WO2011074607A1 - トリテルペン誘導体およびc型慢性肝炎の予防または治療のための薬剤 - Google Patents
トリテルペン誘導体およびc型慢性肝炎の予防または治療のための薬剤 Download PDFInfo
- Publication number
- WO2011074607A1 WO2011074607A1 PCT/JP2010/072564 JP2010072564W WO2011074607A1 WO 2011074607 A1 WO2011074607 A1 WO 2011074607A1 JP 2010072564 W JP2010072564 W JP 2010072564W WO 2011074607 A1 WO2011074607 A1 WO 2011074607A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- formula
- compound
- pharmaceutically acceptable
- triterpene derivative
- Prior art date
Links
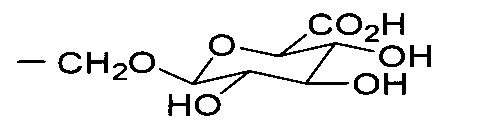
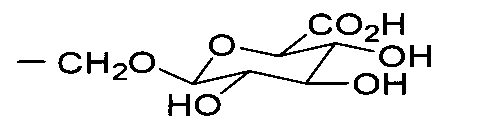
- 0 C*C(C(C(C1O)O)O)OC1C(O)=O Chemical compound C*C(C(C(C1O)O)O)OC1C(O)=O 0.000 description 3
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/075—Ethers or acetals
- A61K31/08—Ethers or acetals acyclic, e.g. paraformaldehyde
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/075—Ethers or acetals
- A61K31/085—Ethers or acetals having an ether linkage to aromatic ring nuclear carbon
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/12—Ketones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
- A61K31/135—Amines having aromatic rings, e.g. ketamine, nortriptyline
- A61K31/136—Amines having aromatic rings, e.g. ketamine, nortriptyline having the amino group directly attached to the aromatic ring, e.g. benzeneamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/56—Compounds containing cyclopenta[a]hydrophenanthrene ring systems; Derivatives thereof, e.g. steroids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/704—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin attached to a condensed carbocyclic ring system, e.g. sennosides, thiocolchicosides, escin, daunorubicin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J63/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton has been modified by expansion of only one ring by one or two atoms
- C07J63/008—Expansion of ring D by one atom, e.g. D homo steroids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J71/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton is condensed with a heterocyclic ring
- C07J71/0005—Oxygen-containing hetero ring
Definitions
- the present invention relates to a novel triterpene derivative or a pharmaceutically acceptable salt thereof, and a drug for preventing or treating chronic hepatitis C comprising them as an active ingredient.
- HCV hepatitis C virus
- the patient who is the target of prevention or treatment of chronic hepatitis C is a patient infected with HCV.
- HCV There are six types of HCV, mainly genotypes, from type 1 to type 6. Particularly, genotype 1 and genotype 2 are known to be widely distributed worldwide (see FIG. Non-Patent Documents 6 and 7).
- genotype 1 and genotype 2 are known to be widely distributed worldwide (see FIG. Non-Patent Documents 6 and 7).
- There are further subtypes in these genotype types. Differences in subtypes can be identified by the base sequence of a specific region of RNA constituting HCV, and the amount of HCV of each subtype can also be measured (see Non-Patent Document 8).
- interferon In the prevention or treatment of chronic hepatitis C, interferon (IFN) is widely used for the purpose of removing HCV.
- IFN is a physiologically active substance that is produced in the innate immune system during viral infection and has antiviral activity that inhibits viral replication.
- IFN is a species-specific protein with high homology, and it is known that four classes of IFN exist in humans (see Non-Patent Document 9).
- IFN ⁇ interferon ⁇
- IFN ⁇ interferon ⁇
- IFN ⁇ interferon ⁇
- IFN ⁇ interferon ⁇
- IFN ⁇ interferon ⁇
- ribavirin in order to expect a high therapeutic effect, it is necessary to use ribavirin in combination, so the problem of side effects caused by ribavirin cannot be avoided. If a useful and highly safe drug is developed for intractable cases of chronic hepatitis C, these medical problems can be solved, and therapeutic agents such as HCV subtypes that are more difficult to treat in the future. Alternatively, it can be expected to be used as a therapeutic agent having a high therapeutic effect that can be used in combination with a combination of a drug that directly inhibits the growth of HCV or an anti-HCV drug including an interferon preparation.
- anti-HCV activity is not known for triterpene derivatives derived from Soyasapogenol B other than 22 ⁇ -methoxyolean-12-ene-3 ⁇ , 24-diol.
- the present invention has been made in view of such circumstances, and an object thereof is to provide a novel triterpene derivative.
- an object of the present invention is to provide a triterpene derivative that exhibits stronger antiviral activity against chronic hepatitis C than conventional triterpene derivatives.
- the present invention aims to provide a triterpene derivative having good pharmacokinetics.
- Another object of the present invention is to provide a drug for the prevention or treatment of chronic hepatitis C, comprising these triterpene derivatives as active ingredients.
- the present inventors have succeeded in synthesizing novel triterpene derivatives having various substituents.
- the anti-HCV action of the synthesized triterpene derivative was investigated, a plurality of compounds showing stronger anti-HCV action than conventional 22 ⁇ -methoxyolean-12-ene-3 ⁇ , 24-diol were found. It was. In addition, many of these compounds also showed good pharmacokinetics. From such characteristics, the present inventors have found that the identified triterpene derivative can be an excellent preventive or therapeutic agent for chronic hepatitis C, and have completed the present invention.
- the present invention provides the following inventions.
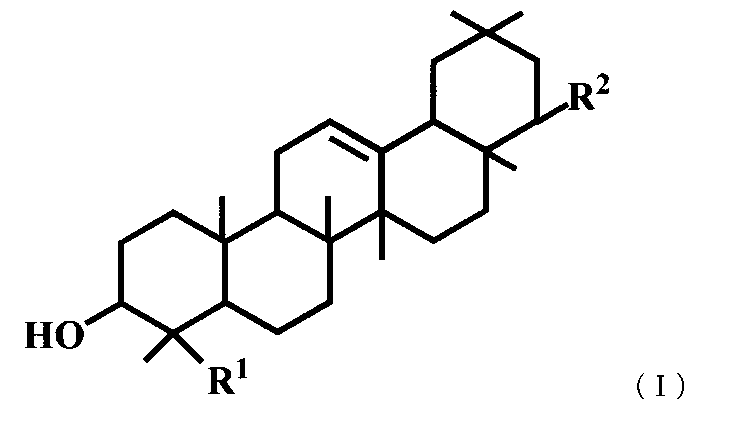
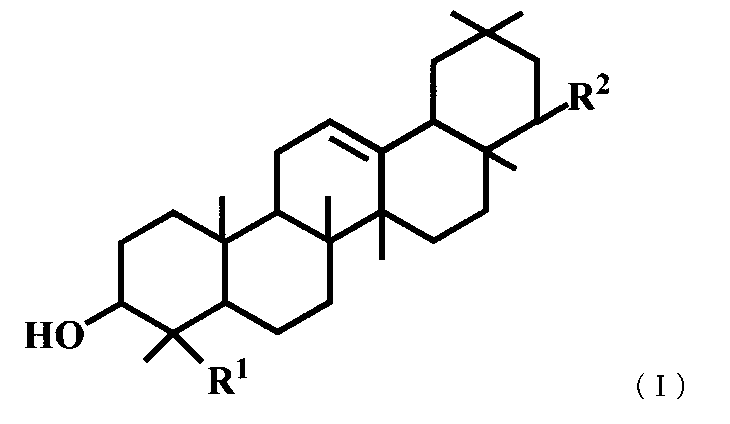
- a triterpene derivative represented by the general formula (I) or a pharmaceutically acceptable salt thereof represented by the general formula (I) or a pharmaceutically acceptable salt thereof.
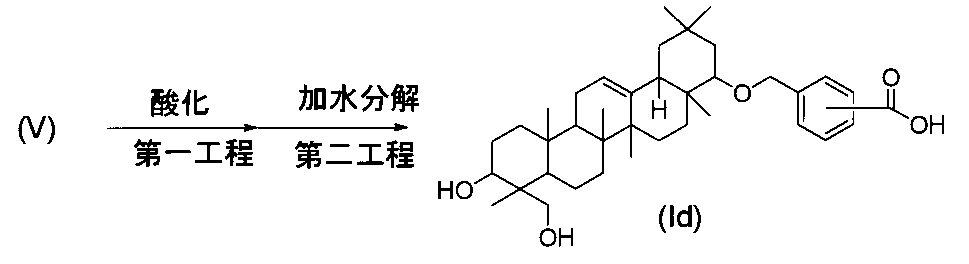
- R 1 represents a carboxyl group, a hydroxymethyl group, —CH 2 OSO 3 H, or
- R 2 represents —OR 3 , or —O— (CH 2 ) m—OR 4 .
- R 3 is a hydroxymethyl group, a dimethylaminomethyl group, a phenylaminomethyl group, a morpholinomethyl group, a benzyl group optionally substituted with a carboxyl group or a formyl group, a C 1-6 alkyl group, a C 2-6 Represents an alkenyl group, a C 2-6 alkynyl group, or a hydroxy C 1-6 alkyl group, R 4 represents a phenyl group which may be substituted with a carboxyl group, and m represents an integer of 1 to 3.
- R 1 is a hydroxymethyl group
- R 2 is —OR 3 and R 3 is a C 1-6 alkyl group, a C 2-6 alkenyl group or a benzyl group is excluded.
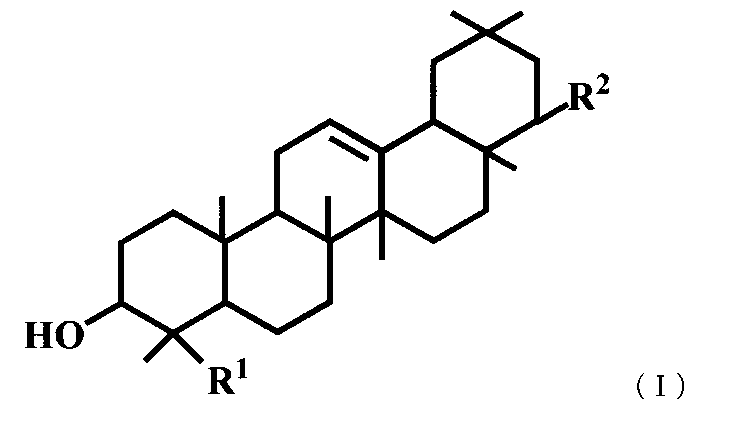
- R 1 represents a carboxyl group
- R 2 represents —OR 3
- R 3 represents a benzyl group, a methyl group or an allyl group.
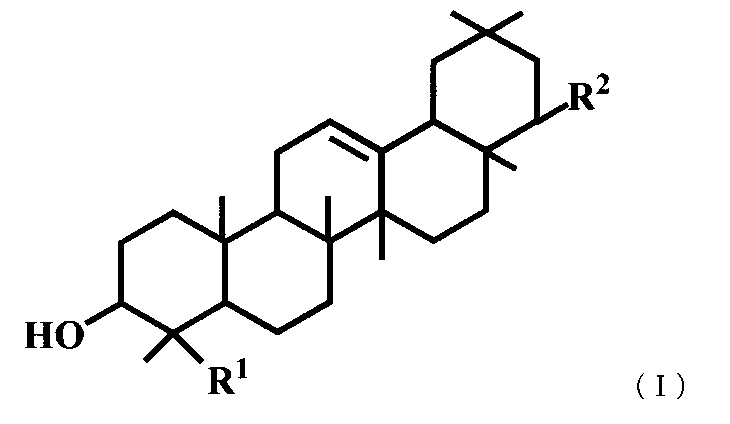
- R 1 represents a hydroxymethyl group
- R 2 represents —OR 3 or —O— (CH 2 ) 3 —OR 4 .
- R 3 is a hydroxymethyl group, a dimethylaminomethyl group, a phenylaminomethyl group, a morpholinomethyl group, a benzyl group substituted with a carboxyl group or a formyl group, a 2-propynyl group, a 3-hydroxypropyl group, or 2 Represents a hydroxypropyl group.
- R 4 represents a phenyl group substituted with a phenyl group or a carboxyl group.
- R 2 represents a methoxy group] (6)
- a drug for the prevention or treatment of chronic hepatitis C comprising as an active ingredient the triterpene derivative according to any one of (1) to (6) or a pharmaceutically acceptable salt thereof.
- (10) Type C comprising administering a prophylactic or therapeutically effective amount of the drug according to any one of (7) to (9) to a patient in need of prevention or treatment of chronic hepatitis C How to prevent or treat chronic hepatitis.
- the prophylactic or therapeutic agent of the present invention comprises a triterpene derivative represented by the general formula (I) or a pharmaceutically acceptable salt thereof as an active ingredient. Since these triterpene derivatives can exhibit excellent anti-HCV activity and excellent pharmacokinetics when administered in an effective amount, a high prophylactic effect or therapeutic effect can be expected for chronic hepatitis C. Further, the preventive agent or therapeutic agent of the present invention can be expected to have a better preventive effect or therapeutic effect even in combination with clinically used IFN or ribavirin, or various anti-HCV agents to be clinically used in the future.
- chronic hepatitis C refers to an inflammatory disease of the liver caused by continuous infection of the hepatitis C virus in the liver. means.
- the triterpene derivative of the present invention can exhibit excellent anti-HCV activity.
- the anti-HCV activity here mainly indicates an activity that inhibits the growth of HCV, and the smaller the concentration at which HCV is inhibited by 50% (IC50 value), the stronger the anti-HCV activity.
- IC50 value concentration at which HCV is inhibited by 50%
- a compound having an IC50 value exceeding 20 ⁇ M cannot be said to have strong anti-HCV activity, and the antiviral effect of a single agent in the prevention or treatment of chronic hepatitis C is considered to be small.
- the triterpene derivative of the present invention preferably has a strong anti-HCV activity, that is, an IC50 value of 20 ⁇ M or less (for example, 15 ⁇ M or less, 10 ⁇ M or less, 8 ⁇ M or less, 6 ⁇ M or less).
- a strong anti-HCV activity that is, an IC50 value of 20 ⁇ M or less (for example, 15 ⁇ M or less, 10 ⁇ M or less, 8 ⁇ M or less, 6 ⁇ M or less).
- those having particularly strong anti-HCV activity include compounds 2 to 10 and compounds 12 to 14 among the compounds shown in Table 1 shown later.
- the triterpene derivative of the present invention preferably has good pharmacokinetics, that is, AUC is 50 ⁇ g ⁇ hr / ml or more (for example, 60 ⁇ g ⁇ hr / ml or more, 70 ⁇ g ⁇ hr / ml or more, 80 ⁇ g ⁇ hr / ml). ml) or AUC of 30-50 ⁇ g ⁇ hr / ml and a half-life (t 1/2 ) of 10 hours or more (eg, 11 hours or more, 12 hours or more, 13 hours or more) is there.
- compounds 2, 12 to 14 are listed as compounds having favorable pharmacokinetics.
- compounds 2, 12 to 14 can be mentioned as compounds having particularly strong anti-HCV activity and good pharmacokinetics.
- Examples of the active ingredient in the prophylactic or therapeutic agent of the present invention include a triterpene derivative represented by the general formula (I) or a pharmaceutically acceptable salt thereof.
- a preferred embodiment of the triterpene derivative of the present invention is that in the formula (I), R 1 is a carboxyl group, R 2 represents —OR 3 , and R 3 represents a hydroxymethyl group, a dimethylaminomethyl group, a phenylaminomethyl group A morpholinomethyl group, a benzyl group, a C 1-6 alkyl group, a C 2-6 alkenyl group, a C 2-6 alkynyl group, or a hydroxy C 1-6 alkyl group which may be substituted with a carboxyl group or a formyl group. It is a compound to represent.
- the “benzyl group optionally substituted with a hydroxymethyl group, dimethylaminomethyl group, phenylaminomethyl group, morpholinomethyl group, carboxyl group or formyl group” represented by R 3 is substituted with each group.
- the substitution position may be any position, but a benzyl group substituted at the 4-position with a hydroxymethyl group, a dimethylaminomethyl group, a phenylaminomethyl group, a morpholinomethyl group, a carboxyl group, or a formyl group is preferable.
- the “C 1-6 alkyl group” represented by R 3 means a linear or branched alkyl group having 1 to 6, preferably 1 to 4 carbon atoms.
- the “C 2-6 alkenyl group” represented by R 3 means a linear or branched alkenyl group having 2 to 6, preferably 2 to 4 carbon atoms.
- the “C 2-6 alkynyl group” represented by R 3 means a linear or branched alkynyl group having 2 to 6, preferably 2 to 4 carbon atoms.
- Specific examples thereof include 2-propynyl group, 1-methyl-2-propynyl group, 2-butynyl group, 3-butynyl group and the like. Of these, a 2-propynyl group is preferable.
- hydroxy C 1-6 alkyl group represented by R 3 may be substituted with any hydroxyl group, and specific examples thereof include a hydroxymethyl group, a hydroxyethyl group, a 2-hydroxypropyl group, A 3-hydroxypropyl group and the like can be mentioned. Among these, 2-hydroxypropyl group or 3-hydroxypropyl group is preferable.
- R 1 is a carboxyl group
- R 2 is —O— (CH 2 ) m—OR 4 (m is an integer of 1 to 3).
- R 4 is a compound representing a phenyl group optionally substituted with a carboxyl group.
- the “phenyl group optionally substituted with a carboxyl group” represented by R 4 may be substituted at any position. Although it may be a position, a phenyl group or a phenyl group substituted at the 4-position with a carboxyl group is preferable.
- a preferred integer m is 3.
- R 1 is a hydroxymethyl group
- R 2 represents —OR 3
- R 3 is a hydroxymethyl group, a dimethylaminomethyl group, a phenyl
- It is a compound representing a benzyl group, a C 2-6 alkynyl group or a hydroxy C 1-6 alkyl group which may be substituted with an aminomethyl group, a morpholinomethyl group, a carboxyl group or a formyl group.
- the “benzyl group optionally substituted with a hydroxymethyl group, a dimethylaminomethyl group, a phenylaminomethyl group, a morpholinomethyl group, a carboxyl group or a formyl group” represented by R 3 can be substituted at any position.
- a benzyl group substituted at the 4-position with a hydroxymethyl group, a dimethylaminomethyl group, a phenylaminomethyl group, a morpholinomethyl group, a carboxyl group or a formyl group is preferred.
- the “C 2-6 alkynyl group” represented by R 3 means a linear or branched alkynyl group having 2 to 6 carbon atoms, preferably 2 to 4 carbon atoms.
- hydroxy C 1-6 alkyl group represented by R 3 may be substituted with any hydroxyl group, and specific examples thereof include a hydroxymethyl group, a hydroxyethyl group, a 2-hydroxypropyl group, A 3-hydroxypropyl group and the like can be mentioned. Among these, 2-hydroxypropyl group or 3-hydroxypropyl group is preferable.
- R 1 is a hydroxymethyl group
- R 2 is —OR 3
- R 3 is a C 1-6 alkyl group, a C 2-6 alkenyl group or a benzyl group Is excluded.
- R 1 is a hydroxymethyl group and R 2 is —O— (CH 2 ) m—OR 4 (m is an integer of 1 to 3)
- R 4 represents a phenyl group which may be substituted with a carboxyl group.
- —O— (CH 2 ) m—OR 4 represented by R 2 the “phenyl group optionally substituted with a carboxyl group” represented by R 4 may be substituted at any position. It may be a position.
- Preferred is a phenyl group or a phenyl group substituted at the 4-position with a carboxyl group.
- a preferred integer m is 3.
- R 1 is —CH 2 OSO 3 H
- R 2 represents —OR 3
- R 3 represents a hydroxymethyl group, dimethylaminomethyl Group, phenylaminomethyl group, morpholinomethyl group, benzyl group optionally substituted by carboxyl group or formyl group, C 1-6 alkyl group, C 2-6 alkenyl group, C 2-6 alkynyl group or hydroxy C 1 It is a compound representing a -6 alkyl group.
- the “benzyl group optionally substituted with a hydroxymethyl group, dimethylaminomethyl group, phenylaminomethyl group, morpholinomethyl group, carboxyl group or formyl group” represented by R 3 is substituted with each group.
- the substitution position may be any position, but a benzyl group substituted at the 4-position with a hydroxymethyl group, dimethylaminomethyl group, phenylaminomethyl group, morpholinomethyl group, carboxyl group or formyl group is preferred.
- the “C 1-6 alkyl group” represented by R 3 means a linear or branched alkyl group having 1 to 6, preferably 1 to 4 carbon atoms.
- the “C 2-6 alkenyl group” represented by R 3 means a linear or branched alkenyl group having 2 to 6, preferably 2 to 4 carbon atoms.
- the “C 2-6 alkynyl group” represented by R 3 means a linear or branched alkynyl group having 2 to 6 carbon atoms, preferably 2 to 4 carbon atoms.
- Specific examples thereof include 2-propynyl group, 1-methyl-2-propynyl group, 2-butynyl group, 3-butynyl group and the like. Of these, a 2-propynyl group is preferable.
- hydroxy C 1-6 alkyl group represented by R 3 may be substituted with any hydroxyl group, and specific examples thereof include a hydroxymethyl group, a hydroxyethyl group, a 2-hydroxypropyl group, A 3-hydroxypropyl group and the like can be mentioned. Among these, 2-hydroxypropyl group or 3-hydroxypropyl group is preferable.
- R 1 is —CH 2 OSO 3 H and R 2 is —O— (CH 2 ) m—OR 4 (m is 1 to R 4 represents a phenyl group which may be substituted with a carboxyl group.
- m is 1 to R 4 represents a phenyl group which may be substituted with a carboxyl group.
- the “phenyl group optionally substituted with a carboxyl group” represented by R 4 may be substituted at any position. It may be a position. Preferably, it may be a phenyl group or a phenyl group substituted at the 4-position with a carboxyl group.
- a preferred integer m is 3.
- R 2 represents —OR 3
- R 3 represents a benzyl group optionally substituted with a hydroxymethyl group, a dimethylaminomethyl group, a phenylaminomethyl group, a morpholinomethyl group, a carboxyl group or a formyl group
- the “benzyl group optionally substituted with a hydroxymethyl group, dimethylaminomethyl group, phenylaminomethyl group, morpholinomethyl group, carboxyl group or formyl group” represented by R 3 is substituted with each group.
- the substitution position may be any position, but a benzyl group substituted at the 4-position with a hydroxymethyl group, dimethylaminomethyl group, phenylaminomethyl group, morpholinomethyl group, carboxyl group or formyl group is preferred.
- the “C 1-6 alkyl group” represented by R 3 means a linear or branched alkyl group having 1 to 6, preferably 1 to 4 carbon atoms. Specific examples thereof include methyl group, ethyl group, propyl group, isopropyl group, n-propyl group, n-butyl group, isobutyl group, t-butyl group and the like. Among them, a methyl group and an ethyl group are preferable.
- the “C 2-6 alkenyl group” represented by R 3 means a linear or branched alkenyl group having 2 to 6, preferably 2 to 4 carbon atoms. Specific examples thereof include vinyl group, allyl group, 1-propenyl group, isopropenyl group, 1-butenyl group, 2-butenyl group, 3-butenyl group, 2-methyl-1-propenyl group and the like. Among these, an allyl group is preferable.
- the “C 2-6 alkynyl group” represented by R 3 means a linear or branched alkynyl group having 2 to 6 carbon atoms, preferably 2 to 4 carbon atoms.
- hydroxy C 1-6 alkyl group represented by R 3 may be substituted with any hydroxyl group, and specific examples thereof include a hydroxymethyl group, a hydroxyethyl group, a 2-hydroxypropyl group, A 3-hydroxypropyl group and the like can be mentioned. Among them, preferred is a 2-hydroxypropyl group or a 3-hydroxypropyl group.
- R 2 represents —O— (CH 2 ) m—OR 4 (m represents an integer of 1 to 3), and R 4 represents a phenyl group which may be substituted with a carboxyl group. is there.
- R 4 represents a phenyl group which may be substituted with a carboxyl group. is there.
- the “phenyl group optionally substituted with a carboxyl group” represented by R 4 may be substituted at any position. It may be a position. Preferably, it may be a phenyl group or a phenyl group substituted at the 4-position with a carboxyl group.
- a preferred integer m is 3.
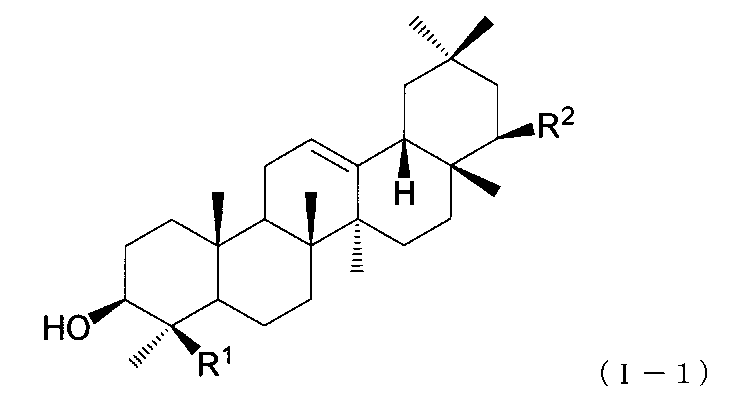
- the triterpene derivative of the present invention preferably has a configuration represented by the following formula (I-1).
- the definitions of R 1 and R 2 in formula (I-1) are the same as described above.
- triterpene derivative of the formula (I) in the present invention include compounds having combinations of substituents shown in Table 1, but the present invention is not limited to these compounds.
- a preferred method for producing the triterpene derivative represented by the general formula (I) of the present invention is as follows. In the following production method, it is desirable that a functional group not involved in the reaction is protected, and a known protective group for protecting the functional group can be used. These facts will be apparent to those skilled in the art.
- a compound of formula (II) (which can be synthesized by the method described in Chem. Pharm. Bull., 36.153 (1988)) and a compound of formula (III) (wherein X and Y Are halogen atoms, which may be the same or different, in the presence of a base to produce a compound of formula (IV).
- the reaction is carried out in a solvent that does not participate in the reaction (for example, chloroform, dichloromethane, diethyl ether, THF, benzene, toluene, DMF, DMSO or the like alone or in a mixed solvent) at a temperature in the range of ⁇ 78 ° C. to 100 ° C.
- the base examples include pyridine, triethylamine, 4-dimethylaminopyridine, sodium hydride, potassium hydride, n-butyllithium, NaCH 2 SOCH 3 , tert-BuOK, tert-BuONa, and the like.
- the base and the compound of the formula (III) are desirably used in the range of 1 to 10 equivalents relative to the compound of the formula (II).
- the compound of formula (IV) is treated with an appropriate base to perform a halogen-metal exchange reaction, and then reacted with DMF to produce compound (V).
- the reaction is carried out in a solvent not involved in the reaction (for example, tetrahydrofuran, diethyl ether, 1,4-dioxane, anisole, dimethoxyethane, dichloromethane, toluene or the like alone or in a mixed solvent) at a temperature in the range of ⁇ 78 ° C. to 30 ° C. To be implemented.
- Examples of the base that can be used include n-butyllithium, sec-butyllithium, tert-butyllithium, ethylmagnesium bromide, isopropylmagnesium bromide and the like.
- the base and DMF are desirably used in the range of 1 to 10 equivalents relative to the compound of formula (IV).
- the compound represented by formula (Ia) can be produced by hydrolyzing the compound represented by formula (V) in the presence of an acid.
- the solvent used in this reaction include methanol, ethanol, propanol, water, dichloromethane, chloroform, THF alone or a mixed solvent.
- the acid include mineral acids such as hydrochloric acid and sulfuric acid, or Lewis acids such as BF 3 ⁇ OEt 2 . The reaction is carried out at a temperature ranging from 0 ° C to 100 ° C.
- Examples of the solvent used in the first step include methanol, ethanol, THF, chloroform, dichloromethane and the like alone or a mixed solvent.
- the reaction is carried out at a temperature in the range of ⁇ 40 ° C. to 30 ° C.
- Examples of the reducing agent include lithium aluminum hydride, sodium borohydride, lithium borohydride and the like, and it is desirable to use in the range of 1 to 10 equivalents relative to the compound of formula (V).
- the second step can be performed according to the method described in the third step of the method (A).
- a compound of the formula (Ic) (wherein R 3c is benzyl substituted with a dimethylaminomethyl group, a phenylaminomethyl group or a morpholinomethyl group)
- R 3c is benzyl substituted with a dimethylaminomethyl group, a phenylaminomethyl group or a morpholinomethyl group
- a compound of formula (V) and a compound of formula (VI) are subjected to conventional reductive aminoalkylation.
- the reaction is carried out according to the method of reaction.
- One equivalent or excess amount of the compound of formula (VI) is used relative to the compound of formula (V), and in the presence of one equivalent or excess amount of acid (eg, acetic acid, trifluoroacetic acid, etc.) , 1,2-dichloroethane, dichloromethane, etc.) at 0 ° C. to 50 ° C.
- the second step can be performed according to the method described in the third step of the method (A).
- Examples of the oxidizing agent that can be used in the oxidation reaction in the first step include pyridinium dichromate, Jones reagent, potassium permanganate, sodium chlorite, and the like, and 1 to 30 equivalents of the compound of formula (V). It is preferably used in a range.
- the oxidation reaction is carried out in a solvent that does not participate in the reaction (eg, DMF, tert-butanol, acetone, water, etc.) at a temperature in the range of 0 ° C. to 60 ° C.
- the second step can be performed according to the method described in the third step of the method (A).
- a compound of formula (II) and a compound of formula (VII): R 3e Y (wherein R 3e is as defined above and Y is a halogen atom) are present in the presence of a base.
- the compound of formula (VIII) is prepared by reacting under: The reaction is carried out in a solvent that does not participate in the reaction (for example, chloroform, dichloromethane, diethyl ether, THF, benzene, toluene, DMF, DMSO, etc. alone or in a mixed solvent) at a temperature in the range of ⁇ 78 ° C. to 100 ° C.
- the base examples include pyridine, triethylamine, 4-dimethylaminopyridine, sodium hydride, potassium hydride, n-butyllithium, NaCH 2 SOCH 3 , tert-BuOK, tert-BuONa, and the like.
- the base and the compound of formula (VII) are desirably used in an amount of 1 to 10 equivalents relative to the compound of formula (II).
- the second step can be performed according to the method described in the third step of the method (A).
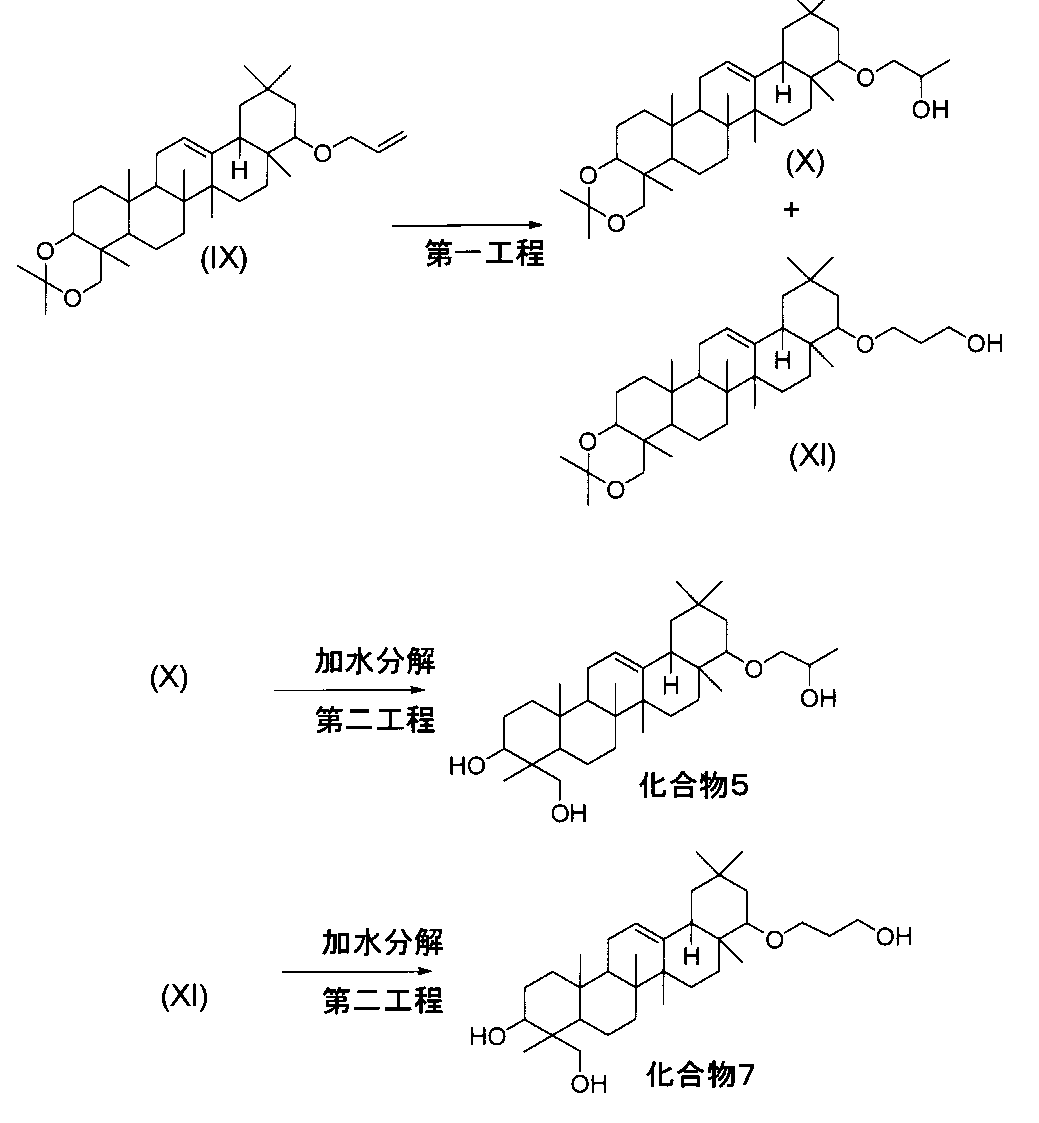
- a compound of the formula (IX) (which can be synthesized by the method described in the specification of Japanese Patent No. 3279574) is reacted with a reagent for hydroboration reaction, and then oxidized, Compounds of formula (X) and formula (XI) are obtained.
- the hydroboration reagent include BH 3 -THF, texylborane, 9-borabicyclo (3,3,1) nonane. This reaction reagent is preferably used in the range of 1 to 10 equivalents relative to the compound of formula (IX).
- the reaction is carried out in a solvent that does not participate in the reaction (eg, diethyl ether, THF, etc.) at a temperature in the range of 0 ° C. to 50 ° C.
- the oxidation reaction is performed at a temperature in the range of 0 ° C. to 40 ° C. by adding an oxidizing agent (for example, sodium hydroxide, 30% hydrogen peroxide solution) to the reaction solution.
- the second step can be performed according to the method described in the third step of the method (A) ⁇ Method (G)>.
- the compound of the formula (Ig) (wherein R 4 and m are as defined above) can be produced by the following method.
- 1 equivalent or excess amount of the compound of formula (XIII) is used with respect to the compound of formula (XII), and 1 equivalent or excess amount of phosphine reagent (for example, triphenylphosphine) and 1 equivalent or excess amount of azo system
- a reagent eg, diethyl azodicarboxylate, 1,1 ′-(azodicarbonyl) dipiperidine, etc.
- a solvent that does not participate in the reaction eg, THF, dichloromethane, toluene, benzene, etc.
- the second step can be performed according to the method described in the third step of the method (A) when R 7 is a hydrogen atom.
- R 7 is a methyl ester of a carboxylic acid
- in the second step first, a hydrolysis reaction of the methyl ester portion with a base is performed, followed by a hydrolysis reaction in the presence of an acid.
- the hydrolysis reaction of the methyl ester with the base is carried out by adding 1 equivalent to an excessive amount of base (for example, sodium hydroxide, potassium hydroxide, lithium hydroxide, barium hydroxide, tert-BuOK, etc.) with a polar solvent (for example, , Methanol, ethanol, propanol, water or the like alone or in a mixed solvent) at a temperature ranging from 0 ° C. to 100 ° C.
- a polar solvent for example, Methanol, ethanol, propanol, water or the like alone or in a mixed solvent
- the subsequent hydrolysis reaction in the presence of an acid is performed in one reaction in a reaction system in which the hydrolysis of the methyl ester is completed (mineral acid such as hydrochloric acid and sulfuric acid, or Lewis acid such as BF 3 ⁇ OEt 2 ). By adding until the reaction system is acidic. In this case, the reaction is carried out at a temperature ranging from 0 ° C to 100 ° C.
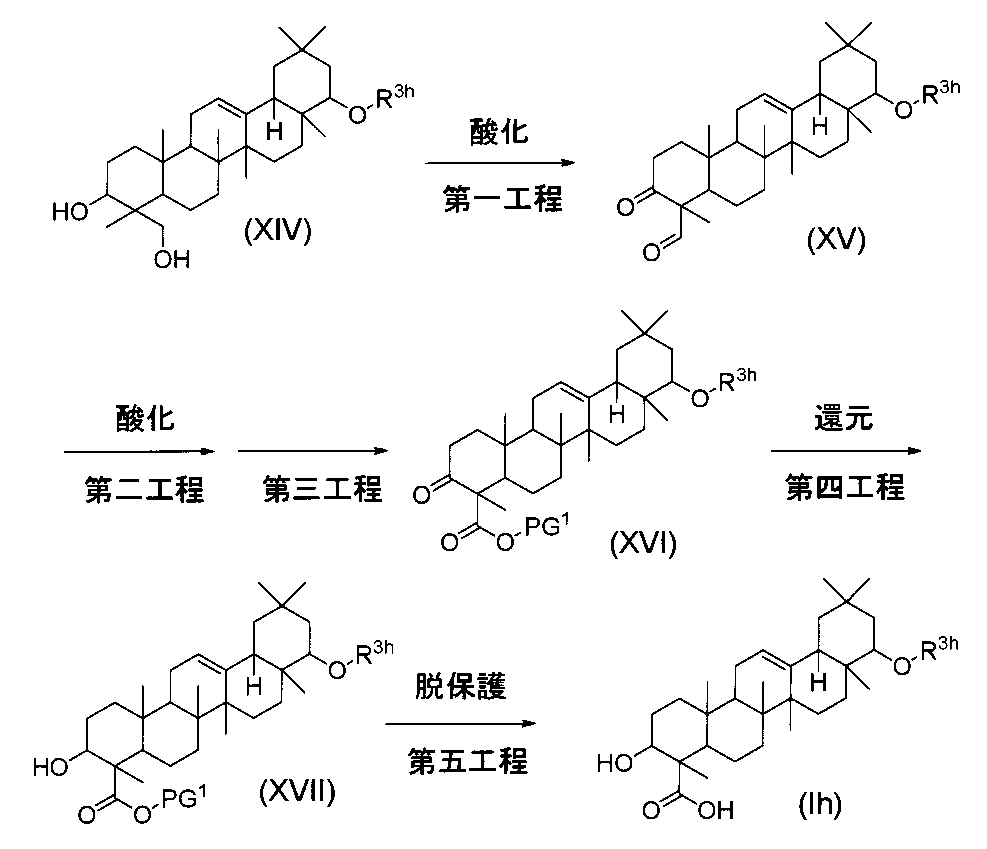
- a compound of the formula (XIV) (synthesized by the method described in the specification of Japanese Patent No. 3279574, the specification of Japanese Patent No. 3727353, or Bioorganic & Medicinal Chemistry, 13, 4900 (2005), etc. Possible) is converted to the compound of formula (XV) by oxidation.
- Usable oxidizing agents are (1) pyridinium chloroformate, (2) pyridinium dichromate, (3) manganese dioxide, (4) tetra-n-propylammonium perruthenate and N-methylmorpholine-N-oxide.
- DMSO oxidation reagents such as a combination of dimethyl sulfoxide (DMSO) and oxalyl chloride, and the like.
- the oxidizing agent is preferably used in the range of 2-10 equivalents relative to the compound of formula (XIV).
- the oxidation reaction can be carried out in a solvent that does not participate in the reaction (for example, dichloromethane, chloroform, diethyl ether, THF, etc.) at a temperature in the range of ⁇ 78 ° C. to 40 ° C.
- the resulting carboxylic acid is protected with a usual protecting group in the third step, whereby the compound of formula (XVI) (wherein PG 1 Represents a normal protecting group for a carboxyl group, where the normal protecting group for a carboxyl group is Protective Groups in Organic Synthesis (Theodora W. Greene, Peter GM Wuts, John Wiley & Sons, Inc.). And is well known to those skilled in the art. More preferably, PG 1 includes methyl, ethyl, benzyl groups, etc.).
- Examples of the oxidizing agent that can be used in the oxidation reaction in the second step include pyridinium dichromate, Jones reagent, potassium permanganate, sodium chlorite, and the like, and 1 to 30 equivalents of the compound of formula (XV). It is preferably used in a range.
- the oxidation reaction is carried out in a solvent that does not participate in the reaction (for example, DMF, tert-butanol, acetone, water or the like alone or in a mixed solvent) at a temperature ranging from 0 ° C to 60 ° C.
- the conditions for protecting the carboxylic acid in the third step vary depending on the type of protecting group used.
- PG 1 is methyl, ethyl, or benzyl
- 1 to 10 equivalents of PG 1 Y (where Y is a halogen atom) with respect to the compound of formula (XV) is used.
- Room temperature to 100 ° C. in a solvent that does not participate in the reaction eg, toluene, benzene, THF, DMF, DMSO, etc.
- an excess amount of base eg, potassium carbonate, sodium carbonate, cesium carbonate, sodium hydride, etc. It can implement by making it react at the temperature of the range of these.
- the formula (XVI) is reduced by a normal reduction reaction and converted into a compound of the formula (XVII).
- the solvent used in the reduction reaction include methanol, ethanol, THF, chloroform, dichloromethane, and the like alone or a mixed solvent.
- the reaction is carried out at a temperature in the range of ⁇ 78 ° C. to 30 ° C.
- the reducing agent include lithium aluminum hydride, sodium borohydride, lithium borohydride, diisobutylaluminum hydride and the like, and should be used in the range of 1 to 10 equivalents relative to the compound of formula (XVI). Is desirable.
- PG 1 of the compound of the formula (XVII) is removed by the usual conditions for deprotecting the protecting group of the carboxyl group to produce the compound of the formula (Ih).
- the deprotection conditions for removing the protecting group vary depending on the kind of the protecting group used.
- a base eg, sodium hydroxide, potassium hydroxide, lithium hydroxide, barium hydroxide, tert-BuOK, etc.
- a polar solvent for example, methanol, ethanol, propanol, water or the like alone or in a mixed solvent
- a polar solvent for example, methanol, ethanol, propanol, water or the like alone or in a mixed solvent
- PG 1 is benzyl
- it can be carried out by catalytic reduction using 0.1 to 0.5 equivalents of a catalyst (for example, palladium carbon, palladium black, palladium hydroxide, etc.).
- the reaction can be carried out in a solvent that does not participate in the reaction (for example, methanol, ethanol, THF, dioxane, dichloromethane, chloroform, water) under a hydrogen atmosphere of usually 1 to 4 atm at room temperature.
- the compound of formula (XVIII) is oxidized and converted to the compound of formula (XIX) by the method described in the first step of method (H).
- PG 2 represents a normal protecting group for a hydroxyl group.
- the “ordinary protecting group for a hydroxyl group” is the one described in Protective Groups in Organic Synthesis (Theodora W. Greene, Peter GM Wuts, John Wiley & Sons, Inc.) and is well known to those skilled in the art. It is.
- PG 2 is preferably benzyl.
- the compound of the formula (XVIII) can be synthesized by the method described in the specification of Japanese Patent No. 3279574, the specification of Japanese Patent No. 3727353, or Bioorganic & Medicinal Chemistry, 13, 4900 (2005).
- the compound is oxidized and converted to the compound of formula (XX) by the method described in the second step of method (H).
- the protecting group is removed under the usual conditions for deprotecting the protecting group of the hydroxyl group to produce the compound of formula (Ii).
- the deprotection conditions for removing the hydroxyl protecting group in the third step vary depending on the type of protecting group used. For example, when PG 2 is benzyl, deprotection can be carried out by catalytic reduction using 0.1 to 0.5 equivalents of a catalyst (eg, palladium carbon, palladium black, palladium hydroxide, etc.). .
- the reaction can be carried out in a solvent that does not participate in the reaction (for example, methanol, ethanol, THF, dioxane, dichloromethane, chloroform, water) under a hydrogen atmosphere of usually 1 to 4 atm at room temperature.
- a solvent that does not participate in the reaction for example, methanol, ethanol, THF, dioxane, dichloromethane, chloroform, water
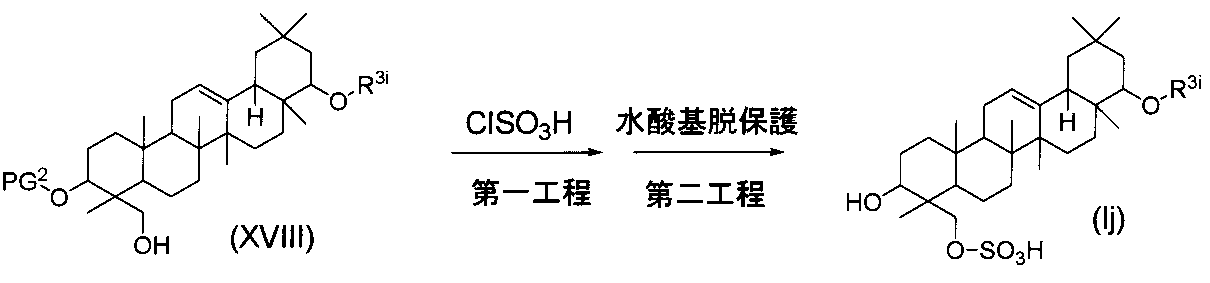
- a compound of formula (XVIII) (wherein PG 2 is as defined above, preferably benzyl in this reaction) and 1 equivalent or an excess amount of chlorosulfonic acid are reacted.
- the reaction is carried out at a temperature in the range of ⁇ 20 ° C. to 50 ° C. in a non-participating solvent (eg THF, dioxane, dichloromethane, toluene, benzene, etc.).
- a non-participating solvent eg THF, dioxane, dichloromethane, toluene, benzene, etc.
- the protecting group is removed under the usual conditions for deprotecting the protecting group of the hydroxyl group to produce the compound of formula (Ij).
- the deprotection conditions for removing the hydroxyl protecting group in the second step vary depending on the type of the protecting group used. For example, when PG 2 is benzyl, deprotection can be carried out by catalytic reduction using 0.1 to 0.5 equivalents of a catalyst (eg, palladium carbon, palladium black, palladium hydroxide, etc.). .
- the reaction may be carried out in a solvent that does not participate in the reaction (eg, methanol, ethanol, THF, dioxane, dichloromethane, chloroform, water, etc., alone or in a mixed solvent) at room temperature under a hydrogen atmosphere of usually 1 to 4 atm. it can.
- a solvent that does not participate in the reaction eg, methanol, ethanol, THF, dioxane, dichloromethane, chloroform, water, etc., alone or in a mixed solvent
- a compound of formula (XVIII) (wherein PG 2 is as defined above and preferably benzyl in this reaction) is converted to a compound of formula (XXI) under the usual glycosylation reaction conditions.
- This first step uses one equivalent or an excess of a compound of formula (XXI) relative to the compound of formula (XVIII) and uses one equivalent or an excess of a bromosugar activator (eg, silver triflate, silver carbonate) Etc.) in the presence of an inert solvent (for example, 1,2-dichloroethane, dichloromethane, benzene, toluene, etc.) at a temperature ranging from 0 ° C. to 100 ° C.
- an inert solvent for example, 1,2-dichloroethane, dichloromethane, benzene, toluene, etc.
- a hydrolysis reaction of the acetyl group and the methyl ester portion is performed using a base.
- This second step is carried out in the presence of 1 equivalent to an excess of a base (eg, sodium hydroxide, potassium hydroxide, lithium hydroxide, barium hydroxide, tert-BuOK, etc.) in a polar solvent (eg, methanol, ethanol, It is carried out by reacting at a temperature in the range of 0 ° C. to 100 ° C. in a single or mixed solvent such as propanol or water.
- a base eg, sodium hydroxide, potassium hydroxide, lithium hydroxide, barium hydroxide, tert-BuOK, etc.
- a polar solvent eg, methanol, ethanol, It is carried out by reacting at a temperature in the range of 0 ° C. to 100 ° C. in a single or mixed solvent such as propanol or water.
- the protecting group is removed under the usual conditions for deprotecting the protecting group for the hydroxyl group to produce the compound of formula (Ik).
- the deprotection conditions for removing the hydroxyl protecting group in the third step vary depending on the type of the protecting group used. For example, when PG 2 is benzyl, deprotection can be carried out by catalytic reduction using 0.1 to 0.5 equivalents of a catalyst (eg, palladium carbon, palladium black, palladium hydroxide, etc.). .
- the reaction can be carried out in a solvent that does not participate in the reaction (for example, methanol, ethanol, THF, dioxane, dichloromethane, chloroform, water) under a hydrogen atmosphere of usually 1 to 4 atm at room temperature.
- a solvent that does not participate in the reaction for example, methanol, ethanol, THF, dioxane, dichloromethane, chloroform, water
- the triterpene derivative of the present invention represented by the above (I) can exist as a salt.
- the triterpene derivative of the present invention can be easily converted into a salt by allowing a pharmaceutically acceptable base to act according to a usual method.
- a pharmaceutically acceptable base for example, an inorganic base such as sodium hydroxide, potassium hydroxide, aluminum hydroxide, sodium carbonate, potassium carbonate, or sodium bicarbonate, or an organic base such as piperazine, morpholine, piperidine, ethylamine, or trimethylamine should be used. Can do.
- the triterpene derivative of the present invention can be used as a prophylactic or therapeutic agent for chronic hepatitis C.
- the prophylactic or therapeutic agent of the present invention can be orally administered as a dosage form such as capsule, microcapsule, tablet, granule, fine granule, powder and the like. It can also be administered parenterally (for example, intravenous injection, intramuscular injection, subcutaneous administration, intraperitoneal administration, rectal administration, transdermal administration) in the form of a conventional pharmaceutical preparation.
- a pharmaceutically acceptable carrier can be added to the above dosage form.
- Pharmaceutically acceptable carriers include, for example, excipients, extenders, binders, wetting agents, disintegrants, surfactants, lubricants, dispersants, buffers, preservatives, solubilizers, preservatives And pharmaceutical additives such as agents, flavoring agents, soothing agents, and stabilizers.
- Each dosage form can be manufactured by a conventional method.
- Specific additives include, for example, lactose, fructose, glucose, starch, gelatin, magnesium carbonate, synthetic magnesium silicate, talc, magnesium stearate, crystalline cellulose, methylcellulose, carboxymethylcellulose or a salt thereof, gum arabic, polyethylene glycol Syrup, petrolatum, glycerin, ethanol, propylene glycol, citric acid, sodium chloride, sodium sulfite, sodium phosphate and the like.
- the dosage form, administration method, dosage, administration period, administration route, etc. of the preventive or therapeutic agent of the present invention can be appropriately set depending on, for example, the patient's weight, age, degree of symptoms, viral load in the patient, etc. .
- the prophylactic or therapeutic agent of the present invention is administered orally or parenterally, for example, in an amount of 1 to 1000 mg per day in one or more divided doses.
- the dose is preferably 5 to 500 mg per day.
- the prophylactic or therapeutic agent of the present invention can be used in combination (that is, used in combination) with other drugs having an antiviral effect such as IFN or ribavirin in some cases.
- the combination of the prophylactic or therapeutic agent of the present invention and another drug may be in the form of a single preparation, or may be a one-drug drug containing the active ingredients of both drugs.
- each drug can be administered at the same time or with a time difference, and the number of administrations of each drug may be the same or different.
- IFN ⁇ Natural type IFN ⁇ (Sumiferon: manufactured by Dainippon Sumitomo Pharma Co., Ltd.), IFN ⁇ -2a, IFN ⁇ -2b (Intron A: manufactured by Schering-Plough) ), Polyethylene glycol (PEG) natural IFN ⁇ , PEGylated IFN ⁇ -2a (Pegasys: Roche, Chugai Pharmaceutical), PEGylated IFN ⁇ -2b (Pegintron A: Schering-Plough), natural IFN ⁇ ( IFN ⁇ Mochida: Mochida Pharmaceutical Co., Ltd., Feron: Toray Industries, Inc.), PEGylated IFN ⁇ , natural IFN ⁇ , consensus IFN (Advaferon: manufactured by Astellas Pharma Inc.) or PEGylated consensus IFN, long-term IFN, etc. IFN derivatives that can be used, but are not limited thereto. Preferably, it is IFN ⁇ 2b or I
- IFN is usually administered subcutaneously, intravenously, or intramuscularly.
- parenteral methods eg, by nasal spray, transdermal, suppository, etc.
- Oral administration is also conceivable when using IFN that is effective by an oral method, such as oral IFN.
- the ratio of each active ingredient (use ratio or combination ratio) in the combination drug is not particularly limited as long as it is effective for the prevention or treatment of chronic hepatitis C cases.
- the dose of the triterpene derivative of the present invention is appropriately determined from the range of 1 mg to 1000 mg per day for the effective dose of IFN.
- the dose of the triterpene derivative of the present invention is preferably in the range of 5 mg to 500 mg per day. Appropriate doses and intervals in the combination of the prophylactic or therapeutic agent of the present invention and other drugs can be determined by controlled clinical trials.
- the aqueous layer was re-extracted with chloroform, the combined organic layers were dried over anhydrous magnesium sulfate, the inorganic salt was filtered off, and the solvent was concentrated under reduced pressure.
- the obtained residue was dissolved in a mixed solution of 5 ml of THF and 1 ml of water, and 0.3 ml of 1N hydrochloric acid aqueous solution was added. The solution was stirred at room temperature for 30 minutes, diluted with saturated brine, and extracted three times with chloroform. The combined organic layers were dried over anhydrous magnesium sulfate, the inorganic salt was filtered off, and the solvent was concentrated under reduced pressure.
- the obtained residue was dissolved in 8.5 ml of methylene chloride, 521 mg of molecular sieves, 408 mg of 4-methylmorpholine N-oxide, and 30 mg of tetra-N-propylammonium perruthenate were added and stirred at room temperature for 20 minutes.
- the aldehyde was dissolved in 4.3 ml of THF and 4.3 ml of 2-methylpropanol, and then 0.45 ml of 2-methyl-2-butene was added.
- a solution of sodium dihydrogen phosphate dihydrate 210 mg and sodium chlorite 147 mg in 2.1 ml of water was added, and the mixture was stirred at room temperature for 40 minutes. Further, 100 mg of sodium dihydrogenphosphate dihydrate and 72 ml of sodium chlorite in 1 ml of water were added, and the mixture was stirred at room temperature for 20 minutes.
- the reaction was stopped with a saturated aqueous sodium thiosulfate solution, and the reaction solution was diluted with dilute hydrochloric acid and ethyl acetate.
- Step 2 551 mg (98%) of the title compound of Step 2 was obtained by a method similar to Step 2 of Example 12 using 557 mg of 22 ⁇ -allyloxy-3-oxoolean-12-ene-24-oic acid methyl ester. 1 H-NMR (CDCl 3 ) ...
- the obtained residue was dissolved in 450 ml of DMF and heated to 45 ° C. After adding 5.0 g of 60% sodium hydride and stirring for 1 hour, 22 g of benzyl bromide was added and stirred for 16 hours. After cooling to room temperature, 2 L of ethyl acetate and 3 L of water were added and the phases were separated. The organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the inorganic salt was filtered off, and the solvent was concentrated under reduced pressure.
- the palladium catalyst was removed by filtration through Celite, and the solvent was concentrated under reduced pressure.
- the obtained crude crystals were washed with water, and the crystals separated by filtration were dissolved in a mixed solution of methanol and THF. Insoluble matters were filtered off and concentrated under reduced pressure.
- the obtained crystal was slurry washed with ethyl acetate and then dried under reduced pressure to obtain 3.35 g (33%) of Compound 15.
- Acetic acid was added to 1.08 g of 2,3,4-tri- O -acetyl-1- O- (3 ⁇ -benzyloxy-22 ⁇ -methoxyolean-12-en-24-yl) - ⁇ -D-glucuronic acid methyl ester.
- the mixture was dissolved in a mixed solvent of 25 ml of ethyl and 16 ml of ethanol, 0.2 g of palladium hydroxide / C was added, and the mixture was stirred at room temperature for 3 hours in a hydrogen atmosphere.
- the catalyst was filtered off, and the filtrate was concentrated under reduced pressure.
- a luciferase gene was introduced as a reporter gene in the HCV genomic gene sequence of Con1 strain. According to the method of Krieger et al. (J. Virol. 75: 4614 (2001)), a luciferase gene was introduced immediately after the IRES (Internal Ribosome Entry Site) of the HCV gene in a form fused with a neomycin resistance gene. The constructed RNA was introduced into Huh7 cells by electroporation and isolated as a G418 resistant clone.
- HCV replicon cells containing a luciferase gene and a neomycin resistance gene are suspended in a culture solution [D-MEM (SIGMA, D6046) containing 10% fetal bovine serum (EQUITECH-BIO, INC.)] And 5000 to 8100 in a 96-well plate. Cells / well were seeded. Test compounds were added to various concentrations, and cultured for 3 days at 37 ° C. with 5% CO 2 . When the test compound was added, the test compound solution dissolved in dimethyl sulfoxide (DMSO) was used and diluted to the above-mentioned culture solution so as to obtain an appropriate concentration. The experiment was performed so that the final concentration of DMSO used for dissolving the test compound in the replicon cell culture was 0.4%. DMSO was added to wells to which no test compound had been added to a final concentration of 0.4%.
- DMSO dimethyl sulfoxide
- the IC50 (50% growth inhibitory concentration) of the drug was calculated with the value of no drug added as 0% inhibition.
- the IC50 value of compound 17 was> 20 ⁇ M, while the IC50 value of the compound of the present invention was as shown in Table 3 below.
- mice Male, 7 to 8 weeks old were used (three or more per group). Test compound suspended in olive oil was orally administered at 20 mg / kg, and blood was collected from the jugular vein after 2, 6 and 24 hours, or after 2, 4, 6, 8, 24 and 48 hours, and liquid chromatography / Plasma drug concentrations were measured using Mass spectrometry / Mass spectroscopy (LC-MS / MS). Further, pharmacokinetic parameters were determined from the obtained plasma concentration transition.
- Test compound suspended in olive oil was orally administered at 20 mg / kg, and blood was collected from the jugular vein after 2, 6 and 24 hours, or after 2, 4, 6, 8, 24 and 48 hours, and liquid chromatography / Plasma drug concentrations were measured using Mass spectrometry / Mass spectroscopy (LC-MS / MS). Further, pharmacokinetic parameters were determined from the obtained plasma concentration transition.
- LC-MS / MS Mass spectrometry / Mass spectroscopy
- Table 4 shows the maximum plasma concentration (C max ), plasma half-life (t 1/2 ), and area under the plasma drug concentration-time curve (AUC) of each compound obtained in this study.
- the compound of the present invention has high absorbency and sustained plasma concentration even when compared with 22 ⁇ -methoxyolean-12-ene-3 ⁇ , 24-diol (compound 17). In terms of sex, it was confirmed to have excellent pharmacokinetics.
- the triterpene derivative of the present invention can exhibit excellent anti-HCV activity and excellent pharmacokinetics when administered in an effective amount, it is expected to have a high preventive or therapeutic effect on chronic hepatitis C. it can. Therefore, the present invention can be used particularly in the medical field.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Virology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Gastroenterology & Hepatology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Steroid Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description
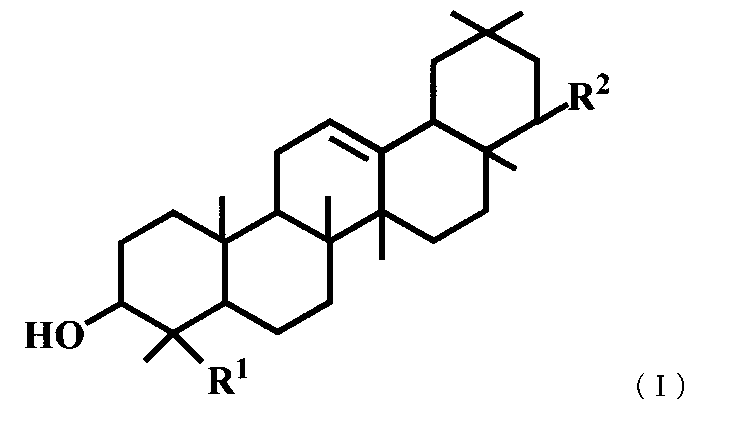

(2) 一般式(I)で表されるトリテルペン誘導体または医薬的に許容されるその塩。
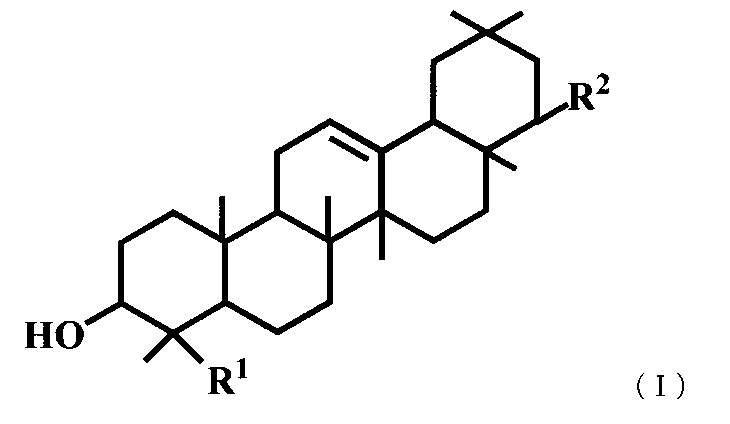
(3) 一般式(I)で表されるトリテルペン誘導体または医薬的に許容されるその塩。
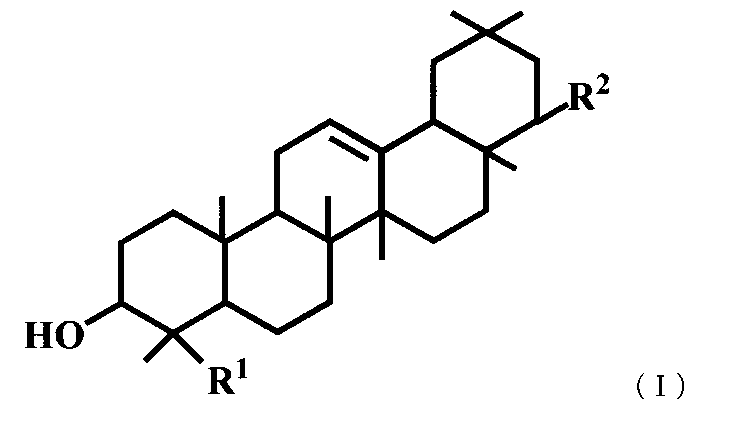
(4) 一般式(I)で表されるトリテルペン誘導体または医薬的に許容されるその塩。

(5) 一般式(I)で表されるトリテルペン誘導体または医薬的に許容されるその塩。
(6) C型肝炎ウイルスの阻害活性のIC50値が20μM以下である、(1)から(5)のいずれかに記載のトリテルペン誘導体または医薬的に許容されるその塩。

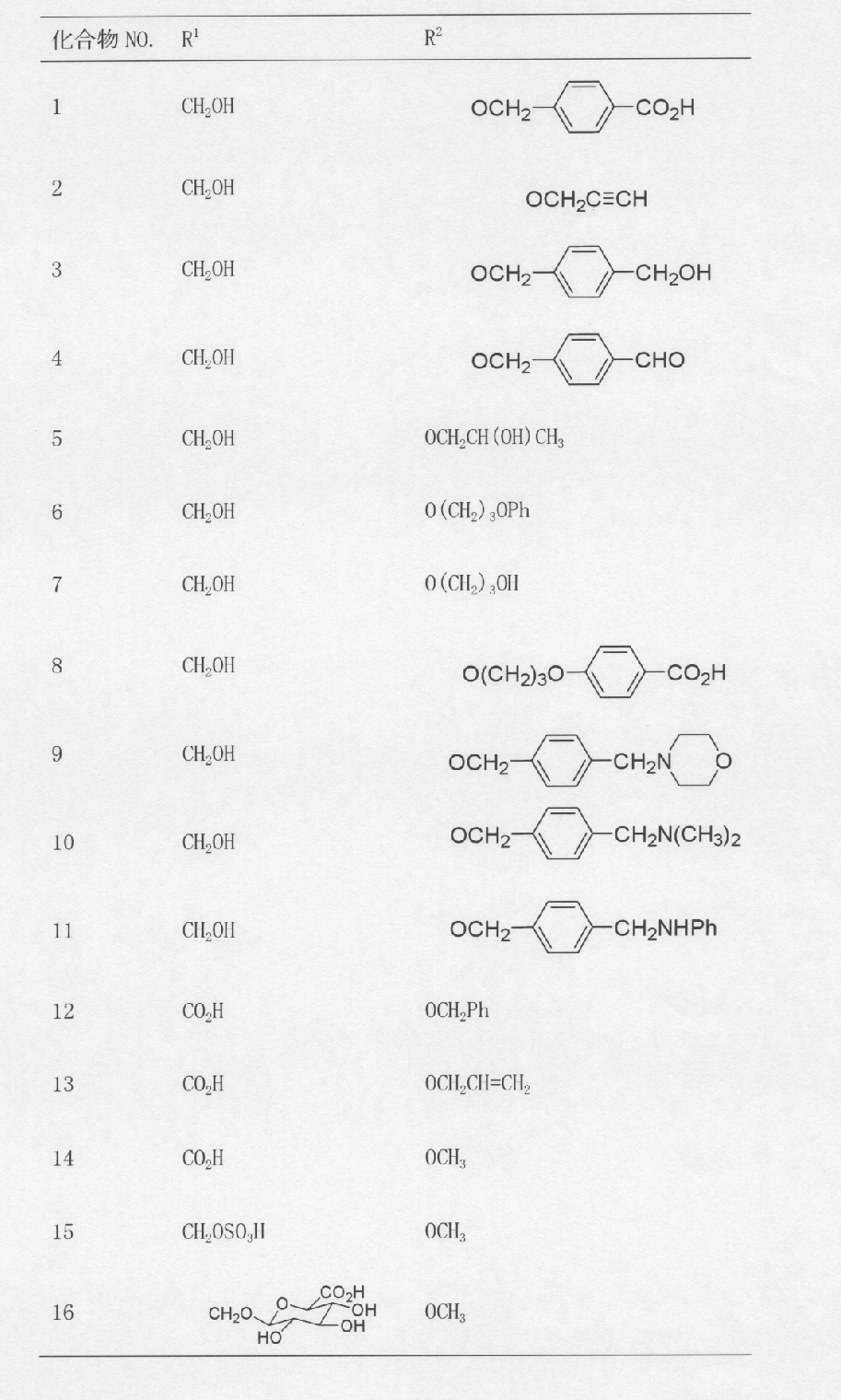
一般式(I)で示されるトリテルペン誘導体のうち、式(Ia)の化合物は、下 記の方法により製造することができる。
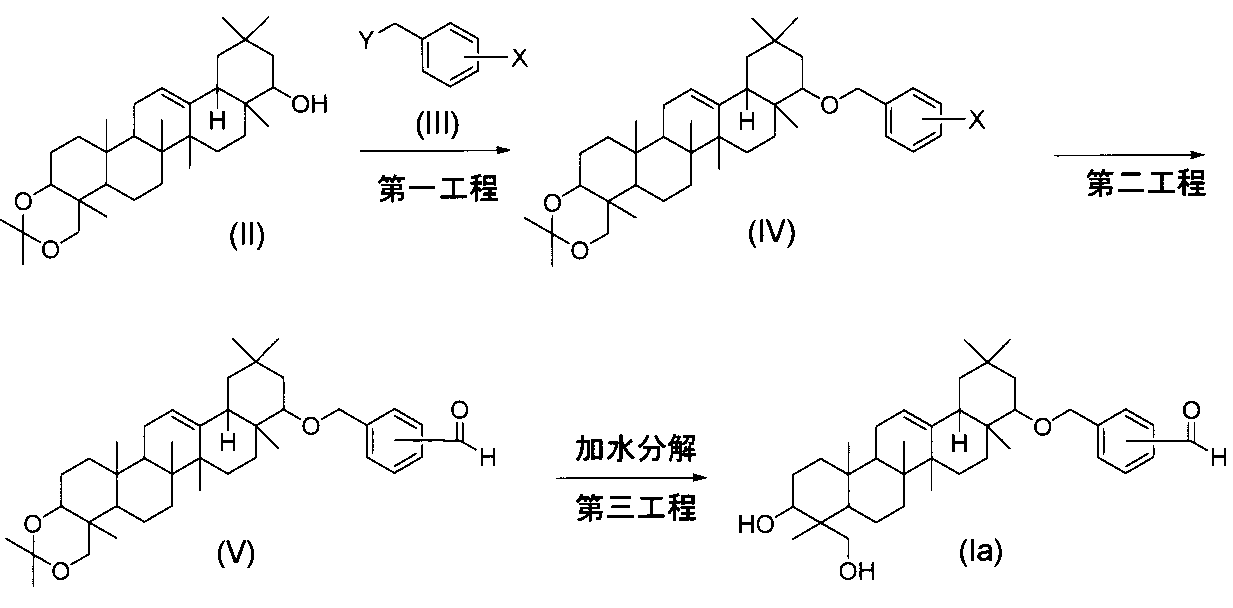
一般式(I)で示されるトリテルペン誘導体のうち、式(Ib)の化合物は、まず、第一工程において、式(V)の化合物を適当な還元剤と反応させ、次いで、第二工程において、加水分解反応を行うことにより製造することができる。

一般式(I)で示されるトリテルペン誘導体のうち、式(Ic)の化合物(式中、R3cはジメチルアミノメチル基、フェニルアミノメチル基またはモルホリノメチル基で置換されているベンジルである)は、まず、第一工程において、式(V)の化合物を、通常の還元的アミノアルキル化反応の方法に従って反応させ、次いで、第二工程において、加水分解反応を行うことにより製造することができる。
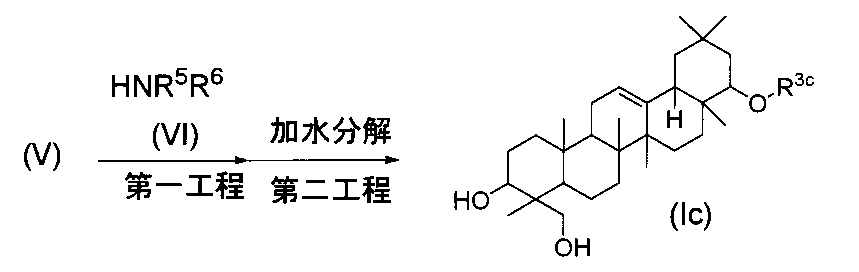
一般式(I)で示されるトリテルペン誘導体のうち、式(Id)の化合物は、まず、第一工程において、式(V)の化合物を、通常の酸化反応の方法に従って反応させ、次いで、第二工程において、加水分解反応を行うことにより製造することができる。
一般式(I)で示されるトリテルペン誘導体のうち、式(Ie)の化合物(式中、R3eはC2-6アルキニル基である)は、下記の方法により製造することができる。
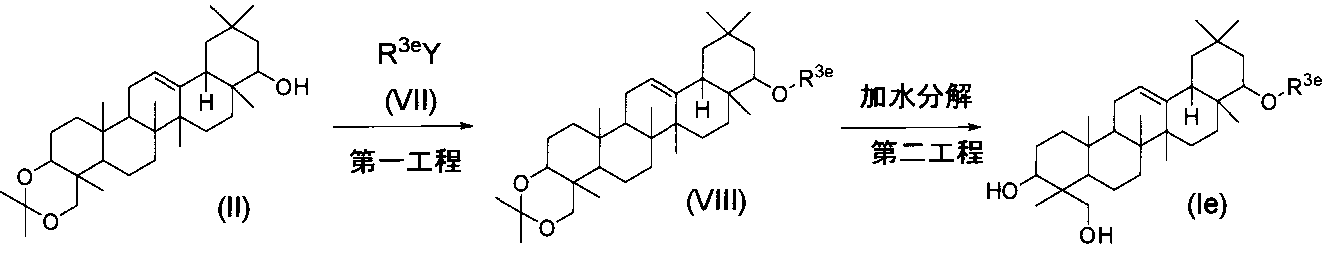
一般式(I)で示されるトリテルペン誘導体のうち、化合物5および化合物7は、下記の方法により製造することができる。
<方法(G)>
一般式(I)で示されるトリテルペン誘導体のうち、式(Ig)の化合物(式中、R4およびmは前記と同義である)は、下記の方法により製造することができる。

一般式(I)で示されるトリテルペン誘導体のうち、式(Ih)の化合物(式中、R3hはC1-6アルキル基、C2-6アルケニル基またはC2-6アルキニル基、またはベンジルである)は、下記の方法により製造することができる。
一般式(I)で示されるトリテルペン誘導体のうち、式(Ii)の化合物(式中、R3iはC1-6アルキル基、C2-6アルケニル基またはC2-6アルキニル基である)は、下記の方法によっても製造することができる。

一般式(I)で示されるトリテルペン誘導体のうち、式(Ij)の化合物(式中、R3iは前記と同義である)は、下記の方法によっても製造することができる。
一般式(I)で示されるトリテルペン誘導体のうち、式(Ik)の化合物(式中、R3iは前記と同義である)は、下記の方法によっても製造することができる。

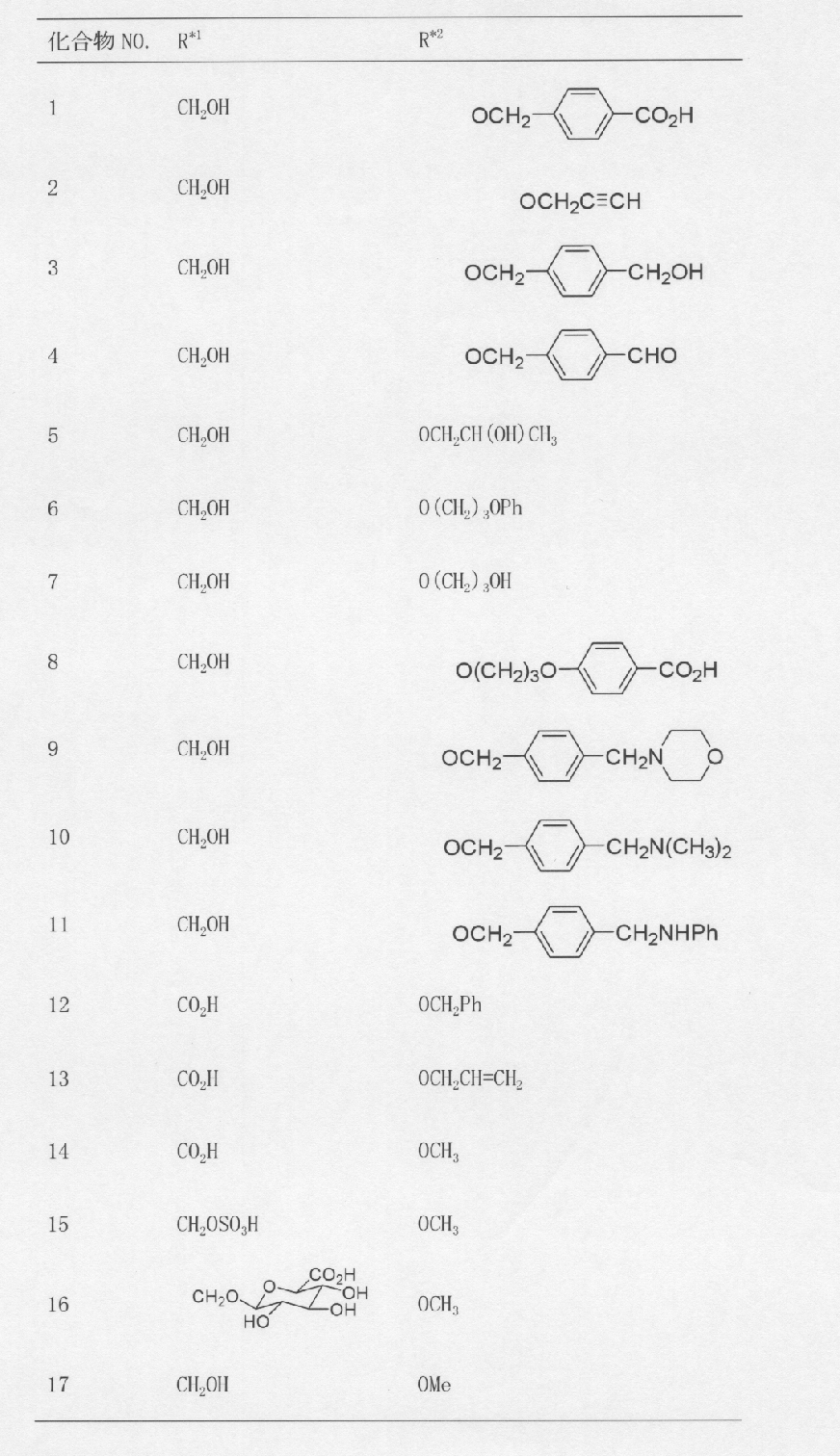
(ステップ1)
22β-(4-ヨードベンジルオキシ)-3β,24-イソプロピリデンジオキシオレアン-12-エン
1H-NMR(CDCl3)...0.89(3H,s),0.92(3H,s),0.99(3H,s),1.03(3H,s),1.12(3H,s),1.15(3H,s),1.22(3H,s),1.38(3H,s),1.44(3H,s),0.87-2.18(21H,m),3.05(1H,dd,J=2.9,6.1Hz),3.23(1H,d,J=11.6Hz),3.46(1H,dd,J=4.6,9.3Hz),4.05(1H,d,J=11.6Hz),4.25(1H,d,J=12.0Hz),4.55(1H,d,J=12.0Hz),5.24(1H,t-like),7.07-7.10(2H,m),7.62-7.66(2H,m).
MS FAB(m/z):715(M++1)
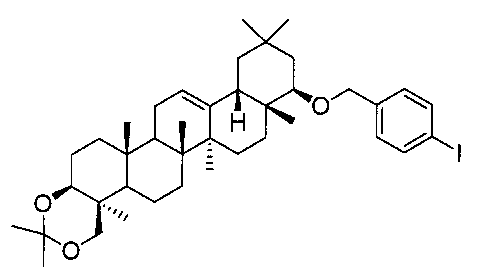
(ステップ2)
22β-(4-ホルミルベンジルオキシ)-3β,24-イソプロピリデンジオキシオレアン-12-エン
1H-NMR(CDCl3)...0.91(3H,s),0.96(3H,s),1.00(3H,s),1.05(3H,s),1.13(3H,s),1.16(3H,s),1.22(3H,s),1.38(3H,s),1.44(3H,s),0.83-2.05(20H,m),2.19(1H,m),3.10(1H,dd,J=2.9,6.1Hz),3.23(1H,d,J=11.5Hz),3.45(1H,dd,J=4.7,9.5Hz),4.07(1H,d,J=11.5Hz),4.40(1H,d,J=12.9Hz),4.70(1H,d,J=12.9Hz),5.26(1H,t-like),7.50(2H,d,J=8.1Hz),7.84(2H,d,J=8.1Hz),10.0(1H,s).
MS FAB(m/z):617(M++1)
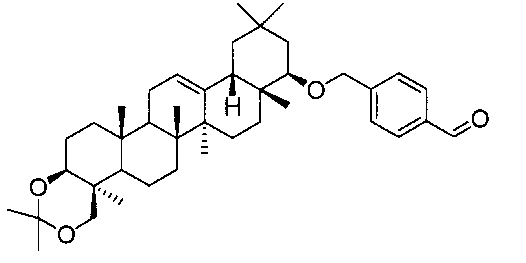
(ステップ3)
22β-(4-カルボキシベンジルオキシ)オレアン-12-エン-3β,24-ジオール(化合物1)
1H-NMR(CDCl3)...0.89(3H,s),0.90(3H,s),0.95(6H,s),1.04(3H,s),1.12(3H,s),1.25(3H,s),0.79-2.20(21H,m),3.09(1H,m),3.36(1H,d,J=11.2Hz),3.45(1H,dd,J=4.4,11.7Hz),4.22(1H,d,J=11.2Hz),4.38(1H,d,J=12.8Hz),4.68(1H,d,J=12.8Hz),5.24(1H,t-like),7.44(2H,d,J=7.9Hz),8.06(2H,d,J=7.9Hz).
MS ES(m/z):593(M++1)
[実施例2] 22β-(2-プロピニルオキシ)オレアン-12-エン-3β,24-ジオール(化合物2)の製造方法
(ステップ1)
22β-(2-プロピニルオキシ)-3β,24-イソプロピリデンジオキシオレアン-12-エン

1H-NMR(CDCl3)...0.88(3H,s),0.91(3H,s),0.99(3H,s),1.01(3H,s),1.12(3H,s),1.16(3H,s),1.22(3H,s),1.38(3H,s),1.44(3H,s),0.82-2.19(21H,m),2.35(1H,t,J=2.4Hz),3.18(1H,dd,J=3.2,6.6Hz),3.23(1H,d,J=11.5Hz),3.46(1H,dd,J=4.4,9.3Hz),4.04(1H,d,J=11.5Hz),4.06(1H,dd,J=2.4,16.1Hz),4.19(1H,dd,J=2.4,16.1Hz),5.24(1H,t-like).
MS FAB(m/z):559(M+Na)+
(ステップ2)
22β-(2-プロピニルオキシ)オレアン-12-エン-3β,24-ジオール(化合物2)
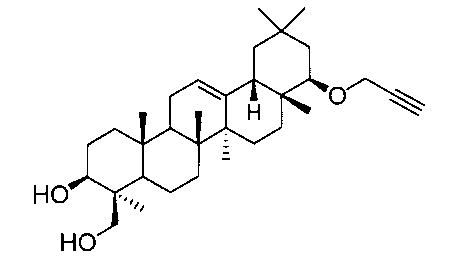
1H-NMR(CDCl3)...0.87(3H,s),0.89(3H,s),0.90(3H,s),0.94(3H,s),1.00(3H,s),1.11(3H,s),1.25(3H,s),0.83-1.88(20H,m),2.11(1H,m),2.34(1H,dd,J=4.2,11.0Hz),2.35(1H,t,J=2.4Hz),2.68(1H,m),3.18(1H,dd,J=3.2,6.8Hz),3.35(1H,m),3.44(1H,m),4.06(1H,dd,J=2.4,15.8Hz),4.17(1H,dd,J=2.4,15.8Hz),4.22(1H,m),5.22(1H,t-like).
MS FAB(m/z):519(M+Na)+
[実施例3] 22β-(4-ヒドロキシメチルベンジルオキシ)オレアン-12-エン-3β,24-ジオール(化合物3)
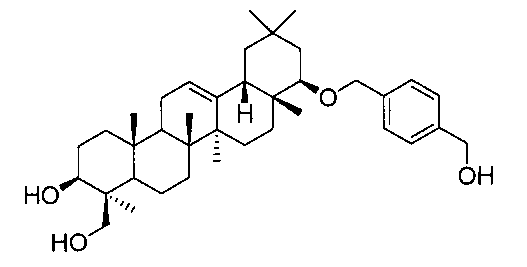
1H-NMR(CDCl3)...0.89(6H,s),0.93(3H,s),0.94(3H,s),1.04(3H,s),1.11(3H,s),1.25(3H,s),0.83-1.88(21H,m),2.15(1H,m),2.35(1H,dd,J=4.2,11.0Hz),2.69(1H,m),3.18(1H,dd,J=2.9,6.1Hz),3.35(1H,m),3.47(1H,m),4.20(1H,m),4.30(1H,d,J=11.9Hz),4.61(1H,d,J=11.9Hz),4.68(2H,d,J=6.1Hz),5.22(1H,t-like),7.33(4H,s).
MS FAB(m/z):601(M+Na)+
[実施例4] 22β-(4-ホルミルベンジルオキシ)オレアン-12-エン-3β,24-ジオール(化合物4)の製造方法
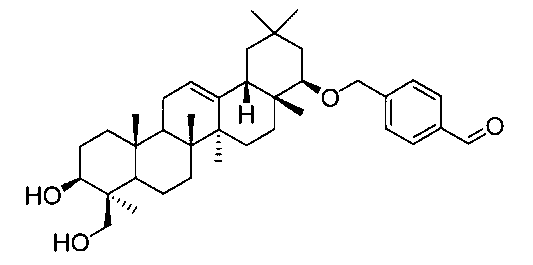
1H-NMR(CDCl3)...0.89(3H,s),0.90(3H,s),0.95(6H,s),1.04(3H,s),1.12(3H,s),1.25(3H,s),0.83-1.88(20H,m),2.17(1H,m),2.35(1H,dd,J=4.2,11.0Hz),2.68(1H,m),3.10(1H,dd,J=2.9,6.1Hz),3.35(1H,m),3.44(1H,m),4.20(1H,m),4.39(1H,d,J=12.9Hz),4.70(1H,d,J=12.9Hz),5.24(1H,t-like),7.50(2H,d,J=8.1Hz),7.84(2H,d,J=8.1Hz),10.0(1H,s).
MS FAB(m/z):599(M+Na)+
[実施例5] 22β-(2-ヒドロキシプロポオキシ)オレアン-12-エン-3β,24-ジオール(化合物5)の製造方法
(ステップ1)
22β-(2-ヒドロキシプロポオキシ)-3β,24-イソプロピリデンジオキシオレアン-12-エン、および22β-(3-ヒドロキシプロポキシ)-3β,24-イソプロピリデンジオキシオレアン-12-エン

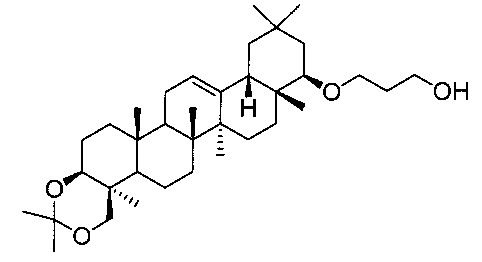
1H-NMR(CDCl3)...0.83-2.38(51H,m),2.37(0.5H,d,J=3.2Hz),2.47(0.5H,d,J=2.8Hz),2.95-3.02(1.5H,m)3.21-3.27(1.5H,m),3.32(0.5H,t,J=9.3Hz),3.45(1H,dd,J=4.6,9.5Hz),3.54(0.5H,dd,J=2.8,9.1Hz),3.91(1H,m),4.05(1H,d,J=11.5Hz),5.25(1H,t-like).
MS FAB(m/z):557(M++1).
22β-(3-ヒドロキシプロポキシ)-3β,24-イソプロピリデンジオキシオレアン-12-エン
1H-NMR(CDCl3)...0.86(3H,s),0.90(3H,s),0.98(3H,s),1.00(3H,s),1.11(3H,s),1.15(3H,s),1.22(3H,s),1.38(3H,s),1.44(3H,s),0.82-2.09(23H,m),2.54(1H,t,J=5.6Hz),2.94(1H,dd,J=2.9,6.6Hz),3.23(1H,d,J=11.6Hz),3.40(1H,m),3.45(1H,m),3.72-3.80(3H,m),4.05(1H,d,J=11.6Hz),5.24(1H,t-like).
MS FAB(m/z):557(M++1).
(ステップ2)
22β-(2-ヒドロキシプロポオキシ)オレアン-12-エン-3β,24-ジオール(化合物5)
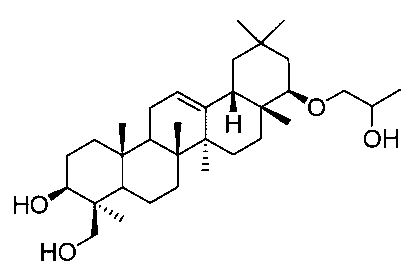
1H-NMR(CDCl3)...0.83-1.88(44H,m),2.11-2.16(1H,m),2.37-2.48(2H,m),2.73(0.5H,m),2.95-3.01(1.5H,m),3.24(0.5H,dd,J=3.2,9.0Hz),3.22-3.38(1.5H,m),3.43-3.46(1.5H,m),3.54(0.5H,dd,J=3.2,9.0Hz),3.88-3.92(1H,m),4.19-4.22(1H,m),5.23(1H,t-like).
MS ES(m/z):517(M++1)
[実施例6] 22β-(3-フェノキシプロポキシ)オレアン-12-エン-3β,24-ジオール(化合物6)の製造方法
(ステップ1)
22β-(3-フェノキシプロポキシ)-3β,24-イソプロピリデンジオキシオレアン-12-エン

1H-NMR(CDCl3)...0.86(3H,s),0.87(3H,s),0.98(3H,s),1.00(3H,s),1.12(3H,s),1.15(3H,s),1.22(3H,s),1.38(3H,s),1.44(3H,s),0.86-2.12(23H,m),2.91(1H,m),3.23(1H,d,J=11.5Hz),3.36(1H,dt,J=9.5,5.8Hz),3.45(1H,dd,J=4.6,9.5Hz),3.70(1H,dt,J=9.0,5.8Hz),4.03-4.08(3H,m),5.24(1H,t-like),6.88-7.00(3H,m),7.25-7.29(2H,m).
MS FAB(m/z):633(M++1).
(ステップ2)
22β-(3-フェノキシプロポキシ)オレアン-12-エン-3β,24-ジオール(化合物6)

1H-NMR(CDCl3)...0.86(3H,s),0.87(3H,s),0.89(3H,s),0.94(3H,s),1.00(3H,s),1.11(3H,s),1.25(3H,s),0.83-1.87(20H,m),1.98-2.09(2H,m),2.12(1H,m),2.35(1H,m),2.71(1H,m),2.91(1H,m),3.29-3.38(2H,m),3.44(1H,m),3.70(1H,m),4.07(2H,t,J=6.3Hz),4.22(1H,d,J=11.0Hz),5.21(1H,t-like),6.88-6.94(2H,m),7.25-7.29(2H,m).
MS FAB(m/z):631(M+K)+
[実施例7] 22β-(3-ヒドロキシプロポキシ)オレアン-12-エン-3β,24-ジオール(化合物7)の製造方法
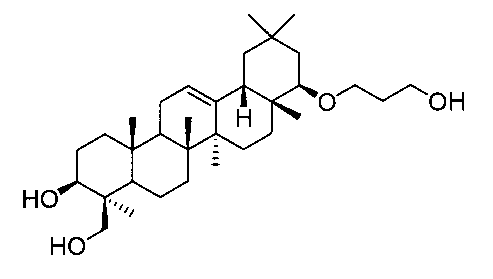
1H-NMR(CDCl3)...0.86(3H,s),0.89(3H,s),0.90(3H,s),0.93(3H,s),1.01(3H,s),1.11(3H,s),1.24(3H,s),0.83-1.91(22H,m),2.07(1H,m),2.64(2H,brs),2.88(1H,brs),2.94(1H,dd,J=2.9,6.6Hz),3.32-3.46(3H,m),3.72-3.79(3H,m),4.21(1H,d,J=11.2Hz),5.21(1H,t-like).
MS FAB(m/z):516(M+)
[実施例8] 22β-(3-(4-カルボキシフェニル)オキシプロポキシ)オレアン-12-エン-3β,24-ジオール(化合物8)の製造方法
(ステップ1)
22β-(3-(4-メトキシカルボニルフェニル)オキシプロポキシ)-3β,24-イソプロピリデンジオキシオレアン-12-エン

1H-NMR(CDCl3)...0.86(3H,s),0.87(3H,s),0.98(3H,s),0.99(3H,s),1.12(3H,s),1.15(3H,s),1.22(3H,s),1.38(3H,s),1.44(3H,s),0.85-2.12(23H,m),2.91(1H,dd,J=2.9,5.5Hz),3.23(1H,d,J=11.5Hz),3.35(1H,dt,J=9.4,6.4Hz),3.45(1H,dd,J=4.2,9.0Hz),3.70(1H,dt,J=9.4,5.5Hz),3.88(3H,s),4.05(1H,d,J=11.7Hz),4.09-4.15(2H,m),5.22(1H,t-like),6.8-7.26(2H,m),7.96-7.99(2H,m).
MS FAB(m/z):691(M++1).
(ステップ2)
22β-(3-(4-カルボキシフェニル)オキシプロポキシ)オレアン-12-エン-3β,24-ジオール(化合物8)
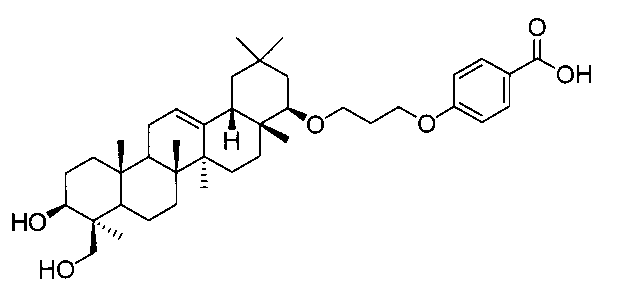
1H-NMR(CDCl3)...0.85(3H,s),0.87(3H,s),0.89(3H,s),0.93(3H,s),0.99(3H,s),1.11(3H,s),1.25(3H,s),0.83-1.87(20H,m),2.00-2.04(2H,m),2.12(1H,m),2.91(1H,m),3.33-3.38(2H,m),3.44(1H,m),3.70(1H,m),4.13-4.16(2H,m),4.21(1H,d,J=11.2Hz),5.21(1H,t-like),6.92-6.94(2H,m),8.02-8.05(2H,m).
MS ES(m/z):637(M++1)
[実施例9] 22β-(4-(4-モルホリルメチル)ベンジルオキシ)オレアン-12-エン-3β,24-ジオール(化合物9)の製造方法
(ステップ1)
22β-(4-(4-モルホリルメチル)ベンジルオキシ)-3β,24-イソプロピリデンジオキシオレアン-12-エン
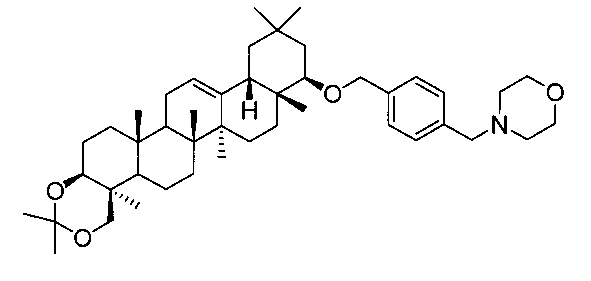
1H-NMR(CDCl3)...0.89(3H,s),0.93(3H,s),0.98(3H,s),1.04(3H,s),1.11(3H,s),1.15(3H,s),1.22(3H,s),1.38(3H,s),1.47(3H,s),0.86-2.01(20H,m),2.15(1H,m),2.43-2.45(4H,m),3.07(1H,dd,J=2.9,6.3Hz),3.23(1H,d,J=11.5Hz),3.44(1H,dd,J=4.4,9.5Hz),3.48(2H,s),3.69-3.72(4H,m),4.05(1H,d,J=11.5Hz),4.30(1H,d,J=11.9Hz),4.59(1H,d,J=11.9Hz),5.23(1H,t-like),7.26-7.37(4H,m).
MS ES(m/z):688(M+)
(ステップ2)
22β-(4-(4-モルホリルメチル)ベンジルオキシ)オレアン-12-エン-3β,24-ジオール(化合物9)

1H-NMR(CDCl3)...0.89(6H,s),0.93(3H,s),0.94(3H,s),1.04(3H,s),1.11(3H,s),1.27(3H,s),0.83-1.86(20H,m),2.17(1H,m),2.42-2.44(5H,m),2.71(1H,m),3.06(1H,dd,J=2.7,6.1Hz),3.33(1H,m),3.43(1H,m),3.48(2H,s),3.69-3.72(4H,m),4.20(1H,d,J=11.2Hz),4.30(1H,d,J=11.7Hz),4.59(1H,d,J=11.7Hz),5.22(1H,t-like),7.23-7.30(4H,m).
MS ES(m/z):648(M++1)
[実施例10] 22β-(4-ジメチルアミノメチルベンジルオキシ)オレアン-12-エン-3β,24-ジオール(化合物10)の製造方法
(ステップ1)
22β-(4-ジメチルアミノメチルベンジルオキシ)-3β,24-イソプロピリデンジオキシオレアン-12-エン
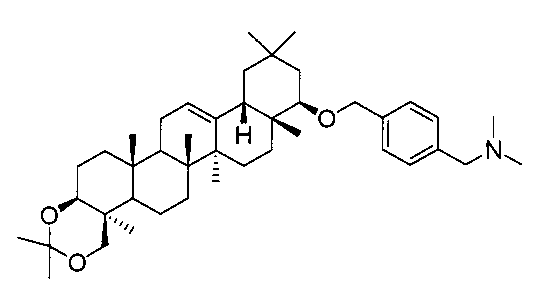
1H-NMR(CDCl3)...0.89(3H,s),0.93(3H,s),0.98(3H,s),1.04(3H,s),1.11(3H,s),1.15(3H,s),1.22(3H,s),1.38(3H,s),1.44(3H,s),0.83-2.04(20H,m),2.13(1H,m),2.23(6H,s),3.06(1H,dd,J=2.9,6.3Hz),3.23(1H,d,J=11.6Hz),3.41(2H,s),3.46(1H,dd,J=4.4,9.3Hz),4.05(1H,d,J=11.6Hz),4.31(1H,d,J=11.9Hz),4.59(1H,d,J=11.9Hz),5.24(1H,t-like),7.26-7.53(4H,m).
MS FAB(m/z):646(M+)
(ステップ2)
22β-(4-ジメチルアミノメチルベンジルオキシ)オレアン-12-エン-3β,24-ジオール(化合物10)

1H-NMR(CDCl3)...0.89(6H,s),0.92(3H,s),0.94(3H,s),1.04(3H,s),1.10(3H,s),1.24(3H,s),0.83-1.86(20H,m),2.17(1H,m),2.23(6H,s),3.06(1H,dd,J=2.9,6.3Hz),3.34(1H,d,J=11.2Hz),3.40(2H,s),3.45(1H,dd,J=4.2,11.5Hz),4.20(1H,d,J=11.2Hz),4.31(1H,d,J=11.9Hz),4.59(1H,d,J=11.9Hz),5.24(1H,t-like),7.25-7.29(4H,m).
MS ES(m/z):606(M++1)
[実施例11] 22β-(4-フェニルアミノメチルベンジルオキシ)オレアン-12-エン-3β,24-ジオール(化合物11)の製造方法

1H-NMR(CDCl3)...0.89(6H,s),0.93(3H,s),0.94(3H,s),1.04(3H,s),1.11(3H,s),1.25(3H,s),0.83-1.86(20H,m),2.05(1H,m),2.41(1H,m),2.72(1H,m),3.06(1H,dd,J=2.9,5.8Hz),3.34(1H,m),3.44(1H,m),4.03(1H,m),4.22(1H,d,J=11.0Hz),4.30(1H,d,J=12.0Hz),4.31(2H,s),4.59(1H,d,J=12.0Hz),5.22(1H,t-like),6.62-6.65(2H,m),6.71(1H,m),7.14-7.20(2H,m),7.29-7.34(4H,m).
MS ES(m/z):654(M++1)
[実施例12] 22β-ベンジルオキシオレアン-12-エン-3β-ヒドロキシ-24―オイックアシッド(化合物12)の製造方法
(ステップ1)
22β-ベンジルオキシ-3-オキソオレアン-12-エン-24―オイックアシッド メチルエステル
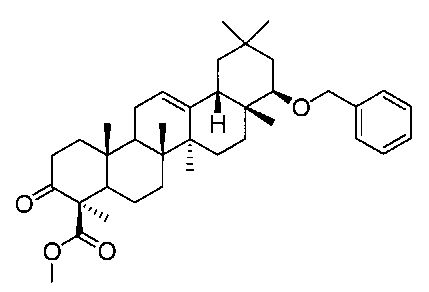
1H-NMR(CDCl3)...0.89(3H,s),0.94(3H,s),1.04(6H,s),1.07(3H,s),1.10(3H,s),1.37(3H,s),0.89-2.05(18H,m),2.17(1H,m),2.36(1H,m),2.95(1H,ddd,J=6.3,14.6,14.6Hz),3.08(1H,dd,J=2.7,6.3Hz),3.68(3H,s),4.32(1H,d,J=11.7Hz),4.62(1H,d,J=11.7Hz),5.24(1H,t-like),7.24-7.35(5H,m).
MS FAB(m/z):575(M++1)
(ステップ2)
22β-ベンジルオキシオレアン-12-エン-3β-ヒドロキシ-24―オイックアシッド メチルエステル

1H-NMR(CDCl3)...0.79(3H,s),0.89(3H,s),0.94(3H,s),0.98(3H,s),1.04(3H,s),1.11(3H,s),1.40(3H,s),0.83-1.89(19H,m),2.03(1H,m),2.15(1H,m),3.06-3.12(2H,m),3.35(1H,d,J=12.0Hz),3.68(3H,s),4.32(1H,d,J=12.0Hz),4.62(1H,d,J=12.0Hz),5.24(1H,t-like),7.23-7.35(5H,m).
MS FAB(m/z):577(M++1)
(ステップ3)
22β-ベンジルオキシオレアン-12-エン-3β-ヒドロキシ-24―オイックアシッド(化合物12)
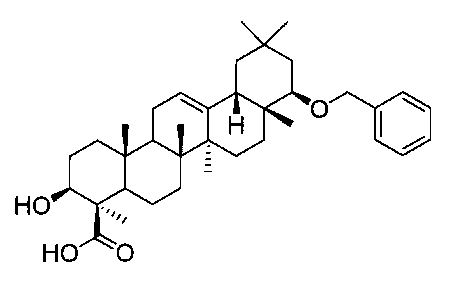
1H-NMR(CDCl3)...0.90(3H,s),0.91(3H,s),0.94(3H,s),1.00(3H,s),1.04(3H,s),1.11(3H,s),1.48(3H,s),0.85-1.90(19H,m),2.00(1H,m),2.15(1H,m),3.08(1H,m),3.15(1H,dd,J=4.2,12.1),4.32(1H,d,J=12.0Hz),4.62(1H,d,J=12.0Hz),5.24(1H,m),7.26-7.35(5H,m).
MS ES(m/z):563(M++1)
[実施例13] 22β-アリルオキシオレアン-12-エン-3β-ヒドロキシ-24―オイックアシッド(化合物13)の製造方法
(ステップ1)
22β-アリルオキシ-3-オキソオレアン-12-エン-24―オイックアシッド メチルエステル

1H-NMR(CDCl3)...0.89(3H,s),0.90(3H,s),1.01(3H,s),1.04(3H,s),1.07(3H,s),1.10(3H,s),1.37(3H,s),0.86-2.05(18H,m),2.12(1H,m),2.38(1H,ddd,J=2.3,4.6,14.6Hz),2.90-3.00(2H,m),3.68(3H,s),3.80(1H,m),4.05(1H,m),5.11(1H,m),5.23-5.29(2H,m),5.90(1H,m).
MS FAB(m/z):525(M++1)
(ステップ2)
22β-アリルオキシオレアン-12-エン-3β-ヒドロキシ-24―オイックアシッド メチルエステル

1H-NMR(CDCl3)...0.79(3H,s),0.89(6H,s),0.98(3H,s),1.01(3H,s),1.11(3H,s),1.41(3H,s),0.82-1.89(19H,m),1.99(1H,m),2.09(1H,m),2.99(1H,dd,J=2.9,6.8Hz),3.09(1H,dt,4.4,12.0Hz),3.35(1H,d,J=12.0Hz),3.68(3H,s),3.79(1H,m),4.06(1H,m),5.11(1H,m),5.23-5.28(2H,m),5.90(1H,m).
MS FAB(m/z):527(M++1)
(ステップ3)
22β-アリルオキシオレアン-12-エン-3β-ヒドロキシ-24―オイックアシッド(化合物13)
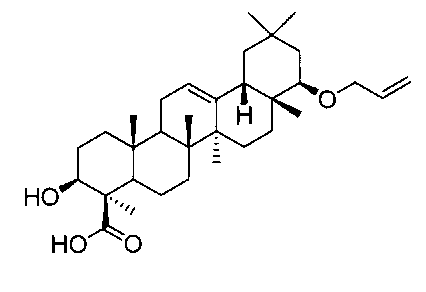
1H-NMR(CDCl3)...0.89(6H,s),0.90(3H,s),1.00(3H,s),1.01(3H,s),1.12(3H,s),1.48(3H,s),0.85-1.90(19H,m),2.01(1H,m),2.12(1H,m),2.99(1H,dd,J=2.9,6.8Hz),3.15(1H,dd,J=4.2,11.7),3.80(1H,dd,J=5.4,13.0Hz),4.06(1H,dd,J=5.1,13.0Hz),5.10(1H,dd,J=1.5,10.5Hz),5.23-5.28(2H,m),5.90(1H,m).
MS ES(m/z):535(M+Na)+
[実施例14] 3β-ヒドロキシ-22β-メトキシオレアン-12-エン-24-オイックアシッド(化合物14)の製造方法
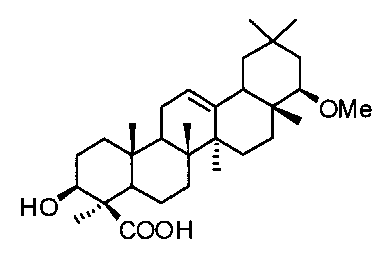
1H-NMR(CDCl3)...0.86(3H,s),0.90(6H,s),1.00(6H,s),1.11(3H,s),1.31(1H,dd,J=3.0,13.9Hz),1.48(3H,s),0.88-2.12(20H,m),2.82(1H,dd,J=2.8,7.0Hz),3.15(1H,dd,J=4.4,12.4Hz),3.28(3H,s),5.24(1H,dd,J=3.6,3.6Hz).
MS FAB(m/z):509(M++Na)
[実施例15] 3β-ヒドロキシ-22β-メトキシオレアン-12-エン-24-イル硫酸 ナトリウム塩(化合物15)の製造方法

1H-NMR(DMSO-d6)...0.81(3H,s),0.87(3H,s),0.91(3H,s),0.94(3H,s),0.97(3H,s),1.01(3H,s),1.08(3H,s),0.68-1.89(20H,m),2.01-2.05(1H,m),2.79-2.81(1H,m),3.01-3.06(1H,m),3.19(3H,s),3.83(1H,d,J=10.4Hz),3.86(1H,d,J=10.4Hz),4.35(1H,d,J=6.3Hz),5.17(1H,t-like).
MS TSP(m/z):551(M+-1)
[実施例16] 1-O-(3β-ヒドロキシ-22β-メトキシオレアン-12-エン-24-イル)-β-D-グルクロン酸(化合物16)の製造方法
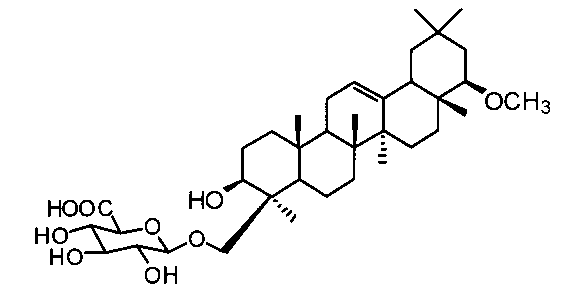
1H-NMR(CD3OD)...0.85(3H,s),0.89(3H,s),0.97(3H,s),0.98(3H,s),1.00(3H,s),1.14(3H,s),1.20(3H,s),0.81-1.91(20H,m),2.09(1H,d,J=10.5Hz),2.84(1H,dd,J=2.9,6.1Hz),3.17-3.23(2H,m),3.26(3H,s),3.37(1H,dd,J=9.0,9.0Hz),3.48(1H,dd,J=9.0,9.0Hz),3.68(1H,d,J=9.8Hz),3.79(1H,d,J=9.8Hz),4.04(1H,d,J=9.5Hz),4.28(1H,d,J=7.8Hz),5.21(1H,t-like).
MS TSP(m/z):647(M+-1)
[実施例17] HCV(Con1株)レプリコンアッセイおよび細胞毒性試験
式(I)で表される化合物について、HCVレプリコンアッセイおよび細胞毒性試験を行った。

Balb/cマウス(雄、7~8週齢)を用いた(一群3匹以上)。オリーブオイルにて懸濁させた試験化合物を20mg/kg経口投与し、2、6および24時間後に、あるいは2、4、6、8、24および48時間後に、頸静脈より採血し、Liquid chromatograph/Mass spectrometry/Mass spectrometry(LC-MS/MS)を用いて血漿中薬物濃度を測定した。また得られた血漿中濃度推移から、薬物動態パラメータを求めた。

Claims (13)
- 一般式(I)で表されるトリテルペン誘導体または医薬的に許容されるその塩。


- 一般式(I)で表されるトリテルペン誘導体または医薬的に許容されるその塩。
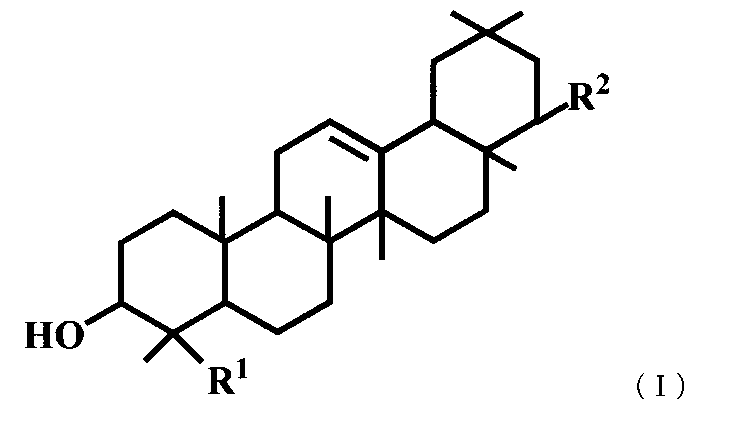
- 一般式(I)で表されるトリテルペン誘導体または医薬的に許容されるその塩。
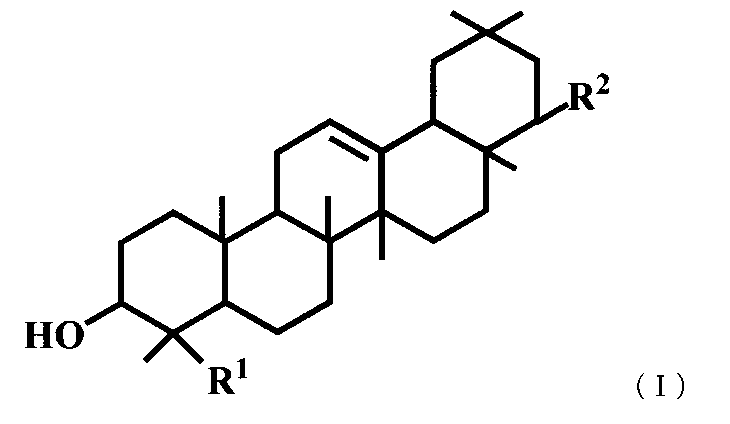
- 一般式(I)で表されるトリテルペン誘導体または医薬的に許容されるその塩。
- 一般式(I)で表されるトリテルペン誘導体または医薬的に許容されるその塩。

- C型肝炎ウイルスの阻害活性のIC50値が20μM以下である、請求項1から5のいずれかに記載のトリテルペン誘導体または医薬的に許容されるその塩。
- 請求項1から6のいずれかに記載のトリテルペン誘導体または医薬的に許容されるその塩を有効成分として含んでなるC型慢性肝炎の予防または治療のための薬剤。
- 医薬的に許容される担体を含む、請求項7に記載の薬剤。
- さらにインターフェロンを組み合わせてなる、請求項7または8に記載の薬剤。
- 請求項7から9のいずれかに記載の薬剤を、C型慢性肝炎の予防または治療が必要な患者に、予防上または治療上の有効量を投与することを含む、C型慢性肝炎の予防または治療の方法。
- C型慢性肝炎の予防または治療のための薬剤の製造のための、請求項1から6のいずれかに記載のトリテルペン誘導体または医薬的に許容される塩の使用方法。
- C型慢性肝炎の予防または治療のための薬剤が、請求項7から9のいずれかに記載の薬剤である、請求項11に記載の使用方法。
- C型慢性肝炎の予防または治療における使用のための、請求項1から6のいずれかに記載のトリテルペン誘導体または医薬的に許容される塩。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10837633.6A EP2514759A4 (en) | 2009-12-15 | 2010-12-15 | TRITERPENE DERIVATIVE AND PROPHYLACTIC OR THERAPEUTIC FOR CHRONIC HEPATITIS C |
| CN2010800569775A CN102656181A (zh) | 2009-12-15 | 2010-12-15 | 三萜衍生物及用于预防或治疗c型慢性肝炎的药剂 |
| US13/516,012 US20120251492A1 (en) | 2009-12-15 | 2010-12-15 | Triterpene derivative and agent for preventing or treating chronic hepatitis c |
| JP2011546149A JPWO2011074607A1 (ja) | 2009-12-15 | 2010-12-15 | トリテルペン誘導体およびc型慢性肝炎の予防または治療のための薬剤 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009284135 | 2009-12-15 | ||
| JP2009-284135 | 2009-12-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011074607A1 true WO2011074607A1 (ja) | 2011-06-23 |
Family
ID=44167359
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2010/072564 WO2011074607A1 (ja) | 2009-12-15 | 2010-12-15 | トリテルペン誘導体およびc型慢性肝炎の予防または治療のための薬剤 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20120251492A1 (ja) |
| EP (1) | EP2514759A4 (ja) |
| JP (1) | JPWO2011074607A1 (ja) |
| CN (1) | CN102656181A (ja) |
| TW (1) | TW201143759A (ja) |
| WO (1) | WO2011074607A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102584930A (zh) * | 2012-01-17 | 2012-07-18 | 北京大学 | 刺囊酸衍生物及其生物转化方法和用途 |
| CN103127135A (zh) * | 2011-11-22 | 2013-06-05 | 北京大学 | 三萜衍生物,其制备方法和用途 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111018938B (zh) * | 2019-12-10 | 2021-05-25 | 中国人民解放军第二军医大学 | 一种五环三萜类甘草次酸衍生物及制备方法与应用 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997003088A1 (fr) * | 1995-07-07 | 1997-01-30 | Meiji Seika Kabushiki Kaisha | Derives triterpenes et medicament pour les maladies du foie |
| WO1997031014A1 (fr) * | 1996-02-26 | 1997-08-28 | Meiji Seika Kabushiki Kaisha | Derives triterpeniques et remedes pour maladies hepatiques |
| WO2008004653A1 (fr) | 2006-07-07 | 2008-01-10 | Meiji Seika Kaisha, Ltd. | Agent prophylactique ou thérapeutique pour une maladie virale |
-
2010
- 2010-12-15 EP EP10837633.6A patent/EP2514759A4/en not_active Withdrawn
- 2010-12-15 US US13/516,012 patent/US20120251492A1/en not_active Abandoned
- 2010-12-15 WO PCT/JP2010/072564 patent/WO2011074607A1/ja active Application Filing
- 2010-12-15 CN CN2010800569775A patent/CN102656181A/zh active Pending
- 2010-12-15 TW TW099143985A patent/TW201143759A/zh unknown
- 2010-12-15 JP JP2011546149A patent/JPWO2011074607A1/ja active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997003088A1 (fr) * | 1995-07-07 | 1997-01-30 | Meiji Seika Kabushiki Kaisha | Derives triterpenes et medicament pour les maladies du foie |
| JP3279574B2 (ja) | 1995-07-07 | 2002-04-30 | 明治製菓株式会社 | トリテルペン誘導体および肝疾患治療剤 |
| WO1997031014A1 (fr) * | 1996-02-26 | 1997-08-28 | Meiji Seika Kabushiki Kaisha | Derives triterpeniques et remedes pour maladies hepatiques |
| JP3727353B2 (ja) | 1996-02-26 | 2005-12-14 | 明治製菓株式会社 | トリテルペン誘導体および肝疾患治療剤 |
| WO2008004653A1 (fr) | 2006-07-07 | 2008-01-10 | Meiji Seika Kaisha, Ltd. | Agent prophylactique ou thérapeutique pour une maladie virale |
Non-Patent Citations (21)
| Title |
|---|
| "Annals of Internal Medicine", vol. 144, 2006, pages: 705 - 765 |
| "B Gata C Gata Knnen Chiryo Ni Okeru Aratana Mondaiten", 2008, MEDICAL JOURNAL SYA. CO., LTD., pages: 19 - 25 |
| "Data on Deaths from Cancer based on Vital Statistics of Japan", 1958, RESEARCH CENTER FOR CANCER PREVENTION AND SCREENING |
| "Mansei Kanen No Chiryo Gaido", 2008 |
| "Mansei Kanen No Chiryo Gaido", vol. 21, 2008, BUNKODO CO., LTD. |
| "Uirusu Kanen No Atarashii Chiryohou", vol. 25, 2008, MEDICAL VIEW CO., LTD., pages: 76 - 81 |
| ANNUAL REVIEW OF BIOCHEMISTRY, vol. 56, 1987, pages 727 - 777 |
| BIOORGANIC & MEDICINAL CHEMISTRY, vol. 13, 2005, pages 4900 |
| CHEM. PHARM. BULL., vol. 36, 1988, pages 153 |
| ELPA, SUMMARY EXPERT RECOMMENDATIONS, 2009 |
| HIASA, Y. ET AL.: "Hepatitis C Virus Replication Is Inhibited by 22p-methoxyolean-12-ene-3p, 24(4p)-diol (ME3738) Through Enhancing Interferon-P", HEPATOLOGY, vol. 48, no. 1, 2008, pages 59 - 69, XP002686099, DOI: doi:10.1002/hep.22289 * |
| IINO SHIRO: "C Gata Kanen Chiryo Gaido Rain (Guideline of Therapy for Hepatitis C", NIPPON RINSHO, vol. 62, no. 7, 2004, pages 342 - 346 |
| JOURNAL OF CLINICAL MICROBIOLOGY, 2006, pages 318 - 323 |
| JOURNAL OF CLINICAL MICROBIOLOGY, vol. 32, no. 4, 1994, pages 884 - 892 |
| KRIEGER ET AL., J. VIROL., vol. 75, 2001, pages 4614 |
| LANCET, vol. 352, no. 9138, 1998, pages 1426 - 1432 |
| NATURE REVIEWS, 2007, pages 991 - 1000 |
| NEW ENGLAND JOURNAL OF MEDICINE, vol. 345, no. 1, 2001, pages 41 - 52 |
| NOBUTO MINOWA: "Daizu Sapogenol o Mochiita C-gata Kan'en Chiryoyaku no Sosei Kenkyu", JOURNAL OF THE PHARMACEUTICAL SOCIETY OF JAPAN, vol. 126, no. SUPP.3, 2006, pages 24 - 27 * |
| See also references of EP2514759A4 |
| THEODORA W. GREENE; PETER G. M. WUTS: "Protective Groups in Organic Synthesis", JOHN WILEY & SONS, INC. |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103127135A (zh) * | 2011-11-22 | 2013-06-05 | 北京大学 | 三萜衍生物,其制备方法和用途 |
| CN102584930A (zh) * | 2012-01-17 | 2012-07-18 | 北京大学 | 刺囊酸衍生物及其生物转化方法和用途 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2514759A4 (en) | 2013-07-03 |
| EP2514759A1 (en) | 2012-10-24 |
| TW201143759A (en) | 2011-12-16 |
| US20120251492A1 (en) | 2012-10-04 |
| JPWO2011074607A1 (ja) | 2013-04-25 |
| CN102656181A (zh) | 2012-09-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8022087B2 (en) | Derivatives of pyridone and use thereof | |
| TW406086B (en) | Propiophenone derivatives and process for preparing the same | |
| JPH05194470A (ja) | 抗−エンドトキシン化合物 | |
| JP2001506267A (ja) | アミノステロールエステル化合物 | |
| AU2017366514B2 (en) | Piperidine-2,6-dione derivative and treatment for ulcerative colitis | |
| WO2011074607A1 (ja) | トリテルペン誘導体およびc型慢性肝炎の予防または治療のための薬剤 | |
| EP0943620A2 (en) | Betulinic acid derivatives for inhibiting cancer growth | |
| US20070197646A1 (en) | Substituted taraxastanes useful for treating viral infections | |
| EP1368014A1 (en) | Use of cyclohexenone derivatives for the manufacture of a medicament in the treatment of dysuria | |
| JPH05208988A (ja) | 3−デオキシマンノサミン誘導体およびそれら誘導体の製法 | |
| WO2011074608A1 (ja) | C型慢性肝炎の予防または治療のための薬剤 | |
| JP4105258B2 (ja) | コレスタノール化合物及びこれを含有する医薬 | |
| KR100564383B1 (ko) | 진세노사이드 유도체의 제조방법 | |
| KR100547253B1 (ko) | 암 예방 및 치료에 유효한 진세노사이드 유도체 | |
| JP3831953B2 (ja) | 7−グリコシロキシベンゾピラン誘導体及びそれを有効成分とする抗アレルギー剤 | |
| JP2675271B2 (ja) | 新規なアクロニシン類似体、その製造方法及びそれを含む医薬組成物 | |
| EP2088152B1 (en) | N'-{n-[3-oxo-lupen-28-oyl]-9-aminononanoyl}-3-amino-3-phenylpropeonic acid and the pharmaceutically acceptable derivatives thereof, a method for the production and the use thereof in the form of a medicinal agent | |
| JP3798043B2 (ja) | 2−チオフラボン誘導体及びその製造方法 | |
| JP4182218B2 (ja) | アポトーシスを誘導する新規なグルコース誘導体、その製造方法及び医薬としてのその用途 | |
| JP2005139066A (ja) | 2、3−ジヒドロ−1h−キノリン−4−オンオキシム誘導体および熱ショック蛋白質発現誘導阻害剤 | |
| WO2012133733A1 (ja) | がん細胞増殖抑制剤 | |
| JP3775513B2 (ja) | 3−グリコシロキシベンゾピラン誘導体及びそれを有効成分とする抗アレルギー剤 | |
| JPS60126221A (ja) | 抗腫瘍組成物 | |
| CN114702544A (zh) | 一种氨基甾体化合物及其制备方法和应用 | |
| JPH05202088A (ja) | グリコシド及びこれを含有する抗肝炎剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 201080056977.5 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10837633 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011546149 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13516012 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010837633 Country of ref document: EP |