WO2011122465A1 - ポリ乳酸系フィルム又はシート、及びその製造方法 - Google Patents
ポリ乳酸系フィルム又はシート、及びその製造方法 Download PDFInfo
- Publication number
- WO2011122465A1 WO2011122465A1 PCT/JP2011/057322 JP2011057322W WO2011122465A1 WO 2011122465 A1 WO2011122465 A1 WO 2011122465A1 JP 2011057322 W JP2011057322 W JP 2011057322W WO 2011122465 A1 WO2011122465 A1 WO 2011122465A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- film
- temperature
- crystallization
- weight
- resin composition
- Prior art date
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/18—Manufacture of films or sheets
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C43/00—Compression moulding, i.e. applying external pressure to flow the moulding material; Apparatus therefor
- B29C43/22—Compression moulding, i.e. applying external pressure to flow the moulding material; Apparatus therefor of articles of indefinite length
- B29C43/24—Calendering
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/03—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the shape of the extruded material at extrusion
- B29C48/07—Flat, e.g. panels
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/25—Component parts, details or accessories; Auxiliary operations
- B29C48/30—Extrusion nozzles or dies
- B29C48/35—Extrusion nozzles or dies with rollers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/25—Component parts, details or accessories; Auxiliary operations
- B29C48/88—Thermal treatment of the stream of extruded material, e.g. cooling
- B29C48/911—Cooling
- B29C48/9135—Cooling of flat articles, e.g. using specially adapted supporting means
- B29C48/914—Cooling of flat articles, e.g. using specially adapted supporting means cooling drums
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L101/00—Compositions of unspecified macromolecular compounds
- C08L101/16—Compositions of unspecified macromolecular compounds the macromolecular compounds being biodegradable
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/26—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers modified by chemical after-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L27/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a halogen; Compositions of derivatives of such polymers
- C08L27/02—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a halogen; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L27/12—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a halogen; Compositions of derivatives of such polymers not modified by chemical after-treatment containing fluorine atoms
- C08L27/18—Homopolymers or copolymers or tetrafluoroethene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L67/00—Compositions of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Compositions of derivatives of such polymers
- C08L67/04—Polyesters derived from hydroxycarboxylic acids, e.g. lactones
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/03—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the shape of the extruded material at extrusion
- B29C48/07—Flat, e.g. panels
- B29C48/08—Flat, e.g. panels flexible, e.g. films
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/03—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor characterised by the shape of the extruded material at extrusion
- B29C48/09—Articles with cross-sections having partially or fully enclosed cavities, e.g. pipes or channels
- B29C48/10—Articles with cross-sections having partially or fully enclosed cavities, e.g. pipes or channels flexible, e.g. blown foils
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29C—SHAPING OR JOINING OF PLASTICS; SHAPING OF MATERIAL IN A PLASTIC STATE, NOT OTHERWISE PROVIDED FOR; AFTER-TREATMENT OF THE SHAPED PRODUCTS, e.g. REPAIRING
- B29C48/00—Extrusion moulding, i.e. expressing the moulding material through a die or nozzle which imparts the desired form; Apparatus therefor
- B29C48/25—Component parts, details or accessories; Auxiliary operations
- B29C48/88—Thermal treatment of the stream of extruded material, e.g. cooling
- B29C48/90—Thermal treatment of the stream of extruded material, e.g. cooling with calibration or sizing, i.e. combined with fixing or setting of the final dimensions of the extruded article
- B29C48/906—Thermal treatment of the stream of extruded material, e.g. cooling with calibration or sizing, i.e. combined with fixing or setting of the final dimensions of the extruded article using roller calibration
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2023/00—Use of polyalkenes or derivatives thereof as moulding material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2027/00—Use of polyvinylhalogenides or derivatives thereof as moulding material
- B29K2027/12—Use of polyvinylhalogenides or derivatives thereof as moulding material containing fluorine
- B29K2027/18—PTFE, i.e. polytetrafluorethene, e.g. ePTFE, i.e. expanded polytetrafluorethene
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2067/00—Use of polyesters or derivatives thereof, as moulding material
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2067/00—Use of polyesters or derivatives thereof, as moulding material
- B29K2067/04—Polyesters derived from hydroxycarboxylic acids
- B29K2067/046—PLA, i.e. polylactic acid or polylactide
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29K—INDEXING SCHEME ASSOCIATED WITH SUBCLASSES B29B, B29C OR B29D, RELATING TO MOULDING MATERIALS OR TO MATERIALS FOR MOULDS, REINFORCEMENTS, FILLERS OR PREFORMED PARTS, e.g. INSERTS
- B29K2105/00—Condition, form or state of moulded material or of the material to be shaped
- B29K2105/0005—Condition, form or state of moulded material or of the material to be shaped containing compounding ingredients
- B29K2105/0038—Plasticisers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2367/00—Characterised by the use of polyesters obtained by reactions forming a carboxylic ester link in the main chain; Derivatives of such polymers
- C08J2367/04—Polyesters derived from hydroxy carboxylic acids, e.g. lactones
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/10—Esters; Ether-esters
- C08K5/11—Esters; Ether-esters of acyclic polycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/10—Esters; Ether-esters
- C08K5/12—Esters; Ether-esters of cyclic polycarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/03—Polymer mixtures characterised by other features containing three or more polymers in a blend
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L23/00—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers
- C08L23/02—Compositions of homopolymers or copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L23/04—Homopolymers or copolymers of ethene
- C08L23/08—Copolymers of ethene
- C08L23/0846—Copolymers of ethene with unsaturated hydrocarbons containing other atoms than carbon or hydrogen atoms
- C08L23/0869—Acids or derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L51/00—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers
- C08L51/06—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers grafted on to homopolymers or copolymers of aliphatic hydrocarbons containing only one carbon-to-carbon double bond
Definitions
- the present invention relates to a polylactic acid film or sheet having heat resistance, roll lubricity and excellent flexibility.
- polylactic acid resin is a biomass polymer, it has attracted attention in recent years against the background of oil depletion and carbon dioxide emission reduction.
- polylactic acid has a low crystallization rate and is hardly crystallized by ordinary film forming means.
- a film made of a resin composition containing polylactic acid has a problem of heat resistance that causes thermal deformation at a temperature of about 60 ° C. or more, which is the glass transition temperature of polylactic acid, and the film shape cannot be maintained.
- Patent Document 2 A method for obtaining a flexible molded article has been proposed (Patent Document 2).
- Patent Document 2 A method for obtaining a flexible molded article has been proposed (Patent Document 2).
- Patent Document 2 it is necessary to add a considerable amount of styrenic copolymer, and the degree of biomass is significantly reduced.
- it has only heat resistance of about 80 ° C., and heat resistance sufficient to cope with heat-resistant parts of home appliances and automobiles is not obtained.
- Patent Document 3 A technique has been proposed (Patent Document 3). However, this method is inefficient because it is cooled and solidified once and then heated again.
- the problem to be solved by the present invention is to provide a polylactic acid film or sheet having excellent heat resistance and flexibility by maintaining high crystallinity, and a method for producing such a film and sheet. It is to be.
- the present inventors have given flexibility by using a specific dicarboxylic acid ester plasticizer that melts in a temperature range in which polylactic acid is melt-kneaded. It has been found that a film or sheet having heat resistance and flexibility can be obtained by blending a polymer and improving the crystallinity of polylactic acid, and has completed the present invention.
- the present invention is as follows.
- seat which consists of a resin composition whose content of the said dicarboxylic ester plasticizer (D) is 8 weight part or more and less than 40 weight part with respect to 100 weight part of polylactic acid (A).
- Aa and Ab are each independently selected from the following (1) to (3) (excluding compounds in which both Aa and Ab are the following (3)): (1) C 6 H 5 — (Ac) —, where Ac is an alkylene group having 1 to 4 carbon atoms; (2) (Ae)-(Ad-O) n- (where Ad is an alkylene group having 1 to 5 carbon atoms, Ae is an alkyl group having 1 to 7 carbon atoms, and n is an integer of 1 to 4) ;as well as (3) An alkyl group having 1 to 14 carbon atoms.
- the dicarboxylic acid ester plasticizer (D) is a phthalic acid ester plasticizer (DP) selected from the compounds represented by the general formulas (2) to (6).
- A1 and A2 are the same or different and each represents an alkylene group having 1 to 4 carbon atoms;
- A3 represents an alkyl group having 1 to 14 carbon atoms;
- A4 represents an alkylene group having 1 to 5 carbon atoms;
- A5 represents an alkyl group having 1 to 7 carbon atoms, and
- n represents an integer of 1 to 4.
- the dicarboxylic acid ester plasticizer (D) is an aliphatic dicarboxylic acid ester plasticizer (DA) selected from the compounds represented by the general formulas (7) to (11), and the aliphatic dicarboxylic acid The film or sheet according to the above [1], wherein the content of the acid ester plasticizer (DA) is 8 to 35 parts by weight with respect to 100 parts by weight of the polylactic acid (A).
- A1 and A2 are the same or different and each represents an alkylene group having 1 to 4 carbon atoms;
- A3 represents an alkyl group having 1 to 14 carbon atoms;
- A4 represents an alkylene group having 1 to 5 carbon atoms;
- A5 represents an alkyl group having 1 to 7 carbon atoms, n represents an integer of 1 to 4, and
- m represents an integer of 0 to 6.
- Relative crystallization rate (%) ( ⁇ Hm ⁇ Hc) / ⁇ Hm ⁇ 100 (I) (In the formula, ⁇ Hc is the calorific value of the exothermic peak accompanying crystallization in the temperature rising process of the film or sheet after film formation, and ⁇ Hm represents the calorific value accompanying melting.) [9] The film or sheet according to any one of [1] to [8] above, wherein the tensile elongation at break is 100% or more and the residual stress rate at 10% elongation is 40% or less .
- the method for producing a film or sheet according to any one of the above [1] to [9], comprising film-forming the resin composition by a melt film-forming method,
- the temperature of the resin composition at the time of melt film formation is a temperature between the crystallization temperature (Tc) + 15 ° C. in the temperature lowering process of the resin composition and the melting temperature (Tm) ⁇ 5 ° C. in the temperature rising process.
- the resin composition that has been melt-formed is subjected to a crystallization promotion step at a temperature between the crystallization temperature (Tc) -25 ° C. and the crystallization temperature (Tc) + 10 ° C.
- the temperature of the resin composition at the time of melt film formation is from the temperature of the crystallization temperature (Tc) + 15 ° C. in the temperature lowering process of the resin composition to the melting temperature (Tm) ⁇ 5 ° C. in the temperature rising process.
- the temperature between and The resin composition that has been melt-formed is subjected to a crystallization promotion step at a temperature between the crystallization temperature (Tc) -25 ° C. and the crystallization temperature (Tc) + 10 ° C.
- the method for producing a film or sheet according to the above [10] wherein the film or sheet is cooled and solidified after passing.
- melt film-forming method is a method of forming a film with a desired thickness by passing a molten resin composition through a gap between two metal rolls.
- a method for producing a film or sheet is a method for producing a film or sheet.
- the film or sheet of the present invention comprises a resin composition containing polylactic acid (A), an acidic functional group-modified olefin polymer (B), a tetrafluoroethylene polymer (C), and a dicarboxylic acid ester plasticizer (D).
- A polylactic acid
- B an acidic functional group-modified olefin polymer
- C a tetrafluoroethylene polymer
- D dicarboxylic acid ester plasticizer
- the film or sheet of the present invention includes transparent, translucent and opaque ones.
- the thickness of the film or sheet of the present invention is not particularly limited, but is usually 10 to 500 ⁇ m, preferably 20 to 400 ⁇ m, more preferably 30 to 300 ⁇ m.
- polylactic acid (A) Since lactic acid, which is a raw material monomer for polylactic acid, has an asymmetric carbon atom, there are L and D isomers of optical isomers.
- the polylactic acid (A) used in the present invention is a polymer composed mainly of L-form lactic acid. The smaller the content of lactic acid in the D-form that is mixed as an impurity during production, the higher the crystalline and high-melting polymer. Therefore, it is preferable to use a L-form having a purity as high as possible, and the L-form purity is 95%. It is more preferable to use the above.
- the polylactic acid (A) used by this invention may contain other copolymerization components other than lactic acid.
- Other monomer units include ethylene glycol, propylene glycol, butanediol, heptanediol, hexanediol, octanediol, nonanediol, decanediol, 1,4-cyclohexanedimethanol, neopentyl glycol, glycerin, pentaerythritol, bisphenol.
- glycol compounds such as polyethylene glycol, polypropylene glycol, polytetramethylene glycol; oxalic acid, adipic acid, sebacic acid, azelaic acid, dodecanedioic acid, malonic acid, glutaric acid, cyclohexanedicarboxylic acid, terephthalic acid, isophthalic acid, phthalate Acid, naphthalenedicarboxylic acid, bis (p-carboxyphenyl) methane, anthracene dicarboxylic acid, 4,4′-diphenyl ether dicarboxylic acid, Dicarboxylic acids such as 5-sodium sulfoisophthalic acid and 5-tetrabutylphosphonium isophthalic acid; hydroxycarboxylic acids such as glycolic acid, hydroxypropionic acid, hydroxybutyric acid, hydroxyvaleric acid, hydroxycaproic acid, hydroxybenzoic acid; caprolactone, valerol
- the weight average molecular weight of the polylactic acid (A) is, for example, 10,000 to 400,000, preferably 50,000 to 300,000, and more preferably 80,000 to 150,000.
- the melt flow rate [JIS K-7210 (Test condition 4)] of polylactic acid (A) at 190 ° C. and a load of 21.2 N is, for example, 0.1 to 50 g / 10 minutes, preferably 0.2 to 20 g. / 10 minutes, more preferably 0.5 to 10 g / 10 minutes, and particularly preferably 1 to 7 g / 10 minutes. If the melt flow rate is too high, the mechanical properties and heat resistance of the film or sheet obtained by film formation may be inferior. On the other hand, if the value of the melt flow rate is too low, the load during film formation may become too high.
- weight average molecular weight refers to that measured by gel permeation chromatography (GPC) (in terms of polystyrene).
- GPC gel permeation chromatography
- the measurement conditions of GPC are as follows. Column: TSKgel SuperHZM-H / HZ2000 / HZ1000 Column size: 4.6 mm I.D. D. ⁇ 150mm Eluent: Chloroform Flow rate: 0.3 ml / min Detector: RI Column temperature: 40 ° C Injection volume: 10 ⁇ l
- the production method of polylactic acid is not particularly limited, but typical production methods include lactide method, direct polymerization method and the like.
- lactide method lactic acid is heated and dehydrated and condensed to give low molecular weight polylactic acid, which is then thermally decomposed under reduced pressure to obtain lactide, which is a cyclic dimer of lactic acid, and this lactide is converted into tin (II) octanoate or the like.
- high molecular weight polylactic acid is obtained by ring-opening polymerization in the presence of a metal salt catalyst.
- the direct polymerization method is a method in which polylactic acid is directly obtained by heating lactic acid in a solvent such as diphenyl ether under reduced pressure and polymerizing it while removing water in order to suppress hydrolysis.
- a commercially available product can be used as polylactic acid (A).
- polylactic acid (A) For example, trade names “Lacia H-400”, “Lacia H-100” (manufactured by Mitsui Chemicals), trade names “Terramac TP-4000”, “Terramac TE-4000” (above, Unitika Ltd.) Manufactured) and the like.
- polylactic acid (A) you may use what was manufactured by the well-known thru
- the acidic functional group-modified olefin polymer (B) contained in the film or sheet of the present invention is a lubricant that imparts desired roll lubricity (that is, peelability from the roll) to the polylactic acid (A) -containing resin composition. As an effect.
- Examples of the acidic functional group of the acidic functional group-modified olefin polymer (B) include a carboxyl group or a derivative group thereof.
- a derivative group of a carboxyl group is chemically derived from a carboxyl group, and examples thereof include an acid anhydride group, an ester group, an amide group, an imide group, and a cyano group of carboxylic acid.
- it is a carboxylic anhydride group.
- the acidic functional group-modified olefin polymer (B) may be abbreviated as an unsaturated compound containing the above “acidic functional group” (hereinafter referred to as an acidic functional group-containing unsaturated compound) in an unmodified polyolefin polymer, for example. Obtained by graft polymerization.
- Examples of the unmodified polyolefin polymer include high density polyethylene, medium density polyethylene, low density polyethylene, polypropylene, polybutene, poly-4-methylpentene-1, a copolymer of ethylene and ⁇ -olefin, and propylene and ⁇ - Polymers of polyolefins such as olefin copolymers or oligomers thereof; ethylene-propylene rubber, ethylene-propylene-diene copolymer rubber, butyl rubber, butadiene rubber, low crystalline ethylene-propylene copolymer, propylene-butene Copolymer, ethylene-vinyl ester copolymer, ethylene-methyl (meth) acrylate copolymer, ethylene-ethyl (meth) acrylate copolymer, ethylene-maleic anhydride copolymer, polypropylene and ethylene-propylene rubber Blend etc.
- Examples include polyolefin elastomers and mixtures of two or more of these.
- Preferable are polypropylene, a copolymer of propylene and ⁇ -olefin, low-density polyethylene and oligomers thereof, and particularly preferable is polypropylene, a copolymer of propylene and ⁇ -olefin and oligomers thereof.
- examples of the “oligomers” include those obtained from a corresponding polymer by a molecular weight degradation method by thermal decomposition. Such oligomers can also be obtained by a polymerization method.
- Examples of the acidic functional group-containing unsaturated compound include a carboxyl group-containing unsaturated compound and a carboxyl group derivative-containing unsaturated compound.
- Examples of the carboxyl group-containing unsaturated compound include maleic acid, itaconic acid, chloroitaconic acid, chloromaleic acid, citraconic acid, and (meth) acrylic acid.
- carboxyl group derivative-containing unsaturated compound examples include, for example, maleic anhydride, itaconic anhydride, chloroitaconic anhydride, chloromaleic anhydride, citraconic anhydride and other carboxylic anhydride group-containing unsaturated compounds; (Meth) acrylate, glycidyl (meth) acrylate, (meth) acrylic acid ester such as 2-hydroxyethyl (meth) acrylate; (meth) acrylamide, maleimide, (meth) acrylonitrile and the like.
- the acidic functional group-modified olefin polymer (B) has a weight average molecular weight of 10,000 to 80,000, preferably 15,000 to 70,000, more preferably 20,000. ⁇ 60,000.
- weight average molecular weight is less than 10,000, it causes bleed out after the film or sheet is formed, and when it exceeds 80,000, it separates from polylactic acid during roll kneading.
- bleed-out refers to a phenomenon in which a low molecular weight component emerges on the film or sheet surface over time after the film or sheet is formed.
- “weight average molecular weight” refers to that measured by gel permeation chromatography (GPC).
- the acidic functional group in the acidic functional group-modified olefin polymer (B) may be bonded to any position of the olefin polymer, and the modification ratio is not particularly limited, but the acidic functional group-modified olefin polymer (B).
- the acid value of is usually 10 to 70 mgKOH / g, preferably 20 to 60 mgKOH / g. If the acid value is less than 10 mgKOH / g, the effect of improving roll lubricity cannot be obtained, and if it exceeds 70 mgKOH / g, plate-out to the roll is caused.
- the plate-out to the roll means that when the resin composition is melt-formed using the metal roll, the components blended in the resin composition or the oxidized, decomposed, combined or deteriorated product is the metal roll. It adheres to or deposits on the surface.
- the “acid value” means that measured in accordance with the neutralization titration method of JISK0070-1992.
- the acidic functional group-modified olefin polymer (B) is obtained by reacting an acidic functional group-containing unsaturated compound with an unmodified polyolefin polymer in the presence of an organic peroxide.
- an organic peroxide those generally used as an initiator in radical polymerization can be used.
- Such a reaction can be performed by either a solution method or a melting method.
- an acidic functional group-modified olefin polymer (B) is obtained by dissolving a mixture of an unmodified polyolefin polymer and an acidic functional group-containing unsaturated compound in an organic solvent together with an organic peroxide and heating. Can do.
- the reaction temperature is preferably about 110 to 170 ° C.
- an acidic functional group-modified olefin polymer (B) is obtained by mixing a mixture of an unmodified polyolefin-based polymer and an acidic functional group-containing unsaturated compound with an organic peroxide, melt-mixing and reacting. be able to. Melt mixing can be performed with various mixers such as an extruder, a plastic bender, a kneader, a Banbury mixer, and the kneading temperature is usually in the temperature range of the melting point of the unmodified polyolefin polymer to 300 ° C.
- the acidic functional group-modified olefin polymer (B) is preferably maleic anhydride group-modified polypropylene.
- “Yumex (registered trademark) 1010” maleic anhydride group-modified polypropylene, acid value: manufactured by Sanyo Chemical Industries, Ltd.) 52 mg KOH / g, weight average molecular weight: 32,000, modification ratio: 10% by weight
- “Yumex (registered trademark) 1001” maleic anhydride group-modified polypropylene, acid value: 26 mg KOH / g, weight average molecular weight: 49,000) , Modification ratio: 5% by weight
- “Yumex (registered trademark) 2000” maleic anhydride group-modified polyethylene, acid value: 30 mg KOH / g, weight average molecular weight: 20,000, modification ratio: 5% by weight
- the content of the acidic functional group-modified olefin polymer (B) is not particularly limited, and is usually 0.1 to 10.0 parts by weight per 100 parts by weight of the polylactic acid (A), and there is no plate-out to the roll. From the viewpoint of sustaining the roll slip effect and maintaining the degree of biomass, the amount is preferably 0.1 to 5.0 parts by weight, particularly preferably 0.3 to 3.0 parts by weight. If the content is less than 0.1 parts by weight, the effect of improving roll lubricity is difficult to obtain, and if it exceeds 10.0 parts by weight, the effect according to the amount added cannot be obtained, and a decrease in the degree of biomass is a problem. It becomes.
- the biomass degree is the ratio of the dry weight of the used biomass to the dry weight of the film or sheet. Biomass is a renewable, organic organic resource excluding fossil resources.
- the tetrafluoroethylene-based polymer (C) contained in the film or sheet of the present invention improves the melt tension of the polylactic acid (A) -containing resin composition and enables oriented crystallization in the flow field during the melt film formation process. By making it, crystallization of polylactic acid (A) can be promoted.
- the tetrafluoroethylene-based polymer (C) has an effect as a crystal nucleating agent for polylactic acid (A), the temperature of the resin composition immediately after film formation is set near the crystallization temperature. The crystallization of (A) can be further promoted. Therefore, the tetrafluoroethylene-based polymer (C) can impart heat resistance to the film or sheet of the present invention by promoting crystallization of the polylactic acid (A).
- the tetrafluoroethylene-based polymer (C) used in the present invention is a tetrafluoroethylene homopolymer or a copolymer of tetrafluoroethylene and another monomer, such as polytetrafluoroethylene, perfluoroalkoxy.
- Alkane copolymer of tetrafluoroethylene and perfluoroalkyl vinyl ether
- perfluoroethylene propene copolymer copolymer of tetrafluoroethylene and hexafluoropropylene
- ethylene-tetrafluoroethylene copolymer tetrafluoroethylene and ethylene Copolymer of tetrafluoroethylene and perfluorodioxole, and the like.
- it is polytetrafluoroethylene.
- the effect of the tetrafluoroethylene polymer (C) as a crystal nucleating agent on the polylactic acid (A) is considered to depend on the crystal structure of the tetrafluoroethylene polymer (C).
- the plane spacing of the polylactic acid (A) crystal lattice was 4.8 angstroms, whereas that of the tetrafluoroethylene polymer was 4.9 angstroms. From this, it is considered that the tetrafluoroethylene-based polymer (C) can act as a crystal nucleating agent for the polylactic acid (A) by having an epitaxy action.
- the epitaxy action means that polylactic acid (A) grows on the surface of the tetrafluoroethylene polymer (C) and aligns with the crystal surface of the crystal surface of the tetrafluoroethylene polymer (C).
- A) refers to the growth pattern arranged.
- the surface spacing of the tetrafluoroethylene-based polymer (C) is governed by the crystal form of the tetrafluoroethylene portion, even if it is a copolymer of tetrafluoroethylene and other monomers. The same. Accordingly, the amount of other monomer components in the copolymer is not particularly limited as long as the crystal form of polytetrafluoroethylene can be maintained and the physical properties are not greatly changed. Usually, a tetrafluoroethylene polymer ( The proportion of other monomer components in C) is desirably 5% by weight or less.
- the method for polymerizing the tetrafluoroethylene-based polymer (C) is not particularly limited, but emulsion polymerization is particularly preferable. Since the tetrafluoroethylene polymer (C) obtained by emulsion polymerization is easy to fiberize, it becomes easy to take a network structure in polylactic acid (A) and improves the melt tension of the resin composition containing polylactic acid (A). It is considered effective for promoting the crystallization of polylactic acid (A) in the flow field during the melt film formation process.
- the weight average molecular weight of the tetrafluoroethylene polymer (C) is not particularly limited, and is usually 1 million to 10 million, preferably 2 million to 8 million.
- the particles of the “tetrafluoroethylene polymer (C)” are converted into polylactic acid (A) such as a (meth) acrylic acid ester polymer.
- a polymer modified with a polymer having good affinity may be used.
- examples of such tetrafluoroethylene-based polymer (C) include acrylic-modified polytetrafluoroethylene.
- a commercially available product may be used. For example, as a commercially available product of polytetrafluoroethylene, “Fluon (registered trademark) CD-014”, “Fluon (registered trademark)” manufactured by Asahi Glass Co., Ltd.
- CD-1 Fullon (registered trademark) CD-145 ", and the like.
- examples of commercially available products of acrylic-modified polytetrafluoroethylene include Metablene (registered trademark) A series (A-3000, A-3800, etc.) manufactured by Mitsubishi Rayon Co., Ltd.
- the content of the tetrafluoroethylene-based polymer (C) is usually 0.5 to 15.0 parts by weight with respect to 100 parts by weight of the polylactic acid (A), the effect of improving the melt tension and maintaining the degree of biomass, and good surface condition. From the viewpoint of obtaining, it is preferably 0.7 to 10.0 parts by weight, particularly preferably 1.0 to 5.0 parts by weight. If the content is less than 0.5 parts by weight, the effect of improving the melt tension is difficult to obtain, and if it exceeds 15.0 parts by weight, the effect according to the amount added cannot be obtained, and the decrease in the degree of biomass is a problem. It becomes.
- the dicarboxylic acid ester plasticizer (D) contained in the film or sheet of the present invention has an effect as a plasticizer that imparts desired flexibility to the resin composition containing polylactic acid (A).
- the dicarboxylic acid ester plasticizer (D) includes a compound represented by the following general formula (1).
- Aa and Ab are each independently selected from the following (1) to (3) (excluding compounds in which both Aa and Ab are the following (3)): (1) C 6 H 5 — (Ac) —, where Ac is an alkylene group having 1 to 4 carbon atoms; (2) (Ae)-(Ad-O) n- (where Ad is an alkylene group having 1 to 5 carbon atoms, Ae is an alkyl group having 1 to 7 carbon atoms, and n is an integer of 1 to 4) ;as well as (3) An alkyl group having 1 to 14 carbon atoms. )
- alkylene group having 1 to 4 carbon atoms examples include a methylene group, an ethylene group, a trimethylene group and a tetramethrene group, and a methylene group is preferred.
- the “alkylene group having 1 to 5 carbon atoms” represented by Ad is a straight chain such as methylene group, ethylene group, trimethylene group, propylene group, tetramethylene group, 1,1′-dimethylethylene group, pentamethylene group, Examples thereof include branched ones, preferably ethylene groups.
- alkyl group having 1 to 7 carbon atoms may be either linear or branched, and examples thereof include a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, an isobutyl group, Examples include sec-butyl group, tert-butyl group, pentyl group, 2-pentyl group, 3-pentyl group, neopentyl group, tertiary pentyl group, hexyl group, isohexyl group, heptyl group and the like.
- C1-C14 alkyl group means a straight-chain or branched saturated hydrocarbon group having 1 to 14 carbon atoms, such as methyl, ethyl, propyl, isopropyl, butyl Group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group, isopentyl group, neopentyl group, 1,2-dimethylpropyl group, 1-ethylpropyl group, hexyl group, isohexyl group, 1,2,2- Trimethylpropyl group, 1,1-dimethylbutyl group, 2,2-dimethylbutyl group, 3,3-dimethylbutyl group, 2-ethylbutyl group, heptyl group, isoheptyl group, 2-ethylhexyl group, octyl group, isooctyl group, Nonyl, isononyl, decyl, isodecy
- a compound in which both Aa and Ab are alkyl groups having 1 to 14 carbon atoms is not included in the dicarboxylic acid ester plasticizer (D) of the present invention.
- at least one of Aa and Ab needs to contain an aryl group such as a phenylene group or an oxyalkylene chain such as an oxyethylene chain.
- the dicarboxylic acid ester plasticizer (D) of the present invention has high hydrolysis resistance and low volatility, thereby preventing the catalytic action of polylactic acid hydrolysis. Flexibility can be imparted. In particular, stress relaxation can be imparted by using a specific blending amount.
- a plasticizer having a low molecular weight in which both Aa and Ab do not contain an aryl group or an oxyalkylene chain has a large plasticizing effect and can obtain flexibility by adding a small amount, but the processing temperature of polylactic acid (approximately 150 to 180 ° C.), the volatility is large and the working environment is deteriorated.
- the blended plasticizer does not remain in the film, and the performance is not stable.
- the dicarboxylic acid ester plasticizer (D) contained in the film or sheet of the present invention is preferably a phthalic acid ester plasticizer (DP) selected from compounds represented by the following general formulas (2) to (6). is there.
- DP phthalic acid ester plasticizer
- A1 and A2 are the same or different and each represents an alkylene group having 1 to 4 carbon atoms;
- A3 represents an alkyl group having 1 to 14 carbon atoms;
- A4 represents an alkylene group having 1 to 5 carbon atoms;
- A5 represents an alkyl group having 1 to 7 carbon atoms.
- n represents an integer of 1 to 4.
- the “C1-C4 alkylene group”, “C1-C5 alkylene group”, “C1-C7 alkyl group”, and “C1-C14 alkyl group” are as described above. .
- Specific examples of the compounds represented by the general formulas (2) to (6) include, for example, ethylbenzyl phthalate, butylbenzyl phthalate, (2-ethylhexyl) benzyl phthalate, (2-ethylhexyl) butyl diglycol phthalate, Examples include butyl (ethyl diglycol) phthalate, bis (butyl diglycol) phthalate, and benzyl (butyl diglycol) phthalate.
- the dicarboxylic acid ester plasticizer (D) contained in the film or sheet of the present invention is preferably an aliphatic dicarboxylic acid ester plasticizer selected from the compounds represented by the following general formulas (7) to (11). (DA).
- A1 and A2 are the same or different and each represents an alkylene group having 1 to 4 carbon atoms;
- A3 represents an alkyl group having 1 to 14 carbon atoms;
- A4 represents an alkylene group having 1 to 5 carbon atoms;
- A5 represents an alkyl group having 1 to 7 carbon atoms.
- n represents an integer of 1 to 4, and
- m represents an integer of 0 to 6.
- the “C1-C4 alkylene group”, “C1-C5 alkylene group”, “C1-C7 alkyl group”, and “C1-C14 alkyl group” are as described above. .
- Specific examples of the compounds represented by the general formulas (7) to (11) include (2-ethylhexyl) benzyl adipate, butylbenzyl adipate, (2-ethylhexyl) benzyl azelate, and (2-ethylhexyl).
- Examples include benzyl sebacate, (2-ethylhexyl) butyl diglycol adipate, benzyl (methoxyethoxyethyl) adipate, bis (butyldiglycol) adipate, bis [2- (2-butoxyethoxy) ethyl] adipate, and the like.
- the dicarboxylic acid ester plasticizer (D) includes the phthalic acid ester plasticizer (DP) and the aliphatic dicarboxylic acid ester plasticizer (DA).
- the content of the dicarboxylic acid ester plasticizer (D) is preferably 8 parts by weight or more and less than 40 parts by weight with respect to 100 parts by weight of the polylactic acid (A).
- the content of the phthalate ester plasticizer (DP) is usually 10 parts by weight or more and less than 40 parts by weight, preferably 12 parts by weight or more and 40 parts by weight with respect to 100 parts by weight of the polylactic acid (A). And more preferably 15 to 38 parts by weight.
- the content of the aliphatic dicarboxylic acid ester plasticizer (DA) is usually 8 to 35 parts by weight, preferably 10 to 30 parts by weight with respect to 100 parts by weight of the polylactic acid (A). More preferably, it is 12 to 27 parts by weight. If the content of the dicarboxylic acid ester plasticizer (D) is less than 8 parts by weight, the effect of imparting flexibility is difficult to obtain, and if it is 40 parts by weight or more, plate-out occurs and crystallization of polylactic acid is inhibited. By doing so, the decrease in heat resistance becomes remarkable.
- the resin composition of the present invention may contain other crystallization accelerator (E) in addition to the tetrafluoroethylene-based polymer (C).
- the crystallization accelerator (E) is not particularly limited as long as the effect of promoting crystallization is recognized, but a substance having a crystal structure having a face spacing close to the face spacing of the crystal lattice of polylactic acid (A). It is desirable to choose. This is because a substance having a crystal lattice spacing close to that of polylactic acid (A) is more effective as a crystal nucleating agent for polylactic acid (A).
- crystallization accelerator (E) examples include organic substances such as melamine polyphosphate, melamine cyanurate, zinc phenylphosphonate, calcium phenylphosphonate, magnesium phenylphosphonate, talc of inorganic substances, and clay. Etc. Among them, zinc phenylphosphonate is most preferable because the plane spacing is most similar to the plane spacing of polylactic acid (A) and a good crystallization promoting effect is obtained.
- a commercial item can be used for the crystallization accelerator (E).
- a commercially available product of zinc phenylphosphonate includes “Eco Promote” manufactured by Nissan Chemical Industries, Ltd.
- the content of the crystallization accelerator (E) is usually 0.1 to 5 parts by weight with respect to 100 parts by weight of the polylactic acid (A), preferably from the viewpoint of better crystallization promoting effect and maintaining the degree of biomass. Is 0.3 to 3 parts by weight. If the content is less than 0.1 parts by weight, the effect of promoting crystallization is difficult to obtain, and if it exceeds 5 parts by weight, the effect according to the amount added cannot be obtained, and a decrease in the degree of biomass becomes a problem. .
- the polylactic acid (A) -containing resin composition may contain various additives as necessary within the range not impairing the object of the present invention.
- additives include known antioxidants, ultraviolet absorbers, plasticizers, stabilizers, mold release agents, antistatic agents, coloring agents, and anti-drip agents.
- the tensile elongation at break measured according to the “plastic-tensile property” test method described in JISK7161 is preferably 100% or more.
- the stress residual rate at the time of 10% displacement measured according to the said test method is 40% or less.
- the film or sheet of the present invention in which the flexibility and the residual stress rate are within the above ranges are polylactic acid (A), acidic functional group-modified olefin polymer (B), tetrafluoroethylene polymer (C), and general formula (1). ),
- the content of the dicarboxylic acid ester plasticizer (D) is within the range specified in the present invention. In particular, it is important that the content of the dicarboxylic acid ester plasticizer (D) is within the range specified in the present invention.
- the heat deformation rate of the film or sheet of the present invention is measured according to the heat deformation test method described in JISC3005.
- the film or sheet of the present invention preferably has a deformation rate of 40% or less when a 10 N load is applied for 30 minutes in a 120 ° C. temperature atmosphere.
- Relative crystallization rate The relative crystallization rate of the film or sheet of the present invention is measured by DSC, and the heat amount ⁇ Hc of the exothermic peak accompanying crystallization in the temperature rising process of the film or sheet sample after film formation, and subsequent melting It calculates from the amount of heat ⁇ Hm using the following formula (I).
- Relative crystallization rate (%) ( ⁇ Hm ⁇ Hc) / ⁇ Hm ⁇ 100 (I)
- the film or sheet of the present invention preferably has a relative crystallization rate of 50% or more. More preferably, according to the heating test method of JISC3005, the film or sheet of the present invention has a deformation rate of 40% or less when a load of 10 N is applied for 30 minutes in a temperature atmosphere of 120 ° C., The relative crystallization rate obtained by the formula (I) is 50% or more.
- the contents of the tetrafluoroethylene-based polymer (C) and the dicarboxylic acid ester-based plasticizer (D) represented by the general formula (1) are set within the ranges specified in the present invention, respectively. It is important that the content of C) and the content of the dicarboxylic acid ester plasticizer (D) are within the ranges specified in the present invention.
- the method includes forming a polylactic acid (A) -containing resin composition by a melt film formation method, and the temperature of the resin composition at the time of melt film formation Is a temperature between the crystallization temperature (Tc) + 15 ° C. in the temperature lowering process of the resin composition and the melting temperature (Tm) ⁇ 5 ° C. in the temperature rising process, and / or melt film formation
- the resin composition thus obtained has a crystallization temperature (Tc) in the process of lowering the temperature of the resin composition of ⁇ 25 ° C. to a crystallization temperature (Tc) + 10 ° C.
- the crystallization temperature (Tc ) ⁇ 10 ° C. using a production method (described later) characterized by being cooled and solidified after undergoing a crystallization promotion step, the deformation rate is 40% or less, and the relative crystallization rate is 50 % Or more is important for realizing the film or sheet of the present invention.
- the film or sheet of the present invention can be used for the same use as a generally used film or sheet, but can be particularly suitably used as a base material for an adhesive film or sheet.
- the method for producing the film or sheet of the present invention is not particularly limited, and a method of forming a polylactic acid (A) -containing resin composition by a melt film forming method is preferable.
- the film or sheet of the present invention is a polylactic acid in which each component is uniformly dispersed by a continuous melt kneader such as a twin screw extruder or a batch type melt kneader such as a pressure kneader, a Banbury mixer, or a roll kneader.
- the resin composition can be prepared by producing a film and cooling and solidifying it by an extrusion method such as a T-die method or an inflation method, a calendar method, a polishing method, or the like.
- a melt film formation method is preferably a method in which a molten resin composition is formed into a desired thickness by passing through a gap between two metal rolls, and particularly preferably a calendar method. This is a polishing method.
- the thickness of the film or sheet of the present invention is appropriately adjusted depending on the application, but is usually 10 to 500 ⁇ m, preferably 20 to 400 ⁇ m, particularly preferably 30 to 300 ⁇ m.
- the temperature of the resin composition at the time of melt film formation (hereinafter referred to as the resin temperature at the time of melt film formation) is not particularly limited, The temperature is preferably between the crystallization temperature (Tc) + 15 ° C. in the temperature-lowering process of the resin composition and the melting temperature (Tm) ⁇ 5 ° C. in the temperature-raising process.
- Tc crystallization temperature
- Tm melting temperature
- the temperature of the resin composition at the time of calender roll rolling (corresponding to the resin temperature at the time of melt film-forming) is reduced in the temperature-decreasing process of the resin composition. Is set to a temperature between the crystallization temperature (Tc) + 15 ° C. and the melting temperature (Tm) ⁇ 5 ° C. in the heating process. By rolling at a temperature below the melting point in this way, oriented crystallization is promoted. This effect of promoting orientation crystallization is markedly improved when the resin composition contains the tetrafluoroethylene-based polymer (C).
- the tetrafluoroethylene-based polymer (C) promotes oriented crystallization by fibrillating and networking in the resin composition and a synergistic effect of its effect as a crystal nucleating agent. Therefore, by rolling in the above temperature range, the film or sheet of the present invention has good heat resistance (that is, a reduction in relative crystallization rate and a heat deformation rate due to the effect of promoting orientational crystallization as well as a smooth surface state. Suppression of the increase in the amount).
- the method for producing a polylactic acid film or sheet of the present invention further comprises a step of controlling the temperature conditions after melt film formation in order to make the crystallization promoting effect of the tetrafluoroethylene polymer (C) more effective. It may be. Specifically, the melt-formed resin composition is heated at a temperature between the crystallization temperature (Tc) ⁇ 25 ° C. and the crystallization temperature (Tc) + 10 ° C. Preferably, it may be cooled and solidified after a step of promoting crystallization by holding at a crystallization temperature (Tc) ⁇ 10 ° C. (hereinafter sometimes simply referred to as “crystallization promotion step”). Good.
- the crystallization promotion step refers to the resin composition that has been melt-deposited from a temperature of crystallization temperature (Tc) of ⁇ 25 ° C. in the temperature lowering process of the resin composition to a temperature of crystallization temperature (Tc) + 10 ° C.
- the resin composition is crystallized while maintaining a smooth surface state after the melt film formation, in which the temperature is controlled to a temperature in the range [preferably, crystallization temperature (Tc) ⁇ 10 ° C.].
- the temperature control method is not particularly limited, and examples thereof include a method in which the resin composition that has been melt-formed is brought into direct contact with a roll or belt that can be heated to a predetermined temperature.
- the time for the crystallization promoting step is preferably as long as possible, and ultimately depends on the degree of crystallization of the resin composition, and thus cannot be specified in general, but usually 2 to 10 seconds, preferably 3 to 8 Seconds.
- the crystallization promotion step even if the crystallization temperature (Tc) in the temperature-decreasing process of the resin composition changes due to the addition of another crystal nucleating agent, etc., the measurement is performed in advance with a differential scanning calorimeter (DSC).
- DSC differential scanning calorimeter
- the method for producing the polylactic acid film or sheet of the present invention is preferably a method comprising forming a polylactic acid (A) -containing resin composition by a melt film formation method,
- the temperature of the resin composition (resin temperature at the time of melt film formation) is from the temperature of the crystallization temperature (Tc) + 15 ° C. in the temperature lowering process of the resin composition to the melting temperature (Tm) ⁇ 5 ° C. in the temperature rising process.
- Tc crystallization temperature
- the method is characterized by cooling and solidifying after passing through a crystallization promoting step at a temperature between [° C.] [preferably, a crystallization temperature (Tc) ⁇ 10 ° C.].
- the highly crystallized film or sheet of the present invention formed by the above production method can maintain the shape up to near the melting point of polylactic acid, and can be used sufficiently even in applications requiring heat resistance that could not be used so far. It becomes possible. Furthermore, since the inefficient process of heating once again after cooling and solidification becomes unnecessary, the above manufacturing method can be said to be a very useful method in terms of economy and productivity.
- a method for producing the polylactic acid-based film or sheet of the present invention including the crystallization promoting step
- a method of continuously performing from the melt film forming step to the crystallization promoting step and the cooling and solidifying step shortens the processing time. Desirable in terms of productivity. Examples of such a method include a method using a calendar film forming machine, a polishing film forming machine or the like.
- FIG. 1 shows a schematic diagram of a calendar film forming machine of one embodiment used in such a manufacturing method.
- the resin composition in a molten state is rolled between four calender rolls, ie, the first roll (1), the second roll (2), the third roll (3), and the fourth roll (4), and gradually thinned. It is prepared to have a desired thickness when it finally passes between the third roll (3) and the fourth roll (4).
- film formation of the resin composition in the first to fourth rolls (1) to (4) corresponds to a “melt film formation step”.
- the temperature of the resin composition in the process of lowering the temperature is between crystallization temperature (Tc) -25 ° C. and crystallization temperature (Tc) + 10 ° C. [preferably, crystallization temperature (Tc) ⁇ 10 ° C.].
- the set take-off roll (5) indicates a roll group to which the resin composition (8) that has been melt-deposited first contacts, and is composed of one or two (three in FIG. 1) roll groups. And it plays the role which peels this resin composition (8) of a molten state from a 4th roll (4). In this way, when the take-off roll (5) is composed of a plurality of rolls and the temperature of each roll can be adjusted, the temperature of each roll is preferably the same, but within the desired temperature range.
- crystallization of the melt-formed resin composition (8) is promoted in the take-off roll (5), so that the resin composition (8) passes through the take-off roll (5).
- the process corresponds to a “crystallization promotion process”.
- the two cooling rolls (6) and (7) allow the resin composition (8) to pass between them to cool and solidify the resin composition (8) and to have a desired shape on the surface. It plays the role of molding.
- one roll for example, cooling roll (6)
- the roll surface is designed to give the surface shape of the resin composition (8)
- the other roll for example, A rubber roll is used as the cooling roll (7).
- the arrow in a figure shows the rotation direction of a roll.
- FIG. 2 shows a schematic diagram of a polishing film forming machine of one embodiment used in such a manufacturing method.
- An extruder tip (10) of an extruder (not shown) is arranged between the heated second roll (2) and third roll (3), and the second roll is set at a preset extrusion speed.
- the molten resin composition (8) is continuously extruded between (2) and the third roll (3).
- the extruded resin composition (8) is rolled and thinned between the second roll (2) and the third roll (3), and finally the third roll (3) and the fourth roll (4). It is prepared to have a desired thickness when passing between them.
- film formation of the resin composition (8) on the second to fourth rolls (2) to (4) corresponds to a “melt film formation step”. Thereafter, a temperature between the crystallization temperature (Tc) -25 ° C. in the temperature lowering process of the resin composition (8) and the crystallization temperature (Tc) + 10 ° C. [Preferably, the crystallization temperature (Tc) ⁇ 10
- the solidified film or sheet is produced by passing through the three take-off rolls (5) set at [° C.] and finally passing through the cooling rolls (6) and (7).
- the step of passing through the take-off roll (5) corresponds to the “crystallization promotion step”.
- the following component (B ′) was examined.
- C1 Polytetrafluoroethylene: Fluon (registered trademark) CD-014 (Asahi Glass Co., Ltd.)
- C2 Acrylic modified polytetrafluoroethylene: Methbrene (registered trademark) A-3000 (manufactured by Mitsubishi Rayon Co., Ltd.)
- DP1 butylbenzyl phthalate: BBP (manufactured by Daihachi Chemical Industry Co., Ltd.), molecular weight 314 DP2: (2-ethylhexyl) benzyl phthalate: Santisizer 261A (manufactured by Ferro Japan), molecular weight 370
- DP1 butylbenzyl phthalate: BBP (manufactured by Daihachi Chemical Industry Co., Ltd.)
- DP2 (2-ethylhexyl) benzyl phthalate: Santisizer 261A (manufactured by Ferro Japan)
- DP ′ phthalester plasticizer
- DP ′ Bis (2-ethylhexyl) phthalate: DOP (manufactured by DIC Corporation), molecular weight 391 [Aliphatic dicarboxylic acid ester plasticizer (DA)] (DA1); bis [2- (2-butoxyethoxy) ethyl] adipate: BXA (manufactured by Daihachi Chemical Industry Co., Ltd.), molecular weight 435 (DA2); benzyl (methoxyethoxyethyl) adipate: DAIFATTY-101 (registered trademark of Daihachi Chemical Industry Co., Ltd.), molecular weight 338
- DA ′ diisononyl adipate: W-242 (manufactured by DIC Corporation), molecular weight 398
- Example 1 After preparing a resin composition in which the above raw materials are blended at the blending ratios shown in Table 1, performing melt kneading with a Banbury mixer, forming a calender film so that the thickness is 100 ⁇ m with an inverted L-shaped four-calendar (Melt film formation) was performed. Next, as shown in FIG. 1, immediately after the melt film formation step, three rolls (take-off rolls) that can be heated to an arbitrary temperature are arranged so that the melt-formed resin composition can pass alternately up and down. By doing so, it was set as the crystallization promotion process. Thereafter, the resin composition was solidified by passing through a cooling roll to prepare a film.
- Table 1 After preparing a resin composition in which the above raw materials are blended at the blending ratios shown in Table 1, performing melt kneading with a Banbury mixer, forming a calender film so that the thickness is 100 ⁇ m with an inverted L-shaped four-calendar (Melt film formation)
- the temperature of the resin composition during melt film formation (the resin temperature during melt film formation) is regarded as the surface temperature of the roll corresponding to the fourth roll (4) in FIG. 1, and the temperature of the resin composition in the crystallization promoting step.
- the surface temperature of the three take-off rolls (5) in FIG. 1 was made substantially the same, and the temperature was taken as the crystallization acceleration temperature.
- the film formation rate was 5 m / min, and the substantial crystallization promotion step time (take-off roll passage time) was about 5 seconds.
- Examples 2-5 Resin compositions were prepared at the blending ratios shown in Table 1 below, and films of Examples 2 to 5 were prepared in the same manner as in Example 1.
- Comparative Examples 1-7 Resin compositions were prepared at the blending ratios shown in Table 1 below, and films of Comparative Examples 1 to 7 were prepared in the same manner as in Example 1. Evaluation of the samples prepared in Examples and Comparative Examples was performed as follows.
- Tm melting temperature
- ⁇ Crystalization temperature> The temperature at the peak top of the exothermic peak accompanying the crystallization of the resin composition after film formation, measured by DSC, during the temperature lowering process from 200 ° C. was defined as the crystallization temperature (Tc; also referred to as crystallization peak temperature). .
- the DSC and measurement conditions used in the measurement of melting temperature (Tm), crystallization temperature (Tc), and relative crystallization rate are as follows. Apparatus: DSC6220 manufactured by SII Nano Technology Co., Ltd. Conditions: Measurement temperature range 20 ° C. ⁇ 200 ° C. ⁇ 0 ° C. ⁇ 200 ° C. (That is, following the measurement in the process of increasing the temperature from 20 ° C to 200 ° C, the measurement in the process of decreasing the temperature from 200 ° C to 0 ° C is performed, and finally the process of increasing the temperature again from 0 ° C to 200 ° C.
- Heat deformation rate It measured according to the heat deformation test method of JISC3005.
- the measurement apparatus and measurement conditions used are as follows.
- Measurement conditions Chuck distance 50mm
- the residual stress rate which is an index of stress relaxation, was measured according to the plastic-tensile property test method of JISK7161.
- the measurement apparatus and measurement conditions used are as follows.
- Apparatus Tensile tester (Autograph AG-20kNG, manufactured by Shimadzu Corporation) Condition: Sample size: 0.1 mm thickness x 10 mm width x 100 mm length The direction parallel to the length direction is the flow direction (MD) during film formation. It cut out so that it might become.
- Distance between chucks 50mm Tensile speed 300mm / min Measurement: When the displacement reaches 10%, the displacement is stopped and held at that position. The stress at that time was defined as 100%, and the residual amount of the stress value after 60 seconds was read to obtain the “stress residual ratio”.
- Acceptance / rejection determination Those having a residual stress rate of 40% or less were determined to be acceptable.
- Table 2 shows the evaluation results for the test pieces of Examples 1 to 5 and Comparative Examples 1 to 7 prepared based on the composition table of Table 1.
- the films of Examples 1 to 5 according to the present invention all have a tensile elongation at break of 100% or more and a residual stress ratio of 40% or less, so that flexibility is secured. It was. Furthermore, it was also confirmed that heat resistance was ensured by a high relative crystallization rate and a suppressed heat deformation rate. In addition, both peelability and volatility resistance were good, and no plate-out to the roll occurred.
- a low molecular weight plasticizer not included in the dicarboxylic acid ester plasticizer (D) of the general formula (1) of the present invention that is, Aa and Ab of the general formula (1) include an aryl group or an oxyalkylene chain.
- Comparative Examples 1 and 2 using a non-plasticizer volatilization of the plasticizer and plate-out to the roll were observed, and the tensile elongation at break and the stress relaxation properties were insufficient.
- the film of the comparative example 3 which does not contain an acidic functional group modified olefin type polymer (B) had inferior peelability from a roll.
- Comparative Examples 4 and 5 in which the blending amount of the dicarboxylic acid ester plasticizer (D) of the general formula (1) was insufficient were insufficient in tensile elongation at break and stress relaxation properties.
- Comparative Examples 6 and 7 in which the amount of the dicarboxylic acid ester plasticizer (D) was excessive were insufficient in heat resistance. That is, in Comparative Examples 1 to 7 which did not contain the component according to the present invention or were not in the blending ratio of the present invention, a film satisfying all the desired flexibility and heat resistance could not be obtained.
- the film or sheet of the present invention can be widely used in the same applications as the generally used film or sheet, but can be particularly suitably used as a substrate for an adhesive film or sheet.
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Mechanical Engineering (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- General Chemical & Material Sciences (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Manufacture Of Macromolecular Shaped Articles (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
しかし、ポリ乳酸は、結晶化速度が遅く、通常の成膜手段では殆ど結晶化しない。例えば、ポリ乳酸を含有する樹脂組成物からなるフィルムは、ポリ乳酸のガラス転移温度である60℃程度以上では熱変形を起こし、フィルム形状を保持できないという耐熱性の問題を有していた。
しかし、一般に、ポリ乳酸と相溶性の良い可塑剤を添加すると、ポリ乳酸のガラス転移温度が低下し、フィルムの耐熱性が、ポリ乳酸単体よりもさらに低下する場合が多いため、ガラス転移温度以上の温度で使用することは困難となる。
特許文献1には、耐熱性を改良する方策として、可塑剤を添加したポリ乳酸を、そのガラス転移温度と融点の間の温度で成形することにより、耐熱性を向上する提案がなされている。しかしながら、結晶化に要する具体的な時間の記載がなく、実用可能な速度条件で連続成膜できるかは不明である。
また、ポリ乳酸に、可塑剤とポリアルキレングリコールさらにはスチレン系共重合体を添加することにより、射出成形性(成形に要する時間)が良好でかつ、ビカット軟化点が高い(耐熱性が高い)、柔軟な成形物を得る手法が提案されている(特許文献2)。
しかし、この手法では、相当量のスチレン系共重合体を添加する必要があり、バイオマス度を著しく低下させてしまう。また、80℃程度の耐熱性しかなく、家電や自動車の耐熱部品に対応できるほどの耐熱性は得られていない。
さらには、ポリ乳酸に特定の可塑剤と結晶核剤を添加した混和物を、押出機によって成膜し、一旦冷却したのちに、さらに60~100℃の工程を通過させることにより、結晶化させる手法が提案されている(特許文献3)。
しかし、この手法では、一度冷却固化してから、再度、加熱するため非効率である。
当該ジカルボン酸エステル系可塑剤(D)の含有量が、ポリ乳酸(A)100重量部に対し、8重量部以上40重量部未満である樹脂組成物からなるフィルム又はシート。
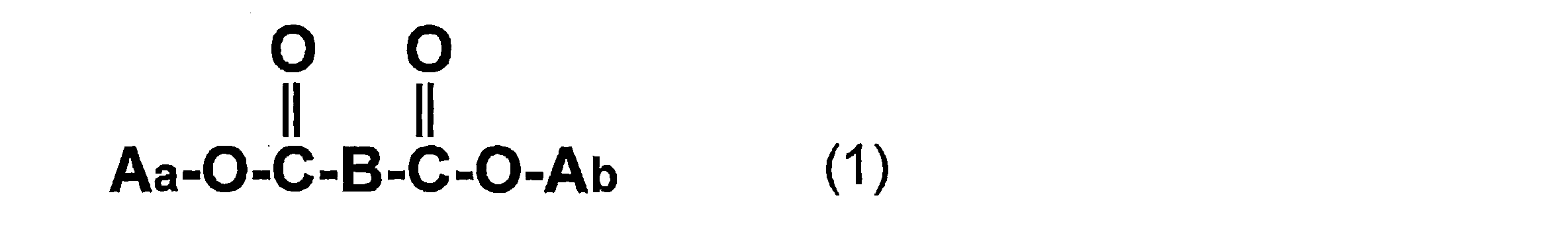
Bは1,2-フェニレン基、または、-CH2(CH2)mCH2-(但し、mは0~6の整数を示す)であり、
Aa及びAbは、それぞれ独立して、下記(1)~(3)から選ばれる(但し、Aa及びAbが共に下記(3)である化合物を除く):
(1)C6H5-(Ac)-、(但し、Acは炭素数1~4のアルキレン基);
(2)(Ae)-(Ad-O)n-、(但し、Adは炭素数1~5のアルキレン基、Aeは炭素数1~7のアルキル基、nは1~4の整数を示す);及び
(3)炭素数1~14のアルキル基。)
[2]当該ジカルボン酸エステル系可塑剤(D)が、一般式(2)~(6)で表される化合物から選ばれるフタル酸エステル系可塑剤(DP)であり、当該フタル酸エステル系可塑剤(DP)の含有量が、ポリ乳酸(A)100重量部に対し10重量部以上40重量部未満である、上記[1]記載のフィルム又はシート。

[3]当該ジカルボン酸エステル系可塑剤(D)が、一般式(7)~(11)で表される化合物から選ばれる脂肪族ジカルボン酸エステル系可塑剤(DA)であり、当該脂肪族ジカルボン酸エステル系可塑剤(DA)の含有量が、ポリ乳酸(A)100重量部に対し8~35重量部である、上記[1]記載のフィルム又はシート。
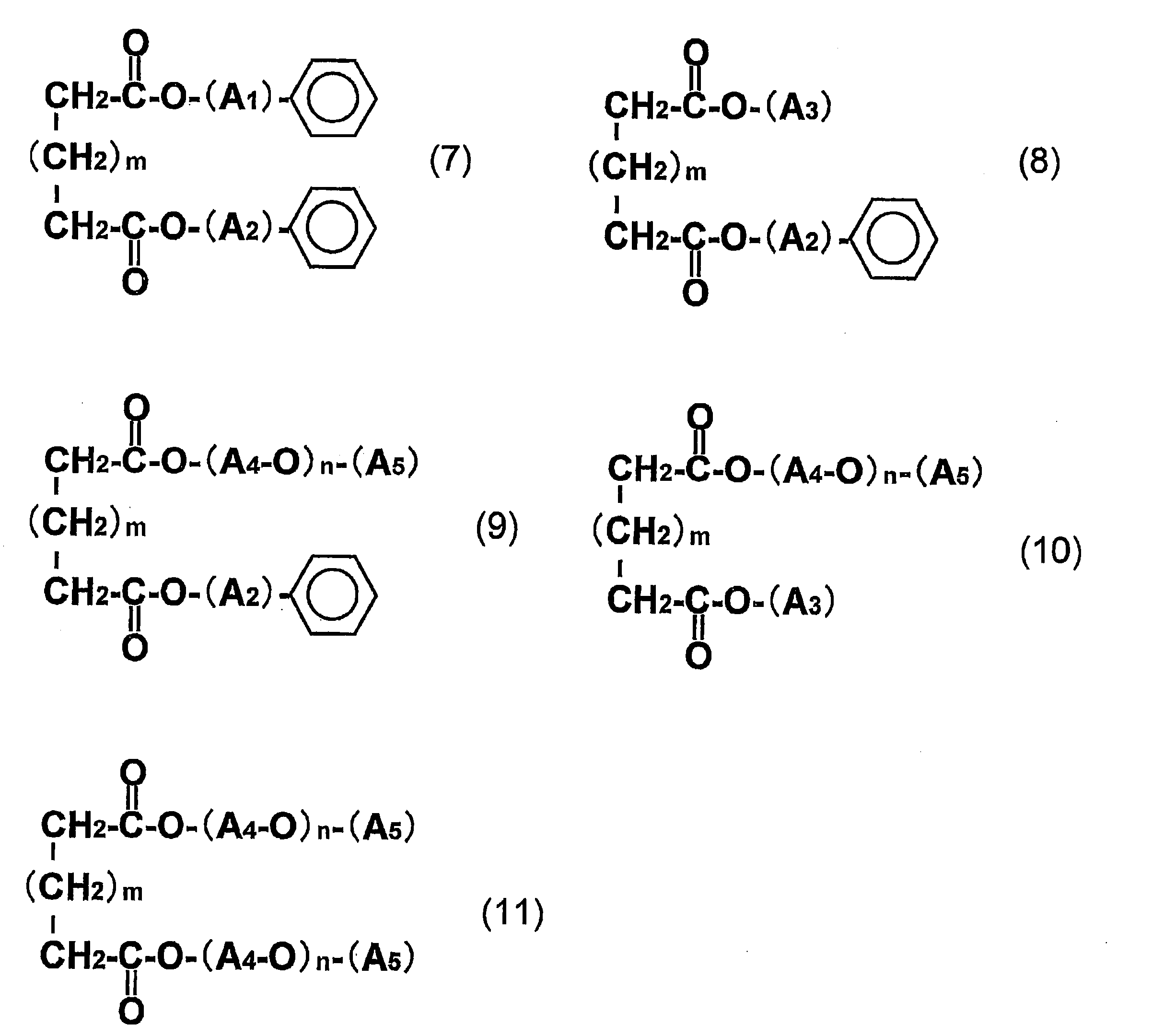
[4]酸性官能基変性オレフィン系ポリマー(B)に含まれる酸性官能基が、カルボン酸無水物基である、上記[1]~[3]のいずれかに記載のフィルム又はシート。
[5]テトラフルオロエチレン系ポリマー(C)の含有量が、ポリ乳酸(A)100重量部に対し、0.5~15.0重量部である、上記[1]~[4]のいずれかに記載のフィルム又はシート。
[6]酸性官能基変性オレフィン系ポリマー(B)の含有量が、ポリ乳酸(A)100重量部に対し、0.1~10.0重量部である、上記[1]~[5]のいずれかに記載のフィルム又はシート。
[7]樹脂組成物が、さらに結晶化促進剤(E)を含有し、当該結晶化促進剤(E)の含有量が、ポリ乳酸(A)100重量部に対し、0.1~5.0重量部である、上記[1]~[6]のいずれかに記載のフィルム又はシート。
[8]JISC3005の加熱変形試験方法に準じて、120℃の温度雰囲気下で30分間、10Nの荷重を加えたときの変形率が、40%以下であり、
下記式(I)で求められる相対結晶化率が50%以上であることを特徴とする、上記[1]~[7]のいずれかに記載のフィルム又はシート。
相対結晶化率(%)=(ΔHm-ΔHc)/ΔHm×100 (I)
(式中、ΔHcは成膜後のフィルム又はシートの昇温過程での結晶化に伴う発熱ピークの熱量であり、ΔHmは融解に伴う熱量を示す。)
[9]引張破断伸びが100%以上であり、10%伸張時の応力残存率が40%以下であることを特徴とする、上記[1]~[8]のいずれかに記載のフィルム又はシート。
[10]樹脂組成物を溶融成膜法により成膜することを含む、上記[1]~[9]のいずれかに記載のフィルム又はシートの製造方法であって、
溶融成膜時の樹脂組成物の温度が、該樹脂組成物の降温過程での結晶化温度(Tc)+15℃の温度から、昇温過程での融解温度(Tm)-5℃の間の温度であるか、又は、
溶融成膜された該樹脂組成物が、該樹脂組成物の降温過程での結晶化温度(Tc)-25℃の温度から結晶化温度(Tc)+10℃の間の温度における結晶化促進工程を経てから冷却固化されることを特徴とする、フィルム又はシートの製造方法。
[11]溶融成膜時の該樹脂組成物の温度が、該樹脂組成物の降温過程での結晶化温度(Tc)+15℃の温度から、昇温過程での融解温度(Tm)-5℃の間の温度であり、かつ、
溶融成膜された該樹脂組成物が、該樹脂組成物の降温過程での結晶化温度(Tc)-25℃の温度から結晶化温度(Tc)+10℃の間の温度における結晶化促進工程を経てから冷却固化されることを特徴とする、上記[10]記載のフィルム又はシートの製造方法。
[12]溶融成膜法が、溶融状態の樹脂組成物を2本の金属ロール間の空隙を通過させることで所望の厚さに成膜する手法である、上記[10]または[11]記載のフィルム又はシートの製造方法。
[13]結晶化促進工程が、金属ロールを用いて行われることを特徴とする、上記[10]~[12]のいずれかに記載のフィルム又はシートの製造方法。
本発明のフィルム又はシートは、ポリ乳酸(A)、酸性官能基変性オレフィン系ポリマー(B)、テトラフルオロエチレン系ポリマー(C)及びジカルボン酸エステル系可塑剤(D)を含有する樹脂組成物からなる。本発明のフィルム又はシートは、透明、半透明及び不透明のものを含む。
本発明のフィルム又はシートの厚さは特に制限されないが、通常、10~500μm、好ましくは、20~400μm、より好ましくは、30~300μmである。
ポリ乳酸の原料モノマーである乳酸は、不斉炭素原子を有するため、光学異性体のL体とD体とが存在する。本発明で使用するポリ乳酸(A)は、L体の乳酸を主成分とした重合物である。製造時に不純物として混入するD体の乳酸の含有量が少ないものほど、高結晶性で高融点の重合物となるため、できるだけL体純度の高いものを用いるのが好ましく、L体純度が95%以上のものを用いるのがより好ましい。また、本発明で使用するポリ乳酸(A)は、乳酸以外の他の共重合成分を含んでいてもよい。他のモノマー単位としては、エチレングリコール、プロピレングリコール、ブタンジオール、ヘプタンジオール、ヘキサンジオール、オクタンジオール、ノナンジオ-ル、デカンジオール、1,4-シクロヘキサンジメタノール、ネオペンチルグリコール、グリセリン、ペンタエリスリトール、ビスフェノールA、ポリエチレングリコール、ポリプロピレングリコール、ポリテトラメチレングリコールなどのグリコール化合物;シュウ酸、アジピン酸、セバシン酸、アゼライン酸、ドデカンジオン酸、マロン酸、グルタル酸、シクロヘキサンジカルボン酸、テレフタル酸、イソフタル酸、フタル酸、ナフタレンジカルボン酸、ビス(p-カルボキシフェニル)メタン、アントラセンジカルボン酸、4,4’-ジフェニルエーテルジカルボン酸、5-ナトリウムスルホイソフタル酸、5-テトラブチルホスホニウムイソフタル酸などのジカルボン酸;グリコール酸、ヒドロキシプロピオン酸、ヒドロキシ酪酸、ヒドロキシ吉草酸、ヒドロキシカプロン酸、ヒドロキシ安息香酸などのヒドロキシカルボン酸;カプロラクトン、バレロラクトン、プロピオラクトン、ウンデカラクトン、1,5-オキセパン-2-オンなどのラクトン類を挙げることができる。このような他の共重合成分は、全モノマー成分に対し、0~30モル%であることが好ましく、0~10モル%であることが好ましい。
カラム:TSKgel SuperHZM-H/HZ2000/HZ1000
カラムサイズ:4.6mmI.D.×150mm
溶離液:クロロホルム
流量:0.3ml/min
検出器:RI
カラム温度:40℃
注入量:10μl
本発明のフィルム又はシートの製造には、例えば、カレンダー成膜機等により、ポリ乳酸(A)含有樹脂組成物を溶融状態にし、金属ロール間の空隙を通過させて成膜することが必要であるため、かかる樹脂組成物は、金属ロール表面から容易に剥離できなくてはならない。本発明のフィルム又はシートに含有される酸性官能基変性オレフィン系ポリマー(B)は、ポリ乳酸(A)含有樹脂組成物に所望のロール滑性(すなわち、ロールからの剥離性)を付与する滑剤としての効果を有する。
酸性官能基変性オレフィン系ポリマー(B)の酸性官能基としては、例えば、カルボキシル基又はその誘導体基等が挙げられる。カルボキシル基の誘導体基とは、カルボキシル基から化学的に誘導されるものであって、例えば、カルボン酸の酸無水物基、エステル基、アミド基、イミド基、シアノ基等が挙げられる。好ましくは、カルボン酸無水物基である。
未変性ポリオレフィン系重合体としては、例えば、高密度ポリエチレン、中密度ポリエチレン、低密度ポリエチレン、ポリプロピレン、ポリブテン、ポリ-4-メチルペンテン-1、エチレンとα-オレフィンの共重合体、プロピレンとα-オレフィンの共重合体等のポリオレフィン類のポリマー又はそれらのオリゴマー類;エチレン-プロピレンゴム、エチレン-プロピレン-ジエン共重合体ゴム、ブチルゴム、ブタジエンゴム、低結晶性エチレン-プロピレン共重合体、プロピレン-ブテン共重合体、エチレン-ビニルエステル共重合体、エチレン-メチル(メタ)アクリレート共重合体、エチレン-エチル(メタ)アクリレート共重合体、エチレン-無水マレイン酸共重合体、ポリプロピレンとエチレン-プロピレンゴムのブレンド等のポリオレフィン系エラストマー類及びこれらの2種以上の混和物等が挙げられる。好ましくは、ポリプロピレン、プロピレンとα-オレフィンの共重合体、低密度ポリエチレン及びそれらのオリゴマー類であり、特に好ましくは、ポリプロピレン、プロピレンとα-オレフィンの共重合体及びそれらのオリゴマー類である。上記「オリゴマー類」としては、対応するポリマーから、熱分解による分子量減成法によって得られるもの等が挙げられる。かかるオリゴマー類は、重合法によっても得ることができる。
溶液法では、未変性ポリオレフィン系重合体及び酸性官能基含有不飽和化合物の混合物を有機過酸化物とともに有機溶媒に溶解し、加熱することにより、酸性官能基変性オレフィン系ポリマー(B)を得ることができる。反応温度は、好ましくは、110~170℃程度である。
溶融法では、未変性ポリオレフィン系重合体及び酸性官能基含有不飽和化合物の混合物を有機過酸化物と混合し、溶融混合して反応させることによって、酸性官能基変性オレフィン系ポリマー(B)を得ることができる。溶融混合は、押し出し機、プラベンダー、ニーダー、バンバリミキサー等の各種混合機で行うことができ、混練温度は通常、未変性ポリオレフィン系重合体の融点~300℃の温度範囲である。
本発明のフィルム又はシートに含有されるテトラフルオロエチレン系ポリマー(C)は、ポリ乳酸(A)含有樹脂組成物の溶融張力を向上させ、溶融成膜過程の流動場での配向結晶化を可能にすることで、ポリ乳酸(A)の結晶化を促進することができる。また、テトラフルオロエチレン系ポリマー(C)はポリ乳酸(A)の結晶核剤としての効果を持ち合わせることから、成膜直後の樹脂組成物の温度を結晶化温度付近に設定することで、ポリ乳酸(A)の結晶化をさらに促進することができる。よって、テトラフルオロエチレン系ポリマー(C)は、ポリ乳酸(A)の結晶化を促進することにより、本発明のフィルム又はシートに耐熱性を付与することが可能である。
テトラフルオロエチレン系ポリマー(C)の重量平均分子量は特に制限されず、通常100万~1000万、好ましくは200万~800万である。
テトラフルオロエチレン系ポリマー(C)は、市販品を用いてもよく、例えば、ポリテトラフルオロエチレンの市販品としては、旭硝子株式会社製の「フルオン(登録商標)CD-014」、「フルオン(登録商標)CD-1」、「フルオン(登録商標)CD-145」等が挙げられる。アクリル変性ポリテトラフルオロエチレンの市販品としては、例えば、三菱レイヨン株式会社製の、メタブレン(登録商標)Aシリーズ(A-3000、A-3800等)が挙げられる。
本発明のフィルム又はシートに含有されるジカルボン酸エステル系可塑剤(D)は、ポリ乳酸(A)を含有する樹脂組成物に所望の柔軟性を付与する可塑剤としての効果を有する。ジカルボン酸エステル系可塑剤(D)は、下記一般式(1)で表される化合物を含む。

Bは1,2-フェニレン基、または、-CH2(CH2)mCH2-(但し、mは0~6の整数を示す。)であり、
Aa及びAbは、それぞれ独立して、下記(1)~(3)から選ばれる(但し、Aa及びAbが共に下記(3)である化合物を除く):
(1)C6H5-(Ac)-、(但し、Acは炭素数1~4のアルキレン基);
(2)(Ae)-(Ad-O)n-、(但し、Adは炭素数1~5のアルキレン基、Aeは炭素数1~7のアルキル基、nは1~4の整数を示す);及び
(3)炭素数1~14のアルキル基。)
Acで示される「炭素数1~4のアルキレン基」とは、例えば、メチレン基、エチレン基、トリメチレン基、テトラメトレン基が挙げられ、好ましくはメチレン基である。
Adで示される「炭素数1~5のアルキレン基」は、メチレン基、エチレン基、トリメチレン基、プロピレン基、テトラメチレン基、1,1’-ジメチルエチレン基、ペンタメチレン基等の直鎖状、分岐鎖状のものが挙げられ、好ましくは、エチレン基である。
Aeで示される「炭素数1~7のアルキル基」は、直鎖または分枝鎖のいずれであってもよく、例えば、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、ペンチル基、2-ペンチル基、3-ペンチル基、ネオペンチル基、第3級ペンチル基、ヘキシル基、イソヘキシル基、ヘプチル基などが挙げられる。
「炭素数1~14のアルキル基」とは、炭素数1~14の直鎖または分枝鎖状の飽和炭化水素基を意味し、例えば、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、ペンチル基、イソペンチル基、ネオペンチル基、1,2-ジメチルプロピル基、1-エチルプロピル基、ヘキシル基、イソヘキシル基、1,2,2-トリメチルプロピル基、1,1-ジメチルブチル基、2,2-ジメチルブチル基、3,3-ジメチルブチル基、2-エチルブチル基、ヘプチル基、イソヘプチル基、2-エチルヘキシル基、オクチル基、イソオクチル基、ノニル基、イソノニル基、デシル基、イソデシル基、ウンデシル基、イソウンデシル基、ドデシル基、イソドデシル基、トリデシル基、イソトリデシル基、テトラデシル基、イソテトラデシル基等が挙げられ、好ましくは、炭素数1~8のアルキル基であり、より好ましくはブチル基、2-エチルヘキシル基である。
mは0~6の整数であり好ましくはm=2である。nは1~4の整数であり好ましくはn=2である。
一方、Aa及びAbが共にアリール基やオキシアルキレン鎖を含まない低分子量の可塑剤は、可塑化効果が大きく、少量添加で柔軟性を得ることができるものの、ポリ乳酸の加工温度(およそ150~180℃)では、揮発性が大きく、作業環境が悪化し、加えて、配合した可塑剤がフィルム中に残存せず性能が安定しないという問題がある。

「炭素数1~4のアルキレン基」、「炭素数1~5のアルキレン基」、「炭素数1~7のアルキル基」及び「炭素数1~14のアルキル基」は、前述の通りである。
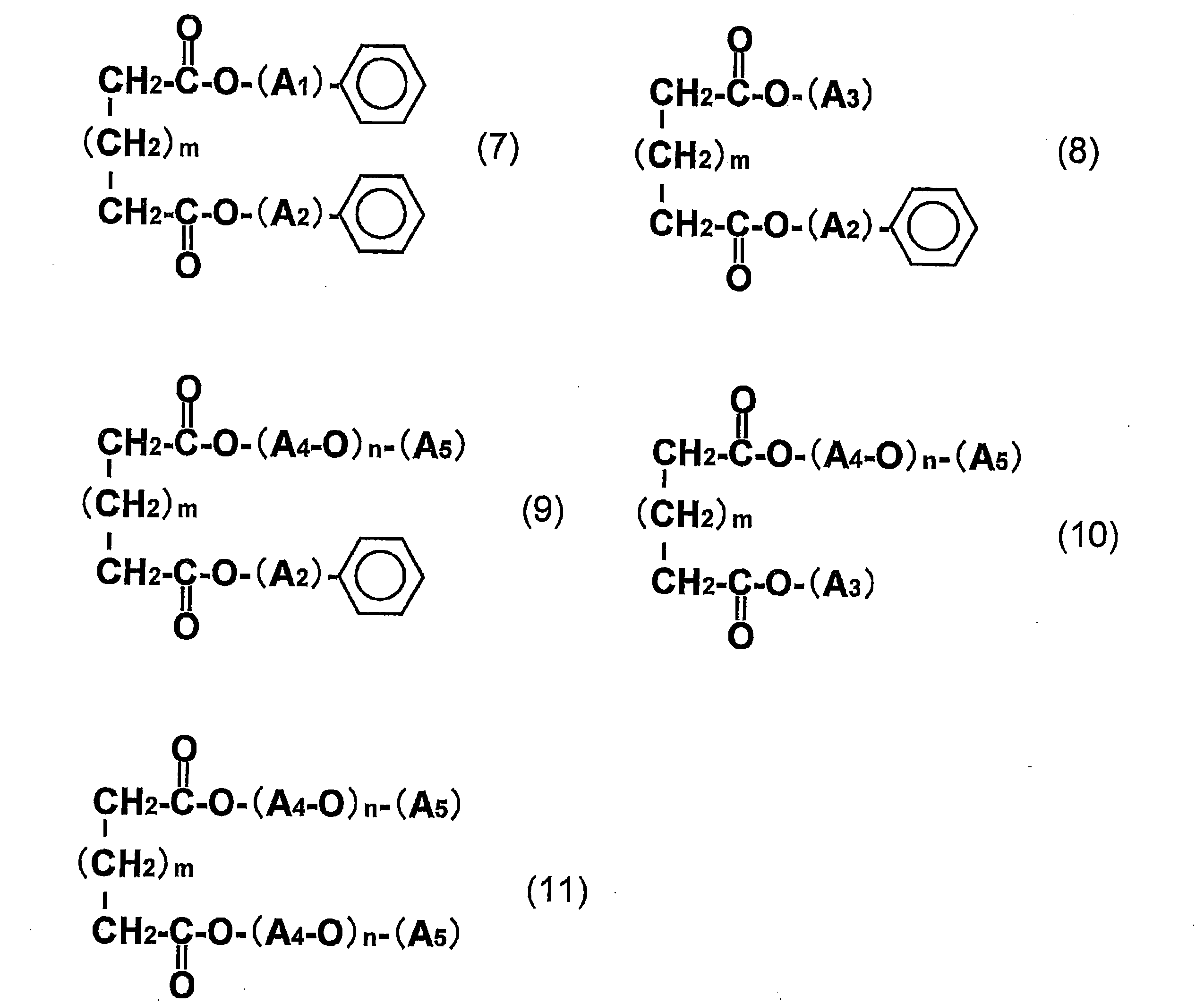
「炭素数1~4のアルキレン基」、「炭素数1~5のアルキレン基」、「炭素数1~7のアルキル基」及び「炭素数1~14のアルキル基」は、前述の通りである。
ジカルボン酸エステル系可塑剤(D)の含有量は、ポリ乳酸(A)100重量部に対して、好ましくは、8重量部以上40重量部未満である。中でも、フタル酸エステル系可塑剤(DP)の含有量は、ポリ乳酸(A)100重量部に対して、通常、10重量部以上40重量部未満であり、好ましくは12重量部以上40重量部未満であり、より好ましくは15~38重量部である。また、脂肪族ジカルボン酸エステル系可塑剤(DA)の含有量は、ポリ乳酸(A)100重量部に対して、通常、8~35重量部であり、好ましくは10~30重量部であり、より好ましくは12~27重量部である。ジカルボン酸エステル系可塑剤(D)の含有量が、8重量部未満では、柔軟性付与効果が得がたく、40重量部以上になると、プレートアウトが発生するとともに、ポリ乳酸の結晶化を阻害することにより耐熱性の低下が顕著となる。
本発明の樹脂組成物は、テトラフルオロエチレン系ポリマー(C)以外に、他の結晶化促進剤(E)を含んでもよい。結晶化促進剤(E)は、結晶化促進の効果が認められるものであれば、特に限定されないが、ポリ乳酸(A)の結晶格子の面間隔に近い面間隔を持つ結晶構造を有する物質を選択することが望ましい。結晶格子の面間隔がポリ乳酸(A)の結晶格子の面間隔に近い物質ほど、ポリ乳酸(A)の結晶核剤としての効果が高いからである。そのような結晶化促進剤(E)としては、例えば、有機系物質であるポリリン酸メラミン、メラミンシアヌレート、フェニルホスホン酸亜鉛、フェニルホスホン酸カルシウム、フェニルホスホン酸マグネシウム、無機系物質のタルク、クレー等が挙げられる。中でも、最も面間隔がポリ乳酸(A)の面間隔に類似し、良好な結晶化促進効果が得られるフェニルホスホン酸亜鉛が好ましい。
結晶化促進剤(E)は、市販品を用いることができる。例えば、フェニルホスホン酸亜鉛の市販品としては、日産化学工業株式会社製の「エコプロモート」等が挙げられる。
本発明のフィルム又はシートの柔軟性の指標として、引張破断伸び及び応力残存率を測定した。
本発明のフィルム又はシートにおいて、JISK7161に記載の「プラスチック-引張特性」の試験方法に準じて測定した引張破断伸びは、100%以上であることが好ましい。また、上記試験方法に準じて測定された10%変位時の応力残存率は40%以下であることが好ましい。引張破断伸び及び応力残存率が上記範囲であれば、柔軟で、かつ、延伸時に応力緩和されるフィルム又はシートを得ることができる。
柔軟性及び応力残存率が上記範囲内である本発明のフィルム又はシートは、ポリ乳酸(A)、酸性官能基変性オレフィン系ポリマー(B)、テトラフルオロエチレン系ポリマー(C)及び一般式(1)で示されるジカルボン酸エステル系可塑剤(D)の含有量をそれぞれ本発明で規定する範囲内にすることで実現できる。特に、ジカルボン酸エステル系可塑剤(D)の含有量を本発明で規定する範囲内とすることが重要である。
本発明のフィルム又はシートの加熱変形率は、JISC3005に記載の加熱変形試験方法に準じて測定する。
本発明のフィルム又はシートは、120℃の温度雰囲気下で30分間、10Nの荷重を加えたときの変形率が40%以下であることが好ましい。
本発明のフィルム又はシートの相対結晶化率は、DSCにて測定した、成膜後のフィルム又はシートのサンプルの昇温過程での結晶化に伴う発熱ピークの熱量ΔHcと、その後の融解に伴う熱量ΔHmから、以下の式(I)を用いて算出する。
相対結晶化率(%)=(ΔHm-ΔHc)/ΔHm×100 (I)
より好ましくは、本発明のフィルム又はシートは、JISC3005の加熱試験方法に準じて、120℃の温度雰囲気下で、30分間、10Nの荷重を加えたときの変形率が40%以下であり、上記式(I)で求められる相対結晶化率が50%以上である。
本発明のフィルム又はシートの製造方法は、特に制限されるものでなく、ポリ乳酸(A)含有樹脂組成物を溶融成膜法により成膜する方法が好ましい。例えば、本発明のフィルム又はシートは、二軸押出機などによる連続溶融混練機、又は加圧ニーダー、バンバリミキサー、ロール混練機などのバッチ式溶融混練機により、各成分を均一分散させたポリ乳酸(A)含有樹脂組成物を作成し、これをTダイ法、インフレーション法などの押出法又はカレンダー法、ポリッシング法などにより成膜、冷却固化することにより製造することができる。かかる溶融成膜法としては、好ましくは、溶融状態の樹脂組成物が2本の金属ロール間の空隙を通過することで所望の厚さに成膜される手法であり、特に好ましくは、カレンダー法、ポリッシング法である。
本発明のフィルム又はシートの厚さはその用途に応じ、適宜調整されるが、通常10~500μm、好ましくは20~400μm、特に好ましくは30~300μmである。
例えば、該樹脂組成物をカレンダー法により溶融成膜する場合は、カレンダーロール圧延時の該樹脂組成物の温度(溶融成膜時の樹脂温度に相当する)を、該樹脂組成物の降温過程での結晶化温度(Tc)+15℃の温度から昇温過程での融解温度(Tm)-5℃の間の温度に設定する。このように融点以下の温度で圧延することにより、配向結晶化が促進される。この配向結晶化促進効果は、該樹脂組成物がテトラフルオロエチレン系ポリマー(C)を含有することにより格段に向上する。テトラフルオロエチレン系ポリマー(C)は、該樹脂組成物中でフィブリル化し、ネットワーク化すること、及びその結晶核剤としての効果の相乗効果により、配向結晶化を促進すると考えられる。従って、上記温度範囲で圧延することにより、本発明のフィルム又はシートは、平滑な表面状態とともに、配向結晶化促進効果により、良好な耐熱性(すなわち、相対結晶化率の低下抑制及び加熱変形率の上昇抑制)を実現することができる。
特に、所定の温度に常に制御する観点から、溶融成膜された該樹脂組成物を、所定の表面温度の金属ロールと接触させることが望ましい。従って、当該工程においても、ポリ乳酸(A)含有樹脂組成物を金属ロールから簡単に剥離できる組成にすることが望ましく、この観点からも上述の酸性官能基変性オレフィン系ポリマー(B)の添加が必要となる。
図1に、かかる製造方法に用いられる一実施形態のカレンダー成膜機の模式図を示す。以下に、図1を詳細に説明する。
第1ロール(1)、第2ロール(2)、第3ロール(3)、第4ロール(4)という、4本のカレンダーロール間で溶融状態の樹脂組成物を圧延して徐々に薄くしていき、最終的に第3ロール(3)と第4ロール(4)の間を通過した時に所望の厚さになるよう調製される。カレンダー成膜の場合、第1~第4ロール(1)~(4)における樹脂組成物の成膜が「溶融成膜工程」に相当する。また、該樹脂組成物の降温過程での結晶化温度(Tc)-25℃の温度から結晶化温度(Tc)+10℃の間の温度[好ましくは、結晶化温度(Tc)±10℃]に設定したテイクオフロール(5)は、溶融成膜された該樹脂組成物(8)が最初に接触するロール群を示し、1つまたは2つ以上(図1では3本)のロール群で構成され、第4ロール(4)から溶融状態の該樹脂組成物(8)を剥離する役割を果たす。このように、テイクオフロール(5)が複数のロールから構成され、各々のロールの温度調節が可能である場合、各々のロールの温度は同じであることが好ましいが、所望の温度範囲内であれば、異なっていても良い。テイクオフロール(5)の本数は多いほうが、等温結晶化時間が長くなり、結晶化を促進するのに有利である。カレンダー成膜の場合、テイクオフロール(5)において、溶融成膜された該樹脂組成物(8)の結晶化が促進されるので、該樹脂組成物(8)がテイクオフロール(5)を通過する工程が「結晶化促進工程」に相当する。
二本の冷却ロール(6)及び(7)は、それらの間に該樹脂組成物(8)を通過させることにより該樹脂組成物(8)を冷却し、固化させるとともにその表面を所望の形状に成形する役割を果たす。そのため、通常は一方のロール(例えば、冷却ロール(6))が金属ロールで、該樹脂組成物(8)の表面形状を出すためにロール表面がデザインされたものであり、他方のロール(例えば、冷却ロール(7))としてゴムロールが使用される。なお、図中の矢印はロールの回転方向を示す。
図2に、かかる製造方法に用いられる一実施形態のポリッシング成膜機の模式図を示す。以下に、図2を詳細に説明する。
押出機(図示せず。)の押出機先端部(10)を、加熱した第2ロール(2)及び第3ロール(3)の間に配置し、予め設定された押出し速度で、第2ロール(2)及び第3ロール(3)の間に溶融状態の樹脂組成物(8)を連続的に押し出す。押し出された樹脂組成物(8)は、第2ロール(2)及び第3ロール(3)の間で圧延されて薄くなり、最終的に第3ロール(3)と第4ロール(4)の間を通過した時に所望の厚さになるよう調製される。ポリッシング成膜の場合、第2~第4ロール(2)~(4)における樹脂組成物(8)の成膜が「溶融成膜工程」に相当する。その後、該樹脂組成物(8)の降温過程での結晶化温度(Tc)-25℃の温度から結晶化温度(Tc)+10℃の間の温度[好ましくは、結晶化温度(Tc)±10℃]に設定された3本のテイクオフロール(5)を通過し、最後に冷却ロール(6)及び(7)を通過することで、固化したフィルム又はシートが作製される。ポリッシング成膜の場合、テイクオフロール(5)を通過する工程が「結晶化促進工程」に相当する。
A1:レイシア(登録商標)H-400(三井化学株式会社製)
B1:無水マレイン酸変性ポリプロピレン(重量平均分子量=49,000、酸価=26mgKOH/g):ユーメックス(登録商標)1001(三洋化成工業株式会社製)
B2:無水マレイン酸変性ポリプロピレン(重量平均分子量=32,000、酸価=52mgKOH/g):ユーメックス(登録商標)1010(三洋化成工業株式会社製)
(B1)成分及び(B2)成分との比較のため、以下の(B’)成分を検討した。
B’:未変性の低分子量ポリプロピレン(重量平均分子量=23,000、酸価=0mgKOH/g):ビスコール(登録商標)440P(三洋化成工業株式会社製)
C1:ポリテトラフルオロエチレン:フルオン(登録商標)CD-014(旭硝子株式会社製)
C2:アクリル変性ポリテトラフルオロエチレン:メタブレン(登録商標)A-3000(三菱レイヨン株式会社製)
DP1:ブチルベンジルフタレート:BBP(大八化学工業株式会社製)、分子量314
DP2:(2-エチルヘキシル)ベンジルフタレート:サンチサイザー261A(フェロ・ジャパン社製)、分子量370
(DP1)及び(DP2)成分との比較のため、以下のフタルエステル系可塑剤(DP’)を対比検討に用いた。
DP’:ビス(2-エチルヘキシル)フタレート:DOP(DIC株式会社製)、分子量391
[脂肪族ジカルボン酸エステル系可塑剤(DA)]
(DA1);ビス[2-(2-ブトキシエトキシ)エチル]アジペート:BXA(大八化学工業株式会社製)、分子量435
(DA2);ベンジル(メトキシエトキシエチル)アジペート:DAIFATTY-101(大八化学工業株式会社 登録商標)、分子量338
(DA1)及び(DA2)成分との比較のため、以下のフタルエステル系可塑剤(DA’)を対比検討に用いた。
(DA’);ジイソノニルアジペート:W-242(DIC株式会社製)、分子量398
E1:フェニルホスホン酸亜鉛:エコプロモート(日産化学工業株式会社製)
上記の原材料が表1に示す配合割合で配合された樹脂組成物を調製し、バンバリミキサーにて溶融混練を行った後、逆L型4本カレンダーにて厚さ100μmになるようにカレンダー成膜(溶融成膜)を行った。次に、図1のように溶融成膜工程の直後に、任意の温度に加熱可能なロール(テイクオフロール)を3本配し、溶融成膜された樹脂組成物が上下交互に通過できるようにすることで結晶化促進工程とした。その後に冷却ロールを通過することで樹脂組成物を固化し、フィルムを作成した。溶融成膜時の樹脂組成物の温度(溶融成膜時の樹脂温度)は、図1における第4ロール(4)に相当するロールの表面温度とみなし、結晶化促進工程における樹脂組成物の温度は、図1の3本のテイクオフロール(5)の表面温度を略同一とし、その温度を結晶化促進温度とした。成膜速度は5m/minとし、実質的な結晶化促進工程の時間(テイクオフロール通過時間)は約5秒間であった。
下記表1に示す配合割合で樹脂組成物を調製し、実施例1と同様の操作により実施例2~5のフィルムをそれぞれ作成した。
下記表1に示す配合割合で樹脂組成物を調製し、実施例1と同様の操作により比較例1~7のフィルムをそれぞれ作成した。
実施例及び比較例で作製した試料の評価は下記のようにして行った。
DSCにて測定した、成膜後の樹脂組成物の再昇温過程での融解に伴う吸熱ピークのトップ時の温度を融解温度(Tm;結晶融解ピーク温度ともいう)とした。
DSCにて測定した、成膜後の樹脂組成物の200℃からの降温過程での結晶化に伴う発熱ピークのピークトップ時の温度を結晶化温度(Tc;結晶化ピーク温度ともいう)とした。
(1)剥離性: 図1の第4ロール(4)からの溶融成膜された樹脂組成物の剥離性により評価し、テイクオフロール(5)で引き取り可能である状態を「○」、テイクオフロール(5)で引き取り不可である状態を「×」と、判定した。
(2)耐揮発性: 成膜時の樹脂組成物を目視で観察し、揮発(白煙)が確認されない状態を「○」(なし)、揮発が確認される状態を「×」(あり)と、判定した。
(3)ロールへのプレートアウト: ロール表面の汚れを目視により評価し、ロール表面の汚れがない状態を「○」、ロール表面の汚れがある状態を「×」と判定した。
なお、比較例3については、剥離しなかったため、ロールへのプレートアウトの評価を行わなかった。
DSC(示差走査熱分析装置)にて測定した、成膜後のフィルムサンプルの昇温過程での結晶化に伴う発熱ピークの熱量ΔHcと、その後の融解に伴う熱量ΔHmから、以下の式(I)を用いて算出した。なお、比較例3については、剥離しなかったため、測定をしていない。
相対結晶化率(%)=(ΔHm-ΔHc)/ΔHm×100 (I)
合否判定:相対結晶化率50%以上を合格とする。
装置:エスアイアイ・ナノテクノロジー株式会社製 DSC6220
条件:測定温度域 20℃→200℃→0℃→200℃
(すなわち、まず20℃から200℃への昇温過程での
測定に続けて、200℃から0℃への降温過程での測
定を行い、最後に0℃から200℃への再昇温過程で
の測定を行った。)
昇温/降温速度:2℃/min
測定雰囲気:窒素雰囲気下(200ml/min)
なお、再昇温過程で、結晶化に伴う発熱ピークが無かったことから、2℃/minの降温速度で結晶化可能領域が100%結晶化するものと判断し、相対結晶化率の算出式の妥当性を確認した。
JISC3005の加熱変形試験方法に準じ測定した。使用した測定装置及び測定条件は、以下の通りである。
装置:テスター産業株式会社製 加熱変形試験機
条件:試料サイズ:厚さ1mm×幅25mm×長さ40mm
(フィルムを総厚1mmになるように重ねた。)
測定温度:120℃
荷重:10N
測定時間:30分(再結晶化を考慮し、エージングなしで試験開始)
加熱変形率算出方法:試験前の厚みT1と試験後の厚みT2を測定し、以下の式(II)を用いて加熱変形率を算出した。なお、比較例3については、剥離しなかったため、測定をしていない。
加熱変形率(%)=(T1-T2)/T1×100 (II)
合否判定:40%以下を合格とする。
JISK7161のプラスチック-引張特性の試験方法に準じて測定した。
使用した測定装置及び測定条件は、以下の通りである。
装置:引張試験機(オートグラフAG-20kNG、(株)島津製作所製)
試料サイズ:厚さ0.1mm×幅10mm×長さ100mm
なお、長さ方向に平行な方向がフィルム成膜時の流れ方向(MD)
となるように切り出した。
測定条件:チャック間距離 50mm
引張速度 300mm/min
合否判定:上記の条件で各試料について測定し、フィルム破断時の伸び値を測定して引張破断伸びを得た。引張破断伸びが100%以上のものを合格と判定した。
応力緩和性の指標となる応力残存率については、JISK7161のプラスチック-引張特性の試験方法に準じて測定した。
使用した測定装置及び測定条件は、以下の通りである。
装置:引張試験機(オートグラフAG-20kNG、(株)島津製作所製)
条件:試料サイズ:厚さ0.1mm×幅10mm×長さ100mm
なお、長さ方向に平行な方向がフィルム成膜時の流れ方向(MD)
となるように切り出した。
チャック間距離 50mm
引張速度 300mm/min
測定:変位が10%となったところで変位を止め、その位置で保持する。
そのときの応力を100%とし、60秒後の応力値の残存量を読み
取り、「応力残存率」を求めた。
合否判定:応力残存率が40%以下のものを合格と判定した。
一方、本発明の一般式(1)のジカルボン酸エステル系可塑剤(D)に含まれない低分子量の可塑剤(すなわち、一般式(1)のAaおよびAbがアリール基やオキシアルキレン鎖を含まない可塑剤)を用いた比較例1及び2のフィルムはいずれも、可塑剤の揮発、及びロールへのプレートアウトが観察され、引張破断伸び及び応力緩和性も不十分であった。また、酸性官能基変性オレフィン系ポリマー(B)を含まない比較例3のフィルムは、ロールからの剥離性が不良であった。また、一般式(1)のジカルボン酸エステル系可塑剤(D)の配合量が不足していた比較例4及び5は引張破断伸び及び応力緩和性が不十分であった。一方、ジカルボン酸エステル系可塑剤(D)の配合量が過剰であった比較例6及び7は、いずれも耐熱性が不十分であった。すなわち、本発明に係る成分を含まないか、又は本発明の配合割合でない比較例1~7では、所望の柔軟性、耐熱性を全て満足するフィルムが得られなかった。


2 第2ロール
3 第3ロール
4 第4ロール
5 テイクオフロール
6 冷却ロール
7 冷却ロール
8 樹脂組成物
9 バンク(樹脂だまり)
10 押出機先端部
Claims (13)
- ポリ乳酸(A)、
酸性官能基を含み、その酸価が10~70mgKOH/gであり、かつ、重量平均分子量が10,000~80,000である、酸性官能基変性オレフィン系ポリマー(B)、
テトラフルオロエチレン系ポリマー(C)、及び、
下記一般式(1)で表されるジカルボン酸エステル系可塑剤(D)を含有し、
当該ジカルボン酸エステル系可塑剤(D)の含有量が、ポリ乳酸(A)100重量部に対し、8重量部以上40重量部未満である樹脂組成物からなるフィルム又はシート。

Bは1,2-フェニレン基、または、-CH2(CH2)mCH2-(但し、mは0~6の整数を示す)であり、
Aa及びAbは、それぞれ独立して、下記(1)~(3)から選ばれる(但し、Aa及びAbが共に下記(3)である化合物を除く):
(1)C6H5-(Ac)-、(但し、Acは炭素数1~4のアルキレン基);
(2)(Ae)-(Ad-O)n-、(但し、Adは炭素数1~5のアルキレン基、Aeは炭素数1~7のアルキル基、nは1~4の整数を示す);及び
(3)炭素数1~14のアルキル基。) - 当該ジカルボン酸エステル系可塑剤(D)が、一般式(2)~(6)で表される化合物から選ばれるフタル酸エステル系可塑剤(DP)であり、当該フタル酸エステル系可塑剤(DP)の含有量が、ポリ乳酸(A)100重量部に対し10重量部以上40重量部未満である、請求項1記載のフィルム又はシート。
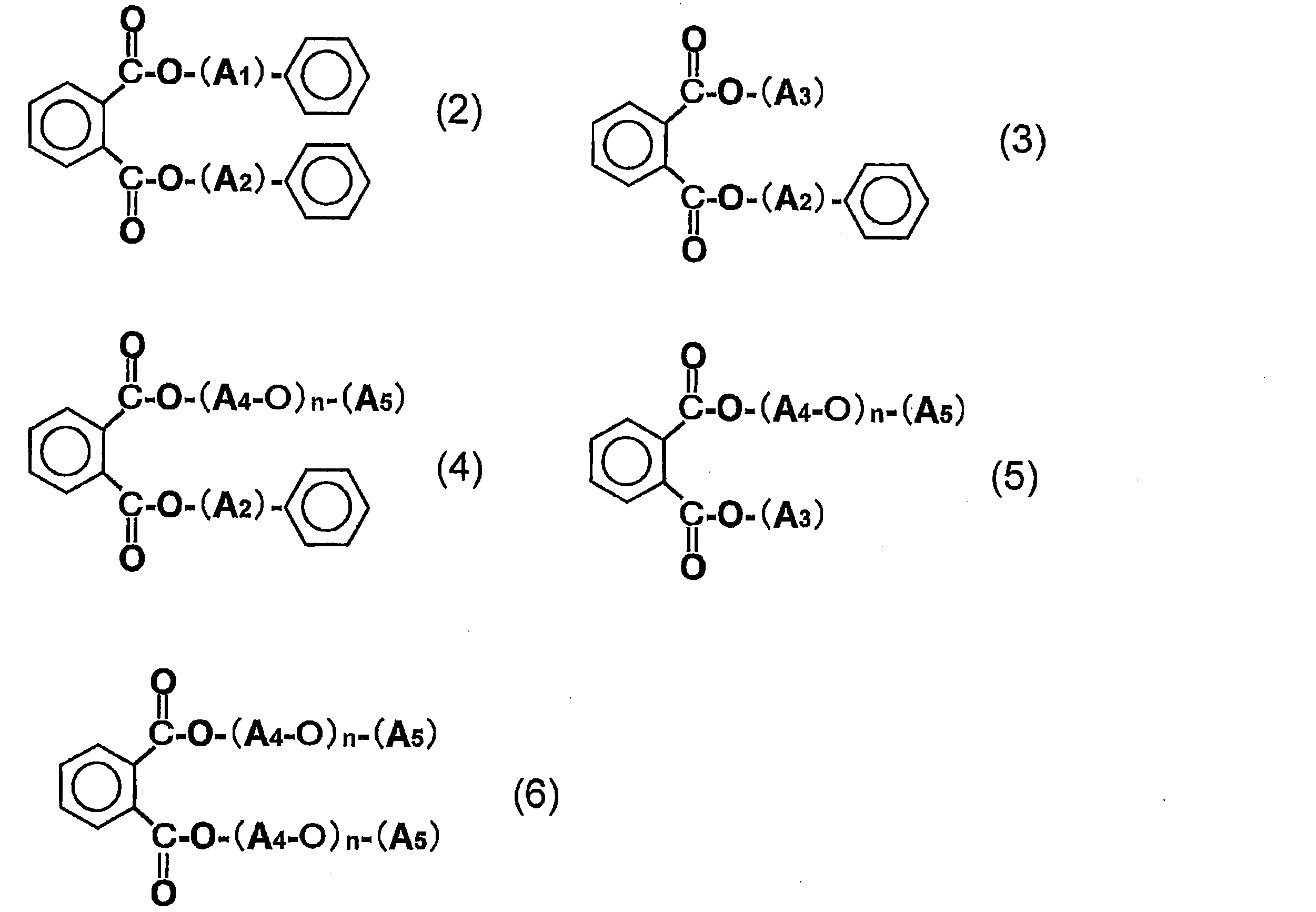
- 当該ジカルボン酸エステル系可塑剤(D)が、一般式(7)~(11)で表される化合物から選ばれる脂肪族ジカルボン酸エステル系可塑剤(DA)であり、当該脂肪族ジカルボン酸エステル系可塑剤(DA)の含有量が、ポリ乳酸(A)100重量部に対し8~35重量部である、請求項1記載のフィルム又はシート。

- 酸性官能基変性オレフィン系ポリマー(B)に含まれる酸性官能基が、カルボン酸無水物基である、請求項1~3のいずれか1項に記載のフィルム又はシート。
- テトラフルオロエチレン系ポリマー(C)の含有量が、ポリ乳酸(A)100重量部に対し、0.5~15.0重量部である、請求項1~4のいずれか1項に記載のフィルム又はシート。
- 酸性官能基変性オレフィン系ポリマー(B)の含有量が、ポリ乳酸(A)100重量部に対し、0.1~10.0重量部である、請求項1~5のいずれか1項に記載のフィルム又はシート。
- 樹脂組成物が、さらに結晶化促進剤(E)を含有し、
当該結晶化促進剤(E)の含有量が、ポリ乳酸(A)100重量部に対し、0.1~5.0重量部である、請求項1~6のいずれか1項に記載のフィルム又はシート。 - JISC3005の加熱変形試験方法に準じて、120℃の温度雰囲気下で30分間、10Nの荷重を加えたときの変形率が、40%以下であり、
下記式(I)で求められる相対結晶化率が50%以上であることを特徴とする、請求項1~7のいずれか1項に記載のフィルム又はシート。
相対結晶化率(%)=(ΔHm-ΔHc)/ΔHm×100 (I)
(式中、ΔHcは成膜後のフィルム又はシートの昇温過程での結晶化に伴う発熱ピークの熱量であり、ΔHmは融解に伴う熱量を示す。) - 引張破断伸びが100%以上であり、10%伸張時の応力残存率が40%以下であることを特徴とする、請求項1~8のいずれか1項に記載のフィルム又はシート。
- 樹脂組成物を溶融成膜法により成膜することを含む、請求項1~9のいずれか1項に記載のフィルム又はシートの製造方法であって、
溶融成膜時の樹脂組成物の温度が、該樹脂組成物の降温過程での結晶化温度(Tc)+15℃の温度から、昇温過程での融解温度(Tm)-5℃の間の温度であるか、又は、
溶融成膜された該樹脂組成物が、該樹脂組成物の降温過程での結晶化温度(Tc)-25℃の温度から結晶化温度(Tc)+10℃の間の温度における結晶化促進工程を経てから冷却固化されることを特徴とする、フィルム又はシートの製造方法。 - 溶融成膜時の該樹脂組成物の温度が、該樹脂組成物の降温過程での結晶化温度(Tc)+15℃の温度から、昇温過程での融解温度(Tm)-5℃の間の温度であり、かつ、
溶融成膜された該樹脂組成物が、該樹脂組成物の降温過程での結晶化温度(Tc)-25℃の温度から結晶化温度(Tc)+10℃の間の温度における結晶化促進工程を経てから冷却固化されることを特徴とする、請求項10記載のフィルム又はシートの製造方法。 - 溶融成膜法が、溶融状態の樹脂組成物を2本の金属ロール間の空隙を通過させることで所望の厚さに成膜する手法である、請求項10又は11に記載のフィルム又はシートの製造方法。
- 結晶化促進工程が、金属ロールを用いて行われることを特徴とする、請求項10~12のいずれか1項に記載のフィルム又はシートの製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201180001491.6A CN102361935B (zh) | 2010-03-30 | 2011-03-25 | 聚乳酸系薄膜或薄片及其制造方法 |
| JP2011526169A JP5726078B2 (ja) | 2010-03-30 | 2011-03-25 | ポリ乳酸系フィルム又はシート、及びその製造方法 |
| EP11762696.0A EP2554596B1 (en) | 2010-03-30 | 2011-03-25 | Poly lactic acid-containing film or sheet, and method for manufacturing thereof |
| US13/266,275 US9403955B2 (en) | 2010-03-30 | 2011-03-25 | Poly lactic acid-containing film or sheet, and method for manufacturing thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2010078661 | 2010-03-30 | ||
| JP2010-078661 | 2010-03-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011122465A1 true WO2011122465A1 (ja) | 2011-10-06 |
Family
ID=44712174
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2011/057322 WO2011122465A1 (ja) | 2010-03-30 | 2011-03-25 | ポリ乳酸系フィルム又はシート、及びその製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US9403955B2 (ja) |
| EP (1) | EP2554596B1 (ja) |
| JP (1) | JP5726078B2 (ja) |
| CN (1) | CN102361935B (ja) |
| WO (1) | WO2011122465A1 (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014001320A (ja) * | 2012-06-19 | 2014-01-09 | Kao Corp | ポリ乳酸樹脂組成物からなる延伸フィルム |
| JP2015224266A (ja) * | 2014-05-26 | 2015-12-14 | 三菱レイヨン株式会社 | 樹脂発泡体及びその製造方法 |
| JP2018062573A (ja) * | 2016-10-13 | 2018-04-19 | 花王株式会社 | ポリ乳酸樹脂組成物 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102171278B (zh) | 2008-10-02 | 2015-01-21 | 日东电工株式会社 | 聚乳酸类膜或片 |
| US20130236723A1 (en) * | 2010-11-26 | 2013-09-12 | Nitto Denko Corporation | Polylactic acid-based film or sheet |
| CN103228713A (zh) * | 2010-11-26 | 2013-07-31 | 日东电工株式会社 | 聚乳酸系薄膜或片 |
| RU2729055C2 (ru) * | 2016-01-20 | 2020-08-04 | Басф Се | Пластифицирующая композиция, которая содержит алифатический сложный эфир дикарбоновой кислоты и сложный диэфир, выбранный из сложных эфиров 1,2-циклогександикарбоновой кислоты и сложных эфиров терефталевой кислоты |
| CN115073898B (zh) * | 2022-04-14 | 2023-12-19 | 万华化学(宁波)有限公司 | 一种高熔体强度pla合金、一种发泡料及其制备方法 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006016447A (ja) * | 2004-06-30 | 2006-01-19 | Toray Ind Inc | 樹脂組成物ならびにそれからなる成形品 |
| JP3865960B2 (ja) | 1999-01-06 | 2007-01-10 | 三井化学株式会社 | 樹脂成形方法 |
| JP2007130894A (ja) * | 2005-11-10 | 2007-05-31 | Kao Corp | 生分解性樹脂成形品の製造法。 |
| JP2007177213A (ja) | 2005-11-29 | 2007-07-12 | Toray Ind Inc | 樹脂組成物およびそれからなる成形品 |
| JP2008013742A (ja) * | 2006-06-08 | 2008-01-24 | Osaka Gas Co Ltd | 耐熱性を向上させた植物由来プラスチック材料及び成形体 |
| JP4246196B2 (ja) | 2005-11-10 | 2009-04-02 | 花王株式会社 | 生分解性樹脂成形品の製造法。 |
| WO2010038833A1 (ja) * | 2008-10-02 | 2010-04-08 | 日東電工株式会社 | ポリ乳酸系フィルムまたはシート |
| JP2011052149A (ja) * | 2009-09-03 | 2011-03-17 | Osaka Gas Co Ltd | ポリ乳酸樹脂組成物 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5502158A (en) * | 1988-08-08 | 1996-03-26 | Ecopol, Llc | Degradable polymer composition |
| US5338822A (en) * | 1992-10-02 | 1994-08-16 | Cargill, Incorporated | Melt-stable lactide polymer composition and process for manufacture thereof |
| WO1999052987A1 (en) * | 1998-04-14 | 1999-10-21 | Sony Chemicals Corp. | Pressure-sensitive adhesive tape and method for producing the same |
| JP4306199B2 (ja) * | 2001-08-03 | 2009-07-29 | 東レ株式会社 | 樹脂組成物ならびにそれからなる成形品、フィルムおよび繊維 |
| WO2003014224A1 (en) * | 2001-08-03 | 2003-02-20 | Toray Industries, Inc. | Resin composition and molded article, film, and fiber each comprising the same |
| JP3962242B2 (ja) * | 2001-11-12 | 2007-08-22 | 三菱レイヨン株式会社 | 熱可塑性樹脂組成物及びそれを用いた成形体 |
| JP4469149B2 (ja) * | 2003-08-07 | 2010-05-26 | ダイセルポリマー株式会社 | 熱可塑性樹脂組成物及び成形品 |
| US20050137304A1 (en) * | 2003-12-18 | 2005-06-23 | Strand Marc A. | Process for calendering of polyesters |
| JP4973848B2 (ja) * | 2004-03-30 | 2012-07-11 | 日産化学工業株式会社 | ポリ乳酸樹脂組成物 |
| ATE509985T1 (de) * | 2004-09-17 | 2011-06-15 | Toray Industries | Harzzusammensetzung und formkörper daraus |
| CN101838425B (zh) * | 2005-08-04 | 2012-07-18 | 东丽株式会社 | 树脂组合物和由该树脂组合物形成的成型品 |
| EP2522696B1 (en) | 2010-01-08 | 2018-03-07 | Nitto Denko Corporation | Flame-retardant poly lactic acid-containing film or sheet, and method for manufacturing thereof |
-
2011
- 2011-03-25 CN CN201180001491.6A patent/CN102361935B/zh active Active
- 2011-03-25 US US13/266,275 patent/US9403955B2/en active Active
- 2011-03-25 JP JP2011526169A patent/JP5726078B2/ja active Active
- 2011-03-25 EP EP11762696.0A patent/EP2554596B1/en active Active
- 2011-03-25 WO PCT/JP2011/057322 patent/WO2011122465A1/ja active Application Filing
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3865960B2 (ja) | 1999-01-06 | 2007-01-10 | 三井化学株式会社 | 樹脂成形方法 |
| JP2006016447A (ja) * | 2004-06-30 | 2006-01-19 | Toray Ind Inc | 樹脂組成物ならびにそれからなる成形品 |
| JP2007130894A (ja) * | 2005-11-10 | 2007-05-31 | Kao Corp | 生分解性樹脂成形品の製造法。 |
| JP4246196B2 (ja) | 2005-11-10 | 2009-04-02 | 花王株式会社 | 生分解性樹脂成形品の製造法。 |
| JP2007177213A (ja) | 2005-11-29 | 2007-07-12 | Toray Ind Inc | 樹脂組成物およびそれからなる成形品 |
| JP2008013742A (ja) * | 2006-06-08 | 2008-01-24 | Osaka Gas Co Ltd | 耐熱性を向上させた植物由来プラスチック材料及び成形体 |
| WO2010038833A1 (ja) * | 2008-10-02 | 2010-04-08 | 日東電工株式会社 | ポリ乳酸系フィルムまたはシート |
| JP2011052149A (ja) * | 2009-09-03 | 2011-03-17 | Osaka Gas Co Ltd | ポリ乳酸樹脂組成物 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2554596A4 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014001320A (ja) * | 2012-06-19 | 2014-01-09 | Kao Corp | ポリ乳酸樹脂組成物からなる延伸フィルム |
| JP2015224266A (ja) * | 2014-05-26 | 2015-12-14 | 三菱レイヨン株式会社 | 樹脂発泡体及びその製造方法 |
| JP2018062573A (ja) * | 2016-10-13 | 2018-04-19 | 花王株式会社 | ポリ乳酸樹脂組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN102361935A (zh) | 2012-02-22 |
| EP2554596A1 (en) | 2013-02-06 |
| EP2554596B1 (en) | 2019-06-12 |
| JP5726078B2 (ja) | 2015-05-27 |
| CN102361935B (zh) | 2015-10-14 |
| JPWO2011122465A1 (ja) | 2013-07-08 |
| US9403955B2 (en) | 2016-08-02 |
| EP2554596A4 (en) | 2013-10-02 |
| US20120046402A1 (en) | 2012-02-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5726078B2 (ja) | ポリ乳酸系フィルム又はシート、及びその製造方法 | |
| JP5543750B2 (ja) | ポリ乳酸系フィルムまたはシート | |
| JP5694165B2 (ja) | 難燃性ポリ乳酸系フィルム又はシート、及びその製造方法 | |
| JP5658667B2 (ja) | 難燃性ポリ乳酸系フィルム又はシート、及びその製造方法 | |
| JP5726077B2 (ja) | 難燃性ポリ乳酸系フィルム又はシート、及びその製造方法 | |
| WO2013118407A1 (ja) | セパレータ | |
| JP6041550B2 (ja) | ポリ乳酸系樹脂組成物及びそれを用いたフィルム又はシート | |
| JP5913798B2 (ja) | ポリ乳酸系フィルム又はシート | |
| JP5731174B2 (ja) | ポリ乳酸系フィルム又はシート | |
| JP2012111914A (ja) | ポリ乳酸系フィルム又はシート | |
| JP2012111205A (ja) | セパレータ | |
| JP2012116887A (ja) | ポリ乳酸系フィルム又はシート | |
| JP2012111204A (ja) | セパレータ | |
| JP2012111207A (ja) | 保護フィルム | |
| JP2012116012A (ja) | セパレータ | |
| JP2012111203A (ja) | セパレータ | |
| JP2012111918A (ja) | 粘着テープ又はシート | |
| JP2012111919A (ja) | 粘着テープ又はシート | |
| JP2012111917A (ja) | 粘着テープ又はシート |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201180001491.6 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011526169 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13266275 Country of ref document: US |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11762696 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011762696 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |