) optionally substituted with 1 to 3 substituents selected from halogen atoms; Etc. When there are two or more substituents, each substituent may be the same or different.
- Ring A is preferably benzene, pyridine or the like which may be further substituted with 1 to 3 halogen atoms.
- ring A is preferably benzene, which may be further substituted with 1 to 3 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom), 1 to 3 (
- halogen atom eg, chlorine atom, fluorine atom
- pyridine which may be further substituted with 1 halogen atom (eg, chlorine atom, fluorine atom), or 1 or 2 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom)
- Benzene which may be further substituted with 1 to 3 (preferably 1) halogen atoms (eg, chlorine atom, fluorine atom).
- It is pyridine or pyrimidine which may be further substituted with 1 to 3 (preferably 1) halogen atom (eg, fluorine atom).
- X 1 represents a bond or O.
- X 1 is preferably O.
- R 1 represents a C 1-6 alkyl group substituted with a 3- to 11-membered cyclic group, an optionally substituted 3- to 11-membered heterocyclic group, or an optionally substituted C 3-11 cycloalkyl group.
- the “C 1-6 alkyl group” in the “C 1-6 alkyl group substituted with a 3- to 11-membered cyclic group” represented by R 1 has 1 to 3 “3- to 11-membered” at substitutable positions. Having a "cyclic group”.
- Examples of the “3- to 11-membered cyclic group” include a 6- to 10-membered aromatic hydrocarbon group, a 5- to 11-membered aromatic heterocyclic group (eg, a 5- to 7-membered monocyclic aromatic heterocyclic group, 8 To 11-membered condensed aromatic heterocyclic group), 3- to 10-membered non-aromatic cyclic hydrocarbon group, 3- to 8-membered non-aromatic heterocyclic group, and the like.
- R 1 '3 to C 1-6 alkyl group substituted by 11-membered cyclic group "preferably 5 to 7-membered monocyclic aromatic C 1-6 alkyl group substituted by a heterocyclic group More preferred is a C 1-6 alkyl group substituted with pyridyl or the like, still more preferred is methyl substituted with pyridyl or the like, and particularly preferred is methyl substituted with pyridyl or the like.
- the “3- to 11-membered heterocyclic group” in the “optionally substituted 3- to 11-membered heterocyclic group” represented by R 1 is, for example, a 5- to 11-membered aromatic heterocyclic group (eg, 5- to 7-membered).
- the “3- to 11-membered heterocyclic group” in the “optionally substituted 3- to 11-membered heterocyclic group” represented by R 1 may have 1 to 3 substituents at substitutable positions. Good.
- substituents examples include those similar to the substituent that the “6-membered aromatic ring” in the “optionally substituted 6-membered aromatic ring” represented by ring A may have. .
- the “3- to 11-membered heterocyclic group” is a non-aromatic heterocyclic group, it may have an oxo group as a substituent.
- each substituent may be the same or different.
- the “optionally substituted 3- to 11-membered heterocyclic group” represented by R 1 is preferably (1) C 1-6 alkoxy group (eg, methoxy), (2) a C 1-6 alkyl group (eg, methyl), and (3) a 5- or 6-membered heterocyclic group (eg, furyl, optionally substituted with 1 to 3 substituents selected from an oxo group) Pyridyl, pyrimidinyl, pyridazinyl, pyrazolyl, thiazolyl, oxazolyl, tetrahydropyranyl, morpholinyl, thiomorpholinyl and the like.
- the “optionally substituted 3- to 11-membered heterocyclic group” represented by R 1 is preferably (A) a C 1-6 alkoxy group (eg, methoxy), (B) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom), (C) a 5- or 6-membered heterocyclic ring optionally substituted with 1 to 3 (preferably 1 or 2) substituents selected from halogen atoms (eg, fluorine atoms) and (D) oxo groups Groups [eg, 5- or 6-membered aromatic heterocyclic groups (eg, furyl, pyridyl, pyrimidinyl, pyridazinyl, pyrazolyl, thiazolyl, oxazolyl, triazolyl, oxadiazolyl, imidazolyl, etc.); 5- or 6-membered non-aromatic heterocyclic groups
- C 3-11 cycloalkyl group in the "optionally substituted C 3-11 cycloalkyl group” represented by R 1, may have 1 to 3 substituents at substitutable positions Good.
- substituents examples include those similar to the substituent which the “6-membered aromatic ring” in the “optionally substituted 6-membered aromatic ring” represented by ring A may have, an oxo group, etc. Is mentioned. When there are two or more substituents, each substituent may be the same or different.
- R 1 is preferably (1) methyl substituted with pyridyl; (2) (A) a C 1-6 alkoxy group (eg, methoxy), (B) a C 1-6 alkyl group (eg, methyl), and (C) a 5- or 6-membered heterocyclic group (eg, furyl, optionally substituted with 1 to 3 substituents selected from an oxo group) Pyridyl, pyrimidinyl, pyridazinyl, pyrazolyl, thiazolyl, oxazolyl, tetrahydropyranyl, morpholinyl, thiomorpholinyl); Etc.
- R 1 is preferably (1) a C 1-6 alkyl group (eg, methyl) substituted with a 5- to 7-membered monocyclic aromatic heterocyclic group (eg, pyridyl), or (2) (A) a C 1-6 alkoxy group ( E.g.
- a C 1-6 alkyl group eg, methyl
- halogen atoms eg, fluorine atom
- C a halogen atom
- D a 5- or 6-membered heterocycle optionally substituted with 1 to 3 (preferably 1 or 2) substituents selected from an oxo group Groups [eg, 5- or 6-membered aromatic heterocyclic groups (eg, furyl, pyridyl, pyrimidinyl, pyridazinyl, pyrazolyl, thiazolyl, oxazolyl, triazolyl, oxadiazolyl, imidazolyl, etc.); 5- or 6-membered non-aromatic heterocyclic groups (eg, , Morpholinyl, dihydropyridyl, dihydropyridazinyl, tetrahydropyranyl, piperaziny
- R 2 represents a C 1-6 alkyl group which may be substituted with a hydrogen atom or a halogen atom.
- the “C 1-6 alkyl group optionally substituted with a halogen atom” for R 2 is preferably a C 1-6 alkyl group, and more preferably methyl.
- R 2 is preferably a hydrogen atom or a C 1-6 alkyl group (eg, methyl), more preferably a hydrogen atom or methyl.
- R 3 is (1) Formula: —X 2 —R 5 (wherein X 2 represents O, S, SO 2 or NR 6 , and R 5 represents an optionally substituted C 1-6 alkyl group or a substituted group) A 3 to 11-membered cyclic group which may be (2) an optionally substituted C 1-6 alkyl group; (3) an optionally substituted C 3-11 cycloalkyl group; or (4) an optionally substituted 3- to 11-membered non-aromatic heterocyclic group.
- R 6 represents a hydrogen atom or an optionally substituted C 1-6 alkyl group.
- the “C 1-6 alkyl group” in the “ optionally substituted C 1-6 alkyl group” for R 6 may have 1 to 3 substituents at substitutable positions.
- substituents for example, similar to the "C 1-6 alkyl group” substituent optionally possessed in the "optionally substituted C 1-6 alkyl group” represented by R 5 described later Things.
- each substituent may be the same or different.
- R 6 is preferably a hydrogen atom.
- X 2 in the "formula the group represented by -X 2 -R 5" is preferably, O, S or NH.
- R 3 Represented by R 3: "R 5" in the "formula the group represented by -X 2 -R 5" is an optionally substituted C 1-6 3 not may be alkyl or substituted to 11-membered A cyclic group is shown.
- C 1-6 alkyl group” in the “ optionally substituted C 1-6 alkyl group” for R 5 is substituted with 1 to 5 (preferably 1 to 3) substituents at substitutable positions. It may have a group.
- substituents examples include: (1) a C 3-10 cycloalkyl group (eg, cyclopropyl, cyclohexyl); (2) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group optionally substituted with 1 to 3 halogen atoms, and (d) a C 6- optionally substituted with 1 to 3 substituents selected from halogen atoms.
- a C 3-10 cycloalkyl group eg, cyclopropyl, cyclohexyl
- substituents include: (1) a C 3-10 cycloalkyl group (eg, cyclopropyl, cyclohexyl); (2) (a) a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group optionally
- aryl groups eg, phenyl, naphthyl
- (3) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms, and (d) 4 to 7 which may be substituted with 1 to 3 substituents selected from halogen atoms.
- aromatic heterocyclic groups eg, thienyl, furyl, pyridyl, pyrazolyl, imidazolyl, tetrazolyl, oxazolyl, thiazolyl, oxadiazolyl, thiadiazolyl; (4) (a) a C 1-6 alkyl group optionally substituted by 1 to 3 halogen atoms, (b) a hydroxy group, (c) a C 1-6 alkoxy group which may be substituted with 1 to 3 halogen atoms, (d) a halogen atom, and (e) a 4- to 7-membered non-aromatic heterocyclic group (eg, tetrahydrofuryl, morpholinyl, thiomorpholinyl, piperidyl) optionally substituted with 1 to 3 substituents selected from oxo groups , Pyrrolidinyl, piperazinyl); (5) (a) a C 1-6 alkyl group
- a C 1-6 alkoxy-carbonyl group (eg, methoxycarbonyl) optionally substituted by 1 to 3 substituents selected from: (8) a C 1-6 alkylsulfonyl group which may be substituted with 1 to 3 halogen atoms (eg, methylsulfonyl, ethylsulfonyl, isopropylsulfonyl); (9) a carbamoyl group which may be mono- or di-substituted with a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atoms; (10) a thiocarbamoyl group optionally mono- or disubstituted with a C 1-6 alkyl group optionally substituted with 1 to 3 halogen atoms; (11) a sulfamoyl group optionally mono- or di-substituted with a C 1-6 alkyl group which may be substituted with 1 to 3 halogen atom
- the “optionally substituted C 1-6 alkyl group” represented by R 5 is preferably (A) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl), (B) a hydroxy group, (C) a C 1-6 alkoxy group (eg, methoxy, ethoxy) optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom), and (D) a halogen atom (eg, fluorine atom)
- the “optionally substituted 3- to 11-membered cyclic group” represented by R 5 is, for example, a 6- to 10-membered aromatic hydrocarbon group, a 5- to 11-membered aromatic heterocyclic group (eg, a 5- to 7-membered monocyclic group). Cyclic aromatic heterocyclic group, 8- to 11-membered condensed aromatic heterocyclic group), 3- to 11-membered non-aromatic cyclic hydrocarbon group (preferably 3- to 10-membered non-aromatic cyclic hydrocarbon group), 3 to An 8-membered non-aromatic heterocyclic group and the like can be mentioned.
- the “3- to 11-membered cyclic group” in the “optionally substituted 3- to 11-membered cyclic group” represented by R 5 represents 1 to 5 (preferably 1 to 3) substitutions at substitutable positions. It may have a group.
- substituents examples include those similar to the substituent that the “6-membered aromatic ring” in the “optionally substituted 6-membered aromatic ring” represented by ring A may have. .
- the “3- to 11-membered cyclic group” is a non-aromatic cyclic hydrocarbon group or a non-aromatic heterocyclic group, it may have an oxo group as a substituent.
- each substituent may be the same or different.
- the “optionally substituted 3- to 11-membered cyclic group” for R 5 is preferably an optionally substituted 3- to 11-membered cyclic group or a C 3-11 cycloalkyl group, and more Preferably, (1) a 3- to 8-membered non-aromatic heterocyclic group, or (2) an optionally substituted C 3-11 cycloalkyl group, and more preferably (1) a 3- to 8-membered non-aromatic group Group C heterocyclic group (eg, oxetanyl) or (2) a C 3-11 cycloalkyl group (eg, cyclobutyl, cyclopentyl, cyclohexyl) optionally substituted with 1 to 5 halogen atoms (eg, fluorine atom) is there.
- a 3- to 8-membered non-aromatic heterocyclic group eg, oxetanyl
- C 3-11 cycloalkyl group eg, cyclobut
- R 3 Represented by R 3: "R 5" in the "formula -X 2 -R 5 in the group represented” is preferably, (1) a 3- to 8-membered non-aromatic heterocyclic group (eg, oxetanyl); or (2) (A) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl), (B) a hydroxy group, (C) C 1-6 alkoxy group (eg, methoxy), and (D) halogen atom (eg, fluorine atom) A C 1-6 alkyl group (eg, methyl, ethyl, propyl) optionally substituted by Etc.
- a 3- to 8-membered non-aromatic heterocyclic group eg, oxetanyl
- a C 1-6 alkylsulfonyl group eg, methylsulfonyl
- C C 1-6 alkoxy group
- R 3 in the "formula the group represented by -X 2 -R 5" is preferably, (1) a 3- to 8-membered non-aromatic heterocyclic group (eg, oxetanyl), (2) (A) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl), (B) a hydroxy group, (C) a C 1-6 alkoxy group (eg, methoxy, ethoxy) optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom), and (D) a halogen atom (eg, fluorine atom)
- a C 1-6 alkyl group eg, methyl, ethyl, propyl, isopropyl, isobutyl
- 1 to 5 selected from: (3) 1 to 5 (preferably 1 to A C 3-11 cycloalkyl group (eg, cyclo
- R 3 Represented by R 3 'wherein: the groups represented by -X 2 -R 5 "is preferably, X 2 is O, S or NH, and R 5 is (1) a 3- to 8-membered non-aromatic heterocyclic group (eg, oxetanyl); or (2) (A) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl), (B) a hydroxy group, (C) C 1-6 alkoxy group (eg, methoxy), and (D) halogen atom (eg, fluorine atom) A C 1-6 alkyl group (eg, methyl, ethyl, propyl) optionally substituted with 1 to 3 substituents selected from: A group represented by the formula: —X 2 —R 5 .
- R 5 is (1) a 3- to 8-membered non-aromatic heterocyclic group (eg, oxetanyl); or (2) (A)
- R 3 In another embodiment, represented by R 3 'wherein: the groups represented by -X 2 -R 5 "is preferably, X 2 is O, S or NH, and R 5 is (1) a 3- to 8-membered non-aromatic heterocyclic group (eg, oxetanyl), (2) (A) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl), (B) a hydroxy group, (C) a C 1-6 alkoxy group (eg, methoxy, ethoxy) optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom), and (D) a halogen atom (eg, fluorine atom) A C 1-6 alkyl group (eg, methyl, ethyl, propyl, isopropyl, isobutyl) optionally substituted with 1 to 5 substituents selected from: (3) 1 to 5 (preferably 1 to C 3-11 cyclo
- the “C 1-6 alkyl group” in the “ optionally substituted C 1-6 alkyl group” represented by R 3 has 1 to 5 (preferably 1 to 3) substituents at substitutable positions. You may have. Examples of the substituent, for example, those similar to the "C 1-6 alkyl group” substituent optionally possessed in the "optionally substituted C 1-6 alkyl group” represented by R 5 Can be mentioned. When there are two or more substituents, each substituent may be the same or different.
- the “optionally substituted C 1-6 alkyl group” represented by R 3 is preferably a C 1-6 alkyl group (eg, propyl).
- the “optionally substituted C 1-6 alkyl group” represented by R 3 is preferably (A) a hydroxy group, (B) a C 1-6 alkoxy group (eg, methoxy, ethoxy) optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom), (C) a halogen atom (eg, fluorine atom), and (D) a C 3-6 cycloalkyl group (eg, cyclopropyl) A C 1-6 alkyl group optionally substituted by 1 to 5 substituents selected from (eg, methyl, ethyl, propyl, butyl, sec-butyl) It is.
- R 3 "C 3-11 cycloalkyl group" of the “optionally substituted C 3-11 cycloalkyl group” may have 1 to 3 substituents at substitutable position .
- substituents include those similar to the substituent which the “6-membered aromatic ring” in the “optionally substituted 6-membered aromatic ring” represented by ring A may have, an oxo group, etc. Is mentioned. When there are two or more substituents, each substituent may be the same or different.
- the “optionally substituted C 3-11 cycloalkyl group” represented by R 3 is preferably (A) C 1-6 alkyl group (eg, methyl), and (B) halogen atom (eg, fluorine atom)
- a C 3-11 cycloalkyl group eg, cyclopropyl, cyclobutyl
- Examples of the “3- to 11-membered non-aromatic heterocyclic group” in the “optionally substituted 3- to 11-membered non-aromatic heterocyclic group” represented by R 3 include the above-mentioned “3- to 8-membered non-aromatic group”. And a group derived from a condensed ring in which the “heterocyclic group” and the above “3- to 8-membered non-aromatic heterocyclic group” are condensed with a benzene ring or the like.
- the “3- to 11-membered non-aromatic heterocyclic group” in the “optionally substituted 3- to 11-membered non-aromatic heterocyclic group” represented by R 3 has 1 to 3 substitutions at substitutable positions. It may have a group.
- substituents include those similar to the substituent which the “6-membered aromatic ring” in the “optionally substituted 6-membered aromatic ring” represented by ring A may have, an oxo group, etc. Is mentioned. When there are two or more substituents, each substituent may be the same or different.
- the “optionally substituted 3- to 11-membered non-aromatic heterocyclic group” represented by R 3 is preferably 1 to 3 (preferably 1 or 2) halogen atoms (eg, fluorine atom).
- a 3- to 11-membered non-aromatic heterocyclic group eg, tetrahydropyranyl, tetrahydrofuryl, pyrrolidinyl which may be substituted with
- R 3 is preferably (1) a group represented by the formula: -X 2 -R 5 X 2 is O, S or NH, and R 5 is (A) a 3- to 8-membered non-aromatic heterocyclic group (eg, oxetanyl); (B) (i) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl), (Ii) a hydroxy group, (Iii) a C 1-6 alkoxy group (eg, methoxy), and (iv) a halogen atom (eg, fluorine atom) A C 1-6 alkyl group (eg, methyl, ethyl, propyl) optionally substituted with 1 to 3 substituents selected from: (2) a C 1-6 alkyl group (eg, propyl); Etc.
- a C 1-6 alkyl group eg, methyl, ethyl, propyl
- Etc a group represented by the
- R 3 is preferably (1) Formula: -X 2 -R 5 [Where: X 2 is O, S or NH, and R 5 is (A) a 3- to 8-membered non-aromatic heterocyclic group (eg, oxetanyl), (B) (i) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl), (Ii) a hydroxy group, (Iii) a C 1-6 alkoxy group (eg, methoxy, ethoxy) optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom), and (iv) a halogen atom (eg, fluorine atom)
- a C 1-6 alkyl group eg, methyl, ethyl, propyl, isopropyl, isobutyl
- 1 to 5 preferably 1 to C 3-11 cyclo
- R 4 represents an optionally substituted C 1-6 alkyl group or an optionally substituted 3- to 11-membered cyclic group.
- the “C 1-6 alkyl group” in the “ optionally substituted C 1-6 alkyl group” represented by R 4 may have 1 to 3 substituents at substitutable positions.
- substituents for example, those similar to the "C 1-6 alkyl group” substituent optionally possessed in the "optionally substituted C 1-6 alkyl group” represented by R 5 Can be mentioned.
- each substituent may be the same or different.
- Substituted optionally a C 1-6 alkyl group represented by R 4, preferably, 1 to 3 halogen atoms (e.g., fluorine atom) with an optionally substituted C 1-6 alkyl group (Eg, ethyl).
- the “optionally substituted 3 to 11-membered cyclic group” in the “optionally substituted 3 to 11-membered cyclic group” represented by R 4 is, for example, a 6 to 10-membered aromatic hydrocarbon group, 5 to 11-membered aromatic heterocyclic group (eg, 5- to 7-membered monocyclic aromatic heterocyclic group, 8- to 11-membered condensed aromatic heterocyclic group), 3- to 10-membered non-aromatic cyclic hydrocarbon group, 3 to 8 Member non-aromatic heterocyclic group and the like.
- the “3- to 11-membered cyclic group” in the “optionally substituted 3- to 11-membered cyclic group” represented by R 4 may have 1 to 3 substituents at substitutable positions.
- substituents include those similar to the substituent which the “6-membered aromatic ring” in the “optionally substituted 6-membered aromatic ring” represented by ring A may have. .
- the “3- to 11-membered cyclic group” is a non-aromatic cyclic hydrocarbon group or a non-aromatic heterocyclic group, it may have an oxo group as a substituent.
- each substituent may be the same or different.
- R 4 is preferably a C 1-6 alkyl group optionally substituted, more preferably, 1 to 3 halogen atoms (e.g., fluorine atom) optionally substituted by C 1-6 An alkyl group (eg, ethyl);
- Preferred examples of compound (I) include the following compounds.
- Ring A is benzene or pyridine which may be further substituted with 1 to 3 halogen atoms;
- X 1 is O;
- R 1 is (1) methyl substituted with pyridyl; or (2) (A) a C 1-6 alkoxy group (eg, methoxy), (B) a C 1-6 alkyl group (eg, methyl), and (C) a 5- or 6-membered heterocyclic group (eg, furyl, optionally substituted with 1 to 3 substituents selected from an oxo group) Pyridyl, pyrimidinyl, pyridazinyl, pyrazolyl, thiazolyl, oxazolyl, tetrahydropyranyl, morpholinyl, thiomorpholinyl) Is;
- R 2 is a hydrogen atom or methyl;
- R 3 is (1) a group represented by the formula: -X 2 -R 5 X 2 is O, S or NH, and R
- Ring A is benzene optionally substituted with 1 to 3 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom), 1 to 3 (preferably 1) halogen Pyridine which may be further substituted with an atom (eg, chlorine atom, fluorine atom), or further substituted with 1 or 2 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom) 1 to 3 (preferably 1 benzene, which may be further substituted with 1 to 3 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom).
- halogen atom eg, chlorine atom, fluorine atom
- 1 to 3 preferably 1 halogen Pyridine which may be further substituted with an atom (eg, chlorine atom, fluorine atom), or further substituted with 1 or 2 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom) 1 to 3 (preferably 1 benzene, which may be further substituted with 1 to
- R 1 is (1) a C 1-6 alkyl group (eg, methyl) substituted with a 5- to 7-membered monocyclic aromatic heterocyclic group (eg, pyridyl) [preferably methyl substituted with pyridyl], (2) (A) a C 1-6 alkoxy group (eg, methoxy), (B) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom), (C) a halogen atom (eg, fluorine atom), and (D) a 5- or 6-membered heterocycle optionally substituted with 1 to 3 (preferably 1 or 2) substituents selected from an oxo group Groups [eg, 5- or 6-membered aromatic heterocyclic groups (eg, furyl, pyridy
- Ring A is benzene optionally substituted with 1 to 3 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom), 1 to 3 (preferably 1) halogen Pyridine which may be further substituted with an atom (eg, chlorine atom, fluorine atom), or further substituted with 1 or 2 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom) 1 to 3 (preferably 1 benzene, which may be further substituted with 1 to 3 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom).
- halogen atom eg, chlorine atom, fluorine atom
- 1 to 3 preferably 1 halogen Pyridine which may be further substituted with an atom (eg, chlorine atom, fluorine atom), or further substituted with 1 or 2 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom) 1 to 3 (preferably 1 benzene, which may be further substituted with 1 to
- a halogen atom eg, fluorine atom
- X 1 is O
- R 1 is (A) a C 1-6 alkoxy group (eg, methoxy), (B) a C 1-6 alkyl group (eg, methyl) optionally substituted with 1 to 3 halogen atoms (eg, fluorine atom), (C) a halogen atom (eg, fluorine atom), and (D) a 5- or 6-membered heterocycle optionally substituted with 1 to 3 (preferably 1 or 2) substituents selected from an oxo group Groups [eg, 5- or 6-membered aromatic heterocyclic groups (eg, furyl, pyridyl, pyrimidinyl, pyridazinyl, pyrazolyl, thiazolyl, oxazolyl, triazolyl, oxadiazolyl, imidazolyl, etc.); 5- or 6-member
- Ring A is benzene optionally substituted with 1 to 3 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom), 1 to 3 (preferably 1) halogen Pyridine which may be further substituted with an atom (eg, chlorine atom, fluorine atom), or further substituted with 1 or 2 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom) 1 to 3 (preferably 1 benzene, which may be further substituted with 1 to 3 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom).
- halogen atom eg, chlorine atom, fluorine atom
- 1 to 3 preferably 1 halogen Pyridine which may be further substituted with an atom (eg, chlorine atom, fluorine atom), or further substituted with 1 or 2 (preferably 1) halogen atom (eg, chlorine atom, fluorine atom) 1 to 3 (preferably 1 benzene, which may be further substituted with 1 to
- a halogen atom eg, fluorine atom
- X 1 is O
- R 1 is a morpholinyl group or a pyrazolyl group optionally substituted by 1 to 3 C 1-6 alkyl groups (eg, methyl)
- R 2 is a hydrogen atom or a C 1-6 alkyl group (eg, methyl);
- R 3 is (1) Formula: -X 2 -R 5 [Where: X 2 is O, S or NH, and R 5 is (A) a 3- to 8-membered non-aromatic heterocyclic group (eg, oxetanyl), (B) (i) a C 1-6 alkylsulfonyl group (eg, methylsulfonyl), (Ii) a hydroxy group, (Iii) a C 1-6 alkoxy group (eg, methoxy, ethoxy) optionally substituted with 1 to 3 halogen atom
- the salt of the compound represented by formula (I) is preferably a pharmacologically acceptable salt.
- a salt with an inorganic base examples include a salt with an inorganic base, a salt with an organic base, and a salt with an inorganic acid.
- the salt with an inorganic base include alkali metal salts such as sodium salt and potassium salt; alkaline earth metal salts such as calcium salt and magnesium salt; aluminum salt; ammonium salt and the like.
- the salt with an organic base include trimethylamine, triethylamine, pyridine, picoline, ethanolamine, diethanolamine, triethanolamine, tromethamine [tris (hydroxymethyl) methylamine], tert-butylamine, cyclohexylamine, benzylamine, And salts with dicyclohexylamine, N, N-dibenzylethylenediamine and the like.
- salt with inorganic acid examples include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like.
- salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, phthalic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, and benzenesulfonic acid And salts with p-toluenesulfonic acid and the like.
- salts with basic amino acids include salts with arginine, lysine, ornithine and the like.
- salt with acidic amino acid include salts with aspartic acid, glutamic acid and the like.
- Compound (I) may be labeled with an isotope (eg, 3 H, 14 C, 35 S, 125 I) or the like.
- Compound (I) may be a solvate (for example, an anhydride) or a solvate (for example, a hydrate).
- a deuterium converter obtained by converting 1 H into 2 H (D) is also encompassed in compound (I).
- Compound (I) may be crystalline or amorphous.
- compound (I) is a crystal, it is included in compound (I) whether it is a single crystal form or a crystal form mixture.
- the crystal can be produced by crystallization by applying a crystallization method known per se.
- the melting point is measured using, for example, a trace melting point measuring device (Yanako, MP-500D type or Buchi, B-545 type) or a DSC (differential scanning calorimetry) apparatus (SEIKO, EXSTAR6000). Mean melting point.
- the melting point may vary depending on measurement equipment, measurement conditions, and the like.
- the crystal in the present specification may be a crystal having a value different from the melting point described in the present specification as long as it is within a normal error range.
- Compound (I) may be a pharmaceutically acceptable cocrystal or cocrystal salt.
- co-crystals or co-crystal salts are two or more unique at room temperature, each having different physical properties (eg structure, melting point, heat of fusion, hygroscopicity, solubility and stability). It means a crystalline substance composed of a simple solid.
- the cocrystal or cocrystal salt can be produced according to a cocrystallization method known per se.
- the crystals of the present invention are excellent in physicochemical properties (eg, melting point, solubility, stability) and biological properties (eg, pharmacokinetics (absorbability, distribution, metabolism, excretion), expression of medicinal properties), and are extremely useful as pharmaceuticals. Useful.
- Compound (I) may be a prodrug.
- the prodrug of compound (I) is a compound that is converted to compound (I) by a reaction with an enzyme or gastric acid under physiological conditions in vivo, that is, compound (I) that undergoes oxidation, reduction, hydrolysis, etc. enzymatically. ), A compound that undergoes hydrolysis or the like due to gastric acid or the like and changes to compound (I).
- the prodrug of compound (I) is a compound that changes to compound (I) under physiological conditions as described in Hirokawa Shoten 1990, “Drug Development”, Volume 7, Molecular Design, pages 163 to 198. It may be.
- each raw material compound may form a salt as long as it does not inhibit the reaction, and examples of the salt include those exemplified as the salt of the compound represented by the aforementioned formula (I). Used.
- the raw material compound can be easily obtained commercially, or can be produced according to a method known per se or a method analogous thereto, unless a specific production method is described.
- the solvent used in the reaction in each of the following schemes is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material to some extent.
- aromatic hydrocarbons such as benzene, toluene and xylene; hexane Aliphatic hydrocarbons such as heptane; ethers such as diethyl ether, diisopropyl ether, tert-butyl methyl ether, tetrahydrofuran, dioxane and 1,2-dimethoxyethane; ketones such as acetone and 2-butanone; acetonitrile, pro Nitriles such as pionitrile; esters such as ethyl acetate, isopropyl acetate and tert-butyl acetate; amides such as N, N-dimethylformamide, N, N-dimethylacetamide and 1-methyl-2-pyrrolidinone; 3-dimethyl-2-imidazolidinone, etc.
- Alcohols such as methanol, ethanol, isopropanol and tert-butanol; halogenated hydrocarbons such as chloroform, dichloromethane, 1,2-dichloroethane and carbon tetrachloride; sulfoxides such as dimethyl sulfoxide; water and the like It is done. These solvents may be mixed and used at an appropriate ratio.
- the reaction temperature is usually carried out below the boiling point of the above-mentioned solvent at ⁇ 100 to 250 ° C., but depending on the case, the reaction may be carried out at a temperature above the boiling point of the solvent using pressure resistant reaction conditions or the like.
- the reaction time is usually 0.5 to 100 hours.
- Rb represents an optionally substituted C 1-6 alkyl group or an optionally substituted 3- to 11-membered cyclic group, and other symbols are as defined above.
- compound (2) obtained in scheme 4 described later is converted into compound (3), and then compound (I) is obtained by a substitution reaction.
- Compound (3) is produced by an S-alkylation reaction using the base of compound (2) and various alkylating agents. Specifically, the reaction is performed using 1.0 to 10.0 mol, preferably 1.0 to 5.0 mol, and 1.0 to 20.0 mol, preferably 1.0 to 10.0 mol, of an S-alkylating agent with respect to 1 mol of compound (2).
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium hydrogen carbonate, sodium carbonate and potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide, hydrogen Examples thereof include metal hydrides such as sodium hydride and potassium hydride, and organic bases such as triethylamine, imidazole and formamidine.
- S-alkylating agent examples include various alkyl halides such as alkyl chloride, alkyl bromide, alkyl iodide and their derivatives, sulfonate esters such as p-toluenesulfonic acid ester and methylsulfonic acid ester, dimethyl sulfate, etc. And the like.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds, but for example, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane and the like.
- Halogenated hydrocarbons alcohols such as methanol, ethanol, propanol and 1,1-dimethylethanol, aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, N , N-dimethylformamide, amides such as N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, water or a mixed solvent thereof are preferable.
- the reaction time is usually 15 minutes to 60 hours, preferably 15 minutes to 24 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the obtained compound (3) can be used in the next reaction as a reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- Compound (4) is produced by an oxidation reaction of compound (3).
- hydrogen peroxide oxone (registered trademark), peracids such as peracetic acid, perbenzoic acid, m-chloroperbenzoic acid and the like, oxoacids such as hypochlorous acid and periodic acid and their salts, chromium, etc.
- peracids such as peracetic acid, perbenzoic acid, m-chloroperbenzoic acid and the like
- oxoacids such as hypochlorous acid and periodic acid and their salts, chromium, etc.
- metal oxo acids such as acids and salts thereof or other oxidizing agents, and are used in an amount of 1.0 to 30.0 mol, preferably 1.0 to 3.0 mol, per 1 mol of compound (3).
- This reaction is preferably carried out using a solvent inert to the reaction.
- Such a solvent is not particularly limited as long as the reaction proceeds, but alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc. , Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide, N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, Organic acids such as trifluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the product is obtained as a single compound of either compound (4a) and compound (4b) or a mixture thereof, and can be used as it is in the reaction solution or as a crude product for the next reaction. It can also be isolated from the mixture and can be easily purified by separation means such as washing, recrystallization, distillation, chromatography and the like.
- Compound (I) can be produced by a substitution reaction of compound (3) or compound (4) using a base and a nucleophile corresponding to R 3 . Specifically, the reaction is performed using 1.0 to 20.0 mol, preferably 1.0 to 10.0 mol, and 1.0 to 100.0 mol, preferably 1.0 to 10.0 mol, of a nucleophile with respect to 1 mol of compound (3) or compound (4).
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen Metal hydrides such as potassium hydride, organic amines such as triethylamine, pyridine, diisopropylethylamine, 1,8-diazabicyclo [5.4.0] undec-7-ene, and the like.
- inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen Metal hydrides such as potassium hydride, organic amines such as triethylamine, pyridine, diisopropylethylamine, 1,8-diazabicyclo [5.4.0] unde
- nucleophile examples include alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, 2,2,2-trifluoroethanol, 2,2,3,3,3-pentafluoro-1-propanol, Various phenol derivatives having an aromatic hydroxyl group, organic thiols such as ethanethiol and thioglycolic acid amide, various aromatic thiol derivatives such as thiophenol, organic bases such as methylamine, ethylamine and propylamine, various fragrances such as aniline Group amines, active methylene compounds such as ⁇ -hydrogen-containing carbonyl compounds, organic Grignard reagents (n-propylmagnesium bromide, n-butylmagnesium bromide), organic lithium reagents (n-propyllithium, n-butyllithium), etc.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, 2,2,2-
- a base can be used as a nucleophile.
- a base is not required for the reaction, it can be produced without using a base.
- This reaction is preferably carried out without solvent or using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol and propanol
- aromatic hydrocarbons such as benzene and toluene
- saturated hydrocarbons such as cyclohexane and hexane
- Ethers such as dioxane and 1,2-dimethoxyethane
- amides such as N, N-dimethylformamide and N, N-dimethylacetamide
- nitriles such as acetonitrile and propionitrile
- ketones such as acetone and methyl ethyl ketone
- a solvent such as water or a mixed solvent thereof is preferred.
- the reaction time is usually 10 minutes to 24 hours, preferably 10 minutes to 12 hours.
- the reaction temperature is usually -100 to 150 ° C, preferably -78 to 100 ° C.
- the obtained compound (I) can be used in the next reaction as a reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- compound (5) obtained in scheme 5 described later is subjected to an O-alkylation reaction using a base and an alkylating agent corresponding to R 5 to obtain compound (Ia) [in compound (I), R 3 Is a compound of the formula: —X 2 —R 5 and X 2 is an oxygen atom].
- O-alkyl is used by using about 1.0 to 3.0 mol, preferably 1.0 to 2.0 mol, and about 1.0 to 20.0 mol, preferably about 1.0 to 10.0 mol, of an O-alkylating agent for 1 mol of compound (5). To do.
- Examples of the base include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, cesium carbonate, and metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, and the like. It is done.
- Examples of the O-alkylating agent include various halogenated alkyls such as methyl iodide, ethyl iodide, and propyl iodide, alkyl sulfates such as dimethyl sulfate and diethyl sulfate, methyl p-toluenesulfonate, methyl methylsulfonate, and the like.
- alkyl sulfonic acid esters This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- a solvent inert for example, alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc. , Aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, and sulfoxides such as dimethyl sulfoxide
- a solvent such as a solvent or a mixed solvent thereof is preferable.
- the reaction time is usually 30 minutes to 60 hours, preferably 30 minutes to 24 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the obtained compound (Ia) can be isolated from the reaction mixture according to a conventional method, and can be easily purified by separation means such as washing, recrystallization, distillation, chromatography and the like.
- R 3 ′ is an optionally substituted C 1-6 alkyl group, an optionally substituted C 3-11 cycloalkyl group, or an optionally substituted 3- to 11-membered non-aromatic heterocycle And other symbols are as defined above.
- compound (Ib) [C 1-6 alkyl optionally substituted with R 3 in compound (I) is obtained by reacting compound (6) obtained in scheme 13 and compound (7), which will be described later.
- about 1.0 to 10.0 mol, preferably about 1.0 to 5.0 mol, of compound (7) is used per 1 mol of compound (6).
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, acetonitrile
- a nitrile such as propionitrile
- a solvent such as sulfoxide such as dimethyl sulfoxide, or a mixed solvent thereof is preferable.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 200 ° C, preferably 0 to 150 ° C.
- This reaction may be performed by adding a base.
- the base include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen Metal hydrides such as potassium hydride, organic amines such as triethylamine, pyridine, diisopropylethylamine, 1,8-diazabicyclo [5.4.0] undec-7-ene, and the like.
- the obtained compound (Ib) can be used in the next reaction as a reaction liquid or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- Compound (7) can be easily obtained as a commercially available product, or can be produced according to a method known per se or a method analogous thereto.
- Ra represents a C 1-6 alkyl group, and other symbols are as defined above.
- the compound (8) obtained in Scheme 6 or 7 described later and the compound (9) obtained in Scheme 11 are subjected to a cyclization reaction in the presence of a base, and the starting compound of the above reaction (Scheme 1) is used.
- a certain compound (2) is obtained.
- the compound (9) is used in an amount of about 1.0 to 5.0 mol, preferably about 1.0 to 2.0 mol, per 1 mol of the compound (8).
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen
- inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen
- metal hydrides such as potassium hydride
- organic bases such as triethylamine, imidazole and formamidine
- This reaction is preferably carried out using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride and 1,2-dichloroethane
- aromatic hydrocarbons such as benzene and toluene
- tetrahydrofuran Ethers such as dioxane, 1,2-dimethoxyethane
- amides such as N, N-dimethylformamide, N, N-dimethylacetamide
- nitriles such as acetonitrile and propionitrile
- sulfoxides such as dimethyl sulfoxide
- hexa Phosphoric amides such as methylphosphoric triamide or a mixed solvent thereof are preferred.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the obtained compound (2) can be used as it is in the reaction solution or as a crude product for the next reaction, but can be isolated from the reaction mixture according to a conventional method, and can be washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- the compound (8) obtained in Scheme 6 or 7 described later and the compound (10) obtained in Scheme 11 are subjected to a ring-closing reaction in the presence of a base, and the starting compound of the reaction (Scheme 2) To obtain the compound (5).
- the compound (10) is used in an amount of about 1.0 to 5.0 mol, preferably about 1.0 to 2.0 mol, per 1 mol of the compound (8).
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen
- inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen
- metal hydrides such as potassium hydride
- organic bases such as triethylamine, imidazole and formamidine
- This reaction is preferably carried out using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride and 1,2-dichloroethane
- aromatic hydrocarbons such as benzene and toluene
- tetrahydrofuran Ethers such as dioxane, 1,2-dimethoxyethane
- amides such as N, N-dimethylformamide, N, N-dimethylacetamide
- nitriles such as acetonitrile and propionitrile
- sulfoxides such as dimethyl sulfoxide
- hexa Phosphoric amides such as methylphosphoric triamide or a mixed solvent thereof are preferred.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the obtained compound (5) can be used as it is in the reaction solution or as a crude product for the next reaction, but can be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- Scheme 6 carries out a dehydration condensation reaction of the compound (11) obtained in Scheme 8, 9 or 10 described later with ammonia or an ammonium salt, and the compound (8) which is the starting compound of the reaction (Schemes 4 and 5) is obtained. To get.
- This reaction can be produced according to a method known per se, for example, the method described in Journal of Medicinal Chemistry (J. Med. Chem), 41, 3186 (1998), or a method analogous thereto.
- ammonia or ammonium salt is used in an amount of about 1.0 to 50.0 mol, preferably about 1.0 to 10.0 mol, per 1 mol of compound (11).
- the ammonium salt include ammonium formate and ammonium acetate.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- a solvent inert such as halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons alcohols such as methanol, ethanol, propanol and 1,1-dimethylethanol, aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, N, Amides such as N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, organic acids such as acetic acid and trifluoroacetic acid, or a mixed solvent thereof Etc. are preferable.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 200 ° C, preferably 0 to 150 ° C.
- an acid catalyst can be used.
- mineral acids such as hydrochloric acid and sulfuric acid
- Lewis acids such as boron trichloride and boron tribromide, acetic acid, trifluoroacetic acid, p-toluenesulfone Organic acids such as acids can be mentioned.
- the obtained compound (8) can be used as it is in the reaction solution or as a crude product for the next reaction, but can be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- Rc represents a C 1-6 alkyl group, and other symbols are as defined above.
- Scheme 7 shows that compound (8a) [compound (8) in which R 1 has a substituent is obtained by a substitution reaction of compound (13) with compound (12), which is a raw material compound of the above reactions (Schemes 4 and 5). And a compound having a 5- to 6-membered cyclic amino group].
- This reaction can be produced according to a method known per se, for example, the method described in Journal of Medicinal Chemistry (J. Med. Chem), 26, 1650 (1983), or a method analogous thereto.
- the compound (13) is used in an amount of about 1.0 to 10.0 mol, preferably about 1.0 to 5.0 mol, per 1 mol of the compound (12).
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- a solvent inert such as halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons alcohols such as methanol, ethanol, propanol and 1,1-dimethylethanol, aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, N, Preference is given to solvents such as amides such as N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, and mixed solvents thereof.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- This reaction may be performed by adding an acid.
- the acid include mineral acids such as hydrochloric acid and sulfuric acid, Lewis acids such as boron trichloride and boron tribromide, and organic acids such as acetic acid, trifluoroacetic acid and p-toluenesulfonic acid.
- an inorganic acid salt or organic acid salt of the compound (12) the reaction is carried out in the presence of an appropriate base such as potassium carbonate, sodium hydrogen carbonate, sodium hydroxide, triethylamine, etc. Good.
- the obtained compound (8a) can be used in the next reaction as a reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- compound (12) and compound (13) commercially available ones can be easily obtained, or they can be produced according to a method known per se or a method analogous thereto.
- Rd represents a C 1-6 alkyl group, and other symbols are as defined above.
- compound (14) and compound (15) are subjected to a Claisen condensation reaction using a base to obtain compound (11) which is a raw material compound of the reaction (Scheme 6).
- the compound (15) is used in an amount of about 1.0 to 10.0 mol, preferably about 1.0 to 5.0 mol, per 1 mol of the compound (14).
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen
- inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen
- metal hydrides such as potassium hydride
- organic bases such as triethylamine, imidazole and formamidine
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons alcohols such as methanol, ethanol, propanol and 1,1-dimethylethanol, aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, N
- solvents such as amides such as N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, and mixed solvents thereof.
- compound (15) may be used as a solvent.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the obtained compound (11) can be used in the next reaction as the reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- compound (14) and compound (15) commercially available ones can be easily obtained, or they can be produced according to a method known per se or a method analogous thereto.
- Scheme 9 is an alternative method in which compound (16) and compound (15) are subjected to a condensation reaction using a base to obtain compound (11) which is a raw material compound of the reaction (Scheme 6).
- the compound (15) is used in an amount of about 1.0 to 10.0 mol, preferably about 1.0 to 5.0 mol, per 1 mol of the compound (16).
- the base include alkyllithiums such as n-butyllithium, sec-butyllithium, tert-butyllithium, etc., and they are used in an equivalent amount or a slight excess with respect to 1 mol of compound (15).
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually ⁇ 100 to 100 ° C., preferably ⁇ 78 to 25 ° C.
- the obtained compound (11) can be used in the next reaction as the reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- Compound (16) can be easily obtained as a commercially available product, or can be produced according to a method known per se or a method analogous thereto.
- M represents potassium or sodium, and other symbols are as defined above.
- Scheme 10 is a reaction of compound (17) with compound (18), followed by condensation reaction with compound (19), followed by decarboxylation to give compound (11) as the starting compound of the above reaction (Scheme 6). Another way to get.
- This reaction can be produced according to a method known per se, for example, the method described in Journal of Medicinal Chemistry (J. Med. Chem), 44, 3978 (2001), or a method analogous thereto.
- 1.0 to 10.0 mol preferably about 1.0 to 5.0 mol, about 1.0 to 10.0 mol, preferably about 1.0 to 5.0 mol of magnesium chloride, and about 1.0 to 5.0 mol of compound (18) with respect to 1 mol of compound (17), and compound (19)
- About 1.0-10.0 mol, preferably about 1.0-5.0 mol is used.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Solvents such as fluorinated hydrocarbons, aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, nitriles such as acetonitrile and propionitrile, and sulfoxides such as dimethyl sulfoxide Or a mixed solvent thereof or the like is preferable.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the obtained compound (11) can be used in the next reaction as the reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- compound (17), compound (18) and compound (19) commercially available ones can be easily obtained, or they can be produced according to a method known per se or a method analogous thereto.
- Scheme 11 is a compound (9) that is a raw material compound of scheme 4 by thioisocyanate formation of compound (20), and compound (10) that is a raw material compound of scheme 5 is obtained by isocyanate formation of compound (20). It is.
- This reaction can be produced according to a method known per se, for example, the method described in Journal of Medicinal Chemistry (J. Med. Chem), 44, 3978 (2001), or a method analogous thereto.
- Compound (9) can be produced by thioisocyanate formation of compound (20). Specifically, the reaction is carried out using about 1.0 to 5.0 mol, preferably about 1.0 to 2.0 mol, of the thioisocyanating agent per 1 mol of compound (20).
- thioisocyanating agent examples include thiophosgene, 1,1′-thiocarbonyldi-2 (1H) -pyridone, di- (2-pyridyl) thionocarbonate, 1,1′-thiocarbonyldiimidazole and the like. It is done.
- thiophosgene is used in this reaction, the reaction can be carried out in the presence of a deoxidizer for the purpose of removing the released hydrogen halide from the reaction system.
- Examples of such a deoxidizer include inorganic bases such as sodium carbonate, potassium carbonate, and sodium bicarbonate, aromatic amines such as pyridine and lutidine, triethylamine, tripropylamine, tributylamine, cyclohexyldimethylamine, 4- Tertiary amines such as dimethylaminopyridine, N, N-dimethylaniline, N-methylpiperidine, N-methylpyrrolidine, and N-methylmorpholine are desirable. This reaction is preferably carried out using a solvent inert to the reaction.
- Such a solvent is not particularly limited as long as the reaction proceeds, but for example, halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, alcohols such as methanol, ethanol, propanol, benzene, Aromatic hydrocarbons such as toluene, saturated hydrocarbons such as cyclohexane and hexane, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide , Nitriles such as acetonitrile and propionitrile, ketones such as acetone and methyl ethyl ketone, sulfoxides such as dimethyl sulfoxide, solvents such as water or a mixed solvent thereof are preferable.
- halogenated hydrocarbons such as dichloromethane, chloroform
- the reaction time is usually 10 minutes to 60 hours, preferably 15 minutes to 12 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the obtained compound (9) can be used in the next reaction as the reaction mixture or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, and separated by recrystallization, distillation, chromatography, etc. It can be easily purified by means.
- Compound (10) can be produced by converting compound (20) to isocyanate. Specifically, the reaction is carried out using about 1.0 to 5.0 moles, preferably about 1.0 to 2.0 moles, of an isocyanate agent per mole of compound (20).
- the isocyanate agent include triphosgene, 1,1′-carbonyldi-2 (1H) -pyridone, di-2-pyridyl carbonate, 1,1′-carbonyldiimidazole and the like.
- triphosgene is used in this reaction, the reaction can be carried out in the presence of a deoxidizing agent for the purpose of removing the released hydrogen halide from the reaction system.
- Examples of such a deoxidizer include basic salts such as sodium carbonate, potassium carbonate and sodium hydrogen carbonate, aromatic amines such as pyridine and lutidine, triethylamine, tripropylamine, tributylamine, cyclohexyldimethylamine, 4- Tertiary amines such as dimethylaminopyridine, N, N-dimethylaniline, N-methylpiperidine, N-methylpyrrolidine, and N-methylmorpholine are desirable. This reaction is preferably carried out using a solvent inert to the reaction.
- Such a solvent is not particularly limited as long as the reaction proceeds, but for example, halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, alcohols such as methanol, ethanol, propanol, benzene, Aromatic hydrocarbons such as toluene, saturated hydrocarbons such as cyclohexane and hexane, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide , Nitriles such as acetonitrile and propionitrile, ketones such as acetone and methyl ethyl ketone, sulfoxides such as dimethyl sulfoxide, solvents such as water or a mixed solvent thereof are preferable.
- halogenated hydrocarbons such as dichloromethane, chloroform
- the reaction time is usually 10 minutes to 60 hours, preferably 15 minutes to 12 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the obtained compound (10) can be used in the next reaction as the reaction solution or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, and separated by recrystallization, distillation, chromatography, etc. It can be easily purified by means.
- Compound (20) may be easily commercially available, or can be produced according to the method shown in Scheme 12 or a method analogous thereto.
- Y represents a leaving group (eg, halogen atom, 4-tolylsulfonyloxy, etc.), and other symbols are as defined above. ]
- compound (20a) [a compound in which X 1 is O in compound (20)] is converted from compound (21) or compound (22) via compound (23) as the starting compound of scheme 11 To get.
- Compound (23) can be produced by a substitution reaction with compound (21) using a phenol or alcohol corresponding to R 4 as a nucleophile under basic conditions. Specifically, about 1.0 to 10.0 mol, preferably about 1.0 to 5.0 mol, and about 1.0 to 100.0 mol, preferably 1.0 to 2.0 mol, of phenols or alcohols corresponding to R 4 with respect to 1 mol of compound (21). Is used.
- the base include inorganic bases such as sodium carbonate and potassium carbonate, and metal hydrides such as sodium hydride and potassium hydride.
- phenols or alcohols examples include ethanol, 2,2,2-trifluoroethanol, cyclopropylmethanol, 2-propanol, 2-methylpropanol, 2,2,3,3,3-pentafluoro-1-propanol, etc. Is mentioned.
- This reaction is preferably carried out using no solvent or a solvent inert to the reaction.
- Such a solvent is not particularly limited as long as the reaction proceeds, but for example, aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, N, N-dimethylformamide, Preference is given to solvents such as amides such as N, N-dimethylacetamide, sulfoxides such as dimethyl sulfoxide, or mixed solvents thereof.
- the reaction time is usually 1 hour to 60 hours, preferably 5 hours to 12 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- compound (23) can also be produced by O-alkylation reaction of compound (22) using a base and an alkylating agent corresponding to R 4 .
- an O-alkylating agent corresponding to R 4 .
- about 1.0 to 5.0 mol, preferably about 1.0 to 2.0 mol, and about 1.0 to 10.0 mol, preferably about 1.0 to 3.0 mol, of an O-alkylating agent are used with respect to 1 mol of compound (22).
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen And metal hydrides such as potassium hydride, and organic bases such as triethylamine, imidazole and formamidine.
- inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen And metal hydrides such as potassium hydride, and organic bases such as triethylamine, imidazole and formamidine.
- O-alkylating agent examples include various alkyl halides such as alkyl chloride, alkyl bromide, and alkyl iodide and derivatives thereof, sulfonic acid esters such as p-toluenesulfonic acid ester and methylsulfonic acid ester, and dimethyl sulfate. And the like.
- This reaction is preferably carried out using a solvent inert to the reaction.
- Such a solvent is not particularly limited as long as the reaction proceeds, but for example, aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, N, N-dimethylformamide, Preference is given to solvents such as amides such as N, N-dimethylacetamide, sulfoxides such as dimethyl sulfoxide, or mixed solvents thereof.
- the reaction time is usually 1 hour to 60 hours, preferably 5 hours to 24 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the obtained compound (23) can be used in the next reaction as a reaction liquid or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- compound (23) can be produced by a method known per se, for example, the method described in Synthesis, page 1 (1981) or the like, or a method analogous thereto. That is, this reaction is usually performed in the presence of an organic phosphorus compound and an azo reagent in a solvent that does not adversely influence the reaction.
- organic phosphorus compound include triphenylphosphine and tri (n-butyl) phosphine.
- the azo reagent include diethyl azodicarboxylate, diisopropyl azodicarboxylate, azodicarbonyldipiperazine, and the like.
- the amount of the organic phosphorus compound and azo reagent used is preferably 1.0 mol to 5.0 mol with respect to 1 mol of compound (22).
- the solvent that does not adversely influence the reaction include ethers such as diethyl ether, tetrahydrofuran and 1,4-dioxane; halogenated hydrocarbons such as chloroform and dichloromethane; aromatic hydrocarbons such as benzene, toluene and xylene Amides such as N, N-dimethylformamide; sulfoxides such as dimethyl sulfoxide; Two or more of these solvents may be mixed and used at an appropriate ratio.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the reaction time is usually about 0.5 to about 20 hours.
- the obtained compound (23) can be used in the next reaction as a reaction liquid or as a crude product, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- Compound (20a) can be synthesized by a reduction reaction of compound (23). Specifically, it is produced by reducing in a hydrogen atmosphere using about 0.01 to 5.0 mol, preferably about 0.01 to 2.0 mol, of a metal catalyst with respect to 1 mol of compound (23).
- a metal catalyst include palladium-carbon, palladium hydroxide-carbon, platinum oxide, platinum and the like. This reaction is preferably carried out using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol and propanol
- aromatic hydrocarbons such as benzene and toluene
- saturated hydrocarbons such as cyclohexane and hexane
- ethers such as dioxane and 1,2-dimethoxyethane
- amides such as N, N-dimethylformamide and N, N-dimethylacetamide
- sulfoxides such as dimethyl sulfoxide
- solvents such as water, or a mixed solvent thereof.
- the reaction time is usually 1 hour to 60 hours, preferably 5 hours to 36 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the pressure is about 1 to 10 atmospheres, preferably about 1 to 5 atmospheres.
- the obtained compound (20a) can be used as it is in the reaction solution or as a crude product for the next reaction, but can be isolated from the reaction mixture according to a conventional method, washing, recrystallization, distillation, chromatography, etc. It can be easily purified by this separation means.
- the reduction reaction of the compound (23) can also be performed using a reduced metal.
- the reducing metal is used in an amount of about 5.0 to 20.0 mol, preferably about 5.0 to 10.0 mol, per 1 mol of compound (23).
- the reduced metal include reduced iron, tin, and zinc.
- hydrochloric acid or a salt such as ammonium chloride or calcium chloride can be added.
- This reaction is preferably carried out using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol and propanol
- aromatic hydrocarbons such as benzene and toluene
- saturated hydrocarbons such as cyclohexane and hexane
- Ethers such as dioxane and 1,2-dimethoxyethane
- amides such as N, N-dimethylformamide and N, N-dimethylacetamide
- ketones such as acetone and methyl ethyl ketone
- sulfoxides such as dimethyl sulfoxide, aqueous ammonia, water Or a mixed solvent thereof.
- the reaction time is usually 1 hour to 60 hours, preferably 5 hours to 36 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the obtained compound (20a) can be used as it is in the reaction solution or as a crude product for the next reaction, but can be isolated from the reaction mixture according to a conventional method, washing, recrystallization, distillation, chromatography, etc. It can be easily purified by this separation means.
- compound (21) and compound (22) commercially available ones can be easily obtained, or they can be produced according to a method known per se or a method analogous thereto.
- compound (6) which is the starting compound of scheme 3, is obtained from compound (11) and compound (20) via compound (24).
- Compound (24) can be produced according to a method known per se, for example, the method described in Journal of Organic Chemistry (J. Org. Chem), 45, 4861 (1980), or a method analogous thereto. Specifically, it is produced by a condensation reaction between the compound (20) and the compound (11).
- Compound (20) is used in an amount of about 1.0 to 5.0 mol, preferably about 1.0 to 3.0 mol, per 1 mol of compound (11). This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons alcohols such as methanol, ethanol, propanol and 1,1-dimethylethanol, aromatic hydrocarbons such as benzene, toluene and xylene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane
- solvents such as amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, or mixed solvents thereof.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 200 ° C, preferably 0 to 150 ° C.
- This reaction may be performed by adding a base.
- the base include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen Metal hydrides such as potassium hydride, organic amines such as triethylamine, pyridine, diisopropylethylamine, 1,8-diazabicyclo [5.4.0] undec-7-ene, and the like.
- the obtained compound (24) can be used as it is in the reaction solution or as a crude product for the next reaction, but can be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- Compound (6) can be produced according to a method known per se, for example, the method described in Journal of Medicinal Chemistry (J. Med. Chem), 41, 3186 (1998), or a method analogous thereto. Specifically, it is produced by a dehydration condensation reaction between the compound (24) and ammonia or an ammonium salt.
- Ammonia or ammonium salt is used in an amount of about 1.0 to 50.0 mol, preferably about 1.0 to 10.0 mol, per 1 mol of compound (24).
- Examples of the ammonium salt include ammonium formate and ammonium acetate. This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons alcohols such as methanol, ethanol, propanol and 1,1-dimethylethanol, aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, N
- Amides such as N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, organic acids such as acetic acid and trifluoroacetic acid, or a mixed solvent thereof Etc.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 200 ° C, preferably 0 to 150 ° C.
- an acid catalyst can be used.
- mineral acids such as hydrochloric acid and sulfuric acid
- Lewis acids such as boron trichloride and boron tribromide
- acetic acid trifluoroacetic acid
- p-toluenesulfone Organic acids such as acids can be mentioned.
- the obtained compound (6) can be used as it is in the reaction solution or as a crude product for the next reaction, but can be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, chromatographed, etc. It can be easily purified by this separation means.
- Scheme 14 is another method of the above-mentioned scheme 3 in which compound (Ib) is obtained via compound (25).
- Compound (25) is produced by cyclization reaction of compound (6) and compound (7).
- compound (7) is used in an amount of 1.0 to 10.0 mol, preferably 1.0 to 5.0 mol, per 1 mol of compound (6).
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, acetonitrile A nitrile such as propionitrile, a solvent such as sulfoxide such as dimethyl sulfoxide, or a mixed solvent thereof is preferable.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 200 ° C, preferably 0 to 150 ° C. This reaction may be performed by adding a base.
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen Metal hydrides such as potassium hydride, organic amines such as triethylamine, pyridine, diisopropylethylamine, 1,8-diazabicyclo [5.4.0] undec-7-ene, and the like.
- inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen Metal hydrides such as potassium hydride, organic amines such as triethylamine, pyridine, diisopropylethylamine, 1,8-diazabicyclo [5.4.0] unde
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (Ib) is produced by a dehydration reaction using compound (25) and an acid.
- the acid is used in an amount of 1.0 to 500.0 mol, preferably 10.0 to 400.0 mol, per 1 mol of compound (25).
- the acid include mineral acids such as hydrochloric acid and sulfuric acid, Lewis acids such as boron trichloride and boron tribromide, and organic acids such as acetic acid, trifluoroacetic acid and p-toluenesulfonic acid.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, acetonitrile
- a solvent such as nitriles such as propionitrile, a sulfoxide such as dimethyl sulfoxide, or a mixed solvent thereof can be used, but when an organic acid is used as the acid, it may also serve as a solvent.
- the reaction time is usually 5 minutes to 60 hours, preferably 15 minutes to 24 hours.
- the reaction temperature is usually 0 to 200 ° C, preferably 25 to 150 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- X 3 represents a chlorine atom, a bromine atom or an iodine atom
- Re represents a C 1-6 alkyl group which may be substituted with a halogen atom
- the other symbols have the same meaning as described above.
- compound (Id) is obtained from compound (Ic) via compound (26).
- Compound (26) is prepared by reaction of a halogenating agent corresponding to the compound (Ic) and X 3.
- Compound (Ic) can be produced according to a method analogous to the method for producing compound (Ib).
- the halogenating agent include N-chlorosuccinimide, N-bromosuccinimide, N-iodosuccinimide, bromine and iodine.
- the halogenating agent is used in an amount of 1.0 to 30.0 mol, preferably 1.0 to 3.0 mol, per 1 mol of compound (Ic).
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- a solvent inert for example, alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc. , Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (Id) is produced by a coupling reaction of compound (26) and an organic boronic acid reagent corresponding to Re.
- the organic boronic acid reagent corresponding to Re is 1.0 to 10.0 mol, preferably 1.0 to 3.0 mol, organometallic reagent 0.01 to 1 mol, preferably 0.05 to 0.2 mol, phosphine ligand, per 1 mol of compound (26). 0.01-1 mol, preferably 0.1-0.5 mol, and base 1.0-10.0 mol, preferably 2.0-6.0 mol are used.
- the reaction can be carried out without using a phosphine ligand.
- Organic boronic acid reagents include methyl boronic acid, 2,4,4,5,5-pentamethyl-1,3,2-dioxaborolane, ethyl boronic acid, N-propyl boronic acid, (1-methylethyl) boronic acid, 4,4 , 5,5-Tetramethyl-2- (1-methylethyl) -1,3,2-dioxaborolane, N-butylboronic acid, 2-butyl-4,4,5,5-tetramethyl-1,3,2 -Dioxaborolane, (2-methylpropyl) boronic acid, 4,4,5,5-tetramethyl-2- (2-methylpropyl) -1,3,2-dioxaborolane and the like.
- organometallic reagents examples include tris (dibenzylideneacetone) dipalladium (0), tetrakis (triphenylphosphine) palladium, [1,1'-bis (diphenylphosphino) ferrocene] palladium (II) dichloride dichloromethane complex, palladium acetate, etc. Is mentioned.
- Bases include sodium hydroxide, potassium hydroxide, barium hydroxide, sodium bicarbonate, sodium carbonate, potassium carbonate, sodium acetate, potassium acetate, cesium carbonate and other basic salts, sodium methoxide, sodium ethoxide, potassium Examples thereof include metal alkoxides such as butoxide, and metal hydrides such as sodium hydride and potassium hydride.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc.
- Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 0 to 300 ° C., preferably 25 to 200 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- compound (If) is obtained from compound (Ie) included in compound (Ib ′) group obtained in scheme 17 described later by an alkylation reaction.
- Compound (If) is produced by an alkylation reaction using a base of compound (Ie) and an alkylating agent corresponding to Re. Specifically, the base is used in an amount of 1.0 to 50.0 mol, preferably 1.0 to 5.0 mol, and the alkylating agent 1.0 to 100.0 mol, preferably 1.0 to 10.0 mol, relative to 1 mol of the compound (Ie).
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium hydrogen carbonate, sodium carbonate and potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide, hydrogen Examples thereof include metal hydrides such as sodium hydride and potassium hydride, and organic bases such as triethylamine, imidazole and formamidine.
- alkylating agent examples include alkyl halides such as alkyl chloride, alkyl bromide and alkyl iodide and derivatives thereof, sulfonic acid esters such as p-toluenesulfonic acid ester and methylsulfonic acid ester, and sulfuric acid ester such as dimethyl sulfate. And the like.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons alcohols such as methanol, ethanol, propanol and 1,1-dimethylethanol, aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, N, Preference is given to amides such as N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, water or a mixed solvent thereof.
- the reaction time is usually 15 minutes to 60 hours, preferably 15 minutes to 24 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- compound (If ′) is obtained by alkylation reaction from compound (Ie ′) included in compound (Ih) group obtained in scheme 22 described later.
- Compound (If ′) is produced by an alkylation reaction using a base of compound (Ie ′) and an alkylating agent corresponding to Re.
- the base is used in an amount of 1.0 to 50.0 mol, preferably 1.0 to 5.0 mol, and the alkylating agent 1.0 to 100.0 mol, preferably 1.0 to 10.0 mol, relative to 1 mol of the compound (Ie ′).
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium hydrogen carbonate, sodium carbonate and potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide, hydrogen Examples thereof include metal hydrides such as sodium hydride and potassium hydride, and organic bases such as triethylamine, imidazole, formamidine and pyridine.
- alkylating agent examples include alkyl halides such as alkyl chloride, alkyl bromide and alkyl iodide and derivatives thereof, sulfonic acid esters such as p-toluenesulfonic acid ester and methylsulfonic acid ester, and sulfuric acid ester such as dimethyl sulfate. And the like.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons alcohols such as methanol, ethanol, propanol and 1,1-dimethylethanol, aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, N, Preference is given to amides such as N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, water or a mixed solvent thereof.
- the reaction time is usually 15 minutes to 60 hours, preferably 15 minutes to 24 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Rf represents a halogen atom or a trifluoromethanesulfonyloxy group, and other symbols are as defined above.
- compound (Ib ′) is obtained from compound (27) obtained in Scheme 18 described later using a coupling reaction.
- Compound (Ib ′) is produced by a coupling reaction between compound (27) and an organic boronic acid reagent or nitrogen-containing reagent corresponding to R 1 .
- the organic boronic acid reagent or nitrogen-containing reagent corresponding to R 1 is 1.0 to 10.0 mol, preferably 1.0 to 3.0 mol, organometallic reagent 0.01 to 1.0 mol, preferably 0.05 to 0.2 mol, per 1 mol of compound (27).
- the phosphine ligand is used in an amount of 0.01 to 1.0 mol, preferably 0.1 to 0.5 mol, and a base 1.0 to 10.0 mol, preferably 2.0 to 6.0 mol.
- a phosphine ligand is not required for the reaction, the reaction can be carried out without using a phosphine ligand.
- Organic boronic acid reagents include 1H-pyrazol-4-ylboronic acid, 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1H-pyrazole, 1-methyl- 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1H-pyrazole, tert-butyl 4- (4,4,5,5-tetramethyl-1, 3,2-dioxaborolan-2-yl) -1H-pyrazole-1-carboxylate, (1-methyl-1H-pyrazol-4-yl) boronic acid, [1- (tert-butoxycarbonyl) -1H-pyrazole- 4-yl] boronic acid, [3- (trifluoromethyl) -1H-pyrazol-4-yl] boronic acid, 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolane-2 -Yl) -3- (tri
- nitrogen-containing reagent examples include morpholine, 3,3-difluoropyrrolidine, 4,4-difluoropiperidine, morpholin-3-one, piperazin-2-one, piperidin-4-one and the like.
- organometallic reagents include tris (dibenzylideneacetone) dipalladium (0), tetrakis (triphenylphosphine) palladium, [1,1'-bis (diphenylphosphino) ferrocene] palladium (II) dichloride dichloromethane complex, palladium acetate, etc. Is mentioned.
- Bases include sodium hydroxide, potassium hydroxide, barium hydroxide, sodium bicarbonate, sodium carbonate, potassium carbonate, sodium acetate, potassium acetate, cesium carbonate and other basic salts, sodium methoxide, sodium ethoxide, potassium Examples thereof include metal alkoxides such as butoxide, and metal hydrides such as sodium hydride and potassium hydride.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc.
- Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 0 to 300 ° C., preferably 25 to 200 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- compound (27) is obtained from compound (28) obtained in scheme 19 via compound (29).
- Compound (29) is produced by a cyclization reaction performed under basic conditions of compound (28) and dialkyl malonate. Specifically, the reaction is carried out using 1.0 to 10.0 mol, preferably 1.0 to 3.0 mol, and 1.0 to 100.0 mol, preferably 2.0 to 10.0 mol, of a base with respect to 1 mol of the compound (28).
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen Metal hydrides such as potassium hydride, organic amines such as triethylamine, pyridine, diisopropylethylamine, 1,8-diazabicyclo [5.4.0] undec-7-ene, and the like.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxy
- alcohols eg, methanol, ethanol, etc.
- esters eg, ethyl acetate, etc.
- aromatic hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- Amides eg, N, N-dimethylformamide, N, N-dimethylacetamide, etc.
- halogenated hydrocarbons eg, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, , Acetonitrile, propionitrile, etc.
- sulfoxides eg, dimethyl sulfoxide, etc.
- organic acids eg, acetic acid, etc.
- the reaction time is usually 10 minutes to 72 hours, preferably 15 minutes to 24 hours.
- the reaction temperature is usually 0 ° C. to 150 ° C., preferably 0 ° C. to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (27) is produced by the substitution reaction of compound (29).
- the halogenating agent is used in an amount of 1.0 to 100.0 mol, preferably 3.0 to 10.0 mol, per 1 mol of compound (29).
- the halogenating agent include phosphorus oxychloride and phosphorus oxybromide. This reaction is advantageously carried out without solvent or using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane, etc.
- esters eg, ethyl acetate, etc.
- aromatics Hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- amides eg, N, N-dimethylformamide, N, N-dimethylacetamide, etc.
- halogenated hydrocarbons Eg, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, acetonitrile, propionitrile, etc.
- sulfoxides eg, dimethyl sulfoxide, etc.
- sulfoxides eg, dimethyl sulfoxide, etc.
- the reaction time is usually 10 minutes to 72 hours, preferably 30 minutes to 3 hours.
- the reaction temperature is usually 0 ° C. to 150 ° C., preferably 0 ° C. to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- the preparation of compound (27) is carried out in the range of 1.0 to 10.0 moles, preferably 1.0 to 3.0 moles, and 1.0 to 20.0 moles, preferably 1.0 to 10.0 moles of trifluoromethanesulfonylating reagent per mole of compound (29). Using moles.
- the trifluoromethanesulfonylation reagent include 1,1,1-trifluoro-N-phenyl-N-[(trifluoromethyl) sulfonyl] methanesulfonamide, 2- [N, N-bis (trifluoromethylsulfonyl) Amine] -5-chloropyridine, trifluoromethanesulfonic anhydride and the like.
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen Metal hydrides such as potassium hydride, organic amines such as triethylamine, pyridine, diisopropylethylamine, 1,8-diazabicyclo [5.4.0] undec-7-ene, and the like.
- This reaction is advantageously carried out without solvent or using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- ethers eg, diethyl ether, tetrahydrofuran, 1,2-dimethoxyethane, etc.
- esters eg, ethyl acetate, etc.
- aromatics Hydrocarbons eg, benzene, toluene, etc.
- aliphatic hydrocarbons eg, hexane, etc.
- amides eg, N, N-dimethylformamide, N, N-dimethylacetamide, etc.
- halogenated hydrocarbons Eg, dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- nitriles eg, acetonitrile, propionitrile, etc.
- sulfoxides eg, dimethyl sulfoxide, etc.
- sulfoxides eg, dimethyl sulfoxide, etc.
- the reaction time is usually 10 minutes to 72 hours, preferably 15 minutes to 24 hours.
- the reaction temperature is usually ⁇ 78 ° C. to 100 ° C., preferably 0 ° C. to 50 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Rg represents an optionally substituted C 1-6 alkyl group, and other symbols are as defined above.
- compound (28), which is the starting material of scheme 18, is obtained from compound (30).
- Compound (30), which is commercially available, can be used as it is, and can also be produced according to a method known per se or a method analogous thereto.
- Compound (31) is produced from compound (30) by a substitution reaction. Specifically, the chlorinating agent is used in an amount of 1.0 to 10.0 mol, preferably 1.0 to 3.0 mol, and 28% aqueous ammonia solution 1.0 to 20.0 mol, preferably 1.0 to 3.0 mol, per 1 mol of compound (30).
- pyridine, dicyclohexylamine, N, N-dimethylformamide, phase transfer catalyst and the like may be used in an amount of 0.001 to 10.0 mol, preferably 0.001 to 3.0 mol, per 1 mol of compound (30).
- the chlorinating agent include oxalyl chloride, thionyl chloride, phosphorus oxychloride and the like.
- the phase transfer catalyst include tetrabutylammonium chloride, tetrabutylammonium bromide, crown ether, and the like. This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, acetonitrile
- a nitrile such as propionitrile
- a solvent such as sulfoxide such as dimethyl sulfoxide, or a mixed solvent thereof is preferable.
- the reaction time is usually 10 minutes to 72 hours, preferably 30 minutes to 24 hours.
- the reaction temperature is usually -78 ° C to 100 ° C, preferably -10 ° C to 25 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- compound (32) is produced by O-alkylation reaction using compound (31) and an alkylating agent corresponding to Rg. Specifically, the reaction is carried out using 1.0 to 50.0 mol, preferably 1.0 to 10.0 mol, and 1.0 to 100.0 mol, preferably 3.0 to 10.0 mol, of a base with respect to 1 mol of the compound (31).
- alkylating agent examples include trimethyloxonium tetrafluoroborate, dimethyl sulfate, methyl trifluoromethanesulfonate, methyl fluorosulfonate, and the like.
- base examples include sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen phosphate, sodium phosphate and other inorganic bases, sodium methoxide, sodium ethoxide, potassium Examples thereof include metal alkoxides such as butoxide.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, acetonitrile
- a nitrile such as propionitrile
- a solvent such as sulfoxide such as dimethyl sulfoxide, or a mixed solvent thereof is preferable.
- the reaction time is usually 10 minutes to 72 hours, preferably 30 minutes to 24 hours.
- the reaction temperature is usually -78 ° C to 100 ° C, preferably -10 ° C to 25 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (33) is produced by a substitution reaction from compound (31).
- the sulfurizing agent is used in an amount of 1.0 to 3.0 mol, preferably 1.0 to 1.30 mol, relative to 1 mol of the compound (31).
- the sulfurizing agent include Lawesson's reagent, phosphorus pentasulfide, and phosphorus pentasulfide.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc.
- Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 72 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually 0 ° C. to 150 ° C., preferably 25 ° C. to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (34) is produced by an S-alkylation reaction using compound (33) and an alkylating agent corresponding to Rg. Specifically, it is carried out using 1.0 to 10.0 moles, preferably 1.0 to 5.0 moles of the S-alkylating agent with respect to 1 mole of the compound (33).
- the alkylating agent include alkyl halides such as alkyl chloride, alkyl bromide and alkyl iodide and derivatives thereof, sulfonic acid esters such as p-toluenesulfonic acid ester and methylsulfonic acid ester, and sulfuric acid ester such as dimethyl sulfate. And the like.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- a solvent inert for example, alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc. , Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 15 minutes to 60 hours, preferably 30 minutes to 24 hours.
- the reaction temperature is usually 0 ° C. to 150 ° C., preferably 25 ° C. to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (28) is produced by a substitution reaction from compound (32) or compound (34). Specifically, compound (20) is used in an amount of 1.0 to 20.0 mol, preferably 1.0 to 2.0 mol, and base 1.0 to 20.0 mol, preferably 1.0 to 10.0 mol, per 1 mol of compound (32) or compound (34). .
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen Metal hydrides such as potassium hydride, organic amines such as triethylamine, pyridine, diisopropylethylamine, 1,8-diazabicyclo [5.4.0] undec-7-ene, and the like.
- a base is not required for the reaction, the reaction can be carried out without using a base. This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc.
- Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 30 minutes to 100 hours, preferably 1 hour to 72 hours.
- the reaction temperature is usually 0 ° C. to 150 ° C., preferably 25 ° C. to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Rj represents an optionally substituted alkyl group, and other symbols have the same meaning as described above.
- compound (Ig) is synthesized via compound (36) using compound (35) obtained in scheme 21 described later as a starting material.
- Compound (36) is produced by the substitution reaction of compound (35).
- the corresponding acetic acid derivative is used in an amount of 1.0 to 10.0 mol, preferably 1.0 to 5.0 mol, per 1 mol of compound (35).
- the acetic acid derivative include 2- (2-chloroalkoxy) acetyl chloride.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, acetonitrile A nitrile such as propionitrile, a solvent such as sulfoxide such as dimethyl sulfoxide, or a mixed solvent thereof is preferable.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 200 ° C, preferably 0 to 150 ° C. This reaction may be performed by adding a base.
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen Metal hydrides such as potassium hydride, organic amines such as triethylamine, pyridine, diisopropylethylamine, 1,8-diazabicyclo [5.4.0] undec-7-ene, and the like.
- inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen Metal hydrides such as potassium hydride, organic amines such as triethylamine, pyridine, diisopropylethylamine, 1,8-diazabicyclo [5.4.0] unde
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (Ig) is produced by the cyclization reaction of compound (36). Specifically, the reaction is carried out using 1.0 to 10.0 mol, preferably 1.0 to 5.0 mol of the base per 1 mol of the compound (36).
- the base include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen Metal hydrides such as potassium hydride, organic amines such as triethylamine, pyridine, diisopropylethylamine, 1,8-diazabicyclo [5.4.0] undec-7-ene, and the like.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- a solvent inert such as halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, acetonitrile A nitrile such as propionitrile, a solvent such as sulfoxide such as dimethyl sulfoxide, or a mixed solvent thereof is preferable.
- the reaction time is usually 1 hour to 60 hours, preferably 1 hour to 24 hours.
- the reaction temperature is usually -50 to 200 ° C, preferably 0 to 150 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Rh represents a benzyl group which may be substituted with an alkyl group or an alkoxy group
- Ri represents a benzophenone imino group which may be substituted with an alkyl group or an alkoxy group, and the other symbols are those described above. Is equivalent to ]
- compound (35) which is the starting material of scheme 20 is obtained from compound (27) obtained in scheme 18.
- Compound (37) or compound (38) is produced by a coupling reaction between compound (27) and an amine reagent corresponding to Rh or an imine reagent corresponding to Ri.
- the amine reagent corresponding to Rh or the imine reagent corresponding to Ri is 1.0 to 10.0 mol, preferably 1.0 to 3.0 mol, the organometallic reagent 0.01 to 1.0 mol, preferably 0.05 to 0.2 mol, per 1 mol of compound (27).
- the phosphine ligand is used in an amount of 0.01 to 1.0 mol, preferably 0.1 to 0.5 mol, and a base 1.0 to 10.0 mol, preferably 2.0 to 6.0 mol.
- a phosphine ligand is not required for the reaction, the reaction can be carried out without using a phosphine ligand.
- Examples of the amine reagent include benzylamine, dibenzylamine, 2-methoxybenzylamine, 3-methoxybenzylamine, 4-methoxybenzylamine, 2,3-dimethoxybenzylamine, 2,4-dimethoxybenzylamine, 3,4- Dimethoxybenzylamine, 2,4,6-trimethoxybenzylamine, 3,4,5-trimethoxybenzylamine, 2,3,4-trimethoxybenzylamine, 2,4,5-trimethoxybenzylamine, 2- Methylbenzylamine, 3-methylbenzylamine, 4-methylbenzylamine, 2,3-dimethylbenzylamine, 2,4-dimethylbenzylamine, 3,4-dimethylbenzylamine, 2,4,6-trimethylbenzylamine, 2,4,5-trimethylbenzylamine is mentioned.
- Examples of the imine reagent include benzophenone imine, 1,1-bis (4-methoxyphenyl) methanimine, 9-iminofluorene and the like.
- organometallic reagents include tris (dibenzylideneacetone) dipalladium (0), tetrakis (triphenylphosphine) palladium, [1,1'-bis (diphenylphosphino) ferrocene] palladium (II) dichloride dichloromethane complex, palladium acetate, etc. Is mentioned.
- Bases include sodium hydroxide, potassium hydroxide, barium hydroxide, sodium bicarbonate, sodium carbonate, potassium carbonate, sodium acetate, potassium acetate, cesium carbonate and other basic salts, sodium methoxide, sodium ethoxide, potassium Examples thereof include metal alkoxides such as butoxide, and metal hydrides such as sodium hydride and potassium hydride.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc.
- Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 0 to 300 ° C., preferably 25 to 200 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (35) is produced by subjecting compound (37) to acid treatment or hydrogenation reaction.
- the acid treatment is performed using 1.0 to 200 mol, preferably 3.0 to 20.0 mol, of an acid reagent per 1 mol of compound (37).
- the acid reagent include Lewis acids such as aluminum chloride, organic acids such as acetic acid and trifluoroacetic acid, and mineral acids such as hydrochloric acid.
- the hydrogenation reaction is performed using a metal reagent in an amount of 5 to 1000% by weight, preferably 10 to 300% by weight, based on the compound (37).
- the metal reagent include palladium carbon, palladium hydroxide, platinum oxide, Raney nickel, Raney cobalt and the like.
- the hydrogen pressure is usually 1 to 100 atm.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- a solvent inert for example, alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc. , Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 25 ° C to 300 ° C, preferably 50 ° C to 200 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (35) is produced by acid-treating compound (38).
- the acid reagent is used in an amount of 1.0 to 200 mol, preferably 3.0 to 20.0 mol, per 1 mol of compound (38).
- the acid reagent include Lewis acids such as aluminum chloride, organic acids such as acetic acid and trifluoroacetic acid, and mineral acids such as hydrochloric acid.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc.
- Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 25 ° C to 300 ° C, preferably 25 ° C to 150 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- compound (Ih) is obtained from compound (27) synthesized in scheme 18 described above via compound (39) and compound (40).
- Compound (39) is produced from compound (27) by a coupling reaction using a metal cyanide reagent.
- the metal cyanide reagent is 1.0 to 10.0 mol, preferably 1.0 to 3.0 mol
- the organometallic reagent 0.01 to 1.0 mol, preferably 0.05 to 0.2 mol
- the phosphine ligand 0.01 to 1.0 mol per 1 mol of the compound (27). Mol, preferably 0.1 to 0.5 mol.
- the reaction can be carried out without using a phosphine ligand.
- metal cyanide reagent examples include zinc cyanide and copper cyanide.
- organometallic reagents include tris (dibenzylideneacetone) dipalladium (0), tetrakis (triphenylphosphine) palladium, [1,1'-bis (diphenylphosphino) ferrocene] palladium (II) dichloride dichloromethane complex, palladium acetate, etc. Is mentioned.
- phosphine ligand 2-dicyclohexylphosphino-2 ', 4', 6'-triisopropylbiphenyl, (9,9-dimethyl-9H-xanthene-4,5-diyl) bis (diphenylphosphane), 2 2,2'-bis (diphenylphosphino) -1,1'-binaphthyl and the like.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc.
- Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 0 to 300 ° C., preferably 25 to 200 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (40) is produced by substitution reaction of compound (39) with hydroxyamine. Specifically, the reaction is carried out using 1.0 to 200 mol, preferably 1.0 to 5.0 mol, and 1.0 to 50.0 mol, preferably 1.0 to 10.0 mol, of a hydroxyamine hydrochloride with respect to 1 mol of the compound (39).
- the base include sodium hydroxide, potassium hydroxide, barium hydroxide, sodium hydrogen carbonate, sodium carbonate, potassium carbonate, sodium acetate, potassium acetate, cesium carbonate and other basic salts, sodium methoxide, sodium ethoxide, potassium Examples thereof include metal alkoxides such as tributoxide, and metal hydrides such as sodium hydride and potassium hydride.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- a solvent inert for example, alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc. , Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 0 to 300 ° C., preferably 25 to 200 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (Ih) is produced from compound (40) by a cyclization reaction using a condensing agent. Specifically, the reaction is carried out using 1.0 to 20.0 mol, preferably 1.0 to 5.0 mol, and 1.0 to 50.0 mol, preferably 1.0 to 10.0 mol, of a base with respect to 1 mol of the compound (40).
- the condensing agent include acetic anhydride, trifluoroacetic anhydride, N, N′-carbonyldiimidazole and the like.
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium hydrogen carbonate, sodium carbonate and potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide, hydrogen Examples thereof include metal hydrides such as sodium hydride and potassium hydride, and organic bases such as triethylamine, imidazole, formamidine and pyridine.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc.
- Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, pyridine and the like
- Organic bases, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 0 to 300 ° C., preferably 25 to 200 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- compound (Ih ′) is obtained from compound (27) synthesized in scheme 18 through compound (41) and compound (42), or further through compound (43).
- Compound (41) is produced from compound (27) by an insertion reaction using an organometallic reagent and carbon monoxide. Specifically, this reaction is carried out in an amount of 0.01 to 1.0 mol, preferably 0.05 to 0.2 mol, phosphine ligand 0.01 to 1.0 mol, preferably 0.1 to 0.5 mol, base 1.0 to 10.0 mol per mol of compound (27).
- the reaction is carried out in an atmosphere of carbon monoxide using 1 mol, preferably 2.0 to 6.0 mol, and 1.0 to 100.0 mol, preferably 1.0 to 10.0 mol, of alcohol corresponding to Rj.
- a phosphine ligand is not required for the reaction, the reaction can be carried out without using a phosphine ligand.
- organometallic reagents include tris (dibenzylideneacetone) dipalladium (0), tetrakis (triphenylphosphine) palladium, [1,1'-bis (diphenylphosphino) ferrocene] palladium (II) dichloride dichloromethane complex, palladium acetate, etc.
- phosphine ligand 2-dicyclohexylphosphino-2 ', 4', 6'-triisopropylbiphenyl, (9,9-dimethyl-9H-xanthene-4,5-diyl) bis (diphenylphosphane), 2 2,2'-bis (diphenylphosphino) -1,1'-binaphthyl and the like.
- Bases include sodium hydroxide, potassium hydroxide, barium hydroxide, sodium bicarbonate, sodium carbonate, potassium carbonate, sodium acetate, potassium acetate, cesium carbonate and other basic salts, sodium methoxide, sodium ethoxide, potassium Examples thereof include metal alkoxides such as butoxide, and metal hydrides such as sodium hydride and potassium hydride.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc.
- Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 0 to 300 ° C., preferably 25 to 200 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (42) is produced by hydrolysis reaction of compound (41). Specifically, water is used in an amount of 1.0 to 100.0 mol, preferably 1.0 to 5.0 mol, and base 1.0 to 100.0 mol, preferably 1.0 to 5.0 mol, per 1 mol of compound (41).
- the base include sodium hydroxide, potassium hydroxide, barium hydroxide, sodium hydrogen carbonate, sodium carbonate, potassium carbonate, sodium acetate, potassium acetate, cesium carbonate and other basic salts, sodium methoxide, sodium ethoxide, potassium Examples thereof include metal alkoxides such as tributoxide, and metal hydrides such as sodium hydride and potassium hydride.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- a solvent inert for example, alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc. , Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 0 to 200 ° C., preferably 25 to 100 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (43) is produced by the substitution reaction of compound (42).
- oxalyl chloride is 1.0 to 2.0 mol, preferably 1.0 to 1.5 mol
- hydrazide corresponding to Rj is 1.0 to 20.0 mol, preferably 1.0 to 5.0 mol, and base 1.0 to 20.0 mol with respect to 1 mol of compound (42).
- Preferably, 1.0 to 10.0 moles are used.
- Examples of the hydrazide corresponding to Rj include acetohydrazide.
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium hydrogen carbonate, sodium carbonate and potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide, hydrogen Examples thereof include metal hydrides such as sodium hydride and potassium hydride, and organic bases such as triethylamine, imidazole, formamidine and pyridine.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, acetonitrile A nitrile such as propionitrile, a solvent such as sulfoxide such as dimethyl sulfoxide, or a mixed solvent thereof is preferable.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 0 to 300 ° C., preferably 25 to 200 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (Ih ′) is produced by the cyclization reaction of compound (42).
- oxalyl chloride is 1.0 to 2.0 mol, preferably 1.0 to 1.5 mol
- N′-hydroxyethaneimidoamide 1.0 to 20.0 mol, preferably 1.0 to 5.0 mol, and base 1.0 to 20.0 mol per mol of compound (42).
- Mol preferably 1.0 to 10.0 mol.
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium hydrogen carbonate, sodium carbonate and potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide, hydrogen Examples thereof include metal hydrides such as sodium hydride and potassium hydride, and organic bases such as triethylamine, imidazole, formamidine and pyridine.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, acetonitrile A nitrile such as propionitrile, a solvent such as sulfoxide such as dimethyl sulfoxide, or a mixed solvent thereof is preferable.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 0 to 300 ° C., preferably 25 to 200 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (Ih ′) can also be produced by cyclization reaction of compound (43). Specifically, (methoxycarbonylsulfamoyl) triethylammonium hydroxide inner salt is used in an amount of 1.0 to 2.0 mol, preferably 1.0 to 1.5 mol, per 1 mol of compound (43).
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc.
- Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 0 to 200 ° C., preferably 25 to 150 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Rk represents an optionally substituted C 1-5 alkyl group
- Rl represents an optionally substituted C 1-6 alkyl group
- other symbols are as defined above.
- compound (Ij) and compound (Ii ′) are obtained from compound (Ii) included in compound (Ib) group of scheme 3 described above.
- Compound (Ij) is produced by subjecting compound (Ii) to an acid treatment or a hydrogenation reaction.
- the acid treatment is performed using 1.0 to 200 mol, preferably 3.0 to 20.0 mol, of an acid reagent per 1 mol of compound (Ii).
- the acid reagent include Lewis acids such as aluminum chloride, organic acids such as acetic acid and trifluoroacetic acid, and mineral acids such as hydrochloric acid.
- the hydrogenation reaction is performed using a metal reagent in an amount of 5 to 1000% by weight, preferably 10 to 300% by weight, based on Compound (Ii).
- the metal reagent examples include palladium carbon, palladium hydroxide, platinum oxide, Raney nickel, Raney cobalt and the like.
- the hydrogen pressure is usually 1 to 100 atm.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol, propanol, 1,1-dimethylethanol, etc.
- Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, acetic acid, tri Organic acids such as fluoroacetic acid, water or a mixed solvent thereof are preferred.
- the reaction time is usually 10 minutes to 50 hours, preferably 30 minutes to 12 hours.
- the reaction temperature is usually 25 ° C to 300 ° C, preferably 50 ° C to 200 ° C. If necessary, the reaction can be carried out under microwave irradiation.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (Ii ′) is produced by an O-alkylation reaction using the base of compound (Ij) and an alkylating agent corresponding to Rl. Specifically, the reaction is performed using 1.0 to 50.0 mol, preferably 1.0 to 5.0 mol, and 1.0 to 100.0 mol, preferably 1.0 to 10.0 mol, of an O-alkylating agent with respect to 1 mol of compound (Ij).
- the base examples include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium hydrogen carbonate, sodium carbonate and potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide and potassium tert-butoxide, hydrogen Examples thereof include metal hydrides such as sodium hydride and potassium hydride, and organic bases such as triethylamine, imidazole and formamidine.
- O-alkylating agent examples include alkyl halides such as alkyl chloride, alkyl bromide and alkyl iodide and derivatives thereof, sulfonates such as p-toluenesulfonic acid ester and methylsulfonic acid ester, and dimethyl sulfate. Examples thereof include sulfate esters.
- This reaction is preferably carried out using a solvent inert to the reaction, and such a solvent is not particularly limited as long as the reaction proceeds.
- halogen such as dichloromethane, chloroform, carbon tetrachloride, 1,2-dichloroethane, etc.
- Hydrocarbons alcohols such as methanol, ethanol, propanol and 1,1-dimethylethanol, aromatic hydrocarbons such as benzene and toluene, ethers such as tetrahydrofuran, dioxane and 1,2-dimethoxyethane, N, Preference is given to amides such as N-dimethylformamide and N, N-dimethylacetamide, nitriles such as acetonitrile and propionitrile, sulfoxides such as dimethyl sulfoxide, water or a mixed solvent thereof.
- the reaction time is usually 15 minutes to 60 hours, preferably 15 minutes to 24 hours.
- the reaction temperature is usually -50 to 150 ° C, preferably 0 to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Rm represents an optionally substituted C 1-2 alkyl group, and other symbols are as defined above.
- compound (Ik) is obtained from compound (3) or compound (4) obtained in scheme 1 described above via compound (Il).
- Compound (Il) is produced by a substitution reaction of Compound (3) or Compound (4) with an acetate derivative corresponding to Rg and a base. Specifically, 1.0 to 20.0 mol, preferably 1.0 to 10.0 mol of the base and 1.0 to 100.0 mol, preferably 1.0 to 10.0 mol, of the acetate derivative corresponding to Rg are added to 1 mol of the compound (3) or the compound (4). To do.
- Examples of the base include inorganic bases such as sodium hydroxide, potassium hydroxide, barium hydroxide, sodium carbonate, potassium carbonate, metal alkoxides such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, sodium hydride, hydrogen Metal hydrides such as potassium hydride, organic amines such as triethylamine, pyridine, diisopropylethylamine, 1,8-diazabicyclo [5.4.0] undec-7-ene, and the like.
- the reaction can be carried out without using a base.
- Examples of the acetate derivative include alkyl hydroxyacetate.
- This reaction is preferably carried out without solvent or using a solvent inert to the reaction.
- a solvent is not particularly limited as long as the reaction proceeds.
- halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride and 1,2-dichloroethane
- aromatic hydrocarbons such as benzene and toluene
- tetrahydrofuran Ethers such as dioxane, 1,2-dimethoxyethane
- amides such as N, N-dimethylformamide, N, N-dimethylacetamide
- nitriles such as acetonitrile and propionitrile
- sulfoxides such as dimethyl sulfoxide, etc.
- the reaction time is usually 10 minutes to 24 hours, preferably 10 minutes to 12 hours.
- the reaction temperature is usually -100 to 150 ° C, preferably -78 to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- Compound (Ik) is produced by nucleophilic substitution reaction of compound (Il) using an alkylating agent corresponding to Rm. Specifically, the reaction is performed using 2.0 to 100.0 mol, preferably 2.0 to 10.0 mol, of an alkylating agent corresponding to Rm with respect to 1 mol of compound (Il).
- the alkylating agent include alkyl magnesium halide (Grignard reagent), alkyl lithium and the like. This reaction is preferably carried out without solvent or using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds.
- halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride and 1,2-dichloroethane
- aromatic hydrocarbons such as benzene and toluene, tetrahydrofuran
- Ethers such as dioxane, 1,2-dimethoxyethane or a mixed solvent thereof are preferable.
- the reaction time is usually 10 minutes to 72 hours, preferably 10 minutes to 24 hours.
- the reaction temperature is usually -100 to 150 ° C, preferably -78 to 100 ° C.
- the product is obtained as a single product or as a mixture and can be used as it is in the reaction solution or as a crude product in the next reaction, but can also be isolated from the reaction mixture according to a conventional method, washed, recrystallized, distilled, It can be easily purified by separation means such as chromatography.
- hydroxy protecting group examples include C 1-6 alkyl (eg, methyl, ethyl, propyl, isopropyl, butyl, tert-butyl), phenyl, trityl, C 7-10 aralkyl (eg, benzyl), formyl, C 1-6 alkyl-carbonyl (eg, acetyl, propionyl), benzoyl, C 7-10 aralkyl-carbonyl (eg, benzylcarbonyl), 2-tetrahydropyranyl, 2-tetrahydrofuranyl, silyl (eg, trimethylsilyl, triethylsilyl, Dimethylphenylsilyl, tert-butyldimethylsilyl, tert-butyldiethylsilyl), C 2-6 alkenyl (eg, 1-allyl) and the like.
- C 1-6 alkyl eg, methyl, ethyl, prop
- These groups include a halogen atom (eg, fluorine atom, chlorine atom, bromine atom, iodine atom), C 1-6 alkyl (eg, methyl, ethyl, propyl), C 1-6 alkoxy (eg, methoxy, ethoxy, It may be substituted with 1 to 3 substituents selected from propoxy), nitro and the like.
- a halogen atom eg, fluorine atom, chlorine atom, bromine atom, iodine atom
- C 1-6 alkyl eg, methyl, ethyl, propyl
- C 1-6 alkoxy eg, methoxy, ethoxy, It may be substituted with 1 to 3 substituents selected from propoxy
- amino protecting groups include formyl, C 1-6 alkyl-carbonyl (eg, acetyl, propionyl), C 1-6 alkoxy-carbonyl (eg, methoxycarbonyl, ethoxycarbonyl, tert-butoxycarbonyl), benzoyl, C 7-10 aralkyl-carbonyl (eg, benzylcarbonyl), C 7-14 aralkyloxy-carbonyl (eg, benzyloxycarbonyl, 9-fluorenylmethoxycarbonyl), C 7-10 aralkyl (eg, benzyl, 4-methoxy) Benzyl), trityl, phthaloyl, N, N-dimethylaminomethylene, silyl (eg, trimethylsilyl, triethylsilyl, dimethylphenylsilyl, tert-butyldimethylsilyl, tert-butyldiethylsilyl), C 2
- These groups include 1 to 3 substituents selected from a halogen atom (eg, fluorine atom, chlorine atom, bromine atom, iodine atom), C 1-6 alkoxy (eg, methoxy, ethoxy, propoxy), nitro and the like. May be substituted.
- a halogen atom eg, fluorine atom, chlorine atom, bromine atom, iodine atom
- C 1-6 alkoxy eg, methoxy, ethoxy, propoxy
- nitro and the like May be substituted.
- carboxy protecting groups include C 1-6 alkyl (eg, methyl, ethyl, propyl, isopropyl, butyl, tert-butyl), C 7-11 aralkyl (eg, benzyl), phenyl, trityl, silyl (eg, Trimethylsilyl, triethylsilyl, dimethylphenylsilyl, tert-butyldimethylsilyl, tert-butyldiethylsilyl, tert-butyldiphenylsilyl), C 2-6 alkenyl (eg, 1-allyl) and the like.
- C 1-6 alkyl eg, methyl, ethyl, propyl, isopropyl, butyl, tert-butyl
- C 7-11 aralkyl eg, benzyl
- phenyl, trityl eg, silyl (eg, Trimethyls
- These groups include 1 to 3 substituents selected from a halogen atom (eg, fluorine atom, chlorine atom, bromine atom, iodine atom), C 1-6 alkoxy (eg, methoxy, ethoxy, propoxy), nitro and the like. May be substituted.
- a halogen atom eg, fluorine atom, chlorine atom, bromine atom, iodine atom
- C 1-6 alkoxy eg, methoxy, ethoxy, propoxy
- nitro and the like May be substituted.
- Examples of the carbonyl protecting group include cyclic acetals (eg, 1,3-dioxane), acyclic acetals (eg, di-C 1-6 alkylacetal) and the like.
- Examples of the mercapto-protecting group include a C 1-6 alkyl group, a phenyl group, a trityl group, a C 7-10 aralkyl group (eg, benzyl), a C 1-6 alkyl-carbonyl group, a benzoyl group, and a C 7-10.
- Aralkyl-carbonyl group eg, benzylcarbonyl
- C 1-6 alkoxy-carbonyl group C 6-14 aryloxy-carbonyl group (eg, phenyloxycarbonyl), C 7-14 aralkyloxy-carbonyl group (eg, benzyl) Oxycarbonyl, 9-fluorenylmethoxycarbonyl), 2-tetrahydropyranyl group, C 1-6 alkylamino-carbonyl group (eg, methylaminocarbonyl, ethylaminocarbonyl) and the like.
- These groups may be substituted with 1 to 3 substituents selected from a halogen atom, a C 1-6 alkyl group, a C 1-6 alkoxy group or a nitro group.
- the above-mentioned protecting group removal method may be carried out according to a method known per se, for example, the method described in Protective Groups in Organic Synthesis, published by John Wiley and Sons (1980). For example, a method using acid, base, ultraviolet light, hydrazine, phenylhydrazine, sodium N-methyldithiocarbamate, tetrabutylammonium fluoride, palladium acetate, trialkylsilyl halide (eg, trimethylsilyl iodide, trimethylsilyl bromide, etc.) A reduction method or the like is used.
- the compound (I) obtained by each of the above production methods can be isolated and purified by known means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, transfer dissolution, chromatography and the like. Moreover, each raw material compound used in each of the above production methods can be isolated and purified by the same known means as described above. On the other hand, you may use these raw material compounds as a reaction mixture as it is as a raw material for the next step without isolation.
- compound (I) has an isomer such as an optical isomer, a stereoisomer, a positional isomer, or a rotational isomer
- any one isomer or a mixture of isomers is included in compound (I). Is done.
- compound (I) has an optical isomer
- the optical isomer resolved from the racemate is also encompassed in compound (I).
- Each of these isomers can be obtained as a single product by a known synthesis method or separation method (concentration, solvent extraction, column chromatography, recrystallization, etc.).
- Compound (I) or a prodrug thereof (hereinafter, sometimes simply abbreviated as the compound of the present invention) has low toxicity and should be used as it is or mixed with a pharmacologically acceptable carrier to form a pharmaceutical composition.
- a pharmaceutical composition e.g, a preventive or therapeutic agent for various diseases described below for mammals (eg, humans, mice, rats, rabbits, dogs, cats, cows, horses, pigs, monkeys).
- the pharmacologically acceptable carrier various organic or inorganic carrier substances commonly used as pharmaceutical materials are used, and excipients, lubricants, binders, disintegrants in solid preparations; solvents in liquid preparations , Solubilizing agents, suspending agents, isotonic agents, buffers, soothing agents and the like. If necessary, preparation additives such as preservatives, antioxidants, colorants, sweeteners and the like can also be used.
- excipients include lactose, sucrose, D-mannitol, D-sorbitol, starch, pregelatinized starch, dextrin, crystalline cellulose, low-substituted hydroxypropylcellulose, sodium carboxymethylcellulose, gum arabic, pullulan, light
- excipients include anhydrous silicic acid, synthetic aluminum silicate, and magnesium aluminate metasilicate.
- lubricant examples include magnesium stearate, calcium stearate, talc and colloidal silica.
- Preferred examples of the binder include pregelatinized starch, sucrose, gelatin, gum arabic, methylcellulose, carboxymethylcellulose, sodium carboxymethylcellulose, crystalline cellulose, sucrose, D-mannitol, trehalose, dextrin, pullulan, hydroxypropylcellulose, hydroxy Examples include propylmethylcellulose and polyvinylpyrrolidone.
- disintegrant examples include lactose, sucrose, starch, carboxymethyl cellulose, carboxymethyl cellulose calcium, croscarmellose sodium, carboxymethyl starch sodium, light anhydrous silicic acid, and low-substituted hydroxypropyl cellulose.
- Suitable examples of the solvent include water for injection, physiological saline, Ringer's solution, alcohol, propylene glycol, polyethylene glycol, sesame oil, corn oil, olive oil, and cottonseed oil.
- solubilizer examples include polyethylene glycol, propylene glycol, D-mannitol, trehalose, benzyl benzoate, ethanol, trisaminomethane, cholesterol, triethanolamine, sodium carbonate, sodium citrate, sodium salicylate, sodium acetate. Is mentioned.
- suspending agent examples include surfactants such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalkonium chloride, benzethonium chloride, glyceryl monostearate; polyvinyl alcohol, polyvinylpyrrolidone , Hydrophilic polymers such as sodium carboxymethylcellulose, methylcellulose, hydroxymethylcellulose, hydroxyethylcellulose, hydroxypropylcellulose; polysorbates, and polyoxyethylene hydrogenated castor oil.
- surfactants such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalkonium chloride, benzethonium chloride, glyceryl monostearate
- polyvinyl alcohol, polyvinylpyrrolidone Hydrophilic polymers such as sodium carboxymethylcellulose, methylcellulose, hydroxymethylcellulose, hydroxyethylcellulose,
- Preferable examples of the isotonic agent include sodium chloride, glycerin, D-mannitol, D-sorbitol and glucose.
- buffers such as phosphate, acetate, carbonate and citrate.
- a preferred example of the soothing agent is benzyl alcohol.
- Preferable examples of the preservative include p-hydroxybenzoates, chlorobutanol, benzyl alcohol, phenethyl alcohol, dehydroacetic acid and sorbic acid.
- Preferable examples of the antioxidant include sulfite and ascorbate.
- the colorant examples include water-soluble edible tar dyes (eg, edible dyes such as edible red Nos. 2 and 3, edible yellows Nos. 4 and 5, edible blue Nos. 1 and 2, etc.), water-insoluble lake dyes (Eg, the aluminum salt of the water-soluble edible tar dye) and natural dyes (eg, ⁇ -carotene, chlorophyll, bengara).
- water-soluble edible tar dyes eg, edible dyes such as edible red Nos. 2 and 3, edible yellows Nos. 4 and 5, edible blue Nos. 1 and 2, etc.
- water-insoluble lake dyes Eg, the aluminum salt of the water-soluble edible tar dye
- natural dyes eg, ⁇ -carotene, chlorophyll, bengara
- Suitable examples of sweeteners include saccharin sodium, dipotassium glycyrrhizinate, aspartame, and stevia.
- the medicament containing the compound of the present invention can be used alone or mixed with a pharmacologically acceptable carrier according to a method known per se as a method for producing a pharmaceutical preparation (eg, a method described in the Japanese Pharmacopoeia).
- tablets including sugar-coated tablets, film-coated tablets, sublingual tablets, orally disintegrating tablets, buccal tablets, etc.
- pills powders, granules, capsules (including soft capsules and microcapsules), troches Agent, syrup, solution, emulsion, suspension, controlled release formulation (eg, immediate release formulation, sustained release formulation, sustained release microcapsule), aerosol, film agent (eg, orally disintegrating film, Oral mucosa adhesive film), injection (eg, subcutaneous injection, intravenous injection, intramuscular injection, intraperitoneal injection), drip, transdermal preparation, ointment, lotion, patch, sitting Suppositories (eg, rectal suppositories) Vaginal suppositories), pellets,
- the pharmaceutical composition can be produced by a method commonly used in the field of pharmaceutical technology, for example, a method described in the Japanese Pharmacopoeia.
- the content of the compound of the present invention in the pharmaceutical composition varies depending on the dosage form, the dose of the compound of the present invention, etc., but is, for example, about 0.1 to 100% by weight.
- coating may be performed for the purpose of taste masking, enteric properties or sustainability.
- coating base used for coating examples include sugar coating base, water-soluble film coating base, enteric film coating base and sustained-release film coating base.
- sucrose is used, and one or more selected from talc, precipitated calcium carbonate, gelatin, gum arabic, pullulan, carnauba wax and the like may be used in combination.
- water-soluble film coating base examples include cellulose polymers such as hydroxypropylcellulose, hydroxypropylmethylcellulose, hydroxyethylcellulose, and methylhydroxyethylcellulose; polyvinyl acetal diethylaminoacetate, aminoalkyl methacrylate copolymer E [Eudragit E (trade name) ], Synthetic polymers such as polyvinylpyrrolidone; polysaccharides such as pullulan.
- enteric film coating bases include cellulose polymers such as hydroxypropylmethylcellulose phthalate, hydroxypropylmethylcellulose acetate succinate, carboxymethylethylcellulose, and cellulose acetate phthalate; methacrylic acid copolymer L [Eudragit L (trade name) ] Acrylic acid polymers such as methacrylic acid copolymer LD [Eudragit L-30D55 (trade name)], methacrylic acid copolymer S [Eudragit S (trade name)]; natural products such as shellac.
- cellulose polymers such as hydroxypropylmethylcellulose phthalate, hydroxypropylmethylcellulose acetate succinate, carboxymethylethylcellulose, and cellulose acetate phthalate
- methacrylic acid copolymer L (Eudragit L (trade name) ]
- Acrylic acid polymers such as methacrylic acid copolymer LD [Eudragit L-30D55 (trade name)], methacrylic acid copolymer
- sustained-release film coating base examples include cellulose polymers such as ethyl cellulose; aminoalkyl methacrylate copolymer RS [Eudragit RS (trade name)], ethyl acrylate-methyl methacrylate copolymer suspension [Eudragit Acrylic polymer such as NE (trade name)].
- cellulose polymers such as ethyl cellulose; aminoalkyl methacrylate copolymer RS [Eudragit RS (trade name)], ethyl acrylate-methyl methacrylate copolymer suspension [Eudragit Acrylic polymer such as NE (trade name)].
- the above-mentioned coating bases may be used by mixing two or more of them in an appropriate ratio. Moreover, you may use light-shielding agents, such as a titanium oxide, ferric oxide, etc. in the case of coating.
- the compound of the present invention has low toxicity (eg, acute toxicity, chronic toxicity, genotoxicity, reproductive toxicity, cardiotoxicity, carcinogenicity), few side effects, and mammals (eg, humans, monkeys, cats, pigs, horses, Cattle, mice, rats, guinea pigs, dogs, rabbits, etc.) can be used as preventive / therapeutic agents or diagnostic agents for various diseases described below.
- toxicity eg, acute toxicity, chronic toxicity, genotoxicity, reproductive toxicity, cardiotoxicity, carcinogenicity
- mammals eg, humans, monkeys, cats, pigs, horses, Cattle, mice, rats, guinea pigs, dogs, rabbits, etc.
- the compound of the present invention has a potent delta-5-saturase inhibitory action, the prevention or prevention of a disease (or a disease whose onset is promoted) associated with an eicosanoid produced via delta-5-desaturase Useful as a therapeutic agent.
- diseases include heart diseases (cardiac hypertrophy, chronic heart failure including acute heart failure and congestion, cardiomyopathy, angina pectoris, myocarditis, arrhythmia, tachycardia, myocardial infarction, etc.), myocardial ischemia, vein Dysfunction, heart failure transition after myocardial infarction, hypertension, pulmonary heart, atherosclerosis including atherosclerosis (aneurysm, coronary sclerosis, cerebral arteriosclerosis, peripheral arteriosclerosis, etc.), vascular thickening, intervention ( Percutaneous coronary angioplasty, stenting, coronary endoscopy, intravascular ultrasound, coronary thrombolysis, etc.) and vascular thickening or occlusion and organ damage after heart transplantation, vascular re-occlusion / restenosis after bypass surgery , Respiratory disease (cold syndrome, pneumonia, asthma, pulmonary hypertension, pulmonary thrombus / pulmonary embolism, etc.), bone disease (fracture, re
- the compound of the present invention is particularly preferably used for the prevention or treatment of arteriosclerosis, diabetes and obesity.
- arteriosclerosis includes ischemic heart disease (unstable angina pectoris, acute myocardial infarction, acute heart failure, heart death) and stroke (transient Prevention of so-called atherothrombosis (including blood) and the delay of their progression, risk of high incidence of cardiovascular events based on arteriosclerosis progression inhibiting action (acute coronary artery disease patients, stroke patients, metabolic disease patients, hypertension / obesity) -Prevention of the onset of cardiovascular events, prevention of recurrence of ischemic heart disease, prevention of primary onset of cardiovascular events, prevention or treatment of peripheral arterial angiopathy, etc.
- ischemic heart disease unstable angina pectoris, acute myocardial infarction, acute heart failure, heart death
- stroke Transient Prevention of so-called atherothrombosis (including blood) and the delay of their progression, risk of high incidence of cardiovascular events based on arteriosclerosis
- diabetes is a fasting blood glucose level (glucose concentration in venous plasma) of 126 mg / dl or higher, and a 75 g oral glucose tolerance test (75 gOGTT) 2-hour value (glucose concentration in venous plasma) of 200 mg / dl or higher.
- 75 gOGTT 75 g oral glucose tolerance test
- a fasting blood glucose level (glucose concentration in venous plasma) is less than 110 mg / dl or a 75 g oral glucose tolerance test (75 g OGTT) 2 hour value (glucose concentration in venous plasma) is 140 mg / dl.
- a state that is not “a state indicating less than dl” (normal type) is referred to as a “boundary type”.
- diabetes is a fasting blood glucose level (glucose concentration in venous plasma) of 126 mg / dl or more, and a 75 g oral glucose tolerance test 2 hour value (glucose concentration in venous plasma) is 200 mg / dl. This is a state showing dl or more.
- glucose intolerance is a fasting blood glucose level (glucose concentration in venous plasma) of less than 126 mg / dl, and a 75-g oral glucose tolerance test 2 hour value (glucose concentration in venous plasma). Is a state showing 140 mg / dl or more and less than 200 mg / dl. Furthermore, according to the report of ADA, the state where the fasting blood glucose level (glucose concentration in venous plasma) is 110 mg / dl or more and less than 126 mg / dl is called IFG (Impaired Fasting Glucose).
- the IFG is a state where the 75 g oral glucose tolerance test 2 hour value (glucose concentration in venous plasma) is less than 140 mg / dl as IFG (Impaired Fasting Glycemia). Call.
- the compound of the present invention is also used as a prophylactic / therapeutic agent for diabetes, borderline type, impaired glucose tolerance, IFG (Impaired Fasting Glucose) and IFG (Impaired Fasting Glycemia) determined by the above criteria. Furthermore, the compound of the present invention can also prevent progression from borderline type, impaired glucose tolerance, IFG (Impaired Fasting Glucose) or IFG (Impaired Fasting Glycemia) to diabetes.
- the compound of the present invention is also used for secondary prevention and progression suppression of the various diseases described above (eg, cardiovascular events such as myocardial infarction).
- cardiovascular events such as myocardial infarction
- the compound of the present invention continuously suppresses the production of eicosanoid over a long period of time, thereby causing an inflammatory disease, for example, asthma, allergic airway hypersensitivity, which has been pointed out to be associated with inflammatory eicosanoids, Fever, pain, thrombosis, cerebral infarction, myocardial infarction, cancer, autoimmune encephalomyelitis, pain, renal failure, rheumatism, osteoarthritis, pruritus, atopic dermatitis, rhinitis, inflammatory bowel disease, clone It can be used for prevention or treatment of diseases and the like.
- an inflammatory disease for example, asthma, allergic airway hypersensitivity, which has been pointed out to be associated with inflammatory eicosanoids, Fever, pain, thrombosis, cerebral infarction, myocardial infarction, cancer, autoimmune encephalomyelitis, pain, renal failure, rheumatism, osteoarthritis, pruritus,
- disorders or abnormalities of biological functions and physiological actions include facial flushing and skin pain (including those associated with administration of nicotinic acid derivative preparations, prostacyclin preparations, etc.), overactive bladder, cerebral circulation / kidney Impaired or abnormal circulatory regulation, circulatory disorders (eg, peripheral, brain, microcirculation, etc.), cerebral blood barrier disorders, salt sensitivity, abnormal coagulation / fibrinolytic system, abnormal blood / blood cell components (eg, sickle cell red blood cells) Disease, increased platelet aggregation, abnormal red blood cell deformability, increased leukocyte adhesion, increased blood viscosity, etc., growth factors and cytokines (eg, PDGF, VEGF, FGF, interleukin, TNF- ⁇ , MCP-1, etc.) ) Production and action,
- Metabolic abnormalities eg, abnormal serum lipids, abnormal blood glucose, etc.
- angiogenesis including abnormal angiogenesis in the formation of abnormal capillary networks of the atherosclerotic epithelium, etc. .
- the compound of the present invention can also be used as an analgesic agent as a prophylactic / therapeutic agent for pain.
- pain disorders include acute pain due to inflammation, pain associated with chronic inflammation, pain associated with acute inflammation, postoperative pain (incisional pain, deep pain, visceral pain, postoperative chronic pain, etc.), muscle pain (chronic) Muscle pain associated with pain, stiff shoulder, etc.), joint pain, toothache, temporomandibular joint pain, headache (migraine, tension headache, fever headache, high blood pressure headache), visceral pain (heart pain, angina pain, abdominal pain) , Kidney pain, ureter pain, bladder pain), gynecological pain (intermediate pain, dysmenorrhea, labor pain), neuralgia (disc herniation, nerve root pain, postherpetic neuralgia, trigeminal neuralgia), cancer Pain, reflex sympathetic atrophy, complex local pain syndrome and the like.
- the compound of the present invention is effective for directly and immediately ameliorating various pains such as neuropathic pain, cancer pain, inflammatory pain, etc., and patients and pathological conditions in which the pain threshold is lowered (eg, hypertension, etc.) , And their complications).
- the content of the compound of the present invention in the pharmaceutical composition is usually about 0.01 to about 99.9% by weight, preferably about 0.1 to about 50% by weight, based on the whole preparation.
- the dose of the compound of the present invention depends on age, body weight, general health condition, sex, meal, administration time, administration method, excretion rate, combination of drugs, and the degree of the medical condition being treated at that time of the patient. It is decided in consideration of these and other factors.
- the dose varies depending on the target disease, symptom, administration subject, administration method, etc.
- the compound of the present invention when it is orally administered to an adult as a therapeutic agent for arteriosclerosis, it is usually about 0.01-100 mg / dose as a single dose.
- kg body weight preferably 0.05 to 30 mg / kg body weight, more preferably 0.5 to 10 mg / kg body weight, and this amount is preferably administered once to 3 times a day.
- the compound of the present invention is excellent in safety, it can be administered over a long period of time.
- the compounds of the present invention include, for example, anti-arteriosclerotic agents, antithrombotic agents, heart failure treatment agents, arrhythmia treatment agents, antihypertensive agents, diabetes treatment agents, diabetic complication treatment agents, HDL increase agents, antihyperlipidemic agents, anti-hyperlipidemic agents, Drugs such as obesity agents, diuretics, anti-inflammatory agents, anti-gout agents, chemotherapeutic agents, immunotherapeutic agents, osteoporosis treatment agents, anti-dementia agents, erectile dysfunction agents, urinary incontinence and dysuria treatment agents (hereinafter, (Abbreviated as a concomitant drug).
- concomitant drugs may be low molecular weight compounds, or may be high molecular weight proteins, polypeptides, antibodies, vaccines, or the like.
- anti-arteriosclerotic agent examples include Lp-PLA2 inhibitor (eg, dalapradiv, rilapradib etc.), FLAP inhibitor (eg, AM-103, AM-803, DG-031 etc.), sPLA2 inhibitor ( E.g., Valespradiv)), 5-lipoxygenase inhibitors (e.g., VIA-2291, etc.), acylcoenzyme A cholesterol acyltransferase (ACAT) inhibitors (e.g., melinamide, abashimive, eflusimib, etc.), lipid rich plaque regression Drugs (eg, compounds described in WO02 / 06264, WO03 / 059900, etc.), HDL preparations (eg, CSL-111, etc.), CTEP inhibitors (eg, torcetrapib, anacetrapib, darcetrapib etc.), MMP inhibitors, chymase inhibitors , SPT inhibitors, ApoA
- antithrombotic agent examples include blood coagulation inhibitors (eg, heparin sodium, heparin calcium, warfarin calcium (warfarin), antithrombin drugs (eg, argatroban, dabigatran), activated blood, and the like.
- blood coagulation inhibitors eg, heparin sodium, heparin calcium, warfarin calcium (warfarin)
- antithrombin drugs eg, argatroban, dabigatran
- Coagulation factor Xa inhibitor eg, rivaroxaban, apixaban, edoxaban, YM-150, WO02 / 06234, WO2004 / 048363, WO2005 / 030740, WO2005 / 058823, WO2005 / 113504 or WO2004 / Etc.
- thrombolytic drugs eg, tPA, urokinase, tisokinase,reteplase, nateplase, monteplase, pamitepase (pamite) plase
- antiplatelet drugs eg, aspirin, sulfinpyrazone (antulan), dipyridamole (persantin), ticlopidine (panaldin), cilostazol (pretal), GPIIb / IIIa antagonists (eg, leopro, etc.), clopidogrel, prasugrel ( prasugrel),
- cardiotonic drugs eg, digitoxin, digoxin, methyldigoxin, lanatoside C, prossilaridin, etc.
- ⁇ , ⁇ stimulants eg, epinephrine, norepinephrine, isoproterenol, dopamine, Docarpamine, dobutamine, denopamine, etc.
- phosphodiesterase inhibitors eg, amrinone, milrinone, olprinone hydrochloride, etc.
- calcium channel sensitivity enhancers eg, pimobentan, etc.
- nitrate drugs eg, nitroglycerin, isosorbide nitrate, etc.
- angiotensin conversion Enzyme inhibitors eg, angiotensin converting enzyme inhibitors described below
- angiotensin II antagonists eg, angiotensin II antagonists described below
- ⁇ -blockers eg, ⁇ blockers described below
- arrhythmic therapeutic agent examples include sodium channel blockers (eg, quinidine, procainamide, disopyramide, ajmarin, cibenzoline, lidocaine, diphenylhydantoin, mexiletine, propafenone, flecainide, pildicinide, phenytoin), ⁇ -blockers (Eg, propranolol, alprenolol, bufetrol, oxprenolol, atenolol, acebutolol, metoprolol, bisoprolol, pindolol, carteolol, arotilolol, etc.), potassium channel blockers (eg, amiodarone, etc.), calcium channel blockers ( Examples, verapamil, diltiazem, etc.).
- sodium channel blockers eg, quinidine, procainamide, disopyramide, ajmarin, cibenzoline, lid
- hypotensive agent examples include angiotensin converting enzyme inhibitors (eg, captopril, enalapril, delapril, etc.), angiotensin II antagonists (eg, candesartan cilexetil, candesartan, losartan, losartan potassium, eprosartan, valsartan, telmisartan, Irbesartan, tasosartan, olmesartan, olmesartan medoxomil, azilsartan, azilsartan medoxomil, etc.), calcium antagonists (eg, manidipine, nifedipine, amlodipine, efonidipine, nicardipine, sinyldipine, etc.), ⁇ -blockers (eg, metoprolol, atenolol protrolol, , Pindolol, etc.), clonidine and the like.
- insulin preparations eg, animal insulin preparations extracted from bovine and porcine pancreas; human insulin preparations synthesized by genetic engineering using Escherichia coli and yeast; insulin zinc; protamine insulin zinc; insulin Fragment or derivative (eg, INS-1), oral insulin preparation
- insulin resistance improving agent eg, pioglitazone or a salt thereof (preferably hydrochloride), rosiglitazone or a salt thereof (preferably maleate)
- Metaglidasen AMG-131, Balaglitazone, MBX-2044, Riboglitazone, Aleglitazar, Chiglitazar, Lobeglitazone, PLX-204, PN-2034 , GFT-505, THR-0921, WO2007 / 013694, WO2007 / 018314, WO2008 / 093639 or WO2008 / 0997 94
- ⁇ -glucosidase inhibitors eg, voglib
- diabetic complication therapeutic agents include aldose reductase inhibitors (eg, tolrestat, epalrestat, zopolrestat, fidarestat, CT-112, ranirestat (AS-3201), ridressat), neurotrophic factor and its increase drug (Eg, NGF, NT-3, BDNF, neurotrophin production / secretion promoter described in WO01 / 14372 (eg, 4- (4-chlorophenyl) -2- (2-methyl-1-imidazolyl) -5- [3- (2-methylphenoxy) propyl] oxazole), compounds described in WO2004 / 039365), PKC inhibitors (eg, ruboxistaurin mesylate), AGE inhibitors (eg, ALT946, N-phenol) Nasyl thiazolium bromide (ALT766), EXO-226, pyridoline (Pyridorin), pyridoxamine), GABA receptor .
- HDL increasing agent examples include squalene synthase inhibitors, CETP inhibitors (eg, torcetrapib, anacetrapib, darcetrapib, etc.), LPL activators, nicotinic acid drugs (eg, nicomol, niceritrol) (niceritrol)), endothelial lipase inhibitors and the like.
- antihyperlipidemic agent examples include, for example, statins that are cholesterol synthesis inhibitors (eg, cerivastatin, pravastatin, simvastatin, lovastatin, rosuvastatin, atorvastatin, fluvastatin, pitavastatin, or salts thereof (eg, sodium) Salt, etc.), squalene synthase inhibitors or triglyceride-lowering fibrate compounds (eg, bezafibrate, clofibrate, synfibrate, clinofibrate, etc.), cholesterol absorption inhibitors (eg, zetia), anion exchange resins (Eg, cholestyramine), probucol, nicotinic acid drugs (eg, nicomol, niceritrol), plant sterols (eg, soysterol, gamma-oryzanol), fish oil Agents (EPA, DHA, omacol, etc.), PPAR ⁇
- anti-obesity agents include monoamine uptake inhibitors (eg, phentermine, sibutramine, mazindol, floxetine, tesofensin), serotonin 2C receptor agonists (eg, lorcaserin), serotonin 6 receptor antagonist, histamine H3 receptor Body, GABA modulator (eg, topiramate), neuropeptide Y antagonist (eg, Berneperit), cannabinoid receptor antagonist (eg, rimonabant, taranaban), ghrelin antagonist, ghrelin receptor antagonist, ghrelin acylating enzyme Inhibitors, opioid receptor antagonists (eg, GSK-1521498), orexin receptor antagonists, melanocortin 4 receptor agonists, 11 ⁇ -hydroxysteroid dehydrogenase inhibitors (eg, AZD-4017), pancreatic lipase inhibitors (eg, , Orlistat, cetilistat), ⁇ 3
- monoamine uptake inhibitors
- FGF21 preparations eg, animals extracted from bovine, porcine pancreas
- FGF21 preparation human FGF21 preparation synthesized by genetic engineering using Escherichia coli and yeast
- antifeedant eg, P-57
- diuretic agent examples include xanthine derivatives (eg, sodium salicylate theobromine, calcium salicylate theobromine), thiazide preparations (eg, etiazide, cyclopenthiazide, trichloromethiazide, hydrochlorothiazide, hydroflumethiazide, benzylhydrochlorothiazide).
- xanthine derivatives eg, sodium salicylate theobromine, calcium salicylate theobromine
- thiazide preparations eg, etiazide, cyclopenthiazide, trichloromethiazide, hydrochlorothiazide, hydroflumethiazide, benzylhydrochlorothiazide.
- Penfluthiazide poly-5thiazide, methiclotiazide, etc.
- anti-aldosterone preparations eg, spironolactone, eplerenone, triamterene, etc.
- carbonic anhydrase inhibitors eg, acetazolamide, etc.
- chlorobenzenesulfonamide preparations eg, chlorthalidone
- azosemide isosorbide, ethacrynic acid, piretanide, bumetanide, furosemide and the like.
- anti-inflammatory agent examples include acetaminophen, phenacetin, etenzamide, sulpyrine, antipyrine, miglenin, aspirin, mefenamic acid, flufenamic acid, diclofenac sodium, loxoprofen sodium, phenylbutazone, indomethacin, ibuprofen, ketoprofen, naproxen.
- anti-gout agent examples include febuxostat, allopurinol, probenecid, colchicine, benzbromarone, febuxostat, citrate and the like.
- chemotherapeutic agent examples include alkylating agents (eg, cyclophosphamide, ifosfamide, etc.), antimetabolites (eg, methotrexate, 5-fluorouracil, etc.), anticancer antibiotics (eg, mitomycin, Adriamycin, etc.), plant-derived anticancer agents (eg, vincristine, vindesine, taxol, etc.), cisplatin, carboplatin, etoposide and the like. Of these, 5-fluorouracil derivatives such as furtulon or neofluturon are preferred.
- alkylating agents eg, cyclophosphamide, ifosfamide, etc.
- antimetabolites eg, methotrexate, 5-fluorouracil, etc.
- anticancer antibiotics eg, mitomycin, Adriamycin, etc.
- plant-derived anticancer agents eg, vincristine, vindesine, taxol, etc
- immunotherapy agent examples include microorganisms or bacterial components (eg, muramyl dipeptide derivatives, picibanil, etc.), polysaccharides having immunopotentiating activity (eg, lentinan, schizophyllan, krestin, etc.), genetic engineering techniques.
- cytokines obtained eg, interferon, interleukin (IL), etc.
- colony stimulating factors eg, granulocyte colony stimulating factor, erythropoietin, etc.
- IL-1 interferon, interleukin (IL), etc.
- IL-12 e.g. preferable.
- osteoporosis therapeutic agent '' examples include, for example, alfacalcidol, calcitriol, elcaltonin, salmon calcitonin (calcitoninolsalmon), estriol, ipriflavone, and pamidronate.
- examples include sodium (pamidronate disodium), alendronate sodium hydrate, minderonate disodium ⁇ , and the like.
- anti-dementia agent examples include tacrine, donepezil, rivastigmine, galantamine, and the like.
- erectile dysfunction ameliorating agent examples include apomorphine, PDE5 (phosphodiesterase 5) inhibitor (eg, sildenafil citrate), and the like.
- urine incontinence therapeutic agent examples include flavoxate hydrochloride, oxybutynin hydrochloride, propiverine hydrochloride and the like.
- examples of the above “difficulty to urinate” include acetylcholinesterase inhibitors (eg, distigmine) and the like.
- concomitant drugs include prostacyclin preparations / derivatives (eg, beraprost, epoprostenol, iloprost, treprostinil, etc.), prostaglandin preparations / derivatives (eg, emprostil, alprostadil, limaprost, misoprostol, orno Prostil, etc.), anti-asthma drugs (eg, salmeterol, fluticasone, montelukast), rheumatoid arthritis drugs (eg, etanercept, infliximab, adalimumab), nerve regeneration promoting drugs (eg, Y-128, VX-853, prosaptide), Antidepressants (eg, desipramine, amitriptyline, imipramine), antiepileptic drugs (eg, lamotrigine), antiarrhythmic drugs (eg, mexiletine), acetylcholine receptor ligands (
- the administration time of the aforementioned concomitant drug is not limited, and the compound of the present invention and the concomitant drug may be administered simultaneously to the administration subject or may be administered with a time difference.
- the dose of the concomitant drug may be determined according to the dose used clinically, and can be appropriately selected depending on the administration subject, administration route, disease, combination and the like.
- these concomitant drugs may be combined in an appropriate ratio of two or more.
- the administration time of the compound of the present invention and the concomitant drug is not limited, and the compound of the present invention and the concomitant drug may be combined at the time of administration.
- Examples of such administration forms include (1) administration of a single preparation obtained by simultaneously formulating the compound of the present invention and a concomitant drug, and (2) formulating the compound of the present invention and the concomitant drug separately. Simultaneous administration of the two obtained preparations by the same administration route, (3) administration of the two preparations obtained by separately formulating the compound of the present invention and the concomitant drug with a time difference in the same administration route, (4) Simultaneous administration by different administration routes of two types of preparations obtained by separately formulating the compound of the present invention and a concomitant drug, (5) 2 obtained by formulating the compound of the present invention and the concomitant drug separately Administration of different types of preparations at different time intervals (for example, administration in the order of the compound of the present invention ⁇ concomitant drug or administration in the reverse order) and the like.
- the dose of the concomitant drug can be appropriately selected based on the clinically used dose.
- the compounding ratio of the compound of the present invention and the concomitant drug can be appropriately selected depending on the administration subject, administration route, target disease, symptom, combination and the like.
- the concomitant drug may be used in an amount of 0.01 to 100 parts by weight per 1 part by weight of the compound of the present invention.
- the compound of the present invention By combining the compound of the present invention and a concomitant drug, (1) The dose can be reduced compared to the case where the compound of the present invention or the concomitant drug is administered alone. (2) By selecting a concomitant drug having a different mechanism of action from the compound of the present invention, the treatment period can be set longer. (3) By selecting a concomitant drug having a different mechanism of action from the compound of the present invention, the therapeutic effect can be sustained. (4) By using the compound of the present invention and a concomitant drug in combination, excellent effects such as a synergistic effect can be obtained.
- HPLC-mass spectrum (LC-MS) was measured under the following conditions.
- Measuring equipment Waters Micromass ZQ-Alliance HT Column: CAPCELL PAK C18UG120, S-3 ⁇ m, 1.5 X 35 mm
- Injection volume 2 ⁇ L
- flow rate 0.5 mL / min
- detection method UV 220 nm
- Ionization method Electron Spray Ionization (ESI)
- Ethyl 3-oxo-3- (pyridin-4-yl) propanoate Ethyl isonicotinate (3.02 g), sodium hydride (60% oil dispersion, 0.53 g) and ethyl acetate (10 mL) Stir at 70 ° C. for 3 hours. A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate / hexane) to give the title compound (1.8 g).
- Example 8 2- (Ethylsulfanyl) -6- (pyridin-3-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one Method according to Example 7 Gave the title compound from ethyl nicotinate.
- Example 14 2- (Methylsulfanyl) -6- (pyridin-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one in I) of Example 1) According to a similar method, 6- (pyridin-4-yl) -2-thioxo-3- [4- (2,2,2-trifluoroethoxy) phenyl] -2,3-dihydropyrimidine-4 (1H)- The title compound was obtained from on and methyl iodide.
- Example 19 2- (Ethylsulfanyl) -6- (pyridazin-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one
- Example 25 2- (Ethylsulfanyl) -6- (6-methoxypyridin-3-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one in Example 7
- the title compound was obtained from 6-methoxypyridine-3-carboxylic acid by a similar method.
- Example 28 2- (Ethylsulfanyl) -5-methyl-6- (2-oxo-1,2-dihydropyridin-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine-4 (3H) -one
- the title compound was obtained from (trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one.
- Example 30 2-Ethoxy-5-methyl-6- (2-oxo-1,2-dihydropyridin-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine-4 (3H) -On by the method according to Example 24, 2- (ethylsulfanyl) -5-methyl-6- (2-oxo-1,2-dihydropyridin-4-yl) -3- [4- (2,2, The title compound was obtained from 2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one.
- Example 31 2- (2-methoxyethoxy) -6- (2-oxo-1,2-dihydropyridin-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine-4 (3H ) -One 2- (ethylsulfanyl) -6- (2-oxo-1,2-dihydropyridin-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine-4 ( A reaction mixture of 3H) -one (120 mg), sodium hydride (60% oil dispersion, 171 mg) and 2-methoxyethanol (3 mL) was stirred at 0 ° C. for 3 hours.
- Example 33 2-Ethoxy-6- (pyridazin-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one According to the method of Example 42, 2 The title compound was obtained from-(ethylsulfanyl) -6- (pyridazin-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one and ethanol .
- Example 35 2-Methoxy-1- [4- (2,2,2-trifluoroethoxy) phenyl] -4,4′-bipyrimidin-6 (1H) -one According to the method of Example 32, 2- (ethylsulfanyl The title compound was obtained from) -1- [4- (2,2,2-trifluoroethoxy) phenyl] -4,4′-bipyrimidin-6 (1H) -one.
- Example 36 2-Ethoxy-1- [4- (2,2,2-trifluoroethoxy) phenyl] -4,4'-bipyrimidin-6 (1H) -one 2- (ethylsulfanyl) -1- [4- (2 , 2,2-Trifluoroethoxy) phenyl] -4,4'-bipyrimidin-6 (1H) -one (150 mg), sodium hydride (60% oil dispersion, 44 mg) and ethanol (3 mL) The mixture was stirred at 70 ° C. for 3 hours, 1N hydrochloric acid was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with 1N aqueous sodium hydroxide solution and saturated brine, and dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure to obtain the title compound (86 mg).
- Example 39 2-propoxy-1- [4- (2,2,2-trifluoroethoxy) phenyl] -4,4'-bipyrimidin-6 (1H) -one 2- (ethylsulfanyl) -1- [4- (2 , 2,2-Trifluoroethoxy) phenyl] -4,4'-bipyrimidin-6 (1H) -one (100 mg), sodium hydride (60% oil dispersion, 59 mg), propan-1-ol (2.2 mL) and THF (5 mL) were stirred at 65 ° C. for 2 h. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- Example 41 [3-Chloro-4- (2,2,2-trifluoroethoxy) phenyl] -2- (ethylsulfanyl) -4,4′-bipyrimidin-6 (1H) -one from Example 20 B) (C)) by a method according to (2Z) -3-amino-3- (pyrimidin-4-yl) prop-2-enoic acid ethyl and 2-chloro-4-isothiocyanato-1- (2,2,2- The title compound was obtained from trifluoroethoxy) benzene.
- Example 43 [3-Chloro-4- (2,2,2-trifluoroethoxy) phenyl] -2-ethoxy-4,4′-bipyrimidin-6 (1H) -one
- the title compound was obtained from-[3-chloro-4- (2,2,2-trifluoroethoxy) phenyl] -2- (ethylsulfanyl) -4,4'-bipyrimidin-6 (1H) -one.
- Example 44 2-Ethoxy-1- [6- (2,2,2-trifluoroethoxy) pyridin-3-yl] -4,4′-bipyrimidin-6 (1H) -one According to a method similar to that in Example 42, 2 The title compound was obtained from-(ethylsulfanyl) -1- [6- (2,2,2-trifluoroethoxy) pyridin-3-yl] -4,4'-bipyrimidin-6 (1H) -one.
- Example 45 2- (Ethylsulfanyl) -1- [4- (2,2,2-trifluoroethoxy) phenyl] -4,5′-bipyrimidin-6 (1H) -one
- pyrimidine- The title compound was obtained from 5-carboxylic acid.
- Example 47 2- (Oxetane-3-yloxy) -1- [4- (2,2,2-trifluoroethoxy) phenyl] -4,4′-bipyrimidin-6 (1H) -one According to the method according to Example 42 2- (ethylsulfanyl) -1- [4- (2,2,2-trifluoroethoxy) phenyl] -4,4′-bipyrimidin-6 (1H) -one and oxetan-3-ol Obtained.
- the extract was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
- the obtained residue was dissolved in trifluoroacetic acid (5 mL) and stirred at 70 ° C. for 8 hours.
- the reaction solvent was evaporated under reduced pressure, saturated aqueous sodium hydrogen carbonate was added, and the mixture was extracted with ethyl acetate.
- the extract was dried over anhydrous magnesium sulfate and the solvent was removed under reduced pressure.
- the residue was purified by silica gel column chromatography (ethyl acetate / hexane) to give the title compound (320 mg).
- reaction solvent was evaporated under reduced pressure, saturated aqueous sodium hydrogen carbonate was added, and the mixture was extracted with ethyl acetate. The extract was dried over anhydrous magnesium sulfate and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate / hexane) and recrystallized from THF / hexane to give the title compound (500 mg).
- Example 70 2-Methoxy-6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one according to Example 69 B) 2- (ethylsulfonyl) -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one and methanol To give the title compound. MS (ESI +): [M + H] + 367.3.
- Example 71 6- (1-Methyl-1H-pyrazol-4-yl) -2- (2,2,2-trifluoroethoxy) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine- 4 (3H) -one
- 6- (1H-pyrazol-4-yl) -2- (2,2,2-trifluoroethoxy) -3- [4- (2,2, 2-Trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one was used to give the title compound.
- Example 72 (1H-pyrazol-4-yl) -2- (tetrahydrofuran-3-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one tetrahydrofuran-
- 3-carboxylic acid 468 mg
- DMF 5 ⁇ L
- oxalyl chloride 0.35 mL
- the reaction solution was (2Z) -3-amino-3- [1- (4-methoxybenzyl) -1H-pyrazol-4-yl] -N- [4- (2,2,2-trifluoroethoxy) phenyl]
- DMA solution 10 mL
- prop-2-enamide 300 mg
- Saturated aqueous sodium hydrogen carbonate was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- the extract was dried over anhydrous magnesium sulfate and the solvent was removed under reduced pressure.
- Example 75 2- (2,2-difluoroethoxy) -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one 2- (Ethylsulfonyl) -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine-4 by a method according to Example 69 B) The title compound was obtained using (3H) -one and 2,2-difluoroethanol. MS (ESI +): [M + H] + 417.2.
- Example 76 2- [2-Fluoro-1- (fluoromethyl) ethoxy] -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine-4 ( 3H) -one 2- (ethylsulfonyl) -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) by a method according to Example 69 B) The title compound was obtained using [phenyl] pyrimidin-4 (3H) -one and 1,3-difluoropropan-2-ol. MS (ESI +): [M + H] + 431.2.
- Example 78 6- (1-Methyl-1H-pyrazol-4-yl) -2- (2,2,2-trifluoroethoxy) -3- [6- (2,2,2-trifluoroethoxy) pyridine-3- Yl] pyrimidin-4 (3H) -one
- silica gel column chromatography ethyl acetate / hexane
- HPLC mobile phase: water / Acetonitrile (TFA-containing system)
- Example 69 The reaction and post-treatment were carried out using 2-methoxyethanol by the method according to (B) of the above, and after purification by silica gel column chromatography (ethyl acetate / hexane), recrystallization from ethyl acetate / hexane gave the title compound. Obtained as a solid. MS (ESI +): [M + H] + 411.1.
- Example 80 2- (2-Fluoroethoxy) -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one
- Example 69 The reaction and post-treatment were carried out using 2-fluoroethanol by the method according to B) of the above, purified by silica gel column chromatography (ethyl acetate / hexane), and recrystallized from ethyl acetate / hexane to give the title compound. Obtained as a solid. MS (ESI +): [M + H] + 399.1.
- Example 81 2- (2,2-Difluoroethoxy) -6- (1-methyl-1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine-4 (3H 2- (2,2-difluoroethoxy) -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) by a method according to Example 3 The title compound was obtained using phenyl] pyrimidin-4 (3H) -one. MS (ESI +): [M + H] + 431.0.
- Example 82 2- (2-Methoxyethoxy) -6- (1-methyl-1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine-4 (3H)- ON 2- (2-Methoxyethoxy) -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine- by the method according to Example 3 Reaction and workup with 4 (3H) -one, purification by aminopropylsilane-bonded silica gel column chromatography (ethyl acetate / hexane), and recrystallization from THF / hexane gave the title compound as a solid . MS (ESI +): [M + H] + 425.2.
- Example 83 2- [2-Fluoro-1- (fluoromethyl) ethoxy] -6- (1-methyl-1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] Pyrimidin-4 (3H) -one
- 2- [2-fluoro-1- (fluoromethyl) ethoxy] -6- (1H-pyrazol-4-yl) -3- [4- ( 2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one was used to give the title compound.
- Example 84 (1-Methyl-1H-pyrazol-4-yl) -2- (propan-2-yloxy) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine-4 (3H) -On 2- (propan-2-yloxy) -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] by a method according to Example 3 The title compound was obtained using pyrimidine-4 (3H) -one. MS (ESI +): [M + H] + 409.1.
- Example 87 2-Ethoxy-6- (1,3-thiazol-5-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one
- Example 69B 2- (ethylsulfonyl) -6- (1,3-thiazol-5-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine-4 (3H ) -One and ethanol were used for the reaction and workup, and the residue was purified by silica gel column chromatography (ethyl acetate / hexane) and recrystallized from ethyl acetate / hexane to give the title compound as a solid.
- Example 92 2-Ethoxy-5-methyl-6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one
- 2- (ethylsulfonyl) -5-methyl-6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine-4 ( Synthesis and purification from 3H) -one and ethanol, followed by recrystallization from ethyl acetate and hexane, gave the title compound. MS (ESI +): [M + H] + 395.3.
- Example 93 2-Ethoxy-6- (1H-pyrazol-3-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one according to Example 69 B) 2- (ethylsulfonyl) -6- (1H-pyrazol-3-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one and The reaction and post-treatment were performed using ethanol, and the residue was purified by silica gel column chromatography (ethyl acetate / hexane), and then recrystallized from THF / hexane to obtain the title compound as a solid. MS (ESI +): [M + H] + 381.2.
- Example 94 (1-Methyl-1H-pyrazol-3-yl) -2- (2,2,2-trifluoroethoxy) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine- 4 (3H) -one
- 6- (1H-pyrazol-3-yl) -2- (2,2,2-trifluoroethoxy) -3- [4- (2,2, 2-Trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one was reacted and worked up, purified by aminopropylsilane-bonded silica gel column chromatography (ethyl acetate / hexane), and then purified from ethyl acetate / hexane. Recrystallization gave the title compound as a solid.
- the reaction mixture was (2Z) -3-amino-3- [1- (4-methoxybenzyl) -1H-pyrazol-4-yl] -N- [4- (2,2,2-trifluoroethoxy) phenyl ]
- acrylamide 200 mg
- DMA dimethyl methacrylate
- Saturated aqueous sodium bicarbonate was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- the extract layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure.
- Example 100 2- (1-methoxypropyl) -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one
- Example 97 The title compound was obtained from 2-methoxybutanoic acid by a method analogous to MS (ESI +): [M + H] + 409.3.
- Example 101 2- (1,1-Difluoropropyl) -5-methyl-6- (1-methyl-1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine -4 (3H) -one 2- (1,1-difluoropropyl) -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine- Sodium hydride (60% oil dispersion, 24.8 mg) and methyl iodide (52 ⁇ L) were added to a DMF solution (15 mL) of 4 (3H) -one (171 mg) at 0 ° C.
- Example 103 6- (1-Methyl-1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] -2- (3,3,3-trifluoropropyl) pyrimidine- 4 (3H) -one
- 1-methyl-4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1H The title compound was obtained as a solid from -pyrazole.
- the reaction mixture was (2Z) -3-amino-3- [1- (4-methoxybenzyl) -1H-pyrazol-4-yl] -N- [4- (2,2,2-trifluoroethoxy) phenyl ]
- acrylamide (1 g) in DMA (15 mL)
- DMA liquid-based polymethyl methacrylate
- Saturated aqueous sodium bicarbonate was added to the reaction mixture, and the mixture was extracted with ethyl acetate.
- the extract layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure.
- Example 105 2- (1-Methylcyclopropyl) -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one
- HPLC HPLC [C18 mobile phase: water / acetonitrile (NH 4 HCO 3 containing system)]
- the obtained residue was dissolved in methanol (50 mL) and 8N aqueous sodium hydroxide solution (80 mL), and the mixture was stirred at room temperature for 7 hr.
- the reaction mixture was concentrated to half volume and washed with diethyl ether.
- the aqueous layer was acidified with 6N hydrochloric acid, and extracted with ethyl acetate.
- the extract layer was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure.
- the residue was purified by silica gel column chromatography (ethyl acetate / hexane) to give the title compound (5.43 g).
- Example 107 6- (morpholin-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] -2- (3,3,3-trifluoropropyl) pyrimidin-4 (3H) -one
- palladium (II) acetate 8.7 mg
- 4,5-bis (diphenylphosphino) -9,9-dimethylxanthene 34 mg
- trifluoromethanesulfonic acid 6-oxo-1- 4- (2,2,2-trifluoroethoxy) phenyl] -2- (3,3,3-trifluoropropyl) -1,6-dihydropyrimidin-4-yl (200 mg)
- cesium carbonate 317 mg
- One Reaction and post-treatment were performed using piperazin-2-one by the method according to Example 107, and purified by aminopropylsilane-bonded silica gel column chromatography (methanol / ethyl acetate), followed by ethyl acetate / hexane. To give the title compound as a solid.
- reaction solution was (2Z) -3-amino-3- (1-methyl-1H-pyrazol-4-yl) -N- [4- (2,2,2-trifluoroethoxy) phenyl] prop-2-enamide (400 mg) in DMA solution (10 mL) and stirred at 150 ° C. for 1 hour under microwave irradiation. Saturated aqueous sodium hydrogen carbonate was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was dried over anhydrous magnesium sulfate and the solvent was removed under reduced pressure.
- Example 110 6- (5-Methyl-1,2,4-oxadiazol-3-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] -2- (3,3,3- (Trifluoropropyl) pyrimidin-4 (3H) -one
- reaction mixture was concentrated under reduced pressure. THF was added to the residue, the insoluble matter was removed by filtration, and the filtrate was concentrated under reduced pressure. 1N Hydrochloric acid (100 mL) was added to a THF (100 mL) solution of the obtained residue at 0 ° C., and the mixture was stirred for 2 hr. The reaction mixture was extracted with ethyl acetate, and the extract was washed with saturated brine, dried over anhydrous magnesium sulfate, and concentrated under reduced pressure.
- Example 112 6- (1-Methyl-1H-pyrazol-4-yl) -2- (pentafluoroethyl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one
- DMF 1,3-bis(trifluoroethoxy) phenyl] pyrimidin-4
- the reaction solution was (2Z) -3-amino-3- (1-methyl-1H-pyrazol-4-yl) -N- [4- (2,2,2-trifluoroethoxy) phenyl] prop-2-enamide (500 mg) in DMA solution (15 mL) and stirred at room temperature for 3 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was dried over anhydrous magnesium sulfate and the solvent was removed under reduced pressure. The obtained residue was purified by silica gel column chromatography (ethyl acetate / hexane), dissolved in trifluoroacetic acid (3 mL), and stirred at 120 ° C.
- Example 116 6- (3-Methyl-1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] -2- (3,3,3-trifluoropropyl) pyrimidine- 4 (3H) -one
- 3-methyl-4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -1H -Pyrazole-1-carboxylate tert-butyl was reacted and worked up, purified by aminopropylsilane-bonded silica gel column chromatography (methanol / ethyl acetate) and recrystallized from ethyl acetate / hexane to give the title compound Was obtained as a solid.
- Example 118 2- (3,3-Difluorocyclobutyl) -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one
- the title compound was obtained from 3,3-difluorocyclobutanecarboxylic acid by a method according to Example 102.
- Example 119 2- (2,2-difluoroethoxy) -3- [3-fluoro-4- (2,2,2-trifluoroethoxy) phenyl] -6- (1H-pyrazol-4-yl) pyrimidine-4 (3H ) -One
- 2- (ethylsulfanyl) -3- [3-fluoro-4- (2,2,2-trifluoroethoxy) phenyl] -6- (1H-pyrazol-4-yl) pyrimidin-4 (3H) -one was obtained.
- Example 120 6- (3,3-Difluoropyrrolidin-1-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] -2- (3,3,3-trifluoropropyl) pyrimidine-4 (3H) -one
- 3H 3,3-difluoropyrrolidine hydrochloride
- Example 123 2- (1,1-Difluoroethyl) -6- (1H-pyrazol-4-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one
- the title compound was obtained as a solid from piperidin-4-one hydrochloride by a method according to Example 107.
- Example 127 3- [3-Fluoro-4- (2,2,2-trifluoroethoxy) phenyl] -6- (1H-pyrazol-4-yl) -2- (pyrrolidin-1-yl) pyrimidine-4 (3H) -On 2- (ethylsulfanyl) -3- [3-fluoro-4- (1) in the same manner as in Example 1 except that 1,2-difluoro-4-nitrobenzene was used instead of 1-fluoro-4-nitrobenzene. 2,2,2-trifluoroethoxy) phenyl] -6- (1H-pyrazol-4-yl) pyrimidin-4 (3H) -one was obtained.
- the title compound was obtained from 2- (2,2,2, -trifluoroethoxy) acetic acid by the method according to Example 97.
- Example 130 6- (1,2,4-oxadiazol-3-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] -2- (3,3,3-trifluoropropyl) Pyrimidin-4 (3H) -one N'-hydroxy-6-oxo-1- [4- (2,2,2-trifluoroethoxy) phenyl] -2- (3,3,3-trifluoropropyl)- Boron trifluoride diethyl etherate (6.09 ⁇ L) was added to a suspension of 1,6-dihydropyrimidine-4-carboximidamide (204 mg) and trimethoxymethane (5 mL) at room temperature and stirred for 3 hours. .
- reaction mixture was diluted with (Z) -3-amino-3- (1-methyl-1H-pyrazol-5-yl) -N- [4- (2,2,2-trifluoroethoxy) phenyl] acrylamide (200 mg ) In DMA (6 mL) and stirred at 50 ° C. for 2 hours. Thereafter, the reaction solution was stirred at 150 ° C. for 30 minutes under microwave irradiation. Saturated aqueous sodium bicarbonate was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The extract layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure.
- Example 132 2- (1,1-Difluoropropyl) -6- (1-methyl-1H-pyrazol-5-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine-4 (3H ) -One 2,2-difluorobutanoic acid (365 mg) and DMF (0.011 mL) were dissolved in THF (1.5 mL), oxalyl chloride (0.205 mL) was added at 0 ° C, and the reaction mixture was stirred at room temperature for 2.5 hours. did.
- the reaction mixture was diluted with (Z) -3-amino-3- (1-methyl-1H-pyrazol-5-yl) -N- [4- (2,2,2-trifluoroethoxy) phenyl] acrylamide (200 mg ) In DMA (6 mL) and stirred at 50 ° C. for 1.5 hours. Saturated aqueous sodium bicarbonate was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The extract layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was dissolved in TFA (10 mL) and stirred at 120 ° C. for 3 hours under microwave irradiation.
- Example 133 2- (Ethylsulfanyl) -6- (5-oxopyrrolidin-3-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidin-4 (3H) -one of Example 1 F) to J), 1- (4-methoxybenzyl) -5-oxopyrrolidine-3-carboxylic acid was used instead of 1- (4-methoxybenzyl) -1H-pyrazole-4-carboxylic acid.
- the title compound was obtained by changing the deprotection conditions of J) as follows: That is, after stirring at 120 ° C. for 2 hours under microwave irradiation, the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate / hexane) to give the title compound. MS (ESI +): [M + H] + 414.1.
- the reaction mixture was concentrated under reduced pressure, saturated aqueous sodium hydrogen carbonate solution was added to the residue, and the mixture was extracted with ethyl acetate. The extract was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was subjected to aminopropylsilane-bonded silica gel column chromatography (ethyl acetate / hexane) to give the title compound (14.8 g) as a solid.
- Example 135 2- [2-Fluoro-1- (fluoromethyl) ethoxy] -6- (1-methyl-1H-pyrazol-5-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] Pyrimidin-4 (3H) -one
- 1,3-difluoropropan-2-ol was used instead of 2,2,2-trifluoroethanol to give the title compound.
- Example 136 2- (3,3-Difluoropyrrolidin-1-yl) -6- (1-methyl-1H-pyrazol-5-yl) -3- [4- (2,2,2-trifluoroethoxy) phenyl] pyrimidine -4 (3H) -one
- 3,3-difluoropyrrolidine instead of 2,2,2-trifluoroethanol, the title compound was obtained.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Diabetes (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Child & Adolescent Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
デルタ-5-デサチュラーゼ阻害作用を有し、動脈硬化、糖尿病、肥満症等の疾患等の予防・治療に有用であり、かつ優れた薬効を有する化合物を提供すること。 本発明は、式 (Ⅰ)〔式中、各記号は明細書記載のとおり〕で表される化合物またはその塩を提供する。
Description
本発明は、医薬として優れた性質を有する新規複素環化合物およびその用途に関する。詳しくは、本発明は、デルタ-5-デサチュラーゼ阻害作用を有し、エイコサノイドの産生を抑制することに基づく種々の薬理作用を示し、良好な結晶性および安定性等の優れた物性を有し、アテローム性動脈硬化症、アテローム血栓症、糖尿病、肥満、喘息、発熱、疼痛、癌、リウマチ、変形性関節症、アトピー性皮膚炎等のエイコサノイド関連疾患の予防・治療剤として有用な、特定構造を有する複素環化合物、またはその塩、またはそのプロドラッグおよびその用途等に関する。
(発明の背景)
プロスタグランジン、ロイコトリエン、トロンボキサン等のエイコサノイドは、各種疾患において重要な役割を果たしていると考えられている。例えば炎症性疾患である動脈硬化、糖尿病、肥満、喘息、リウマチ、変形性関節症および炎症性疼痛等の疾患においては、炎症性エイコサノイドの産生経路が亢進し、疾患の発症、増悪に関わっていると考えられている。
エイコサノイド関連疾患の治療薬としては、プロスタノイド類の産生を抑制するシクロオキシゲナーゼ阻害剤やトロンボキサンA2受容体拮抗剤等の薬剤が臨床応用されている。しかし炎症性疾患等の予防・治療薬のニーズは依然として高く、強力かつ副作用の少ない治療薬の開発が切望されている。
プロスタグランジン、ロイコトリエン、トロンボキサン等のエイコサノイドは、各種疾患において重要な役割を果たしていると考えられている。例えば炎症性疾患である動脈硬化、糖尿病、肥満、喘息、リウマチ、変形性関節症および炎症性疼痛等の疾患においては、炎症性エイコサノイドの産生経路が亢進し、疾患の発症、増悪に関わっていると考えられている。
エイコサノイド関連疾患の治療薬としては、プロスタノイド類の産生を抑制するシクロオキシゲナーゼ阻害剤やトロンボキサンA2受容体拮抗剤等の薬剤が臨床応用されている。しかし炎症性疾患等の予防・治療薬のニーズは依然として高く、強力かつ副作用の少ない治療薬の開発が切望されている。
WO2007/146761(特許文献1)には、下式
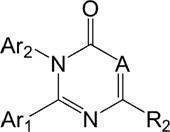
(式中、AはCR1またはNであり;Ar1およびAr2はそれぞれフェニル、ナフチルおよび5ないし10員へテロアリールから選ばれ、それぞれが置換されていてもよく、それぞれが好ましくはRAから独立して選ばれる0ないし6個の置換基で置換されていてもよい;
R1は:
(i) 水素、ハロゲン、ヒドロキシ、シアノ、アミノ、ニトロ、アミノカルボニル、アミノスルフォニルまたは-COOH;
(ii) C1-C6アルキル、C2-C6アルケニル、C2-C6アルキニル、(C3-C8シクロアルキル)C0-C4アルキル、C2-C6アルキルエステル、C1-C6アルコキシ、C1-C6アルキルチオ、C3-C6アルカノン、C1-C6アルコキシカルボニル、 モノ-またはジ-(C1-C6アルキル)アミノC0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノカルボニルC0-C4アルキル、(C1-C6アルキルスルフォニル)C0-C4アルキル、(C1-C6アルキル)スルフォニルアミノC0-C4アルキルまたは(4ないし8員ヘテロサイクル)C0-C4アルキル;それぞれが置換されていてもよく、それぞれが好ましくはRBから独立して選ばれる0ないし6個の置換基で置換されていてもよい;または
(iii) R2と結合して縮合された5ないし8員炭素環またはヘテロサイクルを形成し、それぞれが置換されていてもよく、それぞれが好ましくはRBから独立して選ばれる0ないし4個の置換基で置換されていてもよい;
R2は水素で置換されず; 好ましくは、R2は:
(i) C1-C6アルキル、C2-C6アルケニル、C2-C6アルキニル、(C3-C8シクロアルキル)C0-C4アルキル、C2-C6アルキルエステル、C1-C6アルコキシ、C1-C6アルコキシカルボニル、モノ-またはジ-(C1-C6アルキル)アミノC0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノカルボニルC0-C4アルキル、(C1-C6アルカノイル)アミノC0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノスルフォニルC0-C4アルキル、(C1-C6アルキル)スルフォニルC0-C4アルキル、モノ-またはジ-(C1-C6アルキル) スルフォニルアミノC0-C4アルキル、(C3-C10カルボサイクル) C0-C4アルキルまたは(4-ないし10員 ヘテロサイクル)C0-C4アルキル;それぞれはRBから独立して選ばれる0ないし6個の置換基で置換されていてもよい;
(ii) 式:
R1は:
(i) 水素、ハロゲン、ヒドロキシ、シアノ、アミノ、ニトロ、アミノカルボニル、アミノスルフォニルまたは-COOH;
(ii) C1-C6アルキル、C2-C6アルケニル、C2-C6アルキニル、(C3-C8シクロアルキル)C0-C4アルキル、C2-C6アルキルエステル、C1-C6アルコキシ、C1-C6アルキルチオ、C3-C6アルカノン、C1-C6アルコキシカルボニル、 モノ-またはジ-(C1-C6アルキル)アミノC0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノカルボニルC0-C4アルキル、(C1-C6アルキルスルフォニル)C0-C4アルキル、(C1-C6アルキル)スルフォニルアミノC0-C4アルキルまたは(4ないし8員ヘテロサイクル)C0-C4アルキル;それぞれが置換されていてもよく、それぞれが好ましくはRBから独立して選ばれる0ないし6個の置換基で置換されていてもよい;または
(iii) R2と結合して縮合された5ないし8員炭素環またはヘテロサイクルを形成し、それぞれが置換されていてもよく、それぞれが好ましくはRBから独立して選ばれる0ないし4個の置換基で置換されていてもよい;
R2は水素で置換されず; 好ましくは、R2は:
(i) C1-C6アルキル、C2-C6アルケニル、C2-C6アルキニル、(C3-C8シクロアルキル)C0-C4アルキル、C2-C6アルキルエステル、C1-C6アルコキシ、C1-C6アルコキシカルボニル、モノ-またはジ-(C1-C6アルキル)アミノC0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノカルボニルC0-C4アルキル、(C1-C6アルカノイル)アミノC0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノスルフォニルC0-C4アルキル、(C1-C6アルキル)スルフォニルC0-C4アルキル、モノ-またはジ-(C1-C6アルキル) スルフォニルアミノC0-C4アルキル、(C3-C10カルボサイクル) C0-C4アルキルまたは(4-ないし10員 ヘテロサイクル)C0-C4アルキル;それぞれはRBから独立して選ばれる0ないし6個の置換基で置換されていてもよい;
(ii) 式:

式中、qは0または1;それぞれのnは独立して0、1または2;およびXはO、C(=O)、S、SO、SO2、N(R3)またはC(R4a)(R4b);または
(iii) R1と結合して縮合された5ないし8員の、置換されていてもよい炭素環またはヘテロサイクルを形成し;
R3は:(i) 水素;または(ii) C1-C6アルキル、C2-C6アルケニル、C2-C6アルキニル、C1-C6アルキルスルフォニル、(C3-C8炭素環) C0-C4アルキルまたは(4-ないし8員ヘテロサイクル)C0-C4アルキルであり、それぞれが置換されていてもよく、それぞれが好ましくはRBから独立して選ばれる0ないし6個の置換基で置換されていてもよい;
R4aおよびR4bは独立して:
(i) 水素、ハロゲン、シアノ、-COOHまたはC1-C6アルキルであり;
(ii) -Z-N(R5)(R6)または-Z-O(R7)、Zが存在しない場合、C(=O)
(例、
(iii) R1と結合して縮合された5ないし8員の、置換されていてもよい炭素環またはヘテロサイクルを形成し;
R3は:(i) 水素;または(ii) C1-C6アルキル、C2-C6アルケニル、C2-C6アルキニル、C1-C6アルキルスルフォニル、(C3-C8炭素環) C0-C4アルキルまたは(4-ないし8員ヘテロサイクル)C0-C4アルキルであり、それぞれが置換されていてもよく、それぞれが好ましくはRBから独立して選ばれる0ないし6個の置換基で置換されていてもよい;
R4aおよびR4bは独立して:
(i) 水素、ハロゲン、シアノ、-COOHまたはC1-C6アルキルであり;
(ii) -Z-N(R5)(R6)または-Z-O(R7)、Zが存在しない場合、C(=O)
(例、
)、OC(=O)(例、

)、N(Rw)C(=O)(例、

)、またはSO2(例、
);Rwは水素またはC1-C6アルキル;
(iii) フェニル、ナフチルまたは5ないし10員ヘテロアリールであり、それぞれが置換されていてもよく、それぞれが好ましくはRAから独立して選ばれる0ないし6個の置換基で置換されていてもよい;または
(iv) 結合して置換されていてもよい縮合された4ないし7員シクロアルキルまたはヘテロサイクルを形成し、好ましくはRBから独立して選ばれる0ないし4個の置換基で置換されていてもよい;
R5およびR6は独立して(i) 水素;または(ii) C1-C6アルキル、C2-C6アルケニル、C2-C6アルキニル、C1-C6アルカノイル、C1-C6アルキルカルボニル、C1-C6アルキルスルフォニル、(C3-C8シクロアルキル)C0-C4アルキル、(4ないし8員へテロサイクル)C0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノカルボニルまたはモノ-またはジ-(C1-C6アルキル)アミノスルフォニルであり、それぞれが置換されていてもよく、それぞれが好ましくはRBから独立して選ばれる0ないし6個の置換基で置換されていてもよい;またはR5およびR6は結合して置換されていてもよい4ないし7員ヘテロサイクルを形成してもよく、好ましくはRBから独立して選ばれる0ないし4個の置換基で置換されていてもよい;
R7は水素、C1-C6アルキル、C2-C6アルケニル、C2-C6アルキニル、(C3-C8シクロアルキル)C0-C4アルキルまたは(4ないし8員へテロサイクル)C0-C4アルキルであり、それぞれが置換されていてもよく、それぞれが好ましくはRBから独立して選ばれる0ないし6個の置換基で置換されていてもよい;
それぞれのRAは独立して、
(i) ハロゲン、ヒドロキシ、シアノ、アミノ、ニトロ、アミノカルボニル、アミノスルフォニルおよび-COOH;および
(ii) C1-C6アルキル、C2-C6アルケニル、C2-C6アルキニル、(C3-C8シクロアルキル) C0-C4アルキル、C1-C6ハロアルキル、C1-C6アルコキシ、C1-C6アルキルチオ、C1-C6アルキルスルフォニル、C1-C6アルコキシカルボニル、C1-C6アルキルスルフォニルC0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノC0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノスルフォニルC0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノカルボニルC0-C4アルキル、フェニルC0-C4アルキル、(4ないし8員へテロサイクル)C0-C4アルキルまたは(4ないし8員へテロサイクル)C1-C4アルコキシ;それぞれが置換されていてもよく、それぞれはRBから独立して選ばれる0ないし4個の置換基で置換されていてもよい;から選択され、
およびそれぞれのRBは独立して、オキソ、ハロゲン、ヒドロキシ、シアノ、アミノ、ニトロ、アミノカルボニル、アミノスルフォニル、-COOH、C1-C6アルキル、C1-C6アルケニル、C1-C6アルキニル、C1-C6ハロアルキニル、C1-C6アルコキシ、C1-C6ハロアルコキシ、C1-C6アルキルチオ、C1-C6アルカノイル、C1-C6アルコキシカルボニル、C1-C6アルカノイルオキシ、C3-C6アルカノン、モノ-またはジ-(C1-C6アルキル)アミノ、C1-C6アルカノイルアミノ、C1-C6アルキルスルフォニル、モノ-またはジ-(C1-C6アルキル)アミノスルフォニル、モノ-またはジ-(C1-C6アルキル)アミノカルボニル、(C3-C10炭素環)C0-C4アルキルおよび(4ないし10員へテロサイクル)C0-C4アルキル;
から選択される。)で表される化合物または塩、溶媒和物(例、水和物)、またはエステルが、CB1モデュレーションに関する疾患(肥満等)の治療用途を有することが開示されている。
(iii) フェニル、ナフチルまたは5ないし10員ヘテロアリールであり、それぞれが置換されていてもよく、それぞれが好ましくはRAから独立して選ばれる0ないし6個の置換基で置換されていてもよい;または
(iv) 結合して置換されていてもよい縮合された4ないし7員シクロアルキルまたはヘテロサイクルを形成し、好ましくはRBから独立して選ばれる0ないし4個の置換基で置換されていてもよい;
R5およびR6は独立して(i) 水素;または(ii) C1-C6アルキル、C2-C6アルケニル、C2-C6アルキニル、C1-C6アルカノイル、C1-C6アルキルカルボニル、C1-C6アルキルスルフォニル、(C3-C8シクロアルキル)C0-C4アルキル、(4ないし8員へテロサイクル)C0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノカルボニルまたはモノ-またはジ-(C1-C6アルキル)アミノスルフォニルであり、それぞれが置換されていてもよく、それぞれが好ましくはRBから独立して選ばれる0ないし6個の置換基で置換されていてもよい;またはR5およびR6は結合して置換されていてもよい4ないし7員ヘテロサイクルを形成してもよく、好ましくはRBから独立して選ばれる0ないし4個の置換基で置換されていてもよい;
R7は水素、C1-C6アルキル、C2-C6アルケニル、C2-C6アルキニル、(C3-C8シクロアルキル)C0-C4アルキルまたは(4ないし8員へテロサイクル)C0-C4アルキルであり、それぞれが置換されていてもよく、それぞれが好ましくはRBから独立して選ばれる0ないし6個の置換基で置換されていてもよい;
それぞれのRAは独立して、
(i) ハロゲン、ヒドロキシ、シアノ、アミノ、ニトロ、アミノカルボニル、アミノスルフォニルおよび-COOH;および
(ii) C1-C6アルキル、C2-C6アルケニル、C2-C6アルキニル、(C3-C8シクロアルキル) C0-C4アルキル、C1-C6ハロアルキル、C1-C6アルコキシ、C1-C6アルキルチオ、C1-C6アルキルスルフォニル、C1-C6アルコキシカルボニル、C1-C6アルキルスルフォニルC0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノC0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノスルフォニルC0-C4アルキル、モノ-またはジ-(C1-C6アルキル)アミノカルボニルC0-C4アルキル、フェニルC0-C4アルキル、(4ないし8員へテロサイクル)C0-C4アルキルまたは(4ないし8員へテロサイクル)C1-C4アルコキシ;それぞれが置換されていてもよく、それぞれはRBから独立して選ばれる0ないし4個の置換基で置換されていてもよい;から選択され、
およびそれぞれのRBは独立して、オキソ、ハロゲン、ヒドロキシ、シアノ、アミノ、ニトロ、アミノカルボニル、アミノスルフォニル、-COOH、C1-C6アルキル、C1-C6アルケニル、C1-C6アルキニル、C1-C6ハロアルキニル、C1-C6アルコキシ、C1-C6ハロアルコキシ、C1-C6アルキルチオ、C1-C6アルカノイル、C1-C6アルコキシカルボニル、C1-C6アルカノイルオキシ、C3-C6アルカノン、モノ-またはジ-(C1-C6アルキル)アミノ、C1-C6アルカノイルアミノ、C1-C6アルキルスルフォニル、モノ-またはジ-(C1-C6アルキル)アミノスルフォニル、モノ-またはジ-(C1-C6アルキル)アミノカルボニル、(C3-C10炭素環)C0-C4アルキルおよび(4ないし10員へテロサイクル)C0-C4アルキル;
から選択される。)で表される化合物または塩、溶媒和物(例、水和物)、またはエステルが、CB1モデュレーションに関する疾患(肥満等)の治療用途を有することが開示されている。
Journal of Chemical Society,Perkin Transactions 1(2002),(6),774-784(非特許文献1)には、下式


等の化合物等が開示されている。
Heterocycles(1995), 40(2), 787-800(非特許文献2)には、下式

等の化合物が開示されている。
Tetrahedron Letters (1990), 31(29), 4215-4218(非特許文献3)には、下式

等の化合物が開示されている。
しかし、いずれの化合物についても本願化合物とは区別され、デルタ-5-デサチュラーゼ阻害作用を有する旨の記載も無い。
Journal of Chemical Society,Perkin Transactions 1(2002),(6),774-784
Heterocycles(1995), 40(2), 787-800
Tetrahedron Letters (1990), 31(29), 4215-4218
本発明の目的は、アテローム性動脈硬化症、アテローム血栓症、糖尿病、肥満、喘息、発熱、疼痛、癌、リウマチ、変形性関節症、アトピー性皮膚炎等のエイコサノイド関連疾患の予防・治療に有用であり、かつ優れた薬理作用、物理化学的性質等を有する化合物を提供することにある。
本発明者らは、下記式(I)で表される複素環化合物が、デルタ-5-デサチュラーゼ阻害作用を有し、エイコサノイドの産生を抑制することに基づく種々の薬理作用を示し、良好な結晶性および安定性等の優れた物性を有し、アテローム性動脈硬化症、アテローム血栓症、糖尿病、肥満、喘息、発熱、疼痛、癌、リウマチ、変形性関節症、アトピー性皮膚炎等のエイコサノイド関連疾患の予防・治療に有用であることを初めて見出した。本発明者らは、この知見に基づき、鋭意研究を行った結果、本発明を完成するに至った。
すなわち、本発明は、
[1]化合物が、式(I):
すなわち、本発明は、
[1]化合物が、式(I):

〔式中、
環Aはさらに置換されていてもよい6員芳香環を示し、
X1は結合手またはOを示し、
R1は3ないし11員環状基で置換されたC1-6アルキル基、置換されていてもよい3ないし11員複素環基、または置換されていてもよいC3-11シクロアルキル基を示し、
R2は水素原子またはハロゲン原子で置換されていてもよいC1-6アルキル基を示し、
R3は、
(1)式:-X2-R5(式中、X2はO、S、SO2またはNR6を示し、R5は置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示す)で表される基;
(2)置換されていてもよいC1-6アルキル基;
(3)置換されていてもよいC3-11シクロアルキル基;または
(4)置換されていてもよい3ないし11員非芳香族複素環基
を示し、
R4は置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示し、および
R6は水素原子または置換されていてもよいC1-6アルキル基を示す。〕
で表される化合物またはその塩[以下、化合物(I)と称する場合がある];
[2]X1がOであり、
R4が1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、上記[1]記載の化合物またはその塩;
[3]環Aが、1ないし3個のハロゲン原子でさらに置換されていてもよいベンゼン、1ないし3個のハロゲン原子でさらに置換されていてもよいピリジン、または1もしくは2個のハロゲン原子でさらに置換されていてもよいピリミジンであり;
X1が、Oであり;
R1が、
(1)5ないし7員単環式芳香族複素環基で置換されたC1-6アルキル基、または
(2)(A)C1-6アルコキシ基、
(B)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(C)ハロゲン原子、および
(D)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい5または6員複素環基であり;
R2が、水素原子またはメチル基であり;
R3が、
(1)式:-X2-R5
[式中、
X2が、O、SまたはNHであり、かつ
R5が、
(A)3ないし8員非芳香族複素環基、
(B)(i)C1-6アルキルスルホニル基、
(ii)ヒドロキシ基、
(iii)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(iv)ハロゲン原子
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基、または
(C)1ないし5個のハロゲン原子で置換されていてもよいC3-11シクロアルキル基
である]で表される基、
(2)(A)ヒドロキシ基、
(B)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、
(C)ハロゲン原子、および
(D)C3-6シクロアルキル基
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基、
(3)(A)C1-6アルキル基、および
(B)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC3-11シクロアルキル基、または
(4)1ないし3個のハロゲン原子で置換されていてもよい3ないし11員非芳香族複素環基
であり;
R4が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基
である、上記[1]記載の化合物またはその塩;
[4]R1が、
(A)C1-6アルコキシ基、
(B)C1-6アルキル基、および
(C)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい5または6員複素環基である、上記[3]記載の化合物またはその塩;
[5]R1が、モルホリニル基または1ないし3個のC1-6アルキル基で置換されていてもよいピラゾリル基である、上記[4]記載の化合物またはその塩;
[6]2-(エチルスルファニル)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩;
[7]2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩;
[8]6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩;
[9]2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オンまたはその塩;
[10]上記[1]~[9]のいずれかに記載の化合物またはその塩を含有してなる医薬;
[11]デルタ-5-デサチュラーゼ阻害薬である、上記[10]記載の医薬;
[12]エイコサノイドが媒介する疾患の予防または治療剤である、上記[10]記載の医薬;
[13]動脈硬化症の予防または治療剤である、上記[10]記載の医薬;
[14]糖尿病または肥満症の予防または治療剤である、上記[10]記載の医薬;
[15]上記[1]~[9]のいずれかに記載の化合物またはその塩を哺乳動物に有効量投与することを特徴とする、該哺乳動物における動脈硬化症の予防または治療方法;
[16]上記[1]~[9]のいずれかに記載の化合物またはその塩を哺乳動物に有効量投与することを特徴とする、該哺乳動物における糖尿病または肥満症の予防または治療方法;
[17]動脈硬化症の予防または治療剤を製造するための、上記[1]~[9]のいずれかに記載の化合物またはその塩の使用;
[18]糖尿病または肥満症の予防または治療剤を製造するための、上記[1]~[9]のいずれかに記載の化合物またはその塩の使用;
[19]動脈硬化症の予防または治療のために使用する、上記[1]~[9]のいずれかに記載の化合物またはその塩;
[20]糖尿病または肥満症の予防または治療のために使用する、上記[1]~[9]のいずれかに記載の化合物またはその塩;
等に関する。
環Aはさらに置換されていてもよい6員芳香環を示し、
X1は結合手またはOを示し、
R1は3ないし11員環状基で置換されたC1-6アルキル基、置換されていてもよい3ないし11員複素環基、または置換されていてもよいC3-11シクロアルキル基を示し、
R2は水素原子またはハロゲン原子で置換されていてもよいC1-6アルキル基を示し、
R3は、
(1)式:-X2-R5(式中、X2はO、S、SO2またはNR6を示し、R5は置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示す)で表される基;
(2)置換されていてもよいC1-6アルキル基;
(3)置換されていてもよいC3-11シクロアルキル基;または
(4)置換されていてもよい3ないし11員非芳香族複素環基
を示し、
R4は置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示し、および
R6は水素原子または置換されていてもよいC1-6アルキル基を示す。〕
で表される化合物またはその塩[以下、化合物(I)と称する場合がある];
[2]X1がOであり、
R4が1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、上記[1]記載の化合物またはその塩;
[3]環Aが、1ないし3個のハロゲン原子でさらに置換されていてもよいベンゼン、1ないし3個のハロゲン原子でさらに置換されていてもよいピリジン、または1もしくは2個のハロゲン原子でさらに置換されていてもよいピリミジンであり;
X1が、Oであり;
R1が、
(1)5ないし7員単環式芳香族複素環基で置換されたC1-6アルキル基、または
(2)(A)C1-6アルコキシ基、
(B)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(C)ハロゲン原子、および
(D)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい5または6員複素環基であり;
R2が、水素原子またはメチル基であり;
R3が、
(1)式:-X2-R5
[式中、
X2が、O、SまたはNHであり、かつ
R5が、
(A)3ないし8員非芳香族複素環基、
(B)(i)C1-6アルキルスルホニル基、
(ii)ヒドロキシ基、
(iii)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(iv)ハロゲン原子
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基、または
(C)1ないし5個のハロゲン原子で置換されていてもよいC3-11シクロアルキル基
である]で表される基、
(2)(A)ヒドロキシ基、
(B)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、
(C)ハロゲン原子、および
(D)C3-6シクロアルキル基
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基、
(3)(A)C1-6アルキル基、および
(B)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC3-11シクロアルキル基、または
(4)1ないし3個のハロゲン原子で置換されていてもよい3ないし11員非芳香族複素環基
であり;
R4が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基
である、上記[1]記載の化合物またはその塩;
[4]R1が、
(A)C1-6アルコキシ基、
(B)C1-6アルキル基、および
(C)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい5または6員複素環基である、上記[3]記載の化合物またはその塩;
[5]R1が、モルホリニル基または1ないし3個のC1-6アルキル基で置換されていてもよいピラゾリル基である、上記[4]記載の化合物またはその塩;
[6]2-(エチルスルファニル)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩;
[7]2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩;
[8]6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩;
[9]2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オンまたはその塩;
[10]上記[1]~[9]のいずれかに記載の化合物またはその塩を含有してなる医薬;
[11]デルタ-5-デサチュラーゼ阻害薬である、上記[10]記載の医薬;
[12]エイコサノイドが媒介する疾患の予防または治療剤である、上記[10]記載の医薬;
[13]動脈硬化症の予防または治療剤である、上記[10]記載の医薬;
[14]糖尿病または肥満症の予防または治療剤である、上記[10]記載の医薬;
[15]上記[1]~[9]のいずれかに記載の化合物またはその塩を哺乳動物に有効量投与することを特徴とする、該哺乳動物における動脈硬化症の予防または治療方法;
[16]上記[1]~[9]のいずれかに記載の化合物またはその塩を哺乳動物に有効量投与することを特徴とする、該哺乳動物における糖尿病または肥満症の予防または治療方法;
[17]動脈硬化症の予防または治療剤を製造するための、上記[1]~[9]のいずれかに記載の化合物またはその塩の使用;
[18]糖尿病または肥満症の予防または治療剤を製造するための、上記[1]~[9]のいずれかに記載の化合物またはその塩の使用;
[19]動脈硬化症の予防または治療のために使用する、上記[1]~[9]のいずれかに記載の化合物またはその塩;
[20]糖尿病または肥満症の予防または治療のために使用する、上記[1]~[9]のいずれかに記載の化合物またはその塩;
等に関する。
化合物(I)は、デルタ-5-デサチュラーゼ阻害作用を有し、アテローム性動脈硬化症、アテローム血栓症、糖尿病、肥満、喘息、発熱、疼痛、癌、リウマチ、変形性関節症、アトピー性皮膚炎等のエイコサノイド関連疾患の予防・治療に有用であり、かつ優れた薬効を有する。
(発明の詳細な説明)
(発明の詳細な説明)
以下に、本発明を詳細に説明する。
本明細書中の「ハロゲン原子」としては、例えば、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられる。
本明細書中の「C1-6アルキル(基)」としては、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、イソペンチル、ネオペンチル、tert-ペンチル、ヘキシル、2,2-ジメチルブチル、3,3-ジメチルブチル、2-エチルブチル等が挙げられる。
本明細書中の「C1-6アルコキシ(基)」としては、例えば、メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ、イソブトキシ、sec-ブトキシ、tert-ブトキシ、ペンチルオキシ、イソペンチルオキシ、ネオペンチルオキシ、tert-ペンチルオキシ、ヘキシルオキシ、2-エチルブトキシ等が挙げられる。
本明細書中の「C3-11シクロアルキル(基)」としては、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロノニル、シクロデシル、シクロウンデシル等が挙げられる。
本明細書中の「C3-10シクロアルキル(基)」としては、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロノニル、シクロデシル等が挙げられる。
本明細書中の「C3-10シクロアルキル(基)」としては、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロノニル、シクロデシル等が挙げられる。
本明細書中の「C6-14アリール(基)」としては、例えば、フェニル、ナフチル(例、1-ナフチル、2-ナフチル)、アントリル、フェナントリル等が挙げられる。
本明細書中の「C7-13アラルキル(基)」としては、例えば、ベンジル、フェネチル、ナフチルメチル、ビフェニリルメチル等が挙げられる。
本明細書中の「C2-6アルケニル(基)」としては、例えば、エテニル、1-プロペニル、2-プロペニル、2-メチル-1-プロペニル、1-ブテニル、2-ブテニル、3-ブテニル、3-メチル-2-ブテニル、1-ペンテニル、1-ヘキセニル、3-ヘキセニル、5-ヘキセニル等が挙げられる。
本明細書中の「C7-13アラルキル(基)」としては、例えば、ベンジル、フェネチル、ナフチルメチル、ビフェニリルメチル等が挙げられる。
本明細書中の「C2-6アルケニル(基)」としては、例えば、エテニル、1-プロペニル、2-プロペニル、2-メチル-1-プロペニル、1-ブテニル、2-ブテニル、3-ブテニル、3-メチル-2-ブテニル、1-ペンテニル、1-ヘキセニル、3-ヘキセニル、5-ヘキセニル等が挙げられる。
本明細書中の「6ないし10員芳香族炭化水素基」としては、例えば、フェニル、1-ナフチル、2-ナフチル等が挙げられる。
本明細書中の「5ないし7員単環式芳香族複素環基」としては、例えば、環構成原子として炭素原子以外に酸素原子、硫黄原子(該硫黄原子は酸化されていてもよい)および窒素原子から選ばれるヘテロ原子を1ないし4個含有する、5ないし7員単環式芳香族複素環基等が挙げられ、具体的には、フリル(例、2-フリル、3-フリル)、チエニル(例、2-チエニル、3-チエニル)、ピリジル(例、2-ピリジル、3-ピリジル、4-ピリジル)、ピリミジニル(例、2-ピリミジニル、4-ピリミジニル、5-ピリミジニル、6-ピリミジニル)、ピリダジニル(例、3-ピリダジニル、4-ピリダジニル)、ピラジニル(例、2-ピラジニル)、ピロリル(例、1-ピロリル、2-ピロリル、3-ピロリル)、イミダゾリル(例、1-イミダゾリル、2-イミダゾリル、4-イミダゾリル、5-イミダゾリル)、ピラゾリル(例、1-ピラゾリル、3-ピラゾリル、4-ピラゾリル)、チアゾリル(例、2-チアゾリル、4-チアゾリル、5-チアゾリル)、イソチアゾリル(例、3-イソチアゾリル、4-イソチアゾリル、5-イソチアゾリル)、オキサゾリル(例、2-オキサゾリル、4-オキサゾリル、5-オキサゾリル)、イソオキサゾリル(例、3-イソオキサゾリル、4-イソオキサゾリル、5-イソオキサゾリル)、オキサジアゾリル(例、1,2,4-オキサジアゾール-5-イル、1,2,4-オキサジアゾール-3-イル、1,3,4-オキサジアゾール-2-イル)、チアジアゾリル(例、1,3,4-チアジアゾール-2-イル)、トリアゾリル(例、1,2,4-トリアゾール-1-イル、1,2,4-トリアゾール-3-イル、1,2,3-トリアゾール-1-イル、1,2,3-トリアゾール-2-イル、1,2,3-トリアゾール-4-イル)、テトラゾリル(例、テトラゾール-1-イル、テトラゾール-5-イル)、トリアジニル(例、1,3,5-トリアジン-2-イル)等が挙げられる。
本明細書中の「8ないし11員縮合芳香族複素環基」としては、例えば、環構成原子として炭素原子以外に酸素原子、硫黄原子(該硫黄原子は酸化されていてもよい)および窒素原子から選ばれるヘテロ原子を1ないし4個含有する、5ないし7員単環式芳香族複素環等と、C6-10芳香族炭化水素等が縮合した縮合環から誘導される基;上記5ないし7員単環式芳香族複素環同士が縮合した縮合環から誘導される基等が挙げられる。
該5ないし7員単環式芳香族複素環としては、上述の5ないし7員単環式芳香族複素環基に対応する環が挙げられる。
該C6-10芳香族炭化水素としては、上述のC6-10アリール基に対応する環が挙げられる。
該「8ないし11員縮合芳香族複素環基」としては、具体的には、キノリル(例、2-キノリル、3-キノリル、4-キノリル)、イソキノリル(例、1-イソキノリル、3-イソキノリル、4-イソキノリル)、キナゾリル(例、2-キナゾリル、4-キナゾリル)、キノキサリル(例、2-キノキサリル)、ベンゾフリル(例、2-ベンゾフリル、3-ベンゾフリル)、ベンゾチエニル(例、2-ベンゾチエニル、3-ベンゾチエニル)、ベンズオキサゾリル(例、2-ベンズオキサゾリル)、ベンゾチアゾリル(例、2-ベンゾチアゾリル、5-ベンゾチアゾリル、6-ベンゾチアゾリル)、ベンズイミダゾリル(例、ベンズイミダゾール-1-イル、ベンズイミダゾール-2-イル、ベンズイミダゾール-5-イル)、インドリル(例、インドール-1-イル、インドール-3-イル、インドール-4-イル、インドール-5-イル、インドール-6-イル)、インダゾリル(例、1H-インダゾール-3-イル)、ピロロピラジニル(例、1H-ピロロ[2,3-b]ピラジン-2-イル、1H-ピロロ[2,3-b]ピラジン-6-イル)、イミダゾピリジル(例、1H-イミダゾ[4,5-b]ピリジン-2-イル、1H-イミダゾ[4,5-c]ピリジン-2-イル)、イミダゾピラジニル(例、1H-イミダゾ[4,5-b]ピラジン-2-イル)、ベンズイソオキサゾリル(例、3-ベンズイソオキサゾリル)、ベンゾトリアゾリル(例、1H-1,2,3-ベンゾトリアゾール-5-イル)、ピラゾロピリジル(例、1H-ピラゾロ[4,3-c]ピリジン-3-イル)、ピラゾロチエニル(例、2H-ピラゾロ[3,4-b]チオフェン-2-イル)、ピラゾロトリアジニル(例、ピラゾロ[5,1-c][1,2,4]トリアジン-3-イル)等が挙げられる。
該5ないし7員単環式芳香族複素環としては、上述の5ないし7員単環式芳香族複素環基に対応する環が挙げられる。
該C6-10芳香族炭化水素としては、上述のC6-10アリール基に対応する環が挙げられる。
該「8ないし11員縮合芳香族複素環基」としては、具体的には、キノリル(例、2-キノリル、3-キノリル、4-キノリル)、イソキノリル(例、1-イソキノリル、3-イソキノリル、4-イソキノリル)、キナゾリル(例、2-キナゾリル、4-キナゾリル)、キノキサリル(例、2-キノキサリル)、ベンゾフリル(例、2-ベンゾフリル、3-ベンゾフリル)、ベンゾチエニル(例、2-ベンゾチエニル、3-ベンゾチエニル)、ベンズオキサゾリル(例、2-ベンズオキサゾリル)、ベンゾチアゾリル(例、2-ベンゾチアゾリル、5-ベンゾチアゾリル、6-ベンゾチアゾリル)、ベンズイミダゾリル(例、ベンズイミダゾール-1-イル、ベンズイミダゾール-2-イル、ベンズイミダゾール-5-イル)、インドリル(例、インドール-1-イル、インドール-3-イル、インドール-4-イル、インドール-5-イル、インドール-6-イル)、インダゾリル(例、1H-インダゾール-3-イル)、ピロロピラジニル(例、1H-ピロロ[2,3-b]ピラジン-2-イル、1H-ピロロ[2,3-b]ピラジン-6-イル)、イミダゾピリジル(例、1H-イミダゾ[4,5-b]ピリジン-2-イル、1H-イミダゾ[4,5-c]ピリジン-2-イル)、イミダゾピラジニル(例、1H-イミダゾ[4,5-b]ピラジン-2-イル)、ベンズイソオキサゾリル(例、3-ベンズイソオキサゾリル)、ベンゾトリアゾリル(例、1H-1,2,3-ベンゾトリアゾール-5-イル)、ピラゾロピリジル(例、1H-ピラゾロ[4,3-c]ピリジン-3-イル)、ピラゾロチエニル(例、2H-ピラゾロ[3,4-b]チオフェン-2-イル)、ピラゾロトリアジニル(例、ピラゾロ[5,1-c][1,2,4]トリアジン-3-イル)等が挙げられる。
本明細書中の「3ないし11員非芳香族環状炭化水素基」としては、例えば、C3-11シクロアルキル基、C3-11シクロアルケニル基、C4-11シクロアルカジエニル基等が挙げられ、具体的には、C3-11シクロアルキル基(例、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、シクロノニル、シクロデシル、シクロウンデシル)、C3-11シクロアルケニル基(例、シクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロヘプテニル、シクロオクテニル、シクロノネニル、シクロデセニル、シクロウンデセニル)、C4-11シクロアルカジエニル基(例、シクロブタジエニル、シクロペンタジエニル、シクロヘキサジエニル、シクロヘプタジエニル、シクロオクタジエニル、シクロノナジエニル、シクロデカジエニル、シクロウンデカジエニル)、これらの基とベンゼン環とが縮合した縮合環基(例、インダニル(例、1-インダニル)、テトラヒドロナフチル(例、1,2,3,4-テトラヒドロナフタレン-1-イル))等が挙げられる。
本明細書中の「3ないし10員非芳香族環状炭化水素基」としては、例えば、C3-10シクロアルキル基、C3-10シクロアルケニル基、C4-10シクロアルカジエニル基等が挙げられ、具体的には、C3-10シクロアルキル基(例、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル)、C3-10シクロアルケニル基(例、シクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロヘプテニル、シクロオクテニル)、C4-10シクロアルカジエニル基(例、シクロブタジエニル、シクロペンタジエニル、シクロヘキサジエニル、シクロヘプタジエニル、シクロオクタジエニル、シクロノナジエニル、シクロデカジエニル)、これらの基とベンゼン環とが縮合した縮合環基(例、インダニル(例、1-インダニル)、テトラヒドロナフチル(例、1,2,3,4-テトラヒドロナフタレン-1-イル))等が挙げられる。
本明細書中の「3ないし10員非芳香族環状炭化水素基」としては、例えば、C3-10シクロアルキル基、C3-10シクロアルケニル基、C4-10シクロアルカジエニル基等が挙げられ、具体的には、C3-10シクロアルキル基(例、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル)、C3-10シクロアルケニル基(例、シクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロヘプテニル、シクロオクテニル)、C4-10シクロアルカジエニル基(例、シクロブタジエニル、シクロペンタジエニル、シクロヘキサジエニル、シクロヘプタジエニル、シクロオクタジエニル、シクロノナジエニル、シクロデカジエニル)、これらの基とベンゼン環とが縮合した縮合環基(例、インダニル(例、1-インダニル)、テトラヒドロナフチル(例、1,2,3,4-テトラヒドロナフタレン-1-イル))等が挙げられる。
本明細書中の「3ないし8員非芳香族複素環基」としては、例えば、3ないし8員(好ましくは5または6員)の飽和あるいは不飽和(好ましくは飽和)非芳香族複素環基等が挙げられ、具体的には、オキシラニル(例、2-オキシラニル)、アゼチジニル(例、2-アゼチジニル)、オキセタニル(例、2-オキセタニル、3-オキセタニル)、チエタニル(例、2-チエタニル)、ピロリジニル(例、1-ピロリジニル、2-ピロリジニル、3-ピロリジニル)、テトラヒドロフリル(例、2-テトラヒドロフリル、3-テトラヒドロフリル)、チオラニル(例、2-チオラニル)、ピペリジル(例、1-ピペリジル、2-ピペリジル、3-ピペリジル、4-ピペリジル)、テトラヒドロピラニル(例、2-テトラヒドロピラニル、3-テトラヒドロピラニル、4-テトラヒドロピラニル)、チアニル(例、2-チアニル)、モルホリニル(例、2-モルホリニル、3-モルホリニル、4-モルホリニル)、チオモルホリニル(例、2-チオモルホリニル、3-チオモルホリニル、4-チオモルホリニル)、ピペラジニル(例、1-ピペラジニル、2-ピペラジニル)、アゼパニル(例、2-アゼパニル)、オキセパニル(例、2-オキセパニル)、チエパニル(例、2-チエパニル)、オキサゼパニル(例、1,4-オキサゼパン-5-イル)、チアゼパニル(例、1,4-チアゼパン-5-イル)、アゾカニル(例、2-アゾカニル)、オキソカニル(例、2-オキソカニル)、チオカニル(例、2-チオカニル)、オキサゾカニル(例、1,4-オキサゾカン-5-イル)、チアゾカニル(例、1,4-チアゾカン-5-イル)、ジオキシニル(例、2-ジオキシニル)、ジヒドロピリジル(例、1,2-ジヒドロピリジン-4-イル、1,6-ジヒドロピリジン-3-イル)、ジヒドロピリミダジニル(例、1,6-ジヒドロピリダジン-3-イル)、ジヒドロオキサジアゾリル(例、4,5-ジヒドロ-1,2,4-オキサジアゾール-3-イル)等が挙げられる。
環Aは、さらに置換されていてもよい6員芳香環を示す。
環Aで示される「置換されていてもよい6員芳香環」における「6員芳香環」としては、ベンゼン、ピリジン、ピリダジン、ピリミジン、ピラジン、トリアジン等が挙げられる。好ましくは、ベンゼン、ピリジン、ピリダジン、ピリミジン、ピラジン等であり、より好ましくは、ベンゼン、ピリジン、ピリミジン等であり、さらに好ましくは、ベンゼン、ピリジン等である。
環Aで示される「さらに置換されていてもよい6員芳香環」における「6員芳香環」は、置換可能な位置に1ないし3個の置換基を有していてもよい。
該置換基としては、例えば、
(1)C3-10シクロアルキル基(例、シクロプロピル、シクロヘキシル);
(2)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール基(例、フェニル、ナフチル);
(3)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル);
(4)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、
(d)ハロゲン原子、および
(e)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員非芳香族複素環基(例、テトラヒドロフリル、モルホリニル、チオモルホリニル、ピペリジル、ピロリジニル、ピペラジニル);
(5)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ-カルボニル基、
(d)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル)、
(e)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基、および
(f)4ないし7員芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル)
から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基;
(6)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基;
(7)(a)ハロゲン原子、
(b)C1-6アルコキシ基、
(c)C6-14アリール基(例、フェニル)、および
(d)4ないし7員複素環基(例、テトラヒドロフリル)から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ-カルボニル基(例、メトキシカルボニル);
(8)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル、エチルスルホニル、イソプロピルスルホニル);
(9)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基;
(10)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいチオカルバモイル基;
(11)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいスルファモイル基;
(12)カルボキシ基;
(13)ヒドロキシ基;
(14)(a)ハロゲン原子、
(b)カルボキシ基、
(c)C1-6アルコキシ基、
(d)1ないし3個のC6-14アリール基(例、フェニル)で置換されていてもよいC1-6アルコキシ-カルボニル基、
(e)C1-6アルキル基およびC1-6アルコキシ-カルボニル基から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基、
(f)4ないし7員複素環基(例、テトラヒドロフリル)、および
(g)C3-10シクロアルキル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ基;
(15)1ないし3個のハロゲン原子で置換されていてもよいC2-6アルケニルオキシ基(例、エテニルオキシ);
(16)C7-13アラルキルオキシ基(例、ベンジルオキシ);
(17)C6-14アリールオキシ基(例、フェニルオキシ、ナフチルオキシ);
(18)C1-6アルキル-カルボニルオキシ基(例、アセチルオキシ、tert-ブチルカルボニルオキシ);
(19)(a)ハロゲン原子、および
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール-カルボニル基(例、ベンゾイル);
(20)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員非芳香族複素環カルボニル基(例、ピロリジニルカルボニル、モルホリニルカルボニル);
(21)メルカプト基;
(22)(a)ハロゲン原子、および
(b)C1-6アルコキシ-カルボニル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキルチオ基(例、メチルチオ、エチルチオ);
(23)C7-13アラルキルチオ基(例、ベンジルチオ);
(24)C6-14アリールチオ基(例、フェニルチオ、ナフチルチオ);
(25)シアノ基;
(26)ニトロ基;
(27)ハロゲン原子(例、フッ素原子、塩素原子、臭素原子);
(28)C1-3アルキレンジオキシ基;
(29)C1-3アルキレンオキシ基(例、メチレンオキシ、エチレンオキシ);
(30)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員芳香族複素環カルボニル基(例、ピラゾリルカルボニル、ピラジニルカルボニル、イソキサゾリルカルボニル、ピリジルカルボニル、チアゾリルカルボニル);
(31)(a)ハロゲン原子(例、フッ素原子)、および
(b)C1-6アルコキシ基(例、メトキシ)
から選ばれる1ないし3個の置換基で置換されていてもよいC3-10シクロアルコキシ基(例、シクロプロポキシ、シクロペンチルオキシ);
(32)(a)ハロゲン原子、
(b)カルボキシ基、
(c)ヒドロキシ基、
(d)C1-6アルコキシ-カルボニル基、
(e)C1-6アルコキシ基、および
(f)C1-6アルキル基でモノまたはジ置換されていてもよいアミノ基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基;
(33)(a)ハロゲン原子、
(b)カルボキシ基、
(c)ヒドロキシ基、
(d)C1-6アルコキシ-カルボニル基、
(e)C1-6アルコキシ基、および
(f)C1-6アルキル基でモノまたはジ置換されていてもよいアミノ基
から選ばれる1ないし3個の置換基で置換されていてもよいC2-6アルケニル基(例、エテニル、1-プロペニル);
(34)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)C1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC7-13アラルキル基(例、ベンジル);
等が挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
(1)C3-10シクロアルキル基(例、シクロプロピル、シクロヘキシル);
(2)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール基(例、フェニル、ナフチル);
(3)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル);
(4)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、
(d)ハロゲン原子、および
(e)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員非芳香族複素環基(例、テトラヒドロフリル、モルホリニル、チオモルホリニル、ピペリジル、ピロリジニル、ピペラジニル);
(5)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ-カルボニル基、
(d)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル)、
(e)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基、および
(f)4ないし7員芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル)
から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基;
(6)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基;
(7)(a)ハロゲン原子、
(b)C1-6アルコキシ基、
(c)C6-14アリール基(例、フェニル)、および
(d)4ないし7員複素環基(例、テトラヒドロフリル)から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ-カルボニル基(例、メトキシカルボニル);
(8)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル、エチルスルホニル、イソプロピルスルホニル);
(9)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基;
(10)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいチオカルバモイル基;
(11)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいスルファモイル基;
(12)カルボキシ基;
(13)ヒドロキシ基;
(14)(a)ハロゲン原子、
(b)カルボキシ基、
(c)C1-6アルコキシ基、
(d)1ないし3個のC6-14アリール基(例、フェニル)で置換されていてもよいC1-6アルコキシ-カルボニル基、
(e)C1-6アルキル基およびC1-6アルコキシ-カルボニル基から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基、
(f)4ないし7員複素環基(例、テトラヒドロフリル)、および
(g)C3-10シクロアルキル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ基;
(15)1ないし3個のハロゲン原子で置換されていてもよいC2-6アルケニルオキシ基(例、エテニルオキシ);
(16)C7-13アラルキルオキシ基(例、ベンジルオキシ);
(17)C6-14アリールオキシ基(例、フェニルオキシ、ナフチルオキシ);
(18)C1-6アルキル-カルボニルオキシ基(例、アセチルオキシ、tert-ブチルカルボニルオキシ);
(19)(a)ハロゲン原子、および
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール-カルボニル基(例、ベンゾイル);
(20)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員非芳香族複素環カルボニル基(例、ピロリジニルカルボニル、モルホリニルカルボニル);
(21)メルカプト基;
(22)(a)ハロゲン原子、および
(b)C1-6アルコキシ-カルボニル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキルチオ基(例、メチルチオ、エチルチオ);
(23)C7-13アラルキルチオ基(例、ベンジルチオ);
(24)C6-14アリールチオ基(例、フェニルチオ、ナフチルチオ);
(25)シアノ基;
(26)ニトロ基;
(27)ハロゲン原子(例、フッ素原子、塩素原子、臭素原子);
(28)C1-3アルキレンジオキシ基;
(29)C1-3アルキレンオキシ基(例、メチレンオキシ、エチレンオキシ);
(30)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員芳香族複素環カルボニル基(例、ピラゾリルカルボニル、ピラジニルカルボニル、イソキサゾリルカルボニル、ピリジルカルボニル、チアゾリルカルボニル);
(31)(a)ハロゲン原子(例、フッ素原子)、および
(b)C1-6アルコキシ基(例、メトキシ)
から選ばれる1ないし3個の置換基で置換されていてもよいC3-10シクロアルコキシ基(例、シクロプロポキシ、シクロペンチルオキシ);
(32)(a)ハロゲン原子、
(b)カルボキシ基、
(c)ヒドロキシ基、
(d)C1-6アルコキシ-カルボニル基、
(e)C1-6アルコキシ基、および
(f)C1-6アルキル基でモノまたはジ置換されていてもよいアミノ基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基;
(33)(a)ハロゲン原子、
(b)カルボキシ基、
(c)ヒドロキシ基、
(d)C1-6アルコキシ-カルボニル基、
(e)C1-6アルコキシ基、および
(f)C1-6アルキル基でモノまたはジ置換されていてもよいアミノ基
から選ばれる1ないし3個の置換基で置換されていてもよいC2-6アルケニル基(例、エテニル、1-プロペニル);
(34)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)C1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC7-13アラルキル基(例、ベンジル);
等が挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
環Aは、好ましくは、1ないし3個のハロゲン原子でさらに置換されていてもよいベンゼン、ピリジン等である。
別の態様において、環Aは、好ましくは、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリジン、または1もしくは2個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリミジンであり、より好ましくは、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、フッ素原子)でさらに置換されていてもよいピリジン、またはピリミジンである。
X1は結合手またはOを示す。
X1は、好ましくは、Oである。
R1は3ないし11員環状基で置換されたC1-6アルキル基、置換されていてもよい3ないし11員複素環基、または置換されていてもよいC3-11シクロアルキル基を示す。
R1で示される「3ないし11員環状基で置換されたC1-6アルキル基」における「C1-6アルキル基」は、置換可能な位置に、1ないし3個の「3ないし11員環状基」を有する。
該「3ないし11員環状基」としては、例えば、6ないし10員芳香族炭化水素基、5ないし11員芳香族複素環基(例、5ないし7員単環式芳香族複素環基、8ないし11員縮合芳香族複素環基)、3ないし10員非芳香族環状炭化水素基、3ないし8員非芳香族複素環基等が挙げられる。
R1で示される「3ないし11員環状基で置換されたC1-6アルキル基」は、好ましくは、5ないし7員単環式芳香族複素環基で置換されたC1-6アルキル基であり、より好ましくは、ピリジル等で置換されたC1-6アルキル基であり、さらに好ましくは、ピリジル等で置換されたメチルであり、特に好ましくは、ピリジルで置換されたメチルである。
R1で示される「置換されていてもよい3ないし11員複素環基」における「3ないし11員複素環基」は、例えば、5ないし11員芳香族複素環基(例、5ないし7員単環式芳香族複素環基、8ないし11員縮合芳香族複素環基)、3ないし8員非芳香族複素環基等が挙げられ、中でも5または6員複素環基が好ましい。
R1で示される「置換されていてもよい3ないし11員複素環基」における「3ないし11員複素環基」は、置換可能な位置に1ないし3個の置換基を有していてもよい。
該置換基としては、例えば、環Aで示される「さらに置換されていてもよい6員芳香環」における「6員芳香環」が有していてもよい置換基と同様のもの等が挙げられる。該「3ないし11員複素環基」が非芳香族複素環基である場合は、置換基としてオキソ基を有していてもよい。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
R1で示される「置換されていてもよい3ないし11員複素環基」は、好ましくは、
(1)C1-6アルコキシ基(例、メトキシ)、
(2)C1-6アルキル基(例、メチル)、および
(3)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい5または6員複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、テトラヒドロピラニル、モルホリニル、チオモルホリニル等)である。
(1)C1-6アルコキシ基(例、メトキシ)、
(2)C1-6アルキル基(例、メチル)、および
(3)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい5または6員複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、テトラヒドロピラニル、モルホリニル、チオモルホリニル等)である。
別の態様において、R1で示される「置換されていてもよい3ないし11員複素環基」は、好ましくは、
(A)C1-6アルコキシ基(例、メトキシ)、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)オキソ基
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよい5または6員複素環基[例、5または6員芳香族複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、トリアゾリル、オキサジアゾリル、イミダゾリル等);5または6員非芳香族複素環基(例、モルホリニル、ジヒドロピリジル、ジヒドロピリダジニル、テトラヒドロピラニル、ピペラジニル、ジヒドロオキサジアゾリル、ピロリジニル、ピペリジル等)]である。
(A)C1-6アルコキシ基(例、メトキシ)、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)オキソ基
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよい5または6員複素環基[例、5または6員芳香族複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、トリアゾリル、オキサジアゾリル、イミダゾリル等);5または6員非芳香族複素環基(例、モルホリニル、ジヒドロピリジル、ジヒドロピリダジニル、テトラヒドロピラニル、ピペラジニル、ジヒドロオキサジアゾリル、ピロリジニル、ピペリジル等)]である。
R1で示される「置換されていてもよいC3-11シクロアルキル基」における「C3-11シクロアルキル基」は、置換可能な位置に1ないし3個の置換基を有していてもよい。
該置換基としては、例えば、環Aで示される「さらに置換されていてもよい6員芳香環」における「6員芳香環」が有していてもよい置換基と同様のもの、オキソ基等が挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
R1は、好ましくは、
(1)ピリジルで置換されたメチル;
(2)(A)C1-6アルコキシ基(例、メトキシ)、
(B)C1-6アルキル基(例、メチル)、および
(C)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい5または6員複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、テトラヒドロピラニル、モルホリニル、チオモルホリニル);
等である。
(1)ピリジルで置換されたメチル;
(2)(A)C1-6アルコキシ基(例、メトキシ)、
(B)C1-6アルキル基(例、メチル)、および
(C)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい5または6員複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、テトラヒドロピラニル、モルホリニル、チオモルホリニル);
等である。
別の態様において、R1は、好ましくは、
(1)5ないし7員単環式芳香族複素環基(例、ピリジル)で置換されたC1-6アルキル基(例、メチル)、または
(2)(A)C1-6アルコキシ基(例、メトキシ)、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)オキソ基
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよい5または6員複素環基[例、5または6員芳香族複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、トリアゾリル、オキサジアゾリル、イミダゾリル等);5または6員非芳香族複素環基(例、モルホリニル、ジヒドロピリジル、ジヒドロピリダジニル、テトラヒドロピラニル、ピペラジニル、ジヒドロオキサジアゾリル、ピロリジニル、ピペリジル等)]
であり、より好ましくは、
(A)C1-6アルコキシ基(例、メトキシ)、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)オキソ基
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよい5または6員複素環基[例、5または6員芳香族複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、トリアゾリル、オキサジアゾリル、イミダゾリル等);5または6員非芳香族複素環基(例、モルホリニル、ジヒドロピリジル、ジヒドロピリダジニル、テトラヒドロピラニル、ピペラジニル、ジヒドロオキサジアゾリル、ピロリジニル、ピペリジル等)]
であり、さらに好ましくは、
(A)C1-6アルコキシ基(例、メトキシ)、
(B)C1-6アルキル基(例、メチル)、および
(C)オキソ基
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよい5または6員複素環基[例、5または6員芳香族複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、トリアゾリル、オキサジアゾリル、イミダゾリル等);5または6員非芳香族複素環基(例、モルホリニル、ジヒドロピリジル、ジヒドロピリダジニル、テトラヒドロピラニル、ピペラジニル、ジヒドロオキサジアゾリル、ピロリジニル、ピペリジル等)]
であり、さらにより好ましくは、モルホニリル、または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいピラゾリルである。
(1)5ないし7員単環式芳香族複素環基(例、ピリジル)で置換されたC1-6アルキル基(例、メチル)、または
(2)(A)C1-6アルコキシ基(例、メトキシ)、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)オキソ基
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよい5または6員複素環基[例、5または6員芳香族複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、トリアゾリル、オキサジアゾリル、イミダゾリル等);5または6員非芳香族複素環基(例、モルホリニル、ジヒドロピリジル、ジヒドロピリダジニル、テトラヒドロピラニル、ピペラジニル、ジヒドロオキサジアゾリル、ピロリジニル、ピペリジル等)]
であり、より好ましくは、
(A)C1-6アルコキシ基(例、メトキシ)、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)オキソ基
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよい5または6員複素環基[例、5または6員芳香族複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、トリアゾリル、オキサジアゾリル、イミダゾリル等);5または6員非芳香族複素環基(例、モルホリニル、ジヒドロピリジル、ジヒドロピリダジニル、テトラヒドロピラニル、ピペラジニル、ジヒドロオキサジアゾリル、ピロリジニル、ピペリジル等)]
であり、さらに好ましくは、
(A)C1-6アルコキシ基(例、メトキシ)、
(B)C1-6アルキル基(例、メチル)、および
(C)オキソ基
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよい5または6員複素環基[例、5または6員芳香族複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、トリアゾリル、オキサジアゾリル、イミダゾリル等);5または6員非芳香族複素環基(例、モルホリニル、ジヒドロピリジル、ジヒドロピリダジニル、テトラヒドロピラニル、ピペラジニル、ジヒドロオキサジアゾリル、ピロリジニル、ピペリジル等)]
であり、さらにより好ましくは、モルホニリル、または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいピラゾリルである。
R2は水素原子またはハロゲン原子で置換されていてもよいC1-6アルキル基を示す。
R2で示される「ハロゲン原子で置換されていてもよいC1-6アルキル基」は、好ましくは、C1-6アルキル基であり、より好ましくは、メチルである。
R2は、好ましくは、水素原子またはC1-6アルキル基(例、メチル)であり、より好ましくは、水素原子またはメチルである。
R3は、
(1)式:-X2-R5(式中、X2はO、S、SO2またはNR6を示し、R5は置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示す)で表される基;
(2)置換されていてもよいC1-6アルキル基;
(3)置換されていてもよいC3-11シクロアルキル基;または
(4)置換されていてもよい3ないし11員非芳香族複素環基
を示す。
(1)式:-X2-R5(式中、X2はO、S、SO2またはNR6を示し、R5は置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示す)で表される基;
(2)置換されていてもよいC1-6アルキル基;
(3)置換されていてもよいC3-11シクロアルキル基;または
(4)置換されていてもよい3ないし11員非芳香族複素環基
を示す。
R3で示される「式:-X2-R5で表される基」における「X2」はO、S、SO2またはNR6を示す。
ここで、R6は水素原子または置換されていてもよいC1-6アルキル基を示す。
R6で示される「置換されていてもよいC1-6アルキル基」における「C1-6アルキル基」は置換可能な位置に1ないし3個の置換基を有していてもよい。該置換基としては、例えば、後述のR5で示される「置換されていてもよいC1-6アルキル基」における「C1-6アルキル基」が有していてもよい置換基と同様のものが挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
R6は、好ましくは、水素原子である。
R3で示される「式:-X2-R5で表される基」における「X2」は、好ましくは、O、SまたはNHである。
R3で示される「式:-X2-R5で表される基」における「R5」は、置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示す。
R5で示される「置換されていてもよいC1-6アルキル基」における「C1-6アルキル基」は、置換可能な位置に1ないし5個(好ましくは、1ないし3個)の置換基を有していてもよい。
該置換基としては、例えば、
(1)C3-10シクロアルキル基(例、シクロプロピル、シクロヘキシル);
(2)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール基(例、フェニル、ナフチル);
(3)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル);
(4)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、
(d)ハロゲン原子、および
(e)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員非芳香族複素環基(例、テトラヒドロフリル、モルホリニル、チオモルホリニル、ピペリジル、ピロリジニル、ピペラジニル);
(5)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ-カルボニル基、
(d)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル)、
(e)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基、および
(f)4ないし7員芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル)
から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基;
(6)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基;
(7)(a)ハロゲン原子、
(b)C1-6アルコキシ基、
(c)C6-14アリール基(例、フェニル)、および
(d)4ないし7員複素環基(例、テトラヒドロフリル)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ-カルボニル基(例、メトキシカルボニル);
(8)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル、エチルスルホニル、イソプロピルスルホニル);
(9)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基;
(10)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいチオカルバモイル基;
(11)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいスルファモイル基;
(12)カルボキシ基;
(13)ヒドロキシ基;
(14)(a)ハロゲン原子(例、フッ素原子)、
(b)カルボキシ基、
(c)C1-6アルコキシ基、
(d)1ないし3個のC6-14アリール基(例、フェニル)で置換されていてもよいC1-6アルコキシ-カルボニル基、
(e)C1-6アルキル基およびC1-6アルコキシ-カルボニル基から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基、
(f)4ないし7員複素環基(例、テトラヒドロフリル)、および
(g)C3-10シクロアルキル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ);
(15)1ないし3個のハロゲン原子で置換されていてもよいC2-6アルケニルオキシ基(例、エテニルオキシ);
(16)C7-13アラルキルオキシ基(例、ベンジルオキシ);
(17)C6-14アリールオキシ基(例、フェニルオキシ、ナフチルオキシ);
(18)C1-6アルキル-カルボニルオキシ基(例、アセチルオキシ、tert-ブチルカルボニルオキシ);
(19)(a)ハロゲン原子、および
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール-カルボニル基(例、ベンゾイル);
(20)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員非芳香族複素環カルボニル基(例、ピロリジニルカルボニル、モルホリニルカルボニル);
(21)メルカプト基;
(22)(a)ハロゲン原子、および
(b)C1-6アルコキシ-カルボニル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキルチオ基(例、メチルチオ、エチルチオ);
(23)C7-13アラルキルチオ基(例、ベンジルチオ);
(24)C6-14アリールチオ基(例、フェニルチオ、ナフチルチオ);
(25)シアノ基;
(26)ニトロ基;
(27)ハロゲン原子(例、フッ素原子、塩素原子、臭素原子);
(28)C1-3アルキレンジオキシ基;
(29)C1-3アルキレンオキシ基(例、メチレンオキシ、エチレンオキシ);
(30)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員芳香族複素環カルボニル基(例、ピラゾリルカルボニル、ピラジニルカルボニル、イソキサゾリルカルボニル、ピリジルカルボニル、チアゾリルカルボニル);
等が挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
(1)C3-10シクロアルキル基(例、シクロプロピル、シクロヘキシル);
(2)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール基(例、フェニル、ナフチル);
(3)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(d)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル);
(4)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)ヒドロキシ基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、
(d)ハロゲン原子、および
(e)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員非芳香族複素環基(例、テトラヒドロフリル、モルホリニル、チオモルホリニル、ピペリジル、ピロリジニル、ピペラジニル);
(5)(a)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基、
(c)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ-カルボニル基、
(d)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル)、
(e)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基、および
(f)4ないし7員芳香族複素環基(例、チエニル、フリル、ピリジル、ピラゾリル、イミダゾリル、テトラゾリル、オキサゾリル、チアゾリル、オキサジアゾリル、チアジアゾリル)
から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基;
(6)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル-カルボニル基;
(7)(a)ハロゲン原子、
(b)C1-6アルコキシ基、
(c)C6-14アリール基(例、フェニル)、および
(d)4ないし7員複素環基(例、テトラヒドロフリル)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ-カルボニル基(例、メトキシカルボニル);
(8)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキルスルホニル基(例、メチルスルホニル、エチルスルホニル、イソプロピルスルホニル);
(9)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいカルバモイル基;
(10)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいチオカルバモイル基;
(11)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基でモノまたはジ置換されていてもよいスルファモイル基;
(12)カルボキシ基;
(13)ヒドロキシ基;
(14)(a)ハロゲン原子(例、フッ素原子)、
(b)カルボキシ基、
(c)C1-6アルコキシ基、
(d)1ないし3個のC6-14アリール基(例、フェニル)で置換されていてもよいC1-6アルコキシ-カルボニル基、
(e)C1-6アルキル基およびC1-6アルコキシ-カルボニル基から選ばれる置換基でモノまたはジ置換されていてもよいアミノ基、
(f)4ないし7員複素環基(例、テトラヒドロフリル)、および
(g)C3-10シクロアルキル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ);
(15)1ないし3個のハロゲン原子で置換されていてもよいC2-6アルケニルオキシ基(例、エテニルオキシ);
(16)C7-13アラルキルオキシ基(例、ベンジルオキシ);
(17)C6-14アリールオキシ基(例、フェニルオキシ、ナフチルオキシ);
(18)C1-6アルキル-カルボニルオキシ基(例、アセチルオキシ、tert-ブチルカルボニルオキシ);
(19)(a)ハロゲン原子、および
(b)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基
から選ばれる1ないし3個の置換基で置換されていてもよいC6-14アリール-カルボニル基(例、ベンゾイル);
(20)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員非芳香族複素環カルボニル基(例、ピロリジニルカルボニル、モルホリニルカルボニル);
(21)メルカプト基;
(22)(a)ハロゲン原子、および
(b)C1-6アルコキシ-カルボニル基
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキルチオ基(例、メチルチオ、エチルチオ);
(23)C7-13アラルキルチオ基(例、ベンジルチオ);
(24)C6-14アリールチオ基(例、フェニルチオ、ナフチルチオ);
(25)シアノ基;
(26)ニトロ基;
(27)ハロゲン原子(例、フッ素原子、塩素原子、臭素原子);
(28)C1-3アルキレンジオキシ基;
(29)C1-3アルキレンオキシ基(例、メチレンオキシ、エチレンオキシ);
(30)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基から選ばれる1ないし3個の置換基で置換されていてもよい4ないし7員芳香族複素環カルボニル基(例、ピラゾリルカルボニル、ピラジニルカルボニル、イソキサゾリルカルボニル、ピリジルカルボニル、チアゾリルカルボニル);
等が挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
R5で示される「置換されていてもよいC1-6アルキル基」は、好ましくは、
(A)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(B)ヒドロキシ基、
(C)1~3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(D)ハロゲン原子(例、フッ素原子)
から選ばれる1~5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)
である。
(A)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(B)ヒドロキシ基、
(C)1~3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(D)ハロゲン原子(例、フッ素原子)
から選ばれる1~5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)
である。
R5で示される「置換されていてもよい3ないし11員環状基」は、例えば、6ないし10員芳香族炭化水素基、5ないし11員芳香族複素環基(例、5ないし7員単環式芳香族複素環基、8ないし11員縮合芳香族複素環基)、3ないし11員非芳香族環状炭化水素基(好ましくは、3ないし10員非芳香族環状炭化水素基)、3ないし8員非芳香族複素環基等が挙げられる。
R5で示される「置換されていてもよい3ないし11員環状基」における「3ないし11員環状基」は、置換可能な位置に1ないし5個(好ましくは、1ないし3個)の置換基を有していてもよい。
該置換基としては、例えば、環Aで示される「さらに置換されていてもよい6員芳香環」における「6員芳香環」が有していてもよい置換基と同様のもの等が挙げられる。該「3ないし11員環状基」が非芳香族環状炭化水素基または非芳香族複素環基である場合は、置換基としてオキソ基を有していてもよい。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
R5で示される「置換されていてもよい3ないし11員環状基」は、好ましくは、それぞれ置換されていてもよい、3ないし11員環状基またはC3-11シクロアルキル基であり、より好ましくは、(1)3ないし8員非芳香族複素環基、または(2)置換されていてもよいC3-11シクロアルキル基であり、さらに好ましくは、(1)3ないし8員非芳香族複素環基(例、オキセタニル)または(2)1ないし5個のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-11シクロアルキル基(例、シクロブチル、シクロペンチル、シクロヘキシル)である。
R3で示される「式:-X2-R5で表される基」における「R5」は、好ましくは、
(1)3ないし8員非芳香族複素環基(例、オキセタニル);または
(2)(A)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(B)ヒドロキシ基、
(C)C1-6アルコキシ基(例、メトキシ)、および
(D)ハロゲン原子(例、フッ素原子)
等で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル);
等である。
(1)3ないし8員非芳香族複素環基(例、オキセタニル);または
(2)(A)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(B)ヒドロキシ基、
(C)C1-6アルコキシ基(例、メトキシ)、および
(D)ハロゲン原子(例、フッ素原子)
等で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル);
等である。
別の態様において、R3で示される「式:-X2-R5で表される基」における「R5」は、好ましくは、
(1)3ないし8員非芳香族複素環基(例、オキセタニル)、
(2)(A)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(B)ヒドロキシ基、
(C)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(D)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)、または
(3)1ないし5個(好ましくは1ないし4個)のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-11シクロアルキル基(例、シクロブチル、シクロペンチル、シクロヘキシル)である。
(1)3ないし8員非芳香族複素環基(例、オキセタニル)、
(2)(A)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(B)ヒドロキシ基、
(C)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(D)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)、または
(3)1ないし5個(好ましくは1ないし4個)のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-11シクロアルキル基(例、シクロブチル、シクロペンチル、シクロヘキシル)である。
R3で示される「式:-X2-R5で表される基」は、好ましくは、
X2がO、SまたはNHであり、および
R5が、
(1)3ないし8員非芳香族複素環基(例、オキセタニル);または
(2)(A)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(B)ヒドロキシ基、
(C)C1-6アルコキシ基(例、メトキシ)、および
(D)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル);
である式:-X2-R5で表される基である。
X2がO、SまたはNHであり、および
R5が、
(1)3ないし8員非芳香族複素環基(例、オキセタニル);または
(2)(A)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(B)ヒドロキシ基、
(C)C1-6アルコキシ基(例、メトキシ)、および
(D)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル);
である式:-X2-R5で表される基である。
別の態様において、R3で示される「式:-X2-R5で表される基」は、好ましくは、
X2が、O、SまたはNHであり、かつ
R5が、
(1)3ないし8員非芳香族複素環基(例、オキセタニル)、
(2)(A)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(B)ヒドロキシ基、
(C)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(D)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)、または
(3)1ないし5個(好ましくは1ないし4個)のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-11シクロアルキル基(例、シクロブチル、シクロペンチル、シクロヘキシル)
である式:-X2-R5で表される基である。
X2が、O、SまたはNHであり、かつ
R5が、
(1)3ないし8員非芳香族複素環基(例、オキセタニル)、
(2)(A)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(B)ヒドロキシ基、
(C)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(D)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)、または
(3)1ないし5個(好ましくは1ないし4個)のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-11シクロアルキル基(例、シクロブチル、シクロペンチル、シクロヘキシル)
である式:-X2-R5で表される基である。
R3で示される「置換されていてもよいC1-6アルキル基」における「C1-6アルキル基」は置換可能な位置に1ないし5個(好ましくは、1ないし3個)の置換基を有していてもよい。該置換基としては、例えば、R5で示される「置換されていてもよいC1-6アルキル基」における「C1-6アルキル基」が有していてもよい置換基と同様のものが挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
R3で示される「置換されていてもよいC1-6アルキル基」は、好ましくは、C1-6アルキル基(例、プロピル)である。
別の態様において、R3で示される「置換されていてもよいC1-6アルキル基」は、好ましくは、
(A)ヒドロキシ基、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)C3-6シクロアルキル基(例、シクロプロピル)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、ブチル、sec-ブチル)
である。
(A)ヒドロキシ基、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)C3-6シクロアルキル基(例、シクロプロピル)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、ブチル、sec-ブチル)
である。
R3で示される「置換されていてもよいC3-11シクロアルキル基」における「C3-11シクロアルキル基」は置換可能な位置に1ないし3個の置換基を有していてもよい。該置換基としては、例えば、環Aで示される「さらに置換されていてもよい6員芳香環」における「6員芳香環」が有していてもよい置換基と同様のもの、オキソ基等が挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
R3で示される「置換されていてもよいC3-11シクロアルキル基」は、好ましくは、
(A)C1-6アルキル基(例、メチル)、および
(B)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよいC3-11シクロアルキル基(例、シクロプロピル、シクロブチル)である。
(A)C1-6アルキル基(例、メチル)、および
(B)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよいC3-11シクロアルキル基(例、シクロプロピル、シクロブチル)である。
R3で示される「置換されていてもよい3ないし11員非芳香族複素環基」における「3ないし11員非芳香族複素環基」としては、例えば、上記「3ないし8員非芳香族複素環基」および上記「3ないし8員非芳香族複素環基」がベンゼン環等と縮合した縮合環から誘導される基が挙げられる。
R3で示される、「置換されていてもよい3ないし11員非芳香族複素環基」における「3ないし11員非芳香族複素環基」は、置換可能な位置に1ないし3個の置換基を有していてもよい。該置換基としては、例えば、環Aで示される「さらに置換されていてもよい6員芳香環」における「6員芳香環」が有していてもよい置換基と同様のもの、オキソ基等が挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
R3で示される「置換されていてもよい3ないし11員非芳香族複素環基」は、好ましくは、1ないし3個(好ましくは、1または2個)のハロゲン原子(例、フッ素原子)で置換されていてもよい3ないし11員非芳香族複素環基(例、テトラヒドロピラニル、テトラヒドロフリル、ピロリジニル)である。
R3は、好ましくは、
(1)式:-X2-R5で表される基であり、
X2がO、SまたはNH、および
R5が、
(A)3ないし8員非芳香族複素環基(例、オキセタニル);
(B)(i)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(ii)ヒドロキシ基、
(iii)C1-6アルコキシ基(例、メトキシ)、および
(iv)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル);
(2)C1-6アルキル基(例、プロピル);
等である。
(1)式:-X2-R5で表される基であり、
X2がO、SまたはNH、および
R5が、
(A)3ないし8員非芳香族複素環基(例、オキセタニル);
(B)(i)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(ii)ヒドロキシ基、
(iii)C1-6アルコキシ基(例、メトキシ)、および
(iv)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル);
(2)C1-6アルキル基(例、プロピル);
等である。
別の態様において、R3は、好ましくは、
(1)式:-X2-R5
[式中、
X2が、O、SまたはNHであり、かつ
R5が、
(A)3ないし8員非芳香族複素環基(例、オキセタニル)、
(B)(i)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(ii)ヒドロキシ基、
(iii)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(iv)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)、または
(C)1ないし5個(好ましくは1ないし4個)のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-11シクロアルキル基(例、シクロブチル、シクロペンチル、シクロヘキシル)
である]で表される基、
(2)(A)ヒドロキシ基、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)C3-6シクロアルキル基(例、シクロプロピル)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、ブチル、sec-ブチル)、
(3)(A)C1-6アルキル基(例、メチル)、および
(B)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよいC3-11シクロアルキル基(例、シクロプロピル、シクロブチル)、または
(4)1ないし3個(好ましくは、1または2個)のハロゲン原子(例、フッ素原子)で置換されていてもよい3ないし11員非芳香族複素環基(例、テトラヒドロピラニル、テトラヒドロフリル、ピロリジニル)
である。
(1)式:-X2-R5
[式中、
X2が、O、SまたはNHであり、かつ
R5が、
(A)3ないし8員非芳香族複素環基(例、オキセタニル)、
(B)(i)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(ii)ヒドロキシ基、
(iii)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(iv)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)、または
(C)1ないし5個(好ましくは1ないし4個)のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-11シクロアルキル基(例、シクロブチル、シクロペンチル、シクロヘキシル)
である]で表される基、
(2)(A)ヒドロキシ基、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)C3-6シクロアルキル基(例、シクロプロピル)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、ブチル、sec-ブチル)、
(3)(A)C1-6アルキル基(例、メチル)、および
(B)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよいC3-11シクロアルキル基(例、シクロプロピル、シクロブチル)、または
(4)1ないし3個(好ましくは、1または2個)のハロゲン原子(例、フッ素原子)で置換されていてもよい3ないし11員非芳香族複素環基(例、テトラヒドロピラニル、テトラヒドロフリル、ピロリジニル)
である。
R4は置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示す。
R4で示される「置換されていてもよいC1-6アルキル基」における「C1-6アルキル基」は置換可能な位置に1ないし3個の置換基を有していてもよい。該置換基としては、例えば、R5で示される「置換されていてもよいC1-6アルキル基」における「C1-6アルキル基」が有していてもよい置換基と同様のものが挙げられる。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
R4で示される「置換されていてもよいC1-6アルキル基」は、好ましくは、1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル)である。
R4で示される「置換されていてもよい3ないし11員環状基」における「置換されていてもよい3ないし11員環状基」は、例えば、6ないし10員芳香族炭化水素基、5ないし11員芳香族複素環基(例、5ないし7員単環式芳香族複素環基、8ないし11員縮合芳香族複素環基)、3ないし10員非芳香族環状炭化水素基、3ないし8員非芳香族複素環基等が挙げられる。
R4で示される「置換されていてもよい3ないし11員環状基」における「3ないし11員環状基」は、置換可能な位置に1ないし3個の置換基を有していてもよい。該置換基としては、例えば、環Aで示される「さらに置換されていてもよい6員芳香環」における「6員芳香環」が有していてもよい置換基と同様のもの等が挙げられる。該「3ないし11員環状基」が非芳香族環状炭化水素基または非芳香族複素環基である場合は、置換基としてオキソ基を有していてもよい。置換基が2個以上である場合、各置換基は同一でも異なっていてもよい。
R4は、好ましくは、置換されていてもよいC1-6アルキル基であり、より好ましくは、1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル)である。
化合物(I)の好適な例として、以下の化合物が挙げられる。
[化合物A]
環Aが、1ないし3個のハロゲン原子でさらに置換されていてもよいベンゼンまたはピリジンであり;
X1が、Oであり;
R1が、
(1)ピリジルで置換されたメチル;または
(2)(A)C1-6アルコキシ基(例、メトキシ)、
(B)C1-6アルキル基(例、メチル)、および
(C)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい5または6員複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、テトラヒドロピラニル、モルホリニル、チオモルホリニル)
であり;
R2が、水素原子またはメチルであり;
R3が、
(1)式:-X2-R5で表される基であり、
X2がO、SまたはNH、かつ
R5が、
(A)3ないし8員非芳香族複素環基(例、オキセタニル);または
(B)(i)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(ii)ヒドロキシ基、
(iii)C1-6アルコキシ基(例、メトキシ)、および
(iv)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル);または
(2)C1-6アルキル基(例、プロピル)
であり;
R4が、1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル)である、化合物(I)。
環Aが、1ないし3個のハロゲン原子でさらに置換されていてもよいベンゼンまたはピリジンであり;
X1が、Oであり;
R1が、
(1)ピリジルで置換されたメチル;または
(2)(A)C1-6アルコキシ基(例、メトキシ)、
(B)C1-6アルキル基(例、メチル)、および
(C)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい5または6員複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、テトラヒドロピラニル、モルホリニル、チオモルホリニル)
であり;
R2が、水素原子またはメチルであり;
R3が、
(1)式:-X2-R5で表される基であり、
X2がO、SまたはNH、かつ
R5が、
(A)3ないし8員非芳香族複素環基(例、オキセタニル);または
(B)(i)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(ii)ヒドロキシ基、
(iii)C1-6アルコキシ基(例、メトキシ)、および
(iv)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル);または
(2)C1-6アルキル基(例、プロピル)
であり;
R4が、1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル)である、化合物(I)。
[化合物B]
環Aが、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリジン、または1もしくは2個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリミジン[好ましくは、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、フッ素原子)でさらに置換されていてもよいピリジン、またはピリミジン]であり;
X1が、Oであり;
R1が、
(1)5ないし7員単環式芳香族複素環基(例、ピリジル)で置換されたC1-6アルキル基(例、メチル)[好ましくは、ピリジルで置換されたメチル]、
(2)(A)C1-6アルコキシ基(例、メトキシ)、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)オキソ基
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよい5または6員複素環基[例、5または6員芳香族複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、トリアゾリル、オキサジアゾリル、イミダゾリル等);5または6員非芳香族複素環基(例、モルホリニル、ジヒドロピリジル、ジヒドロピリダジニル、テトラヒドロピラニル、ピペラジニル、ジヒドロオキサジアゾリル、ピロリジニル、ピペリジル等)]
であり;
R2が、水素原子またはC1-6アルキル基(例、メチル)であり;
R3が、
(1)式:-X2-R5
[式中、
X2が、O、SまたはNHであり、かつ
R5が、
(A)3ないし8員非芳香族複素環基(例、オキセタニル)、
(B)(i)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(ii)ヒドロキシ基、
(iii)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(iv)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)、または
(C)1ないし5個(好ましくは1ないし4個)のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-11シクロアルキル基(例、シクロブチル、シクロペンチル、シクロヘキシル)
である]で表される基、
(2)(A)ヒドロキシ基、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)C3-6シクロアルキル基(例、シクロプロピル)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、ブチル、sec-ブチル)、
(3)(A)C1-6アルキル基(例、メチル)、および
(B)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよいC3-11シクロアルキル基(例、シクロプロピル、シクロブチル)、または
(4)1ないし3個(好ましくは、1または2個)のハロゲン原子(例、フッ素原子)で置換されていてもよい3ないし11員非芳香族複素環基(例、テトラヒドロピラニル、テトラヒドロフリル、ピロリジニル)
であり;
R4が、1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル)
である、化合物(I)。
環Aが、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリジン、または1もしくは2個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリミジン[好ましくは、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、フッ素原子)でさらに置換されていてもよいピリジン、またはピリミジン]であり;
X1が、Oであり;
R1が、
(1)5ないし7員単環式芳香族複素環基(例、ピリジル)で置換されたC1-6アルキル基(例、メチル)[好ましくは、ピリジルで置換されたメチル]、
(2)(A)C1-6アルコキシ基(例、メトキシ)、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)オキソ基
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよい5または6員複素環基[例、5または6員芳香族複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、トリアゾリル、オキサジアゾリル、イミダゾリル等);5または6員非芳香族複素環基(例、モルホリニル、ジヒドロピリジル、ジヒドロピリダジニル、テトラヒドロピラニル、ピペラジニル、ジヒドロオキサジアゾリル、ピロリジニル、ピペリジル等)]
であり;
R2が、水素原子またはC1-6アルキル基(例、メチル)であり;
R3が、
(1)式:-X2-R5
[式中、
X2が、O、SまたはNHであり、かつ
R5が、
(A)3ないし8員非芳香族複素環基(例、オキセタニル)、
(B)(i)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(ii)ヒドロキシ基、
(iii)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(iv)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)、または
(C)1ないし5個(好ましくは1ないし4個)のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-11シクロアルキル基(例、シクロブチル、シクロペンチル、シクロヘキシル)
である]で表される基、
(2)(A)ヒドロキシ基、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)C3-6シクロアルキル基(例、シクロプロピル)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、ブチル、sec-ブチル)、
(3)(A)C1-6アルキル基(例、メチル)、および
(B)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよいC3-11シクロアルキル基(例、シクロプロピル、シクロブチル)、または
(4)1ないし3個(好ましくは、1または2個)のハロゲン原子(例、フッ素原子)で置換されていてもよい3ないし11員非芳香族複素環基(例、テトラヒドロピラニル、テトラヒドロフリル、ピロリジニル)
であり;
R4が、1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル)
である、化合物(I)。
[化合物C]
環Aが、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリジン、または1もしくは2個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリミジン[好ましくは、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、フッ素原子)でさらに置換されていてもよいピリジン、またはピリミジン]であり;
X1が、Oであり;
R1が、
(A)C1-6アルコキシ基(例、メトキシ)、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)オキソ基
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよい5または6員複素環基[例、5または6員芳香族複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、トリアゾリル、オキサジアゾリル、イミダゾリル等);5または6員非芳香族複素環基(例、モルホリニル、ジヒドロピリジル、ジヒドロピリダジニル、テトラヒドロピラニル、ピペラジニル、ジヒドロオキサジアゾリル、ピロリジニル、ピペリジル等)]
であり;
R2が、水素原子またはC1-6アルキル基(例、メチル)であり;
R3が、
(1)式:-X2-R5
[式中、
X2が、O、SまたはNHであり、かつ
R5が、
(A)3ないし8員非芳香族複素環基(例、オキセタニル)、
(B)(i)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(ii)ヒドロキシ基、
(iii)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(iv)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)、または
(C)1ないし5個(好ましくは1ないし4個)のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-11シクロアルキル基(例、シクロブチル、シクロペンチル、シクロヘキシル)
である]で表される基、
(2)(A)ヒドロキシ基、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)C3-6シクロアルキル基(例、シクロプロピル)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、ブチル、sec-ブチル)、
(3)(A)C1-6アルキル基(例、メチル)、および
(B)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよいC3-11シクロアルキル基(例、シクロプロピル、シクロブチル)、または
(4)1ないし3個(好ましくは、1または2個)のハロゲン原子(例、フッ素原子)で置換されていてもよい3ないし11員非芳香族複素環基(例、テトラヒドロピラニル、テトラヒドロフリル、ピロリジニル)
であり;
R4が、1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル)
である、化合物(I)。
環Aが、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリジン、または1もしくは2個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリミジン[好ましくは、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、フッ素原子)でさらに置換されていてもよいピリジン、またはピリミジン]であり;
X1が、Oであり;
R1が、
(A)C1-6アルコキシ基(例、メトキシ)、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、メチル)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)オキソ基
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよい5または6員複素環基[例、5または6員芳香族複素環基(例、フリル、ピリジル、ピリミジニル、ピリダジニル、ピラゾリル、チアゾリル、オキサゾリル、トリアゾリル、オキサジアゾリル、イミダゾリル等);5または6員非芳香族複素環基(例、モルホリニル、ジヒドロピリジル、ジヒドロピリダジニル、テトラヒドロピラニル、ピペラジニル、ジヒドロオキサジアゾリル、ピロリジニル、ピペリジル等)]
であり;
R2が、水素原子またはC1-6アルキル基(例、メチル)であり;
R3が、
(1)式:-X2-R5
[式中、
X2が、O、SまたはNHであり、かつ
R5が、
(A)3ないし8員非芳香族複素環基(例、オキセタニル)、
(B)(i)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(ii)ヒドロキシ基、
(iii)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(iv)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)、または
(C)1ないし5個(好ましくは1ないし4個)のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-11シクロアルキル基(例、シクロブチル、シクロペンチル、シクロヘキシル)
である]で表される基、
(2)(A)ヒドロキシ基、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)C3-6シクロアルキル基(例、シクロプロピル)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、ブチル、sec-ブチル)、
(3)(A)C1-6アルキル基(例、メチル)、および
(B)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよいC3-11シクロアルキル基(例、シクロプロピル、シクロブチル)、または
(4)1ないし3個(好ましくは、1または2個)のハロゲン原子(例、フッ素原子)で置換されていてもよい3ないし11員非芳香族複素環基(例、テトラヒドロピラニル、テトラヒドロフリル、ピロリジニル)
であり;
R4が、1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル)
である、化合物(I)。
[化合物D]
環Aが、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリジン、または1もしくは2個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリミジン[好ましくは、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、フッ素原子)でさらに置換されていてもよいピリジン、またはピリミジン]であり;
X1が、Oであり;
R1が、モルホリニル基または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいピラゾリル基であり;
R2が、水素原子またはC1-6アルキル基(例、メチル)であり;
R3が、
(1)式:-X2-R5
[式中、
X2が、O、SまたはNHであり、かつ
R5が、
(A)3ないし8員非芳香族複素環基(例、オキセタニル)、
(B)(i)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(ii)ヒドロキシ基、
(iii)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(iv)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)、または
(C)1ないし5個(好ましくは1ないし4個)のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-11シクロアルキル基(例、シクロブチル、シクロペンチル、シクロヘキシル)
である]で表される基、
(2)(A)ヒドロキシ基、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)C3-6シクロアルキル基(例、シクロプロピル)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、ブチル、sec-ブチル)、
(3)(A)C1-6アルキル基(例、メチル)、および
(B)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよいC3-11シクロアルキル基(例、シクロプロピル、シクロブチル)、または
(4)1ないし3個(好ましくは、1または2個)のハロゲン原子(例、フッ素原子)で置換されていてもよい3ないし11員非芳香族複素環基(例、テトラヒドロピラニル、テトラヒドロフリル、ピロリジニル)
であり;
R4が、1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル)
である、化合物(I)。
環Aが、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリジン、または1もしくは2個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいピリミジン[好ましくは、1ないし3個(好ましくは、1個)のハロゲン原子(例、塩素原子、フッ素原子)でさらに置換されていてもよいベンゼン、1ないし3個(好ましくは、1個)のハロゲン原子(例、フッ素原子)でさらに置換されていてもよいピリジン、またはピリミジン]であり;
X1が、Oであり;
R1が、モルホリニル基または1ないし3個のC1-6アルキル基(例、メチル)で置換されていてもよいピラゾリル基であり;
R2が、水素原子またはC1-6アルキル基(例、メチル)であり;
R3が、
(1)式:-X2-R5
[式中、
X2が、O、SまたはNHであり、かつ
R5が、
(A)3ないし8員非芳香族複素環基(例、オキセタニル)、
(B)(i)C1-6アルキルスルホニル基(例、メチルスルホニル)、
(ii)ヒドロキシ基、
(iii)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、および
(iv)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、イソプロピル、イソブチル)、または
(C)1ないし5個(好ましくは1ないし4個)のハロゲン原子(例、フッ素原子)で置換されていてもよいC3-11シクロアルキル基(例、シクロブチル、シクロペンチル、シクロヘキシル)
である]で表される基、
(2)(A)ヒドロキシ基、
(B)1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルコキシ基(例、メトキシ、エトキシ)、
(C)ハロゲン原子(例、フッ素原子)、および
(D)C3-6シクロアルキル基(例、シクロプロピル)
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基(例、メチル、エチル、プロピル、ブチル、sec-ブチル)、
(3)(A)C1-6アルキル基(例、メチル)、および
(B)ハロゲン原子(例、フッ素原子)
から選ばれる1ないし3個(好ましくは、1または2個)の置換基で置換されていてもよいC3-11シクロアルキル基(例、シクロプロピル、シクロブチル)、または
(4)1ないし3個(好ましくは、1または2個)のハロゲン原子(例、フッ素原子)で置換されていてもよい3ないし11員非芳香族複素環基(例、テトラヒドロピラニル、テトラヒドロフリル、ピロリジニル)
であり;
R4が、1ないし3個のハロゲン原子(例、フッ素原子)で置換されていてもよいC1-6アルキル基(例、エチル)
である、化合物(I)。
[化合物E]
2-(エチルスルファニル)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩。
2-(エチルスルファニル)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩。
[化合物F]
2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩。
2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩。
[化合物G]
6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩。
6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩。
[化合物H]
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オンまたはその塩。
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オンまたはその塩。
式(I)で表される化合物の塩としては、薬理学的に許容される塩が好ましく、このような塩としては、例えば、無機塩基との塩、有機塩基との塩、無機酸との塩、有機酸との塩、塩基性または酸性アミノ酸との塩等が挙げられる。
無機塩基との塩の好適な例としては、ナトリウム塩、カリウム塩等のアルカリ金属塩;カルシウム塩、マグネシウム塩等のアルカリ土類金属塩;アルミニウム塩;アンモニウム塩等が挙げられる。
有機塩基との塩の好適な例としては、トリメチルアミン、トリエチルアミン、ピリジン、ピコリン、エタノールアミン、ジエタノールアミン、トリエタノールアミン、トロメタミン[トリス(ヒドロキシメチル)メチルアミン]、tert-ブチルアミン、シクロヘキシルアミン、ベンジルアミン、ジシクロヘキシルアミン、N,N-ジベンジルエチレンジアミン等との塩が挙げられる。
無機酸との塩の好適な例としては、塩酸、臭化水素酸、硝酸、硫酸、リン酸等との塩が挙げられる。
有機酸との塩の好適な例としては、ギ酸、酢酸、トリフルオロ酢酸、フタル酸、フマル酸、シュウ酸、酒石酸、マレイン酸、クエン酸、コハク酸、リンゴ酸、メタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸等との塩が挙げられる。
塩基性アミノ酸との塩の好適な例としては、アルギニン、リジン、オルニチン等との塩が挙げられる。
酸性アミノ酸との塩の好適な例としては、アスパラギン酸、グルタミン酸等との塩が挙げられる。
化合物(I)は、同位元素(例、3H、14C、35S、125I)等で標識されていてもよい。
また、化合物(I)は、無溶媒和物(例えば、無水物)であっても、溶媒和物(例えば、水和物)であってもよい。
さらに、1Hを2H(D)に変換した重水素変換体も、化合物(I)に包含される。
また、化合物(I)は、無溶媒和物(例えば、無水物)であっても、溶媒和物(例えば、水和物)であってもよい。
さらに、1Hを2H(D)に変換した重水素変換体も、化合物(I)に包含される。
化合物(I)は、結晶であっても無晶形であってもよい。化合物(I)が結晶である場合、結晶形が単一であっても結晶形混合物であっても化合物(I)に包含される。結晶は、自体公知の結晶化法を適用して、結晶化することによって製造することができる。
本明細書中、融点は、例えば、微量融点測定器(ヤナコ、MP-500D型またはBuchi、B-545型)またはDSC(示差走査熱量分析)装置(SEIKO、EXSTAR6000)等を用いて測定される融点を意味する。
一般に、融点は、測定機器、測定条件等によって変動する場合がある。本明細書中の結晶は、通常の誤差範囲内であれば、本明細書に記載の融点と異なる値を示す結晶であってもよい。
本明細書中、融点は、例えば、微量融点測定器(ヤナコ、MP-500D型またはBuchi、B-545型)またはDSC(示差走査熱量分析)装置(SEIKO、EXSTAR6000)等を用いて測定される融点を意味する。
一般に、融点は、測定機器、測定条件等によって変動する場合がある。本明細書中の結晶は、通常の誤差範囲内であれば、本明細書に記載の融点と異なる値を示す結晶であってもよい。
化合物(I)は、薬学的に許容され得る共結晶または共結晶塩であってもよい。ここで、共結晶または共結晶塩とは、各々が異なる物理的特性(例えば、構造、融点、融解熱、吸湿性、溶解性および安定性等)を持つ、室温で二種またはそれ以上の独特な固体から構成される結晶性物質を意味する。共結晶または共結晶塩は、自体公知の共結晶化法に従い製造することができる。本発明の結晶は、物理化学的性質(例、融点、溶解度、安定性)および生物学的性質(例、体内動態(吸収性、分布、代謝、排泄)、薬効発現)に優れ、医薬として極めて有用である。
化合物(I)はプロドラッグであってもよい。化合物(I)のプロドラッグとは、生体内における生理条件下で酵素または胃酸等による反応により化合物(I)に変換する化合物、すなわち酵素的に酸化、還元、加水分解等を起こして化合物(I)に変化する化合物、胃酸等により加水分解等を起こして化合物(I)に変化する化合物である。
化合物(I)のプロドラッグとしては、
化合物(I)のアミノ基がアシル化、アルキル化またはリン酸化された化合物(例、化合物(I)のアミノ基がエイコサノイル化、アラニル化、ペンチルアミノカルボニル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メトキシカルボニル化、テトラヒドロフラニル化、ピロリジルメチル化、ピバロイルオキシメチル化またはtert-ブチル化された化合物);
化合物(I)のヒドロキシ基がアシル化、アルキル化、リン酸化またはホウ酸化された化合物(例、化合物(I)のヒドロキシ基がアセチル化、パルミトイル化、プロパノイル化、ピバロイル化、サクシニル化、フマリル化、アラニル化またはジメチルアミノメチルカルボニル化された化合物);
化合物(I)のカルボキシ基がエステル化またはアミド化された化合物(例、化合物(I)のカルボキシ基がエチルエステル化、フェニルエステル化、カルボキシメチルエステル化、ジメチルアミノメチルエステル化、ピバロイルオキシメチルエステル化、エトキシカルボニルオキシエチルエステル化、フタリジルエステル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メチルエステル化、シクロヘキシルオキシカルボニルエチルエステル化またはメチルアミド化された化合物)等が挙げられる。これらの化合物は自体公知の方法によって化合物(I)から製造することができる。
化合物(I)のアミノ基がアシル化、アルキル化またはリン酸化された化合物(例、化合物(I)のアミノ基がエイコサノイル化、アラニル化、ペンチルアミノカルボニル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メトキシカルボニル化、テトラヒドロフラニル化、ピロリジルメチル化、ピバロイルオキシメチル化またはtert-ブチル化された化合物);
化合物(I)のヒドロキシ基がアシル化、アルキル化、リン酸化またはホウ酸化された化合物(例、化合物(I)のヒドロキシ基がアセチル化、パルミトイル化、プロパノイル化、ピバロイル化、サクシニル化、フマリル化、アラニル化またはジメチルアミノメチルカルボニル化された化合物);
化合物(I)のカルボキシ基がエステル化またはアミド化された化合物(例、化合物(I)のカルボキシ基がエチルエステル化、フェニルエステル化、カルボキシメチルエステル化、ジメチルアミノメチルエステル化、ピバロイルオキシメチルエステル化、エトキシカルボニルオキシエチルエステル化、フタリジルエステル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メチルエステル化、シクロヘキシルオキシカルボニルエチルエステル化またはメチルアミド化された化合物)等が挙げられる。これらの化合物は自体公知の方法によって化合物(I)から製造することができる。
また、化合物(I)のプロドラッグは、広川書店1990年刊「医薬品の開発」第7巻分子設計163頁から198頁に記載されているような、生理的条件で化合物(I)に変化するものであってもよい。
以下、化合物(I)の製造法について説明する。
化合物(I)は、例えば、以下に示す方法またはそれに準ずる方法で製造することができる。
以下の各スキームにおいて、各原料化合物は、反応を阻害しないのであれば、塩を形成していてもよく、該塩としては、前述の式(I)で示される化合物の塩として例示したものが用いられる。
また、以下の各スキームにおいて、原料化合物は、具体的製法を述べない場合、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。
以下の各スキームにおける反応で使用する溶媒としては、反応を阻害せず、出発物質をある程度溶解するものであれば特に限定されないが、例えば、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;ヘキサン、ヘプタン等の脂肪族炭化水素類;ジエチルエーテル、ジイソプロピルエーテル、tert-ブチルメチルエーテル、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類;アセトン、2-ブタノン等のケトン類;アセトニトリル、プロピオニトリル等のニトリル類;酢酸エチル、酢酸イソプロピル、酢酸tert-ブチル等のエステル類;N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、1-メチル-2-ピロリジノン等のアミド類;1,3-ジメチル-2-イミダゾリジノン等のイミド類;例えばメタノール、エタノール、イソプロパノール、tert-ブタノール等のアルコール類;クロロホルム、ジクロロメタン、1,2-ジクロロエタン、四塩化炭素等のハロゲン化炭化水素類;ジメチルスルホキシド等のスルホキシド類;水等が挙げられる。これらの溶媒は、適宜の割合で混合して用いてもよい。反応温度は、通常、-100~250℃の前記した溶媒の沸点以下で行われるが、場合によって、耐圧反応条件等を用いて、溶媒の沸点以上の温度で反応を行っても良い。反応時間は、通常、0.5時間~100時間である。
化合物(I)は、例えば、以下に示す方法またはそれに準ずる方法で製造することができる。
以下の各スキームにおいて、各原料化合物は、反応を阻害しないのであれば、塩を形成していてもよく、該塩としては、前述の式(I)で示される化合物の塩として例示したものが用いられる。
また、以下の各スキームにおいて、原料化合物は、具体的製法を述べない場合、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。
以下の各スキームにおける反応で使用する溶媒としては、反応を阻害せず、出発物質をある程度溶解するものであれば特に限定されないが、例えば、ベンゼン、トルエン、キシレン等の芳香族炭化水素類;ヘキサン、ヘプタン等の脂肪族炭化水素類;ジエチルエーテル、ジイソプロピルエーテル、tert-ブチルメチルエーテル、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類;アセトン、2-ブタノン等のケトン類;アセトニトリル、プロピオニトリル等のニトリル類;酢酸エチル、酢酸イソプロピル、酢酸tert-ブチル等のエステル類;N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、1-メチル-2-ピロリジノン等のアミド類;1,3-ジメチル-2-イミダゾリジノン等のイミド類;例えばメタノール、エタノール、イソプロパノール、tert-ブタノール等のアルコール類;クロロホルム、ジクロロメタン、1,2-ジクロロエタン、四塩化炭素等のハロゲン化炭化水素類;ジメチルスルホキシド等のスルホキシド類;水等が挙げられる。これらの溶媒は、適宜の割合で混合して用いてもよい。反応温度は、通常、-100~250℃の前記した溶媒の沸点以下で行われるが、場合によって、耐圧反応条件等を用いて、溶媒の沸点以上の温度で反応を行っても良い。反応時間は、通常、0.5時間~100時間である。

[式中、Rbは置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示し、その他の記号は前記と同意義を示す。]
スキーム1は、後述するスキーム4で得られる化合物(2)を化合物(3)とした後、置換反応により化合物(I)を得るものである。
化合物(3)は、化合物(2)の塩基と各種アルキル化剤を用いたS-アルキル化反応によって製造される。詳しくは化合物(2)1モルに対して塩基1.0~10.0モル、好ましくは1.0~5.0モル、およびS-アルキル化剤1.0~20.0モル、好ましくは1.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類等が挙げられる。
該S-アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等の各種ハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては、反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常15分~60時間、好ましくは15分~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(3)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(3)は、化合物(2)の塩基と各種アルキル化剤を用いたS-アルキル化反応によって製造される。詳しくは化合物(2)1モルに対して塩基1.0~10.0モル、好ましくは1.0~5.0モル、およびS-アルキル化剤1.0~20.0モル、好ましくは1.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類等が挙げられる。
該S-アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等の各種ハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては、反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常15分~60時間、好ましくは15分~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(3)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(4)は化合物(3)の酸化反応によって製造される。詳しくは酸化剤として過酸化水素、オキソン(登録商標)、過酢酸、過安息香酸、m-クロロ過安息香酸等の過酸類、次亜塩素酸、過ヨウ素酸等のオキソ酸類とその塩、クロム酸等の金属オキソ酸とその塩またはその他の酸化剤が挙げられ、化合物(3)1モルに対して1.0~30.0モル、好ましくは1.0~3.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては、反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
生成物は化合物(4a)および化合物(4b)の何れかの単一化合物またはそれらの混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては、反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
生成物は化合物(4a)および化合物(4b)の何れかの単一化合物またはそれらの混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(I)は化合物(3)または化合物(4)の、塩基とR3に対応する求核剤を用いた置換反応で製造することができる。詳しくは、化合物(3)または化合物(4)1モルに対し、塩基1.0~20.0モル、好ましくは1.0~10.0モル、および求核剤1.0~100.0モル、好ましくは1.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
該求核剤としてはメタノール、エタノール、プロパノール、1,1-ジメチルエタノール、2,2,2-トリフルオロエタノール、2,2,3,3,3-ペンタフルオロ-1-プロパノール等のアルコール類、芳香族水酸基を有する各種フェノール誘導体、エタンチオール、チオグリコール酸アミド等の有機チオール類、チオフェノール等の各種芳香族チオール誘導体、メチルアミン、エチルアミン、プロピルアミン等の有機塩基類、アニリン等の各種芳香族アミン類、α水素を有するカルボニル化合物等の活性メチレン化合物類、有機グリニャール試薬(n-プロピルマグネシウムブロミド、n-ブチルマグネシウムブロミド)、有機リチウム試薬(n-プロピルリチウム、n-ブチルリチウム)等の有機金属試薬類等が挙げられる。なお必要に応じて塩基を求核剤として用いることができる。
反応に塩基が必要でない場合には、塩基を使用せずに製造することができる。
本反応は無溶媒で行うか、反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、シクロヘキサン、ヘキサン等の飽和炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、アセトン、メチルエチルケトン等のケトン類、ジメチルスルホキシド等のスルホキシド類、水等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~24時間、好ましくは10分~12時間である。反応温度は通常-100~150℃、好ましくは-78~100℃である。
得られた化合物(I)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
該求核剤としてはメタノール、エタノール、プロパノール、1,1-ジメチルエタノール、2,2,2-トリフルオロエタノール、2,2,3,3,3-ペンタフルオロ-1-プロパノール等のアルコール類、芳香族水酸基を有する各種フェノール誘導体、エタンチオール、チオグリコール酸アミド等の有機チオール類、チオフェノール等の各種芳香族チオール誘導体、メチルアミン、エチルアミン、プロピルアミン等の有機塩基類、アニリン等の各種芳香族アミン類、α水素を有するカルボニル化合物等の活性メチレン化合物類、有機グリニャール試薬(n-プロピルマグネシウムブロミド、n-ブチルマグネシウムブロミド)、有機リチウム試薬(n-プロピルリチウム、n-ブチルリチウム)等の有機金属試薬類等が挙げられる。なお必要に応じて塩基を求核剤として用いることができる。
反応に塩基が必要でない場合には、塩基を使用せずに製造することができる。
本反応は無溶媒で行うか、反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、シクロヘキサン、ヘキサン等の飽和炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、アセトン、メチルエチルケトン等のケトン類、ジメチルスルホキシド等のスルホキシド類、水等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~24時間、好ましくは10分~12時間である。反応温度は通常-100~150℃、好ましくは-78~100℃である。
得られた化合物(I)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、各記号は前記と同意義を示す。]
スキーム2は、後述するスキーム5で得られる化合物(5)に塩基とR5に対応するアルキル化剤を用いたO-アルキル化反応を行い、化合物(Ia)[化合物(I)において、R3が式:-X2-R5であり、X2が酸素原子である化合物]を得るものである。
詳しくは、化合物(5)1モルに対し、塩基約1.0~3.0、好ましくは1.0~2.0モル、およびO-アルキル化剤約1.0~20.0モル、好ましくは約1.0~10.0モルを用いてO-アルキル化を行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類等が挙げられる。
該O-アルキル化剤としてはヨウ化メチル、ヨウ化エチル、ヨウ化プロピル等の各種ハロゲン化アルキル類、ジメチル硫酸、ジエチル硫酸等のアルキル硫酸類、p-トルエンスルホン酸メチル、メチルスルホン酸メチル等のアルキルスルホン酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常30分~60時間、好ましくは30分~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。得られた化合物(Ia)は常法に従って反応混合物から単離することができ、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(5)1モルに対し、塩基約1.0~3.0、好ましくは1.0~2.0モル、およびO-アルキル化剤約1.0~20.0モル、好ましくは約1.0~10.0モルを用いてO-アルキル化を行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類等が挙げられる。
該O-アルキル化剤としてはヨウ化メチル、ヨウ化エチル、ヨウ化プロピル等の各種ハロゲン化アルキル類、ジメチル硫酸、ジエチル硫酸等のアルキル硫酸類、p-トルエンスルホン酸メチル、メチルスルホン酸メチル等のアルキルスルホン酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常30分~60時間、好ましくは30分~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。得られた化合物(Ia)は常法に従って反応混合物から単離することができ、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、R3’は置換されていてもよいC1-6アルキル基、置換されていてもよいC3-11シクロアルキル基または置換されていてもよい3ないし11員非芳香族複素環基を示し、その他の記号は前記と同意義を示す。]
スキーム3は、後述するスキーム13で得られる化合物(6)と化合物(7)との反応により、化合物(Ib)[化合物(I)において、R3が置換されていてもよいC1-6アルキル基、置換されていてもよいC3-11シクロアルキル基または置換されていてもよい3ないし11員非芳香族複素環基である化合物]を得るものである。
詳しくは、化合物(6)1モルに対し化合物(7)約1.0~10.0モル、好ましくは約1.0~5.0モルを用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
本反応は塩基を加えて行ってもよい。該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
得られた化合物(Ib)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(7)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。
詳しくは、化合物(6)1モルに対し化合物(7)約1.0~10.0モル、好ましくは約1.0~5.0モルを用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
本反応は塩基を加えて行ってもよい。該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
得られた化合物(Ib)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(7)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。

[式中、RaはC1-6アルキル基を示し、その他の記号は前記と同意義を示す。]
スキーム4は、後述するスキーム6または7で得られる化合物(8)とスキーム11で得られる化合物(9)とを、塩基存在下で閉環反応に付し、前記反応(スキーム1)の原料化合物である化合物(2)を得るものである。
詳しくは、化合物(8)1モルに対して化合物(9)を約1.0~5.0モル、好ましくは約1.0~2.0モル用いる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類等が挙げられ、化合物(8)1.0モルに対して約2.0~10.0モル、好ましくは約2.0~5.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、ヘキサメチルリン酸トリアミド等のリン酸アミド類またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(2)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(8)1モルに対して化合物(9)を約1.0~5.0モル、好ましくは約1.0~2.0モル用いる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類等が挙げられ、化合物(8)1.0モルに対して約2.0~10.0モル、好ましくは約2.0~5.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、ヘキサメチルリン酸トリアミド等のリン酸アミド類またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(2)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、各記号は前記と同意義を示す。]
スキーム5は、後述するスキーム6または7で得られる化合物(8)と、スキーム11で得られる化合物(10)とを、塩基存在下で閉環反応に付し、前記反応(スキーム2)の原料化合物である化合物(5)を得るものである。
詳しくは、化合物(8)1モルに対して化合物(10)を約1.0~5.0モル、好ましくは約1.0~2.0モル用いる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類等が挙げられ、化合物(8)1.0モルに対して約2.0~10.0モル、好ましくは約2.0~5.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、ヘキサメチルリン酸トリアミド等のリン酸アミド類またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(5)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(8)1モルに対して化合物(10)を約1.0~5.0モル、好ましくは約1.0~2.0モル用いる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類等が挙げられ、化合物(8)1.0モルに対して約2.0~10.0モル、好ましくは約2.0~5.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、ヘキサメチルリン酸トリアミド等のリン酸アミド類またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(5)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、各記号は前記と同意義を示す。]
スキーム6は、後述するスキーム8、9または10で得られる化合物(11)とアンモニアまたはアンモニウム塩との脱水縮合反応を行い、前記反応(スキーム4および5)の原料化合物である化合物(8)を得るものである。本反応は、自体公知の方法、例えばジャーナル オブ メディシナルケミストリー(J. Med. Chem), 41巻, 3186頁(1998)記載の方法、またはこれらに準じた方法に従って製造することができる。
詳しくは化合物(11)1モルに対しアンモニアまたはアンモニウム塩を約1.0~50.0モル、好ましくは約1.0~10.0モル用いる。
該アンモニウム塩としてはギ酸アンモニウム、酢酸アンモニウム等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
本反応では必要に応じて酸触媒を用いることもでき、酸触媒としては塩酸、硫酸等の鉱酸類、三塩化ホウ素、三臭化ホウ素等のルイス酸類、酢酸、トリフルオロ酢酸、p-トルエンスルホン酸等の有機酸類が挙げられる。
得られた化合物(8)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは化合物(11)1モルに対しアンモニアまたはアンモニウム塩を約1.0~50.0モル、好ましくは約1.0~10.0モル用いる。
該アンモニウム塩としてはギ酸アンモニウム、酢酸アンモニウム等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
本反応では必要に応じて酸触媒を用いることもでき、酸触媒としては塩酸、硫酸等の鉱酸類、三塩化ホウ素、三臭化ホウ素等のルイス酸類、酢酸、トリフルオロ酢酸、p-トルエンスルホン酸等の有機酸類が挙げられる。
得られた化合物(8)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、

は置換基を有していてもよい5ないし6員環状アミノ基を示し、RcはC1-6アルキル基を示し、その他の記号は前記と同意義を示す。]
スキーム7は、化合物(12)に対する化合物(13)の置換反応により、前記反応(スキーム4および5)の原料化合物である化合物(8a)[化合物(8)において、R1が置換基を有していてもよい5ないし6員環状アミノ基である化合物]を得るものである。本反応は、自体公知の方法、例えばジャーナル オブ メディシナルケミストリー(J. Med. Chem), 26巻, 1650頁(1983)記載の方法、またはこれらに準じた方法に従って製造することができる。
詳しくは、化合物(12)1モルに対し化合物(13)を約1.0~10.0モル、好ましくは約1.0~5.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
本反応は酸を加えて行ってもよい。該酸としては塩酸、硫酸等の鉱酸類、三塩化ホウ素、三臭化ホウ素等のルイス酸類、酢酸、トリフルオロ酢酸、p-トルエンスルホン酸等の有機酸類が挙げられる。
化合物(12)の無機酸塩または有機酸塩を用いる場合は、適当な塩基、例えば、炭酸カリウム、炭酸水素ナトリウム、水酸化ナトリウム、トリエチルアミン等を等量または小過剰程度共存させて反応を行うとよい。
得られた化合物(8a)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(12)および化合物(13)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。
詳しくは、化合物(12)1モルに対し化合物(13)を約1.0~10.0モル、好ましくは約1.0~5.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
本反応は酸を加えて行ってもよい。該酸としては塩酸、硫酸等の鉱酸類、三塩化ホウ素、三臭化ホウ素等のルイス酸類、酢酸、トリフルオロ酢酸、p-トルエンスルホン酸等の有機酸類が挙げられる。
化合物(12)の無機酸塩または有機酸塩を用いる場合は、適当な塩基、例えば、炭酸カリウム、炭酸水素ナトリウム、水酸化ナトリウム、トリエチルアミン等を等量または小過剰程度共存させて反応を行うとよい。
得られた化合物(8a)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(12)および化合物(13)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。

[式中、RdはC1-6アルキル基を示し、その他の記号は前記と同意義を示す。]
スキーム8は、化合物(14)と化合物(15)とを塩基を用いたクライゼン縮合反応に付し、前記反応(スキーム6)の原料化合物である化合物(11)を得るものである。
詳しくは化合物(14)1モルに対し化合物(15)を約1.0~10.0モル、好ましくは約1.0~5.0モル用いる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類が挙げられ、化合物(14)1モルに対し約1.0~10.0モル、好ましくは約1.0~5.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。また、化合物(15)を溶媒として用いてもよい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(11)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(14)および化合物(15)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。
詳しくは化合物(14)1モルに対し化合物(15)を約1.0~10.0モル、好ましくは約1.0~5.0モル用いる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類が挙げられ、化合物(14)1モルに対し約1.0~10.0モル、好ましくは約1.0~5.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。また、化合物(15)を溶媒として用いてもよい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(11)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(14)および化合物(15)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。

[式中、各記号は前記と同意義を示す。]
スキーム9は、化合物(16)および化合物(15)を塩基を用いた縮合反応に付し、前記反応(スキーム6)の原料化合物である化合物(11)を得る別法である。
詳しくは化合物(16)1モルに対し化合物(15)を約1.0~10.0モル、好ましくは約1.0~5.0モル用いる。
該塩基としてはn-ブチルリチウム、sec-ブチルリチウム、tert-ブチルリチウム等のアルキルリチウム類が挙げられ、化合物(15)1モルに対し等量または小過剰程度用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-100~100℃、好ましくは-78~25℃である。
得られた化合物(11)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(16)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。
詳しくは化合物(16)1モルに対し化合物(15)を約1.0~10.0モル、好ましくは約1.0~5.0モル用いる。
該塩基としてはn-ブチルリチウム、sec-ブチルリチウム、tert-ブチルリチウム等のアルキルリチウム類が挙げられ、化合物(15)1モルに対し等量または小過剰程度用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-100~100℃、好ましくは-78~25℃である。
得られた化合物(11)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(16)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。

[式中、Mはカリウムまたはナトリウムを示し、その他の記号は前記と同意義を示す。]
スキーム10は、化合物(17)を化合物(18)と作用させた後、化合物(19)との縮合反応、続く脱炭酸反応により、前記反応(スキーム6)の原料化合物である化合物(11)を得る別法である。本反応は、自体公知の方法、例えばジャーナル オブ メディシナルケミストリー(J. Med. Chem), 44巻, 3978頁(2001)記載の方法、またはこれらに準じた方法に従って製造することができる。
詳しくは、化合物(17)1モルに対し化合物(18)約1.0~10.0モル、好ましくは約1.0~5.0モル、塩化マグネシウム約1.0~10.0モル、好ましくは約1.0~5.0モル、および化合物(19)約1.0~10.0モル、好ましくは約1.0~5.0モルを用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(11)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(17)、化合物(18)および化合物(19)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。
詳しくは、化合物(17)1モルに対し化合物(18)約1.0~10.0モル、好ましくは約1.0~5.0モル、塩化マグネシウム約1.0~10.0モル、好ましくは約1.0~5.0モル、および化合物(19)約1.0~10.0モル、好ましくは約1.0~5.0モルを用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(11)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(17)、化合物(18)および化合物(19)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。

[式中、各記号は前記と同意義を示す。]
スキーム11は、化合物(20)のチオイソシアナート化によりスキーム4の原料化合物である化合物(9)を、化合物(20)のイソシアナート化によりスキーム5の原料化合物である化合物(10)を得るものである。本反応は、自体公知の方法、例えばジャーナル オブ メディシナルケミストリー(J. Med. Chem), 44巻, 3978頁(2001)記載の方法、またはこれらに準じた方法に従って製造することができる。
化合物(9)は化合物(20)のチオイソシアナート化により製造することができる。
詳しくは、化合物(20)1モルに対してチオイソシアナート化剤を約1.0~5.0モル、好ましくは約1.0~2.0モル用いて反応を行う。
該チオイソシアナート化剤としてはチオホスゲン、1,1'-チオカルボニルジ-2(1H)-ピリドン、ジ-(2-ピリジル)チオノカルボナート、1,1'-チオカルボニルジイミダゾール等が挙げられる。
本反応においてチオホスゲンを用いる場合は、放出されるハロゲン化水素を反応系内から除去する目的で、脱酸剤の存在下に反応を行うことができる。このような脱酸剤としては、例えば炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム等の無機塩基類、ピリジン、ルチジン等の芳香族アミン類、トリエチルアミン、トリプロピルアミン、トリブチルアミン、シクロヘキシルジメチルアミン、4-ジメチルアミノピリジン、N,N-ジメチルアニリン、N-メチルピペリジン、N-メチルピロリジン、N-メチルモルホリン等の第3級アミン類等が望ましい。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、シクロヘキサン、ヘキサン等の飽和炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、アセトン、メチルエチルケトン等のケトン類、ジメチルスルホキシド等のスルホキシド類、水等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~60時間、好ましくは15分~12時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(9)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(9)は化合物(20)のチオイソシアナート化により製造することができる。
詳しくは、化合物(20)1モルに対してチオイソシアナート化剤を約1.0~5.0モル、好ましくは約1.0~2.0モル用いて反応を行う。
該チオイソシアナート化剤としてはチオホスゲン、1,1'-チオカルボニルジ-2(1H)-ピリドン、ジ-(2-ピリジル)チオノカルボナート、1,1'-チオカルボニルジイミダゾール等が挙げられる。
本反応においてチオホスゲンを用いる場合は、放出されるハロゲン化水素を反応系内から除去する目的で、脱酸剤の存在下に反応を行うことができる。このような脱酸剤としては、例えば炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム等の無機塩基類、ピリジン、ルチジン等の芳香族アミン類、トリエチルアミン、トリプロピルアミン、トリブチルアミン、シクロヘキシルジメチルアミン、4-ジメチルアミノピリジン、N,N-ジメチルアニリン、N-メチルピペリジン、N-メチルピロリジン、N-メチルモルホリン等の第3級アミン類等が望ましい。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、シクロヘキサン、ヘキサン等の飽和炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、アセトン、メチルエチルケトン等のケトン類、ジメチルスルホキシド等のスルホキシド類、水等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~60時間、好ましくは15分~12時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(9)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(10)は化合物(20)のイソシアナート化により製造できる。
詳しくは化合物(20)1モルに対してイソシアナート化剤を約1.0~5.0モル、好ましくは約1.0~2.0モル用いて反応を行う。
該イソシアナート化剤としてはトリホスゲン、1,1'-カルボニルジ-2(1H)-ピリドン、ジ-2-ピリジルカルボナート、1,1'-カルボニルジイミダゾール等が挙げられる。
本反応においてトリホスゲンを用いる場合は、放出されるハロゲン化水素を反応系内から除去する目的で、脱酸剤の存在下に反応を行うことができる。このような脱酸剤としては、例えば炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム等の塩基性塩類、ピリジン、ルチジン等の芳香族アミン類、トリエチルアミン、トリプロピルアミン、トリブチルアミン、シクロヘキシルジメチルアミン、4-ジメチルアミノピリジン、N,N-ジメチルアニリン、N-メチルピペリジン、N-メチルピロリジン、N-メチルモルホリン等の第3級アミン類等が望ましい。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、シクロヘキサン、ヘキサン等の飽和炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、アセトン、メチルエチルケトン等のケトン類、ジメチルスルホキシド等のスルホキシド類、水等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~60時間、好ましくは15分~12時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(10)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(20)は、市販されているものを容易に入手できるか、あるいは、スキーム12に示す方法またはそれに準ずる方法に従って製造することができる。
詳しくは化合物(20)1モルに対してイソシアナート化剤を約1.0~5.0モル、好ましくは約1.0~2.0モル用いて反応を行う。
該イソシアナート化剤としてはトリホスゲン、1,1'-カルボニルジ-2(1H)-ピリドン、ジ-2-ピリジルカルボナート、1,1'-カルボニルジイミダゾール等が挙げられる。
本反応においてトリホスゲンを用いる場合は、放出されるハロゲン化水素を反応系内から除去する目的で、脱酸剤の存在下に反応を行うことができる。このような脱酸剤としては、例えば炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム等の塩基性塩類、ピリジン、ルチジン等の芳香族アミン類、トリエチルアミン、トリプロピルアミン、トリブチルアミン、シクロヘキシルジメチルアミン、4-ジメチルアミノピリジン、N,N-ジメチルアニリン、N-メチルピペリジン、N-メチルピロリジン、N-メチルモルホリン等の第3級アミン類等が望ましい。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、シクロヘキサン、ヘキサン等の飽和炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、アセトン、メチルエチルケトン等のケトン類、ジメチルスルホキシド等のスルホキシド類、水等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~60時間、好ましくは15分~12時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(10)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(20)は、市販されているものを容易に入手できるか、あるいは、スキーム12に示す方法またはそれに準ずる方法に従って製造することができる。

[式中、Yは脱離基(例、ハロゲン原子または4-トリルスルホニルオキシ等)を示し、その他の記号は前記と同意義を示す。]
スキーム12は、化合物(21)または化合物(22)から化合物(23)を経由して、スキーム11の原料化合物である化合物(20a)[化合物(20)において、X1がOである化合物]を得るものである。
化合物(23)は、塩基性条件下、R4に対応するフェノール類またはアルコール類を求核剤とした化合物(21)との置換反応で製造することができる。
詳しくは、化合物(21)1モルに対して塩基約1.0~10.0モル、好ましくは約1.0~5.0モル、およびR4に対応するフェノール類またはアルコール類約1.0~100.0モル、好ましくは1.0~2.0モルを用いる。
該塩基としては炭酸ナトリウム、炭酸カリウム等の無機塩基類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
該フェノール類またはアルコール類としてはエタノール、2,2,2-トリフルオロエタノール、シクロプロピルメタノール、2-プロパノール、2-メチルプロパノール、2,2,3,3,3-ペンタフルオロ-1-プロパノール等が挙げられる。
本反応は無溶媒、または反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは5時間~12時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
化合物(23)は、塩基性条件下、R4に対応するフェノール類またはアルコール類を求核剤とした化合物(21)との置換反応で製造することができる。
詳しくは、化合物(21)1モルに対して塩基約1.0~10.0モル、好ましくは約1.0~5.0モル、およびR4に対応するフェノール類またはアルコール類約1.0~100.0モル、好ましくは1.0~2.0モルを用いる。
該塩基としては炭酸ナトリウム、炭酸カリウム等の無機塩基類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
該フェノール類またはアルコール類としてはエタノール、2,2,2-トリフルオロエタノール、シクロプロピルメタノール、2-プロパノール、2-メチルプロパノール、2,2,3,3,3-ペンタフルオロ-1-プロパノール等が挙げられる。
本反応は無溶媒、または反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは5時間~12時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
また別法として、化合物(23)は塩基とR4に対応するアルキル化剤を用いた化合物(22)のO-アルキル化反応によっても製造できる。
詳しくは化合物(22)1モルに対して塩基約1.0~5.0モル、好ましくは約1.0~2.0モル、およびO-アルキル化剤約1.0~10.0モル、好ましくは約1.0~3.0モルを用いる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類等が挙げられる。
該O-アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等の各種ハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは5時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(23)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは化合物(22)1モルに対して塩基約1.0~5.0モル、好ましくは約1.0~2.0モル、およびO-アルキル化剤約1.0~10.0モル、好ましくは約1.0~3.0モルを用いる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類等が挙げられる。
該O-アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等の各種ハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは5時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(23)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
また別法として化合物(23)は、自体公知の方法、例えば、シンセシス(Synthesis) 1頁(1981年)等に記載の方法、またはこれらに準じた方法により製造できる。すなわち、本反応は、通常、有機リン化合物およびアゾ試薬の存在下、反応に悪影響を及ぼさない溶媒中で行われる。
該有機リン化合物としては、例えばトリフェニルホスフィン、トリ(n-ブチル)ホスフィン等が挙げられる。
該アゾ試薬としては、例えばアゾジカルボン酸ジエチル、アゾジカルボン酸ジイソプロピル、アゾジカルボニルジピペラジン等が挙げられる。有機リン化合物およびアゾ試薬の使用量は、化合物(22)1モルに対し、好ましくは1.0モル~5.0モルである。
該反応に悪影響を及ぼさない溶媒としては、例えばジエチルエーテル、テトラヒドロフラン、1,4-ジオキサン等のエーテル類;クロロホルム、ジクロロメタン等のハロゲン化炭化水素類;ベンゼン、トルエン、キシレン等の芳香族炭化水素類;N,N-ジメチルホルムアミド等のアミド類;ジメチルスルホキシド等のスルホキシド類等が挙げられる。これらの溶媒は、2種以上を適宜の割合で混合して用いてもよい。
反応温度は、通常-50~150℃、好ましくは0~100℃である。反応時間は、通常、約0.5~約20時間である。
得られた化合物(23)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
該有機リン化合物としては、例えばトリフェニルホスフィン、トリ(n-ブチル)ホスフィン等が挙げられる。
該アゾ試薬としては、例えばアゾジカルボン酸ジエチル、アゾジカルボン酸ジイソプロピル、アゾジカルボニルジピペラジン等が挙げられる。有機リン化合物およびアゾ試薬の使用量は、化合物(22)1モルに対し、好ましくは1.0モル~5.0モルである。
該反応に悪影響を及ぼさない溶媒としては、例えばジエチルエーテル、テトラヒドロフラン、1,4-ジオキサン等のエーテル類;クロロホルム、ジクロロメタン等のハロゲン化炭化水素類;ベンゼン、トルエン、キシレン等の芳香族炭化水素類;N,N-ジメチルホルムアミド等のアミド類;ジメチルスルホキシド等のスルホキシド類等が挙げられる。これらの溶媒は、2種以上を適宜の割合で混合して用いてもよい。
反応温度は、通常-50~150℃、好ましくは0~100℃である。反応時間は、通常、約0.5~約20時間である。
得られた化合物(23)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(20a)は化合物(23)の還元反応によって合成することができる。詳しくは化合物(23)1モルに対して金属触媒を約0.01~5.0モル、好ましくは約0.01~2.0モル用いて水素雰囲気下で還元して製造される。
該金属触媒としてはパラジウム-炭素、水酸化パラジウム-炭素、酸化白金、白金等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、シクロヘキサン、ヘキサン等の飽和炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、ジメチルスルホキシド等のスルホキシド類、水等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは5時間~36時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。圧力は約1~10気圧、好ましくは約1~5気圧である。
得られた化合物(20a)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
該金属触媒としてはパラジウム-炭素、水酸化パラジウム-炭素、酸化白金、白金等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、シクロヘキサン、ヘキサン等の飽和炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、ジメチルスルホキシド等のスルホキシド類、水等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは5時間~36時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。圧力は約1~10気圧、好ましくは約1~5気圧である。
得られた化合物(20a)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
また別法として化合物(23)の還元反応は還元金属を用いて行うこともできる。詳しくは化合物(23)1モルに対し還元金属約5.0~20.0モル、好ましくは約5.0~10.0モル用いる。
該還元金属としては還元鉄、スズ、亜鉛等が挙げられる。反応促進の目的で、塩酸または塩化アンモニウム、塩化カルシウム等の塩を添加することができる。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、シクロヘキサン、ヘキサン等の飽和炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトン、メチルエチルケトン等のケトン類、ジメチルスルホキシド等のスルホキシド類、アンモニア水溶液、水等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは5時間~36時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(20a)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(21)および化合物(22)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。
該還元金属としては還元鉄、スズ、亜鉛等が挙げられる。反応促進の目的で、塩酸または塩化アンモニウム、塩化カルシウム等の塩を添加することができる。
本反応は反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、シクロヘキサン、ヘキサン等の飽和炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトン、メチルエチルケトン等のケトン類、ジメチルスルホキシド等のスルホキシド類、アンモニア水溶液、水等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは5時間~36時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
得られた化合物(20a)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(21)および化合物(22)は、市販されているものを容易に入手できるか、あるいは、自体公知の方法またはそれに準ずる方法に従って製造することができる。

[式中、各記号は前記と同意義を示す。]
スキーム13は、化合物(11)および化合物(20)から化合物(24)を経由して、スキーム3の原料化合物である化合物(6)を得るものである。
化合物(24)は自体公知の方法、例えばジャーナル オブ オーガニック ケミストリー(J. Org. Chem), 45巻, 4861頁(1980)記載の方法、またはこれらに準じた方法に従って製造することができる。詳しくは、化合物(20)と化合物(11)との縮合反応によって製造される。化合物(11)1モルに対し化合物(20)を約1.0~5.0モル、好ましくは約1.0~3.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン、キシレン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
本反応は塩基を加えて行ってもよい。該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
得られた化合物(24)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(24)は自体公知の方法、例えばジャーナル オブ オーガニック ケミストリー(J. Org. Chem), 45巻, 4861頁(1980)記載の方法、またはこれらに準じた方法に従って製造することができる。詳しくは、化合物(20)と化合物(11)との縮合反応によって製造される。化合物(11)1モルに対し化合物(20)を約1.0~5.0モル、好ましくは約1.0~3.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン、キシレン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
本反応は塩基を加えて行ってもよい。該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
得られた化合物(24)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(6)は自体公知の方法、例えばジャーナル オブ メディシナルケミストリー(J. Med. Chem), 41巻, 3186頁(1998)記載の方法、またはこれらに準じた方法に従って製造することができる。詳しくは化合物(24)とアンモニアまたはアンモニウム塩との脱水縮合反応で製造される。化合物(24)1モルに対しアンモニアまたはアンモニウム塩を約1.0~50.0モル、好ましくは約1.0~10.0モル用いる。
該アンモニウム塩としてはギ酸アンモニウム、酢酸アンモニウム等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
本反応では必要に応じて酸触媒を用いることもでき、酸触媒としては塩酸、硫酸等の鉱酸類、三塩化ホウ素、三臭化ホウ素等のルイス酸類、酢酸、トリフルオロ酢酸、p-トルエンスルホン酸等の有機酸類が挙げられる。得られた化合物(6)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
該アンモニウム塩としてはギ酸アンモニウム、酢酸アンモニウム等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
本反応では必要に応じて酸触媒を用いることもでき、酸触媒としては塩酸、硫酸等の鉱酸類、三塩化ホウ素、三臭化ホウ素等のルイス酸類、酢酸、トリフルオロ酢酸、p-トルエンスルホン酸等の有機酸類が挙げられる。得られた化合物(6)は反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、各記号は前記と同意義を示す。]
スキーム14は、化合物(25)を経由して化合物(Ib)を得る、前述のスキーム3の別法である。化合物(25)は、化合物(6)と化合物(7)との環化反応により製造される。
詳しくは、化合物(6)1モルに対し化合物(7)を1.0~10.0モル、好ましくは1.0~5.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
本反応は塩基を加えて行ってもよい。該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(6)1モルに対し化合物(7)を1.0~10.0モル、好ましくは1.0~5.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
本反応は塩基を加えて行ってもよい。該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(Ib)は、化合物(25)と酸を用いた脱水反応によって製造される。
詳しくは化合物(25)1モルに対して酸を1.0~500.0モル、好ましくは10.0~400.0モル用いる。
該酸としては塩酸、硫酸等の鉱酸類、三塩化ホウ素、三臭化ホウ素等のルイス酸類、酢酸、トリフルオロ酢酸、p-トルエンスルホン酸等の有機酸類が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等を用いることができるが、酸として有機酸類を用いる場合は溶媒を兼ねてもよい。
反応時間は通常5分~60時間、好ましくは15分~24時間である。反応温度は通常0~200℃、好ましくは25~150℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは化合物(25)1モルに対して酸を1.0~500.0モル、好ましくは10.0~400.0モル用いる。
該酸としては塩酸、硫酸等の鉱酸類、三塩化ホウ素、三臭化ホウ素等のルイス酸類、酢酸、トリフルオロ酢酸、p-トルエンスルホン酸等の有機酸類が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等を用いることができるが、酸として有機酸類を用いる場合は溶媒を兼ねてもよい。
反応時間は通常5分~60時間、好ましくは15分~24時間である。反応温度は通常0~200℃、好ましくは25~150℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、X3は塩素原子、臭素原子またはヨウ素原子を示し、Reはハロゲン原子で置換されていてもよいC1-6アルキル基を示し、その他の記号は前記と同意義を示す。]
スキーム15は、化合物(Ic)から化合物(26)を経由して化合物(Id)を得るものである。
化合物(26)は、化合物(Ic)とX3に対応するハロゲン化剤との反応により製造される。
化合物(Ic)は、化合物(Ib)を製造する方法に準ずる方法に従って製造することができる。
詳しくはハロゲン化剤としてN-クロロスクシンイミド、N-ブロモスクシンイミド、N-ヨウドスクシンイミド、臭素、ヨウ素等が挙げられる。該ハロゲン化剤は、化合物(Ic)1モルに対して1.0~30.0モル、好ましくは1.0~3.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(26)は、化合物(Ic)とX3に対応するハロゲン化剤との反応により製造される。
化合物(Ic)は、化合物(Ib)を製造する方法に準ずる方法に従って製造することができる。
詳しくはハロゲン化剤としてN-クロロスクシンイミド、N-ブロモスクシンイミド、N-ヨウドスクシンイミド、臭素、ヨウ素等が挙げられる。該ハロゲン化剤は、化合物(Ic)1モルに対して1.0~30.0モル、好ましくは1.0~3.0モル用いる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(Id)は、化合物(26)とReに対応する有機ボロン酸試薬のカップリング反応により製造される。
詳しくは化合物(26)1モルに対してReに対応する有機ボロン酸試薬1.0~10.0モル、好ましくは1.0~3.0モル、有機金属試薬0.01~1モル、好ましくは0.05~0.2モル、ホスフィン配位子0.01~1モル、好ましくは0.1~0.5モル、および塩基1.0~10.0モル、好ましくは2.0~6.0モルを用いる。反応にホスフィン配位子が必要でない場合には、ホスフィン配位子を使用せずに行うことができる。
有機ボロン酸試薬としてはメチルボロン酸、2,4,4,5,5-ペンタメチル-1,3,2-ジオキサボロラン、エチルボロン酸、N-プロピルボロン酸、(1-メチルエチル)ボロン酸、4,4,5,5-テトラメチル-2-(1-メチルエチル)-1,3,2-ジオキサボロラン、N-ブチルボロン酸、2-ブチル-4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン、(2-メチルプロピル)ボロン酸、4,4,5,5-テトラメチル-2-(2-メチルプロピル)-1,3,2-ジオキサボロラン等が挙げられる。
有機金属試薬としてはトリス(ジベンジリデンアセトン)ジパラジウム(0)、テトラキス(トリフェニルホスフィン)パラジウム、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリドジクロロメタン錯体、酢酸パラジウム等が挙げられる。
ホスフィン配位子としては2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル、(9,9-ジメチル-9H-キサンテン-4,5-ジイル)ビス(ジフェニルホスファン)、2,2’-ビス(ジフェニルホスフィノ)-1,1’-ビナフチル等が挙げられる。
塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム、酢酸カリウム、炭酸セシウム等の塩基性塩類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは化合物(26)1モルに対してReに対応する有機ボロン酸試薬1.0~10.0モル、好ましくは1.0~3.0モル、有機金属試薬0.01~1モル、好ましくは0.05~0.2モル、ホスフィン配位子0.01~1モル、好ましくは0.1~0.5モル、および塩基1.0~10.0モル、好ましくは2.0~6.0モルを用いる。反応にホスフィン配位子が必要でない場合には、ホスフィン配位子を使用せずに行うことができる。
有機ボロン酸試薬としてはメチルボロン酸、2,4,4,5,5-ペンタメチル-1,3,2-ジオキサボロラン、エチルボロン酸、N-プロピルボロン酸、(1-メチルエチル)ボロン酸、4,4,5,5-テトラメチル-2-(1-メチルエチル)-1,3,2-ジオキサボロラン、N-ブチルボロン酸、2-ブチル-4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン、(2-メチルプロピル)ボロン酸、4,4,5,5-テトラメチル-2-(2-メチルプロピル)-1,3,2-ジオキサボロラン等が挙げられる。
有機金属試薬としてはトリス(ジベンジリデンアセトン)ジパラジウム(0)、テトラキス(トリフェニルホスフィン)パラジウム、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリドジクロロメタン錯体、酢酸パラジウム等が挙げられる。
ホスフィン配位子としては2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル、(9,9-ジメチル-9H-キサンテン-4,5-ジイル)ビス(ジフェニルホスファン)、2,2’-ビス(ジフェニルホスフィノ)-1,1’-ビナフチル等が挙げられる。
塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム、酢酸カリウム、炭酸セシウム等の塩基性塩類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、各記号は前記と同意義を示す。]
スキーム16は、後述するスキーム17で得られる化合物(Ib’)群に含まれる化合物(Ie)から、アルキル化反応により化合物(If)を得るものである。
化合物(If)は、化合物(Ie)の塩基とReに対応するアルキル化剤を用いたアルキル化反応によって製造される。
詳しくは化合物(Ie)1モルに対して塩基1.0~50.0モル、好ましくは1.0~5.0モル、およびアルキル化剤1.0~100.0モル、好ましくは1.0~10.0モルを用いる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類等が挙げられる。
該アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等のハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常15分~60時間、好ましくは15分~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(If)は、化合物(Ie)の塩基とReに対応するアルキル化剤を用いたアルキル化反応によって製造される。
詳しくは化合物(Ie)1モルに対して塩基1.0~50.0モル、好ましくは1.0~5.0モル、およびアルキル化剤1.0~100.0モル、好ましくは1.0~10.0モルを用いる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類等が挙げられる。
該アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等のハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常15分~60時間、好ましくは15分~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

スキーム16’は、後述するスキーム22で得られる化合物(Ih)群に含まれる化合物(Ie’)から、アルキル化反応により化合物(If’)を得るものである。
化合物(If’)は、化合物(Ie’)の塩基とReに対応するアルキル化剤を用いたアルキル化反応によって製造される。
詳しくは化合物(Ie’)1モルに対して塩基1.0~50.0モル、好ましくは1.0~5.0モル、およびアルキル化剤1.0~100.0モル、好ましくは1.0~10.0モルを用いる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン、ピリジン等の有機塩基類等が挙げられる。
該アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等のハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常15分~60時間、好ましくは15分~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(If’)は、化合物(Ie’)の塩基とReに対応するアルキル化剤を用いたアルキル化反応によって製造される。
詳しくは化合物(Ie’)1モルに対して塩基1.0~50.0モル、好ましくは1.0~5.0モル、およびアルキル化剤1.0~100.0モル、好ましくは1.0~10.0モルを用いる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン、ピリジン等の有機塩基類等が挙げられる。
該アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等のハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常15分~60時間、好ましくは15分~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、Rfはハロゲン原子またはトリフルオロメタンスルホニルオキシ基を示し、その他の記号は前記と同意義を示す。]
スキーム17は、後述するスキーム18で得られる化合物(27)から、カップリング反応を用いて化合物(Ib’)を得るものである。
化合物(Ib’)は化合物(27)とR1に対応する有機ボロン酸試薬または含窒素試薬とのカップリング反応により製造される。
詳しくは化合物(27)1モルに対してR1に対応する有機ボロン酸試薬または含窒素試薬1.0~10.0モル、好ましくは1.0~3.0モル、有機金属試薬0.01~1.0モル、好ましくは0.05~0.2モル、ホスフィン配位子0.01~1.0モル、好ましくは0.1~0.5モル、および塩基1.0~10.0モル、好ましくは2.0~6.0モルを用いて行う。反応にホスフィン配位子が必要でない場合には、ホスフィン配位子を使用せずに行うことができる。
有機ボロン酸試薬としては1H-ピラゾール-4-イルボロン酸、4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール、1-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール、tert-ブチル 4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール-1-カルボキシラート、(1-メチル-1H-ピラゾール-4-イル)ボロン酸、[1-(tert-ブトキシカルボニル)-1H-ピラゾール-4-イル]ボロン酸、[3-(トリフルオロメチル)-1H-ピラゾール-4-イル]ボロン酸、4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3-(トリフルオロメチル)-1H-ピラゾール、3-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール、(3-メチル-1H-ピラゾール-4-イル)ボロン酸、tert-ブチル 3-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール-1-カルボキシラート、1H-ピラゾール-5-イルボロン酸、(1-メチル-1H-ピラゾール-5-イル)ボロン酸、1-メチル-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール、1-メチル-5-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール、[1-(tert-ブトキシカルボニル)-1H-ピラゾール-3-イル]ボロン酸、tert-ブチル 3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール-1-カルボキシラート、1-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-イミダゾールtert-ブチル 4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-イミダゾール-1-カルボキシラート等が挙げられる。
含窒素試薬としてはモルホリン、3,3-ジフルオロピロリジン、4,4-ジフルオロピペリジン、モルホリン-3-オン、ピペラジン-2-オン、ピペリジン-4-オン等が挙げられる。
有機金属試薬としてはトリス(ジベンジリデンアセトン)ジパラジウム(0)、テトラキス(トリフェニルホスフィン)パラジウム、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリドジクロロメタン錯体、酢酸パラジウム等が挙げられる。
ホスフィン配位子としては2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル、(9,9-ジメチル-9H-キサンテン-4,5-ジイル)ビス(ジフェニルホスファン)、2,2’-ビス(ジフェニルホスフィノ)-1,1’-ビナフチル等が挙げられる。
塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム、酢酸カリウム、炭酸セシウム等の塩基性塩類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(Ib’)は化合物(27)とR1に対応する有機ボロン酸試薬または含窒素試薬とのカップリング反応により製造される。
詳しくは化合物(27)1モルに対してR1に対応する有機ボロン酸試薬または含窒素試薬1.0~10.0モル、好ましくは1.0~3.0モル、有機金属試薬0.01~1.0モル、好ましくは0.05~0.2モル、ホスフィン配位子0.01~1.0モル、好ましくは0.1~0.5モル、および塩基1.0~10.0モル、好ましくは2.0~6.0モルを用いて行う。反応にホスフィン配位子が必要でない場合には、ホスフィン配位子を使用せずに行うことができる。
有機ボロン酸試薬としては1H-ピラゾール-4-イルボロン酸、4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール、1-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール、tert-ブチル 4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール-1-カルボキシラート、(1-メチル-1H-ピラゾール-4-イル)ボロン酸、[1-(tert-ブトキシカルボニル)-1H-ピラゾール-4-イル]ボロン酸、[3-(トリフルオロメチル)-1H-ピラゾール-4-イル]ボロン酸、4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3-(トリフルオロメチル)-1H-ピラゾール、3-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール、(3-メチル-1H-ピラゾール-4-イル)ボロン酸、tert-ブチル 3-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール-1-カルボキシラート、1H-ピラゾール-5-イルボロン酸、(1-メチル-1H-ピラゾール-5-イル)ボロン酸、1-メチル-3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール、1-メチル-5-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール、[1-(tert-ブトキシカルボニル)-1H-ピラゾール-3-イル]ボロン酸、tert-ブチル 3-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール-1-カルボキシラート、1-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-イミダゾールtert-ブチル 4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-イミダゾール-1-カルボキシラート等が挙げられる。
含窒素試薬としてはモルホリン、3,3-ジフルオロピロリジン、4,4-ジフルオロピペリジン、モルホリン-3-オン、ピペラジン-2-オン、ピペリジン-4-オン等が挙げられる。
有機金属試薬としてはトリス(ジベンジリデンアセトン)ジパラジウム(0)、テトラキス(トリフェニルホスフィン)パラジウム、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリドジクロロメタン錯体、酢酸パラジウム等が挙げられる。
ホスフィン配位子としては2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル、(9,9-ジメチル-9H-キサンテン-4,5-ジイル)ビス(ジフェニルホスファン)、2,2’-ビス(ジフェニルホスフィノ)-1,1’-ビナフチル等が挙げられる。
塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム、酢酸カリウム、炭酸セシウム等の塩基性塩類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、各記号は前記と同意義を示す。]
スキーム18は、スキーム19で得られる化合物(28)から化合物(29)を経由して、化合物(27)を得るものである。
化合物(29)は、化合物(28)とジアルキルマロネートとの塩基性条件化で行う環化反応により製造される。
詳しくは化合物(28)1モルに対してジアルキルマロネート1.0~10.0モル、好ましくは1.0~3.0モル、および塩基1.0~100.0モル、好ましくは2.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、アルコール類(例、メタノール、エタノール等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、有機酸類(例、酢酸等)、水またはこれら二種以上の混合物等が用いられる。
反応時間は通常10分~72時間、好ましくは15分~24時間である。反応温度は通常0℃~150℃、好ましくは0℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(29)は、化合物(28)とジアルキルマロネートとの塩基性条件化で行う環化反応により製造される。
詳しくは化合物(28)1モルに対してジアルキルマロネート1.0~10.0モル、好ましくは1.0~3.0モル、および塩基1.0~100.0モル、好ましくは2.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、アルコール類(例、メタノール、エタノール等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、有機酸類(例、酢酸等)、水またはこれら二種以上の混合物等が用いられる。
反応時間は通常10分~72時間、好ましくは15分~24時間である。反応温度は通常0℃~150℃、好ましくは0℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(27)は化合物(29)の置換反応により製造される。
詳しくは、化合物(29)1モルに対してハロゲン化剤1.0~100.0モル、好ましくは3.0~10.0モルを用いて行う。
ハロゲン化剤としては、オキシ塩化リン、オキシ臭化リン等が挙げられる。
本反応は、無溶媒または反応に不活性な溶媒を用いて行うのが有利である。このような溶媒としては、反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、またはこれら二種以上の混合物等が用いられる。
反応時間は通常10分~72時間、好ましくは30分~3時間である。反応温度は通常0℃~150℃、好ましくは0℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(29)1モルに対してハロゲン化剤1.0~100.0モル、好ましくは3.0~10.0モルを用いて行う。
ハロゲン化剤としては、オキシ塩化リン、オキシ臭化リン等が挙げられる。
本反応は、無溶媒または反応に不活性な溶媒を用いて行うのが有利である。このような溶媒としては、反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、またはこれら二種以上の混合物等が用いられる。
反応時間は通常10分~72時間、好ましくは30分~3時間である。反応温度は通常0℃~150℃、好ましくは0℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
別法として、化合物(27)の製造は、化合物(29)1モルに対してトリフルオロメタンスルホニル化試薬1.0~10.0モル、好ましくは1.0~3.0モル、および塩基1.0~20.0モル、好ましくは1.0~10.0モルを用いて行う。
該トリフルオロメタンスルホニル化試薬としては、1,1,1-トリフルオロ-N-フェニル-N-[(トリフルオロメチル)スルホニル]メタンスルホンアミド、2-[N,N-ビス(トリフルオロメチルスルホニル)アミン]-5-クロロピリジン、トリフルオロメタンスルホン酸 無水物等が挙げられる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
本反応は、無溶媒または反応に不活性な溶媒を用いて行うのが有利である。このような溶媒としては、反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、またはこれら二種以上の混合物等が用いられる。
反応時間は通常10分~72時間、好ましくは15分~24時間である。反応温度は通常-78℃~100℃、好ましくは0℃~50℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
該トリフルオロメタンスルホニル化試薬としては、1,1,1-トリフルオロ-N-フェニル-N-[(トリフルオロメチル)スルホニル]メタンスルホンアミド、2-[N,N-ビス(トリフルオロメチルスルホニル)アミン]-5-クロロピリジン、トリフルオロメタンスルホン酸 無水物等が挙げられる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
本反応は、無溶媒または反応に不活性な溶媒を用いて行うのが有利である。このような溶媒としては、反応が進行する限り特に限定されないが、例えば、エーテル類(例、ジエチルエーテル、テトラヒドロフラン、1,2-ジメトキシエタン等)、エステル類(例、酢酸エチル等)、芳香族炭化水素類(例、ベンゼン、トルエン等)、脂肪族炭化水素類(例、ヘキサン等)、アミド類(例、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等)、ニトリル類(例、アセトニトリル、プロピオニトリル等)、スルホキシド類(例、ジメチルスルホキシド等)、またはこれら二種以上の混合物等が用いられる。
反応時間は通常10分~72時間、好ましくは15分~24時間である。反応温度は通常-78℃~100℃、好ましくは0℃~50℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、Rgは置換されていてもよいC1-6アルキル基を示し、その他の記号は前記と同意義を示す。]
スキーム19は化合物(30)からスキーム18の原料である化合物(28)を得るものである。
化合物(30)は市販されている場合には市販品をそのまま用いることができ、自体公知の方法あるいはこれらに準じた方法に従って製造することもできる。
化合物(31)は化合物(30)から置換反応により製造される。
詳しくは化合物(30)1モルに対してクロル化剤1.0~10.0モル、好ましくは1.0~3.0モル、および28%アンモニア水溶液1.0~20.0モル、好ましくは1.0~3.0モルを用いる。必要に応じてピリジン、ジシクロヘキシルアミン、N,N-ジメチルホルムアミド、相間移動触媒等を化合物(30)1モルに対して0.001~10.0モル、好ましくは0.001~3.0モル用いてもよい。
該クロル化剤としてはオキサリルクロリド、チオニルクロリド、オキシ塩化リン等が挙げられる。
相間移動触媒としてはテトラブチルアンモニウムクロリド、テトラブチルアンモニウムブロミド、クラウンエーテル等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~72時間、好ましくは30分~24時間である。反応温度は通常-78℃~100℃、好ましくは-10℃~25℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(30)は市販されている場合には市販品をそのまま用いることができ、自体公知の方法あるいはこれらに準じた方法に従って製造することもできる。
化合物(31)は化合物(30)から置換反応により製造される。
詳しくは化合物(30)1モルに対してクロル化剤1.0~10.0モル、好ましくは1.0~3.0モル、および28%アンモニア水溶液1.0~20.0モル、好ましくは1.0~3.0モルを用いる。必要に応じてピリジン、ジシクロヘキシルアミン、N,N-ジメチルホルムアミド、相間移動触媒等を化合物(30)1モルに対して0.001~10.0モル、好ましくは0.001~3.0モル用いてもよい。
該クロル化剤としてはオキサリルクロリド、チオニルクロリド、オキシ塩化リン等が挙げられる。
相間移動触媒としてはテトラブチルアンモニウムクロリド、テトラブチルアンモニウムブロミド、クラウンエーテル等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~72時間、好ましくは30分~24時間である。反応温度は通常-78℃~100℃、好ましくは-10℃~25℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(32)は市販されている場合には市販品をそのまま用いることができ、実施例に記載の方法、自体公知の方法あるいはこれらに準じた方法に従って製造することもできる。例えば、化合物(32)は化合物(31)とRgに対応するアルキル化剤を用いたO-アルキル化反応により製造される。
詳しくは、化合物(31)1モルに対し、O-アルキル化剤1.0~50.0モル、好ましくは1.0~10.0モル、および塩基1.0~100.0モル、好ましくは3.0~10.0モルを用いて行う。
該アルキル化剤としてはトリメチルオキソニウム テトラフルオロボラート、ジメチルスルファート、メチル トリフルオロメタンスルホナート、メチル フルオロスルホナート等が挙げられる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、リン酸水素ナトリウム、リン酸ナトリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~72時間、好ましくは30分~24時間である。反応温度は通常-78℃~100℃、好ましくは-10℃~25℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(31)1モルに対し、O-アルキル化剤1.0~50.0モル、好ましくは1.0~10.0モル、および塩基1.0~100.0モル、好ましくは3.0~10.0モルを用いて行う。
該アルキル化剤としてはトリメチルオキソニウム テトラフルオロボラート、ジメチルスルファート、メチル トリフルオロメタンスルホナート、メチル フルオロスルホナート等が挙げられる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、リン酸水素ナトリウム、リン酸ナトリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~72時間、好ましくは30分~24時間である。反応温度は通常-78℃~100℃、好ましくは-10℃~25℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(33)は化合物(31)からの置換反応により製造される。
詳しくは、化合物(31)1モルに対して硫黄化剤1.0~3.0モル、好ましくは1.0~1.30モルを用いて行う。
該硫黄化剤としてはローソン試薬、五硫化リン、五硫化二リン等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~72時間、好ましくは1時間~24時間である。反応温度は通常0℃~150℃、好ましくは25℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(31)1モルに対して硫黄化剤1.0~3.0モル、好ましくは1.0~1.30モルを用いて行う。
該硫黄化剤としてはローソン試薬、五硫化リン、五硫化二リン等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~72時間、好ましくは1時間~24時間である。反応温度は通常0℃~150℃、好ましくは25℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(34)は、化合物(33)とRgに対応するアルキル化剤を用いたS-アルキル化反応によって製造される。
詳しくは化合物(33)1モルに対してS-アルキル化剤1.0~10.0モル、好ましくは1.0~5.0モルを用いて行う。
該アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等のハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常15分~60時間、好ましくは30分~24時間である。反応温度は通常0℃~150℃、好ましくは25℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは化合物(33)1モルに対してS-アルキル化剤1.0~10.0モル、好ましくは1.0~5.0モルを用いて行う。
該アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等のハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常15分~60時間、好ましくは30分~24時間である。反応温度は通常0℃~150℃、好ましくは25℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(28)は化合物(32)または化合物(34)からの置換反応により製造される。
詳しくは、化合物(32)または化合物(34)1モルに対して化合物(20)1.0~20.0モル、好ましくは1.0~2.0モル、および塩基1.0~20.0モル、好ましくは1.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。反応に塩基が必要でない場合には、塩基を使用せずに行うことができる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常30分~100時間、好ましくは1時間~72時間である。反応温度は通常0℃~150℃、好ましくは25℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(32)または化合物(34)1モルに対して化合物(20)1.0~20.0モル、好ましくは1.0~2.0モル、および塩基1.0~20.0モル、好ましくは1.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。反応に塩基が必要でない場合には、塩基を使用せずに行うことができる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常30分~100時間、好ましくは1時間~72時間である。反応温度は通常0℃~150℃、好ましくは25℃~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、Rjは置換されていてもよいアルキル基を示し、その他の記号は前記と同意義を示す。]
スキーム20は後述するスキーム21で得られる化合物(35)を出発原料とし、化合物(36)を経由して化合物(Ig)を合成するものである。
化合物(36)は化合物(35)の置換反応により製造される。
詳しくは、化合物(35)1モルに対し対応する酢酸誘導体1.0~10.0モル、好ましくは1.0~5.0モルを用いる。
該酢酸誘導体としては塩化2-(2-クロロアルコキシ)アセチル等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
本反応は塩基を加えて行ってもよい。該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(36)は化合物(35)の置換反応により製造される。
詳しくは、化合物(35)1モルに対し対応する酢酸誘導体1.0~10.0モル、好ましくは1.0~5.0モルを用いる。
該酢酸誘導体としては塩化2-(2-クロロアルコキシ)アセチル等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
本反応は塩基を加えて行ってもよい。該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(Ig)は化合物(36)の環化反応により製造される。
詳しくは、化合物(36)1モルに対し、塩基1.0~10.0モル、好ましくは1.0~5.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(36)1モルに対し、塩基1.0~10.0モル、好ましくは1.0~5.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常1時間~60時間、好ましくは1時間~24時間である。反応温度は通常-50~200℃、好ましくは0~150℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、Rhはアルキル基またはアルコキシ基で置換されていてもよいベンジル基を示し、Riはアルキル基またはアルコキシ基で置換されていてもよいベンソフェノンイミノ基を示し、その他の記号は前記と同意義を示す。]
スキーム21は、スキーム18で得られる化合物(27)からスキーム20の出発原料である化合物(35)を得るものである。
化合物(37)または化合物(38)は、化合物(27)とRhに対応するアミン試薬またはRiに対応するイミン試薬とのカップリング反応により製造される。
詳しくは化合物(27)1モルに対してRhに対応するアミン試薬またはRiに対応するイミン試薬1.0~10.0モル、好ましくは1.0~3.0モル、有機金属試薬0.01~1.0モル、好ましくは0.05~0.2モル、ホスフィン配位子0.01~1.0モル、好ましくは0.1~0.5モル、および塩基1.0~10.0モル、好ましくは2.0~6.0モルを用いて行う。反応にホスフィン配位子が必要でない場合には、ホスフィン配位子を使用せずに行うことができる。
該アミン試薬としてはベンジルアミン、ジベンジルアミン、2-メトキシベンジルアミン、3-メトキシベンジルアミン、4-メトキシベンジルアミン、2,3-ジメトキシベンジルアミン、2,4-ジメトキシベンジルアミン、3,4-ジメトキシベンジルアミン、2,4,6-トリメトキシベンジルアミン、3,4,5-トリメトキシベンジルアミン、2,3,4-トリメトキシベンジルアミン、2,4,5-トリメトキシベンジルアミン、2-メチルベンジルアミン、3-メチルベンジルアミン、4-メチルベンジルアミン、2,3-ジメチルベンジルアミン、2,4-ジメチルベンジルアミン、3,4-ジメチルベンジルアミン、2,4,6-トリメチルベンジルアミン、2,4,5-トリメチルベンジルアミンが挙げられる。
該イミン試薬としてはベンゾフェノンイミン、1,1-ビス(4-メトキシフェニル)メタンイミン、9-イミノフルオレン等が挙げられる。
有機金属試薬としてはトリス(ジベンジリデンアセトン)ジパラジウム(0)、テトラキス(トリフェニルホスフィン)パラジウム、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリドジクロロメタン錯体、酢酸パラジウム等が挙げられる。
ホスフィン配位子としては2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル、(9,9-ジメチル-9H-キサンテン-4,5-ジイル)ビス(ジフェニルホスファン)、2,2’-ビス(ジフェニルホスフィノ)-1,1’-ビナフチル等が挙げられる。
塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム、酢酸カリウム、炭酸セシウム等の塩基性塩類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(37)または化合物(38)は、化合物(27)とRhに対応するアミン試薬またはRiに対応するイミン試薬とのカップリング反応により製造される。
詳しくは化合物(27)1モルに対してRhに対応するアミン試薬またはRiに対応するイミン試薬1.0~10.0モル、好ましくは1.0~3.0モル、有機金属試薬0.01~1.0モル、好ましくは0.05~0.2モル、ホスフィン配位子0.01~1.0モル、好ましくは0.1~0.5モル、および塩基1.0~10.0モル、好ましくは2.0~6.0モルを用いて行う。反応にホスフィン配位子が必要でない場合には、ホスフィン配位子を使用せずに行うことができる。
該アミン試薬としてはベンジルアミン、ジベンジルアミン、2-メトキシベンジルアミン、3-メトキシベンジルアミン、4-メトキシベンジルアミン、2,3-ジメトキシベンジルアミン、2,4-ジメトキシベンジルアミン、3,4-ジメトキシベンジルアミン、2,4,6-トリメトキシベンジルアミン、3,4,5-トリメトキシベンジルアミン、2,3,4-トリメトキシベンジルアミン、2,4,5-トリメトキシベンジルアミン、2-メチルベンジルアミン、3-メチルベンジルアミン、4-メチルベンジルアミン、2,3-ジメチルベンジルアミン、2,4-ジメチルベンジルアミン、3,4-ジメチルベンジルアミン、2,4,6-トリメチルベンジルアミン、2,4,5-トリメチルベンジルアミンが挙げられる。
該イミン試薬としてはベンゾフェノンイミン、1,1-ビス(4-メトキシフェニル)メタンイミン、9-イミノフルオレン等が挙げられる。
有機金属試薬としてはトリス(ジベンジリデンアセトン)ジパラジウム(0)、テトラキス(トリフェニルホスフィン)パラジウム、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリドジクロロメタン錯体、酢酸パラジウム等が挙げられる。
ホスフィン配位子としては2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル、(9,9-ジメチル-9H-キサンテン-4,5-ジイル)ビス(ジフェニルホスファン)、2,2’-ビス(ジフェニルホスフィノ)-1,1’-ビナフチル等が挙げられる。
塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム、酢酸カリウム、炭酸セシウム等の塩基性塩類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(35)は化合物(37)を酸処理または水素添加反応に付すことにより製造される。
酸処理は、化合物(37)1モルに対して酸試薬1.0~200モル、好ましくは3.0~20.0モルを用いて行う。
該酸試薬としては、塩化アルミニウム等のルイス酸、酢酸、トリフルオロ酢酸等の有機酸、塩酸等の鉱酸が挙げられる。
水素添加反応は、化合物(37)に対して5重量%~1000重量%、好ましくは10重量%~300重量%の金属試薬を用いて行う。該金属試薬としては、パラジウム炭素、水酸化パラジウム、酸化白金、ラネーニッケル、ラネーコバルト等が挙げられる。また水素の圧力は通常1気圧~100気圧である。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常25℃~300℃、好ましくは50℃~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
酸処理は、化合物(37)1モルに対して酸試薬1.0~200モル、好ましくは3.0~20.0モルを用いて行う。
該酸試薬としては、塩化アルミニウム等のルイス酸、酢酸、トリフルオロ酢酸等の有機酸、塩酸等の鉱酸が挙げられる。
水素添加反応は、化合物(37)に対して5重量%~1000重量%、好ましくは10重量%~300重量%の金属試薬を用いて行う。該金属試薬としては、パラジウム炭素、水酸化パラジウム、酸化白金、ラネーニッケル、ラネーコバルト等が挙げられる。また水素の圧力は通常1気圧~100気圧である。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常25℃~300℃、好ましくは50℃~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(35)は化合物(38)を酸処理することによって製造される。
詳しくは、化合物(38)1モルに対して酸試薬1.0~200モル、好ましくは3.0~20.0モルを用いて行う。
該酸試薬としては、塩化アルミニウム等のルイス酸、酢酸、トリフルオロ酢酸等の有機酸、塩酸等の鉱酸が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常25℃~300℃、好ましくは25℃~150℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(38)1モルに対して酸試薬1.0~200モル、好ましくは3.0~20.0モルを用いて行う。
該酸試薬としては、塩化アルミニウム等のルイス酸、酢酸、トリフルオロ酢酸等の有機酸、塩酸等の鉱酸が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常25℃~300℃、好ましくは25℃~150℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、
は置換されていてもよい5員複素環を示し、その他の記号は前記と同意義を示す。]
スキーム22は前述のスキーム18で合成される化合物(27)から化合物(39)および化合物(40)を経由して化合物(Ih)を得るものである。
化合物(39)は化合物(27)から金属シアニド試薬を用いたカップリング反応により製造される。
詳しくは、化合物(27)1モルに対して金属シアニド試薬1.0~10.0モル、好ましくは1.0~3.0モル、有機金属試薬0.01~1.0モル、好ましくは0.05~0.2モル、およびホスフィン配位子0.01~1.0モル、好ましくは0.1~0.5モルを用いて行う。反応にホスフィン配位子が必要でない場合には、ホスフィン配位子を使用せずに行うことができる。
金属シアニド試薬としては、亜鉛シアニド、銅シアニド等が挙げられる。
有機金属試薬としてはトリス(ジベンジリデンアセトン)ジパラジウム(0)、テトラキス(トリフェニルホスフィン)パラジウム、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリドジクロロメタン錯体、酢酸パラジウム等が挙げられる。
ホスフィン配位子としては2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル、(9,9-ジメチル-9H-キサンテン-4,5-ジイル)ビス(ジフェニルホスファン)、2,2’-ビス(ジフェニルホスフィノ)-1,1’-ビナフチル等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(39)は化合物(27)から金属シアニド試薬を用いたカップリング反応により製造される。
詳しくは、化合物(27)1モルに対して金属シアニド試薬1.0~10.0モル、好ましくは1.0~3.0モル、有機金属試薬0.01~1.0モル、好ましくは0.05~0.2モル、およびホスフィン配位子0.01~1.0モル、好ましくは0.1~0.5モルを用いて行う。反応にホスフィン配位子が必要でない場合には、ホスフィン配位子を使用せずに行うことができる。
金属シアニド試薬としては、亜鉛シアニド、銅シアニド等が挙げられる。
有機金属試薬としてはトリス(ジベンジリデンアセトン)ジパラジウム(0)、テトラキス(トリフェニルホスフィン)パラジウム、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリドジクロロメタン錯体、酢酸パラジウム等が挙げられる。
ホスフィン配位子としては2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル、(9,9-ジメチル-9H-キサンテン-4,5-ジイル)ビス(ジフェニルホスファン)、2,2’-ビス(ジフェニルホスフィノ)-1,1’-ビナフチル等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(40)は化合物(39)の、ヒドロキシアミンによる置換反応により製造される。
詳しくは、化合物(39)1モルに対してヒドロキシアミン 塩酸塩1.0~200モル、好ましくは1.0~5.0モル、および塩基1.0~50.0モル、好ましくは1.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム、酢酸カリウム、炭酸セシウム等の塩基性塩類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(39)1モルに対してヒドロキシアミン 塩酸塩1.0~200モル、好ましくは1.0~5.0モル、および塩基1.0~50.0モル、好ましくは1.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム、酢酸カリウム、炭酸セシウム等の塩基性塩類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(Ih)は化合物(40)からの、縮合剤を用いた環化反応により製造される。
詳しくは、化合物(40)1モルに対して縮合剤1.0~20.0モル、好ましくは1.0~5.0モル、および塩基1.0~50.0モル、好ましくは1.0~10.0モルを用いて行う。
該縮合剤としては無水酢酸、トリフルオロ酢酸無水物、N,N’-カルボニルジイミダゾール等が挙げられる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン、ピリジン等の有機塩基類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、ピリジン等の有機塩基類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(40)1モルに対して縮合剤1.0~20.0モル、好ましくは1.0~5.0モル、および塩基1.0~50.0モル、好ましくは1.0~10.0モルを用いて行う。
該縮合剤としては無水酢酸、トリフルオロ酢酸無水物、N,N’-カルボニルジイミダゾール等が挙げられる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン、ピリジン等の有機塩基類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、ピリジン等の有機塩基類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、
は置換されていてもよい5員複素環を示し、その他の記号は前記と同意義を示す。]
スキーム23は前述のスキーム18で合成される化合物(27)から化合物(41)および化合物(42)、あるいはさらに化合物(43)を経由して化合物(Ih’)を得るものである。
化合物(41)は化合物(27)から有機金属試薬と一酸化炭素を用いた挿入反応により製造される。
詳しくは、本反応は化合物(27)1モルに対して有機金属試薬0.01~1.0モル、好ましくは0.05~0.2モル、ホスフィン配位子0.01~1.0モル、好ましくは0.1~0.5モル、塩基1.0~10.0モル、好ましくは2.0~6.0モル、およびRjに対応するアルコール1.0~100.0モル、好ましくは1.0~10.0モルを用いて一酸化炭素雰囲気下で行う。反応にホスフィン配位子が必要でない場合には、ホスフィン配位子を使用せずに行うことができる。
有機金属試薬としてはトリス(ジベンジリデンアセトン)ジパラジウム(0)、テトラキス(トリフェニルホスフィン)パラジウム、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリドジクロロメタン錯体、酢酸パラジウム等が挙げられる。
ホスフィン配位子としては2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル、(9,9-ジメチル-9H-キサンテン-4,5-ジイル)ビス(ジフェニルホスファン)、2,2’-ビス(ジフェニルホスフィノ)-1,1’-ビナフチル等が挙げられる。
塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム、酢酸カリウム、炭酸セシウム等の塩基性塩類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(41)は化合物(27)から有機金属試薬と一酸化炭素を用いた挿入反応により製造される。
詳しくは、本反応は化合物(27)1モルに対して有機金属試薬0.01~1.0モル、好ましくは0.05~0.2モル、ホスフィン配位子0.01~1.0モル、好ましくは0.1~0.5モル、塩基1.0~10.0モル、好ましくは2.0~6.0モル、およびRjに対応するアルコール1.0~100.0モル、好ましくは1.0~10.0モルを用いて一酸化炭素雰囲気下で行う。反応にホスフィン配位子が必要でない場合には、ホスフィン配位子を使用せずに行うことができる。
有機金属試薬としてはトリス(ジベンジリデンアセトン)ジパラジウム(0)、テトラキス(トリフェニルホスフィン)パラジウム、[1,1’-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリドジクロロメタン錯体、酢酸パラジウム等が挙げられる。
ホスフィン配位子としては2-ジシクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル、(9,9-ジメチル-9H-キサンテン-4,5-ジイル)ビス(ジフェニルホスファン)、2,2’-ビス(ジフェニルホスフィノ)-1,1’-ビナフチル等が挙げられる。
塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム、酢酸カリウム、炭酸セシウム等の塩基性塩類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(42)は化合物(41)の加水分解反応により製造される。
詳しくは化合物(41)1モルに対して水1.0~100.0モル、好ましくは1.0~5.0モル、塩基1.0~100.0モル、好ましくは1.0~5.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム、酢酸カリウム、炭酸セシウム等の塩基性塩類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~200℃、好ましくは25~100℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは化合物(41)1モルに対して水1.0~100.0モル、好ましくは1.0~5.0モル、塩基1.0~100.0モル、好ましくは1.0~5.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム、酢酸ナトリウム、酢酸カリウム、炭酸セシウム等の塩基性塩類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~200℃、好ましくは25~100℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(43)は化合物(42)の置換反応により製造される。
詳しくは、化合物(42)1モルに対して塩化オキサリル1.0~2.0モル、好ましくは1.0~1.5モル、Rjに対応するヒドラジド1.0~20.0モル、好ましくは1.0~5.0モル、および塩基1.0~20.0モル、好ましくは1.0~10.0モルを用いて行う。
Rjに対応するヒドラジドとしてはアセトヒドラジドが挙げられる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン、ピリジン等の有機塩基類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(42)1モルに対して塩化オキサリル1.0~2.0モル、好ましくは1.0~1.5モル、Rjに対応するヒドラジド1.0~20.0モル、好ましくは1.0~5.0モル、および塩基1.0~20.0モル、好ましくは1.0~10.0モルを用いて行う。
Rjに対応するヒドラジドとしてはアセトヒドラジドが挙げられる。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン、ピリジン等の有機塩基類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(Ih’)は化合物(42)の環化反応により製造される。
詳しくは、化合物(42)1モルに対して塩化オキサリル1.0~2.0モル、好ましくは1.0~1.5モル、N’-ヒドロキシエタンイミドアミド1.0~20.0モル、好ましくは1.0~5.0モル、および塩基1.0~20.0モル、好ましくは1.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン、ピリジン等の有機塩基類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(42)1モルに対して塩化オキサリル1.0~2.0モル、好ましくは1.0~1.5モル、N’-ヒドロキシエタンイミドアミド1.0~20.0モル、好ましくは1.0~5.0モル、および塩基1.0~20.0モル、好ましくは1.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン、ピリジン等の有機塩基類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~300℃、好ましくは25~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(Ih’)は化合物(43)の環化反応によっても製造される。
詳しくは、化合物(43)1モルに対して(メトキシカルボニルスルファモイル)トリエチルアンモニウム ヒドロキシド 分子内塩1.0~2.0モル、好ましくは1.0~1.5モルを用いて行う。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~200℃、好ましくは25~150℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(43)1モルに対して(メトキシカルボニルスルファモイル)トリエチルアンモニウム ヒドロキシド 分子内塩1.0~2.0モル、好ましくは1.0~1.5モルを用いて行う。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常0~200℃、好ましくは25~150℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、Rkは置換されていてもよいC1-5アルキル基を示し、Rlは置換されていてもよいC1-6アルキル基を示し、その他の記号は前記と同意義を示す。]
スキーム24は前述のスキーム3の化合物(Ib)群に含まれる化合物(Ii)から化合物(Ij)および化合物(Ii’)を得るものである。
化合物(Ij)は化合物(Ii)を酸処理または水素添加反応に付すことにより製造される。
酸処理は、化合物(Ii)1モルに対して酸試薬1.0~200モル、好ましくは3.0~20.0モルを用いて行う。
該酸試薬としては、塩化アルミニウム等のルイス酸、酢酸、トリフルオロ酢酸等の有機酸、塩酸等の鉱酸が挙げられる。
水素添加反応は、化合物(Ii)に対して5重量%~1000重量%、好ましくは10重量%~300重量%の金属試薬を用いて行う。該金属試薬としては、パラジウム炭素、水酸化パラジウム、酸化白金、ラネーニッケル、ラネーコバルト等が挙げられる。また水素の圧力は通常1気圧~100気圧である。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常25℃~300℃、好ましくは50℃~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(Ij)は化合物(Ii)を酸処理または水素添加反応に付すことにより製造される。
酸処理は、化合物(Ii)1モルに対して酸試薬1.0~200モル、好ましくは3.0~20.0モルを用いて行う。
該酸試薬としては、塩化アルミニウム等のルイス酸、酢酸、トリフルオロ酢酸等の有機酸、塩酸等の鉱酸が挙げられる。
水素添加反応は、化合物(Ii)に対して5重量%~1000重量%、好ましくは10重量%~300重量%の金属試薬を用いて行う。該金属試薬としては、パラジウム炭素、水酸化パラジウム、酸化白金、ラネーニッケル、ラネーコバルト等が挙げられる。また水素の圧力は通常1気圧~100気圧である。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、酢酸、トリフルオロ酢酸等の有機酸類、水またはそれらの混合溶媒等が好ましい。
反応時間は、通常10分~50時間、好ましくは30分~12時間である。反応温度は、通常25℃~300℃、好ましくは50℃~200℃である。必要があればマイクロ波照射下で反応を行うことができる。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(Ii’)は、化合物(Ij)の塩基とRlに対応するアルキル化剤を用いたO-アルキル化反応によって製造される。
詳しくは化合物(Ij)1モルに対して塩基1.0~50.0モル、好ましくは1.0~5.0モル、およびO-アルキル化剤1.0~100.0モル、好ましくは1.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類等が挙げられる。
該O-アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等のハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常15分~60時間、好ましくは15分~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは化合物(Ij)1モルに対して塩基1.0~50.0モル、好ましくは1.0~5.0モル、およびO-アルキル化剤1.0~100.0モル、好ましくは1.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸水素ナトリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、イミダゾール、ホルムアミジン等の有機塩基類等が挙げられる。
該O-アルキル化剤としては塩化アルキル、臭化アルキル、ヨウ化アルキル等のハロゲン化アルキル類およびその誘導体、p-トルエンスルホン酸エステル、メチルスルホン酸エステル等のスルホン酸エステル類、ジメチル硫酸等の硫酸エステル類等が挙げられる。
本反応は反応に不活性な溶媒を用いて行うのが好ましく、このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、メタノール、エタノール、プロパノール、1,1-ジメチルエタノール等のアルコール類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類、水またはそれらの混合溶媒等が好ましい。
反応時間は通常15分~60時間、好ましくは15分~24時間である。反応温度は通常-50~150℃、好ましくは0~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。

[式中、Rmは置換されていてもよいC1-2アルキル基を示し、その他の記号は前記と同意義を示す。]
スキーム25は前述のスキーム1で得られる化合物(3)または化合物(4)から化合物(Il)を経由して化合物(Ik)を得るものである。
化合物(Il)は化合物(3)または化合物(4)の、塩基とRgに対応する酢酸エステル誘導体を用いた置換反応で製造される。
詳しくは、化合物(3)または化合物(4)1モルに対し、塩基1.0~20.0モル、好ましくは1.0~10.0モル、およびRgに対応する酢酸エステル誘導体1.0~100.0モル、好ましくは1.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。反応に塩基が必要でない場合には、塩基を使用せずに行うことができる。
該酢酸エステル誘導体としては、アルキル ヒドロキシアセタート等が挙げられる。
本反応は無溶媒で行うか、反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~24時間、好ましくは10分~12時間である。反応温度は通常-100~150℃、好ましくは-78~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(Il)は化合物(3)または化合物(4)の、塩基とRgに対応する酢酸エステル誘導体を用いた置換反応で製造される。
詳しくは、化合物(3)または化合物(4)1モルに対し、塩基1.0~20.0モル、好ましくは1.0~10.0モル、およびRgに対応する酢酸エステル誘導体1.0~100.0モル、好ましくは1.0~10.0モルを用いて行う。
該塩基としては水酸化ナトリウム、水酸化カリウム、水酸化バリウム、炭酸ナトリウム、炭酸カリウム等の無機塩基類、ナトリウムメトキシド、ナトリウムエトキシド、カリウム第三ブトキシド等の金属アルコキシド類、水素化ナトリウム、水素化カリウム等の水素化金属類、トリエチルアミン、ピリジン、ジイソプロピルエチルアミン、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン等の有機アミン等が挙げられる。反応に塩基が必要でない場合には、塩基を使用せずに行うことができる。
該酢酸エステル誘導体としては、アルキル ヒドロキシアセタート等が挙げられる。
本反応は無溶媒で行うか、反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド等のアミド類、アセトニトリル、プロピオニトリル等のニトリル類、ジメチルスルホキシド等のスルホキシド類等の溶媒またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~24時間、好ましくは10分~12時間である。反応温度は通常-100~150℃、好ましくは-78~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
化合物(Ik)は化合物(Il)の、Rmに対応するアルキル化剤を用いた求核置換反応により製造される。
詳しくは、化合物(Il)1モルに対し、Rmに対応するアルキル化剤2.0~100.0モル、好ましくは2.0~10.0モルを用いて行う。
該アルキル化剤としては、アルキルマグネシウムハライド(グリニャール試薬)、アルキルリチウム等が挙げられる。
本反応は無溶媒で行うか、反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~72時間、好ましくは10分~24時間である。反応温度は通常-100~150℃、好ましくは-78~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
詳しくは、化合物(Il)1モルに対し、Rmに対応するアルキル化剤2.0~100.0モル、好ましくは2.0~10.0モルを用いて行う。
該アルキル化剤としては、アルキルマグネシウムハライド(グリニャール試薬)、アルキルリチウム等が挙げられる。
本反応は無溶媒で行うか、反応に不活性な溶媒を用いて行うのが好ましい。このような溶媒としては反応が進行する限り特に限定されないが、例えばジクロロメタン、クロロホルム、四塩化炭素、1,2-ジクロロエタン等のハロゲン化炭化水素類、ベンゼン、トルエン等の芳香族炭化水素類、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタン等のエーテル類またはそれらの混合溶媒等が好ましい。
反応時間は通常10分~72時間、好ましくは10分~24時間である。反応温度は通常-100~150℃、好ましくは-78~100℃である。
生成物は単一または混合物として得られ、反応液のまま、または粗製物として次の反応に用いることもできるが、常法に従って反応混合物から単離することもでき、洗浄、再結晶、蒸留、クロマトグラフィー等の分離手段により容易に精製することができる。
前記のスキーム1~25の各反応において、原料化合物が置換基としてヒドロキシ、アミノ(-NH-、-NH2-を含む)、カルボキシ、カルボニルまたはメルカプトを有する場合、これらの基にペプチド化学等で一般的に用いられるような保護基が導入されていてもよく、反応後に必要に応じて保護基を除去することにより目的化合物を得ることができる。
ヒドロキシの保護基としては、例えば、C1-6アルキル(例、メチル、エチル、プロピル、イソプロピル、ブチル、tert-ブチル)、フェニル、トリチル、C7-10アラルキル(例、ベンジル)、ホルミル、C1-6アルキル-カルボニル(例、アセチル、プロピオニル)、ベンゾイル、C7-10アラルキル-カルボニル(例、ベンジルカルボニル)、2-テトラヒドロピラニル、2-テトラヒドロフラニル、シリル(例、トリメチルシリル、トリエチルシリル、ジメチルフェニルシリル、tert-ブチルジメチルシリル、tert-ブチルジエチルシリル)、C2-6アルケニル(例、1-アリル)等が挙げられる。これらの基は、ハロゲン原子(例、フッ素原子、塩素原子、臭素原子、ヨウ素原子)、C1-6アルキル(例、メチル、エチル、プロピル)、C1-6アルコキシ(例、メトキシ、エトキシ、プロポキシ)、ニトロ等から選ばれる1ないし3個の置換基で置換されていてもよい。
アミノの保護基としては、例えばホルミル、C1-6アルキル-カルボニル(例、アセチル、プロピオニル)、C1-6アルコキシ-カルボニル(例、メトキシカルボニル、エトキシカルボニル、tert-ブトキシカルボニル)、ベンゾイル、C7-10アラルキル-カルボニル(例、ベンジルカルボニル)、C7-14アラルキルオキシ-カルボニル(例、ベンジルオキシカルボニル、9-フルオレニルメトキシカルボニル)、C7-10アラルキル(例、ベンジル、4-メトキシベンジル)、トリチル、フタロイル、N,N-ジメチルアミノメチレン、シリル(例、トリメチルシリル、トリエチルシリル、ジメチルフェニルシリル、tert-ブチルジメチルシリル、tert-ブチルジエチルシリル)、C2-6アルケニル(例、1-アリル)等が挙げられる。これらの基は、ハロゲン原子(例、フッ素原子、塩素原子、臭素原子、ヨウ素原子)、C1-6アルコキシ(例、メトキシ、エトキシ、プロポキシ)、ニトロ等から選ばれる1ないし3個の置換基で置換されていてもよい。
カルボキシの保護基としては、例えば、C1-6アルキル(例、メチル、エチル、プロピル、イソプロピル、ブチル、tert-ブチル)、C7-11アラルキル(例、ベンジル)、フェニル、トリチル、シリル(例、トリメチルシリル、トリエチルシリル、ジメチルフェニルシリル、tert-ブチルジメチルシリル、tert-ブチルジエチルシリル、tert-ブチルジフェニルシリル)、C2-6アルケニル(例、1-アリル)等が挙げられる。これらの基は、ハロゲン原子(例、フッ素原子、塩素原子、臭素原子、ヨウ素原子)、C1-6アルコキシ(例、メトキシ、エトキシ、プロポキシ)、ニトロ等から選ばれる1ないし3個の置換基で置換されていてもよい。
カルボニルの保護基としては、例えば、環状アセタール(例、1,3-ジオキサン)、非環状アセタール(例、ジ-C1-6アルキルアセタール)等が挙げられる。
メルカプトの保護基としては、例えば、C1-6アルキル基、フェニル基、トリチル基、C7-10アラルキル基(例、ベンジル)、C1-6アルキル-カルボニル基、ベンゾイル基、C7-10アラルキル-カルボニル基(例、ベンジルカルボニル)、C1-6アルコキシ-カルボニル基、C6-14アリールオキシ-カルボニル基(例、フェニルオキシカルボニル)、C7-14アラルキルオキシ-カルボニル基(例、ベンジルオキシカルボニル、9-フルオレニルメトキシカルボニル)、2-テトラヒドロピラニル基、C1-6アルキルアミノ-カルボニル基(例、メチルアミノカルボニル、エチルアミノカルボニル)、などが挙げられる。これらの基は、ハロゲン原子、C1-6アルキル基、C1-6アルコキシ基またはニトロ基から選ばれる1ないし3個の置換基で置換されていてもよい。
上記した保護基の除去方法は、自体公知の方法、例えば、プロテクティブ グループス イン オーガニック シンセシス(Protective Groups in Organic Synthesis),JohnWiley and Sons 刊(1980)に記載の方法等に準じて行えばよい。例えば、酸、塩基、紫外光、ヒドラジン、フェニルヒドラジン、N-メチルジチオカルバミン酸ナトリウム、テトラブチルアンモニウムフルオリド、酢酸パラジウム、トリアルキルシリルハライド(例、トリメチルシリルヨージド、トリメチルシリルブロミド等)等を使用する方法、還元法等が用いられる。
上記の各製造法により得られる化合物(I)は、濃縮、減圧濃縮、溶媒抽出、晶出、再結晶、転溶、クロマトグラフィー等の公知の手段により単離精製することができる。また、上記の各製造法において用いられる各原料化合物は、前記と同様の公知の手段によって単離精製することができる。一方、これら原料化合物を単離することなく、そのまま反応混合物として、次の工程の原料として用いてもよい。
化合物(I)が、光学異性体、立体異性体、位置異性体、回転異性体等の異性体を有する場合には、いずれか一方の異性体も、異性体の混合物も化合物(I)に包含される。例えば、化合物(I)に光学異性体が存在する場合には、ラセミ体から分割された光学異性体も化合物(I)に包含される。これらの異性体は、自体公知の合成手法、分離手法(濃縮、溶媒抽出、カラムクロマトグラフィー、再結晶等)によりそれぞれを単品として得ることができる。
化合物(I)またはそのプロドラッグ(以下、単に本発明化合物と略記することがある)は、毒性が低く、そのまま、または薬理学的に許容し得る担体等と混合して医薬組成物とすることにより、哺乳動物(例、ヒト、マウス、ラット、ウサギ、イヌ、ネコ、ウシ、ウマ、ブタ、サル)に対して、後述する各種疾患の予防または治療剤として用いることができる。
ここで、薬理学的に許容し得る担体としては、製剤素材として慣用の各種有機または無機担体物質が用いられ、固形製剤における賦形剤、滑沢剤、結合剤、崩壊剤;液状製剤における溶剤、溶解補助剤、懸濁化剤、等張化剤、緩衝剤、無痛化剤等として配合される。また必要に応じて、防腐剤、抗酸化剤、着色剤、甘味剤等の製剤添加物を用いることもできる。
賦形剤の好適な例としては、乳糖、白糖、D-マンニトール、D-ソルビトール、デンプン、α化デンプン、デキストリン、結晶セルロース、低置換度ヒドロキシプロピルセルロース、カルボキシメチルセルロースナトリウム、アラビアゴム、プルラン、軽質無水ケイ酸、合成ケイ酸アルミニウム、メタケイ酸アルミン酸マグネシウムが挙げられる。
滑沢剤の好適な例としては、ステアリン酸マグネシウム、ステアリン酸カルシウム、タルク、コロイドシリカが挙げられる。
結合剤の好適な例としては、α化デンプン、ショ糖、ゼラチン、アラビアゴム、メチルセルロース、カルボキシメチルセルロース、カルボキシメチルセルロースナトリウム、結晶セルロース、白糖、D-マンニトール、トレハロース、デキストリン、プルラン、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドンが挙げられる。
崩壊剤の好適な例としては、乳糖、白糖、デンプン、カルボキシメチルセルロース、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、カルボキシメチルスターチナトリウム、軽質無水ケイ酸、低置換度ヒドロキシプロピルセルロースが挙げられる。
溶剤の好適な例としては、注射用水、生理的食塩水、リンゲル液、アルコール、プロピレングリコール、ポリエチレングリコール、ゴマ油、トウモロコシ油、オリーブ油、綿実油が挙げられる。
溶解補助剤の好適な例としては、ポリエチレングリコール、プロピレングリコール、D-マンニトール、トレハロース、安息香酸ベンジル、エタノール、トリスアミノメタン、コレステロール、トリエタノールアミン、炭酸ナトリウム、クエン酸ナトリウム、サリチル酸ナトリウム、酢酸ナトリウムが挙げられる。
懸濁化剤の好適な例としては、ステアリルトリエタノールアミン、ラウリル硫酸ナトリウム、ラウリルアミノプロピオン酸、レシチン、塩化ベンザルコニウム、塩化ベンゼトニウム、モノステアリン酸グリセリン等の界面活性剤;ポリビニルアルコール、ポリビニルピロリドン、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース等の親水性高分子;ポリソルベート類、ポリオキシエチレン硬化ヒマシ油が挙げられる。
等張化剤の好適な例としては、塩化ナトリウム、グリセリン、D-マンニトール、D-ソルビトール、ブドウ糖が挙げられる。
緩衝剤の好適な例としては、リン酸塩、酢酸塩、炭酸塩、クエン酸塩等の緩衝液が挙げられる。
無痛化剤の好適な例としては、ベンジルアルコールが挙げられる。
無痛化剤の好適な例としては、ベンジルアルコールが挙げられる。
防腐剤の好適な例としては、パラオキシ安息香酸エステル類、クロロブタノール、ベンジルアルコール、フェネチルアルコール、デヒドロ酢酸、ソルビン酸が挙げられる。
抗酸化剤の好適な例としては、亜硫酸塩、アスコルビン酸塩等が挙げられる。
抗酸化剤の好適な例としては、亜硫酸塩、アスコルビン酸塩等が挙げられる。
着色剤の好適な例としては、水溶性食用タール色素(例、食用赤色2号および3号、食用黄色4号および5号、食用青色1号および2号等の食用色素)、水不溶性レーキ色素(例、前記水溶性食用タール色素のアルミニウム塩)、天然色素(例、β-カロチン、クロロフィル、ベンガラ)が挙げられる。
甘味剤の好適な例としては、サッカリンナトリウム、グリチルリチン酸二カリウム、アスパルテーム、ステビアが挙げられる。
本発明化合物を含有する医薬は、医薬製剤の製造法として自体公知の方法(例、日本薬局方記載の方法等)に従って、本発明化合物を単独で、または薬理学的に許容される担体と混合して、例えば錠剤(糖衣錠、フィルムコーティング錠、舌下錠、口腔内崩壊錠、バッカル錠等を含む)、丸剤、散剤、顆粒剤、カプセル剤(ソフトカプセル剤、マイクロカプセル剤を含む)、トローチ剤、シロップ剤、液剤、乳剤、懸濁剤、放出制御製剤(例、速放性製剤、徐放性製剤、徐放性マイクロカプセル剤)、エアゾール剤、フィルム剤(例、口腔内崩壊フィルム、口腔粘膜貼付フィルム)、注射剤(例、皮下注射剤、静脈内注射剤、筋肉内注射剤、腹腔内注射剤)、点滴剤、経皮吸収型製剤、軟膏剤、ローション剤、貼付剤、坐剤(例、肛門坐剤、膣坐剤)、ペレット、経鼻剤、経肺剤(吸入剤)、点眼剤等として、経口的または非経口的(例、静脈内、筋肉内、皮下、臓器内、鼻腔内、皮内、点眼、脳内、直腸内、膣内、腹腔内、腫瘍内部、腫瘍の近位等への投与および直接的な病巣への投与)に安全に投与することができる。
医薬組成物は、製剤技術分野において慣用の方法、例えば、日本薬局方に記載の方法等により製造することができる。
なお、医薬組成物中の本発明化合物の含量は、剤形、本発明化合物の投与量等により異なるが、例えば、約0.1~100重量%である。
経口剤を製造する際には、必要により、味のマスキング、腸溶性または持続性を目的として、コーティングを行ってもよい。
コーティングに用いられるコーティング基剤としては、例えば、糖衣基剤、水溶性フィルムコーティング基剤、腸溶性フィルムコーティング基剤、徐放性フィルムコーティング基剤が挙げられる。
糖衣基剤としては、白糖が用いられ、さらに、タルク、沈降炭酸カルシウム、ゼラチン、アラビアゴム、プルラン、カルナバロウ等から選ばれる1種または2種以上を併用してもよい。
水溶性フィルムコーティング基剤としては、例えば、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、メチルヒドロキシエチルセルロース等のセルロース系高分子;ポリビニルアセタールジエチルアミノアセテート、アミノアルキルメタアクリレートコポリマーE〔オイドラギットE(商品名)〕、ポリビニルピロリドン等の合成高分子;プルラン等の多糖類が挙げられる。
腸溶性フィルムコーティング基剤としては、例えば、ヒドロキシプロピルメチルセルロース フタレート、ヒドロキシプロピルメチルセルロース アセテートサクシネート、カルボキシメチルエチルセルロース、酢酸フタル酸セルロース等のセルロース系高分子;メタアクリル酸コポリマーL〔オイドラギットL(商品名)〕、メタアクリル酸コポリマーLD〔オイドラギットL-30D55(商品名)〕、メタアクリル酸コポリマーS〔オイドラギットS(商品名)〕等のアクリル酸系高分子;セラック等の天然物が挙げられる。
徐放性フィルムコーティング基剤としては、例えば、エチルセルロース等のセルロース系高分子;アミノアルキルメタアクリレートコポリマーRS〔オイドラギットRS(商品名)〕、アクリル酸エチル-メタクリル酸メチル共重合体懸濁液〔オイドラギットNE(商品名)〕等のアクリル酸系高分子が挙げられる。
上記したコーティング基剤は、その2種以上を適宜の割合で混合して用いてもよい。また、コーティングの際に、例えば、酸化チタン、三二酸化鉄等のような遮光剤を用いてもよい。
本発明化合物は、毒性(例、急性毒性、慢性毒性、遺伝毒性、生殖毒性、心毒性、癌原性)が低く、副作用も少なく、哺乳動物(例、ヒト、サル、ネコ、ブタ、ウマ、ウシ、マウス、ラット、モルモット、イヌ、ウサギ等)に対し、後述する各種疾患の予防・治療剤、または診断薬として用いることができる。
本発明化合物は、強力なデルタ-5-サチュラーゼ阻害作用を有することから、デルタ-5-デサチュラーゼを介して産生されるエイコサノイドが関連して発症する疾患(または発症が促進される疾患)の予防または治療薬として有用である。
このような疾患としては、例えば、心疾患(心肥大、急性心不全およびうっ血性を含む慢性心不全、心筋症、狭心症、心筋炎、不整脈、頻脈、心筋梗塞等)、心筋虚血、静脈機能不全、心筋梗塞後の心不全移行、高血圧症、肺性心、アテローム性を含む動脈硬化症(動脈瘤、冠動脈硬化症、脳動脈硬化症、末梢動脈硬化症等)、血管肥厚、インターベンション(経皮的冠動脈形成術、ステント留置、冠動脈内視鏡、血管内超音波、冠注血栓溶解療法等)および心移植後の血管肥厚または閉塞および臓器障害、バイパス手術後の血管再閉塞・再狭窄、呼吸器疾患(かぜ症候群、肺炎、喘息、肺高血圧症、肺血栓・肺塞栓等)、骨疾患(骨折、再骨折、骨変形・変形脊椎症、骨肉腫、骨髄腫、骨形成不全、側弯症等の非代謝性骨疾患、骨欠損、骨粗鬆症,骨軟化症,くる病、線維性骨炎、腎性骨異栄養症、骨ペーチェット病、硬直性脊髄炎、慢性関節リウマチ、変形性膝関節炎およびそれらの類似疾患における関節組織の破壊等)、炎症性疾患(網膜症、腎症、神経障害、慢性関節リウマチ、変形性関節炎、リウマチ様脊髄炎、骨膜炎等の関節炎、手術・外傷後の炎症、腫脹の緩解、咽頭炎、膀胱炎、アトピー性皮膚炎、クローン病・潰瘍性大腸炎等の炎症性腸疾患、髄膜炎、炎症性眼疾患、肺炎・珪肺・肺サルコイドーシス・肺結核等の炎症性肺疾患等)、アレルギー疾患(アレルギー性鼻炎、結膜炎、消化管アレルギー、花粉症、アナフィラキシー等)、薬物依存、神経変性疾患(アルツハイマー病、パーキンソン病、筋萎縮性側索硬化症、エイズ脳症等)、中枢神経障害(脳出血および脳梗塞等の障害およびその後遺症・合併症、頭部外傷、脊椎損傷、脳浮腫等)、痴呆症、記憶障害、意識障害、健忘症、不安症状、緊張症状、不快精神状態、精神疾患(うつ病、てんかん、アルコール依存症等)、虚血性末梢循環障害、深部静脈血栓症、閉塞性末梢循環障害、閉塞性動脈硬化症、閉塞性血栓性血管炎、糖尿病(1型糖尿病、2型糖尿病、1.5型糖尿病(LADA(Latent Autoimmune Diabetes in Adults))、妊娠糖尿病、インスリン分泌不全型糖尿病、肥満型糖尿病、耐糖能不全(IGT(Impaired Glucose Tolerance))、IFG(Impaired Fasting Glucose)、IFG(Impaired Fasting Glycaemia)等)、糖尿病性合併症(神経障害、腎症、網膜症、白内障、大血管障害、骨減少症、糖尿病性高浸透圧昏睡、感染症(呼吸器感染症、尿路感染症、消化器感染症、皮膚軟部組織感染症、下肢感染症等)、糖尿病性壊疽、口腔乾燥症、聴覚の低下、脳血管障害、末梢血行障害等)、尿失禁、代謝・栄養障害(肥満症(例、悪性肥満細胞(malignant mastocytosis)、外因性肥満(exogenous obesity)、過インシュリン性肥満症(hyperinsulinar obesity)、過血漿性肥満(hyperplasmic obesity)、下垂体性肥満(hypophyseal adiposity)、減血漿性肥満症(hypoplasmic obesity)、甲状腺機能低下肥満症(hypothyroid obesity)、視床下部性肥満(hypothalamic obesity)、症候性肥満症(symptomatic obesity)、小児肥満(infantile obesity)、上半身肥満(upper body obesity)、食事性肥満症(alimentary obesity)、性機能低下性肥満(hypogonadal obesity)、全身性肥満細胞症(systemic mastocytosis)、単純性肥満(simple obesity)、中心性肥満(central obesity)等)、摂食亢進症(hyperphagia)等)、高脂血症、高コレステロール血症、耐糖能異常等)、インスリン抵抗性症候群、シンドロームX、内臓肥満症候群、男性または女性の性機能障害、脳血管障害(無症候性脳血管障害、一過性脳虚血発作、脳卒中、脳血管性痴呆、高血圧性脳症、脳梗塞等)、脳浮腫、脳循環障害、脳血管障害の再発および後遺症(神経症候、精神症候、自覚症状、日常生活動作障害等)、腎疾患(腎炎、糸球体腎炎、糸球体硬化症、腎不全、血栓性微小血管症、糖尿病性ネフロパシー、ネフローゼ症候群、高血圧性腎硬化症、透析の合併症、放射線照射による腎症を含む臓器障害等)、眼疾患(緑内障、高眼圧症等)、血栓症、多臓器不全、内皮機能障害、その他の循環器系疾患(虚血性脳循環障害、レイノー病、バージャー病等)、慢性閉塞性肺疾患、間質性肺炎、カリニ肺炎、膠原病(例、全身性エリテマトーデス、強皮症、多発動脈炎等)、肝臓疾患(慢性を含む肝炎、肝硬変等)、消化器疾患(胃炎、胃潰瘍、胃癌、胃手術後障害、消化不良、食道潰瘍、膵炎、大腸ポリープ、胆石症、痔疾患、食道や胃の静脈瘤破裂等)、血液・造血器疾患(赤血球増加症、血管性紫斑病、自己免疫性溶血性貧血、播種性血管内凝固症候群、多発性骨髄症等)、固形腫瘍、腫瘍(悪性黒色腫、悪性リンパ腫、消化器(例、胃、腸等)癌等)、癌およびそれに伴う悪液質、癌の転移、内分泌疾患(アジソン病、クッシング症候群、褐色細胞種、原発性アルドステロン症等)、泌尿器・男性性器疾患(膀胱炎、前立腺肥大症、前立腺癌、性感染症等)、婦人科疾患(更年期障害、妊娠中毒、子宮内膜症、子宮筋腫、卵巣疾患、乳腺疾患、性感染症等)、感染症(サイトメガルウイルス、インフルエンザウイルス、ヘルペスウイルス等のウイルス感染症、リケッチア感染症、細菌感染症等)、毒血症(敗血症、敗血症性ショック、内毒素性ショック、グラム陰性敗血症、トキシンショック症候群等)、皮膚疾患(ケロイド、血管腫、乾癬等)等が挙げられる。本発明化合物は、特に、動脈硬化症、糖尿病および肥満の予防または治療に使用することが望ましい。ここで動脈硬化症の予防・治療という概念には、動脈硬化プラークの破綻によって生じる虚血性心疾患(不安定狭心症・急性心筋梗塞・急性心不全・心臓死)および脳卒中(一過性脳虚血を含む)等いわゆるアテローム性血栓症の予防およびそれらの重症化遅延、動脈硬化進展抑制作用に基づく心血管イベント高発症リスク患者(急性冠動脈疾患患者、脳卒中患者、代謝性疾患患者、高血圧・肥満・糖尿病・脂質異常症患者等)の心血管イベント発症予防、虚血性心疾患再発予防、心血管イベント一次発症予防、末梢動脈血管症予防または治療等も含まれる。
糖尿病の判定基準については、1999年に日本糖尿病学会から判定基準が報告されている。
この報告によれば、糖尿病とは、空腹時血糖値(静脈血漿におけるグルコース濃度)が126mg/dl以上、75g経口ブドウ糖負荷試験(75gOGTT)2時間値(静脈血漿におけるグルコース濃度)が200mg/dl以上、随時血糖値(静脈血漿におけるグルコース濃度)が200mg/dl以上のいずれかを示す状態である。また、上記糖尿病に該当せず、かつ、「空腹時血糖値(静脈血漿におけるグルコース濃度)が110mg/dl未満または75g経口ブドウ糖負荷試験(75gOGTT)2時間値(静脈血漿におけるグルコース濃度)が140mg/dl未満を示す状態」(正常型)でない状態を、「境界型」と呼ぶ。
また、糖尿病の判定基準については、1997年にADA(米国糖尿病学会)から、1998年にWHOから、判定基準が報告されている。
これらの報告によれば、糖尿病とは、空腹時血糖値(静脈血漿におけるグルコース濃度)が126mg/dl以上であり、かつ、75g経口ブドウ糖負荷試験2時間値(静脈血漿におけるグルコース濃度)が200mg/dl以上を示す状態である。
また、上記報告によれば、耐糖能不全とは、空腹時血糖値(静脈血漿におけるグルコース濃度)が126mg/dl未満であり、かつ、75g経口ブドウ糖負荷試験2時間値(静脈血漿におけるグルコース濃度)が140mg/dl以上200mg/dl未満を示す状態である。さらに、ADAの報告によれば、空腹時血糖値(静脈血漿におけるグルコース濃度)が110mg/dl以上126mg/dl未満の状態をIFG(Impaired Fasting Glucose)と呼ぶ。一方、WHOの報告によれば、該IFG(Impaired Fasting Glucose)のうち、75g経口ブドウ糖負荷試験2時間値(静脈血漿におけるグルコース濃度)が140mg/dl未満である状態をIFG(Impaired Fasting Glycemia)と呼ぶ。
本発明化合物は、上記した判定基準により決定される糖尿病、境界型、耐糖能不全、IFG(Impaired Fasting Glucose)およびIFG(Impaired Fasting Glycemia)の予防・治療剤としても用いられる。さらに、本発明化合物は、境界型、耐糖能不全、IFG(Impaired Fasting Glucose)またはIFG(Impaired Fasting Glycemia)から糖尿病への進展を防止することもできる。
本発明化合物は、上記した各種疾患(例、心筋梗塞等の心血管イベント)の2次予防および進展抑制にも用いられる。
また、本発明化合物は、長時間に亘ってエイコサノイドの産生を持続的に抑制することにより、起炎性エイコサノイドとの関連が指摘されている炎症性疾患、例えば、喘息、アレルギー性気道過敏症、発熱、発痛、血栓症、脳梗塞、心筋梗塞、癌、自己免疫性脳脊髄炎、疼痛、腎不全、リウマチ、変形性関節症、掻痒、アトピー性皮膚炎、鼻炎、炎症性腸疾患、クローン病等の予防または治療に使用することができる。また、炎症反応に伴うさまざまな疾患の原因となる生体機能および生理作用の障害または異常を改善または亢進を抑制し、これらに起因する疾患または病態の一次および二次予防または進展を抑制できる。このような生体機能および生理作用の障害または異常としては、例えば、顔面潮紅および皮膚痛痒感(ニコチン酸誘導体製剤、プロスタサイクリン製剤等の投与に伴うものを含む)、過活動膀胱、脳循環・腎循環自動調節能の障害または異常、循環障害(例、末梢、脳、微小循環等)、脳血液関門の障害、食塩感受性、凝固・線溶系異常、血液・血球成分の性状異常(例、鎌形赤血球症、血小板凝集能亢進、赤血球変形能の異常、白血球粘着能の亢進、血液粘度の上昇等)、増殖因子およびサイトカイン(例、PDGF、VEGF、FGF、インターロイキン、TNF-α、MCP-1等)の産生および作用亢進、炎症系細胞の産生および浸潤亢進、フリーラジカルの産生亢進、脂肪沈着促進、内皮機能障害、内皮、細胞および臓器障害、浮腫、平滑筋等の細胞の形態変化(増殖型等への形態変化)、血管作動性物質および血栓誘発物質(例、カテコラミン、エンドセリン、トロンボキサンA2等)の産生および機能亢進、血管等の異常収縮、代謝異常(例、血清脂質異常、血糖異常等)、細胞等の異常増殖、血管新生(粥状動脈硬化巣外膜の異常毛細血管網形成における異常な脈管形成を含む)等が挙げられる。
また、本発明化合物は鎮痛作用を有することから、鎮痛薬として、疼痛の予防・治療薬として用いることもできる。疼痛疾患としては、例えば、炎症による急性痛、慢性炎症に伴う痛み、急性炎症に伴う痛み、術後痛(切開創の痛み、深部痛、内臓痛、術後慢性痛等)、筋肉痛(慢性痛疾患に伴う筋肉痛、肩こり等)、関節痛、歯痛、顎関節痛、頭痛(偏頭痛、緊張型頭痛、発熱に伴う頭痛、高血圧に伴う頭痛)、内臓痛(心臓痛、狭心痛、腹痛、腎臓の痛み、尿管の痛み、膀胱の痛み)、産婦人科領域の痛み(中間痛、月経困難、陣痛)、神経痛(椎間板ヘルニア、神経根痛、帯状疱疹後神経痛、三叉神経痛)、癌性疼痛、反射性交感神経性萎縮症、複雑局所痛症候群等が挙げられる。本発明化合物は、神経性疼痛、癌性疼痛、炎症性疼痛等の各種疼痛を直接的かつ即効的に鎮めるのに有効であり、痛覚閾値が低下している患者や病態(例、高血圧症等、およびこれらの合併症等)に対して、特に優れた鎮痛効果を示す。
医薬組成物中における本発明化合物の含有量は製剤全体に対して通常、約0.01~約99.9重量%、好ましくは約0.1~約50重量%である。
本発明化合物の投与量は、年令、体重、一般的健康状態、性別、食事、投与時間、投与方法、排泄速度、薬物の組み合わせ、患者のその時に治療を行なっている病状の程度に応じ、それらあるいはその他の要因を考慮して決められる。
投与量は対象疾患、症状、投与対象、投与方法等によって異なるが、例えば、本発明化合物を動脈硬化症治療剤として、成人に経口投与する場合、通常1回量として約0.01~100mg/kg体重、好ましくは0.05~30mg/kg体重、さらに好ましくは0.5~10mg/kg体重であり、この量を1日1回~3回に分けて投与するのが好ましい。
また、本発明化合物は安全性に優れていることから、長期に亘って投与することが可能である。
投与量は対象疾患、症状、投与対象、投与方法等によって異なるが、例えば、本発明化合物を動脈硬化症治療剤として、成人に経口投与する場合、通常1回量として約0.01~100mg/kg体重、好ましくは0.05~30mg/kg体重、さらに好ましくは0.5~10mg/kg体重であり、この量を1日1回~3回に分けて投与するのが好ましい。
また、本発明化合物は安全性に優れていることから、長期に亘って投与することが可能である。
本発明化合物は、例えば、抗動脈硬化剤、抗血栓剤、心不全治療剤、不整脈治療剤、降圧剤、糖尿病治療剤、糖尿病性合併症治療剤、HDL増加剤、抗高脂血症剤、抗肥満剤、利尿剤、抗炎症剤、抗痛風剤、化学療法剤、免疫療法剤、骨粗鬆症治療剤、抗痴呆剤、勃起不全改善剤、尿失禁治療剤・排尿困難治療剤等の薬剤(以下、併用薬剤と略記する)と組み合せて用いることができる。これらの併用薬剤は低分子化合物であっても良く、また高分子の蛋白、ポリペプチド、抗体あるいはワクチン等でも良い。
上記「抗動脈硬化剤」としては、例えば、Lp-PLA2阻害剤 (例、ダラプラディブ、リラプラディブ等)、FLAP阻害剤 (例、AM-103、AM-803、DG-031等)、sPLA2阻害剤(例、バレスプラディブ)、5-リポキシゲナーゼ阻害剤(例、VIA-2291等)、アシルコエンザイムAコレステロールアシル転移酵素(ACAT)阻害薬(例、メリナミド、アバシマイブ、エフルシマイブ等)、リピド・リッチ・プラーク退縮薬(例、WO02/06264、WO03/059900に記載の化合物等)、HDL製剤(例、CSL-111等)、CTEP阻害剤(例、トルセトラピブ、アナセトラピブ、ダルセトラピブ等)、MMP阻害剤、キマーゼ阻害剤、SPT阻害薬、ApoA-1およびその関連分子(例、ApoA-1ミラノ、D-4F、L-4F等)等が挙げられる。
上記「抗血栓剤」としては、例えば、血液凝固阻止薬〔例、ヘパリンナトリウム、ヘパリンカルシウム、ワルファリンカルシウム(ワーファリン)、抗トロンビン薬(例、アルガトロバン(argatroban)、ダビガトラン(dabigatran))、活性化血液凝固第Xa因子阻害薬(例、リバロキサバン(rivaroxaban)、アピキサバン(apixaban)、エドキサバン(edoxaban)、YM-150、WO02/06234、WO2004/048363、WO2005/030740、WO2005/058823、WO2005/113504またはWO2004/048363に記載の化合物等)等〕、血栓溶解薬〔例、tPA、ウロキナーゼ(urokinase)、チソキナーゼ(tisokinase)、アルテプラーゼ(alteplase)、ナテプラーゼ(nateplase)、モンテプラーゼ(monteplase)、パミテプラーゼ(pamiteplase)〕、抗血小板薬〔例、アスピリン、スルフィンピラゾン(アンツーラン)、ジピリダモール(ペルサンチン)、チクロピジン(パナルジン)、シロスタゾール(プレタール)、GPIIb/IIIa拮抗薬(例、レオプロ等)、クロピドグレル、プラスグレル(prasugrel)、ticagrelor 、E5555、SHC530348、イコサペント酸エチル、ベラプロストナトリウム(beraprost sodium)、塩酸サルポグレラート(sarpogrelate hydrochloride)等〕等が挙げられる。
上記「心不全治療剤」としては、例えば、強心薬(例、ジギトキシン、ジゴキシン、メチルジゴキシン、ラナトシドC、プロスシラリジン等)、α、β刺激薬(例、エピネフリン、ノルエピネフリン、イソプロテレノール、ドパミン、ドカルパミン、ドブタミン、デノパミン等)、ホスホジエステラーゼ阻害薬(例、アムリノン、ミルリノン、塩酸オルプリノン等)、カルシウムチャンネル感受性増強薬(例、ピモベンタン等)、硝酸薬(例、ニトログリセリン、硝酸イソソルビド等)、アンジオテンシン変換酵素阻害剤(例えば後述のアンジオテンシン変換酵素阻害剤等)、アンジオテンシンII拮抗剤(例えば後述のアンジオテンシンII拮抗剤等)、βブロッカー(例えば後述のβブロッカー等)、利尿薬(例えば後述の利尿薬等)、ANP、sGC活性化剤、ミオシン感受性増強剤、カルペリチド、ユビデカレノン、ベスナリノン、アミノフィリン等が挙げられる。
上記「不整脈治療剤」としては、例えば、ナトリウムチャンネル遮断薬(例、キニジン、プロカインアミド、ジソピラミド、アジマリン、シベンゾリン、リドカイン、ジフェニルヒダントイン、メキシレチン、プロパフェノン、フレカイニド、ピルジカイニド、フェニトイン等)、β遮断薬(例、プロプラノロール、アルプレノロール、ブフェトロール、オクスプレノロール、アテノロール、アセブトロール、メトプロロール、ビソプロロール、ピンドロール、カルテオロール、アロチロール等)、カリウムチャンネル遮断薬(例、アミオダロン等)、カルシウムチャンネル遮断薬(例、ベラパミル、ジルチアゼム等)等が挙げられる。
上記「降圧剤」としては、例えば、アンジオテンシン変換酵素阻害剤(例、カプトプリル、エナラプリル、デラプリル等)、アンジオテンシンII拮抗剤(例、カンデサルタン シレキセチル、カンデサルタン、ロサルタン、ロサルタン カリウム、エプロサルタン、バルサルタン、テルミサルタン、イルベサルタン、タソサルタン、オルメサルタン、オルメサルタン メドキソミル、アジルサルタン、アジルサルタン メドキソミル等)、カルシウム拮抗剤(例、マニジピン、ニフェジピン、アムロジピン、エホニジピン、ニカルジピン、シニルジピン等)、βブロッカー(例、メトプロロール、アテノロール、プロプラノロール、カルベジロール、ピンドロール等)、クロニジン等が挙げられる。
上記「糖尿病治療剤」としては、インスリン製剤(例、ウシ、ブタの膵臓から抽出された動物インスリン製剤;大腸菌、イーストを用い遺伝子工学的に合成したヒトインスリン製剤;インスリン亜鉛;プロタミンインスリン亜鉛;インスリンのフラグメントまたは誘導体(例、INS-1)、経口インスリン製剤)、インスリン抵抗性改善剤(例、ピオグリタゾンまたはその塩(好ましくは、塩酸塩)、ロシグリタゾンまたはその塩(好ましくは、マレイン酸塩)、メタグリダセン(Metaglidasen)、AMG-131、バラグリタゾン(Balaglitazone)、MBX-2044、リボグリタゾン(Rivoglitazone)、アレグリタザール(Aleglitazar)、チグリタザール(Chiglitazar)、ロベグリタゾン(Lobeglitazone)、PLX-204、PN-2034、GFT-505、THR-0921、WO2007/013694、WO2007/018314、WO2008/093639またはWO2008/099794記載の化合物)、α-グルコシダーゼ阻害剤(例、ボグリボース、アカルボース、ミグリトール、エミグリテート)、ビグアナイド剤(例、メトホルミン、ブホルミンまたはそれらの塩(例、塩酸塩、フマル酸塩、コハク酸塩))、インスリン分泌促進剤(例、スルホニルウレア剤(例、トルブタミド、グリベンクラミド、グリクラジド、クロルプロパミド、トラザミド、アセトヘキサミド、グリクロピラミド、グリメピリド、グリピザイド、グリブゾール)、レパグリニド、ナテグリニド、ミチグリニドまたはそのカルシウム塩水和物)、ジペプチジルペプチダーゼIV阻害剤(例、アログリプチン(Alogliptin)またはその塩(好ましくは、安息香酸塩)、ヴィルダグリプチン(Vildagliptin)、シタグリプチン(Sitagliptin)、サクサグリプチン(Saxagliptin)、BI1356、GRC8200、MP-513、PF-00734200、PHX1149、SK-0403、ALS2-0426、TA-6666、TS-021、KRP-104、2-[[6-[(3R)-3-アミノ-1-ピペリジル]-3,4-ジヒドロ-3-メチル-2,4-ジオキソ-1(2H)-ピリミジニル]メチル]-4-フルオロベンゾニトリルまたはその塩)、β3アゴニスト(例、N-5984)、GPR40アゴニスト(例、WO2004/041266、WO2004/106276、WO2005/063729、WO2005/063725、WO2005/087710、WO2005/095338、WO2007/013689またはWO2008/001931記載の化合物)、GLP-1受容体アゴニスト(例、GLP-1、GLP-1MR剤、リラグルチド(Liraglutide)、エキセナチド(Exenatide)、AVE-0010、BIM-51077、Aib(8,35)hGLP-1(7,37)NH2、CJC-1131、Albiglutide)、アミリンアゴニスト(例、プラムリンチド)、ホスホチロシンホスファターゼ阻害剤(例、バナジン酸ナトリウム)、糖新生阻害剤(例、グリコーゲンホスホリラーゼ阻害剤、グルコース-6-ホスファターゼ阻害剤、グルカゴン拮抗剤、FBPase阻害薬)、SGLT2(sodium-glucose cotransporter 2)阻害剤(例、Depagliflozin、AVE2268、TS-033、YM543、TA-7284、Remogliflozin、ASP1941)、SGLT1阻害薬、11β-ヒドロキシステロイドデヒドロゲナーゼ阻害薬(例、BVT-3498、INCB-13739)、アジポネクチンまたはその作動薬、IKK阻害薬(例、AS-2868)、レプチン抵抗性改善薬、ソマトスタチン受容体作動薬、グルコキナーゼ活性化薬(例、Piragliatin、AZD1656、AZD6370、TTP-355、WO2006/112549、WO2007/028135、WO2008/047821、WO2008/050821、WO2008/136428またはWO2008/156757記載の化合物)、GIP(Glucose-dependent insulinotropic peptide)、GPR119アゴニスト(例、PSN821)、FGF21、FGFアナログ等が挙げられる。
上記「糖尿病合併症治療剤」としては、アルドース還元酵素阻害剤(例、トルレスタット、エパルレスタット、ゾポルレスタット、フィダレスタット、CT-112、ラニレスタット(AS-3201)、リドレスタット)、神経栄養因子およびその増加薬(例、NGF、NT-3、BDNF、WO01/14372に記載のニューロトロフィン産生・分泌促進剤(例、4-(4-クロロフェニル)-2-(2-メチル-1-イミダゾリル)-5-[3-(2-メチルフェノキシ)プロピル]オキサゾール)、WO2004/039365記載の化合物)、PKC阻害剤(例、ルボキシスタウリン メシレート(ruboxistaurin mesylate))、AGE阻害剤(例、ALT946、N-フェナシルチアゾリウム ブロマイド(ALT766)、EXO-226、ピリドリン(Pyridorin)、ピリドキサミン)、GABA受容体作動薬(例、ギャバペンチン、プレギャバリン)、セロトニン・ノルアドレナリン再取込み阻害薬(例、デュロキセチン)、ナトリウムチャンネル阻害薬(例、ラコサミド)、活性酸素消去薬(例、チオクト酸)、脳血管拡張剤(例、チアプリド、メキシレチン)、ソマトスタチン受容体作動薬(例、BIM23190)、アポトーシスシグナルレギュレーティングキナーゼ-1(ASK-1)阻害薬等が挙げられる。
上記「HDL増加剤」としては、例えば、スクワレン合成酵素阻害薬、CETP阻害薬 (例、トルセトラピブ、アナセトラピブ、ダルセトラピブ等)、LPL活性化薬、ニコチン酸系薬剤(例、ニコモール(nicomol)、ニセリトロール(niceritrol))、内皮リパーゼ阻害薬等が挙げられる。
上記「抗高脂血症剤」としては、例えば、コレステロール合成阻害剤であるスタチン系化合物(例、セリバスタチン、プラバスタチン、シンバスタチン、ロバスタチン、ロスバスタチン、アトルバスタチン、フルバスタチン、ピタバスタチンまたはそれらの塩(例、ナトリウム塩等)等)、スクアレン合成酵素阻害剤あるいはトリグリセリド低下作用を有するフィブラート系化合物(例、ベザフィブラート、クロフィブラート、シンフィブラート、クリノフィブラート等)、コレステロール吸収阻害剤(例、ゼチア)、陰イオン交換樹脂(例、コレスチラミン)、プロブコール、ニコチン酸系薬剤(例、ニコモール(nicomol)、ニセリトロール(niceritrol))、植物ステロール(例、ソイステロール(soysterol)、ガンマオリザノール(γ-oryzanol))、魚油製剤(EPA、DHA、オマコール等)、PPARα作動薬、PPARγ作動薬、PPARδ作動薬、LXR作動薬、FXR拮抗薬、FXR作動薬、DGAT阻害薬、MGAT阻害薬、MTP阻害薬(例、lomitapide)、ApoBアンチセンス(例、mipomersen)およびPCSK9 siRNAアンチセンスオリゴヌクレオチドを含む核酸医薬等が挙げられる。
上記「抗肥満剤」としては、モノアミン取り込み阻害薬(例、フェンテルミン、シブトラミン、マジンドール、フロキセチン、テソフェンシン)、セロトニン2C受容体作動薬(例、ロルカセリン)、セロトニン6受容体拮抗薬、ヒスタミンH3受容体、GABA調節薬(例、トピラメイト)、ニューロペプチドY拮抗薬(例、ベルネペリット)、カンナビノイド受容体拮抗薬(例、リモナバン、タラナバン)、グレリン拮抗薬、グレリン受容体拮抗薬、グレリンアシル化酵素阻害薬、オピオイド受容体拮抗薬(例、GSK-1521498)、オレキシン受容体拮抗薬、メラノコルチン4受容体作動薬、11β-ヒドロキシステロイドデヒドロゲナーゼ阻害薬(例、AZD-4017)、膵リパーゼ阻害薬(例、オルリスタット、セティリスタット(cetilistat))、β3アゴニスト(例、N-5984)、ジアシルグリセロールアシルトランスフェラーゼ1(DGAT1)阻害薬、アセチルCoAカルボキシラーゼ(ACC)阻害薬、ステアリン酸CoA脱飽和酵素阻害、ミクロソームトリグリセリド転送蛋白阻害薬(例、R-256918)、Na-グルコース共輸送担体阻害薬(例、JNJ-28431754、レモグリフロジン)、NFκ阻害(例、HE-3286)、PPARアゴニスト(例、GFT-505、DRF-11605)、ホスホチロシンホスファターゼ阻害剤(例、バナジン酸ナトリウム、トロダスケミン(Trodusquemin))、GPR119作動薬(例、PSN-821)、グルコキナーゼ活性化薬(例、AZD-1656)、レプチン、レプチン誘導体(例、メトレレプチン)、CNTF(毛様体神経栄養因子)、BDNF(脳由来神経栄養因子)、コレシストキニンアゴニスト、グルカゴン様ペプチド-1(GLP-1)製剤(例、ウシ、ブタの膵臓から抽出された動物GLP-1製剤;大腸菌、イーストを用い遺伝子工学的に合成したヒトGLP-1製剤;GLP-1のフラグメントまたは誘導体(例、エクセナチド、リラグルチド))、アミリン製剤(例、プラムリンタイド、AC-2307)、ニューロペプチドYアゴニスト(例、PYY3-36、PYY3-36の誘導体、オビネプタイド、TM-30339、TM-30335)、オキシントモジュリン製剤:FGF21製剤(例、ウシ、ブタの膵臓から抽出された動物FGF21製剤;大腸菌、イーストを用い遺伝子工学的に合成したヒトFGF21製剤;FGF21のフラグメントまたは誘導体))、摂食抑制薬(例、P-57)等が挙げられる。
上記「利尿剤」としては、例えば、キサンチン誘導体(例、サリチル酸ナトリウムテオブロミン、サリチル酸カルシウムテオブロミン等)、チアジド系製剤(例、エチアジド、シクロペンチアジド、トリクロルメチアジド、ヒドロクロロチアジド、ヒドロフルメチアジド、ベンチルヒドロクロロチアジド、ペンフルチアジド、ポリ5チアジド、メチクロチアジド等)、抗アルドステロン製剤(例、スピロノラクトン、エプレレノン、トリアムテレン等)、炭酸脱水酵素阻害剤(例、アセタゾラミド等)、クロルベンゼンスルホンアミド系製剤(例、クロルタリドン、メフルシド、インダパミド等)、アゾセミド、イソソルビド、エタクリン酸、ピレタニド、ブメタニド、フロセミド等が挙げられる。
上記「抗炎症剤」としては、例えば、アセトアミノフェン、フェナセチン、エテンザミド、スルピリン、アンチピリン、ミグレニン、アスピリン、メフェナム酸、フルフェナム酸、ジクロフェナックナトリウム、ロキソプロフェンナトリウム、フェニルブタゾン、インドメタシン、イブプロフェン、ケトプロフェン、ナプロキセン、オキサプロジン、フルルビプロフェン、フェンブフェン、プラノプロフェン、フロクタフェニン、エピリゾール、塩酸チアラミド、ザルトプロフェン、メシル酸ガベキサート、メシル酸カモスタット、ウリナスタチン、コルヒチン、プロベネジド、スルフィンピラゾン、ベンズブロマロン、アロプリノール、金チオリンゴ酸ナトリウム、ヒアルロン酸ナトリウム、サリチル酸ナトリウム、塩酸モルヒネ、サリチル酸、アトロピン、スコポラミン、モルヒネ、ペチジン、レボルファイノール、ケトプロフェン、ナプロキセン、オキシモルフォンまたはその塩等の非ステロイド性抗炎症剤等が挙げられる。
上記「抗痛風剤」としては、例えば、フェブキソスタット(febuxostat)、アロプリノール、プロベネシド、コルヒチン、ベンズブロマロン、フェブキソスタット、クエン酸塩等が挙げられる。
上記「化学療法剤」としては、例えば、アルキル化剤(例、サイクロフォスファミド、イフォスファミド等)、代謝拮抗剤(例、メソトレキセート、5-フルオロウラシル等)、抗癌性抗生物質(例、マイトマイシン、アドリアマイシン等)、植物由来抗癌剤(例、ビンクリスチン、ビンデシン、タキソール等)、シスプラチン、カルボプラチン、エトポシド等が挙げられる。中でも、5-フルオロウラシル誘導体であるフルツロンあるいはネオフルツロン等が好ましい。
上記「免疫療法剤」としては、例えば、微生物または細菌成分(例、ムラミルジペプチド誘導体、ピシバニール等)、免疫増強活性のある多糖類(例、レンチナン、シゾフィラン、クレスチン等)、遺伝子工学的手法で得られるサイトカイン(例、インターフェロン、インターロイキン(IL)等)、コロニー刺激因子(例、顆粒球コロニー刺激因子、エリスロポエチン等)等が挙げられ、中でもIL-1、IL-2、IL-12等が好ましい。
上記「骨粗鬆症治療剤」としては、例えば、アルファカルシドール(alfacalcidol)、カルシトリオール(calcitriol)、エルカルトニン(elcaltonin)、サケカルシトニン(calcitonin salmon)、エストリオール(estriol)、イプリフラボン(ipriflavone)、パミドロン酸二ナトリウム(pamidronate disodium)、アレンドロン酸ナトリウム水和物(alendronate sodium hydrate)、インカドロン酸二ナトリウム(incadronate disodium) 等が挙げられる。
上記「抗痴呆剤」としては、例えば、タクリン(tacrine)、ドネペジル(donepezil)、リバスチグミン(rivastigmine)、ガランタミン(galantamine) 等が挙げられる。
上記「勃起不全改善剤」としては、例えば、アポモルフィン(apomorphine)、PDE5(ホスホジエステラーゼ5)阻害剤(例、クエン酸シルデナフィル(sildenafil citrate))等が挙げられる。
上記「尿失禁治療剤」としては、例えば、塩酸フラボキサート(flavoxate hydrochloride)、塩酸オキシブチニン(oxybutynin hydrochloride)、塩酸プロピベリン(propiverine hydrochloride)等が挙げられる。
上記「排尿困難治療剤」としては、例えばアセチルコリンエステラーゼ阻害薬(例、ジスチグミン)等が挙げられる。
上記「排尿困難治療剤」としては、例えばアセチルコリンエステラーゼ阻害薬(例、ジスチグミン)等が挙げられる。
さらに、併用薬剤としては、プロスタサイクリン製剤・誘導体(例、ベラプロスト、エポプロステノール、イロプロスト、トレプロスチニル等)、プロスタグランジン製剤・誘導体 (例、エンプロスチル、アルプロスタジル、リマプロスト、ミソプロストール、オルノプロスチル等)、抗喘息薬(例、サルメテロール、フルチカゾン、モンテルカスト)、関節リウマチ治療剤(例、エタネルセプト、インフリキシマブ、アダリムマブ)、神経再生促進薬(例、Y-128、VX-853、prosaptide)、抗うつ薬(例、デシプラミン、アミトリプチリン、イミプラミン)、抗てんかん薬(例、ラモトリジン)、抗不整脈薬(例、メキシレチン)、アセチルコリン受容体リガンド(例、ABT-594)、エンドセリン受容体拮抗薬(例、ボセンタン、ABT-627)、モノアミン取り込み阻害薬(例、トラマドル)、麻薬性鎮痛薬(例、モルヒネ)、GABA受容体作動薬(例、ギャバペンチン)、α2受容体作動薬(例、クロニジン)、局所鎮痛薬(例、カプサイシン)、抗不安薬(例、ベンゾジアゼピン)、ドーパミン作動薬(例、アポモルフィン)、ミダゾラム、ケトコナゾール等も挙げられる。
前記した併用薬剤の投与時期は限定されず、本発明化合物と併用薬剤とを、投与対象に対し、同時に投与してもよいし、時間差をおいて投与してもよい。併用薬剤の投与量は、臨床上用いられている投与量に準ずればよく、投与対象、投与ルート、疾患、組み合わせ等により適宜選択することができる。
また、これらの併用薬剤は、2種類以上を適宜の割合で組み合わせても良い。この場合、本発明化合物と併用薬剤の投与時期は限定されず、投与時に本発明化合物と併用薬剤とが組み合わされていればよい。
このような投与形態としては、例えば、(1)本発明化合物と併用薬剤とを同時に製剤化して得られる単一の製剤の投与、(2)本発明化合物と併用薬剤とを別々に製剤化して得られる2種の製剤の同一投与経路での同時投与、(3)本発明化合物と併用薬剤とを別々に製剤化して得られる2種の製剤の同一投与経路での時間差をおいての投与、(4)本発明化合物と併用薬剤とを別々に製剤化して得られる2種の製剤の異なる投与経路での同時投与、(5)本発明化合物と併用薬剤とを別々に製剤化して得られる2種の製剤の異なる投与経路での時間差をおいての投与(例えば、本発明化合物→併用薬剤の順序での投与、あるいは逆の順序での投与)等が挙げられる。併用薬剤の投与量は、臨床上用いられている用量を基準として適宜選択することができる。また、本発明化合物と併用薬剤の配合比は、投与対象、投与ルート、対象疾患、症状、組み合わせ等により適宜選択することができる。例えば、投与対象がヒトである場合、本発明化合物1重量部に対し、併用薬剤を0.01~100重量部用いればよい。
本発明化合物と併用薬剤とを組み合わせることにより、
(1)本発明化合物または併用薬剤を単独で投与する場合に比べて、その投与量を軽減することができる、
(2)本発明化合物と作用機序が異なる併用薬剤を選択することにより、治療期間を長く設定することができる、
(3)本発明化合物と作用機序が異なる併用薬剤を選択することにより、治療効果の持続を図ることができる、
(4)本発明化合物と併用薬剤とを併用することにより、相乗効果が得られる、等の優れた効果を得ることができる。
(1)本発明化合物または併用薬剤を単独で投与する場合に比べて、その投与量を軽減することができる、
(2)本発明化合物と作用機序が異なる併用薬剤を選択することにより、治療期間を長く設定することができる、
(3)本発明化合物と作用機序が異なる併用薬剤を選択することにより、治療効果の持続を図ることができる、
(4)本発明化合物と併用薬剤とを併用することにより、相乗効果が得られる、等の優れた効果を得ることができる。
本発明は、更に以下の実施例、試験例および製剤例によって詳しく説明されるが、これらは本発明を限定するものではなく、また本発明の範囲を逸脱しない範囲で変化させてもよい。
以下の実施例中の「室温」は通常約10℃ないし約35℃を示す。混合溶媒において示した比は、特に断らない限り容量比を示す。%は、特に断らない限り重量%を示す。
シリカゲルカラムクロマトグラフィーにおいて、NHと記載した場合は、アミノプロピルシラン結合シリカゲルを用いた。HPLC(高速液体クロマトグラフィー)において、C18と記載した場合は、オクタデシル結合シリカゲルを用いた。溶出溶媒の比は、特に断らない限り容量比を示す。
以下の実施例中の「室温」は通常約10℃ないし約35℃を示す。混合溶媒において示した比は、特に断らない限り容量比を示す。%は、特に断らない限り重量%を示す。
シリカゲルカラムクロマトグラフィーにおいて、NHと記載した場合は、アミノプロピルシラン結合シリカゲルを用いた。HPLC(高速液体クロマトグラフィー)において、C18と記載した場合は、オクタデシル結合シリカゲルを用いた。溶出溶媒の比は、特に断らない限り容量比を示す。
実施例で用いる略号は、次のような意義を有する。
s :シングレット
d :ダブレット
t :トリプレット
q :カルテット
dd :ダブルダブレット
ddd :ダブルダブルダブレット
dt :ダブルトリプレット
td :トリプルダブレット
tt :トリプルトリプレット
tq :トリプルカルテット
spt :セプテット
sxt :セクステット
br. s.:ブロード(幅広い)シングレット
m :マルチプレット
J :カップリング定数
Hz :ヘルツ
CHLOROFORM-d:重クロロホルム
DMSO-d6:重ジメチルスルホキシド
1H NMR:プロトン核磁気共鳴
THF:テトラヒドロフラン
DMF:N,N-ジメチルホルムアミド
DMA:N,N-ジメチルアセトアミド
NMP:N-メチル-2-ピロリドン
DMSO:ジメチルスルホキシド
s :シングレット
d :ダブレット
t :トリプレット
q :カルテット
dd :ダブルダブレット
ddd :ダブルダブルダブレット
dt :ダブルトリプレット
td :トリプルダブレット
tt :トリプルトリプレット
tq :トリプルカルテット
spt :セプテット
sxt :セクステット
br. s.:ブロード(幅広い)シングレット
m :マルチプレット
J :カップリング定数
Hz :ヘルツ
CHLOROFORM-d:重クロロホルム
DMSO-d6:重ジメチルスルホキシド
1H NMR:プロトン核磁気共鳴
THF:テトラヒドロフラン
DMF:N,N-ジメチルホルムアミド
DMA:N,N-ジメチルアセトアミド
NMP:N-メチル-2-ピロリドン
DMSO:ジメチルスルホキシド
1H NMR(プロトン核磁気共鳴スペクトル)はフーリエ変換型NMRで測定した。解析にはACD/SpecManager(商品名)などを用いた。水酸基やアミノ基などのプロトンが非常に緩やかなピークについては記載していない。
MS(マススペクトル)は、LC/MS(液体クロマトグラフ質量分析計)により測定した。
イオン化法としては、ESI(ElectroSpray Ionization、エレクトロスプレーイオン化)法、または、APCI(Atomospheric Pressure Cheimcal Ionization、大気圧化学イオン化)法を用いた。データは実測値(found)を記載した。塩の場合は、通常、フリー体の分子イオンピークもしくはフラグメントイオンピークが観測される。
MS(マススペクトル)は、LC/MS(液体クロマトグラフ質量分析計)により測定した。
イオン化法としては、ESI(ElectroSpray Ionization、エレクトロスプレーイオン化)法、または、APCI(Atomospheric Pressure Cheimcal Ionization、大気圧化学イオン化)法を用いた。データは実測値(found)を記載した。塩の場合は、通常、フリー体の分子イオンピークもしくはフラグメントイオンピークが観測される。
下記の実施例においてHPLC-マススペクトル(LC-MS)は以下の条件により測定した。
測定機器:ウォーターズ社 Micromass ZQ-Alliance HT
カラム:CAPCELL PAK C18UG120, S-3μm, 1.5 X 35 mm
溶媒:A液;0.05%トリフルオロ酢酸含有水、B液;0.04%トリフルオロ酢酸含有アセトニトリル
グラジエントサイクル:0.00分(A液/B液=90/10), 2.00分(A液/B液=5/95), 2.75分(A液/B液=5/95), 2.76分(A液/B液=90/10), 3.45分(A液/B液=90/10)
注入量:2μL、流速:0.5 mL/min、検出法:UV 220 nm
イオン化法:電子衝撃イオン化法 (Electron Spray Ionization: ESI)
測定機器:ウォーターズ社 Micromass ZQ-Alliance HT
カラム:CAPCELL PAK C18UG120, S-3μm, 1.5 X 35 mm
溶媒:A液;0.05%トリフルオロ酢酸含有水、B液;0.04%トリフルオロ酢酸含有アセトニトリル
グラジエントサイクル:0.00分(A液/B液=90/10), 2.00分(A液/B液=5/95), 2.75分(A液/B液=5/95), 2.76分(A液/B液=90/10), 3.45分(A液/B液=90/10)
注入量:2μL、流速:0.5 mL/min、検出法:UV 220 nm
イオン化法:電子衝撃イオン化法 (Electron Spray Ionization: ESI)
実施例1
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 1-ニトロ-4-(2,2,2-トリフルオロエトキシ)ベンゼン
1-フルオロ-4-ニトロベンゼン(10.6 g)および2,2,2-トリフルオロエタノール(12.0 g)をN,N-ジメチルホルムアミド(80 mL)に溶かし、そこへ炭酸カリウム(15.5 g)を加えて80℃で2時間攪拌した。放冷後、反応混合物に酢酸エチル(100 mL)を加えて、白色沈殿をろ別し、濾液を減圧濃縮して橙色の残渣を得た。得られた残渣を再び酢酸エチル(400 mL)に溶かし、水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して橙色の粗製固体を得た。得られた固体を10%ジエチルエーテル/ヘキサン混合溶媒で洗浄し、黄白色、針状結晶の標題化合物(15.8 g)を得た。
1H NMR (400 MHz, DMSO-d6) δ 4.99 (2H, q, J=8.8 Hz), 7.30 (2H, d, J=9.3 Hz), 8.26 (2H, d, J=9.3 Hz).
1-フルオロ-4-ニトロベンゼン(10.6 g)および2,2,2-トリフルオロエタノール(12.0 g)をN,N-ジメチルホルムアミド(80 mL)に溶かし、そこへ炭酸カリウム(15.5 g)を加えて80℃で2時間攪拌した。放冷後、反応混合物に酢酸エチル(100 mL)を加えて、白色沈殿をろ別し、濾液を減圧濃縮して橙色の残渣を得た。得られた残渣を再び酢酸エチル(400 mL)に溶かし、水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して橙色の粗製固体を得た。得られた固体を10%ジエチルエーテル/ヘキサン混合溶媒で洗浄し、黄白色、針状結晶の標題化合物(15.8 g)を得た。
1H NMR (400 MHz, DMSO-d6) δ 4.99 (2H, q, J=8.8 Hz), 7.30 (2H, d, J=9.3 Hz), 8.26 (2H, d, J=9.3 Hz).
B) 4-(2,2,2-トリフルオロエトキシ)アニリン
1-ニトロ-4-(2,2,2-トリフルオロエトキシ)ベンゼン(5.5 g)をメタノール(100 mL)に溶かし、そこへ10%パラジウム/活性炭素(50% 含水,2.5 g)を加え、水素雰囲気下室温で24時間攪拌した。その後パラジウム/活性炭をろ別し、濾液を減圧濃縮して濃橙色油状の残渣を得た。得られた残渣を酢酸エチル(200 mL)に溶かし、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して得られた濃橙色油状の残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して橙色油状物の標題化合物(4.5 g)を得た。
1H NMR (400 MHz, DMSO-d6) δ4.53 (2H, q, J=9.0 Hz), 4.76 (2H, s), 6.52 (2H, d, J=8.9 Hz), 6.75 (2H, d, J=8.9 Hz).
1-ニトロ-4-(2,2,2-トリフルオロエトキシ)ベンゼン(5.5 g)をメタノール(100 mL)に溶かし、そこへ10%パラジウム/活性炭素(50% 含水,2.5 g)を加え、水素雰囲気下室温で24時間攪拌した。その後パラジウム/活性炭をろ別し、濾液を減圧濃縮して濃橙色油状の残渣を得た。得られた残渣を酢酸エチル(200 mL)に溶かし、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して得られた濃橙色油状の残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して橙色油状物の標題化合物(4.5 g)を得た。
1H NMR (400 MHz, DMSO-d6) δ4.53 (2H, q, J=9.0 Hz), 4.76 (2H, s), 6.52 (2H, d, J=8.9 Hz), 6.75 (2H, d, J=8.9 Hz).
C) 1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン
4-(2,2,2-トリフルオロエトキシ)アニリン(10 g)をテトラヒドロフラン(100 mL)に溶かし、6M塩酸(9 mL)を加えた後、-5℃に冷却した。そこへチオホスゲン(4.01 mL)のテトラヒドロフラン(20mL)溶液を5分間かけて滴下した。-5℃で10分間撹拌後、飽和炭酸水素ナトリウム水溶液(125 mL)を注ぎ、酢酸エチル(200 mL)で2回抽出した。抽出層を合わせ、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた褐色の粗製物をシリカゲルカラムクロマトグラフィーで精製し、得られた薄黄色固体をヘキサンで洗浄して標題化合物(10.7 g)を白色結晶として得た。
1H NMR (300 MHz, DMSO-d6) δ4.81 (2H, q, J=8.8 Hz), 7.13 (2H, d, J=9.1 Hz), 7.45 (2H, d, J=9.1 Hz).
4-(2,2,2-トリフルオロエトキシ)アニリン(10 g)をテトラヒドロフラン(100 mL)に溶かし、6M塩酸(9 mL)を加えた後、-5℃に冷却した。そこへチオホスゲン(4.01 mL)のテトラヒドロフラン(20mL)溶液を5分間かけて滴下した。-5℃で10分間撹拌後、飽和炭酸水素ナトリウム水溶液(125 mL)を注ぎ、酢酸エチル(200 mL)で2回抽出した。抽出層を合わせ、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去して得られた褐色の粗製物をシリカゲルカラムクロマトグラフィーで精製し、得られた薄黄色固体をヘキサンで洗浄して標題化合物(10.7 g)を白色結晶として得た。
1H NMR (300 MHz, DMSO-d6) δ4.81 (2H, q, J=8.8 Hz), 7.13 (2H, d, J=9.1 Hz), 7.45 (2H, d, J=9.1 Hz).
D) 1-(4-メトキシベンジル)-1H-ピラゾール-4-カルボン酸エチル
1H-ピラゾール-4-カルボン酸エチル(10 g)、1-(クロロメチル)-4-メトキシベンゼン(12.3 g)、炭酸カリウム(14.8 g)およびTHF(200 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(10 g)を得た。
MS (ESI+): [M+H]+ 261.2.
1H-ピラゾール-4-カルボン酸エチル(10 g)、1-(クロロメチル)-4-メトキシベンゼン(12.3 g)、炭酸カリウム(14.8 g)およびTHF(200 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(10 g)を得た。
MS (ESI+): [M+H]+ 261.2.
E) 1-(4-メトキシベンジル)-1H-ピラゾール-4-カルボン酸
1-(4-メトキシベンジル)-1H-ピラゾール-4-カルボン酸エチル(10 g)、4規定水酸化ナトリウム(50 mL)、メタノール(50 mL)およびTHF(50 mL)の反応混合物を70℃で3時間撹拌した。反応混合物を減圧下濃縮し、得られた残渣に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をジイソプロピルエーテルを用いて固化させ、標題化合物(7.36 g)を得た。
MS (API-): [M-H]- 231.2.
1-(4-メトキシベンジル)-1H-ピラゾール-4-カルボン酸エチル(10 g)、4規定水酸化ナトリウム(50 mL)、メタノール(50 mL)およびTHF(50 mL)の反応混合物を70℃で3時間撹拌した。反応混合物を減圧下濃縮し、得られた残渣に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をジイソプロピルエーテルを用いて固化させ、標題化合物(7.36 g)を得た。
MS (API-): [M-H]- 231.2.
F) 3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-オキソプロパン酸エチル
1-(4-メトキシベンジル)-1H-ピラゾール-4-カルボン酸(7.36 g)のTHF溶液(50 mL)にカルボニルジイミダゾール(5.14 g)を0℃で加えた。反応混合物を室温で1時間撹拌後、塩化マグネシウム(3.02 g)、マロン酸モノエチルカリウム塩(8.09 g)およびTHF(50 mL)の混合物に加えた。反応混合物を50℃で終夜撹拌後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(8.1 g)を得た。
MS (ESI+): [M+H]+ 303.1.
1-(4-メトキシベンジル)-1H-ピラゾール-4-カルボン酸(7.36 g)のTHF溶液(50 mL)にカルボニルジイミダゾール(5.14 g)を0℃で加えた。反応混合物を室温で1時間撹拌後、塩化マグネシウム(3.02 g)、マロン酸モノエチルカリウム塩(8.09 g)およびTHF(50 mL)の混合物に加えた。反応混合物を50℃で終夜撹拌後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(8.1 g)を得た。
MS (ESI+): [M+H]+ 303.1.
G) (2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]プロパ-2-エン酸エチル
3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-オキソプロパン酸エチル(5 g)、ギ酸アンモニウム(5.21 g)およびエタノール(70 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(NH、酢酸エチル/ヘキサン)で精製して標題化合物(3.9 g)を得た。
MS (ESI+): [M+H]+ 302.3.
3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-オキソプロパン酸エチル(5 g)、ギ酸アンモニウム(5.21 g)およびエタノール(70 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(NH、酢酸エチル/ヘキサン)で精製して標題化合物(3.9 g)を得た。
MS (ESI+): [M+H]+ 302.3.
H) 6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]プロパ-2-エン酸エチル(3 g)および1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(2.79 g)のDMF溶液(50 mL)に、水素化ナトリウム(60% oil dispersion、876 mg)を0℃で加えた。反応混合物を0℃で2時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(2.2 g)を得た。
MS (ESI+): [M+H]+ 489.1.
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]プロパ-2-エン酸エチル(3 g)および1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(2.79 g)のDMF溶液(50 mL)に、水素化ナトリウム(60% oil dispersion、876 mg)を0℃で加えた。反応混合物を0℃で2時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(2.2 g)を得た。
MS (ESI+): [M+H]+ 489.1.
I) 2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(2.2 g)、ヨードエタン(0.558 mL)、炭酸カリウム(0.934 g)およびTHF(40 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(2.15 g)を得た。
MS (ESI+): [M+H]+ 517.3.
6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(2.2 g)、ヨードエタン(0.558 mL)、炭酸カリウム(0.934 g)およびTHF(40 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(2.15 g)を得た。
MS (ESI+): [M+H]+ 517.3.
J) 2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(2.15 g)のトリフルオロ酢酸溶液(15 mL)を70℃で終夜撹拌した。反応混合物にトルエンを加えた後、減圧下濃縮し、得られた残渣をジイソプロピルエーテルを用いて固化させ、標題化合物(1.65 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.28 (3H, t, J = 7.3 Hz), 3.13 (2H, q, J = 7.4 Hz), 4.86 (2H, q, J = 9.0 Hz), 6.58 (1H, s), 7.16-7.23 (2H, m), 7.29-7.36 (2H, m), 8.11 (1H, brs), 8.39 (1H, brs), 13.26 (1H, brs).
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(2.15 g)のトリフルオロ酢酸溶液(15 mL)を70℃で終夜撹拌した。反応混合物にトルエンを加えた後、減圧下濃縮し、得られた残渣をジイソプロピルエーテルを用いて固化させ、標題化合物(1.65 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.28 (3H, t, J = 7.3 Hz), 3.13 (2H, q, J = 7.4 Hz), 4.86 (2H, q, J = 9.0 Hz), 6.58 (1H, s), 7.16-7.23 (2H, m), 7.29-7.36 (2H, m), 8.11 (1H, brs), 8.39 (1H, brs), 13.26 (1H, brs).
実施例2
2-エトキシ-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(40 mg)およびエタノール(0.029 mL)のDMF溶液(1 mL)に水素化ナトリウム(60% oil dispersion、40.4 mg)を室温で加えた。反応混合物を室温で終夜撹拌した後、1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(6 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.18 (3H, t, J = 7.0 Hz), 4.43 (2H, q, J = 7.2 Hz), 4.83 (2H, q, J = 9.1 Hz), 6.48 (1H, s), 7.11-7.17 (2H, m), 7.24-7.29 (2H, m), 8.08 (1H, brs), 8.37 (1H, brs), 13.22 (1H, brs).
2-エトキシ-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(40 mg)およびエタノール(0.029 mL)のDMF溶液(1 mL)に水素化ナトリウム(60% oil dispersion、40.4 mg)を室温で加えた。反応混合物を室温で終夜撹拌した後、1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(6 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.18 (3H, t, J = 7.0 Hz), 4.43 (2H, q, J = 7.2 Hz), 4.83 (2H, q, J = 9.1 Hz), 6.48 (1H, s), 7.11-7.17 (2H, m), 7.24-7.29 (2H, m), 8.08 (1H, brs), 8.37 (1H, brs), 13.22 (1H, brs).
実施例3
2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(800 mg)のDMF溶液(10 mL)に水素化ナトリウム(60% oil dispersion、58.1 mg)を0℃で加えた。反応混合物を0℃で10分間撹拌した後、ヨードメタン(0.151 mL)を加え、さらに室温で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(630 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.28 (3H, t, J = 7.4 Hz), 3.12 (2H, q, J = 7.2 Hz), 3.91 (3H, s), 4.86 (2H, q, J = 9.1 Hz), 6.52 (1H, s), 7.15-7.22 (2H, m), 7.29-7.38 (2H, m), 8.06 (1H, s), 8.34 (1H, s).
2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(800 mg)のDMF溶液(10 mL)に水素化ナトリウム(60% oil dispersion、58.1 mg)を0℃で加えた。反応混合物を0℃で10分間撹拌した後、ヨードメタン(0.151 mL)を加え、さらに室温で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(630 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.28 (3H, t, J = 7.4 Hz), 3.12 (2H, q, J = 7.2 Hz), 3.91 (3H, s), 4.86 (2H, q, J = 9.1 Hz), 6.52 (1H, s), 7.15-7.22 (2H, m), 7.29-7.38 (2H, m), 8.06 (1H, s), 8.34 (1H, s).
実施例4
2-エトキシ-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-エトキシ-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-(1-メチル-1H-ピラゾール-4-イル)-3-オキソプロパン酸エチル
1-メチル-1H-ピラゾール-4-カルボン酸(1 g)および塩化チオニル(10 mL)の反応混合物を80℃で3時間撹拌した。反応混合物にトルエンを加え、減圧下濃縮し、塩化1-メチル-1H-ピラゾール-4-カルボニルを得た。リチウムジイソプロピルアミド(0.6 M THF溶液、26.5 mL)を-78℃に冷却し、酢酸エチル(0.77 mL)を加えた。同温度で30分間撹拌した後、上述の塩化1-メチル-1H-ピラゾール-4-カルボニルのTHF溶液(10 mL)を加えた。反応混合物を室温まで昇温した後、1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(780 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.18 (3H, t, J = 7.2 Hz), 3.88 (5H, s), 4.10 (2H, q, J = 7.2 Hz), 7.95 (1H, s), 8.41 (1H, s).
1-メチル-1H-ピラゾール-4-カルボン酸(1 g)および塩化チオニル(10 mL)の反応混合物を80℃で3時間撹拌した。反応混合物にトルエンを加え、減圧下濃縮し、塩化1-メチル-1H-ピラゾール-4-カルボニルを得た。リチウムジイソプロピルアミド(0.6 M THF溶液、26.5 mL)を-78℃に冷却し、酢酸エチル(0.77 mL)を加えた。同温度で30分間撹拌した後、上述の塩化1-メチル-1H-ピラゾール-4-カルボニルのTHF溶液(10 mL)を加えた。反応混合物を室温まで昇温した後、1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(780 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.18 (3H, t, J = 7.2 Hz), 3.88 (5H, s), 4.10 (2H, q, J = 7.2 Hz), 7.95 (1H, s), 8.41 (1H, s).
B) (2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-4-イル)プロパ-2-エン酸エチル
3-(1-メチル-1H-ピラゾール-4-イル)-3-オキソプロパン酸エチル(780 mg)、ギ酸アンモニウム(7.5 g)およびエタノール(20 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(770 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.17 (3H, t, J = 7.0 Hz), 3.84 (3H, s), 4.01 (2H, q, J = 6.9 Hz), 4.84 (1H, s), 7.86 (1H, s), 8.17 (1H, s).
3-(1-メチル-1H-ピラゾール-4-イル)-3-オキソプロパン酸エチル(780 mg)、ギ酸アンモニウム(7.5 g)およびエタノール(20 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(770 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.17 (3H, t, J = 7.0 Hz), 3.84 (3H, s), 4.01 (2H, q, J = 6.9 Hz), 4.84 (1H, s), 7.86 (1H, s), 8.17 (1H, s).
C) 6-(1-メチル-1H-ピラゾール-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン
(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-4-イル)プロパ-2-エン酸エチル(770 mg)、1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(960 mg)およびジメチルアミノピリジン(48 mg)のDMF溶液(10 mL)に、水素化ナトリウム(60% oil dispersion、333 mg)を室温で加えた。反応混合物を室温で3時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(380 mg)を得た。
MS (ESI+): [M+H]+ 411.1.
(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-4-イル)プロパ-2-エン酸エチル(770 mg)、1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(960 mg)およびジメチルアミノピリジン(48 mg)のDMF溶液(10 mL)に、水素化ナトリウム(60% oil dispersion、333 mg)を室温で加えた。反応混合物を室温で3時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(380 mg)を得た。
MS (ESI+): [M+H]+ 411.1.
D) 2-エトキシ-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1-メチル-1H-ピラゾール-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(180 mg)、ヨードエタン(0.058 mL)、炭酸カリウム(0.13 g)およびTHF(5 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をエタノール(3 mL)に溶解し、水素化ナトリウム(60% oil dispersion、94 mg)を室温で加えた。反応混合物を70℃で16時間撹拌した後、1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をHPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物(29 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.18 (3H, t, J = 7.2 Hz), 3.89 (3H, s), 4.42 (2H, q, J = 7.1 Hz), 4.83 (2H, q, J = 8.7 Hz), 6.42 (1H, s), 7.10-7.17 (2H, m), 7.23-7.30 (2H, m), 8.03 (1H, s), 8.32 (1H, s).
6-(1-メチル-1H-ピラゾール-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(180 mg)、ヨードエタン(0.058 mL)、炭酸カリウム(0.13 g)およびTHF(5 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をエタノール(3 mL)に溶解し、水素化ナトリウム(60% oil dispersion、94 mg)を室温で加えた。反応混合物を70℃で16時間撹拌した後、1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をHPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物(29 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.18 (3H, t, J = 7.2 Hz), 3.89 (3H, s), 4.42 (2H, q, J = 7.1 Hz), 4.83 (2H, q, J = 8.7 Hz), 6.42 (1H, s), 7.10-7.17 (2H, m), 7.23-7.30 (2H, m), 8.03 (1H, s), 8.32 (1H, s).
実施例5
2-(エチルスルファニル)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) (2E)-3-アミノ-3-(モルホリン-4-イル)プロパ-2-エン酸エチル
3-エトキシ-3-イミノプロパン酸エチル塩酸塩(5 g)、モルホリン(11 g)およびエタノール(70 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(4.8 g)を得た。
MS (ESI+): [M+H]+ 201.4.
3-エトキシ-3-イミノプロパン酸エチル塩酸塩(5 g)、モルホリン(11 g)およびエタノール(70 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(4.8 g)を得た。
MS (ESI+): [M+H]+ 201.4.
B) 6-(モルホリン-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン
(2E)-3-アミノ-3-(モルホリン-4-イル)プロパ-2-エン酸エチル(4 g)および1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(5.12 g)のDMF溶液(60 mL)に、水素化ナトリウム(60% oil dispersion、1.76 g)を0℃で加えた。反応混合物を0℃で2時間撹拌後、反応混合物を1規定塩酸に加えた。1規定水酸化ナトリウム水溶液でpH7に調整した後、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(NH、酢酸/酢酸エチル)で精製して標題化合物(1.85 g)を得た。
MS (ESI+): [M+H]+ 388.3.
(2E)-3-アミノ-3-(モルホリン-4-イル)プロパ-2-エン酸エチル(4 g)および1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(5.12 g)のDMF溶液(60 mL)に、水素化ナトリウム(60% oil dispersion、1.76 g)を0℃で加えた。反応混合物を0℃で2時間撹拌後、反応混合物を1規定塩酸に加えた。1規定水酸化ナトリウム水溶液でpH7に調整した後、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(NH、酢酸/酢酸エチル)で精製して標題化合物(1.85 g)を得た。
MS (ESI+): [M+H]+ 388.3.
C) 2-(エチルスルファニル)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(モルホリン-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(1 g)、ヨードエタン(0.32 mL)、炭酸カリウム(0.714 g)およびTHF(20 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣を酢酸エチル/ヘキサンから再結晶して標題化合物(710 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.22 (3H, t, J = 7.2 Hz), 2.98 (2H, q, J = 7.3 Hz), 3.48-3.56 (4H, m), 3.63-3.71 (4H, m), 4.84 (2H, q, J = 9.1 Hz), 5.28 (1H, s), 7.11-7.27 (4H, m).
6-(モルホリン-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(1 g)、ヨードエタン(0.32 mL)、炭酸カリウム(0.714 g)およびTHF(20 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣を酢酸エチル/ヘキサンから再結晶して標題化合物(710 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.22 (3H, t, J = 7.2 Hz), 2.98 (2H, q, J = 7.3 Hz), 3.48-3.56 (4H, m), 3.63-3.71 (4H, m), 4.84 (2H, q, J = 9.1 Hz), 5.28 (1H, s), 7.11-7.27 (4H, m).
実施例6
2-エトキシ-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(100 mg)およびエタノール(2 mL)の反応混合物に、水素化ナトリウム(60% oil dispersion、28 mg)を加えた。反応混合物を70℃で終夜撹拌した後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去し、標題化合物(52 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.14 (3H, t, J = 7.2 Hz), 3.43-3.55 (4H, m), 3.62-3.72 (4H, m), 4.29 (2H, q, J = 7.1 Hz), 4.81 (2H, q, J = 9.1 Hz), 5.19 (1H, s), 7.02-7.25 (4H, m).
2-エトキシ-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(100 mg)およびエタノール(2 mL)の反応混合物に、水素化ナトリウム(60% oil dispersion、28 mg)を加えた。反応混合物を70℃で終夜撹拌した後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去し、標題化合物(52 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.14 (3H, t, J = 7.2 Hz), 3.43-3.55 (4H, m), 3.62-3.72 (4H, m), 4.29 (2H, q, J = 7.1 Hz), 4.81 (2H, q, J = 9.1 Hz), 5.19 (1H, s), 7.02-7.25 (4H, m).
実施例7
2-(エチルスルファニル)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-オキソ-3-(ピリジン-4-イル)プロパン酸エチル
イソニコチン酸エチル(3.02 g)、水素化ナトリウム(60% oil dispersion、0.53g)および酢酸エチル(10 mL)の反応混合物を70℃で3時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(1.8 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.17 (3H, t, J = 7.0 Hz), 4.12 (2H, q, J = 7.2 Hz), 4.27 (2H, s), 7.78-7.85 (2H, m), 8.82-8.87 (2H, m).
イソニコチン酸エチル(3.02 g)、水素化ナトリウム(60% oil dispersion、0.53g)および酢酸エチル(10 mL)の反応混合物を70℃で3時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(1.8 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.17 (3H, t, J = 7.0 Hz), 4.12 (2H, q, J = 7.2 Hz), 4.27 (2H, s), 7.78-7.85 (2H, m), 8.82-8.87 (2H, m).
B) (2Z)-3-アミノ-3-(ピリジン-4-イル)プロパ-2-エン酸エチル
実施例1のG)に準じた方法により、3-オキソ-3-(ピリジン-4-イル)プロパン酸エチルから標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.20 (3H, t, J = 7.2 Hz), 4.07 (2H, q, J = 6.9 Hz), 4.90 (1H, s), 7.56-7.61 (2H, m), 8.64-8.68 (2H, m).
実施例1のG)に準じた方法により、3-オキソ-3-(ピリジン-4-イル)プロパン酸エチルから標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.20 (3H, t, J = 7.2 Hz), 4.07 (2H, q, J = 6.9 Hz), 4.90 (1H, s), 7.56-7.61 (2H, m), 8.64-8.68 (2H, m).
C) 6-(ピリジン-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-(ピリジン-4-イル)プロパ-2-エン酸エチルから標題化合物を得た。
MS (ESI+): [M+H]+ 379.9.
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-(ピリジン-4-イル)プロパ-2-エン酸エチルから標題化合物を得た。
MS (ESI+): [M+H]+ 379.9.
D) 2-(エチルスルファニル)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により、6-(ピリジン-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンから標題化合物を得た。
実施例1のI)に準じた方法により、6-(ピリジン-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンから標題化合物を得た。
実施例8
2-(エチルスルファニル)-6-(ピリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例7に準じた方法により、ニコチン酸エチルから標題化合物を得た。
2-(エチルスルファニル)-6-(ピリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例7に準じた方法により、ニコチン酸エチルから標題化合物を得た。
実施例9
2-(エチルスルファニル)-6-(2-メチルピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(2-メチルピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 2-メチルピリジン-4-カルボン酸エチル
2-メチルイソニコチン酸(680 mg)、硫酸(2.5 mL)およびエタノール(50 mL)の反応混合物を70℃で16時間撹拌した。反応混合物に飽和重曹水を加えた後、減圧下濃縮し、得られた残渣を酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥、溶媒を減圧下留去し、標題化合物(1.8 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.33 (3H, t, J = 7.0 Hz), 2.56 (3H, s), 4.35 (2H, q, J = 7.1 Hz), 7.63 (1H, d, J = 5.3 Hz), 7.71 (1H, s), 8.65 (1H, d, J = 5.3 Hz).
2-メチルイソニコチン酸(680 mg)、硫酸(2.5 mL)およびエタノール(50 mL)の反応混合物を70℃で16時間撹拌した。反応混合物に飽和重曹水を加えた後、減圧下濃縮し、得られた残渣を酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥、溶媒を減圧下留去し、標題化合物(1.8 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.33 (3H, t, J = 7.0 Hz), 2.56 (3H, s), 4.35 (2H, q, J = 7.1 Hz), 7.63 (1H, d, J = 5.3 Hz), 7.71 (1H, s), 8.65 (1H, d, J = 5.3 Hz).
B) 2-(エチルスルファニル)-6-(2-メチルピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例7に準じた方法により、2-メチルピリジン-4-カルボン酸エチルから標題化合物を得た。
MS (ESI+): [M+H]+ 422.0.
実施例7に準じた方法により、2-メチルピリジン-4-カルボン酸エチルから標題化合物を得た。
MS (ESI+): [M+H]+ 422.0.
実施例10
2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 1-メチル-1H-ピラゾール-5-カルボン酸エチル
1-メチル-1H-ピラゾール-5-カルボン酸(1.50 g)、硫酸(1.0 mL)およびエタノール(20 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に飽和重曹水を加えた後、減圧下濃縮し、得られた残渣を酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥、溶媒を減圧下留去し、標題化合物(1.42 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.31 (3H, t, J = 7.0 Hz), 4.08 (3H, s), 4.30 (2H, q, J = 7.1 Hz), 6.86 (1H, d, J = 1.9 Hz), 7.53 (1H, d, J = 2.3 Hz).
1-メチル-1H-ピラゾール-5-カルボン酸(1.50 g)、硫酸(1.0 mL)およびエタノール(20 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に飽和重曹水を加えた後、減圧下濃縮し、得られた残渣を酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥、溶媒を減圧下留去し、標題化合物(1.42 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.31 (3H, t, J = 7.0 Hz), 4.08 (3H, s), 4.30 (2H, q, J = 7.1 Hz), 6.86 (1H, d, J = 1.9 Hz), 7.53 (1H, d, J = 2.3 Hz).
B) 3-(1-メチル-1H-ピラゾール-5-イル)-3-オキソプロパン酸エチル
1-メチル-1H-ピラゾール-5-カルボン酸エチル(1.40 g)、水素化ナトリウム(60% oil dispersion、363 mg)および酢酸エチル(10 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(662 mg)を得た。
MS (ESI+): [M+H]+ 197.0.
1-メチル-1H-ピラゾール-5-カルボン酸エチル(1.40 g)、水素化ナトリウム(60% oil dispersion、363 mg)および酢酸エチル(10 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(662 mg)を得た。
MS (ESI+): [M+H]+ 197.0.
C) (2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)プロパ-2-エン酸エチル
3-(1-メチル-1H-ピラゾール-5-イル)-3-オキソプロパン酸エチル(662 mg)、ギ酸アンモニウム(2.13 g)およびエタノール(15 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(317 mg)を得た。
MS (ESI+): [M+H]+ 196.3.
3-(1-メチル-1H-ピラゾール-5-イル)-3-オキソプロパン酸エチル(662 mg)、ギ酸アンモニウム(2.13 g)およびエタノール(15 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(317 mg)を得た。
MS (ESI+): [M+H]+ 196.3.
D) 6-(1-メチル-1H-ピラゾール-5-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン
1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(383 mg)のDMF溶液(3 mL)に、水素化ナトリウム(60% oil dispersion、134 mg)を0℃で加え、5分間撹拌した後、(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)プロパ-2-エン酸エチル(310 mg)を加えた。反応混合物を0℃で1.5時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(202 mg)を得た。
MS (ESI+): [M+H]+ 383.2.
1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(383 mg)のDMF溶液(3 mL)に、水素化ナトリウム(60% oil dispersion、134 mg)を0℃で加え、5分間撹拌した後、(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)プロパ-2-エン酸エチル(310 mg)を加えた。反応混合物を0℃で1.5時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(202 mg)を得た。
MS (ESI+): [M+H]+ 383.2.
E) 2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1-メチル-1H-ピラゾール-5-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(202 mg)、ヨードエタン(85μL)、炭酸カリウム(146 mg)およびTHF(5 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(116 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.25 (3H, t, J = 7.2 Hz), 3.11 (2H, q, J = 7.6 Hz), 4.18 (3H, s), 4.87 (2H, q, J = 8.7 Hz), 6.70 (1H, s), 6.93 (1H, d, J = 1.9Hz), 7.18-7.26 (2H, m), 7.34-7.43 (2H, m), 7.52 (1H, d, J = 2.3 Hz).
6-(1-メチル-1H-ピラゾール-5-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(202 mg)、ヨードエタン(85μL)、炭酸カリウム(146 mg)およびTHF(5 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(116 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.25 (3H, t, J = 7.2 Hz), 3.11 (2H, q, J = 7.6 Hz), 4.18 (3H, s), 4.87 (2H, q, J = 8.7 Hz), 6.70 (1H, s), 6.93 (1H, d, J = 1.9Hz), 7.18-7.26 (2H, m), 7.34-7.43 (2H, m), 7.52 (1H, d, J = 2.3 Hz).
実施例11
2-エトキシ-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(40 mg)およびエタノール(0.028 mL)のTHF溶液(2 mL)に水素化ナトリウム(60% oil dispersion、40.4 mg)を室温で加えた。反応混合物を室温で4時間撹拌した後、1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をジイソプロピルエーテルを用いて固化させ、標題化合物(27 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.16-1.26 (3H, m), 4.19 (3H, s), 4.42 (2H, q, J = 6.9 Hz), 4.84 (2H, q, J = 8.7 Hz), 6.60 (1H, s), 6.92 (1H, d, J = 1.9 Hz), 7.12-7.23 (2H, m), 7.27-7.39 (2H, m), 7.50 (1H, d, J = 1.9 Hz).
2-エトキシ-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(40 mg)およびエタノール(0.028 mL)のTHF溶液(2 mL)に水素化ナトリウム(60% oil dispersion、40.4 mg)を室温で加えた。反応混合物を室温で4時間撹拌した後、1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をジイソプロピルエーテルを用いて固化させ、標題化合物(27 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.16-1.26 (3H, m), 4.19 (3H, s), 4.42 (2H, q, J = 6.9 Hz), 4.84 (2H, q, J = 8.7 Hz), 6.60 (1H, s), 6.92 (1H, d, J = 1.9 Hz), 7.12-7.23 (2H, m), 7.27-7.39 (2H, m), 7.50 (1H, d, J = 1.9 Hz).
実施例12
2-(エチルスルファニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 1-(4-メトキシベンジル)-1H-ピラゾール-3-カルボン酸メチル
1H-ピラゾール-3-カルボン酸メチル(2.75 g)、1-(クロロメチル)-4-メトキシベンゼン(5.12 g)、炭酸カリウム(9.04 g)およびTHF(73 mL)の反応混合物を70℃で終夜撹拌した。反応混合物を濾過し、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(5.37 g)を得た。
MS (ESI+): [M+H]+ 247.3.
1H-ピラゾール-3-カルボン酸メチル(2.75 g)、1-(クロロメチル)-4-メトキシベンゼン(5.12 g)、炭酸カリウム(9.04 g)およびTHF(73 mL)の反応混合物を70℃で終夜撹拌した。反応混合物を濾過し、減圧下濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(5.37 g)を得た。
MS (ESI+): [M+H]+ 247.3.
B) 1-(4-メトキシベンジル)-1H-ピラゾール-3-カルボン酸
1-(4-メトキシベンジル)-1H-ピラゾール-3-カルボン酸メチル(5.37 g)、8規定水酸化ナトリウム(13.6 mL)、メタノール(10 mL)およびTHF(10 mL)の反応混合物を60℃で3時間撹拌した。反応混合物を減圧下濃縮し、得られた残渣に1規定塩酸を加え、沈殿物を濾取した。水で洗浄し、減圧下乾燥させ、標題化合物(3.95 g)を得た。
MS (ESI+): [M+H]+ 233.0.
1-(4-メトキシベンジル)-1H-ピラゾール-3-カルボン酸メチル(5.37 g)、8規定水酸化ナトリウム(13.6 mL)、メタノール(10 mL)およびTHF(10 mL)の反応混合物を60℃で3時間撹拌した。反応混合物を減圧下濃縮し、得られた残渣に1規定塩酸を加え、沈殿物を濾取した。水で洗浄し、減圧下乾燥させ、標題化合物(3.95 g)を得た。
MS (ESI+): [M+H]+ 233.0.
C) 3-[1-(4-メトキシベンジル)-1H-ピラゾール-3-イル]-3-オキソプロパン酸エチル
1-(4-メトキシベンジル)-1H-ピラゾール-3-カルボン酸(3.15 g)のTHF溶液(40 mL)にカルボニルジイミダゾール(2.00 g)を0℃で加えた。反応混合物を室温で1時間撹拌後、塩化マグネシウム(1.30 g)、マロン酸モノエチルカリウム塩(3.46 g)およびTHF(40 mL)の混合物に加えた。反応混合物を50℃で終夜撹拌後、反応混合物を水に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(1.95 g)を得た。
MS (ESI+): found: 322.1.
1-(4-メトキシベンジル)-1H-ピラゾール-3-カルボン酸(3.15 g)のTHF溶液(40 mL)にカルボニルジイミダゾール(2.00 g)を0℃で加えた。反応混合物を室温で1時間撹拌後、塩化マグネシウム(1.30 g)、マロン酸モノエチルカリウム塩(3.46 g)およびTHF(40 mL)の混合物に加えた。反応混合物を50℃で終夜撹拌後、反応混合物を水に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(1.95 g)を得た。
MS (ESI+): found: 322.1.
D) (2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-3-イル]プロパ-2-エン酸エチル
3-[1-(4-メトキシベンジル)-1H-ピラゾール-3-イル]-3-オキソプロパン酸エチル(1.95 g)、ギ酸アンモニウム(2.03 g)およびエタノール(20 mL)の反応混合物を70℃で終夜撹拌した。反応混合物を濃縮し、得られた残渣に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して標題化合物(1.9 g)を得た。
MS (ESI+):found: 433.0.
3-[1-(4-メトキシベンジル)-1H-ピラゾール-3-イル]-3-オキソプロパン酸エチル(1.95 g)、ギ酸アンモニウム(2.03 g)およびエタノール(20 mL)の反応混合物を70℃で終夜撹拌した。反応混合物を濃縮し、得られた残渣に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して標題化合物(1.9 g)を得た。
MS (ESI+):found: 433.0.
E) 2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-3-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-3-イル]プロパ-2-エン酸エチル(1.4 g)および1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(1.08g)のDMF溶液(15 mL)に、水素化ナトリウム(60% oil dispersion、372 mg)を0℃で加えた。反応混合物を室温で2時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して6-[1-(4-メトキシベンジル)-1H-ピラゾール-3-イル]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(1.63 g)を得た。得られた6-[1-(4-メトキシベンジル)-1H-ピラゾール-3-イル]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(1.63 g)、ヨードエタン(0.517 mL)、炭酸カリウム(0.922 g)およびTHF(30 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(538 mg)を得た。
MS (ESI+): [M+H]+ 517.0.
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-3-イル]プロパ-2-エン酸エチル(1.4 g)および1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(1.08g)のDMF溶液(15 mL)に、水素化ナトリウム(60% oil dispersion、372 mg)を0℃で加えた。反応混合物を室温で2時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して6-[1-(4-メトキシベンジル)-1H-ピラゾール-3-イル]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(1.63 g)を得た。得られた6-[1-(4-メトキシベンジル)-1H-ピラゾール-3-イル]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(1.63 g)、ヨードエタン(0.517 mL)、炭酸カリウム(0.922 g)およびTHF(30 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(538 mg)を得た。
MS (ESI+): [M+H]+ 517.0.
F) 2-(エチルスルファニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-3-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(336 mg)のトリフルオロ酢酸溶液(3 mL)を70℃で終夜撹拌した。反応混合物にトルエンを加えた後、減圧下濃縮し、得られた残渣をジイソプロピルエーテルを用いて固化させ、得られた粗結晶をTHF-ヘキサンから再結晶し、標題化合物(50.9 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.19-1.36 (3H, m), 3.05-3.28 (2H, m), 4.87 (2H, q, J = 8.9 Hz), 6.60-6.78 (1H, m), 6.81-7.01 (1H, m), 7.20 (2H, d, J = 9.1 Hz), 7.36 (2H, d, J = 8.7 Hz), 7.59-7.93 (1H, m), 13.20-13.63 (1H, m).
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-3-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(336 mg)のトリフルオロ酢酸溶液(3 mL)を70℃で終夜撹拌した。反応混合物にトルエンを加えた後、減圧下濃縮し、得られた残渣をジイソプロピルエーテルを用いて固化させ、得られた粗結晶をTHF-ヘキサンから再結晶し、標題化合物(50.9 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.19-1.36 (3H, m), 3.05-3.28 (2H, m), 4.87 (2H, q, J = 8.9 Hz), 6.60-6.78 (1H, m), 6.81-7.01 (1H, m), 7.20 (2H, d, J = 9.1 Hz), 7.36 (2H, d, J = 8.7 Hz), 7.59-7.93 (1H, m), 13.20-13.63 (1H, m).
実施例13
2-(エチルスルファニル)-6-(2-メトキシピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(2-メトキシピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) (2Z)-3-アミノ-3-(2-メトキシピリジン-4-イル)プロパ-2-エン酸エチル
実施例1のE)からG)に準じた方法により、2-メトキシピリジン-4-カルボン酸から標題化合物を得た。
MS (ESI+): [M+H]+ 223.3.
実施例1のE)からG)に準じた方法により、2-メトキシピリジン-4-カルボン酸から標題化合物を得た。
MS (ESI+): [M+H]+ 223.3.
B) 6-(2-メトキシピリジン-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-(2-メトキシピリジン-4-イル)プロパ-2-エン酸エチルから標題化合物を得た。
MS (ESI+): [M+H]+ 410.2.
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-(2-メトキシピリジン-4-イル)プロパ-2-エン酸エチルから標題化合物を得た。
MS (ESI+): [M+H]+ 410.2.
C) 2-(エチルスルファニル)-6-(2-メトキシピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により、6-(2-メトキシピリジン-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンから標題化合物を得た。
実施例1のI)に準じた方法により、6-(2-メトキシピリジン-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンから標題化合物を得た。
実施例14
2-(メチルスルファニル)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により、6-(ピリジン-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンおよびヨウ化メチルから標題化合物を得た。
2-(メチルスルファニル)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により、6-(ピリジン-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンおよびヨウ化メチルから標題化合物を得た。
実施例15
2-(エチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(2-メトキシピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(100 mg)、ピリジン塩酸塩(264 mg)およびDMF(5 mL)の反応混合物を100℃で3時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥、溶媒を減圧下留去し、標題化合物(60 mg)を得た。
2-(エチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(2-メトキシピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(100 mg)、ピリジン塩酸塩(264 mg)およびDMF(5 mL)の反応混合物を100℃で3時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加えた後、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥、溶媒を減圧下留去し、標題化合物(60 mg)を得た。
実施例16
2-(エチルスルファニル)-6-(フラン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(フラン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) (2Z)-3-アミノ-3-(フラン-3-イル)プロパ-2-エン酸エチル
実施例1のG)に準じた方法により、3-(フラン-3-イル)-3-オキソプロパン酸エチルから標題化合物を得た。
MS (ESI+): [M+H]+ 201.1.
実施例1のG)に準じた方法により、3-(フラン-3-イル)-3-オキソプロパン酸エチルから標題化合物を得た。
MS (ESI+): [M+H]+ 201.1.
B) 6-(フラン-3-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン
実施例4のC)に準じた方法により、(2Z)-3-アミノ-3-(フラン-3-イル)プロパ-2-エン酸エチルから標題化合物を得た。
MS (ESI+): [M+H]+ 369.2.
実施例4のC)に準じた方法により、(2Z)-3-アミノ-3-(フラン-3-イル)プロパ-2-エン酸エチルから標題化合物を得た。
MS (ESI+): [M+H]+ 369.2.
C) 2-(エチルスルファニル)-6-(フラン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により、6-(フラン-3-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンから標題化合物を得た。
実施例1のI)に準じた方法により、6-(フラン-3-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンから標題化合物を得た。
実施例17
2-エトキシ-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-エトキシ-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 2-(メチルスルフィニル)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(メチルスルファニル)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(360 mg)の酢酸溶液(4 mL)に、オキソン(登録商標)(842 mg)の水溶液(1 mL)を室温で加えた。反応混合物を室温で3時間撹拌後、飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥、溶媒を減圧下留去し、標題化合物(280 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 2.79 (3H, s), 4.88 (2H, q, J = 9.0 Hz), 7.21-7.29 (2H, m), 7.46 (1H, s), 8.16 (2H, d, J = 6.1 Hz), 8.77-8.82 (2H, m).
2-(メチルスルファニル)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(360 mg)の酢酸溶液(4 mL)に、オキソン(登録商標)(842 mg)の水溶液(1 mL)を室温で加えた。反応混合物を室温で3時間撹拌後、飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥、溶媒を減圧下留去し、標題化合物(280 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 2.79 (3H, s), 4.88 (2H, q, J = 9.0 Hz), 7.21-7.29 (2H, m), 7.46 (1H, s), 8.16 (2H, d, J = 6.1 Hz), 8.77-8.82 (2H, m).
B) 2-エトキシ-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(メチルスルフィニル)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(70 mg)、ナトリウムエトキシド(20%エタノール溶液、0.23 mL)およびエタノール(3 mL)の反応混合物を室温で3時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(30 mg)を得た。
2-(メチルスルフィニル)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(70 mg)、ナトリウムエトキシド(20%エタノール溶液、0.23 mL)およびエタノール(3 mL)の反応混合物を室温で3時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(30 mg)を得た。
実施例18
2-(2,2-ジフルオロ-3-ヒドロキシプロポキシ)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(メチルスルフィニル)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(100 mg)および2,2-ジフルオロプロパン-1,3-ジオール(100 mg)のTHF溶液(5 mL)に1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン(0.044 mL)を0℃で加えた。反応混合物を0℃で0.5時間撹拌後、1規定水酸化ナトリウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(42 mg)を得た。
2-(2,2-ジフルオロ-3-ヒドロキシプロポキシ)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(メチルスルフィニル)-6-(ピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(100 mg)および2,2-ジフルオロプロパン-1,3-ジオール(100 mg)のTHF溶液(5 mL)に1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン(0.044 mL)を0℃で加えた。反応混合物を0℃で0.5時間撹拌後、1規定水酸化ナトリウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(42 mg)を得た。
実施例19
2-(エチルスルファニル)-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例16に準じた方法により、ピリダジン-4-カルボン酸から標題化合物を得た。
2-(エチルスルファニル)-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例16に準じた方法により、ピリダジン-4-カルボン酸から標題化合物を得た。
実施例20
2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
A) (2Z)-3-アミノ-3-(ピリミジン-4-イル)プロパ-2-エン酸エチル
実施例10のA)からC)に準じた方法により、ピリミジン-4-カルボン酸から標題化合物を得た。
MS (ESI+): [M+H]+ 194.3.
実施例10のA)からC)に準じた方法により、ピリミジン-4-カルボン酸から標題化合物を得た。
MS (ESI+): [M+H]+ 194.3.
B) 2-チオキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロ-4,4'-ビピリミジン-6(1H)-オン
(2Z)-3-アミノ-3-(ピリミジン-4-イル)プロパ-2-エン酸エチル(4.00 g)のDMF溶液(35 mL)に、水素化ナトリウム(60% oil dispersion、1.74 g)を0℃で加え、10分間撹拌した後、1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(5.00 g)のDMF溶液(5 mL)を0℃で加えた。反応混合物を0℃で3時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(1.38 g)を得た。
MS (ESI+): [M+H]+ 380.9.
(2Z)-3-アミノ-3-(ピリミジン-4-イル)プロパ-2-エン酸エチル(4.00 g)のDMF溶液(35 mL)に、水素化ナトリウム(60% oil dispersion、1.74 g)を0℃で加え、10分間撹拌した後、1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(5.00 g)のDMF溶液(5 mL)を0℃で加えた。反応混合物を0℃で3時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(1.38 g)を得た。
MS (ESI+): [M+H]+ 380.9.
C) 2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
実施例1のI)に準じた方法により、2-チオキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロ-4,4'-ビピリミジン-6(1H)-オンから標題化合物を得た。
実施例1のI)に準じた方法により、2-チオキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロ-4,4'-ビピリミジン-6(1H)-オンから標題化合物を得た。
実施例21
3-[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(エチルスルファニル)-6-(ピリジン-4-イル)ピリミジン-4(3H)-オン
3-[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(エチルスルファニル)-6-(ピリジン-4-イル)ピリミジン-4(3H)-オン
A) (3-クロロ-4-ヒドロキシフェニル)カルバミン酸tert-ブチル
4-アミノ-2-クロロフェノール(5.00 g)のTHF溶液(20 mL)に、トリエチルアミン(12 mL)および二炭酸ジ-tert-ブチル(9.65 mL)を室温で加え、終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(7.63 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.38-1.53 (9H, m), 6.84 (1H, d, J = 8.7 Hz), 7.15 (1H, dd, J = 8.7, 2.3 Hz), 7.47 (1H, brs), 9.19 (1H, brs), 9.72 (1H, s).
4-アミノ-2-クロロフェノール(5.00 g)のTHF溶液(20 mL)に、トリエチルアミン(12 mL)および二炭酸ジ-tert-ブチル(9.65 mL)を室温で加え、終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(7.63 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.38-1.53 (9H, m), 6.84 (1H, d, J = 8.7 Hz), 7.15 (1H, dd, J = 8.7, 2.3 Hz), 7.47 (1H, brs), 9.19 (1H, brs), 9.72 (1H, s).
B) [3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]カルバミン酸tert-ブチル
(3-クロロ-4-ヒドロキシフェニル)カルバミン酸tert-ブチル(7.63 g)、炭酸セシウム(30.0 g)およびDMF(60 mL)の反応混合物に2,2,2-トリフルオロエタノール(7.99 g)を室温で加え、室温で3時間撹拌した。反応混合物に水を加え酢酸エチルで抽出し、抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、標題化合物(7.81 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.47 (9H, s), 4.77 (2H, q, J = 9.1 Hz), 7.19 (1H, d, J = 9.1 Hz), 7.34 (1H, dd, J = 9.1, 2.3 Hz), 7.63 (1H, d, J = 2.6 Hz), 9.44 (1H, s).
(3-クロロ-4-ヒドロキシフェニル)カルバミン酸tert-ブチル(7.63 g)、炭酸セシウム(30.0 g)およびDMF(60 mL)の反応混合物に2,2,2-トリフルオロエタノール(7.99 g)を室温で加え、室温で3時間撹拌した。反応混合物に水を加え酢酸エチルで抽出し、抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、標題化合物(7.81 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.47 (9H, s), 4.77 (2H, q, J = 9.1 Hz), 7.19 (1H, d, J = 9.1 Hz), 7.34 (1H, dd, J = 9.1, 2.3 Hz), 7.63 (1H, d, J = 2.6 Hz), 9.44 (1H, s).
C) 2-クロロ-4-イソチオシアナト-1-(2,2,2-トリフルオロエトキシ)ベンゼン
[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]カルバミン酸tert-ブチル(7.81 g)を4規定塩酸(酢酸エチル溶液、40 mL)に溶解し、終夜撹拌した。生じた固体を濾取し、酢酸エチルで洗浄後、THF(15 mL)に溶解した。得られた3-クロロ-4-(2,2,2-トリフルオロエトキシ)アニリン塩酸塩のTHF溶液に、6規定塩酸(2.4 mL)およびチオホスゲン(1.08 mL)のTHF溶液(2.5 mL)を-5℃で滴下し、同温度で10分間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(1.16 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 4.92 (2H, q, J = 8.7 Hz), 7.34 (1H, d, J = 9.0 Hz), 7.45-7.53 (1H, m), 7.70 (1H, d, J = 2.6 Hz).
[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]カルバミン酸tert-ブチル(7.81 g)を4規定塩酸(酢酸エチル溶液、40 mL)に溶解し、終夜撹拌した。生じた固体を濾取し、酢酸エチルで洗浄後、THF(15 mL)に溶解した。得られた3-クロロ-4-(2,2,2-トリフルオロエトキシ)アニリン塩酸塩のTHF溶液に、6規定塩酸(2.4 mL)およびチオホスゲン(1.08 mL)のTHF溶液(2.5 mL)を-5℃で滴下し、同温度で10分間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(1.16 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 4.92 (2H, q, J = 8.7 Hz), 7.34 (1H, d, J = 9.0 Hz), 7.45-7.53 (1H, m), 7.70 (1H, d, J = 2.6 Hz).
D) 3-[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(ピリジン-4-イル)-2-チオキソ-2,3-ジヒドロピリミジン-4(1H)-オン
実施例4のC)に準じた方法により、3-オキソ-3-(ピリジン-4-イル)プロパン酸エチルおよび2-クロロ-4-イソチオシアナト-1-(2,2,2-トリフルオロエトキシ)ベンゼンから標題化合物を得た。
MS (ESI+): [M+H]+ 414.2.
実施例4のC)に準じた方法により、3-オキソ-3-(ピリジン-4-イル)プロパン酸エチルおよび2-クロロ-4-イソチオシアナト-1-(2,2,2-トリフルオロエトキシ)ベンゼンから標題化合物を得た。
MS (ESI+): [M+H]+ 414.2.
E) 3-[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(エチルスルファニル)-6-(ピリジン-4-イル)ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により、3-[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(ピリジン-4-イル)-2-チオキソ-2,3-ジヒドロピリミジン-4(1H)-オンから標題化合物を得た。
実施例1のI)に準じた方法により、3-[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(ピリジン-4-イル)-2-チオキソ-2,3-ジヒドロピリミジン-4(1H)-オンから標題化合物を得た。
実施例22
6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 6-(2-メトキシピリジン-4-イル)-2-(メチルスルフィニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例17のA)に準じた方法により、6-(2-メトキシピリジン-4-イル)-2-(メチルチオ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 2.77 (3H, s), 3.93 (3H, s), 4.80-4.95 (2H, m), 7.11-7.29 (3H, m), 7.43 (1H, s), 7.47-7.64 (2H, m), 7.75 (1H, dd, J = 5.3, 1.5 Hz), 8.29-8.40 (1H, m).
実施例17のA)に準じた方法により、6-(2-メトキシピリジン-4-イル)-2-(メチルチオ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 2.77 (3H, s), 3.93 (3H, s), 4.80-4.95 (2H, m), 7.11-7.29 (3H, m), 7.43 (1H, s), 7.47-7.64 (2H, m), 7.75 (1H, dd, J = 5.3, 1.5 Hz), 8.29-8.40 (1H, m).
B) 6-(2-メトキシピリジン-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(2-メトキシピリジン-4-イル)-2-(メチルスルフィニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン、2,2,2-トリフルオロエタノール(60μL)、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン(40μL)およびTHF(3 mL)の反応混合物を0℃で30分間撹拌した。反応混合物に1規定水酸化ナトリウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(50 mg)を得た。
MS (ESI+): [M+H]+ 476.2.
6-(2-メトキシピリジン-4-イル)-2-(メチルスルフィニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン、2,2,2-トリフルオロエタノール(60μL)、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン(40μL)およびTHF(3 mL)の反応混合物を0℃で30分間撹拌した。反応混合物に1規定水酸化ナトリウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(50 mg)を得た。
MS (ESI+): [M+H]+ 476.2.
C) 6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例15に準じた方法により、6-(2-メトキシピリジン-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
実施例15に準じた方法により、6-(2-メトキシピリジン-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
実施例23
2-(メチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(メチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 6-(2-メトキシピリジン-4-イル)-2-(メチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により、6-(2-メトキシピリジン-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンおよびヨウ化メチルから標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 2.54 (3H, s), 3.91 (3H, s), 4.88 (2H, q, J = 8.7 Hz), 7.07 (1H, s), 7.19-7.26 (2H, m), 7.37-7.44 (2H, m), 7.53 (1H, s), 7.69 (1H, dd, J = 5.3, 1.5 Hz), 8.32 (1H, d, J = 5.3 Hz).
実施例1のI)に準じた方法により、6-(2-メトキシピリジン-4-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンおよびヨウ化メチルから標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 2.54 (3H, s), 3.91 (3H, s), 4.88 (2H, q, J = 8.7 Hz), 7.07 (1H, s), 7.19-7.26 (2H, m), 7.37-7.44 (2H, m), 7.53 (1H, s), 7.69 (1H, dd, J = 5.3, 1.5 Hz), 8.32 (1H, d, J = 5.3 Hz).
B) 2-(メチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例15に準じた方法により、6-(2-メトキシピリジン-4-イル)-2-(メチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
実施例15に準じた方法により、6-(2-メトキシピリジン-4-イル)-2-(メチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
実施例24
2-エトキシ-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(メチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(100 mg)、ナトリウムエトキシド(20%エタノール溶液、1.91 mL)およびエタノール(10 mL)の反応混合物を70℃で16時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をHPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物(29 mg)を得た。
2-エトキシ-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(メチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(100 mg)、ナトリウムエトキシド(20%エタノール溶液、1.91 mL)およびエタノール(10 mL)の反応混合物を70℃で16時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をHPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物(29 mg)を得た。
実施例25
2-(エチルスルファニル)-6-(6-メトキシピリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例7に準じた方法により、6-メトキシピリジン-3-カルボン酸から標題化合物を得た。
2-(エチルスルファニル)-6-(6-メトキシピリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例7に準じた方法により、6-メトキシピリジン-3-カルボン酸から標題化合物を得た。
実施例26
2-(エチルスルファニル)-6-(テトラヒドロ-2H-ピラン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(テトラヒドロ-2H-ピラン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-オキソ-3-(テトラヒドロ-2H-ピラン-4-イル)プロパン酸エチル
テトラヒドロ-2H-ピラン-4-カルボン酸(1 g)および塩化チオニル(3 mL)の反応混合物を80℃で3時間撹拌した。反応混合物を減圧下濃縮して得られた残渣をTHF(5 mL)に溶解し、塩化テトラヒドロ-2H-ピラン-4-カルボニルのTHF溶液とした。
ジイソプロピルアミン(1.08 mL)のTHF溶液(5 mL)にn-ブチルリチウム(4.8 mL)を0℃で加えた。同温度で15分間撹拌した後、反応混合物を-78℃に冷却し、酢酸エチル(0.75mL)を加え、30分間撹拌した。塩化テトラヒドロ-2H-ピラン-4-カルボニルのTHF溶液を-78℃で加え、1時間撹拌した後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(800 mg)を得た。
MS (ESI+): [M+H]+ 201.1.
テトラヒドロ-2H-ピラン-4-カルボン酸(1 g)および塩化チオニル(3 mL)の反応混合物を80℃で3時間撹拌した。反応混合物を減圧下濃縮して得られた残渣をTHF(5 mL)に溶解し、塩化テトラヒドロ-2H-ピラン-4-カルボニルのTHF溶液とした。
ジイソプロピルアミン(1.08 mL)のTHF溶液(5 mL)にn-ブチルリチウム(4.8 mL)を0℃で加えた。同温度で15分間撹拌した後、反応混合物を-78℃に冷却し、酢酸エチル(0.75mL)を加え、30分間撹拌した。塩化テトラヒドロ-2H-ピラン-4-カルボニルのTHF溶液を-78℃で加え、1時間撹拌した後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(800 mg)を得た。
MS (ESI+): [M+H]+ 201.1.
B) 2-(エチルスルファニル)-6-(テトラヒドロ-2H-ピラン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のG)からI)に準じた方法により、3-オキソ-3-(テトラヒドロ-2H-ピラン-4-イル)プロパン酸エチルから標題化合物を得た。
実施例1のG)からI)に準じた方法により、3-オキソ-3-(テトラヒドロ-2H-ピラン-4-イル)プロパン酸エチルから標題化合物を得た。
実施例27
2-(エチルスルファニル)-6-(2-メトキシピリジン-4-イル)-5-メチル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(2-メトキシピリジン-4-イル)-5-メチル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-(2-メトキシピリジン-4-イル)-2-メチル-3-オキソプロパン酸エチル
2-メトキシピリジン-4-カルボン酸エチル(830 mg)、水素化ナトリウム(60% oil dispersion、183 mg)およびプロピオン酸エチル(8 mL)の反応混合物を90℃で3時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(473 mg)を得た。
MS (ESI+): [M+H]+ 238.1.
2-メトキシピリジン-4-カルボン酸エチル(830 mg)、水素化ナトリウム(60% oil dispersion、183 mg)およびプロピオン酸エチル(8 mL)の反応混合物を90℃で3時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(473 mg)を得た。
MS (ESI+): [M+H]+ 238.1.
B) 2-(エチルスルファニル)-6-(2-メトキシピリジン-4-イル)-5-メチル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例10のD)からE)に準じた方法により、3-(2-メトキシピリジン-4-イル)-2-メチル-3-オキソプロパン酸エチルから標題化合物を得た。
実施例10のD)からE)に準じた方法により、3-(2-メトキシピリジン-4-イル)-2-メチル-3-オキソプロパン酸エチルから標題化合物を得た。
実施例28
2-(エチルスルファニル)-5-メチル-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例15に準じた方法により、2-(エチルスルファニル)-6-(2-メトキシピリジン-4-イル)-5-メチル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
2-(エチルスルファニル)-5-メチル-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例15に準じた方法により、2-(エチルスルファニル)-6-(2-メトキシピリジン-4-イル)-5-メチル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
実施例29
2-メトキシ-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(150 mg)、ナトリウムメトキシド(191 mg)およびメタノール(8 mL)の反応混合物を65℃で1時間半撹拌した。その後、ナトリウムメトキシド(764 mg)を加え、65℃にて5時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をHPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物(51 mg)を得た。
2-メトキシ-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(150 mg)、ナトリウムメトキシド(191 mg)およびメタノール(8 mL)の反応混合物を65℃で1時間半撹拌した。その後、ナトリウムメトキシド(764 mg)を加え、65℃にて5時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をHPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物(51 mg)を得た。
実施例30
2-エトキシ-5-メチル-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例24に準じた方法により、2-(エチルスルファニル)-5-メチル-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
2-エトキシ-5-メチル-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例24に準じた方法により、2-(エチルスルファニル)-5-メチル-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
実施例31
2-(2-メトキシエトキシ)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(120 mg)、水素化ナトリウム(60% oil dispersion、171 mg)および2-メトキシエタノール(3 mL)の反応混合物を0℃で3時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(28 mg)を得た。
2-(2-メトキシエトキシ)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(120 mg)、水素化ナトリウム(60% oil dispersion、171 mg)および2-メトキシエタノール(3 mL)の反応混合物を0℃で3時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(28 mg)を得た。
実施例32
2-メトキシ-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(100 mg)、ナトリウムメトキシド(265 mg)およびメタノール(5 mL)の反応混合物を65℃で1時間撹拌し、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を1規定水酸化ナトリウム水溶液および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去し、標題化合物(23 mg)を得た。
2-メトキシ-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(100 mg)、ナトリウムメトキシド(265 mg)およびメタノール(5 mL)の反応混合物を65℃で1時間撹拌し、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を1規定水酸化ナトリウム水溶液および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去し、標題化合物(23 mg)を得た。
実施例33
2-エトキシ-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例42に準じた方法により、2-(エチルスルファニル)-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよびエタノールから標題化合物を得た。
2-エトキシ-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例42に準じた方法により、2-(エチルスルファニル)-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよびエタノールから標題化合物を得た。
実施例34
2-(エチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
A) 6-(2,2,2-トリフルオロエトキシ)ピリジン-3-アミン
水素化ナトリウム(60% oil dispersion、4.51 g)のDMF懸濁液(250 mL)に2,2,2-トリフルオロエタノール(13 mL)を0℃で加えた。同温度で10分間撹拌した後、2-クロロ-5-ニトロピリジン(20 g)を加え、得られた反応混合物を室温で2時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をメタノール(250 mL)に溶解し、10%パラジウム/活性炭(50% 含水,3 g)を加え、水素雰囲気下、室温で3時間撹拌した。不溶物をセライトで濾過した後、濾液を減圧下留去して得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、標題化合物(20 g)を得た。
MS (ESI+): [M+H]+ 193.0.
水素化ナトリウム(60% oil dispersion、4.51 g)のDMF懸濁液(250 mL)に2,2,2-トリフルオロエタノール(13 mL)を0℃で加えた。同温度で10分間撹拌した後、2-クロロ-5-ニトロピリジン(20 g)を加え、得られた反応混合物を室温で2時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をメタノール(250 mL)に溶解し、10%パラジウム/活性炭(50% 含水,3 g)を加え、水素雰囲気下、室温で3時間撹拌した。不溶物をセライトで濾過した後、濾液を減圧下留去して得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、標題化合物(20 g)を得た。
MS (ESI+): [M+H]+ 193.0.
B) 5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジン
6-(2,2,2-トリフルオロエトキシ)ピリジン-3-アミン(20 g)のTHF溶液(300 mL)に0℃でチオホスゲン(18 g)を滴下し、得られた反応混合物を0℃で1時間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(18.5 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 5.01 (2H, q, J = 9.1 Hz), 7.09 (1H, d, J = 9.4 Hz), 7.94 (1H, dd, J = 8.7, 2.6 Hz), 8.38 (1H, d, J = 2.6 Hz).
6-(2,2,2-トリフルオロエトキシ)ピリジン-3-アミン(20 g)のTHF溶液(300 mL)に0℃でチオホスゲン(18 g)を滴下し、得られた反応混合物を0℃で1時間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(18.5 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 5.01 (2H, q, J = 9.1 Hz), 7.09 (1H, d, J = 9.4 Hz), 7.94 (1H, dd, J = 8.7, 2.6 Hz), 8.38 (1H, d, J = 2.6 Hz).
C) 2-(エチルスルファニル)-6-(2-メトキシピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例10のD)からE)に準じた方法により、(2Z)-3-アミノ-3-(2-メトキシピリジン-4-イル)プロパ-2-エン酸エチルおよび5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジンから標題化合物を得た。
MS (ESI+): [M+H]+ 493.3.
実施例10のD)からE)に準じた方法により、(2Z)-3-アミノ-3-(2-メトキシピリジン-4-イル)プロパ-2-エン酸エチルおよび5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジンから標題化合物を得た。
MS (ESI+): [M+H]+ 493.3.
D) 2-(エチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
実施例15に準じた方法により、2-(エチルスルファニル)-6-(2-メトキシピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
実施例15に準じた方法により、2-(エチルスルファニル)-6-(2-メトキシピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
実施例35
2-メトキシ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
実施例32に準じた方法により、2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オンから標題化合物を得た。
2-メトキシ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
実施例32に準じた方法により、2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オンから標題化合物を得た。
実施例36
2-エトキシ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン(150 mg)、水素化ナトリウム(60% oil dispersion、44 mg)およびエタノール(3 mL)の反応混合物を70℃で3時間撹拌し、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を1規定水酸化ナトリウム水溶液および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去し、標題化合物(86 mg)を得た。
2-エトキシ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン(150 mg)、水素化ナトリウム(60% oil dispersion、44 mg)およびエタノール(3 mL)の反応混合物を70℃で3時間撹拌し、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を1規定水酸化ナトリウム水溶液および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去し、標題化合物(86 mg)を得た。
実施例37
6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-2-プロポキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(150 mg)の1-プロパノール懸濁液(3 mL)に、水素化ナトリウム(60% oil dispersion、42 mg)を加え、得られた反応混合物を70℃で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をHPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物(20 mg)を得た。
6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-2-プロポキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(150 mg)の1-プロパノール懸濁液(3 mL)に、水素化ナトリウム(60% oil dispersion、42 mg)を加え、得られた反応混合物を70℃で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をHPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物(20 mg)を得た。
実施例38
2-(エチルスルファニル)-6-(1,3-オキサゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1,3-オキサゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-(1,3-オキサゾール-4-イル)-3-オキソプロパン酸エチル
1,3-オキサゾール-4-カルボン酸(1 g)および塩化チオニル(4 mL)の反応混合物を室温で5時間撹拌した。反応混合物を減圧下濃縮して得られた残渣をTHF(5 mL)に溶解し、塩化1,3-オキサゾール-4-カルボニルのTHF溶液とした。
ジイソプロピルアミン(1.24 mL)のTHF溶液(5 mL)にn-ブチルリチウム(5.5 mL)を0℃で加えた。同温度で15分間撹拌した後、反応混合物を-78℃に冷却し、酢酸エチル(0.87 mL)を加え、30分間撹拌した。塩化1,3-オキサゾール-4-カルボニルのTHF溶液を-78℃で加え、2時間撹拌した後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(578 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.17 (3H, t, J = 7.2 Hz), 3.97 (2H, s), 4.11 (2H, q, J = 7.1 Hz), 8.57 (1H, s), 9.00 (1H, s).
1,3-オキサゾール-4-カルボン酸(1 g)および塩化チオニル(4 mL)の反応混合物を室温で5時間撹拌した。反応混合物を減圧下濃縮して得られた残渣をTHF(5 mL)に溶解し、塩化1,3-オキサゾール-4-カルボニルのTHF溶液とした。
ジイソプロピルアミン(1.24 mL)のTHF溶液(5 mL)にn-ブチルリチウム(5.5 mL)を0℃で加えた。同温度で15分間撹拌した後、反応混合物を-78℃に冷却し、酢酸エチル(0.87 mL)を加え、30分間撹拌した。塩化1,3-オキサゾール-4-カルボニルのTHF溶液を-78℃で加え、2時間撹拌した後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(578 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.17 (3H, t, J = 7.2 Hz), 3.97 (2H, s), 4.11 (2H, q, J = 7.1 Hz), 8.57 (1H, s), 9.00 (1H, s).
B) 2-(エチルスルファニル)-6-(1,3-オキサゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例10に準じた方法により、3-(1,3-オキサゾール-4-イル)-3-オキソプロパン酸エチルから標題化合物を得た。
実施例10に準じた方法により、3-(1,3-オキサゾール-4-イル)-3-オキソプロパン酸エチルから標題化合物を得た。
実施例39
2-プロポキシ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン(100 mg)、水素化ナトリウム(60% oil dispersion、59 mg)、プロパン-1-オール(2.2 mL)およびTHF(5 mL)の反応混合物を65℃で2時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(77 mg)を得た。
2-プロポキシ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン(100 mg)、水素化ナトリウム(60% oil dispersion、59 mg)、プロパン-1-オール(2.2 mL)およびTHF(5 mL)の反応混合物を65℃で2時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(77 mg)を得た。
実施例40
2-(エチルスルファニル)-1-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-4,4'-ビピリミジン-6(1H)-オン
2-(エチルスルファニル)-1-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-4,4'-ビピリミジン-6(1H)-オン
A) 2-チオキソ-1-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロ-4,4'-ビピリミジン-6(1H)-オン
実施例20のB)に準じた方法により、(2Z)-3-アミノ-3-(ピリミジン-4-イル)プロパ-2-エン酸エチルおよび5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジンから標題化合物を得た。
MS (ESI+): [M+H]+ 382.0.
実施例20のB)に準じた方法により、(2Z)-3-アミノ-3-(ピリミジン-4-イル)プロパ-2-エン酸エチルおよび5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジンから標題化合物を得た。
MS (ESI+): [M+H]+ 382.0.
B) 2-(エチルスルファニル)-1-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-4,4'-ビピリミジン-6(1H)-オン
実施例1のI)に準じた方法により、2-チオキソ-1-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロ-4,4'-ビピリミジン-6(1H)-オンから標題化合物を得た。
実施例1のI)に準じた方法により、2-チオキソ-1-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロ-4,4'-ビピリミジン-6(1H)-オンから標題化合物を得た。
実施例41
1-[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(エチルスルファニル)-4,4'-ビピリミジン-6(1H)-オン
実施例20のB)からC)に準じた方法により、(2Z)-3-アミノ-3-(ピリミジン-4-イル)プロパ-2-エン酸エチルおよび2-クロロ-4-イソチオシアナト-1-(2,2,2-トリフルオロエトキシ)ベンゼンから標題化合物を得た。
1-[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(エチルスルファニル)-4,4'-ビピリミジン-6(1H)-オン
実施例20のB)からC)に準じた方法により、(2Z)-3-アミノ-3-(ピリミジン-4-イル)プロパ-2-エン酸エチルおよび2-クロロ-4-イソチオシアナト-1-(2,2,2-トリフルオロエトキシ)ベンゼンから標題化合物を得た。
実施例42
2-エトキシ-6-(1,3-オキサゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1,3-オキサゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(45 mg)、水素化ナトリウム(60% oil dispersion、27 mg)、エタノール(17μL)およびテトラヒドロフラン(2 mL)の反応混合物を室温で終夜撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去し、標題化合物(10 mg)を得た。
2-エトキシ-6-(1,3-オキサゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1,3-オキサゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(45 mg)、水素化ナトリウム(60% oil dispersion、27 mg)、エタノール(17μL)およびテトラヒドロフラン(2 mL)の反応混合物を室温で終夜撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去し、標題化合物(10 mg)を得た。
実施例43
1-[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-2-エトキシ-4,4'-ビピリミジン-6(1H)-オン
実施例42に準じた方法により、1-[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(エチルスルファニル)-4,4'-ビピリミジン-6(1H)-オンから標題化合物を得た。
1-[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-2-エトキシ-4,4'-ビピリミジン-6(1H)-オン
実施例42に準じた方法により、1-[3-クロロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(エチルスルファニル)-4,4'-ビピリミジン-6(1H)-オンから標題化合物を得た。
実施例44
2-エトキシ-1-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-4,4'-ビピリミジン-6(1H)-オン
実施例42に準じた方法により、2-(エチルスルファニル)-1-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-4,4'-ビピリミジン-6(1H)-オンから標題化合物を得た。
2-エトキシ-1-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-4,4'-ビピリミジン-6(1H)-オン
実施例42に準じた方法により、2-(エチルスルファニル)-1-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-4,4'-ビピリミジン-6(1H)-オンから標題化合物を得た。
実施例45
2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,5'-ビピリミジン-6(1H)-オン
実施例7に準じた方法により、ピリミジン-5-カルボン酸から標題化合物を得た。
2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,5'-ビピリミジン-6(1H)-オン
実施例7に準じた方法により、ピリミジン-5-カルボン酸から標題化合物を得た。
実施例46
2-エトキシ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,5'-ビピリミジン-6(1H)-オン
6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-2-プロポキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(100 mg)およびエタノール(0.043 mL)のTHF溶液(3 mL)に、水素化ナトリウム(60% oil dispersion、58.8 mg)を加え、得られた反応混合物を室温で3時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(69 mg)を得た。
2-エトキシ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,5'-ビピリミジン-6(1H)-オン
6-(2-オキソ-1,2-ジヒドロピリジン-4-イル)-2-プロポキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(100 mg)およびエタノール(0.043 mL)のTHF溶液(3 mL)に、水素化ナトリウム(60% oil dispersion、58.8 mg)を加え、得られた反応混合物を室温で3時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(69 mg)を得た。
実施例47
2-(オキセタン-3-イルオキシ)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
実施例42に準じた方法により、2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オンおよびオキセタン-3-オールから標題化合物を得た。
2-(オキセタン-3-イルオキシ)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
実施例42に準じた方法により、2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オンおよびオキセタン-3-オールから標題化合物を得た。
実施例48
2-(エチルスルファニル)-6-(1,3-チアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1,3-チアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-オキソ-3-(1,3-チアゾール-5-イル)プロパン酸エチル
1,3-チアゾール-5-カルボン酸(350 mg)のTHF溶液(5 mL)にカルボニルジイミダゾール(439 mg)を0℃で加えた。反応混合物を室温で1時間撹拌後、塩化マグネシウム(258 mg)、マロン酸モノエチルカリウム塩(692 mg)およびTHF(5 mL)の混合物に加えた。反応混合物を50℃で終夜攪拌した後、反応混合物を水に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(290 mg)を得た。
MS (ESI+): [M+H]+ 200.0.
1,3-チアゾール-5-カルボン酸(350 mg)のTHF溶液(5 mL)にカルボニルジイミダゾール(439 mg)を0℃で加えた。反応混合物を室温で1時間撹拌後、塩化マグネシウム(258 mg)、マロン酸モノエチルカリウム塩(692 mg)およびTHF(5 mL)の混合物に加えた。反応混合物を50℃で終夜攪拌した後、反応混合物を水に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(290 mg)を得た。
MS (ESI+): [M+H]+ 200.0.
B) (2Z)-3-アミノ-3-(1,3-チアゾール-5-イル)プロパ-2-エン酸エチル
3-オキソ-3-(1,3-チアゾール-5-イル)プロパン酸エチル(290 mg)、ギ酸アンモニウム(459 mg)およびエタノール(5 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をジイソプロピルエーテルで固化させ標題化合物(195 mg)を得た。
MS (ESI+): [M+H]+ 199.1.
3-オキソ-3-(1,3-チアゾール-5-イル)プロパン酸エチル(290 mg)、ギ酸アンモニウム(459 mg)およびエタノール(5 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をジイソプロピルエーテルで固化させ標題化合物(195 mg)を得た。
MS (ESI+): [M+H]+ 199.1.
C) 6-(1,3-チアゾール-5-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン
(2Z)-3-アミノ-3-(1,3-チアゾール-5-イル)プロパ-2-エン酸エチル(195 mg)および1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(275 mg)のDMF溶液(3 mL)に、水素化ナトリウム(60% oil dispersion、98 mg)を0℃で加えた。反応混合物を0℃で2時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(190 mg)を得た。
MS (ESI+): [M+H]+ 384.1.
(2Z)-3-アミノ-3-(1,3-チアゾール-5-イル)プロパ-2-エン酸エチル(195 mg)および1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(275 mg)のDMF溶液(3 mL)に、水素化ナトリウム(60% oil dispersion、98 mg)を0℃で加えた。反応混合物を0℃で2時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(190 mg)を得た。
MS (ESI+): [M+H]+ 384.1.
D) 2-(エチルスルファニル)-6-(1,3-チアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1,3-チアゾール-5-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(190 mg)、ヨードエタン(0.061 mL)、炭酸カリウム(102 mg)およびTHF(10 mL)の反応混合物を70℃で4時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン) で精製して標題化合物(115 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.30 (3H, t, J = 7.4 Hz), 3.10 (2H, q, J = 7.3 Hz), 4.87 (2H, q, J = 9.1 Hz), 6.94 (1H, s), 7.17-7.26 (2H, m), 7.34-7.42 (2H, m), 8.71 (1H, s), 9.27 (1H, s).
6-(1,3-チアゾール-5-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(190 mg)、ヨードエタン(0.061 mL)、炭酸カリウム(102 mg)およびTHF(10 mL)の反応混合物を70℃で4時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン) で精製して標題化合物(115 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.30 (3H, t, J = 7.4 Hz), 3.10 (2H, q, J = 7.3 Hz), 4.87 (2H, q, J = 9.1 Hz), 6.94 (1H, s), 7.17-7.26 (2H, m), 7.34-7.42 (2H, m), 8.71 (1H, s), 9.27 (1H, s).
実施例49
2-(エチルスルファニル)-6-(6-オキソ-1,6-ジヒドロピリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(6-オキソ-1,6-ジヒドロピリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 6-オキソ-1,6-ジヒドロピリジン-3-カルボン酸メチル
6-オキソ-1,6-ジヒドロピリジン-3-カルボン酸(1.23 g)のメタノール溶液(20 mL)に、塩化チオニル(0.774 mL)を0℃で加えた。反応混合物を60℃で4時間撹拌後、濃縮し、得られた残渣に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して標題化合物(1.35 g)を得た。
MS (ESI+): [M+H]+ 154.0.
6-オキソ-1,6-ジヒドロピリジン-3-カルボン酸(1.23 g)のメタノール溶液(20 mL)に、塩化チオニル(0.774 mL)を0℃で加えた。反応混合物を60℃で4時間撹拌後、濃縮し、得られた残渣に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して標題化合物(1.35 g)を得た。
MS (ESI+): [M+H]+ 154.0.
B) 1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-カルボン酸
6-オキソ-1,6-ジヒドロピリジン-3-カルボン酸メチル(1.35 g)のDMF溶液(15 mL)に水素化ナトリウム(60% oil dispersion、388 mg)を0℃で加えた。反応混合物を室温で1時間撹拌後、反応混合物に1-(クロロメチル)-4-メトキシベンゼン(1.52 g)を加え、反応混合物を50℃で4時間攪拌した。反応混合物を水に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-カルボン酸メチルを含む混合物を得た。得られた混合物、8規定水酸化ナトリウム(5 mL)、メタノール(10 mL)およびTHF(10 mL)の反応混合物を60℃で3時間撹拌した。反応混合物を減圧下濃縮し、得られた残渣に1規定塩酸を加え、沈殿物を濾取した。水で洗浄し、減圧下乾燥させ、標題化合物(1.89 g)を得た。
MS (API-): [M-H]- 258.1.
6-オキソ-1,6-ジヒドロピリジン-3-カルボン酸メチル(1.35 g)のDMF溶液(15 mL)に水素化ナトリウム(60% oil dispersion、388 mg)を0℃で加えた。反応混合物を室温で1時間撹拌後、反応混合物に1-(クロロメチル)-4-メトキシベンゼン(1.52 g)を加え、反応混合物を50℃で4時間攪拌した。反応混合物を水に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-カルボン酸メチルを含む混合物を得た。得られた混合物、8規定水酸化ナトリウム(5 mL)、メタノール(10 mL)およびTHF(10 mL)の反応混合物を60℃で3時間撹拌した。反応混合物を減圧下濃縮し、得られた残渣に1規定塩酸を加え、沈殿物を濾取した。水で洗浄し、減圧下乾燥させ、標題化合物(1.89 g)を得た。
MS (API-): [M-H]- 258.1.
C) 3-[1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-イル]-3-オキソプロパン酸エチル
1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-カルボン酸(1.89 g)のTHF溶液(30 mL)にカルボニルジイミダゾール(1.24 g)を0℃で加えた。反応混合物を室温で1時間撹拌後、塩化マグネシウム(0.701 g)、マロン酸モノエチルカリウム塩(1.86 g)およびTHF(20 mL)の混合物に加えた。反応混合物を50℃で2時間攪拌し、室温で終夜撹拌後、反応混合物を水に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(424 mg)を得た。
MS (ESI+): [M+H]+ 330.4.
1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-カルボン酸(1.89 g)のTHF溶液(30 mL)にカルボニルジイミダゾール(1.24 g)を0℃で加えた。反応混合物を室温で1時間撹拌後、塩化マグネシウム(0.701 g)、マロン酸モノエチルカリウム塩(1.86 g)およびTHF(20 mL)の混合物に加えた。反応混合物を50℃で2時間攪拌し、室温で終夜撹拌後、反応混合物を水に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(424 mg)を得た。
MS (ESI+): [M+H]+ 330.4.
D) (2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-イル]プロパ-2-エン酸エチル
3-[1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-イル]-3-オキソプロパン酸エチル(424 mg)、ギ酸アンモニウム(406 mg)およびエタノール(3 mL)の反応混合物を70℃で終夜撹拌した。反応混合物を濃縮し、得られた残渣を水に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して標題化合物(389 mg)を得た。
MS (ESI+): [M+H]+ 329.3.
3-[1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-イル]-3-オキソプロパン酸エチル(424 mg)、ギ酸アンモニウム(406 mg)およびエタノール(3 mL)の反応混合物を70℃で終夜撹拌した。反応混合物を濃縮し、得られた残渣を水に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して標題化合物(389 mg)を得た。
MS (ESI+): [M+H]+ 329.3.
E) 6-[1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-イル]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-イル]プロパ-2-エン酸エチル(389 mg)および1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(304 mg)のDMF溶液(5 mL)に、水素化ナトリウム(60% oil dispersion、56.8 mg)を0℃で加えた。反応混合物を室温で4時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して標題化合物を得た。
MS (ESI+): [M+H]+ 516.2.
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-イル]プロパ-2-エン酸エチル(389 mg)および1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(304 mg)のDMF溶液(5 mL)に、水素化ナトリウム(60% oil dispersion、56.8 mg)を0℃で加えた。反応混合物を室温で4時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して標題化合物を得た。
MS (ESI+): [M+H]+ 516.2.
F)2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-[1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-イル]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(344 mg)、ヨードエタン(0.104 mL)、炭酸カリウム(185 mg)およびTHF(5 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(222 mg)を得た。
MS (ESI+): [M+H]+ 544.1.
6-[1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-イル]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(344 mg)、ヨードエタン(0.104 mL)、炭酸カリウム(185 mg)およびTHF(5 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(222 mg)を得た。
MS (ESI+): [M+H]+ 544.1.
G) 2-(エチルスルファニル)-6-(6-オキソ-1,6-ジヒドロピリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(222 mg)のトリフルオロ酢酸溶液(3 mL)をマイクロウェーブ照射下150℃で1時間撹拌した。反応混合物にトルエンを加えた後、減圧下濃縮し、得られた残渣をジイソプロピルエーテルを用いて固化させ、得られた粗結晶をTHF/ヘキサンから再結晶し、標題化合物(53.3 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.27 (3H, t, J = 7.4 Hz), 3.11 (2H, q, J = 7.4 Hz), 4.87 (2H, q, J = 8.7 Hz), 6.44 (1H, d, J = 9.8 Hz), 6.70 (1H, s), 7.20 (2H, d, J = 9.1 Hz), 7.34 (2H, d, J = 9.1 Hz), 8.13 (1H, dd, J = 9.6, 2.5 Hz), 8.25 (1H, d, J = 2.3 Hz), 12.05 (1H, brs).
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-6-オキソ-1,6-ジヒドロピリジン-3-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(222 mg)のトリフルオロ酢酸溶液(3 mL)をマイクロウェーブ照射下150℃で1時間撹拌した。反応混合物にトルエンを加えた後、減圧下濃縮し、得られた残渣をジイソプロピルエーテルを用いて固化させ、得られた粗結晶をTHF/ヘキサンから再結晶し、標題化合物(53.3 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.27 (3H, t, J = 7.4 Hz), 3.11 (2H, q, J = 7.4 Hz), 4.87 (2H, q, J = 8.7 Hz), 6.44 (1H, d, J = 9.8 Hz), 6.70 (1H, s), 7.20 (2H, d, J = 9.1 Hz), 7.34 (2H, d, J = 9.1 Hz), 8.13 (1H, dd, J = 9.6, 2.5 Hz), 8.25 (1H, d, J = 2.3 Hz), 12.05 (1H, brs).
実施例50
2-[3-(メチルスルホニル)プロポキシ]-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
2-[3-(メチルスルホニル)プロポキシ]-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
A) 2-[3-(メチルスルファニル)プロポキシ]-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
実施例42に準じた方法により、2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オンおよび3-(メチルスルファニル)プロパン-1-オールを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 453.2.
実施例42に準じた方法により、2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オンおよび3-(メチルスルファニル)プロパン-1-オールを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 453.2.
B) 2-[3-(メチルスルホニル)プロポキシ]-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
2-[3-(メチルスルファニル)プロポキシ]-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オンの酢酸エチル溶液(2 mL)にm-クロロ過安息香酸(106 mg)を0℃で加え、反応混合物を室温で1時間撹拌した。反応混合物にm-クロロ過安息香酸(106 mg)を加え、反応混合物を室温で3時間撹拌した。反応混合物を飽和炭酸水素ナトリウム溶液に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(21.7 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ2.00-2.13 (2H, m), 2.93 (3H, s), 3.01-3.12 (2H, m),4.59 (2H, t, J = 6.4 Hz), 4.84 (2H, q, J = 9.1 Hz), 7.12-7.26 (3H, m), 7.33-7.43 (2H, m), 8.34 (1H, dd, J = 5.3, 1.5 Hz), 9.08 (1H, d, J = 5.3 Hz), 9.36 (1H, d, J = 1.5 Hz).
2-[3-(メチルスルファニル)プロポキシ]-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オンの酢酸エチル溶液(2 mL)にm-クロロ過安息香酸(106 mg)を0℃で加え、反応混合物を室温で1時間撹拌した。反応混合物にm-クロロ過安息香酸(106 mg)を加え、反応混合物を室温で3時間撹拌した。反応混合物を飽和炭酸水素ナトリウム溶液に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(21.7 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ2.00-2.13 (2H, m), 2.93 (3H, s), 3.01-3.12 (2H, m),4.59 (2H, t, J = 6.4 Hz), 4.84 (2H, q, J = 9.1 Hz), 7.12-7.26 (3H, m), 7.33-7.43 (2H, m), 8.34 (1H, dd, J = 5.3, 1.5 Hz), 9.08 (1H, d, J = 5.3 Hz), 9.36 (1H, d, J = 1.5 Hz).
実施例51
2-(エチルスルファニル)-6-(1,3-オキサゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1,3-オキサゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-(1,3-オキサゾール-5-イル)-3-オキソプロパン酸エチル
1,3-オキサゾール-5-カルボン酸(1.00 g)のTHF溶液(24 mL)にカルボニルジイミダゾール(1.51 g)を0℃で加えた。反応混合物を室温で1時間撹拌後、塩化マグネシウム(850 mg)、マロン酸モノエチルカリウム塩(2.26 g)およびTHF(20 mL)の混合物に加えた。反応混合物を50℃で2時間撹拌した後、室温にて終夜撹拌後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(656 mg)を得た。
MS (ESI+): [M+H]+ 184.1.
1,3-オキサゾール-5-カルボン酸(1.00 g)のTHF溶液(24 mL)にカルボニルジイミダゾール(1.51 g)を0℃で加えた。反応混合物を室温で1時間撹拌後、塩化マグネシウム(850 mg)、マロン酸モノエチルカリウム塩(2.26 g)およびTHF(20 mL)の混合物に加えた。反応混合物を50℃で2時間撹拌した後、室温にて終夜撹拌後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(656 mg)を得た。
MS (ESI+): [M+H]+ 184.1.
B) (2Z)-3-アミノ-3-(1,3-オキサゾール-5-イル)プロパ-2-エン酸エチル
3-(1,3-オキサゾール-5-イル)-3-オキソプロパン酸エチル(655 mg)、ギ酸アンモニウム(2.26 g)およびエタノール(5 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(NH、酢酸エチル/ヘキサン)で精製して標題化合物(484 mg)を得た。
MS (ESI+): [M+H]+ 183.2.
3-(1,3-オキサゾール-5-イル)-3-オキソプロパン酸エチル(655 mg)、ギ酸アンモニウム(2.26 g)およびエタノール(5 mL)の反応混合物を70℃で2時間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(NH、酢酸エチル/ヘキサン)で精製して標題化合物(484 mg)を得た。
MS (ESI+): [M+H]+ 183.2.
C) 6-(1,3-オキサゾール-5-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン
1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(644 mg)のDMF溶液(3 mL)に、水素化ナトリウム(60% oil dispersion、134 mg)を0℃で加え、5分間撹拌した。得られた反応混合物に、(2Z)-3-アミノ-3-(1,3-オキサゾール-5-イル)プロパ-2-エン酸エチル(484 mg)を加え0℃で2時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(252 mg)を得た。
MS (ESI+): [M+H]+ 370.1.
1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(644 mg)のDMF溶液(3 mL)に、水素化ナトリウム(60% oil dispersion、134 mg)を0℃で加え、5分間撹拌した。得られた反応混合物に、(2Z)-3-アミノ-3-(1,3-オキサゾール-5-イル)プロパ-2-エン酸エチル(484 mg)を加え0℃で2時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(252 mg)を得た。
MS (ESI+): [M+H]+ 370.1.
D) 2-(エチルスルファニル)-6-(1,3-オキサゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1,3-オキサゾール-5-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(252 mg)、ヨードエタン(0.11 mL)、炭酸カリウム(189 mg)およびTHF(5 mL)の反応混合物を70℃で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。続いて溶媒を減圧下留去し、標題化合物(199 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.27 (3H, t, J = 7.4 Hz), 3.12 (2H, q, J = 7.2 Hz), 4.87 (2H, q, J = 8.9 Hz), 6.53 (1H, s), 7.16-7.28 (2H, m), 7.31-7.45 (2H, m), 7.92 (1H, s), 8.65 (1H, s).
6-(1,3-オキサゾール-5-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン(252 mg)、ヨードエタン(0.11 mL)、炭酸カリウム(189 mg)およびTHF(5 mL)の反応混合物を70℃で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。続いて溶媒を減圧下留去し、標題化合物(199 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.27 (3H, t, J = 7.4 Hz), 3.12 (2H, q, J = 7.2 Hz), 4.87 (2H, q, J = 8.9 Hz), 6.53 (1H, s), 7.16-7.28 (2H, m), 7.31-7.45 (2H, m), 7.92 (1H, s), 8.65 (1H, s).
実施例52
2-(2,2,2-トリフルオロエトキシ)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
2-(2,2,2-トリフルオロエトキシ)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
A) 1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-2,6(1H,3H)-ジオン
実施例20で得られた2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン(120 mg)および2-メチルプロパン-1,2-ジオール(53.0 mg)のTHF溶液(1.5 mL)に、水素化ナトリウム(60% oil dispersion、118 mg)を0℃で加えた。反応混合物を室温で6時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して標題化合物(58.9 mg)を得た。
MS (ESI+): [M+H]+ 363.2.
実施例20で得られた2-(エチルスルファニル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン(120 mg)および2-メチルプロパン-1,2-ジオール(53.0 mg)のTHF溶液(1.5 mL)に、水素化ナトリウム(60% oil dispersion、118 mg)を0℃で加えた。反応混合物を室温で6時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去して標題化合物(58.9 mg)を得た。
MS (ESI+): [M+H]+ 363.2.
B) 2-(2,2,2-トリフルオロエトキシ)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-6(1H)-オン
1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-2,6(1H,3H)-ジオン(58.9 mg)および炭酸セシウム(55.3 mg)のDMF溶液に2,2,2-トリフルオロエチル トリフルオロメタンスルホナート(24μL)を0℃で加えた。反応混合物を0℃で1時間、および室温で2時間撹拌後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、得られた粗結晶をTHF及びヘキサンより再結晶し標題化合物(12.5 mg)を得た。
1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-4,4'-ビピリミジン-2,6(1H,3H)-ジオン(58.9 mg)および炭酸セシウム(55.3 mg)のDMF溶液に2,2,2-トリフルオロエチル トリフルオロメタンスルホナート(24μL)を0℃で加えた。反応混合物を0℃で1時間、および室温で2時間撹拌後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、得られた粗結晶をTHF及びヘキサンより再結晶し標題化合物(12.5 mg)を得た。
実施例53
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
A) 6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロピリミジン-4(1H)-オン
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]プロパ-2-エン酸エチル(585 mg)および5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジン(500 mg)のDMF溶液(3 mL)に、水素化ナトリウム(60% oil dispersion、170 mg)を0℃で加えた。反応混合物を0℃で3時間撹拌後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(400 mg)を得た。
MS (ESI+): [M+H]+ 490.1.
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]プロパ-2-エン酸エチル(585 mg)および5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジン(500 mg)のDMF溶液(3 mL)に、水素化ナトリウム(60% oil dispersion、170 mg)を0℃で加えた。反応混合物を0℃で3時間撹拌後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(400 mg)を得た。
MS (ESI+): [M+H]+ 490.1.
B) 2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロピリミジン-4(1H)-オン(400 mg)、ヨードエタン(0.078 mL)、炭酸カリウム(226 mg)およびTHF(10 mL)の反応混合物を70℃で3時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(320 mg)を得た。
MS (ESI+): [M+H]+ 518.3.
6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロピリミジン-4(1H)-オン(400 mg)、ヨードエタン(0.078 mL)、炭酸カリウム(226 mg)およびTHF(10 mL)の反応混合物を70℃で3時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(320 mg)を得た。
MS (ESI+): [M+H]+ 518.3.
C) 2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(320 mg)のトリフルオロ酢酸溶液(5 mL)を70℃で終夜撹拌した。反応混合物にトルエンを加えた後、減圧下濃縮し、飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をジイソプロピルエーテルを用いて固化させ、標題化合物(170 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.30 (3H, t, J = 7.2 Hz), 3.17 (2H, q, J = 7.4 Hz), 5.07 (2H, qd, J = 9.0, 1.7 Hz), 6.61 (1H, s), 7.17 (1H, d, J = 8.7 Hz), 7.91 (1H, dd, J = 8.7, 2.7 Hz), 8.12 (1H, brs), 8.27 (1H, d, J = 2.7 Hz), 8.41 (1H, brs), 13.28 (1H, brs).
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(320 mg)のトリフルオロ酢酸溶液(5 mL)を70℃で終夜撹拌した。反応混合物にトルエンを加えた後、減圧下濃縮し、飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をジイソプロピルエーテルを用いて固化させ、標題化合物(170 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.30 (3H, t, J = 7.2 Hz), 3.17 (2H, q, J = 7.4 Hz), 5.07 (2H, qd, J = 9.0, 1.7 Hz), 6.61 (1H, s), 7.17 (1H, d, J = 8.7 Hz), 7.91 (1H, dd, J = 8.7, 2.7 Hz), 8.12 (1H, brs), 8.27 (1H, d, J = 2.7 Hz), 8.41 (1H, brs), 13.28 (1H, brs).
実施例54
2-(エチルスルファニル)-6-(ピリジン-4-イルメチル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(ピリジン-4-イルメチル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-オキソ-4-(ピリジン-4-イル)ブタン酸エチル
ピリジン-4-イル酢酸塩酸塩(2.30 g)のTHF溶液(50 mL)にトリエチルアミン(1.85 mL)およびカルボニルジイミダゾール(2.15 g)を0℃で加えた。反応混合物を室温で1時間撹拌後、反応溶液に、塩化マグネシウム(1.26 g)およびマロン酸モノエチルカリウム塩(3.50 g)のTHF(50 mL)懸濁液を加えた。反応混合物を50℃で18時間撹拌した後、室温まで冷却し、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣を酢酸エチルで洗浄して標題化合物(1.58 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.04 (3H, t, J = 7.2 Hz), 3.24 (2H, s), 3.82 (2H, q, J = 7.2 Hz), 4.50 (1H, s), 7.26 (2H, d, J = 6.1 Hz), 8.38 (2H, d, J = 5.7 Hz).
ピリジン-4-イル酢酸塩酸塩(2.30 g)のTHF溶液(50 mL)にトリエチルアミン(1.85 mL)およびカルボニルジイミダゾール(2.15 g)を0℃で加えた。反応混合物を室温で1時間撹拌後、反応溶液に、塩化マグネシウム(1.26 g)およびマロン酸モノエチルカリウム塩(3.50 g)のTHF(50 mL)懸濁液を加えた。反応混合物を50℃で18時間撹拌した後、室温まで冷却し、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣を酢酸エチルで洗浄して標題化合物(1.58 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.04 (3H, t, J = 7.2 Hz), 3.24 (2H, s), 3.82 (2H, q, J = 7.2 Hz), 4.50 (1H, s), 7.26 (2H, d, J = 6.1 Hz), 8.38 (2H, d, J = 5.7 Hz).
B) (2Z)-3-アミノ-4-(ピリジン-4-イル)ブタ-2-エン酸エチル
3-オキソ-4-(ピリジン-4-イル)ブタン酸エチル(650 mg)、ギ酸アンモニウム(4.00 g)およびエタノール(45 mL)の反応混合物を70℃で3時間撹拌した。反応溶媒を減圧下留去し、飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(NH、酢酸エチル/ヘキサン)で精製して標題化合物(381 mg)を得た。
MS (ESI+): [M+H]+ 207.4
3-オキソ-4-(ピリジン-4-イル)ブタン酸エチル(650 mg)、ギ酸アンモニウム(4.00 g)およびエタノール(45 mL)の反応混合物を70℃で3時間撹拌した。反応溶媒を減圧下留去し、飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(NH、酢酸エチル/ヘキサン)で精製して標題化合物(381 mg)を得た。
MS (ESI+): [M+H]+ 207.4
C) 2-(エチルスルファニル)-6-(ピリジン-4-イルメチル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
(2Z)-3-アミノ-4-(ピリジン-4-イル)ブタ-2-エン酸エチル(380 mg)および1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(516 mg)のDMF溶液(15 mL)に、水素化ナトリウム(60% oil dispersion、162 mg)を0℃で加えた。反応混合物を0℃で2時間撹拌後、1規定塩酸および酢酸エチルを加え、有機層と水層を分離した。水層に飽和重曹水を加え中和後、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製後、得られた化合物にヨードエタン(0.045 mL)、炭酸カリウム(196 mg)およびTHF(15 mL)を加え、混合物を80℃で30分間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(19.3 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.10 (3H, t, J = 7.4 Hz), 2.94 (2H, q, J = 7.2 Hz), 3.86 (2H, s), 4.84 (2H, q, J = 9.1 Hz), 6.23 (1H, s), 7.08-7.21 (2H, m), 7.25-7.33 (2H, m), 7.38 (2H, d, J = 6.1 Hz), 8.52 (2H, d, J = 6.1 Hz).
(2Z)-3-アミノ-4-(ピリジン-4-イル)ブタ-2-エン酸エチル(380 mg)および1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン(516 mg)のDMF溶液(15 mL)に、水素化ナトリウム(60% oil dispersion、162 mg)を0℃で加えた。反応混合物を0℃で2時間撹拌後、1規定塩酸および酢酸エチルを加え、有機層と水層を分離した。水層に飽和重曹水を加え中和後、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製後、得られた化合物にヨードエタン(0.045 mL)、炭酸カリウム(196 mg)およびTHF(15 mL)を加え、混合物を80℃で30分間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(19.3 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.10 (3H, t, J = 7.4 Hz), 2.94 (2H, q, J = 7.2 Hz), 3.86 (2H, s), 4.84 (2H, q, J = 9.1 Hz), 6.23 (1H, s), 7.08-7.21 (2H, m), 7.25-7.33 (2H, m), 7.38 (2H, d, J = 6.1 Hz), 8.52 (2H, d, J = 6.1 Hz).
実施例55
2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-オキソ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパンアミド
3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-オキソプロパン酸エチル(2.8 g)および4-(2,2,2-トリフルオロエトキシ)アニリン(1.95 g)のm-キシレン溶液(30 mL)を140℃で終夜撹拌した。反応混合物を室温に冷却し、析出した固体を濾取、ジイソプロピルエーテルで洗浄して標題化合物(2.67g)を得た。
MS (ESI+): [M+H]+ 448.3
3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-オキソプロパン酸エチル(2.8 g)および4-(2,2,2-トリフルオロエトキシ)アニリン(1.95 g)のm-キシレン溶液(30 mL)を140℃で終夜撹拌した。反応混合物を室温に冷却し、析出した固体を濾取、ジイソプロピルエーテルで洗浄して標題化合物(2.67g)を得た。
MS (ESI+): [M+H]+ 448.3
B) (2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド
3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-オキソ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパンアミド(2 g)、ギ酸アンモニウム(1.42 g)およびエタノール(30 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥、溶媒を減圧下留去し標題化合物(1.80 g)を得た。
MS (ESI+): [M+H]+ 447.1.
3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-オキソ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパンアミド(2 g)、ギ酸アンモニウム(1.42 g)およびエタノール(30 mL)の反応混合物を70℃で終夜撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥、溶媒を減圧下留去し標題化合物(1.80 g)を得た。
MS (ESI+): [M+H]+ 447.1.
C) 2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(800 mg)のDMA溶液(10 mL)に、ブチリルクロリド(0.21 mL)を加え、100℃で3時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をトリフルオロ酢酸(5 mL)に溶解し、70℃で8時間撹拌した。反応溶媒を減圧下留去し、飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(320 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.82 (3H, t, J = 7.4 Hz), 1.66 (2H, d, J = 7.6 Hz), 2.28 (2H, t, J = 7.6 Hz), 4.86 (2H, d, J = 9.1 Hz), 6.67 (1H, s), 7.15-7.24 (2H, m), 7.29-7.39 (2H, m), 8.05 (1H, brs), 8.34 (1H, brs), 13.19 (1H, brs).
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(800 mg)のDMA溶液(10 mL)に、ブチリルクロリド(0.21 mL)を加え、100℃で3時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をトリフルオロ酢酸(5 mL)に溶解し、70℃で8時間撹拌した。反応溶媒を減圧下留去し、飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(320 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.82 (3H, t, J = 7.4 Hz), 1.66 (2H, d, J = 7.6 Hz), 2.28 (2H, t, J = 7.6 Hz), 4.86 (2H, d, J = 9.1 Hz), 6.67 (1H, s), 7.15-7.24 (2H, m), 7.29-7.39 (2H, m), 8.05 (1H, brs), 8.34 (1H, brs), 13.19 (1H, brs).
実施例56
2-(プロピルアミノ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(プロピルアミノ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-(プロピルアミノ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(930 mg)のアセトニトリル溶液(20 mL)にm-クロロ過安息香酸(621 mg)を0℃で加えた。反応混合物を室温で3時間撹拌した後、プロパン-1-アミン(523 mg)を加え、さらに1時間撹拌した。反応溶媒を減圧下留去し、飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(NH、酢酸エチル/ヘキサン)で精製して標題化合物(630 mg)を得た。
MS (ESI+): [M+H]+ 514.1.
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(930 mg)のアセトニトリル溶液(20 mL)にm-クロロ過安息香酸(621 mg)を0℃で加えた。反応混合物を室温で3時間撹拌した後、プロパン-1-アミン(523 mg)を加え、さらに1時間撹拌した。反応溶媒を減圧下留去し、飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(NH、酢酸エチル/ヘキサン)で精製して標題化合物(630 mg)を得た。
MS (ESI+): [M+H]+ 514.1.
B) 2-(プロピルアミノ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-(プロピルアミノ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(880 mg)をトリフルオロ酢酸(5 mL)に溶解し、70℃で終夜撹拌した。反応溶媒を減圧下留去し、飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、THF/ヘキサンから再結晶して標題化合物(500 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.82 (3H, t, J = 7.4 Hz), 1.43-1.58 (2H, m), 3.21-3.30 (2H, m), 4.84 (2H, q, J = 9.1 Hz), 6.02 (1H, t, J = 5.5 Hz), 6.07 (1H, s), 7.17-7.27 (4H, m), 7.98 (1H, brs), 8.24 (1H, brs), 13.10 (1H, brs).
6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-(プロピルアミノ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(880 mg)をトリフルオロ酢酸(5 mL)に溶解し、70℃で終夜撹拌した。反応溶媒を減圧下留去し、飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、THF/ヘキサンから再結晶して標題化合物(500 mg)を得た。
1H NMR (300 MHz, DMSO-d6) δ 0.82 (3H, t, J = 7.4 Hz), 1.43-1.58 (2H, m), 3.21-3.30 (2H, m), 4.84 (2H, q, J = 9.1 Hz), 6.02 (1H, t, J = 5.5 Hz), 6.07 (1H, s), 7.17-7.27 (4H, m), 7.98 (1H, brs), 8.24 (1H, brs), 13.10 (1H, brs).
実施例57
2-[3-(メチルスルホニル)プロポキシ]-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-[3-(メチルスルホニル)プロポキシ]-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-[3-(メチルスルファニル)プロポキシ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例42に準じた方法により、2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび3-(メチルチオ)プロパン-1-オールを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 561.1.
実施例42に準じた方法により、2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび3-(メチルチオ)プロパン-1-オールを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 561.1.
B) 6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-[3-(メチルスルホニル)プロポキシ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例50のB)に準じた方法により、6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-[3-(メチルスルファニル)プロポキシ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 593.0.
実施例50のB)に準じた方法により、6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-[3-(メチルスルファニル)プロポキシ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 593.0.
C) 2-[3-(メチルスルホニル)プロポキシ]-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-[3-(メチルスルホニル)プロポキシ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(790 mg)の酢酸溶液(20 mL)に、水酸化パラジウム(20 wt%、1.1 g)を加え、室温で2日間撹拌した。不溶物を濾去後、溶媒を減圧下留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(350 mg)を得た。
MS (ESI+): [M+H]+ 473.0.
6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-[3-(メチルスルホニル)プロポキシ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(790 mg)の酢酸溶液(20 mL)に、水酸化パラジウム(20 wt%、1.1 g)を加え、室温で2日間撹拌した。不溶物を濾去後、溶媒を減圧下留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(350 mg)を得た。
MS (ESI+): [M+H]+ 473.0.
実施例58
2-(エチルスルファニル)-4-(1H-ピラゾール-4-イル)-2'-(2,2,2-トリフルオロエトキシ)-6H-1,5'-ビピリミジン-6-オン
2-(エチルスルファニル)-4-(1H-ピラゾール-4-イル)-2'-(2,2,2-トリフルオロエトキシ)-6H-1,5'-ビピリミジン-6-オン
A) 2-(2,2,2-トリフルオロエトキシ)ピリミジン-5-アミン
2,2,2-トリフルオロエタノール(11.1 g)のDMF溶液(160 mL)に水素化ナトリウム(60% oil dispersion、3.32 g)を0℃で加えた。同温度で10分間撹拌した後、2-クロロ-5-ニトロピリミジン(8.82 g)を加え、得られた反応混合物を同温度で1時間撹拌した。反応混合物を水に加え、酢酸エチルで抽出した。抽出液を10%クエン酸水溶液および水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をメタノール(150 mL)に溶解し、10%パラジウム/活性炭(50% 含水,1.50 g)を加え、水素雰囲気下、室温で5時間撹拌した。不溶物を濾去後、濾液を減圧下留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、標題化合物(2.70 g)を得た。
MS (ESI+): [M+H]+ 194.1.
2,2,2-トリフルオロエタノール(11.1 g)のDMF溶液(160 mL)に水素化ナトリウム(60% oil dispersion、3.32 g)を0℃で加えた。同温度で10分間撹拌した後、2-クロロ-5-ニトロピリミジン(8.82 g)を加え、得られた反応混合物を同温度で1時間撹拌した。反応混合物を水に加え、酢酸エチルで抽出した。抽出液を10%クエン酸水溶液および水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をメタノール(150 mL)に溶解し、10%パラジウム/活性炭(50% 含水,1.50 g)を加え、水素雰囲気下、室温で5時間撹拌した。不溶物を濾去後、濾液を減圧下留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、標題化合物(2.70 g)を得た。
MS (ESI+): [M+H]+ 194.1.
B) 5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリミジン
2-(2,2,2-トリフルオロエトキシ)ピリミジン-5-アミン(2.70 g)のTHF溶液(25 mL)に0℃でチオホスゲン(2.41 g)を滴下し、得られた反応混合物を室温で4時間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(2.92 g)を得た。
MS (ESI+): [M+H]+ 236.1.
2-(2,2,2-トリフルオロエトキシ)ピリミジン-5-アミン(2.70 g)のTHF溶液(25 mL)に0℃でチオホスゲン(2.41 g)を滴下し、得られた反応混合物を室温で4時間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(2.92 g)を得た。
MS (ESI+): [M+H]+ 236.1.
C) 2-(エチルスルファニル)-4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2'-(2,2,2-トリフルオロエトキシ)-6H-1,5'-ビピリミジン-6-オン
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]プロパ-2-エン酸エチルおよび5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリミジンを用いて反応を行い、4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-2'-(2,2,2-トリフルオロエトキシ)-2,3-ジヒドロ-6H-1,5'-ビピリミジン-6-オンを得た(MS (ESI+): [M+H]+ 519.2)。また副生成物として2'-エトキシ-4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-2,3-ジヒドロ-6H-1,5'-ビピリミジン-6-オンを得た(MS (ESI+): [M+H]+ 465.2)。ついで4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-2'-(2,2,2-トリフルオロエトキシ)-2,3-ジヒドロ-6H-1,5'-ビピリミジン-6-オンを用い、実施例1のI)に準じた方法により標題化合物を得た。
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]プロパ-2-エン酸エチルおよび5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリミジンを用いて反応を行い、4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-2'-(2,2,2-トリフルオロエトキシ)-2,3-ジヒドロ-6H-1,5'-ビピリミジン-6-オンを得た(MS (ESI+): [M+H]+ 519.2)。また副生成物として2'-エトキシ-4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-2,3-ジヒドロ-6H-1,5'-ビピリミジン-6-オンを得た(MS (ESI+): [M+H]+ 465.2)。ついで4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-2'-(2,2,2-トリフルオロエトキシ)-2,3-ジヒドロ-6H-1,5'-ビピリミジン-6-オンを用い、実施例1のI)に準じた方法により標題化合物を得た。
D) 2-(エチルスルファニル)-4-(1H-ピラゾール-4-イル)-2'-(2,2,2-トリフルオロエトキシ)-6H-1,5'-ビピリミジン-6-オン
実施例1のJ)に準じた方法により、2-(エチルスルファニル)-4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2'-(2,2,2-トリフルオロエトキシ)-6H-1,5'-ビピリミジン-6-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 398.9.
実施例1のJ)に準じた方法により、2-(エチルスルファニル)-4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2'-(2,2,2-トリフルオロエトキシ)-6H-1,5'-ビピリミジン-6-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 398.9.
実施例59
2'-エトキシ-2-(エチルスルファニル)-4-(1H-ピラゾール-4-イル)-6H-1,5'-ビピリミジン-6-オン
2'-エトキシ-2-(エチルスルファニル)-4-(1H-ピラゾール-4-イル)-6H-1,5'-ビピリミジン-6-オン
A) 2'-エトキシ-2-(エチルスルファニル)-4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-6H-1,5'-ビピリミジン-6-オン
実施例58のC)で得られた2'-エトキシ-4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-2,3-ジヒドロ-6H-1,5'-ビピリミジン-6-オンを用い、実施例1のI)に準じた方法により標題化合物を得た。
MS (ESI+): [M+H]+ 465.2.
実施例58のC)で得られた2'-エトキシ-4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-2,3-ジヒドロ-6H-1,5'-ビピリミジン-6-オンを用い、実施例1のI)に準じた方法により標題化合物を得た。
MS (ESI+): [M+H]+ 465.2.
B) 2'-エトキシ-2-(エチルスルファニル)-4-(1H-ピラゾール-4-イル)-6H-1,5'-ビピリミジン-6-オン
実施例1のJ)に準じた方法により、2'-エトキシ-2-(エチルスルファニル)-4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-6H-1,5'-ビピリミジン-6-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 345.1.
実施例1のJ)に準じた方法により、2'-エトキシ-2-(エチルスルファニル)-4-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-6H-1,5'-ビピリミジン-6-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 345.1.
実施例60
2-ブチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-ブチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-オキソ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパンアミド
3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-オキソプロパン酸エチル(10 g)および4-(2,2,2-トリフルオロエトキシ)アニリン(6.95 g)のm-キシレン溶液(100 mL)を140℃で終夜撹拌した。反応混合物を室温まで冷却後、得られた沈殿物を濾取し、標題化合物を得た。
MS (ESI+): [M+H]+ 448.0.
3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-オキソプロパン酸エチル(10 g)および4-(2,2,2-トリフルオロエトキシ)アニリン(6.95 g)のm-キシレン溶液(100 mL)を140℃で終夜撹拌した。反応混合物を室温まで冷却後、得られた沈殿物を濾取し、標題化合物を得た。
MS (ESI+): [M+H]+ 448.0.
B) (2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド
実施例1のG)に準じた方法により、3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-オキソ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパンアミドを用いて標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 3.74 (3H, s), 4.67 (2H, q, J = 8.8 Hz), 4.97 (1H, s), 5.27 (2H, s), 6.89-6.98 (4H, m), 7.22-7.29 (2H, m), 7.49-7.57 (2H, m), 7.74 (1H, s), 8.07 (1H, s), 9.23 (1H, s).
実施例1のG)に準じた方法により、3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-オキソ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパンアミドを用いて標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 3.74 (3H, s), 4.67 (2H, q, J = 8.8 Hz), 4.97 (1H, s), 5.27 (2H, s), 6.89-6.98 (4H, m), 7.22-7.29 (2H, m), 7.49-7.57 (2H, m), 7.74 (1H, s), 8.07 (1H, s), 9.23 (1H, s).
C) 2-ブチル-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(2 g)および塩化ペンタノイル(2.7 g)のDMA溶液(30 mL)を100℃で終夜撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(1.72 g)を得た。
MS (ESI+): [M+H]+ 513.3.
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(2 g)および塩化ペンタノイル(2.7 g)のDMA溶液(30 mL)を100℃で終夜撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(1.72 g)を得た。
MS (ESI+): [M+H]+ 513.3.
D) 2-ブチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のJ)に準じた方法により、2-ブチル-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 393.1.
実施例1のJ)に準じた方法により、2-ブチル-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 393.1.
実施例61
2-(メトキシメチル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(メトキシメチル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-(メトキシメチル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例60のC)に準じた方法により、(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミドおよび塩化2-メトキシアセチルを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 501.4.
実施例60のC)に準じた方法により、(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミドおよび塩化2-メトキシアセチルを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 501.4.
B) 2-(メトキシメチル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のJ)に準じた方法により、6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-(メトキシメチル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 381.1.
実施例1のJ)に準じた方法により、6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-(メトキシメチル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 381.1.
実施例62
6-(1H-ピラゾール-4-イル)-2-(テトラヒドロ-2H-ピラン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1H-ピラゾール-4-イル)-2-(テトラヒドロ-2H-ピラン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-(テトラヒドロ-2H-ピラン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例60のC)に準じた方法により、(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミドおよび塩化テトラヒドロ-2H-ピラン-4-カルボニルを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 541.1.
実施例60のC)に準じた方法により、(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミドおよび塩化テトラヒドロ-2H-ピラン-4-カルボニルを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 541.1.
B) 6-(1H-ピラゾール-4-イル)-2-(テトラヒドロ-2H-ピラン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のJ)に準じた方法により、6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-(テトラヒドロ-2H-ピラン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 421.1.
実施例1のJ)に準じた方法により、6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-(テトラヒドロ-2H-ピラン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 421.1.
実施例63
6-(1-メチル-1H-ピラゾール-5-イル)-2-プロピル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1-メチル-1H-ピラゾール-5-イル)-2-プロピル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-(1-メチル-1H-ピラゾール-5-イル)-3-オキソ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパンアミド
実施例55のA)に準じた方法により、3-(1-メチル-1H-ピラゾール-5-イル)-3-オキソプロパン酸エチルから標題化合物を得た。
MS (API-): [M-H]- 340.1.
実施例55のA)に準じた方法により、3-(1-メチル-1H-ピラゾール-5-イル)-3-オキソプロパン酸エチルから標題化合物を得た。
MS (API-): [M-H]- 340.1.
B) (2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド
実施例55のB)に準じた方法により、3-(1-メチル-1H-ピラゾール-5-イル)-3-オキソ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパンアミドから標題化合物を得た。
MS (ESI+): [M+H]+ 341.0.
実施例55のB)に準じた方法により、3-(1-メチル-1H-ピラゾール-5-イル)-3-オキソ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパンアミドから標題化合物を得た。
MS (ESI+): [M+H]+ 341.0.
C) 6-(1-メチル-1H-ピラゾール-5-イル)-2-プロピル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(200 mg)のDMA溶液(2 mL)に、塩化ブチリル(0.31 mL)を加え、マイクロ波照射下150℃で15分間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(35.1 mg)を得た。
MS (ESI+): [M+H]+ 393.1.
(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(200 mg)のDMA溶液(2 mL)に、塩化ブチリル(0.31 mL)を加え、マイクロ波照射下150℃で15分間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(35.1 mg)を得た。
MS (ESI+): [M+H]+ 393.1.
実施例64
2-(メトキシメチル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(メトキシメチル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例63のC)に準じた方法により、(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミドおよび塩化メトキシアセチルから標題化合物を得た。
MS (ESI+): [M+H]+ 395.3.
MS (ESI+): [M+H]+ 395.3.
実施例65
2-エチル-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(300 mg)のDMA溶液(3 mL)に、塩化プロパノイル(0.39 mL)を加え、100℃で4.5時間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(85.0 mg)を得た。
MS (ESI+): [M+H]+ 379.3.
2-エチル-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(300 mg)のDMA溶液(3 mL)に、塩化プロパノイル(0.39 mL)を加え、100℃で4.5時間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(85.0 mg)を得た。
MS (ESI+): [M+H]+ 379.3.
実施例66
2-エトキシ-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
2-エトキシ-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
A) 6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロピリミジン-4(1H)-オン
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]プロパ-2-エン酸エチルおよび5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 490.0.
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]プロパ-2-エン酸エチルおよび5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 490.0.
B) 2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロピリミジン-4(1H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 518.0.
実施例1のI)に準じた方法により6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロピリミジン-4(1H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 518.0.
C) 2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
実施例1のJ)に準じた方法により2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 396.1.
実施例1のJ)に準じた方法により2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 396.1.
D) 2-エトキシ-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(150 mg)、アセトニトリル(4 mL)および酢酸エチル溶液(4 mL)の混合物に、m-クロロ過安息香酸(261 mg)を0℃で加え、反応混合物を室温で5時間撹拌した。反応混合物を飽和炭酸水素ナトリウム溶液に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をTHF(10 mL)に溶解し、エタノール(4 mL)および水素化ナトリウム(60% oil dispersion、18 mg)を0℃で加えた。反応混合物を0℃で3時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(17 mg)を得た。
MS (ESI+): [M+H]+ 382.0.
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(150 mg)、アセトニトリル(4 mL)および酢酸エチル溶液(4 mL)の混合物に、m-クロロ過安息香酸(261 mg)を0℃で加え、反応混合物を室温で5時間撹拌した。反応混合物を飽和炭酸水素ナトリウム溶液に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をTHF(10 mL)に溶解し、エタノール(4 mL)および水素化ナトリウム(60% oil dispersion、18 mg)を0℃で加えた。反応混合物を0℃で3時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(17 mg)を得た。
MS (ESI+): [M+H]+ 382.0.
実施例67
2-エチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(1.77 g)のDMA溶液(10 mL)に、塩化プロパノイル(1.74 mL)を加え、100℃で1時間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製後、トリフルオロ酢酸(15 mL)に溶解し、マイクロ波照射下100℃で30分間撹拌した。反応溶媒を減圧下留去し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(370 mg)を得た。
MS (ESI+): [M+H]+ 365.2.
2-エチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(1.77 g)のDMA溶液(10 mL)に、塩化プロパノイル(1.74 mL)を加え、100℃で1時間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製後、トリフルオロ酢酸(15 mL)に溶解し、マイクロ波照射下100℃で30分間撹拌した。反応溶媒を減圧下留去し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(370 mg)を得た。
MS (ESI+): [M+H]+ 365.2.
実施例68
2-(1-メチルプロピル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(250 mg)のDMA溶液(4 mL)に、塩化2-メチルブタノイル(0.35 mL)を加え、マイクロ波照射下150℃で30分間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、得られた化合物をトリフルオロ酢酸(4 mL)に溶解後、マイクロ波照射下100℃で30分間撹拌した。反応溶媒を減圧下留去し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(20.1 mg)を得た。
MS (ESI+): [M+H]+ 393.1.
2-(1-メチルプロピル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(250 mg)のDMA溶液(4 mL)に、塩化2-メチルブタノイル(0.35 mL)を加え、マイクロ波照射下150℃で30分間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、得られた化合物をトリフルオロ酢酸(4 mL)に溶解後、マイクロ波照射下100℃で30分間撹拌した。反応溶媒を減圧下留去し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(20.1 mg)を得た。
MS (ESI+): [M+H]+ 393.1.
実施例69
6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(1 g)、アセトニトリル(10 mL)および酢酸エチル溶液(10 mL)の混合物に、m-クロロ過安息香酸(2.61 g)を0℃で加え、反応混合物を室温で5時間撹拌した。反応混合物を飽和炭酸水素ナトリウム溶液に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた固体を濾取し、標題化合物(1.05 g)を得た。
MS (API-): [M-H]- 427.3.
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(1 g)、アセトニトリル(10 mL)および酢酸エチル溶液(10 mL)の混合物に、m-クロロ過安息香酸(2.61 g)を0℃で加え、反応混合物を室温で5時間撹拌した。反応混合物を飽和炭酸水素ナトリウム溶液に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた固体を濾取し、標題化合物(1.05 g)を得た。
MS (API-): [M-H]- 427.3.
B) 6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(190 mg)および2,2,2-トリフルオロエタノール(222 mg)のTHF溶液(10 mL)に、水素化ナトリウム(60% oil dispersion、21 mg)を加えた。反応混合物を室温で1時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(130 mg)を得た。
MS (ESI+): [M+H]+ 434.8.
1H NMR (300 MHz, DMSO-d6) δ 4.84 (2H, q, J = 8.9 Hz), 5.14 (2H, q, J = 8.7 Hz), 6.59 (1H, s), 7.12-7.22 (2H, m), 7.27-7.35 (2H, m), 8.04-8.72 (2H, m), 13.27 (1H, brs).
2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(190 mg)および2,2,2-トリフルオロエタノール(222 mg)のTHF溶液(10 mL)に、水素化ナトリウム(60% oil dispersion、21 mg)を加えた。反応混合物を室温で1時間撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(130 mg)を得た。
MS (ESI+): [M+H]+ 434.8.
1H NMR (300 MHz, DMSO-d6) δ 4.84 (2H, q, J = 8.9 Hz), 5.14 (2H, q, J = 8.7 Hz), 6.59 (1H, s), 7.12-7.22 (2H, m), 7.27-7.35 (2H, m), 8.04-8.72 (2H, m), 13.27 (1H, brs).
実施例70
2-メトキシ-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよびメタノールを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 367.3.
2-メトキシ-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよびメタノールを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 367.3.
実施例71
6-(1-メチル-1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 449.3.
6-(1-メチル-1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 449.3.
実施例72
6-(1H-ピラゾール-4-イル)-2-(テトラヒドロフラン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
テトラヒドロフラン-3-カルボン酸(468 mg)のTHF溶液(2 mL)にDMF(5μL)と塩化オキサリル(0.35 mL)を0℃で加え、室温で30分間撹拌した。反応溶液を(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(300 mg)のDMA溶液(10 mL)に加え、マイクロ波照射下150℃で30分間攪拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、得られた化合物をトリフルオロ酢酸(4 mL)に溶解し、マイクロ波照射下100℃で30分間攪拌した。反応溶媒を減圧下留去し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(68.6 mg)を得た。
MS (ESI+): [M+H]+ 407.0.
6-(1H-ピラゾール-4-イル)-2-(テトラヒドロフラン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
テトラヒドロフラン-3-カルボン酸(468 mg)のTHF溶液(2 mL)にDMF(5μL)と塩化オキサリル(0.35 mL)を0℃で加え、室温で30分間撹拌した。反応溶液を(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(300 mg)のDMA溶液(10 mL)に加え、マイクロ波照射下150℃で30分間攪拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、得られた化合物をトリフルオロ酢酸(4 mL)に溶解し、マイクロ波照射下100℃で30分間攪拌した。反応溶媒を減圧下留去し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(68.6 mg)を得た。
MS (ESI+): [M+H]+ 407.0.
実施例73
6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のA)に準じた方法により2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 442.9.
実施例69のA)に準じた方法により2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 442.9.
B) 6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 449.0.
実施例69のB)に準じた方法により2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 449.0.
実施例74
6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
A) 2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により、6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロピリミジン-4(1H)-オンより標題化合物を得た。
MS (ESI+): [M+H]+ 518.3.
実施例1のI)に準じた方法により、6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-チオキソ-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロピリミジン-4(1H)-オンより標題化合物を得た。
MS (ESI+): [M+H]+ 518.3.
B) 2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(1.07 g)をトリフルオロ酢酸(5 mL)に溶かし、マイクロウェーブ照射下100℃で30分間攪拌した。溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(576 mg)を得た。
MS (ESI+): [M+H]+ 398.2.
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(1.07 g)をトリフルオロ酢酸(5 mL)に溶かし、マイクロウェーブ照射下100℃で30分間攪拌した。溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(576 mg)を得た。
MS (ESI+): [M+H]+ 398.2.
C) 2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(522 mg)を酢酸エチル(10 mL)およびアセトニトリル(10 mL)に溶かし、m-クロロ過安息香酸(1.36 g)を加え、室温で3時間半攪拌した。反応混合物に、2-メチル-2-ブテンを0℃で加え攪拌した。反応混合物を濃縮し、残渣を酢酸エチルに溶かし、飽和重曹水で三回洗浄した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去することで標題化合物(576 mg)を粗生成物として得た。
MS (API-): [M-H]- 428.0.
1H NMR (300 MHz, DMSO-d6) δ 1.32 (3H, t, J = 7.2 Hz), 3.83 (2H, qd, J = 7.2, 2.8 Hz), 4.83-5.20 (2H, m), 7.04 (1H, s), 7.14 (1H, d, J = 8.7 Hz), 7.90 (1H, dd, J = 9.1, 2.6 Hz), 8.07-8.38 (2H, m), 8.39-8.74 (1H, m), 13.42 (1H, brs).
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(522 mg)を酢酸エチル(10 mL)およびアセトニトリル(10 mL)に溶かし、m-クロロ過安息香酸(1.36 g)を加え、室温で3時間半攪拌した。反応混合物に、2-メチル-2-ブテンを0℃で加え攪拌した。反応混合物を濃縮し、残渣を酢酸エチルに溶かし、飽和重曹水で三回洗浄した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去することで標題化合物(576 mg)を粗生成物として得た。
MS (API-): [M-H]- 428.0.
1H NMR (300 MHz, DMSO-d6) δ 1.32 (3H, t, J = 7.2 Hz), 3.83 (2H, qd, J = 7.2, 2.8 Hz), 4.83-5.20 (2H, m), 7.04 (1H, s), 7.14 (1H, d, J = 8.7 Hz), 7.90 (1H, dd, J = 9.1, 2.6 Hz), 8.07-8.38 (2H, m), 8.39-8.74 (1H, m), 13.42 (1H, brs).
D) 6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
実施例2に準じた方法により2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(349 mg)および2,2,2-トリフルオロエタノール(0.292 mL)から合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物(174 mg)を得た。
MS (API-): [M-H]- 434.2.
1H NMR (300 MHz, DMSO-d6) δ 4.93-5.29 (4H, m), 6.62 (1H, s), 7.15 (1H, d, J = 8.7 Hz), 7.88 (1H, dd, J = 8.7, 2.6 Hz), 8.23 (1H, d, J = 2.6 Hz), 8.35 (2H, brs), 13.29 (1H, brs).
実施例2に準じた方法により2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(349 mg)および2,2,2-トリフルオロエタノール(0.292 mL)から合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物(174 mg)を得た。
MS (API-): [M-H]- 434.2.
1H NMR (300 MHz, DMSO-d6) δ 4.93-5.29 (4H, m), 6.62 (1H, s), 7.15 (1H, d, J = 8.7 Hz), 7.88 (1H, dd, J = 8.7, 2.6 Hz), 8.23 (1H, d, J = 2.6 Hz), 8.35 (2H, brs), 13.29 (1H, brs).
実施例75
2-(2,2-ジフルオロエトキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび2,2-ジフルオロエタノールを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 417.2.
2-(2,2-ジフルオロエトキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび2,2-ジフルオロエタノールを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 417.2.
実施例76
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび1,3-ジフルオロプロパン-2-オールを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 431.2.
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび1,3-ジフルオロプロパン-2-オールを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 431.2.
実施例77
2-(プロパン-2-イルオキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよびプロパン-2-オールを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 395.3.
2-(プロパン-2-イルオキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により2-(エチルスルホニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよびプロパン-2-オールを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 395.3.
実施例78
6-(1-メチル-1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
実施例3に準じた方法により、6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オンを用いて反応後、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)およびHPLC[C18、移動相:水/アセトニトリル(TFA含有系)]にて精製した。得られた固体を酢酸エチルおよびヘキサンより再結晶を行うことで標題化合物を得た。
MS (ESI+): [M+H]+ 450.2.
6-(1-メチル-1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
実施例3に準じた方法により、6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オンを用いて反応後、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)およびHPLC[C18、移動相:水/アセトニトリル(TFA含有系)]にて精製した。得られた固体を酢酸エチルおよびヘキサンより再結晶を行うことで標題化合物を得た。
MS (ESI+): [M+H]+ 450.2.
実施例79
2-(2-メトキシエトキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-メトキシエタノールを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 411.1.
2-(2-メトキシエトキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-メトキシエタノールを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 411.1.
実施例80
2-(2-フルオロエトキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-フルオロエタノールを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 399.1.
2-(2-フルオロエトキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-フルオロエタノールを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 399.1.
実施例81
2-(2,2-ジフルオロエトキシ)-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-(2,2-ジフルオロエトキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 431.0.
2-(2,2-ジフルオロエトキシ)-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-(2,2-ジフルオロエトキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 431.0.
実施例82
2-(2-メトキシエトキシ)-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-(2-メトキシエトキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、THF/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 425.2.
2-(2-メトキシエトキシ)-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-(2-メトキシエトキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、THF/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 425.2.
実施例83
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 445.0.
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 445.0.
実施例84
6-(1-メチル-1H-ピラゾール-4-イル)-2-(プロパン-2-イルオキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-(プロパン-2-イルオキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 409.1.
6-(1-メチル-1H-ピラゾール-4-イル)-2-(プロパン-2-イルオキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-(プロパン-2-イルオキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 409.1.
実施例85
2-(2-フルオロエトキシ)-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-(2-フルオロエトキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、THF/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 413.1.
2-(2-フルオロエトキシ)-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-(2-フルオロエトキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、THF/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 413.1.
実施例86
6-(1,3-チアゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1,3-チアゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 2-(エチルスルホニル)-6-(1,3-チアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1,3-チアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(802 mg)の酢酸エチル(20 mL)/アセトニトリル(20 mL)溶液にm-クロロ過安息香酸(2.87 g)を室温で加え、5時間撹拌した。反応混合物に2-メチル-2-ブテンおよび飽和炭酸水素ナトリウム水溶液を0℃で加え、酢酸エチルで抽出した。抽出液を飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄後、硫酸マグネシウムで乾燥し、減圧濃縮した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(756 mg)を固体として得た。
MS (ESI+): [M+H]+ 446.1.
2-(エチルスルファニル)-6-(1,3-チアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(802 mg)の酢酸エチル(20 mL)/アセトニトリル(20 mL)溶液にm-クロロ過安息香酸(2.87 g)を室温で加え、5時間撹拌した。反応混合物に2-メチル-2-ブテンおよび飽和炭酸水素ナトリウム水溶液を0℃で加え、酢酸エチルで抽出した。抽出液を飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄後、硫酸マグネシウムで乾燥し、減圧濃縮した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(756 mg)を固体として得た。
MS (ESI+): [M+H]+ 446.1.
B) 6-(1,3-チアゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1,3-チアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 452.1.
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1,3-チアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 452.1.
実施例87
2-エトキシ-6-(1,3-チアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1,3-チアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよびエタノールを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 398.1.
2-エトキシ-6-(1,3-チアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1,3-チアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよびエタノールを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 398.1.
実施例88
2-(2-ヒドロキシ-2-メチルプロポキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(2-ヒドロキシ-2-メチルプロポキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) ({6-オキソ-4-(1H-ピラゾール-4-イル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,6-ジヒドロピリミジン-2-イル}オキシ)酢酸エチル
実施例69のB)に準じた方法により、ヒドロキシ酢酸エチルを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 439.1.
実施例69のB)に準じた方法により、ヒドロキシ酢酸エチルを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 439.1.
B) 2-(2-ヒドロキシ-2-メチルプロポキシ)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
({6-オキソ-4-(1H-ピラゾール-4-イル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,6-ジヒドロピリミジン-2-イル}オキシ)酢酸エチル(63.0 mg)のTHF(3 mL)溶液にメチルマグネシウム ブロミドのTHF溶液(1.0M, 0.998 mL)を0℃で加え、室温で4時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を1規定塩酸および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、THF/ヘキサンから再結晶して標題化合物(26.0 mg)を固体として得た。
MS (ESI+): [M+H]+ 425.2.
({6-オキソ-4-(1H-ピラゾール-4-イル)-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,6-ジヒドロピリミジン-2-イル}オキシ)酢酸エチル(63.0 mg)のTHF(3 mL)溶液にメチルマグネシウム ブロミドのTHF溶液(1.0M, 0.998 mL)を0℃で加え、室温で4時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を1規定塩酸および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、THF/ヘキサンから再結晶して標題化合物(26.0 mg)を固体として得た。
MS (ESI+): [M+H]+ 425.2.
実施例89
6-(1H-ピラゾール-3-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1H-ピラゾール-3-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 2-(エチルスルホニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例86のA)に準じた方法により、2-(エチルスルファニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 429.1.
実施例86のA)に準じた方法により、2-(エチルスルファニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 429.1.
B) 6-(1H-ピラゾール-3-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 435.1.
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 435.1.
実施例90
5-メチル-2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
5-メチル-2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 5-ブロモ-2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(210 mg)および酢酸ナトリウム(50 mg)の酢酸溶液(2 mL)に、臭素(49 mg)の酢酸溶液(1 mL)を加えた。反応混合物を室温で30分間撹拌した後、反応混合物を飽和炭酸水素ナトリウム溶液に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた固体を濾取し、標題化合物(190 mg)を得た。
MS (ESI+): [M+H]+ 457.1.
2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(210 mg)および酢酸ナトリウム(50 mg)の酢酸溶液(2 mL)に、臭素(49 mg)の酢酸溶液(1 mL)を加えた。反応混合物を室温で30分間撹拌した後、反応混合物を飽和炭酸水素ナトリウム溶液に加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた固体を濾取し、標題化合物(190 mg)を得た。
MS (ESI+): [M+H]+ 457.1.
B) 5-ブロモ-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-プロピル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
5-ブロモ-2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(162 mg)および1-(クロロメチル)-4-メトキシベンゼン(67 mg)のDMF溶液(3 mL)に、水素化ナトリウム(60% oil dispersion、10 mg)を加えた。反応混合物を室温で終夜撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(140 mg)を得た。
MS (ESI+): [M+H]+ 577.3.
5-ブロモ-2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(162 mg)および1-(クロロメチル)-4-メトキシベンゼン(67 mg)のDMF溶液(3 mL)に、水素化ナトリウム(60% oil dispersion、10 mg)を加えた。反応混合物を室温で終夜撹拌後、反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(140 mg)を得た。
MS (ESI+): [M+H]+ 577.3.
C) 6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-5-メチル-2-プロピル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
5-ブロモ-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-プロピル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(140 mg)、メチルボロン酸(29 mg)、炭酸カリウム(101 mg)、ジシクロヘキシル(2’,4’,6’-トリイソプロピルビフェニル-2-イル)ホスフィン(12 mg)、トリス(ジベンジリデンアセトン)ジパラジウム(0)(6 mg)、THF(4 mL)および水(2 mL)の反応混合物をマイクロウェーブ照射下120℃で2時間攪拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物(70 mg)を得た。
MS (ESI+): [M+H]+ 513.2.
5-ブロモ-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-2-プロピル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(140 mg)、メチルボロン酸(29 mg)、炭酸カリウム(101 mg)、ジシクロヘキシル(2’,4’,6’-トリイソプロピルビフェニル-2-イル)ホスフィン(12 mg)、トリス(ジベンジリデンアセトン)ジパラジウム(0)(6 mg)、THF(4 mL)および水(2 mL)の反応混合物をマイクロウェーブ照射下120℃で2時間攪拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物(70 mg)を得た。
MS (ESI+): [M+H]+ 513.2.
D) 5-メチル-2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のJ)に準じた方法により6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-5-メチル-2-プロピル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 393.1.
実施例1のJ)に準じた方法により6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-5-メチル-2-プロピル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 393.1.
実施例91
5-メチル-6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
5-メチル-6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 5-ブロモ-2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(800 mg)および酢酸ナトリウム(174 mg)を酢酸(15 mL)に溶かし、そこへ臭素(0.109 mL)を加えて室温で40分間攪拌した。その後、酢酸ナトリウム(83 mg)および臭素(0.052 mL)を加えて、再び室温で30分間攪拌した。反応混合物に飽和チオ硫酸ナトリウム水溶液を加えて激しく攪拌した後、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(739 mg)を得た。
MS (ESI+): [M+H]+ 475.0.
2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(800 mg)および酢酸ナトリウム(174 mg)を酢酸(15 mL)に溶かし、そこへ臭素(0.109 mL)を加えて室温で40分間攪拌した。その後、酢酸ナトリウム(83 mg)および臭素(0.052 mL)を加えて、再び室温で30分間攪拌した。反応混合物に飽和チオ硫酸ナトリウム水溶液を加えて激しく攪拌した後、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(739 mg)を得た。
MS (ESI+): [M+H]+ 475.0.
B) 5-ブロモ-2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
5-ブロモ-2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(735 mg)および炭酸カリウム(321 mg)をDMF(7 mL)に溶かし、そこへ4-メトキシベンジルクロリド(0.231 mL)を加えて50℃で3時間半攪拌した。反応混合物に水を加えて、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣を酢酸エチルおよびヘキサンから再結晶し、標題化合物(529 mg)を得た。
MS (ESI+): [M+H]+ 595.0.
5-ブロモ-2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(735 mg)および炭酸カリウム(321 mg)をDMF(7 mL)に溶かし、そこへ4-メトキシベンジルクロリド(0.231 mL)を加えて50℃で3時間半攪拌した。反応混合物に水を加えて、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、減圧濃縮した。残渣を酢酸エチルおよびヘキサンから再結晶し、標題化合物(529 mg)を得た。
MS (ESI+): [M+H]+ 595.0.
C) 2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル)-5-メチル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
5-ブロモ-2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(537 mg)、メチルボロン酸(108 mg)、炭酸カリウム(374 mg)、ジシクロヘキシル(2’,4’,6’-トリイソプロピルビフェニル-2-イル)ホスフィン(172 mg)、トリス(ジベンジリデンアセトン)ジパラジウム(0)(83 mg)、THF(8 mL)および水(2 mL)の反応混合物をマイクロウェーブ照射下100℃で30分間攪拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物(363 mg)を得た。
MS (ESI+): [M+H]+ 531.2.
5-ブロモ-2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(537 mg)、メチルボロン酸(108 mg)、炭酸カリウム(374 mg)、ジシクロヘキシル(2’,4’,6’-トリイソプロピルビフェニル-2-イル)ホスフィン(172 mg)、トリス(ジベンジリデンアセトン)ジパラジウム(0)(83 mg)、THF(8 mL)および水(2 mL)の反応混合物をマイクロウェーブ照射下100℃で30分間攪拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物(363 mg)を得た。
MS (ESI+): [M+H]+ 531.2.
D) 2-(エチルスルファニル)-5-メチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル)-5-メチル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(356 mg)をトリフルオロ酢酸(2.5 mL)に溶かして、マイクロウェーブ照射下100℃で30分間攪拌した。溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(188 mg)を得た。
MS (ESI+): [M+H]+ 411.1.
2-(エチルスルファニル)-6-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル)-5-メチル-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(356 mg)をトリフルオロ酢酸(2.5 mL)に溶かして、マイクロウェーブ照射下100℃で30分間攪拌した。溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(188 mg)を得た。
MS (ESI+): [M+H]+ 411.1.
E) 2-(エチルスルホニル)-5-メチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例74のC)に準じた方法により、2-(エチルスルファニル)-5-メチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
MS (API-): [M-H]- 441.2.
1H NMR (300 MHz, CDCl3) δ 1.44-1.53 (3H, m), 2.29-2.42 (3H, m), 3.58-3.75 (2H, m), 4.31-4.45 (2H, m), 6.96-7.16 (2H, m), 7.28-7.41 (2H, m), 8.02-8.11 (2H, m).
実施例74のC)に準じた方法により、2-(エチルスルファニル)-5-メチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンから標題化合物を得た。
MS (API-): [M-H]- 441.2.
1H NMR (300 MHz, CDCl3) δ 1.44-1.53 (3H, m), 2.29-2.42 (3H, m), 3.58-3.75 (2H, m), 4.31-4.45 (2H, m), 6.96-7.16 (2H, m), 7.28-7.41 (2H, m), 8.02-8.11 (2H, m).
F) 5-メチル-6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例2に準じた方法により、2-(エチルスルホニル)-5-メチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび2,2,2-トリフルオロエタノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (API-): [M-H]- 447.1.
1H NMR (300 MHz, CDCl3) δ 2.29 (3H, s), 4.40 (2H, q, J = 7.9 Hz), 4.78 (2H, q, J = 8.3 Hz), 7.02-7.10 (2H, m), 7.14-7.23 (2H, m), 8.12 (2H, s), 10.39 (1H, brs).
実施例2に準じた方法により、2-(エチルスルホニル)-5-メチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび2,2,2-トリフルオロエタノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (API-): [M-H]- 447.1.
1H NMR (300 MHz, CDCl3) δ 2.29 (3H, s), 4.40 (2H, q, J = 7.9 Hz), 4.78 (2H, q, J = 8.3 Hz), 7.02-7.10 (2H, m), 7.14-7.23 (2H, m), 8.12 (2H, s), 10.39 (1H, brs).
実施例92
2-エトキシ-5-メチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例2に準じた方法により、2-(エチルスルホニル)-5-メチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよびエタノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (ESI+): [M+H]+ 395.3.
2-エトキシ-5-メチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例2に準じた方法により、2-(エチルスルホニル)-5-メチル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよびエタノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (ESI+): [M+H]+ 395.3.
実施例93
2-エトキシ-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよびエタノールを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、THF/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 381.2.
2-エトキシ-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよびエタノールを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、THF/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 381.2.
実施例94
6-(1-メチル-1H-ピラゾール-3-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により6-(1H-ピラゾール-3-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 449.1.
6-(1-メチル-1H-ピラゾール-3-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により6-(1H-ピラゾール-3-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 449.1.
実施例95
2-エトキシ-6-(1-メチル-1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-エトキシ-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 395.2.
2-エトキシ-6-(1-メチル-1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-エトキシ-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 395.2.
実施例96
2-(2,2-ジフルオロエトキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(2,2-ジフルオロエトキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(5.00 g)のアセトニトリル(60 mL)/酢酸エチル(60 mL)溶液にm-クロロ過安息香酸(21.0 g)を0℃で加え、室温で4.5時間撹拌した後、さらにm-クロロ過安息香酸(10.5 g)を0℃で加え、室温で4時間撹拌した。反応混合物に2-メチル-2-ブテンおよび飽和炭酸水素ナトリウム水溶液を0℃で加えた。同様に、2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(5.00 g)のアセトニトリル(60 mL)/酢酸エチル(60 mL)溶液にm-クロロ過安息香酸(21.0 g)を0℃で加え、室温で4.5時間撹拌した後、さらにm-クロロ過安息香酸(10.5 g)を0℃で加え、室温で4時間撹拌した。反応混合物に2-メチル-2-ブテンおよび飽和炭酸水素ナトリウム水溶液を0℃で加えた。各々を混合し、酢酸エチルで抽出した。抽出液を飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(10.1 g)を固体として得た。
MS (ESI+): [M+H]+ 443.0.
1H NMR (300 MHz, DMSO-d6) δ 1.28 (3H, t, J = 7.4 Hz), 3.69 (2H, q, J = 7.6 Hz), 4.14 (3H, s), 4.86 (2H, q, J = 9.1 Hz), 7.01 (1H, d, J = 1.9 Hz), 7.08-7.27 (3H, m), 7.31-7.48 (2H, m), 7.56 (1H, d, J = 1.9 Hz).
2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(5.00 g)のアセトニトリル(60 mL)/酢酸エチル(60 mL)溶液にm-クロロ過安息香酸(21.0 g)を0℃で加え、室温で4.5時間撹拌した後、さらにm-クロロ過安息香酸(10.5 g)を0℃で加え、室温で4時間撹拌した。反応混合物に2-メチル-2-ブテンおよび飽和炭酸水素ナトリウム水溶液を0℃で加えた。同様に、2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(5.00 g)のアセトニトリル(60 mL)/酢酸エチル(60 mL)溶液にm-クロロ過安息香酸(21.0 g)を0℃で加え、室温で4.5時間撹拌した後、さらにm-クロロ過安息香酸(10.5 g)を0℃で加え、室温で4時間撹拌した。反応混合物に2-メチル-2-ブテンおよび飽和炭酸水素ナトリウム水溶液を0℃で加えた。各々を混合し、酢酸エチルで抽出した。抽出液を飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(10.1 g)を固体として得た。
MS (ESI+): [M+H]+ 443.0.
1H NMR (300 MHz, DMSO-d6) δ 1.28 (3H, t, J = 7.4 Hz), 3.69 (2H, q, J = 7.6 Hz), 4.14 (3H, s), 4.86 (2H, q, J = 9.1 Hz), 7.01 (1H, d, J = 1.9 Hz), 7.08-7.27 (3H, m), 7.31-7.48 (2H, m), 7.56 (1H, d, J = 1.9 Hz).
B) 2-(2,2-ジフルオロエトキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび2,2-ジフルオロエタノールを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 431.2.
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび2,2-ジフルオロエタノールを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 431.2.
実施例97
2-(1,1-ジフルオロプロピル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2,2-ジフルオロブタン酸およびDMF(0.017 mL)をTHF(1 mL)に溶かし、塩化オキサリルを0℃で加え、反応混合物を室温で1時間撹拌した。反応混合液を(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]アクリルアミド(200 mg)のDMA(3 mL)溶液に加えて、50℃で終夜攪拌後、反応溶液をマイクロウェーブ照射下150℃で30分間攪拌した。反応混合物に飽和重曹水を加えて、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して黄色固体を得た。得られた黄色固体をTFA(3 mL)に溶かし、マイクロウェーブ照射下100℃で30分間攪拌した。反応液を濃縮し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物(27.3 mg)を得た。
MS (ESI+): [M+H]+ 415.0.
2-(1,1-ジフルオロプロピル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2,2-ジフルオロブタン酸およびDMF(0.017 mL)をTHF(1 mL)に溶かし、塩化オキサリルを0℃で加え、反応混合物を室温で1時間撹拌した。反応混合液を(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]アクリルアミド(200 mg)のDMA(3 mL)溶液に加えて、50℃で終夜攪拌後、反応溶液をマイクロウェーブ照射下150℃で30分間攪拌した。反応混合物に飽和重曹水を加えて、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して黄色固体を得た。得られた黄色固体をTFA(3 mL)に溶かし、マイクロウェーブ照射下100℃で30分間攪拌した。反応液を濃縮し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物(27.3 mg)を得た。
MS (ESI+): [M+H]+ 415.0.
実施例98
2-(エチルスルファニル)-6-(1H-1,2,4-トリアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1H-1,2,4-トリアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-オキソ-3-(1-トリチル-1H-1,2,4-トリアゾール-3-イル)プロパン酸エチル
実施例1のF)に準じた方法により1-トリチル-1H-1,2,4-トリアゾール-3-カルボン酸を用いて標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.08 (3H, t, J = 7.2 Hz), 3.95-4.07 (4H, m), 7.02-7.12 (6H, m), 7.36-7.45 (9H, m), 8.41 (1H, s).
実施例1のF)に準じた方法により1-トリチル-1H-1,2,4-トリアゾール-3-カルボン酸を用いて標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.08 (3H, t, J = 7.2 Hz), 3.95-4.07 (4H, m), 7.02-7.12 (6H, m), 7.36-7.45 (9H, m), 8.41 (1H, s).
B) (2Z)-3-アミノ-3-(1-トリチル-1H-1,2,4-トリアゾール-3-イル)プロパ-2-エン酸エチル
実施例1のG)に準じた方法により3-オキソ-3-(1-トリチル-1H-1,2,4-トリアゾール-3-イル)プロパン酸エチルを用いて標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ1.18 (3H, t, J = 6.8 Hz), 4.05 (2H, q, J = 7.1 Hz), 5.28 (1H, s), 6.92 (1H, brs), 7.05-7.13 (6H, m), 7.36-7.45 (9H, m), 7.62 (1H, brs), 8.28 (1H, s).
実施例1のG)に準じた方法により3-オキソ-3-(1-トリチル-1H-1,2,4-トリアゾール-3-イル)プロパン酸エチルを用いて標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ1.18 (3H, t, J = 6.8 Hz), 4.05 (2H, q, J = 7.1 Hz), 5.28 (1H, s), 6.92 (1H, brs), 7.05-7.13 (6H, m), 7.36-7.45 (9H, m), 7.62 (1H, brs), 8.28 (1H, s).
C) 2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-トリチル-1H-1,2,4-トリアゾール-3-イル)-2,3-ジヒドロピリミジン-4(1H)-オン
実施例1のH)に準じた方法により(2Z)-3-アミノ-3-(1-トリチル-1H-1,2,4-トリアゾール-3-イル)プロパ-2-エン酸エチルを用いて標題化合物を得た。
MS (API-): [M-H]- 610.3.
実施例1のH)に準じた方法により(2Z)-3-アミノ-3-(1-トリチル-1H-1,2,4-トリアゾール-3-イル)プロパ-2-エン酸エチルを用いて標題化合物を得た。
MS (API-): [M-H]- 610.3.
D) 2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-トリチル-1H-1,2,4-トリアゾール-3-イル)ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-トリチル-1H-1,2,4-トリアゾール-3-イル)-2,3-ジヒドロピリミジン-4(1H)-オンを用いて標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ1.24 (3H, t, J = 7.4 Hz), 3.06 (2H, q, J = 7.2 Hz), 4.86 (2H, q, J = 8.9 Hz), 6.76 (1H, s), 7.08-7.16 (6H, m), 7.20 (2H, d, J = 8.7 Hz), 7.32-7.46 (11H, m), 8.31 (1H, s).
実施例1のI)に準じた方法により2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-トリチル-1H-1,2,4-トリアゾール-3-イル)-2,3-ジヒドロピリミジン-4(1H)-オンを用いて標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ1.24 (3H, t, J = 7.4 Hz), 3.06 (2H, q, J = 7.2 Hz), 4.86 (2H, q, J = 8.9 Hz), 6.76 (1H, s), 7.08-7.16 (6H, m), 7.20 (2H, d, J = 8.7 Hz), 7.32-7.46 (11H, m), 8.31 (1H, s).
E) 2-(エチルスルファニル)-6-(1H-1,2,4-トリアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のJ)に準じた方法により2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-トリチル-1H-1,2,4-トリアゾール-3-イル)ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 398.3.
実施例1のJ)に準じた方法により2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-トリチル-1H-1,2,4-トリアゾール-3-イル)ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 398.3.
実施例99
6-(1H-1,2,4-トリアゾール-3-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1H-1,2,4-トリアゾール-3-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 2-(エチルスルフィニル)-6-(1H-1,2,4-トリアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のA)に準じた方法により2-(エチルスルファニル)-6-(1H-1,2,4-トリアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 414.1
実施例69のA)に準じた方法により2-(エチルスルファニル)-6-(1H-1,2,4-トリアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 414.1
B) 6-(1H-1,2,4-トリアゾール-3-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により2-(エチルスルフィニル)-6-(1H-1,2,4-トリアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (API-): [M-H]- 434.2.
実施例69のB)に準じた方法により2-(エチルスルフィニル)-6-(1H-1,2,4-トリアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (API-): [M-H]- 434.2.
実施例100
2-(1-メトキシプロピル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例97に準じた方法により2-メトキシブタン酸から標題化合物を得た。
MS (ESI+): [M+H]+ 409.3.
2-(1-メトキシプロピル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例97に準じた方法により2-メトキシブタン酸から標題化合物を得た。
MS (ESI+): [M+H]+ 409.3.
実施例101
2-(1,1-ジフルオロプロピル)-5-メチル-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(1,1-ジフルオロプロピル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(171 mg)のDMF溶液(15 mL)に水素化ナトリウム(60% oil dispersion、24.8 mg)およびヨウ化メチル(52μL)を0℃で加えた。反応混合物を0℃で1.5時間撹拌後、反応混合物に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(34.1 mg)を得た。
MS (ESI+): [M+H]+ 443.2.
2-(1,1-ジフルオロプロピル)-5-メチル-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(1,1-ジフルオロプロピル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(171 mg)のDMF溶液(15 mL)に水素化ナトリウム(60% oil dispersion、24.8 mg)およびヨウ化メチル(52μL)を0℃で加えた。反応混合物を0℃で1.5時間撹拌後、反応混合物に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(34.1 mg)を得た。
MS (ESI+): [M+H]+ 443.2.
実施例102
6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
A) 4,4,4-トリフルオロブタンアミド
4,4,4-トリフルオロブタン酸(19.9 g)のTHF(200 mL)溶液に塩化オキサリル(30.5 mL)およびDMF(0.54 mL)を0℃で加え、4時間撹拌した。得られた反応混合物を28%アンモニア水(150 mL)に0℃で加え、1時間撹拌した。反応混合物を濾過した後、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(17.9 g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.27-2.35 (2H, m), 2.37-2.55 (2H, m), 6.96 (1H, brs), 7.44 (1H, brs).
4,4,4-トリフルオロブタン酸(19.9 g)のTHF(200 mL)溶液に塩化オキサリル(30.5 mL)およびDMF(0.54 mL)を0℃で加え、4時間撹拌した。得られた反応混合物を28%アンモニア水(150 mL)に0℃で加え、1時間撹拌した。反応混合物を濾過した後、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(17.9 g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.27-2.35 (2H, m), 2.37-2.55 (2H, m), 6.96 (1H, brs), 7.44 (1H, brs).
B) 4,4,4-トリフルオロブタンチオアミド
4,4,4-トリフルオロブタンアミド(17.9 g)、2,4-ビス(4-メトキシフェニル)-1,3,2,4-ジチアジホスフェタン 2,4-ジスルフィド(51.3 g)およびトルエン(400 mL)からなる混合物を80℃で3時間撹拌した。反応混合物をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル)に付した後、再度アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)にて精製し、標題化合物(14.6 g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.59-2.79 (4H, m), 9.37 (1H, brs), 9.60 (1H, brs).
4,4,4-トリフルオロブタンアミド(17.9 g)、2,4-ビス(4-メトキシフェニル)-1,3,2,4-ジチアジホスフェタン 2,4-ジスルフィド(51.3 g)およびトルエン(400 mL)からなる混合物を80℃で3時間撹拌した。反応混合物をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル)に付した後、再度アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)にて精製し、標題化合物(14.6 g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.59-2.79 (4H, m), 9.37 (1H, brs), 9.60 (1H, brs).
C) 4,4,4-トリフルオロブタンイミドチオ酸メチル ヨウ素酸塩
4,4,4-トリフルオロブタンチオアミド(8.27 g)のアセトン(160 mL)溶液にヨードメタン(16.4 mL)を室温で加え、40℃で3時間撹拌した。反応混合物を減圧濃縮することにより標題化合物(15.4 g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.68 (3H, s), 2.72-2.87 (2H, m), 3.02-3.10 (2H, m), 11.64 (2H, brs).
4,4,4-トリフルオロブタンチオアミド(8.27 g)のアセトン(160 mL)溶液にヨードメタン(16.4 mL)を室温で加え、40℃で3時間撹拌した。反応混合物を減圧濃縮することにより標題化合物(15.4 g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.68 (3H, s), 2.72-2.87 (2H, m), 3.02-3.10 (2H, m), 11.64 (2H, brs).
D) 4,4,4-トリフルオロ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]ブタンイミドアミド
4,4,4-トリフルオロブタンイミドチオ酸メチル ヨウ素酸塩(15.3 g)、4-(2,2,2-トリフルオロエトキシ)アニリン(8.90 g)およびエタノール(150 mL)からなる混合物を室温で2時間撹拌した。反応混合物に酢酸エチルおよび飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(10.9 g)を固体として得た。
MS (ESI+): [M+H]+ 315.1.
4,4,4-トリフルオロブタンイミドチオ酸メチル ヨウ素酸塩(15.3 g)、4-(2,2,2-トリフルオロエトキシ)アニリン(8.90 g)およびエタノール(150 mL)からなる混合物を室温で2時間撹拌した。反応混合物に酢酸エチルおよび飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(10.9 g)を固体として得た。
MS (ESI+): [M+H]+ 315.1.
E) 6-ヒドロキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
4,4,4-トリフルオロ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]ブタンイミドアミド(20.9 g)、マロン酸ジエチル(20.2 mL)、ナトリウムメトキシド(10.8 g)および2-メトキシエタノール(400 mL)からなる混合物を終夜加熱還流し、反応混合物を減圧濃縮した。同様に、4,4,4-トリフルオロ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]ブタンイミドアミド(22.6 g)、マロン酸ジエチル(21.8 mL)、ナトリウムメトキシド(11.7 g)および2-メトキシエタノール(400 mL)からなる混合物を終夜加熱還流し、反応混合物を減圧濃縮した。各々から得られた残渣を混合し、ジエチルエーテルと水で分配した。水層を分離後、有機層に1規定塩酸を加え酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(41.6 g)を固体として得た。
MS (ESI+): [M+H]+ 383.1.
4,4,4-トリフルオロ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]ブタンイミドアミド(20.9 g)、マロン酸ジエチル(20.2 mL)、ナトリウムメトキシド(10.8 g)および2-メトキシエタノール(400 mL)からなる混合物を終夜加熱還流し、反応混合物を減圧濃縮した。同様に、4,4,4-トリフルオロ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]ブタンイミドアミド(22.6 g)、マロン酸ジエチル(21.8 mL)、ナトリウムメトキシド(11.7 g)および2-メトキシエタノール(400 mL)からなる混合物を終夜加熱還流し、反応混合物を減圧濃縮した。各々から得られた残渣を混合し、ジエチルエーテルと水で分配した。水層を分離後、有機層に1規定塩酸を加え酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(41.6 g)を固体として得た。
MS (ESI+): [M+H]+ 383.1.
F) トリフルオロメタンスルホン酸6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル
6-ヒドロキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン(2.63 g)およびトリエチルアミン(1.44 mL)のTHF(50 mL)溶液に1,1,1-トリフルオロ-N-フェニル-N-[(トリフルオロメチル)スルホニル]メタンスルホンアミド(2.46 g)を0℃で加え、室温で終夜撹拌した。反応混合物に氷冷した飽和食塩水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(3.21 g)を固体として得た。
MS (ESI+): [M+H]+ 515.2.
6-ヒドロキシ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン(2.63 g)およびトリエチルアミン(1.44 mL)のTHF(50 mL)溶液に1,1,1-トリフルオロ-N-フェニル-N-[(トリフルオロメチル)スルホニル]メタンスルホンアミド(2.46 g)を0℃で加え、室温で終夜撹拌した。反応混合物に氷冷した飽和食塩水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(3.21 g)を固体として得た。
MS (ESI+): [M+H]+ 515.2.
G) 6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
トリフルオロメタンスルホン酸6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル(506 mg)、4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール-1-カルボン酸tert-ブチル(434 mg)、1,1'-ビス(ジフェニルホスフィノ)フェロセンパラジウム(II) ジクロリドジクロロメタン錯体(81 mg)、炭酸ナトリウム水溶液(2M, 1.48 mL)およびDMF(5 mL)からなる混合物をマイクロウェーブ照射下100℃で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(71 mg)を固体として得た。
MS (ESI+): [M+H]+ 433.2.
トリフルオロメタンスルホン酸6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル(506 mg)、4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール-1-カルボン酸tert-ブチル(434 mg)、1,1'-ビス(ジフェニルホスフィノ)フェロセンパラジウム(II) ジクロリドジクロロメタン錯体(81 mg)、炭酸ナトリウム水溶液(2M, 1.48 mL)およびDMF(5 mL)からなる混合物をマイクロウェーブ照射下100℃で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(71 mg)を固体として得た。
MS (ESI+): [M+H]+ 433.2.
実施例103
6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例102のG)に準じた方法により、1-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾールから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 447.2.
6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例102のG)に準じた方法により、1-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾールから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 447.2.
実施例104
2-(シクロプロピルメチル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-シクロプロピル酢酸(1.12 g)およびDMF(0.087 mL)をTHF(3 mL)に溶かし、塩化オキサリルを0℃で加え、反応混合物を室温で1時間撹拌した。反応混合液を(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]アクリルアミド(1 g)のDMA(15 mL)溶液に加えて、50℃で6時間攪拌した。反応混合物に飽和重曹水を加えて、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して黄色油状物を得た。得られた油状物をTFA(10 mL)に溶かし、マイクロウェーブ照射下100℃で30分間攪拌した。反応液を濃縮し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した。その後、HPLC[C18、移動相:水/アセトニトリル(NH4HCO3含有系)]にて分取し、得られた画分を濃縮した後、酢酸エチルおよびヘキサンより再結晶を行うことで標題化合物(18.4 mg)を得た。
MS (ESI+): [M+H]+ 391.1.
2-(シクロプロピルメチル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-シクロプロピル酢酸(1.12 g)およびDMF(0.087 mL)をTHF(3 mL)に溶かし、塩化オキサリルを0℃で加え、反応混合物を室温で1時間撹拌した。反応混合液を(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]アクリルアミド(1 g)のDMA(15 mL)溶液に加えて、50℃で6時間攪拌した。反応混合物に飽和重曹水を加えて、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して黄色油状物を得た。得られた油状物をTFA(10 mL)に溶かし、マイクロウェーブ照射下100℃で30分間攪拌した。反応液を濃縮し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した。その後、HPLC[C18、移動相:水/アセトニトリル(NH4HCO3含有系)]にて分取し、得られた画分を濃縮した後、酢酸エチルおよびヘキサンより再結晶を行うことで標題化合物(18.4 mg)を得た。
MS (ESI+): [M+H]+ 391.1.
実施例105
2-(1-メチルシクロプロピル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例97に準じた方法により1-メチルシクロプロパンカルボン酸から合成および精製した後、HPLC[C18移動相:水/アセトニトリル(NH4HCO3含有系)]にて分取し、得られた画分を濃縮した。残渣を酢酸エチルおよびヘキサンより再結晶することで標題化合物を得た。
MS (ESI+): [M+H]+ 390.95.
2-(1-メチルシクロプロピル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例97に準じた方法により1-メチルシクロプロパンカルボン酸から合成および精製した後、HPLC[C18移動相:水/アセトニトリル(NH4HCO3含有系)]にて分取し、得られた画分を濃縮した。残渣を酢酸エチルおよびヘキサンより再結晶することで標題化合物を得た。
MS (ESI+): [M+H]+ 390.95.
実施例106
2-(1-ヒドロキシエチル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(1-ヒドロキシエチル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 2-(ベンジルオキシ)プロパン酸
乳酸エチル(5 g)をDMF(150 mL)に溶かし、水素化ナトリウム(60% oil dispersion、2.03 g)を0℃で加えて室温で30分間攪拌した。反応溶液にベンジルブロミドを加えて、室温で4時間攪拌した。反応混合物に水を加えて酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。得られた残渣をメタノール(50 mL)および8規定水酸化ナトリウム水溶液(80 mL)に溶かし、室温で7時間攪拌した。反応混合液を半分量まで濃縮した後、ジエチルエーテルで洗浄した。水層を6規定塩酸で酸性にした後、酢酸エチルを加えて抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(5.43 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.31 (3H, d, J = 6.8 Hz), 3.77-4.16 (1H, m), 4.29-4.48 (1H, m), 4.59 (1H, d, J = 11.7 Hz), 6.85-7.58 (5H, m), 12.70 (1H, brs).
乳酸エチル(5 g)をDMF(150 mL)に溶かし、水素化ナトリウム(60% oil dispersion、2.03 g)を0℃で加えて室温で30分間攪拌した。反応溶液にベンジルブロミドを加えて、室温で4時間攪拌した。反応混合物に水を加えて酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。得られた残渣をメタノール(50 mL)および8規定水酸化ナトリウム水溶液(80 mL)に溶かし、室温で7時間攪拌した。反応混合液を半分量まで濃縮した後、ジエチルエーテルで洗浄した。水層を6規定塩酸で酸性にした後、酢酸エチルを加えて抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(5.43 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.31 (3H, d, J = 6.8 Hz), 3.77-4.16 (1H, m), 4.29-4.48 (1H, m), 4.59 (1H, d, J = 11.7 Hz), 6.85-7.58 (5H, m), 12.70 (1H, brs).
B) 2-(1-ヒドロキシエチル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例97に準じた方法により2-(ベンジルオキシ)プロパン酸から、標題化合物を得た。
MS (ESI+): [M+H]+ 381.4.
実施例97に準じた方法により2-(ベンジルオキシ)プロパン酸から、標題化合物を得た。
MS (ESI+): [M+H]+ 381.4.
実施例107
6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
酢酸パラジウム(II)(8.7 mg)および4,5-ビス(ジフェニルホスフィノ)-9,9-ジメチルキサンテン(34 mg)のトルエン(3 mL)溶液にトリフルオロメタンスルホン酸6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル(200 mg)、モルホリン(0.051 mL)および炭酸セシウム(317 mg)を室温で加え、100℃で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(メタノール/酢酸エチル)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(123 mg)を固体として得た。
MS (ESI+): [M+H]+ 452.2.
6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
酢酸パラジウム(II)(8.7 mg)および4,5-ビス(ジフェニルホスフィノ)-9,9-ジメチルキサンテン(34 mg)のトルエン(3 mL)溶液にトリフルオロメタンスルホン酸6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル(200 mg)、モルホリン(0.051 mL)および炭酸セシウム(317 mg)を室温で加え、100℃で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(メタノール/酢酸エチル)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(123 mg)を固体として得た。
MS (ESI+): [M+H]+ 452.2.
実施例108
6-(3-オキソピペラジン-1-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例107に準じた方法により、ピペラジン-2-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(メタノール/酢酸エチル)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 465.2.
6-(3-オキソピペラジン-1-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例107に準じた方法により、ピペラジン-2-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(メタノール/酢酸エチル)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 465.2.
実施例109
2-(1,1-ジフルオロプロピル)-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(1,1-ジフルオロプロピル)-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) (2Z)-3-ヒドロキシ-3-(1-メチル-1H-ピラゾール-4-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド
実施例55のA)に準じた方法により、1-メチル-1H-ピラゾール-4-カルボン酸を用いて標題化合物を得た。
MS (ESI+): [M+H]+ 342.2.
実施例55のA)に準じた方法により、1-メチル-1H-ピラゾール-4-カルボン酸を用いて標題化合物を得た。
MS (ESI+): [M+H]+ 342.2.
B) (2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-4-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド
実施例55のB)に準じた方法により、(2Z)-3-ヒドロキシ-3-(1-メチル-1H-ピラゾール-4-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミドを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 341.3.
実施例55のB)に準じた方法により、(2Z)-3-ヒドロキシ-3-(1-メチル-1H-ピラゾール-4-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミドを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 341.3.
C) 2-(1,1-ジフルオロプロピル)-6-(1-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2,2-ジフルオロブタン酸(729 mg)のTHF溶液(2 mL)にDMF(1μL)および塩化オキサリル(0.50 mL)を0℃で加え、室温で10分間撹拌した。反応溶液を(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-4-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(400 mg)のDMA溶液(10 mL)に加え、マイクロ波照射下150℃で1時間攪拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、再結晶(酢酸エチル/ジイソプロピルエーテル)により標題化合物(69.0 mg)を得た。
MS (ESI+): [M+H]+ 429.4.
2,2-ジフルオロブタン酸(729 mg)のTHF溶液(2 mL)にDMF(1μL)および塩化オキサリル(0.50 mL)を0℃で加え、室温で10分間撹拌した。反応溶液を(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-4-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(400 mg)のDMA溶液(10 mL)に加え、マイクロ波照射下150℃で1時間攪拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、再結晶(酢酸エチル/ジイソプロピルエーテル)により標題化合物(69.0 mg)を得た。
MS (ESI+): [M+H]+ 429.4.
実施例110
6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
A) 6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボニトリル
トリフルオロメタンスルホン酸6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル(1.09 g)のNMP(12 mL)溶液に亜鉛シアニド(248 mg)およびテトラキス(トリフェニルホスフィン)パラジウム(0)(244 mg)を室温で加え、100℃で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(723 mg)を固体として得た。
MS (API-): [M-H]- 390.0.
トリフルオロメタンスルホン酸6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル(1.09 g)のNMP(12 mL)溶液に亜鉛シアニド(248 mg)およびテトラキス(トリフェニルホスフィン)パラジウム(0)(244 mg)を室温で加え、100℃で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(723 mg)を固体として得た。
MS (API-): [M-H]- 390.0.
B) N'-ヒドロキシ-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボキシミドアミド
ヒドロキシルアミン 塩酸塩(1.28 g)、炭酸水素ナトリウム(2.32 g)およびDMSO(10 mL)からなる混合物を50℃で30分間撹拌した後、反応混合物に6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボニトリル(720 mg)のDMSO溶液を加え、90℃で3時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去することで標題化合物(732 mg)を固体として得た。
MS (ESI+): [M+H]+ 425.1.
ヒドロキシルアミン 塩酸塩(1.28 g)、炭酸水素ナトリウム(2.32 g)およびDMSO(10 mL)からなる混合物を50℃で30分間撹拌した後、反応混合物に6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボニトリル(720 mg)のDMSO溶液を加え、90℃で3時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去することで標題化合物(732 mg)を固体として得た。
MS (ESI+): [M+H]+ 425.1.
C) 6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
N'-ヒドロキシ-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボキシミドアミド(100 mg)、無水酢酸(0.033 mL)およびピリジン(2 mL)からなる混合物を終夜加熱還流し、反応混合物を減圧濃縮した。残渣に1規定塩酸を加え酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(72.0 mg)を固体として得た。
MS (ESI+): [M+H]+ 449.2.
N'-ヒドロキシ-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボキシミドアミド(100 mg)、無水酢酸(0.033 mL)およびピリジン(2 mL)からなる混合物を終夜加熱還流し、反応混合物を減圧濃縮した。残渣に1規定塩酸を加え酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(72.0 mg)を固体として得た。
MS (ESI+): [M+H]+ 449.2.
実施例111
4-{6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル}モルホリン-3-オン
4-{6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル}モルホリン-3-オン
A) 6-アミノ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
トリス(ジベンジリデンアセトン)ジパラジウム(0)(537 mg)および4,5-ビス(ジフェニルホスフィノ)-9,9-ジメチルキサンテン(679 mg)のトルエン(50 mL)溶液にトリフルオロメタンスルホン酸6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル(5.03 g)、1,1-ジフェニルメタンイミン(1.97 mL)および炭酸セシウム(7.97 g)を室温で加え、100℃で終夜撹拌した。反応混合物を減圧濃縮した。残渣にTHFを加え、濾過して不溶物を取り除き、濾液を減圧濃縮した。得られた残留物のTHF(100 mL)溶液に1規定塩酸(100 mL)を0℃で加え、2時間撹拌した。反応混合物を酢酸エチルで抽出し、抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)およびシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(2.64 g)を固体として得た。
MS (ESI+): [M+H]+ 382.1.
トリス(ジベンジリデンアセトン)ジパラジウム(0)(537 mg)および4,5-ビス(ジフェニルホスフィノ)-9,9-ジメチルキサンテン(679 mg)のトルエン(50 mL)溶液にトリフルオロメタンスルホン酸6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル(5.03 g)、1,1-ジフェニルメタンイミン(1.97 mL)および炭酸セシウム(7.97 g)を室温で加え、100℃で終夜撹拌した。反応混合物を減圧濃縮した。残渣にTHFを加え、濾過して不溶物を取り除き、濾液を減圧濃縮した。得られた残留物のTHF(100 mL)溶液に1規定塩酸(100 mL)を0℃で加え、2時間撹拌した。反応混合物を酢酸エチルで抽出し、抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)およびシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(2.64 g)を固体として得た。
MS (ESI+): [M+H]+ 382.1.
B) 2-(2-クロロエトキシ)-N-{6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル}アセトアミド
6-アミノ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン(95 mg)のDMA(2 mL)溶液に2-(2-クロロエトキシ)アセチル クロリド(47 mg)を室温で加え、終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(111 mg)を固体として得た。
MS (ESI+): [M+H]+ 502.2.
6-アミノ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン(95 mg)のDMA(2 mL)溶液に2-(2-クロロエトキシ)アセチル クロリド(47 mg)を室温で加え、終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(111 mg)を固体として得た。
MS (ESI+): [M+H]+ 502.2.
C) 4-{6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル}モルホリン-3-オン
2-(2-クロロエトキシ)-N-{6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル}アセトアミド(109 mg)のTHF(2 mL)/DMF(1 mL)溶液に水素化ナトリウム(60% oil dispersion、17 mg)を0℃で加え、室温で3時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(12 mg)を固体として得た。
MS (ESI+): [M+H]+ 466.2.
2-(2-クロロエトキシ)-N-{6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル}アセトアミド(109 mg)のTHF(2 mL)/DMF(1 mL)溶液に水素化ナトリウム(60% oil dispersion、17 mg)を0℃で加え、室温で3時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(12 mg)を固体として得た。
MS (ESI+): [M+H]+ 466.2.
実施例112
6-(1-メチル-1H-ピラゾール-4-イル)-2-(ペンタフルオロエチル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
3,3,4,4,4-ペンタフルオロプロピオン酸(1.21 g)のTHF溶液(2 mL)にDMF(1μL)および塩化オキサリル(0.63 mL)を0℃で加え、室温で30分間撹拌した。反応溶液を(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-4-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(500 mg)のDMA溶液(15 mL)に加え、室温で3時間攪拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、トリフルオロ酢酸(3 mL)に溶解後、マイクロ波照射下120℃で1時間撹拌した。反応溶媒を減圧下留去し、残渣を再結晶(酢酸エチル/ジイソプロピルエーテル)することにより標題化合物(36.8 mg)を得た。
MS (ESI+): [M+H]+ 469.0.
6-(1-メチル-1H-ピラゾール-4-イル)-2-(ペンタフルオロエチル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
3,3,4,4,4-ペンタフルオロプロピオン酸(1.21 g)のTHF溶液(2 mL)にDMF(1μL)および塩化オキサリル(0.63 mL)を0℃で加え、室温で30分間撹拌した。反応溶液を(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-4-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド(500 mg)のDMA溶液(15 mL)に加え、室温で3時間攪拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製し、トリフルオロ酢酸(3 mL)に溶解後、マイクロ波照射下120℃で1時間撹拌した。反応溶媒を減圧下留去し、残渣を再結晶(酢酸エチル/ジイソプロピルエーテル)することにより標題化合物(36.8 mg)を得た。
MS (ESI+): [M+H]+ 469.0.
実施例113
6-(モルホリン-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(モルホリン-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 2-[(4-メトキシベンジル)オキシ]-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例11に準じた方法により、2-(エチルスルファニル)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび4-メトキシベンジルアルコールから標題化合物を得た。
MS (ESI+): [M+H]+ 492.2.
実施例11に準じた方法により、2-(エチルスルファニル)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび4-メトキシベンジルアルコールから標題化合物を得た。
MS (ESI+): [M+H]+ 492.2.
B) 6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-2,4(1H,3H)-ジオン
2-[(4-メトキシベンジル)オキシ]-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(700 mg)のトリフルオロ酢酸溶液(13 mL)を、マイクロ波照射下100℃で30分間撹拌した。反応溶媒を減圧下留去し、残渣をジイソプロピルエーテルにて洗浄することにより標題化合物(491 mg)を得た。
MS (ESI+): [M+H]+ 372.2.
2-[(4-メトキシベンジル)オキシ]-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(700 mg)のトリフルオロ酢酸溶液(13 mL)を、マイクロ波照射下100℃で30分間撹拌した。反応溶媒を減圧下留去し、残渣をジイソプロピルエーテルにて洗浄することにより標題化合物(491 mg)を得た。
MS (ESI+): [M+H]+ 372.2.
C) 6-(モルホリン-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-2,4(1H,3H)-ジオン(400 mg)のTHF(20 mL)およびDMF(4 mL)の混合溶液に、水素化ナトリウム(60% oil dispersion、259 mg)を0℃で加え、室温で5分間撹拌した。反応溶液にトリフルオロメタンスルホン酸2,2,2-トリフルオロエチル(0.62 mL)を室温で加え、終夜撹拌後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(87.9 mg)を得た。
MS (ESI+): [M+H]+ 454.4.
6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-2,4(1H,3H)-ジオン(400 mg)のTHF(20 mL)およびDMF(4 mL)の混合溶液に、水素化ナトリウム(60% oil dispersion、259 mg)を0℃で加え、室温で5分間撹拌した。反応溶液にトリフルオロメタンスルホン酸2,2,2-トリフルオロエチル(0.62 mL)を室温で加え、終夜撹拌後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(87.9 mg)を得た。
MS (ESI+): [M+H]+ 454.4.
実施例114
6-(4-メチル-5-オキソ-4,5-ジヒドロ-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
6-(4-メチル-5-オキソ-4,5-ジヒドロ-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
A) 6-(5-オキソ-4,5-ジヒドロ-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
N'-ヒドロキシ-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボキシミドアミド(168 mg)および1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン(0.118 mL)のTHF(5 mL)溶液にN,N'-カルボニルジイミダゾール(128 mg)を室温で加え、2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(174 mg)を固体として得た。
MS (ESI+): [M+H]+ 451.2.
N'-ヒドロキシ-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボキシミドアミド(168 mg)および1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン(0.118 mL)のTHF(5 mL)溶液にN,N'-カルボニルジイミダゾール(128 mg)を室温で加え、2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(174 mg)を固体として得た。
MS (ESI+): [M+H]+ 451.2.
B) 6-(4-メチル-5-オキソ-4,5-ジヒドロ-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
6-(5-オキソ-4,5-ジヒドロ-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン(96 mg)のDMF(2 mL)溶液に水素化ナトリウム(60% oil dispersion、13 mg)を0℃で加え、10分間撹拌した。反応混合物にヨウ化メチル(0.020 mL)を加え、室温で3時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(44 mg)を固体として得た。
MS (API-): [M-H]- 463.0.
6-(5-オキソ-4,5-ジヒドロ-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン(96 mg)のDMF(2 mL)溶液に水素化ナトリウム(60% oil dispersion、13 mg)を0℃で加え、10分間撹拌した。反応混合物にヨウ化メチル(0.020 mL)を加え、室温で3時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(44 mg)を固体として得た。
MS (API-): [M-H]- 463.0.
実施例115
3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-[5-(トリフルオロメチル)-1,2,4-オキサジアゾール-3-イル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
N'-ヒドロキシ-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボキシミドアミド(100 mg)のピリジン(2 mL)溶液にトリフルオロ酢酸無水物(0.100 mL)を室温で加え、10分間撹拌した。反応混合物を減圧濃縮後、残渣に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(69 mg)を固体として得た。
MS (ESI+): [M+H]+ 503.1.
3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-[5-(トリフルオロメチル)-1,2,4-オキサジアゾール-3-イル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
N'-ヒドロキシ-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボキシミドアミド(100 mg)のピリジン(2 mL)溶液にトリフルオロ酢酸無水物(0.100 mL)を室温で加え、10分間撹拌した。反応混合物を減圧濃縮後、残渣に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(69 mg)を固体として得た。
MS (ESI+): [M+H]+ 503.1.
実施例116
6-(3-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例102のG)に準じた方法により、3-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール-1-カルボン酸tert-ブチルを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(メタノール/酢酸エチル)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 447.1.
6-(3-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例102のG)に準じた方法により、3-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾール-1-カルボン酸tert-ブチルを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(メタノール/酢酸エチル)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 447.1.
実施例117
2-(エチルスルファニル)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)ピリミジン-4(3H)-オン
実施例1に準じた方法で、1-フルオロ-4-ニトロベンゼンの代わりに1,2-ジフルオロ-4-ニトロベンゼンを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 415.0.
2-(エチルスルファニル)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)ピリミジン-4(3H)-オン
実施例1に準じた方法で、1-フルオロ-4-ニトロベンゼンの代わりに1,2-ジフルオロ-4-ニトロベンゼンを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 415.0.
実施例118
2-(3,3-ジフルオロシクロブチル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例102に準じた方法により、3,3-ジフルオロシクロブタンカルボン酸より標題化合物を得た。
MS (ESI+): [M+H]+ 427.0.
2-(3,3-ジフルオロシクロブチル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例102に準じた方法により、3,3-ジフルオロシクロブタンカルボン酸より標題化合物を得た。
MS (ESI+): [M+H]+ 427.0.
実施例119
2-(2,2-ジフルオロエトキシ)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)ピリミジン-4(3H)-オン
実施例1に準じた方法で、1-フルオロ-4-ニトロベンゼンの代わりに1,2-ジフルオロ-4-ニトロベンゼンを用いて2-(エチルスルファニル)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)ピリミジン-4(3H)-オンを得た。実施例69に準じた方法で、2-(エチルスルファニル)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)ピリミジン-4(3H)-オンを用い、2,2,2-トリフルオロエタノールの代わりに2,2-ジフルオロエタノールを用いて、標題化合物を得た。
MS (ESI+): [M+H]+ 435.1.
2-(2,2-ジフルオロエトキシ)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)ピリミジン-4(3H)-オン
実施例1に準じた方法で、1-フルオロ-4-ニトロベンゼンの代わりに1,2-ジフルオロ-4-ニトロベンゼンを用いて2-(エチルスルファニル)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)ピリミジン-4(3H)-オンを得た。実施例69に準じた方法で、2-(エチルスルファニル)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)ピリミジン-4(3H)-オンを用い、2,2,2-トリフルオロエタノールの代わりに2,2-ジフルオロエタノールを用いて、標題化合物を得た。
MS (ESI+): [M+H]+ 435.1.
実施例120
6-(3,3-ジフルオロピロリジン-1-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例107に準じた方法により、3,3-ジフルオロピロリジン 塩酸塩を用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 472.2.
6-(3,3-ジフルオロピロリジン-1-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例107に準じた方法により、3,3-ジフルオロピロリジン 塩酸塩を用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 472.2.
実施例121
6-(3-メチル-1,2,4-オキサジアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
6-(3-メチル-1,2,4-オキサジアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
A) 6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボン酸メチル
トリフルオロメタンスルホン酸6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル(350 mg)およびトリエチルアミン(0.142 mL)のメタノール(4 mL)/THF(2 mL)溶液に1,1'-ビス(ジフェニルホスフィノ)フェロセンパラジウム(II) ジクロリドジクロロメタン錯体(56.0 mg)を室温で加え、一酸化炭素雰囲気下80℃で4時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(229 mg)を固体として得た。
MS (ESI+): [M+H]+ 425.1.
トリフルオロメタンスルホン酸6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-イル(350 mg)およびトリエチルアミン(0.142 mL)のメタノール(4 mL)/THF(2 mL)溶液に1,1'-ビス(ジフェニルホスフィノ)フェロセンパラジウム(II) ジクロリドジクロロメタン錯体(56.0 mg)を室温で加え、一酸化炭素雰囲気下80℃で4時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(229 mg)を固体として得た。
MS (ESI+): [M+H]+ 425.1.
B) 6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボン酸
6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボン酸メチル(226 mg)のメタノール(4.5 mL)/THF(1.5 mL)溶液に1規定水酸化ナトリウム水溶液(1.07 mL)を0℃で加え、2時間撹拌した。反応混合物に1規定塩酸を加えた後、溶媒を減圧下留去した。残渣に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去することで標題化合物(220 mg)を固体として得た。
MS (ESI+): [M+H]+ 411.1.
6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボン酸メチル(226 mg)のメタノール(4.5 mL)/THF(1.5 mL)溶液に1規定水酸化ナトリウム水溶液(1.07 mL)を0℃で加え、2時間撹拌した。反応混合物に1規定塩酸を加えた後、溶媒を減圧下留去した。残渣に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去することで標題化合物(220 mg)を固体として得た。
MS (ESI+): [M+H]+ 411.1.
C) 6-(3-メチル-1,2,4-オキサジアゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボン酸(117 mg)のTHF(2 mL)溶液に塩化オキサリル(0.062 mL)およびDMFを0℃で加え、室温で2時間撹拌した後、反応混合物を減圧濃縮した。得られた固体、(1Z)-N'-ヒドロキシエタンイミドアミド(32 mg)およびピリジン(2 mL)からなる混合物を室温で1時間、および100℃で4時間撹拌した。反応混合物を減圧濃縮後、残渣に1規定塩酸を加え酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(28 mg)を固体として得た。
MS (API-): [M-H]- 447.1.
6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボン酸(117 mg)のTHF(2 mL)溶液に塩化オキサリル(0.062 mL)およびDMFを0℃で加え、室温で2時間撹拌した後、反応混合物を減圧濃縮した。得られた固体、(1Z)-N'-ヒドロキシエタンイミドアミド(32 mg)およびピリジン(2 mL)からなる混合物を室温で1時間、および100℃で4時間撹拌した。反応混合物を減圧濃縮後、残渣に1規定塩酸を加え酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(28 mg)を固体として得た。
MS (API-): [M-H]- 447.1.
実施例123
2-(1,1-ジフルオロエチル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例97に準じた方法により2,2-ジフルオロプロパン酸を用いて、標題化合物を得た。
MS (ESI+): [M+H]+ 401.0.
2-(1,1-ジフルオロエチル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例97に準じた方法により2,2-ジフルオロプロパン酸を用いて、標題化合物を得た。
MS (ESI+): [M+H]+ 401.0.
実施例124
2-(1,1-ジフルオロプロピル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-[3-(トリフルオロメチル)-1H-ピラゾール-4-イル]ピリミジン-4(3H)-オン
2-(1,1-ジフルオロプロピル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-[3-(トリフルオロメチル)-1H-ピラゾール-4-イル]ピリミジン-4(3H)-オン
A) 1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-カルボン酸エチル
実施例1のD)に準じた方法により、3-(トリフルオロメチル)-1H-ピラゾール-4-カルボン酸エチルを用いて、標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.25 (3H, t, J = 7.0 Hz), 3.73 (3H, s), 4.22 (2H, q, J = 6.9 Hz), 5.36 (2H, s), 6.92 (2H, d, J = 8.7 Hz), 7.31 (2H, d, J = 8.7 Hz), 8.66 (1H, s).
実施例1のD)に準じた方法により、3-(トリフルオロメチル)-1H-ピラゾール-4-カルボン酸エチルを用いて、標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.25 (3H, t, J = 7.0 Hz), 3.73 (3H, s), 4.22 (2H, q, J = 6.9 Hz), 5.36 (2H, s), 6.92 (2H, d, J = 8.7 Hz), 7.31 (2H, d, J = 8.7 Hz), 8.66 (1H, s).
B) 1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-カルボン酸
実施例1のE)に準じた方法により、1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-カルボン酸エチルを用いて標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 3.74 (3H, s), 5.35 (2H, s), 6.69-7.10 (2H, m), 7.31 (2H, d, J = 8.7 Hz), 8.57 (1H, s), 12.92 (1H, brs).
実施例1のE)に準じた方法により、1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-カルボン酸エチルを用いて標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 3.74 (3H, s), 5.35 (2H, s), 6.69-7.10 (2H, m), 7.31 (2H, d, J = 8.7 Hz), 8.57 (1H, s), 12.92 (1H, brs).
C) 3-[1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-イル]-3-オキソプロパン酸エチル
実施例1のF)に準じた方法により、1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-カルボン酸を用いて標題化合物を得た。
MS (ESI+): [M+H]+ 371.2.
実施例1のF)に準じた方法により、1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-カルボン酸を用いて標題化合物を得た。
MS (ESI+): [M+H]+ 371.2.
D) 3-[1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-イル]-3-オキソ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパンアミド
実施例55のA)に準じた方法により、3-[1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-イル]-3-オキソプロパン酸エチルを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 516.1.
実施例55のA)に準じた方法により、3-[1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-イル]-3-オキソプロパン酸エチルを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 516.1.
E) (2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド
実施例55のB)に準じた方法により、3-[1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-イル]-3-オキソ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパンアミドを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 515.0.
実施例55のB)に準じた方法により、3-[1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-イル]-3-オキソ-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパンアミドを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 515.0.
F) 2-(1,1-ジフルオロプロピル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-[3-(トリフルオロメチル)-1H-ピラゾール-4-イル]ピリミジン-4(3H)-オン
実施例112に準じた方法により、(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミドを用い、3,3,4,4,4-ペンタフルオロプロピオン酸の代わりに2,2-ジフルオロブタン酸を用いることにより標題化合物を得た。
MS (API-): [M-H]- 481.0.
実施例112に準じた方法により、(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-3-(トリフルオロメチル)-1H-ピラゾール-4-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミドを用い、3,3,4,4,4-ペンタフルオロプロピオン酸の代わりに2,2-ジフルオロブタン酸を用いることにより標題化合物を得た。
MS (API-): [M-H]- 481.0.
実施例125
6-(4-オキソピペリジン-1-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例107に準じた方法により、ピペリジン-4-オン 塩酸塩から標題化合物を固体として得た。
MS (ESI+): [M+H]+ 464.1.
6-(4-オキソピペリジン-1-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例107に準じた方法により、ピペリジン-4-オン 塩酸塩から標題化合物を固体として得た。
MS (ESI+): [M+H]+ 464.1.
実施例126
6-(5-メチル-1,3,4-オキサジアゾール-2-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
6-(5-メチル-1,3,4-オキサジアゾール-2-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
A) N'-アセチル-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボヒドラジド
6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボン酸(211 mg)のTHF(4 mL)溶液に塩化オキサリル(0.112 mL)およびDMFを0℃で加え、室温で2時間撹拌した後、反応混合物を減圧濃縮した。得られた固体のTHF(5 mL)溶液にアセトヒドラジド(57 mg)およびトリエチルアミン(0.143 mL)を0℃で加え、室温で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(200 mg)を固体として得た。
MS (ESI+): [M+H]+ 467.1.
6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボン酸(211 mg)のTHF(4 mL)溶液に塩化オキサリル(0.112 mL)およびDMFを0℃で加え、室温で2時間撹拌した後、反応混合物を減圧濃縮した。得られた固体のTHF(5 mL)溶液にアセトヒドラジド(57 mg)およびトリエチルアミン(0.143 mL)を0℃で加え、室温で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(200 mg)を固体として得た。
MS (ESI+): [M+H]+ 467.1.
B) 6-(5-メチル-1,3,4-オキサジアゾール-2-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
N'-アセチル-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボヒドラジド(199 mg)、(メトキシカルボニルスルファモイル)トリエチルアンモニウム ヒドロキシド 分子内塩(305 mg)およびTHF(3 mL)からなる混合物をマイクロウェーブ照射下100℃で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(56 mg)を固体として得た。
MS (ESI+): [M+H]+ 449.1.
N'-アセチル-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボヒドラジド(199 mg)、(メトキシカルボニルスルファモイル)トリエチルアンモニウム ヒドロキシド 分子内塩(305 mg)およびTHF(3 mL)からなる混合物をマイクロウェーブ照射下100℃で1時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(56 mg)を固体として得た。
MS (ESI+): [M+H]+ 449.1.
実施例127
3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)-2-(ピロリジン-1-イル)ピリミジン-4(3H)-オン
実施例1に準じた方法で、1-フルオロ-4-ニトロベンゼンの代わりに1,2-ジフルオロ-4-ニトロベンゼンを用いて2-(エチルスルファニル)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)ピリミジン-4(3H)-オンを得た。実施例69に準じた方法で、2-(エチルスルファニル)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)ピリミジン-4(3H)-オンを用い、2,2,2-トリフルオロエタノールの代わりにピロリジンを用いて、標題化合物を得た。
MS (ESI+): [M+H]+ 424.1.
3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)-2-(ピロリジン-1-イル)ピリミジン-4(3H)-オン
実施例1に準じた方法で、1-フルオロ-4-ニトロベンゼンの代わりに1,2-ジフルオロ-4-ニトロベンゼンを用いて2-(エチルスルファニル)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)ピリミジン-4(3H)-オンを得た。実施例69に準じた方法で、2-(エチルスルファニル)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1H-ピラゾール-4-イル)ピリミジン-4(3H)-オンを用い、2,2,2-トリフルオロエタノールの代わりにピロリジンを用いて、標題化合物を得た。
MS (ESI+): [M+H]+ 424.1.
実施例128
6-(1H-ピラゾール-4-イル)-2-[(2,2,2-トリフルオロエトキシ)メチル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例97に準じた方法により2-(2,2,2,-トリフルオロエトキシ)酢酸から標題化合物を得た。
MS (ESI+): [M+H]+ 449.0.
6-(1H-ピラゾール-4-イル)-2-[(2,2,2-トリフルオロエトキシ)メチル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例97に準じた方法により2-(2,2,2,-トリフルオロエトキシ)酢酸から標題化合物を得た。
MS (ESI+): [M+H]+ 449.0.
実施例129
2-(1,1-ジフルオロプロピル)-6-(5-オキソピロリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(1,1-ジフルオロプロピル)-6-(5-オキソピロリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-[1-(4-メトキシベンジル)-5-オキソピロリジン-3-イル]-3-オキソプロパン酸エチル
実施例1のF)に準じた方法により、1-(4-メトキシベンジル)-1H-ピラゾール-4-カルボン酸の代わりに1-(4-メトキシベンジル)-5-オキソピロリジン-3-カルボン酸を用い、標題化合物を得た。
MS (ESI+): [M+H]+ 320.2.
実施例1のF)に準じた方法により、1-(4-メトキシベンジル)-1H-ピラゾール-4-カルボン酸の代わりに1-(4-メトキシベンジル)-5-オキソピロリジン-3-カルボン酸を用い、標題化合物を得た。
MS (ESI+): [M+H]+ 320.2.
B) (2Z)-3-ヒドロキシ-3-[1-(4-メトキシベンジル)-5-オキソピロリジン-3-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド
実施例55のA)に準じた方法により、3-[1-(4-メトキシベンジル)-5-オキソピロリジン-3-イル]-3-オキソプロパン酸エチルを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 465.1.
実施例55のA)に準じた方法により、3-[1-(4-メトキシベンジル)-5-オキソピロリジン-3-イル]-3-オキソプロパン酸エチルを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 465.1.
C) (2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-5-オキソピロリジン-3-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミド
実施例55のB)に準じた方法により、(2Z)-3-ヒドロキシ-3-[1-(4-メトキシベンジル)-5-オキソピロリジン-3-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミドおよび2,2-ジフルオロブタン酸を用いて標題化合物を得た。
MS (ESI+): [M+H]+ 464.0.
実施例55のB)に準じた方法により、(2Z)-3-ヒドロキシ-3-[1-(4-メトキシベンジル)-5-オキソピロリジン-3-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミドおよび2,2-ジフルオロブタン酸を用いて標題化合物を得た。
MS (ESI+): [M+H]+ 464.0.
D) 2-(1,1-ジフルオロプロピル)-6-(5-オキソピロリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例112に準じた方法により、(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-5-オキソピロリジン-3-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミドを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 432.1.
実施例112に準じた方法により、(2Z)-3-アミノ-3-[1-(4-メトキシベンジル)-5-オキソピロリジン-3-イル]-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]プロパ-2-エンアミドを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 432.1.
実施例130
6-(1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
N'-ヒドロキシ-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボキシミドアミド(204 mg)およびトリメトキシメタン(5 mL)からなる懸濁液に三フッ化ホウ素 ジエチルエーテラート(6.09μL)を室温で加え、3時間撹拌した。反応混合物を減圧濃縮後、残渣に水を加え酢酸エチルで抽出した。抽出液を飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(109 mg)を固体として得た。
MS (ESI+): [M+H]+ 435.1.
6-(1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
N'-ヒドロキシ-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)-1,6-ジヒドロピリミジン-4-カルボキシミドアミド(204 mg)およびトリメトキシメタン(5 mL)からなる懸濁液に三フッ化ホウ素 ジエチルエーテラート(6.09μL)を室温で加え、3時間撹拌した。反応混合物を減圧濃縮後、残渣に水を加え酢酸エチルで抽出した。抽出液を飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(109 mg)を固体として得た。
MS (ESI+): [M+H]+ 435.1.
実施例131
2-(1,1-ジフルオロエチル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2,2-ジフルオロプロピオン酸(323 mg)およびDMF(0.011 mL)をTHF(1.5 mL)に溶かし、塩化オキサリル(0.205 mL)を0℃で加え、反応混合物を室温で2時間半撹拌した。反応混合液を(Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]アクリルアミド(200 mg)のDMA(6 mL)溶液に加えて、50℃で2時間攪拌した。その後、反応溶液をマイクロウェーブ照射下150℃で30分間攪拌した。反応混合物に飽和重曹水を加えて、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。得られた残渣をTFA(10 mL)に溶かし、マイクロウェーブ照射下100℃で60分間、および120℃で1時間半攪拌した。反応液を濃縮し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチルおよびヘキサンより再結晶を行うことで標題化合物(112 mg)を得た。
MS (ESI+): [M+H]+ 415.0.
2-(1,1-ジフルオロエチル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2,2-ジフルオロプロピオン酸(323 mg)およびDMF(0.011 mL)をTHF(1.5 mL)に溶かし、塩化オキサリル(0.205 mL)を0℃で加え、反応混合物を室温で2時間半撹拌した。反応混合液を(Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]アクリルアミド(200 mg)のDMA(6 mL)溶液に加えて、50℃で2時間攪拌した。その後、反応溶液をマイクロウェーブ照射下150℃で30分間攪拌した。反応混合物に飽和重曹水を加えて、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。得られた残渣をTFA(10 mL)に溶かし、マイクロウェーブ照射下100℃で60分間、および120℃で1時間半攪拌した。反応液を濃縮し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチルおよびヘキサンより再結晶を行うことで標題化合物(112 mg)を得た。
MS (ESI+): [M+H]+ 415.0.
実施例132
2-(1,1-ジフルオロプロピル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2,2-ジフルオロブタン酸(365 mg)およびDMF(0.011 mL)をTHF(1.5 mL)に溶かし、塩化オキサリル(0.205 mL)を0℃で加え、反応混合物を室温で2時間半撹拌した。反応混合液を(Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]アクリルアミド(200 mg)のDMA(6 mL)溶液に加えて、50℃で1時間半攪拌した。反応混合物に飽和重曹水を加えて、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。得られた残渣をTFA(10 mL)に溶かし、マイクロウェーブ照射下120℃で3時間攪拌した。反応液を濃縮し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチルおよびヘキサンより再結晶を行うことで標題化合物(116 mg)を得た。
MS (ESI+): [M+H]+ 429.1.
2-(1,1-ジフルオロプロピル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2,2-ジフルオロブタン酸(365 mg)およびDMF(0.011 mL)をTHF(1.5 mL)に溶かし、塩化オキサリル(0.205 mL)を0℃で加え、反応混合物を室温で2時間半撹拌した。反応混合液を(Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]アクリルアミド(200 mg)のDMA(6 mL)溶液に加えて、50℃で1時間半攪拌した。反応混合物に飽和重曹水を加えて、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。得られた残渣をTFA(10 mL)に溶かし、マイクロウェーブ照射下120℃で3時間攪拌した。反応液を濃縮し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチルおよびヘキサンより再結晶を行うことで標題化合物(116 mg)を得た。
MS (ESI+): [M+H]+ 429.1.
実施例133
2-(エチルスルファニル)-6-(5-オキソピロリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のF)からJ)に準じた方法で、1-(4-メトキシベンジル)-1H-ピラゾール-4-カルボン酸の代わりに1-(4-メトキシベンジル)-5-オキソピロリジン-3-カルボン酸を用い、J)の脱保護の条件を以下のように変更して表題化合物を得た。すなわちマイクロ波照射下120℃で2時間攪拌後、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物を得た。
MS (ESI+): [M+H]+ 414.1.
2-(エチルスルファニル)-6-(5-オキソピロリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のF)からJ)に準じた方法で、1-(4-メトキシベンジル)-1H-ピラゾール-4-カルボン酸の代わりに1-(4-メトキシベンジル)-5-オキソピロリジン-3-カルボン酸を用い、J)の脱保護の条件を以下のように変更して表題化合物を得た。すなわちマイクロ波照射下120℃で2時間攪拌後、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物を得た。
MS (ESI+): [M+H]+ 414.1.
実施例134
2-(エチルスルファニル)-6-(6-オキソピペリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(6-オキソピペリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 1-(2,4-ジメトキシベンジル)-6-オキソ-1,4,5,6-テトラヒドロピリジン-3-カルボン酸メチル
プロパ-2-イン酸メチル(8.71 g)のジエチルエーテル(120 mL)溶液に1-(2,4-ジメトキシフェニル)メタンアミン(15.7 g)のジエチルエーテル(30 mL)溶液を0℃で加え、室温で終夜撹拌した後、反応混合物を減圧濃縮した。得られた油状物、塩化アクリロイル(9.18 mL)およびTHF(300 mL)からなる混合物を2時間加熱還流した。反応混合物を減圧濃縮後、残渣に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(14.8 g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.55-2.62 (4H, m), 3.72 (3H, s), 3.80 (3H, s), 3.82 (3H, s), 4.66 (2H, s), 6.42-6.47 (2H, m), 7.17-7.22 (1H, m), 7.46 (1H, s).
プロパ-2-イン酸メチル(8.71 g)のジエチルエーテル(120 mL)溶液に1-(2,4-ジメトキシフェニル)メタンアミン(15.7 g)のジエチルエーテル(30 mL)溶液を0℃で加え、室温で終夜撹拌した後、反応混合物を減圧濃縮した。得られた油状物、塩化アクリロイル(9.18 mL)およびTHF(300 mL)からなる混合物を2時間加熱還流した。反応混合物を減圧濃縮後、残渣に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(14.8 g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.55-2.62 (4H, m), 3.72 (3H, s), 3.80 (3H, s), 3.82 (3H, s), 4.66 (2H, s), 6.42-6.47 (2H, m), 7.17-7.22 (1H, m), 7.46 (1H, s).
B) 1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-カルボン酸メチル
1-(2,4-ジメトキシベンジル)-6-オキソ-1,4,5,6-テトラヒドロピリジン-3-カルボン酸メチル(1.05 g)、炭酸ナトリウム(1.09 g)、10%パラジウム/活性炭(50% 含水,365 mg)およびエタノール(20 mL)からなる混合物を水素雰囲気下、室温で3時間撹拌した。不溶物をセライトで濾過した後、濾液に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去することで標題化合物(1.04 g)を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.91-2.02 (1H, m), 2.07-2.17 (1H, m), 2.38-2.61 (2H, m), 2.72-2.83 (1H, m), 3.44 (2H, d, J = 7.2 Hz), 3.67 (3H, s), 3.80 (3H, s), 3.80 (3H, s), 4.52 (1H, d, J = 14.7 Hz), 4.61 (1H, d, J = 14.7 Hz), 6.43-6.47 (2H, m), 7.14-7.18 (1H, m).
1-(2,4-ジメトキシベンジル)-6-オキソ-1,4,5,6-テトラヒドロピリジン-3-カルボン酸メチル(1.05 g)、炭酸ナトリウム(1.09 g)、10%パラジウム/活性炭(50% 含水,365 mg)およびエタノール(20 mL)からなる混合物を水素雰囲気下、室温で3時間撹拌した。不溶物をセライトで濾過した後、濾液に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去することで標題化合物(1.04 g)を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.91-2.02 (1H, m), 2.07-2.17 (1H, m), 2.38-2.61 (2H, m), 2.72-2.83 (1H, m), 3.44 (2H, d, J = 7.2 Hz), 3.67 (3H, s), 3.80 (3H, s), 3.80 (3H, s), 4.52 (1H, d, J = 14.7 Hz), 4.61 (1H, d, J = 14.7 Hz), 6.43-6.47 (2H, m), 7.14-7.18 (1H, m).
C) 1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-カルボン酸
1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-カルボン酸メチル(1.04 g)のメタノール(15 mL)溶液に1規定水酸化ナトリウム水溶液(6.78 mL)を室温で加え、3時間撹拌した。反応混合物に1規定塩酸および水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去することで標題化合物(901 mg)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.76-1.91 (1H, m), 1.95-2.06 (1H, m), 2.23-2.43 (2H, m), 2.75-2.85 (1H, m), 3.27-3.34 (2H, m), 3.74 (3H, s), 3.78 (3H, s), 4.34 (1H, d, J = 15.1 Hz), 4.43 (1H, d, J = 15.1 Hz), 6.47 (1H, dd, J = 8.7, 2.3 Hz), 6.56 (1H, d, J = 2.3 Hz), 7.00 (1H, d, J = 8.7 Hz), 12.60 (1H, brs).
1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-カルボン酸メチル(1.04 g)のメタノール(15 mL)溶液に1規定水酸化ナトリウム水溶液(6.78 mL)を室温で加え、3時間撹拌した。反応混合物に1規定塩酸および水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去することで標題化合物(901 mg)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.76-1.91 (1H, m), 1.95-2.06 (1H, m), 2.23-2.43 (2H, m), 2.75-2.85 (1H, m), 3.27-3.34 (2H, m), 3.74 (3H, s), 3.78 (3H, s), 4.34 (1H, d, J = 15.1 Hz), 4.43 (1H, d, J = 15.1 Hz), 6.47 (1H, dd, J = 8.7, 2.3 Hz), 6.56 (1H, d, J = 2.3 Hz), 7.00 (1H, d, J = 8.7 Hz), 12.60 (1H, brs).
D) 3-[1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-イル]-3-オキソプロパン酸エチル
実施例1のF)に準じた方法により、1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-カルボン酸から標題化合物を油状物として得た。
MS (ESI+): [M+H]+ 364.2.
実施例1のF)に準じた方法により、1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-カルボン酸から標題化合物を油状物として得た。
MS (ESI+): [M+H]+ 364.2.
E) (2Z)-3-アミノ-3-[1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-イル]プロパ-2-エン酸エチル
実施例55のB)に準じた方法により、3-[1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-イル]-3-オキソプロパン酸エチルから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 363.2.
実施例55のB)に準じた方法により、3-[1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-イル]-3-オキソプロパン酸エチルから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 363.2.
F) 6-[1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-イル]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-[1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-イル]プロパ-2-エン酸エチルを用いて反応および後処理を行い、得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物を固体として得た。
MS (ESI+): [M+H]+ 550.2.
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-[1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-イル]プロパ-2-エン酸エチルを用いて反応および後処理を行い、得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物を固体として得た。
MS (ESI+): [M+H]+ 550.2.
G) 6-[1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-イル]-2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により、6-[1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-イル]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンを用いて反応および後処理を行い、残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物を固体として得た。
MS (ESI+): [M+H]+ 578.2.
実施例1のI)に準じた方法により、6-[1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-イル]-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンを用いて反応および後処理を行い、残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物を固体として得た。
MS (ESI+): [M+H]+ 578.2.
H) 2-(エチルスルファニル)-6-(6-オキソピペリジン-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-[1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-イル]-2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(200 mg)のトリフルオロ酢酸(3 mL)溶液をマイクロウェーブ照射下100℃で1時間撹拌した後、反応混合物を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(メタノール/酢酸エチル)に付し、得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(125 mg)を固体として得た。
MS (ESI+): [M+H]+ 428.1.
6-[1-(2,4-ジメトキシベンジル)-6-オキソピペリジン-3-イル]-2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(200 mg)のトリフルオロ酢酸(3 mL)溶液をマイクロウェーブ照射下100℃で1時間撹拌した後、反応混合物を減圧濃縮した。残渣をシリカゲルカラムクロマトグラフィー(メタノール/酢酸エチル)に付し、得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(125 mg)を固体として得た。
MS (ESI+): [M+H]+ 428.1.
実施例135
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例73に準じた方法で、2,2,2-トリフルオロエタノールの代わりに1,3-ジフルオロプロパン-2-オールを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 445.1.
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例73に準じた方法で、2,2,2-トリフルオロエタノールの代わりに1,3-ジフルオロプロパン-2-オールを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 445.1.
実施例136
2-(3,3-ジフルオロピロリジン-1-イル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例73に準じた方法で、2,2,2-トリフルオロエタノールの代わりに3,3-ジフルオロピロリジンを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 456.1.
2-(3,3-ジフルオロピロリジン-1-イル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例73に準じた方法で、2,2,2-トリフルオロエタノールの代わりに3,3-ジフルオロピロリジンを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 456.1.
実施例137
2-(2,2-ジフルオロエトキシ)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび2,2-ジフルオロエタノールを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 417.1.
2-(2,2-ジフルオロエトキシ)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび2,2-ジフルオロエタノールを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 417.1.
実施例138
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび1,3-ジフルオロプロパン-2-オールを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 431.1.
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび1,3-ジフルオロプロパン-2-オールを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 431.1.
実施例139
2-(シクロペンチルオキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例11に準じた方法で、エタノールの代わりにシクロペンタノールを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 435.1.
2-(シクロペンチルオキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例11に準じた方法で、エタノールの代わりにシクロペンタノールを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 435.1.
実施例140
6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロ-2-ヒドロキシプロポキシ)ピリミジン-4(3H)-オン
6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロ-2-ヒドロキシプロポキシ)ピリミジン-4(3H)-オン
A) 3-{[tert-ブチル(ジフェニル)シリル]オキシ}-1,1,1-トリフルオロプロパン-2-オール
3,3,3-トリフルオロプロパン-1,2-ジオール(187 mg)およびイミダゾール(117 mg)のDMF(20 mL)溶液にtert-ブチル(クロロ)ジフェニルシラン(0.392 mL)を0℃で加え、室温で終夜撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(353 mg)を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.00 (9H, s), 3.68-3.81 (2H, m), 4.06-4.16 (1H, m), 6.38 (1H, br. s), 7.40-7.52 (6H, m), 7.61-7.67 (4H, m).
3,3,3-トリフルオロプロパン-1,2-ジオール(187 mg)およびイミダゾール(117 mg)のDMF(20 mL)溶液にtert-ブチル(クロロ)ジフェニルシラン(0.392 mL)を0℃で加え、室温で終夜撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(353 mg)を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.00 (9H, s), 3.68-3.81 (2H, m), 4.06-4.16 (1H, m), 6.38 (1H, br. s), 7.40-7.52 (6H, m), 7.61-7.67 (4H, m).
B) 2-(2-{[tert-ブチル(ジフェニル)シリル]オキシ}-3,3,3-トリフルオロプロポキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび3-{[tert-ブチル(ジフェニル)シリル]オキシ}-1,1,1-トリフルオロプロパン-2-オールから標題化合物を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.88 (9H, s), 3.93-4.07 (2H, m), 4.04 (3H, s), 4.73-4.87 (2H, m), 5.90-6.03 (1H, m), 6.70 (1H, s), 6.93 (1H, d, J = 1.9 Hz), 7.07-7.53 (15H, m).
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび3-{[tert-ブチル(ジフェニル)シリル]オキシ}-1,1,1-トリフルオロプロパン-2-オールから標題化合物を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 0.88 (9H, s), 3.93-4.07 (2H, m), 4.04 (3H, s), 4.73-4.87 (2H, m), 5.90-6.03 (1H, m), 6.70 (1H, s), 6.93 (1H, d, J = 1.9 Hz), 7.07-7.53 (15H, m).
C) 6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロ-2-ヒドロキシプロポキシ)ピリミジン-4(3H)-オン
2-(2-{[tert-ブチル(ジフェニル)シリル]オキシ}-3,3,3-トリフルオロプロポキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(138 mg)のTHF(3 mL)溶液にテトラブチルアンモニウム フルオリドのTHF溶液(1.0M, 0.385 mL)を0℃で加え、室温で2時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(53 mg)を固体として得た。
MS (ESI+): [M+H]+ 479.2.
2-(2-{[tert-ブチル(ジフェニル)シリル]オキシ}-3,3,3-トリフルオロプロポキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(138 mg)のTHF(3 mL)溶液にテトラブチルアンモニウム フルオリドのTHF溶液(1.0M, 0.385 mL)を0℃で加え、室温で2時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(53 mg)を固体として得た。
MS (ESI+): [M+H]+ 479.2.
実施例141
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1-メチル-1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付した後、HPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物を固体として得た。
MS (ESI+): [M+H]+ 445.1.
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1-メチル-1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付した後、HPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物を固体として得た。
MS (ESI+): [M+H]+ 445.1.
実施例142
2-(2,2-ジフルオロエトキシ)-6-(1-メチル-1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-(2,2-ジフルオロエトキシ)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付した後、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製後、ジエチルエーテル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 431.1.
2-(2,2-ジフルオロエトキシ)-6-(1-メチル-1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法により2-(2,2-ジフルオロエトキシ)-6-(1H-ピラゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付した後、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製後、ジエチルエーテル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 431.1.
実施例143
2-(エチルスルファニル)-6-(1H-1,2,3-トリアゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1H-1,2,3-トリアゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 1-トリチル-1H-1,2,3-トリアゾール-4-カルボン酸
1-トリチル-1H-1,2,3-トリアゾール-4-カルボン酸エチル(17.9 g)を4規定水酸化ナトリウム水溶液(200 mL)およびメタノール(200 mL)に溶かし、1時間加熱還流した。反応混合液を0℃に冷却し、水(300 mL)および6規定塩酸を加え、酢酸エチルおよびTHFで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥させ、溶媒を減圧下留去した。得られた残渣をジイソプロピルエーテルで洗浄して、標題化合物(16.4 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 6.78-7.21 (6H, m), 7.28-7.73 (9H, m), 8.07 (1H, s), 13.25 (1H, brs).
1-トリチル-1H-1,2,3-トリアゾール-4-カルボン酸エチル(17.9 g)を4規定水酸化ナトリウム水溶液(200 mL)およびメタノール(200 mL)に溶かし、1時間加熱還流した。反応混合液を0℃に冷却し、水(300 mL)および6規定塩酸を加え、酢酸エチルおよびTHFで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥させ、溶媒を減圧下留去した。得られた残渣をジイソプロピルエーテルで洗浄して、標題化合物(16.4 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 6.78-7.21 (6H, m), 7.28-7.73 (9H, m), 8.07 (1H, s), 13.25 (1H, brs).
B) 3-オキソ-3-(1-トリチル-1H-1,2,3-トリアゾール-4-イル)プロパン酸エチル
実施例1のF)に準じた方法により、1-トリチル-1H-1,2,3-トリアゾール-4-カルボン酸より標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ1.26 (3H, t, J = 7.2 Hz), 4.16 (2H, s), 4.22 (2H, d, J = 7.2 Hz), 7.02-7.19 (6H, m), 7.28-7.48 (9H, m), 8.08 (1H, s).
実施例1のF)に準じた方法により、1-トリチル-1H-1,2,3-トリアゾール-4-カルボン酸より標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ1.26 (3H, t, J = 7.2 Hz), 4.16 (2H, s), 4.22 (2H, d, J = 7.2 Hz), 7.02-7.19 (6H, m), 7.28-7.48 (9H, m), 8.08 (1H, s).
C)(2Z)-3-アミノ-3-(1-トリチル-1H-1,2,3-トリアゾール-4-イル)プロパ-2-エン酸エチル
実施例1のG)に準じた方法により3-オキソ-3-(1-トリチル-1H-1,2,3-トリアゾール-4-イル)プロパン酸エチルより標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ1.18 (3H, t, J = 7.2 Hz), 4.04 (2H, q, J = 7.2 Hz), 5.31 (1H, s), 6.91-7.16 (6H, m), 7.57 (2H, br. s), 7.28-7.56 (9H, m), 8.48 (1H, s).
実施例1のG)に準じた方法により3-オキソ-3-(1-トリチル-1H-1,2,3-トリアゾール-4-イル)プロパン酸エチルより標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ1.18 (3H, t, J = 7.2 Hz), 4.04 (2H, q, J = 7.2 Hz), 5.31 (1H, s), 6.91-7.16 (6H, m), 7.57 (2H, br. s), 7.28-7.56 (9H, m), 8.48 (1H, s).
D) 2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-トリチル-1H-1,2,3-トリアゾール-4-イル)-2,3-ジヒドロピリミジン-4(1H)-オン
実施例1のH)に準じた方法により(2Z)-3-アミノ-3-(1-トリチル-1H-1,2,3-トリアゾール-4-イル)プロパ-2-エン酸エチルより標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 4.38 (2H, q, J = 8.3 Hz), 6.16 (1H, s), 6.88-7.51 (19H, m), 7.87 (1H, s), 9.73-10.72 (1H, m).
実施例1のH)に準じた方法により(2Z)-3-アミノ-3-(1-トリチル-1H-1,2,3-トリアゾール-4-イル)プロパ-2-エン酸エチルより標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ 4.38 (2H, q, J = 8.3 Hz), 6.16 (1H, s), 6.88-7.51 (19H, m), 7.87 (1H, s), 9.73-10.72 (1H, m).
E) 2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-トリチル-1H-1,2,3-トリアゾール-4-イル)-ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-トリチル-1H-1,2,3-トリアゾール-4-イル)-2,3-ジヒドロピリミジン-4(1H)-オンより標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.10 (3H, t, J = 7.2 Hz), 2.87-2.97 (2H, m), 4.86 (2H, q, J = 9.2 Hz), 6.79 (1H, s), 7.06-7.16 (6H, m), 7.20 (2H, d, J = 9.1 Hz), 7.32 (2H, s), 7.39-7.49 (9H, m), 8.08 (1H, s).
実施例1のI)に準じた方法により2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-トリチル-1H-1,2,3-トリアゾール-4-イル)-2,3-ジヒドロピリミジン-4(1H)-オンより標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.10 (3H, t, J = 7.2 Hz), 2.87-2.97 (2H, m), 4.86 (2H, q, J = 9.2 Hz), 6.79 (1H, s), 7.06-7.16 (6H, m), 7.20 (2H, d, J = 9.1 Hz), 7.32 (2H, s), 7.39-7.49 (9H, m), 8.08 (1H, s).
F) 2-(エチルスルファニル)-6-(1H-1,2,3-トリアゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-トリチル-1H-1,2,3-トリアゾール-4-イル)-ピリミジン-4(3H)-オン(3.31 g)を酢酸(30 mL)および水(15 mL)に溶かして、70℃で1時間半攪拌した。反応混合物を濃縮し、残渣を酢酸エチルに溶かした。続いて飽和重曹水および飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(1.62 g)を得た。
MS (ESI+): [M+H]+ 398.0.
2-(エチルスルファニル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-トリチル-1H-1,2,3-トリアゾール-4-イル)-ピリミジン-4(3H)-オン(3.31 g)を酢酸(30 mL)および水(15 mL)に溶かして、70℃で1時間半攪拌した。反応混合物を濃縮し、残渣を酢酸エチルに溶かした。続いて飽和重曹水および飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(1.62 g)を得た。
MS (ESI+): [M+H]+ 398.0.
実施例144
6-(1H-1,2,3-トリアゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1H-1,2,3-トリアゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 2-(エチルスルホニル)-6-(1H-1,2,3-トリアゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例74のC)に準じた方法により2-(エチルスルファニル)-6-(1H-1,2,3-トリアゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(1.5 g)から標題化合物(1.17 g)を得た。
MS (API-): [M-H]- 428.0.
1H NMR (300 MHz, DMSO-d6) δ 1.31 (3H, t, J = 7.3 Hz), 3.80 (2H, q, J = 7.3 Hz), 4.85 (2H, q, J = 8.9 Hz), 6.97-7.26 (3H, m), 7.30-7.48 (2H, m), 8.67 (1H, brs).
実施例74のC)に準じた方法により2-(エチルスルファニル)-6-(1H-1,2,3-トリアゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(1.5 g)から標題化合物(1.17 g)を得た。
MS (API-): [M-H]- 428.0.
1H NMR (300 MHz, DMSO-d6) δ 1.31 (3H, t, J = 7.3 Hz), 3.80 (2H, q, J = 7.3 Hz), 4.85 (2H, q, J = 8.9 Hz), 6.97-7.26 (3H, m), 7.30-7.48 (2H, m), 8.67 (1H, brs).
B) 6-(1H-1,2,3-トリアゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例2に準じた方法により2-(エチルスルホニル)-6-(1H-1,2,3-トリアゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(1 g)およびトリフルオロエタノール(0.339 mL)から合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物(727 mg)を得た。
MS (API-): [M-H]-434.0.
1H NMR (300 MHz, DMSO-d6) δ 4.85 (2H, q, J = 8.7 Hz), 5.17 (2H, q, J = 9.1 Hz), 6.76 (1H, s), 7.03-7.26 (2H, m), 7.26-7.40 (2H, m), 8.63 (1H, s), 15.60 (1H, brs).
実施例2に準じた方法により2-(エチルスルホニル)-6-(1H-1,2,3-トリアゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(1 g)およびトリフルオロエタノール(0.339 mL)から合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物(727 mg)を得た。
MS (API-): [M-H]-434.0.
1H NMR (300 MHz, DMSO-d6) δ 4.85 (2H, q, J = 8.7 Hz), 5.17 (2H, q, J = 9.1 Hz), 6.76 (1H, s), 7.03-7.26 (2H, m), 7.26-7.40 (2H, m), 8.63 (1H, s), 15.60 (1H, brs).
実施例145
2-(エチルスルファニル)-6-(1-メチル-1H-1,2,4-トリアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法で、2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンの代わりに2-(エチルスルファニル)-6-(1H-1,2,4-トリアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 412.1.
2-(エチルスルファニル)-6-(1-メチル-1H-1,2,4-トリアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例3に準じた方法で、2-(エチルスルファニル)-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンの代わりに2-(エチルスルファニル)-6-(1H-1,2,4-トリアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 412.1.
実施例146
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-2,4(1H,3H)-ジオン(400 mg)およびオキシ塩化リン(4 mL)の混合溶液を80℃で2時間撹拌した。反応溶液を減圧下留去した後、得られた残渣、THF溶液および1,3-ジフルオロプロパン-2-オール(400μL)の混合物に、水素化ナトリウム(60% oil dispersion、130 mg)を0℃で加えた。反応混合物を室温で終夜撹拌した後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(36.2 mg)を得た。
MS (ESI+): [M+H]+ 450.4.
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-2,4(1H,3H)-ジオン(400 mg)およびオキシ塩化リン(4 mL)の混合溶液を80℃で2時間撹拌した。反応溶液を減圧下留去した後、得られた残渣、THF溶液および1,3-ジフルオロプロパン-2-オール(400μL)の混合物に、水素化ナトリウム(60% oil dispersion、130 mg)を0℃で加えた。反応混合物を室温で終夜撹拌した後、反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去し、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(36.2 mg)を得た。
MS (ESI+): [M+H]+ 450.4.
実施例147
6-{2-(エチルスルファニル)-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,6-ジヒドロピリミジン-4-イル}ピリダジン-3(2H)-オン
6-{2-(エチルスルファニル)-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,6-ジヒドロピリミジン-4-イル}ピリダジン-3(2H)-オン
A) 6-[(4-メトキシベンジル)オキシ]ピリダジン-3-カルボン酸メチル
6-ヒドロキシピリダジン-3-カルボン酸メチル(9.51 g)のDMF(200 mL)溶液に水素化ナトリウム(60% oil dispersion、3.70 g)を0℃で加え、室温で1時間撹拌した。反応混合物に1-(クロロメチル)-4-メトキシベンゼン(10.0 mL)を0℃で加え、室温で4時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(8.63 g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 3.72 (3H, s), 3.86 (3H, s), 5.25 (2H, s), 6.87-6.92 (2H, m), 7.04 (1H, d, J = 9.8 Hz), 7.23-7.29 (2H, m), 7.85 (1H, d, J = 9.8 Hz).
6-ヒドロキシピリダジン-3-カルボン酸メチル(9.51 g)のDMF(200 mL)溶液に水素化ナトリウム(60% oil dispersion、3.70 g)を0℃で加え、室温で1時間撹拌した。反応混合物に1-(クロロメチル)-4-メトキシベンゼン(10.0 mL)を0℃で加え、室温で4時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(8.63 g)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 3.72 (3H, s), 3.86 (3H, s), 5.25 (2H, s), 6.87-6.92 (2H, m), 7.04 (1H, d, J = 9.8 Hz), 7.23-7.29 (2H, m), 7.85 (1H, d, J = 9.8 Hz).
B) 6-[(4-メトキシベンジル)オキシ]ピリダジン-3-カルボン酸
6-[(4-メトキシベンジル)オキシ]ピリダジン-3-カルボン酸メチル(8.62 g)のメタノール(30 mL)/THF(30 mL)/水(10 mL)溶液に8規定水酸化ナトリウム水溶液(11.8 mL)を室温で加え、70℃で2時間撹拌した。反応混合物を減圧下濃縮し、得られた残渣に6規定塩酸を0℃で加え、酢酸エチル/THFで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去することにより標題化合物(8.18 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 3.72 (3H, s), 5.23 (2H, s), 6.87-6.93 (2H, m), 7.01 (1H, d, J = 9.8 Hz), 7.24-7.30 (2H, m), 7.84 (1H, d, J = 9.8 Hz), 13.61 (1H, brs).
6-[(4-メトキシベンジル)オキシ]ピリダジン-3-カルボン酸メチル(8.62 g)のメタノール(30 mL)/THF(30 mL)/水(10 mL)溶液に8規定水酸化ナトリウム水溶液(11.8 mL)を室温で加え、70℃で2時間撹拌した。反応混合物を減圧下濃縮し、得られた残渣に6規定塩酸を0℃で加え、酢酸エチル/THFで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去することにより標題化合物(8.18 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 3.72 (3H, s), 5.23 (2H, s), 6.87-6.93 (2H, m), 7.01 (1H, d, J = 9.8 Hz), 7.24-7.30 (2H, m), 7.84 (1H, d, J = 9.8 Hz), 13.61 (1H, brs).
C) 3-{6-[(4-メトキシベンジル)オキシ]ピリダジン-3-イル}-3-オキソプロパン酸エチル
実施例1のF)に準じた方法により、6-[(4-メトキシベンジル)オキシ]ピリダジン-3-カルボン酸から標題化合物を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.13 (3H, t, J = 7.0 Hz), 3.73 (3H, s), 4.02 (2H, s), 4.07 (2H, q, J = 6.9 Hz), 5.22 (2H, s), 6.87-6.93 (2H, m), 7.05 (1H, d, J = 9.5 Hz), 7.28-7.34 (2H, m), 7.84 (1H, d, J = 9.5 Hz).
実施例1のF)に準じた方法により、6-[(4-メトキシベンジル)オキシ]ピリダジン-3-カルボン酸から標題化合物を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.13 (3H, t, J = 7.0 Hz), 3.73 (3H, s), 4.02 (2H, s), 4.07 (2H, q, J = 6.9 Hz), 5.22 (2H, s), 6.87-6.93 (2H, m), 7.05 (1H, d, J = 9.5 Hz), 7.28-7.34 (2H, m), 7.84 (1H, d, J = 9.5 Hz).
D) (2Z)-3-アミノ-3-{6-[(4-メトキシベンジル)オキシ]ピリダジン-3-イル}プロパ-2-エン酸エチル
実施例55のB)に準じた方法により、3-{6-[(4-メトキシベンジル)オキシ]ピリダジン-3-イル}-3-オキソプロパン酸エチルから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 330.1.
実施例55のB)に準じた方法により、3-{6-[(4-メトキシベンジル)オキシ]ピリダジン-3-イル}-3-オキソプロパン酸エチルから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 330.1.
E) 6-{6-オキソ-2-チオキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,2,3,6-テトラヒドロピリミジン-4-イル}ピリダジン-3(2H)-オン
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-{6-[(4-メトキシベンジル)オキシ]ピリダジン-3-イル}プロパ-2-エン酸エチルを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)にて精製した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物を固体として得た。
MS (ESI+): [M+H]+ 397.1.
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-{6-[(4-メトキシベンジル)オキシ]ピリダジン-3-イル}プロパ-2-エン酸エチルを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)にて精製した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物を固体として得た。
MS (ESI+): [M+H]+ 397.1.
F) 6-{2-(エチルスルファニル)-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,6-ジヒドロピリミジン-4-イル}ピリダジン-3(2H)-オン
実施例1のI)に準じた方法により、6-{6-オキソ-2-チオキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,2,3,6-テトラヒドロピリミジン-4-イル}ピリダジン-3(2H)-オンを用いて反応および後処理を行い、残渣を酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 425.1.
実施例1のI)に準じた方法により、6-{6-オキソ-2-チオキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,2,3,6-テトラヒドロピリミジン-4-イル}ピリダジン-3(2H)-オンを用いて反応および後処理を行い、残渣を酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 425.1.
実施例148、実施例150、実施例155
実施例148
6-(2-メチル-2H-1,2,3-トリアゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例150
6-(1-メチル-1H-1,2,3-トリアゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例155
6-(1-メチル-1H-1,2,3-トリアゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1H-1,2,3-トリアゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(622 mg)をDMF(10 mL)に溶かし、水素化ナトリウム(60% oil dispersion、62.9 mg)を加えて0℃で40分間攪拌した。その後、ヨウ化メチル(0.098 mL)を加えて、さらに0℃で1時間半攪拌した。反応混合物に水を加えて、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して酢酸エチルおよびヘキサンより再結晶を行い実施例148の化合物(230 mg)を得た。シリカゲルカラムクロマトグラフィー精製時に同時に得られた他の2種の混合物を酢酸エチルおよびヘキサンより再結晶することで、実施例150の化合物(180 mg)を得た。母液を濃縮し、酢酸エチルおよびヘキサンより再結晶することで実施例155の化合物(31 mg)を得た。
実施例148: MS (ESI+): [M+H]+ 450.1.
実施例150: MS (ESI+): [M+H]+ 450.1.
実施例155: MS (ESI+): [M+H]+ 450.1.
実施例148
6-(2-メチル-2H-1,2,3-トリアゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例150
6-(1-メチル-1H-1,2,3-トリアゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例155
6-(1-メチル-1H-1,2,3-トリアゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1H-1,2,3-トリアゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(622 mg)をDMF(10 mL)に溶かし、水素化ナトリウム(60% oil dispersion、62.9 mg)を加えて0℃で40分間攪拌した。その後、ヨウ化メチル(0.098 mL)を加えて、さらに0℃で1時間半攪拌した。反応混合物に水を加えて、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して酢酸エチルおよびヘキサンより再結晶を行い実施例148の化合物(230 mg)を得た。シリカゲルカラムクロマトグラフィー精製時に同時に得られた他の2種の混合物を酢酸エチルおよびヘキサンより再結晶することで、実施例150の化合物(180 mg)を得た。母液を濃縮し、酢酸エチルおよびヘキサンより再結晶することで実施例155の化合物(31 mg)を得た。
実施例148: MS (ESI+): [M+H]+ 450.1.
実施例150: MS (ESI+): [M+H]+ 450.1.
実施例155: MS (ESI+): [M+H]+ 450.1.
実施例149
6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例102のG)に準じた方法により1-メチル-5-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾールから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 447.1.
6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例102のG)に準じた方法により1-メチル-5-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-ピラゾールから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 447.1.
実施例151
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1H-1,2,3-トリアゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例2に準じた方法により2-(エチルスルホニル)-6-(1H-1,2,3-トリアゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび1,3-ジフルオロ-2-プロパノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (ESI+): [M+H]+ 432.1.
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1H-1,2,3-トリアゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例2に準じた方法により2-(エチルスルホニル)-6-(1H-1,2,3-トリアゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび1,3-ジフルオロ-2-プロパノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (ESI+): [M+H]+ 432.1.
実施例152
2-(エチルスルファニル)-6-(5-オキソピロリジン-2-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例133に準じた方法で、1-(4-メトキシベンジル)-5-オキソピロリジン-3-カルボン酸の代わりに1-(4-メトキシベンジル)-5-オキソプロリンを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 414.1.
2-(エチルスルファニル)-6-(5-オキソピロリジン-2-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例133に準じた方法で、1-(4-メトキシベンジル)-5-オキソピロリジン-3-カルボン酸の代わりに1-(4-メトキシベンジル)-5-オキソプロリンを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 414.1.
実施例153
6-(1-メチル-1H-ピラゾール-5-イル)-2-[2-(2,2,2-トリフルオロエトキシ)エトキシ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例73に準じた方法で、2,2,2-トリフルオロエタノールの代わりに2-(2,2,2-トリフルオロエトキシ)エタノールを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 493.1.
6-(1-メチル-1H-ピラゾール-5-イル)-2-[2-(2,2,2-トリフルオロエトキシ)エトキシ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例73に準じた方法で、2,2,2-トリフルオロエタノールの代わりに2-(2,2,2-トリフルオロエトキシ)エタノールを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 493.1.
実施例154
2-(エチルスルファニル)-6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 3-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-オキソプロパン酸エチル
実施例1のF)に準じた方法により、5-メチル-1,2,4-オキサジアゾール-3-カルボン酸から標題化合物を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.19 (3H, t, J = 7.2 Hz), 2.17 (3H, s), 3.58 (2H, s), 4.09 (2H, q, J = 7.2 Hz).
実施例1のF)に準じた方法により、5-メチル-1,2,4-オキサジアゾール-3-カルボン酸から標題化合物を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.19 (3H, t, J = 7.2 Hz), 2.17 (3H, s), 3.58 (2H, s), 4.09 (2H, q, J = 7.2 Hz).
B) (2Z)-3-アミノ-3-(5-メチル-1,2,4-オキサジアゾール-3-イル)プロパ-2-エン酸エチル
実施例55のB)に準じた方法により、3-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-オキソプロパン酸エチルから標題化合物を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.13 (3H, t, J = 7.0 Hz), 1.81 (3H, s), 3.95 (2H, q, J = 6.9 Hz), 4.29 (1H, s), 6.96 (1H, brs), 7.71 (1H, brs).
実施例55のB)に準じた方法により、3-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-オキソプロパン酸エチルから標題化合物を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.13 (3H, t, J = 7.0 Hz), 1.81 (3H, s), 3.95 (2H, q, J = 6.9 Hz), 4.29 (1H, s), 6.96 (1H, brs), 7.71 (1H, brs).
C) 6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オン
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-(5-メチル-1,2,4-オキサジアゾール-3-イル)プロパ-2-エン酸エチルを用いて反応および後処理を行い、得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.15 (3H, s), 4.81 (2H, q, J = 9.1 Hz), 5.91 (1H, s), 7.06-7.16 (4H, m), 12.60 (1H, s).
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-(5-メチル-1,2,4-オキサジアゾール-3-イル)プロパ-2-エン酸エチルを用いて反応および後処理を行い、得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.15 (3H, s), 4.81 (2H, q, J = 9.1 Hz), 5.91 (1H, s), 7.06-7.16 (4H, m), 12.60 (1H, s).
D) 2-(エチルスルファニル)-6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により、6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.21 (3H, t, J = 7.6 Hz), 2.23 (3H, s), 3.01 (2H, q, J = 7.6 Hz), 4.85 (2H, q, J = 8.7 Hz), 6.13 (1H, d, J = 0.8 Hz), 7.15-7.21 (2H, m), 7.26-7.32 (2H, m).
実施例1のI)に準じた方法により、6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-2-チオキソ-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2,3-ジヒドロピリミジン-4(1H)-オンを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 1.21 (3H, t, J = 7.6 Hz), 2.23 (3H, s), 3.01 (2H, q, J = 7.6 Hz), 4.85 (2H, q, J = 8.7 Hz), 6.13 (1H, d, J = 0.8 Hz), 7.15-7.21 (2H, m), 7.26-7.32 (2H, m).
実施例156
6-(1-メチル-1H-ピラゾール-5-イル)-2-[(2,2,3,3-テトラフルオロシクロブチル)オキシ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび2,2,3,3-テトラフルオロシクロブタノールを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 493.1.
6-(1-メチル-1H-ピラゾール-5-イル)-2-[(2,2,3,3-テトラフルオロシクロブチル)オキシ]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび2,2,3,3-テトラフルオロシクロブタノールを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 493.1.
実施例157
6-(1-メチル-1H-ピラゾール-5-イル)-2-[(2,2,2-トリフルオロエトキシ)メチル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(2,2,2-トリフルオロエトキシ)酢酸(700 mg)をTHF(3 mL)に溶かし、室温で3時間攪拌する。反応溶液を(Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]アクリルアミド(301 mg)のDMA(8 mL)溶液に0℃で加える。50℃で1時間攪拌した後、マイクロウェーブ照射下150℃で30分攪拌した。反応混合物に飽和重曹水を加えて、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、HPLC[C18、(移動相:水/アセトニトリル(NH4HCO3含有系)]にて分取した。得られた画分を濃縮した後、酢酸エチルおよびヘキサンより再結晶を行うことで標題化合物(142 mg)を得た。
MS (ESI+): [M+H]+ 463.1.
6-(1-メチル-1H-ピラゾール-5-イル)-2-[(2,2,2-トリフルオロエトキシ)メチル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(2,2,2-トリフルオロエトキシ)酢酸(700 mg)をTHF(3 mL)に溶かし、室温で3時間攪拌する。反応溶液を(Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]アクリルアミド(301 mg)のDMA(8 mL)溶液に0℃で加える。50℃で1時間攪拌した後、マイクロウェーブ照射下150℃で30分攪拌した。反応混合物に飽和重曹水を加えて、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、HPLC[C18、(移動相:水/アセトニトリル(NH4HCO3含有系)]にて分取した。得られた画分を濃縮した後、酢酸エチルおよびヘキサンより再結晶を行うことで標題化合物(142 mg)を得た。
MS (ESI+): [M+H]+ 463.1.
実施例160
6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-2,4(1H,3H)-ジオン
2-(エチルスルファニル)-6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(403 mg)の酢酸エチル(7 mL)/アセトニトリル(7 mL)溶液にm-クロロ過安息香酸(2.41 g)を0℃で加え、室温で5時間撹拌した。反応混合物に2-メチル-2-ブテン、飽和炭酸水素ナトリウム水溶液を0℃で加え、酢酸エチルで抽出した。抽出液を飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄後、硫酸マグネシウムで乾燥し、減圧濃縮した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(157 mg)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.09 (3H, s), 4.80 (2H, q, J = 8.8 Hz), 5.55 (1H, d, J = 0.8 Hz), 7.06-7.19 (4H, m), 11.21 (1H, brs).
2-(エチルスルファニル)-6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(403 mg)の酢酸エチル(7 mL)/アセトニトリル(7 mL)溶液にm-クロロ過安息香酸(2.41 g)を0℃で加え、室温で5時間撹拌した。反応混合物に2-メチル-2-ブテン、飽和炭酸水素ナトリウム水溶液を0℃で加え、酢酸エチルで抽出した。抽出液を飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄後、硫酸マグネシウムで乾燥し、減圧濃縮した。得られた固体をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物(157 mg)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.09 (3H, s), 4.80 (2H, q, J = 8.8 Hz), 5.55 (1H, d, J = 0.8 Hz), 7.06-7.19 (4H, m), 11.21 (1H, brs).
B) 6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(236 mg)および2,2,2-トリフルオロエチル トリフルオロメタンスルホナート(0.370 mL)のDMF(5 mL)溶液に水素化ナトリウム(60% oil dispersion、128 mg)を0℃で加え、室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和塩化アンモニウム水溶液および飽和食塩水で洗浄後、硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(51 mg)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.21 (3H, d, J = 0.8 Hz), 4.83 (2H, q, J = 8.7 Hz), 4.97 (2H, q, J = 8.7 Hz), 6.14 (1H, d, J = 0.8 Hz), 7.12-7.19 (2H, m), 7.23-7.29 (2H, m).
2-(エチルスルファニル)-6-(5-メチル-1,2,4-オキサジアゾール-3-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(236 mg)および2,2,2-トリフルオロエチル トリフルオロメタンスルホナート(0.370 mL)のDMF(5 mL)溶液に水素化ナトリウム(60% oil dispersion、128 mg)を0℃で加え、室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和塩化アンモニウム水溶液および飽和食塩水で洗浄後、硫酸マグネシウムで乾燥し、減圧濃縮した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(51 mg)を固体として得た。
1H NMR (300 MHz, DMSO-d6) δ 2.21 (3H, d, J = 0.8 Hz), 4.83 (2H, q, J = 8.7 Hz), 4.97 (2H, q, J = 8.7 Hz), 6.14 (1H, d, J = 0.8 Hz), 7.12-7.19 (2H, m), 7.23-7.29 (2H, m).
実施例161
6-(1-メチル-1H-イミダゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例102のG)に準じた方法により1-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-イミダゾールから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 447.1.
6-(1-メチル-1H-イミダゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-(3,3,3-トリフルオロプロピル)ピリミジン-4(3H)-オン
実施例102のG)に準じた方法により1-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-1H-イミダゾールから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 447.1.
実施例162
2-(2-ヒドロキシ-2-メチルプロポキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(2-ヒドロキシ-2-メチルプロポキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) ({4-(1-メチル-1H-ピラゾール-5-イル)-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,6-ジヒドロピリミジン-2-イル}オキシ)酢酸エチル
実施例73に準じた方法で、2,2,2-トリフルオロエタノールの代わりにヒドロキシ酢酸エチルを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 453.1.
実施例73に準じた方法で、2,2,2-トリフルオロエタノールの代わりにヒドロキシ酢酸エチルを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 453.1.
B) 2-(2-ヒドロキシ-2-メチルプロポキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
({4-(1-メチル-1H-ピラゾール-5-イル)-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,6-ジヒドロピリミジン-2-イル}オキシ)酢酸エチル(700 mg)のTHF溶液(20 mL)にブロモ(メチル)マグネシウムのTHF溶液(1 mol/L溶液、15 mL)を0℃で滴下し、室温で終夜撹拌した。反応混合物に飽和アンモニウム水溶液を0℃で加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去後、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(195 mg)を得た。
MS (ESI+): [M+H]+ 439.1.
({4-(1-メチル-1H-ピラゾール-5-イル)-6-オキソ-1-[4-(2,2,2-トリフルオロエトキシ)フェニル]-1,6-ジヒドロピリミジン-2-イル}オキシ)酢酸エチル(700 mg)のTHF溶液(20 mL)にブロモ(メチル)マグネシウムのTHF溶液(1 mol/L溶液、15 mL)を0℃で滴下し、室温で終夜撹拌した。反応混合物に飽和アンモニウム水溶液を0℃で加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去後、残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(195 mg)を得た。
MS (ESI+): [M+H]+ 439.1.
実施例163
2-(2,2-ジフルオロエトキシ)-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(2,2-ジフルオロエトキシ)-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 2-[(4-メトキシベンジル)オキシ]-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(2.00 g)および(4-メトキシフェニル)メタノール(3.05 mL)のTHF(50 mL)溶液に水素化ナトリウム(60% oil dispersion、979 mg)を0℃で加え、室温で3時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(1.83 g)を固体として得た。
MS (ESI+): [M+H]+ 485.2.
2-(エチルスルファニル)-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(2.00 g)および(4-メトキシフェニル)メタノール(3.05 mL)のTHF(50 mL)溶液に水素化ナトリウム(60% oil dispersion、979 mg)を0℃で加え、室温で3時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(1.83 g)を固体として得た。
MS (ESI+): [M+H]+ 485.2.
B) 6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-2,4(1H,3H)-ジオン
2-[(4-メトキシベンジル)オキシ]-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(1.83 g)のトリフルオロ酢酸(20 mL)溶液をマイクロウェーブ照射下100℃で30分間撹拌した後、反応混合物を減圧濃縮した。残渣をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物を含む混合物(1.77 g)を固体として得た。
MS (ESI+): [M+H]+ 365.0.
2-[(4-メトキシベンジル)オキシ]-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(1.83 g)のトリフルオロ酢酸(20 mL)溶液をマイクロウェーブ照射下100℃で30分間撹拌した後、反応混合物を減圧濃縮した。残渣をジイソプロピルエーテルに懸濁させた。沈殿物を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物を含む混合物(1.77 g)を固体として得た。
MS (ESI+): [M+H]+ 365.0.
C) 2-(2,2-ジフルオロエトキシ)-6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-2,4(1H,3H)-ジオン(250 mg)、炭酸セシウム(893 mg)およびDMF(7 mL)からなる混合物にトリフルオロメタンスルホン酸2,2-ジフルオロエチル(440 mg)を0℃で加え、室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和塩化アンモニウム水溶液および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(87 mg)を固体として得た。
MS (ESI+): [M+H]+ 429.1.
6-(ピリダジン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-2,4(1H,3H)-ジオン(250 mg)、炭酸セシウム(893 mg)およびDMF(7 mL)からなる混合物にトリフルオロメタンスルホン酸2,2-ジフルオロエチル(440 mg)を0℃で加え、室温で終夜撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を飽和塩化アンモニウム水溶液および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物(87 mg)を固体として得た。
MS (ESI+): [M+H]+ 429.1.
実施例164
6-(ピリダジン-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例163のC)に準じた方法によりトリフルオロメタンスルホン酸2,2,2-トリフルオロエチルから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 447.1.
6-(ピリダジン-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例163のC)に準じた方法によりトリフルオロメタンスルホン酸2,2,2-トリフルオロエチルから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 447.1.
実施例165
2-(2,2-ジフルオロエトキシ)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例113に準じた方法で、トリフルオロメタンスルホン酸2,2,2-トリフルオロエチルの代わりにトリフルオロメタンスルホン酸2,2-ジフルオロエチルを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 436.1.
2-(2,2-ジフルオロエトキシ)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例113に準じた方法で、トリフルオロメタンスルホン酸2,2,2-トリフルオロエチルの代わりにトリフルオロメタンスルホン酸2,2-ジフルオロエチルを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 436.1.
実施例166
6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
A) 2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン3-イル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(3.08 g)を酢酸エチル(30 mL)およびアセトニトリル(30 mL)に溶かし、m-クロロ過安息香酸(11.1 g)を加えて室温で3時間攪拌した。m-クロロ過安息香酸(11.1 g)を加えて室温でさらに2時間攪拌した。反応混合物に、2-メチル-2-ブテンを0℃で加えた後、析出物を濾過して標題化合物(1.12 g)を得た。さらに、濾液を飽和重曹水で5回洗浄後、飽和食塩水で洗浄した。抽出層を無水硫酸ナトリウムで乾燥させ、溶媒を減圧下留去し標題化合物(1.81 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.30 (3H, t, J = 7.3 Hz), 3.75 (2H, q, J = 7.4 Hz), 4.14 (3H, s), 4.84-5.29 (2H, m), 7.03 (1H, d, J = 2.3 Hz), 7.10-7.22 (2H, m), 7.57 (1H, d, J = 1.9 Hz), 7.92 (1H, dd, J = 8.7, 2.6 Hz), 8.29 (1H, d, J = 2.6 Hz).
2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(3.08 g)を酢酸エチル(30 mL)およびアセトニトリル(30 mL)に溶かし、m-クロロ過安息香酸(11.1 g)を加えて室温で3時間攪拌した。m-クロロ過安息香酸(11.1 g)を加えて室温でさらに2時間攪拌した。反応混合物に、2-メチル-2-ブテンを0℃で加えた後、析出物を濾過して標題化合物(1.12 g)を得た。さらに、濾液を飽和重曹水で5回洗浄後、飽和食塩水で洗浄した。抽出層を無水硫酸ナトリウムで乾燥させ、溶媒を減圧下留去し標題化合物(1.81 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.30 (3H, t, J = 7.3 Hz), 3.75 (2H, q, J = 7.4 Hz), 4.14 (3H, s), 4.84-5.29 (2H, m), 7.03 (1H, d, J = 2.3 Hz), 7.10-7.22 (2H, m), 7.57 (1H, d, J = 1.9 Hz), 7.92 (1H, dd, J = 8.7, 2.6 Hz), 8.29 (1H, d, J = 2.6 Hz).
B) 6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
実施例2に準じた方法により2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オンおよび2,2,2-トリフルオロエタノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (ESI+): [M+H]+ 450.09.
実施例2に準じた方法により2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オンおよび2,2,2-トリフルオロエタノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (ESI+): [M+H]+ 450.09.
実施例167
2-(2,2-ジフルオロエトキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
実施例2に準じた方法により2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オンおよび2,2-ジフルオロエタノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (ESI+): [M+H]+ 431.98.
2-(2,2-ジフルオロエトキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
実施例2に準じた方法により2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オンおよび2,2-ジフルオロエタノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (ESI+): [M+H]+ 431.98.
実施例168
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(250 mg)および1,3-ジフルオロ-2-プロパノール(0.131 mL)をDMF(5 mL)に溶かし、0℃で水素化ナトリウム(60% oil dispersion、 56.4 mg)を加えて室温で5時間攪拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物(164 mg)を得た。
MS (ESI+): [M+H]+ 446.2.
1H NMR (300 MHz, DMSO-d6) δ 4.14 (3H, s), 4.43-4.60 (1H, m), 4.70 (2H, dd, J = 10.9, 5.3 Hz), 4.77-4.91 (1H, m), 5.06 (2H, q, J = 9.2 Hz), 5.48-5.84 (1H, m), 6.69 (1H, s), 6.95 (1H, d, J = 1.9 Hz), 7.17 (1H, d, J = 8.7 Hz), 7.51 (1H, d, J = 1.9 Hz), 7.88 (1H, dd, J = 8.7, 2.6 Hz), 8.24 (1H, d, J = 1.9 Hz).
2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン(250 mg)および1,3-ジフルオロ-2-プロパノール(0.131 mL)をDMF(5 mL)に溶かし、0℃で水素化ナトリウム(60% oil dispersion、 56.4 mg)を加えて室温で5時間攪拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物(164 mg)を得た。
MS (ESI+): [M+H]+ 446.2.
1H NMR (300 MHz, DMSO-d6) δ 4.14 (3H, s), 4.43-4.60 (1H, m), 4.70 (2H, dd, J = 10.9, 5.3 Hz), 4.77-4.91 (1H, m), 5.06 (2H, q, J = 9.2 Hz), 5.48-5.84 (1H, m), 6.69 (1H, s), 6.95 (1H, d, J = 1.9 Hz), 7.17 (1H, d, J = 8.7 Hz), 7.51 (1H, d, J = 1.9 Hz), 7.88 (1H, dd, J = 8.7, 2.6 Hz), 8.24 (1H, d, J = 1.9 Hz).
実施例169
2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
A) 6-(1-メチル-1H-ピラゾール-5-イル)-2-チオキソ-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロピリミジン-4(1H)-オン
(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)プロパ-2-エン酸エチル(3.0 g)および5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジン(4.32 g)のDMF溶液(40 mL)に、水素化ナトリウム(60% oil dispersion、1.35 g)を0℃で加えた。反応混合物を室温で終夜撹拌した後、水に注ぎ、ついで1規定塩酸を加えて中和後、酢酸エチルを加えた。沈殿物を濾取し、水、ついで2-プロパノールで洗浄後、減圧下加熱乾燥し、沈殿物を濾取して標題化合物(2.35 g)を得た。得られた濾液より酢酸エチル層を分離し、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣を酢酸エチル/ジイソプロピルエーテルで結晶化させ、濾取して標題化合物(1.25 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ3.97 (3H, s), 5.04 (2H, q, J = 8.8 Hz), 6.34 (1H, brs), 6.74 (1H, d, J = 1.9 Hz), 7.12 (1H, d, J = 8.7 Hz), 7.57 (1H, d, J = 1.9 Hz), 7.76 (1H, dd, J = 8.5, 2.4 Hz), 8.10 (1H, d, J = 2.6 Hz), 12.98 (1H, brs).
(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)プロパ-2-エン酸エチル(3.0 g)および5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジン(4.32 g)のDMF溶液(40 mL)に、水素化ナトリウム(60% oil dispersion、1.35 g)を0℃で加えた。反応混合物を室温で終夜撹拌した後、水に注ぎ、ついで1規定塩酸を加えて中和後、酢酸エチルを加えた。沈殿物を濾取し、水、ついで2-プロパノールで洗浄後、減圧下加熱乾燥し、沈殿物を濾取して標題化合物(2.35 g)を得た。得られた濾液より酢酸エチル層を分離し、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣を酢酸エチル/ジイソプロピルエーテルで結晶化させ、濾取して標題化合物(1.25 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ3.97 (3H, s), 5.04 (2H, q, J = 8.8 Hz), 6.34 (1H, brs), 6.74 (1H, d, J = 1.9 Hz), 7.12 (1H, d, J = 8.7 Hz), 7.57 (1H, d, J = 1.9 Hz), 7.76 (1H, dd, J = 8.5, 2.4 Hz), 8.10 (1H, d, J = 2.6 Hz), 12.98 (1H, brs).
B) 2-(エチルスルファニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オン
6-(1-メチル-1H-ピラゾール-5-イル)-2-チオキソ-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロピリミジン-4(1H)-オン(3.60 g)、ヨードエタン(0.90 mL)、炭酸カリウム(1.95 g)およびTHF(100 mL)の反応混合物を70℃で2時間撹拌した。反応混合物を飽和塩化アンモニウム水溶液に注ぎ、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた固体をジイソプロピルエーテルで洗浄して、標題化合物(3.23 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.27 (3H, t, J = 7.2 Hz), 3.15 (2H, q, J = 7.4 Hz), 4.18 (3H, s), 4.83-5.34 (2H, m), 6.74 (1H, s), 6.95 (1H, d, J = 1.9 Hz), 7.20 (1H, d, J =9.1 Hz), 7.52 (1H, d, J = 1.9 Hz), 7.95 (1H, dd, J = 8.7, 2.7 Hz), 8.31 (1H, d, J = 2.3 Hz).
6-(1-メチル-1H-ピラゾール-5-イル)-2-チオキソ-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2,3-ジヒドロピリミジン-4(1H)-オン(3.60 g)、ヨードエタン(0.90 mL)、炭酸カリウム(1.95 g)およびTHF(100 mL)の反応混合物を70℃で2時間撹拌した。反応混合物を飽和塩化アンモニウム水溶液に注ぎ、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた固体をジイソプロピルエーテルで洗浄して、標題化合物(3.23 g)を得た。
1H NMR (300 MHz, DMSO-d6) δ1.27 (3H, t, J = 7.2 Hz), 3.15 (2H, q, J = 7.4 Hz), 4.18 (3H, s), 4.83-5.34 (2H, m), 6.74 (1H, s), 6.95 (1H, d, J = 1.9 Hz), 7.20 (1H, d, J =9.1 Hz), 7.52 (1H, d, J = 1.9 Hz), 7.95 (1H, dd, J = 8.7, 2.7 Hz), 8.31 (1H, d, J = 2.3 Hz).
実施例170
2-(2,2-ジフルオロエトキシ)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オン
2-(2,2-ジフルオロエトキシ)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オン
A) 2-(エチルスルファニル)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オン
実施例1のA)~C)に準じた方法で、1-フルオロ-4-ニトロベンゼンの代わりに1,2-ジフルオロ-4-ニトロベンゼンを用いて3-フルオロ-1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼンを得た。実施例10に準じた方法で、3-フルオロ-1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 429.1.
実施例1のA)~C)に準じた方法で、1-フルオロ-4-ニトロベンゼンの代わりに1,2-ジフルオロ-4-ニトロベンゼンを用いて3-フルオロ-1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼンを得た。実施例10に準じた方法で、3-フルオロ-1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 429.1.
B) 3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-2,4(1H,3H)-ジオン
2-(エチルスルファニル)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オン(1.50 g)のアセトニトリル(100 mL)および酢酸エチル(100 mL)の混合溶液に、m-クロロ過安息香酸(8.63 g)を0℃で加え、室温で2時間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(328 mg)を得た。
MS (ESI+): [M+H]+ 384.8.
2-(エチルスルファニル)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オン(1.50 g)のアセトニトリル(100 mL)および酢酸エチル(100 mL)の混合溶液に、m-クロロ過安息香酸(8.63 g)を0℃で加え、室温で2時間撹拌した。反応混合物に飽和重曹水を加え、酢酸エチルで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(328 mg)を得た。
MS (ESI+): [M+H]+ 384.8.
C) 2-(2,2-ジフルオロエトキシ)-3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オン
実施例113のC)に準じた方法で、3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-2,4(1H,3H)-ジオンを用い、トリフルオロメタンスルホン酸2,2,2-トリフルオロエチルの代わりにトリフルオロメタンスルホン酸2,2-ジフルオロエチルを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 449.9.
実施例113のC)に準じた方法で、3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-2,4(1H,3H)-ジオンを用い、トリフルオロメタンスルホン酸2,2,2-トリフルオロエチルの代わりにトリフルオロメタンスルホン酸2,2-ジフルオロエチルを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 449.9.
実施例171
6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,3,3,3-ペンタフルオロプロポキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例73に準じた方法で、2,2,2-トリフルオロエタノールの代わりに2,2,3,3,3-ペンタフルオロプロパン-1-オールを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 499.0.
6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,3,3,3-ペンタフルオロプロポキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例73に準じた方法で、2,2,2-トリフルオロエタノールの代わりに2,2,3,3,3-ペンタフルオロプロパン-1-オールを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 499.0.
実施例172
2-(2,2-ジフルオロエトキシ)-6-(3-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法で、2-(エチルスルホニル)-6-(3-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび2,2,2-トリフルオロエタノールの代わりに2,2-ジフルオロエタノールを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 431.0.
2-(2,2-ジフルオロエトキシ)-6-(3-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法で、2-(エチルスルホニル)-6-(3-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび2,2,2-トリフルオロエタノールの代わりに2,2-ジフルオロエタノールを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 431.0.
実施例173
6-(3-メチル-1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69に準じた方法により、2-(エチルスルホニル)-6-(3-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 449.0.
6-(3-メチル-1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例69に準じた方法により、2-(エチルスルホニル)-6-(3-メチル-1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 449.0.
実施例174
6-(1-メチル-1H-ピラゾール-5-イル)-2-[1-(2,2,2-トリフルオロエトキシ)エチル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
6-(1-メチル-1H-ピラゾール-5-イル)-2-[1-(2,2,2-トリフルオロエトキシ)エチル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
A) 2-アセチル-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)オン
ピルビン酸(1.29 g)およびDMF(0.023 mL)をTHF(4 mL)に溶かし、塩化オキサリル(1 mL)を0℃で加え、反応混合物を室温で2時間半撹拌した。反応混合液を(Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]アクリルアミド(1 g)のDMA(8 mL)溶液に加えて、50℃で2時間攪拌した。反応混合物に飽和重曹水を加えて、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。得られた残渣を二つの容器に分けて、それぞれTFA(15 mL)に溶かし、マイクロウェーブ照射下100℃で1時間攪拌した。各々の反応液を混合し濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(819 mg)を得た。
MS (ESI+): [M+H]+ 393.1.
ピルビン酸(1.29 g)およびDMF(0.023 mL)をTHF(4 mL)に溶かし、塩化オキサリル(1 mL)を0℃で加え、反応混合物を室温で2時間半撹拌した。反応混合液を(Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)-N-[4-(2,2,2-トリフルオロエトキシ)フェニル]アクリルアミド(1 g)のDMA(8 mL)溶液に加えて、50℃で2時間攪拌した。反応混合物に飽和重曹水を加えて、酢酸エチルで抽出した。抽出層を水および飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。得られた残渣を二つの容器に分けて、それぞれTFA(15 mL)に溶かし、マイクロウェーブ照射下100℃で1時間攪拌した。各々の反応液を混合し濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(819 mg)を得た。
MS (ESI+): [M+H]+ 393.1.
B) 2-(1-ヒドロキシエチル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-アセチル-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)オン(50 mg)およびセリウムクロリド 7水和物(95 mg)をメタノール(2 mL)に溶かし、水素化ホウ素ナトリウム(7.2 mg)を加えて0℃で1時間攪拌した。反応混合物に飽和塩化アンモニウム水溶液を加えて、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥して、溶媒を減圧下留去した。同様に、2-アセチル-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)オン(770 mg)およびセリウムクロリド 7水和物(1.46 g)をメタノール(20 mL)に溶かし、水素化ホウ素ナトリウム(111 mg)を加えて0℃で30分間攪拌した。反応混合物に飽和塩化アンモニウム水溶液を加えて、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥して、溶媒を減圧下留去した。各々から得られた残渣を混合し、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(628 mg)を得た。
MS (ESI+): [M+H]+ 395.0.
2-アセチル-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)オン(50 mg)およびセリウムクロリド 7水和物(95 mg)をメタノール(2 mL)に溶かし、水素化ホウ素ナトリウム(7.2 mg)を加えて0℃で1時間攪拌した。反応混合物に飽和塩化アンモニウム水溶液を加えて、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥して、溶媒を減圧下留去した。同様に、2-アセチル-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)オン(770 mg)およびセリウムクロリド 7水和物(1.46 g)をメタノール(20 mL)に溶かし、水素化ホウ素ナトリウム(111 mg)を加えて0℃で30分間攪拌した。反応混合物に飽和塩化アンモニウム水溶液を加えて、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥して、溶媒を減圧下留去した。各々から得られた残渣を混合し、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製して標題化合物(628 mg)を得た。
MS (ESI+): [M+H]+ 395.0.
C) 6-(1-メチル-1H-ピラゾール-5-イル)-2-[1-(2,2,2-トリフルオロエトキシ)エチル]-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(1-ヒドロキシエチル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(300 mg)をDMF(5 mL)に溶かし、水素化ナトリウム(60% oil dispersion、152 mg)を加えて0℃で20分間攪拌した。反応混合液にトリフルオロメタンスルホン酸2,2,2-トリフルオロエチルを0℃で加えて、室温で1時間攪拌した。反応溶液に水を加えて、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥させ、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、HPLC[C18、移動相:水/アセトニトリル(TFA含有系)]にて分取した。得られた画分に飽和重曹水を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物(49.6 mg)を得た。
MS (ESI+): [M+H]+ 477.1.
2-(1-ヒドロキシエチル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(300 mg)をDMF(5 mL)に溶かし、水素化ナトリウム(60% oil dispersion、152 mg)を加えて0℃で20分間攪拌した。反応混合液にトリフルオロメタンスルホン酸2,2,2-トリフルオロエチルを0℃で加えて、室温で1時間攪拌した。反応溶液に水を加えて、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥させ、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、HPLC[C18、移動相:水/アセトニトリル(TFA含有系)]にて分取した。得られた画分に飽和重曹水を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮し、標題化合物(49.6 mg)を得た。
MS (ESI+): [M+H]+ 477.1.
実施例175
3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)ピリミジン-4(3H)-オン
実施例170に準じた方法により、トリフルオロメタンスルホン酸2,2-ジフルオロエチルの代わりにトリフルオロメタンスルホン酸2,2,2-トリフルオロエチルを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 467.0.
3-[3-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)ピリミジン-4(3H)-オン
実施例170に準じた方法により、トリフルオロメタンスルホン酸2,2-ジフルオロエチルの代わりにトリフルオロメタンスルホン酸2,2,2-トリフルオロエチルを用いて標題化合物を得た。
MS (ESI+): [M+H]+ 467.0.
実施例176
2-[1-(2,2-ジフルオロエトキシ)エチル]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(1-ヒドロキシエチル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(319 mg)をDMF(5 mL)に溶かし、水素化ナトリウム(60% oil dispersion、97 mg)を加えて0℃で5分間攪拌した。反応混合液にトリフルオロメタンスルホン酸2,2-ジフルオロエチル(520 mg)を加えて、0℃で1時間攪拌した。反応溶液に水を加えて、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥させ、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、標題化合物(149 mg)を得た。
MS (ESI+): [M+H]+ 459.2.
2-[1-(2,2-ジフルオロエトキシ)エチル]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
2-(1-ヒドロキシエチル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(319 mg)をDMF(5 mL)に溶かし、水素化ナトリウム(60% oil dispersion、97 mg)を加えて0℃で5分間攪拌した。反応混合液にトリフルオロメタンスルホン酸2,2-ジフルオロエチル(520 mg)を加えて、0℃で1時間攪拌した。反応溶液に水を加えて、酢酸エチルで抽出した。抽出層を飽和食塩水で洗浄後、無水硫酸ナトリウムで乾燥させ、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、標題化合物(149 mg)を得た。
MS (ESI+): [M+H]+ 459.2.
実施例177
3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)ピリミジン-4-(3H)-オン
3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)ピリミジン-4-(3H)-オン
A) 2-フルオロ-1-ニトロ-4-(2,2,2-トリフルオロエトキシ)ベンゼン
3-フルオロ-4-ニトロフェノール(15.2 g)、炭酸カリウム(10.2 g)およびN,N-ジメチルホルムアミド(80 mL)の混合物を氷浴で冷却し、トリフルオロメタンスルホン酸2,2,2-トリフルオロエチル(14.7 mL)を滴下した。反応混合物を室温で66時間攪拌した後、水で希釈し、6規定塩酸で中和した。反応混合物を酢酸エチルで抽出し、酢酸エチル層を10%クエン酸水溶液、水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して標題化合物(23.1 g)を得た。
1H NMR (300 MHz, CDCl3) δ4.44 (2H, q, J = 7.7 Hz), 6.71-7.00 (2H, m), 8.00-8.30 (1H, m).
3-フルオロ-4-ニトロフェノール(15.2 g)、炭酸カリウム(10.2 g)およびN,N-ジメチルホルムアミド(80 mL)の混合物を氷浴で冷却し、トリフルオロメタンスルホン酸2,2,2-トリフルオロエチル(14.7 mL)を滴下した。反応混合物を室温で66時間攪拌した後、水で希釈し、6規定塩酸で中和した。反応混合物を酢酸エチルで抽出し、酢酸エチル層を10%クエン酸水溶液、水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して標題化合物(23.1 g)を得た。
1H NMR (300 MHz, CDCl3) δ4.44 (2H, q, J = 7.7 Hz), 6.71-7.00 (2H, m), 8.00-8.30 (1H, m).
B) 2-フルオロ-4-(2,2,2-トリフルオロエトキシ)アニリン
2-フルオロ-1-ニトロ-4-(2,2,2-トリフルオロエトキシ)ベンゼン(23.1 g)をエタノール(500 mL)に溶かし、そこへ10%パラジウム/活性炭素(50% 含水,6.0 g)を加え、水素雰囲気下室温で5時間攪拌した。その後パラジウム/活性炭を濾去し、濾液を減圧濃縮して濃橙色油状の残渣を得た。得られた残渣を酢酸エチルに溶かし、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して標題化合物(19.7 g)を得た。
MS (ESI+): [M+H]+ 210.2.
2-フルオロ-1-ニトロ-4-(2,2,2-トリフルオロエトキシ)ベンゼン(23.1 g)をエタノール(500 mL)に溶かし、そこへ10%パラジウム/活性炭素(50% 含水,6.0 g)を加え、水素雰囲気下室温で5時間攪拌した。その後パラジウム/活性炭を濾去し、濾液を減圧濃縮して濃橙色油状の残渣を得た。得られた残渣を酢酸エチルに溶かし、飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、減圧濃縮して標題化合物(19.7 g)を得た。
MS (ESI+): [M+H]+ 210.2.
C) 2-フルオロ-1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼン
実施例58のB)に準じた方法により、2-フルオロ-4-(2,2,2-トリフルオロエトキシ)アニリンから標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ4.34 (2H, q, J = 8.0 Hz), 6.58-6.89 (2H, m), 7.15 (1H, t, J = 8.7 Hz).
実施例58のB)に準じた方法により、2-フルオロ-4-(2,2,2-トリフルオロエトキシ)アニリンから標題化合物を得た。
1H NMR (300 MHz, CDCl3) δ4.34 (2H, q, J = 8.0 Hz), 6.58-6.89 (2H, m), 7.15 (1H, t, J = 8.7 Hz).
D) 3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)-2-チオキソ-2,3-ジヒドロピリミジン-4(1H)-オン
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)プロパ-2-エン酸エチルおよび2-フルオロ-1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼンから標題化合物を得た。
MS (API-): [M-H]- 399.1.
1H NMR (300 MHz, DMSO-d6) δ3.92-4.03 (3H, m), 4.87 (2H, q, J = 9.0 Hz), 6.27 (1H, s), 6.78 (1H, d, J = 1.9 Hz), 7.01 (1H, dd, J = 8.9, 2.1 Hz), 7.18 (1H, dd, J = 11.7, 3.0 Hz), 7.37 (1H, t, J = 8.9 Hz), 7.57 (1H, s), 13.01 (1H, brs).
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)プロパ-2-エン酸エチルおよび2-フルオロ-1-イソチオシアナト-4-(2,2,2-トリフルオロエトキシ)ベンゼンから標題化合物を得た。
MS (API-): [M-H]- 399.1.
1H NMR (300 MHz, DMSO-d6) δ3.92-4.03 (3H, m), 4.87 (2H, q, J = 9.0 Hz), 6.27 (1H, s), 6.78 (1H, d, J = 1.9 Hz), 7.01 (1H, dd, J = 8.9, 2.1 Hz), 7.18 (1H, dd, J = 11.7, 3.0 Hz), 7.37 (1H, t, J = 8.9 Hz), 7.57 (1H, s), 13.01 (1H, brs).
E) 2-(エチルスルファニル)-3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により、3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)-2-チオキソ-2,3-ジヒドロピリミジン-4(1H)-オンから標題化合物を得た。
MS (ESI+): [M+H]+ 429.1
実施例1のI)に準じた方法により、3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)-2-チオキソ-2,3-ジヒドロピリミジン-4(1H)-オンから標題化合物を得た。
MS (ESI+): [M+H]+ 429.1
F) 2-(エチルスルホニル)-3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オン
実施例74のC)に準じた方法により、2-(エチルスルファニル)-3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オンから標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.30 (3H, t, J = 7.2 Hz), 3.78 (2H, qd, J = 7.3, 2.6 Hz), 4.15 (3H, s), 4.90 (2H, q, J = 8.9 Hz), 6.97-7.10 (2H, m), 7.16-7.30 (2H, m), 7.44-7.63 (2H, m).
実施例74のC)に準じた方法により、2-(エチルスルファニル)-3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オンから標題化合物を得た。
1H NMR (300 MHz, DMSO-d6) δ 1.30 (3H, t, J = 7.2 Hz), 3.78 (2H, qd, J = 7.3, 2.6 Hz), 4.15 (3H, s), 4.90 (2H, q, J = 8.9 Hz), 6.97-7.10 (2H, m), 7.16-7.30 (2H, m), 7.44-7.63 (2H, m).
G) 3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-ピリミジン-4-(3H)-オン
実施例2に準じた方法により2-(エチルスルホニル)-3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オンおよび2,2,2-トリフルオロエタノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (ESI+): [M+H]+ 467.0.
実施例2に準じた方法により2-(エチルスルホニル)-3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オンおよび2,2,2-トリフルオロエタノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (ESI+): [M+H]+ 467.0.
実施例178
2-(2,2-ジフルオロエトキシ)-3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4-(3H)-オン
実施例2に準じた方法により、2-(エチルスルホニル)-3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オンおよび2,2-ジフルオロエタノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (ESI+): [M+H]+ 449.0.
2-(2,2-ジフルオロエトキシ)-3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4-(3H)-オン
実施例2に準じた方法により、2-(エチルスルホニル)-3-[2-フルオロ-4-(2,2,2-トリフルオロエトキシ)フェニル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オンおよび2,2-ジフルオロエタノールから合成および精製した後、酢酸エチルおよびヘキサンから再結晶して標題化合物を得た。
MS (ESI+): [M+H]+ 449.0.
実施例179
2-(2,2-ジフルオロエトキシ)-3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オン
2-(2,2-ジフルオロエトキシ)-3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オン
A) 3-フルオロ-5-ニトロ-2-(2,2,2-トリフルオロエトキシ)ピリジン
2-クロロ-3-フルオロ-5-ニトロピリジン(8.17 g)、2,2,2-トリフルオロエタノール(3.99 mL)、炭酸カリウム(7.68 g)およびDMF(50 mL)からなる混合物を80℃で2時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(10.6 g)を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 5.24 (2H, q, J = 8.7 Hz), 8.73 (1H, dd, J = 9.6, 2.3 Hz), 8.98 (1H, d, J = 2.3 Hz).
2-クロロ-3-フルオロ-5-ニトロピリジン(8.17 g)、2,2,2-トリフルオロエタノール(3.99 mL)、炭酸カリウム(7.68 g)およびDMF(50 mL)からなる混合物を80℃で2時間撹拌した。反応混合物に水を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(10.6 g)を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 5.24 (2H, q, J = 8.7 Hz), 8.73 (1H, dd, J = 9.6, 2.3 Hz), 8.98 (1H, d, J = 2.3 Hz).
B) 5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-アミン
3-フルオロ-5-ニトロ-2-(2,2,2-トリフルオロエトキシ)ピリジン(10.6 g)、10%パラジウム/活性炭(50% 含水,1.50 g)およびメタノール(150 mL)からなる混合物を水素雰囲気下、室温で3時間撹拌した。不溶物をセライトで濾去した後、濾液を減圧濃縮した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(8.96 g)を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 4.88 (2H, q, J = 9.1 Hz), 5.22 (2H, s), 6.98 (1H, dd, J = 12.5, 2.3 Hz), 7.33 (1H, d, J = 2.3 Hz).
3-フルオロ-5-ニトロ-2-(2,2,2-トリフルオロエトキシ)ピリジン(10.6 g)、10%パラジウム/活性炭(50% 含水,1.50 g)およびメタノール(150 mL)からなる混合物を水素雰囲気下、室温で3時間撹拌した。不溶物をセライトで濾去した後、濾液を減圧濃縮した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物(8.96 g)を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 4.88 (2H, q, J = 9.1 Hz), 5.22 (2H, s), 6.98 (1H, dd, J = 12.5, 2.3 Hz), 7.33 (1H, d, J = 2.3 Hz).
C) 3-フルオロ-5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジン
5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-アミン(8.92 g)のTHF(150 mL)溶液にトリホスゲン(4.86 mL)を0℃で加え、3時間撹拌した。反応混合物に炭酸水素ナトリウム水溶液を0℃で加え酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン)に付し、標題化合物(9.64 g)を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 5.10 (2H, q, J = 8.8 Hz), 8.14 (1H, dd, J = 10.6, 2.3 Hz), 8.24 (1H, d, J = 2.3 Hz).
5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-アミン(8.92 g)のTHF(150 mL)溶液にトリホスゲン(4.86 mL)を0℃で加え、3時間撹拌した。反応混合物に炭酸水素ナトリウム水溶液を0℃で加え酢酸エチルで抽出した。抽出液を飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン)に付し、標題化合物(9.64 g)を油状物として得た。
1H NMR (300 MHz, DMSO-d6) δ 5.10 (2H, q, J = 8.8 Hz), 8.14 (1H, dd, J = 10.6, 2.3 Hz), 8.24 (1H, d, J = 2.3 Hz).
D) 3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-6-(1-メチル-1H-ピラゾール-5-イル)-2-チオキソ-2,3-ジヒドロピリミジン-4(1H)-オン
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)プロパ-2-エン酸エチルおよび3-フルオロ-5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジンを用いて反応および後処理を行い、得られた固体をジイソプロピルエーテルに懸濁させた。沈殿を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物を固体として得た。
MS (API-): [M-H]- 400.0.
実施例1のH)に準じた方法により、(2Z)-3-アミノ-3-(1-メチル-1H-ピラゾール-5-イル)プロパ-2-エン酸エチルおよび3-フルオロ-5-イソチオシアナト-2-(2,2,2-トリフルオロエトキシ)ピリジンを用いて反応および後処理を行い、得られた固体をジイソプロピルエーテルに懸濁させた。沈殿を濾取し、ジイソプロピルエーテルで洗浄後、乾燥することにより標題化合物を固体として得た。
MS (API-): [M-H]- 400.0.
E) 2-(エチルスルファニル)-3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オン
実施例1のI)に準じた方法により、3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-6-(1-メチル-1H-ピラゾール-5-イル)-2-チオキソ-2,3-ジヒドロピリミジン-4(1H)-オンを用いて反応および後処理を行い、残渣を酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 430.1.
実施例1のI)に準じた方法により、3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-6-(1-メチル-1H-ピラゾール-5-イル)-2-チオキソ-2,3-ジヒドロピリミジン-4(1H)-オンを用いて反応および後処理を行い、残渣を酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 430.1.
F) 3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2-[(4-メトキシベンジル)オキシ]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オン
実施例163のA)に準じた方法により、2-(エチルスルファニル)-3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オンを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物を得た。
MS (ESI+): [M+H]+ 506.1.
実施例163のA)に準じた方法により、2-(エチルスルファニル)-3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オンを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付し、標題化合物を得た。
MS (ESI+): [M+H]+ 506.1.
G) 3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-2,4(1H,3H)-ジオン
実施例163のB)に準じた方法により、3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2-[(4-メトキシベンジル)オキシ]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オンから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 386.1.
実施例163のB)に準じた方法により、3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-2-[(4-メトキシベンジル)オキシ]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オンから標題化合物を固体として得た。
MS (ESI+): [M+H]+ 386.1.
H) 2-(2,2-ジフルオロエトキシ)-3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-4(3H)-オン
実施例160のB)に準じた方法により、3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-2,4(1H,3H)-ジオンおよびトリフルオロメタンスルホン酸2,2-ジフルオロエチルを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付した後、HPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮した。得られた固体を酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 450.0.
実施例160のB)に準じた方法により、3-[5-フルオロ-6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]-6-(1-メチル-1H-ピラゾール-5-イル)ピリミジン-2,4(1H,3H)-ジオンおよびトリフルオロメタンスルホン酸2,2-ジフルオロエチルを用いて反応および後処理を行い、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)に付した後、HPLC(C18、移動相:水/アセトニトリル(0.1% TFA含有系))にて分取し、得られた画分に飽和炭酸水素ナトリウム水溶液を加え、酢酸エチルで抽出した。無水硫酸マグネシウムで乾燥後、減圧下濃縮した。得られた固体を酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 450.0.
実施例180
2-[(4,4-ジフルオロシクロヘキシル)オキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例73に準じた方法で、2,2,2-トリフルオロエタノールの代わりに4,4-ジフルオロシクロヘキサノールを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 485.2.
2-[(4,4-ジフルオロシクロヘキシル)オキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン
実施例73に準じた方法で、2,2,2-トリフルオロエタノールの代わりに4,4-ジフルオロシクロヘキサノールを用い、標題化合物を得た。
MS (ESI+): [M+H]+ 485.2.
実施例181
6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン 塩酸塩
6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを酢酸エチルおよび6規定塩酸水溶液を用いて再結晶することで、標題化合物を得た。
MS (ESI+): [M+H-HCl]+ 449.2.
6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン 塩酸塩
6-(1-メチル-1H-ピラゾール-5-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンを酢酸エチルおよび6規定塩酸水溶液を用いて再結晶することで、標題化合物を得た。
MS (ESI+): [M+H-HCl]+ 449.2.
実施例182
6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-[(1S)-2,2,2-トリフルオロ-1-メチルエトキシ]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび(2S)-1,1,1-トリフルオロプロパン-2-オールを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 463.1.
6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-[(1S)-2,2,2-トリフルオロ-1-メチルエトキシ]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび(2S)-1,1,1-トリフルオロプロパン-2-オールを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 463.1.
実施例183
6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-[(1R)-2,2,2-トリフルオロ-1-メチルエトキシ]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび(2R)-1,1,1-トリフルオロプロパン-2-オールを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 463.1.
6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]-2-[(1R)-2,2,2-トリフルオロ-1-メチルエトキシ]ピリミジン-4(3H)-オン
実施例69のB)に準じた方法により、2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンおよび(2R)-1,1,1-トリフルオロプロパン-2-オールを用いて反応および後処理を行い、アミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した後、酢酸エチル/ヘキサンから再結晶して標題化合物を固体として得た。
MS (ESI+): [M+H]+ 463.1.
実施例184-187
実施例184: 2-[(1R or S)-2-フルオロ-1-メチルエトキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(保持時間小)
実施例185: 2-[(1S or R)-2-フルオロ-1-メチルエトキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(保持時間大)
実施例186: 2-{[(2R or S)-2-フルオロプロピル]オキシ}-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(保持時間小)
実施例187: 2-{[(2S or R)-2-フルオロプロピル]オキシ}-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(保持時間大)
2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(3.28 g)、1-フルオロプロパン-2-オールと2-フルオロプロパン-1-オール(4.53 mL)の混合物をTHF(120 mL)に溶解し、水素化ナトリウム(60% oil dispersion、740 mg)を0℃で加え、2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した。得られた2-(2-フルオロ-1-メチルエトキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンのラセミ体および2-(2-フルオロプロポキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンのラセミ体の混合物をHPLC(カラム:CHIRALPAK AD (AK001)、50 mmID×500 mmL、ダイセル化学工業製、移動相:メタノール = 100)にて分離精製し、保持時間の小さいほうから順に実施例184の化合物(799 mg)、実施例185の化合物(788 mg)、実施例186の化合物(472 mg)、実施例187の化合物(502 mg)を得た。得られた固体を各々酢酸エチル/ヘキサンから再結晶して実施例184の化合物(559 mg)、実施例185の化合物(470 mg)、実施例186の化合物(387 mg)、実施例187の化合物(397 mg)を固体として得た。
実施例184: 1H NMR (300 MHz, DMSO-d6) δ 1.24 (3H, dd, J = 6.4, 1.1 Hz), 4.16 (3H, s), 4.24-4.68 (2H, m), 4.84 (2H, q, J = 9.1 Hz), 5.37-5.55 (1H, m), 6.61 (1H, s), 6.92 (1H, d, J = 2.3 Hz), 7.13-7.20 (2H, m), 7.26-7.34 (2H, m), 7.50 (1H, d, J = 2.3 Hz).
MS (ESI+): [M+H]+ 427.1.
実施例185: 1H NMR (300 MHz, DMSO-d6) δ 1.24 (3H, dd, J = 6.4, 1.1 Hz), 4.16 (3H, s), 4.24-4.68 (2H, m), 4.84 (2H, q, J = 9.1 Hz), 5.37-5.55 (1H, m), 6.61 (1H, s), 6.92 (1H, d, J = 2.3 Hz), 7.13-7.20 (2H, m), 7.26-7.33 (2H, m), 7.50 (1H, d, J = 2.3 Hz).
MS (ESI+): [M+H]+ 427.1.
実施例186: 1H NMR (300 MHz, DMSO-d6) δ 1.17 (3H, dd, J = 23.8, 6.4 Hz), 4.18 (3H, s), 4.34-4.65 (2H, m), 4.75-5.02 (3H, m), 6.62 (1H, s), 6.93 (1H, d, J = 1.9 Hz), 7.15-7.21 (2H, m), 7.29-7.37 (2H, m), 7.50 (1H, d, J = 1.9 Hz).
MS (ESI+): [M+H]+ 427.1.
実施例187: 1H NMR (300 MHz, DMSO-d6) δ 1.17 (3H, dd, J = 23.8, 6.4 Hz), 4.18 (3H, s), 4.33-4.65 (2H, m), 4.75-5.02 (3H, m), 6.62 (1H, s), 6.93 (1H, d, J = 1.9 Hz), 7.14-7.22 (2H, m), 7.30-7.36 (2H, m), 7.50 (1H, d, J = 1.9 Hz).
MS (ESI+): [M+H]+ 427.1.
実施例184: 2-[(1R or S)-2-フルオロ-1-メチルエトキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(保持時間小)
実施例185: 2-[(1S or R)-2-フルオロ-1-メチルエトキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(保持時間大)
実施例186: 2-{[(2R or S)-2-フルオロプロピル]オキシ}-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(保持時間小)
実施例187: 2-{[(2S or R)-2-フルオロプロピル]オキシ}-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(保持時間大)
2-(エチルスルホニル)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オン(3.28 g)、1-フルオロプロパン-2-オールと2-フルオロプロパン-1-オール(4.53 mL)の混合物をTHF(120 mL)に溶解し、水素化ナトリウム(60% oil dispersion、740 mg)を0℃で加え、2時間撹拌した。反応混合物に1規定塩酸を加え、酢酸エチルで抽出した。抽出液を水および飽和食塩水で洗浄後、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をアミノプロピルシラン結合シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン)で精製した。得られた2-(2-フルオロ-1-メチルエトキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンのラセミ体および2-(2-フルオロプロポキシ)-6-(1-メチル-1H-ピラゾール-5-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンのラセミ体の混合物をHPLC(カラム:CHIRALPAK AD (AK001)、50 mmID×500 mmL、ダイセル化学工業製、移動相:メタノール = 100)にて分離精製し、保持時間の小さいほうから順に実施例184の化合物(799 mg)、実施例185の化合物(788 mg)、実施例186の化合物(472 mg)、実施例187の化合物(502 mg)を得た。得られた固体を各々酢酸エチル/ヘキサンから再結晶して実施例184の化合物(559 mg)、実施例185の化合物(470 mg)、実施例186の化合物(387 mg)、実施例187の化合物(397 mg)を固体として得た。
実施例184: 1H NMR (300 MHz, DMSO-d6) δ 1.24 (3H, dd, J = 6.4, 1.1 Hz), 4.16 (3H, s), 4.24-4.68 (2H, m), 4.84 (2H, q, J = 9.1 Hz), 5.37-5.55 (1H, m), 6.61 (1H, s), 6.92 (1H, d, J = 2.3 Hz), 7.13-7.20 (2H, m), 7.26-7.34 (2H, m), 7.50 (1H, d, J = 2.3 Hz).
MS (ESI+): [M+H]+ 427.1.
実施例185: 1H NMR (300 MHz, DMSO-d6) δ 1.24 (3H, dd, J = 6.4, 1.1 Hz), 4.16 (3H, s), 4.24-4.68 (2H, m), 4.84 (2H, q, J = 9.1 Hz), 5.37-5.55 (1H, m), 6.61 (1H, s), 6.92 (1H, d, J = 2.3 Hz), 7.13-7.20 (2H, m), 7.26-7.33 (2H, m), 7.50 (1H, d, J = 2.3 Hz).
MS (ESI+): [M+H]+ 427.1.
実施例186: 1H NMR (300 MHz, DMSO-d6) δ 1.17 (3H, dd, J = 23.8, 6.4 Hz), 4.18 (3H, s), 4.34-4.65 (2H, m), 4.75-5.02 (3H, m), 6.62 (1H, s), 6.93 (1H, d, J = 1.9 Hz), 7.15-7.21 (2H, m), 7.29-7.37 (2H, m), 7.50 (1H, d, J = 1.9 Hz).
MS (ESI+): [M+H]+ 427.1.
実施例187: 1H NMR (300 MHz, DMSO-d6) δ 1.17 (3H, dd, J = 23.8, 6.4 Hz), 4.18 (3H, s), 4.33-4.65 (2H, m), 4.75-5.02 (3H, m), 6.62 (1H, s), 6.93 (1H, d, J = 1.9 Hz), 7.14-7.22 (2H, m), 7.30-7.36 (2H, m), 7.50 (1H, d, J = 1.9 Hz).
MS (ESI+): [M+H]+ 427.1.
表1~表21に上記の実施例化合物の化学名、構造式およびMSの実測値を示す。





















試験例1
デルタ-5-デサチュラーゼに対する試験化合物の阻害活性を、以下に記述する方法で測定した。あらかじめ調製した試験化合物のDMSO溶液に対してバッファー(300mM NaH2PO4[pH7.4]、450mM KCl、30mM NaF、9mM MgCl2、4.5mM glutathione[reduced form]、0.3%BSA[fatty acid free、SIGMA])を用いて二次希釈を行い、アッセイバッファーとした。このアッセイバッファー10μLをポリプロピレン製の96穴ディープウェルブロックに分注した後、ミクロソームバッファー(10mM Tris-HCl[pH7.4]、1mM EDTA、250mM sucrose)を用いて3mg/mLに希釈したラット肝ミクロソーム画分10μLを添加した。酵素反応は、9mM NADH、9mM ATP、0.9mM CoA、10μCi/mL(8E,11E,14E)-(1-14C)eicosa-8,11,14-trienoic acid(PerkinElmer Inc.)を10μL添加することで開始した。120分間、室温で酵素反応を行い、10μLの2.5M NaOHの添加によって酵素反応を停止した。反応停止後プレートシールを施したうえで55℃に設定した乾熱器で一晩インキュベーションしてケン化を行った。脂肪酸の溶媒抽出はカナディアン ジャーナル オブ バイオケミストリー アンド フィジオロジー(Can.J.Biochem.Physiol.)、第37巻、911頁(1959)に記載のBligh & Dyer法に基づき、ギ酸:メタノール:クロロホルム(1:6:3)200μLを添加して一層状態を保ち、十分な撹拌を行った後に純水120μLを添加して、二層への分離を行った。下層のクロロホルム層10μLを逆相TLCプレート(RP-18、1154230001、Merck Japan, Ltd.)にスポットして、アセトニトリル:純水:酢酸(95:4.5:0.5)で展開し、乾燥後のTLCプレートをImaging Plate(Fuji Photo Film Co.,Ltd.)に5時間以上転写させた。検出はBAS-5000(Fuji Photo Film Co.,Ltd)を用いて行い、得られたスポット画像をMulti Gauge Ver2.3(Fuji Photo Film Co.,Ltd)を用いて数値化し、試験化合物の10μMでのデルタ-5-デサチュラーゼ阻害率(%)を求めた。結果を表22~30に示す。
表22~30から明らかなように、本発明化合物は優れたデルタ-5-デサチュラーゼ阻害作用を示した。
デルタ-5-デサチュラーゼに対する試験化合物の阻害活性を、以下に記述する方法で測定した。あらかじめ調製した試験化合物のDMSO溶液に対してバッファー(300mM NaH2PO4[pH7.4]、450mM KCl、30mM NaF、9mM MgCl2、4.5mM glutathione[reduced form]、0.3%BSA[fatty acid free、SIGMA])を用いて二次希釈を行い、アッセイバッファーとした。このアッセイバッファー10μLをポリプロピレン製の96穴ディープウェルブロックに分注した後、ミクロソームバッファー(10mM Tris-HCl[pH7.4]、1mM EDTA、250mM sucrose)を用いて3mg/mLに希釈したラット肝ミクロソーム画分10μLを添加した。酵素反応は、9mM NADH、9mM ATP、0.9mM CoA、10μCi/mL(8E,11E,14E)-(1-14C)eicosa-8,11,14-trienoic acid(PerkinElmer Inc.)を10μL添加することで開始した。120分間、室温で酵素反応を行い、10μLの2.5M NaOHの添加によって酵素反応を停止した。反応停止後プレートシールを施したうえで55℃に設定した乾熱器で一晩インキュベーションしてケン化を行った。脂肪酸の溶媒抽出はカナディアン ジャーナル オブ バイオケミストリー アンド フィジオロジー(Can.J.Biochem.Physiol.)、第37巻、911頁(1959)に記載のBligh & Dyer法に基づき、ギ酸:メタノール:クロロホルム(1:6:3)200μLを添加して一層状態を保ち、十分な撹拌を行った後に純水120μLを添加して、二層への分離を行った。下層のクロロホルム層10μLを逆相TLCプレート(RP-18、1154230001、Merck Japan, Ltd.)にスポットして、アセトニトリル:純水:酢酸(95:4.5:0.5)で展開し、乾燥後のTLCプレートをImaging Plate(Fuji Photo Film Co.,Ltd.)に5時間以上転写させた。検出はBAS-5000(Fuji Photo Film Co.,Ltd)を用いて行い、得られたスポット画像をMulti Gauge Ver2.3(Fuji Photo Film Co.,Ltd)を用いて数値化し、試験化合物の10μMでのデルタ-5-デサチュラーゼ阻害率(%)を求めた。結果を表22~30に示す。
表22~30から明らかなように、本発明化合物は優れたデルタ-5-デサチュラーゼ阻害作用を示した。
試験例2(HepG2細胞を用いたデルタ-5-デサチュラーゼ阻害活性)
96穴プレートに1×105cells/ウェルとなるようにHepG2を播種し、10% FBS(ウシ胎児血清)を含むDMEM(ダルベッコ改変イーグル培地)で一晩培養を行った。リン酸緩衝生理食塩水(PBS)(200μL×2)を用いて洗浄後、試験化合物を含む反応培地(DMEM、penicillin/streptomycin、0.3% BSA)を40μL添加し、CO2インキュベータにて37℃で30分間のプレインキュベーションを行った。反応は、0.1μCiの[14C]Eicosatrienoic acidを含む反応培地20μLの添加によって開始し、37℃に設定したCO2インキュベータにてインキュベーションを行った。インキュベーションを3時間行った後、200μLのPBSで2回洗浄し、50μLのトリプシン-EDTAで剥離した細胞を96穴ディープウェルブロックに移した。さらに、20μLの2.5N NaOHを添加し、プレートシールを施した上で、55℃に設定した乾熱器で一晩インキュベーションし、ケン化を行った。脂肪酸抽出は、10μLのギ酸添加で酸性化を行った後、クロロホルム:メタノール(1:4)を添加して攪拌し、さらに200μLの純水を添加して二層化することにより行った。TLC展開以降の操作は、試験例1におけるTLCを用いた検出系の手法に従った。試験化合物の1μMでのデルタ-5-デサチュラーゼ阻害率(%)を求めた。結果を表22~30に示す。
表22~30から明らかなように、本発明化合物は優れたデルタ-5-デサチュラーゼ阻害作用を示した。
96穴プレートに1×105cells/ウェルとなるようにHepG2を播種し、10% FBS(ウシ胎児血清)を含むDMEM(ダルベッコ改変イーグル培地)で一晩培養を行った。リン酸緩衝生理食塩水(PBS)(200μL×2)を用いて洗浄後、試験化合物を含む反応培地(DMEM、penicillin/streptomycin、0.3% BSA)を40μL添加し、CO2インキュベータにて37℃で30分間のプレインキュベーションを行った。反応は、0.1μCiの[14C]Eicosatrienoic acidを含む反応培地20μLの添加によって開始し、37℃に設定したCO2インキュベータにてインキュベーションを行った。インキュベーションを3時間行った後、200μLのPBSで2回洗浄し、50μLのトリプシン-EDTAで剥離した細胞を96穴ディープウェルブロックに移した。さらに、20μLの2.5N NaOHを添加し、プレートシールを施した上で、55℃に設定した乾熱器で一晩インキュベーションし、ケン化を行った。脂肪酸抽出は、10μLのギ酸添加で酸性化を行った後、クロロホルム:メタノール(1:4)を添加して攪拌し、さらに200μLの純水を添加して二層化することにより行った。TLC展開以降の操作は、試験例1におけるTLCを用いた検出系の手法に従った。試験化合物の1μMでのデルタ-5-デサチュラーゼ阻害率(%)を求めた。結果を表22~30に示す。
表22~30から明らかなように、本発明化合物は優れたデルタ-5-デサチュラーゼ阻害作用を示した。









試験例3
試験化合物のin vivoにおけるデルタ-5-デサチュラーゼ阻害作用の評価は以下の方法で行った。個別ケージを用いて一般飼育飼料(CE-2、日本クレア社)にて馴化した7-9週齢の雄性正常マウス(C57BL/6J, 日本クレア社)に、試験化合物3mg/kgを0.5%のメチルセルロース溶液に懸濁し、10mL/kgで4日間、毎朝強制経口投与した。最終投与翌日の朝に、マウスを麻酔し、肝臓を摘出した。約40 mgの肝臓サンプルから、ヘキサン-プロパノール溶液を用いて総脂質を抽出し、固相抽出カラム(Sep-Pak Vac NH2, Waters社)を用いてリン脂質画分を抽出した後、YMC社エステル型脂肪酸ラベル化試薬(YMC社)を用いてラベル化した。ラベル化後のサンプル中のアラキドン酸およびジホモ-γ-リノレン酸含有量は高速液体クロマトグラフィー(Agilent 1200、アジレント・テクノロジー社)を用いて測定した。アラキドン酸(AA)含有量をジホモ-γ-リノレン酸(DGLA)含有量で除することで、AA/DGLA比を算出し、溶媒投与群のAA/DGLA比を100%としたときの試験化合物投与群の低下率を算出することで、in vivoにおける肝臓デルタ-5-デサチュラーゼ阻害作用の指標とした。結果を表31に示す。
試験化合物のin vivoにおけるデルタ-5-デサチュラーゼ阻害作用の評価は以下の方法で行った。個別ケージを用いて一般飼育飼料(CE-2、日本クレア社)にて馴化した7-9週齢の雄性正常マウス(C57BL/6J, 日本クレア社)に、試験化合物3mg/kgを0.5%のメチルセルロース溶液に懸濁し、10mL/kgで4日間、毎朝強制経口投与した。最終投与翌日の朝に、マウスを麻酔し、肝臓を摘出した。約40 mgの肝臓サンプルから、ヘキサン-プロパノール溶液を用いて総脂質を抽出し、固相抽出カラム(Sep-Pak Vac NH2, Waters社)を用いてリン脂質画分を抽出した後、YMC社エステル型脂肪酸ラベル化試薬(YMC社)を用いてラベル化した。ラベル化後のサンプル中のアラキドン酸およびジホモ-γ-リノレン酸含有量は高速液体クロマトグラフィー(Agilent 1200、アジレント・テクノロジー社)を用いて測定した。アラキドン酸(AA)含有量をジホモ-γ-リノレン酸(DGLA)含有量で除することで、AA/DGLA比を算出し、溶媒投与群のAA/DGLA比を100%としたときの試験化合物投与群の低下率を算出することで、in vivoにおける肝臓デルタ-5-デサチュラーゼ阻害作用の指標とした。結果を表31に示す。

表31から明らかなように、本発明化合物は優れたin vivoデルタ-5-デサチュラーゼ阻害作用を示した。
製剤例1
(1)実施例1の化合物 10.0g
(2)乳糖 70.0g
(3)コーンスターチ 50.0g
(4)可溶性デンプン 7.0g
(5)ステアリン酸マグネシウム 3.0g
実施例1の化合物10.0gとステアリン酸マグネシウム3.0gを可溶性デンプンの水溶液70ml(可溶性デンプンとして7.0g)で顆粒化した後、乾燥し、乳糖70.0gおよびコーンスターチ50.0gと混合する(乳糖、コーンスターチ、可溶性デンプンおよびステアリン酸マグネシウムはいずれも第十四改正日本薬局方適合品)。混合物を圧縮して錠剤を得る。
(1)実施例1の化合物 10.0g
(2)乳糖 70.0g
(3)コーンスターチ 50.0g
(4)可溶性デンプン 7.0g
(5)ステアリン酸マグネシウム 3.0g
実施例1の化合物10.0gとステアリン酸マグネシウム3.0gを可溶性デンプンの水溶液70ml(可溶性デンプンとして7.0g)で顆粒化した後、乾燥し、乳糖70.0gおよびコーンスターチ50.0gと混合する(乳糖、コーンスターチ、可溶性デンプンおよびステアリン酸マグネシウムはいずれも第十四改正日本薬局方適合品)。混合物を圧縮して錠剤を得る。
本発明化合物は、デルタ-5-デサチュラーゼ阻害作用を有し、動脈硬化症、アテローム血栓症、糖尿病、肥満、喘息、発熱、疼痛、癌、リウマチ、変形性関節症またはアトピー性皮膚炎等の予防・治療に有用である。
本出願は、日本で出願された特願2010-166260および特願2010-167918を基礎としており、その内容は本明細書にすべて包含されるものである。
Claims (20)
- 式(I):

環Aはさらに置換されていてもよい6員芳香環を示し、
X1は結合手またはOを示し、
R1は3ないし11員環状基で置換されたC1-6アルキル基、置換されていてもよい3ないし11員複素環基、または置換されていてもよいC3-11シクロアルキル基を示し、
R2は水素原子またはハロゲン原子で置換されていてもよいC1-6アルキル基を示し、
R3は、
(1)式:-X2-R5(式中、X2はO、S、SO2またはNR6を示し、R5は置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示す)で表される基;
(2)置換されていてもよいC1-6アルキル基;
(3)置換されていてもよいC3-11シクロアルキル基;および
(4)置換されていてもよい3ないし11員非芳香族複素環基
を示し、
R4は置換されていてもよいC1-6アルキル基または置換されていてもよい3ないし11員環状基を示し、または
R6は水素原子または置換されていてもよいC1-6アルキル基を示す。〕
で表される化合物またはその塩。 - X1がOであり、
R4が1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基である、請求項1記載の化合物またはその塩。 - 環Aが、1ないし3個のハロゲン原子でさらに置換されていてもよいベンゼン、1ないし3個のハロゲン原子でさらに置換されていてもよいピリジン、または1もしくは2個のハロゲン原子でさらに置換されていてもよいピリミジンであり;
X1が、Oであり;
R1が、
(1)5ないし7員単環式芳香族複素環基で置換されたC1-6アルキル基、または
(2)(A)C1-6アルコキシ基、
(B)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基、
(C)ハロゲン原子、および
(D)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい5または6員複素環基であり;
R2が、水素原子またはメチル基であり;
R3が、
(1)式:-X2-R5
[式中、
X2が、O、SまたはNHであり、かつ
R5が、
(A)3ないし8員非芳香族複素環基、
(B)(i)C1-6アルキルスルホニル基、
(ii)ヒドロキシ基、
(iii)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、および
(iv)ハロゲン原子
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基、または
(C)1ないし5個のハロゲン原子で置換されていてもよいC3-11シクロアルキル基
である]で表される基、
(2)(A)ヒドロキシ基、
(B)1ないし3個のハロゲン原子で置換されていてもよいC1-6アルコキシ基、
(C)ハロゲン原子、および
(D)C3-6シクロアルキル基
から選ばれる1ないし5個の置換基で置換されていてもよいC1-6アルキル基、
(3)(A)C1-6アルキル基、および
(B)ハロゲン原子
から選ばれる1ないし3個の置換基で置換されていてもよいC3-11シクロアルキル基、または
(4)1ないし3個のハロゲン原子で置換されていてもよい3ないし11員非芳香族複素環基
であり;
R4が、1ないし3個のハロゲン原子で置換されていてもよいC1-6アルキル基
である、請求項1記載の化合物またはその塩。 - R1が、
(A)C1-6アルコキシ基、
(B)C1-6アルキル基、および
(C)オキソ基
から選ばれる1ないし3個の置換基で置換されていてもよい5または6員複素環基である、請求項3記載の化合物またはその塩。 - R1が、モルホリニル基または1ないし3個のC1-6アルキル基で置換されていてもよいピラゾリル基である、請求項4記載の化合物またはその塩。
- 2-(エチルスルファニル)-6-(モルホリン-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩。
- 2-プロピル-6-(1H-ピラゾール-4-イル)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩。
- 6-(1H-ピラゾール-4-イル)-2-(2,2,2-トリフルオロエトキシ)-3-[4-(2,2,2-トリフルオロエトキシ)フェニル]ピリミジン-4(3H)-オンまたはその塩。
- 2-[2-フルオロ-1-(フルオロメチル)エトキシ]-6-(1-メチル-1H-ピラゾール-5-イル)-3-[6-(2,2,2-トリフルオロエトキシ)ピリジン-3-イル]ピリミジン-4(3H)-オンまたはその塩。
- 請求項1記載の化合物またはその塩を含有してなる医薬。
- デルタ-5-デサチュラーゼ阻害薬である、請求項10記載の医薬。
- エイコサノイドが媒介する疾患の予防または治療剤である、請求項10記載の医薬。
- 動脈硬化症の予防または治療剤である、請求項10記載の医薬。
- 糖尿病または肥満症の予防または治療剤である、請求項10記載の医薬。
- 請求項1記載の化合物またはその塩を哺乳動物に有効量投与することを特徴とする、該哺乳動物における動脈硬化症の予防または治療方法。
- 請求項1記載の化合物またはその塩を哺乳動物に有効量投与することを特徴とする、該哺乳動物における糖尿病または肥満症の予防または治療方法。
- 動脈硬化症の予防または治療剤を製造するための、請求項1記載の化合物またはその塩の使用。
- 糖尿病または肥満症の予防または治療剤を製造するための、請求項1記載の化合物またはその塩の使用。
- 動脈硬化症の予防または治療のために使用する、請求項1記載の化合物またはその塩。
- 糖尿病または肥満症の予防または治療のために使用する、請求項1記載の化合物またはその塩。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012525457A JPWO2012011592A1 (ja) | 2010-07-23 | 2011-07-22 | 複素環化合物およびその用途 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010166260 | 2010-07-23 | ||
| JP2010-166260 | 2010-07-23 | ||
| JP2010167918 | 2010-07-27 | ||
| JP2010-167918 | 2010-07-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012011592A1 true WO2012011592A1 (ja) | 2012-01-26 |
Family
ID=45497009
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/066767 WO2012011592A1 (ja) | 2010-07-23 | 2011-07-22 | 複素環化合物およびその用途 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JPWO2012011592A1 (ja) |
| WO (1) | WO2012011592A1 (ja) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9056843B2 (en) | 2011-07-08 | 2015-06-16 | Novartis Ag | Trifluoromethyl-oxadiazole derivatives and their use in the treatment of disease |
| US9434695B2 (en) | 2012-07-18 | 2016-09-06 | Sunshine Lake Pharma Co., Ltd | Nitrogenous heterocyclic derivatives and their application in drugs |
| US9670193B2 (en) | 2011-11-28 | 2017-06-06 | Novartis Ag | Trifluoromethyl-oxadiazole derivatives and their use in the treatment of disease |
| WO2017222952A1 (en) * | 2016-06-23 | 2017-12-28 | Merck Sharp & Dohme Corp. | 3- heteroaryl substituted 5-trifluoromethyl oxadiazoles as histone deacetylase 6 (hdac6) inhibitors |
| US9902712B2 (en) | 2013-12-19 | 2018-02-27 | Sunshine Lake Pharma Co., Ltd. | Nitrogenous heterocyclic derivatives and their application in drugs |
| TWI729443B (zh) * | 2018-07-26 | 2021-06-01 | 韓商鐘根堂股份有限公司 | 做為組蛋白去乙醯酶6抑制劑之1,3,4-㗁二唑衍生物及包含彼之醫藥組合物 |
| CN113651762A (zh) * | 2021-09-03 | 2021-11-16 | 上海晋鲁医药科技有限公司 | 一种1-甲基-1h-1,2,4-三氮唑-3-甲酸甲酯的制备方法 |
| US11512097B2 (en) | 2019-11-25 | 2022-11-29 | Amgen Inc. | Heterocyclic compounds as Delta-5 desaturase inhibitors and methods of use |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007146761A2 (en) * | 2006-06-12 | 2007-12-21 | Neurogen Corporation | Diaryl pyrimidinones and related compounds |
| WO2008089307A2 (en) * | 2007-01-18 | 2008-07-24 | Lexicon Pharmaceuticals, Inc. | Delta 5 desaturase inhibitors for the treatment of pain, inflammation and cancer |
| WO2008156607A1 (en) * | 2007-06-12 | 2008-12-24 | Neurogen Corporation | Substituted pyrimidinones |
| JP2010510962A (ja) * | 2006-11-24 | 2010-04-08 | 武田薬品工業株式会社 | 複素単環化合物およびその用途 |
| WO2010087467A1 (en) * | 2009-01-27 | 2010-08-05 | Takeda Pharmaceutical Company Limited | Delta-5-desaturase inhibitors |
-
2011
- 2011-07-22 WO PCT/JP2011/066767 patent/WO2012011592A1/ja active Application Filing
- 2011-07-22 JP JP2012525457A patent/JPWO2012011592A1/ja not_active Withdrawn
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007146761A2 (en) * | 2006-06-12 | 2007-12-21 | Neurogen Corporation | Diaryl pyrimidinones and related compounds |
| JP2010510962A (ja) * | 2006-11-24 | 2010-04-08 | 武田薬品工業株式会社 | 複素単環化合物およびその用途 |
| WO2008089307A2 (en) * | 2007-01-18 | 2008-07-24 | Lexicon Pharmaceuticals, Inc. | Delta 5 desaturase inhibitors for the treatment of pain, inflammation and cancer |
| WO2008156607A1 (en) * | 2007-06-12 | 2008-12-24 | Neurogen Corporation | Substituted pyrimidinones |
| WO2010087467A1 (en) * | 2009-01-27 | 2010-08-05 | Takeda Pharmaceutical Company Limited | Delta-5-desaturase inhibitors |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9056843B2 (en) | 2011-07-08 | 2015-06-16 | Novartis Ag | Trifluoromethyl-oxadiazole derivatives and their use in the treatment of disease |
| US9670193B2 (en) | 2011-11-28 | 2017-06-06 | Novartis Ag | Trifluoromethyl-oxadiazole derivatives and their use in the treatment of disease |
| US9434695B2 (en) | 2012-07-18 | 2016-09-06 | Sunshine Lake Pharma Co., Ltd | Nitrogenous heterocyclic derivatives and their application in drugs |
| US9902712B2 (en) | 2013-12-19 | 2018-02-27 | Sunshine Lake Pharma Co., Ltd. | Nitrogenous heterocyclic derivatives and their application in drugs |
| WO2017222952A1 (en) * | 2016-06-23 | 2017-12-28 | Merck Sharp & Dohme Corp. | 3- heteroaryl substituted 5-trifluoromethyl oxadiazoles as histone deacetylase 6 (hdac6) inhibitors |
| TWI729443B (zh) * | 2018-07-26 | 2021-06-01 | 韓商鐘根堂股份有限公司 | 做為組蛋白去乙醯酶6抑制劑之1,3,4-㗁二唑衍生物及包含彼之醫藥組合物 |
| US11958844B2 (en) | 2018-07-26 | 2024-04-16 | Chong Kun Dang Pharmaceutical Corp. | 1,3,4-oxadiazole derivative compounds as histone deacetylase 6 inhibitor, and pharmaceutical composition comprising the same |
| US11512097B2 (en) | 2019-11-25 | 2022-11-29 | Amgen Inc. | Heterocyclic compounds as Delta-5 desaturase inhibitors and methods of use |
| CN113651762A (zh) * | 2021-09-03 | 2021-11-16 | 上海晋鲁医药科技有限公司 | 一种1-甲基-1h-1,2,4-三氮唑-3-甲酸甲酯的制备方法 |
| CN113651762B (zh) * | 2021-09-03 | 2023-12-26 | 上海晋鲁医药科技有限公司 | 一种1-甲基-1h-1,2,4-三氮唑-3-甲酸甲酯的制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2012011592A1 (ja) | 2013-09-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5824517B2 (ja) | 二環性化合物 | |
| RU2757457C2 (ru) | Пирролопиримидины в качестве потенциаторов мвтр | |
| CN106103446B (zh) | 作为自分泌运动因子(atx)和溶血磷脂酸(lpa)生产抑制剂的二环化合物 | |
| WO2012011592A1 (ja) | 複素環化合物およびその用途 | |
| TWI609018B (zh) | 新穎雙環衍生物 | |
| KR101181194B1 (ko) | 바이아릴 에터 우레아 화합물 | |
| KR101544624B1 (ko) | 피리미디닐 피리다지논 유도체 | |
| TWI433845B (zh) | 嗒酮衍生物 | |
| JP5800814B2 (ja) | 複素環化合物およびその用途 | |
| JP2021534139A (ja) | モノアシルグリセロールリパーゼ阻害剤としての新規の複素環化合物 | |
| JP2021533093A (ja) | モノアシルグリセロールリパーゼ阻害剤としての新規複素環化合物 | |
| KR101661777B1 (ko) | Met 키나아제 저해제로서의 3-(3-피리미딘-2-일-벤질)-〔1,2,4〕트리아졸로〔4,3-b〕피리다진 유도체 | |
| JP2019535689A (ja) | 4,5−縮環1,2,4−トリアゾロン | |
| JP2009544616A (ja) | アミド化合物 | |
| US20120264735A1 (en) | Tyrosine kinase inhibitors | |
| WO2014030743A1 (ja) | 複素環化合物 | |
| JP2015508051A (ja) | 芳香環化合物 | |
| TW201217362A (en) | Heteroaryls and uses thereof | |
| WO2010021381A1 (ja) | 縮合複素環誘導体およびその用途 | |
| US9023858B2 (en) | Substituted pyrido[2,3-d]pyrimidines as delta-5-desaturase inhibitors | |
| JP2022530097A (ja) | がんを処置するためのアシルスルホンアミド類 | |
| JP5722892B2 (ja) | 複素環化合物 | |
| WO2010001869A1 (ja) | 4置換ベンゼン化合物およびその用途 | |
| WO2013147026A1 (ja) | 芳香環化合物 | |
| TW201444825A (zh) | 嗒□酮化合物的製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11809762 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012525457 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 11809762 Country of ref document: EP Kind code of ref document: A1 |