WO2008132057A1 - Verfahren zur herstellung optisch aktiver carbonylverbindungen - Google Patents
Verfahren zur herstellung optisch aktiver carbonylverbindungen Download PDFInfo
- Publication number
- WO2008132057A1 WO2008132057A1 PCT/EP2008/054644 EP2008054644W WO2008132057A1 WO 2008132057 A1 WO2008132057 A1 WO 2008132057A1 EP 2008054644 W EP2008054644 W EP 2008054644W WO 2008132057 A1 WO2008132057 A1 WO 2008132057A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optically active
- carbon monoxide
- hydrogen
- asymmetric hydrogenation
- aryl
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/17—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by hydrogenation of carbon-to-carbon double or triple bonds
- C07C29/172—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by hydrogenation of carbon-to-carbon double or triple bonds with the obtention of a fully saturated alcohol
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B53/00—Asymmetric syntheses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/56—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by isomerisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/62—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by hydrogenation of carbon-to-carbon double or triple bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Definitions
- the present invention relates to a process for the preparation of optically active carbonyl compounds by asymmetric hydrogenation of ⁇ , ß-unsaturated carbonyl compounds in the presence of soluble in the reaction mixture, optically active transition metal catalysts having at least one carbon monoxide ligand.
- the present invention relates to a process for producing optically active al-dehyde or ketones, in particular citronellal by asymmetric hydrogenation of the corresponding optically active ⁇ , ß-unsaturated aldehydes or ketones in the presence of carbon monoxide.
- EP-A 0 000 315 relates to a process for the preparation of optically active citronellal by hydrogenation of geranial or neral in the presence of a catalyst complex of rhodium and a chiral phosphine dissolved in the reaction system.
- a problem in carrying out (homogeneous) catalytic reactions catalyzed by soluble catalysts is the often inadequate stability of the catalyst complexes used or of the catalytically active metal or transition metal complex compound formed therefrom.
- homogeneous catalytic reactions with complex transition metal catalysts can only be used on an industrial scale in an economical manner in special cases.
- JP-A 52078812 describes a process for the hydrogenation of ⁇ , ⁇ -unsaturated aldehydes such as crotonaldehyde, cinnamic aldehyde or ⁇ -methylcinnamaldehyde on homogeneous Rh catalysts under hydroformylation conditions in the presence of a triarylphosphine, a tertiary amine in stoichiometric amount and carbon monoxide.
- WO 2006/040096 discloses a process for the preparation of optically active carbonyl compounds by asymmetric hydrogenation of ⁇ , ⁇ -unsaturated carbonyl compounds in the presence of optically active transition metal catalysts which are soluble in the reaction mixture and have at least one carbon monoxide ligand which characterized in that the catalyst is pretreated with a gas mixture containing carbon monoxide and hydrogen and / or the asymmetric hydrogenation in the presence of additionally supplied carbon monoxide in the presence of the reaction mixture.
- the reaction is carried out in such a way that the catalyst is pretreated with a gas mixture containing carbon monoxide and hydrogen and the subsequent hydrogenation additionally carried out in the presence of carbon monoxide, the carbon monoxide concentration of the reaction mixture during the hydrogenation is often difficult to control.
- the pretreatment is usually carried out using significantly higher carbon monoxide concentrations than the asymmetric hydrogenation, so that large amounts of carbon monoxide, which originate from the pretreatment of the catalyst, can be introduced into the hydrogenation and have unfavorable effects there.
- the object has been achieved by providing a process for the preparation of optically active carbonyl compounds by asymmetric hydrogenation of ⁇ , ß-unsaturated carbonyl compounds in the presence of soluble in the reaction mixture, optically active transition metal catalysts having at least one carbon monoxide ligand, being used for the preparation of each optically active, at least one carbon monoxide ligand catalyst having a catalyst precursor with a carbon monoxide and hydrogen-containing gas mixture pre-treated and performs the asymmetric hydrogenation in the presence of the reaction mixture additionally supplied carbon monoxide, which is characterized in that
- Carbon monoxide content of 100 to 1200 ppm performs.
- the process according to the invention is suitable for preparing optically active carbonyl compounds, such as aldehydes, ketones, esters, lactones or lactams, by a-symmetric, i. Enantioselective hydrogenation of the corresponding carbonyl compounds which have an ethylenic double bond in ⁇ , ß-position to the carbonyl group.
- asymmetric hydrogenation is to be understood as meaning a hydrogenation in which the two enantiomeric forms of the hydrogenation product are not obtained in equal parts.
- the respective optically active catalyst having at least one carbon monoxide ligand pretreatment of a catalyst precursor with a gas mixture containing carbon monoxide and hydrogen is carried out.
- the asymmetric hydrogenation according to the invention is carried out in the presence of the reaction mixture additionally supplied carbon monoxide.
- the inventive method is characterized in that the said pretreatment of the catalyst precursor with a gas mixture comprising 20 to 90 vol .-% carbon monoxide, 10 to 80% by volume of hydrogen and 0 to 5 vol .-% of further gases, wherein supplement the stated proportions by volume to 100% by volume, at a pressure of from 5 to 100 bar, and remove excess carbon monoxide from the catalyst thus obtained before use in the asymmetric hydrogenation and the a-symmetric hydrogenation in the presence of hydrogen with a carbon monoxide content from 100 to 1200 ppm.
- a gas mixture comprising 20 to 90 vol .-% carbon monoxide, 10 to 80% by volume of hydrogen and 0 to 5 vol .-% of further gases, wherein supplement the stated proportions by volume to 100% by volume, at a pressure of from 5 to 100 bar, and remove excess carbon monoxide from the catalyst thus obtained before use in the asymmetric hydrogenation and the a-symmetric hydrogenation in the presence of hydrogen with a carbon monoxide content from 100 to 1200 ppm.
- transition metal catalysts which are to be used according to the invention and which are soluble in the reaction mixture have at least one CO ligand at least in a catalytic cycle or in a preform upstream of the actual catalytic cycle, it being immaterial whether these have at least one CO 2 ligand.
- Ligand-containing catalyst form represents the actual catalytically active catalyst form.
- R 1 , R 2 are each an unbranched, branched or cyclic alkyl radical having 1 to 25 carbon atoms, optionally one or more, usually 1 to about 5, preferably 1 to 3, particularly preferably 1 or 2, ethylenic double bonds and / or one or more, usually 1 to about 5, preferably 1 to 3, more preferably 1 or 2 identical or different
- Substituents selected from the group of the substituents OR 4 , NR 5 R 6 , halogen, C ⁇ -C-io-Arvl and Cs-Cg-hetaryl can carry and together with R 3 can form a 5 to 25-membered ring, stand, with the proviso that R 1 and R 2 are different,
- R 3 is hydrogen or an unbranched, branched or cyclic alkyl radical having 1 to 25 carbon atoms, optionally one or more, usually 1 to about 5, preferably 1 to 3, particularly preferably 1 or 2, ethylenic double bonds and / or one or several, usually 1 to about 5, preferably 1 to 3, particularly preferably 1 or 2, identical or different substituents selected from the group of the substituents OR 4 , NR 5 R 6 , halogen, Ce-do-aryl and C 3 - C 9 -hetaryl, or is OR 7 or NR 8 R 9 ,
- R 4 , R 5 , R 6 are each, independently of one another, hydrogen, C 1 -C 6 -alkyl, C 3 -C 4 -ione
- R 5 and R 6 together may also denote an alkylene chain of 2 to 5 carbon atoms, which may be interrupted by N or O, and
- R 7 is an unbranched, branched or cyclic alkyl radical having 1 to
- Substituents selected from the group of the substituents OR 4 , NR 5 R 6 , Halogen, C ⁇ -do-Ary! and Cs-Cg-hetaryl, and together with R 1 or R 2 can form a 5- to 25-membered ring and
- R 8 is an unbranched, branched or cyclic alkyl radical having 1 to 25 carbon atoms, optionally one or more, preferably
- R 1 , R 2 or R 9 is a 5- to 25-membered
- R 9 represents hydrogen, Ci -C 6 -alkyl, C 6 -C -aryl, C 7 -C 2 -aralkyl or C 7 -C 2 -
- Alkylaryl or together with R 8 can form a 5 to 25-membered ring and can
- radicals R 1 to R 3 have the abovementioned meaning.
- C 1 -C 6 -alkyl is, for example, methyl, ethyl, propyl, 1-methylethyl, butyl, 1-methylpropyl, 2-methylpropyl, 1, 1-dimethylethyl, pentyl, 1-methylbutyl, 2-methylbutyl, 3-methylbutyl , 2,2-dimethylpropyl, 1-ethylpropyl, hexyl, 1, 1-dimethylpropyl, 1, 2-dimethylpropyl, 1-methylpentyl, 2-methylpentyl, 3-methylpentyl, 4-methylpentyl, 1, 1-dimethylbutyl, 1 , 2-Dimethylbutyl, 1, 3-dimethylbutyl, 2,2-dimethylbutyl, 2,3-dimethylbutyl, 3,3-dimethylbutyl, 1-ethylbutyl, 2-ethylbutyl, 1, 1, 2-trimethylpropyl, 1, 2,2 Trimethylpropyl, 1-ethy
- C ⁇ -Cio-aryl is, for example, phenyl or naphthyl.
- C 7 -C 12 Aralkyl is, for example, phenylmethyl, 1-phenylethyl, 2-phenylethyl, 1-phenylpropyl, 2-phenylpropyl or 3-phenylpropyl.
- C 3 -C 9 -hetaryl is, for example, 2-furyl, 3-furyl, 2-pyrroyl, 3-pyrroyl, 2-thienyl, 3-thienyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-indolyl, 3 indolyl, 4-indolyl, 5-indolyl, 6-indolyl, 7-indolyl.
- C 7 -C 12 -alkylaryl is, for example, 1-methylphenyl, 2-methylphenyl, 3-methylphenyl, 1-ethylphenyl, 2-ethylphenyl, 3-ethylphenyl, 1-propylphenyl, 2-propylphenyl, 3-propylphenyl, 1-iso-propylphenyl, 2-iso-propylphenyl, 3-iso-propylphenyl, 1-butylphenyl, 2-butylphenyl, 3-butylphenyl, 1-iso-butylphenyl, 2-iso-butylphenyl, 3-iso-butylphenyl, 1-sec-butylphenyl, 2 sec-butylphenyl, 3-sec-butylphenyl, 1-tert-butylphenyl, 2-tert-butylphenyl, 3-tert-butylphenyl, 1- (1-pentenyl) phenyl, 2- (1
- halogen is to be understood as meaning fluorine, chlorine, bromine or iodine, preferably fluorine, chlorine or bromine.
- the process according to the invention is suitable for the preparation of optically active aldehydes or ketones by asymmetric hydrogenation of ⁇ , ⁇ -unsaturated aldehydes or ketones. Accordingly, it is particularly suitable for the preparation of optically active compounds of the formula (I ')
- R 1 ', R 2' may have the same meanings as above R 1 and R 2 and
- R 3 ' is hydrogen or an unbranched, branched or cyclic
- Alkyl radical having 1 to 25 carbon atoms, which optionally one or more, usually 1 to about 5, preferably 1 to 3, particularly preferably
- R 4 , R 5 and R 6 may have the meanings given above,
- the process according to the invention is preferably suitable for preparing optically active aldehydes of the formula (III) which have a methyl group in the ⁇ -position to the carbonyl group
- R 10 is an unbranched or branched alkyl radical having 2 to 25 carbon atoms, which may optionally have 1 to 5, preferably 1 to 3, particularly preferably 1 or 2, ethylenic double bonds, and denotes an asymmetric carbon atom,
- aldehydes or ketones of the formulas (I ') or (III) which can be prepared according to the invention in optically active form, the following compounds may be mentioned:
- aldehydes of the formula (III) are according to the invention by asymmetric, i. enantioselective hydrogenation of the corresponding ⁇ , ⁇ -unsaturated aldehydes of the formulas
- mixtures of the two double bond isomers can also be reacted in accordance with the invention. This gives mixtures of the two enantiomers of the desired target compound.
- the process according to the invention is particularly preferably suitable for the preparation of optically active citronellal of the formula (VI)
- mixtures of geranial and neral in accordance with the invention, wherein, as described above, mixtures of D- or L-citronellal are present which are optically active if the two enantiomers are not present in equal parts therein.
- the preparation according to the invention is particularly preferably D-citronellal by asymmetric hydrogenation of neral or geranial.
- the preparation process according to the invention is carried out in the presence of an optically active transition metal catalyst which has at least one carbon monoxide ligand and is soluble in the reaction mixture.
- Such catalysts are obtainable, for example, by reaction of at least one suitable transition metal compound soluble in the reaction mixture with an optically active ligand which has at least one phosphorus and / or arsenic atom.
- Preferred transition metal compounds are those of the metals of VIII.
- Subgroup of the Periodic Table of the Elements in particular Ru, Rh, Pd, Ir and Pt.
- Subgroup of the periodic table according to the invention are Rh and Ir.
- Suitable compounds of said transition metals are, in particular, those which are soluble in the chosen reaction medium, for example salts or complex compounds with suitable ligands such as carbonyl, acetylacetonate, hydroxy, cyclooctadiene, norbornadiene, cyclooctene, methoxy, acetyl or other aliphatic or aromatic carboxylates.
- suitable ligands such as carbonyl, acetylacetonate, hydroxy, cyclooctadiene, norbornadiene, cyclooctene, methoxy, acetyl or other aliphatic or aromatic carboxylates.
- Transition metal compounds which are preferred in the process according to the invention are Rh (I) and Rh (III) and Rh (O) compounds, Ir (I), Ir (III), Ir (IV) and Ir (0) compounds , Ru (II), Ru (III), Ru (IV) - and Ru (0) compounds, Pd (II) -, Pd (IV) - and Pd (0) compounds and Pt (II) -, Pt (IV) and Pt (0) compounds.
- Leave aside Transition-metal compounds which have no CO ligand can also be used in the context of the process according to the invention as starting compound for the preparation of the catalysts to be used according to the invention. These are converted into the desired catalysts under the conditions of the preforming or the hydrogenation conditions according to the invention optionally to be carried out with the addition of carbon monoxide.
- transition metal compounds which can be used according to the invention are: RhCl 3 , Rh 2 (OAc) 4 , [Rh (cod) Cl] 2 , Rh (CO) 2 acac, [Rh (cod) OH] 2 , [Rh (cod) OMe ] 2 , Rh 4 (CO) I 2 , Rh 6 (CO) -I 6 or Ir 4 (CO) -I 2 , [Ir (cod) Cl] 2 , wherein "acac” for a Acetylaceto- nat- and "Cod” stands for a cyclooctadiene ligand.
- the transition metal compounds mentioned are used according to the invention usually in an amount of from about 0.01 to about 1 mol%, preferably from about 0.05 to about 0.5 mol%, in particular from about 0.02 to about 0.2 mol%. % (based on the transition metal atoms contained) in relation to the amount of substrate to be hydrogenated.
- the ratio of the amount of transition metal compound used as precursor of the homogeneous catalyst according to the invention to the amount of substrate to be hydrogenated is advantageously chosen such that a catalyst concentration in the range from about 100 ppm to 10000 ppm, in particular in the range of about 200 ppm up to 5000 ppm is maintained.
- the said transition metal compounds are contacted according to the invention with another compound which is optically active, preferably substantially enantiomerically pure (i.e., has an enantiomeric excess of at least about 99%) and at least one phosphorus and / or arsenic atom, preferably at least one phosphorus atom.
- This compound referred to as the chiral ligand, forms in the reaction mixture or in the preforming mixture with the transition metal compound used, the transition metal catalyst to be used according to the invention.
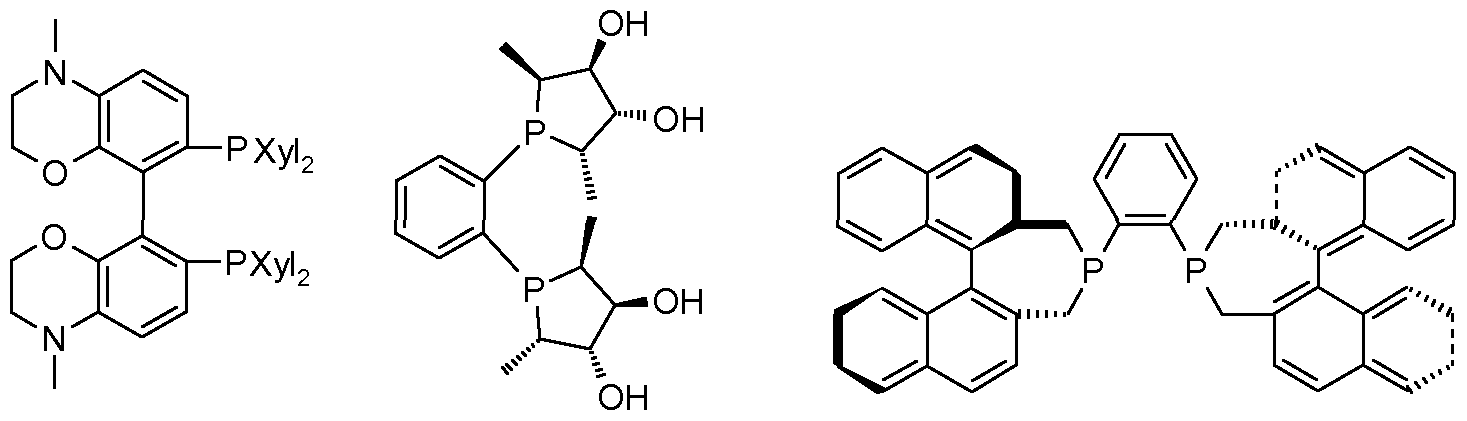
- Suitable chiral ligands in the context of the present invention are those compounds as described, for example, in: I. Ojima (ed.), Catalytic Asymmetry Synthesis, Wiley-VCh, 2nd edition, 2000 or in EN Jacobsen, A. Pfaltz, H. Yamamoto (ed.), Comprehensive Asymmetry Catalysis, 2000, Springer or in W. Tang, X. Zhang, Chem. Rev. 2003, 103, 3029-3069.
- Ph is phenyl
- Cy is cyclohexyl
- XyI is XyIyI
- ToI is p-tolyl
- Bn is benzyl.
- Particularly preferred ligands according to the invention are those of the structural formulas (1) to (13) and (37), (38), (41), (43), (49), (50), (51), (52), (65 ), (66), (67), (68), (69), (71), (72), (73), (74), (75), (83), (84), (85), (86), (87).
- Particularly preferred ligands are those of the general formulas (IX) to (XI)
- R 11 , R 12 are each, independently of one another, an unbranched, branched or cyclic alkyl radical having 1 to 20 carbon atoms which optionally has one or more, generally 1 to about 4, ethylenic double bonds and / or one or more, in the Rule 1 to about 4, identical or different substituents selected from the group of the substituents OR 19 , NR 20 R 21 , halogen, C ⁇ -Cio-aryl and Cs-Cg-hetaryl can carry and R 11 and R 12 together a 4- to 20-membered ring, which may include one or more, usually 1 or 2 O atoms, and
- R 13 , R 14 each independently of one another denote hydrogen or straight-chain or branched d- to C 4 -alkyl and
- R 15 , R 16 , R 17 , R 18 in each case C 1 to C 10 aryl optionally having one or more, generally from 1 to 8, preferably from 1 to 4, substituents selected from the group of the substituents d to C 4 -Al kyl, C ⁇ - to Cio-aryl, C 1 to C 4 alkoxy and amino can carry, stand and
- R 19 , R 20 , R 21 each independently of one another are hydrogen, C 1 -C 4 -alkyl, C 6 -C 10 -aryl, C 7 -C 12 -aralkyl or C 7 -C 12 -alkylaryl, where
- R 20 , R 21 : together may also denote an alkylene chain having 2 to 5 carbon atoms which may be interrupted by N or O.
- Particularly preferred ligands within the scope of the process according to the invention are those of the general formula (IX), in particular the compounds of the formula (1) which are referred to below as “chibrosses".
- the selected chiral ligands can be used according to the invention in each case in the form of their two enantiomers.
- a particularly preferred ligand according to the invention is (R, R) -chiraphos (ent (1)).
- chiral ligands having two phosphorus atoms these are advantageously used in an amount of about 1 to about 10 mol, preferably about 1 to about 4 mol, very particularly preferably 1 to 2 mol per mol equivalent of metal in the transition metal used Connection is included.
- the actual precatalysts containing at least one carbon monoxide ligand can be obtained by contacting and then pretreating with a mixture of hydrogen and carbon monoxide as described below.
- At least one carbon monoxide ligand-containing catalyst is treated according to step a) of the process according to the invention, a catalyst precursor with a gas mixture comprising 20 to 90 vol .-% carbon monoxide, 10 to 80% by volume of hydrogen and 0 to 5 vol .-% of other gases, wherein said volume percentages to 100 vol .-% complement, at a pressure of 5 to 100 bar ago.
- This pretreatment is also referred to below as preforming as in the context of the present invention.
- catalyst precursor is to be understood as meaning those compounds which are obtainable by contacting or reacting at least one transition metal compound which is soluble in the reaction mixture as described above with an optically active ligand as mentioned above which contains at least one phosphorus and / or or arsenic atom.
- the substrate to be asymmetrically hydrogenated in a suitable inert solvent or solvent medium, for example ether, tetrahydrofuran, toluene, chlorobenzene, octadecanol, under the reaction conditions.
- a suitable inert solvent or solvent medium for example ether, tetrahydrofuran, toluene, chlorobenzene, octadecanol
- the solution medium may also be the substrate to be reacted, the product or any high-boiling by-products obtained during the reaction.
- the resulting solution is, advantageously in a suitable pressure reactor or autoclave, at a pressure in the range of 5 to 100 bar (absolute), preferably at a pressure of 10 to 100 bar, more preferably at a pressure of 20 to 95 bar and in particular preferably compressed at a pressure of 50 to 90 bar (each absolute), a gas mixture containing hydrogen and carbon monoxide as described above.
- a particularly preferred gas mixture for preforming is so-called synthesis gas, which usually consists of about 35 to 55% by volume of carbon monoxide and about 45 to 65% by volume of hydrogen and optionally traces of other gases.
- the preformation of the catalyst according to the invention is usually at temperatures of about 25 ° C to about 100 0 C, preferably at about 40 0 C to about 80 ° C, particularly preferably carried out at 50-70 0 C. If the preformation is carried out in the presence of the substrate to be asymmetrically hydrogenated, the temperature is advantageously chosen so that no isomerization of the double bond to be hydrogenated occurs to any extent.
- the preforming is usually completed after about 1 to about 24 hours, often after about 1 to about 12 hours.
- step b) of the process according to the invention excess carbon monoxide is separated from the catalyst obtained by preforming or pretreatment with said gas mixture before use in the asymmetric hydrogenation.
- excess carbon monoxide is to be understood as meaning carbon monoxide which is present in gaseous or dissolved form in the reaction mixture obtained by preforming in step a) and is not bound to the transition metal catalyst or its precursor. Accordingly, excess excess carbon monoxide not bound to the catalyst is at least widely removed. ie, to the extent that any residual amounts of dissolved carbon monoxide do not interfere in the subsequent hydrogenation. This is usually ensured when about 90%, preferably about 95% or more, of the carbon monoxide used for preformation is removed in step b) of the process according to the invention. Preferably, according to step b) of the process according to the invention, excess carbon monoxide is completely removed from the catalyst obtained by preforming.
- the separation of the excess carbon monoxide from the catalyst obtained according to step a) or from the catalyst-containing reaction mixture according to step b) of the process according to the invention can be carried out in various ways.
- the catalyst or the mixture containing the catalyst obtained by preforming according to step a) is preferably expanded to a pressure of up to about 5 bar (absolute), preferably, especially, when preforming is carried out in the pressure range from 5 to 10 bar , to a pressure of less than 5 bar (absolute), preferably to a pressure in the range of about 1 bar to about 5 bar, preferably 1 to less than 5 bar, more preferably to a pressure in the range of 1 to 3 bar, completely particularly preferably to a pressure in the range of about 1 to about 2 bar, more preferably to atmospheric pressure, so that gaseous, unbound carbon monoxide escapes from the product of the preforming.
- the abovementioned expansion of the preformed catalyst can be carried out, for example, by using a high-pressure separator, as is known per se to the person skilled in the art.
- a high-pressure separator as is known per se to the person skilled in the art.
- Such separators in which the liquid is in the continuous phase are described, for example, in: Perry's Chemical Engineers' Handbook, 1997, 7th ed., McGraw-Hill, pp. 14.95 and 14.96; the prevention of a possible drop rash is described on pages 14.87 to 14.90.
- the relaxation of the preformed catalyst can be carried out in one or two stages until reaching the desired pressure in the range of 1 bar to about 5 bar, wherein the temperature usually drops to 10 to 40 0 C.
- the removal of excess carbon monoxide in step b) can also be achieved by so-called stripping of the catalyst or of the mixture containing the catalyst with a gas, advantageously with a gas which is inert under the reaction conditions.
- stripping the expert understands the introduction of a gas into the catalyst or the reaction mixture containing the catalyst such as in WRA Vauck, HA Müller, basic operations chemical engineering, German Verlag für Grundstoffchemie für, Stuttgart, 10th edition, 1984, page 800 described.
- suitable inert gases include: hydrogen, helium, neon, argon, xenon, nitrogen and / or CO 2, preferably hydrogen, nitrogen, argon.
- step c) the asymmetric hydrogenation of the selected substrate in the presence of hydrogen having a carbon monoxide content of 100 to 1200 ppm is carried out in step c) of the process according to the invention.
- the addition of additional carbon monoxide to the reaction mixture of the asymmetric hydrogenation can be carried out in various ways:
- the carbon monoxide can be added to the hydrogen used for asymmetric hydrogenation or metered directly into the reaction solution in gaseous form.
- Another possibility is, for example, to add to the reaction mixture compounds that easily release carbon monoxide, such as formates or oxalyl compounds.
- the proportion of carbon monoxide in the hydrogen used is within the scope of a preferred embodiment of the method according to the invention about 300 to 1000 ppm, more preferably 400 to 800 ppm.
- the asymmetric hydrogenation according to the invention is advantageously carried out at a pressure of about 1 to about 300 bar, preferably from about 10 to about 100 bar, in particular at about 50 to about 100 bar, more preferably at about 60 to about 100 bar and a temperature of usually about 0 0 C to about 100 0 C, preferably about 0 0 C to about 30 0 C, especially at about 10 ° C to about 30 0 C before.
- Suitable solvents are, for example, those mentioned for carrying out the preforming according to the invention.
- the asymmetric hydrogenation is carried out in the same solvent as the optionally previously performed preforming.
- Suitable reaction vessels for carrying out the asymmetric hydrogenation according to the invention are in principle all those which permit reactions under the conditions mentioned, in particular pressure and temperature, and are suitable for hydrogenation reactions, such as, for example, autoclaves, tubular reactors, bubble columns, etc.
- step c) of the process according to the invention using high-boiling, usually viscous solvents, as described, for example, above for use in the pretreatment of the catalyst according to step a) of the method according to the invention (such as the Solvent octadecanol, biphenyl ether, Texanol, Marlotherm ® , Oxoöl 9N) or the hydrogenation is carried out without additional use of solvent but with the accumulation of by-products to a small extent high boiling point (As dimers or trimers, which are formed by reactions of the reactants or products and subsequent subsequent reactions), it may be advantageous to ensure good gas input and good mixing of the gas phase and condensed phase.
- high-boiling usually viscous solvents
- Gas circulation reactors are known per se to the person skilled in the art and are described, for example, in P. Trambouze, J. -P. Euzen, Chemical Reactors, Ed. Technip, 2004, pp. 280-283 and P. Zehner, R. Benfer, Chem. Eng. Be. 1996, 51, 1735-1744 and z. As described in EP 1 140 349.
- the gas or gas mixture carbon monoxide-containing hydrogen
- the two-fluid nozzle is characterized in that to be introduced into the reactor
- Liquid and gas pass through two separate nested tubes under pressure to the nozzle mouth where they are combined.
- the amount of amine used may be between 0.5 and 500 molar equivalents based on the amount of metal used, but preferably 1 to 100 molar equivalents based on the amount of metal used.
- the choice of tertiary amine is not critical. In addition to short-chain alkylamines, such as triethylamine and long-chain alkyl amines, such as tridodecylamine, can be used.
- the hydrogenation process according to the invention is carried out in the presence of a tertiary amine, preferably tridodecylamine, in an amount of about 2 to 30 molar equivalents, preferably about 5 to 20 molar equivalents and more preferably 5 to 15 molar equivalents the amount of transition metal used.
- a tertiary amine preferably tridodecylamine
- the reaction is stopped when the target compound in the desired yield and optical activity, i. is present in the reaction mixture with the desired enantiomeric excess (ee), as can be ascertained by the person skilled in the art by routine examinations, for example by means of chromatographic methods.
- the hydrogenation is completed after about 1 to about 150 hours, often after about 2 to about 24 hours.
- the process according to the invention makes it possible to prepare optically active carbonyl compounds, in particular optically active aldehydes in high yields and enantiomeric compounds. to provide surpluses.
- the desired asymmetrically hydrogenated compounds are obtained in an enantiomeric excess of at least 80% ee, often with an enantiomeric excess of from about 85 to about 99% ee. It should be noted that the maximum achievable enantiomeric excess may depend on the purity of the substrate used, in particular with regard to the isomeric purity of the double bond to be hydrogenated.
- suitable starting substances in particular those having an isomer ratio of at least about 90:10, preferably at least about 95: 5 with respect to the E / Z double bond isomers.
- the inventive method is characterized by the fact that the homogeneous catalysts used are stabilized by the additionally introduced into the reaction system carbon monoxide, whereby on the one hand the service life of the catalysts is significantly increased and on the other the reusability of the homogeneous catalysts is made possible.
- the resulting reaction product can be prepared by methods known to those skilled in the art, e.g. Remove more from the reaction mixture by distillation, for example by means of a fine film evaporator, Sambay or the like and use the remaining catalyst, optionally after repeated, as described above preforming in the context of further reactions.
- the process according to the invention can be operated batchwise or semicontinuously as well as continuously and is particularly suitable for reactions on an industrial scale.
- the process is carried out continuously.
- step a) The pretreatment of the catalyst precursor (preformation) according to step a) to be carried out according to the invention and the actual asymmetric hydrogenation in step c) are advantageously carried out in separate reaction vessels.
- the excess carbon monoxide can then be removed from the catalyst, for example by depressurizing the pressure used for the preforming.
- the hydrogenation can also take place in several, preferably in two or three, particularly preferably in two, hydrogenation reactors connected in series. Different or identical reactor types can be used.
- the asymmetric hydrogenation is carried out, for example, in a cascade of two gas circulation reactors, one acting as the main reactor and the second as the postreactor.
- To transfer the reaction mixture from Main reactor in the secondary reactor can be used, for example, if desired, to be set pressure gradient.
- neral or geranial preferably neral, which contains up to about 5 mol%, preferably up to about 2 mol% of the respective double bond isomer, is converted to optically active citronellal.
- a rhodium compound which is soluble in the reaction mixture preference is given to using a rhodium compound which is soluble in the reaction mixture, in particular Rh 2 (OAc) 4 , [Rh (COd) Cl] 2 , Rh (CO) 2 acac, [Rh (cod) OH] 2 , [Rh (cod) OMe] 2 , Rh 4 (CO) i 2 or Rh 6 (CO) i 6 and as chiral ligands (R, R) -chiraphos or (S, S) -chiraphos ((2R, 3R) - ( +) - 2,3-bis (diphenylphosphino) butane or (2S, 3S) - (-) - 2,3-bis (diphenylphosphino) butane) in a molar ratio of about 1: 1 to about 1: 4 relative to rhodium one.
- Rh 2 (OAc) 4 Rh (COd) Cl] 2
- neral containing up to about 5 mol%, preferably up to about 2 mol%, of geranial is used in the presence of Rh (OAc) 3 , [Rh (COd) Cl] 2 , Rh (CO) 2 acac, [Rh (cod) OH] 2 , [Rh (cod) OMe] 2 , Rh 4 (CO) I 2 or Rh 6 (CO) -I 6 and (R, R) -chiraphos to D-Citronellal order.
- a further aspect of the present invention relates to a process for the preparation of optically active menthol using optically active citronellal prepared by the process according to the invention.
- the preparation of optically active menthol starting from optically active citronellal is known.
- a key step here is the cyclization of optically active citronellal to optically active isopulegol, as described for example in EP-A 1 225 163.
- optically active citronellal prepared according to the invention can be cyclized, as shown schematically below for the preparation of L-menthol of the formula (XIII), in the presence of a suitable acid, in particular a Lewis acid, to form L-isopulegol of the formula (XII) and subsequently methods known to the person skilled in the art, for example by catalytic hydrogenation, as described, for example, in J. Am. Chem. Soc. 1984, 106, 5208-5217 or Synthesis 1991, 665-680. hydrogenated to L-menthol.
- Another aspect of the present invention accordingly relates to a process for producing optically active menthol comprising the steps
- R-citronellal is prepared by hydrogenation of geranial or neral, preferably neral, according to the invention, the resulting R-citronellal is cyclized according to step ii) to give L-isopulegol and hydrogenated in step iii) obtained L-isopulegol to L-menthol.
- the yield of D-citronellal was based on the cis-citral used only 86%.
- the ratio of hydrogen to carbon monoxide was set to 1: 1 in the preforming reactor at a pressure of 80 bar and a temperature of 60 ° C.
- the discharge of the preforming reactor was expanded to atmospheric pressure in a high-pressure separator and then compressed to 80 bar in the hydrogenation reactor.
- 60 Nl / h of H2 (with 245 ppm CO) were introduced into the hydrogenation reactors at 20 ° C., so that a CO value of about 600 ppm was established in the offgas of the hydrogenation reactor.
- the hydrogenation reactor was charged with 60.3 Nl / h Hb with 265 ppm CO, wherein in the exhaust gas of the hydrogenation catalyst, a CO value of about 1300 ppm was established. In this way 2.87 moles (441.9 g) of D-citronellal were isolated within 5 days. The yield of D-citronellal was based on the cis-citral used 90%.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
Description




























Claims






Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2008800136150A CN101675020B (zh) | 2007-04-25 | 2008-04-17 | 合成光学活性羰基化合物的方法 |
| JP2010504637A JP5479320B2 (ja) | 2007-04-25 | 2008-04-17 | 光学活性カルボニル化合物の調製方法 |
| EP08736312.3A EP2139835B1 (de) | 2007-04-25 | 2008-04-17 | Verfahren zur herstellung optisch aktiver carbonylverbindungen |
| ES08736312.3T ES2636840T3 (es) | 2007-04-25 | 2008-04-17 | Procedimiento para la preparación de compuestos carbonílicos ópticamente activos |
| US12/597,025 US7973198B2 (en) | 2007-04-25 | 2008-04-17 | Method for synthesizing optically active carbonyl compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07106922.3 | 2007-04-25 | ||
| EP07106922 | 2007-04-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008132057A1 true WO2008132057A1 (de) | 2008-11-06 |
Family
ID=39592078
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2008/054644 WO2008132057A1 (de) | 2007-04-25 | 2008-04-17 | Verfahren zur herstellung optisch aktiver carbonylverbindungen |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7973198B2 (de) |
| EP (1) | EP2139835B1 (de) |
| JP (1) | JP5479320B2 (de) |
| CN (1) | CN101675020B (de) |
| ES (1) | ES2636840T3 (de) |
| WO (1) | WO2008132057A1 (de) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20130089493A1 (en) * | 2010-06-15 | 2013-04-11 | Youl Chon Chemical Co., Ltd. | Device and method for generating hydrogen from an ammonia borane-based compound hydrogen reservoir, a catalyst used with the same, and a device for using emitted hydrogen |
| WO2014167014A1 (de) * | 2013-04-10 | 2014-10-16 | Basf Se | Verfahren zur herstellung von citronellal |
| WO2016097242A1 (de) * | 2014-12-19 | 2016-06-23 | Basf Se | Verfahren zur herstellung optisch aktiver carbonylverbindungen |
| WO2020048998A1 (de) | 2018-09-05 | 2020-03-12 | Basf Se | Verfahren zur durchführung einer gas/flüssig-zweiphasigen hochdruckreaktion |
| WO2020048975A1 (de) | 2018-09-05 | 2020-03-12 | Basf Se | Kontinuierliche herstellung einer optisch aktiven carbonylverbindung durch asymmetrische hydrierung |
| WO2020048991A1 (de) | 2018-09-05 | 2020-03-12 | Basf Se | Reaktor zur durchführung einer gas/flüssig-zweiphasigen hochdruckreaktion mit einem schäumenden medium |
| WO2020048986A1 (de) | 2018-09-05 | 2020-03-12 | Basf Se | Reaktor zur durchführung einer reaktion zwischen zwei nicht mischbaren fluiden unterschiedlicher dichte |
| US10919921B2 (en) | 2016-05-06 | 2021-02-16 | Basf Se | P-chiral phosphine ligands and use thereof for asymmetric synthesis |
| WO2021114021A1 (zh) * | 2019-12-09 | 2021-06-17 | 万华化学集团股份有限公司 | 一种光学活性的香茅醛的制备方法 |
| WO2021209415A1 (en) | 2020-04-14 | 2021-10-21 | Basf Se | Hydrogenation of l-sorbose |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102008015773A1 (de) * | 2008-03-26 | 2009-10-01 | Albert-Ludwigs-Universität Freiburg | Verfahren zur decarboxylativen Hydroformylierung alpha,beta-ungesättigter Carbonsäuren |
| WO2012074128A1 (en) * | 2010-11-29 | 2012-06-07 | Takasago International Corporation | Catalyst for asymmetric hydrogenation and method for manufacturing optically active carbonyl compound using the same |
| JP5560464B2 (ja) * | 2010-11-29 | 2014-07-30 | 高砂香料工業株式会社 | 不斉水素化触媒 |
| CN105218335B (zh) * | 2015-10-20 | 2017-06-16 | 万华化学集团股份有限公司 | 一种由柠檬醛不对称催化氢化制备手性香茅醛的方法 |
| CN105541579B (zh) * | 2015-12-30 | 2018-07-17 | 浙江新和成股份有限公司 | 一种制备光学活性羰基化合物的方法 |
| CN111056932A (zh) * | 2019-12-09 | 2020-04-24 | 万华化学集团股份有限公司 | 一种制备光学活性香茅醛的方法 |
| CN110872217A (zh) * | 2019-12-09 | 2020-03-10 | 万华化学集团股份有限公司 | 一种光学活性的香茅醛的制备方法 |
| CN111004102B (zh) * | 2019-12-23 | 2022-11-04 | 万华化学集团股份有限公司 | 一种制备光学活性香茅醛的方法及用于该方法的催化剂 |
| CN111056933B (zh) * | 2019-12-24 | 2022-11-08 | 万华化学集团股份有限公司 | 一种制备光学活性香茅醛的方法及用于该方法的催化剂体系 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006040096A1 (de) * | 2004-10-11 | 2006-04-20 | Basf Aktiengesellschaft | Verfahren zur herstellung optisch aktiver carbonylverbindungen |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5278812A (en) * | 1975-12-26 | 1977-07-02 | Nikki Chem Co Ltd | Selective production method of saturated aldehyde |
| FR2396735A1 (fr) * | 1977-07-04 | 1979-02-02 | Rhone Poulenc Ind | Procede de preparation de citronellal optiquement actif |
| JPH05278812A (ja) | 1992-04-06 | 1993-10-26 | Murata Mach Ltd | スタッカクレーン |
| DE19854637A1 (de) | 1998-11-26 | 2000-05-31 | Basf Ag | Reaktor zur kontinuierlichen Durchführung von Gas-Flüssig-, Flüssig-Flüssig- oder Gas-Flüssig-Fest-Reaktionen |
| JP4676617B2 (ja) | 2001-01-18 | 2011-04-27 | 高砂香料工業株式会社 | イソプレゴールの製造方法 |
| DE102005036340A1 (de) | 2005-07-29 | 2007-02-01 | Basf Ag | Verfahren zur Herstellung optisch aktiver Bis-Phosphinylalkane |
| DE102005040655A1 (de) | 2005-08-26 | 2007-03-01 | Basf Ag | Verfahren zur Herstellung von angereichertem Isopulegol |
-
2008
- 2008-04-17 WO PCT/EP2008/054644 patent/WO2008132057A1/de active Application Filing
- 2008-04-17 EP EP08736312.3A patent/EP2139835B1/de active Active
- 2008-04-17 CN CN2008800136150A patent/CN101675020B/zh active Active
- 2008-04-17 JP JP2010504637A patent/JP5479320B2/ja active Active
- 2008-04-17 ES ES08736312.3T patent/ES2636840T3/es active Active
- 2008-04-17 US US12/597,025 patent/US7973198B2/en active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006040096A1 (de) * | 2004-10-11 | 2006-04-20 | Basf Aktiengesellschaft | Verfahren zur herstellung optisch aktiver carbonylverbindungen |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8926940B2 (en) * | 2010-06-15 | 2015-01-06 | Youl Chon Chemical Co. Ltd. | Device and method for generating hydrogen from an ammonia borane-based compound hydrogen reservoir, a catalyst used with the same, and a device for using emitted hydrogen |
| US20130089493A1 (en) * | 2010-06-15 | 2013-04-11 | Youl Chon Chemical Co., Ltd. | Device and method for generating hydrogen from an ammonia borane-based compound hydrogen reservoir, a catalyst used with the same, and a device for using emitted hydrogen |
| WO2014167014A1 (de) * | 2013-04-10 | 2014-10-16 | Basf Se | Verfahren zur herstellung von citronellal |
| WO2016097242A1 (de) * | 2014-12-19 | 2016-06-23 | Basf Se | Verfahren zur herstellung optisch aktiver carbonylverbindungen |
| US9975837B2 (en) | 2014-12-19 | 2018-05-22 | Basf Se | Method for synthesizing optically active carbonyl compounds |
| US10301244B2 (en) | 2014-12-19 | 2019-05-28 | Basf Se | Method for synthesizing optically active carbonyl compounds |
| EP3489213A1 (de) | 2014-12-19 | 2019-05-29 | Basf Se | Zusammensetzung zur verwendung in einem verfahren zur herstellung optisch aktiver carbonylverbindungen |
| USRE49036E1 (en) | 2014-12-19 | 2022-04-19 | Basf Se | Method for synthesizing optically active carbonyl compounds |
| US10919921B2 (en) | 2016-05-06 | 2021-02-16 | Basf Se | P-chiral phosphine ligands and use thereof for asymmetric synthesis |
| WO2020048975A1 (de) | 2018-09-05 | 2020-03-12 | Basf Se | Kontinuierliche herstellung einer optisch aktiven carbonylverbindung durch asymmetrische hydrierung |
| WO2020048986A1 (de) | 2018-09-05 | 2020-03-12 | Basf Se | Reaktor zur durchführung einer reaktion zwischen zwei nicht mischbaren fluiden unterschiedlicher dichte |
| WO2020048991A1 (de) | 2018-09-05 | 2020-03-12 | Basf Se | Reaktor zur durchführung einer gas/flüssig-zweiphasigen hochdruckreaktion mit einem schäumenden medium |
| WO2020048998A1 (de) | 2018-09-05 | 2020-03-12 | Basf Se | Verfahren zur durchführung einer gas/flüssig-zweiphasigen hochdruckreaktion |
| US11766653B2 (en) | 2018-09-05 | 2023-09-26 | Basf Se | Method for carrying out a gas/fluid two-phase high-pressure reaction |
| US11884611B2 (en) | 2018-09-05 | 2024-01-30 | Basf Se | Reactor for carrying out a gas-liquid two-phase high-pressure reaction with a foaming medium |
| WO2021114021A1 (zh) * | 2019-12-09 | 2021-06-17 | 万华化学集团股份有限公司 | 一种光学活性的香茅醛的制备方法 |
| WO2021209415A1 (en) | 2020-04-14 | 2021-10-21 | Basf Se | Hydrogenation of l-sorbose |
Also Published As
| Publication number | Publication date |
|---|---|
| US20100152494A1 (en) | 2010-06-17 |
| JP5479320B2 (ja) | 2014-04-23 |
| CN101675020B (zh) | 2013-11-06 |
| ES2636840T3 (es) | 2017-10-09 |
| US7973198B2 (en) | 2011-07-05 |
| EP2139835A1 (de) | 2010-01-06 |
| EP2139835B1 (de) | 2017-05-10 |
| JP2010525008A (ja) | 2010-07-22 |
| CN101675020A (zh) | 2010-03-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2139835B1 (de) | Verfahren zur herstellung optisch aktiver carbonylverbindungen | |
| EP1802561B1 (de) | Verfahren zur herstellung optisch aktiver carbonylverbindungen | |
| EP3233778B1 (de) | Verfahren zur herstellung optisch aktiver carbonylverbindungen | |
| EP3419954B1 (de) | Verfahren zur herstellung von terpinen-4-ol | |
| WO2011110249A1 (de) | Verfahren zur herstellung von linearen alpha,omega-dicarbonsäurediestern | |
| EP2459579B1 (de) | Imidazolgruppenhaltige phosphinoboran-verbindungen und verfahren zur herstellung von imidazolgruppenhaltige phosphorverbindungen | |
| EP3452488A1 (de) | P-chirale phosphinliganden und deren verwendung zur asymmetrischen synthese | |
| DE2943098C2 (de) | ||
| WO2009101162A1 (de) | Imidazolgruppenhaltige phosphorverbindungen | |
| EP1694621A1 (de) | Verfahren zur herstellung von tricyclodecandialdehyd | |
| EP0780157B1 (de) | Rutheniumkomplexe mit einem chiralen, zweizähnigen Phosphinoxazolin-Liganden zur enantioselektiven Transferhydrierung von prochiralen Ketonen | |
| EP3994117B1 (de) | Hydrierung von estern zu alkoholen in gegenwart eines ru-pnn-komplexes | |
| DE10349399A1 (de) | Verfahren zur Reduktion von Binaphthylderivaten | |
| DE69505226T2 (de) | Verfahren zur herstellung von (+)-(1r)-cis-3-oxo-2-pentyl-1-cyclopentanessigsäure | |
| EP1595885A2 (de) | Chirale Diphosphorverbindungen und deren Übergangsmetallkomplexe | |
| WO2001009147A1 (de) | Neue chirale phosphorliganden und ihre verwendung in der herstellung optisch aktiver produkte | |
| EP2838872B1 (de) | Verfahren zur herstellung von verzweigten alkoholen | |
| EP1861352B1 (de) | Verfahren zur herstellung von optisch aktiven 3-phenylpropionsäurederivaten und folgeprodukte davon | |
| WO2014012908A1 (de) | Übergangsmetallcarbenkomplex- katalysiertes verfahren zur herstellung von carbonsäureestern aus alkoholen unter dehydrierung | |
| EP3847151B1 (de) | Kontinuierliche herstellung einer optisch aktiven carbonylverbindung durch asymmetrische hydrierung | |
| EP2657216B1 (de) | Verfahren zum Umsetzen von Farnesol zu Nerolidol in Gegenwart von alpha-Bisabolol | |
| EP2855409B1 (de) | Verfahren zur aufarbeitung von gemischen | |
| DE68908450T2 (de) | Organische Phosphorverbindungen und Verfahren zu ihrer Herstellung. | |
| WO2006002999A2 (de) | Verfahren zur herstellung von optisch aktiven alkylbernsteinsäuremonoalkylestern |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200880013615.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08736312 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| REEP | Request for entry into the european phase |
Ref document number: 2008736312 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008736312 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12597025 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010504637 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 6938/CHENP/2009 Country of ref document: IN |