WO2008003378A1 - Sulfamat-benzothiophen-derivate - Google Patents
Sulfamat-benzothiophen-derivate Download PDFInfo
- Publication number
- WO2008003378A1 WO2008003378A1 PCT/EP2007/004962 EP2007004962W WO2008003378A1 WO 2008003378 A1 WO2008003378 A1 WO 2008003378A1 EP 2007004962 W EP2007004962 W EP 2007004962W WO 2008003378 A1 WO2008003378 A1 WO 2008003378A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- salts
- compounds
- solvates
- tautomers
- compound
- Prior art date
Links
- 0 COc1ccc(cc(*=N)[s]2)c2c1 Chemical compound COc1ccc(cc(*=N)[s]2)c2c1 0.000 description 8
- VHVRCXIIFVHBFI-UHFFFAOYSA-N COc1ccc(cc(C=O)[s]2)c2c1 Chemical compound COc1ccc(cc(C=O)[s]2)c2c1 VHVRCXIIFVHBFI-UHFFFAOYSA-N 0.000 description 2
- KMKHDPYOFHIPAW-UHFFFAOYSA-N CC(C)(C)CC(S(c1c2)(=O)=O)=Cc1ccc2OC Chemical compound CC(C)(C)CC(S(c1c2)(=O)=O)=Cc1ccc2OC KMKHDPYOFHIPAW-UHFFFAOYSA-N 0.000 description 1
- RWQWMBHCANFIBV-UHFFFAOYSA-N CC(c([s]c1c2)cc1ccc2OC)O Chemical compound CC(c([s]c1c2)cc1ccc2OC)O RWQWMBHCANFIBV-UHFFFAOYSA-N 0.000 description 1
- DNIIXTNQTJQIKY-UHFFFAOYSA-N CCCC(S(c1c2)(=O)=O)=Cc1ccc2OC Chemical compound CCCC(S(c1c2)(=O)=O)=Cc1ccc2OC DNIIXTNQTJQIKY-UHFFFAOYSA-N 0.000 description 1
- YLTVECJKMOXCFU-UHFFFAOYSA-N CCCC1=Cc(ccc(O)c2)c2S1(=O)=O Chemical compound CCCC1=Cc(ccc(O)c2)c2S1(=O)=O YLTVECJKMOXCFU-UHFFFAOYSA-N 0.000 description 1
- BRKKCWMGFHIKGU-UHFFFAOYSA-N CCCC1=Cc(ccc(OS(N)(=O)=O)c2)c2S1(=O)=O Chemical compound CCCC1=Cc(ccc(OS(N)(=O)=O)c2)c2S1(=O)=O BRKKCWMGFHIKGU-UHFFFAOYSA-N 0.000 description 1
- WGDVDMKNSDCNGB-UHFFFAOYSA-N COc1cc([s]cc2)c2cc1 Chemical compound COc1cc([s]cc2)c2cc1 WGDVDMKNSDCNGB-UHFFFAOYSA-N 0.000 description 1
- ZPGUHASFFYDIDX-UHFFFAOYSA-N COc1ccc(cc([s]2)Br)c2c1 Chemical compound COc1ccc(cc([s]2)Br)c2c1 ZPGUHASFFYDIDX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/54—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/50—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D333/52—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes
- C07D333/54—Benzo[b]thiophenes; Hydrogenated benzo[b]thiophenes with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to carbon atoms of the hetero ring
- C07D333/56—Radicals substituted by oxygen atoms
Definitions
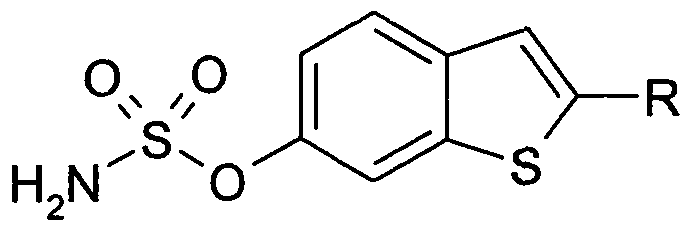
- the invention relates to novel compounds of the formula (I)
- R is cycloalkyl- (CH 2 ) m-, (AA ' R 1 ) C- (CH 2 ) n-, cycloalkylidene- (CH 2 ) n -,
- a 1 A 1 are each independently of the other alkyl of 1 to 4 carbon atoms m is 2, 3 or 4, n is 1, 2, 3 or 4, o is 0, 1, 2 or 3
- Tautomers including mixtures thereof in all proportions.
- the object of the invention was to find new compounds with valuable properties, in particular those used for the production of medicaments.
- WO2004 / 101545 A1 wherein all compounds are inhibitors of steroid sulphatase.
- the enzyme steroid sulphatase (EC 3.1.6.2., STS) catalyzes the hydrolysis of estrone sulfate to estrone and of DHEA sulphate to DHEA (Dibbelt L, Biol. Chem, Hoppe-Seyler, 1991, 372, 173-185 and Stein C, J Biol. Chem., 1989, 264, 13865 13872).
- Aromatase inhibitors used to prevent estrogen synthesis were used to prevent estrogen synthesis. Clinical studies, however, showed a comparatively lack of efficacy in patients with estrogen receptor-positive tumors (Castiglione-Gertsch M, Eur. J. Cancer, 1996, 32A, 393-395 and Jonat W. Eur. J. Cancer, 1996, 32A, 404 -412). This could be explained by the steroid sulfatase pathway being another important pathway for estrogen formation in breast tumors.
- estrone-3-sulfamate is the classic standard steroid sulfatase inhibitor, but with the main drawback of being estrogenic due to its inhibitory mechanism is: the sulfamate unit is cleaved during the enzyme inactivation process, whereby E 1 does not consist of E 1 S, but is released from EMATE itself (Ahmed SJ Steroid Biochem., Mol. Biol., 2002, 80, 429-440).
- non-steroid sulfamate compounds that release derivatives without estrogenic properties are presented as acceptable drug candidates, notably 6,6,7-COUMATE, a non-estrogenic standard sulfatase inhibitor from the literature (Purohit A, Cancer Res., 2000 , 60, 3394-3396).
- the invention also relates to the hydrates and solvates of these compounds.
- Solvates of the compounds are understood to mean additions of inert solvent molecules to the compounds which form due to their mutual attraction. Solvates are e.g. Mono or dihydrate or alcoholates.
- compositions are understood, for example, as the salts of the compounds according to the invention as well as so-called prodrug compounds. Under prodrug derivatives is understood with z.
- prodrug derivatives is understood with z.
- sugars or oligopeptides are understood, for example, as the salts of the compounds according to the invention as well as so-called prodrug compounds.
- prodrug derivatives is understood with z.
- modified alkyl or acyl groups sugars or oligopeptides
- an effective amount means the amount of a drug or pharmaceutical agent that elicits a biological or medical response in a tissue, system, animal, or human, such as is sought or sought by a researcher or physician.
- an effective amount means an amount that, compared to a corresponding subject who has not received this amount, results in: improved curative treatment, cure, prevention or elimination of a disease, a disease, a disease state, a condition, a disorder, or of The term “therapeutically effective amount” also includes the amounts that are effective to increase normal physiological function, or even the reduction of the progression of a disease, condition or disorder.
- the invention relates to the compounds of the formula (I) and their salts, and to a process for the preparation of compounds of the formula (I) and their pharmaceutically usable derivatives, salts and solvates, characterized in that
- R has the meaning given in the general formula (I) according to claim 1, reacted with sulfamoyl chloride (H 2 N-SO 2 -CI); or
- A, A 1 independently of one another, are alkyl having 1, 2, 3 or 4
- C atoms is unbranched (linear) or branched, and is preferred
- Cycloalkyl has 3 to 8 C atoms and is cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl or cyclooctyl, preferably cyclopentyl, cyclohexyl or cycloheptyl, particularly preferably cycloheptyl or cyclohexyl.
- Cycloalkylidene has 3 to 8 carbon atoms and is cyclopropylidene
- n 1, 2, 3 or 4, preferably 1 or 2 and most preferably 1.
- o is 0, 1, 2, or 3, preferably 0, 1 or 2 and most preferably 0.
- the invention relates in particular to those compounds of the formula (I) in which at least one of the radicals mentioned has one of the preferred meanings given above.
- Some preferred groups of compounds can be expressed by the following partial formulas Ia to Ik which correspond to the formula (I) and in which the unspecified radicals have the meaning given in the formula (I) but in which
- R is cyclohexyl- (CH 2 ) m - or cycloheptyl- (CH 2 ) m- and m is 2 or 3;
- A, A ' are each independently of the other alkyl of 1 and / or 2 C atoms, n is 1 or 2;
- Ic R is cyclohexylidene- (CH 2 ) n- or cycloheptylidene- (CH 2 ) n - and n is 1 or 2;
- R is cyclohexyl- (CH 2 ) O CO- or cycloheptyl- (CH 2 ) O CO- and o is 0 or 1;
- Compounds of formula (I) may preferably be obtained by reacting compounds of formula (II) with sulfamoyl chloride or oxidizing compounds of formula (III).
- the compounds of the formula (II) and of the formula (III) are generally known. If they are new, they can be produced by methods known per se.
- reaction of the compounds of the formula (II) with sulfamoyl chloride is carried out in an inert solvent.
- reaction time is between a few minutes and 14 days depending on the conditions used, the reaction temperature between about
- Suitable inert solvents are, for example, hydrocarbons such as hexane, petroleum ether, benzene, toluene or xylene; chlorinated hydrocarbons such as trichlorethylene, 1, 2-dichloroethane, carbon tetrachloride, chloroform or dichloromethane; Alcohols such as methanol, ethanol, isopropanol, n-propanol, n-butanol or tert-butanol; Ethers, such as diethyl ether, diisopropyl ether, tetrahydrofuran (THF) or dioxane; Glycol ethers, such as ethylene glycol monomethyl or monoethyl ether (methyl glycol or ethyl glycol), ethylene glycol dimethyl ether (diglyme); Ketones such as acetone or butanone; amides such as acetamide, dimethylacetamide (DMA) or dimethyl
- Oxidations, in particular the oxidation of compounds of the formula (III) in compounds of the formula (I) are carried out by methods known to the person skilled in the art.
- a standard method is the oxidation with hydrogen peroxide in trifluoroacetic acid (TFA), for example under conditions as described by Grivas and Rönne (Acta Chemica Scandinavia, 49, 225-229 (1995)).
- BBr 3 boron tribromide
- the abovementioned compounds according to the invention can be used in their final non-salt form.
- the present invention also encompasses the use of these compounds in the form of their pharmaceutically acceptable salts, which may be derived from various organic and inorganic acids and bases by art-known procedures.
- Pharmaceutically acceptable salt forms of the compounds of formula (I) are for the most part prepared conventionally.
- acid addition salts can be formed by reacting these compounds with pharmaceutically acceptable organic and inorganic acids, e.g. Hydrogen halides such as hydrogen chloride, hydrogen bromide or
- Hydrogen iodide other mineral acids and their corresponding salts such as sulfate, nitrate or phosphate and the like, and alkyl and monoaryl sulfonates such as ethane sulfonate, toluenesulfonate and benzenesulfonate, as well as other organic acids and their corresponding salts such as acetate, trifluoroacetate, tartrate, maleate, succinate, citrate, benzoate, salicylate, ascorbate and the like.
- pharmaceutically acceptable acid addition salts of the compounds of formula (I) include the following: acetate, adipate, alginate, arginate, aspartate, benzoate, benzenesulfonate (besylate), bisulfate, bisulfite, bromide, butyrate, camphorate, camphorsulfonate, caprylate, chloride, chlorobenzoate , Citrate,
- the base salts of the compounds according to the invention include aluminum, ammonium, calcium, copper, iron (III), iron (II), lithium, magnesium, manganese (III), manganese (II) , Potassium,
- Salts of the compounds of formula (I), the oU be derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary and tertiary amines, substituted amines, also including naturally occurring substituted amines, cyclic amines, and basic ion exchanger resins, for example, 35 Arginine, betaine, caffeine, chloroprocaine, choline, N.N'-dibenzylethylenediamine (Benzathine), dicyclohexylamine, diethanolamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine,
- Compounds of the present invention containing basic nitrogen-containing groups can be reacted with agents such as (C 1 -C 4 ) alkyl halides, eg methyl, ethyl, isopropyl and tert-butyl chloride, bromide and iodide; Di (C r C 4 ) alkyl sulfates, for example dimethyl, diethyl and diamylsulfate; (Ci 0 -
- C 18 alkyl halides, eg decyl, dodecyl, lauryl, myristyl and
- compositions which are preferred include acetate, trifluoroacetate, besylate, citrate, fumarate, gluconate, hemisuccinate, hippurate, hydrochloride, hydrobromide, isethionate, mandelate,
- the amount of the desired acid brings into contact, which is the salt in a conventional manner.
- the free base can be regenerated by contacting the salt form with a base and isolating the free base in a conventional manner.
- the free base forms differ in some sense from their corresponding salt forms in relation to particular ones physical properties such as solubility in polar solvents; however, in the context of the invention, the salts otherwise correspond to their respective free base forms.
- the pharmaceutically acceptable base addition salts of the compounds of the formula (I) are formed with metals or amines such as alkali metals and alkaline earth metals or organic amines.
- metals are sodium, potassium, magnesium and calcium.
- Preferred organic amines are N, N'-dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, N-methyl-D-glucamine and procaine.
- the base addition salts of acidic compounds of the invention are prepared by contacting the free acid form with a sufficient amount of the desired base to form the salt in a conventional manner.
- the free acid can be regenerated by contacting the salt form with an acid and isolating the free acid in a conventional manner.
- the free acid forms in some sense differ from their corresponding salt forms in terms of certain physical properties such as solubility in polar solvents; However, in the context of the invention, the salts otherwise correspond to their respective free acid forms.
- Invention also multiple salts.
- Typical multiple salt forms include, for example, bitartrate, diacetate, difumarate, dimeglumine, diphosphate, disodium and trihydrochloride, but are not limiting SO
- the term "pharmaceutically acceptable salt” as used herein means an active ingredient which contains a compound of formula (I) in the form of one of its salts, especially if this salt form the Drug provides improved pharmacokinetic properties compared to the free form of the drug or any other salt form of the drug previously used.
- the pharmaceutically acceptable salt form of the active substance may also first impart a desired pharmacokinetic property to this active ingredient which it has not previously possessed, and may even positively influence the pharmacodynamics of this active ingredient in terms of its therapeutic activity in the body.
- the invention furthermore relates to medicaments comprising at least one compound according to the invention and / or pharmaceutically usable derivatives, salts, solvates and tautomers thereof, including mixtures thereof in all ratios, and optionally carrier and / or
- compositions may be administered in the form of dosage units containing a predetermined amount of active ingredient per unit dose.
- a moiety may contain, for example, from 0.1 mg to 3 g, preferably from 1 mg to 700 mg, more preferably from 5 mg to 100 mg of a compound of the invention, depending on the condition being treated, the route of administration and the age, weight and condition of the patient, or pharmaceutical formulations can be used in
- dosage unit included, to be presented.
- Preferred dosage unit formulations are those containing a daily or partial dose as indicated above or a corresponding fraction of an active ingredient.
- such pharmaceutical formulations can be prepared by any of the methods well known in the pharmaceutical art.
- compositions may be administered by any suitable route, for example, oral (including buccal or sublingual), rectal, nasal, topical (including buccal, sublingual or transdermal), vaginal or parenteral (including subcutaneous, intramuscular, intravenous or intradermal) routes.
- oral including buccal or sublingual
- rectal including buccal or sublingual
- nasal including buccal, sublingual or transdermal
- vaginal or parenteral including subcutaneous, intramuscular, intravenous or intradermal
- Carrier (s) or excipient (s) is brought together.
- compositions adapted for oral administration may be presented as separate entities, such as capsules or tablets; Powder - ) 0 or granules; Solutions or suspensions in aqueous or non-aqueous liquids; edible foams or foam foods; or oil-in-water liquid emulsions or water-in-oil liquid emulsions.
- the active ingredient component may be reconstituted with an oral, non-toxic and pharmaceutically acceptable inert carrier, e.g. Ethanol, glycerin, water and the like. combine. Powders are produced,
- Capsules are made by preparing a powder mix as described above and filling shaped gelatin casings therewith.
- Lubricants and lubricants such as finely divided silica, talc, magnesium stearate, calcium stearate or polyethylene glycol in solid form can be added to the powder mixture before the filling process.
- a disintegrants or solubilizers such as agar-agar, calcium carbonate or sodium carbonate may also be added to improve the availability of the drug after ingestion of the capsule. 35
- suitable binding, lubricating and disintegrants as well as dyes can also be incorporated into the mixture.
- Suitable binders include starch, gelatin, natural sugars such as glucose or beta-lactose, corn sweeteners, natural and synthetic gums such as acacia, tragacanth or sodium alginate, carboxymethyl cellulose, polyethylene glycol, waxes, etc.
- Lubricants include sodium oleate, sodium stearate, magnesium stearate, sodium benzoate, sodium acetate, sodium chloride, etc.
- the disintegrators include, but are not limited to, starch, methyl cellulose, agar, bentonite,
- the tablets are formulated by, for example, preparing a powder mix, granulating or dry pressing, adding a lubricant and disintegrant, and compressing the whole into tablets.
- a powder mixture is prepared by dissolving the appropriately comminuted compound with a diluent or a base as described above and optionally with a binder such as carboxymethylcellulose, an alginate, gelatin or polyvinylpyrrolidone, a dissolution reducer such as paraffin, a resorption accelerator, such as a quaternary salt and / or an absorbent, such as bentonite, kaolin or dicalcium phosphate.
- a binder such as carboxymethylcellulose, an alginate, gelatin or polyvinylpyrrolidone
- a dissolution reducer such as paraffin
- a resorption accelerator such as a quaternary salt and / or an absorbent, such as bentonite, kaolin or dicalcium phosphate.
- the powder mixture can be granulated by wetting it with a binder such as syrup, starch paste, Acadia slime, or solutions of cellulosic or polymeric materials and pressing it through a sieve.
- a binder such as syrup, starch paste, Acadia slime, or solutions of cellulosic or polymeric materials and pressing it through a sieve.
- the powder mixture can be run through a tabletting machine to produce non-uniformly shaped lumps which are broken up into granules.
- the granules may be greased by the addition of stearic acid, a stearate salt, talc or mineral oil to prevent sticking to the tablet molds.
- the greased mixture is then compressed into tablets.
- the compounds according to the invention can also be combined with a free-flowing inert carrier and then directly without carrying out the granulation or dry-pressing steps be compressed into tablets.
- a transparent or opaque protective layer consisting of a shellac sealant, a layer of sugar
- Oral fluids e.g. Solution, syrups and elixirs may be prepared in unit dosage form such that a given quantity contains a predetermined amount of the compound.
- Syrups can be prepared by dissolving the compound in an aqueous solution of suitable taste, while elixirs are prepared using a non-toxic alcoholic vehicle.
- Suspensions may be formulated by dispersing the compound in a non-toxic vehicle.
- Solubilizers and emulsifiers e.g. ethoxylated isostearyl alcohols and polyoxyethylene sorbitol ethers, preservatives, flavoring additives such as e.g. Peppermint oil or natural sweeteners or saccharin or other artificial sweeteners, i.a. can also be added.
- the unit dosage formulations for oral administration may optionally be encapsulated in microcapsules.
- the formulation may also be prepared to prolong or retard the release, such as by coating or embedding particulate material in polymers, wax, and the like.
- the compounds of the invention can also be administered in the form of liposome delivery systems, e.g. small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles.
- liposomes can be prepared from various phospholipids, such as e.g.
- Cholesterol, stearylamine or phosphatidylcholines can also be delivered using monoclonal antibodies as individual carriers to which the compound molecules are coupled.
- Drug carriers are coupled.
- Such polymers may include polyvinylpyrrolidone, pyran copolymer, polyhydroxypropylmethacrylamidephenol, polyhydroxyethylaspartamidephenol or polyethyleneoxidepolylysine substituted with palmitoyl radicals.
- compositions adapted for transdermal administration may be presented as discrete patches for prolonged, close contact with the epidermis of the recipient.
- the drug may be delivered from the patch by iontophoresis as generally described in Pharmaceutical Research, 3 (6), 318 (1986).
- Pharmaceutical compounds adapted for topical administration may be formulated as ointments, creams, suspensions, lotions, powders, solutions, pastes, gels, sprays, aerosols or oils.
- the formulations are preferably applied as a topical ointment or cream.
- the active ingredient may be either paraffinic or water-miscible
- Cream base can be used.
- the active ingredient may be added to a 35 Cream can be formulated with an oil-in-water cream base or a water-in-oil base.
- eye drops wherein the active ingredient is dissolved or suspended in a suitable carrier, especially an aqueous solvent.
- Formulations include lozenges, lozenges and mouthwashes.
- compositions adapted for rectal administration may be presented in the form of suppositories or enemas.
- compositions adapted for nasal administration in which the vehicle is a solid contain a coarse powder having a particle size, for example in the range of 20-500 microns, which is administered in the manner in which snuff is received, i. by rapid inhalation via the nasal passages from a container held close to the nose with the powder.
- Suitable formulations for administration as a nasal spray or nasal drops with a liquid carrier include drug solutions in water or oil.
- Fine particulate dusts or mists which may be generated by various types of pressurized dosing dispensers with aerosols, nebulizers or insufflators.
- Formulations can be used as pessaries, tampons, creams, gels, pastes,
- Foams or spray formulations are presented.
- compositions adapted for parenteral administration include aqueous and non-aqueous sterile injection solutions containing antioxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the recipient to be treated; and aqueous and non-aqueous sterile suspensions which may contain suspending agents and thickeners.
- Formulations may be used in single or multiple dose containers, e.g. 5 sealed ampoules and vials, presented and stored in freeze-dried (lyophilized) condition so that only the addition of the sterile carrier liquid, e.g. Water for injections, needed immediately before use.
- sterile carrier liquid e.g. Water for injections
- Formulated solutions and suspensions can be prepared from sterile powders, granules and tablets.
- formulations in addition to the above particularly mentioned ingredients, may include other conventional means with reference to Figure 15, the particular type of formulation; for example, formulations suitable for oral administration may contain flavorings.
- a therapeutically effective amount of a compound of the invention depends on a number of factors, including e.g. the age and weight of the individual or animal, the exact condition of the disease requiring treatment, and the severity of the condition, the nature of the disease
- Amount of a compound of the invention for the treatment generally in the range of 0.1 to 100 mg / kg body weight of the recipient (mammal) per day and more typically in the range of 1 to
- the actual amount per day will usually be between 70 and 700 mg, this amount being administered as a single dose per day or more commonly in a number of divided doses (such as two, three, four, five or more)
- O5 six) per day can be given, so that the total daily dose the same is.
- An effective amount of a salt or solvate or a physiologically functional derivative thereof can be determined as a proportion of the effective amount of the compound of the invention per se. It can be assumed that similar dosages are suitable for the treatment of the other, above-mentioned disease states.
- the invention furthermore relates to medicaments comprising at least one compound according to the invention and / or pharmaceutically usable derivatives, salts, solvates and tautomers, including mixtures thereof in all ratios, and at least one further active pharmaceutical ingredient.
- the invention is also a set (kit) comprising separate packages of
- the kit contains suitable containers, such as boxes or boxes, individual bottles, bags or ampoules.
- suitable containers such as boxes or boxes, individual bottles, bags or ampoules.
- the set may e.g. separate
- Ampoules contain, in each of which an effective amount of a compound of the invention and / or its pharmaceutically acceptable derivatives, solvates and tautomers, including mixtures thereof in all proportions, and an effective amount of another drug or dissolved in lyophilized form.
- the instant compounds are useful as pharmaceutical agents for mammals, particularly humans, in the treatment of diseases in which steroid sulphatase plays a role.
- the invention thus relates to the use of compounds according to the invention, and their pharmaceutically usable derivatives, solvates and tautomers, including mixtures thereof in all ratios, for the preparation of a medicament for
- the compounds of this invention may be used alone or in combination with one or more other sex hormone therapeutics / therapeutics, such as anti-estrogens, SERMs (Selective Estrogen Receptor Modulators). , Antiaromatases,
- the compounds of the invention may also be useful for the control or management of estrogen-driven reproductive functions, such as male or female
- the compounds of the invention may be more benign for treatment or prevention
- the compounds of the invention may be used alone or in combination with one or more other sex hormone therapeutics / therapeutics such as those mentioned above.
- the invention therefore also provides the use of the compounds of the formula (I) and their pharmaceutically usable derivatives, salts, solvates and tautomers, including mixtures thereof in all ratios, for the preparation of a medicament for the treatment or prevention of benign or malignant diseases of the breast, of the uterus or ovaries, optionally also in combination with one or more active ingredients selected from the group of antiestrogens, SERMs, aromatase inhibitors, antiandrogens, lyase inhibitors, progestogens and LH-RH agonists and antagonists.
- the compounds of the invention may be used to treat or prevent androgen dependent disorders such as androgenic alopecia (Hoffman R et al. J. Invest. Dermatol., 2001, 117, 1342-1348) or acne (Billich A et al., 1999, WO 9952890), benign or malignant diseases of the prostate or testes (Reed MJ 1 Rev. Endocr., Relat. Cancer , 1993, 45, 51-62), alone or in combination with one or more other sex hormones.
- Therapeutics / therapeutics such as antiandrogens, anti-estrogens, SERMs, anti-aromatase, progestins, lyase inhibitors or LH-RH agonists or antagonists. Subject of the
- the invention therefore further relates to the use of compounds of the formula
- Tautomers including mixtures thereof in all proportions, for the manufacture of a medicament for the treatment or prevention of benign or malignant diseases of the prostate or the testes optionally also in combination with one or more active substances selected from the group of antiestrogens, SERMs, aromatase inhibitors, antiandrogens, lyase inhibitors, progestogens and LH-RH agonists and antagonists.
- active substances selected from the group of antiestrogens, SERMs, aromatase inhibitors, antiandrogens, lyase inhibitors, progestogens and LH-RH agonists and antagonists.
- steroid sulfatase inhibitors may potentially be included for the treatment of cognitive dysfunction because they are capable of enhancing learning and spatial memory in the rat (Johnson DA, Brain Res, 2000, 865, 286-290).
- DHEA sulfate acts as a neurosteroid on a number of neurotransmitter systems, including those involving acetylcholine, glutamate and GABA, leading to increased neuronal excitability (Wolf OT, Brain Res. Rev, 1999, 30, 264-288).
- the invention therefore also relates to the use of the compounds of formula (I) and their pharmaceutically acceptable
- estrogens are involved in the regulation of the balance between the major immune functions Thi and Th 2 , and may therefore be useful for the treatment or prevention of sexually-dependent autoimmune diseases such as lupus erythematosus, multiple sclerosis, rheumatoid arthritis and the like (Daynes RA, J. Exp.
- the invention therefore also relates to the use of the compounds of the formula (I) and their pharmaceutically usable derivatives, salts, solvates and tautomers, including mixtures thereof in all ratios, for the preparation of a medicament for the treatment or prevention of immune diseases.
- the method comprises administering a therapeutically effective amount of a compound of formula (I) to an individual (human or animal) in need thereof.
- the human choriocarcinoma cell line JEG3 constitutively expresses high levels of steroid sulfatase and can therefore be used to determine the inhibition of cellular sulfatase activity.
- the substrate of sulfatase, estrogen sulfate in a defined physiological concentration is added to the cells and the amount of the product formed, the estrone and Estradiolkonzentration measured.
- JEG3 cells are seeded in 96 well plates at a density of approx. 1x10 5 cells / well in MEM plus 10% FCS. At about 80%
- the cells are washed with PBS and the test substances are added in a concentration series and 5 nM radioactive 3 H-EiS in DMEM. After an incubation period of 4 hours at 37 ° C., 100 ⁇ l of the incubation medium are removed and transferred to another 96 well plate. To extract the radioactive products E1 and E2 formed, 300 ⁇ l of toluene are added. After shaking for 30 seconds and centrifuging, the toluene phase is removed and evaporated overnight with liquid stock. The next day 100 .mu.l of ethanol is added, shaken and added to 150 .mu.l scintillation and determines the radioactivity.
- Alkaline phosphatase gene via the estrogen receptor and thus via estrogens.
- the addition of substances with estrogenic activity causes an induction of alkaline phosphatase and thus an increase in the activity, which is determined by the conversion of a substrate into an optically measurable product.
- Ishikawa cells are seeded in 96 well plates at a density of approximately 1x10 4 cells / well in DMEM plus 10% FCS. The next day, the medium is changed to DMEM with 5% estrogen-free FCS. Again, 24 hours later, the test substances are added in a concentration series in DMEM with 5% estrogen-free FCS. After incubation for 4 days at 37 ° C., the activity of the alkaline phosphatase is determined. For this purpose, the cells are washed twice with PBS, residual PBS, and the cells removed by 15 minutes of freezing at - 80 0 C lysed. After a 10 minute soak at room temperature, the substrate buffer (5 mM p.p.
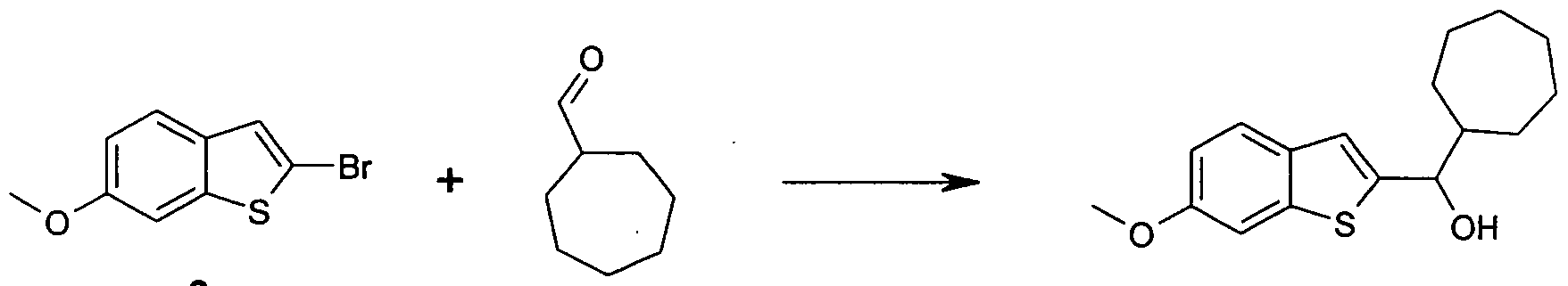
- the intermediates for the preparation of the -JO compounds according to the invention can be prepared by the known general methods of the prior art, preferably they are as shown below:
- the building block 2 (73 g, 0.445 mol) is in 1 L at room temperature
- R cycloalkyl- (CH 2 ) O C (OH) -.
- the following examples show individual embodiments of the compounds according to the invention.
- Trifluoroacetic acid (0.694 mL, 9.01 mmol) is added dropwise to a solution of 8a (668 mg, 2.43 mmol) in 5 mL dichloromethane at 5 ° C, followed by the addition of 0.795 mL (7.79 mmol) hydrogen peroxide (30% in water). at 10 0 C.
- the mixture is stirred overnight, poured onto ice-water, adjusted to pH 10-11 with sodium hydroxide solution (1 N) and extracted with dichloromethane.
- the organic phase is washed with a 10% strength iron (II) sulfate solution, dried with sodium sulfate and concentrated under reduced pressure.
- the moist residue is purified by chromatography.
- Trifluoroacetic added dropwise. After stirring for 5 minutes with cooling, the mixture is allowed to come to room temperature, stirring is continued for 1 h, two
Abstract
Description



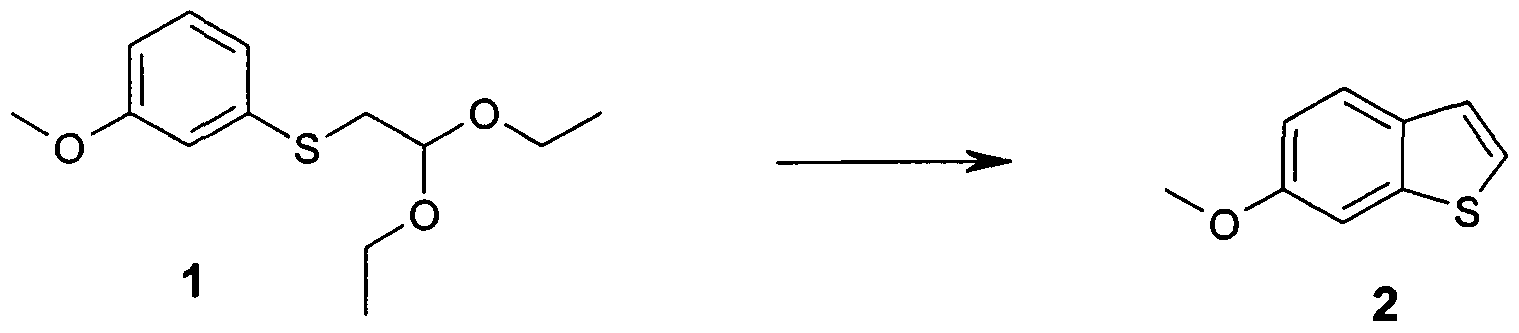























Claims





Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002656627A CA2656627A1 (en) | 2006-07-05 | 2007-06-05 | Sulfamatobenzothiophene derivatives |
| US12/307,311 US20090286863A1 (en) | 2006-07-05 | 2007-06-05 | Sulfamatobenzothiophene derivatives |
| AU2007271486A AU2007271486A1 (en) | 2006-07-05 | 2007-06-05 | Sulfamate benzothiophene derivatives |
| EP07725830A EP2035405A1 (de) | 2006-07-05 | 2007-06-05 | Sulfamat-benzothiophen-derivate |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06013922.7 | 2006-07-05 | ||
| EP06013922 | 2006-07-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008003378A1 true WO2008003378A1 (de) | 2008-01-10 |
Family
ID=38452540
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/004962 WO2008003378A1 (de) | 2006-07-05 | 2007-06-05 | Sulfamat-benzothiophen-derivate |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20090286863A1 (de) |
| EP (1) | EP2035405A1 (de) |
| AR (1) | AR061810A1 (de) |
| AU (1) | AU2007271486A1 (de) |
| CA (1) | CA2656627A1 (de) |
| WO (1) | WO2008003378A1 (de) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008067892A1 (de) * | 2006-12-04 | 2008-06-12 | Merck Patent Gmbh | Sulfamat-benzothiophen-derivate |
| WO2010014666A3 (en) * | 2008-07-31 | 2010-04-22 | Senomyx, Inc. | Processes and intermediates for making sweet taste enhancers |
| WO2012122969A1 (de) | 2011-03-16 | 2012-09-20 | Creative Therapeutics Gmbh | Substituierte diphenylderivate |
| US8877922B2 (en) | 2012-08-06 | 2014-11-04 | Senomyx, Inc. | Sweet flavor modifier |
| US9000151B2 (en) | 2013-02-19 | 2015-04-07 | Senomyx, Inc. | Sweet flavor modifier |
| US9561201B2 (en) | 2006-07-05 | 2017-02-07 | Fibrotech Therapeutics Pty Ltd | Therapeutic compounds |
| US9603848B2 (en) | 2007-06-08 | 2017-03-28 | Senomyx, Inc. | Modulation of chemosensory receptors and ligands associated therewith |
| US9951087B2 (en) | 2009-10-22 | 2018-04-24 | Fibrotech Therapeutics Pty Ltd | Fused ring analogues of anti-fibrotic agents |
| US11014873B2 (en) | 2017-02-03 | 2021-05-25 | Certa Therapeutics Pty Ltd. | Anti-fibrotic compounds |
| US11945813B2 (en) | 2018-08-07 | 2024-04-02 | Firmenich Incorporated | 5-substituted 4-amino-1H-benzo[c][1,2,6]thiadiazine 2,2-dioxides and formulations and uses thereof |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110240552A (zh) * | 2019-07-16 | 2019-09-17 | 重庆医药高等专科学校 | 氨基磺酸甲酯的制备方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004101545A1 (en) * | 2003-05-16 | 2004-11-25 | Laboratoire Theramex | Sulfamate benzothiophene derivatives as steroid sulfatase inhibitors |
| WO2005058842A1 (en) * | 2003-12-15 | 2005-06-30 | Laboratoire Theramex | 1-n-phenyl-amino-1h-imidazole derivatives and pharmaceutical compositions containing them |
-
2007
- 2007-06-05 EP EP07725830A patent/EP2035405A1/de not_active Withdrawn
- 2007-06-05 AU AU2007271486A patent/AU2007271486A1/en not_active Abandoned
- 2007-06-05 CA CA002656627A patent/CA2656627A1/en not_active Abandoned
- 2007-06-05 WO PCT/EP2007/004962 patent/WO2008003378A1/de active Application Filing
- 2007-06-05 US US12/307,311 patent/US20090286863A1/en not_active Abandoned
- 2007-07-04 AR ARP070102979A patent/AR061810A1/es unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004101545A1 (en) * | 2003-05-16 | 2004-11-25 | Laboratoire Theramex | Sulfamate benzothiophene derivatives as steroid sulfatase inhibitors |
| WO2005058842A1 (en) * | 2003-12-15 | 2005-06-30 | Laboratoire Theramex | 1-n-phenyl-amino-1h-imidazole derivatives and pharmaceutical compositions containing them |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2035405A1 * |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9561201B2 (en) | 2006-07-05 | 2017-02-07 | Fibrotech Therapeutics Pty Ltd | Therapeutic compounds |
| WO2008067892A1 (de) * | 2006-12-04 | 2008-06-12 | Merck Patent Gmbh | Sulfamat-benzothiophen-derivate |
| US9603848B2 (en) | 2007-06-08 | 2017-03-28 | Senomyx, Inc. | Modulation of chemosensory receptors and ligands associated therewith |
| US9732052B2 (en) | 2008-07-31 | 2017-08-15 | Senomyx, Inc. | Processes and intermediates for making sweet taste enhancers |
| WO2010014666A3 (en) * | 2008-07-31 | 2010-04-22 | Senomyx, Inc. | Processes and intermediates for making sweet taste enhancers |
| CN102171197A (zh) * | 2008-07-31 | 2011-08-31 | 西诺米克斯公司 | 制备增甜剂的方法和中间体 |
| US8586733B2 (en) | 2008-07-31 | 2013-11-19 | Senomyx, Inc. | Processes and intermediates for making sweet taste enhancers |
| US10570105B2 (en) | 2008-07-31 | 2020-02-25 | Firmenich Incorporated | Processes and intermediates for making sweet taste enhancers |
| CN102171197B (zh) * | 2008-07-31 | 2014-12-10 | 西诺米克斯公司 | 制备增甜剂的方法和中间体 |
| US10308621B2 (en) | 2008-07-31 | 2019-06-04 | Senomyx, Inc. | Processes and intermediates for making sweet taste enhancers |
| US10087154B2 (en) | 2008-07-31 | 2018-10-02 | Senomyx, Inc. | Processes and intermediates for making sweet taste enhancers |
| US9382196B2 (en) | 2008-07-31 | 2016-07-05 | Senomyx, Inc. | Processes and intermediates for making sweet taste enhancers |
| US9951087B2 (en) | 2009-10-22 | 2018-04-24 | Fibrotech Therapeutics Pty Ltd | Fused ring analogues of anti-fibrotic agents |
| WO2012122969A1 (de) | 2011-03-16 | 2012-09-20 | Creative Therapeutics Gmbh | Substituierte diphenylderivate |
| US9745293B2 (en) | 2012-08-06 | 2017-08-29 | Senomyx, Inc. | Sweet flavor modifier |
| US9687015B2 (en) | 2012-08-06 | 2017-06-27 | Senomyx, Inc. | Sweet flavor modifier |
| US9420814B2 (en) | 2012-08-06 | 2016-08-23 | Senomyx, Inc. | Sweet flavor modifier |
| US9138013B2 (en) | 2012-08-06 | 2015-09-22 | Senomyx, Inc. | Sweet flavor modifier |
| US8877922B2 (en) | 2012-08-06 | 2014-11-04 | Senomyx, Inc. | Sweet flavor modifier |
| US9695162B2 (en) | 2013-02-19 | 2017-07-04 | Senomyx, Inc. | Sweet flavor modifier |
| US9475803B2 (en) | 2013-02-19 | 2016-10-25 | Senomyx, Inc. | Sweet flavor modifier |
| US9371317B2 (en) | 2013-02-19 | 2016-06-21 | Senomyx, Inc. | Sweet flavor modifier |
| US9000151B2 (en) | 2013-02-19 | 2015-04-07 | Senomyx, Inc. | Sweet flavor modifier |
| US11014873B2 (en) | 2017-02-03 | 2021-05-25 | Certa Therapeutics Pty Ltd. | Anti-fibrotic compounds |
| US11603349B2 (en) | 2017-02-03 | 2023-03-14 | Certa Therapeutics Pty Ltd | Anti-fibrotic compounds |
| US11945813B2 (en) | 2018-08-07 | 2024-04-02 | Firmenich Incorporated | 5-substituted 4-amino-1H-benzo[c][1,2,6]thiadiazine 2,2-dioxides and formulations and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2007271486A1 (en) | 2008-01-10 |
| AR061810A1 (es) | 2008-09-24 |
| EP2035405A1 (de) | 2009-03-18 |
| CA2656627A1 (en) | 2008-01-10 |
| US20090286863A1 (en) | 2009-11-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008003378A1 (de) | Sulfamat-benzothiophen-derivate | |
| CN107405327B (zh) | 大麻素化合物的混合物、其制备和应用 | |
| EP3083603B1 (de) | Benzimidazolderivate als ep4-liganden | |
| DE102005008310A1 (de) | Verwendung von CDKII Inhibitoren zur Fertilitätskontrolle | |
| DE60005493T2 (de) | Pyrimido[6,1-a]isochinolinonderivate | |
| EP0204349A2 (de) | Neue heteroaromatische Aminderivate, diese Verbindungen enthaltende Arzneimittel und Verfahren zu ihrer Herstellung | |
| KR19980702086A (ko) | 티에노피리미딘 유도체, 그의 제조 방법 및 용도 | |
| EP0058341B1 (de) | Azepinderivate, ihre Herstellung und diese enthaltende Arzneimittel | |
| DD209624A5 (de) | Verfahren zur herstellung von benzimidazolverbindungen | |
| EP1529041B1 (de) | Neue prodrugs von 1-methyl-2-(4-amidinophenylaminomethyl)-benzimidazol-5-yl-carbonsäure-(n -2-pyridil-n-2-hydroxycarbonylethyl)-amid, ihre herstellung und ihre verwendung als arzneimittel | |
| DE60222286T2 (de) | Verfahren zur erhöhung des testosteronspiegels | |
| EP0408509A2 (de) | Substituierte Benzonitrile | |
| CH643565A5 (de) | Derivate von androstan-17-aethern. | |
| EP0490816A2 (de) | Fluorverbindungen | |
| EP0445073A1 (de) | Benzofurane | |
| EP0411735B1 (de) | Cycloakylenazole, Verfahren zu deren Herstellung, pharmazeutische Präparate, die diese enthalten sowie ihre Verwendung zur Herstellung von Arzneimitteln | |
| EP0103158B1 (de) | Neue Thieno-thiazol-Derivate, Verfahren zu ihrer Herstellung und diese Verbindungen enthaltende Arzneimittel | |
| AT400845B (de) | Neue thienothiazinderivate, ein verfahren zu ihrer herstellung und ihre verwendung | |
| EP0455596B1 (de) | Substituierte Indole | |
| EP0171645A1 (de) | Neue 2H-1-Benzopyran-2-on-Derivate, Verfahren zu ihrer Herstellung und Arzneimittel, die diese Verbindungen enthalten | |
| DE602004011914T2 (de) | Benzimidazole derivate und ihre anwendung als gaba a rezeptor komplex modulatoren | |
| DE60305069T2 (de) | 1-n-phenylamino-1h-imidazolderivate und deren verwendung als aromatase inhibitoren | |
| EP2086957A1 (de) | Sulfamat-benzothiophen-derivate | |
| DE19707656A1 (de) | Sulfonamid-substituierte anellierte 7-Ring-Verbindungen, Verfahren zu ihrer Herstellung, ihre Verwendung als Medikament oder Diagnostikum sowie sie enthaltende pharmazeutische Zubereitungen | |
| DE2551879A1 (de) | Neue heterozyklische verbindungen, verfahren zu deren herstellung und dieselben enthaltende pharmazeutische praeparate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07725830 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007725830 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2656627 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12307311 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007271486 Country of ref document: AU |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| ENP | Entry into the national phase |
Ref document number: 2007271486 Country of ref document: AU Date of ref document: 20070605 Kind code of ref document: A |