WO2007118964A1 - Derives de n-(phenethyl)benzamide substitues, preparation et utilisations - Google Patents
Derives de n-(phenethyl)benzamide substitues, preparation et utilisations Download PDFInfo
- Publication number
- WO2007118964A1 WO2007118964A1 PCT/FR2007/000513 FR2007000513W WO2007118964A1 WO 2007118964 A1 WO2007118964 A1 WO 2007118964A1 FR 2007000513 W FR2007000513 W FR 2007000513W WO 2007118964 A1 WO2007118964 A1 WO 2007118964A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenoxy
- methoxybenzamido
- methylpropanoic acid
- propan
- radical
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(*)(*)c1ccccc1 Chemical compound CC(*)(*)c1ccccc1 0.000 description 2
- AAPBEBAVSDSBKZ-UHFFFAOYSA-N CC(C)(C(O)=O)Oc1ccc(CCNC(c(cc(C)cc2)c2OC)=O)cc1 Chemical compound CC(C)(C(O)=O)Oc1ccc(CCNC(c(cc(C)cc2)c2OC)=O)cc1 AAPBEBAVSDSBKZ-UHFFFAOYSA-N 0.000 description 1
- MZBUFGZOFNTGJQ-UHFFFAOYSA-N CC(C)(C)c(cc1C(NCCC(CC2)=CC=C2OC(C)(C)C(O)=O)=O)ccc1OC Chemical compound CC(C)(C)c(cc1C(NCCC(CC2)=CC=C2OC(C)(C)C(O)=O)=O)ccc1OC MZBUFGZOFNTGJQ-UHFFFAOYSA-N 0.000 description 1
- YOOOLMYSBLFBBX-UHFFFAOYSA-N CC(CNC(c(cc(cc1)-c(ccc(F)c2)c2F)c1O)=O)c(cc1)ccc1OC(C)(C)C(OC(C)(C)C)=O Chemical compound CC(CNC(c(cc(cc1)-c(ccc(F)c2)c2F)c1O)=O)c(cc1)ccc1OC(C)(C)C(OC(C)(C)C)=O YOOOLMYSBLFBBX-UHFFFAOYSA-N 0.000 description 1
- SASCEGHIKNIBRS-UHFFFAOYSA-N CC(CNC(c1cc(-c(ccc(F)c2)c2F)ccc1OC)=O)c(cc1)ccc1OC(C)(C)C(O)=O Chemical compound CC(CNC(c1cc(-c(ccc(F)c2)c2F)ccc1OC)=O)c(cc1)ccc1OC(C)(C)C(O)=O SASCEGHIKNIBRS-UHFFFAOYSA-N 0.000 description 1
- BOCGTDHBNPBWAE-UHFFFAOYSA-N CC(CNC(c1cc(Cl)ccc1OC)=O)c(cc1)ccc1O Chemical compound CC(CNC(c1cc(Cl)ccc1OC)=O)c(cc1)ccc1O BOCGTDHBNPBWAE-UHFFFAOYSA-N 0.000 description 1
- FETJELBMGASUIK-UHFFFAOYSA-N CC(CNC(c1cc(Cl)ccc1OC)=O)c(cc1)ccc1OC(C)(C)C(NN(C)C)=O Chemical compound CC(CNC(c1cc(Cl)ccc1OC)=O)c(cc1)ccc1OC(C)(C)C(NN(C)C)=O FETJELBMGASUIK-UHFFFAOYSA-N 0.000 description 1
- RLYCGTIDDGWSKD-UHFFFAOYSA-N CC(Cc(cc1)ccc1O)NC(c(cc(cc1)OC)c1OC)=O Chemical compound CC(Cc(cc1)ccc1O)NC(c(cc(cc1)OC)c1OC)=O RLYCGTIDDGWSKD-UHFFFAOYSA-N 0.000 description 1
- RSKOKRROEGCTPS-UHFFFAOYSA-N CC(Cc(cc1)ccc1OC(C)(C)C(O)=O)NC(c(cc(cc1)-c2ccccc2)c1OC)=O Chemical compound CC(Cc(cc1)ccc1OC(C)(C)C(O)=O)NC(c(cc(cc1)-c2ccccc2)c1OC)=O RSKOKRROEGCTPS-UHFFFAOYSA-N 0.000 description 1
- JRLPEMVDPFPYPJ-UHFFFAOYSA-N CCc1ccc(C)cc1 Chemical compound CCc1ccc(C)cc1 JRLPEMVDPFPYPJ-UHFFFAOYSA-N 0.000 description 1
- JFJPGCYHZYQWKQ-UHFFFAOYSA-N COc(c(C(O)=O)c1)ccc1-[n]1cccc1 Chemical compound COc(c(C(O)=O)c1)ccc1-[n]1cccc1 JFJPGCYHZYQWKQ-UHFFFAOYSA-N 0.000 description 1
- NJLHBBFVHSDGIB-UHFFFAOYSA-N COc(ccc(-c1ccccc1)c1)c1C(NCCc(cc1)ccc1O)=O Chemical compound COc(ccc(-c1ccccc1)c1)c1C(NCCc(cc1)ccc1O)=O NJLHBBFVHSDGIB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/22—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with hetero atoms directly attached to ring nitrogen atoms
- C07D295/28—Nitrogen atoms
- C07D295/32—Nitrogen atoms acylated with carboxylic or carbonic acids, or their nitrogen or sulfur analogues
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C215/00—Compounds containing amino and hydroxy groups bound to the same carbon skeleton
- C07C215/46—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C215/48—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by hydroxy groups
- C07C215/52—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by hydroxy groups linked by carbon chains having two carbon atoms between the amino groups and the six-membered aromatic ring or the condensed ring system containing that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/56—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms
- C07C217/60—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains not further substituted by singly-bound oxygen atoms linked by carbon chains having two carbon atoms between the amino groups and the six-membered aromatic ring or the condensed ring system containing that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/64—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings
- C07C233/67—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms
- C07C233/68—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom
- C07C233/73—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by an acyclic carbon atom of a carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/42—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/44—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring
- C07C235/58—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring with carbon atoms of carboxamide groups and singly-bound oxygen atoms, bound in ortho-position to carbon atoms of the same non-condensed six-membered aromatic ring
- C07C235/60—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring with carbon atoms of carboxamide groups and singly-bound oxygen atoms, bound in ortho-position to carbon atoms of the same non-condensed six-membered aromatic ring having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/42—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/66—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings being part of condensed ring systems and singly-bound oxygen atoms, bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/50—Compounds containing any of the groups, X being a hetero atom, Y being any atom
- C07C311/51—Y being a hydrogen or a carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/50—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton
- C07C323/62—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and carboxyl groups bound to the same carbon skeleton having the sulfur atom of at least one of the thio groups bound to a carbon atom of a six-membered aromatic ring of the carbon skeleton
Definitions
- the present invention relates to poly-substituted N- (phenethyl) benzamide derivatives, the pharmaceutical compositions comprising them and their therapeutic applications, in particular in the fields of human and animal health.
- the present invention also relates to a process for the preparation of these derivatives.
- the compounds according to the invention possess activating properties of PPAR (Peroxisome Proliferator-Activated Receptor), in particular PPAR ⁇ and / or PPAR ⁇ .
- PPAR Peroxisome Proliferator-Activated Receptor
- compounds according to the invention have insulinosecretory properties: the compounds according to the invention can therefore act by at least two complementary and independent pathways for treating pathologies related to lipid and / or carbohydrate disorders.
- the molecules described in the invention are of particular interest for treating the complications associated with the metabolic syndrome, insulin resistance, diabetes, dyslipidemias, atherosclerosis, cardiovascular diseases, obesity, hypertension, inflammatory diseases (asthma, etc.), etc., as well as to reduce the overall risk for cardiovascular diseases.
- the compounds according to the invention can be used in particular for the treatment of inflammatory pathologies and / or atherosclerosis.
- the compounds according to the invention can be used for the treatment of type 2 diabetes and / or dyslipidemias.
- cardiovascular disease is the leading cause of death in industrialized countries and is becoming increasingly common in developing countries. These diseases include cardiac and vascular diseases, hypertension, atherosclerosis, myocardial infarction and cerebral ischemia (Muscat G and Dressel U, 2005). Atherosclerosis is a condition characterized by deregulation of glucose and lipid metabolism, oxidative stress and inflammation.
- Diabetes, obesity and dyslipidemia are some of the clearly identified cardiovascular risk factors that predispose an individual to develop cardiovascular disease (Mensah M, 2004 ). These risk factors add up to lifestyle risk factors such as smoking, physical inactivity and unbalanced diets. A synergistic effect exists between these different factors: the concomitant presence of several of them leads to a dramatic worsening of the cardiovascular risk and it is then appropriate to speak of global risk (“global risk”) for cardiovascular diseases.
- the prevalence of dyslipidemia was 43.6% in 2004 in the main developed countries.
- the prevalence of diabetes, currently rising sharply, is becoming increasingly significant in the epidemiology of cardiovascular diseases: it is estimated at 7.6% for 2010 (Fox-Tucker J, 2005).
- ATP-dependent potassium channels of ⁇ -pancreatic cells involved in the regulation of insulin secretion.
- the compounds according to the invention therefore act by at least two complementary and independent pathways for treating pathologies related to lipid and carbohydrate disorders.
- the molecules described in the invention are of particular interest for the treatment of type 2 diabetes, this pathology being currently treated either by insulin-secreting molecules (eg sulphonylureas), or by agonist molecules of PPAR (eg thiazolidinediones), or by a combination of such molecules (Gin H and Rigalleau V, 2002).
- insulin-secreting molecules eg sulphonylureas
- agonist molecules of PPAR eg thiazolidinediones
- the molecules described in the invention are particularly interesting for the treatment of atherosclerosis, dyslipidemias, obesity and vascular inflammation.
- the compounds according to the invention represent an advantageous therapeutic means for treating, in addition to diabetes and dyslipidemias, the complications associated with the metabolic syndrome (whose characteristics are obesity). , especially abdominal obesity, an abnormal concentration of blood lipids (high triglyceride and / or low HDL cholesterol (dyslipidemia)), high blood glucose and / or insulin resistance, hypertension), atherosclerosis, cardiovascular disease, obesity , hypertension, inflammatory diseases, etc.
- the compounds according to the invention represent an advantageous therapeutic tool for treating several cardiovascular risk factors related to disorders of lipid and / or carbohydrate metabolism (hyperlipidemia, hyperglycemia, etc.). They allow the reduction of the overall risk for cardiovascular diseases.
- PPARs ( ⁇ , ⁇ and ⁇ ) are known to be involved in pathologies of lipid and / or carbohydrate metabolism (Kota BP et al., 2005): ligands of these receptors are therefore marketed to treat such pathologies (Lefebvre P and al., 2006) and many PPAR modulators, agonists or antagonists, selective or not, are currently in advanced development for the treatment of such pathologies.
- a PPAR modulator with beneficial effects on insulin resistance, obesity, dyslipidemia, hypertension and / or inflammation could be used in the treatment of metabolic syndrome (or syndrome X) (Liu Y and Miller A, 2005).
- the PPAR family comprises three distinct members, designated ⁇ , ⁇ and ⁇
- PPAR Peroxisome Proliferator Response Element
- PPAR ⁇ controls the lipid metabolism (hepatic and skeletal muscle), influences the intracellular lipid metabolism by a direct control of the transcription of genes encoding proteins involved in lipid homeostasis, exerts anti-inflammatory and anti-proliferative effects, and prevents the pro-atherogenic effects of cholesterol accumulation in macrophages by stimulating cholesterol efflux (also known as reverse cholesterol transport) (Lefebvre P, Chinetti G, Fruchart JC and Staels B, 2006).
- fibrates (fenofibrate, bezafibrate, ciprofibrate, gemfibrozil), via PPAR ⁇ , are thus used clinically in the treatment of certain dyslipidemias by lowering triglycerides and increasing the HDL (High Density Lipoprotein) levels.
- PPAR ⁇ is a key regulator of adipogenesis. In addition, it is involved in the lipid metabolism of mature adipocytes, glucose homeostasis, including insulin resistance, inflammation, macrophage cholesterol accumulation, and cell proliferation. (Lehrke M and Lazar MA, 2005). PPAR ⁇ therefore plays a role in the pathogenesis of obesity, insulin resistance and diabetes.
- the thiazolidinediones (Rosiglitazone, Troglitazone, etc.) are ligands of the PPAR ⁇ receptor used in the treatment of type 2 diabetes.
- the pharmacological interest of PPAR ⁇ agonists in the treatment of cardiovascular and metabolic pathologies and their risk factors has been clearly highlighted (Muscat G and Dressel U, 2005).
- PPAR ⁇ is involved in the control of lipid and carbohydrate metabolism, energy balance, neurodegeneration, obesity, foam cell formation and inflammation (Gross B et al., 2005): this receptor therefore an attractive target for the development of drugs useful in the treatment of dyslipidemia, atherosclerosis, obesity, insulin resistance, vascular inflammation (Gross B et al., 2005) and the type 2 diabetes (Luquet S et al., 2005).
- ligands of PPAR ⁇ L-165041, GW501516 currently in clinical development
- no PPAR ⁇ ligand is currently used as a drug.
- PPARs therapeutic targets of interest for the treatment of diseases such as atherosclerosis, cerebral ischemia, hypertension, diseases related to neovascularization (diabetic retinopathies, etc.), inflammatory and autoimmune diseases (Crohn's disease, psoriasis, multiple sclerosis, asthma, etc.), neoplastic diseases (carcinogenesis, etc.), neurodegenerative diseases, complications associated with metabolic syndrome, insulin resistance, diabetes, dyslipidemias, cardiovascular diseases, obesity, etc., as well as to reduce overall risk.
- diseases such as atherosclerosis, cerebral ischemia, hypertension, diseases related to neovascularization (diabetic retinopathies, etc.), inflammatory and autoimmune diseases (Crohn's disease, psoriasis, multiple sclerosis, asthma, etc.), neoplastic diseases (carcinogenesis, etc.), neurodegenerative diseases, complications associated with metabolic syndrome, insulin resistance, diabetes, dyslipidemias, cardiovascular diseases, obesity, etc., as well
- the inventors have also shown that the compounds according to the invention stimulate the secretion of insulin, a property which makes it possible to regulate blood glucose and which is used in the context of major pathologies such as type 2 diabetes.
- sulphonylureas and meglitinides also called glinides
- the main targets of insulin-secreting molecules are the ATP-dependent potassium channels of ⁇ -pancreatic cells, which play an important role in controlling the membrane potential of these cells (Proks P et al., 2002). Closure of this glucose-induced channel and / or insulin secretory agent (via membrane receptors, for example "Sulphonyl Urea Receptors" or SUR) induces depolarization of the plasma membrane which causes the opening of voltage-gated calcium channels. The influx of calcium ions (Ca ++ ) generated by this phenomenon causes the secretion of insulin.
- Meglitinides are a recent class of drugs represented by repaglinide, nateglinide and mitiglinide.
- repaglinide (described in WO93 / 00337) is a benzoic acid derivative. Numerous molecules containing a benzoic acid function have been described in the literature during the last 25 years, following the discovery of meglitinide (HB699) by chemical modification of glibenclamide (Rufer C and Losert W, 1979). glibenclamide
- the compounds according to the invention by their PPAR and insulin secretory agonist properties, represent an advantageous therapeutic tool for the amelioration of pathologies related to disorders of lipid and / or carbohydrate metabolism, in particular dyslipidemias and / or type 2 diabetes, as well as for the reduction of the global cardiovascular risk.
- the activating properties of PPAR ⁇ and / or PPAR ⁇ of the compounds according to the invention make them targets of interest for the treatment of atherosclerosis, dyslipidemias, type 2 diabetes, obesity, and vascular inflammation.
- the present invention relates to poly-substituted N- (phenethyl) benzamide derivatives of general formula (I):
- G represents - a radical -OR d) -SR d ;
- R d represents an alkyl, alkenyl, aryl, aralkyl radical or a heterocycle; G possibly forming a heterocycle with X1;
- R1, R2, R3 and R4 independently represent a hydrogen atom or an alkyl, alkenyl, aryl or aralkyl radical
- R1 and R4 may each independently be bound to the molecular backbone by a double bond, R2 or R3 then being absent;
- Y represents an oxygen atom, a sulfur atom (optionally oxidized to sulfoxide or sulfone function), a selenium atom (optionally oxidized to selenoxide or selenone function) or an NR type amine group;
- R representing a hydrogen atom or an alkyl, alkenyl, aryl or aralkyl radical, preferentially a hydrogen atom or an alkyl radical;
- R5 represents a halogen atom or an alkyl, alkenyl, aryl, aralkyl, alkyloxy or alkylthio radical
- R6 represents a hydrogen atom, halogen atom or an alkyl, alkenyl, aryl, aralkyl, alkyloxy or alkylthio radical; or R5 being able to form a ring with R6, m and n independently represent an integer from 0 to 10; Y1 and Y2 independently represent a covalent bond or a heteroatom selected from oxygen, sulfur or nitrogen (thus forming a radical of NR type, R representing a hydrogen atom or a radical of alkyl, alkenyl, aryl or aralkyl, preferably a hydrogen atom or an alkyl radical); Z represents a covalent bond or an alkyl, alkenyl, aryl or aralkyl radical; if Z is a covalent bond then Y1 and / or Y2 is a covalent bond;
- W represents:
- a carboxyl radical or a derivative preferably of the type -COOR a , -COSR 3 , -CONR 3 Rb, -CSNR a R b ; or
- an isosteric group of the carboxyl radical preferentially an acylsulfonamide radical (-CONHSO 2 Rc), hydrazide (-CONHNR 3 Rb) or tetrazolyl;
- R 3 and R b identical or different, substituted or unsubstituted, representing a hydrogen atom or an alkyl, alkenyl, aryl, aralkyl or heterocycle radical; or R 3 and R b may together form a heterocycle with the nitrogen atom; R 0 , substituted or unsubstituted, representing an alkyl, alkenyl, aryl, aralkyl radical or a heterocycle;
- X1, X2, X3 and X4 independently represent a hydrogen or halogen atom, a -NO 2 or -CN function, an alkyl, alkenyl, aryl, aralkyl, alkyloxy or alkylthio radical, -NR 3 R 0 , -NR a COR b , -NR a COOR c , -NR a CONR b R c , -NR a CSR b , -NR 3 COSR C , -NRaCSORc, -NR has CSSR c , -NR has CSNR b Rc, -SOR 0 , -SO 2 R 0 , -COR 9 ,
- R 3 , R b and R 0 being as defined above;
- R a , Rb and / or R 0 may together form a heterocycle with the nitrogen atom
- X1 and X3 each being able to form a ring (aromatic or otherwise, heterocyclic or not) with X2 and X4 respectively;
- R1, R2, R3, R4, X1, X2, X3 and X4 representing an alkyl, alkenyl, aryl or aralkyl radical, stereoisomers (diastereoisomers, enantiomers), pure or in mixture, as well as racemic mixtures, geometric isomers, tautomers, salts, hydrates, solvates, solid forms, prodrugs and mixtures.
- alkyl denotes a saturated hydrocarbon radical, linear, branched or cyclic, substituted or unsubstituted, with more particularly from 1 to 24, preferably 1 to 10, carbon atoms. Mention may be made, for example, of the methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, sec-butyl, pentyl, neopentyl, n-hexyl or cyclohexyl radicals.
- alkenyl refers to an unsaturated hydrocarbon radical, linear, branched or cyclic, substituted or unsubstituted, with more particularly from 2 to 24, preferably 2 to 10 carbon atoms.
- alkenyl refers to an unsaturated hydrocarbon radical, linear, branched or cyclic, substituted or unsubstituted, with more particularly from 2 to 24, preferably 2 to 10 carbon atoms.
- alkyloxy refers to an alkyl chain attached to the molecule through an oxygen atom (ether linkage).
- the alkyl chain meets the previously stated definition.
- alkylthio refers to an alkyl chain bonded to the molecule via a sulfur atom (thioether bond).
- the alkyl chain meets the previously stated definition.
- aryl refers to an aromatic hydrocarbon radical, substituted or unsubstituted, preferably having 6 to 14 carbon atoms.
- the aryl radical will be preferably substituted by at least one halogen atom, an alkyl, hydroxyl, thiol, alkyloxy, alkylthio or a nitro group.
- the aryl radicals according to the present invention are chosen from phenyl, naphthyl (for example 1-naphthyl or 2-naphthyl), biphenyl (for example, 2-, 3- or 4-biphenyl), anthryl or fluorenyl. Phenyl groups, substituted or unsubstituted, are very particularly preferred.
- aralkyl refers to an alkyl radical substituted by an aryl group, substituted or unsubstituted.
- benzyl and phenethyl groups are particularly preferred.
- heterocycle refers to a mono- or poly-cyclic radical, saturated, unsaturated or aromatic, comprising one or more heteroatoms, such as nitrogen, sulfur or oxygen. They may be advantageously substituted by at least one alkyl, alkenyl, aryl, alkyloxy or alkylthio group as defined above or a halogen atom.
- ring is meant more particularly a hydrocarbon ring, optionally having at least one heteroatom (especially a nitrogen, sulfur or oxygen), saturated, unsaturated or aromatic.
- the rings include especially aryl groups or heterocycles, as defined above.
- the radicals thus defined may be substituted, in particular by at least one halogen atom, an alkyl, hydroxyl, thiol, alkyloxy or alkylthio radical, or a nitro function.
- the alkyl radical may be a perhaloalkyl radical, in particular perfluoroalkyl, such as in particular -CF 3 .
- the halogen atom is selected from a chlorine, bromine, iodine or fluorine atom.
- n and n independently represent an integer from 0 to 10. More specifically, they may be independently 0, 1, 2, 3, 4, 5, 6, 7, 8, 9 or 10.
- the compounds of formula (I) have a group G in position ortho (relative to the -CONH- motif) of the phenyl radical to which it is attached.
- the compounds of formula (I) have a Y-E group in para position (with respect to the -CR3R4- motif) of the phenyl radical to which it is attached.
- the compounds of formula (I) have a group G in the ortho position (with respect to the -CONH- motif) of the phenyl radical to which it is attached and a Y-E group in the para position (with respect to the -
- Another particular object of the invention relates to the compounds of general formula (I), advantageously (II), in which G represents a radical -OR d or -SRd, Rd representing an alkyl, alkenyl, aryl, aralkyl radical or a heterocycle .
- G represents a radical -OR d or -SRd
- Rd representing an alkyl, alkenyl, aryl, aralkyl radical or a heterocycle .
- the alkyl, alkenyl, aryl, aralkyl or heterocycle radical preferably contains 1 to 10 carbon atoms.
- Another particular object of the invention relates to the compounds of general formula (I), advantageously (II), in which R 1, R 2, R 3 and / or R 4 represent an alkyl, alkenyl, aryl or aralkyl radical, preferably comprising 1 to 6 carbon atoms. Even more preferentially, R1,
- R2, R3 and / or R4 represent an alkyl radical.
- Another particular object of the invention relates to the compounds of general formula (I), advantageously (II), in which Y represents an oxygen or sulfur atom. Even more preferably, Y represents an oxygen atom.
- Another particular object of the invention relates to the compounds of general formula (I), advantageously (II), in which E represents an alkyl or alkenyl chain, substituted with a group -CR 5 R 6 -W, in which: R 5 represents a halogen atom or an alkyl, alkenyl, aryl, aralkyl, alkyloxy or alkylthio radical; R6 represents a hydrogen atom, halogen atom or an alkyl, alkenyl, aryl, aralkyl, alkyloxy or alkylthio radical; or R5 can form a ring with R6, W represents:
- a carboxyl radical or a derivative preferably of the type -COOR a , -COSR 3 , -CONR 3 Rb, -CSNR a R b ; or
- an isosteric group of the carboxyl radical preferentially an acylsulfonamide radical (-CONHSO 2 Rc), hydrazide (-CONHNR 3 Rb) or tetrazolyl;
- R 3 , R b and R c being as defined above.
- the compounds of general formula (I), advantageously (II) have a group E representing a chain corresponding to the formula - (CH 2 ) n -CR 5 R 6 -W, with n, R 5, R 6 and W such as defined above.
- the compounds of general formula (I), advantageously (II) have the group E representing a chain corresponding to the formula -CR 5 R 6 -W with R 5 , R 6 and W as defined above. (In this case, m and n are 0 and Y1 -Z-Y2 represents a covalent bond).
- the compounds of general formula (I), advantageously (II), have a group W representing a carboxyl radical (-COOH), alkoxycarbonyl (-COOR 3 ), hydrazide (-CONHNR 3 Rb) or acylsulfonamide (-CONHSO 2 Rc ), Ra, Rb and R c being as defined above.
- W represents a carboxyl (-COOH) or alkoxycarbonyl (-COOR 3 ) radical, R 3 being as defined above.
- the compounds of general formula (I), advantageously (II), have R5 and / or R6 radicals representing an alkyl radical, preferably a methyl radical.
- the compounds of general formula (II) have an X 1 group in the para position (with respect to the radical unit G) of the phenyl radical to which it is attached.
- a group E representing a chain corresponding to the formula -CR 5 R 6 -W with R 5 , R 6 and W as defined above (and where m and n are equal to O and the unit Y1-Z-Y2 represents a covalent bond).
- the compounds of general formula (I), advantageously (II) or (III), have a group YE of the type -OC (CHa) 2 COOH.
- Another particular object of the invention relates to the compounds of general formula (I), advantageously (II) or (III), in which X 1 represents a halogen, preferably chlorine, or a radical -COR a , alkyl, alkenyl, aryl or substituted or unsubstituted aralkyl, R a being as defined above.
- X 1 represents a halogen, preferably chlorine, or a radical -COR a , alkyl, alkenyl, aryl or substituted or unsubstituted aralkyl, R a being as defined above.
- Another particular object of the invention relates to the compounds of general formula (I), advantageously (II) or (III), in which X 2, X 3 and / or X 4 represent a hydrogen atom.
- X3 and X4 represent a hydrogen atom.
- a particularly preferred object of the invention relates to the compounds of general formula (I), advantageously (II) or (III), in which R 1 represents an alkyl, alkenyl, aryl or aralkyl radical and R 2, R 3 and R 4 are carbon atoms. 'hydrogen.
- the compounds of general formula (I), advantageously (II) or (III) have groups R 1 and X 1 independently representing an alkyl, alkenyl, aryl or aralkyl radical.
- Another particular object of the invention relates to the compounds of general formula (I), advantageously (II) or (III), wherein X1 forms a ring with X2 and / or X3 forms a ring with X4.
- the resulting ring forms with the adjacent phenyl ring a naphthyl, 1,2,3,4-tetrahydronaphthyl, indenyl or indanyl radical.
- the ring formed by X1 and X2 and / or X3 and X4 may be substituted, in particular by at least one halogen atom, an alkyl, hydroxyl, thiol, alkyloxy, alkylthio or a nitro group.
- Another particular object of the invention relates to the compounds of general formula (I), advantageously (II) or (III), in which X 1 forms a ring with G.
- the resulting ring forms with the adjacent phenyl ring a radical of type 2H-chromene, 3,4-dihydro-2H-chromene, 2H-thiochromene, 3,4-dihydro-2H-thiochromene, benzofuran, 2,3-dihydrobenzofuran, benzothiophene, 2,3-dihydrobenzothiophene, benzoxazole or benzothiazole.
- the ring formed by X1 and G may be substituted, in particular by at least one halogen atom, an alkyl, hydroxyl, thiol, alkyloxy, alkylthio, hydroxyl, thiol or nitro group.
- Another particular object of the invention relates to the compounds of general formula (I), advantageously (II) or (III), in which R5 forms with R6 (and the carbon to which they are attached) a cycle of cyclopropyl or cyclobutyl type, cyclopentyl or cyclohexyl.
- the subject of the invention is the compounds of general formula (I), advantageously (II) or (III), in which at least one of the following conditions, preferably all the conditions, is fulfilled:
- G represents a radical -OR d or -SR d , the radical R d being preferably selected from methyl, trifluoromethyl, ethyl, n-propyl, isopropyl, cyclopropyl, n-butyl, isobutyl, tert-butyl, sec-butyl, cyclobutyl, pentyl, neopentyl, cyclopentyl, n-hexyl, vinyl, allyl, homoallyl, propargyl, cyclohexyl, phenyl, benzyl or phenethyl; and or E represents an alkyl or alkenyl chain, substituted with a -CR 5 R 6 -W group,
- - W representing a carboxyl radical (-COOH), alkoxycarbonyl (-COOR a ), hydrazide (-CONHNR 3 Rb) or acylsulfonamide (-CONHSO 2 Rc),
- R 3 , Rb and R 0 being as defined above;
- R1, R2, R3, R4 and R6 independently represent a hydrogen atom or a methyl, ethyl, n-propyl, isopropyl, cyclopropyl, n-butyl, isobutyl, tert-butyl, sec-butyl, cyclobutyl, pentyl, neopentyl, cyclopentyl radical; , n-hexyl, cyclohexyl, vinyl, allyl, homoallyl, propargyl, phenyl, benzyl or phenethyl; and or
- R5 is selected from methyl, ethyl, n-propyl, isopropyl, cyclopropyl, n-butyl, isobutyl, tert-butyl, sec-butyl, cyclobutyl, pentyl, neopentyl, cyclopentyl, n-hexyl, cyclohexyl, vinyl, allyl, homoallyl, propargyl, phenyl, benzyl or phenethyl; and or
- Y represents an oxygen or sulfur atom
- X1, X2, X3 and X4 independently represent a hydrogen or halogen atom, a methyl, trifluoromethyl, ethyl, n-propyl, isopropyl, cyclopropyl, n-butyl, isobutyl, tert-butyl, sec-butyl, cyclobutyl, pentyl radical; , neopentyl, cyclopentyl, n-hexyl, cyclohexyl, vinyl, allyl, homoallyl, propargyl, phenyl, benzyl, phenethyl, methoxy, trifluoromethoxy, thiomethoxy, nitro, methylamino, dimethylamino, cyano, formyl, acetyl, propanoyl, butanoyl, pentanoyl, hexanoyl
- R1, R2, R3, R4, X1, X2, X3 and X4 representing an alkyl, alkenyl, aryl or aralkyl radical.
- Compound 1 2- [4- [2- (5-chloro-2-methoxybenzamido) propyl] phenoxy] -2-methylpropanoic acid;
- Compound 1 2- [4- [2- (5-chloro-2-methoxybenzamido) propyl] phenoxy] -2-methylpropanoic acid;
- the compounds according to the invention may contain one or more asymmetric centers.
- the present invention includes stereoisomers (diastereoisomers, enantiomers), pure or in admixture, as well as racemic mixtures and geometric isomers, tautomers, salts, hydrates, solvates, solid forms, prodrugs of compounds according to the present invention. invention and mixtures thereof.
- an enantiomerically pure (or enriched) mixture it may be obtained either by purification of the final product or chiral intermediates, or by asymmetric synthesis according to methods known to those skilled in the art (using, for example, reagents and catalysts chiral).
- Some compounds according to the invention may have different stable tautomeric forms and all such forms and mixtures thereof are included in the invention.
- the present invention also relates to the "pharmaceutically acceptable" salts of the compounds according to the invention.
- this term refers to the low or non-toxic salts obtained from bases or acids, organic or inorganic. These salts can be obtained during the final purification step of the compound according to the invention or by incorporation of the salt on the already purified compound.
- the group E as described above can have an acidic character.
- the corresponding salts are chosen from metal salts (for example, aluminum, zinc, chromium), alkaline salts (lithium, sodium, potassium) or alkaline earth (calcium, magnesium).
- organic salts such as ammonium and non-toxic amine derivatives: ammonium, quaternary ammonium (tetramethylammonium, tetraethylammonium, etc.), alkylamines (methylamine, dimethylamine, trimethylamine, triethylamine, ethylamine, etc.), hydroxyalkylamines (2-hydroxyethylamine, bis- (2-hydroxyethyl) amine, tri- (2-hydroxyethyl) amine, etc.), cycloalkylamines (bicyclohexylamine, glucamine, etc.), pyridines and the like (collidine, quinine, quinoline, etc.) of basic amino acid salts (lysine, arginine, etc.).
- alkylamines methylamine, dimethylamine, trimethylamine, triethylamine, ethylamine, etc.
- hydroxyalkylamines (2-hydroxyethylamine, bis-
- Certain compounds according to the invention and their salts could be stable in several solid forms.
- the present invention includes all solid forms of the compounds according to the invention which includes amorphous, polymorphic, mono- and polycrystalline forms.
- the compounds according to the invention may exist in free form or in solvated form, for example with pharmaceutically acceptable solvents such as water (hydrates) or ethanol.
- the present invention also includes the prodrugs of the compounds according to the invention which, after administration in a subject, are transformed into compounds as described in the invention or their metabolites which have therapeutic activities comparable to the compounds according to the invention.
- the compounds according to the invention labeled with one or more isotopes are also included in the invention: these compounds are structurally identical but differ in that at least one atom of the structure is replaced by an isotope (radioactive or not).
- isotopes that can be included in the structure of the compounds of the invention may be selected from hydrogen, carbon, nitrogen, oxygen, sulfur such as 2 H, 3 H, 13 C, 14 C, 15 N, 18 O, 17 O, 35 S, respectively.
- the 3 H and 14 C radioactive isotopes are particularly preferred because they are easy to prepare and detect in vivo in vivo bioavailability studies.
- Heavy isotopes (such as H 2) are particularly preferred because they are used as internal standards in analytical studies.
- the subject of the present invention is also the compounds as described above, as medicaments.
- the present invention also relates to a pharmaceutical composition
- a pharmaceutical composition comprising, in a pharmaceutically acceptable carrier, at least one compound as described above, optionally in combination with one or more other therapeutic and / or cosmetic active ingredients.
- a pharmaceutical composition for the treatment of type 2 diabetes, dyslipidemias, insulin resistance, pathologies associated with the metabolic syndrome, atherosclerosis, cardiovascular diseases, obesity, hypertension, inflammatory diseases, etc. Inflammatory pathologies are particularly important for asthma.
- It is preferably a pharmaceutical composition for treating cardiovascular risk factors related to disorders of lipid and / or carbohydrate metabolism (hyperlipidemia, type 2 diabetes, obesity, etc.), while reducing the overall risk.
- the pharmaceutical composition according to the invention is preferably used for the treatment of type 2 diabetes and / or dyslipidemias.
- Another subject of the invention relates to a nutritional composition
- a nutritional composition comprising at least one compound as described above.
- Another subject of the invention resides in the use of at least one compound as described above for the preparation of pharmaceutical compositions intended for the treatment of various pathologies, in particular related to metabolic disorders among which mention may be made of dyslipidemias, type 2 diabetes, insulin resistance, metabolic syndrome, atherosclerosis, cardiovascular diseases, obesity, hypertension, inflammatory diseases, etc. More generally, the subject of the invention is the use of at least one compound as described above for the preparation of pharmaceutical compositions for treating risk factors for cardiovascular diseases related to disorders of the metabolism of lipids and / or carbohydrates and intended to reduce the overall risk.
- the molecules according to the invention may advantageously be administered in combination with other therapeutic and / or cosmetic agents, commercialized or in development, such as:
- insulinosecretors sulfonylureas (glibenclamide, glimepiride, gliclazide, etc.) and glinides (repaglinide, nateglinide, etc.)
- alpha-glucosidase inhibitors PPAR ⁇ agonists (thiazolidinediones such as rosiglitazone, pioglitazone), mixed agonists PPAR ⁇ / PPAR ⁇ (tesaglitazar, muraglitazar), pan-PPAR (compounds that simultaneously activate the 3 PPAR isoforms), biguanides (metformin), dipeptidyl peptidase IV inhibitors (sitagliptin, vildagliptin), agonists Glucagon-Like Peptide-1 (GLP-1) (exenatide), etc.
- PPAR ⁇ agonists thiazolidinediones such as rosiglitazone, pioglitazone
- hypolipidemic and / or hypocholesterolemic molecules fibrates (fenofibrate, gemfibrozil), HMG CoA reductase inhibitors or hydroxylmethylglutaryl Coenzyme A reductase inhibitors (statins such as atorvastatin, simvastatin, fluvastatin), cholesterol absorption inhibitors ( ezetimibe, phytosterols), inhibitors of CETP or Cholesteryl Ester Transfer Protein (torcetrapib), inhibitors of Acyl-CoA: Cholesterol O-Acyl Transferase (ACAT) (avasimibe, eflucimibe), inhibitors MTP (Microsomal Triglyceride Transfer Protein ), sequestering agents of bile acids (cholestyramine), vitamin E, polyunsaturated fatty acids, omega 3 fatty acids, nicotinic acid derivatives (niacin), etc.
- fibrates fibrates
- angiotensin-converting enzyme (ACE) inhibitors captopril, enalapril, ramipril or quinapril
- angiotensin II receptor antagonists leukin, renin, renin, renin, renin, renin, renin, renin, renin, renin, renin, renin, renin, renin, renin, renin, renin II receptor antagonists
- beta-blockers atenolol, metoprolol, labetalol, propranolol
- thiazide diuretics and non-thiazides thiazide diuretics and non-thiazides (furosemide, indapamide, hydrochlorothiazide, anti-aldosterone), vasodilators, calcium channel blockers (nifedipine, felodipine or amlodipine, diltizem or verapamil), etc
- anti-platelet agents aspirin, ticlopidine, dipyridamol, clopidogrel, flurbiprofen, etc.
- anti-obesity agents sibutramine, lipase inhibitors (orlistat), PPAR ⁇ agonists and antagonists, cannabinoid CB1 receptor antagonists (rimonabant), etc.
- anti-inflammatory agents for example, corticosteroids (prednisone, betamethazone, dexamethazone, prednisolone, methylprednisolone, hydrocortisone, etc.), NSAIDs or non-steroidal anti-inflammatory drugs derived from indole (indomethacin, sulindac), NSAIDs from the arylcarboxylic group (thiaprofenic acid, diclofenac, etodolac, flurbiprofen, ibuprofen, ketoprofen, naproxen, nabumetone, alminoprofen), NSAIDs derived from oxicam (meloxicam, piroxicam, tenoxicam), NSAIDs of the fenamate group, selective inhibitors COX2 (celecoxib, rofecoxib), etc.
- corticosteroids prednisone, betamethazone, dexamethazone, prednisol
- anti-oxidizing agents for example probucol, etc.
- agents used in the treatment of heart failure thiazide or non-thiazide diuretics (furosemide, indapamide, hydrochlorthiazide, anti-aldosterone), ACE (Angiotensin Converting Enzyme) inhibitors (captopril, enalapril, ramipril or quinapril) ), digitalis (digoxin, digitoxin), beta blockers (atenolol, metoprolol, labetalol, propranolol), phosphodiesterase inhibitors (enoximone, milrinone), etc.
- beta blockers atenolol, metoprolol, labetalol, propranolol
- calcium channel blockers nifedipine, felodipine or amlodipine, bepridil, diltiazem or verapamil
- NO donor agents trinitrin, isosorbide dinitrate, molsidomine
- Amiodarone etc.
- anticancer agents cytotoxic agents (DNA interacting agents, alkylating agents, cisplatin and derivatives), cytostatic agents (GnRH analogs, somatostatin analogues, progestins, anti-estrogens, aromatase inhibitors, etc.), modulators of the immune response (interferons, IL2, etc.), etc.
- cytotoxic agents DNA interacting agents, alkylating agents, cisplatin and derivatives
- cytostatic agents GnRH analogs, somatostatin analogues, progestins, anti-estrogens, aromatase inhibitors, etc.
- modulators of the immune response interferons, IL2, etc.
- anti-asthmatics such as bronchodilators (beta 2 -agonist agonists), corticosteroids, cromoglycate, leukotriene receptor antagonists (montelukast), etc.
- corticosteroids used in the treatment of skin pathologies such as psoriasis and dermatitis
- vasodilators and / or anti-ischemic agents (buflomedil, Ginkgo Biloba extract, naftidrofuryl, pentoxifylline, piribedil), etc.
- the invention also relates to a method for treating pathologies related to the metabolism of lipids and / or carbohydrates comprising the administration to a subject, in particular a human subject, of an effective amount of a compound or a pharmaceutical composition as defined above.
- an effective amount refers to an amount of the compound sufficient to produce the desired biological result, preferably nontoxic.
- subject means a mammal and more particularly a human.
- treatment refers to curative, symptomatic or preventive treatment.
- the compounds of the present invention can thus be used in humans with a declared disease.
- the compounds of the present invention can also be used to delay or slow progression or prevent further progression of the disease, thereby improving the condition of patients.
- the compounds of the present invention may finally be administered to non-diseased individuals who may develop the disease normally or who have a significant risk of developing the disease.
- compositions according to the invention advantageously comprise one or more excipients or vehicles, which are pharmaceutically acceptable.
- excipients or vehicles for example, saline, physiological, isotonic, buffered, etc., solutions compatible with a pharmaceutical use and known to those skilled in the art may be mentioned.
- the compositions may contain one or several agents or vehicles chosen from dispersants, solubilizers, stabilizers, preservatives, etc.
- Agents or vehicles that can be used in formulations include methylcellulose, hydroxymethylcellulose, carboxymethylcellulose, polysorbate 80, mannitol, gelatin, lactose, vegetable oils, and the like. acacia, liposomes, etc.
- compositions may be formulated as injectable suspensions, gels, oils, tablets, suppositories, powders, capsules, aerosols, etc., optionally using dosage forms or devices providing sustained and / or delayed release.
- an agent such as cellulose, carbonates or starches is advantageously used.
- the compounds or compositions according to the invention can be administered in different ways and in different forms. Thus, they may be, for example, administered systemically, orally, parenterally, by inhalation or by injection, for example intravenously, intramuscularly, subcutaneously, trans-dermally, intra-arterially, etc.
- the compounds are generally packaged as liquid suspensions, which can be injected by means of syringes or infusions, for example. It is understood that the flow rate and / or the injected dose can be adapted by those skilled in the art depending on the patient, the pathology, the mode of administration, etc.
- the compounds are administered at doses ranging between 1 ⁇ g and 2 g per administration, preferably from 0.1 mg to 1 g per administration. Administrations can be daily or repeated several times a day, if necessary.
- the compositions according to the invention may comprise, in addition, other agents or active principles.
- the invention also relates to processes for the preparation of substituted N- (phenethyl) benzamide compounds according to the invention.
- the compounds of the invention can be prepared from commercial products, by implementing a combination of chemical reactions. More particularly, several synthesis steps are necessary to obtain the compounds according to the invention.
- the key step is the formation of the amide bond of the substituted N- (phenethyl) benzamide derivative compounds which are the subject of the invention, advantageously by condensation of a mono- or poly-substituted 2-phenylethanamine derivative with a derivative of mono- or poly-substituted benzoic acid (or derivative) type.
- This condensation can be carried out according to the methods known to those skilled in the art, and in particular those which have been developed in the context of peptide synthesis.
- the subject of the present invention is thus a process for the preparation of the compounds according to the invention as described above, comprising:
- the process for preparing the compounds according to the invention makes it possible to obtain compounds in optically pure (or enriched) form.
- the process for the preparation of the compounds according to the invention makes it possible to prepare compounds referred to below as intermediate compounds.
- the present invention also relates to certain raw materials and intermediate compounds obtained in the context of the present invention. These intermediate compounds are more particularly chosen from: Example 2-6: Ethyl 2- [4- (1-aminopropan-2-yl) phenoxy] -2-methylpropanoate;
- Example 2-7 4- (1-aminohexan-2-yl) phenol
- Example 2-8 Tertiary butyl 2- [4- (1-aminopropan-2-yl) -3-methyl-phenoxy] -2-methylpropanoate;
- Example 2-9 tert-butyl 2- [4- (3-aminobutan-2-yl) phenoxy] -2-methylpropanoate;
- Example 2-10 tert-butyl 2- [4- (2-aminopentyl) phenoxy] -2-methylpropanoate;
- Example 2-11 tert-butyl 2- [4- (1-aminopropan-2-yl) phenoxy] -2-methylpropanoate;
- Example 2-12 tert-butyl 2- [3- (1-aminopropan-2-yl) phenoxy] -2-methylpropanoate;
- Example 3-1 5-methyl-2-octyloxybenzoic acid
- Example 4-1 4- [2- (5-chloro-2-methoxybenzamido) propyl] phenol;
- Example 4-2 4- [2- (5-Chloro-2-methoxybenzamido) -2-methylpropyl] phenol;
- Example 4-3 4- [1- (5-Chloro-2-methoxybenzamido) -2-methylpropan-2-yl] phenol;
- Example 4-4 4- [1- (5-Chloro-2-methoxybenzamido) propan-2-yl] phenol;
- Example 4-5 4- [1- (5-Chloro-2-methoxybenzamido) hexan-2-yl] phenol;
- Example 4-6 4- [2- (2-methoxy-5-methylbenzamido) ethyl] phenol;
- Example 4-7 4- [2- (5-methyl-2-octyloxybenzamido) ethyl] phenol;
- Example 4-8 4- [2- (2-Methoxy-5-phenylbenzamido) ethyl] phenol
- Example 4-9 4- [2 - ((3-methoxy-2-naphthoyl) amino) propyl] phenol;
- Example 4-11 4- [2- (5-tert-butyl-2-methoxybenzamido) propyl] phenol;
- Example 4-12 4- [2- (5-Butyryl-2-methoxybenzamido) propyl] phenol;
- Example 4-13 4- [2- (5-phenyl-2-methoxybenzamido) propyl] phenol
- Example 4-14 4- [2- (5- (2,4-difluorophenyl) -2-methoxybenzamido) propyl] phenol;
- Example 4-15 4- [2- (5- (pyrrol-1-yl) -2-methoxybenzamido) propyl] phenol;
- Example 5-1 Ethyl 2- [4- [2- (2-fluorobenzamido) propyl] phenoxy] -2-methylpropanoate;
- Example 5-2 Ethyl 2- [4- [1 - ((3-hydroxy-2-naphthoyl) amino) propan-2-yl] phenoxy] -2-methylpropanoate;
- Example 5-3 Ethyl 2- [4- [2- (2-fluoro-5-trifluoromethylbenzamido) propyl] phenoxy] -2-methylpropanoate;
- Example 5-4 Ethyl 2- [4- [1 ⁇ ((2-hydroxy-1-naphthoyl) amino) propan-2-yl] phenoxy] -2-methylpropanoate;
- Example 5-5 tert-butyl 2- [4- [1 - ((3-hydroxy-7-methoxy-2-naphthoyl) amino) propan-2-yl] phenoxy] -2-methylpropanoate;
- Example 5-6 tert-Butyl 2- [4- [1- (5- (2,4-difluorophenyl) -2-hydroxybenzamido) propan-2-yl] phenoxy] propanoate.
- the compounds of general formula (I) according to the invention are preferably obtained by condensation between a carboxylic acid (VI) and an amine (VII).
- the condensation reaction can be carried out by multiple routes, known to those skilled in the art.
- coupling agents Py-BOP, DCC 1 EDC, HBTU, CDI 1, etc.
- activated acid derivatives in this case, the acid is first converted into active derivative of the acyl chloride type, activated ester, mixed anhydride, etc.
- the condensation in mass bringing into contact with the two entities hot, without solvent
- the condensation by azeotropic distillation of the water formed during the reaction etc.
- a preferred route is to work in a solvent such as dichloromethane, chloroform, diethyl ether, diisopropyl ether, methyl tert-butyl ether, tetrahydrofuran, dioxane, toluene, acetonitrile or dimethylformamide.
- a solvent such as dichloromethane, chloroform, diethyl ether, diisopropyl ether, methyl tert-butyl ether, tetrahydrofuran, dioxane, toluene, acetonitrile or dimethylformamide.
- Such reactions are carried out in the presence of agents that activate the acid function (DCC / HOBt, Py-BOP, etc.) or in the presence of a preactivated form of the acid (acyl chloride, mixed anhydride, etc.).
- a base is often necessary: it may be inorganic bases such as carbonates (sodium, cesium) or potash; organic bases such as trialkylamines (triethylamine, diisopropylethylamine, etc.) or pyridine may also be employed. These reactions can be carried out at temperatures of between -25 ° C. and 250 ° C., preferably between -10 ° C. and the boiling point of the envisaged solvent.
- Another way is to work in the absence of solvent.
- the reactions are carried out by eliminating the water formed as the condensation progresses. These reactions can be carried out at temperatures of between 25 ° C. and 250 ° C.
- the water can be removed by evaporation (reaction under reduced pressure, for example).
- reaction is in this case carried out at the reflux of a solvent such as toluene.
- Method B The compounds of general formula (I) according to the invention are preferably obtained by hydrolysis, thermolysis, hydrogenolysis or functionalization of the intermediate (I ').
- Groups G 1 X 1 , X 2, X 3, X 4, R 1, R 2, R 3, R 4, Y, E are as defined above.
- the group E ' is by definition a group which by hydrolysis, thermolysis, hydrogenolysis or functionalization makes it possible to generate the group E.
- E contains at least one carboxylic acid function.
- E ' is in this case a group containing a chemical function that can be converted into a carboxylic derivative by hydrolysis, thermolysis or hydrogenolysis.
- carboxylic acid-hydrolysable chemical functions are acid derivatives (esters, thioesters, orthoesters, etc.) and nitrile, tetrazolyl, 1,3-oxazol-2-yl, 1,3-oxazolin-2- functions. yle, etc.
- thermolysis generates an acid function
- tertiary alkyl esters preferably tert-butyl esters.
- Examples of chemical functions whose hydrogenolysis generates an acid function are the aralkyl esters, preferably the benzyl esters.
- the hydrolysis reactions may advantageously be carried out in the presence of an organic acid (eg trifluoroacetic acid) or an inorganic acid (eg hydrochloric acid) or in the presence of a base (eg sodium hydroxide) in water or a solvent mixture containing water (water / methanol, water / ethanol, water / THF (TetraHydroFuran), water / dioxane, etc.). They are carried out at temperatures between -10 ° C. and 120 ° C., preferably between 20 ° C. and the reflux temperature of the solvent used.
- an organic acid eg trifluoroacetic acid
- an inorganic acid eg hydrochloric acid
- a base eg sodium hydroxide
- thermolysis reactions are preferably carried out in the absence of solvent (molten mixture) or in an inert solvent such as dichloromethane, chloroform, toluene, tetrahydrofuran or dioxane.
- thermolysis The addition of catalytic amounts of strong acids such as para-toluenesulfonic acid is generally necessary for thermolysis. These reactions are preferably carried out hot, advantageously at the boiling point of the solvent.
- the hydrogenolysis reactions are preferably carried out in the presence of a metal catalyst (Pd / C, Pt, etc.) in a suitable solvent such as methanol, ethanol, tetrahydrofuran (THF), acetic acid ethyl acetate, etc. They are carried out at temperatures between 0 0 C and
- the compounds (I 1 ) correspond to the esterified form of the compounds (I).
- different methods are applicable to regenerate E acid:
- E contains at least one acid derived function (COOR a , -COSR 3 , -CONR a R b , -CSNR a R b ) or isostere (an acylsulfonamide, hydrazide or tetrazolyl radical). ).
- E ' is in this case a group containing a chemical function that can be converted into acid derivative or isosteric acid by functionalization.
- E 'contains a carboxylic acid function This function can easily be converted into an acid derivative according to the methods known to those skilled in the art: by way of example, the preparation of ester from carboxylic acid is feasible according to many well-documented methods.
- the carboxylic acid function will be functionalized by condensation according to the methodologies described in the preceding paragraph (method A). This type of methodology will also be preferentially applied for the preparation of acylsulfonamide and hydrazide isosteres.
- This function can easily be converted into a tetrazolyl derivative according to the methods known to those skilled in the art: for example, action of an azide derivative in an anhydrous solvent (toluene, tetrahydrofuran (THF), etc.) at temperatures comprised between 0 0 C and the boiling point of the solvent in question.
- anhydrous solvent toluene, tetrahydrofuran (THF), etc.
- the compounds according to the invention of general formula (I) are preferably obtained by functionalization of the intermediate (VIII).
- Group X is:
- a leaving group for example, a halogen atom or a triflate group
- a nucleophilic group for example, a hydroxyl, thiol or amino radical. If X is a leaving group, it may advantageously be substituted by various methods known to those skilled in the art.
- a preferred pathway is aromatic nucleophilic substitution. This type of reaction proceeds hot in the presence of a large excess of nucleophile
- Bases are often used to activate the nucleophile (for example, cesium carbonate, sodium tert-butylate, etc.). This type of reaction is preferably carried out in common solvents such as dimethylformamide, acetonitrile, tetrahydrofuran (THF), dioxane or toluene. It can be carried out at temperatures between
- the molecules of general formula (I) may be obtained by a palladium catalyzed coupling reaction (such as the Buchwald-Hartwig reaction and its variants). These reactions are well known to those skilled in the art.
- This type of reaction requires the presence of a palladium catalyst (for example, Pd (OAc) 2 , Pd 2 (dba) 3 ), a ligand (for example BINAP, X-Phos, tri-fe / t- butylphosphine) and a base (e.g., Cs 2 CO 3 , sodium t-butylate). It can be carried out at temperatures of between 20 ° C. and the reflux temperature of the chosen solvent.
- Various solvents are usable, for example toluene, dimethylformamide, tetrahydrofuran (THF), N-methyl-2-pyrrolidone or dioxane.
- X is a nucleophilic group
- various methods known to those skilled in the art can be envisaged.
- X may advantageously be alkylated to obtain a compound of formula (I).
- X is a nitrogen atom (optionally substituted alkyl)
- it can be functionalized as a secondary (or tertiary) amine function by nucleophilic substitution of a halogen derivative.
- X is an oxygen atom (or sulfur)
- it can be functionalized in ether function (or thioether) by nucleophilic substitution of a halogen derivative.
- a base is often necessary: it can be inorganic bases such as carbonates (for example sodium, cesium) or potash; organic bases such as trialkylamines (triethylamine, diisopropylethylamine, etc.) or pyridine may also be employed.
- These reactions can be carried out at temperatures between -25 and 250 ° C., preferably between -10 ° C. and the boiling point of the envisaged solvent.
- X is a nitrogen atom (optionally substituted alkyl)
- it can be functionalized by reductive alkylation: condensation of an aldehyde or a ketone on the amine (formation of an imine) which can be reduced in situ (or after isolation) with a reducing agent (eg, NaBH 3 CN,
- X is an oxygen or sulfur atom
- it may be functionalized according to the conditions of Mitsunobu (triphenylphosphine, diethyl azodicarboxylate).
- Mitsunobu triphenylphosphine, diethyl azodicarboxylate
- X is an oxygen atom
- a preferred strategy consists in treating the phenolic derivative in the presence of sodium hydroxide in acetone with chloroform at a temperature of between 20 ° C. and 50 ° C.
- the thioether function (-SE) may be oxidized by methods known to those skilled in the art.
- oxone ® may be used in a solvent such as water, methanol or dichloromethane at room temperature.
- the compounds according to the invention of general formula (I) are preferably obtained by functionalization of the intermediate (IX).
- Group X is:
- a leaving group for example, a halogen atom or a triflate group
- a nucleophilic group for example, a hydroxyl or thiol radical.
- Group G may be introduced according to the methods proposed in the preceding paragraph (method C).
- the compounds according to the invention are preferably obtained by functionalization of the intermediate (X).
- Group X is: - Let a leaving group: for example, a halogen atom or a triflate group;
- a nucleophilic group for example, a hydroxyl, thiol or amino radical.
- Group X1 can be introduced according to the methods proposed in the previous paragraph (method D).
- X is a nucleophilic group
- another preferred route consists in condensing a carboxylic acid derivative (or activated derivative), chloroformate or isocyanate derivative (and sulfur analogs: isothiocyanate, etc.): for example, if X is an atom of nitrogen, these strategies make it possible to respectively generate amide, carbamate or ureido groups respectively. These reactions are preferably carried out in dichloromethane, chloroform, diethyl ether, diisopropyl ether, methyl tert-butyl ether, tetrahydrofuran (THF), dioxane, toluene, acetonitrile or dimethylformamide.
- THF tetrahydrofuran
- a base is often necessary: it may be inorganic bases such as carbonates (sodium, cesium) or potash; organic bases such as trialkylamines (triethylamine, diisopropylethylamine, etc.) or pyridine may also be employed. These reactions can be carried out at temperatures between -25 ° C and
- the molecules of general formula (I) may be obtained by a palladium-catalyzed coupling reaction chosen from the reactions of Suzuki-Miyaura, Heck, StNIe,
- the Suzuki reaction involves coupling an organoborated derivative (eg, a boronic acid) to the derivative bearing the leaving group (e.g., a brominated derivative) in the presence of a palladium catalyst (eg, Pd (PPh 3 ) 4 , Pd (OAc) 2 ), a ligand (eg BINAP) and a base (for example, Cs 2 CO 3 , CsF).
- a palladium catalyst eg, Pd (PPh 3 ) 4 , Pd (OAc) 2
- a ligand eg BINAP
- a base for example, Cs 2 CO 3 , CsF.
- Various solvents are usable, for example dimethylformamide, toluene, tetrahydrofuran, N-methyl-2-pyrrolidone or dioxane.
- the compounds according to the invention of general formula (I) are preferably obtained by functionalization of the intermediate (Xl).
- Group X is:
- a leaving group for example, a halogen atom or a triflate group
- a nucleophilic group for example, a hydroxyl, thiol or amino radical.
- Group X3 can be introduced according to the methods proposed in the previous paragraph (method E).
- a preferred route is to functionalize the ketone function according to methods well known to those skilled in the art such as the Wittig reaction or one of its variants (for example, the Homer-Emmons reaction).
- the ketone reacts with a phosphorus ylide (commercial or prepared according to methods known to those skilled in the art) in the presence of a base.
- phosphorus ylide commercial or prepared according to methods known to those skilled in the art
- These reactions can be carried out in different solvents depending on the stability of the ylides used: they are preferably carried out in water, diethyl ether, diisopropyl ether, methyl tertiary butyl ether, tetrahydrofuran (THF), dioxane, toluene. acetonitrile or dimethylformamide.
- the bases preferentially used are hydroxides (sodium, potassium), carbonates (potassium, cesium), sodium hydride, potassium bis (trimethylsilyl) amide (KHMDS), etc. These reactions can be carried out at temperatures of between -78 ° C. and 250 ° C., preferably between -10 ° C. and the boiling point of the envisaged solvent.
- chiral reducing agents for example, the use of transition metals in the presence of chiral ligands
- transition metals in the presence of chiral ligands
- chiral reducing agents may preferentially be envisaged to preferentially synthesize one or other of the stereoisomers at the level of R4.
- a reduction catalyzed by rhodium in the presence of a chiral phosphine ligand such as BINAP.
- the compounds according to the invention of general formula (I) are preferably obtained by functionalization of the intermediate (XIII).
- a preferential access route consists in deprotonating the substrate of formula (XIII) with a strong base. This activation allows the substitution of an R3-X derivative,
- X denoting a leaving group and R3 being as defined above.
- These reactions can be carried out in various anhydrous solvents such as methyl tertiobutyl ether, tetrahydrofuran (THF), dioxane, toluene, acetonitrile.
- the bases preferentially used are those derived from lithium (tBuLi, secBu ⁇ , LDA, LiHMDS, etc.). These reactions can be carried out at temperatures of between -78 ° C. and 50 ° C.
- the leaving group X is chosen from the numerous leaving groups known to those skilled in the art: halogen atom, tosyl or mesyl groups , triflate group, etc.
- this nucleophilic substitution can be carried out in the presence of a chiral ligand.
- This type of approach allows an enantioselective synthesis of the derivatives of formula (I) by controlling the stereochemistry of the C-R3 bond formed.
- Many chiral ligands known to those skilled in the art can be used for this type of reaction.
- the optically pure compounds when the molecule comprises one or more chiral centers, the optically pure compounds (or enriched mixtures) can be prepared or purified according to usual methods known to those skilled in the art: asymmetric synthesis using agents chiral (catalysts, reagents); purification of the compounds or intermediates by stereoselective methods (chromatography on chiral phases, precipitation of a salt formed with a chiral counterion); etc.
- the synthesis of the substituted N- (phenethyl) benzamide derivative compounds according to the invention preferably comprises a step of condensation of a mono- or poly-substituted 2-phenylethanamine type derivative with a mono benzoic acid derivative (or derivative) derivative. - or poly-substituted.
- these two reagents will be synthesized or purified in optically pure (or enriched) forms before condensation.
- a preferred route is to purify the amine or acid by a chromatographic technique (for example, on a chiral phase column).
- Another preferred route is to purify the amine or acid by crystallization of the salts formed with respectively chiral carboxylic acids enantiopurs (tartaric acid, etc.) or chiral organic enantiopure bases (ephedrine, etc.).
- Another preferred route is to protect the amine or acid with a protecting group containing an asymmetric center. The mixture can then be enriched in one or other of the diastereoisomers by crystallization, which after deprotection allows isolation of the amine or optically pure (or enriched) acid.
- a particularly preferred object of the invention relates to the compounds of general formula (I), advantageously (II) or (III), in which R 1 represents an alkyl, alkenyl, aryl or aralkyl radical and R 2, R 3 and R 4 are carbon atoms. 'hydrogen.
- R 1 represents an alkyl, alkenyl, aryl or aralkyl radical
- R 2 represents carbon atoms. 'hydrogen.
- the inventors have developed a process for synthesizing these preferred compounds in enantiopure form. This methodology is given by way of example and is in no way limiting as to how to prepare the optically pure compounds according to the invention.
- the intermediate (VIII ') will be easily obtained by condensation of the amine (XV) on the acid (VI) according to the methods known to those skilled in the art (see
- the intermediate (XV) can be advantageously synthesized in 2 key steps from an ⁇ -amino acid, the absolute configuration of the latter being preserved.
- Friedel-Craft acylation is a reaction well known to those skilled in the art. It is carried out in the presence of an acidic catalyst (for example, AlCl 3 or FeCl 3), an activated form of the protected ⁇ -amino acid (for example, acyl chloride), and an aryl derivative. These reactions are preferably carried out in anhydrous organic solvents such as dichloromethane, chloroform, diethyl ether, diisopropyl ether, methyl tert-butyl ether or tetrahydrofuran (THF). They can be carried out at temperatures between -78 ° C. and 100 ° C., preferably between -20 ° C. and the point boiling of the envisaged solvent. Pacylation orientation (ortho / meta / para) may vary depending on the substituents present on the aryl ring according to rules well known to those skilled in the art.
- an acidic catalyst for example, AlCl 3 or FeCl 3
- the Grignard reaction is again a well-known reaction.
- the proposed method is not limited to organomagnesium agents but more generally to the numerous organometallic derivatives that can be prepared from an aryl halide (magnesium, lithian, zinc, etc.).
- the aryl substrate previously activated in the form of an organometallic ion is reacted with the previously protected protected ⁇ -amino acid.
- the activation of the acid functional function "Weinreb amide" is particularly relevant to achieve this type of reaction.
- reaction are preferably carried out in anhydrous organic solvents such as dichloromethane, chloroform, diethyl ether, diisopropyl ether, methyl tert-butyl ether, tetrahydrofuran (THF), dioxane, or toluene. They can be carried out at temperatures of between -100 ° C. and + 100 ° C., preferably between -78 ° C. and the boiling point of the envisaged solvent. The reaction will preferably be carried out under mild conditions (especially at low temperature) so as not to favor racemization.
- anhydrous organic solvents such as dichloromethane, chloroform, diethyl ether, diisopropyl ether, methyl tert-butyl ether, tetrahydrofuran (THF), dioxane, or toluene. They can be carried out at temperatures of between -100 ° C. and + 100 ° C., preferably between -78 ° C
- the reduction of the intermediate (XIV) to (XV) can be envisaged in various ways known to those skilled in the art.
- this reduction will be carried out in an acid medium (for example, trifluoroacetic acid) with trialkyl silane (for example, triethylsilane).
- the acid used can act as a solvent.
- solvents may be added such as acetonitrile, carbon tetrachloride or water. These reactions can be carried out at temperatures between -78 ° C. and 100 ° C., preferably between 0 ° C. and the boiling point of the envisaged solvent.
- This reduction can be envisaged directly on the intermediate of type (XIV) or after a partial reduction of the ketone function as a function of alcohol (by prior action of a hydride such as NaBH 4 for example).
- This general method makes it possible to obtain (XV) optically pure, without significant racemization of the starting ⁇ -amino acid. It is described in the examples and it is a preferred access route to the compounds according to the invention.
- the compounds of formula (XV) according to the invention or X is a hydroxyl radical (-OH) located para to the - CH2- motif are here named (XV). They are preferably obtained according to the following process:
- the ⁇ -amino acid is protected by a Cbz (benzyloxycarbonyl) group and then activated in the form of Weinreb amide.
- the action of the organomagnesium makes it possible to form a ketonic intermediate which is then reduced and then deprotected, which makes it possible to isolate the intermediates of formula (XV) in enantiopure form.
- Another methodology consists in purifying (or enriching) a racemic mixture of compounds of formula (I) according to the invention.
- a preferred route is to purify the compounds by chromatography.
- Some of the compounds according to the invention may contain an acid function (in particular at the group E) and / or a basic function. They can therefore advantageously be purified by crystallization as described above: formation of salts or protection of the molecule by chiral agents. If the compound is desired in the form of a salt, the latter will be obtained in a last step known to those skilled in the art, not mentioned in the synthesis routes presented above. For example, a preferred route is to use an ion exchange resin to obtain the desired salts.
- FIG. 1 In Vitro Evaluation of the Activating Properties of PPARs of the Compounds According to the Invention at 10 ⁇ M
- the activation of the PPARs is evaluated in vitro on a monkey kidney fibroblast (COS-7) line, by measuring the transcriptional activity of chimeras consisting of the DNA binding domain of the Gal4 transcription factor of the yeast and the ligand binding domain of the different PPARs.
- COS-7 line monkey kidney fibroblast
- the compounds are tested at 10 ⁇ M on Gal4-PPAR ⁇ , ⁇ , ⁇ chimeras.
- the induction factor i.e., the ratio of the luminescence induced by the compound to the luminescence induced by the control, is measured for each condition. It is then normalized with respect to an internal reference compound and the results are expressed in percentages: the higher the percentage of activation, the more the compound has an activating character of the PPARs.
- FIG. 2 In Vitro Evaluation of the Activating Properties of the PPARs of the Compounds According to the Invention as a Function of the Dose
- the activation of the PPARs is evaluated in vitro on a monkey kidney fibroblast (COS-7) line, by measuring the transcriptional activity of chimeras consisting of the DNA binding domain of the Gal4 transcription factor of the yeast and the ligand binding domain of the different PPARs.
- the compounds are tested at doses of between 0.01 and 10 ⁇ M on GaW-PPAR ⁇ , ⁇ and ⁇ chimeras.
- the induction factor i.e., the ratio of the luminescence induced by the compound to the luminescence induced by the control, is measured for each condition. It is then normalized with respect to an internal reference compound and the results are expressed in percentages: the higher the percentage of activation, the more the compound has a character activator PPARs.
- FIG. 3 In Vitro Evaluation of the Affinity of the Compounds According to the Invention for 100 ⁇ M ATP-Dependent Potassium Channels
- the results presented reflect the specific affinity of the compounds according to the invention for the ATP-dependent potassium channels (K + A TP).
- the compounds according to the invention were tested at 100 ⁇ M: the higher the measured percentage, the greater the The affinity of the compound for the ATP-dependent potassium channels is high.
- FIG. 4 In vitro Evaluation of the Insulinosecreating Character of the Compounds According to the Invention
- the activation of insulin secretion is evaluated in vitro on a rat pancreatic cell line (INS-1) by measuring the concentration of insulin secreted in the culture medium in the presence of a low glucose concentration. (2.8 mM).
- the compounds are tested at doses of between 1 and 100 ⁇ M.
- the results are represented by the induction factor for each condition, that is to say the ratio between the insulin concentration induced by the compound according to the invention and the concentration induced by glucose 2.8 mM alone: the higher this factor, the more the compound according to the invention has an insulin-secretory character.
- FIG. 5 In Vivo Evaluation in the Rat of the Hypoglycemic Character of the Compounds
- the object of this study is to evaluate in vivo the insulinosecreating nature of the compounds according to the invention.
- the oral administration of insulin-secretory compounds results in a significant increase in the plasma insulin level.
- This induced hyperinsulinemia causes a drop in blood sugar.
- the curve presented represents the blood glucose of the animal over time and shows the hypoglycemic effect of the compounds according to the invention (administered orally in fasted rats).
- FIGS. 6a, 6b, 6c In vivo evaluation of the lipid-lowering properties of the compounds according to the invention and measurement of the regulation of the expression of genes involved in lipid and glucidic metabolism.
- the lipid-lowering effect of the compounds according to the invention is evaluated in vivo in the humanized mouse for the E2 isoform of apolipoprotein E.
- the induced mutation prevents the hepatic clearance of lipids and leads to a plasma accumulation of triglycerides and cholesterol in this animal.
- the treatment of these animals with a PPAR ⁇ agonist must induce a decrease in plasma lipid levels and must be expressed in the liver by a modification of the expression of the target genes whose expression is under the control of the PPAR ⁇ receptor.
- Figure (6a) shows the plasma cholesterol levels after 7 days of treatment with compound 5, administered at 300 mg / kg / day. These levels are compared with those obtained with control animals (not treated with the compounds according to the invention): the measured difference reflects the lipid-lowering effect of the compounds according to the invention.
- the effectiveness of the compounds according to the invention is also evaluated by measuring, in the liver tissues, the expression of genes involved in lipid and carbohydrate metabolism. The higher the induction factor measured, the more the compound has a character of gene expression activator.
- Figures (6b) and (6c) represent the hepatic expression levels of PDK4 (pyruvate dehydrogenase kinase isoform 4, enzyme carbohydrate metabolism) and ACO (acyl coenzyme A oxidase, a key enzyme involved in the mechanism of ⁇ -oxidation of fatty acids) after 7 days of treatment with compound 5 (300 mg / kg / day).
- PDK4 pyruvate dehydrogenase kinase isoform 4, enzyme carbohydrate metabolism
- ACO acyl coenzyme A oxidase, a key enzyme involved in the mechanism of ⁇ -oxidation of fatty acids
- FIGS. 7a to 7e In Vivo Evaluation, in the E2 / E2 Mouse, of the Lipid and Lipidic Properties of Hypolipidemic and Stimulating Factors of HDL-Cholesterol Synthesis of the Compounds According to the Invention by Lipid Assays and Measurement of the Expression of Genes Involved in Lipid Metabolism and carbohydrate and energy dissipation.
- the lipid-lowering effect of the compounds according to the invention is evaluated in vivo in the mouse E2 / E2 (humanized for the E2 isoform of apolipoprotein E) by analyzing the distribution of cholesterol in the various plasma lipoprotein fractions and by measuring plasma triglyceride levels after 7 days of oral treatment. These levels are compared with those obtained with control animals (not treated with the compounds according to the invention): the measured difference reflects the lipid-lowering effect of the compounds according to the invention.
- FIG. 7a shows the distribution of cholesterol in the various plasma lipoprotein fractions after 7 days of treatment with compound 29, administered at 50 mg / kg / day.
- the effectiveness of the compounds according to the invention is also evaluated by measuring, in the liver and muscle tissues (skeletal), the expression of genes involved in lipid and carbohydrate metabolism and energy dissipation. The higher the induction factor measured, the more the compound has a character of gene expression activator.
- FIG. 7c shows the expression of PDK4 (pyruvate dehydrogenase kinase isoform 4, carbohydrate metabolism enzyme) in liver tissue, in E2 / E2 mice, after 7 days of treatment with compound 29 (50 mg / kg /day).
- PDK4 pyruvate dehydrogenase kinase isoform 4, carbohydrate metabolism enzyme
- FIG. 7d represents the expression of ACO (acyl coenzyme A oxidase, a key enzyme involved in the mechanism of ⁇ -oxidation of fatty acids) in liver tissue, in E2 / E2 mice, after 7 days of treatment with compound 29 (50 mg / kg / day).
- ACO acyl coenzyme A oxidase, a key enzyme involved in the mechanism of ⁇ -oxidation of fatty acids
- FIG. 7e represents the expression of UCP3 (uncoupling protein 3, implicated in energy dissipation) in skeletal muscle, in E2 / E2 mice, after 7 days of treatment with compound 29 (50 mg / kg / day).
- Figures 8a to 8e In vivo evaluation, in C57BI6 mice, of the properties hypolipidemic and stimulating the synthesis of HDL-cholesterol compounds according to the invention by lipid assays and measuring the expression of genes involved in lipid and carbohydrate metabolism.
- the effect of the compounds according to the invention is evaluated in vivo in C57BI6 mice by measuring the plasma levels of HDL-cholesterol, plasma triglycerides and free fatty acids after 14 days of oral treatment; these levels are compared with those obtained with control animals (not treated with the compounds according to the invention): the measured difference indicates the effect of the compounds according to the invention.
- FIG. (8a) represents plasma HDL-cholesterol levels after 14 days of treatment with compound 29, administered at 50 mg / kg / day.
- FIG. 8b shows the levels of plasma triglycerides after 14 days of treatment with compound 29, administered at 50 mg / kg / day.
- - Figure (8c) represents plasma free fatty acid levels after
- the effectiveness of the compounds according to the invention is also evaluated by measuring, in the liver tissue, the expression of genes involved in lipid and carbohydrate metabolism metabolism. The higher the induction factor measured, the more the compound has a character of gene expression activator.
- FIG. 8d represents the expression of PDK4 (pyruvate dehydrogenase kinase isoform 4, carbohydrate metabolism enzyme) in liver tissue, in C57BI6 mice, after 14 days of treatment with compound 29 (50 mg / kg / day) ).
- PDK4 pyruvate dehydrogenase kinase isoform 4, carbohydrate metabolism enzyme
- FIG. 8 (e) represents the expression of ACO (acyl coenzyme A oxidase, a key enzyme involved in the mechanism of ⁇ -oxidation of fatty acids) in liver tissue, in the C57BI6 mouse, after 14 days of treatment with 29 (50 mg / kg / day).
- ACO acyl coenzyme A oxidase, a key enzyme involved in the mechanism of ⁇ -oxidation of fatty acids
- FIG. 9 In vitro evaluation of the properties activating the reverse transport of cholesterol of the compounds according to the invention by measuring the expression of the ABCA1 gene in macrophages. The effect of the compounds according to the invention on the reverse transport of cholesterol was evaluated by measuring the expression of the ABCA1 gene in macrophages. The results are shown in FIG. 9: the higher the expression of ABCA1 (high induction factor), the more the compound according to the invention stimulates the reverse transport of cholesterol.
- Figure 10 In vitro evaluation of the anti-inflammatory properties of the compounds according to the invention by measuring the secretion of IL- ⁇ by macrophages pretreated with the compounds according to the invention and stimulated with LPS.
- the anti-inflammatory effects of the compounds according to the invention were evaluated by measuring the secretion of interleukin-6 (IL-6) by macrophages pretreated for 24 hours with the compounds according to the invention and stimulated for 6 hours with LPS (LipoPolySaccharide, which causes an inflammatory response of the cells).
- IL-6 interleukin-6
- LPS LipoPolySaccharide, which causes an inflammatory response of the cells.
- Figure 11 In vitro evaluation of the activating properties of the oxidation of fatty acids of the compounds according to the invention.
- the effect of the compounds according to the invention on the oxidation of fatty acids was evaluated in mouse muscle cells (CaC 12 ) by measuring the amount of tritiated water formed during the mitochondrial oxidation of labeled fatty acids. at 3 H.
- the results are represented in FIG. 11 by the induction factor, that is to say the ratio between the beta-oxidation of the fatty acids induced by the compound according to the invention and that induced by the DMSO alone: the higher this factor, the more the compound according to the invention activates the oxidation of fatty acids.
- organometallic (organomagnesium) derivatives used are commercially available or can be synthesized from the corresponding halides according to methods known to those skilled in the art.
- the washes are carried out with saturated aqueous solutions of sodium chloride (NaCl.sub.3), molar solutions of sodium hydroxide (1M NaOH) or hydrochloric acid (1M HCl).
- the aryl halide derivative (1 eq) is dissolved in acetonitrile (0.1 to 5 mol / l) and potassium carbonate (3 to 4 eq) is added.
- the nucleophile to be substituted (amine or thiol, 1 to 3 eq) is added.
- the mixture is refluxed and stirred for 16 h.
- the salts are filtered and the solvent is partially removed by evaporation under reduced pressure.
- the residue is acidified with an aqueous hydrochloric acid solution and extracted with ethyl acetate (3 times).
- the organic phases are combined and the resulting solution is successively washed with NaCIsat, dried over magnesium sulphate (MgSO 4 ), filtered and evaporated.
- the expected product is purified by chromatography on silica gel or by recrystallization of the corresponding salt (for example, by preparation of the hydrochloride in diethyl ether).
- Protocol B LDA-mediated alkylation
- the substrate to be alkylated (1 eq) is dissolved in THF (0.1 to 1 mol / l) and cooled to -78 ° C.
- the LDA (1 to 3 eq, 2M solution in THF) is added dropwise and the medium is stirred for 30 min at -78 ° C.
- the halogenated derivative (1 to 3 eq) is then added and the reaction medium is brought slowly to room temperature. After a few hours, the reaction medium is cooled to 0 ° C., hydrolyzed with a saturated solution of ammonium chloride, and extracted with ethyl acetate (3 times).
- Protocol C rearrangement of Curtius Under an anhydrous atmosphere, the acid (1 eq) is dissolved in toluene (0.1 to 1 mol / l). Triethylamine (1, 1 eq) and diphenylphosphoryl azide (DPPA, 1eq) are successively added. The solution is stirred at room temperature for 30 minutes and then gently brought to reflux. The reflux is maintained for 1 to 2 hours then the reaction medium is brought to 50 ° C. The benzyl alcohol (3 eq) is added and the mixture is refluxed and stirred for 16 hours. The medium is cooled to room temperature and hydrolyzed with a solution of sodium hydrogencarbonate. It is extracted with ethyl acetate (3 times).
- Protocol D substitution of a halogen derivative with a phenol derivative
- the phenol derivative (1 eq) is dissolved in acetonitrile (0.1 to 1 mol / l) and potassium carbonate (3 to 4 eq) is added.
- the suspension is stirred for 30 minutes under reflux and then the halogenated derivative (1 to 2 eq) is added dropwise.
- the heating is maintained for 16 h and then the solvent is evaporated and taken up in ethyl acetate.
- the residue is filtered and the filtrate is washed successively with 1M HCl, 1M NaOH and NaCl.
- the crude reaction product is then dried over magnesium sulphate (MgSO 4 ), filtered and evaporated.
- the expected product is purified by chromatography on silica gel or by recrystallization.
- Protocol E catalytic hydrogenation
- the derivative to be reduced (alkene, benzyl ether, CBz, etc.) (1 eq) is solubilized in methanol (0.1 to 1 mol / l) and added with a catalytic amount of palladium on carbon (Pd / C 10 %).
- the reaction medium is placed under hydrogen at atmospheric pressure for 16 hours.
- the crude is then filtered on celite ® and the filtrate evaporated.
- the expected product is purified by chromatography on silica gel or by recrystallization.
- Protocol F condensation with PyBQP
- the amine (1 eq) and the acid (1 eq) are mixed in dichloromethane (0.1 to 1 mol / l).
- Triethylamine (1 to 3 eq) is added, followed by PyBOP (1 eq).
- the resulting solution is stirred for 1 hour at room temperature.
- the reaction crude is successively washed with 1M HCl (3 times), 1M NaOH (3 times) and NaCl 3 (3 times).
- the organic phase is dried over magnesium sulphate (MgSO 4 ), filtered and evaporated.
- the expected product is purified by chromatography on silica gel or by recrystallization.
- Protocol G acid hydrolysis of the tert-butyl ester functional groups The ester (1 eq) is solubilized in dichloromethane (1 to 5 mol / l) and then trifluoroacetic acid (1 to 5 eq) is added. The reaction mixture is stirred at room temperature for 2 hours and then evaporated to dryness. Depending on the case, the expected product is purified by washing, by chromatography on silica gel or by recrystallization. Protocol H: saponification
- the ester (1 eq) is dissolved in an equivolumetric mixture ethanol / 2M sodium hydroxide (0.1 to 1 mol / l) and the mixture is stirred vigorously for 3 h at room temperature.
- the crude reaction product is acidified by adding an aqueous solution of hydrochloric acid, concentrated, and then extracted with dichloromethane (3 times).
- the resulting organic phase is washed with NaCIsat, dried over magnesium sulphate (MgSO 4 ), filtered and concentrated.
- the expected product is purified by chromatography on silica gel or by recrystallization.
- Protocol I HBTU / HOBt condensation
- Phenol (1 eq) is solubilized in acetone (36 to 72 eq). Soda (9 to 25 eq) is added and the resulting suspension is heated at 35 ° C for 30 min. Chloroform (4.5 eq) is added dropwise to the medium with stirring at 35 ° C.
- Protocol L Condensation of a sulfonamide derivative by EDC / DMAP
- the sulfonamide (1 eq) is solubilized in dichloromethane (0.1 to 1 mol / l) and successively added DMAP (1 to 2 eq) and the acid to be condensed (1 eq).
- the reaction crude is cooled to 0 ° C. and the EDC (1 eq) is added. Stirring is maintained overnight at room temperature.
- the reaction crude is successively washed with 1M HCl (3 times) and NaCl 3 (3 times).
- the organic phase is dried over magnesium sulphate (MgSO 4 ), filtered and evaporated.
- the expected product is purified by chromatography on silica gel or by recrystallization.
- Step 1 1- (4- (benzyloxy) phenyl) propan-2-one
- the 4-hydroxyphenylacetone is benzylated with benzyl bromide according to protocol D.
- the crude is purified by chromatography on silica gel (cyclohexane / ethyl acetate 8/2). Yield: 90% Appearance: pale yellow powder
- Step 2 N-benzyl-1- (4- (benzyloxy) phenyl) propan-2-amine
- This compound is obtained by hydrogenation of the above intermediate according to protocol E. It is purified by chromatography on silica gel (dichloromethane / methanol 8/2, supplemented with 0.1% of ammonia).
- Step 1 (S) -2- (benzyloxycarbonylamino) propanoic acid.
- L-alanine (1.0 g, 11 mmol) is solubilized in distilled water (20 ml) and then sodium carbonate (4.0 g, 36 mmol) is added.
- the reaction medium is stirred at ambient temperature, and then the benzyl chloroformate (2.0 ml, 11 mmol) is added dropwise. Stirring is maintained at room temperature for 18 h. Then the reaction medium is cooled to 0 ° C. and acidified by addition of citric acid until a pH of about 4 is reached. It is extracted with dichloromethane (3 times).
- Step 2 (S) -2- (benzyloxycarbonylamino) -N-methoxy-N-methylpropionamide.
- Step 3 (S) -1 - [(4- (Benzyloxy) phenyl)] - 2 - [(benzyloxycarbonylamino)] propan-1-one
- magnesium 1.1 g, 44 mmol
- An iodine crystal is added to the reaction medium and the mixture is refluxed.
- 1- (benzyloxy) -4-bromobenzene 13 g, 48 mmol
- solubilized in 50 ml of freshly distilled THF is added dropwise and the reaction mixture is stirred under reflux of THF for 3 hours.
- Step 4 (S) -1 - [(4- (Benzyloxy) phenyl)] - 2 - [(benzyloxycarbonylamino)] propan-1-ol.
- the preceding intermediate (4.0 g, 10 mmol) is solubilized in methanol (40 ml) and sodium tetraborohydride (1.0 g, 31 mmol) is added to the reaction medium. After stirring for 3 hours at room temperature, the reaction medium is cooled to 0 ° C., it is hydrolyzed with a 1M solution of HCl and extracted with ethyl acetate (3 times).
- Step 5 (S) - [I - [4- (2- (benzyloxycarbonylamino) propyl) phenoxy] methyl] benzene.
- the previous intermediate (3.16 g, 8.08 mmol) is solubilized in trifluoroacetic acid (24 ml).
- Triethylsilane (2.82 g, 24.2 mmol) is added to the previously cooled solution by an ice bath. After stirring for 5 hours at 0 ° C., the reaction medium is diluted with dichloromethane, hydrolyzed with aqueous ammonia solution (28%) and extracted with dichloromethane (3 times).
- Step 6 (S) -4- (2-aminopropyl) phenol.
- This amine is synthesized according to the same procedures from D-alanine.
- This amine is synthesized in 3 steps. In this example, it is isolated as trifluoroacetate salt.
- Step 1 4- (2-N-FeAf-butoxycarbonyl-aminoethyl) phenol 4- (2-Aminoethyl) phenol hydrochloride (10 g, 57.59 mmol) is dissolved in 1M sodium hydroxide solution (75 ml) and Fe-butanol (75 ml). Di-tert-butyl dicarbonate (12.6 g, 57.59 mmol) is added portionwise over 15 minutes.
- Step 2 Ethyl 2 - [4- (2-N-fe-butoxycarbonyl-aminoethyl) phenoxy] -2-methylpropanoate
- the preceding phenolic intermediate (8.5 g, 35.8 mmol) is solubilized in dimethylformamide (50 ml). Potassium carbonate (14.85 g, 107 mmol) is added and the suspension is stirred for 30 minutes at room temperature. The ethyl bromoisobutyrate (5.6 ml, 39.4 mmol) is then added dropwise and the mixture is stirred for 16 hours at room temperature. The salts are filtered on frit and rinsed with ethyl acetate (300 ml). The filtrate is washed with water (100 ml), 1 M NaOH (3 x 100 ml), 1 M HCl (3 x 100 ml) and NaCl 3 (3 x 100 ml).
- Step 3 Ethyl 2- [4- (2-aminoethyl) phenoxy] -2-methylpropanoate trifluoroacetate
- Methyl 3- (4-hydroxyphenyl) propionate is benzylated according to Protocol D.
- Step 2 methyl 3- (4- (benzyloxy) phenyl) -2,2-dimethylpropionate
- the preceding methyl ester is alkylated with methyl iodide according to protocol B. It is purified by chromatography on silica gel (cyclohexane ethyl acetate 95/5). Yield: 53%
- Step 4 Benzyl 1- (4- (benzyloxy) phenyl) -2-methylpropan-2-ylcarbamate
- Step 5 4- (2-Amino-2-methylpropyl) phenol
- the previous intermediate is hydrogenated according to protocol E. No purification is necessary except the filtration of the catalyst.
- Step 1 2- (4- (benzyloxy) phenyl) -2-methylpropanenitrile
- 2- (4- (benzyloxy) phenyl) acetonitrile is alkylated with methyl iodide according to protocol B. It is purified by chromatography on silica gel (cyclohexane / ethyl acetate 95/5). Yield: 40% Appearance: 1 H NMR solid (300 MHz, CDCl 3 , ⁇ in ppm): 1.72 (s, 6H); 5.09 (s, 2H); 7.00 (d, 8.8 Hz, 2H); from 7.35 to 7.50 (m, 7H).
- the preceding intermediate (4.5 g, 17.9 mmol) is solubilized in methanol previously saturated with ammonia (100 ml). Raney nickel (catalytic amount) is added and the reaction mixture is stirred overnight at 80 0 C under a hydrogen pressure of 80 bar. The medium is cooled and filtered on celite ® . The resulting amine is purified by chromatography on silica gel
- Step 3 4- (1-Amino-2-methylpropan-2-yl) phenol
- the previous intermediate is hydrogenated according to protocol E. No purification is necessary except the fitration of the catalyst.
- Step 1 2- (4- (benzyloxy) phenyl) propanenitrile
- Step 2 2- (4- (Benzyloxy) phenyl) propan-1-amine
- the preceding intermediate (5.2 g, 22.1 mmol) is solubilized in methanol previously saturated with ammonia (100 ml). Raney nickel (catalytic amount) is added and the reaction medium is stirred overnight at 70 ° C. under a hydrogen pressure of 70 bar. The medium is cooled and filtered on celite ® . The resulting amine is purified by chromatography on silica gel (95/5 dichloromethane / methanol). Yield: 74% Appearance: Solid
- Step 3 4- (1-Aminopropan-2-yl) phenol
- Example 2-6 Ethyl 2- [4- (1-aminopropan-2-yl) phenoxy] -2-methylpropanoate
- This amine is synthesized in 3 steps from amine 2-5 following procedures identical to those described in Example 2-2.
- the amine is isolated as trifluoroacetate salt.
- Step 1 4- (1-N-Fe / f-butoxycarbonyl-aminopropan-2-yl) phenol Yield: 83%
- Step 2 Ethyl 2- [4- (1-N-Fe / f-butoxycarbonyl-aminopropan-2-yl) phenoxy] -2-methylpropanoate
- Step 3 Ethyl 2- [4- (1-aminopropan-2-yl) phenoxy] -2-methylpropanoate trifluoroacetate
- Step 1 2- (4- (benzyloxy) phenyl) hexanenitrile
- the preceding intermediate (4.9 g, 17.5 mmol) is solubilized in methanol previously saturated with ammonia (100 ml). Raney nickel (catalytic amount) is added and the reaction medium is stirred overnight at 70 ° C. under a hydrogen pressure of 70 bar. The medium is cooled and filtered on celite ® . The resulting crude is used as it is for the next step, without analysis.
- Step 3 4- (1-aminohexan-2-yl) phenol
- Example 2-8 Fert -butyl 2- [4- (1-aminopropan-2-yl) -3-methyl-phenoxy] -2-methylpropanoate
- Step 1 tert-butyl 2- [4-acetyl-3-methyl-phenoxy] -2-methylpropanoate 4-hydroxy-2-methylacetophenone is alkylated according to protocol D.
- the crude is purified by chromatography on silica (cyclohexane / ethyl acetate 9/1). Yield: 62% Appearance: white powder
- Step 2 Ferf-butyl 2- [4- (1- (ethoxycarbonyl) prop-2-en-2-yl) -3-methylphenoxy] -2-methylpropanoate
- the preceding intermediate is functionalized with triethyl phosphonoacetate according to protocol J.
- the crude is purified by chromatography on silica gel
- Step 3 Fe ⁇ f-butyl 2- [4- (1- (ethoxycarbonyl) propan-2-yl) -3-methylphenoxy] -2-methylpropanoate
- Step 4 Fe / f-butyl 2- [4- (1- (hydroxycarbonyl) propan-2-yl) -3-methylphenoxy] -2-methylpropanoate
- Step 5 Fe / f-butyl 2- [4- (1-benzyloxycarbonylamino-propan-2-yl) -3-methylphenoxy] -2-methylpropanoate
- the above acid is functionalized according to protocol C.
- the crude is purified by chromatography on silica gel (cyclohexane / ethyl acetate 8/2).
- Step 6 Fe / l-butyl 2- [4- (1-aminopropan-2-yl) -3-methyl-phenoxy] -2-methylpropanoate
- Example 2-9 tert-butyl 2- [4- (3-aminobutan-2-yl) phenoxy] -2-methylpropanoate
- Step 1 4- (3- (ethoxycarbonyl) but-2-en-2-yl) phenyl benzyl ether
- the 4-benzyloxyacetophenone is functionalized with triethyl 2-phosphonopropionate according to protocol J.
- the crude is purified by chromatography on silica gel (cyclohexane / ethyl acetate 9/1). Yield: 65% Appearance: pale yellow oil
- Step 2 4- (3- (ethoxycarbonyl) butan-2-yl) phenol!
- Step 3 tert-butyl 2- [4- (3- (ethoxycarbonyl) butan-2-yl) phenoxy] -2-methylpropanoate
- the phenolic intermediate is alkylated according to protocol D.
- the crude is purified by gel chromatography. silica (cyclohexane / ethyl acetate 95/5). Yield: 79% Appearance: colorless oil
- Step 4 tert-Butyl 2- [4- (3- (hydroxycarbonyl) butan-2-yl) phenoxy] -2-methylpropanoate
- the previous intermediate is saponified according to protocol H.
- the residue is purified by chromatography on silica gel (95/5 dichloromethane / methanol).
- Step 5 tert-butyl 2- [4- (3-benzyloxycarbonylamino-butan-2-yl) phenoxy] -2-methylpropanoate
- Step 6 Terf-butyl 2- [4- (3-aminobutan-2-yl) phenoxy] -2-methylpropanoate
- protocol E No purification is necessary except for filtration of the catalyst. Yield: 97% Appearance: colorless oil
- the 4-hydroxybenzaldehyde is alkylated according to protocol D.
- the crude is purified by chromatography on silica gel (cyclohexane / ethyl acetate 85/15). Yield: 49%
- Step 2 tert-butyl 2- [4- (2- (ethoxycarbonyl) pentenyl) phenoxy] -2-methylpropanoate
- Step 4 tert-butyl 2- [4- (2- (hydroxycarbonyl) pentyl) phenoxy] -2-methylpropanoate
- Step 5 Fe / f-butyl 2- [4- (2- (benzyloxycarbonylamino) pentyl) phenoxy] -2-methylpropanoate
- the above acid is functionalized according to protocol C.
- the crude is purified by chromatography on silica gel (cyclohexane / ethyl acetate 8/2).
- Step 6 Fe / f-butyl 2- [4- (2-aminopentyl) phenoxy] -2-methylpropanoate
- Step 1 4- (1- (ethoxycarbonyl) prop-1-en-2-yl) phenyl benzyl ether
- Step 2 4- (1- (ethoxycarbonyl) propan-2-yl) phenol
- the previous intermediate is reduced according to protocol E.
- the crude is purified by chromatography on silica gel (cyclohexane / acetone 9/1).
- Step 3 Fe / f-butyl 2- [4- (1- (ethoxycarbonyl) propan-2-yl) phenoxy] -2-methylpropanoate
- Step 4 tert-butyl 2- [4- (1- (hydroxycarbonyl) propan-2-yl) phenoxy] -2-methylpropanoate
- Step 5 Ferf-butyl 2- [4- (1-benzyloxycarbonylamino-propan-2-yl) phenoxy] -2-methylpropanoate
- the above acid is functionalized according to protocol C.
- the crude is purified by chromatography on silica gel (cyclohexane / ethyl acetate 8/2).
- Step 6 Ferf-butyl 2- [4- (1-aminopropan-2-yl) phenoxy] -2-methylpropanoate
- protocol E No purification is necessary except for filtration of the catalyst.
- Step 1 tert-butyl 2- (3-acetylphenoxy) -2-methylpropanoate
- the 3-hydroxyacetophenone is alkylated according to protocol D.
- the crude is purified by filtration on silica gel (elution with dichloromethane) and precipitation with petroleum ether. Yield: 81%
- Step 2 tert-butyl 2- (3- (1- (ethoxycarbonyl) prop-1-en-2-yl) phenoxy] -2-methylpropanoate
- the previous intermediate is functionalized with triethyl 2-phosphonoacetate according to protocol J.
- the crude is used as such for the next step, without analyzes.
- Step 3 tert-butyl 2- [3- (1- (ethoxycarbonyl) propan-2-yl) phenoxy] -2-methylpropanoate
- Step 4 Fe / f-butyl 2- [3- (1- (hydroxycarbonyl) propan-2-yl) phenoxy] -2-methylpropanoate
- Step 5 Ferf-butyl 2- [3- (1-benzyloxycarbonylamino-propan-2-yl) phenoxy] -2-methylpropanoate
- Step 6 tert-butyl 2- [3- (1-aminopropan-2-yl) phenoxy] -2-methylpropanoate
- Methyl 5-methylsalicylate is alkylated with 1-iodooctane according to the protocol
- the previous intermediate is saponified according to protocol H. No purification is necessary after washing.
- Step 1 methyl 2-methoxy-5-phenylbenzoate
- Methyl 5-bromo-2-methoxybenzoate (9.0 g, 37 mmol)
- n-tetrabutylammonium bromide 44 g, 137 mmol
- tetrakis (triphenylphosphine) palladium 0.4 g, 0.4 mmol
- the previous intermediate is saponified according to protocol H. No purification is necessary after washing.
- Step 1 methyl 5-butanoyl-2-hydroxybenzoate
- the previous intermediate is alkylated with methyl iodide according to protocol D.
- This compound is obtained by reducing the acid 3-3 according to the following procedure:
- the 3-3 acid (1.0 g, 4.5 mmol) is dissolved in TFA (10 ml).
- Triethylsilane (719 ⁇ l, 4.5 mmol) is added dropwise.
- the reaction mixture is stirred at 55 ° C for 6 hours.
- the medium is then evaporated under reduced pressure and the residue purified by chromatography on silica gel (cyclohexane / ethyl acetate 9/1). Yield: 66% Appearance: pale yellow oil
- Step 2 5- (2,4-difluorophenyl) -2-methoxybenzoic acid
- Example 4-1 4- [2- (5-chloro-2-methoxybenzamido) propyl] phenol
- This intermediate is obtained by condensation between amine 2-1 and 5-chloro-2-methoxybenzoic acid according to protocol F. It is purified by chromatography on silica gel (cyclohexane / ethyl acetate 6/4).
- This intermediate is obtained by condensation between tyramine and 2-methoxy-5-methylbenzoic acid according to protocol I. It is purified by chromatography on silica gel (heptane / ethyl acetate 1/1).
- This intermediate is obtained by condensation between tyramine and 3-2 acid according to protocol I. It is purified by chromatography on silica gel (heptane / ethyl acetate 55/45) and then recrystallized from toluene.
- This intermediate is obtained by condensation between amine 2-1 and 2,5-dimethoxybenzoic acid according to protocol F. It is purified by chromatography on silica gel (dichloromethane / ethyl acetate 9/1).
- This intermediate is obtained by condensation between amine 2-1 and 5-tert-butyl-2-methoxybenzoic acid according to protocol F. It is purified by chromatography on silica gel (dichloromethane / ethyl acetate 9/1).
- This intermediate is obtained by condensation between amine 2-1 and acid 3-5 according to protocol F. It is purified by chromatography on silica gel
- Example 5-1 Ethyl 2- [4- [2- (2-fluorobenzamido) propyl] phenoxy] -2-methylpropanoate
- This intermediate is obtained by condensation between amine 2-1 and 2-fluorobenzoic acid according to protocol F. It is purified by chromatography on silica gel (cyclohexane / ethyl acetate 7/3).
- Example 5-2 Ethyl 2- [4- [1 - ((3-hydroxy-2-naphthoyl) amino) propan-2-yl] phenoxy] -2-methylpropanoate
- Step 1 4- [2- (2-fluoro-5-trifluoromethylbenzamido) propyl] phenol
- This intermediate is obtained by condensation between the amine 2-1 and the 2-fluorinated acid.
- 5-trifluoromethylbenzoic according to protocol F. It is purified by chromatography on silica gel (dichloromethane / ethyl acetate 9/1). Yield: 64% Appearance: white powder
- Step 2 Ethyl 2- [4- [2- (2-fluoro-5-trifluoromethylbenzamido) propyl] phenoxy] -2-methylpropanoate
- This intermediate is obtained by substituting ethyl bromoisobutyrate with the above phenol derivative according to protocol D. It is purified by chromatography on silica gel (cyclohexane / ethyl acetate 8/2).
- Example 5-4 Ethyl 2- [4- [1 - ((2-hydroxy-1-naphthoyl) amino) propan-2-yl] phenoxy] -2-methylpropanoate
- Example 5-6 Fert -butyl 2- [4- [1- (5- (2,4-difluorophenyl) -2-hydroxybenzamido) propan-2-yl] phenoxy] propanoate
- Step 1 Ethyl 2- [4- [2- (5-chloro-2-methoxybenzamido) propyl] phenoxy] -2-methylpropanoate
- Step 2 2- [4- [2- (5-chloro-2-methoxybenzamido) propyl] phenoxy] -2-methylpropanoic acid This compound is obtained by saponification of the previous intermediate according to protocol H. It is purified by chromatography on silica gel (9/1 dichloromethane / methanol). Yield: 48% Appearance: white powder
- Step 1 Ethyl 2- [4- [2 - ((3-methoxy-2-naphthoyl) amino) ethyl] phenoxy] -2-methylpropanoate
- This intermediate is obtained by condensation between amine 2-2 and 3-methoxy-2-naphthoic acid according to protocol F. It is purified by chromatography on silica gel (cyclohexane / ethyl acetate 8/2).
- Step 2 2- [4- [2 - ((3-methoxy-2-naphthoyl) amino) ethyl] phenoxy] -2-methylpropanoic acid
- This compound is obtained by functionalization of the phenol derivative 4-2 according to the protocol K. It is purified by chromatography on silica gel
- This compound is obtained by functionalization of the phenol derivative 4-4 according to the protocol K. It is purified by chromatography on silica gel
- Step 1 Ethyl 2- [4- [1 - ((3-methoxy-2-naphthoyl) amino) propan-2-yl] phenoxy] -2-methylpropanoate
- This intermediate is obtained by substitution of methyl iodide with the phenol derivative 5-2 according to protocol D (using cesium carbonate). It is purified by chromatography on silica gel (cyclohexane / ethyl acetate 75/25). Yield: 97% Appearance: colorless oil
- Step 2 2- [4- [1 - ((3-methoxy-2-naphthoyl) amino) propan-2-yl] phenoxy] -2-methylpropanoic acid
- This compound is obtained by saponification of the preceding intermediate according to the H protocol. It is purified by silica gel chromatography.
- Step 1 Ethyl 2- [4- [1 - ((3-isobutoxy-2-naphthoyl) amino) propan-2-yl] phenoxy] -2-methylpropanoate
- Step 2 2- [4- [1 - ((3-Isobutoxy-2-naphthoyl) amino) propan-2-yl] phenoxy] -2-methylpropanoic acid
- Step 1 Ethyl 2- [4- [2- (2 ⁇ (phenylthio) -5-trifluoromethylbenzamido) propyl] phenoxy] -2-methylpropanoate
- This intermediate is obtained by substitution of the 5-3 fluorinated derivative with thiophenol according to protocol A. It is purified by silica gel chromatography.
- Step 2 2- [4- [2- (2- (phenylthio) -5-trifluoromethylbenzamido) propyl] phenoxy] -2-methylpropanoic acid
- This compound is obtained by saponification of the previous intermediate according to protocol H. It is purified by chromatography on silica gel (9/1 dichloromethane / methanol). Yield: 29% Appearance: white powder
- Step 1 Ethyl 2- [4- [1 - ((2-methoxy-1-naphthoyl) amino) propan-2-yl] phenoxy] -2-methylpropanoate
- This intermediate is obtained by substitution of methyl iodide with the phenol derivative 5-4 according to protocol D (using cesium carbonate). It is purified by chromatography on silica gel (cyclohexane / ethyl acetate 6/4).
- Step 2 2- [4- [1 - ((2-methoxy-1-naphthoyl) amino) propan-2-yl] phenoxy] -2-methylpropanoic acid
- This intermediate is obtained by substituting ethyl bromoisobutyrate with the phenol derivative 4-6 according to protocol D. It is purified by chromatography on silica gel (heptane / ethyl acetate 65/35).
- Step 2 2- [4- [2- (2-Methoxy-5-methylbenzamido) ethyl] phenoxy] -2-methylpropanoic acid
- This compound is obtained by saponification of the previous intermediate according to protocol H. No purification is necessary to obtain the pure product after acidification of the medium and extraction with diethyl ether.
- Step 1 2- [4- [1- (5-chloro-2-methoxybenzamido) propan-2-yl] -3-methyl-phenoxy]
- Step 2 2- [4- [1- (5-chloro-2-methoxybenzamido) propan-2-yl] -3-methylphenoxy] -2-methylpropanoic acid
- Step 1 tert-butyl 2- [4- [3- (5-chloro-2-methoxybenzamido) butan-2-yl] phenoxy] -2-methylpropanoate
- Step 2 2- [4- [3- (5-chloro-2-methoxybenzamido) butan-2-yl] phenoxy] -2-methylpropanoic acid
- This compound is obtained by acid hydrolysis of the tert-butyl ester according to Protocol G. It is purified by chromatography on silica gel (9/1 dichloromethane / methanol).
- This compound is obtained by condensation between compound 6 and methanesulfonamide according to protocol L. It is purified by chromatography on silica gel (95/5 dichloromethane / methanol).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Obesity (AREA)
- Diabetes (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Hematology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
La présente invention concerne des dérivés du type N-(phénéthyl)benzamide poly-substitués, les compositions pharmaceutiques les comprenant ainsi que leurs applications thérapeutiques, notamment dans les domaines de la santé humaine et animale. La présente invention a également trait à un procédé de préparation de ces dérivés.
Description
DERIVES DE N-(PHENETHYL)BENZAMIDE SUBSTITUES, PREPARATION ET UTILISATIONS
La présente invention concerne des dérivés du type N- (phénéthyl)benzamide poly-substitués, les compositions pharmaceutiques les comprenant ainsi que leurs applications thérapeutiques, notamment dans les domaines de la santé humaine et animale. La présente invention a également trait à un procédé de préparation de ces dérivés.
Les inventeurs ont mis en évidence, de manière surprenante, que les composés selon l'invention possèdent des propriétés activatrices de PPAR (Peroxisome Proliferator-Activated Receptor), notamment PPARα et/ou PPARδ. De plus, des composés selon l'invention présentent des propriétés insulinosécrétrices : les composés selon l'invention peuvent donc agir par au moins deux voies complémentaires et indépendantes pour soigner les pathologies liées à des désordres lipidiques et/ou glucidiques.
Les molécules décrites dans l'invention sont d'un intérêt particulier pour traiter les complications associées au syndrome métabolique, l'insulino-résistance, le diabète, les dyslipidémies, l'athérosclérose, les maladies cardiovasculaires, l'obésité, l'hypertension, les maladies inflammatoires (asthme, etc.), etc., ainsi que pour permettre la diminution du risque global pour les maladies cardiovasculaires. Les composés selon l'invention sont utilisables notamment pour le traitement des pathologies inflammatoires et/ou de l'athérosclérose. Préférentiellement, les composés selon l'invention sont utilisables pour le traitement du diabète de type 2 et/ou des dyslipidémies.
Selon l'International Atherosclerosis Society (International Atherosclerosis Society, 2003), les maladies cardiovasculaires représentent la première cause de mortalité dans les pays industrialisés et deviennent de plus en plus fréquentes dans les pays en voie de développement. Ces maladies sont notamment les pathologies cardiaques et vasculaires, l'hypertension, l'athérosclérose, l'infarctus du myocarde et l'ischémie cérébrale (Muscat G and Dressel U, 2005).
L'athérosclérose est une pathologie caractérisée par une dérégulation du métabolisme du glucose et des lipides, par un stress oxydatif et par l'inflammation.
Le diabète, l'obésité et les dyslipidémies (taux plasmatiques de cholestérol LDL et de triglycérides élevés, cholestérol HDL faible, etc.) font partie des facteurs de risque cardiovasculaire clairement identifiés qui prédisposent un individu à développer une pathologie cardiovasculaire (Mensah M, 2004). Ces facteurs de risque s'additionnent aux facteurs de risque liés au mode de vie tels que le tabagisme, l'inactivité physique et les régimes alimentaires déséquilibrés. Un effet synergique existe entre ces différents facteurs : la présence concomitante de plusieurs d'entre eux conduit à une aggravation dramatique du risque cardiovasculaire et il convient alors de parler de risque global (« global risk ») pour les maladies cardiovasculaires. La prévalence des dyslipidémies atteignait 43.6% en 2004 dans les principaux pays développés. La prévalence du diabète, actuellement en nette augmentation, est en passe de devenir de plus en plus significative dans l'épidémiologie des maladies cardiovasculaires : elle est en effet estimée à 7,6% pour 2010 (Fox-Tucker J, 2005).
Ces données justifient donc l'adoption de mesures énergiques pour réduire significativement la morbidité et la mortalité cardiovasculaires et la nécessité de trouver des traitements efficaces, complémentaires d'une modification de l'hygiène de vie, agissant sur les facteurs de risque des maladies cardiovasculaires et sur leurs conséquences devient une urgence mondiale.
Les stratégies thérapeutiques actuelles consistent à associer plusieurs médicaments afin de réduire individuellement les différents facteurs de risque. Néanmoins, la combinaison de drogues peut parfois engendrer des réactions secondaires graves : par exemple, l'administration simultanée de fibrates et de statines augmente le risque de myopathie (Denke M, 2003).
II existe donc aujourd'hui un réel besoin de médicaments dont le mécanisme d'action « multimodal » présente des avantages en terme de compliance, de tolérance, de pharmacocinétique et de pharmacodynamie, liés à
l'administration d'un seul principe actif. De tels produits permettraient de diminuer le risque de maladie cardiovasculaire et de traiter chaque dysfonctionnement et/ou ses conséquences pris indépendamment (dyslipidémies, diabète, etc.).
De manière inattendue, les inventeurs ont découvert une famille de molécules originales ayant un mécanisme d'action « multimodal ». Les composés selon l'invention ont notamment montré qu'ils avaient un effet sur au moins deux types de récepteurs impliqués dans des processus de régulation majeurs :
- Les récepteurs nucléaires PPAR, impliqués dans de nombreux processus biologiques : régulation des lipides et des glucides, inflammation, etc.;
Les canaux potassiques ATP-dépendants des cellules β- pancréatiques, impliqués dans la régulation de la sécrétion d'insuline.
Les composés selon l'invention agissent donc par au moins deux voies complémentaires et indépendantes pour soigner les pathologies liées à des désordres lipidiques et glucidiques.
Les molécules décrites dans l'invention, par leurs propriétés insulinosécrétrices et agonistes de PPAR, sont d'un intérêt particulier pour le traitement du diabète de type 2, cette pathologie étant traitée actuellement soit par des molécules insulinosécrétrices (ex : sulphonylurées), soit par des molécules agonistes de PPAR (ex: thiazolidinediones), soit par par une combinaison de telles molécules (Gin H and Rigalleau V, 2002).
De plus, les molécules décrites dans l'invention, notamment par leurs propriétés activatrices de PPARα et/ou de PPARδ, sont particulièrement intéressantes pour le traitement de l'athérosclérose, des dyslipidémies, de l'obésité et de l'inflammation vasculaire.
Plus généralement, en agissant de manière simultanée sur plusieurs processus de régulation, les composés selon l'invention représentent un moyen thérapeutique avantageux pour traiter, en plus du diabète et des dyslipidémies, les complications associées au syndrome métabolique (dont les caractéristiques sont l'obésité, en particulier l'obésité abdominale, une concentration anormale de
lipides sanguins (taux élevé de triglycérides et/ou faible taux de cholestérol HDL (dyslipidémie)), une glycémie élevée et/ou une résistance à l'insuline, de l'hypertension), l'athérosclérose, les maladies cardiovasculaires, l'obésité, l'hypertension, les maladies inflammatoires, etc. Enfin, les composés selon l'invention représentent un outil thérapeutique avantageux pour traiter plusieurs facteurs de risque cardiovasculaire liés aux dérèglements du métabolisme lipidique et/ou glucidique (hyperlipidémie, hyperglycémie, etc.). Ils permettent la diminution du risque global pour les maladies cardiovasculaires.
Les PPARs (α, γ et δ) sont connus comme étant impliqués dans les pathologies du métabolisme lipidique et/ou glucidique (Kota BP et ai, 2005) : des ligands de ces récepteurs sont donc commercialisés pour traiter de telles pathologies (Lefebvre P et al., 2006) et de nombreux modulateurs de PPAR, agonistes ou antagonistes, sélectifs ou non, sont actuellement en développement avancé pour le traitement de telles pathologies. Un modulateur de PPAR ayant des effets bénéfiques sur la résistance à l'insuline, l'obésité, les dyslipidémies, l'hypertension et/ou l'inflammation pourrait être utilisé dans le traitement du syndrome métabolique (ou syndrome X) (Liu Y and Miller A, 2005). La famille des PPAR comprend trois membres distincts, désignés α, γ et δ
(encore appelé β), chacun codé par un gène différent. Ces récepteurs font partie de la superfamille des récepteurs nucléaires et des facteurs de transcription qui sont activés par la liaison de certains acides gras et/ou de leurs métabolites lipidiques. Les PPARs activés forment des hétérodimères avec les récepteurs de l'acide rétinoïque 9-cis (RXR ou Retinoid X Receptor) et se fixent sur des éléments de réponse spécifiques (PPRE ou Peroxisome Proliferator Response Elément) au niveau du promoteur de leurs gènes cibles, permettant ainsi le contrôle de la transcription de gènes cibles.
PPARα contrôle le métabolisme lipidique (hépatique et du muscle squelettique), influence le métabolisme intracellulaire des lipides par un contrôle direct de la transcription de gènes codant pour des protéines impliquées dans l'homéostasie lipidique, exerce des effets anti-inflammatoires et anti-prolifératifs, et prévient les effets pro-athérogéniques de l'accumulation du cholestérol dans les
macrophages en stimulant l'efflux du cholestérol (appelé aussi transport inverse du cholestérol) (Lefebvre P, Chinetti G, Fruchart JC and Staels B, 2006). Les fibrates (fénofibrate, bézafibrate, ciprofibrate, gemfibrozil), par l'intermédiaire de PPARα, sont ainsi utilisés en clinique dans le traitement de certaines dyslipidémies en baissant les triglycérides et en augmentant les taux de HDL (High Density Lipoprotein).
PPARγ est un régulateur-clé de l'adipogenèse. De plus, il est impliqué dans le métabolisme lipidique des adipocytes matures, dans l'homéostasie du glucose, notamment dans la résistance à l'insuline, dans l'inflammation, dans l'accumulation de cholestérol au niveau des macrophages et dans la prolifération cellulaire (Lehrke M and Lazar MA, 2005). PPARγ joue par conséquent un rôle dans la pathogenèse de l'obésité, de l'insulino-résistance et du diabète. Les thiazolidinediones (Rosiglitazone, Troglitazone, etc.) sont des ligands du récepteur PPARγ utilisés dans le traitement du diabète de type 2. L'intérêt pharmacologique des agonistes PPARδ dans le traitement des pathologies cardiovasculaires et métaboliques et de leurs facteurs de risque a été clairement mis en évidence (Muscat G and Dressel U, 2005). PPARδ est impliqué dans le contrôle du métabolisme lipidique et glucidique, dans la balance énergétique, dans la neurodégénération, dans l'obésité, dans la formation des cellules spumeuses et dans l'inflammation (Gross B et al., 2005) : ce récepteur constitue donc une cible attractive pour le développement de drogues utilisables dans le traitement des dyslipidémies, de l'athérosclérose, de l'obésité, de la résistance à l'insuline, de l'inflammation vasculaire (Gross B et al., 2005) et du diabète de type 2 (Luquet S et al., 2005). Il existe des ligands de PPARδ (L- 165041 , GW501516 actuellement en développement clinique) mais aucun ligand PPARδ n'est actuellement utilisé comme médicament.
Au-delà du rôle direct joué par les ligands de PPAR sur la régulation du métabolisme des lipides et des glucides, ces molécules ont un spectre d'action pléiotropique dû à la grande diversité des gènes cibles des PPARs. Ces multiples propriétés font des PPARs des cibles thérapeutiques d'intérêt pour le traitement de maladies comme l'athérosclérose, l'ischémie cérébrale, l'hypertension, les maladies liées à une néo-vascularisation (rétinopathies diabétiques, etc.), les
maladies inflammatoires et auto-immunes (maladie de Crohn, psoriasis, sclérose en plaques, asthme, etc), les maladies néoplasiques (carcinogenèse, etc.), les maladies neurodégénératives, les complications associées au syndrome métabolique, l'insulino-résistance, le diabète, les dyslipidémies, les maladies cardiovasculaires, l'obésité, etc., ainsi que pour permettre la diminution du risque global.
Les inventeurs ont également montré que les composés selon l'invention stimulent la sécrétion d'insuline, propriété qui permet de réguler la glycémie et qui est utilisée dans le cadre de pathologies majeures telles que le diabète de type 2.
Principalement deux familles de médicaments insulinosécréteurs sont actuellement commercialisées : les sulphonylurées et les méglitinides (appelés aussi glinides) (McCormick M and Quinn L, 2002). Les principales cibles des molécules insulinosécrétrices sont les canaux potassiques ATP-dépendants des cellules β-pancréatiques, qui jouent un rôle important dans le contrôle du potentiel membranaire de ces cellules (Proks P et al., 2002). La fermeture de ce canal induite par le glucose et/ou un agent insulinosécréteur (via des récepteurs membranaires, par exemple les « SulphonylUrea Receptors » ou SUR) induit une dépolarisation de la membrane plasmique qui provoque l'ouverture des canaux calciques voltage-dépendants. L'influx d'ions calciques (Ca++) engendré par ce phénomène provoque la sécrétion d'insuline. De nombreuses sulphonylurées sont ou ont été commercialisées : par exemple le tolbutamide, le glibenclamide, le glipizide, le gliclazide, le glimepiride, etc. Les méglitinides sont une classe de médicaments récente représentée par le répaglinide, le natéglinide et le mitiglinide.
Parmi les composés insulinosécréteurs, le répaglinide (décrit dans WO93/00337) est un dérivé de type acide benzoïque. De nombreuses molécules contenant une fonction acide benzoïque ont été décrites dans la littérature durant les 25 dernières années, suite à la découverte du méglitinide (HB699) par modification chimique du glibenclamide (Rufer C and Losert W, 1979).
Glibenclamide
Méglitinide
Répaglinide
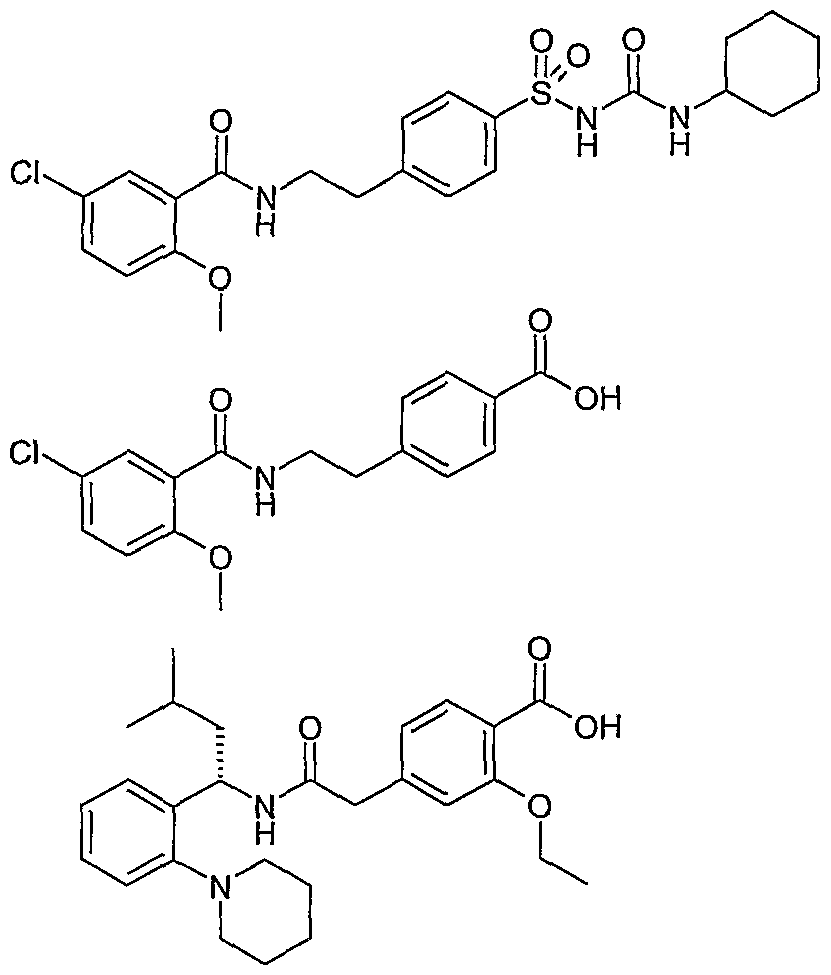
II a notamment été montré que les analogues du méglitinide dans lesquels la fonction acide benzoïque est remplacée par d'autres fonctions (phénol, aniline, chlorophényl, etc.) n'avaient plus de caractère insulinosécréteur (Brown G and Foubister A, 1984). De même, lors des travaux ayant mené à la découverte du répaglinide (Grell W et al., 1998), de nombreux composés ont été synthétisés et évalués : il a été démontré que le remplacement de la fonction acide par un groupe isostère tel que tétrazolyle inhibe l'activité, rappelant ainsi le rôle essentiel de la fonction acide benzoïque.
Les composés selon l'invention, par leurs propriétés agonistes de PPAR et insulinosécrétrices, représentent un outil thérapeutique avantageux pour l'amélioration des pathologies liées aux dérèglements du métabolisme lipidique et/ou glucidique, notamment les dyslipidémies et/ou le diabète de type 2, ainsi que pour la diminution du risque cardiovasculaire global. En particulier, les propriétés activatrices de PPARα et/ou de PPARδ des composés selon l'invention en font des cibles d'intérêt pour le traitement de l'athérosclérose, des dyslipidémies, du diabète de type 2, de l'obésité et de l'inflammation vasculaire.
La présente invention a pour objet des dérivés de N-(phénéthyl)benzamide poly-substitués de formule générale (I) :

Formule (I)
Dans laquelle : G représente - Un radical -0Rd) -SRd ; ou
- Un radical -SORd ou -SO2Rd;
Rd représentant un radical alkyle, alkényle, aryle, aralkyle ou un hétérocycle; G pouvant éventuellement former un hétérocycle avec X1 ;
R1 , R2, R3 et R4 représentent indépendamment un atome d'hydrogène ou un radical de type alkyle, alkényle, aryle ou aralkyle;
R1 et R4 pouvant chacun, indépendamment, être liés au squelette moléculaire par une double liaison, R2 ou R3 étant alors absents;
Y représente un atome d'oxygène, un atome de soufre (éventuellement oxydé en fonction sulfoxyde ou sulfone), un atome de sélénium (éventuellement oxydé en fonction sélénoxyde ou sélénone) ou un groupe aminé de type NR ; R représentant un atome d'hydrogène ou un radical de type alkyle, alkényle, aryle ou aralkyle, préférentiellement un atome d'hydrogène ou un radical alkyle ;
E représente :
- Une chaîne alkyle ou alkényle, substituée par un groupement -CRsRβ-W; ou
- Une chaîne répondant à la formule -(CH2)m-Y1-Z-Y2-(CH2)n-CR5R6-W; dans lesquelles :
R5 représente un atome d'halogène ou un radical alkyle, alkényle, aryle, aralkyle, alkyloxy ou alkylthio;
R6 représente un atome d'hydrogène, d'halogène ou un radical alkyle, alkényle, aryle, aralkyle, alkyloxy ou alkylthio; ou
R5 pouvant former un cycle avec R6, m et n représentent indépendamment un nombre entier compris entre 0 et 10; Y1 et Y2 représentent indépendamment une liaison covalente ou un hétéroatome choisi parmi l'oxygène, le soufre ou l'azote (formant ainsi un radical de type NR, R représentant un atome d'hydrogène ou un radical de type alkyle, alkényle, aryle ou aralkyle, préférentiellement un atome d'hydrogène ou un radical alkyle); Z représente une liaison covalente ou un radical alkyle, alkényle, aryle, aralkyle; si Z représente une liaison covalente alors Y1 et/ou Y2 représente une liaison covalente ;
W représente :
- un radical carboxyle ou un dérivé, préférentiellement de type -COORa, -COSR3, -CONR3Rb, -CSNRaRb; ou
- un groupement isostère du radical carboxyle, préférentiellement un radical acylsulfonamide (-CONHSO2Rc), hydrazide (-CONHNR3Rb) ou tétrazolyle;
R3 et Rb identiques ou différents, substitués ou non, représentant un atome d'hydrogène ou un radical alkyle, alkényle, aryle, aralkyle ou un hétérocycle; ou R3 et Rb pouvant former ensemble un hétérocycle avec l'atome d'azote; R0, substitué ou non, représentant un radical alkyle, alkényle, aryle, aralkyle ou un hétérocycle;
X1, X2, X3 et X4 représentent indépendamment un atome d'hydrogène ou d'halogène, une fonction -NO2 ou -CN, un radical alkyle, alkényle, aryle, aralkyle, alkyloxy, alkylthio, -NR3R0, -NRaCORb, -NRaCOORc, -NRaCONRbRc, -NRaCSRb, -NR3COSRC, -NRaCSORc, -NRaCSSRc, -NRaCSNRbRc, -SOR0, -SO2R0, -COR9,
-SO2NR3Rb, -CONR3Rb ou un hétérocycle;
R3, Rb et R0 étant tels que définis précédemment; ou
Ra, Rb et/ou R0 pouvant former ensemble un hétérocycle avec l'atome d'azote ;
X1 et X3 pouvant chacun former un cycle (aromatique ou non, hétérocyclique ou non) avec X2 et X4 respectivement ;
avec au moins un des groupements choisis parmi R1 , R2, R3, R4, X1 , X2, X3 et X4 représentant un radical alkyle, alkényle, aryle ou aralkyle,
les stéréoisomères (diastéréoisomères, énantiomères), purs ou en mélange, ainsi que les mélanges racémiques, les isomères géométriques, les tautomères, les sels, les hydrates, les solvates, les formes solides, les prodrogues et les mélanges.
Dans le cadre de la présente invention, le terme « alkyle » désigne un radical hydrocarboné saturé, linéaire, ramifié ou cyclique, substitué ou non, ayant plus particulièrement de 1 à 24, de préférence 1 à 10, atomes de carbone. On peut citer, par exemple, le radical méthyle, éthyle, n-propyle, isopropyle, n-butyle, isobutyle, tertiobutyle, sec-butyle, pentyle, néopentyle, n-hexyle ou cyclohexyle.
Le terme « alkényle » désigne un radical hydrocarboné insaturé, linéaire, ramifié ou cyclique, substitué ou non, ayant plus particulièrement de 2 à 24, de préférence 2 à 10 atomes de carbone. On peut citer, par exemple, le radical éthényle, 1-propényle, 2-propényle, 1-butényle, 2-butényle, 1-pentényle, 2- pentényle, 3-méthyl-3-butényle, éthynyle, 1-propynyle, 2-propynyle, 1-butynyle, 2- butynyle, 1-pentynyle ou 2-pentynyle.
Le terme « alkyloxy » fait référence à une chaîne alkyle liée à la molécule par l'intermédiaire d'un atome d'oxygène (liaison éther). La chaîne alkyle répond à la définition précédemment énoncée. A titre d'exemple, on peut citer les radicaux méthoxy, éthoxy, n-propyloxy, isopropyloxy, n-butoxy, iso-butoxy, tertio-butoxy, sec-butoxy ou hexyloxy.
Le terme « alkylthio » fait référence à une chaîne alkyle liée à la molécule par l'intermédiaire d'un atome de soufre (liaison thioéther). La chaîne alkyle répond à la définition précédemment énoncée. A titre d'exemple, on peut citer les radicaux méthylthio, éthylthio, n-propylthio, isopropylthio, n-butylthio, iso-butylthio, tertio-butylthio, sec-butylthio ou hexylthio.
Le terme « aryle » désigne un radical hydrocarboné aromatique, substitué ou non, ayant de préférence 6 à 14 atomes de carbone. Le radical aryle sera
préférentieilement substitué par au moins un atome d'halogène, un radical alkyle, hydroxyle, thiol, alkyloxy, alkylthio ou une fonction nitro. De préférence, les radicaux aryles selon la présente invention sont choisis parmi le phényle, le naphtyle (par exemple 1-naphtyle ou 2-naphtyle), le biphényle (par exemple, 2-, 3- ou 4- biphényle), l'anthryle ou le fluorényle. Les groupes phényles, substitués ou non, sont tout particulièrement préférés.
Le terme « aralkyle » désigne un radical du type alkyle substitué par un groupement aryle, substitué ou non. Les groupes benzyles et phénéthyles éventuellement substitués sont tout particulièrement préférés.
Le terme « hétérocycle » désigne un radical mono- ou poly-cyclique, saturé, insaturé ou aromatique, comprenant un ou plusieurs hétéroatomes, tels que l'azote, le soufre ou l'oxygène. Ils peuvent être substitués avantageusement par au moins un groupement alkyle, alkényle, aryle, alkyloxy, alkylthio tels que définis précédemment ou un atome d'halogène. Les radicaux pyridyle, furyle, thiényle, isoxazolyle, pyrrolidine, oxadiazolyle, oxazolyle, benzimidazolyle, indolyle, benzofuranyle, morpholinyle, pipéridinyle, pipérazinyle, 2-oxo-pypéridin-1-yle, 2- oxo-pyrrolidin-1-yle, cyclohexaméthylèneimino et tétrazolyle sont particulièrement préférés.
Par le terme « cycle », on entend plus particulièrement un cycle hydrocarboné, présentant éventuellement au moins un hétéroatome (notamment un atome d'azote, de soufre ou d'oxygène), saturé, insaturé ou aromatique. Les cycles incluent notamment les groupes aryles ou hétérocycles, tels que définis ci- dessus.
Les radicaux ainsi définis peuvent être substitués, en particulier par au moins un atome d'halogène, un radical alkyle, hydroxyle, thiol, alkyloxy, alkylthio, ou une fonction nitro. Ainsi, le radical alkyle peut être un radical perhalogénoalkyle, en particulier perfluoroalkyle, tel que notamment -CF3.
L'atome d'halogène est choisi parmi un atome de chlore, brome, iode ou fluor.
Tels que définis ci-dessus, m et n représentent indépendamment un nombre entier compris entre 0 et 10. Plus spécifiquement, ils peuvent être indépendamment 0, 1 , 2, 3, 4, 5, 6, 7, 8, 9 ou 10.
Selon un aspect particulier de l'invention, les composés de formule (I) présentent un groupement G en position ortho (par rapport au motif -CONH-) du radical phényle auquel il est attaché.
Selon un autre aspect particulier de l'invention, les composés de formule (I) présentent un groupement Y-E en position para (par rapport au motif -CR3R4-) du radical phényle auquel il est attaché.
De manière préférentielle, les composés de formule (I) présentent un groupement G en position ortho (par rapport au motif -CONH-) du radical phényle auquel il est attaché et un groupement Y-E en position para (par rapport au motif -
CR3R4-) du radical phényle auquel il est attaché. Ces composés sont représentés par la formule générale (II) :

Formule (II)
Dans laquelle G, X1 , X2, X3, X4, R1 , R2, R3, R4, Y, E sont tels que définis ci- dessus.
Un autre objet particulier de l'invention concerne les composés de formule générale (I), avantageusement (II), dans laquelle G représente un radical -ORd ou -SRd, Rd représentant un radical alkyle, alkényle, aryle, aralkyle ou un hétérocycle.
Le radical alkyle, alkényle, aryle, aralkyle ou l'hétérocycle comporte préférentiellement 1 à 10 atomes de carbone.
Un autre objet particulier de l'invention concerne les composés de formule générale (I), avantageusement (II), dans laquelle R1 , R2, R3 et/ou R4 représentent un radical alkyle, alkényle, aryle ou aralkyle, comportant préférentiellement 1 à 6 atomes de carbone. Encore plus préférentiellement, R1 ,
R2, R3 et/ou R4 représentent un radical alkyle.
Un autre objet particulier de l'invention concerne les composés de formule générale (I), avantageusement (II), dans laquelle Y représente un atome d'oxygène ou de soufre. Encore plus préférentiellement, Y représente un atome d'oxygène.
Un autre objet particulier de l'invention concerne les composés de formule générale (I), avantageusement (II), dans laquelle E représente une chaîne alkyle ou alkényle, substituée par un groupement -CR5R6-W, dans laquelle : R5 représente un atome d'halogène ou un radical alkyle, alkényle, aryle, aralkyle, alkyloxy ou alkylthio; R6 représente un atome d'hydrogène, d'halogène ou un radical alkyle, alkényle, aryle, aralkyle, alkyloxy ou alkylthio; ou R5 pouvant former un cycle avec R6, W représente :
- un radical carboxyle ou un dérivé, préférentiellement de type -COORa, -COSR3, -CONR3Rb, -CSNRaRb; ou
- un groupement isostère du radical carboxyle, préférentiellement un radical acylsulfonamide (-CONHSO2Rc), hydrazide (-CONHNR3Rb) ou tétrazolyle;
R3, Rb et Rc étant tels que définis ci-avant.
Préférentiellement, les composés de formule générale (I), avantageusement (II), ont un groupement E représentant une chaîne répondant à la formule -(CH2)n-CR5R6-W, avec n, R5, R6 et W tels que définis ci-avant.
Encore plus préférentiellement, les composés de formule générale (I), avantageusement (II), ont le groupement E représentant une chaîne répondant à la formule -CR5R6-W avec R5, R6 et W tels que définis ci-avant (dans ce cas, m et n sont égaux à 0 et le motif Y1 -Z- Y2 représente une liaison covalente).
Préférentiellement, les composés de formule générale (I), avantageusement (II), ont un groupement W représentant un radical carboxyle (-COOH), alkoxycarbonyle (-COOR3), hydrazide (-CONHNR3Rb) ou acylsulfonamide (-CONHSO2Rc), Ra, Rb et Rc étant tels que définis ci-avant. Encore plus préférentiellement, W représente un radical carboxyle (-COOH) ou alkoxycarbonyle (-COOR3), R3 étant tel que défini ci-avant.
Préférentiellement, les composés de formule générale (I), avantageusement (II), présentent des radicaux R5 et /ou R6 représentant un radical alkyle, préférentiellement un méthyle.
Selon un aspect préféré, les composés de formule générale (II) présentent un groupement X1 en position para (par rapport au motif radical G) du radical phényle auquel il est attaché.
De manière encore plus préférentielle, l'invention concerne les composés de formule générale (I) présentant :
Un groupement G en position ortho (par rapport au motif -CONH-) du radical phényle auquel il est attaché ; et Un groupement X1 en position para (par rapport au motif radical G) du radical phényle auquel il est attaché ; et
Un groupement Y-E en position para (par rapport au motif -CR3R4-) du radical phényle auquel il est attaché ; et
Un groupement E représentant une chaîne répondant à la formule -CR5R6- W avec R5, R6 et W tels que définis ci-avant (et où m et n sont égaux à O et le motif Y1-Z-Y2 représente une liaison covalente).
Ces composés sont représentés par la formule générale (III) :

Formule (III)
Dans laquelle G, X1 , X2, X3, X4, R1, R2, R3, R4, R5, R6, Y et W sont tels que définis ci-dessus.
Préférentiellement, les composés de formule générale (I), avantageusement (II) ou (III), présentent un groupement Y-E du type -OC(CHa)2COOH.
Un autre objet particulier de l'invention concerne les composés de formule générale (I), avantageusement (II) ou (III), dans laquelle X1 représente un halogène, préférentiellement le chlore, ou un radical -CORa, alkyle, alkényle, aryle ou aralkyle substitué ou non, Ra étant tel que défini ci-avant.
Un autre objet particulier de l'invention concerne les composés de formule générale (I), avantageusement (II) ou (III), dans laquelle X2, X3 et/ou X4 représentent un atome d'hydrogène. Préférentiellement, X3 et X4 représentent un atome d'hydrogène.
Un objet particulièrement préféré de l'invention concerne les composés de formule générale (I), avantageusement (II) ou (III), dans laquelle R1 représente un radical alkyle, alkényle, aryle ou aralkyle et R2, R3 et R4 sont des atomes d'hydrogène.
Encore plus préférentiellement, les composés de formule générale (I), avantageusement (II) ou (III), ont des groupements R1 et X1 représentant indépendamment un radical alkyle, alkényle, aryle ou aralkyle.
Un autre objet particulier de l'invention concerne les composés de formule générale (I), avantageusement (II) ou (III), dans laquelle X1 forme un cycle avec X2 et/ou X3 forme un cycle avec X4. Préférentiellement, le cycle résultant forme avec le noyau phényle adjacent un radical de type naphtyle, 1 ,2,3,4- tetrahydronaphtyle, indènyle ou indanyle. Le cycle formé par X1 et X2 et/ou X3 et X4 peut être substitué, en particulier par au moins un atome d'halogène, un radical alkyle, hydroxyle, thiol, alkyloxy, alkylthio ou une fonction nitro.
Un autre objet particulier de l'invention concerne les composés de formule générale (I), avantageusement (II) ou (III), dans laquelle X1 forme un cycle avec G. Préférentiellement, le cycle résultant forme avec le noyau phényle adjacent un radical de type 2H-chromène, 3,4-dihydro-2H-chromène, 2/-/-thiochromène, 3,4- dihydro-2H-thiochromène, benzofurane, 2,3-dihydrobenzofurane, benzothiophène, 2,3-dihydrobenzothiophène, benzoxazole ou benzothiazole. Le cycle formé par X1 et G peut être substitué, en particulier par au moins un atome d'halogène, un radical alkyle, hydroxyle, thiol, alkyloxy, alkylthio, hydroxyle, thiol ou une fonction nitro.
Un autre objet particulier de l'invention concerne les composés de formule générale (I), avantageusement (II) ou (III), dans laquelle R5 forme avec R6 (et le carbone auquel ils sont rattachés) un cycle de type cyclopropyle, cyclobutyle, cyclopentyle ou cyclohexyle.
De manière encore plus préférentielle, l'invention a pour objet les composés de formule générale (I), avantageusement (II) ou (III), dans laquelle au moins l'une des conditions suivantes, de préférence toutes les conditions, est remplie :
G représente un radical -ORd ou -SRd, le radical Rd étant préférentiellement choisi parmi les radicaux méthyle, trifluorométhyle, éthyle, n-propyle, isopropyle, cyclopropyle, n-butyle, isobutyle, tertiobutyle, sec-butyle, cyclobutyle, pentyle, néopentyle, cyclopentyle, n-hexyle, vinyle, allyle, homoallyle, propargyle, cyclohexyle, phényle, benzyle ou phénéthyle; et/ou
E représente une chaîne alkyle ou alkényle, substituée par un groupement -CR5R6-W,
- W représentant un radical carboxyle (-COOH), alkoxycarbonyle (-COORa), hydrazide (-CONHNR3Rb) ou acylsulfonamide (-CONHSO2Rc),
R3, Rb et R0 étant tels que définis ci-avant; et/ou
R1, R2, R3, R4 et R6 représentent indépendamment un atome d'hydrogène ou un radical méthyle, éthyle, n-propyle, isopropyle, cyclopropyle, n-butyle, isobutyle, tertiobutyle, sec-butyle, cyclobutyle, pentyle, néopentyle, cyclopentyle, n-hexyle, cyclohexyle, vinyle, allyle, homoallyle, propargyle, phényle, benzyle ou phénéthyle; et/ou
R5 est choisi parmi les groupements méthyle, éthyle, n-propyle, isopropyle, cyclopropyle, n-butyle, isobutyle, tertiobutyle, sec-butyle, cyclobutyle, pentyle, néopentyle, cyclopentyle, n-hexyle, cyclohexyle, vinyle, allyle, homoallyle, propargyle, phényle, benzyle ou phénéthyle; et/ou
Y représente un atome d'oxygène ou de soufre; et/ou
X1 , X2, X3 et X4 représentent indépendamment un atome d'hydrogène ou d'halogène, un radical méthyle, trifluorométhyle, éthyle, n-propyle, isopropyle, cyclopropyle, n-butyle, isobutyle, tertiobutyle, sec-butyle, cyclobutyle, pentyle, néopentyle, cyclopentyle, n-hexyle, cyclohexyle, vinyle, allyle, homoallyle, propargyle, phényle, benzyle, phénéthyle, méthoxy, trifluorométhoxy, thiométhoxy, nitro, méthylamino, diméthylamino, cyano, formyle, acétyle, propanoyle, butanoyle, pentanoyle, hexanoyle
Avec au moins un des groupements choisis parmi R1 , R2, R3, R4, X1 , X2, X3 et X4 représentant un radical alkyle, alkényle, aryle ou aralkyle.
Selon un mode particulier de l'invention, les composés préférés sont indiqués ci-dessous :
Composé 1 : Acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
Composé 2 : Acide 2-[4-[2-((3-méthoxy-2-naphtoyl)amino)éthyl]phénoxy]-2- méthylpropanoïque ; Composé 3 : Acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)-2- méthylpropyl]phénoxy]-2-méthylpropanoïque ;
Composé 4 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)-2-méthylpropan-2- yl]phénoxy]-2-méthylpropanoïque ;
Composé 5 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]- 2-méthylpropanoïque ;
Composé 6 : Acide 2-[4-[1-((3-méthoxy-2-naphtoyl)amino)propan-2-yl]phénoxy]-2- méthylpropanoïque ;
Composé 7 : Acide 2-[4-[1-((3-isobutoxy-2-naphtoyl)amino)propan-2-yl]phénoxy]-
2-méthyipropanoïque ; Composé 8 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)hexan-2-yl]phénoxy]-2- méthylpropanoïque ;
Composé 9 : Acide 2-[4-[2-(2-(phénylthio)-5- trifluorométhylberizamido)propyl]phénoxy]-2-méthylpropanoïque ;
Composé 10 : Acide 2-[4-[1-((2-méthoxy-1-naphtoyl)amino)propan-2-yl]phénoxy]- 2-méthylpropanoïque ;
Composé 11 : Acide 2-[4-[2-(2-méthoxy-5-méthylbenzamido)éthyl]phénoxy]-2- méthylpropanoïque ;
Composé 12 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]-3-méthyl- phénoxy]-2-méthylpropanoïque ; Composé 13 : Acide 2-[4-[3-(5-chloro-2-méthoxybenzamido)butan-2-yl]phénoxy]-
2-méthylpropanoïque ;
Composé 14 : Acide 2-[4-[1-(5-fert-butyl-2-méthoxybenzamido)éthyl]phénoxy]-2- méthylpropanoïque ;
Composé 15 : N-[méthylsulfonyl]-2-[4-[1-((3-méthoxy-2-naphtoyl)amino)propan-2- yl]phénoxy]-2-méthylpropanamide ;
Composé 16 : Acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)pentyl]phénoxy]-2- méthylpropanoïque ;
Composé 17 : Acide 2-[4-[2-(5-méthyl-2-octyloxybenzamido)éthyl]phénoxy]-2- méthylpropanoïque ;
Composé 18 : Acide 2-[4-[2-(2-méthoxy-5-phénylbenzamido)éthyl]phénoxy]-2- méthylpropanoïque ; Composé 19 : Acide 2-[4-[2-(5-butyl-2~méthoxybenzamido)éthyl]phénoxy]-2- méthylpropanoïque ;
Composé 20 : Acide 2-[4-[1-(3-chloro-4-méthoxybenzamido)propan-2-yl]phénoxy]-
2-méthylpropanoïque ;
Composé 21 : Acide 2-[4-[1-(4-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]- 2-méthylpropanoïque ;
Composé 22 : Acide 2-[4-[2-((3-méthoxy-2-naphtoyl)amino)propyl]phénoxy]-2- méthylpropanoïque ;
Composé 23 : Acide 2-[4-[1-((3,7-diméthoxy-2-naphtoyl)amino)propan-2- yl]phénoxy]-2-méthylpropanoïque ; Composé 24 : N-[phénylsulfonyl]-2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanamide ;
Composé 25 : N-[pipéridin-1-yl]-2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanamide ;
Composé 26 : Acide 2-[4-[2-(2,5-diméthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
Composé 27 : Acide 2-[4-[2-(5-tertiobutyl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
Composé 28 : Acide 2-[4-[2-(5-butyryl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ; Composé 29 : Acide 2-[4-[2-(5-phényl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
Composé 30 : N-[méthylsulfonyl]-2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanamide ;
Composé 31 : N-[benzylsulfonyl]-2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanamide ;
Composé 32 : N-[diméthylamino]-2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-
2-yl]phénoxy]-2-méthylpropanamide ;
Composé 33 : Acide 2-[3-[1-(5-chloro-2-méthoxybenzamido)propan-2-ylJphénoxy]-
2-méthylpropanoïque ;
Composé 34 : Acide 2-[4-[2-(5-butyl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ; Composé 35 : Acide 2-[4-[1-(5-phényl-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanoïque ;
Composé 36 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]propanoïque ;
Composé 37 : Acide 2-[4-[1-(5-(2,4-difluorophényl)-2-méthoxybenzamido)propan- 2-yl]phénoxy]propanoïque ;
Composé 38 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-
3-méthylbutanoïque ;
Composé 39 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]pentanoïque ; Composé 40 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-
2-phényléthanoïque ;
Composé 41 : Acide 5-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-
2,2-diméthylpentanoïque ;
Composé 42 : Acide 2-[4-[2-(5-(2,4-difluorophényl)-2- méthoxybenzamido)propyl]phénoxy]-2-méthylpropanoïque ;
Composé 43 : Acide 2-[4-[2-(5-(pyrrol-1-yl)-2-méthoxybenzamido)propyl]phénoxy]-
2-méthylpropanoïque.
Selon la présente invention, les composés encore plus préférés sont : Composé 1 : Acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
Composé 16 : Acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)pentyl]phénoxy]-2- méthylpropanoïque ;
Composé 22 : Acide 2-[4-[2-((3-méthoxy-2-naphtoyl)amino)propyl]phénoxy]-2- méthylpropanoïque ;
Composé 27 : Acide 2-[4-[2-(5-tertiobutyl-2-méthoxybenzamido)ρropyl]phénoxy]-2- méthylpropanoïque ;
Composé 28 : Acide 2-[4-[2-(5-butyryl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
Composé 29 : Acide 2-[4-[2-(5-phényl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ; Composé 34 : Acide 2-[4-[2-(5-butyl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
Composé 42 : Acide 2-[4-[2-(5-(2,4-difluorophényl)-2- méthoxybenzamido)propyl]phénoxy]-2-méthylpropanoïque ; Composé 43 : Acide 2-[4-[2-(5-(pyrrol-1-yl)-2-méthoxybenzamido)propyl]phénoxy]- 2-méthylpropanoïque.
Les composés selon l'invention peuvent contenir un ou plusieurs centres asymétriques. La présente invention inclut les stéréoisomères (diastéréoisomères, énantiomères), purs ou en mélange, ainsi que les mélanges racémiques et les isomères géométriques, les tautomères, les sels, les hydrates, les solvates, les formes solides, les prodrogues des composés selon l'invention ainsi que leurs mélanges. Quand un mélange énantiomériquement pur (ou enrichi) est souhaité, il pourra être obtenu soit par purification du produit final ou d'intermédiaires chiraux, soit par synthèse asymétrique suivant des méthodes connues de l'homme du métier (utilisant par exemple des réactifs et catalyseurs chiraux). Certains composés selon l'invention peuvent avoir différentes formes tautomères stables et toutes ces formes ainsi que leurs mélanges sont inclus dans l'invention.
La présente invention concerne également les sels « pharmaceutiquement acceptables » des composés selon l'invention. D'une manière générale, ce terme désigne les sels peu ou non toxiques obtenus à partir de bases ou d'acides, organiques ou inorganiques. Ces sels peuvent être obtenus lors de l'étape de purification finale du composé selon l'invention ou par incorporation du sel sur le composé déjà purifié.
Plus particulièrement, le groupement E tel que décrit précédemment peut avoir un caractère acide. Les sels correspondants sont choisis parmi les sels métaux (par exemple, aluminium, zinc, chrome), les sels alcalins (lithium, sodium,
potassium) ou alcalino-terreux (calcium, magnésium). Il peut s'agir par exemple de sels organiques tels que des dérivés ammonium et aminés non toxiques : ammonium, ammonium quaternaire (tétraméthylammonium, tétraéthylammonium, etc.), alkylamines (méthylamine, diméthylamine, triméthylamine, triéthylamine, éthylamine, etc.), hydroxyalkylamines (2-hydroxyéthylamine, bis-(2- hydroxyéthyl)amine, tri-(2-hydroxyéthyl)amine, etc.), cycloalkylamines (bicyclohexylamine, glucamine, etc.), pyridines et analogues (collidine, quinine, quinoline, etc.), de sels d'acides aminés à caractère basique (lysine, arginine, etc.).
Certains composés selon l'invention et leurs sels pourraient être stables sous plusieurs formes solides. La présente invention inclut toutes les formes solides des composés selon l'invention ce qui inclut les formes amorphes, polymorphes, mono- et poly-cristallines.
Les composés selon l'invention peuvent exister sous forme libre ou sous forme solvatée, par exemple avec des solvants pharmaceutiquement acceptables tels que l'eau (hydrates) ou l'éthanol.
La présente invention inclut également les prodrogues des composés selon l'invention qui, après administration chez un sujet, se transforment en composés tels que décrits dans l'invention ou en leurs métabolites qui présentent des activités thérapeutiques comparables aux composés selon l'invention.
Les composés selon l'invention marqués par un ou des isotopes sont également inclus dans l'invention : ces composés sont structurellement identiques mais diffèrent par le fait qu'au moins un atome de la structure est remplacé par un isotope (radioactif ou non). Des exemples d'isotopes pouvant être inclus dans la structure des composés selon l'invention peuvent être choisis parmi l'hydrogène, le carbone, l'azote, l'oxygène, le soufre tels que 2H, 3H, 13C, 14C, 15N, 18O, 17O, 35S respectivement. Les isotopes radioactifs 3H et 14C sont particulièrement préférés car faciles à préparer et à détecter dans le cadre d'études de biodisponibilité in vivo des substances. Les isotopes lourds (tels que 2H) sont particulièrement
préférés car ils sont utilisés comme standards internes dans des études analytiques.
La présente invention a aussi pour objet les composés tels que décrits ci- avant, à titre de médicaments.
La présente invention a également pour objet une composition pharmaceutique comprenant, dans un support acceptable sur le plan pharmaceutique, au moins un composé tel que décrit ci-dessus, éventuellement en association avec un ou plusieurs autres principes actifs thérapeutiques et/ou cosmétiques. Il s'agit avantageusement d'une composition pharmaceutique pour le traitement du diabète de type 2, des dyslipidémies, de l'insulino-résistance, des pathologies associées au syndrome métabolique, de l'athérosclérose, des maladies cardiovasculaires, de l'obésité, de l'hypertension, des maladies inflammatoires, etc. Les pathologies inflammatoires désignent de manière particulière l'asthme. Il s'agit préférentiellement d'une composition pharmaceutique pour traiter les facteurs de risque cardiovasculaire liés aux dérèglements du métabolisme lipidique et/ou glucidique (hyperlipidémie, diabète de type 2, obésité etc.) en permettant la diminution du risque global. La composition pharmaceutique selon l'invention est utilisable préférentiellement pour le traitement du diabète de type 2 et/ou des dyslipidémies.
Un autre objet de l'invention concerne une composition nutritionnelle comprenant au moins un composé tel que décrit ci-dessus.
Un autre objet de l'invention réside dans l'utilisation d'au moins un composé tel que décrit ci-avant pour la préparation de compositions pharmaceutiques destinées au traitement de diverses pathologies, notamment liées à des troubles du métabolisme parmi lesquelles on peut citer les dyslipidémies, le diabète de type 2, l'insulino-résistance, les pathologies associées au syndrome métabolique, l'athérosclérose, les maladies cardiovasculaires, l'obésité, l'hypertension, les maladies inflammatoires, etc. Plus généralement, l'invention a pour objet l'utilisation d'au moins un composé tel que décrit ci-avant pour la préparation de
compositions pharmaceutiques destinées à traiter les facteurs de risques pour les maladies cardiovasculaires liés aux dérèglements du métabolisme des lipides et/ou des glucides et destinées à diminuer ainsi le risque global.
A titre d'exemple (et de manière non limitative), les molécules selon l'invention pourront de manière avantageuse être administrées en combinaison avec d'autres agents thérapeutiques et/ou cosmétiques, commercialisés ou en développement, tels que :
- des anti-diabétiques : les insulinosécréteurs (sulfonylurées (glibenclamide, glimépiride, gliclazide, etc.) et glinides (répaglinide, natéglinide, etc.)), les inhibiteurs de l'alpha-glucosidase, les agonistes PPARγ (thiazolidinediones telles que rosiglitazone, pioglitazone), les agonistes mixtes PPARα/PPARγ (tésaglitazar, muraglitazar), les pan-PPAR (composés activant simultanément les 3 isoformes PPAR), des biguanides (metformine), les inhibiteurs de la Dipeptidyl Peptidase IV (sitagliptin, vildagliptin), les agonistes du Glucagon-Like Peptide-1 (GLP-1) (exenatide), etc.
- l'insuline
- des molécules hypolipémiantes et/ou hypocholestérolémiantes : les fibrates (fénofibrate, gemfibrozil), les inhibiteurs de la HMG CoA réductase ou hydroxylméthylglutaryl Coenzyme A réductase (les statines telles que atorvastatine, simvastatine, fluvastatine), les inhibiteurs de l'absorption du cholestérol (ézétimibe, phytostérols), les inhibiteurs de la CETP ou Cholesteryl Ester Transfer Protein (torcetrapib), les inhibiteurs de l'Acyl- CoA:Cholesterol O-Acyl Transferase (ACAT) (avasimibe, éflucimibe), les inhibiteurs MTP (Microsomal Triglycéride Transfer Protein), les agents séquestrants des acides biliaires (cholestyramine), la vitamine E, les acides gras poly-insaturés, les acides gras oméga 3, les dérivés de type acide nicotinique (niacine), etc.
- des agents anti-hypertenseurs et les agents hypotenseurs : les inhibiteurs ACE (Angiotensin-Converting Enzyme) (captopril, enalapril, ramipril ou quinapril), les antagonistes du récepteur de l'angiotensine II (losartan, valsartan, telmisartan, éprosartan, irbésartan, etc.), les bêta-bloquants (atenolol, metoprolol, labetalol, propranolol), les diurétiques thiazidiques et
non thiazidiques (furosemide, indapamide, hydrochlorthiazide, anti- aldostérone), les vasodilatateurs, les bloquants des canaux calciques (nifédipine, félodipine or amlodipine, diltizem or vérapamil), etc.
- des agents anti-plaquettaires : aspirine, ticlopidine, dipyridamol, clopidogrel, flurbiprofène, etc.
- des agents anti-obésité : sibutramine, les inhibiteurs de lipases (orlistat), les agonistes et antagonistes PPARδ, les antagonistes du récepteur cannabinoïde CB1 (rimonabant) etc.
- des agents anti-inflammatoires : par exemple, les corticoïdes (prednisone, bêtaméthazone, dexaméthazone, prednisolone, méthylprednisolone, hydrocortisone, etc.), les AINS ou Anti-Inflammatoires Non Stéroidiens dérivés de l'indole (indométhacine, sulindac), les AINS du groupe des arylcarboxyliques (acide thiaprofénique, diclofénac, étodolac, flurbiprofène, ibuprofène, kétoprofène, naproxène, nabumétone, alminoprofène), les AINS dérivés de l'oxicam (meloxicam, piroxicam, tenoxicam), les AINS du groupe des fénamates, les inhibiteurs sélectifs de la COX2 (célécoxib, rofécoxib), etc.
- des agents anti-oxydants : par exemple le probucol, etc.
- des agents utilisés dans le traitement de l'insuffisance cardiaque : les diurétiques thiazidiques ou non thiazidiques (furosemide, indapamide, hydrochlorthiazide, anti-aldosterone), les inhibiteurs de l'ACE (Angiotensin Converting Enzyme) (captopril, enalapril, ramipril or quinapril), les digitaliques (digoxin, digitoxin), les bêta bloquants (atenolol, metoprolol, labetalol, propranolol), les inhibiteurs de phosphodiestérases (enoximone, milrinone), etc.
- des agents utilisés pour le traitement de l'insuffisance coronaire : les bêta bloquants (atenolol, metoprolol, labetalol, propranolol), les bloquants des canaux calciques (nifédipine, félodipine ou amlodipine, bepridil, diltiazem ou vérapamil), les agents donneurs de NO (trinitrine, isosorbide dinitrate, molsidomine), l'Amiodarone, etc.
- des anticancéreux : les agents cytotoxiques (agents intéragissant avec PADN, agents alkylants, cisplatine et dérivés), les agents cytostatiques (les analogues GnRH, les analogues de la somatostatine, les progestatifs, les
anti-oestrogènes, les inhibiteurs de l'aromatase, etc.), les modulateurs de la réponse immunitaire (interférons, IL2, etc.), etc.
- des anti-asthmatiques tels que des bronchodilatateurs (agonistes des récepteurs bêta 2), des corticoïdes, le cromoglycate, les antagonistes du récepteur aux leucotriènes (montelukast), etc.
- des corticoïdes utilisés dans le traitement des pathologies de la peau telles que le psoriasis et les dermatites
- des vasodilatateurs et/ou des agents anti-ischémiques (buflomedil, extrait de Ginkgo Biloba, naftidrofuryl, pentoxifylline, piribédil), etc.
L'invention concerne également une méthode de traitement des pathologies liées au métabolisme des lipides et/ou des glucides comprenant l'administration à un sujet, notamment humain, d'une quantité efficace d'un composé ou d'une composition pharmaceutique tels que définis ci-avant. Au sens de l'invention le terme «une quantité efficace » se réfère à une quantité du composé suffisante pour produire le résultat biologique désiré, de préférence non toxique. Au sens de la présente invention le terme « sujet » signifie un mammifère et plus particulièrement un humain.
Le terme « traitement » désigne le traitement curatif, symptomatique ou préventif. Les composés de la présente invention peuvent ainsi être utilisés chez des humains atteints d'une maladie déclarée. Les composés de la présente invention peuvent aussi être utilisés pour retarder ou ralentir la progression ou prévenir une progression plus en avant de la maladie, améliorant ainsi la condition des patients. Les composés de la présente invention peuvent enfin être administrés aux personnes non malades, mais qui pourraient développer normalement la maladie ou qui ont un risque important de développer la maladie.
Les compositions pharmaceutiques selon l'invention comprennent avantageusement un ou plusieurs excipients ou véhicules, acceptables sur le plan pharmaceutique. On peut citer par exemple des solutions salines, physiologiques, isotoniques, tamponnées, etc., compatibles avec un usage pharmaceutique et connues de l'homme du métier. Les compositions peuvent contenir un ou
plusieurs agents ou véhicules choisis parmi les dispersants, solubilisants, stabilisants, conservateurs, etc. Des agents ou véhicules utilisables dans des formulations (liquides et/ou injectables et/ou solides) sont notamment la méthylcellulose, l'hydroxyméthylcellulose, la carboxyméthylcellulose, le polysorbate 80, le mannitol, la gélatine, le lactose, des huiles végétales, l'acacia, les liposomes, etc. Les compositions peuvent être formulées sous forme de suspensions injectables, gels, huiles, comprimés, suppositoires, poudres, gélules, capsules, aérosols, etc., éventuellement au moyen de formes galéniques ou de dispositifs assurant une libération prolongée et/ou retardée. Pour ce type de formulation, on utilise avantageusement un agent tel que la cellulose, des carbonates ou des amidons.
Les composés ou compositions selon l'invention peuvent être administrés de différentes manières et sous différentes formes. Ainsi, ils peuvent être par exemple administrés de manière systémique, par voie orale, parentérale, par inhalation ou par injection, comme par exemple par voie intraveineuse, intramusculaire, sous-cutanée, trans-dermique, intra-artérielle, etc. Pour les injections, les composés sont généralement conditionnés sous forme de suspensions liquides, qui peuvent être injectées au moyen de seringues ou de perfusions, par exemple. II est entendu que le débit et/ou la dose injectée peuvent être adaptés par l'homme du métier en fonction du patient, de la pathologie, du mode d'administration, etc. Typiquement, les composés sont administrés à des doses pouvant varier entre 1 μg et 2 g par administration, préférentiellement de 0,1 mg à 1 g par administration. Les administrations peuvent être quotidiennes voire répétées plusieurs fois par jour, le cas échéant. D'autre part, les compositions selon l'invention peuvent comprendre, en outre, d'autres agents ou principes actifs.
L'invention a également pour objet des procédés de préparation des composés dérivés de N-(phénéthyl)benzamide substitués selon l'invention.
Les composés de l'invention peuvent être préparés à partir de produits du commerce, en mettant en œuvre une combinaison de réactions chimiques.
Plus particulièrement, plusieurs étapes de synthèse sont nécessaires à l'obtention des composés selon l'invention.
L'étape clef est la formation de la liaison amide des composés dérivés de N-(phénéthyl)benzamide substitués objets de l'invention, avantageusement par condensation d'un dérivé de type 2-phényléthanamine mono- ou poly-substitué avec un dérivé de type acide (ou dérivé) benzoïque mono- ou poly-substitué.
Cette condensation peut être réalisée suivant les méthodes connues de l'homme du métier, et notamment celles qui ont été développées dans le cadre de la synthèse peptid iq ue .
D'autres étapes consistent à incorporer ou à transformer différents groupes fonctionnels, ce qui peut intervenir avant et/ou après l'étape de condensation comme illustré dans les méthodes présentées ci-après.
La présente invention a ainsi pour objet un procédé de préparation des composés selon l'invention tels que décrits ci-avant comprenant:
(i) Une étape de condensation, préférentiellement d'un dérivé de type
2-phényléthanamine mono- ou poly-substitué avec un dérivé de type acide (ou dérivé) benzoïque mono- ou poly-substitué; et éventuellement, avant et/ou après l'étape (i),
(ii) Une ou plusieurs étapes d'insertion et/ou de transformation de groupements fonctionnels.
De manière préférentielle, le procédé de préparation des composés selon l'invention permet d'obtenir des composés sous forme optiquement pure (ou enrichie).
Le procédé de préparation des composés selon l'invention permet de préparer des composés appelés ci-dessous composés intermédiaires. La présente invention a également pour objet certaines matières premières et composés intermédiaires obtenus dans le cadre de la présente invention. Ces composés intermédiaires sont plus particulièrement choisis parmi :
Exemple 2-6 : 2-[4-(1-aminopropan-2-yl)phénoxy]-2-méthylpropanoate d'éthyle ;
Exemple 2-7 : 4-(1-aminohexan-2-yl)phénol ;
Exemple 2-8 : 2-[4-(1-aminopropan-2-yl)-3-méthyl-phénoxy]-2-méthylpropanoate de terf-butyle ; Exemple 2-9 : 2-[4-(3-aminobutan-2-yl)phénoxy]-2-méthylpropanoate de tert- butyle ;
Exemple 2-10 : 2-[4-(2-aminopentyl)phénoxy]-2-méthylpropanoate de tert-buty\e ;
Exemple 2-11 : 2-[4-(1-aminopropan-2-yl)phénoxy]-2-méthylpropanoate de tert- butyle ; Exemple 2-12 : 2-[3-(1-aminopropan-2-yl)phénoxy]-2-méthylpropanoate de tert- butyle ;
Exemple 3-1 : acide 5-méthyl-2-octyloxybenzoïque ;
Exemple 3-4 : acide 5-butyl-2-méthoxybenzoïque ;
Exemple 4-1 : 4-[2-(5-chloro-2-méthoxybenzamido)propyl]phénol ; Exemple 4-2 : 4-[2-(5-chloro-2-méthoxybenzamido)-2-méthylpropyl]phénol ;
Exemple 4-3 : 4-[1-(5-chloro-2-méthoxybenzamido)-2-méthylpropan-2-yl]phénol ;
Exemple 4-4 : 4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénol ;
Exemple 4-5 : 4-[1-(5-chloro-2-méthoxybenzamido)hexan-2-yl]phénol ;
Exemple 4-6 : 4-[2-(2-méthoxy-5-méthylbenzamido)éthyl]phénol ; Exemple 4-7 : 4-[2-(5-méthyl-2-octyloxybenzamido)éthyl]phénol ;
Exemple 4-8 : 4-[2-(2-méthoxy-5-phénylbenzamido)éthyl]phénol ;
Exemple 4-9 : 4-[2-((3-méthoxy-2-naphtoyl)amino)propyl]phénol ;
Exemple 4-10 : 4-[2-(2,5-diméthoxybenzamido)propyl]phénol ;
Exemple 4-11 : 4-[2-(5-tertiobutyl-2-méthoxybenzamido)propyl]phénol ; Exemple 4-12 : 4-[2-(5-butyryl-2-méthoxybenzamido)propyl]phénol ;
Exemple 4-13 : 4-[2-(5-phényl-2-méthoxybenzamido)propyl]phénol ;
Exemple 4-14 : 4-[2-(5-(2,4-difluorophényl)-2-méthoxybenzamido)propyl]phénol ;
Exemple 4-15 : 4-[2-(5-(pyrrol-1-yl)-2-méthoxybenzamido)propyl]phénol ;
Exemple 5-1 : 2-[4-[2-(2-fluorobenzamido)propyl]phénoxy]-2-méthylpropanoate d'éthyle ;
Exemple 5-2 : 2-[4-[1-((3-hydroxy-2-naphtoyl)amino)propan-2-yl]phénoxy]-2- méthylpropanoate d'éthyle ;
Exemple 5-3 : 2-[4-[2-(2-fluoro-5-trifluorométhylbenzamido)propyl]phénoxy]-2- méthylpropanoate d'éthyle ;
Exemple 5-4 : 2-[4-[1~((2-hydroxy-1-naphtoyl)amino)propan-2-yi]phénoxy]-2- méthylpropanoate d'éthyle ; Exemple 5-5 : 2-[4-[1-((3-hydroxy-7-méthoxy-2-naphtoyl)amino)propan-2- yl]phénoxy]-2-méthylpropanoate de tert-butyle ;
Exemple 5-6 : 2-[4-[1-(5-(2,4-difluorophényl)-2-hydroxybenzamido)propan-2- yl]phénoxy]propanoate de tert-butyle.
Les procédés de préparation présentés ci-après sont donnés à titre d'exemples et ne sont en aucun cas limitatifs quant à la manière de préparer les composés selon l'invention.
Méthode A Les composés de formule générale (I) selon l'invention sont obtenus de préférence par condensation entre un acide carboxylique (Vl) et une aminé (VII).

VI VII I
Les groupes G, X1 , X2, X3, X4, R1 , R2, R3, R4, Y, E sont tels que définis précédemment.
La réaction de condensation peut être réalisée par de multiples voies, connues de l'homme du métier. A titre d'exemple, on peut citer l'utilisation d'agents de couplages (Py-BOP, DCC1 EDC, HBTU, CDI1 etc.), l'utilisation de dérivés d'acide activés (dans ce cas, l'acide est d'abord transformé en dérivé actif du type chlorure d'acyle, ester activé, anhydride mixte, etc.), la condensation en masse (mise en présence des deux entités à chaud, sans solvant), la condensation par distillation azéotropique de l'eau formée en cours de réaction, etc.
Une voie préférée consiste à travailler dans un solvant comme le dichlorométhane, le chloroforme, l'éther diéthylique, l'éther diisopropylique, le méthyl tertio-butyl éther, le tétrahydrofuranne, le dioxanne, le toluène, l'acétonitrile ou le diméthylformamide. De telles réactions sont réalisées en présence d'agents activant la fonction acide (DCC/HOBt, Py-BOP, etc.) ou en présence d'une forme préactivée de l'acide (chlorure d'acyle, anhydride mixte, etc.). Une base est souvent nécessaire : il peut s'agir de bases inorganiques telles que les carbonates (de sodium, de césium) ou la potasse ; des bases organiques telles que des trialkylamines (triéthylamine, diisopropyléthylamine, etc.) ou la pyridine peuvent aussi être employées. Ces réactions peuvent être réalisées à des températures comprises entre -25°C et 2500C, préférentiellement entre -100C et le point d'ébullition du solvant envisagé.
Une autre voie consiste à travailler en l'absence de solvant. Dans ce cas, les réactions sont réalisées par élimination de l'eau formée au fur et à mesure de l'avancement de la condensation. Ces réactions peuvent être réalisées à des températures comprises entre 25°C et 2500C. L'eau peut être éliminée par évaporation (réaction sous pression réduite par exemple).
Une autre voie consiste encore à éliminer l'eau formée au fur et à mesure de l'avancement de la condensation par distillation azéotropique : la réaction est dans ce cas réalisée au reflux d'un solvant tel que le toluène.
Méthode B Les composés de formule générale (I) selon l'invention sont obtenus de préférence par hydrolyse, thermolyse, hydrogénolyse ou fonctionnalisation de l'intermédiaire (I').
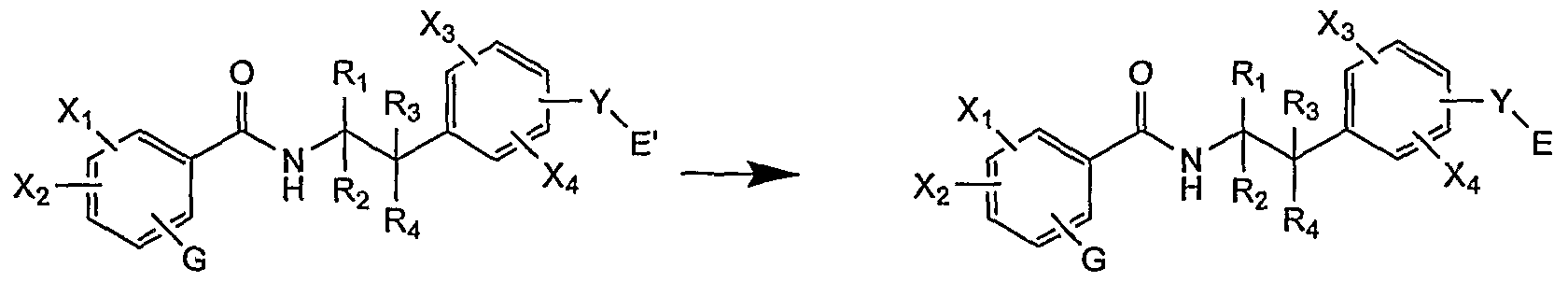
Selon un aspect préférentiel de l'invention, E contient au moins une fonction acide carboxylique. E' est dans ce cas un groupe contenant une fonction chimique pouvant être transformée en dérivé carboxylique par hydrolyse, thermolyse ou hydrogénolyse.
Des exemples de fonctions chimiques hydrolysables en acide carboxylique sont les dérivés d'acide (esters, thioesters, orthoesters, etc.) et les fonctions nitrile, tétrazolyle, 1 ,3-oxazol-2-yle, 1 ,3-oxazolin-2-yle, etc.
Des exemples de fonctions chimiques dont la thermolyse génère une fonction acide sont les esters d'alkyles tertiaires, de préférence les esters tertiobutyliques.
Des exemples de fonctions chimiques dont l'hydrogénolyse génère une fonction acide sont les esters d'aralkyle, de préférence les esters benzyliques.
Les réactions d'hydrolyse peuvent être avantageusement réalisées en présence d'un acide organique (ex : acide trifluoroacétique) ou inorganique (ex : acide chlorhydrique) ou en présence d'une base (ex : hydroxyde de sodium) dans l'eau ou un mélange de solvants contenant de l'eau (eau/méthanol, eau/éthanol, eau/THF (TetraHydroFuranne), eau/dioxanne, etc.). Elles sont menées à des températures comprises entre -1O0C et 1200C, préférentiellement entre 200C et la température de reflux du solvant utilisé.
Les réactions de thermolyse sont préférentiellement réalisées en l'absence de solvant (mélange en fusion) ou dans un solvant inerte tel que le dichlorométhane, le chloroforme, le toluène, le tétrahydrofuranne ou le dioxanne.
L'ajout de quantités catalytiques d'acides forts tels que l'acide paratoluènesulfonique est en général nécessaire à la thermolyse. Ces réactions
sont menées préférentiellement à chaud, avantageusement à la température d'ébullition du solvant.
Les réactions d'hydrogénolyse sont préférentiellement réalisées en présence d'un catalyseur métallique (Pd/C, Pt, etc.) dans un solvant adapté tel que le méthanol, l'éthanol, le tétrahydrofuranne (THF), l'acide acétique, l'acétate d'éthyle, etc. Elles sont réalisées à des températures comprises entre O0C et
600C, préférentiellement à température ambiante, sous une pression d'hydrogène comprise entre 1 et 6 bars. Une alternative consiste à libérer l'hydrogène in situ grâce au formiate d'ammonium.
De manière préférentielle, E' contient la fonction acide sous forme protégée. Il appartient à l'homme de l'art de choisir les groupes protecteurs les plus appropriés en fonction de la nature des différents substituants (Greene TW and Wuts PGM, 1999). Selon un mode préféré de l'invention, les composés (I1) correspondent à la forme estérifiée des composés (I). Selon la nature de la fonction ester contenue dans le groupe E', différentes méthodes sont applicables pour régénérer l'acide E :
- Hydrolyse basique : cette méthodologie est applicable aux esters d'alkyle tels que les esters de méthyle et d'éthyle;
- Hydrolyse acide : cette méthodologie est applicable aux esters d'alkyle tels que les esters de tertio-butyle;
- Hydrogénolyse : cette méthodologie est applicable aux esters de type benzylique et analogues.
Selon un autre aspect préférentiel de l'invention, E contient au moins une fonction dérivée d'acide (COORa, -COSR3, -CONRaRb, -CSNRaRb) ou isostère (un radical acylsulfonamide, hydrazide ou tétrazolyle). E' est dans ce cas un groupe contenant une fonction chimique pouvant être transformée en dérivé d'acide ou isostère d'acide par fonctionnalisation.
De manière préférentielle, E' contient une fonction acide carboxylique. Cette fonction pourra aisément être convertie en dérivé d'acide selon les
méthodes connues de l'homme de l'art : à titre d'exemple, la préparation d'ester à partir d'acide carboxylique est réalisable suivant de nombreuses méthodes très documentées. Selon un aspect préféré, la fonction acide carboxylique sera fonctionnalisée par condensation suivant les méthodologies décrites dans le paragraphe précédent (méthode A). Ce type de méthodologie sera aussi préférentiellement appliqué pour la préparation d'isostères de type acylsulfonamide et hydrazide.
De manière préférentielle, E' contient une fonction nitrile. Cette fonction pourra être aisément convertie en dérivé tétrazolyle suivant les méthodes connues de l'homme de l'art : par exemple, action d'un dérivé azoture dans un solvant anhydre (toluène, tétrahydrofuranne (THF), etc.) à des températures comprises entre 00C et la température d'ébullition du solvant considéré.
Méthode C
Les composés selon l'invention de formule générale (I) sont obtenus de préférence par fonctionnalisation de l'intermédiaire (VIII).

VIII I Les groupes G, X1 , X2, X3, X4, R1 , R2, R3, R4, Y1 E sont tels que définis précédemment.
Le groupe X est :
- Soit un groupe partant : par exemple, un atome d'halogène ou un groupement triflate;
- Soit un groupe nucléophile : par exemple, un radical hydroxyle, thiol ou amino.
Si X est un groupe partant, il pourra avantageusement être substitué par différentes méthodes connues de l'homme de l'art.
Une voie d'accès préférentielle est la substitution nucléophile aromatique. Ce type de réaction procède à chaud en présence d'un large excès de nucléophile
(qui peut le cas échéant faire office de solvant). Des bases sont souvent utilisées pour activer le nucléophile (par exemple, carbonate de césium, tertio-butylate de sodium, etc.). Ce type de réaction est préférentiellement réalisé dans des solvants usuels tels que le diméthyformamide, l'acétonitrile, le tétrahydrofuranne (THF), le dioxanne ou le toluène. Elle peut être réalisée à des températures comprises entre
200C et la température de reflux du solvant choisi.
Une autre voie d'accès préférentielle est le couplage catalysé par des métaux de transition. Selon un mode particulier de l'invention, les molécules de formule générale (I) pourront être obtenues par une réaction de couplage catalysée par le palladium (telle que la réaction de Buchwald-Hartwig et ses variantes). Ces réactions sont bien connues de l'homme de l'art. Ce type de réaction nécessite la présence d'un catalyseur au palladium (par exemple, Pd(OAc)2, Pd2(dba)3), d'un ligand (par exemple BINAP, X-Phos, tri-fe/t- butylphosphine) et d'une base (par exemple, Cs2CO3, tertiobutylate de sodium). Elle peut être réalisée à des températures comprises entre 200C et la température de reflux du solvant choisi. Différents solvants sont utilisables, par exemple le toluène, le diméthylformamide, le tétrahydrofuranne (THF), la N-méthyl-2- pyrrolidone ou le dioxanne.
Si X est un groupe nucléophile, différentes méthodes connues de l'homme de l'art pourront être envisagées.
Dans ce cas, X pourra avantageusement être alkylé pour obtenir un composé de formule (I). Par exemple, si X est un atome d'azote (éventuellement alkyle substitué), il pourra être fonctionnalisé en fonction aminé secondaire (ou tertiaire) par substitution nucléophile d'un dérivé halogène. De même, si X est un atome d'oxygène (ou de soufre), il pourra être fonctionnalisé en fonction éther (ou
thioéther) par substitution nucléophile d'un dérivé halogène. Ces réactions sont préférentiellement réalisées dans le dichlorométhane, le chloroforme, l'éther diéthylique, l'éther diisopropylique, le méthyl tertiobutyle éther, le tétrahydrofuranne (THF), le dioxanne, le toluène, l'acétonitrile ou le diméthylformamide. Une base est souvent nécessaire : il peut s'agir de bases inorganiques telles que les carbonates (par exemple de sodium, de césium) ou la potasse ; des bases organiques telles que des trialkylamines (triéthylamine, diisopropyléthylamine, etc.) ou la pyridine peuvent aussi être employées. Ces réactions peuvent être réalisées à des températures comprises entre -25 et 2500C, préférentiellement entre -100C et le point d'ébullition du solvant envisagé.
Selon un mode préféré, si X est un atome d'azote (éventuellement alkyle substitué), il pourra être fonctionnalisé par alkylation réductive : condensation d'un aldéhyde ou d'une cétone sur l'aminé (formation d'une imine) qui peut être réduite in situ (ou après isolement) par un agent réducteur (par exemple, NaBH3CN,
NaBH(OAc)3).
Selon un mode préféré, si X est un atome d'oxygène ou de soufre, il pourra être fonctionnalisé suivant les conditions de Mitsunobu (triphénylphosphine, azodicarboxylate de diéthyle). Cette méthodologie permet de générer une fonction éther à partir de l'intermédiaire (VIII) et d'un dérivé de type alcool.
Selon un mode particulier, si X est un atome d'oxygène, il pourra être fonctionnalisé sous forme d'acide isobutyrique (E = -C(Me^COOH) à partir du 2- trichlorométhyl-2-propanol dans l'acétone en présence d'hydroxyde de sodium. Alternativement, une stratégie préférentielle consiste à traiter le dérivé phénolique mis en présence d'hydroxyde de sodium dans l'acétone par du chloroforme à une température comprise entre 2O0C et 500C.
Si l'on souhaite obtenir des dérivés sulfoxyde ou sulfone, la fonction thioéther (-S-E) pourra être oxydée par les méthodes connues de l'homme de l'art. A titre d'exemple, l'oxone® pourra être utilisée dans un solvant tel que l'eau, le méthanol ou le dichlorométhane à température ambiante.
Méthode D
Les composés selon l'invention de formule générale (I) sont obtenus de préférence par fonctionnalisation de l'intermédiaire (IX).
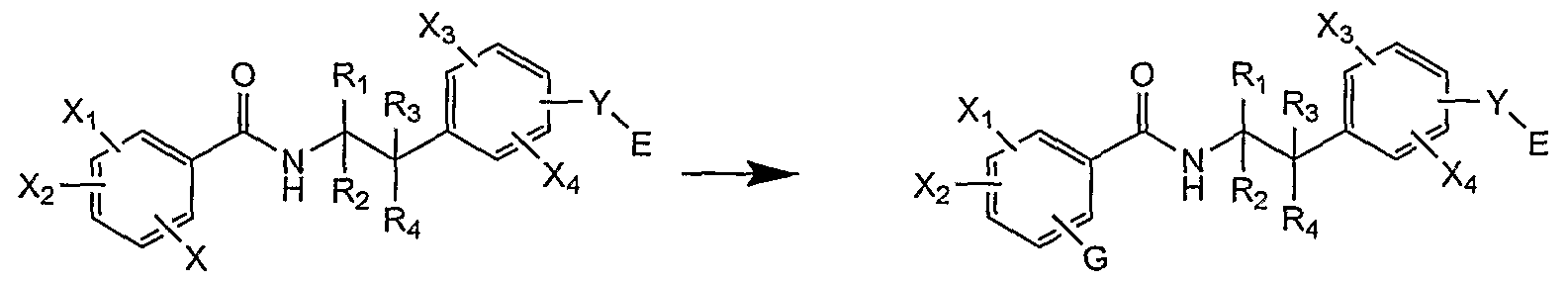
IX I
Les groupes G, X1 , X2, X3, X4, R1 , R2, R3, R4, Y, E sont tels que définis précédemment.
Le groupe X est :
- Soit un groupe partant : par exemple, un atome d'halogène ou un groupement triflate ;
- Soit un groupe nucléophile : par exemple, un radical hydroxyle ou thiol.
Le groupe G pourra être introduit suivant les méthodes proposées dans le paragraphe précédent (méthode C).
Méthode E
Les composés suivant l'invention sont obtenus de préférence par fonctionnalisation de l'intermédiaire (X).
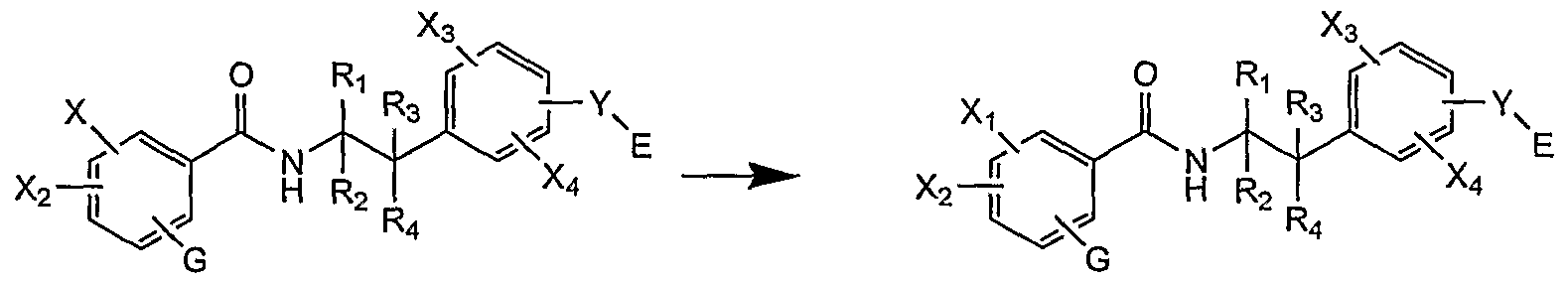
X I
Les groupes G, X1 , X2, X3, X4, R1 , R2, R3, R4, Y, E sont tels que définis précédemment.
Le groupe X est :
- Soit un groupe partant : par exemple, un atome d'halogène ou un groupement triflate;
- Soit un groupe nucléophile : par exemple, un radical hydroxyle, thiol ou amino.
Le groupe X1 pourra être introduit suivant les méthodes proposées dans le paragraphe précédent (méthode D).
Dans le cas où X est un groupe nucléophile, une autre voie préférentielle consiste à condenser un dérivé de type acide carboxylique (ou dérivé activé), chloroformiate ou isocyanate (et analogues soufrés : isothiocyanate, etc.) : par exemple si X est un atome d'azote, ces stratégies permettent de générer respectivement des groupements de type amide, carbamate ou uréido respectivement. Ces réactions sont préférentiellement réalisées dans le dichlorométhane, le chloroforme, l'éther diéthylique, l'éther diisopropylique, le méthyl tertiobutyle éther, le tétrahydrofuranne (THF), le dioxanne, le toluène, l'acétonitrile ou le diméthylformamide. Une base est souvent nécessaire : il peut s'agir de bases inorganiques telles que les carbonates (de sodium, de césium) ou la potasse ; des bases organiques telles que des trialkylamines (triéthylamine, diisopropyléthylamine, etc.) ou la pyridine peuvent aussi être employées. Ces réactions peuvent être réalisées à des températures comprises entre -25°C et
2500C, préférentiellement entre -100C et le point d'ébullition du solvant envisagé.
Dans le cas où X est un groupe partant, une autre voie préférentielle consiste à réaliser un couplage carbone-carbone catalysé par des métaux de transition. Selon un mode particulier de l'invention, les molécules de formule générale (I) pourront être obtenues par une réaction de couplage catalysée par le palladium choisie parmi les réactions de Suzuki-Miyaura, Heck, StNIe,
Sonogashira, etc. Ces réactions sont bien connues de l'homme de l'art. Par exemple, la réaction de Suzuki consiste à coupler un dérivé organoboré (par exemple, un acide boronique) au dérivé portant le groupement partant (par exemple un dérivé brome) en présence d'un catalyseur au palladium (par exemple, Pd(PPh3)4, Pd(OAc)2), d'un ligand (par exemple le BINAP) et d'une base
(par exemple, Cs2CO3, CsF). Elle peut être réalisée à des températures comprises entre 200C et la température de reflux du solvant choisi. Différents solvants sont utilisables, par exemple le diméthylformamide, le toluène, le tétrahydrofuranne, la N-méthyl-2-pyτrolidone ou le dioxanne.
Méthode F
Les composés selon l'invention de formule générale (I) sont obtenus de préférence par fonctionnalisation de l'intermédiaire (Xl).

XI I Les groupes G, X1 , X2, X3, X4, R1 , R2, R3, R4, Y, E sont tels que définis précédemment.
Le groupe X est :
- Soit un groupe partant : par exemple, un atome d'halogène ou un groupement triflate;
- Soit un groupe nucléophile : par exemple, un radical hydroxyle, thiol ou amino.
Le groupe X3 pourra être introduit suivant les méthodes proposées dans le paragraphe précédent (méthode E).
Méthode G
Les composés selon l'invention de formule générale (I) pour laquelle R4 forme une double liaison avec le squelette de la molécule sont ici nommés (I"). Ils sont obtenus de préférence par fonctionnalisation des dérivés de formule (XII).

XII I"
Les groupes G, X1 , X2, X3, X4, R1 , R2, R4, Y, E sont tels que définis précédemment.
Une voie d'accès préférentielle consiste à fonctionnaliser la fonction cétone suivant des méthodes bien connues de l'homme de l'art telles que la réaction de Wittig ou une de ses variantes (par exemple, la réaction de Homer-Emmons). Typiquement, la cétone réagit avec un ylure de phosphore (commercial ou préparé suivant les méthodes connues de l'homme de l'art) en présence d'une base. Ces réactions peuvent être réalisées dans différents solvants selon la stabilité des ylures utilisés : elles sont préférentiellement réalisées dans l'eau, l'éther diéthylique, l'éther diisopropylique, le méthyl tertiobutyle éther, le tétrahydrofuranne (THF), le dioxanne, le toluène, l'acétonitrile ou le diméthylformamide. Les bases préférentiellement utilisées sont les hydroxydes (de sodium, de potassium), les carbonates (de potassium, de césium), l'hydrure de sodium, le bis(triméthylsilyl)amide de potassium (KHMDS), etc. Ces réactions peuvent être réalisées à des températures comprises entre -78°C et 2500C, préférentiellement entre -100C et le point d'ébullition du solvant envisagé.
II est bien entendu que les composés insaturés de formule (I") obtenus suivant cette voie peuvent être aisément réduits lors d'une étape supplémentaire pour générer des composés de formule générale (I) selon l'invention (pour lesquels R3 = H).

I"
Diverses méthodologies sont applicables pour réduire une double liaison. On peut citer par exemple la réduction chimique (en catalyse homogène par exemple), l'hydrogénation catalytique (en présence de Pd/C par exemple), etc.
L'utilisation d'agents réducteurs chiraux (par exemple, l'utilisation de métaux de transition en présence de ligands chiraux) pourra préférentiellement être envisagée pour synthétiser préférentiellement l'un ou l'autre des stéréoisomères au niveau de R4. A titre d'exemple, on peut envisager une réduction catalysée par le rhodium en présence d'un ligand phosphine chiral tel que le BINAP.
Méthode H
Les composés selon l'invention de formule générale (I) sont obtenus de préférence par fonctionnalisation de l'intermédiaire (XIII).

Les groupes G, X1 , X2, X3, X4, R1 , R3, R2, R4, Y, E sont tels que définis précédemment.
Une voie d'accès préférentielle consiste à déprotoner le substrat de formule (XIII) par une base forte. Cette activation permet la substitution d'un dérivé R3-X,
X désignant un groupe partant et R3 étant tel que défini précédemment. Ces réactions peuvent être réalisées dans différents solvants anhydres tels que le méthyl tertiobutyle éther, le tétrahydrofuranne (THF), le dioxanne, le toluène, l'acétonitrile. Les bases préférentiellement utilisées sont celles dérivées du lithium (tBuLi, secBuϋ, LDA, LiHMDS, etc.). Ces réactions peuvent être réalisées à des températures comprises entre -780C et 5O0C. Le groupe partant X est à choisir parmi les nombreux groupes partants connus de l'homme de l'art : atome d'halogène, groupements tosyle ou mésyle, groupement triflate, etc.
Selon un mode préféré, cette substitution nucléophile peut être réalisée en présence d'un ligand chiral. Ce type d'approche permet une synthèse énantiosélective des dérivés de formule (I) en contrôlant la stéréochimie de la liaison C-R3 formée. De nombreux ligand chiraux connus de l'homme de l'art peuvent être utilisés pour ce type de réaction.
Dans le cadre de l'invention, lorsque la molécule comporte un ou plusieurs centres chiraux, les composés optiquement purs (ou les mélanges enrichis) peuvent être préparés ou purifiés suivant des méthodes usuelles connues de l'homme du métier : synthèse asymétrique utilisant des agents chiraux (catalyseurs, réactifs) ; purification des composés ou intermédiaires par des méthodes stéréosélectives (chromatographies sur phases chirales, précipitation d'un sel formé avec un contre-ion chiral) ; etc.
La synthèse des composés dérivés de N-(phénéthyl)benzamide substitués selon l'invention comprend préférentiellement une étape de condensation d'un dérivé de type 2-phényléthanamine mono- ou poly-substitué avec un dérivé de type acide (ou dérivé) benzoïque mono- ou poly-substitué. De manière préférentielle, ces deux réactifs seront synthétisés ou purifiés sous formes optiquement pures (ou enrichies) avant la condensation. Une voie privilégiée consiste à purifier l'aminé ou l'acide par une technique chromatographique (par exemple, sur colonne à phase chirale). Une autre voie privilégiée consiste à purifier l'aminé ou l'acide par cristallisation des sels formés avec respectivement des acides carboxyliques chiraux énantiopurs (acide tartrique, etc.) ou des bases organiques chirales énantiopures (éphédrine, etc.). Une autre voie privilégiée consiste à protéger l'aminé ou l'acide par un groupe protecteur contenant un centre asymétrique. Le mélange pourra alors être enrichi en l'un ou l'autre des diastéréoisomères par cristallisation, ce qui permet après déprotection d'isoler l'aminé ou l'acide optiquement pur (ou enrichi).
Plus généralement, l'ensemble des méthodes présentées (A à H) peut être réalisé à partir de réactifs (Vl à XIII, I', I") optiquement purs (ou enrichis). Ces
méthodes permettent alors d'isoler les composés selon l'invention sous forme optiquement pure (ou enrichie).
Un objet particulièrement préféré de l'invention concerne les composés de formule générale (I), avantageusement (II) ou (III), dans laquelle R1 représente un radical alkyle, alkényle, aryle ou aralkyle et R2, R3 et R4 sont des atomes d'hydrogène. Les inventeurs ont mis au point un procédé de synthèse de ces composés préférés sous forme énantiopure. Cette méthodologie est donnée à titre d'exemple et n'est en aucun cas limitative quant à la manière de préparer les composés selon l'invention optiquement purs.
Méthode 1
Les composés de formule générale (I), avantageusement (II) ou (III), dans laquelle R1 représente un radical alkyle, alkényle, aryle ou aralkyle et R2, R3 et R4 sont des atomes d'hydrogène sont ici nommés (I1"). Ces composés chiraux sont obtenus de préférence par fonction nalisation des dérivés de formule (XV) optiquement purs selon les méthodes précédemment citées.
A titre d'exemple et de manière non limitative, les méthodes A et C peuvent être utilisées et permettent de préserver la pureté optique du synthon (XV).
Méthode A


L'intermédiaire (VIII') sera aisément obtenu par condensation de l'aminé (XV) sur l'acide (Vl) selon les méthodes connues de l'homme de métier (lire
« méthode A »). L'intermédiaire (VII') sera aisément obtenu par fonctionnalisation de l'intermédiaire (XV) selon les méthodes connues de l'homme de métier (lire
« méthode C »). Une protection préalable de la fonction aminé de (XV) par un groupement protecteur pourra le cas échéant être nécessaire. De nombreux exemples illustrant ces voies d'accès sont décrits dans la partie expérimentale, à partir d'intermédiaires de formule (XV) racémiques et/ou énantiopurs.
L'intermédiaire (XV) peut être avantageusement synthétisé en 2 étapes clefs à partir d'un acide α-aminé, la configuration absolue de ce dernier étant préservée.

XIV XV II est bien entendu que l'homme de l'art doit adapter ce schéma réactionnel en fonction du substrat. Notamment, une protection préalable de la fonction aminé de l'acide α-aminé est souvent nécessaire. De nombreux groupes protecteurs sont décrits dans la littérature pour la protection / déprotection d'acide aminés (sans racémisation). A titre d'exemple et de manière non limitative les groupements protecteurs « Boc », « Fmoc », « Cbz », et trifluoroacétamide sont particulièrement préférés.
L'obtention de l'intermédiaire (XIV) est envisageable selon 2 méthodologies.
- La réaction d'acylation de Friedel-Craft - La réaction de Grignard (ou réaction dérivée utilisant un agent organométallique)
L'acylation de Friedel-Craft est une réaction très connue de l'homme de métier. Elle est réalisée en présence de catalyseur acide (par exemple, AICI3 ou FeCI3), d'une forme activée de l'acide α-aminé protégé (par exemple, chlorure d'acyle), et d'un dérivé aryle. Ces réactions sont préférentiellement réalisées dans des solvants organiques anhydres tels que le dichlorométhane, le chloroforme, l'éther diéthylique, l'éther diisopropylique, le méthyl tertiobutyle éther, ou le tétrahydrofuranne (THF). Elles peuvent être réalisées à des températures comprises entre -78°C et 1000C, préférentiellement entre -200C et le point
d'ébullition du solvant envisagé. L'orientation de Pacylation (ortho/méta/para) pourra varier selon les substituants présents sur le noyau aryle selon des règles bien connues de l'homme de métier.
La réaction de Grignard est là encore une réaction très connue. Aussi la méthode proposée ne se restreint pas aux agents organomagnésiens mais plus généralement aux nombreux dérivés organométalliques pouvant être préparés à partir d'un halogénure d'aryle (magnésien, lithien, zincique etc.). Le substrat aryle préalablement activé sous forme d'ion organométaliique est mis en réaction avec l'acide α-aminé protégé préalablement activé. A titre d'exemple, l'activation de la fonction acide en fonction « amide de Weinreb » est particulièrement pertinente pour réaliser ce type de réaction. Ces réactions sont préférentiellement réalisées dans des solvants organiques anhydres tels que le dichlorométhane, le chloroforme, l'éther diéthylique, l'éther diisopropylique, le méthyl tertiobutyle éther, le tétrahydrofuranne (THF), le dioxanne, ou le toluène. Elles peuvent être réalisées à des températures comprises entre -1000C et +1000C, préférentiellement entre -78°C et le point d'ébullition du solvant envisagé. La réaction sera de manière préférentielle réalisée dans des conditions douces (notamment à basse température) pour ne pas favoriser la racémisation.
La réduction de l'intermédiaire (XIV) en (XV) peut être envisagée de différentes manières connues de l'homme de l'art. De manière préférentielle, cette réduction sera réalisée en milieu acide (par exemple, l'acide trifluoroacétique) par du trialkyle silane (par exemple, triéthylsilane). L'acide utilisé peut jouer le rôle de solvant. Si nécessaire, des solvants peuvent être ajoutés tels que l'acétonitrile, le tétrachlorure de carbone ou l'eau. Ces réactions peuvent être réalisées à des températures comprises entre -78°C et 1000C, préférentiellement entre 00C et le point d'ébullition du solvant envisagé. Cette réduction est envisageable directement sur l'intermédiaire de type (XIV) ou après une réduction partielle de la fonction cétone en fonction alcool (par action préalable d'un hydrure tel que NaBH4 par exemple).
Cette méthode générale permet d'obtenir (XV) optiquement pur, sans racemisation notable de l'acide α-aminé de départ. Elle est décrite dans les exemples et il s'agit d'une voie d'accès préférée aux composés selon l'invention.
De manière encore plus préférentielle, les composés de formule (XV) selon l'invention ou X est un radical hydroxyle (-OH) situé en para par rapport au motif - CH2- sont ici nommés (XV). Ils sont préférentiellement obtenus suivant le procédé suivant :
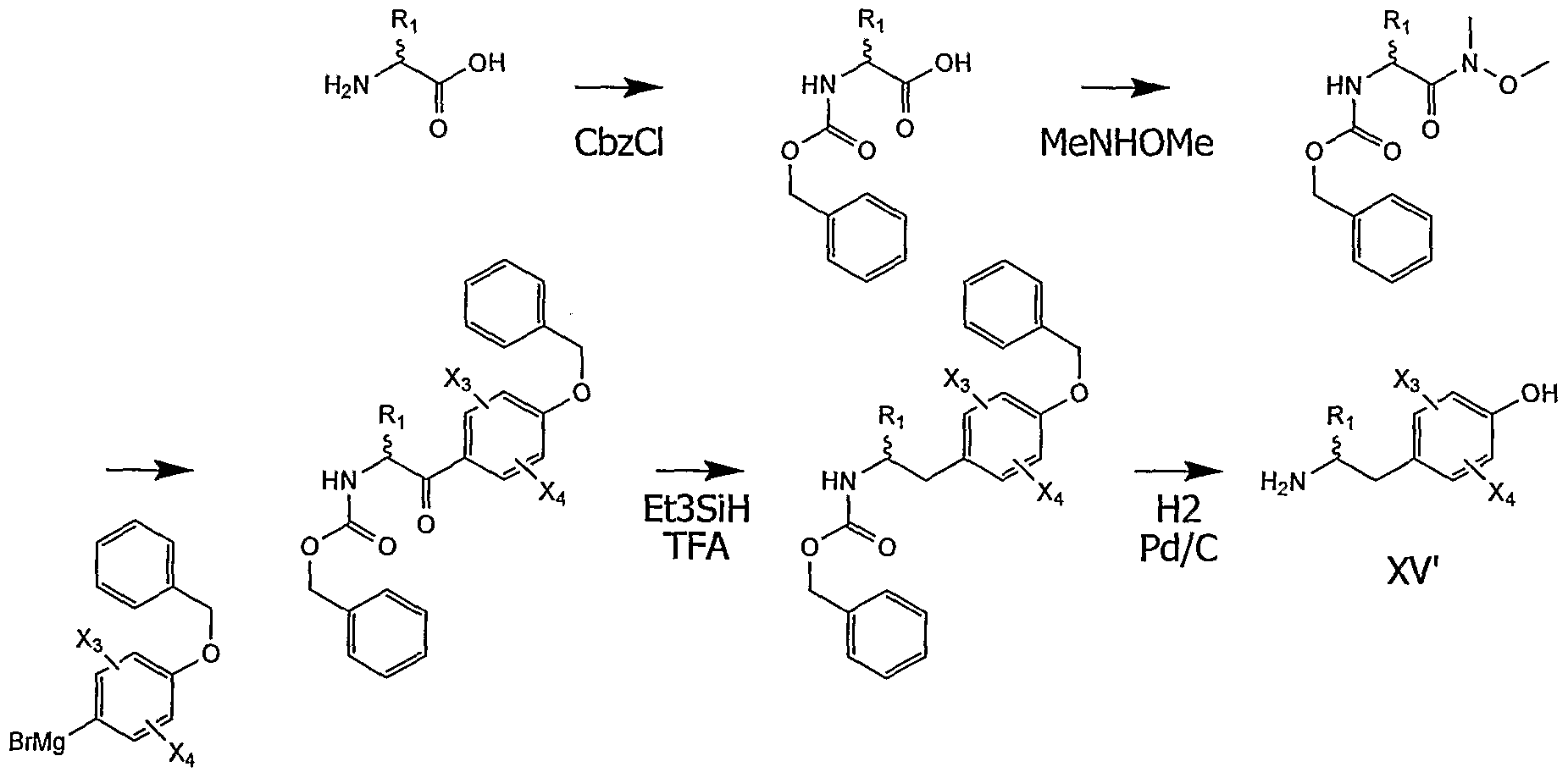
Une autre méthodologie consiste à purifier (ou enrichir) un mélange racémique de composés de formule (I) selon l'invention. Une voie privilégiée consiste à purifier les composés par chromatographie. Certains des composés selon l'invention peuvent contenir une fonction acide (notamment au niveau du groupe E) et/ou une fonction basique. Ils pourront donc avantageusement être purifiés par cristallisation comme décrit ci-avant : formation de sels ou protection de la molécule par des agents chiraux.
Si le composé est souhaité sous forme d'un sel, ce dernier sera obtenu lors d'une dernière étape connue de l'homme du métier, non évoquée dans les voies de synthèse présentées ci-dessus. A titre d'exemple, une voie préférentielle consiste à utiliser une résine échangeuse d'ions pour obtenir les sels souhaités.
Les procédés de préparation indiqués ci-avant sont donnés à titre illustratif, et tout autre procédé équivalent pourra naturellement être également mis en œuvre.
LEGENDES DES FIGURES
Au niveau des figures :
- Cpd = Composé - Ctrl = Contrôle
- mpk = mg/kg/jour
- HDL = High Density Lipoprotein
- LDL = Low Density Lipoprotein
- VLDL = Very Low Density Lipoprotein
Figure 1 : Evaluation in vitro des propriétés activatrices des PPARs des composés selon l'invention à 10 μM
L'activation des PPARs est évaluée in vitro sur une lignée de fibroblastes de rein de singe (COS-7), par la mesure de l'activité transcriptionnelle de chimères constituées du domaine de liaison à l'ADN du facteur de transcription Gal4 de la levure et du domaine de liaison au ligand des différents PPARs.
Les composés sont testés à 10 μM sur les chimères Gal4-PPARα, γ, δ. Le facteur d'induction, c'est-à-dire le rapport entre la luminescence induite par le composé et la luminescence induite par le contrôle, est mesuré pour chaque condition. Il est ensuite normalisé par rapport à un composé de référence interne et les résultats sont exprimés en pourcentages : plus le pourcentage d'activation est élevé, plus le composé a un caractère activateur des PPARs.
Figure 2 : Evaluation in vitro des propriétés activatrices des PPARs des composés selon l'invention en fonction de la dose
L'activation des PPARs est évaluée in vitro sur une lignée de fibroblastes de rein de singe (COS-7), par la mesure de l'activité transcriptionnelle de chimères constituées du domaine de liaison à l'ADN du facteur de transcription Gal4 de la levure et du domaine de liaison au ligand des différents PPARs. Les composés sont testés à des doses comprises entre 0,01 et 10 μM sur les chimères GaW-PPAR α, γ, δ. Le facteur d'induction, c'est-à-dire le rapport entre la luminescence induite par le composé et la luminescence induite par le contrôle, est mesuré pour chaque condition. Il est ensuite normalisé par rapport à
un composé de référence interne et les résultats sont exprimés en pourcentages : plus le pourcentage d'activation est élevé, plus le composé a un caractère activateur des PPARs.
Figure 3 : Evaluation in vitro de l'affinité des composés selon l'invention pour les canaux potassiques ATP-dépendants à 100 μM
Les résultats présentés reflètent l'affinité spécifique des composés selon l'invention pour les canaux potassiques ATP-dépendants (K+ ATP)- Les composés selon l'invention ont été testés à 100 μM : plus le pourcentage mesuré est élevé, plus l'affinité du composé pour les canaux potassiques ATP-dépendants est forte.
Figure 4 : Evaluation in vitro du caractère insulinosécréteur des composés selon l'invention
L'activation de la sécrétion d'insuline est évaluée in vitro sur une lignée de cellules pancréatiques de rat (INS-1) par la mesure de la concentration d'insuline sécrétée dans le milieu de culture en présence d'une faible concentration de glucose (2,8 mM). Les composés sont testés à des doses comprises entre 1 et 100 μM. Les résultats sont représentés par le facteur d'induction pour chaque condition, c'est- à-dire le rapport entre la concentration d'insuline induite par le composé selon l'invention et la concentration induite par le glucose 2,8 mM seul : plus ce facteur est élevé, plus le composé selon l'invention a un caractère insulino-sécréteur.
Figure 5 : Evaluation in vivo chez le rat du caractère hypoglycémiant des composés L'objet de cette étude est d'évaluer in vivo le caractère insulinosécréteur des composés selon l'invention. Chez des animaux normaux (comme chez l'homme), l'administration orale de composés insulino-sécréteurs a pour conséquence une augmentation significative du taux plasmatique d'insuline. Cette hyper-insulinémie induite provoque une baisse de la glycémie. La courbe présentée représente la glycémie de l'animal au cours du temps et témoigne de l'effet hypoglycémiant des composés selon l'invention (administrés par voie orale chez le rat à jeun).
Figures 6a, 6b, 6c : Evaluation in vivo des propriétés hypolipémiantes des composés selon l'invention et mesure de la régulation de l'expression de gènes impliqués dans le métabolisme lipidique et glucidigue.
L'effet hypolipémiant des composés selon l'invention est évalué in vivo chez la souris humanisée pour l'isoforme E2 de l'apolipoproteine E. La mutation induite empêche la clairance hépatique des lipides et conduit à une accumulation plasmatique des triglycérides et du cholestérol chez cet animal. Le traitement de ces animaux par un agoniste de PPARα doit induire une baisse des taux plasmatiques de lipides et doit se traduire au niveau hépatique par une modification de l'expression des gènes cibles dont l'expression est sous le contrôle du récepteur PPARα.
La figure (6a) présentée relate les taux de cholestérol plasmatique après 7 jours de traitement avec le composé 5, administré à 300 mg/kg/jour. Ces taux sont comparés à ceux obtenus avec des animaux contrôles (non traités par les composés selon l'invention) : la différence mesurée témoigne de l'effet hypolipémiant des composés selon l'invention.
L'efficacité des composés selon l'invention est aussi évaluée par mesure, dans les tissus hépatiques, de l'expression de gènes impliqués dans le métabolisme lipidique et glucidique. Plus le facteur d'induction mesuré est élevé, plus le composé a un caractère activateur d'expression génique.
Les figures (6b) et (6c) représentent respectivement les niveaux d'expression hépatiques de PDK4 (Pyruvate Deshydrogenase Kinase isoforme 4, enzyme du métabolisme glucidique) et de l'ACO (acyl coenzyme A oxydase, une enzyme clé impliquée dans le mécanisme de la β-oxydation des acides gras) après 7 jours de traitement avec le composé 5 (300 mg/kg/jour).
Figures 7a à 7e : Evaluation in vivo, chez la souris E2/E2, des propriétés hypolipémiantes et stimulatrices de la synthèse de HDL-cholestérol des composés selon l'invention par dosages lipidiques et mesure de l'expression de gènes impliqués dans le métabolisme lipidique et glucidique et la dissipation d'énergie. L'effet hypolipémiant des composés selon l'invention est évalué in vivo chez la
souris E2/E2 (humanisée pour l'isoforme E2 de l'apolipoproteine E) par l'analyse de la répartition du cholestérol dans les différentes fractions lipoprotéiques plasmatiques et par la mesure des taux de triglycérides plasmatiques après 7 jours de traitement par voie orale. Ces taux sont comparés à ceux obtenus avec des animaux contrôles (non traités par les composés selon l'invention) : la différence mesurée témoigne de l'effet hypolipémiant des composés selon l'invention.
- La figure (7a) représente la répartition du cholestérol dans les différentes fractions lipoprotéiques plasmatiques après 7 jours de traitement avec le composé 29, administré à 50 mg/kg/jour.
- La figure (7b) relate les taux de triglycérides plasmatiques après 7 jours de traitement avec le composé 29, administré à 50 mg/kg/jour.
L'efficacité des composés selon l'invention est aussi évaluée par mesure, dans les tissus hépatiques et musculaires (squelettiques), de l'expression de gènes impliqués dans le métabolisme lipidique et glucidique et la dissipation d'énergie. Plus le facteur d'induction mesuré est élevé, plus le composé a un caractère activateur d'expression génique.
- La figure (7c) représente l'expression de PDK4 (Pyruvate Deshydrogenase Kinase isoforme 4, enzyme du métabolisme glucidique) dans le tissu hépatique, chez la souris E2/E2, après 7 jours de traitement avec le composé 29 (50 mg/kg/jour).
- La figure (7d) représente l'expression de l'ACO (acyl coenzyme A oxydase, une enzyme clé impliquée dans le mécanisme de la β-oxydation des acides gras) dans le tissu hépatique, chez la souris E2/E2, après 7 jours de traitement avec le composé 29 (50 mg/kg/jour).
- La figure (7e) représente l'expression d'UCP3 (uncoupling protein 3, impliqiée dans la dissipation d'énergie) dans le muscle squelettique, chez la souris E2/E2, après 7 jours de traitement avec le composé 29 (50 mg/kg/jour).
Figures 8a à 8e : Evaluation in vivo, chez la souris C57BI6, des propriétés
hypolipémiantes et stimulatrices de la synthèse de HDL-cholestérol des composés selon l'invention par dosages lipidiques et mesure de l'expression de gènes impliqués dans le métabolisme lipidique et glucidique. L'effet des composés selon l'invention est évalué in vivo chez la souris C57BI6 par la mesure des taux plasmatiques de HDL-cholestérol, de triglycérides plasmatiques et d'acides gras libres après 14 jours de traitement par voie orale; ces taux sont comparés à ceux obtenus avec des animaux contrôles (non traités par les composés selon l'invention) : la différence mesurée témoigne de l'effet des composés selon l'invention.
- La figure (8a) représente les taux de HDL-cholestérol plasmatique après 14 jours de traitement avec le composé 29, administré à 50 mg/kg/jour.
- La figure (8b) représente les taux de triglycérides plasmatiques après 14 jours de traitement avec le composé 29, administré à 50 mg/kg/jour. - La figure (8c) représente les taux d'acides gras libres plasmatiques après
14 jours de traitement avec le composé 29, administré à 50 mg/kg/jour.
L'efficacité des composés selon l'invention est aussi évaluée par mesure, dans le tissus hépatique, de l'expression de gènes impliqués dans le métabolisme métabolisme lipidique et glucidique. Plus le facteur d'induction mesuré est élevé, plus le composé a un caractère activateur d'expression génique.
- La figure (8d) représente l'expression de PDK4 (Pyruvate Deshydrogénase Kinase isoforme 4, enzyme du métabolisme glucidique) dans le tissu hépatique, chez la souris C57BI6, après 14 jours de traitement avec le composé 29 (50 mg/kg/jour).
- La figure (8e) représente l'expression de l'ACO (acyl coenzyme A oxydase, une enzyme clé impliquée dans le mécanisme de la β-oxydation des acides gras) dans le tissu hépatique, chez la souris C57BI6, après 14 jours de traitement avec le composé 29 (50 mg/kg/jour).
Figure 9 : Evaluation in vitro des propriétés activatrices du transport inverse du cholestérol des composés selon l'invention par mesure de l'expression du gène ABCA1 dans les macrophages.
L'effet des composés selon l'invention sur le transport inverse de cholestérol a été évalué par la mesure de l'expression du gène ABCA1 dans des macrophages. Les résultats sont présentés sur la figure (9) : plus l'expression d'ABCAl est augmentée (facteur d'induction élevé), plus le composé selon l'invention stimule le transport inverse du cholestérol.
Figure 10 : Evaluation in vitro des propriétés anti-inflammatoires des composés selon l'invention par mesure de la sécrétion d'IL-β par des macrophages prétraités avec les composés selon l'invention et stimulés avec du LPS.
Les effets anti-inflammatoires des composés selon l'invention ont été évalués par la mesure de la sécrétion d'interleukine 6 (IL-6) par des macrophages prétraités pendant 24 heures avec les composés selon l'invention et stimulés pendant 6 heures avec du LPS (LipoPolySaccharide, qui provoque une réponse inflammatoire des cellules). Les résultats sont présentés sur la figure (10) : plus la quantité d'IL-6 sécrétée est diminuée, plus le composé selon l'invention inhibe la réaction inflammatoire.
Figure 11 : Evaluation in vitro des propriétés activatrices de l'oxydation des acides gras des composés selon l'invention.
L'effet des composés selon l'invention sur l'oxydation des acides gras a été évalué dans des cellules musculaires de souris (CaC12) par la mesure de la quantité d'eau tritiée formée durant l'oxydation mitochondriale d'acides gras marqués au 3H. Les résultats sont représentés sur la figure (11) par le facteur d'induction c'est-à-dire le rapport entre la bêta-oxydation des acides gras induite par le composé selon l'invention et celle induite par le DMSO seul : plus ce facteur est élevé, plus le composé selon l'invention active l'oxydation des acides gras.
ABREVIATIONS
BINAP 2,2'-Bis(diphénylphosphιno)-1 , 1 '-binaphthyle
Cbz Benzyloxycarbonyl
CDI N,N-Carbonyldiimidazole
DCC N , N-Dicyclohexylcarbodiimide
DCU N,N-Dicyclohexylurée
DIPEA Diisopropyléthylamine
DMAP Diméthylaminopyridine
EDC Chlorure de N-éthyl-N-(3-diméthylaminopropyl)carbodiimide
DMF Diméthylformamide
HBTU Hexafluorophosphate de 2-(1H-benzotriazol-1-yl)-1 , 1 ,3,3 tetraméthyluronium
HOBt 1 -Hydroxybenzotriazole
LDA Lithium diisopropylamide
Pd Palladium
Pd(OAc)2 Acétate de palladium (II)
Pd2(dba)3 Tris(dibenzylidèneacétone)dipalladium (0)
PPTS Para-toluènesulfonate de pyridinium
PyBOP Hexafluorophosphate de benzotriazol-1-yl oxytrispyrrolidinophosphonium TBTU Tétrafluoroborate de O-(Benzotriazol-1-yl)-1 , 1 ,3,3- tétraméthyluronium THF Tétrahydrofuranne
X-Phos 2-Dicyclohexylphosphino-2'-4'-6'-triisopropylbiphényle
D'autres avantages et aspects de l'invention apparaîtront à la lecture des exemples qui suivent, qui doivent être considérés comme illustratifs et non limitatifs.
EXEMPLES
Les réactifs et catalyseurs usuels sont disponibles commercialement (Aldrich, Alfa Aesar, Acros, Fluka ou Lancaster selon les cas).
Les dérivés organométalliques (organomagnésiens) utilisés sont disponibles commercialement ou peuvent être synthétisés à partir des halogénures correspondants suivant des méthodes connues de l'homme du métier.
Les lavages sont réalisés par des solutions aqueuses saturées en chlorure de sodium (NaCIsat), des solutions molaires de soude (NaOH 1M) ou d'acide chlorhydrique (HCI 1M).
Les spectres de Résonance Magnétique Nucléaire du Proton (RMN 1H) ont été enregistrés sur un spectromètre Bruker AC300P. Les déplacements chimiques sont exprimés en ppm (partie par million) et les multiplicités par les abréviations usuelles.
Exemple 1 : Description des protocoles généraux de synthèse selon l'invention
Protocole A : substitution nucléophile aromatique
Le dérivé du type halogénure d'aryle (1 eq) est mis en solution dans l'acétonitrile (0,1 à 5 mol/l) et du carbonate de potassium (3 à 4 eq) est additionné. Le nucléophile à substituer (aminé ou thiol, 1 à 3 eq) est ajouté. Le mélange est porté au reflux et agité pendant 16 h. Les sels sont filtrés et le solvant est partiellement éliminé par évaporation sous pression réduite. Le résidu est acidifié par une solution aqueuse d'acide chlorhydrique et extrait par l'acétate d'éthyle (3 fois). Les phases organiques sont rassemblées et la solution résultante est successivement
lavée par NaCIsat, séchée sur sulfate de magnésium (MgSO4), filtrée, et évaporée. Selon les cas, le produit attendu est purifié par chromatographie sur gel de silice ou par recristallisation du sel correspondant (par exemple, par préparation du chlorhydrate dans l'éther diéthylique).
Protocole B : alkylation médiée par la LDA
Sous atmosphère anhydre, le substrat à alkyler (1 eq) est mis en solution dans le THF (0,1 à 1 mol/l) et refroidi à -78°C. La LDA (1 à 3 eq, solution 2M dans le THF) est ajoutée goutte à goutte et le milieu est agité pendant 30 min à -78°C. Le dérivé halogène (1 à 3 eq) est alors additionné et le milieu réactionnel est ramené lentement à température ambiante. Après quelques heures, le milieu réactionnel est refroidi à 00C, hydrolyse par une solution saturée en chlorure d'ammonium, et extrait par l'acétate d'éthyle (3 fois). Les phases organiques sont rassemblées et la phase résultante est lavée par une solution saturée en chlorure de sodium (3 fois), séchée sur sulfate de magnésium, filtrée et concentrée. Selon les cas, le produit attendu est purifié par chromatographie sur gel de silice ou par recristaliisation.
Protocole C : réarrangement de Curtius Sous atmosphère anhydre, l'acide (1 eq) est mis en solution dans le toluène (0,1 à 1 mol/l). La triéthylamine (1 ,1 eq) et l'azoture de diphénylphosphoryle (DPPA, 1eq) sont successivement ajoutés. La solution est agitée à température ambiante pendant 30 minutes puis portée doucement à reflux. Le reflux est maintenu pendant 1 à 2 heures puis le milieu réactionnel est ramené à 500C. L'alcool benzylique (3 eq) est ajouté et le milieu est porté à reflux et agité pendant 16 heures. Le milieu est ramené à température ambiante et hydrolyse par une solution d'hydrogénocarbonate de sodium. Il est extrait par l'acétate d'éthyle (3 fois). Les phases organiques sont rassemblées et la phase résultante est lavée par une solution saturée en chlorure de sodium (3 fois), séchée sur sulfate de magnésium, filtrée et concentrée. Selon les cas, le produit attendu est purifié par chromatographie sur gel de silice ou par recristallisation.
Protocole D : substitution d'un dérivé halogène par un dérivé phénolique
Le dérivé du type phénol (1 eq) est mis en solution dans l'acétonitrile (0,1 à 1 mol/l) et du carbonate de potassium (3 à 4 eq) est additionné. La suspension est agitée 30 min au reflux puis le dérivé halogène (1 à 2 eq) est ajouté goutte à goutte. Le chauffage est maintenu pendant 16 h puis le solvant est évaporé et repris dans l'acétate d'éthyle. Le résidu est filtré et le filtrat est lavé successivement par HCI 1M, NaOH 1M et NaCIsat. Le brut réactionnel est alors séché sur sulfate de magnésium (MgSO4), filtré et évaporé. Selon les cas, le produit attendu est purifié par chromatographie sur gel de silice ou par recristallisation.
Protocole E : hydrogénation catalytique
Le dérivé à réduire (alcène, benzyl éther, CBz, etc.) (1 eq) est solubilisé dans le méthanol (0,1 à 1 mol/l) et additionné d'une quantité catalytique de palladium sur charbon (Pd/C 10%). Le milieu réactionnel est placé sous hydrogène à pression atmosphérique pendant 16 heures. Le brut est ensuite filtré sur célite® et le filtrat évaporé. Selon les cas, le produit attendu est purifié par chromatographie sur gel de silice ou par recristallisation.
Protocole F : condensation au PyBQP L'aminé (1 eq) et l'acide (1 eq) sont mélangés dans le dichlorométhane (0,1 à 1 mol/l). La triéthylamine (1 à 3 eq) est additionnée, suivie du PyBOP (1 eq). La solution résultante est agitée pendant 1 heure à température ambiante. Le brut réactionnel est successivement lavé par HCI 1M (3 fois), NaOH 1M (3 fois) et NaCIsat (3 fois). La phase organique est séchée sur sulfate de magnésium (MgSO4), filtrée et évaporée. Selon les cas, le produit attendu est purifié par chromatographie sur gel de silice ou par recristallisation.
Protocole G : hydrolyse acide des fonctions ester tert-butyliques L'ester (1 eq) est solubilisé dans le dichlorométhane (1 à 5 mol/l) puis l'acide trifluoroacétique (1 à 5 eq) est ajouté. Le mélange réactionnel est agité à température ambiante pendant 2 heures puis évaporé à sec. Selon les cas, le produit attendu est purifié par lavages, par chromatographie sur gel de silice ou par recristallisation.
Protocole H : saponification
L'ester (1 eq) est mis en solution dans un mélange équivolumétrique éthanol/soude 2M (0,1 à 1 mol/l) et le mélange est agité vigoureusement pendant 3 h à température ambiante. Le brut réactionnel est acidifié par adjonction d'une solution aqueuse d'acide chlorhydrique, concentré, puis extrait au dichlorométhane (3 fois). La phase organique résultante est lavée avec NaCIsat, séchée sur sulfate de magnésium (MgSO4), filtrée et concentrée. Selon les cas, le produit attendu est purifié par chromatographie sur gel de silice ou par recristallisation.
Protocole I : condensation par HBTU/HOBt
L'acide (1 eq), la diisopropyléthylamine (1 à 3 eq), le HBTU (1 à 2 eq) et le HOBt
(0,1 à 1 eq) sont mis en solution dans le dichlorométhane (0,1 à 5 mol/l). Le mélange est agité 30 minutes à température ambiante puis l'aminé à condenser (1 eq) est additionnée. La solution résultante est agitée pendant quelques heures à température ambiante. Le brut réactionnel est successivement lavé par HCI 1M (3 fois), NaOH 1M (3 fois) et NaCIsat (3 fois). La phase organique est séchée sur sulfate de magnésium (MgSO4), filtrée et évaporée. Selon les cas, le produit attendu est purifié par chromatographie sur gel de silice ou par recristallisation.
Protocole J : Réaction type Homer-Emmons
Sous atmosphère anhydre, l'hydrure de sodium (1 eq) est mis en suspension dans le THF anhydre (0,1 à 5 mol/l). Le milieu est refroidi à O0C et le phosphonate (1 eq) est additionné lentement. Le mileu est ramené à température ambiante et agité pendant 30 minutes. Le dérivé carbonyle (1 eq) est ajouté puis le milieu réactionnel est porté à reflux pendant 16 heures. Le milieu est refroidi à température ambiante et hydrolyse par adjonction d'eau (éventuellement acide). Le milieu est extrait par l'acétate d'éthyle (3 fois). La phase organique résultante est lavée avec NaCIsat, séchée sur sulfate de magnésium (MgSO4), filtrée et concentrée. Selon les cas, le produit attendu est purifié par chromatographie sur gel de silice ou par recristallisation.
Protocole K : conversion d'un dérivé phénolique en dérivé acide de type 2-méthyl- phénoxy-propanoïque
Le phénol (1 eq) est solubilisé dans l'acétone (36 à 72 eq). La soude (9 à 25 eq) est ajoutée et la suspension qui en résulte est chauffée à 35°C pendant 30 min. Le chloroforme (4,5 eq) est additionné goutte à goutte au milieu sous agitation à 350C
(réaction fortement exothermique). La réaction est maintenue pendant 1 heure, refroidie à température ambiante, acidifiée par addition de HCI, et extraite par le dichlorométhane (3 fois). Les phases organiques sont rassemblées et la solution résultante est successivement lavée par NaCIsat, séchée sur sulfate de magnésium (MgSO4), filtrée, et évaporée. Selon les cas, le produit attendu est purifié par chromatographie sur gel de silice ou par recristallisation.
Protocole L : condensation d'un dérivé sulfonamide par EDC/DMAP
A température ambiante, le sulfonamide (1 eq) est solubilisé dans le dichlorométhane (0,1 à 1 mol/l) et additionné successivement de DMAP (1 à 2 eq) et de l'acide à condenser (1 eq). Le brut réactionnel est refroidi à O0C et l'EDC (1 eq) est ajoutée. L'agitation est maintenue pendant une nuit à température ambiante. Le brut réactionnel est successivement lavé par HCI 1 M (3 fois) et NaCIsat (3 fois). La phase organique est séchée sur sulfate de magnésium (MgSO4), filtrée, et évaporée. Selon les cas, le produit attendu est purifié par chromatographie sur gel de silice ou par recristallisation.
Exemple 2 : Synthèse des intermédiaires de type aminé selon l'invention
Exemple 2-1 : 4-(2-aminopropyl)phénol

Cette aminé est synthétisée en 3 étapes : Etape 1 : 1-(4-(benzyloxy)phényl)propan-2-one
La 4-hydroxyphénylacétone est benzylée par le bromure de benzyle selon le protocole D. Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2). Rendement : 90 %
Aspect : poudre jaune pâle
RMN 1H (300MHz, CDCI3, δ en ppm) : 2,16 (s, 3H) ; 3,65 (s, 2H) ; 5,07 (s, 2H) ;
6,96 (d, 8,6 Hz, 2H) ; 7,13 (d, 8,6 Hz, 2H) ; de 7,32 à 7,47 (m, 5H).
Etape 2 : N-benzyl-1-(4-(benzyloxy)phényl)propan-2-amine
La cétone précédemment isolée (6,8 g, 28,1 mmol) et la benzylamine (4,6 ml, 42,2 mmol) sont solubilisés dans le méthanol (50 ml) additionné d'acide acétique (1 ,6 ml, 28,1 mmol). Le milieu est refroidi sous atmosphère anhydre et le borohydrure de sodium (1 ,6 g, 42,2 mmol) est ajouté par portions. Le milieu réactionnel est agité pendant une nuit à température ambiante. Il est ensuite refroidi par un bain de glace et hydrolyse par adjonction d'eau (50 ml). Le composé attendu est extrait au dichlorométhane (2 x 100 ml) et la phase organique résultante est séchée sur sulfate de magnésium, filtrée et concentrée. Le résidu obtenu est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5). Rendement : 62 % Aspect : huile
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,11 (d, 6,3 Hz1 3H) ; 1 ,52 (s(large), 1 H) ; de 2,59 à 2,76 (m, 2H) ; de 2,87 à 2,97 (m, 1 H) ; 3,75 (d, 13,2 Hz, 1 H) ; 3,88 (d, 13,2 Hz, 1H) ; 5,08 (s, 2H) ; 6,92 (d, 8,6 Hz, 2H) ; 7,10 (d, 8,6 Hz, 2H) ; de 7,21 à 7,48 (m, 10H).
Etape 3 : 4-(2-aminopropyl)phénol
Ce composé est obtenu par hydrogénation de l'intermédiaire précédent selon le protocole E. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 8/2, additionné de 0,1% d'ammoniaque).
Rendement : 87 %
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,19 (d, 6,3 Hz, 3H) ; 2,45 (dd, 13,6 Hz et
8,7 Hz, 1H) ; 2,73 (dd, 13,6 Hz et 4,7 Hz, 1 H) ; 3,19 (m, 1H) ; 3,61 (m, 3H) ; 6,74 (d, 8,2 Hz, 2H) ; 7,02 (d, 8,2 Hz, 2H).
Exemple 2-1(S) : (S)-4-(2-aminopropyl)phénol
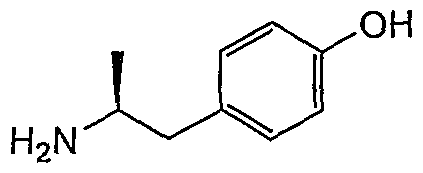
Cette aminé est synthétisée en 6 étapes : Etape 1 : acide (S)-2-(benzyloxycarbonylamino)propanoïque. La L-alanine (1,0 g, 11 mmol) est solubilisée dans de l'eau distillée (20 ml) puis du carbonate de sodium (4,0 g, 36 mmol) est additionné. Le milieu réactionnel est agité à température ambiante, puis le chloroformiate de benzyle (2,0 ml, 11 mmol) est additionné goutte à goutte. L'agitation est maintenue à température ambiante pendant 18 h. Ensuite le milieu réactionnel est refroidi à 00C et acidifié par ajout d'acide citrique jusqu'à atteindre un pH voisin de 4. Il est extrait par le dichlorométhane (3 fois). Les phases organiques sont rassemblées et la phase résultante est lavée par NaCIsat (3 fois), séchée sur sulfate de magnésium, filtrée et concentrée. Le résidu est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5). Rendement : 74 % Aspect : solide blanc
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,48 (d, 7,3 Hz, 3H) ; de 4,33 à 4,49 (m, 1H) ; de 5,14 à 5,18 (m, 2H) ; 5,37 (d, 7,3 Hz, 0,7H) ; 6,89 (s(large), 0,3H) ; de 7,33 à 7,37 (m, 5H) ; 8,76 (s (large), 1H).
Etape 2 : (S)-2-(benzyloxycarbonylamino)-N-méthoxy-N-méthylpropionamide.
Le dérivé acide précédent (407 mg, 1 ,82 mmol) est solubilisé dans le DMF (5 ml), sous atmosphère inerte. A 00C, le chlorure de N,O-diméthylhydroxylamine (213 mg, 2,19 mmol), le tétrafluoroborate de 2-(1H-benzotriazol-1-yl)-1 , 1 ,3,3- tétramethyluronium (TBTU, 877 mg, 2,73 mmol), puis la N, N- diisopropyléthylamine (DIPEA, 0,9 ml, 5,47 mmol) sont additionnés au milieu réactionnel. Après 2 heures d'agitation à température ambiante, le brut est hydrolyse par ajout de NaCIsat et extrait par l'acétate d'éthyle (3 fois). Les phases organiques sont rassemblées et la phase résultante est lavée par NaCIsat (3 fois), séchée sur sulfate de magnésium (MgSO4), filtrée et concentrée. Le résidu est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5). Rendement : 93 % Aspect : huile transparente
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,34 (d, 7 Hz, 3H) ; 3,2 (s, 3H) ; 3,77 (s, 3H) ; de 4,68 à 4,77 (m, 1H) ; de 5,05 à 5,15 (m, 2H) ; 5,67 (d, 8,2 Hz, 1 H) ; de 7,30 à 7,36 (m, 5H).
Etape 3 : (S)-1-[(4-(benzyloxy)phényl)]-2-[(benzyloxycarbonylamino)]propan-1-one Sous atmosphère inerte à température ambiante, le magnésium (1 ,1 g, 44 mmol) est mis en suspension dans du THF fraîchement distillé (50 ml). Un cristal d'iode est additionné au milieu réactionnel puis le mélange est porté à reflux. Ensuite le 1-(benzyloxy)-4-bromobenzène (13 g, 48 mmol), solubilisé dans 50 ml de THF fraîchement distillé, est additionné goutte à goutte et le milieu réactionnel est agité au reflux du THF pendant 3 heures. La solution est refroidie à température ambiante, puis le réactif de Grignard ainsi formé est additionné goutte à goutte sur le (S)-2-(benzyloxycarbonylamino)-N-méthoxy-N-méthylpropionamide (3,9 g, 15 mmol) solubilisé dans du THF fraîchement distillé (50 ml). Après 1 heure d'agitation à température ambiante, le milieu réactionnel est hydrolyse par une solution de HCI 1M et extrait au dichlorométhane (3 fois). Les phases organiques sont rassemblées et la phase résultante est lavée par NaCIsat (3 fois), séchée sur sulfate de magnésium (MgSO4), filtrée et concentrée. Le résidu est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 3/7). Rendement : 84 % Aspect : solide blanc
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,46 (d, 7 Hz, 3H) ; 5,16 (s, 4H) ; de 5,30 à 5,35 (m, 1 H) ; 5,97 (d, 7,6 Hz, 1H) ; 7,06 (d, 9,1 Hz, 2H) ; de 7,34 à 7,47 (m, 10H) ; 7,98 (d, 9,1 Hz, 2H).
Etape 4 : (S)-1-[(4-(benzyloxy)phényl)]-2-[(benzyloxycarbonylamino)]propan-1-ol. L'intermédiaire précédent (4,0 g, 10 mmol) est solubilisé dans du méthanol (40 ml) et du tétraborohydrure de sodium (1 ,0 g, 31 mmol) est additionné au milieu réactionnel. Après 3 heures d'agitation à température ambiante, le milieu réactionnel est refroidi à 00C, il est hydrolyse par une solution de HCI 1M et extrait par l'acétate d'éthyle (3 fois). Les phases organiques sont rassemblées et la phase résultante est lavée par une solution saturée en chlorure de sodium (3 fois),
séchée sur sulfate de magnésium, filtrée et concentrée. Le résidu est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 2/8). Rendement : 90 % Aspect : solide blanc.
Etape 5 : (S)-[I -[4-(2-(benzyloxycarbonylamino)propyl)phénoxy]méthyl]benzène. L'intermédiaire précédent (3,16 g, 8,08 mmol) est solubilisé dans l'acide trifluoroacétique (24 ml). Du triéthylsilane (2,82 g, 24,2 mmol) est additionné à la solution préalablement refroidie par un bain de glace. Après 5 heures d'agitation à 00C, le milieu réactionnel est dilué par du dichlorométhane, hydrolyse par une solution aqueuse d'ammoniaque (28%), et extrait au dichlorométhane (3 fois). Les phases organiques sont rassemblées et la phase résultante est lavée par NaCIsat (3 fois), séchée sur sulfate de magnésium, filtrée et concentrée. Le résidu est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 3/7). Rendement : 69 % Aspect : solide blanc
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,14 (d, 6,7 Hz, 3H) ; de 2,64 à 2,71 (m, 1H) ; de 2,78 à 2,84 (m, 1 H) ; de 3,99 à 4,04 (m, 1H) ; 4,67 (s (large), 1H) ; 5,07 (s, 2H) ; 5,12 (s, 2H) ; 6,93 (d, 8,5 Hz, 2H) ; 7,11 (d, 8,5 Hz, 2H) ; de 7,34 à 7,48 (m, 10H).
Etape 6 : (S)-4-(2-aminopropyl)phénol.
L'intermédiaire précédent (2,1 g, 5,5 mmol) est solubilisé dans le méthanol (100 ml) puis du palladium activé sur charbon (Pd/C 10%, 1 ,7 g) est additionné au milieu réactionnel. La solution est agitée à température ambiante sous pression atmosphérique d'hydrogène. Après 48 heures d'agitation à température ambiante, le milieu réactionnel est filtré sur célite et un lavage abondant au méthanol est effectué. Le filtrat ainsi obtenu est concentré sous pression réduite. Rendement : 100 % Aspect : huile beige
RMN 1H (300MHz, CDCI3, δ en ppm) : conforme avec le spectre obtenu pour 2-1.
Exemple 2-1(R) : (R)-4-(2-aminopropyl)ρhénol
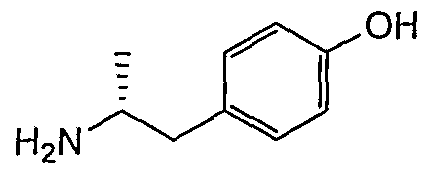
Cette aminé est synthétisée suivant les mêmes procédures à partir de la D- alanine.
Exemple 2-2 : 2-[4-(2-aminoéthyl)phénoxy]-2-méthylpropanoate d'éthyle

Cette aminé est synthétisée en 3 étapes. Dans cet exemple, elle est isolée sous forme de sel trifluoroacétate.
Etape 1 : 4-(2-N-feAf-butoxycarbonyl-aminoéthyl)phénol Le chlorhydrate de 4-(2-aminoéthyl)phénol (10 g, 57,59 mmol) est dissous dans un mélange de soude 1M (75 ml) et de feAf-butanol (75 ml). Le di-tert-butyl dicarbonate (12,6 g, 57,59 mmol) est ajouté par portions durant 15 minutes.
L'agitation est maintenue 1 h à température ambiante. Le brut réactionnel est alors extrait par l'acétate d'éthyle (2 x 150 ml). La phase organique est successivement lavée par NaOH 1 M (3 x 100 ml), HCI 1M (3 x 100 ml), et NaCIsat (3 x 100 ml). La solution est séchée sur sulfate de magnésium, filtrée et évaporée à sec.
Rendement : 70 %
Aspect : solide blanc
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,46 (s, 9H) ; 2,71 (t, 7,1 Hz, 2H) ; 3,33 (m, 2H) ; 4,65 (m, 1 H) ; 6,79 (d, 8,0 Hz, 2H) ; 7,01 (d, 8,0 Hz, 2H).
Etape 2 : 2~[4-(2-N-fe/f-butoxycarbonyl-aminoéthyl)phénoxy]-2-méthylpropanoate d'éthyle
L'intermédiaire phénolique précédent (8,5 g, 35,8 mmol) est solubilisé dans le diméthylformamide (50 ml). Le carbonate de potassium (14,85 g, 107 mmol) est ajouté et la suspension est agitée pendant 30 minutes à température ambiante. Le bromoisobutyrate d'éthyle (5,6 ml, 39,4 mmol) est alors ajouté goutte à goutte et le mélange est agité pendant 16 h à température ambiante. Les sels sont filtrés sur fritte et rincés par l'acétate d'éthyle (300 ml). Le filtrat est lavé par l'eau (100 ml), NaOH 1 M (3 x 100 ml), HCI 1M (3 x 100 ml) et NaCIsat (3 x 100 ml). La phase
organique est séchée sur sulfate de magnésium, filtrée et évaporée à sec. L'huile obtenue est purifiée par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2). Rendement : 29 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,26 (t, 7,0 Hz, 3H) ; 1 ,44 (s, 9H) ; 1 ,59 (s, 6H) ; 2,72 (t, 7,0 Hz, 2H) ; 3,33 (m, 2H) ; 4,24 (q, 7,0 Hz, 2H) ; 4,52 (m, 1 H) ; 6,79 (d, 8,5 Hz, 2H) ; 7,05 (d, 8,5 Hz, 2H).
Etape 3 : trifluoroacétate de 2-[4-(2-aminoéthyl)phénoxy]-2-méthylpropanoate d'éthyle
L'intermédiaire précédent (3,3 g, 9,39 mmol) est solubilisé dans le dichlorométhane (15 ml). L'acide trifluoroacétique (15 ml) est ajouté et le mélange est agité pendant 1 h à température ambiante. Les solvants sont évaporés sous pression réduite. Aucune purification supplémentaire n'est nécessaire. Rendement : quantitatif Aspect : huile jaune pâle
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,28 (t, 7,1 Hz, 3H) ; 1 ,55 (s, 6H) ; 2,89 (t, 7,3 Hz, 2H) ; 3,19 (m, 2H) ; 4,24 (q, 7,1 Hz, 2H) ; 6,76 (d, 8,5 Hz, 2H) ; 7,06 (d, 8,5 Hz, 2H) ; 7,49 (s(large), 3H).
Exemple 2-3 : 4-(2-amino-2-méthylpropyl)phénol

Cette aminé est synthétisée en 5 étapes : Etape 1 : 3-(4-(benzyloxy)phényl)propionate de méthyle
Le 3-(4-hydroxyphényl)propionate de méthyle est benzylé selon le protocole D.
Rendement : 93 %
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 2,62 (t, 7,7 Hz, 2H) ; 2,92 (t, 7,7 Hz, 2H) ; 3,69 (s, 3H) ; 5,06 (s, 2H) ; 6,92 (d, 8,5 Hz, 2H) ; 7,14 (d, 8,5 Hz, 2H) ; de 7,34 à
7,47 (m, 5H).
Etape 2 : 3-(4-(benzyloxy)phényl)-2,2-diméthylpropionate de méthyle L'ester méthylique précédent est alkylé par l'iodure de méthyle selon le protocole B. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 95/5). Rendement : 53%
Aspect : huile jaune pâle
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,19 (s, 6H) ; 2,81 (s, 2H) ; 3,67 (s, 3H) ;
5,05 (s, 2H) ; 6,89 (d, 8,5 Hz, 2H) ; 7,03 (d, 8,5 Hz, 2H) ; de 7,34 à 7,46 (m, 5H).
Etape 3 : acide 3-(4-(benzyloxy)phényl)-2,2-diméthylpropionique
L'ester méthylique précédent est saponifié selon le protocole H.
Rendement : 95%
Aspect : poudre beige
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,22 (s, 6H) ; 2,86 (s, 2H) ; 5,05 (s, 2H) ; 6,91 (d, 8,5 Hz, 2H) ; 7,11 (d, 8,5 Hz, 2H) ; de 7,31 à 7,46 (m, 5H).
Etape 4 : 1-(4-(benzyloxy)phényl)-2-méthylpropan-2-ylcarbamate de benzyle
L'acide carboxylique précédent est réarrangé suivant le protocole C. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 95/5). Rendement : 82%
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,30 (s, 6H) ; 2,93 (s, 2H) ; 4,53 (s, 1 H) ;
5,05 (s, 2H) ; 5,11 (s, 2H) ; 6,84 (d, 8,5 Hz, 2H) ; 6,99 (d, 8,5 Hz, 2H) ; de 7,35 à
7,47 (m, 10H).
Etape 5 : 4-(2-amino-2-méthylpropyl)phénol
L'intermédiaire précédent est hydrogéné selon le protocole E. Aucune purification n'est nécessaire hormis la filtration du catalyseur.
Rendement : quantitatif Aspect : poudre beige
RMN 1H (300MHz, DMSO-c/6, δ en ppm) : 0,95 (s, 6H) ; 1,73 (s(large), 2H) ; 2,45
(s, 2H) ; 6,65 (d, 8,5 Hz, 2H) ; 6,95 (d, 8,5 Hz, 2H) ; 9,17 (s(large), 1H).
Exemple 2-4 : 4-(1-amino-2-méthylpropan-2-yl)phénol

Cette aminé est synthétisée en 3 étapes : Etape 1 : 2-(4-(benzyloxy)phényl)-2-méthylpropanenitrile Le 2-(4-(benzyloxy)phényl)acétonitrile est alkylé par l'iodure de méthyle selon le protocole B. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 95/5). Rendement : 40% Aspect : solide RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,72 (s, 6H) ; 5,09 (s, 2H) ; 7,00 (d, 8,8 Hz, 2H) ; de 7,35 à 7,50 (m, 7H).
Etape 2 : 2-(4-(benzyloxy)phényl)-2-méthylpropan-1 -aminé
L'intermédiaire précédent (4,5 g, 17,9 mmol) est solubilisé dans le méthanol préalablement saturé en ammoniaque (100 ml). Le nickel de Raney (quantité catalytique) est ajouté et le milieu réactionnel est agité une nuit à 800C sous une pression d'hydrogène de 80 bars. Le milieu est refroidi et filtré sur célite®. L'aminé résultante est purifiée par chromatographie sur gel de silice
(dichlorométhane/méthanol 95/5). Rendement : 70%
Aspect : solide brun
RMN 1H (300MHz, CDCI3/D2O, δ en ppm) : 1 ,30 (s, 6H) ; 2,76 (s, 2H) ; 5,07 (s,
2H) ; 6,96 (d, 8,8 Hz, 2H) ; de 7,26 à 7,49 (m, 7H).
Etape 3 : 4-(1-amino-2-méthylpropan-2-yl)phénol
L'intermédiaire précédent est hydrogéné selon le protocole E. Aucune purification n'est nécessaire hormis la fitration du catalyseur.
Rendement : 95 %
Aspect : poudre beige RMN 1H (300MHz, CDCVD2O, δ en ppm) : 1 ,30 (s, 6H) ; 2,80 (s, 2H) ; 6,72 (d, 8,5
Hz, 2H) ; 7,16 (d, 8,5 Hz, 2H).
Exemple 2-5 : 4-(1-aminopropan-2-yl)phénol

Cette aminé est synthétisée en 3 étapes : Etape 1 : 2-(4-(benzyloxy)phényl)propanenitrile
Le 2-(4-(benzyloxy)phényl)acétonitrile est alkylé par l'iodure de méthyle selon le protocole B. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 95/5). Rendement : 55% Aspect : solide
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,63 (d, 7,3 Hz, 3H) ; 3,87 (q, 7,3 Hz, 1H) ; 5,09 (s, 2H) ; 6,99 (d, 8,5 Hz, 2H) ; 7,29 (d, 8,5 Hz, 2H) ; de 7,32 à 7,49 (m, 5H).
Etape 2 : 2-(4-(benzyloxy)phényl)propan-1 -aminé L'intermédiaire précédent (5,2 g, 22,1 mmol) est solubilisé dans le méthanol préalablement saturé en ammoniaque (100 ml). Le nickel de Raney (quantité catalytique) est ajouté et le milieu réactionnel est agité une nuit à 700C sous une pression d'hydrogène de 70 bars. Le milieu est refroidi et filtré sur célite®. L'aminé résultante est purifiée par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5). Rendement : 74% Aspect : solide
RMN 1H (300MHz, CDCI3/D2O, δ en ppm) : 1 ,24 (d, 6,7 Hz, 3H) ; de 2,69 à 2,85 (m, 3H) ; 5,07 (s, 2H) ; 6,96 (d, 8,8 Hz, 2H) ; 7,15 (d, 8,8 Hz1 2H) ; de 7,32 à 7,49 (m, 5H).
Etape 3 : 4-(1-aminopropan-2-yl)phénol
L'intermédiaire précédent est hydrogéné selon le protocole E. Aucune purification n'est nécessaire hormis la fitration du catalyseur. Rendement : 97 % Aspect : poudre beige
RMN 1H (300MHz1 DMSO d6/D2O, δ en ppm) : 1 ,12 (d, 6,2 Hz1 3H) ; 2,57 (m, 3H) 6,68 (d, 8,5 Hz, 2H) ; 6,98 (d, 8,5 Hz1 2H).
Exemple 2-6 : 2-[4-(1-aminopropan-2-yl)phénoxy]-2-méthylpropanoate d'éthyle

Cette aminé est synthétisée en 3 étapes à partir de l'aminé 2-5 suivant des procédures identiques à celles décrites dans l'exemple 2-2. L'aminé est isolée sous forme de sel trifluoroacétate.
Etape 1 : 4-(1-N-fe/f-butoxycarbonyl-aminopropan-2-yl)phénol Rendement : 83 %
Aspect : huile jaune pâle
RMN 1H (300MHz1 CDCI3, δ en ppm) : 1 ,24 (d, 7,0 Hz, 3H) ; 1 ,44 (s, 9H) ; 2,86 (m,
1H) ; 3,15 (m, 1 H) ; 3,35 (m, 1H) ; 4,45 (m, 1H) ; 6,81 (d, 8,5 Hz, 2H) ; 7,08 (d, 8,5
Hz, 2H).
Etape 2 : 2-[4-(1-N-fe/f-butoxycarbonyl-aminopropan-2-yl)phénoxy]-2- méthylpropanoate d'éthyle
Rendement : 69 %
Aspect : huile incolore RMN 1H (300MHz, CDCI3, δ en ppm) : de 1 ,22 à 1 ,29 (m, 6H) ; 1 ,43 (s, 9H) ; 1 ,60
(s, 6H) ; 2,86 (m, 1H) ; 3,13 (m, 1 H) ; 3,35 (m, 1 H) ; 4,25 (q, 7,0 Hz, 2H) ; 4,41 (m,
1H) ; 6,81 (d, 8,5 Hz, 2H) ; 7,07 (d, 8,5 Hz1 2H).
Etape 3 : trifluoroacétate de 2-[4-(1-aminopropan-2-yl)phénoxy]-2- méthylpropanoate d'éthyle
Rendement : quantitatif
Aspect : huile jaune pâle
RMN 1H (300MHz1 CDCI3, δ en ppm) : 1 ,31 (m, 6H) ; 1 ,58 (s, 6H) ; 3,06 (m, 2H) ;
3,21 (m, 1H) ; 4,26 (q, 7,0 Hz, 2H) ; 4,33 (s(large), 3H) ; 6,77 (d, 8,5 Hz1 2H) ; 7,09 (d, 8,5 Hz, 2H).
Exemple 2-7 : 4~(1-aminohexan-2-yl)phénol

Cette aminé est synthétisée en 3 étapes : Etape 1 : 2-(4-(benzyloxy)phényl)hexanenitrιïe
Le 2-(4-(benzyloxy)phényl)acétonitrile est alkylé par le bromure de butyle selon le protocole B. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 98/2). Rendement : 33% Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 0,92 (t, 7,2 Hz, 3H) ; de 1 ,31 à 1,55 (m, 4H) ; de 1 ,78 à 1 ,99 (m, 2H) ; 3,73 (t, 7,5 Hz, 1H) ; 5,08 (s, 2H) ; 6,99 (d, 8,5 Hz, 2H) ; 7,25 (d, 8,5 Hz, 2H) ; de 7,33 à 7,46 (m, 5H).
Etape 2 : 2-(4-(benzyloxy)phényl)hexan-1 -aminé
L'intermédiaire précédent (4,9 g, 17,5 mmol) est solubilisé dans le méthanol préalablement saturé en ammoniaque (100 ml). Le nickel de Raney (quantité catalytique) est ajouté et le milieu réactionnel est agité une nuit à 700C sous une pression d'hydrogène de 70 bars. Le milieu est refroidi et filtré sur célite®. Le brut résultant est utilisé tel quel pour l'étape suivante, sans analyse.
Etape 3 : 4-(1-aminohexan-2-yl)phénol
L'intermédiaire précédent est hydrogéné selon le protocole E. Aucune purification n'est nécessaire hormis la fitration du catalyseur. Rendement : 91 %
Aspect : poudre beige
RMN 1H (300MHz, CDCI3, δ en ppm) : 0,84 (t, 7,0 Hz, 3H) ; de 1 ,10 à 1 ,34 (m,
4H) ; de 1 ,44 à 1 ,66 (m, 2H) ; 2,55 (m, 1H) ; 2,81 (m, 1 H) ; 2,96 (m, 1H) ; 3,37
(s(large), 3H) ; 6,72 (d, 8,5 Hz, 2H) ; 7,00 (d, 8,5 Hz, 2H).
Exemple 2-8 : 2-[4-(1-aminopropan-2-yl)-3-méthyiphénoxy]-2-méthylpropanoate de fert-butyle

Cette aminé est synthétisée en 6 étapes :
Etape 1 : 2-[4-acétyl-3-méthyl-phénoxy]-2-méthylpropanoate de te/f-butyle La 4-hydroxy-2-méthylacétophénone est alkylée selon le protocole D. Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 9/1). Rendement : 62 % Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,43 (s, 9H) ; 1 ,60 (s, 6H) ; 2,51 (s, 3H) ; 2,53 (s, 3H) ; de 6,63 à 6,66 (m, 2H) ; 7,69 (d, 9,4 Hz, 1H).
Etape 2 : 2-[4-(1-(éthoxycarbonyl)prop-2-èn-2-yl)-3-méthylphénoxy]-2- méthylpropanoate de ferf-butyle
L'intermédiaire précédent est fonctionnalisé par le phosphonoacétate de triéthyle selon le protocole J. Le brut est purifié par chromatographie sur gel de silice
(cyclohexane/acétate d'éthyle 8/2).
Rendement : 38 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) ; mélange équimolaire de 2 diastéréoisomères : 1,31 (t, 7,0 Hz, 3H) ; 1 ,46 (s, 9H) ; 1,58 (s, 6H) ; 2,24 (s, 3H) ;
2,43 et 2,44 (s et s, 3H) ; 4,21 (q, 7,0 Hz1 2H) ; 5,74 et 5,75 (s et s, 1H) ; de 6,62 à
6,70 (m, 2H) ; 6,96 (d, 8,5 Hz, 1H).
Etape 3 : 2-[4-(1-(éthoxycarbonyl)propan-2-yl)-3-méthylphénoxy]-2- méthylpropanoate de feπf-butyle
L'intermédiaire précédent est réduit selon le protocole E. Aucune purification n'est nécessaire hormis la filtration du catalyseur. Rendement : 91 %
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,16 (t, 7,0 Hz, 3H) ; 1 ,21 (d, 6,7 Hz, 3H) ; 1 ,44 (s, 9H) ; 1,54 (s, 6H) ; 2,30 (s, 3H) ; de 2,43 à 2,60 (m, 2H) ; de 3,40 à 3,48 (m, 1H) ; 4,06 (q, 7,0 Hz, 2H) ; de 6,64 à 6,70 (m, 2H) ; 7,01 (d, 9,4 Hz, 1H).
Etape 4 : 2-[4-(1-(hydroxycarbonyl)propan-2-yl)~3-méthylphénoxy]-2- méthylpropanoate de fe/f-butyle
L'intermédiaire précédent est saponifié selon le protocole H. Aucune purification n'est nécessaire hormis les lavages (HCI 1M puis NaCIsat). Rendement : 93 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,25 (d, 7,0 Hz, 3H) ; 1 ,44 (s, 9H) ; 1,56 (s, 6H) ; 2,30 (s, 3H) ; de 2,48 à 2,67 (m, 2H) ; 3,43 (m, 1H) ; de 6,65 à 6,67 (m, 2H) ; 7,02 (d, 9,3 Hz, 1H).
Etape 5 : 2-[4-(1-benzyloxycarbonylamino-propan-2-yl)-3-méthylphénoxy]-2- méthylpropanoate de fe/f-butyle
L'acide précédent est fonctionnalisé selon le protocole C. Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2).
Rendement : 76 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,20 (d, 6,7 Hz, 3H) ; 1,45 (s, 9H) ; 1,56 (s,
6H) ; 2,25 (s, 3H) ; de 3,13 à 3,44 (m, 3H) ; 4,69 (m, 1H) ; de 5,04 à 5,13 (m, 2H) ; de 6,67 à 6,69 (m, 2H) ; 7,01 (d, 9,4 Hz, 1H) ; de 7,34 à 7,37 (m, 5H).
Etape 6 : 2-[4-(1-aminopropan-2-yl)-3-méthyl-phénoxy]-2-méthylpropanoate de fe/ï-butyle
L'intermédiaire précédent est réduit selon le protocole E. Aucune purification n'est nécessaire hormis la filtration du catalyseur.
Rendement : quantitatif Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,18 (d, 6,7 Hz, 3H) ; 1,45 (s, 9H) ; 1,55 (s,
6H) ; 2,27 (s, 3H) ; 2,51 (s(large), 2H) ; de 2,77 à 2,90 (m, 2H) ; de 2,98 à 3,05 (m,
1 H) ; de 6,67 à 6,69 (m, 2H) ; 7,00 (d, 9,1 Hz, 1 H).
Exemple 2-9 : 2-[4-(3-aminobutan-2-y!)phénoxy]-2-méthylpropanoate de tert- butyle

Cette aminé est synthétisée en 6 étapes :
Etape 1 : 4-(3-(éthoxycarbonyl)but-2-èn-2-yl)phényl benzyl éther
La 4-benzyloxyacétophénone est fonctionnalisée par le 2-phosphonopropionate de triéthyle selon le protocole J. Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 9/1). Rendement : 65 % Aspect : huile jaune pâle
RMN 1H (300MHz, CDCI3, δ en ppm) ; mélange 60/40 des 2 diastéréoisomères : 0,90 et 1,39 (t et t, 7,0 Hz, 3H) ; 1,81 et 2,02 et 2,09 et 2,25 (m, 6H) ; 3,89 et 4,28 (q, 7,0 Hz, 2H) ; 5,08 et 5,09 (s et s, 2H) ; de 6,91 à 7,01 (m, 2H) ; de 7,07 à 7,14 (m, 2H) ; de 7,33 à 7,49 (m, 5H).
Etape 2 : 4-(3-(éthoxycarbonyl)butan-2-yl)phéno!
L'intermédiaire précédent est réduit selon le protocole E. Aucune purification n'est nécessaire hormis la filtration du catalyseur.
Rendement : 99 %
Aspect : huile jaune pâle
RMN 1H (300MHz, CDCI3, δ en ppm) ; mélange 60/40 des 2 diastéréoisomères :
0,94 (d, 7,0 Hz, 1,2/3H) ; 1 ,07 (t, 7,0 Hz, 1 ,8/3H) ; de 1 ,18 à 1 ,33 (m, 6H) ; de 2,48 à 2,66 (m, 1H) ; de 2,82 à 3,01 (m, 1H) ; 3,52 (s, 1H) ; 3,95 et 4,20 (q et q, 7,0 Hz,
2H) ; de 6,71 à 6,81 (m, 2H) ; de 7,03 à 7,07 (m, 2H).
Etape 3 : 2-[4-(3-(éthoxycarbonyl)butan-2-yl)phénoxy]-2-méthylpropanoate de tert- butyle L'intermédiaire phénolique est alkylé selon le protocole D. Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 95/5).
Rendement : 79 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) ; mélange 60/40 des 2 diastéréoisomères : 0,91 et 1 ,16 (d et d, 7,0 Hz, 3H) ; 1,04 et 1,29 (t et t, 7,0 Hz, 3H) ; de 1,22 à 1 ,25 (m, 3H) ; 1,44 et 1 ,45 (s et s, 9H) ; 1,55 et 1,57 (s et s, 6H) ; de 2,46 à 2,64 (m, 1 H) ; de 2,80 à 3,03 (m, 1 H) ; 3,92 et 4,18 (q et q, 7,0 Hz, 2H) ; 6,80 (m, 2H) ; 7,05 (m, 2H).
Etape 4 : 2-[4-(3-(hydroxycarbonyl)butan-2-yl)phénoxy]-2-méthylpropanoate de tert-butyle
L'intermédiaire précédent est saponifié selon le protocole H. Le résidu est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5).
Rendement : 81 %
Aspect : huile jaune pâle RMN 1H (300MHz, CDCI3, δ en ppm) ; mélange 60/40 des 2 diastéréoisomères :
0,95 et 1 ,15 (d et d, 7,0 Hz1 3H) ; 1 ,28 (m, 3H) ; 1 ,44 et 1 ,45 (s et s, 9H) ; 1 ,56 et
1 ,57 (s et s, 6H) ; de 2,48 à 2,71 (m, 1 H) ; de 2,80 à 3,16 (m, 1H) ; 6,80 (m, 2H) ;
7,06 (m, 2H).
Etape 5 : 2-[4-(3-benzyloxycarbonylamino-butan-2-yl)phénoxy]-2- méthylpropanoate de te/f-butyle
L'acide précédent est fonctionnalisé selon le protocole C. Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 9/1). Rendement : 81 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) ; mélange 60/40 des 2 diastéréoisomères : 0,95 et 1 ,06 (d et d, 6,7 Hz, 3H) ; 1 ,27 (m, 3H) ; 1 ,45 (s, 9H) ; 1 ,57 (s, 6H) ; de 2,72 à 2,87 (m, 1H) ; 3,89 (m, 1H) ; 4,51 (m, 1H) ; de 5,01 à 5,12 (m, 2H) ; 6,81 (m, 2H) ; 7,04 (m, 2H), de 7,33 à 7,39 (m, 5H).
Etape 6 : 2-[4-(3-aminobutan-2-yl)phénoxy]-2-méthylpropanoate de terf-butyle L'intermédiaire précédent est réduit selon le protocole E. Aucune purification n'est nécessaire hormis la filtration du catalyseur.
Rendement : 97% Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) ; mélange 60/40 des 2 diastéréoisomères : 0,98 et 1 ,14 (d et d, 6,4 Hz, 3H) ; de 1 ,22 à 1 ,30 (m, 3H) ; 1 ,45 (s, 9H) ; 1 ,57 (s, 6H) ; 1 ,92 (s(large), 2H) ; de 2,48 à 2,64 (m, 1 H) ; 2,99 (m, 1 H) ; 6,81 (m, 2H) ; 7,07 (m, 2H).
Exemple 2-10 : 2-[4-(2-aminopentyl)phénoxy]-2-méthylpropanoate de terî-butyle

Cette aminé est synthétisée en 6 étapes :
Etape 1 : 2-[4-formylphénoxy]-2-méthylpropanoate de ferî-butyle
Le 4-hydroxybenzaldehyde est alkylé selon le protocole D. Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 85/15). Rendement : 49 %
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,40 (s, 9H) ; 1 ,62 (s, 6H) ; 6,89 (d, 8,5 Hz,
2H) ; 7,77 (d, 8,5 Hz, 2H) ; 9,86 (s, 1H).
Etape 2 : 2-[4-(2-(éthoxycarbonyl)pentényl)phénoxy]-2-méthylpropanoate de tert- butyle
L'intermédiaire précédent est fonctionnalisé par le phosphonopentanoate de triéthyle selon le protocole J. Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 9/1). Rendement : 81 %
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 0,99 (t, 7,3 Hz, 3H) ; 1,35 (t, 7,0 Hz1 3H) ;
1 ,44 (s, 9H) ; de 1 ,54 à 1 ,64 (m, 8H) ; de 2,48 à 2,54 (m, 2H) ; 4,26 (q, 7,0 Hz, 2H)
; 6,86 (d, 8,8 Hz, 2H) ; 7,28 (d, 8,8 Hz1 1 H) ; 7,60 (s, 1 H).
Etape 3 : 2-[4-(2-(éthoxycarbonyl)pentyl)phénoxy]-2-méthylpropanoate de tert- butyle
L'intermédiaire précédent est réduit selon le protocole E. Aucune purification n'est nécessaire hormis la filtration du catalyseur. Rendement : 95 %
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 0,89 (t, 7,3 Hz, 3H) ; 1 ,13 (t, 7,0 Hz, 3H) ; de 1,26 à 1 ,64 (m, 19H) ; de 2,56 à 2,88 (m, 3H) ; 4,04 (q, 7,0 Hz, 2H) ; 6,77 (d,
8,5 Hz, 2H) ; 7,02 (d, 8,5 Hz, 1H).
Etape 4 : 2-[4-(2-(hydroxycarbonyl)pentyl)phénoxy]-2-méthylpropanoate de tert- butyle
L'intermédiaire précédent est saponifié selon le protocole H. Aucune purification n'est nécessaire hormis les lavages (HCI 1M puis NaCIsat). Rendement : quantitatif
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 0,87 (t, 7,3 Hz, 3H) ; de 1 ,25 à 1 ,64 (m,
19H) ; de 2,58 à 2,95 (m, 3H) ; 6,79 (d, 8,5 Hz, 2H) ; 7,05 (d, 8,5 Hz, 1 H).
Etape 5 : 2-[4-(2-(benzyloxycarbonylamino)pentyl)phénoxy]-2-méthylpropanoate de fe/f-butyle
L'acide précédent est fonctionnalisé selon le protocole C. Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2).
Rendement : 46 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 0,88 (t, 6,4 Hz, 3H) ; de 1 ,27 à 1,65 (m,
19H) ; 2,73 (m, 2H) ; 3,86 (m, 1 H) ; 4,54 (d, 8,8 Hz, 1 H) ; 5,08 (s, 2H) ; 6,79 (d, 8,5
Hz, 2H) ; 7,02 (d, 8,5 Hz, 1H) ; de 7,29 à 7,40 (m, 5H).
Etape 6 : 2-[4-(2-aminopentyl)phénoxy]-2-méthylpropanoate de fe/f-butyle
L'intermédiaire précédent est réduit selon le protocole E. Aucune purification n'est nécessaire hormis la filtration du catalyseur. Rendement : 94 %
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 0,92 (t, 5,6 Hz, 3H) ; de 1,25 à 1,66 (m, 19H) ; 2,29 (s(large), 2H) ; 2,45 (m, 1H) ; 2,74 (m, 1 H) ; 2,99 (m, 1H) ; 6,81 (d, 8,5 Hz, 2H) ; 7,06 (d, 8,5 Hz, 1 H).
Exemple 2-11 : 2-[4-(1-aminopropan-2-yl)phénoxy]-2-méthylpropanoate de tert- butyle

Cette aminé est synthétisée en 6 étapes : Etape 1 : 4-(1-(éthoxycarbonyl)prop-1-èn-2-yl)phényl benzyl éther
La 4-benzyloxyacétophénone est fonctionnalisée par le 2-phosphonoacétate de triéthyle selon le protocole J. Le brut est utilisé tel quel pour l'étape suivante, sans analyses.
Etape 2 : 4-(1-(éthoxycarbonyl)propan-2-yl)phénol
L'intermédiaire précédent est réduit selon le protocole E. Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétone 9/1).
Rendement : 26 %
Aspect : huile incolore RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,20 (t, 7,1 Hz, 3H) ; 1 ,28 (d, 7,0 Hz, 3H),
2,57 (m, 2H) ; 3,21 (m, 1 H) ; 4,09 (q, 7,1 Hz, 2H) ; 5,29 (s(large), 1H) ; 6,74 (d, 8,5
Hz, 2H) ; 7,09 (d, 8,5 Hz, 2H).
Etape 3 : 2-[4-(1-(éthoxycarbonyl)propan-2-yl)phénoxy]-2-méthylpropanoate de fe/f-butyle
L'intermédiaire phénolique est alkylé selon le protocole D. Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 95/5). Rendement : 80 % Aspect : huile incolore
RMN 1H (300MHz1 CDCI3, δ en ppm) : 1 ,17 (t, 7,0 Hz, 3H) ; 1 ,27 (d, 7,0 Hz, 3H), 1,45 (s, 9H) ; 1 ,55 (s, 6H) ; 2,53 (m, 2H) ; 3,22 (m, 1H) ; 4,06 (q, 7,0 Hz, 2H) ; 6,79 (d, 8,8 Hz, 2H) ; 7,07 (d, 8,8 Hz, 2H).
Etape 4 : 2-[4-(1-(hydroxycarbonyl)propan-2-yl)phénoxy]-2-méthylpropanoate de tert-butyle
L'intermédiaire précédent est saponifié selon le protocole H. Le résidu est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 9/1). Rendement : quantitatif Aspect : huile jaune
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,29 (d, 7,0 Hz, 3H), 1 ,44 (s, 9H) ; 1 ,56 (s, 6H) ; 2,60 (m, 2H) ; 3,21 (m, 1H) ; 6,80 (d, 8,8 Hz, 2H) ; 7,08 (d, 8,8 Hz, 2H).
Etape 5 : 2-[4-(1-benzyloxycarbonylamino-propan-2-yl)phénoxy]-2- méthylpropanoate de ferf-butyle
L'acide précédent est fonctionnalisé selon le protocole C. Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2).
Rendement : 71 %
Aspect : huile incolore RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,24 (d, 7,0 Hz, 3H), 1 ,45 (s, 9H) ; 1 ,57 (s,
6H) ; 2,87 (m, 1 H) ; 3,21 (m, 1H) ; 3,46 (m, 1 H) ; 4,64 (m, 1 H) ; 5,08 (s, 2H) ; 6,81
(d, 8,8 Hz, 2H) ; 7,05 (d, 8,8 Hz, 2H) ; 7,35 (m, 5H).
Etape 6 : 2-[4-(1-aminopropan-2-yl)phénoxy]-2-méthylpropanoate de ferf-butyle L'intermédiaire précédent est réduit selon le protocole E. Aucune purification n'est nécessaire hormis la filtration du catalyseur.
Rendement : 98%
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,22 (d, 6,4 Hz1 3H), 1 ,45 (s, 9H) ; 1 ,56 (s, 6H) ; 1 ,77 (s(large, 2H) ; de 2,68 à 2,83 (m, 3H) ; 6,82 (d, 8,5 Hz, 2H) ; 7,06 (d, 8,5
Hz, 2H).
Exemple 2-12 : 2-[3-(1-aminopropan-2-yl)phénoxy]-2-méthylpropanoate de tert- butyle

Cette aminé est synthétisée en 6 étapes :
Etape 1 : 2-(3-acétylphénoxy]-2-méthylpropanoate de te/f-butyle
La 3-hydroxyacétophénone est alkylée selon le protocole D. Le brut est purifié par filtration sur gel de silice (élution par le dichlorométhane) et précipitation par l'éther de pétrole. Rendement : 81 %
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,47 (s, 9H) ; 1 ,61 (s, 6H) ; 2.58 (s, 3H) ;
7.08 (dd, 7.9 Hz, 2.0 Hz) ; 7.34 (t, 7.9 Hz, 1 H) ; 7.46 (m, 1 H), 7.57 (dd, 7.9 Hz, 2.0
Hz).
Etape 2 : 2-(3-(1-(éthoxycarbonyl)prop-1-èn-2-yl)phénoxy]-2-méthylpropanoate de te/f-butyle
L'intermédiaire précédent est fonctionnalisé par le 2-phosphonoacétate de triéthyle selon le protocole J. Le brut est utilisé tel quel pour l'étape suivante, sans analyses.
Rendement : quantitatif
Aspect : huile jaune pâle
Etape 3 : 2-[3-(1-(éthoxycarbonyl)propan-2-yl)phénoxy]-2-méthylpropanoate de te/f-butyle
L'intermédiaire précédent est réduit selon le protocole E. Le brut est utilisé tel quel pour l'étape suivante, après filtration du catalyseur et évaporation. Rendement : quantitatif Aspect : huile jaune pâle
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,20 (t, 7,1 Hz, 3H) ; 1 ,28 (d, 7,0 Hz, 3H), 1.46 (s, 9H) ; 1.57 (s, 6H) ; 2,57 (m, 2H) ; 3,21 (m, 1 H) ; 4,09 (q, 7,1 Hz, 2H) ; 6,68 (dd, 8.0 Hz, 2.0 Hz, 1 H) ; 6,76 (m, 1 H) ; 6,84 (d, 8.0 Hz, 1 H) ; 7,15 (t, 8.0 Hz, 1 H).
Etape 4 : 2-[3-(1-(hydroxycarbonyl)propan-2-yl)phénoxy]-2-méthylpropanoate de fe/f-butyle
L'intermédiaire précédent est saponifié selon le protocole H. Le résidu est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 9/1). Rendement : 17 % Aspect : solide blanc
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,29 (d, 7,0 Hz, 3H), 1 ,46 (s, 9H) ; 1 ,57 (s, 6H) ; 2,60 (m, 2H) ; 3,21 (m, 1 H) ; 6,70 (dd, 8,0 Hz, 2.0 Hz, 2H) ; 6.76 (m, 1 H) ; 6.84 (d, 8.0 Hz, 1H) ; 7.17 (t, 8.0 Hz).
Etape 5 : 2-[3-(1-benzyloxycarbonylamino-propan-2-yl)phénoxy]-2- méthylpropanoate de ferf-butyle
L'acide précédent est fonctionnalisé selon le protocole C. Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2). Rendement : 97 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,24 (d, 7,0 Hz, 3H), 1 ,45 (s, 9H) ; 1 ,58 (s, 6H) ; 2,88 (m, 1 H) ; 3,21 (m, 1H) ; 3,49 (m, 1H) ; 4,64 (m, 1H) ; 5,08 (s, 2H) ; de 6,68 à 6,74 (m, 2H) ; 6,81 (d, 7.9 Hz, 1H) ; 7.17 (t, 7.9 Hz, 1H) ; 7,35 (m, 5H).
Etape 6 : 2-[3-(1-aminopropan-2-yl)phénoxy]-2-méthylpropanoate de te/f-butyle
L'intermédiaire précédent est réduit selon le protocole E. Aucune purification n'est nécessaire hormis la filtration du catalyseur.
Rendement : 99%
Aspect : huile incolore RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,34 (d, 6,1 Hz, 3H), 1 ,46 (s, 9H) ; 1 ,55 (s,
6H) ; de 2,95 à 3,15 (m, 3H) ; 6,31 (sflarge, 2H) ; 6,70 (dd, 7.9 Hz, 2.0 Hz, 1H) ;
6,80 (m, 1 H) ; 6.87 (d, 7.9 Hz, 1 H) ; 7.19 (t, 7.9 Hz, 1H).
Exemple 3 : Synthèse des intermédiaires de type acide selon l'invention
Exemple 3-1 : acide 5-méthyl-2-octyloxybenzoïque

Cet acide est synthétisé en 2 étapes :
Etape 1 : 5-méthyl-2-octyloxybenzoate de méthyle
Le 5-méthylsalicylate de méthyle est alkylé par le 1-iodooctane selon le protocole
D. Aucune purification n'est nécessaire après les lavages. Rendement : quantitatif
Aspect : huile jaune
RMN 1H (300MHz, CDCI3, δ en ppm) : 0,89 (t, 6,9 Hz, 3H) ; 1 ,31 (m, 8H) ; 1 ,46 (m,
2H) ; 1 ,81 (m, 2H) ; 2,29 (s, 3H) ; 3,87 (s, 3H) ; 3,99 (t, 6,4 Hz, 2H) ; 6,84 (d, 8,5
Hz, 1 H) ; 7,22 (dd, 8,5 Hz et 2,3 Hz, 1 H) ; 7,59 (d, 2,3 Hz, 1 H).
Etape 2 : acide 5-méthyl-2-octyloxybenzoïque
L'intermédiaire précédent est saponifié selon le protocole H. Aucune purification n'est nécessaire après les lavages.
Rendement : 96 % Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 0,89 (t, 6,9 Hz, 3H) ; 1 ,32 (m, 8H) ; 1 ,48 (m,
2H) ; 1 ,88 (m, 2H) ; 2,34 (s, 3H) ; 4,22 (t, 6,7 Hz, 2H) ; 6,95 (d, 8,5 Hz, 1 H) ; 7,34
(dd, 8,5 Hz et 2,3 Hz, 1 H) ; 7,99 (d, 2,3 Hz, 1 H) ; 11 ,12 (s, 1 H).
Exemple 3-2 : acide 2-méthoxy-5-phénylbenzoïque

Cet acide est synthétisé en 2 étapes : Etape 1 : 2-méthoxy-5-phénylbenzoate de méthyle
Le 5-bromo-2-méthoxybenzoate de méthyle (9,0 g, 37 mmol), le bromure de n- tétrabutylammonium (44 g, 137 mmol) et le tétrakis(triphénylphosphine)palladium (0,4 g, 0,4 mmol) sont chauffés à 1200C jusqu'à ce que le milieu se liquéfie et prenne une coloration marron clair. La solution aqueuse de carbonate de potassium 2M (41 ml, 81 mmol) puis l'acide phénylboronique (5,4 g, 44 mmol) sont alors ajoutés et le milieu est agité 1 ,5 h à 1200C. Après retour à température ambiante, le milieu est extrait par de l'éther diéthylique (3 x 80 ml). La phase organique est séchée sur MgSO4 et évaporée. L'huile ainsi obtenue est purifiée par chromatographie sur gel de silice (heptane/acétate d'éthyle 9/1). Rendement : 70 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 3,89 (s, 3H) ; 3,95 (s, 3H) ; 7,06 (d, 8,8 Hz, 1 H) ; 7,36 (m, 1H) ; 7,43 (m, 2H) ; 7,59 (m, 2H) ; 7,71 (dd, 8,8 Hz et 2,3 Hz, 1 H) ; 8,07 (d, 2,3 Hz, 1 H).
Etape 2 : acide 2-méthoxy-5-phénylbenzoïque
L'intermédiaire précédent est saponifié selon le protocole H. Aucune purification n'est nécessaire après les lavages.
Rendement : 75 % Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 4,15 (s, 3H) ; 7,16 (d, 8,5 Hz, 1 H) ; de 7,36 à 7,49 (m, 3H) ; 7,61 (m, 2H) ; 7,82 (dd, 8,5 Hz et 2,6 Hz, 1 H) ; 8,47 (d, 2,6 Hz, 1 H) ; 10,81 (s(large), 1H).
Exemple 3-3 : acide 5-butanoyl-2-méthoxybenzoïque

Cet acide est synthétisé en 3 étapes : Etape 1 : 5-butanoyl-2-hydroxybenzoate de méthyle
Le chlorure d'aluminium (28,0 g, 210 mmol) est mis en suspension dans le dichlorométhane (500 ml). Le salicylate de méthyle (10,0 g, 65,7 mmol) est additionné et la solution est refroidie à O0C. Le chlorure de butanoyle est
additionné goutte à goutte au mélange et le milieu est agité pendant 16 heures à température ambiante. Le brut est neutralisé par adjonction de glace et l'attendu est extrait par du dichlorométhane (200 ml). Le brut est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 9/1). Rendement : 48 %
Aspect : poudre blanche
RMN 1 H (300MHz, CDCI3, δ en ppm) : 1 ,01 (t, 7,3 Hz, 3H) ; 1 ,76 (m, 2H) ; 2,90 (t,
7,3 Hz, 2H) ; 4,00 (s, 3H) ; 7,03 (d, 8,6 Hz, 1H) ; 8,09 (dd, 8,6 Hz et 2,3 Hz, 1H) ;
8,49 (d, 2,3 Hz, 1H) ; 11 ,21 (s, 1H).
Etape 2 : 5-butanoyl-2-méthoxybenzoate de méthyle
L'intermédiaire précédent est alkylé par l'iodure de méthyle selon le protocole D.
Le brut est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 9/1). Rendement : 96 %
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1,00 (t, 7,6 Hz, 3H) ; 1,76 (m, 2H) ; 2,92 (t,
7,3 Hz, 2H) ; 3,92 (s, 3H) ; 3,97 (s, 3H) ; 7,03 (d, 8,8 Hz, 1 H) ; 8,11 (dd, 8,8 Hz et
2,3 Hz, 1 H) ; 8,40 (d, 2,3 Hz, 1 H).
Etape 3 : acide 5-butanoyl-2-méthoxybenzoïque
L'intermédiaire précédent saponifié selon le protocole H. Aucune purification n'est nécessaire après les lavages.
Rendement : 93 % Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,02 (t, 7,6 Hz, 3H) ; 1 ,78 (m, 2H) ; 2,98 (t,
7,3 Hz, 2H) ; 4,17 (s, 3H) ; 7,16 (d, 8,8 Hz, 1H) ; 8,26 (dd, 8,8 Hz et 2,3 Hz, 1 H) ;
8,76 (d, 2,3 Hz, 1H).
Exemple 3-4 : acide 5-butyl-2-méthoxybenzoïque

RMN 1H (300MHz, CDCI3, δ en ppm) : 0,93 (t, 7,3 Hz, 3H) ; 1 ,34 (m, 2H) ; 1 ,59 (m, 2H) ; 2,61 (t, 7,6 Hz, 2H) ; 4,07 (s, 3H) ; 6,99 (d, 8,5 Hz, 1H) ; 7,38 (dd, 8,5 Hz et 2,3 Hz, 1 H) ; 8,01 (d, 2,3 Hz, 1 H).
Exemple 3-5 : acide 5-(2,4-difluorophényl)-2-méthoxybenzoïque
Cet acide est synthétisé en 2 étapes :
Etape 1 : 5-(2,4-difluorophényl)-2-méthoxybenzoate de méthyle
L'acide 5-(2,4-difluorophényl)-2-hydroxybenzoïque (5,0 g, 20 mmol) est solubilisé dans l'acétonitrile (100 ml). Le carbonate de potassium est ajouté (13,9 g, 100 mmol) et le milieu est chauffé à 60 0C. L'iodure de méthyle est additionné (3,0 ml, 48 mmol) et le milieu est agité vigoureusement pendant 16 heures. Le milieu est refroidi, repris par l'eau (50 ml) et extrait par l'acétate d'éthyle (160 ml). La phase organique est lavée par NaCIsat, séchée sur sulfate de magnésium (MgSO4), filtrée et évaporée. Il est utilisé tel quel pour l'étape suivante. Rendement : quantitatif Aspect : solide jaune pâle
RMN 1H (300MHz, CDCI3, δ en ppm) : 3,94 (s, 3H) ; 3,97 (s, 3H) ; de 6,85 à 7,00 (m, 2H) ; 7,06 (d, 8,8 Hz, 1 H) ; 7,40 (m, 1 H) ; 7,64 (d, 8,8 Hz, 1 H) ; 7,95 (s, 1 H).
Etape 2 : acide 5-(2,4-difluorophényl)-2-méthoxybenzoïque
L'intermédiaire précédent saponifié selon le protocole H. Aucune purification n'est nécessaire après les lavages.
Rendement : 87 %
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 4,15 (s, 3H) ; de 6,90 à 7,05 (m, 2H) ; 7,16
(d, 8,8 Hz, 1 H) ; 7,43 (m, 1 H) ; 7,76 (d, 8,8 Hz, 1 H) ; 8,33 (s, 1 H) ; 10,76 (s(large),
1 H).
Exemple 3-6 : acide 5-(pyrrol-1-yl)-2-méthoxybenzoïque

Cet acide est synthétisé en 2 étapes :
Etape 1 : 5-(pyrrol-1-yl)-2-méthoxybenzoate de méthyle
L'acide 5-(pyrrol-1-yl)-2-hydroxybenzoïque (2,0 g, 9,8 mmol) est solubilisé dans l'acétonitrile (100 ml). Le carbonate de potassium est ajouté (4,4 g, 31 mmol) et le milieu est chauffé à 60 0C. L'iodure de méthyle est additionné (1 ,5 ml, 24 mmol) et le milieu est agité vigoureusement pendant 3 jours. Le milieu est refroidi, repris par l'eau (50 ml) et extrait par l'acétate d'éthyle (160 ml). La phase organique est lavée par NaCIsat, séchée sur sulfate de magnésium (MgSO4), filtrée et évaporée. Il est utilisé tel quel pour l'étape suivante. Rendement : quantitatif
Aspect : solide blanc
RMN 1H (300MHz, CDCI3, δ en ppm) : 3,94 (s, 3H) ; 3,96 (s, 3H) ; 6,36 (m, 2H) ;
7,04 (m, 3H) ; 7,51 (dd, 9,1 Hz1 2,6 Hz, 1 H) ; 7,85 (d, 2,6 Hz, 1 H).
Etape 2 : acide 5-(pyrrol-1-yl)-2-méthoxybenzoïque
L'intermédiaire précédent saponifié selon le protocole H. Aucune purification n'est nécessaire après les lavages. Rendement : 79 % Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 4,14 (s, 3H) ; 6,38 (m, 2H) ; 7,09 (m, 2H) ; 7,15 (d, 9,1 Hz, 1 H) ; 7,61 (dd, 9,1 Hz, 2,6 Hz, 1 H) ; 8,25 (d, 2,6 Hz, 1 H) ; 10,78 (s, 1 H).
Exemple 4 : Synthèse des intermédiaires de formule (VIII) selon l'invention
Exemple 4-1 : 4-[2-(5-chloro-2-méthoxybenzamido)propyl]phénol

Rendement : 82 %
Aspect : huile incolore RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,22 (d, 6,6 Hz, 3H) ; de 2,74 à 2,89 (m,
2H) ; 3,88 (s, 3H) ; 4,45 (m, 1 H) ; 5,45 (s, 1 H) ; 6,79 (d, 8,3 Hz, 2H) ; 6,89 (d, 8,8
Hz, 1 H) ; 7,09 (d, 8,3 Hz, 2H) ; 7,38 (dd, 8,8 Hz et 2,8 Hz, 1 H) ; 7,74 (d, 7,4 Hz,
1 H) ; 8,17 (d, 2,8 Hz, 1H).
Exemple 4-2 : 4-[2-(5-chloro-2-méthoxybenzamido)-2-méthylpropyl]phénol
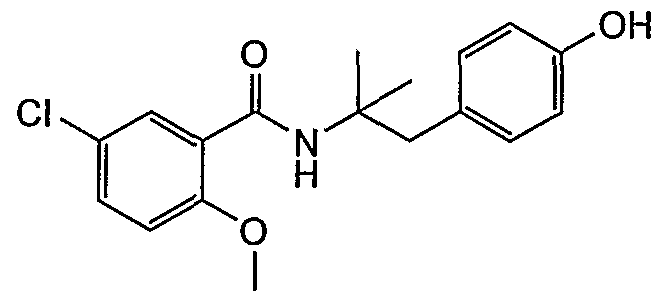
Cet intermédiaire est obtenu par condensation entre l'aminé 2-3 et l'acide 5- chloro-2-méthoxybenzoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 95/5). Rendement : 81 % Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,43 (s, 6H) ; 3,08 (s, 2H) ; 3,80 (s, 3H) ; 5,18 (s(large), 1H) ; 6,74 (d, 8,5 Hz, 2H) ; 6,87 (d, 9,1 Hz, 1H) ; 7,03 (d, 8,5 Hz, 2H) ; 7,37 (dd, 9, 1 Hz et 2,6 Hz, 1 H) ; 7,50 (s, 1 H) ; 8, 15 (d, 2,6 Hz, 1 H).
Exemple 4-3 : 4-[1-(5-chloro-2-méthoxybenzamido)-2-méthylpropan-2-yl]phénol

Cet intermédiaire est obtenu par condensation entre l'aminé 2-4 et l'acide 5- chloro-2-méthoxybenzoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 98/2). Rendement : 68 % Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 1 ,25 (s, 6H) ; 3,46 (d, 5,9 Hz, 2H) ; 3,68 (s, 3H) ; 6,75 (d, 8,5 Hz, 2H) ; 7,12 (d, 9,1 Hz, 1 H) ; 7,22 (d, 8,5 Hz, 2H) ; 7,50 (dd, 9,1 Hz et 2,9 Hz, 1 H) ; 7,70 (d, 2,9 Hz, 1 H) ; 7,78 (t, 5,9 Hz, 1 H) ; 9,27 (s, 1 H).
Exemple 4-4 : 4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénol

Cet intermédiaire est obtenu par condensation entre l'aminé 2-5 et l'acide 5- chloro-2-méthoxybenzoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 95/5). Rendement : 65 % Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 1 ,19 (d, 7,0 Hz, 3H) ; 2,89 (m, 1 H) ; 3,31 (m, 1 H) ; 3,44 (m, 1H) ; 3,75 (s, 3H) ; 6,71 (d, 8,5 Hz, 2H) ; 7,07 (d, 8,5 Hz, 2H) ; 7,13 (d, 9,1 Hz, 1 H) ; 7,49 (dd, 9,1 Hz et 2,6 Hz, 1 H) ; 7,62 (d, 2,6 Hz, 1 H) ; 8,04 (m, 1H) , 9,22 (s, 1 H).
Exemple 4-5 : 4-[1-(5-chloro-2-méthoxybenzamido)hexan-2-yl]phénol

Cet intermédiaire est obtenu par condensation entre l'aminé 2-7 et l'acide 5- chloro-2-méthoxybenzoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 95/5). Rendement : 33 % Aspect : poudre blanche
RMN 1H (300MHz, DMSO-tf6, δ en ppm) : 0,80 (t, 7,3 Hz, 3H) ; de 1 ,02 à 1 ,65 (m, 6H) ; 2,72 (m, 1 H) ; 3,27 (m, 1 H) ; 3,53 (m, 1H) ; 3,72 (s, 3H) ; 6,72 (d, 8,5 Hz, 2H) ; 7,03 (d, 8,5 Hz, 2H) ; 7,11 (d, 9,1 Hz, 1 H) ; 7,48 (dd, 9,1 Hz et 2,9 Hz, 1 H) ; 7,62 (d, 2,9 Hz, 1H) ; 7,96 (t, 5,4 Hz, 1H) , 9,23 (s, 1H).
Exemple 4-6 : 4-[2-(2-méthoxy-5-méthylbenzamido)éthyl]phénol

Rendement : 68 %
Aspect : poudre blanche RMN 1H (300MHz, CDCI3, δ en ppm) : 2,32 (s, 3H) ; 2,85 (t, 6,9 Hz, 2H) ; de 3,69 à 3,77 (m, 5H) ; 6,67 (s(large), 1 H) ; de 6,81 à 6,87 (m, 3H) ; 7,09 (d, 8,5 Hz, 2H) ;
7,23 (dd, 8,5 Hz, 1 ,7 Hz, 1H) ; de 8,01 à 8,07 (m, 2H).
Exemple 4-7 : 4-[2-(5-méthyl-2-octyloxybenzamido)éthyl]phénol

RMN 1H (300MHz, CDCI3, δ en ppm) : 0,89 (t, 6,7 Hz, 3H) ; de 1 ,29 à 1 ,43 (m, 10H) ; 1 ,68 (m, 2H) ; 2,32 (s, 3H) ; 2,85 (t, 7,3 Hz, 2H) ; 3,71 (m, 2H) ; 4,01 (t, 6,7 Hz, 2H) ; 6,83 (m, 3H) ; 7,06 (d, 8,5 Hz, 2H) ; 7,21 (dd, 8,5 Hz, 2,3 Hz, 1H) ; 8,05 (d, 2,3 Hz, 1H) ; 8,28 (t, 5,5 Hz, 1 H).
Exemple 4-8 : 4-[2-(2-méthoxy-5-phénylbenzamido)éthyl]phénol
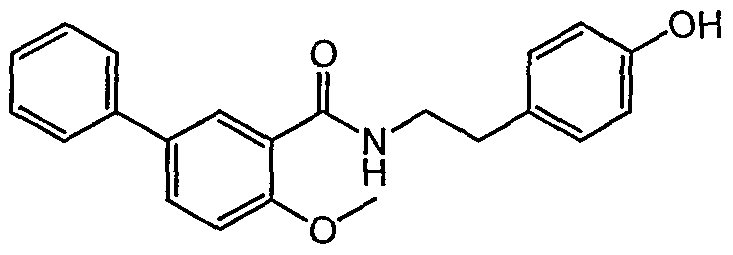
Cet intermédiaire est obtenu par condensation entre la tyramine et l'acide 3-2 selon le protocole I. Il est purifié par chromatographie sur gel de silice (heptane/acétate d'éthyle 55/45) puis recristallisé dans le toluène.
Rendement : 46 %
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 2,89 (t, 6,8 Hz, 2H) ; 3,77 (m, 2H) ; 3,85 (s,
3H) ; 5,19 (s(large), 1H) ; 6,84 (d, 8,5 Hz, 2H) ; 7,02 (d, 8,5 Hz, 1 H) ; 7,16 (d, 8,5 Hz, 2H) ; 7,36 (m, 1H) ; 7,43 (m, 2H) ; 7,62 (m, 2H) ; 7,68 (dd, 8,5 Hz et 2,3 Hz,
1H) ; 7,97 (m, 1 H) ; 8,51 (d, 2,3 Hz, 1H).
Exemple 4-9 : 4-[2-((3-méthoxy-2-naphtoyl)amino)propyl]phénol

RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,26 (d, 6,4 Hz, 3H) ; de 2,75 à 2,90 (m, 2H) ; 3,94 (s, 3H) ; 4,52 (m, 1H) ; 5,31 (s, 1 H) ; 6,84 (d, 8,2 Hz, 2H) ; 7,08 (d, 8,2 Hz, 2H) ; 7,16 (s, 1H) ; 7,37 (m, 1H) ; 7,51 (m, 1H) ; 7,72 (d, 8,2 Hz, 1 H) ; 7,86 (d, 8,2 Hz, 1 H) ; 8,02 (d, 7,9 Hz, 1H) ; 8,73 (s, 1H).
Exemple 4-10 : 4-[2-(2,5-diméthoxybenzamido)propyl]phénol
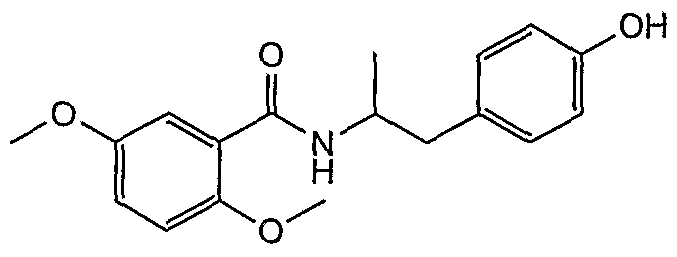
Cet intermédiaire est obtenu par condensation entre l'aminé 2-1 et l'acide 2,5- diméthoxybenzoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 9/1).
Rendement : 91 %
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,23 (d, 6,5 Hz, 3H) ; de 2,72 à 2,87 (m, 2H) ; 3,79 (s, 3H) ; 3,84 (s, 3H) ; 4,45 (m, 1 H) ; 6,72 (s, 1 H) ; 6,78 (d, 8,5 Hz, 2H) ;
6,90 (d, 8,8 Hz, 1 H) ; 6,99 (dd, 8,8 Hz et 3,2 Hz, 1H) ; 7,06 (d, 8,5 Hz, 2H) ; 7,74
(d, 3,2 Hz, 1 H) ; 8,03 (d, 7,9 Hz, 1 H).
Exemple 4-11 : 4-[2-(5-tertiobutyl-2-méthoxybenzamido)propyl]phénol

Cet intermédiaire est obtenu par condensation entre l'aminé 2-1 et l'acide 5- tertiobutyl-2-méthoxybenzoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 9/1).
Rendement : 54 %
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,22 (d, 6,7 Hz, 3H) ; 1 ,31 (s, 9H) ; de 2,71 à 2,88 (m, 2H) ; 3,86 (s, 3H) ; 4,45 (m, 1 H) ; 6,78 (d, 8,5 Hz, 2H) ; 6,89 (d, 8,8 Hz,
1 H) ; 6,91 (s, 1 H) ; 7,06 (d, 8,5 Hz, 2H) ; 7,45 (dd, 8,8 Hz, 2,6 Hz, 1 H) ; 7,95 (d, 7,9 Hz, 1 H) ; 8,25 (d, 2,6 Hz, 1 H).
Exemple 4-12 : 4-[2-(5-butyryl-2-méthoxybenzamido)propyl]phénol

Cet intermédiaire est obtenu par condensation entre l'aminé 2-1 et l'acide 3-3 selon le protocole F. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 8/2). Rendement : 71 % Aspect : solide blanc
RMN 1H (300MHz, CDCI3, δ en ppm) : 0,99 (t, 7,3 Hz, 3H) ; 1,25 (d, 6,7 Hz1 3H) ; de 2,75 à 2,89 (m, 2H) ; 2,97 (t, 7,3 Hz, 2H) ; 3,96 (s, 3H) ; de 4,43 à 4,52 (m, 1H) ; 6,24 (s, 1H) ; 6,79 (d, 8,5 Hz1 2H) ; 7,03 (d, 8,8 Hz, 1 H) ; 7,08 (d, 8,5 Hz1 2H) ; 7,72 (d, 7,6 Hz, 1H) ; 8,12 (dd, 8,8 Hz, 2,6 Hz, 1H) ; 8,79 (d, 2,6 Hz, 1H).
Exemple 4-13 : 4-[2-(5-phényl-2-méthoxybenzamido)propyl]phénol

Cet intermédiaire est obtenu par condensation entre l'aminé 2-1 et l'acide 3-2 selon le protocole F. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 8/2). Rendement : quantitatif Aspect : solide blanc
RMN 1H (300MHz, CDCI3, δ en ppm) : 1,24 (d, 6,4 Hz, 3H) ; de 2,74 à 2,88 (m, 2H) ; 3,89 (s, 3H) ; 4,45 (m, 1H) ; 6,82 (d, 8,5 Hz, 2H) ; 7,00 (d, 8,8 Hz, 1H) ; 7,07 (d, 8,5 Hz, 2H) ; de 7,29 à 7,44 (m, 3H) ; 7,59 (d, 7,0 Hz, 2H) ; 7,66 (dd, 8,8 Hz, 2,3 Hz, 1 H) ; 7,92 (d, 7,9 Hz, 1 H) ; 8,46 (d, 2,3 Hz, 1H).
Exemple 4-13(S) : (S)-4-[2-(5-phényl-2-méthoxybenzamido)propyl]phénol

Cet intermédiaire est obtenu par condensation entre l'aminé 2-1(S) et l'acide 3-2 selon le protocole F. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 8/2). Rendement : 91 % Aspect : solide blanc RMN 1H (300MHz, CDCI3, δ en ppm) : conforme avec le spectre obtenu pour 4-13.
Exemple 4-13(R) : (R)-4-[2-(5-phényl-2-méthoxybenzamido)propyl]phénol

Cet intermédiaire est obtenu suivant la même procédure à partir de l'aminé 2-1(R).
Exemple 4-14 : 4-[2-(5-(2,4-difluorophényl)-2-méthoxybenzamido)propyl]phénol

(dichlorométhane/méthanol 98/2).
Rendement : quantitatif
Aspect : huile incolore RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,24 (d, 6,4 Hz, 3H) ; 2,82 (m, 2H) ; 3,92 (s,
3H) ; 4,48 (m, 1H) ; 6,55 (s, 1 H) ; 6,78 (d, 8,2 Hz, 2H) ; de 6,85 à 6,95 (m, 2H) ;
7,02 (d, 8,5 Hz, 1H) ; 7,07 (d, 8,2 Hz, 2H) ; 7,40 (m, 1 H) ; 7,60 (dd, 8,5 Hz, 2,1 Hz,
2H) ; 7,86 (d, 7,9 Hz, 1 H) ; 8,32 (d, 2,1 Hz, 1H).
Exemple 4-15 : 4-[2-(5-(pyrrol-1-yl)-2-méthoxybenzamido)propyl]phénol

Cet intermédiaire est obtenu par condensation entre l'aminé 2-1 et l'acide 3-6 selon le protocole F. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 98/2). Rendement : quantitatif Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,24 (d, 6,7 Hz, 3H) ; 2,83 (m, 2H) ; 3,91 (s, 3H) ; 4,46 (m, 1H) ; 6,34 (m, 2H) ; 6,80 (d, 8,5 Hz, 2H) ; 7,00 (d, 9,1 Hz, 1 H) ; de 7,05 à 7,20 (m, 4H) ; 7,45 (dd, 9,1 Hz, 2,9 Hz, 1H) ; 7,88 (d, 7,9 Hz, 1 H) ; 8,25 (d, 2,9 Hz, 1H).
Exemple 5 : Synthèse des intermédiaires de formule (IX) selon l'invention
Exemple 5-1 : 2-[4-[2-(2-fluorobenzamido)propyl]phénoxy]-2-méthylpropanoate d'éthyle

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 4-[2-(2-fluorobenzamido)propyl]phénol
Cet intermédiaire est obtenu par condensation entre l'aminé 2-1 et l'acide 2- fluorobenzoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 7/3).
Rendement : 46 %
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,24 (d, 6,7 Hz, 3H) ; de 2,73 à 2,92 (m,
2H) ; 4,47 (m, 1 H) ; 5,57 (s, 1H) ; 6,66 (m, 1H) ; 6,78 (d, 8,5 Hz, 2H) ; 7,11 (m,
3H) ; 7,26 (m, 1 H) ; de 7,43 à 7,51 (m, 1 H) ; 8,08 (m, 1 H).
Etape 2 : 2-[4-[2-(2-fluorobenzamido)propyl]phénoxy]-2-méthylpropanoate d'éthyle Cet intermédiaire est obtenu par substitution du bromoisobutyrate d'éthyle par le dérivé phénolique précédent selon le protocole D. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 7/3). Rendement : 64 % Aspect : huile incolore
RMN 1H (300MHz1 CDCI3, δ en ppm) : 1 ,23 (m, 6H) ; 1,59 (s, 6H) ; de 2,74 à 2,93 (m, 2H) ; 4,23 (q, 7,0 Hz, 2H) ; 4,45 (m, 1H) ; 6,59 (m, 1 H) ; 6,79 (d, 8,5 Hz, 2H) ; 7,09 (m, 3H) ; 7,26 (m, 1H) ; 7,46 (m, 1 H) ; 8,08 (m, 1 H).
Exemple 5-2 : 2-[4-[1-((3-hydroxy-2-naphtoyl)amino)propan-2-yl]phénoxy]-2- méthylpropanoate d'éthyle

Exemple 5-3 : 2-[4-[2-(2-fluoro-5-trifluorométhylbenzamido)propyl]phénoxy]-2- méthylpropanoate d'éthyle

RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,25 (d, 6,4 Hz, 3H) ; de 2,75 à 2,90 (m, 2H) ; 4,46 (m, 1 H) ; 6,63 (m, 1H) ; 6,78 (d, 8,5 Hz, 2H) ; 7,07 (d, 8,5 Hz, 2H) ; 7,23 (m, 1 H) ; 7,73 (m, 1 H) ; 8,37 (dd, 7,0 Hz et 2,3 Hz, 1 H).
Etape 2 : 2-[4-[2-(2-fluoro-5-trifluorométhylbenzamido)propyl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par substitution du bromoisobutyrate d'éthyle par le dérivé phénolique précédent selon le protocole D. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2).
Rendement : 49 %
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : de 1 ,21 à 1 ,26 (m, 6H) ; 1 ,59 (s, 6H) ; de 2,76 à 2,92 (m, 2H) ; 4,23 (q, 7,3 Hz, 2H) ; 4,46 (m, 1 H) ; 6,55 (m, 1 H) ; 6,81 (d,
8,5 Hz, 2H) ; 7,10 (d, 8,5 Hz, 2H) ; 7,23 (m, 1H) ; 7,72 (m, 1H) ; 8,39 (dd, 7,0 Hz et
2,3 Hz, 1 H).
Exemple 5-4 : 2-[4-[1-((2-hydroxy-1-naphtoyl)amino)propan-2-yl]phénoxy]-2- méthylpropanoate d'éthyle

Cet intermédiaire est obtenu par condensation entre l'aminé 2-6 et l'acide 2- hydroxy-1-naphtoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2).
Rendement : 44 %
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,21 (t, 7,3 Hz, 3H) ; 1,37 (d, 6,7 Hz, 3H) ;
1 ,60 (s, 6H) ; 3,14 (m, 1H) ; 3,53 (m, 1H) ; 3,94 (m, 1H) ; 4,21 (q, 7,3 Hz, 2H) ;
6,24 (m, 1H) ; 6,86 (d, 8,8 Hz1 2H) ; de 7,13 à 7,21 (m, 3H) ; de 7,26 à 7,33 (m,
2H) ; 7,58 (m, 1 H) ; de 7,73 à 7,79 (m, 2H).
Exemple 5-5 : 2-[4-[1-((3-hydroxy-7-méthoxy-2-naphtoyl)amino)propan-2- yl]phénoxy]-2-méthylpropanoate de terf-butyle

Cet intermédiaire est obtenu par condensation entre l'aminé 2-11 et l'acide 3- hydroxy-7-méthoxy-2-naphtoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 9/1). Rendement : 27 % Aspect : poudre jaune pâle
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,36 (d, 7,0 Hz, 3H) ; 1 ,43 (s, 9H) ; 1 ,58 (s, 6H) ; 3,07 (m, 1 H) ; 3,39 (m, 1H) ; de 3,83 à 3,91 (m, 4H) ; 6,24 (s, 1H) ; 6,41 (m, 1H) ; 6,89 (d, 8,8 Hz, 2H) ; 6,98 (d, 2,3 Hz, 1 H) ; 7,15 (m, 3H) ; 7,26 (s, 1H) ; de 7,56 à 7,59 (m, 2H).
Exemple 5-6 : 2-[4-[1-(5-(2,4-difluorophényl)-2-hydroxybenzamido)propan-2- yl]phénoxy]propanoate de fert-butyle

Cet intermédiaire est obtenu par condensation entre l'aminé 2-11 et l'acide 5-(2,4- difluorophényl)-2-hydroxybenzoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2).
Rendement : 70 %
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1,33 (d, 7,0 Hz, 3H) ; 1,42 (s, 9H) ; 1,55 (s,
6H) ; 3,01 (m, 1H) ; 3,35 (m, 1H) ; 3,80 (m, 1 H) ; 6,24 (s(large), 1 H) ; 6,84 (d, 8,5
Hz, 2H) ; de 6,90 à 7,00 (m, 2H) ; 7,04 (d, 8,8 Hz, 1H) ; 7,12 (d, 8,5 Hz, 2H) ; de
7,20 à 7,35 (m, 2H) ; 7,50 (d, 8,8 Hz, 1 H).
Exemple 6 : Synthèse des composés selon l'invention
Composé 1 : Acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)propyl]phénoxy]- 2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes : Etape 1 : 2-[4-[2-(5-chloro-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par substitution du bromoisobutyrate d'éthyle par le dérivé phénolique 4-1 selon le protocole D. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 6/4). Rendement : 80 %
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : de 1 ,19 à 1,27 (m, 6H) ; 1 ,59 (s, 6H) ; de
2,75 à 2,90 (m, 2H) ; 3,86 (s, 3H) ; 4,24 (q, 7,0 Hz, 2H) ; de 4,38 à 4,51 (m, 1 H) ;
6,80 (d, 8,5 Hz, 2H) ; 6,89 (d, 8,8 Hz, 1H) ; 7,11 (d, 8,5 Hz, 2H) ; 7,37 (dd, 8,8 Hz et 2,8 Hz, 1H) ; 7,68 (d, 7,6 Hz, 1H) ; 8,17 (d, 2,8 Hz, 1H).
Etape 2 : acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 9/1). Rendement : 48 % Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 1 ,10 (d, 6,4 Hz, 3H) ; 1 ,43 (s, 6H) ; de 2,64 à 2,80 (m, 2H) ; 3,82 (s, 3H) ; 4,13 (m, 1H) ; 6,76 (d, 8,5 Hz, 2H) ; de 7,07 à 7,15 (m, 3H) ; de 7,47 à 7,54 (m, 2H) ; 8,02 (d, 7,6 Hz, 1H).
Composé 2 : Acide 2-[4-[2-((3-méthoxy-2-naphtoyl)amino)éthyI]phénoxy]-2- méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[2-((3-méthoxy-2-naphtoyl)amino)éthyl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par condensation entre l'aminé 2-2 et l'acide 3- méthoxy-2-naphthoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2).
Rendement : 27 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,22 (t, 7,0 Hz, 3H) ; 1 ,58 (s, 6H) ; 2,86 (t,
6,7 Hz, 2H) ; de 3,71 à 3,80 (m, 5H) ; 4,21 (q, 7,0 Hz, 2H) ; 6,83 (d, 8,5 Hz, 2H) ; de 7,09 à 7,15 (m, 3H) ; 7,33 (m, 1H) ; 7,45 (m, 1H) ; 7,67 (d, 8,2Hz, 1 H) ; 7,84 (d,
7,9 Hz, 1 H) ; 7,99 (m, 1H) ; 8,73 (s, 1 H).
Etape 2 : acide 2-[4-[2-((3-méthoxy-2-naphtoyl)amino)éthyl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5).
Rendement : 49 %
Aspect : poudre blanche
RMN 1H (300MHz1 CDCI3, δ en ppm) : 1 ,63 (s, 6H) ; 2,89 (t, 7,0 Hz, 2H) ; 3,76 (m,
2H) ; 3,86 (s, 3H) ; 6,92 (d, 8,5 Hz, 2H) ; 7,14 (m, 3H) ; 7,40 (m, 1H) ; 7,52 (m,
1 H) ; 7,70 (d, 8,2Hz, 1 H) ; 7,90 (d, 8,2 Hz, 1H) ; 8,09 (m, 1H) ; 8,77 (s, 1H).
Composé 3 : Acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)-2- méthylpropyI]phénoxy]-2-méthylpropanoïque

(dichlorométhane/méthanol 95/5).
Rendement : 41 %
Aspect : poudre blanche RMN 1H (300MHz, DMSO-CZ6, δ en ppm) : 1 ,30 (s, 6H) ; 1 ,46 (s, 6H) ; 2,97 (s, 2H) ;
3,80 (s, 3H) ; 6,73 (d, 8,5 Hz, 2H) ; 7,06 (d, 8,5 Hz, 2H) ; 7,13 (d, 9,1 Hz, 1H) ;
7,47 (dd, 9,1 Hz et 2,6 Hz, 1 H) ; 7,56 (d, 2,6 Hz, 1H) ; 7,66 (s, 1H).
Composé 4 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)-2-méthylpropan- 2-yl]phénoxy]-2-méthylpropanoïque

Ce composé est obtenu par fonctionnalisation du dérivé phénolique 4-3 selon le protocole K. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5). Rendement : 28 % Aspect : poudre blanche
RMN 1H (300MHz, DMSO-cfe, δ en ppm) : 1 ,26 (s, 6H) ; 1,48 (s, 6H) ; 3,49 (d, 5,9 Hz, 2H) ; 3,66 (s, 3H) ; 6,81 (d, 8,8 Hz, 2H) ; 7,11 (d, 9,1 Hz, 1 H) ; 7,29 (d, 8,8 Hz, 2H) ; 7,49 (dd, 9,1 Hz et 2,6 Hz, 1H) ; 7,70 (d, 2,6 Hz, 1H) ; 7,79 (t, 5,9 Hz, 1H).
Composé 5 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanoïque

Ce composé est obtenu par fonction nalisation du dérivé phénolique 4-4 selon le protocole K. Il est purifié par chromatographie sur gel de silice
(dichlorométhane/méthanol 96/4).
Rendement : 18 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-cfe, δ en ppm) : 1 ,19 (d, 7,0 Hz, 3H) ; 1 ,49 (s, 6H) ; 2,94 (m, 1H) ; de 3,29 à 3,52 (m, 2H) ; 3,73 (s, 3H) ; 6,79 (d, 8,8 Hz, 2H) ; 7,15 (m,
3H) ; 7,49 (dd, 9,1 Hz et 2,6 Hz, 1H) ; 7,62 (d, 2,6 Hz, 1 H) ; 8,07 (t, 5,3 Hz1 1H) ,
13,03 (s, 1 H).
Composé 6 : Acide 2-[4-[1-((3-méthoxy-2-naphtoyl)amino)propan-2- yl]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[1-((3-méthoxy-2-naphtoyl)amino)propan-2-yl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par substitution de l'iodure de méthyle par le dérivé phénoϋque 5-2 selon le protocole D (en utilisant le carbonate de césium). Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 75/25). Rendement : 97 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,27 (t, 7,0 Hz, 3H) ; 1 ,34 (d, 6,7 Hz, 3H) ; 1 ,61 (s, 6H) ; de 3,01 à 3,11 (m, 1H) ; de 3,43 à 3,54 (m, 1H) ; 3,79 (s, 3H) ; de 3,89 à 3,98 (m, 1H) ; 4,25 (q, 7,0 Hz, 2H) ; 6,87 (d, 8,5 Hz, 2H) ; 7,17 (m, 3H) ; 7,39 (m, 1H) ; 7,51 (m, 1H) ; 7,73 (d, 8,2 Hz, 1 H) ; 7,92 (m, 2H) ; 8,75 (s, 1H).
Etape 2 : acide 2-[4-[1-((3-méthoxy-2-naphtoyl)amino)propan-2-yI]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par chromatographie sur gel de silice
(dichlorométhane/méthanol 95/5).
Rendement : 86 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-ofe, δ en ppm) : 1 ,23 (d, 6,7 Hz, 3H) ; 1 ,50 (s, 6H) ; 2,99
(m, 1H) ; 3,38 (m, 1H) ; 3,52 (m, 1H) ; 3,83 (s, 3H) ; 6,81 (d, 8,5 Hz, 2H) ; 7,21 (d,
8,5 Hz1 2H) ; 7,39 (m, 2H) ; 7,52 (m, 1 H) ; 7,84 (d, 8,2 Hz, 1 H) ; 7,92 (d, 8,2 Hz,
1H) ; de 8,16 à 8,21 (m, 2H) ; 13,07 (s(large), 1H).
Composé 7 : Acide 2-[4-[1-((3-isobutoxy-2-naphtoyl)amino)propan-2- yl]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[1 -((3-isobutoxy-2-naphtoyl)amino)propan-2-yl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par substitution du bromure d'isobutyle par le dérivé phénolique 5-2 selon le protocole D (en utilisant le carbonate de césium). Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2). Rendement : 88 % Aspect : huile incolore
RMN 1H (300MHz1 CDCI3, δ en ppm) : 1 ,00 (d, 6,7 Hz, 6H) ; 1,24 (t, 7,3 Hz, 3H) ; 1,33 (d, 7,0 Hz, 3H) ; 1 ,59 (s, 6H) ; de 1 ,92 à 2,05 (m, 1 H) ; de 3,01 à 3,13 (m, 1 H) ; de 3,53 à 3,62 (m, 1 H) ; de 3,75 à 3,82 (m, 1 H) ; 3,89 (d, 6,7 Hz, 2H) ; 4,23 (q, 7,3 Hz1 2H) ; 6,81 (d, 8,5 Hz, 2H) ; 7,15 (m, 3H) ; 7,39 (m, 1 H) ; 7,51 (m, 1 H) ; 7,71 (d, 8,2 Hz, 1 H) ; 7,91 (d, 8,2 Hz, 1 H) ; 8,13 (m, 1H) ; 8,78 (s, 1H).
Etape 2 : acide 2-[4-[1 -((3-isobutoxy-2-naphtoyl)amino)propan-2-yl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5). Rendement : 82 % Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 0,95 (d, 6,4 Hz, 6H) ; 1,22 (d, 6,7 Hz, 3H) ; 1 ,49 (s, 6H) ; 1 ,99 (m, 1H) ; 2,97 (m, 1H) ; de 3,38 à 3,53 (m, 2H) ; 3,90 (d, 6,4 Hz, 2H) ; 6,78 (d, 8,5 Hz, 2H) ; 7,17 (d, 8,5 Hz, 2H) ; 7,39 (m, 2H) ; 7,52 (m, 1H) ; 7,83 (d, 8,2 Hz, 1H) ; 7,92 (d, 7,9 Hz, 1H) ; 8,23 (m, 2H) ; 13,05 (s(large), 1H).
Composé 8 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)hexan-2- yl]phénoxy]-2-méthylpropanoïque

RMN 1H (300MHz, DMSO-d6l δ en ppm) : 0,80 (t, 7,3 Hz, 3H) ; de 1 ,02 à 1 ,30 (m, 4H) ; de 1 ,49 à 1 ,72 (m, 8H) ; 2,77 (m, 1 H) ; 3,30 (m, 1 H) ; 3,57 (m, 1H) ; 3,69 (s, 3H) ; 6,78 (d, 8,5 Hz, 2H) ; 7,13 (m, 3H) ; 7,48 (dd, 9,1 Hz et 2,9 Hz, 1H) ; 7,60 (d, 2,9 Hz, 1 H) ; 7,98 (t, 5,3 Hz, 1 H) , 13,03 (s, 1H).
Composé 9 : Acide 2-[4-[2-(2-(phénylthio)-5- trifluorométhylbenzamido)propyl]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes : Etape 1 : 2-[4-[2-(2~(phénylthio)-5-trifluorométhylbenzamido)propyl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par substitution du dérivé fluoré 5-3 par le thiophénol selon le protocole A. Il est purifié par chromatographie sur gel de silice
(dichlorométhane/méthanol 95/5). Rendement : 80 %
Aspect : huile jaune pâle
RMN 1H (300MHz, CDCI3, δ en ppm) : de 1 ,22 à 1 ,27 (m, 6H) ; 1 ,58 (s, 6H) ; de
2,76 à 2,94 (m, 2H) ; 4,22 (q, 7,0 Hz, 2H) ; 4,42 (m, 1H) ; 6,05 (d, 7,9 Hz, 1H) ;
6,80 (d, 8,5 Hz, 2H) ; 7,03 (d, 8,5 Hz, 1H) ; 7,13 (d, 8,5 Hz, 2H) ; de 7,41 à 7,49 (m, 6H) ; 7,68 (d, 2,0 Hz, 1 H).
Etape 2 : acide 2-[4-[2-(2-(phénylthio)-5- trifluorométhylbenzamido)propyl]phénoxy]-2-méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 9/1). Rendement : 29 % Aspect : poudre blanche
RMN 1H (300MHz, DMSO-c/6, δ en ppm) : 1 ,15 (d, 6,5 Hz, 3H) ; 1 ,44 (s, 6H) ; de 2,67 à 2,83 (m, 2H) ; 4,14 (m, 1 H) ; 6,75 (d, 8,5 Hz, 2H) ; 6,95 (d, 8,2 Hz, 1 H) ; 7,15 (d, 8,5 Hz, 2H) ; 7,50 (m, 5H) ; 7,61 (m, 2H) ; 8,57 (d, 7,9 Hz, 1H).
Composé 10 : Acide 2-[4-[1-((2-méthoxy-1-naphtoyl)amino)propan-2- yl]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[1-((2-méthoxy-1-naphtoyl)amino)propan-2-yl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par substitution de l'iodure de méthyle par le dérivé phénolique 5-4 selon le protocole D (en utilisant le carbonate de césium). Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 6/4).
Rendement : 90 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,21 (t, 7,3 Hz, 3H) ; 1 ,37 (d, 7,0 Hz1 3H) ;
1 ,58 (s, 6H) ; 3,10 (m, 1H) ; de 3,53 à 3,62 (m, 1H) ; 3,90 (m, 4H) ; 4,21 (q, 7,3 Hz,
2H) ; 5,89 (m, 1 H) ; 6,80 (d, 8,5 Hz, 2H) ; 7,16 (d, 8,5 Hz1 2H) ; de 7,22 à 7,28 (m,
1 H) ; 7,35 (m, 1H) ; 7,46 (m, 1H) ; de 7,75 à 7,87 (m, 3H).
Etape 2 : acide 2-[4-[1-((2-méthoxy-1-naphtoyl)amino)propan-2-yl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5).
Rendement : 67 % Aspect : poudre blanche
RMN 1H (300MHz, DMSO-ûfe, δ en ppm) : 1 ,24 (d, 7,0 Hz1 3H) ; 1 ,50 (s, 6H) ; 3,02 (m, 1 H) ; 3,35 (m, 1H) ; 3,54 (m, 1H) ; 3,85 (s, 3H) ; 6,79 (d, 8,5 Hz, 2H) ; 7,20 (d, 8,5 Hz, 2H) ; de 7,30 à 7,46 (m, 4H) ; 7,85 (d, 7,6 Hz, 1H) ; 7,94 (d, 9,1 Hz, 1 H) ; 8,38 (t, 5,6 Hz, 1 H) ; 13,06 (s(large), 1H).
Composé 11 : Acide 2-[4-[2-(2-méthoxy-5-méthylbenzamido)éthyl]phénoxy]- 2-méthylpropanoïque
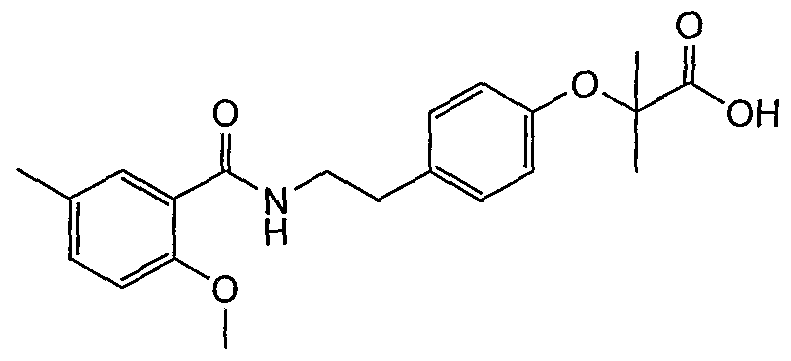
La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[2-(2-méthoxy-5-méthylbenzamido)éthyl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par substitution du bromoisobutyrate d'éthyle par le dérivé phénolique 4-6 selon le protocole D. Il est purifié par chromatographie sur gel de silice (heptane/acétate d'éthyle 65/35).
Rendement : 93 %
Aspect : huile jaune pâle
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,26 (t, 7,0 Hz, 3H) ; 1 ,59 (s, 6H) ; 2,32 (s, 3H) ; 2,85 (t, 6,7 Hz, 2H) ; de 3,69 à 3,75 (m, 5H) ; 4,24 (q, 7,0 Hz, 2H) ; 6,82 (m,
3H) ; 7,13 (d, 8,5 Hz1 2H) ; 7,22 (dd, 8,4 Hz, 2,3 Hz, 1H) ; 7,93 (m, 1H) ; 8,01 (d,
2,3 Hz, 1H).
Etape 2 : acide 2-[4-[2-(2-méthoxy-5-méthylbenzamido)éthyl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Aucune purification n'est nécessaire à l'obtention du produit pur après acidification du milieu et extraction à l'éther diéthylique.
Rendement : 69 % Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1,62 (s, 6H) ; 2,32 (s, 3H) ; 2,85 (t, 6,7 Hz, 2H) ; de 3,68 à 3,74 (m, 5H) ; 6,79 (d, 8,5 Hz, 1 H) ; 6,90 (d, 8,5 Hz1 2H) ; 7,13 (d, 8,5 Hz, 2H) ; 7,21 (dd, 8,5 Hz, 2,3 Hz, 1H) ; de 8,01 à 8,07 (m, 2H) ; 9,66 (s(large), 1 H).
Composé 12 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]-3- méthyl-phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes : Etape 1 : 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]-3-méthyl-phénoxy]-
2-méthylpropanoate de terf-butyle
Cet intermédiaire est obtenu par condensation entre l'aminé 2-8 et l'acide 5- chloro-2-méthoxybenzoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2). Rendement : 68 %
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,26 (d, 6,7 Hz, 3H) ; 1,46 (s, 9H) ; 1,56 (s,
6H) ; 2,29 (s, 3H) ; de 3,24 à 3,49 (m, 2H) ; 3,66 (s, 3H) ; de 3,78 à 3,87 (m, 1H) ; de 6,71 à 6,75 (m, 2H) ; 6,82 (d, 8,8 Hz, 1H) ; 7,12 (d, 8,5 Hz, 1H) ; 7,34 (dd, 8,8 Hz et 2,9 Hz, 1H) ; 7,75 (m, 1H) ; 8,17 (d, 2,9 Hz1 1H).
Etape 2 : acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]-3-méthyl- phénoxy]-2-méthylpropanoïque
Ce composé est obtenu par hydrolyse acide de l'ester terf-butylique selon le protocole G. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 9/1). Rendement : 43 % Aspect : poudre blanche
RMN 1H (300MHz1 DMSO-ûfe, δ en ppm) : 1,16 (d, 6,7 Hz, 3H) ; 1,48 (s, 6H) ; 2,24 (s, 3H) ; de 3,16 à 3,23 (m, 1 H) ; de 3,34 à 3,42 (m, 2H) ; 3,73 (s, 3H) ; de 6,63 à 6,65 (m, 2H) ; de 7,11 à 7,16 (m, 2H) ; 7,49 (dd, 8,8 Hz et 2,9 Hz, 1H) ; 7,62 (d, 2,9 Hz, 1H) ; 8,12 (t, 5,3 Hz, 1 H) ; 13,02 (s, 1H).
Composé 13 : Acide 2-[4-[3~(5-chloro-2-méthoxybenzamido)butan-2- yI]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes : Etape 1 : 2-[4-[3-(5-chloro-2-méthoxybenzamido)butan-2-yl]phénoxy]-2- méthylpropanoate de ferf-butyle
Cet intermédiaire est obtenu par condensation entre l'aminé 2-9 et l'acide 5- chloro-2-méthoxybenzoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 9/1). Rendement : 97 %
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) ; mélange 60/40 de diastéréoisomères : 1,05 et 1 ,15 (d et d, 6,7 Hz, 3H) ; de 1 ,31 à 1 ,35 (m, 3H) ; 1 ,43 et 1 ,44 (s et s, 9H) ;
1,56 et 1,57 (s et s, 6H) ; de 2,88 à 2,96 (m, 1H) ; 3,81 et 3,86 (s et s, 3H) ; 4,42 (m, 1H) ; de 6,81 à 6,91 (m, 3H) ; 7,12 (m, 2H) ; de 7,34 à 7,40 (m, 1 H) ; 7,64 (m,
1H) ; de 8,16 à 8,19 (m, 1H).
Etape 2 : acide 2-[4-[3-(5-chloro-2-méthoxybenzamido)butan-2-yl]phénoxy]-2- méthylpropanoïque Ce composé est obtenu par hydrolyse acide de l'ester te/f-butylique selon le protocole G. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 9/1). Rendement : 48 % Aspect : poudre blanche
RMN 1H (300MHz1 DMSO-cfe, δ en ppm) ; mélange 60/40 de diastéréoisomères : 0,91 et 1 ,08 (d et d, 6,6 Hz, 3H) ; 1 ,22 (m, 3H) ; 1 ,47 et 1 ,49 (s et s, 6H) ; de 2,77 à 2,93 (m, 1 H) ; 3,79 et 3,85 (s et s, 3H) ; de 4,07 à 4,16 (m, 1 H) ; 6,78 et 6,79 (d et d, 8,5 Hz, 2H) ; de 7,11 à 7,18 (m, 3H) ; de 7,46 à 7,60 (m, 2H) ; 7,87 et 8,01 (d et d, 8,9 Hz, 1 H) ; 13,05 (s, 1H).
Composé 14 : Acide 2-[4-[1-(5-ferf-butyl-2- méthoxybenzamido)éthyl]phénoxy]-2-méthylpropanoïque

Ce composé est synthétisé suivant la même méthodologie que le composé 11. RMN 1H (300MHz, CDCI3, δ en ppm) : 1,28 (s, 9H) ; 1 ,59 (s, 6H) ; 2,88 (t, 6,9 Hz, 2H) ; de 3,68 à 3,79 (m, 5H) ; de 6,85 à 6,92 (m, 3H) ; 7,16 (d, 8,5 Hz, 2H) ; 7,45 (dd, 8,8 Hz, 2,6 Hz, 1H) ; 8,02 (m, 1 H) ; 8,26 (d, 2,6 Hz, 1 H).
Composé 15 : N-[méthylsulfonyl]-2-[4-[1-((3-méthoxy-2-naphtoyl)amîno) propan-2-yl]phénoxy]-2-méthylpropanamide

Ce composé est obtenu par condensation entre le composé 6 et le méthanesulfonamide selon le protocole L. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5).
Rendement : 20 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 1,24 (d, 7,0 Hz, 3H) ; 1,47 (s, 6H) ; 3,02
(m, 1H) ; 3,24 (s, 3H) ; de 3,38 à 3,52 (m, 2H) ; 3,85 (s, 3H) ; 6,86 (d, 8,5 Hz, 2H) ;
7,24 (d, 8,5 Hz, 2H) ; de 7,36 à 7,42 (m, 2H) ; 7,52 (m, 1H) ; 7,83 (d, 8,2 Hz, 1 H) ; 7,92 (d, 8,2 Hz, 1 H) ; 8,20 (m, 2H) ; 11 ,99 (s(large), 1 H).
Composé 16 : Acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)pentyl]phénoxyj- 2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[2-(5-chloro-2-méthoxybenzamido)pentyl]phénoxy]-2- méthylpropanoate de ferf-butyle
Cet intermédiaire est obtenu par condensation entre l'aminé 2-10 et l'acide 5- chloro-2-méthoxybenzoïque selon le protocole F. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 9/1).
Rendement : quantitatif Aspect : huile incolore
RMN 1H (300MHz1 CDCI3, δ en ppm) : 0,91 (t, 6,7 Hz, 3H) ; de 1 ,37 à 1 ,56 (m,
19H) ; 2,83 (d, 5,9 Hz, 2H) ; 3,85 (s, 3H) ; 4,38 (m, 1H) ; 6,80 (d, 8,5 Hz, 2H) ; 6,89
(d, 8,8 Hz, 1 H) ; 7,09 (d, 8,5 Hz, 2H) ; 7,37 (dd, 8,8 Hz et 2,9 Hz, 1H) ; 7,60 (d, 8,8
Hz, 1H) ; 8,16 (d, 2,9 Hz, 1H).
Etape 2 : acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)pentyl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par hydrolyse acide de l'ester ferf-butylique selon le protocole G. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5).
Rendement : 48 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-Cf6, δ en ppm) : 0,86 (t, 7,0 Hz, 3H) ; de 1,25 à 1 ,47 (m,
10H) ; 2,70 (d, 6,7 Hz, 2H) ; 3,81 (s, 3H) ; 4,09 (m, 1H) ; 6,74 (d, 8,5 Hz, 2H) ; de
7,09 à 7,14 (m, 3H) ; 7,41 (d, 2,9 Hz, 1H) ; 7,46 (dd, 8,8 Hz et 2,9 Hz, 1 H) ; 7,92 (d, 8,5 Hz, 1H) ; 12,99 (s, 1 H).
Composé 17 : Acide 2-[4-[2-(5-méthyl-2-octyloxybenzamido)éthyl]phénoxy]- 2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[2-(5-méthyl-2-octyloxybenzamido)éthyl]phénoxy]-2- méthylpropanoate d'éthyle Cet intermédiaire est obtenu par substitution du bromoisobutyrate d'éthyle par le dérivé phénolique 4-7 selon le protocole D. Il est purifié par chromatographie sur gel de silice (heptane/acétate d'éthyle 8/2).
Rendement : 80 %
Aspect : huile incolore RMN 1H (300MHz, CDCI3, δ en ppm) : 0,90 (t, 6,7 Hz, 3H) ; 1 ,25 (t, 7,3 Hz, 3H) ;
1 ,30 (m, 10H) ; 1 ,59 (s, 6H) ; 1 ,67 (m, 2H) ; 2,33 (s, 3H) ; 2,86 (t, 7,3 Hz, 2H) ;
3,70 (m, 2H) ; 4,01 (t, 6,4 Hz, 2H) ; 4,24 (q, 7,3 Hz, 2H) ; 6,81 (m, 3H) ; 7,12 (d,
8,5 Hz, 2H) ; 7,21 (dd, 8,2 Hz, 2,0 Hz, 1 H) ; 8,03 (d, 2,0 Hz, 1 H) ; 8,14 (m, 1H).
Etape 2 : acide 2-[4-[2-(5-méthyl-2-octyloxybenzamido)éthyl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. L'attendu est purifié par recristallisation dans l'acétonitrile.
Rendement : 34 % Aspect : poudre blanche
RMN 1H (300MHz, DMSO-^6, δ en ppm) : 0,83 (t, 7,0 Hz, 3H) ; 1 ,24 (m, 10H) ;
1 ,47 (s, 6H) ; 1 ,62 (m, 2H) ; 2,25 (s, 3H) ; 2,74 (t, 7,0 Hz, 2H) ; 3,49 (m, 2H) ; 3,99
(t, 6,4 Hz, 2H) ; 6,75 (d, 8,5 Hz, 2H) ; 7,00 (d, 8,2 Hz, 1 H) ; 7,12 (d, 8,5 Hz, 2H) ;
7,23 (dd, 8,2 Hz, 2,0 Hz, 1 H) ; 7,59 (d, 2,0 Hz, 1H) ; 8,13 (t, 5,3 Hz, 1 H) ; 13,02 (s(large), 1H).
Composé 18 : Acide 2-[4-[2-(2-méthoxy-5-phény!benzamido)éthyI]phénoxy]- 2-méthylpropanoïque

Etape 1 : 2-[4-[2-(2-méthoxy-5-phénylbenzamido)éthyl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par substitution du bromoisobutyrate d'éthyle par le dérivé phénolique 4-8 selon le protocole D. Il est purifié par chromatographie sur gel de silice (heptane/acétate d'éthyle 1/1).
Rendement : 91 %
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1,28 (t, 7,0 Hz, 3H) ; 1,61 (s, 6H) ; 2,89 (t,
6,7 Hz, 2H) ; 3,75 (m, 2H) ; 3,83 (s, 3H) ; 4,25 (q, 7,0 Hz, 2H) ; 6,85 (d, 8,7 Hz, 2H) ; 7,02 (d, 8,8 Hz, 1 H) ; 7,17 (d, 8,7 Hz, 2H) ; 7,34 (m, 1H) ; 7,44 (m, 2H) ; de
7,60 à 7,70 (m, 3H) ; 7,93 (m, 1H) ; 8,51 (d, 2,6 Hz, 1H).
Etape 2 : acide 2-[4-[2-(2-méthoxy-5-phénylbenzamido)éthyl]phénoxy]-2- méthylpropanoïque Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. L'attendu est purifié par recristallisation dans l'acétonitrile.
Rendement : 26 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 1 ,48 (s, 6H) ; 2,77 (t, 7,0 Hz, 2H) ; 3,50 (m, 2H) ; 3,84 (s, 3H) ; 6,78 (d, 8,5 Hz, 2H) ; de 7,16 à 7,22 (m, 3H) ; 7,34 (m,
1H) ; 7,46 (m, 2H) ; 7,62 (d, 8,5 Hz, 2H) ; 7,76 (dd, 8,5 Hz et 2,3 Hz, 1H) ; 7,98 (d,
2,3 Hz, 1H) ; 8,23 (t, 5,3 Hz, 1H) ; 13,02 (s(large), 1 H).
Composé 19 : Acide 2-[4-[2-(5-butyI-2-méthoxybenzamido)éthyl]phénoxy]-2- méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes.
Etape 1 : 2-[4-[2-(5-butyl-2-méthoxybenzamido)éthyl]phénoxy]-2- méthylpropanoate d'éthyle Ce composé est obtenu par condensation entre l'acide 3-4 et l'aminé 2-2 suivant le protocole F. L'attendu est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 9/1). Rendement : 12 % Aspect : huile incolore RMN 1H (300MHz, CDCI3, δ en ppm) : 0,89 (t, 7,3 Hz, 3H) ; de 1,21 à 1 ,37 (m, 5H) ; 1 ,57 (m, 8H) ; 2,57 (t, 7,3 Hz, 2H) ; 2,84 (t, 6,7 Hz, 2H) ; 3,71 (m, 5H) ; 4,22 (q, 7,0 Hz, 2H) ; 6,81 (m, 3H) ; 7,12 (d, 7,0 Hz, 2H) ; 7,20 (d, 8,2 Hz, 1H) ; 7,94 (m, 1 H) ; 8,02 (s, 1H).
Etape 2 : acide 2-[4-[2-(5-butyl-2-méthoxybenzamido)éthyl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. L'attendu est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 9/1). Rendement : 31 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 0,88 (t, 7,3 Hz, 3H) ; 1 ,27 (m, 2H) ; de 1 ,46 à 1 ,55 (m, 8H) ; 2,53 (t, 7,3 Hz, 2H) ; 2,74 (t, 7,3 Hz, 2H) ; 3,47 (m, 2H) ; 3,76 (s, 3H) ; 6,78 (d, 8,5 Hz, 2H) ; 7,01 (d, 8,5 Hz, 1H) ; 7,13 (d, 8,5 Hz, 2H) ; 7,26 (dd, 8,5 Hz, 2,3 Hz, 1H) ; 7,57 (d, 2,3 Hz, 1H) ; 8,13 (t, 5,6 Hz, 1H).
Composé 20 : Acide 2-[4-[1-(3-chloro-4-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[1-(3-chloro-4-méthoxybenzamido)propan-2-yl]phénoxy]-2- méthylpropanoate de ferf-butyle
Ce composé est obtenu par condensation entre l'acide 3-chloro-4- méthoxybenzoïque et l'aminé 2-11 suivant le protocole F. L'attendu est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 9/1).
Rendement : 89 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,28 (d, 6,7 Hz, 3H) ; 1 ,44 (s, 9H) ; 1 ,57 (s,
6H) ; 3,01 (m, 1 H) ; 3,32 (m, 1H) ; 3,78 (m, 1H) ; 3,93 (s, 3H) ; 5,89 (m, 1 H) ; de
6,83 à 6,91 (m, 3H) ; 7,11 (d, 8,5 Hz, 2H) ; 7,50 (dd, 8,8 Hz et 2,0 Hz, 1 H) ; 7,67
(d, 2,0 Hz, 1 H).
Etape 2 : acide 2-[4-[1-(3-chloro-4-méthoxybenzamido)propan-2-yl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par hydrolyse acide de l'ester ferf-butylique selon le protocole G. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5).
Rendement : 81 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-cfe, δ en ppm) : 1 ,17 (d, 7,0 Hz, 3H) ; 1 ,47 (s, 6H) ; 2,98
(m, 1H) ; de 3,29 à 3,35 (m, 2H) ; 3,90 (s, 3H) ; 6,75 (d, 8,8 Hz, 2H) ; 7,13 (d, 8,8 Hz, 2H) ; 7,21 (d, 8,5 Hz, 1H) ; 7,78 (dd, 8,5 Hz et 2,0 Hz, 1 H) ; 7,87 (d, 2,0 Hz,
1 H) ; 8,48 (t, 5,6 Hz, 1H) , 12,98 (s, 1 H).
Composé 21 : Acide 2-[4-[1-(4-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[1-(4-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-2- méthylpropanoate de tert-butyle
Ce composé est obtenu par condensation entre l'acide 4-chloro-2- méthoxybenzoïque et l'aminé 2-11 suivant le protocole F. L'attendu est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 95/5).
Rendement : 95 %
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,30 (d, 6,7 Hz, 3H) ; 1 ,44 (s, 9H) ; 1,57 (s,
6H) ; 2,98 (m, 1 H) ; 3,39 (m, 1H) ; 3,67 (s, 3H) ; 3,87 (m, 1H) ; de 6,84 à 6,87 (m,
3H) ; 7,03 (dd, 8,5 Hz et 1 ,8 Hz, 1 H) ; 7,13 (d, 8,8 Hz, 2H) ; 7,65 (m, 1 H) ; 8,13 (d,
8,5 Hz, 1 H).
Etape 2 : acide 2-[4-[1-(4-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par hydrolyse acide de l'ester ferf-butylique selon le protocole G. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5). Rendement : 59 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 1 ,19 (d, 6,7 Hz, 3H) ; 1 ,49 (s, 6H) ; 2,94 (m, 1H) ; de 3,28 à 3,51 (m, 2H) ; 3,76 (s, 3H) ; 6,89 (d, 8,5 Hz, 2H) ; 7,07 (dd, 8,2 Hz et 1 ,8 Hz, 1H) ; 7,17 (m, 3H) ; 7,69 (d, 8,2 Hz, 1H) ; 7,97 (t, 5,6 Hz, 1H) , 13,04 (s, 1H).
Composé 22 : Acide 2-[4-[2-((3-méthoxy-2-naphtoyl)amino)propyl]phénoxy]- 2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[2-((3-méthoxy-2-naphtoyl)amino)propyl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par substitution du bromoisobutyrate d'éthyle par le dérivé phénolique 4-9 selon le protocole D. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 9/1).
Rendement : 99 %
Aspect : huile incolore
RMN 1H (300MHz1 CDCI3, δ en ppm) : de 1 ,21 à 1 ,25 (m, 6H) ; 1 ,59 (s, 6H) ; de
2,79 à 2,96 (m, 2H) ; 3,97 (s, 3H) ; 4,22 (q, 7,0 Hz, 2H) ; 4,52 (m, 1 H) ; 6,81 (d, 8,5
Hz, 2H) ; 7,14 (d, 8,5 Hz, 2H) ; 7,20 (s, 1H) ; 7,40 (m, 1 H) ; 7,52 (m, 1H) ; 7,74 (d,
8,2 Hz, 1 H) ; de 7,85 à 7,92 (m, 2H) ; 8,75 (s, 1 H).
Etape 2 : 2-[4-[2-((3-méthoxy-2-naphtoyl)amino)propyl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par chromatographie sur gel de silice
(dichlorométhane/méthanol 9/1). Rendement : 12 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 1 ,14 (d, 6,4 Hz, 3H) ; 1 ,48 (s, 6H) ; de
2,67 à 2,84 (m, 2H) ; 3,91 (s, 3H) ; de 4,15 à 4,24 (m, 1H) ; 6,78 (d, 8,5 Hz, 2H) ;
7,16 (d, 8,5 Hz, 2H) ; 7,39 (m, 2H) ; 7,52 (m, 1 H) ; 7,84 (d, 8,2 Hz1 1 H) ; 7,91 (d, 8,2 Hz, 1 H) ; de 8,09 à 8,14 (m, 2H) ; 13,01 (s(large), 1H).
Composé 23 : Acide 2-[4-[1-((3,7-diméthoxy-2-naphtoyl)amino)propan-2- yl]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[1-((3,7-diméthoxy-2-naphtoyl)amino)propan-2-yl]phénoxy]-2- méthylpropanoate de fe/Y-butyle Cet intermédiaire est obtenu par substitution de l'iodure de méthyle par le dérivé phénolique 5-5 selon le protocole D. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 9/1). Rendement : 74 % Aspect : huile incolore RMN 1H (300MHz, CDCI3, δ en ppm) : 1,33 (d, 7,0 Hz1 3H) ; 1,45 (s, 9H) ; 1 ,57 (s, 6H) ; 3,04 (m, 1H) ; 3,47 (m, 1H) ; 3,74 (s, 3H) ; de 3,88 à 3,98 (m, 4H) ; 6,87 (d, 8,8 Hz, 2H) ; 7,08 (s, 1H) ; de 7,15 à 7,19 (m, 4H) ; 7,61 (d, 8,8 Hz, 1 H) ; 8,06 (m, 1H) ; 8,64 (s, 1H).
Etape 2 : acide 2-[4-[1-((3,7-diméthoxy-2-naphtoyl)amino)propan-2-yl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par hydrolyse acide de l'ester te/f-butylique selon le protocole G. Il est purifié par chromatographie sur gel de silice
(dichlorométhane/méthanol 95/5). Rendement : 35 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 1 ,23 (d, 7,0 Hz, 3H) ; 1,50 (s, 6H) ; 2,98
(m, 1 H) ; de 3,35 à 3,57 (m, 2H) ; 3,79 (s, 3H) ; 3,84 (s, 3H) ; 6,81 (d, 8,8 Hz, 2H) ; de 7,16 à 7,22 (m, 3H) ; 7,37 (m, 2H) ; 7,76 (d, 9,1 Hz, 1 H) ; de 8,15 à 8,18 (m, 2H) ; 13,03 (s(large), 1H).
Composé 24 : N-[phénylsulfonyl]-2-[4-[1-(5-chloro-2- méthoxybenzamido)propan-2-yl]phénoxy]-2-méthylpropanamide

Ce composé est obtenu par condensation entre le composé 5 et le benzènesulfonamide selon le protocole L. Il est purifié par recristallisation dans l'acétonitrile.
Rendement : 35 %
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,31 (d, 7,0 Hz1 3H) ; 1,44 (m, 6H) ; 3,02
(m, 1 H) ; 3,46 (m, 1H) ; de 3,74 à 3,85 (m, 4H); 6,75 (d, 8,5 Hz, 2H) ; 6,85 (d, 8,8
H, 1H) ; 7,12 (d, 8,5 Hz, 2H) ; 7,36 (dd, 8,8 Hz et 2,9 Hz, 1 H) ; 7,58 (m, 2H) ; 7,71
(m, 2H) ; 8,07 (m, 2H) , 8,17 (d, 2,9 Hz, 1H) ; 9,10 (s, 1 H).
Composé 25 : N-[pipéridin-1-yl]-2-[4-[1-(5-chloro-2- méthoxybenzamîdo)propan-2-yl]phénoxy]-2-méthylpropanamîde

Rendement : 60 %
Aspect : poudre blanche RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,32 (d, 7,0 Hz1 3H) ; 1 ,41 (m, 2H) ; 1 ,52
(m, 6H) ; 1,72 (m, 4H) ; 2,76 (m, 4H) ; 3,03 (m, 1H) ; 3,50 (m, 1 H) ; de 3,74 à 3,83
(m, 4H); 6,85 (d, 8,8 H, 1H) ; 6,92 (d, 8,5 Hz, 2H) ; 7,17 (d, 8,5 Hz, 2H) ; 7,36 (m,
2H) ; 7,74 (m, 1H) ; 8,16 (d, 2,9 Hz, 1 H).
Composé 26 : Acide 2-[4-[2-(2,5-diméthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[2-(2,5-diméthoxybenzamido)propyl]phénoxy]-2-méthylpropanoate d'éthyle Cet intermédiaire est obtenu par substitution du bromoisobutyrate d'éthyle par le dérivé phénolique 4-10 selon le protocole D. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 9/1). Rendement : 35 % Aspect : huile incolore RMN 1 H (300MHz, CDCI3, δ en ppm) : de 1 ,19 à 1 ,27 (m, 6H) ; 1 ,59 (s, 6H) ; de 2,73 à 2,92 (m, 2H) ; 3,82 (s, 3H) ; 3,83 (s, 3H) ; 4,23 (q, 7,3 Hz, 2H) ; 4,44 (m, 1H) ; 6,80 (d, 8,5 Hz, 2H) ; 6,88 (d, 9,0 Hz, 1 H) ; 6,98 (dd, 9,0 Hz et 3,2 Hz, 1 H) ; 7,12 (d, 8,5 Hz, 2H) ; 7,78 (d, 3,2 Hz, 1 H) ; 7,90 (d, 7,6 Hz, 1H).
Etape 2 : acide 2-[4-[2-(2,5-diméthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 9/1). Rendement : 65 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 1 ,12 (d, 6,7 Hz, 3H) ; 1,47 (s, 6H) ; de 2,65 à 2,81 (m, 2H) ; 3,71 (s, 3H) ; 3,77 (s, 3H) ; de 4,11 à 4,20 (m, 1 H) ; 6,77 (d, 8,5 Hz, 2H) ; de 6,98 à 7,06 (m, 2H) ; 7,13 (d, 8,5 Hz, 2H) ; 7,24 (d, 2,6 Hz) ; 8,01 (d, 7,9 Hz, 1H).
Composé 27 : Acide 2-[4-[2-(5-tertiobutyl-2- méthoxybenzamido)propyl]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[2-(5-tertiobutyl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par substitution du bromoisobutyrate d'éthyle par le dérivé phénolique 4-11 selon le protocole D. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 95/5).
Rendement : 52 % Aspect : huile incolore
RMN 1 H (300MHz, CDCI3, δ en ppm) : de 1 ,19 à 1 ,27 (m, 6H) ; 1 ,33 (s, 9H) ; 1 ,59
(s, 6H) ; de 2,73 à 2,93 (m, 2H) ; 3,85 (s, 3H) ; 4,23 (q, 7,3 Hz, 2H) ; de 4,43 à
4,47 (m, 1H) ; 6,80 (d, 8,5 Hz, 2H) ; 6,88 (d, 8,5 Hz, 1 H) ; 7,11 (d, 8,5 Hz, 2H) ;
7,45 (dd, 8,5 Hz et 2,6 Hz, 1H) ; 7,83 (d, 7,6 Hz, 1H) ; 8,27 (d, 2,6 Hz, 1 H).
Etape 2 : acide 2-[4-[2-(5-tertiobutyl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 9/1).
Rendement : 71 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 1 ,11 (d, 6,4 Hz, 3H) ; 1 ,25 (s, 9H) ; 1 ,47
(s, 6H) ; de 2,65 à 2,80 (m, 2H) ; 3,80 (s, 3H) ; de 4,13 à 4,17 (m, 1H) ; 6,75 (d, 8,5 Hz, 2H) ; 7,02 (d, 8,8 Hz, 1H) ; 7,13 (d, 8,5 Hz, 2H) ; 7,45 (dd, 8,8 Hz, 2,6 Hz,
1H) ; 7,69 (d, 2,6 Hz) ; 7,95 (d, 7,9 Hz, 1H).
Composé 28 : Acide 2-[4-[2-(5-butyryl-2- méthoxybenzamido)propyl]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[2-(5-butyryl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoate d'éthyle Cet intermédiaire est obtenu par substitution du bromoisobutyrate d'éthyle par le dérivé phénolique 4-12 selon le protocole D. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 9/1).
Rendement : 97 %
Aspect : huile incolore RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,00 (t, 7,3 Hz, 3H) ; de 1 ,21 à 1 ,29 (m,
6H) ; 1 ,58 (s, 6H) ; de 1,70 à 1 ,82 (m, 2H) ; de 2,75 à 2,92 (m, 2H) ; 2,99 (t, 7,0
Hz, 2H) ; 3,94 (s, 3H) ; 4,23 (q, 7,3 Hz, 2H) ; de 4,42 à 4,46 (m, 1H) ; 6,80 (d, 8,5
Hz, 2H) ; 7,03 (d, 8,8 Hz, 1 H) ; 7,12 (d, 8,5 Hz, 2H) ; 7,64 (d, 7,9 Hz, 1H) ; 8,12
(dd, 8,8 Hz et 2,3 Hz, 1H) ; 8,80 (d, 2,3 Hz, 1H).
Etape 2 : acide 2-[4-[2-(5-butyryl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 9/1).
Rendement : 49 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-cfe, δ en ppm) : 0,92 (t, 7,6 Hz, 3H) ; 1,12 (d, 6,4 Hz,
3H) ; 1 ,47 (s, 6H) ; 1 ,63 (m, 2H) ; de 2,65 à 2,81 (m, 2H) ; 2,93 (t, 7,3 Hz, 2H) ; 3,89 (s, 3H) ; de 4,11 à 4,20 (m, 1H) ; 6,76 (d, 8,5 Hz1 2H) ; 7,13 (d, 8,5 Hz, 2H) ;
7,20 (d, 8,8 Hz, 1 H) ; de 8,00 à 8,06 (m, 2H) ; 8,14 (d, 2,3 Hz, 1 H) ; 13,04 (s, 1H).
Composé 29 : Acide 2-[4-[2-(5-phényl-2- méthoxybenzamido)propyl]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[2-(5-phényl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par substitution du bromoisobutyrate d'éthyle par le dérivé phénolique 4-13 selon le protocole D. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 9/1).
Rendement : 97 % Aspect : huile incolore
RMN 1 H (300MHz, CDCI3, δ en ppm) : de 1 ,21 à 1 ,29 (m, 6H) ; 1 ,59 (s, 6H) ; de
2,77 à 2,94 (m, 2H) ; 3,90 (s, 3H) ; 4,23 (q, 7,0 Hz, 2H) ; de 4,44 à 4,53 (m, 1 H) ;
6,81 (d, 8,5 Hz, 2H) ; 7,03 (d, 8,8 Hz, 1 H) ; 7,12 (d, 8,5 Hz, 2H) ; de 7,30 à 7,46
(m, 3H) ; 7,62 (d, 8,5 Hz, 2H) ; 7,67 (dd, 8,8 Hz, 2,6 Hz, 1H) ; 7,79 (d, 7,6 Hz, 1H) ; 8,49 (d, 2,6 Hz, 1 H).
Etape 2 : acide 2-[4-[2-(5-phényl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par chromatographie sur gel de silice
(dichlorométhane/méthanol 9/1).
Rendement : 8 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-cfe, δ en ppm) : 1 ,14 (d, 6,5 Hz, 3H) ; 1 ,46 (s, 6H) ; de 2,67 à 2,82 (m, 2H) ; 3,86 (s, 3H) ; de 4,14 à 4,20 (m, 1H) ; 6,76 (d, 8,5 Hz, 2H) ; de 7,14 à 7,21 (m, 3H) ; de 7,31 à 7,48 (m, 3H) ; 7,61 (d, 7,3 Hz, 2H) ; 7,73 (dd,
8,8 Hz, 2,3 Hz, 1H) ; 7,85 (d, 2,3 Hz, 1H) ; 8,03 (d, 7,9 Hz, 1H) ; 12,70 (s, 1H).
Composés 29A et 29B (respectivement (-)29 et (+)29) :

Méthode 1 : séparation chirale
Les deux énantiomères du composé 29 sont séparés par HPLC semi-préparative sur colonne chirale Chiralpak®AD (250*2.5mm, 20μm, Chiral Technologies Europe) à température ambiante. L'élution est réalisée en mode isocratique avec une phase mobile n-heptane-isopropanol (75-25) additionnée de 0.1% d'acide trifluoroacétique au débit de 30 ml/min.
La pureté énantiomérique de chacun des deux énantiomères ainsi obtenus est contrôlée par HPLC analytique : colonne Chiralpak®AD-H (250*4.6mm, 5μm, Daicel Chemical Industries, LTD) à 300C ; élution isocratique avec phase mobile n-heptane-isopropanol (75-25) additionnée de 0.1% d'acide trifluoroacétique ; débit 1 ml/min ; détection UV à 230 nm. Composé 29A : tR = 10,9 min ((-)29, ee > 98 %) Composé 29B : tR = 21,5 min ((+)29, ee > 98 %)
Méthode 2 : synthèse énantiosélective
Les deux énantiomères du composé 29 sont synthétisés sous forme optiquement pure selon la méthodologie mise au point par les inventeurs.
Les intermédiaires énantiopurs 4-13 ((R) et (S)) décris ci-avant permettent d'isoler les composés 29A et 29B énantiopurs en deux étapes similaires à celles qui permettent l'obtention du composé 29 racémique à partir du phénol 4-13 racémique : substitution du bromoisobutyrate d'éthyle par le dérivé phénolique selon le protocole D puis saponification suivant le protocole H.
La pureté énantiomérique de chacun des deux énantiomères ainsi obtenus est contrôlée par HPLC analytique : colonne Chiralpak®AD-H (250*4.6mm, 5μm, Daicel Chemical Industries, LTD) à 3O0C ; élution isocratique avec phase mobile
n-heptane-isopropanol (75-25) additionnée de 0.1% d'acide trifluoroacétique ; débit 1 ml/min ; détection UV à 230 nm.
Composé 29A : tR = 10,9 min ((-)29, ee > 98 %)
Composé 29B : tR = 21,5 min ((+)29, ee > 98 %)
Aucune racémisation notable n'est observée au cours du procédé de synthèse.
Composé 30 : N-[méthylsulfonyl]-2-[4-[1-(5-chloro-2- méthoxybenzamido)propan-2-yI]phénoxy]-2-méthylpropanamide

Ce composé est obtenu par condensation entre le composé 5 et le méthanesulfonamide selon le protocole L. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 8/2). Rendement : 15 % Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,33 (d, 7,0 Hz, 3H) ; 1 ,55 (s, 6H) ; 3,05 (m, 1H) ; 3,36 (s, 3H) ; 3,50 (m, 1H) ; 3,75 (m et s, 4H) ; 6,90 (m, 3H) ; 7,22 (d, 8,5 Hz, 2H) ; 7,38 (dd, 8,8 Hz et 2,6 Hz, 1H) ; 7,75 (s(large), 1 H) ; 8,16 (d, 2,6 Hz, 1 H) ; 9,00 (s, 1H).
Composé 31 : N-[benzylsulfonyl]-2-[4-[1-(5-chloro-2- méthoxybenzamido)propan-2-yl]phénoxy]-2-méthyIpropanamide

Ce composé est obtenu par condensation entre le composé 5 et le benzylsulfonamide selon le protocole L. Il est purifié par recristallisations successives dans l'éthanol puis l'acétonitrile.
Rendement : 59 %
Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,30 (d, 7,0 Hz, 3H) ; 1 ,48 (s, 6H) ; 3,04 (m,
1 H) ; 3,45 (m, 1 H) ; 3,69 (s, 3H) ; 3,79 (m, 1H) ; 4,75 (s, 2H) ; 6,81 (m, 3H) ; 7,16
(d, 8,5 Hz, 2H) ; 7,35 (dd, 8,8 Hz et 2,9 Hz1 1H) ; 7,42 (m, 5H) ; 7,73 (s(large),
1 H) ; 8,16 (d, 2,9 Hz, 1H) ; 8,68 (s, 1H).
Composé 32 : N-[diméthylamino]-2-[4-[1-(5-chloro-2- méthoxybenzamido)propan-2-yI]phénoxy]-2-méthylpropanamide

Ce composé est obtenu par condensation entre le composé 5 et la N, N- diméthylhydrazine selon le protocole I. Il est purifié par chromatographie sur gel de silice (dichlorométhane/acétate d'éthyle 1/1). Il est isolé sous forme de chlohydrate après traitement par une solution d'acide chlorhydrique dans l'éther diéthylique, filtration et séchage. Rendement : 58 % Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6 / D2O, δ en ppm) : 1,20 (d, 7,0 Hz, 3H) ; 1,46 (s, 6H) ; 2,86 (s, 6H) ; 3,00 (m, 1 H) ; 3,40 (m, 2H) ; 3,77 (s, 3H) ; 6,87 (d, 8,6 Hz, 2H) ; 7,12 (d, 9,1 Hz, 1 H) ; 7,20 (d, 8,6 Hz, 2H) ; 7,47 (dd, 9,1 Hz et 2,6 Hz, 1 H) ; 7,57 (d, 2,6 Hz, 1H).
Composé 33 : Acide 2-[3-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[3-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-2- méthylpropanoate de tert-butyle
Ce composé est obtenu par condensation entre l'acide 5-chloro-2- méthoxybenzoïque et l'aminé 2-12 suivant le protocole F. L'attendu est purifié par chromatographie sur gel de silice (acétate d'éthyle). Rendement : 80 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1,32 (d, 7,0 Hz, 3H) ; 1,44 (s, 9H) ; 1,63 (s, 6H) ; 3,00 (m, 1 H) ; 3,41 (m, 1H) ; 3,66 (s, 3H) ; 3,85 (m, 1H) ; 6,73 (dd, 8,2 Hz et 1 ,8 Hz, 1H) ; 6,82 (m, 2H) ; 6,89 (d, 7,9 Hz, 1 H) ; 7,22 (m, 1H) ; 7,34 (dd, 9,0 Hz et 2,6 Hz, 1H) ; 7,75 (s(large), 1 H) ; 8,16 (d, 2,6 Hz, 1H).
Etape 2 : acide 2-[3-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-2- méthylpropanoïque Ce composé est obtenu par hydrolyse acide de l'ester tert-butylique selon le protocole G. Il est purifié par chromatographie sur gel de silice
(dichlorométhane/méthanol 9/1).
Rendement : 52 %
Aspect : poudre blanche RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,32 (d, 7,0 Hz, 3H) ; 1 ,63 (s, 6H) ; 3,00 (m,
1 H) ; 3,41 (m, 1H) ; 3,66 (s, 3H) ; 3,85 (m, 1H) ; de 6,75 à 6,90 (m, 3H) ; 6,95 (d,
7,6 Hz, 1H) ; de 7,20 à 7,35 (m, 2H) ; 7,80 (s(large), 1H) ; 8,10 (d, 2,6 Hz, 1H).
Composé 34 : Acide 2-[4-[2-(5-butyl-2-méthoxybenzamido)propyl]phénoxy]- 2-méthylpropanoïque

Ce composé est obtenu par réduction du composé 28 :
Le composé 28 (0,9 g, 2,0 mmol) est dissous dans le TFA (5 ml). Le triéthylsilane (358 μl, 2,2 mmol) est additionné goutte à goutte. Le mélange réactionnel est
ensuite agité à 55°C pendant 6 heures. Il est alors refroidi, dilué par l'eau (300 ml), et extrait par l'acétate d'éthyle (100 ml). Après évaporation sous pression réduite, le résidu est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 9/1). Rendement : 16 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 0,88 (t, 7,3 Hz, 3H) ; 1 ,11 (d, 6,4 Hz, 3H) ; 1,27 (m, 2H) ; de 1 ,47 à 1 ,54 (m, 8H) ; 2,51 (t, 7,3 Hz, 2H) ; de 2,64 à 2,80 (m, 2H) ; 3,79 (s, 3H) ; 4,15 (m, 1H) ; 6,76 (d, 8,5 Hz, 2H) ; 7,00 (d, 8,5 Hz, 1H) ; 7,12 (d, 8,5 Hz, 2H) ; 7,24 (dd, 8,5 Hz, 2,3 Hz, 1 H) ; 7,47 (d, 2,3 Hz, 1H) ; 7,94 (d, 7,6 Hz, 1H) ; 13,08 (s, 1 H).
Composé 35 : Acide 2-[4-[1-(5-phényl-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[1-(5-phényl-2-méthoxybenzamido)propan-2-yl]phénoxy]-2- méthylpropanoate de te/f-butyle
Ce composé est obtenu par condensation entre l'acide 3-2 et l'aminé 2-11 suivant le protocole F. L'attendu est purifié par chromatographie sur gel de silice
(cyclohexane/acétate d'éthyle 7/3).
Rendement : 90 %
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1,33 (d, 7,0 Hz, 3H) ; 1 ,46 (s, 9H) ; 1,58 (s, 6H) ; 3,02 (m, 1H) ; 3,45 (m, 1H) ; 3,73 (s, 3H) ; 3,90 (m, 1H) ; 6,88 (d, 8,5 Hz,
2H) ; 6,97 (d, 8,5 Hz, 1 H) ; 7,17 (d, 8,5 Hz, 2H) ; de 7,30 à 7,46 (m, 3H) ; de 7,60 à
7,67 (m, 3H) ; 7,84 (s(large), 1 H) ; 8,47 (d, 2,3 Hz, 1H).
Etape 2 : acide 2-[4-[1-(5-phényl-2-méthoxybenzamido)propan-2-yl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par hydrolyse acide de l'ester tert-butylique selon le protocole G. Il est purifié par chromatographie sur gel de silice (acétate d'éthyle). Rendement : 45 % Aspect : poudre blanche
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,33 (d, 7,0 Hz, 3H) ; 1 ,62 (s, 6H) ; 3,02 (m, 1H) ; 3,45 (m, 1 H) ; 3,73 (s, 3H) ; 3,87 (m, 1H) ; 6,94 (m, 3H) ; 7,19 (d, 8,5 Hz, 2H) ; de 7,30 à 7,46 (m, 3H) ; de 7,60 à 7,66 (m, 3H) ; 7,84 (s(large), 1 H) ; 8,47 (d, 2,3 Hz, 1H).
Composés 35A et 35B (respectivement (+)35 et (-)35) :

Les deux énantiomères du composé 35 sont séparés par HPLC semi-préparative sur colonne chirale Chiralpak®AD (250*2.5mm, 20μm, Chiral Technologies Europe) à température ambiante. L'élution est réalisée en mode isocratique avec une phase mobile n-heptane-isopropanol (80-20) additionnée de 0.1% d'acide trifluoroacétique au débit de 30 ml/min.
La pureté énantiomérique de chacun des deux énantiomères ainsi obtenus est contrôlée par HPLC analytique : colonne Chiralpak®AD-H (250*4.6mm, 5μm, Daicel Chemical Industries, LTD) à 300C ; élution isocratique avec phase mobile n-heptane-isopropanol (75-25) additionnée de 0.1% d'acide trifluoroacétique ; débit 1 ml/min ; détection UV à 206 nm. Composé 35A : tR = 12,5 min ((+)35, ee > 98 %) Composé 35B : tR = 22,7 min ((-)35, ee > 98 %)
Composé 36 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]propanoïque

Etape 1 : 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]propanoate d'éthyle Cet intermédiaire est obtenu par substitution du 2-bromopropanoate d'éthyle par le dérivé phénolique 4-4 selon le protocole D. Il est purifié par chromatographie sur gel de silice (cyclohexa ne/acétate d'éthyle 8/2). Rendement : 69 % Aspect : huile incolore RMN 1H (300MHz, CDCI3, δ en ppm) : 1,28 (m, 6H) ; 1,63 (d, 6,7 Hz, 3H) ; 2,99 (m, 1H) ; 3,38 (m, 1 H) ; 3,65 (s, 3H) ; 3,89 (m, 1H) ; 4,17 (q, 7,0 Hz, 2H) ; 4,72 (q, 6,7 Hz, 1H) ; de 6,81 à 6,89 (m, 3H) ; 7,17 (d, 8,5 Hz, 2H) ; 7,34 (dd, 8,8 Hz et 2,6 Hz, 1 H) ; 7,72 (s(large), 1 H) ; 8,17 (d, 2,6 Hz, 1H).
Etape 2 : acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]propanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié filtration et lavages des cristaux obtenus lors de l'ajout d'acide chlorhydrique dans le mieu réactionnel en fin de réaction. Rendement : 52 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-cfe, δ en ppm) : 1 ,19 (d, 7,0 Hz, 3H) ; 1,48 (d, 6,7 Hz, 3H) ; 2,96 (m, 1H) ; 3,34 (m, 1H) ; 3,45 (m, 1H) ; 3,73 (s, 3H) ; 4,77 (q, 6,7 Hz, 1 H) ; 6,82 (d, 8,5 Hz, 2H) ; 7,17 (m, 3H) ; 7,49 (dd, 9,1 Hz et 2,6 Hz, 1H) ; 7,63 (d, 2,6 Hz, 1 H) ; 8,05 (s(large), 1 H).
Composé 37 : Acide 2-[4-[1-(5-(2,4-difluorophényl)-2- méthoxybenzamido)propan-2-yl]phénoxy]propanoïque
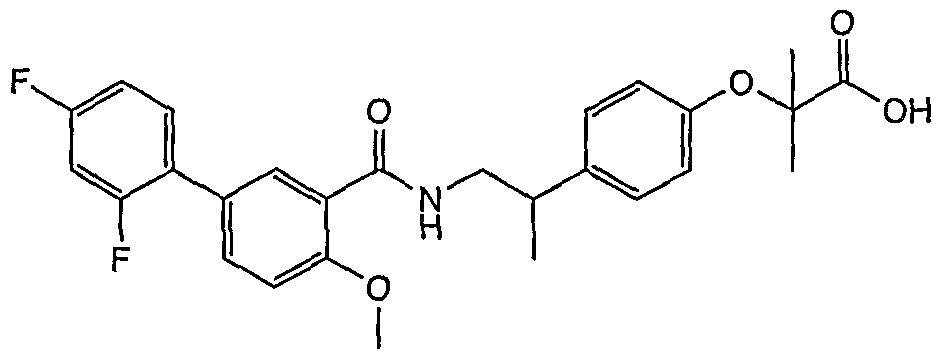
Cet intermédiaire est obtenu par substitution de l'iodure de méthyle par le dérivé phénolique 5-6 selon le protocole D. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2). Rendement : 78 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,33 (d, 7,0 Hz, 3H) ; 1,47 (s, 9H) ; 1,58 (s, 6H) ; 3,01 (m, 1H) ; 3,44 (m, 1H) ; 3,73 (s, 3H) ; 3,90 (m, 1 H) ; de 6,85 à 6,98 (m, 5H) ; 7,16 (d, 8,5 Hz, 2H) ; 7,42 (dd, 8,8 Hz et 2,3 Hz, 1H) ; 7,58 (d, 8,8 Hz, 1H) ; 7,81 (s(large), 1H) ; 8,33 (d, 2,3 Hz, 1 H).
Etape 2 : acide 2-[4-[1-(5-(2,4-difluorophényl)-2-méthoxybenzamido)propan-2- yl]phénoxy]propanoïque Ce composé est obtenu par hydrolyse acide de l'ester te/f-butylique selon le protocole G. Il est purifié par chromatographie sur gel de silice
(dichlorométhane/méthanol 95/5).
Rendement : 87 %
Aspect : poudre blanche RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,31 (d, 7,0 Hz, 3H) ; 1,61 (s, 6H) ; 3,02 (m,
1 H) ; 3,44 (m, 1 H) ; 3,72 (s, 3H) ; 3,86 (m, 1H) ; de 6,80 à 7,00 (m, 5H) ; 7,18 (d,
8,5 Hz, 2H) ; 7,42 (m, 1 H) ; 7,58 (d, 8,8 Hz, 1 H) ; 7,84 (s(large), 1H) ; 8,30 (d, 2,3
Hz, 1H).
Composé 38 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]-3-méthylbutanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-3- méthylbutanoate d'éthyle
Cet intermédiaire est obtenu par substitution du 2-bromo-3-méthylbutanoate d'éthyle par le dérivé phénolique 4-4 selon le protocole D. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2). Il est utilisé tel quel, sans analyses. Rendement : 63 % Aspect : huile incolore
Etape 2 : acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-3- méthylbutanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 9/1). Rendement : 71 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 0,99 (d, 6,7 Hz, 6H) ; 1 ,18 (d, 7,0 Hz1 3H) ; 2,15 (m, 1 H) ; 2,91 (m, 1H) ; 3,33 (m, 1H) ; 3,45 (m, 1 H) ; 3,72 (s, 3H) ; 4,40 (d, 5,0 Hz, 1 H) ; 6,82 (d, 8,5 Hz, 2H) ; 7,11 (m, 3H) ; 7,47 (dd, 8,8 Hz et 2,9 Hz, 1 H) ; 7,64 (d, 2,9 Hz, 1H) ; 8,05 (s(large), 1 H) ; 13,01 (s, 1H).
Composé 39 : Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]pentanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]pentanoate d'éthyle
Cet intermédiaire est obtenu par substitution du 2-bromopentanoate d'éthyle par le dérivé phénolique 4-4 selon le protocole D. Il est purifié par chromatographie sur
gel de silice (cyclohexane/acétate d'éthyle 8/2). Il est utilisé tel quel, sans analyses.
Rendement : 50 %
Aspect : huile incolore
Etape 2 : acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]pentanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par recristallisation dans l'acétonitrile. Rendement : 32 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 0,91 (t, 7,3 Hz, 3H) ; 1 ,18 (d, 7,0 Hz,
3H) ; 1 ,47 (m, 2H) ; 1 ,81 (m, 2H) ; 2,94 (m, 1H) ; 3,33 (m, 1H) ; 3,47 (m, 1H) ; 3,72
(s, 3H) ; 4,63 (t, 6,2 Hz, 1H) ; 6,83 (d, 8,8 Hz, 2H) ; 7,14 (m, 3H) ; 7,49 (dd, 8,9 Hz et 2,9 Hz, 1 H) ; 7,63 (d, 2,9 Hz, 1 H) ; 8,05 (s(large), 1 H) ; 13,07 (s, 1 H).
Composé 40 : Acide 2-[4-[1-(5-ch!oro-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-phényléthanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-2- phényléthanoate d'éthyle
Cet intermédiaire est obtenu par substitution du 2-bromo-2-phénylacétate d'éthyle par le dérivé phénolique 4-4 selon le protocole D. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2). Il est utilisé tel quel, sans analyses.
Rendement : 86 %
Aspect : huile incolore
Etape 2 : acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-2- phényléthanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par recristallisations successives dans l'acétate d'éthyle, Pacétonitrile, puis Féthanol. Rendement : 21 % Aspect : poudre blanche
RMN 1H (300MHz, DMSO-Cf6, δ en ppm) : 1 ,19 (d, 6,7 Hz, 3H) ; 2,96 (m, 1H) ; 3,33 (m, 1 H) ; 3,46 (m, 1 H) ; 3,68 (s, 3H) ; 5,79 (s, 1 H) ; 6,92 (d, 8,8 Hz, 2H) ; 7,12 (d, 8,8 Hz, 1H) ; 7,21 (d, 8,8 Hz, 2H) ; de 7,37 à 7,57 (m, 6H) ; 7,63 (d, 2,6 Hz, 1 H) ; 8,04 (s(large), 1 H) ; 13,22 (s, 1 H).
Composé 41 : Acide 5-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]-2,2-diméthylpentanoïque

La synthèse de ce produit nécessite 2 étapes :
Etape 1 : 5-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-2,2- diméthylpentanoate de méthyle Cet intermédiaire est obtenu par substitution du 5-iodo-2,2-diméthylpentanoate de méthyle par le dérivé phénolique 4-4 selon le protocole D. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2).
Rendement : 76 %
Aspect : huile incolore RMN 1H (300MHz, CDCI3, δ en ppm) : 1 ,23 (s, 6H) ; 1 ,32 (d, 7,0 Hz, 3H) ; 1 ,68 (m,
4H) ; 2,98 (m, 1 H) ; 3,41 (m, 1H) ; 3,67 (s, 6H) ; de 3,82 à 3,96 (m, 3H) ; de 6,81 à
6,90 (m, 3H) ; 7,19 (d, 8,5 Hz, 2H) ; 7,36 (dd, 8,8 Hz et 2,9 Hz, 1H) ; 7,71 (s(large),
1 H) ; 8,17 (d, 2,9 Hz1 1H).
Etape 2 : acide 5-[4-[1-(5-chloro-2-méthoxybenzamido)proρan-2-yl]phénoxy]-2,2- diméthylpentanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par filtration et lavage par l'éther diéthylique. Rendement : 27 %
Aspect : poudre blanche
RMN 1H (300MHz, DMSO-d6, δ en ppm) : 1,10 (s, 6H) ; 1 ,19 (d, 6,7 Hz, 3H) ; 1 ,61 (m, 4H) ; 2,94 (m, 1 H) ; de 3,33 à 3,46 (m, 2H) ; 3,75 (s, 3H) ; 3,91 (t, 5,9 Hz, 2H) ; 6,87 (d, 8,8 Hz, 2H) ; de 7,11 à 7,19 (m, 3H) ; 7,49 (dd, 9,0 Hz, 2,9 Hz, 1H) ; 7,60 (d, 2,9 Hz, 1 H) ; 8,05 (s(large), 1H) ; 12,13 (s, 1H).
Composé 42 : Acide 2-[4-[2-(5-(2,4-difluorophényl)-2- méthoxybenzamido)propyl]phénoxy]-2-méthylpropanoïque

Etape 1 : 2-[4-[2-(5-(2,4-difluorophényl)-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par substitution du bromoisobutyrate d'éthyle par le dérivé phénolique 4-14 selon le protocole D. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 8/2).
Rendement : 65 %
Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : de 1 ,21 à 1 ,27 (m, 6H) ; 1 ,59 (s, 6H) ; de
2,76 à 2,93 (m, 2H) ; 3,91 (s, 3H) ; 4,22 (q, 7,0 Hz1 2H) ; de 4,43 à 4,52 (m, 1 H) ; 6,80 (d, 8,8 Hz1 2H) ; de 6,85 à 7,00 (m, 2H) ; 7,02 (d, 8,8 Hz, 1H) ; 7,12 (d, 8,8
Hz, 2H) ; 7,43 (m, 1 H) ; 7,60 (dd, 8,8 Hz, 2,6 Hz, 1 H) ; 7,76 (d, 7,9 Hz, 1 H) ; 8,33
(d, 2,6 Hz, 1 H).
Etape 2 : acide 2-[4-[2-(5-(2,4-difluorophényl)-2- méthoxybenzamido)propyl]phénoxy]~2-méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. Il est purifié par chromatographie sur gel de silice (dichlorométhane/méthanol 95/5). Rendement : 78 % Aspect : poudre blanche
RMN 1H (300MHz1 CDCI3, δ en ppm) : 1 ,21 (d, 6,7 Hz, 3H) ; 1 ,59 (s, 6H) ; de 2,75 à 2,94 (m, 2H) ; 3,90 (s, 3H) ; 4,48 (m, 1H) ; de 6,87 à 7,02 (m, 5H) ; 7,12 (d, 8,8 Hz, 2H) ; 7,42 (m, 1H) ; 7,60 (dd, 8,8 Hz, 2,0 Hz, 1 H) ; 7,81 (d, 7,9 Hz, 1H) ; 8,33 (d, 2,0 Hz, 1 H).
Composé 43 : Acide 2-[4-[2-(5-(pyrrol-1-yl)-2- méthoxybenzamido)propyl]phénoxy]-2-méthylpropanoïque

La synthèse de ce produit nécessite 2 étapes : Etape 1 : 2-[4-[2-(5-(pyrrol-1-yl)-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoate d'éthyle
Cet intermédiaire est obtenu par substitution du bromoisobutyrate d'éthyle par le dérivé phénolique 4-15 selon le protocole D. Il est purifié par chromatographie sur gel de silice (cyclohexane/acétate d'éthyle 7/3). Rendement : 75 % Aspect : huile incolore
RMN 1H (300MHz, CDCI3, δ en ppm) : de 1,22 à 1 ,30 (m, 6H) ; 1,59 (s, 6H) ; de 2,78 à 2,93 (m, 2H) ; 3,91 (s, 3H) ; 4,25 (q, 7,0 Hz, 2H) ; 4,46 (m, 1H) ; 6,34 (m, 2H) ; 6,80 (d, 8,5 Hz, 2H) ; 7,01 (d, 8,8 Hz, 1H) ; de 7,05 à 7,20 (m, 4H) ; 7,45 (dd, 8,8 Hz, 2,9 Hz, 1 H) ; 7,80 (d, 7,9 Hz, 1 H) ; 8,27 (d, 2,9 Hz, 1 H).
Etape 2 : acide 2-[4-[2-(5-(pyrrol-1-yl)-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque
Ce composé est obtenu par saponification de l'intermédiaire précédent suivant le protocole H. En fin de réaction, le solvant est évaporé, le milieu repris par l'eau et
lavé à l'éther diéthylique. Après acidification de la phase aqueuse par HCI 6M, le composé attendu est extrait par le dichlorométhane, lavé par NaCIsat, séché sur sulfate de magnésium (MgSO4), filtré et évaporé. Rendement : 52 % Aspect : poudre blanche
RMN 1 H (300MHz, CDCI3, δ en ppm) : 1 ,24 (d, 6,5 Hz, 3H) ; 1 ,59 (s, 6H) ; de 2,76 à 2,96 (m, 2H) ; 3,91 (s, 3H) ; 4,46 (m, 1H) ; 6,34 (m, 2H) ; 6,90 (d, 8,5 Hz, 2H) ; 7,01 (d, 8,8 Hz, 1H) ; 7,09 (m, 2H) ; 7,16 (d, 8,5 Hz, 2H) ; 7,45 (dd, 8,8 Hz, 2,9 Hz, 1H) ; 7,83 (d, 7,9 Hz, 1 H) ; 8,27 (d, 2,9 Hz, 1H).
Exemple 7 : Evaluation in vitro des propriétés activatrices des PPAR des composés selon l'invention
Les propriétés activatrices des PPARs des composés selon l'invention sont évaluées in vitro.
Principe
L'activation des PPARs est évaluée in vitro sur une lignée de fibroblastes de rein de singe (COS-7) par la mesure de l'activité transcriptionnelle de chimères constituées du domaine de liaison à l'ADN du facteur de transcription Gal4 de la levure et du domaine de liaison au ligand des différents PPAR. Les composés sont testés à des doses comprises entre 0,01 et 10 μM sur les chimères Gal4- PPARα, γ, δ. Le facteur d'induction (rapport entre la luminescence induite par le composé et la luminescence induite par le contrôle) est mesuré pour chaque condition. Il est ensuite normalisé par rapport à un composé de référence interne et les résultats sont exprimés en pourcentages : plus le pourcentage d'activation est élevé, plus le composé a un caractère activateur des PPARs.
Protocole a- Culture des cellules
Les cellules COS-7 proviennent de l'ATCC et sont cultivées dans du milieu DMEM supplémenté de 10% (vol/vol) de sérum de veau fœtal, 1% de
pénicilline/streptomycine (Biochrom, AG), 1% d'acides aminés (Gibco) et 1% de pyruvate de sodium (Gibco). Les cellules sont incubées à 37°C dans une atmosphère humide contenant 5% de CO2.
b- Description des plasmides utilisés en transfection
Les plasmides Gal4(RE)_TkpGL3, pGal4-hPPARα, pGal4-hPPARγ, pGal4- hPPARδ et pGal4-φ ont été décrits dans la littérature (Raspe E et al., 1999). Les constructions pGal4-hPPARα, pGal4-hPPARγ et pGal4-hPPARδ ont été obtenues par clonage dans le vecteur pGal4-φ de fragments d'ADN amplifiés par PCR correspondants aux domaines DEF des récepteurs nucléaires PPARα, PPARγ et PPARδ humains.
c- Transfection
Les cellules COS-7 en suspension sont transfectées avec 150 ng d'ADN par puits, avec un ratio pGal4-PPAR / Gal4(RE)_TkpGL3 de 1/10, en présence de 10% de sérum de veau fœtal. Les cellules sont ensuite ensemencées dans des plaques de 96 puits (4x104 cellules/puits) puis incubées pendant 24 heures à 37°C.
L'activation avec les composés à tester s'effectue pendant 24h à 37°C dans du milieu sans sérum. A l'issue de l'expérience, les cellules sont lysées et l'activité luciférase est déterminée à l'aide du Steady-Lite™ HTS (Perkin Elmer) selon les recommandations du fournisseur.
Résultats
De manière inattendue, les données expérimentales présentées ci-après montrent que les composés selon l'invention lient les PPARs in vitro et induisent une activation de l'activité transcriptionnelle.
Exemple 7-1 : Transactivation de pGal4-hPPAR (α/γ/δ) à 10 μM Les composés selon l'invention ont été testés à 10 μM sur les 3 isoformes de PPAR. Les résultats obtenus sont détaillés sur la figure (1). La transactivation mesurée est exprimée en pourcentage de réponse par rapport à un composé de référence interne pour chacun des isoformes.
L'analyse de cette figure montre que les composés selon l'invention sont agonistes des récepteurs nucléaires PPAR : les composés selon l'invention lient et activent hPPARα, hPPARγ, et/ou hPPARδ de manière significative. Les niveaux de transactivation obtenus grâce aux composés selon l'invention sont variables selon le sous-type de PPAR étudié et différents pour chaque composé.
Aussi, on observe de manière surprenante parmi les composés selon l'invention plus ou moins de sélectivité vis-à-vis des isoformes de PPAR à 10 μM :
- Certains composés selon l'invention sont sélectifs par rapport à un sous- type de PPAR : c'est par exemple le cas des composés 14 et 26 sélectifs de hPPARα ; ces derniers n'activent pas significativement hPPARγ ou hPPARδ à iO μM.
- Certains composés selon l'invention sont simultanément activateurs de deux sous-types de PPAR : à titre d'exemple, les composés 15 et 31 activent hPPARα et hPPARγ à 10μM, mais n'activent pas hPPARδ.
- Certains composés selon l'invention sont simultanément activateurs des trois sous-types de PPAR : à titre d'exemple, les composés 2 et 19 sont à la fois ligands de hPPARα, hPPARγ et hPPARδ.
Exemple 7-2 : Transactivation de pGal4-hPPAR (α/γ/δ) en fonction de la dose
Les composés selon l'invention ont été testés à des doses comprises entre 0,01 et 10 μM sur les 3 isoformes de PPAR. Les résultats obtenus sont détaillés sur la figure (2) : évolution du niveau de transactivation en fonction de la dose pour les trois sous-types de PPAR (α, γ, et δ).
Les inventeurs mettent en évidence une augmentation significative et dose- dépendante de l'activité luciférase dans les cellules transfectées avec les plasmides pGal4-hPPAR (α/γ/δ) et pGal4(RE) traitées avec les composés selon l'invention. Aussi, les résultats présentés sur la figure (2) montrent que les EC50 et les niveaux d'activation sont très différents selon le sous-type de PPAR étudié : à titre d'exemple, le composé 29 active hPPARα à près de 50 % avec une EC50
de l'ordre de 30 nM alors que le même composé active hPPARδ à près de 100 % avec une EC50 de l'ordre de 300 nM.
Comme illustré dans l'exemple 7-1 , la sélectivité des composés est étroitement associée à leur structure. De manière intéressante et complémentaire, les résultats présentés sur la figure (2) montrent que les propriétés des composés sont aussi « énantiomère-dépendantes » : l'énantiomère 29A active préférentiellement les isoformes α et δ de hPPAR alors que 29B active spécifiquement hPPARα.
Conclusion
Ces résultats montrent que les composés selon l'invention lient et activent les récepteurs hPPARα, et/ou hPPARγ, et/ou hPPARδ de manière significative. Les niveaux de transactivation obtenus grâce aux composés selon l'invention sont variables selon la structure du composé testé et selon le sous-type de PPAR étudié.
Exemple 8 : Evaluation in vitro de l'affinité des composés selon l'invention pour les canaux potassiques ATP-dépendants
L'objet de cette étude est d'évaluer in vitro l'interaction des composés selon l'invention avec les canaux potassiques ATP-dépendants (K+ ATP), cible connue des médicaments insulino-sécréteurs actuellement commercialisés.
Principe
Les résultats présentés reflètent l'affinité spécifique des composés selon l'invention pour les canaux potassiques ATP-dépendants (K+ ATP)- Le test de « binding » a été réalisé par MDS Pharma Services (Taiwan) sur des cellules pancréatiques de hamster HIT-T15. Le composé de référence est le [3H]glibenclamide à 5 nM. Les composés selon l'invention ont été testés à 100 μM.
Résultats
Les résultats présentés sur la figure (3) montrent l'affinité des composés selon l'invention à 100 μM pour les canaux potassiques ATP-dépendants. Le ligand radiomarqué est déplacé de manière spécifique par les composés selon l'invention à des niveaux significatifs (exprimés en %). Plus le pourcentage mesuré est élevé, plus l'affinité du composé pour les canaux potassiques ATP-dépendants est forte.
Conclusion De manière inattendue, les inventeurs ont mis en évidence une affinité spécifique des composés selon l'invention pour les canaux potassiques ATP-dépendants (K+
ATP)-
Exemple 9 : Evaluation in vitro du caractère insulinosécréteur des composés selon l'invention
Les propriétés insulinosécrétrices des composés selon l'invention sont évaluées in vitro.
Principe
L'activation de la sécrétion d'insuline est évaluée in vitro sur une lignée de cellules pancréatiques de rat (INS-1) par la mesure de la concentration d'insuline sécrétée dans le milieu de culture en présence d'une faible concentration de glucose (2,8 mM). Les composés sont testés à des doses comprises entre 1 et 100 μM. Les résultats sont représentés par le facteur d'induction pour chaque condition, c'est- à-dire le rapport entre la concentration d'insuline induite par le composé selon l'invention et la concentration induite par le glucose 2,8 mM seul : plus ce facteur est élevé, plus le composé a un caractère insulinosécréteur.
Protocole
Les cellules INS-1 sont cultivées dans des plaques 24 puits (3x105 cellules/puits) pendant 72 heures dans du milieu RPMI 1640 supplémenté avec les additifs
suivants : glucose (5mM) ; SVF Biowest décomplémenté (10%) ; glutamine (1%) ; Pénicilline-Streptomycine (1%) ; Pyruvate de Sodium (1%); Tampon Hepes (1OmM) ; 2-mercaptoéthanol (50μM) ; hydrochloride d'aminoguanidine (1OmM) ; G418 (15 μL/50 ml_ de milieu). Le milieu est ensuite changé par du RPMI avec 2,8 mM glucose (mêmes additifs) pendant 24 heures. Après un lavage au tampon Krebs [NaCI (14OmM); KCI (3,6mM); CaCI2 (1 ,5mM); NaH2PO4 (0,5mM) ; MgSO4 (0,5mM) ; NaHCO3 (2mM) ; Hépès (10 mM) ; BSA (0.1%) ; pH 7.4], une incubation de 30 min à 37°C dans ce tampon est réalisée. Le traitement avec différentes doses de composés selon l'invention est alors effectué durant 20 min à 370C. Suite à ce traitement, la concentration d'insuline sécrétée dans le milieu de culture est mesurée dans chaque puits à l'aide d'une trousse ELISA (Insulin Elisa Kit - Crystal Chem., USA).
Résultats Les résultats présentés sur la figure (4) mettent en évidence une augmentation de la sécrétion d'insuline dans les cellules INS-1 traitées avec les composés selon l'invention : les facteurs d'induction mesurés sont significatifs, variables d'un composé à l'autre, et dose-dépendants.
Conclusion
De manière inattendue, les données expérimentales montrent que les composés selon l'invention stimulent la sécrétion d'insuline in vitro.
Exemple 10 : Evaluation in vivo du caractère hvpoqlvcémiant des composés
L'objet de cette étude est d'évaluer in vivo le caractère hypoglycémiant des composés selon l'invention.
Principe :
Chez des animaux normaux (comme chez l'homme), l'administration de composés insulino-sécréteurs a pour conséquence une augmentation significative du taux plasmatique d'insuline. Cette hyper-insulinémie induite provoque une baisse de la
glycémie. Cet exemple étudie l'évolution de la glycémie après administration orale des composés selon l'invention chez le rat : l'action insulino-sécrétrice des composés in vivo doit notamment se traduire par une chute de la glycémie après administration.
Protocole :
Des rats de sexe mâle (300 - 320 g) Sprague-Dawley (CERJ - Le Genest St IsIe- France) sont utilisés pour réaliser cette expérience. Les rats sont alimentés avec un régime standard en granulés ; ils ont libre accès à la nourriture et à la boisson et sont hébergés avec un rythme lumière/obscurité de 12h/12h dans des cages individuelles ventilées. Les animaux sont privés de nourriture 16 heures avant l'expérience.
Le composé selon l'invention est suspendu dans une solution aqueuse de carboxyméthylcellulose (Sigma C4888) à 1% contenant 0,1% de TweenδO (Sigma P8074). Chaque groupe est constitué de 5 animaux. Un 1er recueil de sang est effectué sous anesthésie volatile à l'isoflurane par une ponction au sinus rétro- orbital des animaux: cet échantillon constitue le temps initial de l'expérience. La substance ou le véhicule est ensuite immédiatement administré aux animaux par gavage et des prélèvements sanguins sont effectués au fil du temps. Les animaux du groupe contrôle reçoivent le véhicule seul alors que les animaux traités reçoivent une administration unique du composé (la suspension est administrée par gavage à raison de 10ml/kg).
La glycémie des animaux est mesurée en temps réel à l'aide d'un glucomètre (Glucotrend2-Roche Diagnostic-France). Les insulinémies sont mesurées à l'aide d'une trousse ELISA (Insulin Elisa Kit- Crystal Chem. USA).
Résultats :
Les résultats présentés sur la figure (5) montrent que les composés selon l'invention ont un effet hypoglycémiant : le composé 5 administré à 300 mg/kg/jour induit une diminution significative de la glycémie dès 30 min après l'administration. De plus, l'effet observé est dose-dépendant : le même composé administré à 30 mg/kg/jour n'induit pas d'effet significatif.
Conclusion
Les inventeurs ont mis en évidence que les composés selon l'invention permettent de réguler la glycémie. De telles molécules présentent donc un intérêt majeur dans le cadre du diabète de type 2 et des pathologies associées.
Exemple 11 : Evaluation in vivo des propriétés hypolipémiantes des composés selon l'invention
Cette étude a pour objectif d'évaluer le potentiel hypolipémiant des composés selon l'invention in vivo.
Principe :
L'effet hypolipémiant des composés selon l'invention est évalué in vivo chez la souris hypercholestérolémique E2/E2 : cette souris est humanisée pour l'isoforme e2 de l'apolipoproteine E ce qui affecte la clairance hépatique des lipides et conduit à une hyperlipidémie. Les taux de cholestérol plasmatique sont mesurés après 7 jours de traitement par voie orale par les composés selon l'invention ; ces taux sont comparés à ceux obtenus avec des animaux contrôles (non traités par les composés selon l'invention) : la différence mesurée témoigne de l'effet hypolipémiant des composés selon l'invention.
Le sacrifice de ces animaux permet en plus d'évaluer la capacité des composés selon l'invention à induire une activité transcriptionnelle. Les propriétés agonistes de PPARα préalablement mesurées in vitro doivent se traduire au niveau hépatique par une sur-expression des gènes cibles dont l'expression est sous le contrôle du récepteur PPARα. Les gènes que nous étudions dans cette expérience sont PDK4 (Pyruvate Deshydrogénase Kinase isoforme 4, enzyme du métabolisme glucidique) et l'ACO (acyl Co-enzymeA oxydase, une enzyme clé impliquée dans le mécanisme de la β-oxydation des acides gras). Plus le facteur d'induction mesuré est élevé, plus le composé testé augmente l'expression du gène étudié.
Protocole
a- Traitement des animaux
Des souris E2/E2 (Sullivan PM et al., 1998) femelles âgées de 24 semaines au début de l'expérience ont été rassemblées par groupes de 6 animaux, sélectionnés de telle sorte que la distribution de leur taux de lipides plasmatiques déterminés une première fois avant l'expérience soit uniforme. Les souris reçoivent un régime standard A04 (SAFE-Augy-France). Elles sont maintenues sous un cycle lumière/obscurité de 12 heures/12 heures à une température constante de 20 ± 3°C et ont libre accès à l'eau et à la nourriture. La prise de nourriture et la prise de poids sont enregistrées. Les composés testés sont suspendus dans la carboxyméthylcellulose (Sigma C4888) à 1% contenant 0,1% de TweenδO (Sigma P8074) et administrés quotidiennement par gavage intra- gastrique pendant 7 jours à la dose de 300 mg/kg/jour. A l'issue de l'expérience, les animaux sont anesthésiés après un jeun de 5 heures et un prélèvement sanguin est effectué sur anticoagulant (EDTA). Les souris sont ensuite pesées puis euthanasiées. Le plasma est préparé par centrifugation à 3000 tours/minutes pendant 20 minutes, les échantillons sont conservés à + 4°C. Les échantillons de foie sont prélevés, congelés dans l'azote liquide et conservés à - 800C pour des analyses ultérieures.
b- Dosage des lipides
Les concentrations plasmatiques de cholestérol total sont mesurées par dosages enzymatiques (bioMérieux-Lyon-France) selon les recommandations du fournisseur.
c- Analyse d'expression qénique par RT-PCR quantitative
L'ARN total est extrait à partir de fragments de foie en utilisant le kit NucleoSpin® 96 RNA (Macherey Nagel, Hoerdt, France) selon les instructions du constructeur. 1 μg d'ARN total (quantifié en utilisant le Ribogreen RNA quantification kit (Molecular Probes)) est ensuite transcrit de manière reverse en ADN complémentaire par une réaction d'une heure à 370C dans un volume total de 20 μl contenant du tampon 1X (Sigma), 1.5 mM de DTT, 0,18 mM de dNTPs (Promega), 200 ng de pdN6 (Amersham), 3OU d'inhibiteur de RNase (Sigma) et 1 μl de MMLV-RT (Sigma). Les expériences de PCR quantitative ont été effectuées
en utilisant le MyiQ Single-Color Real-Time PCR Détection System (Biorad, Marnes-la-Coquette, France). Brièvement, les réactions de PCR ont été réalisées, en utilisant le kit iQ SYBR Green Supermix selon les recommandations du fournisseur, en plaques 96 puits sur 5 μl de réactions de reverse transcription dilué avec une température d'hybridation de 55°C. Des paires d'amorces spécifiques des gènes étudiés ont été utilisées. Les séquences de ces dernières pour l'étude de PDK4 sont pour l'amorce sens : 5'- TACTCCACTGCTCCAACACCTG-3' (SEQ ID No : 1) et pour l'amorce antisens 5'- GTTCTTCGGTTCCCTGCTTG-3' (SEQ ID No : 2). Les séquences des amorces spécifiques pour ACO sont pour l'amorce sens : 5'- GAAGCCAGCGTTACGAGGTG-3' (SEQ ID No : 3) et pour l'amorce antisens 5'- TGGAGTTCTTGGGACGGGTG-3' (SEQ ID No : 4). La quantité de fluorescence émise est directement proportionnelle à la quantité d'ADN complémentaire présent au début de la réaction et amplifiée au cours de la PCR. Pour chaque cible étudiée, une gamme est réalisée par des dilutions successives d'un pool constitué de quelques μl de différentes réactions de reverse transcription. Les niveaux d'expression relatifs de chaque cible sont ainsi déterminés en utilisant les courbes d'efficacité obtenues avec les points de gamme. Les niveaux d'expression des gènes d'intérêt sont ensuite normalisés par rapport au niveau d'expression du gène de référence 36B4 (dont les amorces spécifiques utilisées ont pour séquences amorce sens : 5'-
CATGCTCAACATCTCCCCCTTCTCC-S1 (SEQ ID No : 7); amorce antisens : 5'- GGGAAGGTGTAATCCGTCTCCACAG -3' (SEQ ID No : 8)). Le facteur d'induction, c'est-à-dire le rapport entre le signal relatif (induit par le composé selon l'invention) et la moyenne des valeurs relatives du groupe contrôle, est ensuite calculé pour chaque échantillon. Plus ce facteur est élevé, plus le composé a un caractère activateur d'expression génique. Le résultat final est représenté comme moyenne des valeurs d'induction dans chaque groupe expérimental.
Résultats
La figure (6a) relate l'effet du composé 5 administré à 300 mg/kg/jour pendant 7 jours chez la souris hypercholestérolémique. De manière inattendue, les taux de
cholestérol plasmatique sont très significativement diminués par le traitement (plus de 50 % de diminution par rapport aux animaux contrôle).
Aussi, les inventeurs ont mis en évidence que les composés selon l'invention sont des régulateurs de l'expression de gènes in vivo. Les résultats présentés sur les figures (6b) et (6c) montrent que les composés selon l'invention induisent une augmentation significative de l'expression hépatique des gènes codant pour PDK4 et ACO. Ces gènes (dont le niveau d'expression est connu pour être régulé par
PPARα) codent pour des enzymes fortement impliquées dans le métabolisme des lipides et glucides et leur sur-expression renforce l'idée que les composés selon l'invention présentent un intérêt potentiel majeur dans le cadre des maladies métaboliques.
Conclusion Les inventeurs ont mis en évidence que les composés selon l'invention ont des propriétés hypolipémiantes. Ces résultats obtenus in vivo témoignent du potentiel thérapeutique des composés selon l'invention vis-à-vis de pathologies majeures telles que les dyslipidémies.
Exemple 12 : Evaluation in vivo, chez la souris E2/E2, des propriétés hypolipémiantes et stimulatrices de la synthèse du HDL-cholestérol des composés selon l'invention
Principe
Un traitement par les activateurs de PPARδ se traduit chez l'homme comme chez le rongeur, par une augmentation du taux de HDL-cholestérol plasmatique. Les propriétés agonistes de PPARδ des molécules selon l'invention préalablement mesurées in vitro devraient aussi se traduire au niveau du muscle squelettique par une sur-expression des gènes cibles connus pour être directement sous le contrôle du récepteur PPARδ comme par exemple les gènes codant pour UCP2 et UCP3 (Uncoupling Protein 2 et 3, des transporteurs mitochondriaux impliqués dans la thermorégulation).
Les propriétés hypolipémiantes et stimulatrices de la synthèse du HDL-cholestérol des composés selon l'invention ont été évaluées in vivo par dosage des lipides plasmatiques, analyse de la répartition du cholestérol dans les différentes fractions lipoprotéiques plasmatiques et par mesure de l'expression génique de gènes cibles des PPAR dans le foie et le muscle squelettique après traitement, par les composés selon l'invention, de la souris E2/E2 dyslipidémique.
Protocole a- Traitement des animaux Des souris transgéniques E2/E2 ont été maintenues sous un cycle lumière/obscurité de 12/12 heures à une température constante de 20 ± 3°C. Après une acclimatation d'une semaine, les souris ont été pesées et rassemblées par groupes de 6 animaux sélectionnés de telle sorte que la distribution de leurs poids corporels et de leurs taux de lipides plasmatiques déterminés une première fois avant l'expérience soient uniformes. Les composés testés ont été suspendus dans la carboxyméthylcellulose (Sigma C4888) et administrés par gavage intra- gastrique, à raison d'une fois par jour pendant 7 jours à la dose choisie. Les animaux ont eu un accès libre à l'eau et à la nourriture (régime standard). A l'issue de l'expérience, les animaux ont été anesthésiés après un jeûne de 4 heures, un prélèvement sanguin a été effectué sur anticoagulant (EDTA) puis les souris ont été pesées et euthanasiées. Le plasma a été séparé par centrifugation à 3000 tours/minutes pendant 20 minutes, les échantillons ont été conservés à + 4°C. Des échantillons de foie et de tissu musculaire squelettique (muscle gastrocnémien) ont été prélevés et congelés immédiatement dans de l'azote liquide puis conservés à -800C pour les analyses ultérieures.
b- Analyse de la répartition du cholestérol dans les fractions lipoprotéiques plasmatiques.
Les différentes fractions lipidiques (VLDL, LDL, HDL) du plasma ont été séparées par une chromatographie Gel - Filtration. Les concentrations de cholestérol ont ensuite été mesurées dans chaque fraction par dosages enzymatiques (bioMérieux-Lyon-France) selon les recommandations du fournisseur.
c- Mesure des lipides plasmatiques
Les concentrations plasmatiques de triglycérides ont été mesurées par dosages enzymatiques (bioMérieux-Lyon-France) selon les recommandations du fournisseur.
d- Analyse d'expression génique par RT-PCR quantitative
L1ARN total hépatique est extrait à partir de fragments de foie en utilisant le kit NucleoSpin® 96 RNA (Macherey Nagel, Hoerdt, France) selon les instructions du fabricant. Pour le tissu squelettique, l'ARN total est extrait à partir de fragments de muscle squelettique gastrocnémien en utilisant le kit RNeasy® Fibrous Tissue kit (Qiagen) selon les instructions du fabricant.
1 μg d'ARN total (quantifié par spectrophotométrie) est ensuite « reverse transcrit » en ADN complémentaire par une réaction d'1 heure à 37°C dans un volume total de 20 μl contenant du tampon 1X (Sigma), 1 ,5mM de DTT, 0,18mM de dNTPs (Promega), 200ng de pdN6 (Amersham), 3OU d'inhibiteur de RNase (Sigma) et 1 μl de MMLV-RT (Sigma).
Les expériences de PCR quantitative sont effectuées en utilisant le MyiQ Single- Color Real-Time PCR Détection System (Biorad, Mames-la-Coquette, France) et sont réalisées en utilisant le kit iQ SYBR Green Supermix selon les recommandations du fournisseur, en plaques 96 puits, sur 5 μl de réaction de reverse transcription diluée avec une température d'hybridation de 55°C. Des paires d'amorces spécifiques des gènes étudiés sont utilisées : PDK4 (amorce sens : 5'- TACTCCACTGCTCCAACACCTG-3' (SEQ ID No : 1) ; amorce antisens 5'- GTTCTTCGGTTCCCTGCTTG-3' (SEQ ID No : 2)) , ACO (amorce sens : 5'- GAAGCCAGCGTTACGAGGTG-3' (SEQ ID No : 3) ; amorce antisens 5'- TGGAGTTCTTGGGACGGGTG-3' (SEQ ID No : 4)), et UCP3 (amorce sens : 51- GCACCGCCAGATGAGTTTTG-S1 (SEQ ID NO : 5) ; amorce antisens : 51- GACGCTGGAGTGGTCCGCTC-3' (SEQ ID NO : 6)).
Dans les deux cas (tissu hépatique et tissu musculaire squelettique), la quantité de fluorescence émise est directement proportionnelle à la quantité d'ADN complémentaire présent au début de la réaction et amplifié au cours de la PCR. Pour chaque cible étudiée, une gamme est réalisée par des dilutions successives
d'un pool constitué de quelques microlitres de différentes réactions de « reverse transcription ». Les niveaux d'expression relatifs de chaque cible sont ainsi déterminés en utilisant les courbes d'efficacité obtenues avec les points de gamme. Les niveaux d'expression des gènes d'intérêt ont ensuite été normalisés, dans le tissu hépatique par rapport au niveau d'expression du gène de référence 36B4 et, dans le tissu musculaire squelettique par rapport au niveau d'expression du gène de référence 18S (dont les amorces spécifiques utilisées ont pour séquences amorce sens : 5>-CGGACACGGACAGGATTGACAG-3I (SEQ ID NO : 9) et amorce antisens : 5I-AATCTCGGGTGGCTGAACGC-3I (SEQ ID NO : 1O)). Le facteur d'induction, c'est-à-dire le rapport entre le signal relatif (induit par le composé selon l'invention) et la moyenne des valeurs relatives du groupe contrôle, a ensuite été calculé pour chaque échantillon. Plus ce facteur est élevé, plus le composé a un caractère activateur d'expression génique. Le résultat final est représenté comme moyenne des valeurs d'induction dans chaque groupe expérimental.
Résultats
La figure (7a) présente la répartition du cholestérol dans les différentes fractions lipoprotéiques plasmatiques des souris E2/E2 contrôles ou traitées pendant 7 jours avec le composé 29 à 50 mg/kg/jour. De manière inattendue, les taux plasmatiques de LDL- et VLDL-cholestérol ont été diminués et le taux de HDL- cholestérol plasmatique a été augmenté par le traitement avec le composé 29, administré à la dose de 50 mg/kg/jour.
La figure (7b) compare les taux de triglycérides plasmatiques après 7 jours de traitement avec le composé 29 à 50 mg/kg/jour aux taux obtenus avec les animaux contrôle. De manière inattendue, les taux de triglycérides plasmatiques ont été significativement diminués par le traitement.
Les inventeurs ont aussi mis en évidence que les composés selon l'invention sont, in vivo, des régulateurs de l'expression de gènes cibles des PPARs. Les résultats présentés sur les figures (7c), (7d), (7e) montrent que le composé 29 selon l'invention, administré à 50 mg/kg/jour pendant 7 jours à des souris E2/E2, induit une augmentation significative de l'expression hépatique des gènes codant pour
PDK4 (figure (7c)), l'ACO (figure (7d)), ainsi qu' une augmentation significative dans le muscle squelettique de l'expression du gène codant pour UCP3 (figure (7f)). L'ensemble de ces gènes codent pour des enzymes fortement impliquées dans le métabolisme des lipides et des glucides ainsi que la dissipation d'énergie. La modulation de leur expression par les composés selon l'invention renforce l'idée que ces composés présentent un intérêt potentiel majeur dans le cadre des pathologies métaboliques.
Conclusion De manière inattendue, les données expérimentales présentées montrent que les composés selon l'invention stimulent in vivo la synthèse de HDL-cholestérol en parallèle d'un effet hypolipémiant (diminution des taux plasmatiques de cholestérol total et de triglycérides). De plus, les données expérimentales présentées montrent que les composés selon l'invention modulent l'expression de gènes (régulés par l'activation des PPARs) qui codent pour des enzymes fortement impliquées dans le métabolisme des lipides et des glucides et dans la dissipation d'énergie.
Exemple 13 : Evaluation in vivo, chez la souris C57BI6, des propriétés hvpolipémiantes et stimulatrices de la synthèse de HDL cholestérol des composés selon l'invention
Principe Les propriétés hypolipémiantes et stimulatrices de la synthèse du HDL-cholestérol des composés selon l'invention ont été évaluées in vivo par dosage des lipides plasmatiques et par mesure de l'expression génique de gènes cibles des PPARs dans le tissu hépatique après traitement par voie orale, par les composés selon l'invention, de la souris « normale » C57BI6.
Protocole a- Traitement des animaux
Des souris C57BI6 femelles ont été maintenues sous un cycle lumière/obscurité de 12/12 heures à une température constante de 20 ± 30C. Après une acclimatation d'une semaine, les souris ont été pesées et rassemblées par groupes de 6 animaux sélectionnés de telle sorte que la distribution de leurs poids corporel et de leurs taux de lipides plasmatiques déterminés une première fois avant l'expérience soient uniformes. Les composés testés ont été suspendus dans la carboxyméthylcellulose (Sigma C4888) et administrés par gavage intra- gastrique, à raison d'une fois par jour pendant 14 jours à la dose choisie. Les animaux ont eu un accès libre à l'eau et à la nourriture (régime standard). A l'issue de l'expérience, les animaux ont été anesthésiés après un jeûne de 4 heures, un prélèvement sanguin a été effectué sur anticoagulant (EDTA) puis les souris ont été pesées et euthanasiées. Le plasma a été séparé par centrifugation à 3000 tours/minutes pendant 20 minutes, les échantillons ont été conservés à + 4°C. Des échantillons de tissu hépatique ont été prélevés et congelés immédiatement dans de l'azote liquide puis conservés à -800C pour les analyses ultérieures.
b- Mesure du HDL-cholestérol
Les lipoprotéines de basse densité (VLDL et LDL) sont précipitées par phosphotungstate. Le précipité est éliminé par centrifugation. Le HDL-cholestérol présent dans le surnageant est quantifié par dosages enzymatiques (bioMérieux- Lyon-France) selon les recommandations du fournisseur.
c- Mesure des triglycérides plasmatiques
Les concentrations plasmatiques de triglycérides ont été mesurées par dosages enzymatiques (bioMérieux-Lyon-France) selon les recommandations du fournisseur.
d- Analyse d'expression qénique par RT-PCR quantitative L'ARN total hépatique est extrait à partir de fragments de foie en utilisant le kit NucleoSpin® 96 RNA (Macherey Nagel, Hoerdt, France) selon les instructions du fabricant.
1 μg d'ARN total (quantifié par spectrophotométrie) est ensuite « reverse transcrit » en ADN complémentaire par une réaction d'1 heure à 370C dans un
volume total de 20 μl contenant du tampon 1X (Sigma), 1 ,5mM de DTT, 0,18mM de dNTPs (Promega), 200ng de pdN6 (Amersham), 3OU d'inhibiteur de RNase (Sigma) et 1 μl de MMLV-RT (Sigma).
Les expériences de PCR quantitative sont effectuées en utilisant le MyiQ Single- Color Real-Time PCR Détection System (Biorad, Marnes-la-Coquette, France) et sont réalisées en utilisant le kit iQ SYBR Green Supermix selon les recommandations du fournisseur, en plaques 96 puits, sur 5 μl de réaction de reverse transcription diluée avec une température d'hybridation de 55°C. Des paires d'amorces spécifiques des gènes PDK4 (amorce sens : 5'- TACTCCACTGCTCCAACACCTG-3' (SEQ ID No : 1) ; amorce antisens 5'- GTTCTTCGGTTCCCTGCTTG-3' (SEQ ID No : 2)), ACO (amorce sens : 5'- GAAGCCAGCGTTACGAGGTG-3' (SEQ ID No : 3) ; amorce antisens 5'- TGGAGTTCTTGGGACGGGTG-3' (SEQ ID No : 4)) étudiés ont été utilisées.
La quantité de fluorescence émise est directement proportionnelle à la quantité d'ADN complémentaire présent au début de la réaction et amplifié au cours de la PCR. Pour chaque cible étudiée, une gamme est réalisée par des dilutions successives d'un pool constitué de quelques microlitres de différentes réactions de reverse transcription. Les niveaux d'expression relatifs de chaque cible sont ainsi déterminés en utilisant les courbes d'efficacité obtenues avec les points de gamme. Les niveaux d'expression des gènes d'intérêt ont ensuite été normalisés, par rapport au niveau d'expression du gène de référence 36B4. Le facteur d'induction, a ensuite été calculé pour chaque échantillon. Plus ce facteur est élevé, plus le composé a un caractère activateur d'expression génique. Le résultat final est représenté comme moyenne des valeurs d'induction dans chaque groupe expérimental.
Résultats
La figure (8a) compare les taux plasmatiques de HDL-cholestérol après 14 jours de traitement avec le composé 29 à 50 mg/kg/jour aux taux obtenus avec les animaux contrôle. De manière inattendue, les taux circulants de HDL-cholestérol sont très significativement augmentés par le traitement.
Les figures (8b) et (8c) comparent les taux plasmatiques de triglycérides et d'acides gras libres après 14 jours de traitement avec le composé 29 à 50 mg/kg/jour aux taux obtenus avec les animaux contrôle. De manière inattendue, les taux circulants de triglycérides et d'acides gras libres ont été très significativement diminués par le traitement.
Les inventeurs ont aussi mis en évidence que les composés selon l'invention sont, in vivo, des régulateurs de l'expression de gènes cibles des PPARs. Les résultats présentés sur les figures (8d) et (8e) montrent que le composé 29 selon l'invention, administré à 50 mg/kg/jour pendant 14 jours à des souris C57BI6, induit une augmentation significative de l'expression hépatique des gènes codant pour PDK4 (figure (8d)) et l'ACO (figure (8e)). L'ensemble de ces gènes codent pour des enzymes fortement impliquées dans le métabolisme des lipides, des glucides et le fait que leur expression soit modulée par les composés selon l'invention renforce l'idée que ces composés présentent un intérêt potentiel majeur dans le cadre des pathologies métaboliques.
Conclusion
De manière inattendue, les données expérimentales présentées montrent que les composés selon l'invention stimulent in vivo la synthèse de HDL-cholestérol en parallèle d'un effet hypolipémiant marqué (diminution des taux plasmatiques de triglycérides et d'acides gras libres). De plus, les données expérimentales présentées montrent que les composés selon l'invention modulent l'expression de gènes régulés par l'activation des PPARs qui codent pour des enzymes fortement impliquées dans le métabolisme des lipides et des glucides.
Exemple 14 : Evaluation in vitro des propriétés activatrices du transport inverse du cholestérol des composés selon l'invention
Principe
L'effet des composés selon l'invention sur le transport inverse de cholestérol a été évalué par la mesure de l'expression du gène ABCA1 dans des macrophages.
Plus l'expression d'ABCAl est augmentée, plus le composé selon l'invention stimule le transport inverse du cholestérol.
Protocole a- Différenciation des cellules THP1 en macrophages.
La lignée de monocytes humains THP1 (provenance ATCC) est cultivée dans du milieu RPMI1640 additionné de 25mM Hepes (Gibco ; 42401-018), 1% glutamine (Gibco ; 25030-24), 1% pénicilline/streptomycine (Biochrom AG ; A 2213) et 10% de sérum de veau fœtal décomplémenté (SVF. Gibco ; 26050-088). Les cellules sont ensemencées sur des plaques de 24 puits (Primaria BD Falcon) à la densité de 3.105 cellules/puit puis incubées à 37°C et 5% de CO2 pendant 72h en présence de 30 ng/ml de phorbol 12-myristate 13-acétate (PMA) afin de les différencier en macrophages.
b- Traitement
Le milieu de différenciation est aspiré et remplacé par le milieu de traitement
(même composition que le milieu de culture mais sans sérum de veau fœtal et avec 1% d'ultroser (PaII Life Science ; P/N267051)).
Les composés selon l'invention sont dissous dans du diméthyl sulfoxide (DMSO, Fluka ; 41640). Les composés selon l'invention ont été testés à la dose de 10μM.
Les cellules sont traitées pendant 24h à 370C1 5% CO2. L'effet des composés selon l'invention est comparé à l'effet du DMSO seul.
c- Extraction des ARN, transcription inverse et PCR quantitative. Après traitement, l'ARN total est extrait des cellules en utilisant le kit NucleoSpin® 96 RNA (Macherey Nagel, Hoerdt, France) selon les instructions du fabricant. 1 μg d'ARN total (quantifié par lecture au spectrophotomètre) a ensuite été reverse transcrit en ADN complémentaire par une réaction d'1 heure à 37°C dans un volume total de 20 microlitres contenant du tampon 1X (Sigma), 1 ,5mM de DTT, 0,18mM de dNTPs (Promega), 200ng de pdN6 (Amersham), 3OU d'inhibiteur de RNase (Sigma) et 1 μl de MMLV-RT (Sigma).
Les expériences de PCR quantitative sont effectuées en utilisant le MyiQ Single- Color Real-Time PCR Détection System (Biorad, Marnes-la-Coquette, France) et
sont réalisées en utilisant le kit iQ SYBR Green Supermix selon les recommandations du fournisseur, en plaques 96 puits, sur 5 μl de réactions de reverse transcription diluées avec une température d'hybridation de 55°C. Des paires d'amorces spécifiques d'ABCAl sont utilisées : amorce sens : 5'- CTGAGGTTGCTGCTGTGGAAG-3' (SEQ ID NO : 11) et amorce antisens : 5'- CATCTGAGAACAGGCGAGCC-3' (SEQ ID NO : 12).
La quantité de fluorescence émise est directement proportionnelle à la quantité d'ADN complémentaire présent au début de la réaction et amplifié au cours de la PCR. Une gamme est réalisée par des dilutions successives d'un pool constitué de quelques microlitres de différentes réactions de reverse transcription. Les niveaux d'expression relatifs d'ABCAl sont ainsi déterminés en utilisant les courbes d'efficacité obtenues avec les points de gamme.
Les niveaux d'expression ont ensuite été normalisés, par rapport au niveau d'expression du gène de référence 36B4. Le facteur d'induction, c'est-à-dire le rapport entre le signal relatif (induit par le composé selon l'invention) et la moyenne des valeurs relatives du groupe contrôle, a ensuite été calculé. Plus ce facteur est élevé, plus le composé a un caractère activateur d'expression génique. Le résultat final est représenté comme moyenne des valeurs d'induction dans chaque groupe expérimental.
Résultats
Les inventeurs ont mis en évidence que les composés selon l'invention sont, in vitro dans les macrophages, des stimulateurs du transport inverse du cholestérol. Les résultats présentés sur la figure (9) montrent que les composés 29 et 42 selon l'invention induisent, à 1μM, une augmentation significative de l'expression du gène codant pour ABCA1 dans les macrophages.
Conclusion
De manière inattendue, les données expérimentales présentées montrent que les composés selon l'invention stimulent le transport inverse du cholestérol.
Exemple 15 : Evaluation in vitro des propriétés anti-inflammatoires des composés selon l'invention
Principe Les effets anti-inflammatoires des composés selon l'invention ont été évalués par la mesure de la sécrétion d'interleukine 6 (IL-6) par des macrophages prétraités pendant 24 heures avec les composés selon l'invention et stimulés pendant 6 heures avec du LPS (lipopolysaccharide, provoque une réponse inflammatoire des cellules). Plus la quantité d'IL-6 sécrétée est diminuée, plus le composé selon l'invention inhibe la réaction inflammatoire.
Protocole a- Différenciation des cellules THP1 en macrophages.
La lignée de monocytes humains THP 1 (provenance ATCC) est cultivée dans du milieu RPMI1640 additionné de 25mM Hepes (Gibco ; 42401-018), 1% glutamine
(Gibco ; 25030-24) 1% pénicilline/streptomycine (Biochrom AG ; A 2213) et 10% sérum de veau foetal décomplémenté (SVF. Gibco ; 26050-088).
Les cellules ensemencées sur des plaques de 24 puits (Primaria BD Falcon) à la densité de 3.105 cellules/puit puis incubées à 37°C et 5% de CO2 pendant 72h en présence de 30 ng/ml de phorbol 12-myristate 13-acetate (PMA) afin de les différencier en macrophages.
b- Traitement
Le milieu de différenciation est aspiré et remplacé par le milieu de traitement (même composition que le milieu de culture mais sans sérum de veau fœtal et avec 1% d'ultroser (PaII Life Science ; P/N267051)).
Les composés selon l'invention ont été testés aux doses de 1 et 10μM. Les composés selon l'invention sont dissous dans du diméthyl sulfoxide (DMSO1
Fluka ; 41640). Les cellules sont traitées pendant 24h à 37°C, 5% CO2. Après 24h d'incubation, la réponse inflammatoire est induite par l'ajout de 1μg/ml de lipopolysaccharide (LPS, Sigma ; L4005) pendant 6 heures. L'effet des composés selon l'invention est comparé à l'effet du DMSO seul.
c- Mesure de la sécrétion d'IL-6
Le milieu de traitement est récupéré et la concentration d'IL-6 est mesurée en utilisant la trousse Elisa « Human IL-6 Elisa set » (BD OptEIA ; 555220) selon les recommandations du fabricant. Le facteur d'induction, c'est-à-dire le rapport entre le signal induit par le composé selon l'invention et le signal du groupe contrôle, a ensuite été calculé. Plus ce facteur est faible, plus le composé a un caractère inhibiteur de la sécrétion d'IL-6.
Le résultat final est représenté comme moyenne des valeurs d'induction de chaque groupe expérimental.
Résultats
Les inventeurs ont aussi mis en évidence, sur des macrophages in vitro, que les composés selon l'invention ont des effets anti-inflammatoires. Les résultats présentés sur la figure (10) montrent que les composés 29 et 42 selon l'invention induisent, à 10μM, une diminution significative de la sécrétion d'IL-6 dans les macrophages.
Conclusion
De manière inattendue, les données expérimentales présentées montrent que les composés selon l'invention ont une action anti-inflammatoire dans les macrophages stimulés avec du LPS.
Exemple 16 : Evaluation in vitro des propriétés activatrices de l'oxydation des acides gras des composés selon l'invention.
L'oxydation des acides gras est un des mécanismes physiologiques permettant de diminuer les taux plasmatiques en lipides et le contenu hépatique en triglycérides.
Principe
L'effet des composés selon l'invention sur l'oxydation des acides gras a été évalué dans des cellules musculaires de souris (C2Ci2) par la mesure de la quantité d'eau tritiée formée durant l'oxydation mitochondriale d'acides gras marqués au 3H.
Protocole a- Culture et différenciation des cellules :
Les cellules musculaires murines C2Ci2 (provenance ECACC) sont cultivées en flasks T225 dans du milieu DMEM 4,5g/L 10% sérum de veau fœtal 1 % pénicilline/streptomycine 1% L-glutamine. Après un lavage au PBS 1X, les cellules sont trypsinisées avec 2ml_ de trypsine 0,05% puis reprises dans du milieu de culture. Une centrifugation de 5 min à 1300 tr/min permet l'élimination de la trypsine. Les cellules sont ensuite platées (à raison de 25000 cellules par puits) en plaques 48 puits préalablement « coatées » à la gélatine (0,2% en concentration finale). Une fois la confluence des cellules atteinte, celles-ci sont différenciées dans du DMEM 4,5g/L 2% sérum de cheval 1% pénicilline/streptomycine 1% L- glutamine. Le milieu de différenciation est changé tous les deux jours. Les cellules différenciées sont traitées 24h (du jour 4 de la différenciation au jour 5) dans le DMEM 4,5g/L 2% sérum de cheval 1% pénicilline/streptomycine 1% L- glutamine.
La cinétique de beta-oxydation des acides gras est réalisée dans le milieu FRM (Fatty acid Oxidation Reaction Médium : DMEM low glucose 1g/L 1% pénicilline/streptomycine 1% L-glutamine sans sérum +carnitine 1mM).
b- Préparation de la solution de BSA:
Préparer une solution de BSA à 2mM dans du DMEM low glucose 1g/L 1% pénicilline/streptomycine 1% L-glutamine sans sérum. Filtrer la solution sur un filtre 0,22μm puis la conserver à +4°C.
c- Préparation de la solution d'acide palmitique froid 5OmM :
La solution d'acide palmitique est préparée dans de l'éthanol 100% puis conservée à -200C
d- Saponification et production du complexe 3H palmitate/ BSA (1mM/0.2mM):
Le ratio à respecter entre le chaud : froid est de 1 : 10 000 et la concentration finale de palmitate (chaud+froid) désirée est de 1mM. Pour cela : 0,5 nmole d'acide palmitique [9,10-3H] sont mises en présence de 5 μmoles d'acide
palmitique froid ainsi que de NaOH 1N à raison de 6μl_ d'acide palmitique froid pourlOOμL.
Le mélange est vortexé puis placé sur un bloc chauffant à 900C pour éliminer la totalité de l'éthanol. Le tout est resuspendu dans 500μL d'eau ppi puis chauffé à 900C jusqu'à dissolution complète du palmitate. Une fois la solution limpide, celle- ci est injectée rapidement dans 500μL de solution BSA à 2mM refroidie dans la glace.
Du milieu DMEM low glucose 1g/L 1% pénicilline/streptomycine 1% L-glutamine sans sérum est ensuite ajouté pour obtenir un volume final de 5 ml. Cette solution est filtrée à travers un filtre de 0,22μm.
e- Cinétique de beta oxydation des acides gras :
La solution stock de 3H palmitate/ BSA (1mM/0.2mM) est diluée au 1/10 dans le milieu FRM (DMEM 4,5g/L 1%P/S 1%L-glutamine 1mM camitine sans sérum) puis incubée à 37°C. Les cellules sont incubées avec le complexe 3H palmitate 0.1mM/ BSA (150μL/ puits) à 37°C 5% CO2. La réaction est stoppée à 1h, 2h et 3h pour s'assurer de la bonne linéarité de la réaction dans le temps. Les protéines sont précipitées avec 100μL de TCA 10% pendant 5 min à température ambiante, puis les surnageants centrifugés 5 min à 14000 tr/min à 1O0C. Le comptage en 3H est tout d'abord réalisé sur 100μL de milieu/TCA puis, dans un deuxième temps 50 μL de milieu/TCA sont mis en évaporation pendant 2 jours puis comptés en 3H afin d'évaluer la quantité de palmitate 3H qui n'a pas été converti en 3H2O. Une courbe standard est réalisée par dilutions successives au 1/5ème du complexe 3H palmitate 0.1mM/BSA dans le FRM (DMEM low glucose 1g/L 1%P/S 1% L-glutamine 1mM carnitine sans sérum).
d- Analyse des résultats :
Pour chaque temps de cinétique 1h, 2h, 3h la quantité en nanomoles de palmitate oxydé est calculée par régression linéaire. Les aires sous les courbes sont calculées et les résultats sont exprimés en induction versus le DMSO 0.1%.
Résultats
Les inventeurs ont mis en évidence que les composés selon l'invention sont, in vitro dans des cellules musculaires, des stimulateurs de l'oxydation des acides gras. Les résultats présentés sur la figure (11) montrent que les composés 29 et 42 selon l'invention induisent, à 1μM, une augmentation significative de l'oxydation mitochondriale d'acides gras marqués au 3H.
Conclusion
De manière inattendue, les données expérimentales présentées montrent que les composés selon l'invention stimulent l'oxydation des acides gras dans des cellules musculaires.
BIBLIOGRAPHIE
Brown G and Foubister A, Receptor binding sites of hypoglycémie sulfonylureas and related [(acylamino)alkyl]benzoic acids., J Med Chem, 1984, 27 (1), 79-81
Denke M, Combination therapy., J Manag Care Pharm, 2003, 9 (1 Suppl), 17-9
Fox-Tucker J, The Cardiovasular Market Outlook to 2010, BUSINESS INSIGHTS REPORTS, 2005, 1-174
Gin H and Rigalleau V, Oral anti diabetic polychemotherapy in type 2 diabètes mellitus., Diabètes Metab, 2002, 28 (5), 350-3
Greene TW and Wuts PGM, Protective Groups in Organic Synthesis, 1999, (3rd édition), 816
Grell W, et al., Repaglinide and related hypoglycémie benzoic acid derivatives, J Med Chem, 1998, 41 (26), 5219-46.
Gross B, et al., Peroxisome Proliferator-Activated Receptor b/d: A novel target for the réduction of atherosclerosis, DRUG DISCOVERY TODAY: THERAPEUTIC STRATEGIES, 2005, 2 (3), 237-243
International Atherosclerosis Society, Harmonized Clinical. Guidelines on Prévention of Atherosclerotic Vascular Disease, 2003,
Kota BP, et al., An overview on biological mechanisms of PPARs, Pharmacol Res, 2005, 51 (2), 85-94
Lefebvre P, et al., Sorting out the rôles of PPARalpha in energy metabolism and vascular homeostasis, J Clin Invest, 2006, 116 (3), 571-580
Lehrke M and Lazar MA1 777e many faces of PPARgamma, CeII, 2005, 123 (6), 993-9
Liu Y and Miller A, Ligands to peroxisome proliferator-activated receptors as therapeutic options for metabolic syndrome, DRUG DISCOVERY TODAY: THERAPEUTIC STRATEGIES, 2005, 2 (3), 165-169
McCormick M and Quinn L1 Treatment of type 2 diabètes mellitus: pharmacologie intervention, J Cardiovasc Nurs, 2002, 16 (2), 55-67.
Mensah M, The Atlas of Heart Disease and Stroke, 2004,
Morphy R and Rankovic Z1 Designed multiple ligands. An emerging drug discovery paradigm, J Med Chem, 2005, 48 (21), 6523-43
Proks P, et al., Sulfonylurea stimulation of insulin sécrétion, Diabètes, 2002, 51 Suppl 3 S368-76.
Raspe E, et al., Modulation of rat liver apolipoprotein gène expression and sérum lipid levels by tetradecylthioacetic acid (TTA) via PPAR{alpha} activation, J. Lipid Res., 1999, 40 (11), 2099-2110
Rufer C and Losert W, Blood glucose lowering sulfonamides with asymmetric carbon atoms. 3. Related N-substituted carbamoylbenzoic acids., J Med Chem, 1979, 22 (6), 750-2
Sullivan PM, et al., Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gène replacement of mouse Apoe with human Apoe*2, J Clin Invest, 1998, 102 (1), 130-5
Claims
REVENDICATIONS
1- Composés dérivés de N-(phénéthyl)benzamide poly-substitués de formule générale (I) :

Formule (I)
Dans laquelle : G représente
- Un radical -ORd, -SRd ; ou
- Un radical -SORd ou -SO2Rd!
Rd représentant un radical alkyle, alkényle, aryle, aralkyle ou un hétérocycle; G pouvant éventuellement former un hétérocycle avec X1 ;
R1 , R2, R3 et R4 représentent indépendamment un atome d'hydrogène ou un radical de type alkyle, alkényle, aryle ou aralkyle;
R1 et R4 pouvant chacun, indépendamment, être liés au squelette moléculaire par une double liaison, R2 ou R3 étant alors absents;
Y représente un atome d'oxygène, un atome de soufre (éventuellement oxydé en fonction sulfoxyde ou sulfone), un atome de sélénium (éventuellement oxydé en fonction sélénoxyde ou sélénone) ou un groupe aminé de type NR ; R représentant un atome d'hydrogène ou un radical de type alkyle, alkényle, aryle ou aralkyle, préférentiellement un atome d'hydrogène ou un radical alkyle ;
E représente :
- Une chaîne alkyle ou alkényle, substituée par un groupement -CR5Re-W; ou
- Une chaîne répondant à la formule -(CH2)m-Y1-Z-Y2-(CH2)n-CR5R6-W;
dans lesquelles :
R5 représente un atome d'halogène ou un radical alkyle, alkényle, aryle, aralkyle, alkyloxy ou alkylthio;
R6 représente un atome d'hydrogène, d'halogène ou un radical alkyle, alkényle, aryle, aralkyle, alkyloxy ou alkylthio; ou
R5 pouvant former un cycle avec R6, m et n représentent indépendamment un nombre entier compris entre 0 et 10;
Y1 et Y2 représentent indépendamment une liaison covalente ou un hétéroatome choisi parmi l'oxygène, le soufre ou l'azote (formant ainsi un radical de type NR, R représentant un atome d'hydrogène ou un radical de type alkyle, alkényle, aryle ou aralkyle, préférentiellement un atome d'hydrogène ou un radical alkyle);
Z représente une liaison covalente ou un radical alkyle, alkényle, aryle, aralkyle; si Z représente une liaison covalente alors Yl et/ou Y2 représente une liaison covalente.W représente : - un radical carboxyle ou un dérivé, préférentiellement de type -COORa,
-COSR3, -CONR9Rb, -CSNR3Rb! ou
- un groupement isostère du radical carboxyle, préférentiellement un radical acylsulfonamide (-CONHSO2Rc), hydrazide (-CONHNR3Rb) ou tétrazolyle;
R3 et Rb identiques ou différents, substitués ou non, représentant un atome d'hydrogène ou un radical alkyle, alkényle, aryle, aralkyle ou un hétérocycle; ou
R3 et Rb pouvant former ensemble un hétérocycle avec l'atome d'azote;
Rc, substitué ou non, représentant un radical alkyle, alkényle, aryle, aralkyle ou un hétérocycle;
X1, X2, X3 et X4 représentent indépendamment un atome d'hydrogène ou d'halogène, une fonction -NO2 ou -CN, un radical alkyle, alkényle, aryle, aralkyle, alkyloxy, alkylthio, -NRaRc, -NRaCORb, -NRaCOORc, -NRaCONRbRc, -NRaCSRb, -NRaCOSR0, -NR3CSORc, -NRaCSSRc, -NRaCSNRbRc, -SOR0, -SO2R0, -COR3, -SO2NR3Rb, -CONRaRb ou un hétérocycle; Ra, Rb et Rc étant tels que définis précédemment; ou
R3, Rb et/ou R0 pouvant former ensemble un hétérocycle avec l'atome d'azote ; X1 et X3 pouvant chacun former un cycle (aromatique ou non, hétérocyclique ou non) avec X2 et X4 respectivement ;
avec au moins un des groupements choisis parmi R1 , R2, R3, R4, X1 , X2, X3 et X4 représentant un radical alkyle, aikényle, aryle ou aralkyle,
les stéréoisomères, purs ou en mélange, les mélanges racémiques, les isomères géométriques, les tautomères, les sels, les hydrates, les solvates, les formes solides, les prodrogues et les mélanges de ceux-ci.
2- Composés selon la revendication 1 , caractérisés en ce qu'ils présentent un groupement G en position ortho (par rapport au motif -CONH-) du radical phényle auquel il est attaché.
3- Composés selon la revendication 1 ou 2, caractérisés en ce qu'ils présentent un groupement Y-E en position para (par rapport au motif -CR3R4-) du radical phényle auquel il est attaché.
4- Composés selon la revendication 2 ou 3, caractérisés en ce qu'ils présentent un groupement X1 en position para (par rapport au radical G) du radical phényle auquel il est attaché.
5- Composés selon la revendication 1 à 4, caractérisés en ce que le groupement G représente un radical -ORd ou -SRdi Ra représentant un radical alkyle, aikényle, aryle, aralkyle ou un hétérocycle.
6- Composés selon l'une quelconque des revendications précédentes, caractérisés en ce que les groupements R1, R2, R3 et/ou R4 représentent un radical alkyle, aikényle, aryle ou aralkyle.
7- Composés selon l'une quelconque des revendications précédentes, caractérisés en ce que Y représente un atome d'oxygène ou de soufre.
8- Composés selon l'une quelconque des revendications précédentes, caractérisés en ce que le groupement E représente une chaîne alkyle ou alkényle, substituée par un groupement -CR5R6-W, dans laquelle :
R5 représente un atome d'halogène ou un radical alkyle, alkényle, aryle, aralkyle, alkyloxy ou alkylthio;
R6 représente un atome d'hydrogène, d'halogène ou un radical alkyle, alkényle, aryle, aralkyle, alkyloxy ou alkylthio; ou R5 pouvant former un cycle avec R6, W représente : - un radical carboxyle ou un dérivé, préférentiellement de type -COORa,
-COSR3, -CONR9Rb, -CSNR3Rb; ou - un groupement isostère du radical carboxyle, préférentiellement un radical acylsulfonamide (-CONHSO2Rc), hydrazide (-CONHNRaRb) ou tétrazolyle; Ra, Rb et R0 étant tels que définis ci-avant.
9- Composés selon l'une quelconque des revendications précédentes, caractérisés en ce que le groupement E représente une chaîne répondant à la formule -(CH2)P-CR5R6-W, avec n, R5, R6 et W tels que définis dans la revendication 1.
10- Composés selon l'une quelconque des revendications précédentes, caractérisés en ce que le groupement E représente une chaîne répondant à la formule -CR5R6-W avec R5, R6 et W tels que définis dans la revendication 1.
11- Composés selon l'une quelconque des revendications précédentes, caractérisés en ce que W représente un radical carboxyle (-COOH) ou alkoxycarbonyle (-COORa), Ra étant tel que défini dans la revendication 1.
12- Composés selon l'une quelconque des revendications précédentes, caractérisés en ce que les radicaux R5 et/ou R6 représentent un radical alkyle.
13- Composés selon l'une quelconque des revendications précédentes, caractérisés en ce que le groupement Y-E représente -OC(CH3)2COOH.
14- Composés selon l'une quelconque des revendications précédentes, caractérisés en ce que le groupement X1 représente un halogène ou un radical - COR3, alkyle, alkényle, aryle ou aralkyle substitué ou non, R3 étant tel que défini dans la revendication 1.
15- Composés selon l'une quelconque des revendications précédentes caractérisés en ce que R1 représente un radical alkyle, alkényle, aryle ou aralkyle et R2, R3 et R4 sont des atomes d'hydrogène.
16- Composés selon l'une quelconque des revendications précédentes caractérisés en ce que R1 et X1 représentent indépendamment un radical alkyle, alkényle, aryle ou aralkyle.
17- Composés selon l'une quelconque des revendications précédentes, caractérisés en ce que X1 forme un cycle avec X2 et/ou X3 forme un cycle avec X4.
18- Composés selon l'une quelconque des revendications précédentes, caractérisés en ce qu'ils sont choisis parmi :
L'Acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[2-((3-méthoxy-2-naphtoyl)amino)éthyl]phénoxy]-2- méthylpropanoïque ; L'Acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)-2-méthylpropyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[1-(5~chloro-2-méthoxybenzamido)-2-méthylpropan-2-yl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2~yl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[1-((3-méthoxy-2-naphtoyl)amino)propan-2-yl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[1-((3-isobutoxy-2-naphtoyl)aminσ)propan-2-yl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)hexan-2-yl]phénoxy]-2- méthylpropanoïque ; L'Acide 2-[4-[2-(2-(phénylthio)-5-trifluorométhylbenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[1 -((2-méthoxy-1 -naphtoyl)amino)propan-2-yl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[2-(2-méthoxy-5-méthylbenzamido)éthyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)-propan-2-yl]-3-méthylphénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[3-(5-chloro-2-méthoxybenzamido)-butan-2-yl]phénoxy]-2- méthylpropanoïque ; L'Acide 2-[4-[1-(5-ferf-butyl-2-méthoxybenzamido)éthyl]phénoxy]-2- méthylpropanoïque ;
Le N-[méthylsulfonyl]-2-[4-[1-((3-méthoxy-2-naphtoyl)amino)propan-2-yl]phénoxy]-
2-méthylpropanamide ;
L'Acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)pentyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[2-(5-méthyl-2-octyloxybenzamido)éthyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[2-(2-méthoxy-5-phénylbenzamido)éthyl]phénoxy]-2- méthylpropanoïque ; L'Acide 2-[4-[2-(5-butyi-2-méthoxybenzamido)éthyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[1-(3-chloro-4-méthoxybenzamido)propan-2-yl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[1-(4-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[2-((3-méthoxy-2-naphtoyl)amino)propyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[1-((3,7-diméthoxy-2-naphtoyl)amino)proρan-2-yl]phénoxy]-2- méthylpropanoïque ;
Le N-[phénylsulfonyl]-2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanamide ; Le N-[pipéridin-1-yl]-2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]-
2-méthylpropanamide.
L'Acide 2-[4-[2-(2,5-diméthoxybenzamido)propyl]phénoxy]-2-méthylpropanoïque ;
L'Acide 2-[4-[2-(5-tertiobutyl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ; L'Acide 2-[4-[2-(5-butyryl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[2-(5-phényl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
Le N-[méthylsulfonyl]-2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanamide ;
Le N-[benzylsulfonyl]-2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanamide ;
Le N-[diméthylamino]-2-[4-[1-(5-chloro-2-méthoxybenzamido)propan-2- yl]phénoxy]-2-méthylpropanamide ; L'Acide 2-[3-[1 -(5-chloro-2-méthoxybenzamido)propan-2-yi]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[2-(5-butyl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[1-(5-phényl-2-méthoxybenzamido)propan-2-yl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[1 -(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]propanoïque ;
L'Acide 2-[4-[1-(5-(2,4-difluorophényl)-2-méthoxybenzamido)propan-2- yl]phénoxy]propanoïque ;
L'Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)-propan-2-yl]phénoxy]-3- méthylbutanoïque ;
L'Acide 2-[4-[1 -(5-chloro-2-méthoxybenzamido)propan-2-yl]phénoxy]pentanoïque ;
L'Acide 2-[4-[1-(5-chloro-2-méthoxybenzamido)-propan-2-yl]phénoxy]-2- phényléthanoïque ;
L'Acide 5-[4-[1-(5-chloro-2-méthoxybenzamido)-propan-2-yl]phénoxy]-2,2- diméthylpentanoïque ;
L'Acide 2-[4-[2-(5-(2,4-difluorophényl)-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ; L'Acide 2-[4-[2-(5-(pyrrol-1 -yl)-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque.
19- Composés selon l'une quelconque des revendications précédentes, caractérisés en ce qu'ils sont choisis parmi : L'Acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[2-(5-chloro-2-méthoxybenzamido)pentyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[2-((3-méthoxy-2-naphtoyl)amino)propyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[2-(5-tertiobutyl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[2-(5-butyry!-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ; L'Acide 2-[4-[2-(5-phényl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4~[2-(5-butyl-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[2-(5-(2,4-difluorophényl)-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque ;
L'Acide 2-[4-[2-(5-(pyrrol-1-yl)-2-méthoxybenzamido)propyl]phénoxy]-2- méthylpropanoïque.
20- Procédé de préparation des composés tels que définis dans l'une des revendications 1 à 19 comprenant:
(i) Une étape de condensation, préférentiellement d'un dérivé de type 2- phényléthanamine mono- ou poly-substitué avec un dérivé de type
acide (ou dérivé) benzoïque mono- ou poly-substitué; et éventuellement, avant et/ou après l'étape (i), (ii) Une ou plusieurs étapes d'insertion et/ou de transformation de groupements fonctionnels.
21- Procédé de préparation des composés tel que défini dans la revendication 20 permettant l'obtention des composés sous forme optiquement pure partant de substrats chiraux énantiopurs, préférentiellement des acides examinés.
22- Composés caractérisés en ce qu'ils sont choisis parmi : Le 2-[4-(1-aminopropan-2-yl)phénoxy]-2-méthylpropanoate d'éthyle ; Le 4-(1-aminohexan-2-yl)phénol ;
Le 2-[4-(1-aminopropan-2-yl)-3-méthyl-phénoxy]-2-méthylpropanoate de tert- butyle ;
Le 2-[4-(3-aminobutan-2-yl)phénoxy]-2-méthylpropanoate de fert-butyle ;
Le 2-[4-(2-aminopentyl)phénoxy]-2-méthylpropanoate de terf-butyle ;
Le 2-[4-(1-aminopropan-2-yl)phénoxy]-2-méthylpropanoate de fe/f-butyle ;
Le 2-[3-(1-aminopropan-2-yl)phénoxy]-2-méthylpropanoate de tert-butyle ; L' acide 5-méthyl-2-octyloxybenzoïque ;
L' acide 5-butyl-2-méthoxybenzoïque ;
Le 4-[2-(5-chloro-2-méthoxybenzamido)propyl]phénol ;
Le 4-[2-(5-chloro-2-méthoxybenzamido)-2-méthylpropyl]phénol ;
Le 4-[1 -(5-chloro-2-méthoxybenzamido)-2-méthylpropan-2-yl]phénol ; Le 4-[1-(5-chloro-2-méthoxybenzamido)propan-2-yl]phénol ;
Le 4-[1-(5-chloro-2-méthoxybenzamido)hexan-2-yl]phénol ;
Le 4-[2-(2-méthoxy-5-méthylbenzamido)éthyl]phénol ;
Le 4-[2-(5-méthyl-2-octyloxybenzamido)éthyl]phénol ;
Le 4-[2-(2-méthoxy-5-phénylbenzamido)éthyl]phénol ; Le 4-[2-((3-méthoxy-2-naphtoyl)amino)propyl]phénol ;
Le 4-[2-(2,5-diméthoxybenzamido)propyl]phénol ;
Le 4-[2-(5-tertiobutyl-2-méthoxybenzamido)propyl]phénol ;
Le 4-[2-(5-butyryl-2-méthoxybenzamido)propyl]phénol ;
Le 4-[2-(5-phényl-2-méthoxybenzamido)propyl]phénol ;
Le 4-[2-(5-(2,4-difluorophényl)-2-méthoxybenzamido)propyl]phénol ;
Le 4-[2-(5-(pyrrol-1-yl)-2-méthoxybenzamido)propyl]phénol ;
Le 2-[4-[2-(2-fluorobenzamido)propyl]phénoxy]-2-méthylpropanoate d'éthyle ; Le 2-[4-[1-((3-hydroxy-2-naphtoyl)amino)propan-2-yl]phénoxy]-2-méthyl- propanoate d'éthyle ;
Le 2-[4-[2-(2-fluoro-5-trifluorométhylbenzamido)propyl]phénoxy]-2-méthyl- propanoate d'éthyle ;
Le 2-[4-[1 -((2-hydroxy-1 -naphtoyl)amino)propan-2-yl]phénoxy]-2-méthyl- propanoate d'éthyle ;
Le 2-[4-[1-((3-hydroxy-7-méthoxy-2-naphtoyl)amino)propan-2-yl]phénoxy]-2- méthyl-propanoate de tert-butyle ;
Le 2-[4-[1-(5-(2,4-difluorophényl)-2-hydroxybenzamido)propan-2-yl]phénoxy]- propanoate de tert-butyle.
23- Composés selon l'une quelconque des revendications 1 à 19 à titre de médicaments.
24- Composition pharmaceutique comprenant, dans un support acceptable sur le plan pharmaceutique, au moins un composé tel que défini dans l'une des revendications 1 à 19, éventuellement en association avec un ou plusieurs autres actifs thérapeutiques et/ou cosmétiques.
25- Composition pharmaceutique comprenant, dans un support acceptable sur le plan pharmaceutique, au moins un composé tel que défini dans l'une des revendications 1 à 19 en association avec un ou plusieurs composés sélectionnés dans la liste ci-dessous :
- un anti-diabétique
- l'insuline - une molécule hypolipémiante et/ou hypocholestérolémiante
- un agent anti-hypertenseur ou hypotenseur
- un agent anti-plaquettaire
- un agent anti-obésité
- un agent anti-inflammatoire
- un agent anti-oxydant
- un agent utilisé dans le traitement de l'insuffisance cardiaque
- un agent utilisé pour le traitement de l'insuffisance coronaire - un agent anticancéreux
- un anti-asthmatique
- un corticoïde utilisé dans le traitement des pathologies de la peau
- un vasodilatateur et/ou un agent anti-ischémique.
26- Composition pharmaceutique selon la revendication 24 ou 25 pour le traitement du diabète de type 2, des dyslipidémies, de l'insulino-résistance, des pathologies associées au syndrome métabolique, de l'athérosclérose, des maladies cardiovasculaires, de l'obésité, de l'hypertension et/ou des maladies inflammatoires.
27- Composition pharmaceutique selon la revendication 26 pour le traitement de l'athérosclérose et/ou des maladies inflammatoires.
28- Composition pharmaceutique selon la revendication 24 ou 25 pour traiter les facteurs de risque cardiovasculaire liés aux dérèglements du métabolisme lipidique et/ou glucidique.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0602586A FR2898892A1 (fr) | 2006-03-24 | 2006-03-24 | Derives de n-(phenethyl)benzamide substitues,preparation et utilisations |
| FR0602586 | 2006-03-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007118964A1 true WO2007118964A1 (fr) | 2007-10-25 |
Family
ID=37400977
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR2007/000513 Ceased WO2007118964A1 (fr) | 2006-03-24 | 2007-03-23 | Derives de n-(phenethyl)benzamide substitues, preparation et utilisations |
Country Status (2)
| Country | Link |
|---|---|
| FR (1) | FR2898892A1 (fr) |
| WO (1) | WO2007118964A1 (fr) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (fr) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Nouvelles tétrahydronaphtalines substituées, leurs procédés de préparation et leur utilisation comme médicaments |
| WO2011107494A1 (fr) | 2010-03-03 | 2011-09-09 | Sanofi | Nouveaux dérivés aromatiques de glycoside, médicaments contenants ces composés, et leur utilisation |
| WO2011157827A1 (fr) | 2010-06-18 | 2011-12-22 | Sanofi | Dérivés d'azolopyridin-3-one en tant qu'inhibiteurs de lipases et de phospholipases |
| WO2011161030A1 (fr) | 2010-06-21 | 2011-12-29 | Sanofi | Dérivés de méthoxyphényle à substitution hétérocyclique par un groupe oxo, leur procédé de production et leur utilisation comme modulateurs du récepteur gpr40 |
| WO2012004269A1 (fr) | 2010-07-05 | 2012-01-12 | Sanofi | Dérivés d'acide ( 2 -aryloxy -acétylamino) - phényl - propionique, procédé de production et utilisation comme médicament |
| WO2012004270A1 (fr) | 2010-07-05 | 2012-01-12 | Sanofi | Dérivés 1,3-propanedioxyde à substitution spirocyclique, procédé de préparation et utilisation comme médicament |
| WO2012010413A1 (fr) | 2010-07-05 | 2012-01-26 | Sanofi | Acides hydroxy-phényl-hexiniques substitués par aryloxy-alkylène, procédé de production et utilisation comme médicament |
| WO2012120056A1 (fr) | 2011-03-08 | 2012-09-13 | Sanofi | Dérivés oxathiazine tétra-substitués, procédé pour leur préparation, utilisation en tant que médicament, agent pharmaceutique contenant ces dérivés et utilisation |
| WO2012120054A1 (fr) | 2011-03-08 | 2012-09-13 | Sanofi | Dérivés oxathiazine di- et tri-substitués, procédé pour leur préparation, utilisation en tant que médicament, agent pharmaceutique contenant ces dérivés et utilisation |
| WO2012120055A1 (fr) | 2011-03-08 | 2012-09-13 | Sanofi | Dérivés oxathiazine di- et tri-substitués, procédé pour leur préparation, utilisation en tant que médicament, agent pharmaceutique contenant ces dérivés et utilisation |
| WO2012120052A1 (fr) | 2011-03-08 | 2012-09-13 | Sanofi | Dérivés d'oxathiazine substitués par des carbocycles ou des hétérocycles, leur procédé de préparation, médicaments contenant ces composés et leur utilisation |
| WO2012120053A1 (fr) | 2011-03-08 | 2012-09-13 | Sanofi | Dérivés oxathiazine ramifiés, procédé pour leur préparation, utilisation en tant que médicament, agents pharmaceutiques contenant ces dérivés et leur utilisation |
| WO2013037390A1 (fr) | 2011-09-12 | 2013-03-21 | Sanofi | Dérivés amides d'acide 6-(4-hydroxyphényl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylique en tant qu'inhibiteurs de kinase |
| WO2013045413A1 (fr) | 2011-09-27 | 2013-04-04 | Sanofi | Dérivés d'amide d'acide 6-(4-hydroxyphényl)-3-alkyl-1h-pyrazolo[3,4-b] pyridine-4-carboxylique utilisés comme inhibiteurs de kinase |
| CN111148742A (zh) * | 2017-11-30 | 2020-05-12 | 四川科伦博泰生物医药股份有限公司 | 芳香族化合物及其药物组合物和用途 |
| CN113631542A (zh) * | 2019-05-24 | 2021-11-09 | 四川科伦博泰生物医药股份有限公司 | 一种芳香族化合物的固体形式及其制备方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2154739A1 (fr) * | 1971-10-01 | 1973-05-11 | Boehringer Mannheim Gmbh | |
| FR2328461A1 (fr) * | 1975-10-21 | 1977-05-20 | Boehringer Mannheim Gmbh | Nouveaux acides phenoxyalcanecarboxyliques et leur procede de preparation |
| DE2558719A1 (de) * | 1975-12-24 | 1977-06-30 | Boehringer Mannheim Gmbh | Phenoxyalkylcarbonsaeurederivate und verfahren zur herstellung derselben |
| EP1184366A1 (fr) * | 1999-06-09 | 2002-03-06 | Kyorin Pharmaceutical Co., Ltd. | DERIVES D'ACIDE PHENYLPROPIONIQUE SUBSTITUES COMME AGONISTES DU RECEPTEUR HUMAIN ACTIVE DE LA PROLIFERATION DES PEROXYSOMES (PPAR) $g(a) |
-
2006
- 2006-03-24 FR FR0602586A patent/FR2898892A1/fr not_active Withdrawn
-
2007
- 2007-03-23 WO PCT/FR2007/000513 patent/WO2007118964A1/fr not_active Ceased
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2154739A1 (fr) * | 1971-10-01 | 1973-05-11 | Boehringer Mannheim Gmbh | |
| FR2328461A1 (fr) * | 1975-10-21 | 1977-05-20 | Boehringer Mannheim Gmbh | Nouveaux acides phenoxyalcanecarboxyliques et leur procede de preparation |
| DE2558719A1 (de) * | 1975-12-24 | 1977-06-30 | Boehringer Mannheim Gmbh | Phenoxyalkylcarbonsaeurederivate und verfahren zur herstellung derselben |
| EP1184366A1 (fr) * | 1999-06-09 | 2002-03-06 | Kyorin Pharmaceutical Co., Ltd. | DERIVES D'ACIDE PHENYLPROPIONIQUE SUBSTITUES COMME AGONISTES DU RECEPTEUR HUMAIN ACTIVE DE LA PROLIFERATION DES PEROXYSOMES (PPAR) $g(a) |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (fr) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Nouvelles tétrahydronaphtalines substituées, leurs procédés de préparation et leur utilisation comme médicaments |
| WO2011107494A1 (fr) | 2010-03-03 | 2011-09-09 | Sanofi | Nouveaux dérivés aromatiques de glycoside, médicaments contenants ces composés, et leur utilisation |
| WO2011157827A1 (fr) | 2010-06-18 | 2011-12-22 | Sanofi | Dérivés d'azolopyridin-3-one en tant qu'inhibiteurs de lipases et de phospholipases |
| WO2011161030A1 (fr) | 2010-06-21 | 2011-12-29 | Sanofi | Dérivés de méthoxyphényle à substitution hétérocyclique par un groupe oxo, leur procédé de production et leur utilisation comme modulateurs du récepteur gpr40 |
| WO2012004269A1 (fr) | 2010-07-05 | 2012-01-12 | Sanofi | Dérivés d'acide ( 2 -aryloxy -acétylamino) - phényl - propionique, procédé de production et utilisation comme médicament |
| WO2012004270A1 (fr) | 2010-07-05 | 2012-01-12 | Sanofi | Dérivés 1,3-propanedioxyde à substitution spirocyclique, procédé de préparation et utilisation comme médicament |
| WO2012010413A1 (fr) | 2010-07-05 | 2012-01-26 | Sanofi | Acides hydroxy-phényl-hexiniques substitués par aryloxy-alkylène, procédé de production et utilisation comme médicament |
| WO2012120052A1 (fr) | 2011-03-08 | 2012-09-13 | Sanofi | Dérivés d'oxathiazine substitués par des carbocycles ou des hétérocycles, leur procédé de préparation, médicaments contenant ces composés et leur utilisation |
| WO2012120054A1 (fr) | 2011-03-08 | 2012-09-13 | Sanofi | Dérivés oxathiazine di- et tri-substitués, procédé pour leur préparation, utilisation en tant que médicament, agent pharmaceutique contenant ces dérivés et utilisation |
| WO2012120055A1 (fr) | 2011-03-08 | 2012-09-13 | Sanofi | Dérivés oxathiazine di- et tri-substitués, procédé pour leur préparation, utilisation en tant que médicament, agent pharmaceutique contenant ces dérivés et utilisation |
| WO2012120056A1 (fr) | 2011-03-08 | 2012-09-13 | Sanofi | Dérivés oxathiazine tétra-substitués, procédé pour leur préparation, utilisation en tant que médicament, agent pharmaceutique contenant ces dérivés et utilisation |
| WO2012120053A1 (fr) | 2011-03-08 | 2012-09-13 | Sanofi | Dérivés oxathiazine ramifiés, procédé pour leur préparation, utilisation en tant que médicament, agents pharmaceutiques contenant ces dérivés et leur utilisation |
| WO2013037390A1 (fr) | 2011-09-12 | 2013-03-21 | Sanofi | Dérivés amides d'acide 6-(4-hydroxyphényl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylique en tant qu'inhibiteurs de kinase |
| WO2013045413A1 (fr) | 2011-09-27 | 2013-04-04 | Sanofi | Dérivés d'amide d'acide 6-(4-hydroxyphényl)-3-alkyl-1h-pyrazolo[3,4-b] pyridine-4-carboxylique utilisés comme inhibiteurs de kinase |
| CN111148742A (zh) * | 2017-11-30 | 2020-05-12 | 四川科伦博泰生物医药股份有限公司 | 芳香族化合物及其药物组合物和用途 |
| JP2021504298A (ja) * | 2017-11-30 | 2021-02-15 | シチュアン ケルン−バイオテック バイオファーマシューティカル カンパニー リミテッド | 芳香族化合物、薬学的組成物及びその使用 |
| EP3719010A4 (fr) * | 2017-11-30 | 2021-08-04 | Sichuan Kelun-Biotech Biopharmaceutical Co., Ltd. | Composé aromatique, composition pharmaceutique et utilisation associées |
| US11261170B2 (en) | 2017-11-30 | 2022-03-01 | Sichuan Kelun-Biotech Biopharmaceutical Co., Ltd. | Aromatic compound, pharmaceutical composition and use thereof |
| US11332457B2 (en) | 2017-11-30 | 2022-05-17 | Sichuan Kelun-Biotech Biopharmaceutical Co., Ltd. | Aromatic compound, pharmaceutical composition and use thereof |
| JP7259850B2 (ja) | 2017-11-30 | 2023-04-18 | シチュアン ケルン-バイオテック バイオファーマシューティカル カンパニー リミテッド | 芳香族化合物、薬学的組成物及びその使用 |
| CN113631542A (zh) * | 2019-05-24 | 2021-11-09 | 四川科伦博泰生物医药股份有限公司 | 一种芳香族化合物的固体形式及其制备方法 |
| CN113631542B (zh) * | 2019-05-24 | 2023-07-07 | 四川科伦博泰生物医药股份有限公司 | 一种芳香族化合物的固体形式及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| FR2898892A1 (fr) | 2007-09-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007118964A1 (fr) | Derives de n-(phenethyl)benzamide substitues, preparation et utilisations | |
| WO2007118963A2 (fr) | Composes derives de n-(phenethyl)benzamide substitues, preparation et utilisations | |
| EP2046716B1 (fr) | Derives de 1,3-diphenylpropane substitues, preparations et utilisations | |
| CA2673761C (fr) | Derives de 3-phenyl-1-(phenylthienyl)propan-1-one et de 3-phenyl-1-(phenylfuranyl)propan-1-one substitues, preparation et utilisation | |
| WO2008087367A2 (fr) | Derives de (phenylthiazolyl)-phenyl-propan-1-one et de (phenyloxazodyl)-phenyl-propan-1-one substitues, preparations et utilisations | |
| EP2310371B1 (fr) | Composés agonistes ppar, préparation et utilisations pour le traitement du diabète et/ou des dyslipidémies | |
| WO2008087365A2 (fr) | Derives de 1,3-diphenylpropane substitues, preparations et utilisations | |
| FR2734816A1 (fr) | Nouveaux aryl (alkyl) propylamides, leur procede de preparation et les compositions pharmaceutiques qui les contiennent | |
| WO2006035157A2 (fr) | Composes derives de n- (benzyl) phenylacetamide substitues , preparation et utilisations comme ligands des ppar dans le traitement des dereglements lipidiques et/ou glucidiques | |
| WO2007141423A1 (fr) | Derives activateurs de ppars, procede de preparation et application en therapeutique. | |
| KR100969979B1 (ko) | 페닐(알킬)카르복시산 유도체 및 디이온성페닐알킬헤테로사이클릭 유도체, 및 혈청 글루코스및/또는 혈청 지질 강하 활성을 갖는 약제로서의 그의 용도 | |
| EP1347950A1 (fr) | Derives de l'acide 4-hydroxybutano que et de son homologue superieur comme ligands des recepteurs du gamma-hydroxybutyrate (ghb) compositions pharmaceutiques les contenant et utilisations pharmaceutiques | |
| FR2890072A1 (fr) | Nouveaux composesde pyrrolopyridine | |
| FR2883000A1 (fr) | Derives de trifluoromethylbenzamide et leurs utilisations en therapeutique | |
| FR2707638A1 (fr) | Nouveaux dérivés d'amino-acides, leurs procédés de préparation et leur application thérapeutique. | |
| BE885303A (fr) | Glycinamides | |
| FR2757159A1 (fr) | Nouveaux derives nitres analgesiques, anti-inflammatoires et anti-thrombotiques, leur procede de preparation, leur application comme medicaments | |
| FR2817257A1 (fr) | Cyclohexyl(alkyl)-propanolamines, leur preparation et compositions pharmaceutiques en contenant | |
| FR2862964A1 (fr) | Derives de la diphenylamine. | |
| FR2886293A1 (fr) | Nouveaux composes de l'indoline | |
| FR2726268A1 (fr) | Nouvelles o-arylmethyl n-(thio)acyl hydroxylamines, leur procede de preparation, et les compositions pharmaceutiques qui les contiennent | |
| WO1997020827A1 (fr) | Derives de fluorophenyl-triazines et pyrimidines comme composes actifs sur le snc | |
| FR2677016A1 (fr) | Nouveaux derives d'amide antagonistes des recepteurs a l'angiotensine ii; leurs procedes de preparation, compositions pharmaceutiques les contenant. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07731196 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07731196 Country of ref document: EP Kind code of ref document: A1 |