WO2007043638A1 - 縮合へテロ環化合物 - Google Patents
縮合へテロ環化合物 Download PDFInfo
- Publication number
- WO2007043638A1 WO2007043638A1 PCT/JP2006/320434 JP2006320434W WO2007043638A1 WO 2007043638 A1 WO2007043638 A1 WO 2007043638A1 JP 2006320434 W JP2006320434 W JP 2006320434W WO 2007043638 A1 WO2007043638 A1 WO 2007043638A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- substituted
- same
- methyl
- different
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention relates to a novel condensed heterocyclic compound useful as a pharmaceutical, particularly as a therapeutic agent for diabetes.
- GK (Dalcokinase (ATP: D-hexose 6-phosphotransferase, EC2.7.1.1)) is an enzyme that phosphorylates 6-carbon sugars expressed in the spleen and liver. It has been. This enzyme belongs to the hexokinase family and is also known as hexokinase IV. Compared to other hexokinases, GK has 1) low affinity for glucose as a substrate and a Km value close to blood glucose concentration, 2) no inhibition by enzymatic reaction product glucose 6-phosphate, 3 ) It has characteristics such as 50 kDa, which is about half the molecular weight.
- the human dalcokinase gene is located on chromosome 7 pl3 as a single gene, and is controlled by different tissue-specific promoters separated by 30 kb or more in spleen
- GK acts as a glucose sensor in spleen j8 cells and plays an important role in the control of insulin secretion. Even in the liver, GK acts as a glucose sensor, reacting to an increase in blood glucose level and converting glucose to glucose 6-phosphate. As a result, production of glycogen increases, glycolysis is activated, and gluconeogenesis in the liver is suppressed.
- Hyperglycemia occurs frequently in patients with impaired glucose phosphate due to GK gene mutations, and juvenile diabetes develops (MODY2).
- Km value of GK activity by gene mutation In patients with low values, hypoglycemia is observed after meals and on an empty stomach. That is, even in humans, GK acts as a glucose sensor and plays an important role in maintaining normal glucose levels in the blood.
- drugs that activate GK enhance glucose-dependent insulin secretion from spleen j8 cells, correct postprandial hyperglycemia, and suppress the release of sugar from the liver.
- GK which exists in the brain, is a spleen type and is highly expressed in the nerve of VMH (Ventromedial hypothalamus), the feeding center.
- Glucose-sensitive nerves are classified into GE (Glucose Exited) -neurons that are excitable to dalcose and GI (Glucose Inhibited) -neurons that are inhibitory to glucose.
- GK mRNA and protein are present in about 70% of GE-neurons and in about 40% of GI-neurons.
- GK senses an increase in intracellular glucose, activates the glycolysis, and increases the intracellular ATP / ADP ratio.
- GK senses an increase in intracellular glucose, activates the glycolysis, and increases the intracellular ATP / ADP ratio.
- Yannel closes-Euron action potential frequency increases and neurotransmitters are released.
- GI-neuron is thought to involve the C1-channel. In rats with increased GK mRNA expression in VMH, the compensation for glucose deficiency is reduced.
- Glucose-sensitive nerves also have receptors for levtin and insulin that are involved in feeding behavior. In GE-neurons under high glucose conditions, leptin and insulin open K channels and reduce action potential frequency. In addition to the ARC
- NPY Neuropeptide Y
- POMC Proopiomelanocortin
- A is substituted with a selected group of halogen, carboxy and C alkyl forces.
- Patent Document 1 International Publication WO2004 / 046139 Pamphlet
- An object of the present invention is to provide a novel compound useful as a pharmaceutical having GK activity and particularly as a therapeutic agent for diabetes.
- the present inventors have a group represented by -XB and a force rubamoyl group on the benzene ring of the condensed heterocyclic ring, and the nitrogen atom of the carbamoyl group has Is pyridyl, birazinyl or thio which may be substituted with a specific substituent. It was confirmed that a compound characterized by binding an azolyl group has a good GK activity, and the present invention was completed.
- the present invention relates to a condensed heterocyclic compound represented by the general formula (I) or a salt thereof.
- ⁇ a single bond, optionally substituted lower alkylene, -R. . -0- R. . -,-R. . -CON (R G )-,-R
- V may be lower alkylene) -N (R G ) CO-,
- R ° the same or different from each other, lower alkyl
- R °° the same or different from each other, lower alkylene
- R G the same or different from each other, H or R °,
- R 1 and R 2 the same or different from each other and the following (0 or (ii) force is also selected
- R A the same or different, -H, -R °, -halogeno lower alkyl or -R °° -aryl
- R B -CO H, -CO R °, -CO-NR C R D , -CO-NR C -OR D , -R °° -NR C R D , -R °° -OR A , -R. 0 -C
- R C and R D the same or different from each other,-H,-R °,-R °° -N (R A ),-R °° -0R A ,-R °° -C0 H
- R 3 and R 4 same or different from each other, - H, - halogen, - R °, - the halogeno-lower alkyl or - R 00 - 0H,
- R 5 -H, -R °, -halogeno lower alkyl,-(optionally substituted aryl) or -R °°-(substituted, optionally aryl).
- B represents an aryl substituted with at least one -SO R °.
- the present application also relates to a pharmaceutical comprising the condensed heterocyclic compound represented by the general formula (I) or a salt thereof as an active ingredient, particularly a GK activator and a therapeutic agent for diabetes.
- the present application further relates to the use of the condensed heterocyclic compound represented by the general formula (I) or a salt thereof for the manufacture of a therapeutic agent for diabetes and a method for treating diabetes.
- the compound of the present invention has a GK activation action, it is useful as a therapeutic and prophylactic agent for diabetes, particularly type 2 diabetes. It is also useful for the treatment and prevention of diabetic complications such as nephropathy, retinopathy, neuropathy, peripheral circulatory disorder, cerebrovascular disorder, ischemic heart disease, and arteriosclerosis. Furthermore, by suppressing overeating, it is useful as a therapeutic and prophylactic agent for obesity and metabolic syndrome.
- alkyl In the present specification, “alkyl”, “alkylene” and “alkyl” mean a linear or branched hydrocarbon chain. “Lower alkyl” is a C 1-6 (hereinafter referred to as C) alkyl.
- Lower alkylene means a divalent group formed by removing any one hydrogen atom of the above “lower alkyl”, preferably C alkylene, more preferably methylene, ethyl. Len, methylmethylene and propylene.
- a “lower alkali” is a C alkali having one or more double bonds, preferably a C alkaline, and more preferably.
- Halogen is F, Cl, Br and I.
- Halogeno lower alkyl means C alkyl substituted with one or more (preferably 1-5, more preferably 1-3) halogens.
- Cycloalkyl is a C cycloalkyl, which forms a bridged ring such as adamantyl.
- cycloalkyl more preferably cyclopropyl.
- Cyclobutyl Cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl.
- a “cycloalkyl” is a C ring group having 1 or 2 double bonds.
- “Areel” is C
- hydrocarbon ring includes the above “cycloalkyl”, “cycloalkenyl” and “aryl”.
- a “heterocyclic group” is a 3- to 7-membered monocyclic or bicyclic heterocyclic group containing 1 to 4 heteroatoms selected from 0, S and N forces, and a saturated ring , Aromatic rings, and partially hydrogenated ring groups thereof.
- Optionally substituted means “unsubstituted” or “having 1 to 5 substituents which are the same or different”. In the case of having a plurality of substituents, these substituents may be the same or different from each other! /, May! /.
- Substituents in “substituted and optionally aryl”, “substituted and optionally heterocyclic” and “optionally substituted cycloalkyl” are preferably —R °, halogeno lower alkyl Kill, halogen, -OH, -R °° -OH, -N, -OR °, -R °° -OR °, -O-hydrocarbon ring, -O-hete
- hydrocarbon ring and the “heterocycle” include 1 to 5 —R Q , halogeno Alkyl, halogen, -OH, -OR ° and -N (R G ) forces optionally substituted with selected groups
- halogeno lower alkyl Preferably 1 to 5 -R °, halogeno lower alkyl, halogen, -OH and -OR. May be substituted with a group selected from:
- A is preferably S or N (R 5 ).
- R 5 is preferably H, lower alkyl or substituted phenyl, more preferably C alkyl or 4-alkyl at C alkyl-SO.
- the file is replaced with-.
- (2) X is preferably 0.
- (3) B is preferably substituted with 1 or 2 substituents !, may! /,
- At least one is a phenyl having a substituent at the 4-position, and more preferably a phenyl having one substituent at the 4-position.
- the optionally substituted “aryl”, “heteroaryl” or “cycloalkyl” in B is preferably a halogeno lower alkyl, halogen, —OR 0 , —CN, —NO, -CHO, -CO H, -CO
- 0-heterocycle more preferably halogeno lower alkyl, halogen, -NO, -CO-R °,
- 2 2 is -so-ethyl, and more preferably -so-methyl.
- D is thiazole (ie thiazol-2-yl), pyridine (ie 2-pyridyl) or virazine (ie pyrazine-2-) which is bonded to the nitrogen atom of the amide group at the 2-position. It is).
- R 1 and R 2 are preferably both H; another preferred embodiment of R 1 and R 2 is preferably as follows: one is H and the other is (0 and GO force selected A group other than H, more preferably R 1 is H and R 2 is a group other than H (0 and (ii) force selected, where
- Groups in which 0 and GO force are also selected are preferably -CO H, -CO R °, -CH (OR A )-R B ,-C
- O—NR C R D more preferably ⁇ —CH (OH) —CH OH, —CH (OR °) —CH OH, —CH (OR.) —
- R 3 and R 4 are preferably H or R °.
- a compound comprising a combination of each preferred group described in the above (1) to (6) is preferred. That is,
- B is R °,-(optionally substituted aryl) or -R °°-(optionally substituted aryl), and R 1 and R 2 are the same or different from each other.
- the compound according to (5) which is a group selected from (0 or GO).
- R 1 is H, R 2 CH (OH) -CH OH, -CH (OR °) -CH OH, -CH (OR.)-CH OR.
- the compound of the present invention may have other tautomers and geometric isomers depending on the type of substituent. In the present specification, only one form of these isomers may be described, but the present invention includes these isomers, and also includes a separated isomer or a mixture thereof.
- compound (I) may have asymmetric carbon atoms or axial asymmetry, and optical isomers such as (R) and (S) isomers may exist based on this.
- optical isomers such as (R) and (S) isomers may exist based on this.
- the present invention is a mixture of these optical isomers or a simple one. Includes everything that has been released.
- the present invention includes pharmacologically acceptable prodrugs of Compound (I).
- a pharmacologically acceptable prodrug is a compound having a group that can be converted into an amino group, —OH, —CO 2 H, etc. of the present invention by solvolysis or under physiological conditions.
- Examples of the group to be formed include groups described in Prog. Med., 5, 2157-2161 (1985) and “Drug Development” (Yodogawa Shoten, 1990), No. 7 Molecular Design 163-198.
- the compound of the present invention may form a salt with a base depending on the type of the acid addition salt or the substituent, and the present invention is not limited as long as the earning salt is a pharmaceutically acceptable salt.
- inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, fumaric acid, maleic acid Acid, lactic acid, malic acid, tartaric acid, citrate, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, aspartic acid, acid addition salts with organic acids such as glutamic acid, sodium, potassium, magnesium, calcium, aluminum Inorganic bases such as methylamine, ethylamine, ethanolamine, lysine, and ortho salts, and ammonium salts
- the present invention also includes various hydrates and solvates of the compound of the present invention and pharmaceutically acceptable salts thereof, and substances having crystalline polymorphs.
- the compound of the present invention and a pharmaceutically acceptable salt thereof can be produced by applying various known synthetic methods utilizing characteristics based on the basic skeleton or the type of substituent.
- an appropriate protecting group a group that can be easily converted into the functional group
- functional groups include amino groups, hydroxyl groups, and carboxyl groups, and examples of their protecting groups include Greene and Wuts, “Protective Groups in Organic Synthesis (3rd edition, 1999). And the like, and these may be appropriately selected and used according to the reaction conditions.
- the desired compound after carrying out the reaction by introducing the protecting group, the desired compound can be obtained by removing the protecting group as necessary.
- the prodrug of compound (I) can be produced by introducing a specific group at the raw material or intermediate stage, or reacting with the obtained compound (I).
- the reaction can be carried out by applying a method known by those skilled in the art, such as ordinary esterification, amidation, dehydration and the like.
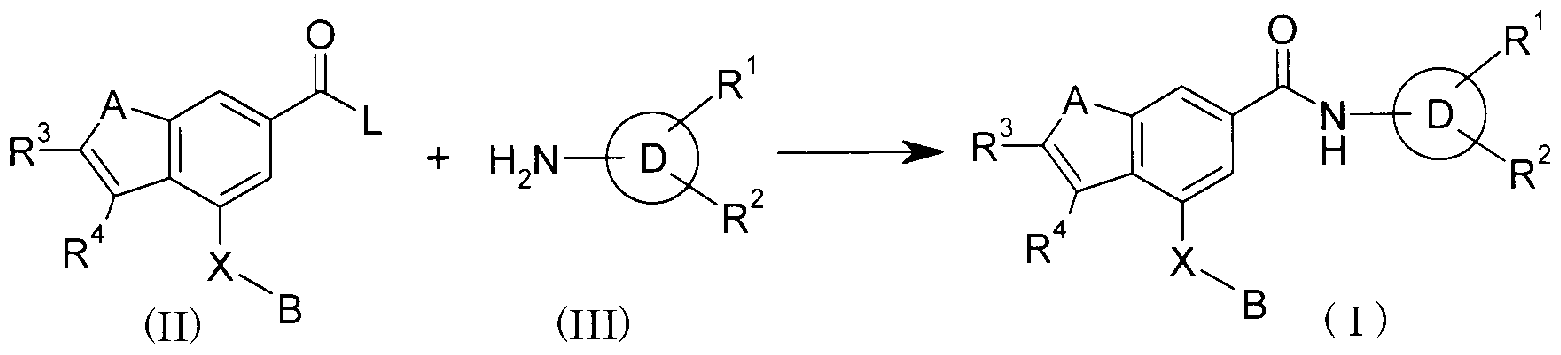
- This production method is a method for obtaining the compound of the present invention represented by the formula (I) by subjecting the carboxylic acid derivative (II) and the amino compound (III) to an amidy reaction.
- the leaving group for L include an organic sulfonic acid group such as methanesulfonyloxy or P-toluenesulfo-oxy, halogen, and the like.
- various acid anhydrides can be used as (II).
- ⁇ ⁇ , ⁇ '-dicyclohexylcarbodiimide (DCC), 1-ethyl-3- (3'-dimethylaminopropyl) carbodiimide (WSC), 1,1'-carbodidiimidazole ( CDI), diphenylphosphoryl azide (DPPA), phosphorus oxychloride / pyridine, triphenylphosphine / ⁇ -promosuccinimide and other condensing agents can be used.
- DCC 1-ethyl-3- (3'-dimethylaminopropyl) carbodiimide
- CDI 1,1'-carbodidiimidazole
- DPPA diphenylphosphoryl azide
- DPPA diphenylphosphoryl azide
- DPPA phosphorus oxychloride / pyridine
- triphenylphosphine / ⁇ -promosuccinimide and other condensing agents can be used.
- polystyrene resin carrying an isocyanate for the purpose of removing excess amine after completion of the reaction such as PS-isocyanate (manufactured by Argonaut), PL-isocyanate resin (PL-isocyanate). NCO Resin) (Polymer Laboratories, UK) is preferably used.
- polystyrene resin supporting quaternary ammonium salt for example, MP-force MP-Carbonate (manufactured by Biotage) or polystyrene-terminated resin loaded with amine, such as PS-Trisamine (manufactured by Biotage), PL-DETA Resin Resin (PL-DETA Resin) ) (Polymer Laboratories, UK) may be preferred.
- the reaction may be carried out in the presence of an inorganic base such as sodium carbonate, potassium carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate, or an organic base such as triethylamine, diisopropylethylamine, pyridine. It may be preferable.
- an inorganic base such as sodium carbonate, potassium carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate, or an organic base such as triethylamine, diisopropylethylamine, pyridine. It may be preferable.
- Solvents include aromatic hydrocarbons such as benzene, toluene and xylene, ethers such as jetyl ether, tetrahydrofuran (THF), dioxane, diglyme, 1,2-dimethoxyethane, 2-methoxydiethyl ether, dichloromethane, 1, Halogenated hydrocarbons such as 2-dichloroethane and chloroform, dimethylformamide (DMF), dimethylacetamide (DMA), acetonitrile, ethyl acetate, and other solvents inert to the reaction, or a mixture of two or more Can be used.
- Compound (II) and compound (III) are used in an equimolar to excess amount depending on the reaction and the compound.
- Compound (la) can be produced by the etherification / thioether reaction of the corresponding alcohol or thiol compound (IV) with compound (V).
- halogen or P-toluenesulfoxy group is preferred.
- B is 4-substituted phenol, F is preferred as the same.
- the reaction can be carried out in the presence of a base using DMF, DMA, N-methylpyrrolidone or the like as a solvent from room temperature under heating conditions.
- a reduction reaction of ketone or ester an oxidation reaction of olefin (such as osmium tetroxide), and the like can be appropriately selected in consideration of the structure of the compound.
- olefin such as osmium tetroxide
- carboxylic acid as a substituent on B can be converted to various amides.
- P 1 represents a protecting group for carboxylic acid such as lower alkyl.
- the starting compound (lib) was etherified / thioether using Compound (VI) in the same manner as in the second production method described above.
- a raw material compound (IXa) in which A force N (R 5 ) and R 5 is other than H can be produced by deprotecting the carboxylic acid in the following step: It can be manufactured by this method.
- alkylation conditions can be applied to the alkylation reaction of the compound (Vllb).
- an alkylating agent using (tributylphosphor-lidene) acetonitrile as a reactant in the presence of the corresponding alcohol may be effective.
- P 2 is preferably a benzyl group!
- the raw material compound (IX) in which X 1 is 0 can be produced by the following method.
- the production method of the starting compound (IVa) is described below.
- Compound (XII) was obtained through protection of the hydroxyl group of compound (IX) and deprotection of carboxylic acid.
- a compound (XIII) is obtained by amidation with amin (III)
- a raw material compound (IVa) is obtained by deprotection of the hydroxyl group.
- P 4 is preferably a methoxymethyl group.
- the compound of the present invention is isolated and purified as a free compound, a pharmaceutically acceptable salt, hydrate, solvate, or crystalline polymorphic substance.
- the pharmaceutically acceptable salt of the compound (I) of the present invention can also be produced by subjecting it to a conventional salt formation reaction.
- Isolation and purification are performed by applying ordinary chemical operations such as extraction, fractional crystallization, and various fractional chromatography.
- optical isomers can be separated by selecting an appropriate raw material compound or by utilizing the difference in physical and physical properties between isomers.
- optical isomers can be obtained by stereochemically pure isomers by general optical resolution methods (for example, fractional crystallization leading to diastereomeric salts with optically active bases or acids, chromatography using chiral columns, etc.). Can lead to. It can also be produced from a suitable optically active raw material compound.
- GK activity is the change in absorbance due to NADPH directly produced by NADP (nicotinamide adenine dinucleotide phosphate) when glucose-6-phosphate is dehydrogenated by glucose-6-phosphate dehydrogenase. It was measured.
- the recombinant human liver GK (GST-hGK2) used in this assay was expressed in E. coli as GST (Glutathi one S transferase) -fusion protein and purified with Glutathione Sepharose 7 fume.
- Glucokinase isoform 2 was based on AK122876.1 (accession number), and ORF (open reading frame) was cloned on the following Tagawa page.
- pME18S- FL3- Glu cokinase isoform 2 as a saddle, 5'— TAGAATTCATGGCGATGGATGTCACAAG— 3 ′ (SEQ ID NO: 1) as 5 ′ primer, 5′-ATCTCGAGTCACTGGCCCAGCATACAG-3-3 (SEQ ID NO: 2) as 3 ′ primer PCR (polimerase chain reaction) was performed, and the PCR product was TA-cloned into the pGEM-T easy vector. The sequence of this clone was confirmed by sequencing. Thereafter, the fragment cleaved with EcoRI and Xhol was ligated to the similarly cleaved vector pGE X-5X-1 to prepare pGEX-human Glucokinase 2.
- the enzyme reaction was measured at 27 ° C using a 96-well flat bottom plate. 25 mM HEPES pH 7.4; 25 mM KC1; 2 mM MgCl; 1 mM ATP; 0.1% BSA; 1 mM DTT;
- GK activation (%) [(A OD)-(A OD)] / (A OD) X100
- a OD A OD in 10 tons of test drug
- the compound of Example 2 showed 178% and the compound of Example 7 showed 158% GK activity.
- Test Example 2 Blood glucose lowering effect in C57BL6 mice
- the test compound was dissolved in Cremophor (registered trademark) solvent (Cremophor: DMSO: Water 5: 5: 90, v / v / v). Mice were orally dosed with 10 ml / kg of drug solution or 10 ml / kg of solvent control.
- Approximately 60 1 blood was collected from the orbital venous plexus immediately before administration of the test compound. Blood was collected in the same manner 1 to 4 hours after administration of the test compound. The collected blood was separated from plasma, and the blood glucose level was measured. After administration of the test compound, the blood glucose level after 4 hours was compared with the blood glucose level of the solvent control group at the same time.
- the compound of Example 2 showed 21.6% inhibition (30 mg / kg) of hypoglycemic action.
- Test Example 3 Effects on hyperglycemia after oral glucose load in ICR mice
- the test compound was dissolved in Cremophor solvent (Cremophor: DMSO: Water 5: 5: 90, v / v / v).
- Mice were orally dosed with 10 ml / kg of drug solution or 10 ml / kg of solvent control.
- 10 ml / kg of drug solution or 10 ml / kg of solvent control.
- Approximately 60 1 blood was collected from the orbital venous plexus immediately before administration of the test compound.
- an aqueous solution of 200 mg / ml glucose was orally administered at 10 ml / kg (corresponding to 2 g / kg).
- Approximately 60 L of blood was collected from the orbital venous plexus just before glucose administration.
- Blood was collected in the same manner 0.5, 1, and 2 hours after glucose administration.
- the collected blood was separated from plasma and the blood glucose level was measured.
- the AUC of the blood glucose level from the administration of the test compound to 2 hours after the addition of sugar and the AUC of the solvent control group at the same time were compared.
- Test Example 4 Effect on hyperglycemia after oral glucose load in db / db mice
- the test compound was dissolved in Cremophor solvent (Cremophor: DMSO: Water 5: 5: 90, v / v / v).
- the drug solution was orally administered to db / db mice at 10 ml / kg or solvent control at 10 ml / kg. Covered Approximately 60 1 blood was collected from the orbital venous plexus immediately before administration of the test compound.
- an aqueous solution of 200 mg / ml glucose was orally administered at 10 ml / kg (corresponding to 2 g / kg).
- Approximately 60 1 blood was collected from the orbital venous plexus just before glucose administration. Blood was collected in the same manner 0.5, 1, and 2 hours after glucose administration. The collected blood was separated from plasma and the blood glucose level was measured. The AUC of the blood glucose level from the administration of the test compound to 2 hours after the addition of sugar and the AUC of the solvent control group at the same time were compared.
- the compound of the present invention has a good GK activation action.
- various compounds with improved side effects (effects on hERG and CYP) and Z or solubility have been found, the compounds of the present invention are useful as preventive or therapeutic agents for diabetes and the like. it is obvious.
- a preparation containing one or more of the compounds (I) of the present invention or a salt thereof as an active ingredient is usually used using a pharmaceutical carrier, excipient, etc. that are usually used in the art.
- Administration is oral by tablets, pills, capsules, granules, powders, solutions, etc., or injections such as intraarticular, intravenous, intramuscular, suppositories, eye drops, eye ointments, transdermal solutions.
- Any form of parenteral administration such as an ointment, a transdermal patch, a transmucosal liquid, a transmucosal patch, and an inhalant may be used.
- solid composition for oral administration tablets, powders, granules and the like are used.
- one or more active ingredients are combined with at least one inert excipient such as lactose, mannitol, glucose, hydroxypropyl cellulose, microcrystalline cellulose, starch. , Polybulurpyrrolidone, and / or magnesium aluminate metasilicate.
- the composition may contain an inert additive, for example, a lubricant such as magnesium stearate, a disintegrant such as carboxymethyl starch sodium, a stabilizer, and a solubilizing agent according to a conventional method. Good.
- Liquid compositions for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups or elixirs, etc., commonly used inert diluents such as purified Contains water or ethanol.
- the liquid composition is solubilized in addition to the inert diluent It may contain adjuvants such as humectants, wetting agents and suspending agents, sweeteners, flavors, fragrances and preservatives.
- Injections for parenteral administration contain sterile aqueous or non-aqueous solutions, suspensions or emulsions.
- aqueous solvent include distilled water for injection or physiological saline.
- water-insoluble solvent include propylene glycol, polyethylene glycol or vegetable oil such as olive oil, alcohols such as ethanol, or polysorbate 80 (Pharmacopeia name).
- Such compositions may further contain isotonic agents, preservatives, wetting agents, emollients, dispersants, stabilizers, or solubilizing agents. These are sterilized by, for example, filtration through a pacteria retention filter, blending of a bactericide or irradiation. These can also be used by producing a sterile solid composition and dissolving or suspending it in sterile water or a sterile solvent for injection before use.
- Transmucosal agents such as inhalants and nasal agents are used in solid, liquid or semi-solid form and can be produced according to conventionally known methods.
- known excipients, and further pH adjusters, preservatives, surfactants, lubricants, stabilizers, thickeners and the like may be appropriately added.
- an appropriate device for inhalation or insufflation can be used.
- a known device such as a metered dose inhalation device or a nebulizer
- the compound is administered alone or as a powder in a formulated mixture or as a solution or suspension in combination with a pharmaceutically acceptable carrier. be able to.
- the dry powder inhaler or the like can use a dry powder or a powder-containing capsule which can be used for single or multiple administrations.
- a dry powder or a powder-containing capsule which can be used for single or multiple administrations.
- it may be in the form of an appropriate propellant such as a pressurized aerosol spray using a suitable gas such as black mouth fluoroalkane, hydrofluoroalkane or carbon dioxide.
- the appropriate daily dose is about 0.001 to 100 mg / kg, preferably 0.1 to 30 mg / kg, more preferably 0.1 to 10 mg / kg per body weight. Or administer in 2 to 4 divided doses.
- the appropriate daily dose is about 0.0001 to 10 mg / kg per body weight, and should be administered once to several times a day.
- a transmucosal agent administer about 0.001 to 100 mg / kg per body weight once or multiple times a day.
- the dosage is appropriately determined according to the individual case in consideration of symptoms, age, sex, etc.
- Jetyl ether (40 ml) and water (30 ml) were added, and after liquid separation, saturated aqueous sodium bicarbonate (40 ml) was added to the organic layer, and the pH of the aqueous layer was adjusted to about 8.
- the aqueous layer was washed with jetyl ether (30 ml), and then concentrated hydrochloric acid (6 ml) was added to adjust the pH of the aqueous layer to about 3.
- the aqueous layer was extracted with ethyl acetate (40 ml), and the obtained organic layer was washed with saturated brine (30 ml) and dried over anhydrous magnesium sulfate.
- Methyl 4- (acetyloxy) -2-methyl-1-benzothiophene-6-carboxylate (1.0 g) A 28% sodium methoxide / methanol solution (3.3 ml) was added to the ethanol (32 ml) solution, and the mixture was stirred at 75 ° C overnight. Under ice-cooling, ethyl acetate (40 ml) was added for dilution, and 1M hydrochloric acid (18 ml) was added to adjust the pH of the aqueous layer to about 3. The organic layer obtained by the liquid separation operation was washed successively with water (30 ml) and saturated brine (30 ml), and then dried over anhydrous magnesium sulfate. The solvent was distilled off under reduced pressure to obtain methyl 4-hydroxy-2-methyl-1-benzothiophene-6-carboxylate (823 mg) as a pale yellow solid.
- Bromomethylbenzene (320 ⁇ 1) was added dropwise at -10 ° C to a DMF (10 ml) suspension of methyl 4-hydroxy-1H-indole-6-carboxylate (500 mg) and potassium carbonate (361 mg). The mixture was stirred for 30 minutes, and further stirred at room temperature for 5 hours. To the reaction mixture are added water (30 ml) and sodium acetyl acetate (30 ml), and the mixture is separated. The organic layer is washed successively with saturated aqueous sodium hydrogen carbonate (30 ml) and saturated brine (30 ml), and dried over anhydrous magnesium sulfate. did.
- N-bromosuccinimide (6.6 g) was added to a methylene chloride solution (250 ml) of triphenylphosphine (9.6 g) under ice cooling. After stirring for 20 minutes under ice-cooling, a methylene chloride suspension (250 ml) of 4- (methoxymethoxy) -2-methyl-tobenzothiophene-6-carboxylic acid (5.5 g) was added. After stirring for 20 minutes under ice-cooling, 2-aminothiazole (5.5 g) was stirred at the same temperature for 20 minutes and at room temperature overnight.
- the organic layer obtained by the liquid separation operation was washed successively with 1M hydrochloric acid (30 ml), saturated aqueous sodium hydrogen carbonate (30 ml) and saturated brine (30 ml), and dried over anhydrous magnesium sulfate.
- the residue obtained by distilling off the solvent under reduced pressure was purified by silica gel column chromatography (hexane / ethyl acetate) to give 1-methyl-4- [4- (methylsulfol) phenoxy] -N- (l , 3-Thiazole-2-yl) -1H-indole-6-carboxamide (44 mg) was obtained as a colorless solid.
- Tables 19 and 20 show the structures of other compounds of the present invention. These can be easily synthesized by using the above-described production methods, the methods described in the examples, methods obvious to those skilled in the art, or variations thereof.
- the compound of the present invention has a GK activation action, it is useful as a therapeutic and prophylactic agent for diabetes, particularly type 2 diabetes. It is also useful for the treatment and prevention of diabetic complications such as nephropathy, retinopathy, neuropathy, peripheral circulatory disorder, cerebrovascular disorder, ischemic heart disease, and arteriosclerosis. Furthermore, by suppressing overeating, it is useful as a therapeutic and prophylactic agent for obesity and metabolic syndrome.
Abstract
【課題】GK活性化剤として有用な化合物を提供する。
【解決手段】発明者等は、縮合へテロ環について鋭意検討した結果、縮合へテロ環のベンゼン環上に-X-Bで示される基及びカルバモイル基を有し、当該カルバモイル基の窒素原子には、特定の置換基で置換されていてもよいピリジル、ピラジニル又はチアゾリル基が結合することを特徴とする化合物が、良好なGK活性化作用を有することを確認し、本発明を完成した。
本発明化合物は、良好なGK活性化作用を有することから、糖尿病、特に2型糖尿病の治療剤として有用である。
Description
明 細 書
縮合へテロ環化合物
技術分野
[0001] 本発明は、医薬、殊に糖尿病治療剤として有用な新規な縮合へテロ環化合物に関 する。
背景技術
[0002] GK (ダルコキナーゼ (ATP:D- hexose 6- phosphotransferase, EC2.7.1.1))は脾臓、 肝臓に発現する 6炭糖をリン酸化する酵素で、近年脳にも存在することが明らかにさ れている。本酵素はへキソキナーゼファミリーに属し、別名へキソキナーゼ IVともよば れる。 GKは他のへキソキナーゼと比較し 1)基質であるグルコースに対する親和性 が低く血糖濃度に近い Km値を示す、 2)酵素反応生成物のグルコース 6-リン酸によ る阻害を受けない、 3)分子量が約半分の 50 kDaである等の特徴を持つ。
ヒト—ダルコキナーゼ遺伝子は、単一遺伝子として第 7染色体 pl3に位置し、脾 |8細 胞と肝細胞では 30 kb以上離れた組織特異的な異なるプロモーターで制御され、異 なる第 1ェクソンを用いる力 残りのェクソン 2-10は共通である。そのため、脾型と肝 型の GK蛋白では N末 15残基が異なるのみである。
[0003] 血糖値の上昇に伴!、、脾 j8細胞内のグルコース濃度は糖輸送担体である GLUT2 を介し速やかに平衡に達し、 GKが細胞内のグルコース濃度変化を察知し解糖系を 活性化する。この結果脾 β細胞内の ATP/ADP比が上昇し Κ チャンネルが閉鎖し、
ΑΤΡ
これを電位依存性の Caチャンネルが察知し細胞内カルシウム濃度が上昇しインスリ ンの放出が起こる。即ち脾 j8細胞において GKはグルコースセンサーとして働きインス リン分泌の制御に重要な役割を果たして 、る。肝臓でも GKはグルコースセンサーとし て働き、血糖値の上昇に反応し、グルコースをグルコース 6-リン酸に変換する。この 結果グリコーゲンの産生が上昇すると共に、解糖系が活性化され肝臓での糖新生が 抑制される。
GKの遺伝子変異によりグルコースのリン酸ィ匕能が低下した患者では高血糖が頻発 し、若年性の糖尿病を発症する (MODY2)。一方遺伝子変異により GK活性の Km値
が低値を示す患者では、食後並びに空腹時に低血糖が認められる。即ち、ヒトにお いても GKはグルコースセンサーとして働き、血中のグルコースレベルを正常に維持 する上で重要な役割を演じている。これらの事実から、 GKを活性ィ匕する薬剤は、脾 j8細胞内からのグルコース依存的なインスリン分泌を亢進させ食後高血糖を是正す ると共に、肝臓からの糖放出を抑制する優れた 2型糖尿病の治療薬となると期待され る。更に食後の高血糖状態で肝臓への糖取り込みが促進され、余分なインスリン分 泌亢進が起こらず、従来スルホ -ルゥレア (SU)剤で問題となる膝疲弊を回避できる可 能性もある。また、近年マウス培養脾細胞(MIN6N8)を高グルコース下で培養すると アポトーシスが誘発されることが報告された。更にこの細胞にダルコキナーゼを過剰 発現させると、 MIN6N8細胞のアポトーシスが抑制されたことから (Diabetes 54:2 2602 -2611(2005》 GK活性化剤は脾保護作用を示すことが期待される。
脳に存在する GKは脾臓タイプであり、摂食中枢である VMH (視床下部腹内側部: V entromedial hypothalamus)の神経に多く発現する。グルコース感受性神経はダルコ ースに対し興奮性の GE (Glucose Exited)- neuronと、グルコースに対し抑制性の GI ( Glucose Inhibited)- neuronに分類される。 GKの mRNAや蛋白は、 GE- neuronの約 70% に、 GI-neuronの約 40%にその存在が認められる。
これらグルコース感受性神経では、 GKが細胞内グルコースの上昇を察知し、解糖 系が活性化され、細胞内の ATP/ADP比が上昇する。この結果 GE-neuronでは K チ
ATP
ヤンネルが閉鎖し-ユーロンの活動電位頻度が高まり、神経伝達物質が放出される。 一方、 GI-neuronでは C1—チャンネルが関与すると考えられている。 VMHにおいて GK mRNAの発現を上昇させたラットでは、グルコース欠乏状態に対する補償作用が低下 する。
グルコース感受性神経には摂食行動に関与するレブチンやインスリンに対する受 容体も存在する。高グルコース条件下の GE- neuronにおいて、レプチンやインスリン は、 K チャンネルを開口させ活動電位頻度を減少させる。さらに ARC (弓状核)にお
ATP
いて食欲増進に働く NPY (Neuroeptide Y)- neuronはグルコースに対し抑制性であり、 食欲抑制に働く POMC (Proopiomelanocortin)- neuronはグルコースに対し興奮性で ある(Diabetes 53: 2521-2528 (2004))。これらの事実から、中枢の GKを活性化するこ
とで摂食行動が抑制され、肥満やメタボリックシンドロームの治療に有効であることが 期待される。
[0005] GK活性ィ匕作用を有する化合物は多数報告されているものの、現在までのところ臨 床での有効性が確認された化合物の報告は無い。また、種々の副作用(hERGや CY Pに対する作用)の軽減や溶解性にお!、ても良好なプロファイルを有する新規な GK 活性化剤が切望されている。
GK活性ィ匕作用を有する下記べンゾフラン誘導体が報告されている (例えば、特許 文献 1)。
[化 1]

(式中、 Aはハロゲン、カルボキシ及び C アルキル力 選択される基で置換されてい
1-4
てもよい、ピリジン- 2-ィル又はチアゾール -2-ィルを示し、 R3は、それぞれハロゲン、
C アルキル、 C アルコキシ、モノ又はジアルキルアミ入ヘテロ環、炭化水素環- 0
1-4 1-4
、ヘテロ環- 0又はカルボシクリリデュルで置換されていてもよい、 C アルキル、 C
1-4 1-4 アルコキシ、炭化水素環、ヘテロ環、炭化水素環- 0又はへテロ環- 0を示す。その他 は当該公報参照。 )
[0006] 特許文献 1:国際公開 WO2004/046139号パンフレット
発明の開示
発明が解決しょうとする課題
[0007] 本発明の課題は、 GK活性ィ匕作用を有する医薬、特に糖尿病治療剤として有用な、 新規な化合物の提供である。
課題を解決するための手段
[0008] 本発明者等は、縮合へテロ環化合物について鋭意検討した結果、縮合へテロ環の ベンゼン環上に- X-Bで示される基及び力ルバモイル基を有し、当該カルバモイル基 の窒素原子には、特定の置換基で置換されていてもよいピリジル、ビラジニル又はチ
ァゾリル基が結合することを特徴とする化合物が、良好な GK活性ィ匕作用を有すること を確認し、本発明を完成した。
即ち、本発明は、一般式 (I)で示される縮合へテロ環化合物又はその塩に関する。
[化 2]

(式中の記号は以下の意味を示す。
A: S、 0又は N(R5)、
X : 0、 Sゝ SO又は SO、
2
B :- R°、 -低級ァルケ-ル、 - R°°- CN、 - R°°- C0N(RG)、 - R°°- 0- RG、 - R°°- N(RG)CO- R。
2
、 -Y- (置換されていてもよいァリール)、 -Y- (置換されていてもよいへテロ環)又は- Y-( 置換されて 、てもよ ヽシクロアルキル)、
Υ:単結合、置換されていてもよい低級アルキレン、 - R。。- 0- R。。-、 - R。。- CON(RG) -、 - R
°°- N(RG)CO-、 -(置換されて 、てもよ 、低級アルキレン)- CON(RG)-又は- (置換されて
V、てもよ 、低級アルキレン)- N(RG)CO-、
R°:同一又は互いに異なって、低級アルキル、
R°°:同一又は互いに異なって、低級アルキレン、
RG:同一又は互いに異なって、 H又は R°、
D :チアゾール、ピリジン又はビラジン、
R1及び R2 :同一又は互いに異なって、下記 (0又は (ii)力も選択される基、
(0:- CH(0RA)- RB、 - C(R°)(0RA)- RB、 - CO- CO- NRCRD、 - CO- CO- NRC- 0RD、 - C(0 RE)(0RF)- RB、 - C(0RE)(0RF)- R°、 -CH(0RE)-CH(0RF)-RC及び/又は- CH(0RE)-CH (0RF)-RB、
(ii) :— Hゝ -ハロゲン、 -CO H、 -CO R。、 -NO、 - CN、 - R。、 -低級アルケニル、 CO - C
2 2 2
0 H、 - C0-C0-0R°、 -ハロゲノ低級アルキル及び/又は-低級アルキレン -NRCRD、
2
RA:同一又は互いに異なって、 - H、 - R°、 -ハロゲノ低級アルキル又は- R°°-ァリール、
RB :- CO H、 -CO R°、 - CO- NRCRD、 - CO- NRC- ORD、 - R°°- NRCRD、 - R°°- ORA、 - R。0- C
2 2
O R°、 -低級アルキレン- CO- NRCRD又は- R°°- CO- Nl - ORD、
2
RC及び RD:同一又は互いに異なって、 - H、 - R°、 - R°°-N(RA)、 - R°°-0RA、 - R°°-C0 H
2 2
、— R°°— C〇 R°又は— R°°— C〇— N(RA)、
2 2
RE及び :同一又は互いに異なって、 RAに記載の基、或いは、 RE及び RFがー体とな つて、 R00、
R3及び R4:同一又は互いに異なって、 - H、 -ハロゲン、 - R°、 -ハロゲノ低級アルキル又 は- R00- 0H、
R5 :-H、 - R°、 -ハロゲノ低級アルキル、 -(置換されていてもよいァリール)又は- R°°- (置 換されて 、てもよ 、ァリール)。
但し、 Aが 0のとき、 Bは、少なくとも 1個の- SO R°で置換されたァリールを示す。 )
2
[0009] 更に本願は、一般式 (I)で示される縮合へテロ環化合物又はその塩を有効成分とす る医薬、殊に GK活性化剤及び糖尿病治療剤にも関する。
更に本願は、糖尿病治療剤の製造のための、一般式 (I)で示される縮合へテロ環化 合物又はその塩の使用、並びに、糖尿病の治療方法にも関する。
発明の効果
[0010] 本発明化合物は、 GK活性化作用を有することから、糖尿病、特に 2型糖尿病の治 療並びに予防薬として有用である。また糖尿病の合併症である腎症、網膜症、神経 障害、末梢循環障害、脳血管障害、虚血性心疾患、動脈硬化症の治療並びに予防 薬として有用である。更に過食を抑制することにより、肥満、メタボリックシンドロームの 治療並びに予防薬としても有用である。
発明を実施するための最良の形態
[0011] 以下、本発明を詳細に説明する。
本明細書中、「アルキル」、「アルキレン」及び「ァルケ-ル」とは、直鎖状又は分枝 状の炭化水素鎖を意味する。「低級アルキル」とは、炭素数 1〜6個(以下、 C )のァ
1-6 ルキル基であり、好ましくはメチル、ェチル、 n-プロピル、 2-プロピル、へキシル等で ある。「低級アルキレン」は、上記「低級アルキル」の任意の水素原子 1個を除去して なる二価基を意味し、好ましくは C のアルキレンであり、より好ましくはメチレン、ェチ
レン、メチルメチレン及びプロピレンである。「低級ァルケ-ル」とは、一個以上の二重 結合を有する C のァルケ-ルであり、好ましくは C のァルケ-ルであり、より好まし
2-6 2-4
くはビニル、 1-プロぺニル及びァリルである。
「ハロゲン」とは、 F、 Cl、 Br及び Iである。「ハロゲノ低級アルキル」とは、 1個以上(好 ましくは 1〜5個、より好ましくは 1〜3個)のハロゲンで置換された C アルキルを意味
1-6
し、好ましくは 1個以上の Fで置換された C アルキルであり、より好ましくは、 1〜3個
1-6
の Fで置換された C アルキルであり、より更に好ましくは、フルォロメチル、ジフルォ
1-6
ロメチル、トリフルォロメチル及びトリフルォロェチルである。
[0012] 「シクロアルキル」は、 C のシクロアルキルであり、ァダマンチル等の架橋環を形成
3-10
して 、てもよ 、。好ましくはじ のシクロアルキルであり、より好ましくはシクロプロピル
3-7
、シクロブチル、シクロペンチル、シクロへキシル及びシクロへプチルである。「シクロ ァルケ-ル」は、 1又は 2個の二重結合を有する C の環基である。「ァリール」は、 C
3-7 6- の芳香族炭化水素基を意味し、「シクロアルケ-ル」と縮環したフエニル基、例えば
14
インデュル、テトラヒドロナフチル、フルォレニル基を含む。好ましくはフエ-ル及びナ フチルであり、より好ましくはフエ-ルである。
「炭化水素環」とは、前記「シクロアルキル」、「シクロアルケニル」及び「ァリール」を 包含する。
[0013] 「ヘテロ環基」とは、 0、 S及び N力 選択されるへテロ原子を 1〜4個含有する 3〜7 員の単環又は二環式へテロ環基であり、飽和環、芳香環 (ヘテロァリール)、及びそ の部分的に水素化された環基を包含する。例えば、ピリジル、ピラジニル、ピリミジ- ル、ピリダジ -ル、イミダゾリル、ベンゾイミダゾリル、ベンゾフラ -ル、ベンゾチェ-ル 、ベンゾチアジアゾリノレ、ベンゾチアゾリノレ、ベンゾイソチアゾリノレ、ベンゾ才キサゾリ ル、ベンゾイソォキサゾリル、ピロリル、ピロリジニル、チェニル、フリル、ジォキサニル 、ジォキソラニル、トリアジニル、トリァゾリル、チアゾリル、チアジアゾリル、ォキサジァ ゾリル、ピラゾリル、ビラゾリジニル、イソチアゾリル、ォキサゾリル、イソォキサゾリル、 キノリル、イソキノリル、テトラヒドロキノリル、テトラヒドロイソキノリル、キナゾリニル、キノ キサリニル、フタラジニル、ピペリジル、ピペラジニル、ァゼパニル、ジァゼパニル、テ トラヒドロフラニル、テトラヒドロビラニル、モルホリニル、メチレンジォキシフエニル、ェ
チレンジォキシフエ-ル、トリチアニル、インドリル、イソインドリル、インドリ-ル、インダ ゾリル、テトラヒドロべンゾイミダゾリル、クロマ-ル、クロモ-ル(4-ォキソ - 4H- 1-ベン ゾビラ-ル)、ベンゾイミダゾ口-ル、チェノビラゾリル及び(2,3-ジヒドロ- 2-ォキソベン ゾイミダゾリル)が挙げられる。好ましくは、 5乃至 6員単環式へテロァリールであり、更 に好ましくは、フリル、チェ-ル、イミダゾリル、チアゾリル、又はピリジルである。
「置換されていてもよい」とは、「無置換」あるいは「同一又は異なる置換基を 1〜5個 有していること」を示す。なお、複数個の置換基を有する場合、それらの置換基は同 一でも互!ヽに異なって!/、てもよ!/、。
「置換されて 、てもよ 、ァリール」、「置換されて 、てもよ 、ヘテロ環」及び「置換され ていてもよいシクロアルキル」における置換基は、好ましくは、 - R°、ハロゲノ低級アル キル、ハロゲン、 - OH、 - R°°- OH、 - N、 -OR°, - R°°- OR°、 -O-炭化水素環、 - O-へテ
3
口環、 - CN、 -NO、 - CHO、 -CO H、 -CO R°、 - R°°- CO H、 - R°°- CO R°、 - CO- R°、 - C
2 2 2 2 2
O-炭化水素環、 -CO-ヘテロ環、 - CO- NH、 - CO- NH- R°、 - CO- N(R°)、 - CO- N(RG)
2 2
- R°°- ORG、 - CO- N(R°)- R°°- N(RG)、 - CO- NRG-炭化水素環、 - CO- NRG-ヘテロ環、 -
2
CO- N(RG)- R。。-ヘテロ環、 - NHCO- R°、 - N(R°)CO- R°、 - NHCO-炭化水素環、 - NHC O-ヘテロ環、 -SH、 -SR°、 -S-炭化水素環、 -S-ヘテロ環、 -SO- R°、 -SO-炭化水素環 、 -SO-ヘテロ環、 -SO R°、 -SO -炭化水素環、 -SO -ヘテロ環、 -SO H、 -SO NH、 - S
2 2 2 3 2 2
O NH- R°、 -SO N(R°)、 -SO NH-炭化水素環、 -SO NH-ヘテロ環、 - NHSO - R°、 - N(
2 2 2 2 2 2
R°)-SO - R°、 -NHSO -炭化水素環、 -NHSO -ヘテロ環であり、より好ましくは、 - R°、
2 2 2
ハロゲノ低級アルキル、ハロゲン、 -。 H、 - R°°-〇H、 - N、 -OR°, - R°°-〇R°、 -〇-炭化
3
水素環、 〇—ヘテロ環、 CN、 -NO、 - CH〇、 -CO H、 -CO R°、 - R°°- CO H、 - R°°- C
2 2 2 2
〇 R°、 - C〇- R°、 - C〇-炭化水素環、 - C〇-ヘテロ環、 - C〇- NH、 - C〇- NH- R°、 - C〇-
2 2
N(R°)、 - C〇- NH-炭化水素環、 - C〇- NH-ヘテロ環、 - NHCO- R°、 - N(R°)C〇- R°、 - N
2
HC〇-炭化水素環、 -NHCO-ヘテロ環、 -SH、 -SRG、 -S-炭化水素環、 -S-ヘテロ環、 -S〇- RG、 -S〇-炭化水素環、 -SO-ヘテロ環、 -SO RG、 -SO -炭化水素環、 -SO -へテ
2 2 2 口環、 -SO H、 -SO NH、 -SO NH- R°、 -SO N(R°)、 -SO NH-炭化水素環、 -SO NH-
3 2 2 2 2 2 2 2 ヘテロ環、 -NHSO - R°、 -N(R°)-SO - R°、 -NHSO -炭化水素環、 -NHSO -ヘテロ環で
2 2 2 2
あり、ここに、上記「炭化水素環」及び「ヘテロ環」は、 1〜5個の- RQ、ハロゲノ低級ァ
ルキル、ハロゲン、 - OH、 - OR°及び- N(RG)力 選択される基で置換されていてもよく
2
、好ましくは 1〜5個の- R°、ハロゲノ低級アルキル、ハロゲン、 -OH及び- OR。から選 択される基で置換されて 、てもよ 、。
また、「置換されていてもよい低級アルキレン」における置換基としては- R°又は- R°° - ORGが好ましい。
本発明の好ましい態様を以下に示す。
(1) Aとしては、好ましくは S又は N(R5)である。ここに R5として好ましくは H、低級アルキ ル又は置換されたフエ-ル、より好ましくは C アルキル又は 4位が C アルキル- SO
1-4 1-2 2
-で置換されたフエ-ルである。
(2) Xとしては、好ましくは 0である。
(3) Bとしては、好ましくは 1又は 2個の置換基で置換されて!、てもよ!/、フエ-ルであり
、より好ましくは少なくとも 1個は 4位に置換基を有するフ ニルであり、更に好ましくは 4位に 1個の置換基を有するフエ-ルである。ここに、 Bにおけるそれぞれ置換されて いてもよい「ァリール」、「ヘテロァリール」又は「シクロアルキル」の置換基としては、好 ましくはハロゲノ低級アルキル、ハロゲン、 -OR0, - CN、 -NO、 - CHO、 -CO H、 -CO
2 2 2
R°、 - CO- R°、 -CO-炭化水素環、 -CO-ヘテロ環、 -SO R°、 -SO -炭化水素環又は- S
2 2
0 -ヘテロ環であり、より好ましくはハロゲノ低級アルキル、ハロゲン、 -NO、 - CO-R°、
2 2
-CO-炭化水素環、 -CO-ヘテロ環又は- SO RQであり、更に好ましくは- SO -メチル又
2 2 は- so -ェチルであり、より更に好ましくは- so -メチルである。
2 2
(4) Dとしては、アミド基の窒素原子とそれぞれ 2位で結合する、チアゾール (即ち、チ ァゾール -2-ィル)、ピリジン(即ち、 2-ピリジル)又はビラジン(即ち、ピラジン- 2-ィル) である。
(5) R1及び R2としては、好ましくはともに Hである; R1及び R2の別の好ま 、態様として は、好ましくは一方が Hであり、他方が (0及び GO力 選択される H以外の基であり、よ り好ましくは R1が Hであり、 R2が (0及び (ii)力 選択される H以外の基である。ここで、
(0は、 - CH(ORA)- RB、 - CO- CO- NRCRD、 - CO- CO- NRC- ORD、 - C(ORE)(ORF)- RB、 - C (ORE)(ORF)- R°、 -CH(ORE)-CH(ORF)-RC及び/又は- CH(ORE)-CH(ORF)-RBであり、 (ii)は、 - Hゝ -ハロゲン、 -CO H、 -CO R°、 -NO、 - CNゝ - R°、 -低級ァルケ-ル、 -CO-
CO H、 - CO-CO-OR°、 -ハロゲノ低級アルキル及び/又は-低級アルキレン -NR D
2
である。
(0及び GO力も選択される基としては、好ましくは- CO H、 -CO R°、 -CH(ORA)- RB、 - C
2 2
O- CO- NRCRD、 - C(ORE)(ORF)- RB、 - C(ORE)(ORF)- R。又は- CH(ORE)- CH(ORF)- RCで あり、より好ましく ίま— CH(OH)— CH OH、 -CH(OR°)-CH OH、— CH(OR°)— CH OR0、— C
2 2 2
H(OH)-CO H、 -CH(OR°)-CO H、 - CH(OH)- CO R。、 - CH(OR°)-CO R。又は- CO- C
2 2 2 2
O— NRCRDであり、更に好ましく ίま— CH(OH)— CH OH、— CH(OR°)— CH OH、— CH(OR。)—
2 2
CH OR°、— CH(OH)— CO R°、— CH(OR°)— CO R°、— CO— CO— NH、— CO— CO— NH— R0、
2 2 2 2
- CO- CO- N(R°)、 - CO- CO- NH-低級アルキレン- O- R°、又は- CO- CO- NH-低級ァ
2
ルキレン- OHである。
(6) R3及び R4としては、好ましくは H又は R°である。
更に別の好まし 、態様としては、上記(1)〜(6)に記載の各好ま ヽ基の組合せか らなる化合物が好ましい。即ち、
(7) Bが、 R°、 -(置換されていてもよいァリール)又は- R°°- (置換されていてもよいァリー ル)であり、 R1及び R2が、同一又は互いに異なって、上記(5)に記載の (0又は GOから 選択される基である化合物。
(8) Xが 0である、上記(7)の化合物。
(9) R1及び R2が Hである上記(7)又は(8)の化合物。
(10) R1が Hであり、 R2カ CH(OH)- CH OH、 - CH(OR°)- CH OH、 - CH(OR。)- CH OR。
2 2 2
、— CH(OH)— CO R。、 -CH(OR°)-CO R。、— CO— CO— NH、— CO— CO— NH— R。、— CO— C
2 2 2
O- N(R°)、 - CO- CO- NH-低級アルキレン- O- R°、又は- CO- CO- NH-低級アルキレ
2
ン- OHである、上記(7)又は(8)の化合物。
本発明の化合物は、置換基の種類によっては他の互変異性体や幾何異性体が存 在する場合もある。本明細書中、それら異性体の一形態のみで記載することがあるが 、本発明にはこれらの異性体も包含し、異性体の分離したもの、あるいは混合物も包 含する。
また、化合物 (I)は不斉炭素原子や軸不斉を有する場合があり、これに基づく (R)体 、(S)体などの光学異性体が存在しうる。本発明はこれらの光学異性体の混合物や単
離されたものを全て包含する。
更に、本発明には、化合物 (I)の薬理学的に許容されるプロドラッグも含まれる。薬 理学的に許容されるプロドラッグとは、加溶媒分解により又は生理学的条件下で本発 明のアミノ基、 - OH、 -CO H等に変換できる基を有する化合物である。プロドラッグを
2
形成する基としては、例えば、 Prog. Med., 5, 2157-2161 (1985)や「医薬品の開発」( 廣川書店、 1990年)第 7卷 分子設計 163-198に記載の基が挙げられる。
[0017] 更に、本発明化合物は、酸付加塩又は置換基の種類によっては塩基との塩を形成 する場合もあり、カゝかる塩が製薬学的に許容され得る塩である限りにおいて本発明に 包含される。具体的には、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、リン酸等 の無機酸や、ギ酸、酢酸、プロピオン酸、シユウ酸、マロン酸、コハク酸、フマル酸、マ レイン酸、乳酸、リンゴ酸、酒石酸、クェン酸、メタンスルホン酸、エタンスルホン酸、 p -トルエンスルホン酸、ァスパラギン酸、又はグルタミン酸等の有機酸との酸付加塩、 ナトリウム、カリウム、マグネシウム、カルシウム、アルミニウム等の無機塩基、メチルァ ミン、ェチルァミン、エタノールァミン、リシン、オル-チン等の有機塩基との塩やアン モ-ゥム塩等が挙げられる。
本発明は、本発明化合物及びその製薬学的に許容され得る塩の各種の水和物や 溶媒和物、及び結晶多形を有する物質も包含する。
[0018] (製造法)
本発明化合物及びその製薬学的に許容され得る塩は、その基本骨格あるいは置 換基の種類に基づく特徴を利用し、種々の公知の合成法を適用して製造することが できる。その際、官能基の種類によっては、当該官能基を原料乃至中間体の段階で 適当な保護基 (容易に当該官能基に転化可能な基)に置き換えておくことが製造技 術上効果的な場合がある。このような官能基としては例えばアミノ基、水酸基、カルボ キシル基等であり、それらの保護基としては例えばグリーン (Greene)及びウッツ (Wuts) 著、「Protective Groups in Organic Synthesis (第 3版、 1999年)」に記載の保護基等を 挙げることができ、これらを反応条件に応じて適宜選択して用いればよい。このような 方法では、当該保護基を導入して反応を行った後、必要に応じて保護基を除去する ことにより、所望の化合物を得ることができる。
また、化合物 (I)のプロドラッグは上記保護基と同様、原料乃至中間体の段階で特 定の基を導入、あるいは得られた化合物 (I)を用い反応を行うことで製造できる。反応 は通常のエステル化、アミド化、脱水等、当業者により公知の方法を適用することによ り行うことができる。
以下、本発明化合物の代表的な製造法を説明する。なお、本発明の製造法は以 下に示した例には限定されな!、。
第一製法
[化 3]
(式中、 Lは脱離基又は OHを示す。以下同様。 )
本製法は、カルボン酸誘導体 (II)とァミノ化合物(III)とをアミドィ匕反応に付して、式( I)で示される本発明化合物を得る方法である。 Lの脱離基としては、メタンスルホニル ォキシもしくは P-トルエンスルホ-ルォキシ等の有機スルホン酸基、ハロゲン等が挙 げられる。或いは、(II)として、種々の酸無水物が使用できる。
Lが水酸基である場合は Ν,Ν'-ジシクロへキシルカルボジイミド(DCC)、 1-ェチル -3 -(3'-ジメチルァミノプロピル)カルボジイミド (WSC)、 1,1 '-カルボ-ルジイミダゾール( CDI)、ジフエ-ルホスホリルアジド(DPPA)、ォキシ塩化リン/ピリジン、トリフエ-ルホ スフイン/ Ν-プロモスクシンイミド等の縮合剤の存在下反応を行うことができ、場合によ つては、更に添加剤(例えば、 Ν-ヒドロキシスクシンイミド(HONSu)、 1-ヒドロキシベン ゾトリアゾール (HOBt)等)の存在下行うことができる。また、場合によっては、更に反 応終了後の過剰なアミンを除去する目的でイソシァネートを担持したポリスチレン榭 脂、例えば PS-イソシァネート(PS- Isocyanate) (Argonaut社製)、 PL-イソシァネート レジン(PL- NCO Resin) (ポリマ^ ~ ·ラボラトリーズ社(Polymer Laboratories)、英国)等 を用いることが好ましい。また、更に反応終了後の過剰なカルボン酸、前述の添加剤 等を除去する目的で 4級アンモ-ゥム塩を担持したポリスチレン榭脂、例えば MP-力
ルボネート(MP- Carbonate) (バイオタージ社製),あるいはアミンを担持したポリスチ レン榭脂、例えば PS-トリスァミン(PS- Trisamine) (バイオタージ社製)、 PL-ジェチレ ントリアミノメチル(PL- DETA Resin) (ポリマ^ ~ ·ラボラトリーズ社(Polymer Laboratorie s)、英国)等を用いることが好ましい場合がある。
Lが脱離基である場合は、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸 水素カリウム等の無機塩基、或いは、トリェチルァミン、ジイソプロピルェチルァミン、 ピリジン等の有機塩基の存在下反応させるのが好ましい場合がある。
溶媒は、ベンゼン、トルエン、キシレン等の芳香族炭化水素類、ジェチルエーテル 、テトラヒドロフラン(THF)、ジォキサン、ジグリム、 1,2-ジメトキシェタン、 2-メトキシジ ェチルエーテル等のエーテル類、ジクロロメタン、 1,2-ジクロロェタン、クロ口ホルム等 のハロゲン化炭化水素類、ジメチルホルムアミド(DMF)、ジメチルァセトアミド(DMA) 、ァセトニトリル、酢酸ェチル等の反応に不活性な溶媒を単独で、又は 2種以上混合 して用いることができる。また、化合物(II)及びィ匕合物(III)は、等モル乃至過剰量を、 反応や化合物に応じて適宜使用する。
第二製法
[化 4]

化合物(la)は対応するアルコール又はチオールィ匕合物(IV)と化合物 (V)とのエー テル化/チォエーテルィ匕反応により製造することができる。じとしてはハロゲン又は P- トルエンスルホ -ルォキシ基等が好ましぐ Bが 4-置換フエ-ルの場合、じとしては F が好ましい。反応は、塩基存在下、 DMF、 DMA又は N-メチルピロリドン等を溶媒とし て用い、室温から加熱条件下で行うことができる。
また、原料ィ匕合物 (la)のうち、 X1が Sの化合物を酸ィ匕反応に付すことにより、 X1が SO 又は SOの化合物を得ることができる。
[0021] 式 (I)における基 R1及び R2、或 、は B上の種々の置換基は、本発明化合物 (I)を原 料として、当業者にとって自明である反応、又はこれらの変法を用いることにより、他 の官能基へと容易に変換することができる。例えば、アルキル化、ァシル化、酸化、 還元、加水分解、アミドィ匕等、当業者が通常採用し得る工程を任意に組み合わせて 行うことができる。例えば、水酸基を有する化合物の製造においては、ケトンやエステ ルの還元反応ゃォレフインの酸化反応(四酸化オスミウム等)等を、化合物の構造を 考慮して適宜選択できる。また、 B上の置換基としてのカルボン酸を種々アミドに変換 することちでさる。
(原料合成)
[0022] [化 5]

(式中、 P1は低級アルキルなどのカルボン酸の保護基を示す。以下同様。 ) 原料化合物 (lib)は、化合物 (VI)を用いて前述の第二製法と同様にエーテル化/チ ォエーテル化反応を行な 、、次 、でカルボン酸の脱保護を行うことにより製造できる 更に、 A力N(R5)であり、 R5が H以外の原料ィ匕合物(IXa)は、以下の方法で製造でき る。
[0023] [化 6]

(式中、 P2は水酸基の保護基を示す。以下同様。 )
化合物(Vllb)のアルキル化反応は、種々のアルキル化の条件が適用できる。種々 のアルキル化の条件のうち、対応するアルコール存在下に (トリブチルホスホラ-リデ ン)ァセトニトリルを反応剤として用いるアルキル化剤が効果的であることがある。 P2と してはべンジル基が好まし!/、。
また、 X1が 0である原料ィ匕合物 (IX)は、以下の方法で製造できる。
[化 7]

(式中、 P3は水酸基の保護基を示す。以下同様。 )
本合成は J.Chem.Soc.(C), 3171-3173 (1971)に記載の方法に準拠して行うことがで きる。すなわち、ベンズアルデヒド誘導体 (X)に対し、コハク酸エステルを作用させる ことにより(XI)とし、次いで、酸無水物存在下の環化反応に付し環化体 (VIII)を得る 。その後、脱保護することにより、原料ィ匕合物 (IX)を得ることができる。 P3としてはァセ
チル基が好ましい
[0025] [化 8]

(式中、 P4は水酸基の保護基を示す。以下同様。 )
原料化合物(IVa)の製法を以下に述べる。化合物(IX)の水酸基の保護及びカルボ ン酸の脱保護を経て化合物 (XII)とした。次いで、前述の第一製法のアミド化方法に 従って、ァミン (III)とのアミドィ匕によりィ匕合物 (XIII)とし、最後に水酸基の脱保護により 、原料ィ匕合物(IVa)を得ることができる。 P4としてはメトキシメチル基が好ましい。
[0026] 本発明化合物は、遊離化合物、その製薬学的に許容される塩、水和物、溶媒和物 、あるいは結晶多形の物質として単離され、精製される。本発明化合物 (I)の製薬学 的に許容される塩は、常法の造塩反応に付すことにより製造することもできる。
単離、精製は、抽出、分別結晶化、各種分画クロマトグラフィー等通常の化学操作 を適用して行われる。
各種の異性体は、適当な原料化合物を選択することにより、あるいは異性体間の物 理ィ匕学的性質の差を利用して分離することができる。例えば、光学異性体は一般的 な光学分割法 (例えば、光学活性な塩基又は酸とのジァステレオマー塩に導く分別 結晶化やキラルカラム等を用いたクロマトグラフィー等)により、立体化学的に純粋な 異性体に導くことができる。また、適当な光学活性な原料ィ匕合物より製造することもで きる。
[0027] 本発明化合物の薬理活性は以下の試験により確認した。
試験例 1 : GK活性化の測定
被検薬剤の GK活性化測定は、 Science 301: 370-373, 2003に記載された方法に準 拠し、一部これを改変して実施した。 GK活性は、 glucoseを基質とし GKにより産生され glucose— 6— phosphateを glucose— 6— phosphate dehydrogenaseにより脱水素する際に 、 NADP (nicotinamide adenine dinucleotide phosphate)より転換される NADPH直に づく吸光度の変化として測定した。
本アツセィで使用する recombinant human liver GK (GST- hGK2)は、 GST (Glutathi one S transferase)— fusion proteinとして E. coliに発現させ、 Glutathione Sepharose7フ ムで精製したものを使用した。
Glucokinase isoform 2の配列は、 AK122876.1 (ァクセッションナンバー)に基づき、 以下の手川頁で ORF (open reading frame)のクロー-ングを行った。 pME18S- FL3- Glu cokinase isoform 2を铸型として、 5'— TAGAATTCATGGCGATGGATGTCACAAG— 3' (配列番号 1)を 5'プライマーに、 5'- ATCTCGAGTCACTGGCCCAGCATACAG- 3' ( 配列番号 2)を 3'プライマーに使って PCR (polimerase chain reaction)を行い、 PCR産 物を pGEM- T easy vectorに TAクローユングした。このクローンの配列はシークェンス して確認した。その後、 EcoRIと Xholで切断した断片を、同様に切断したベクター pGE X- 5X- 1にライゲーシヨンして pGEX- human Glucokinase 2を作製した。
酵素反応は 96穴の平底プレートを用い、 27°Cで測定を行った。酵素混合液として、 25 mM HEPES pH7.4 ; 25 mM KC1 ; 2 mM MgCl ; 1 mM ATP ; 0.1% BSA; 1 mM DTT ;
2
0.8 mM NADP ; 2.5 U/ml glucose- 6- phosphate dehydrogenase ; GST- hGK2 (いずれ も終末濃度。但し、 GST-hGK2量は DMSOコントロールの 10分間の吸光度の増カロ(Δ OD)が 0.12程度となるよう調製)を調製した。上記プレートに、酵素混合液 89 1を分 取し、 DMSOに溶解させた被検薬剤もしくは DMSOコントロールを 1 1加えた。つい で基質溶液として glucose (終末濃度 5 mM)を 10 μ 1加え 27°Cで反応を開始させた。 反応の開始後、 340 nMの波長で約 30秒ごとに 15分間吸光度を測定し、最初の 10 分間の吸光度の増加 ( Δ OD)力 化合物の GK活性ィ匕を計算した。被検薬剤の GK活 性化の指標は、 GK活性化 (%)として下記の式力も算出した。
GK活性化(%) = [ ( A OD ) - ( A OD ) ] / ( A OD ) X100
A OD :被検薬剤 10 Μにおける A OD
Test
Δ〇D : DMS〇コントロール Δ〇D
Cont
上記測定の結果、実施例 2の化合物は 178%、実施例 7の化合物は 158%の GK活性を 示した。
[0028] 試験例 2 : C57BL6マウスにおける血糖低下作用
自由摂食させた C57BL6マウス (N=5)の体重を測定した。被験化合物をクレモフォ ール(Cremophor:登録商標)溶媒 (Cremophor: DMSO: Water 5: 5: 90, v/v/v)に 溶解した。マウスに薬液を 10 ml/kgもしくは溶媒コントロール 10 ml/kgを経口投与した 。被験化合物投与直前に眼窩静脈叢からキヤビラリ一で血液約 60 1を採取した。被 験化合物投与後 1,4時間後に同様にして血液を採取した。採取した血液は血漿を分 離して血糖値を測定した。被験化合物投与後、ほたは 4時間後の血糖値と、同一時 間における溶媒対照群の血糖値を比較した。
上記試験の結果、実施例 2の化合物は 21.6%抑制(30 mg/kg)の血糖低下作用を示 した。
[0029] 試験例 3 : ICRマウスにおける経口糖負荷後の高血糖に対する作用
ICRマウス (N=5)を一晩絶食した後、体重を測定した。被験化合物を Cremophor溶 媒 (Cremophor: DMSO: Water 5: 5: 90, v/v/v)に溶解した。マウスに薬液を 10 ml/ kgもしくは溶媒コントロール 10 ml/kgを経口投与した。被験化合物投与直前に眼窩 静脈叢からキヤビラリ一で血液約 60 1を採取した。被験化合物を投与後 30分後に 2 00 mg/mlのグルコースの水溶液を 10 ml/kg (2 g/kg相当)経口投与した。グルコース 投与直前に眼窩静脈叢からキヤビラリ一で血液約 60 Lを採取した。グルコース投 与後 0.5, 1, 2時間後に同様にして血液を採取した。採取した血液は血漿を分離して 血糖値を測定した。被験化合物投与後から糖付加後 2時間までの血糖値の AUCと同 一時間における溶媒対照群の AUCを比較した。
[0030] 試験例 4 : db/dbマウスにおける経口糖負荷後の高血糖に対する作用
db/db(C57BL/KsJ- db/db)マウス (N=5)をー晚絶食した後、体重を測定した。被験 ィ匕合物を Cremophor溶媒 (Cremophor: DMSO: Water 5: 5: 90, v/v/v)に溶解した。 db/dbマウスに薬液を 10 ml/kgもしくは溶媒コントロール 10 ml/kgを経口投与した。被
験化合物投与直前に眼窩静脈叢からキヤビラリ一で血液約 60 1を採取した。 GK活 性化剤を投与後 30分後に 200 mg/mlのグルコースの水溶液を 10 ml/kg (2 g/kg相当) 経口投与した。グルコース投与直前に眼窩静脈叢からキヤビラリ一で血液約 60 1を 採取した。グルコース投与後 0.5, 1, 2時間後に同様にして血液を採取した。採取した 血液は血漿を分離して血糖値を測定した。被験化合物投与後から糖付加後 2時間ま での血糖値の AUCと同一時間における溶媒対照群の AUCを比較した。
以上の試験結果より、本発明化合物は、良好な GK活性化作用を有することが確認 された。また、種々の副作用(hERGや CYPに対する作用)及び Z又は溶解性が改善 された化合物も見出されていることから、本発明化合物は糖尿病等の予防'治療薬と して有用であることは明らかである。
本発明化合物 (I)又はその塩の 1種又は 2種以上を有効成分として含有する製剤 は、当分野において通常用いられている薬剤用担体、賦形剤等を用いて通常使用さ れて 、る方法によって調製することができる。
投与は錠剤、丸剤、カプセル剤、顆粒剤、散剤、液剤等による経口投与、又は、関 節内、静脈内、筋肉内等の注射剤、坐剤、点眼剤、眼軟膏、経皮用液剤、軟膏剤、 経皮用貼付剤、経粘膜液剤、経粘膜貼付剤、吸入剤等による非経口投与のいずれ の形態であってもよい。
本発明による経口投与のための固体組成物としては、錠剤、散剤、顆粒剤等が用 いられる。このような固体組成物においては、 1種又は 2種以上の有効成分を、少なく とも 1種の不活性な賦形剤、例えば乳糖、マン-トール、ブドウ糖、ヒドロキシプロピル セルロース、微結晶セルロース、デンプン、ポリビュルピロリドン、及び/又はメタケイ 酸アルミン酸マグネシウム等と混合される。組成物は、常法に従って、不活性な添カロ 剤、例えばステアリン酸マグネシウムのような潤滑剤やカルボキシメチルスターチナト リウム等のような崩壊剤、安定化剤、溶解補助剤を含有していてもよい。錠剤又は丸 剤は必要により糖衣又は胃溶性若しくは腸溶性物質のフィルムで被膜してもよ 、。 経口投与のための液体組成物は、薬剤的に許容される乳濁剤、溶液剤、懸濁剤、 シロップ剤又はエリキシル剤等を含み、一般的に用いられる不活性な希釈剤、例え ば精製水又はエタノールを含む。当該液体組成物は不活性な希釈剤以外に可溶化
剤、湿潤剤、懸濁剤のような補助剤、甘味剤、風味剤、芳香剤、防腐剤を含有してい てもよい。
非経口投与のための注射剤は、無菌の水性又は非水性の溶液剤、懸濁剤又は乳 濁剤を含有する。水性の溶剤としては、例えば注射用蒸留水又は生理食塩液が含ま れる。非水溶性の溶剤としては、例えばプロピレングリコール、ポリエチレングリコール 又はオリーブ油のような植物油、エタノールのようなアルコール類、又はポリソルベー ト 80 (局方名)等がある。このような組成物は、さらに等張化剤、防腐剤、湿潤剤、乳 ィ匕剤、分散剤、安定化剤、又は溶解補助剤を含んでもよい。これらは例えばパクテリ ァ保留フィルターを通す濾過、殺菌剤の配合又は照射によって無菌化される。また、 これらは無菌の固体組成物を製造し、使用前に無菌水又は無菌の注射用溶媒に溶 解又は懸濁して使用することもできる。
吸入剤や経鼻剤等の経粘膜剤は固体、液体又は半固体状のものが用いられ、従 来公知の方法に従って製造することができる。例えば公知の賦形剤や、更に、 pH調 整剤、防腐剤、界面活性剤、滑沢剤、安定剤や増粘剤等が適宜添加されていてもよ い。投与は、適当な吸入又は吹送のためのデバイスを使用することができる。例えば 、計量投与吸入デバイス等の公知のデバイスや噴霧器を使用して、化合物を単独で 又は処方された混合物の粉末として、もしくは医薬的に許容し得る担体と組み合わせ て溶液又は懸濁液として投与することができる。乾燥粉末吸入器等は、単回又は多 数回の投与用のものであってもよぐ乾燥粉末又は粉末含有カプセルを利用すること ができる。あるいは、適当な駆出剤、例えば、クロ口フルォロアルカン、ヒドロフルォロ アルカン又は二酸ィ匕炭素等の好適な気体を使用した加圧エアゾールスプレー等の 形態であってもよい。
通常経口投与の場合、 1日の投与量は、体重当たり約 0.001〜100 mg/kg、好ましく は 0.1〜30 mg/kg、更に好ましくは 0.1〜10 mg/kgが適当であり、これを 1回であるい は 2乃至 4回に分けて投与する。静脈内投与される場合は、 1日の投与量は、体重当 たり約 0.0001〜10 mg/kgが適当で、 1日 1回乃至複数回に分けて投与する。また、経 粘膜剤としては、体重当たり約 0.001〜100 mg/kgを 1日 1回乃至複数回に分けて投与 する。投与量は症状、年令、性別等を考慮して個々の場合に応じて適宜決定される
実施例
[0033] 以下、実施例に基づき本発明化合物 (I)の製法を更に詳細に説明する。本発明化 合物は下記実施例に記載の化合物に限定されるものではな 、。また原料化合物の 製法を参考例に示す。
参考例 1
カリウム t-ブトキシド (1.3 g)を t-ブチルアルコール (5.6 ml)に溶解し、 70°Cで 1時間撹 拌した。反応液に 5-メチル -2-チォフェンカルボアルデヒド (1.0 g)とコハク酸ジメチル( 1.2 ml)の t-ブチルアルコール (1.2 ml)溶液を滴下し、 70°Cにて 1時間撹拌した。氷冷 下、酢酸 (1.0 ml)を加え pHを約 5にし、 10分間撹拌した。ジェチルエーテル (40 ml)と 水 (30 ml)をカ卩え、分液後、有機層に飽和重曹水 (40 ml)を力卩ぇ水層の pHを約 8にした 。水層をジェチルエーテル (30 ml)で洗浄した後、濃塩酸 (6 ml)を力卩ぇ水層の pHを約 3 にした。水層を酢酸ェチル (40 ml)で抽出後、得られた有機層を飽和食塩水 (30 ml)で 洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去した後、得られた残渣 をシリカゲルカラムクロマトグラフィー(へキサン/酢酸ェチル)にて精製し、 3- (メトキシ カルボ-ル)- 4-(5-メチル -2-チェ-ル)- 3-ブテンカルボン酸 (1.3 g)を淡黄色固体とし て得た。
[0034] 参考例 2
3- (メトキシカルボ-ル)- 4-(5-メチル -2-チェ-ル)- 3-ブテンカルボン酸 (1.2 g)のト ルェン (48 ml)溶液に、酢酸ナトリウム (410 mg)および無水酢酸 (4.8 ml)を順次加え、 7 0°Cにて 3時間撹拌した。反応液を室温まで放冷し、飽和重曹水 (30 ml)をカ卩ぇ 10分間 撹拌した後、酢酸ェチル (40 ml)で希釈した。得られた有機層を水 (30 ml)及び飽和食 塩水 (30 ml)にて順次洗浄し、無水硫酸マグネシウムで乾燥した。溶媒を減圧下留去 し、得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン/酢酸ェチル)にて精 製し、 4- (ァセチルォキシ) -2-メチル -1-ベンゾチォフェン- 6-カルボン酸メチル (1.1 g) を淡黄色固体として得た。
参考例 3
4- (ァセチルォキシ) -2-メチル -1-ベンゾチォフェン- 6-カルボン酸メチル (1.0 g)のメ
タノール (32 ml)溶液に、 28%ナトリウムメトキシド /メタノール溶液 (3.3 ml)を加え、 75°C にて終夜撹拌した。氷冷下、酢酸ェチル (40 ml)を加えて希釈した後、 1M塩酸 (18 ml) を加え水層の pHを約 3にした。分液操作により得られた有機層を水 (30 ml),飽和食 塩水 (30 ml)にて順次洗浄した後、無水硫酸マグネシウムで乾燥した。溶媒を減圧下 留去し、 4-ヒドロキシ -2-メチル -1-ベンゾチォフェン- 6-カルボン酸メチル (823 mg)を 淡黄色固体として得た。
[0035] 参考例 4
4-ヒドロキシ -1H-インドール- 6-カルボン酸メチル (500 mg)及び炭酸カリウム (361 mg )の DMF (10 ml)懸濁液にブロモメチルベンゼン (320 μ 1)を- 10°Cにて滴下し、 30分間 撹拌後、室温で更に 5時間撹拌した。反応混合物に水 (30 ml)と酢酸ヱチル (30 ml)を 加え、分液し、有機層を飽和重曹水 (30 ml),飽和食塩水 (30 ml)で順次洗浄し、無水 硫酸マグネシウムで乾燥した。減圧下溶媒を留去して得られた残渣をシリカゲルカラ ムクロマトグラフィー (へキサン/酢酸ェチル)で精製して、 4- (ベンジルォキシ) -1H-ィ ンドール- 6-カルボン酸メチル (543 mg)を黄色固体として得た。
参考例 5
4-ヒドロキシ- 1-ベンゾチォフェン- 6-カルボン酸メチル (150 mg)の DMF (3 ml)溶液 に炭酸カリウム (200 mg)及び 2-ョードイソプロパン (100 μ 1)をカ卩え、終夜撹拌した。反 応液に水 (30 ml)と酢酸ェチル (40 ml)をカ卩え、分液操作により得られた有機層を水 (30 mi x 3)、飽和重曹水 (30 ml)及び飽和食塩水 (30 ml)にて順次洗浄した。有機層を無 水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。得られた残渣をシリカゲル力 ラムクロマトグラフィー (へキサン/酢酸メチル)にて精製し、 4-イソプロポキシー卜ベンゾ チォフェン- 6-カルボン酸メチル (142 mg)を黄土色固体として得た。
[0036] 参考例 6
4- (ベンジルォキシ) -1H-インドール- 6-カルボン酸メチル (150 mg)及びメタノール (6 1 μ 1)のトルエン (3 ml)溶液に (トリブチルホスホラ-リデン)ァセトニトリル (193 mg)のト ルェン (0.6 ml)溶液を氷冷下にて滴下した。反応液を氷冷下で 30分間撹拌した後、 5 0°Cで終夜撹拌した。反応溶液を室温まで放冷した後、減圧下溶媒を留去して得ら れた残渣をシリカゲルカラムクロマトグラフィー (へキサン/酢酸ェチル)で精製して、 4-
(ベンジルォキシ) -1-メチル -1H-インドール- 6-カルボン酸メチル (139 mg)を赤褐色固 体として得た。
参考例 7
20%水酸化パラジウム/炭素 (58 mg)及び 4- (ベンジルォキシ) -1-メチル -1H-インド ール- 6-カルボン酸メチル (130 mg)のメタノール (4 ml)懸濁液を、 4気圧の水素雰囲気 下、室温で 2時間撹拌した。反応液をセライトろ過後、減圧下溶媒を留去し、 4-ヒドロ キシ -1-メチル -1H-インドール- 6-カルボン酸メチル (78 mg)を無色固体として得た。
[0037] 参考例 8
4-ヒドロキシ -1-メチル -1H-インドール- 6-カルボン酸メチル (80 mg)の DMA(1.6 ml) 溶液に、炭酸カリウム (81 mg)及び 1-フルォロ- 4- (メチルスルホ -ル)ベンゼン (102 mg) を順次加え、 150°Cで終夜撹拌した。反応混合物を室温まで放冷し、水 (30 ml)及び 酢酸ェチル (30 ml)を加えた。分液操作により得られた有機層を飽和重曹水 (30 ml)及 び飽和食塩水 (30 ml)で順次洗浄し、無水硫酸マグネシウムで乾燥した。減圧下溶媒 を留去して得られた残渣をシリカゲルカラムクロマトグラフィー (へキサン/酢酸ェチル) で精製して、 1-メチル - 4-[4- (メチルスルホ -ル)フエノキシ] -1H-インドール- 6-カルボ ン酸メチル (94 mg)を無色固体として得た。
参考例 9
1-メチル - 4-[4- (メチルスルホ -ル)フエノキシ] -1H-インドール- 6-カルボン酸メチル (90 mg)のメタノール (2 ml)、 THF(2 ml)混合溶液に 1M水酸化ナトリウム水溶液 (4 ml) を加え、室温で 6時間撹拌した。反応溶液に水 (20 ml)及びジェチルエーテル (20 ml) を加え、分液操作により得られた水層に 1M塩酸 (4.5 ml)を加え pHを約 3とした。酢酸 ェチル (30 ml)を加え、分液操作により得られた有機層を、飽和食塩水 (20 ml)で洗浄 し、無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去し、 1-メチル -4-[4- (メチル スルホ -ル)フエノキシ] -1H-インドール- 6-カルボン酸 (83 mg)を無色固体として得た。
[0038] 参考例 10〜27
上記参考例 1と同様にして参考例 10の化合物を、参考例 2と同様にして参考例 11 の化合物を、参考例 3と同様にして参考例 12及び 13の化合物を、参考例 6と同様に して参考例 14及び 15の化合物を、参考例 7と同様にして参考例 16及び 17の化合
物を、参考例 8と同様にして参考例 18〜22の化合物を、参考例 9と同様にして参考 例 23〜27の化合物をそれぞれ合成した。
[0039] 参考例 28
4-ヒドロキシ -2-メチル -1-ベンゾチォフェン- 6-カルボン酸メチル (6 g)及び Ν,Ν-ジィ ソプロピルェチルァミン (11.4 ml)のジクロロメタン (150 ml)溶液に、氷冷下にてクロ口 (メ トキシ)メタン (2.4 ml)を滴下し、室温まで昇温後 16時間撹拌した。反応溶液に水 (150 ml)及びクロ口ホルム (150 ml)を力卩ぇ分液を行った。得られた有機層を飽和炭酸水素 ナトリウム水溶液 (150 ml)及び飽和食塩水 (150 ml)にて順次洗浄後、無水硫酸マグネ シゥムにて乾燥し、溶媒を減圧下留去することにより 4- (メトキシメトキシ) -2-メチル -1- ベンゾチォフェン- 6-カルボン酸メチル (5.9 g)を淡黄色固体として得た。
参考例 29
4- (メトキシメトキシ) -2-メチル -1-ベンゾチォフェン- 6-カルボン酸メチル (5.9 g)の TH F (31 ml)及びメタノール (31 ml)の混合溶液に、 1M水酸化ナトリウム (66 ml)を加え室 温で 2時間、 50°Cで 1時間撹拌した。反応液を減圧下濃縮し、ジェチルエーテル (50 ml)及び水 (50 ml)を力卩ぇ分液操作を行った。水層を 1M塩酸で pHを約 7〖こし、酢酸ェ チル (100 ml)で抽出した。得られた有機層を飽和食塩水 (70 ml)で洗浄後、無水硫酸 マグネシウムで乾燥した。溶媒を減圧下留去し、 4- (メトキシメトキシ)- 2-メチル -卜べ ンゾチォフェン- 6-カルボン酸 (5.5 g)を淡黄色固体として得た。
[0040] 参考例 30
トリフエ-ルホスフィン (9.6 g)の塩化メチレン溶液 (250 ml)に氷冷下にて N-ブロモス クシンイミド (6.6 g)を加えた。氷冷下で 20分撹拌後、 4- (メトキシメトキシ) -2-メチル -ト ベンゾチォフェン- 6-カルボン酸 (5.5 g)の塩化メチレン懸濁液 (250 ml)を加えた。氷冷 下にて 20分間撹拌した後、 2-ァミノチアゾール (5.5 g)を力卩ぇ同温にて 20分、室温にて 終夜撹拌した。反応液を減圧下濃縮して得られた残渣に酢酸ェチル (200 ml)を加え 、水 (200 ml),飽和重曹水 (150 ml)及び飽和食塩水 (150 ml)で順次洗浄した。無水硫 酸マグネシウムで乾燥した後、溶媒を減圧下留去し、得られた残渣をシリカゲルカラ ムクロマトグラフィー(へキサン:酢酸ェチル =80/20→ 60/40)で精製し、 4- (メトキシ メトキシ) -2-メチル -N- 1,3-チアゾール -2-ィル- 1-ベンゾチォフェン- 6-カルボキサミド
(6.1 g)を淡黄色固体として得た。
参考例 31
4- (メトキシメトキシ)- 2-メチル -N- 1,3-チアゾール -2-ィル- 1-ベンゾチォフェン- 6-力 ルボキサミド (6.1 g)の THF (191 ml)溶液に、氷冷下にて 3M塩酸 (64 ml)をカ卩ぇ室温で 60時間撹拌した。反応液を減圧下濃縮し、得られた固体を 80°Cでメタノール (140 ml) に溶解し、室温まで放冷した。溶媒を減圧下留去し、全量が 20 gになったところで析 出した固体を濾取し、 4-ヒドロキシ -2-メチル -N- 1,3-チアゾール -2-ィル- 1-ベンゾチ ォフェン- 6-カルボキサミド (4.3 g)を黄土色固体として得た。
[0041] 参考例 32
4-ヒドロキシ -2-メチル -N- 1,3-チアゾール -2-ィル- 1-ベンゾチォフェン- 6-カルボキ サミド (841 mg)の N-メチルピロリドン (20 ml)溶液にェチル 4-フルォロベンゾエート(58 5 mg)及び炭酸カリウム (602 mg)を加え、 150°Cで一終夜、 200°Cで 4時間撹拌した。酢 酸ェチル (30 ml)及び水 (30 ml)を加え、分液後有機層を水 (30 ml)で洗浄した。水層を 酢酸ェチルで抽出し、有機層を硫酸マグネシウムで乾燥後、減圧下濃縮した。得ら れた残渣をシリカゲルカラムクロマトグラフィーで精製し、ェチル 4-({2-メチル -6-[(1,3 -チアゾール -2-ィルァミノ)カルボ二ル]- 1-ベンゾチェン- 4-ィル }ォキシ)ベンゾエート (318 mg)を得た。
参考例 33
ェチル 4-({2-メチル -6-[(1 ,3-チアゾール -2-ィルァミノ)カルボ-ル]- 1-ベンゾチェ ン- 4-ィル }ォキシ)ベンゾエート (318 mg)のエタノーノレ (20 ml)溶液に、 1M水酸化ナトリ ゥム水溶液 (1.80 ml)をカ卩え、 60°Cで終夜撹拌した。更に 1M水酸化ナトリウム水溶液( 1.80 ml)をカ卩え、 80°Cにて 4時間撹拌した。 1M塩酸 (3.6 ml)を加え、生じた固体をろ 取し、水で洗浄した。得られた固体を DMF-酢酸ェチルで結晶化し、 4-({2-メチル -6- [(1,3-チアゾール -2-ィルァミノ)カルボ-ル]- 1-ベンゾチェン- 4-ィル }ォキシ)安息香 酸 (142 mg)を得た。
各参考例化合物の構造及び物理化学的データを後記表 1〜5に示す。
[0042] 実施例 1
1-メチル - 4-[4- (メチルスルホ -ル)フエノキシ] -1H-インドール- 6-カルボン酸 (75 mg
)と 1,3-チアゾール -2-ァミン (24 mg)のピリジン (2.5 ml)溶液にォキシ塩化リン (20 μ 1)を - 10°Cにて滴下した。反応液を- 10°Cで 30分間撹拌した後、 50°Cで終夜撹拌した。反 応溶液を室温まで放冷後、クロ口ホルム (30 ml)で希釈し、水 (30 ml)をカ卩ぇ 10分間撹 拌した。分液操作により得られた有機層を 1M塩酸(30 ml),飽和重曹水 (30 ml),飽和 食塩水 (30 ml)で順次洗浄し、無水硫酸マグネシウムで乾燥した。減圧下溶媒を留去 して得られた残渣をシリカゲルカラムクロマトグラフィー (へキサン/酢酸ェチル)で精製 して、 1-メチル - 4-[4- (メチルスルホ -ル)フエノキシ]- N-(l ,3-チアゾール -2-ィル) -1H -インドール- 6-カルボキサミド (44 mg)を無色固体として得た。
実施例 2
4-メチルモルホリン- 4-ォキシド (75 mg)の THF(5 ml)及び水 (3 ml)溶液に、氷冷下で 四酸化オスミウム (0.08 M 2-メチル -2-プロパノール溶液) (0.800 ml)及び 2_メチル -4- [4- (メチルスルホ -ル)フエノキシ]- N-(5-ビュルピラジン- 2-ィル) -1-ベンゾチォフェン -6-カルボキサミド (280 mg)の THF(7 ml)溶液を順次カ卩えた。氷冷下で 1時間撹拌後、 室温で 15時間撹拌した。反応溶液に酢酸ェチル (30 ml), 10%チォ硫酸ナトリウム水 溶液 (30 ml)を加え、分液により得られた有機層を飽和食塩水 (20 ml)で洗浄後、無水 硫酸マグネシウムで乾燥した。減圧下溶媒を留去して得られた残渣をシリカゲルカラ ムクロマトグラフィー (クロ口ホルム/メタノール)で精製して、 N-[5-(l,2-ジヒドロキシェ チル)ピラジン- 2-ィル] -2-メチル -4-[4- (メチルスルホ -ル)フエノキシ ]-1-ベンゾチォ フェン- 6-カルボキサミド (244 mg)を薄茶色アモルファスとして得た。
実施例 3〜10
実施例 1の方法と同様にして後記表に示す実施例 3〜10の化合物をそれぞれ対 応する原料を使用して製造した。
実施例 11
1-(4-フルォロ- 1-ナフチル)エタノン (35 μ mol)及び 4-ヒドロキシ -2-メチル - Ν-1,3- チアゾール -2-ィル- 1-ベンゾチォフェン- 6-カルボキサミド (30 μ mol)の Ν-メチルピロ リドン (1 ml)溶液に、炭酸カリウム (40 mol)を加え、 150°Cにて終夜撹拌した。反応液 に水 (2 ml)及びクロ口ホルム(3 ml)をカ卩ぇ撹拌した。 IPS (Whatman社)にて有機層を 回収し、減圧下濃縮後、分取高速液体クロマトグラフィー(ODS ;メタノール- 0.1%ギ酸
水溶液)で精製し、 4-[(4-ァセチル- 1-ナフチル)ォキシ ]-2-メチル - N-1,3-チアゾー ル -2-ィル- 1-ベンゾチォフェン- 6-カルボキサミド (5.5 mg)を得た。
実施例 12
1-ブロモブタン (40 μ mol)及びメチル 4-ヒドロキシ -2-メチル -1-ベンゾチォフェン- 6 -カルボキシレート (30 mol)の N-メチルピロリドン (1 ml)溶液に炭酸カリウム (40 mol )及びヨウ化カリウム (3 μ mol)をカ卩え、 80°Cで終夜撹拌した。反応液に水 (2 ml)及びク ロロホルム (3 ml)をカ卩ぇ撹拌し、 IPS (Whatman社)にて有機層を回収し、減圧下濃縮 した。得られた残渣にエタノール (2 ml), 1M水酸化ナトリウム水溶液 (0.1 ml)をカ卩え、 8 0°Cにて終夜撹拌した。 1M塩酸水溶液 (0.1 ml)を加え、室温で 2時間撹拌し、減圧下 濃縮した。塩化ォキザリル (70 μ mol)のジクロロェタン (1 ml)溶液及び DMF (0.25 mg) のジクロロェタン (0.5 ml)溶液をカ卩え、 60°Cにて 3時間撹拌した。 2-ァミノチアゾール (7 0 mol)のジクロ口エタン (1 ml)溶液、 Ν,Ν-ジイソプロピルェチルァミン (210 mol)を 加え、室温で 16時間撹拌した。水 (2 ml)およびクロ口ホルム (2 ml)を加え撹拌後、 IPS (Whatman社)にて有機層を回収した。減圧下濃縮後、分取高速液体クロマトグラフィ 一(ODS;メタノール- 0.1%ギ酸水溶液)で精製し、 4-ブトキシ -2-メチル -N- 1,3-チアゾ ール -2-ィル- 1-ベンゾチォフェン- 6-カルボキサミド (5.9 mg)を得た。
[0044] 実施例 13
4-({2-メチル -6-[(1,3-チアゾール -2-ィルァミノ)カルボ-ル]- 1-ベンゾチェン- 4-ィ ル}ォキシ)安息香酸 (30 μ mol)の DMF (1 ml)溶液、 HOBt (30 μ mol)及びジメチルァ ミン (40 mol)の N-メチルピロリドン (80 1)溶液並びに PS- Carbodiimide (50 mg, Arg onaut社)の混合物を室温で終夜振とうした。 PS-Trisamine (50 mg, Argonaut社)及び PS-Isocyanate (50 mg, Argonaut社)をカ卩え、室温で更に 4時間振とうした。フィルター チューブにてろ過し、更にろ液を減圧下濃縮して、 4-{4- [(ジメチルァミノ)カルボ-ル] フエノキシ }-2-メチル -N- 1,3-チアゾール -2-ィル- 1-ベンゾチォフェン- 6-カルボキサ ミド (13.1 mg)を得た。
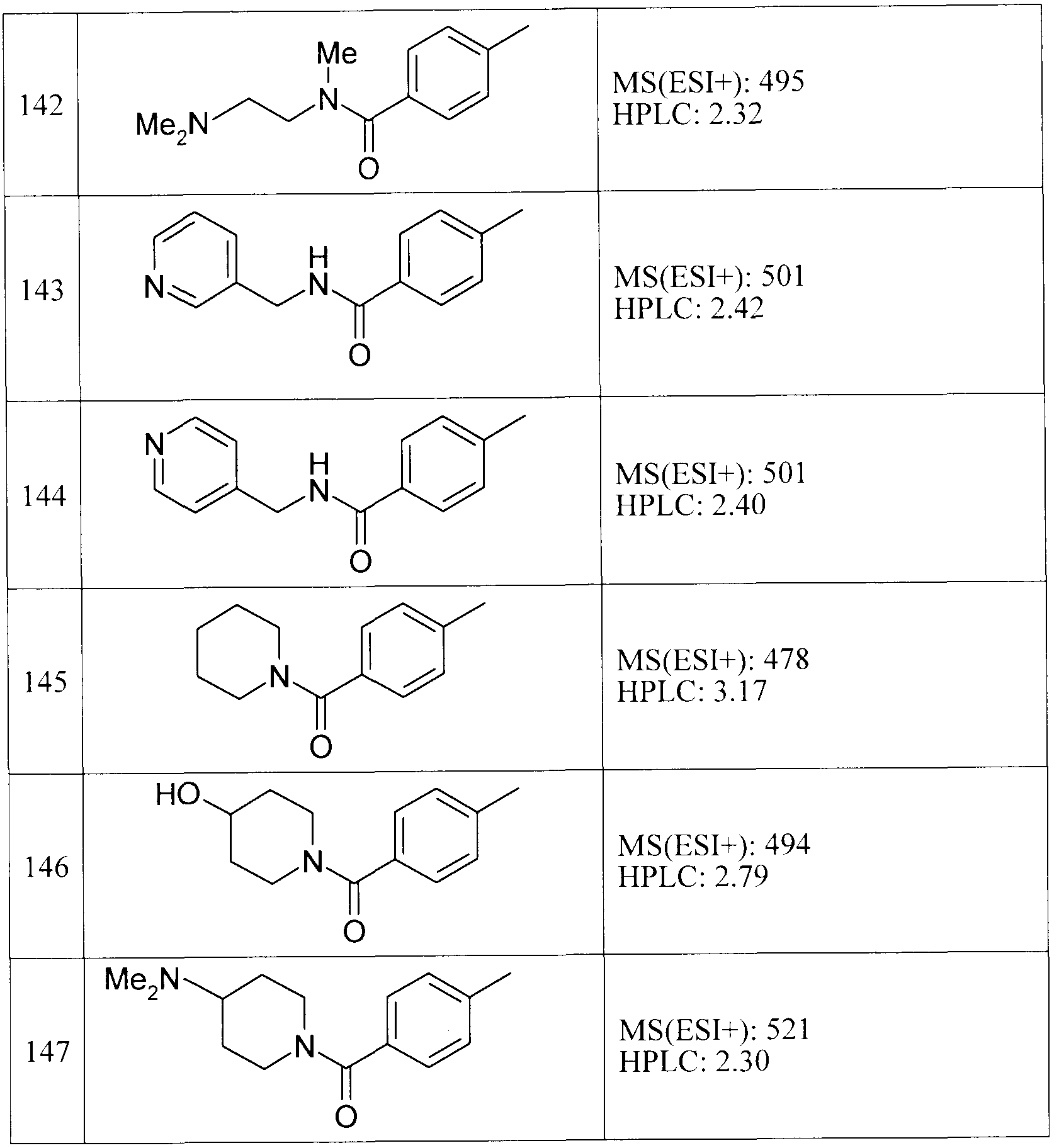
[0045] 実施例 11の方法と同様にして後記表に示す実施例 14〜20及び実施例 22〜31 の化合物を、実施例 12の方法と同様にして後記表に示す実施例 32〜 138の化合 物を、実施例 13の方法と同様にして後記表に示す実施例 21及び実施例 139〜147
の化合物を、それぞれ対応する原料を使用して製造した。各実施例化合物の構造及 び物理化学的データを表 6〜 18に示す。
また、表 19及び 20に本発明の別の化合物の構造を示す。これらは、上記の製造法 や実施例に記載の方法及び当業者にとって自明である方法、又はこれらの変法を用 いることにより、容易に合成することができる。
[0046] 後記表中以下の略号を用いる。 REx:参考例番号、 Ex:実施例番号、 No :化合物番 号、 Str:構造式、 Dat :物理ィ匕学的データ (MS :質量分析における m/z値 (+:陽イオン 、—:陰イオン)、 NMRl : DMSO— d中の NMRにおける δ (ppm)、 NMR2 : CDC1中の1
6 3
H NMRにおける δ (ppm)、 HPLC :高速液体クロマトグラフィーにおける保持時間(min ) (HPLCの測定条件は、カラム: WakosiHI (和光純薬製) 5C18 (ODS, 5micron) AR; 4 .6 x 30mm,展開溶媒:メタノール /5 mMトリフルォロ酢酸水溶液 = 10/90 (0 min)-10 0/0 (4.0 min)- 100/0 (4.5 min)ゝ流速: 1.2 ml/minゝ波長: 254 nmである。))、 Me :メチ ル、 Et:ェチル、 iPr: 2-プロピル、 cPr:シクロプロピル、 nBu:ノノレマノレブチノレ、 tBu:tert -ブチノレ、 cBu:シクロブチル、 nPen:ノルマルペンチル、 cHex:シクロへキシル、 Ms :メ チルスルホ -ル、 A ァセチル、 Ph:フエ-ル、 Bn:ベンジル。また、置換基の前の数 字は置換位置を示し、例えば 4-Ms-Phは 4-メチルスルホユルフェ-ルを示す。
[0047] [表 1]
O 82εさ-oozAV:/v>d sさ s900zfcl


Ks 00
師] [6濯]

^et0ZC900idfX3J 62 8e9e oz.ooz O


£ 0Z£/900Zd£/13d οε 8£9£嫁 ΟΟί OAV
[S峯] [Ϊ500]




Ex R3 B Dat
NMRl : 1.41 (6H, d, J=5.9 Hz). 4.90-5.00 (IH, m). 7.28 (IH, d, .1=3.7 Hz), 7.49 (IH. d, J=5.5 Hz), 7.58 (IH, d, J=3.5 Hz), 7.63
5 H iPr
( I H, s), 7.89 (IH. d. J=5.5 Hz), 8.35 (IH, s). 12.69 (IH, s); MS (ESI+): 319
NMRl : 3.23 (3H, s), 7.25-7.31 (3H, m), 7.42 (IH. d. J=5.6
6 H 4-Ms-Ph Hz), 7.55 (IH, d, J=3.6 Hz), 7.82 ( I H, s), 7.96 (2H, d. J=8.8
Hz), 8.06 (IH, d, J=5.5 Hz), 8.74 ( I H, s); MS (HSI+): 431
NMRl : 2.60 (3H, s), 3.22 (3H, s), 7.12-7.15 (IH. m), 7.24 (2H, d, J=9.0 Hz), 7.28 (III, d, J=3.6 Hz), 7.56 (I H, d, J=3.6 Hz),
7 Me 4-Ms-Ph 7.78-7.82 (IH, m), 7.94 (211, d, J=8.7 Hz), 8.63 ( I H, s);
MS (ESI十): 445
NMR2: 1.39 (3H, t, J=7.5 Hz). 2.97 (2H, q, J=7.5 Hz), 3.09 (3H, s), 6.97-7.01 (I H, m), 7.07 (IH. s), 7.10-7.17 (2H, m).
8 Et 4-Ms-Ph
7.22-7.29 (I H. m), 7.55-7.59 ( I H. m). 7.88-7.97 (2H, m).
8.25-8.26 ( IH. m); MS (ESI+): 459
[0054] [表 8]


[0055] [表 9]


£ 0Ζ£/900Ζάΐ/13ά 9ε 8£9£滅00 OAV


t£t0l£/900Zdr/13d 9S 8£9£嫁 00Z ΟΛ\
〕〔〕〔9005

〔〕


[ΐ9οο]

£ 0Z£/900ZdT/13d 6S 8£9£滅00 OAV
 16]
16]

[0063] [表 17]
[8ΐ挲] 900]

nt0l£/900ZdT/L3d zv 8f 9£嫁 00Z ΟΛ\
 19]
19]
ΘΙΛΙ SIAI oz ΛΙ L\
HO^ 厂 SIAI 31
N
H 0£d 61 H 91
HO N
H ΘΙΛΙ 81 9ΙΛΙ H SI
ON ON

[9900]



WW / 900Zdf/ェ:) d ャャ 8£9£滅00 OAV
産業上の利用可能性
[0067] 本発明化合物は、 GK活性化作用を有することから、糖尿病、特に 2型糖尿病の治 療並びに予防薬として有用である。また糖尿病の合併症である腎症、網膜症、神経 障害、末梢循環障害、脳血管障害、虚血性心疾患、動脈硬化症の治療並びに予防 薬として有用である。更に過食を抑制することにより、肥満、メタボリックシンドロームの 治療並びに予防薬としても有用である。
配列表フリーテキスト
[0068] 以下の配列表の数字見出し < 223 >には、「Artificial Sequenceの説明を記載す る。具体的には、配列表の配列番号 1及び 2の配列で表される塩基配列は、人工的 に合成したプライマー配列である。
Claims
請求の範囲
下記式 (I)で示される縮合へテロ環化合物又はその製薬学的に許容される塩。
[化 9]

(式中の記号は以下の意味を示す。
A: S、 0又は N(R5)、
X : 0、 Sゝ SO又は SO、
2
B :- R°、 -低級ァルケ-ル、 - R°°- CN、 - R°°- CON(RG)、 - R°°- O- RG、 - R°°- N(RG)CO- R。
2
、 -Y- (置換されていてもよいァリール)、 -Y- (置換されていてもよいへテロ環)又は- Y-( 置換されて 、てもよ ヽシクロアルキル)、
Υ:単結合、置換されていてもよい低級アルキレン、 - R。。- 0- R。。-、 - R。。- CON(RG) -、 - R
°°- N(RG)CO-、 -(置換されて 、てもよ 、低級アルキレン)- CON(RG)-又は- (置換されて
V、てもよ 、低級アルキレン)- N(RG)CO-、
R°:同一又は互いに異なって、低級アルキル、
R°°:同一又は互いに異なって、低級アルキレン、
RG:同一又は互いに異なって、 H又は R°、
D :チアゾール、ピリジン又はビラジン、
R1及び R2 :同一又は互いに異なって、下記 (0又は (ii)力も選択される基、
(0:- CH(0RA)- RB、 - C(R°)(0RA)- RB、 - CO- CO- NRCRD、 - CO- CO- NRC- 0RD、 - C(0 RE)(0RF)- RB、 - C(0RE)(0RF)- R°、 -CH(0RE)-CH(0RF)-RC及び/又は- CH(0RE)-CH (0RF)-RB、
(ii) :- H、 -ハロゲン、 -CO H、 -CO R°、 -NO、 - CN、 - R°、 -低級ァルケ-ル、 - CO- C
2 2 2
0 H、 - C0-C0-0R°、 -ハロゲノ低級アルキル及び/又は-低級アルキレン -NR D、
2
RA:同一又は互いに異なって、 - H、 - R°、 -ハロゲノ低級アルキル又は- R°°-ァリール、
RB :- CO H、 -CO R°、 - CO- NRCRD、 - CO- NRC- ORD、 - R°°- NRCRD、 - R°°- ORA、 - R。0- C
2 2
O R°、 -低級アルキレン- CO- NRCRD又は- R°°- CO- Nl - ORD、
2
RC及び RD:同一又は互いに異なって、 - H、 - R°、 - R°°-N(RA)、 - R°°-0RA、 - R°°-C0 H
2 2
、— R°°— C〇 R°又は— R°°— C〇— N(RA)、
2 2
RE及び :同一又は互いに異なって、 RAに記載の基、或いは、 RE及び RFがー体とな つて、 R00、
R3及び R4:同一又は互いに異なって、 - H、 -ハロゲン、 - R°、 -ハロゲノ低級アルキル又 は- R00- 0H、
R5 :-H、 - R°、 -ハロゲノ低級アルキル、 -(置換されていてもよいァリール)又は- R°°- (置 換されて 、てもよ 、ァリール)。
但し、 Aが 0のとき、 Bは、少なくとも 1個の- SO R°で置換されたァリールを示す。 )
2
[2] B力 R°、 -(置換されて 、てもよ 、ァリール)又は-低級アルキレン- (置換されて ヽても よいァリール)
R1及び R2が、同一又は互いに異なって、下記 (0又は GO力 選択される基、
(0 :- CH(0RA)- RB、 - CO- CO- NRCRDゝ - CO- CO- NRC- OR。、 - C(ORE)(ORF)- RBゝ - C(0
RE)(ORF)- R°、 -CH(ORE)-CH(ORF)-RC及び/又は- CH(ORE)-CH(ORF)-RB、
(ii) :- H、 -ハロゲン、 -CO H、 -CO R°、 -NO、 - CN、 - R°、 -低級ァルケ-ル、 -CO- CO
2 2 2
H、 - CO-CO-OR°、 -ハロゲノ低級アルキル及び/又は-低級アルキレン- NRCRD、
2
である、請求の範囲 1記載の化合物又はその製薬学的に許容される塩。
[3] 請求の範囲 1記載の式 (I)で示される縮合へテロ環化合物又はその製薬学的に許容 される塩と、製薬学的に許容される担体とからなる医薬組成物。
[4] 糖尿病治療剤である請求の範囲 3記載の医薬組成物。
[5] 請求の範囲 1記載の式 (I)で示される縮合へテロ環化合物又はその製薬学的に許容 される塩を含有するダルコキナーゼ阻害剤。
[6] 糖尿病治療剤の製造のための、請求の範囲 1記載の式 (I)で示される化合物又はそ の製薬学的に許容される塩の使用。
[7] 請求の範囲 1記載の式 (I)で示される化合物又はその製薬学的に許容される塩の有 効量を患者に投与することからなる、糖尿病の治療方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005300584A JP2009013065A (ja) | 2005-10-14 | 2005-10-14 | 縮合へテロ環化合物 |
| JP2005-300584 | 2005-10-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007043638A1 true WO2007043638A1 (ja) | 2007-04-19 |
Family
ID=37942859
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2006/320434 WO2007043638A1 (ja) | 2005-10-14 | 2006-10-13 | 縮合へテロ環化合物 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP2009013065A (ja) |
| WO (1) | WO2007043638A1 (ja) |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (de) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2009082152A2 (en) | 2007-12-20 | 2009-07-02 | Lg Life Sciences Ltd. | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
| JP2009534369A (ja) * | 2006-04-20 | 2009-09-24 | ファイザー・プロダクツ・インク | グルコキナーゼ仲介疾患を予防および治療するための縮合フェニルアミド複素環化合物 |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2010084428A1 (en) | 2009-01-20 | 2010-07-29 | Pfizer Inc. | Substituted pyrazinone amides |
| WO2010103438A1 (en) | 2009-03-11 | 2010-09-16 | Pfizer Inc. | Substituted indazole amides and their use as glucokinase activators |
| WO2010103437A1 (en) | 2009-03-11 | 2010-09-16 | Pfizer Inc. | Benzofuranyl derivatives used as glucokinase inhibitors |
| US7842713B2 (en) | 2006-04-20 | 2010-11-30 | Pfizer Inc | Fused phenyl amido heterocyclic compounds |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| US7977367B2 (en) | 2008-09-11 | 2011-07-12 | Pfizer Inc | Substituted imidazole propanamide glucokinase activators |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011158149A1 (en) | 2010-06-18 | 2011-12-22 | Pfizer Inc. | 2-(3,5-disubstitutedphenyl)pyrimidin-4(3h)-one derivatives |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1067128A1 (en) * | 1998-04-01 | 2001-01-10 | Ono Pharmaceutical Co., Ltd. | Fused thiophene derivatives and drugs containing the same as the active ingredient |
| WO2004031179A1 (en) * | 2002-10-03 | 2004-04-15 | F. Hoffmann-La Roche Ag | Indole-3-carboxamides as glucokinase (gk) activators |
| WO2004046139A1 (en) * | 2002-11-19 | 2004-06-03 | Astrazeneca Ab | Benzofuran derivates, process for their preparation and intermediates thereof |
| WO2005049019A1 (en) * | 2003-11-24 | 2005-06-02 | Novo Nordisk A/S | N-heteroaryl indole carboxamides and analogues thereof, for use as glucokinase activators in the treatment of diabetes |
-
2005
- 2005-10-14 JP JP2005300584A patent/JP2009013065A/ja not_active Withdrawn
-
2006
- 2006-10-13 WO PCT/JP2006/320434 patent/WO2007043638A1/ja active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1067128A1 (en) * | 1998-04-01 | 2001-01-10 | Ono Pharmaceutical Co., Ltd. | Fused thiophene derivatives and drugs containing the same as the active ingredient |
| WO2004031179A1 (en) * | 2002-10-03 | 2004-04-15 | F. Hoffmann-La Roche Ag | Indole-3-carboxamides as glucokinase (gk) activators |
| WO2004046139A1 (en) * | 2002-11-19 | 2004-06-03 | Astrazeneca Ab | Benzofuran derivates, process for their preparation and intermediates thereof |
| WO2005049019A1 (en) * | 2003-11-24 | 2005-06-02 | Novo Nordisk A/S | N-heteroaryl indole carboxamides and analogues thereof, for use as glucokinase activators in the treatment of diabetes |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7842713B2 (en) | 2006-04-20 | 2010-11-30 | Pfizer Inc | Fused phenyl amido heterocyclic compounds |
| JP2009534369A (ja) * | 2006-04-20 | 2009-09-24 | ファイザー・プロダクツ・インク | グルコキナーゼ仲介疾患を予防および治療するための縮合フェニルアミド複素環化合物 |
| US8119624B2 (en) | 2006-04-20 | 2012-02-21 | Pfizer Inc. | Fused phenyl amido heterocyclic compounds |
| WO2008017381A1 (de) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2009082152A2 (en) | 2007-12-20 | 2009-07-02 | Lg Life Sciences Ltd. | Glucokinase activators and pharmaceutical compositions containing the same as an active ingredient |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| US8329920B2 (en) | 2008-09-11 | 2012-12-11 | Pfizer Inc. | Substituted imidazoles useful for treating type II diabetes |
| US8389552B2 (en) | 2008-09-11 | 2013-03-05 | Pfizer Inc. | (S)-6-(2-(4-(cyclobutylsulfonyl)-1H-imidazol-1-yl)-3-cyclopentylpropanamido)nicotinic acid useful as a glucokinase activator |
| US7977367B2 (en) | 2008-09-11 | 2011-07-12 | Pfizer Inc | Substituted imidazole propanamide glucokinase activators |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2010084428A1 (en) | 2009-01-20 | 2010-07-29 | Pfizer Inc. | Substituted pyrazinone amides |
| US8071606B2 (en) | 2009-01-20 | 2011-12-06 | Pfizer Inc. | Substituted pyrazinone amides useful for activation of glucokinase |
| WO2010103438A1 (en) | 2009-03-11 | 2010-09-16 | Pfizer Inc. | Substituted indazole amides and their use as glucokinase activators |
| JP2012520286A (ja) * | 2009-03-11 | 2012-09-06 | ファイザー・インク | グルコキナーゼ阻害剤として使用されるベンゾフラニル誘導体 |
| WO2010103437A1 (en) | 2009-03-11 | 2010-09-16 | Pfizer Inc. | Benzofuranyl derivatives used as glucokinase inhibitors |
| US20110319379A1 (en) * | 2009-03-11 | 2011-12-29 | Corbett Jeffrey W | Substituted Indazole Amides And Their Use As Glucokinase Activators |
| US8455496B2 (en) | 2009-03-11 | 2013-06-04 | Pfizer Inc. | Benzofuranyl derivatives |
| EP2604604A1 (en) * | 2009-03-11 | 2013-06-19 | Pfizer Inc | Benzofuranyl derivatives used as glucokinase inhibitors |
| US8735396B2 (en) | 2009-03-11 | 2014-05-27 | Pfizer Inc. | Benzofuranyl derivatives |
| CN102388038B (zh) * | 2009-03-11 | 2014-04-23 | 辉瑞大药厂 | 用作葡糖激酶活化剂的苯并呋喃基衍生物 |
| EA018894B1 (ru) * | 2009-03-11 | 2013-11-29 | Пфайзер Инк. | N,n-диметил-5-(2-метил-6-((5-метилпиразин-2-ил)карбамоил)бензо-фуран-4-илокси)пиримидин-2-карбоксамид, используемый в качестве активатора глюкокиназы, и содержащая его фармацев-тическая композиция |
| CN102388038A (zh) * | 2009-03-11 | 2012-03-21 | 辉瑞大药厂 | 用作葡糖激酶抑制剂的苯并呋喃基衍生物 |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011158149A1 (en) | 2010-06-18 | 2011-12-22 | Pfizer Inc. | 2-(3,5-disubstitutedphenyl)pyrimidin-4(3h)-one derivatives |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009013065A (ja) | 2009-01-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007043638A1 (ja) | 縮合へテロ環化合物 | |
| JP4800964B2 (ja) | 肥満症の治療においてグルコキナーゼ活性化剤として使用するための、ヘテロアリールインドールカルボキサミドおよびその類似体 | |
| US20090281142A1 (en) | Thiazole derivative | |
| JP5292100B2 (ja) | Gk活性化作用を有する2−ピリジンカルボキサミド誘導体 | |
| US20100179137A1 (en) | Pyridone compound | |
| JP5287730B2 (ja) | フェニルアセトアミド誘導体 | |
| CN101712657A (zh) | 杂芳基氨基甲酰基苯衍生物 | |
| JP2008519083A (ja) | アミノキナゾリン化合物 | |
| JP2010138073A (ja) | ピコリン酸アミド化合物 | |
| WO2007072782A1 (ja) | カルボン酸誘導体又はその塩 | |
| US20070149514A1 (en) | Indole-phenylsulfonamide derivatives used as ppar-delta activating compounds | |
| TW201522330A (zh) | 2-醯胺噻唑衍生物或其鹽 | |
| WO2009081782A1 (ja) | N-ピラゾール-2-ピリジンカルボキサミド誘導体 | |
| US9889130B2 (en) | Substituted 3-benzofuranyl-indol-2-one-3-acetamididopiperazine derivatives, preparation thereof, and therapeutic use thereof | |
| US20120277208A1 (en) | Benzamide compound | |
| JP5694959B2 (ja) | 三位で置換された3−ベンゾフラニル−インドール−2−オン誘導体、この調製およびこの治療的使用 | |
| CN100591671C (zh) | 杂芳基氨基甲酰基苯衍生物 | |
| TW200410957A (en) | Acyloxypyrrolidine derivatives, preparation thereof and application thereof in therapeutics | |
| JPWO2009041475A1 (ja) | ピラゾール−3−イル−ベンズアミド誘導体の製造方法 | |
| AU2015232269A1 (en) | Compound binding to PPARG but not acting as promoter and pharmaceutical composition for treating PPARG-related diseases containing same as active ingredient | |
| US20240076272A1 (en) | Activators of Heme Regulated Inhibitor Kinase (HRI) | |
| WO2021070957A1 (ja) | ベンゼン縮合環化合物、およびそれを含有する医薬組成物 | |
| CN114057716A (zh) | 苯并氮杂卓酮类化合物及其盐、其制备方法及医药用途 | |
| TW201617077A (zh) | 用以治療及/或預防呼吸器官疾病之醫藥組成物 | |
| KR20150064727A (ko) | 신규한 페닐아세트아미드 화합물 및 이를 함유하는 의약 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06811722 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |