正常細胞と比較してがん細胞は一般的に増殖が活発であるとされており _、多くの場 合'、細胞周期制御機構の異常による無秩序 増殖力 §.がんの原因であると考え-ちれてい-
― る。 細胞周期において有糸分裂期 (_Μ期) は、― _色^き良龜胸に均.等に分配す _る テ . ップであり、 この過程の厳密な制御は細胞の増殖、 生存に不可欠である。 従って Μ 期の進行を阻害することは細嗨増殖の抑制に有効な手段であると考えられ、実際にタ キソールゃピンクリスチン等の Μ期を標的をした抗がん剤が臨床で有効な成績を収 めている。 . -,
Μ期の進行の多くのステップがタンパク質のリン酸化を行うプロテイン ·キナーゼ によって制御されていることが知.られている。 PLK (ポロ ライク キナーゼ ο 1 o l i k e , k i n a s e s)) フアミリーは M期を含む細胞周期の制御に重 要な役割を果たしているセリンスレオニンキナーゼであり、 PLK1、 PLK 2、 P LK3、 SAKの 4つの類似したタンパク質から構成される(ネイチヤー'レビュー ' モレキュラー'セル'バイオロジー(Na t . R e v. Mo 1. C e 1 1 B i o 1.)、 第 5卷、 429 頁、 (2004年))。 その中でも P L K 1は哺乳動物細胞の M期の複 数の重要な段階に関与していることが知られている。即ち、 PLK1は M期への進入、 · 中心体の制御、 染色体の分離、 細胞質分裂等の各ステップに、 様々な基質をリン酸ィ匕 することによって関与していることが報告されている (ネイチヤー'レビュー 'モレ キュラー ·セル ·バイオロジー (Na t. Re v. Mo 1 - Ce l 1 B i o l .), 第 5巻、 429 頁、 (2004年))。
さらに、 PLK1はヒトの様々ながん組織において過剰発現しているという報告が 多数ある。 例えば、 非小細胞肺がん (オンコジーン (On c o g e n e)、 第 14巻、 5 43頁、 (1997年))、頭頸部がん (キャンサー ·リサーチ (C a n c e r R e s e a r c h)、 第 15卷、 2794頁、 (1999年)) において P LK 1の過剰発現が 認められ、また P L K 1の過剰発現はこれら患者の予後と相関するというデータが得 られている。 他に、 大腸がん、 食道がん、 卵巣がん、 メラノーマといったがん種にお
いても PLK;iの 現が上昇しているという報告がある。 以上の報告は、 PLK1の 過剰発現が細胞 .瘅化になんらかの形で関与していること、また P LK 1の機能が特 にがん細胞における M期の進行に重要である、 ということを示唆している。
これらの事実より、 PLK1を標的とした抗がんアプローチとしての可能性が考え られる。 実際に、様々な実験手法を用いてがん細胞に対する P LK 1の機能阻害効果 を調べる実験が複数報告されている。例えば p LK 1の機能阻害変異体をウィルスべ クタ一を用いて細胞内に発現させる実験では、 P LK 1阻害によるがん細胞選択的な 細胞死の誘導が報^されている (セル ·グロウス ·アンド ·ディファレンシエーショ ン(Ce l 1 g r owt h &D i f f -)、 第 1 1卷、 615頁、 (2000年))。 ま た P LK 1に対する s i RNA (ジャーナル ·ォブ ·ナショナル ·キャンサー 'イン スティチュート (J. Na t l . Ca n c e r I n s t.)、第 94巻、 1863頁、
(2002年)) 力 がん細胞に増殖阻害やアポトーシスを誘導するという報告もあ る。 さらに、 PLT iに対する s hRNA (ジャーナル'ォブ'ナショナル'キャン サー ·ィンスティチュート ( J . Na t l . Ca n c e r I n s t.)、 第 96卷、 862頁、 ( 2004年)) ゃァンチセンスオリゴヌクレオチド (オンコジーン(On c o g e n e)、 第 21卷、 3 , 162頁、 (2002年)) がマウス X e n o g r a f tモデルで抗腫瘍効果を示すことが報告されている。 これらの実験結果は、 PLK1 活性の阻害ががん細胞に増殖抑制や細胞死の誘導をもたらすことを示しており、 P L K 1阻害剤が抗がん剤とし有効であることを強く示唆している。
過去に P LK阻害作用を有する化合物に関する特許出願はなされている (国際公開 第 2004/043936号パンフレツト、同第 2004/014899号パンフレ ットなど)。 しかしながら、 優れた PLK1阻害作用を有する置換イミダゾール誘導 体については、 今まで報告されていない。 発明の開示
PLK1阻害作用に基づく抗腫瘍剤を開発すべく、 PLK1阻害作用を示し、 それ に基づく細胞増殖抑制作用が優れた新規な置換ィミダゾール誘導体を創製すること が本発明の解決課題である。
本発明者等は、 上記課題を解決すべく、 置換イミダゾール誘導体を広く合成し、一 般式 [I] で表される化合物が、 優れた PLK1阻害作用、 及ぴそれに基づく細胞増 殖抑制作用を示すことを見いだして本努明を完成した。
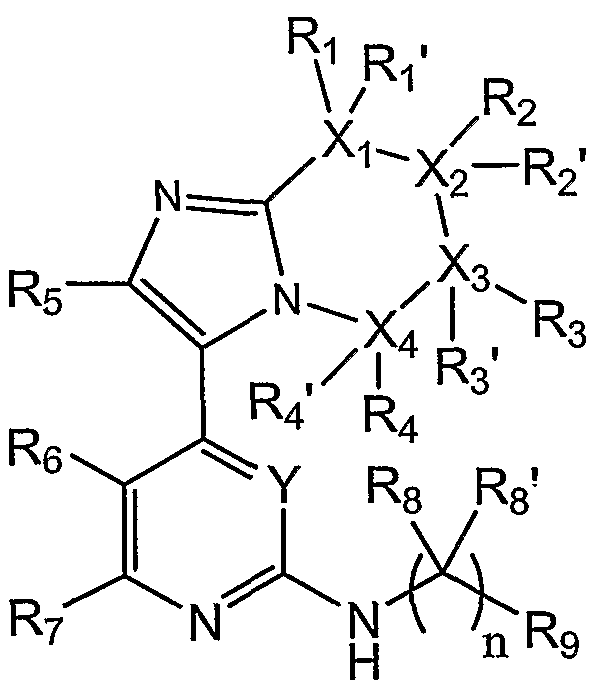
即ち、 本発明は、
[式中、
x1 , x2 、 x3及ぴ x, は、 同一若しくは異なって、 C又は Nであり (但し、 X ! Χ2 , X3及ぴ X4のうち、 Nであるものは、 0個ないし 2個である。)、
Yは、 CH又は Nであり、 — ― "
R1 、 R1 ,、 R2 、 R2 ,、 R3 、 R3 ,、 R4、 及ぴ1 4 , は、 同一若しくは異な つて、 水素原子、 く置換基群 から選択される置換基、 <置換基群 α>から選択さ れる置換基で 1個若しくは 2侮以上置換されてもよい低級アルキル基、シクロアルキ ル基、 ァリール基、 又はへテロアリール基 (ここで、 前記ァリール基及ぴヘテロァリ ール基は、 互いに独立して、 下記 1) ないし 3) :
_ 1) 低級アルキル基、
2) く置換基群ひ >から選択される置換基、 及び
3) <置換基群 α>から選択される置換基で置換される低級アルキル基: から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。) である力、、
或いは、 Raであり (ここで、 Raは、 一 — Z2— Z 3で表され、
Z 1 は、 O又は NHCOであり ;
Z2 は、 単結合又は (CHWi) nlであり (ここで、. は、 1ないし 3のいず れかの整数であり、 iは、 1ないし nェのいずれかの整数であり、 (CHWi) nlは、 のとき (CHWJ を表し、 1^=2のとき (CHWJ — (CHW2) を表し、
1^=3のとき (CHWJ —(CHW2) — (CHW3) を表し、 Wい W2、及ぴ W3は、 同一若しくは異なって、 水素原子又は炭素数 1個ないし 2個の低級アルキル基であ る。) ;
Z3は、低級アルコキシ基又はフエニル基であり(ここで、前記フエニル基は、 く置換基群 α〉から選択される置換基で 1個若しくは 2個以上置換されていてもよ い。)、
但し、 X 、 X2 、 X3 及ぴ X4のいずれかが Nであるとき、 Nである当 該 Xi (ここで、 iは、 1ないし 4 のいずれかの整数である。) に結合する Ri及び
R.i , のいずれか一方は、 当該: X; と一緒になつて Nとなり、 他方の Ri又は Ri ' は、 前記に定義したとおりであり、
また、 及び!^ェ , のいずれか一方と、 R2及び R2, のいずれか一方 力 一緒になつて、 ェ -X2の結合において 2重結合を形成してもよく、
また、 R3及び R3 , のいずれか一方と、 R4及ぴ R4, のいずれか一方 力 一緒になつて、 X3 ~X4の結合において 2重結合を形成してもよく、
R 5は、 水素原子又はメチル基であり、
R6及び R7は、 同一若しくは異なって、 水素原子、 <置換基群 >から選択され る置換基、 く置換基群 >から選択される置換基で 1個若しくは 2個以上置換されて もよい低級アルキル基、 又は、 ピロリジニル基、 ピペリジニル基、 及ぴピペラジニル 基から選択される 5員ないし 6員の脂肪族複素環基 (ここで、 前記 5員ないし 6員の ' 脂肪族複素環基は、 下記 1) ないし 3) :
1) 低級アルキル基、
2) <置換基群 ]3 >から選択される置換基、 及び
3) <置換基群 >から選択される置換基で置換される低級アルキル基: から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。) であるか、
或いは、 R6及び R7は一緒になつて、 これらが結合する環に縮合した、 下記環:
を形成し、
R8及ぴ1 8 ' は、 同一若しくは異なって、 水素原子又は <置換基群 α〉から選択 される置換基で 1個若しくは 2個以上置換されていてもよい、低級アルキル基であり、
R 9は、 ァリール基又はへテロアリール基 (ここで、 前記ァリ一ル基及ぴへテ口了 リール基は、 互いに独立して、 下記 1) ないし 9) :
1) 低級アルキル基、
2) く置換基群ひ >から選択される置換基、
3) <置換基群0; >から選択される置換基で置換される低級アルキル基、
4)ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 ピペラジニル基、 1, 4—ペルヒドロジァゼピエル基、及びモルホリノ基から選択される 4員ないし 7員の 脂肪族複素環基 (ここで、 前記 4員ないし 7員の脂肪族複素環基は、 下記 a) ないし c) :
a) 低級アルキル基、
b) ぐ置換基群 α>から選択される置換基、 及び
c ) く置換基群 a >から選択される置換基で置換される低級アルキル基: 力 ら選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。 また、 前記 4員ないし 7員の脂肪族複素環基は、 同一炭素原子に結合する水 素原子 2個がォキソ基で置換されていてもよく、当該複素環を構成する隣接する炭素 原子間の結合が 2重結合であってもよく、当該複素環を構成する非隣接炭素原子が架 橋を形成してもよい。)、 '
5)前記 4)の 4員ないし 7員の脂肪族複素環基で置換される低級アルキル基、
6) — (CH2) ∞1— NR10R10, (ここで、
rn^は、 0ないし 3のいずれかの整数であり、
Ri。は、 水素原子又は低級アルキル基であり、
R10' は、
a) <置換基群 o;〉から選択される置換基で置換される低級アルキル基; b) ァゼチジュル基、 ピロリジニル基、 ピペリジニル基、 ピペラジ-ル基、 及びモルホリノ基から選択される 4員ないし 6員の脂肪族複素環基 (ここで、 前記 4 員ないし 6員の脂肪族複素環基は、 下記 a a) ないし f f ) :
a a ) 低級アルキ 基、
b b) <置換基群 α>から選択される置換基、
c c) <置換基群 α>から選択される置換基で置換される低級アルキル基、 d d) 置換されてもよい、 ピロリジ -ル基、 ピベリジ-ル基、 及ぴピペラ ジニル基から選択される 5員ないし 6員の脂肪族複素環基、
e e) 前記 d d) の 5員ないし 6員の脂肪族複素環基で置換される低級ァ ルキル基、 及ぴ
f f ) 置換されてもよい、 ピリジル基、 ピラジュル基、 及びピリミジニル 基から選択される芳香族複素環で置換される低級アルキル基:
力 ^選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。) ;
c) 前記 b) の 4員ないし 6員の脂肪族複素環基で置換される低級アルキル 基;
d) 置換されていてもよい、 ピラゾリル基、 ピリジル基、 ピラジュル基、 ピ リミジニル基、及びテトラヒドロビラニル基から選択される 5員ないし 6員の芳香族 若しくは脂肪族複素環基; 又は
e) 置換されていてもよい、 炭素数 5個ないし 6個のシクロアルキル基 (こ こで、前記シクロアルキル基の同一炭素原子に結合する水素原子 2個がォキソ基で置 換されていてもよい。) である。)、
7) -ORl x (ここで、
R,,は、
a) <置換基群 a〉から選択される置換基で置換される低級アルキル基; b) ピロリジニル基、 ピペリジニル基、 ピペラジニル基、 及びモルホリノ基 力 選択される 5員ないし 6員の脂肪族複素環基 (ここで、 前記 5員ないし 6員の脂 肪族複素環基は、 下記 a a) ないし c c) :
a a) 低級アルキル基、
b b) <置換基群 α>から選択される置換基、 及ぴ
c c) <置換基群 α >から選択される置換基で置換される低級アルキル 基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。) ;
c) 前記 b) 5員ないし 6員の脂肪族複素環基で置換される低級アルキル基 である。);又は
d) ピロリル基、 ィミダゾリル基、 ビラゾリル基、 ピリジル基、 ビラジニル 基、 及びピリミジニル基から選択される 5員ないし 6員の芳香族複素環基である。)、 8) ― (CH2) m2-NHCOR13 (ここで、
m2は、 0ないし 3のいずれかの整数であり、
R13は、
a ) 低級アルキル基;
b) く置換基群ひ >から選択される置換基;
c) <置換基群 α>から選択される置換基で 1個若しくは 2個以上置換され る低級アルキル基;
d) 置換されてもよい、 炭素数 3個ないし 6個のシクロアルキル基; e) 置換されてもよい、 ピロリル基、 イミダゾリル基、 及ぴピラゾリル基か ら選択される 5員の芳香族複素環基で置換される低級アルキル基;又は
f )置換されてもよい、ピロリジニル基、ピペリジニル基、ピペラジニル基、 及びモルホリノ基から選択される 5員ないし 6員の脂肪族複素環基である。)、 及び 9) ― (CH2) m3— CONR14R14, (ここで、
m3は、 0ないし 3のいずれかの整数であり、
R14及び R14, は、 同一若しくは異なって、
a ) 水素原子;
b ) 低級アルキル基;
c) <置換基群 α>から選択される置換基; 又は
d ) <置換基群 α >から選択される置換基で 1個若しくは 2個以上置換され る低級アルキル基であるか;
或いは、 1 14及ぴ1 14' 1S その結合する窒素原子と一緒になつて、 置換 されていてもよい、 ピロリジニル基、 ピペリジニノレ基、 及びピペラジニル基から選択
される 5員ないし 6員の脂肪族複素環を形成する。) :
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。) であり、
nは、 1ないし 3のいずれかの整数であり、
く置換基群ひ〉及びく置換基群 >は、 下記のように定義される。
<置換基群 α〉: '
ハロゲン原子、 ヒ ドロキシ基、 ニトロ基、 シァノ基、 アミノ基、 カルパモイル基、 ァ ミノスルホニル基、 低級アルキルァミノ基、 ヒ ドロキシ低級アルキルァミノ基、 ジ低 級アルキルアミノ基、 イミノ基、 低級アルキルスルホニル基、 低級アルキルスルホ二 ルァミノ基、ハロゲン原子で 1個ないし 3個置換されていてもよい低級アルコキシ基、 低級アルコキシカルボニル基、 低級アルコキシカルボニルァミノ基、 ハロゲン原子で 1個ないし 3個置換されていてもよ V、低級アル力ノィル基、 及び力ルポキシル基 く置換基群 j8 >:
ハロゲン原子、 ヒドロキシ基、 シァノ基、 'アミノ基、 ホルミル基、 力ルバモイル基、 ァミノスルホニル基、 低級アルキルァミノ基、 ジ低級アルキルァミノ基、 ヒ ドロキシ イミノメチル基、 メ トキシイミノメチル基、 低級アルコキシ基、 低級アルコキシカル ボニル基、 低級アルコキシカルボニルァミノ基、 低級アルカノィル基、 低級アルカノ ィルォキシ基、 低級アルキルチオ基、 低級アルキルスルホ-ル基、 カルボキシル基、 及びテトラゾリル基
]で示される化合物又はその薬学的に許容される塩若しくはエステル、 に関する。 な お、 上記式 (I ) で表される化合物には、 当該化合物のラセミ体のみならず、 存在可 能なすべてのェナンチォマー及ぴジァステレオマーを含む。 次に、 本明細書に記載された記号及び用語について説明する。
上記式 (I ) 中の 「低級アルキル基」 とは、 炭素数 1ないし 6個の直鎖状又は分岐 状のアルキル基をいい、 例えばメチル基、 ェチル基、 プロピル基、 イソプロピル基、 ブチル基、 ィソブチル基、 s e c一プチル基、 t e r t—ブチル基、 ペンチル基、 へ キシル基等が挙げられる。
上記式 (I ) 中の 「シクロアルキル基」 とは、 3員ないし 8員の脂肪族環状基であ り、 例えば、 シクロプロピル基、 シクロプチル基、 シクロペンチル基、 シクロへキシ ル基、 シクロへプチル基、 シクロォクチル基等が挙げられる。
上記式 (I ) 中の 「ァリール基」 とは、 炭素数 6ないし 1 4個の単環式、 二環式、 又は三環式の芳香族炭化水素基などをいい、 具体的には、 フヱ-ル基、 ナフチル基、 インデニル基、 アントラニル基などが挙げられる。
上記式 (I ) 中の 「ヘテロァリール基」 とは、 炭素原子以外に、 窒素原子、 酸素原 子、 及ぴ硫黄原子から選ばれる少なくも 1個の原子を含む、 芳香族複秦環基をいい、
例えば、 5員ないし 7員の単環式複素環基、 及び、 これに 3員ないし 8員の環が縮合 した縮環式複素環基などであり、 具体的には、 チェニル基、 ピロリル基、 フリル基、 チアゾリル基、 イミダゾリル基、 ピラゾリル基、 ォキサゾリル基、 ピリジル基、 ビラ ジニル基、 ピリミジニル基、 ピリダジニル基、 ィソォキサゾリル基、 イソキノリル基 、 イソインドリル基、 インダゾリル基、 インドリル基、 キノキサリニル基、 キノルル 基、 ベンゾィミダゾリノレ基、'ベンゾフラニル基などが挙げられる。
上記式 (I ) 中の 「ハロゲン原子」 としては、 例えばフッ素原子、 塩素原子、 臭素 原子、 ヨウ素原子等が挙げられ、 中でも例えばフッ素原子、 塩素原子、 又は臭素原子 が好ましい。
上記式 (I ) 中の 「低級アルキルアミノ基」 とは、 ァミノ基に上記 「低級アルキル 基」 が N—置換した置換基をいい、 例えば N—メチルァミノ基、 N—ェチルァミノ 基、 N—プロピルアミソ基、 N—イソプロピルアミノ基、 N—ブチルァミノ基、 N 一イソプチルァミノ基、 N— t e r t—ブチルァミノ基、 N—ペンチルァミノ基、 N—へキシルァミノ基等が挙げられる。
上記式 (I ) 中の 「ヒドロキシ低級アルキルアミノ基」 とは、 上記 「低級アルキル アミノ基」 にヒドロキシ基が 1個ないし 2個以上置換した置換基をいい、 例えば N _ ヒ ドロキシェチルァミノ基、 N—ヒ ドロキシプロピルアミノ基、 N—ヒ ドロキシィ ソプロピルアミノ基、 N—ヒドロキシブチルァミノ基、 N—ヒドロキシイソブチル アミノ基、 N—ヒドロキシー t e r t—プチルァミノ基、 N—ヒドロキシペンチル アミノ基、 N—ヒドロキシへキシルァミノ基等が挙げられる。
上記式 (I ) 中の 「ジ低級アルキルアミノ基」 とは、 ァミノ基に上記 「低級アルキ ル基」 が N, N—ジ置換した置換基をレ、い、例えば N, N—ジメチルァミノ基、 N, N—ジェチルァミノ基、 N, N—ジプロピルアミノ基、 N , N—ジイソプロピルァ ミノ基、 N, N—ジブチルァミノ基、 N, N—ジイソブチルァミノ基、 N, N— ジ t e r t—ブチルァミノ基、 N, N—ジペンチルァミノ基、 N, N—ジへキシル アミノ基、 N - ェチル一N—メチルァミノ基、 N —メチル一 N—プロピルアミノ基 等が挙げら る。
上記式 (I ) 中の 「低級アルキルスルホニル基」 とは、 スルホニル基の硫黄原子に 上記 「低級アルキル」 が結合した置換基をいい、 例えばメチルスルホニル基、 ェチ ルスホニル基、 プチルスルホニル基等が挙げられる。
上記式 (I ) 中の 「低級アルキルスルホニルァミノ基」 とは、 ァミノ基に上記 「低 級アルキルスルホニル基」 が N—置換した置換基をいい、 例えばメチルスルホニルァ ミノ基、 ェチルスホニルァミノ基、 プチルスルホニルァミノ基等が挙げられる。 上記式 (I ) 中の 「低級アルコキシ基」 とは、 酸素原子に 「低級アルキル基」 が結 合した基をいい、例えばメ トキシ基、エトキシ基、プロポキシ基、ィソプロポキシ基、 ブトキシ基、 イソブトキシ基、 s e c—ブトキシ基、 t e r t—ブトキシ基、 ペンチ
ルォキシ基、 ネオペンチルォキシ基、 へキシルォキシ基、 ィソへキシルォキシ基等が 挙げられる。
上記式 (I) 中の 「低級アルコキシカルボニル基」 とは、 カルボニル基に上記 「低 級アルコキシ基」 が結合した基をいい、 具体的には例えばメ トキシカルボニル基、 ェ トキシカルボニル基、 プロポキシカルボ-ル基、 イソプロポキシカルボニル基、 ブト キシカルボ-ル基、 イソプ'トキシカルボニル基、 s e c—ブトキシカルボニル基、 t e r tープトキシカルボ-ル基、 ペンチルォキシカルポニル基、 ネオペンチルォキシ カルボニル基、 へキシルォキシカルボ-ル基、 ィソへキシルォキシカルボニル基等が 挙げられる。
上記式( I ) 中の「低級アルコキシカルボニルァミノ基」 とは、ァミノ基に上記「低 . 級アルコキシカルボニル基」 が N—置換した基をいい、 具体的には例えばメ トキシカ ルボニルァミノ基、 エトキシカルボニルァミノ基、 プロポキシカルボニルァミノ基、 ィソプロポキシカルボニルァミノ基、 ブトキシカルボニルァミノ基、 ィソブトキシカ ルポニルァミノ基、 s e c—プトキシカルボニルァミノ基、 t e r t—ブトキシカル ボニルァミノ基、 ペンチルォキシカルボニルァミノ基、 ネオペンチノレォキシカノレポ二 ルァミノ基、 へキシルォキシカルボニルァミノ基、 ィソへキシルォキシカルボニルァ ミノ基等が挙げられる。
上記式 (I) 中の 「低級アルカノィル基」 とは、 カルボニル基に上記 「低級アルキ ル基」 が結合した基をいい、 カルボニル基に炭素数 1ないし 5個のアルキル基が結合 した基が好ましく、 例えばァセチル基、 プロピオニル基、 プチリル基、 イソブチリル 基、 バレリル基、 イソパレリル基、 ビバロイル基、 ペンタノィル基等が挙げられる。 上記式 (I) 中の 「低級アルカノィルォキシ基」 とは、 酸素原子に上記 「低級アル カノィル基」が結合した基をレ、い、例えばァセチルォキシ基、プロピオニルォキシ基、 プチリルォキシ基、 イソプチリルォキシ基、 バレリルォキシ基、 イソパレリルォキシ 基、 ビバロイルォキシ基、 ペンタノィルォキシ基等が挙げられる。
上記式 (I) 中の 「低級アルキルチオ基」 とは、 硫黄原子に上記 「低級アルキル」 が結合した置換基をいい、 例えばメチルチオ基、 ェチルチオ基、 プチルチオ基等が 挙げられる。
「PLK」 とは、 ポロ ライク キナーゼ (p o l o l i k e k i n a s e) を表す。
「PLK1」 とは、 PLK (ポロ ライク キナーゼ (p o l o l i k e k i n a s e)) フアミリーは PLK 1、 PLK2、 PLK3、 SAKから構成されるが、 その中の一つである。
「PLK1阻害剤」 とは、 ポロ ライク キナーゼ 1を阻害する薬剤である。
「その薬学的に許容される塩若しくはエステル」、 及び 「薬学的に許容できる担体 又は希釈剤」 の説明は後述する。
上記式 ( I ) で示される化合物の実施の形態についてさらに詳しく説明 する。
Xx , X2、 X3及ぴ X4 は、 同一若しくは異なって、 C又は Nであり (但し、 X 、 X2、 X3及ぴ X4 のうち、 Nであるものは、 0個ないし 2個である。)、 好ましくは、 、 X2、' X3及ぴ X4 は、 すべて Cであるか、 又は x2、 x3及 び X4のいずれか 1個のみが Nであり、 残りがすべて。であり、
さらに好ましくは、 、 x2、 x3及ぴ x4 は、 すべて cである。
Yは、 CH又は Nであり、 好ましくは、 Yは、 Nである。
R1、 R1 '、 R2、 R2 ,、 R3、 R3 ,、 R4 、 及び R4 , は、 同一若しくは異な つて、 水素原子; ハロゲン原子、 ヒドロキシ基、 ニトロ基、 シァノ基、 アミノ基、 カルパモイル基、 アミノスルホ -ル基、 低級アルキルアミノ基、 ヒドロキシ低級アル キルアミノ基、 ジ低級アルキルアミノ基、 イミノ基、 低級アルキルスルホニル基、 ハ 口ゲン原子で 1個ないし 3個置換されていてもよい低級アルコキシ基、低級アルコキ シカルボニル基、 低級アルコキシカルボニルァミノ基、 ハロゲン原子で 1個ないし 3 個置換されていてもよい低級 Tルカノィル基、 及ぴカルボキシル基 (以下、 これを < 置換基群《〉という。) 力 ら選択される置換基; <置換基群 >から選択される置 換基で 1 個若しくは 2個以上置換されてもよい低級アルキル基; シクロアルキル 基; ァリール基; 又はへテロアリール基である力 或いは、 Raであり (ここで、 Raは、 一 Z — Z2— Z 3で表され、 は、 O又は NHCOであり ; Z 2 は、 単 結合又は (CHW;) nlであり (ここで、 は、 1ないし 3のいずれかの整数であ り、 iは、 1ないし n のいずれかの整数であり、 (CHWi) nlは、 1^= 1のとき (CHW,) を表し、 のとき (CHWJ -(CHW2) を表し、 1^= 3のと き (CHW ) -(CHW2) -(CHW3) を表し、 Wい W2、 及び W3は、 同一若し くは異なって、 水素原子又は炭素数 1個ないし 2個の低級アルキル基である。) ; Z
3 は、 低級アルコキシ基又はフエニル基であり (ここで、 前記フエニル基は、 く置換 基群ひ>から選択される置換基で 1個若しくは 2個以上置換されていてもよい。)、 また、 前記ァリール基及びへテロアリール基は、 互いに独立して、 下記 1) ないし 3) :
1) 低級アルキル基、
2) <置換基群ひ >から選択される置換基、 及び
3) <置換基群 o; >から選択される置換基で置換される低級アルキル基: 力 ら選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて あよい。
但し、 、 X2、 X3及ぴ X4 のいずれかが Nであるとき、 Nである当該 Xi (こ こで、 iは、 1ないし 4 のいずれかの整数である。) に結合する Ri 及ぴ Ri ' のい
ずれか一方は、 当該 Xi と一緒になつて Nとなり、 他方の Ri 又は Ri ' は、 前記に 定義した通りである。
また、 及ぴ1^ , のいずれか一方と、 R2及び R2 ' のいずれか一方が、 一緒 になって、 -X2の結合において 2重結合を形成してもよく、 また、 R3及び R a ' のいずれか一方と、 R4及び R4 ' のいずれか一方が、 一緒になつて、 X3 — X
4の結合において 2重結合を形成してもよい。
R1 、 Rx ,、 R2、 R2 \ R3、 R3 '、 R4、及ぴ R4, に関して、好ましくは、 Rj及ぴ1 1 ,のいずれか一方と、 R2及び R2 ,のいずれか一方が、一緒になつて、 Xx -X2の結合において 2重結合を形成し;
R3及ぴ R3 ' のいずれか一方と、 R4及ぴ R4 , のいずれか一方が、 一緒になつ て、 X3 — X4の結合において 2重結合を形成し;
2重結合の形成に関与しない 1^及ぴ1^ , のいずれか一方が、 <置換基群 α>か ら選択される置換基、 <置換基群 α >から選択される置換基で 1個若しくは 2個以上 置換されてもよい低級アルキル基、 シクロアルキル基、 ァリール基、 又はへテロァリ ール基であり ;
2重結合の形成に関与しない R2及び R 2 , のいずれか一方が、 水素原子、 <置換 基群ひ >から選択される置換基、又はく置換基群ひ 〉から選択される置換基で 1個若 しくは 2個以上置換されてもよい低級アルキル基である力、 或いは、 Nである X2 と 一緒になつて Nとなり ;
2重結合の形成に関与しない R3及び R3 ' のいずれか一方が、 水素原子、 く置換 基群ひ 〉から選択される置換基、又は <置換基群 a〉から選択される置換基で 1個若 しくは 2個以上置換されてもよい低級アルキル基である力、 或いは、 Nである X3 と 一緒になつて Nとなり ;及び、
2重結合の形成に関与しない R4及び R4 , のいずれか一方が、 水素原子である 力、 或いは、 Nである X4 と一緒になって Nとなる。
また、 、 Rj ,、 R2、 R2 ,、 R3、 R3 ,、 R4、 及び R4 , に関して、 好ま しくは、 及ぴ1^ , のいずれか一方が、 水素原子であり、 他方が、 く置換基群 a >から選択される置換基、 <置換基群 a >から選択される置換基で 1個若しくは 2個 以上置換されてもよい低級アルキル基、 ァリール基、 又はへテロアリール基であり ; 及ぴ、 R2、 R2 '、 R3、 R3 '、 R4、 及ぴ R4 ' 力 すべて水素原子である。 さらに、 、 R1 ,、 R2、 R2 '、 R3、 R3 '、 R4、 及ぴ R4 , に関して、 よ り好ましくは、 2重結合の形成に関与しない 及ぴ!^ ' のいずれか一方が、 ハロ ゲン原子; シァノ基; ハロゲン原子で 1個ないし 3個置換されてもよい炭素数 1 個ないし 2個の低級アルキル基; ハロゲン原子で 1個ないし 3個置換されてもよい 炭素数 1個ないし 2個の低級アルコキシ基; ヒドロキシ基で置換されてもよい炭素 数 1個ないし 3個の低級アルキル基; 又はシク口プロピル基であり ;
2重結合の形成に関与しない、 R2及び R2 , のいずれか一方、 並びに R3及び R 3 , のいずれか一方が、 同一若しくは異なって、 水素原子; ハロゲン原子; シァ ノ基; ハロゲン原子で 1個ないし 3個置換されてもょ 、炭素数 1個ないし 2個の低 級アルキル基; ハロゲン原子で 1個ないし 3個置換されてもよい炭素数 1個ないし 2個の低級アルコキシ基; 又は、 ヒドロキシ基で置換されてもよい炭素数 1個ない し 3個の低級アルキル基であり ;
2重結合の形成に関与しない R4及び R4 ' のいずれか一方が、水素原子である。 また、 1^ 、 R, R2、 R2 '、 R3、 R3 '、 R4 、 及ぴ R4 , に関して、 さら に好ましくは、 2重結合の形成に関与しない 及ぴ!^ ' のいずれか一方が、 フッ 素原子;塩素原子; 臭素原子; シァノ基; ハロゲン原子で 1個ないし 3個置換 されてもよいメチル基; ハロゲン原子で 1個ないし 3個置換されてもよいメ トキシ 基; ヒ ドロキシ基で置換されてもよい炭素数 1個ないし 3個の低級アルキル基; 又はシクロプロピノレ基であり ;
2重結合の形成に関与しない、 R2及び R2 ' のいずれか一方、 R3及び R3 , の いずれか一方、並びに R4及ぴ1 4 , のいずれか一方が、いずれも水素原子である。
R5 は、 水素原子又はメチノ,レ基であり、 好ましくは、 水素原子である。
R6及ぴ R7 は、 同一若しくは異なって、 水素原子; ハロゲン原子、 ヒドロキシ 基、 シァノ基、 アミノ基、 ホルミル基、 カルパモイル基、 アミノスルホニル基、 低級 アルキルァミノ基、 ジ低級アルキルァミノ基、 ヒドロキシイミノメチル基、 メ トキシ イミノメチル基、 低級アルコキシ基、 低級アルコキシカルボ二ル基、 低級アルコキシ カルボニルァミノ基、 低級アルカノィル基、 低級アルカノィルォキシ基、 低級アルキ ルチオ基、低級アルキルスルホニル基、カルボキシル基、及ぴテトラゾリル基(以下、 <置換基群 >という。) 力 ら選択される置換基; く置換基群 |3 >から選択される 置換基で 1個若しくは 2個以上置換されてもよい低級アルキル基; 又は、 ピロリジ ニル基、 ピペリジニル基、 及ぴピペラジニル基から選択される 5員ないし 6員の脂肪 族複素環基 (ここで、前記 5員ないし 6員の脂肪族複素環基は、 下記 1) ないし 3) :
1) 低級アルキル基、
2) <置換基群] 3 >から選択される置換基、 及び '
3) く置換基群 >から選択される置換基で置換される低級アルキル基: 力^選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよレヽ。) である力、、 或いは、
R6及ぴ 7 は一緒になつて、 これから結合する環に縮合した、 下記環:
R6及び R7 に関して、 好ましくは、 R6 1S 水素原子又はく置換基群.] 3 >から選 択される置換基であり、 <置換基群 /3>が、 ハロゲン原子、 シァノ基、 及び低級アル コキシ基であり、 かつ、 R7が水素原子である。
R6及び R7 に関して、 さらに好ましくは、 R6力 シァノ基であり、 かつ、 R7 が水素原子である。 '
R8及ぴ1 8 ' は、 同一若しくは異なって、 水素原子又は <置換基群 α>から選択 される置換基で 1個若しくは 2個以上置換されていてもよい、低級アルキル基であり、 好ましくは、 R8及び R8 , の一方が、 水素原子であり、 他方がメチル基又はェチル 基であり ;さらに好ましくは、 R8及ぴ1 8 , の一方が、 水素原子であり、 他方がメ チル基である。 なお、 R8及び R8 ' が結合する炭素原子が不斉炭素であるとき、 S 体であることが好ましい。
R9は、 ァリール基又はへテロアリール基 (ここで、 前記ァリール基及ぴヘテロァ リール基は、 互いに独立して、 下記 1) ないし 9) :
1) 低級アルキル基、
2) <置換基群 α>から選択される置換基、
3) く置換基群 α〉から選択される置換基で置換される低級アルキル基、
4)ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 ピペラジニル基、 1, 4一ペルヒドロジァゼピニル基、及びモルホリノ基から選択される 4員ないし 7員の 脂肪族複素環基 (ここで、 前記 4員ないし 7員の脂肪族複素環基は、 下記 a) ないし c) :
a) 低級アルキル基、
b) <置換基群 a >から選択される置換基、 及ぴ
c ) く置換基群 α >から選択される置換基で置換される低級アルキル基: から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。 また、 前記 4員ないし 7員の脂肪族複素環基は、 同一炭素原子に結合する水 素原子 2個がォキソ基で置換されていてもよく、当該複素環を構成する Ρ舞接する炭素 原子間の結合が 2重結合であってもよく、当該複素環を構成する非隣接炭素原子が架 橋を形成してもよい。)、
5)前記 4)の 4員ないし 7員の脂肪族複素環基で置換される低級アルキル基、
6) — (CH2) ml-NR10R10' (ここで、
rr^は、 0ないし 3のいずれかの整数であり、
。は、 水素原子又は低級アルキル基であり、
R10' は、
a) <置換基群 〉から選択される置換基で置換される低級アルキル基; b) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 ピペラジニル基、
及びモルホリノ基から選択される 4員ないし 6員の脂肪族複素環基(ここで、 前記 4 員ないし 6員の脂肪族複素環基は、 下記 a a) ないし f f ) :
a a ) 低級アルキル基、
b b) <置換基群 α >から選択される置換基、
c c) <置換基群 a >から選択される置換基で置換される低級アルキル基、 d d) 置換されてもよい、 ピロリジニル基、 ピペリジニル基、 及ぴピペラ ジニル基から選択される 5員ないし 6員の脂肪族複素環基、
e e) 前記 d d) の 5員ないし 6員の脂肪族複素環基で置換される低級ァ ルキル基、 及び
f f ) 置換されてもよい、 ピリジル基、 ピラジュル基、 及びピリミジェル 基から選択される芳香族複素環で置換される低級アルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて あよレヽ。) ;
c) 前記 b) の 4員ないし 6員の脂肪族複素環基で置換される低級アルキル 基;
d) 置換されていてもよい、 ピラゾリル基、 ピリジル基、 ビラジニル基、 ピ リミジニル基、及ぴテトラヒドロビラニル基から選択される 5員ないし 6員の芳香族 若しくは脂肪族複素環基; 又は
e) 置換されていてもよい、 炭素数 5個ないし 6個のシクロアルキル基 (こ こで、前記シクロアルキル基の同一炭素原子に結合する水素原子 2個がォキソ基で置 換されていてもよい。) である。)、
7) -ORx l (ここで、
Rェ iは、
a) <置換基群 α>から選択される置換基で置換される低級アルキル基; b) ピロリジニル基、 ピペリジニル基、 ピペラジニル基、 及びモルホリノ基 力、ら選択される 5員ないし 6員の脂肪族複素環基 (ここで、前記 5員ないし 6員の脂 肪族複素環基は、 下記 a a) ないし c c) :
a a ) 低級アルキル基、
b b) <置換基群ひ >から選択される置換基、 及び
c c) <置換基群 α >から選択される置換基で置換される低級アルキル 基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。) ;
c) 前記 b) 5員ないし 6員の脂肪族複素環基で置換される低級アルキル基 である。) ;又は '
d) ピロリル基、 イミダゾリル基、 ピラゾリル基、 ピリジル基、 ピラジュル
基、 及びピリミジニル基から選択される 5員ないし 6員の芳香族複素環基である。)、
8) 一 (CH2) ra2-NHCOR13 (ここで、
ηι2は、 0ないし 3のいずれかの整数であり、
R13は、
a ) 低級アルキル基;
b) <置換基群ひ' >から選択される置換基;
c ) <置換基群ひ >から選択される置換基で 1個若しくは 2個以上置換され る低級アルキル基;
d) 置換されてもよレ、、 炭素数 3個ないし 6個のシクロアルキル基; e) 置換されてもよい、 ピロリル基、 イミダゾリル基、 及びピラゾリル基か ら選択される 5員の芳香族複素環基で置換される低級アルキル基;又は
f)置換されてもよい、ピロリジニル基、ピペリジニル基、ピペラジニル基、 及びモルホリノ基から選択される 5員ないし 6員の脂肪族複素環基である。)、 及び
9) - (CH2) m3— CONR14R14, (ここで、
m3は、 0ないし 3のいずれかの整数であり、
R14及ぴ1 14' は、 同一若しくは異なって、
a ) 水素原子;
b ) 低級アルキル基;
c) <置換基群ひ >から選択される置換基; 又は
d) <置換基群 α>から選択される置換基で 1個若しくは 2個以上置換され る低級アルキル基であるか;
或いは、 R14及び R14' 、 その結合する窒素原子と一緒になつて、 置換 されていてもよい、 ピロリジニル基、 ピペリジニル基、 及ぴピペラジニル基から選択 される 5員ないし 6員の脂肪族複素環を形成する。) :
力 ら選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。) である。 '
ここで、 前記 4) の 4員ないし 7員の脂肪族複素環基中、 「当該複素環を構成する 非隣接炭素原子が架橋を形成してもよい」 とは、 具体的には、 当該複素環を構成する 非隣接炭素原子が一 (CH2)
r- (ここで、 rは、 1又は 2の整数である。) で表さ れる架橋を形成してもよいことを意味し、 一例を挙げると下記の場合である。
R9 は、 好ましくは、 置換されてもよいフエ-ル基である。 即ち、 R9 は、 フエェ ル基 (ここで、 前記フエニル基は、 下記 1) ないし 8) :
1) 低級アルキル基、
2) く置換基群 α>から選択される置換基、
3) <置換基群 α >から選択される置換基で置換される低級アルキル基、
4) ァゼチジニル基、 ピロリジ -ル基、 ピペリジ-ル基、 ピペラジニル基、 及 ぴモルホリノ基から選択される 4員ないし 6員の脂肪族複素環基 (ここで、前記 4員 ないし 6員の脂肪族複素環基は、 下記 a) ないし c) :
a) 低級アルキル基、
b) く置換基群 α>から選択される置換基、 及び
c) く置換基群 α〉から選択される置換基で置換される低級アルキル基: 力 ら選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。 また、 前記 4員ないし 6員の脂肪族複素環基は、 同一炭素原子に結合する水 素原子 2個がォキソ基で置換されていてもよく、当該複素環を構成する隣接する炭素 原子間の結合が 2重結合であってもよく、当該複素環を構成する非隣接炭素原子が架 橋を形成してもよい。)、
5)前記 4)の 4員ないし 6員の脂肪族複素環基で置換される低級アルキル基、
6) — NR10R10, 若レくは一 CH2NR10R10, (ここで、
R i。は、 水素原子又は低級アルキル基であり、
R10, は、
a ) く置換基群ひ >から選択される置換基で置換される低級アルキル基; b) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 ピペラジニル基、 及びモルホリノ基から選択される 4員ないし 6員の脂肪族複素環基 (ここで、 前記 4 員ないし 6員の脂肪族複素環基は、 下記 a a) ないし f f ) :
a a ) 低級アルキル基、
b b) く置換基群 a >から選択される置換基、
c c) <置換基群 a >から選択される置換基で置換される低級アルキル基、 d d) 置換されてもよレ、、 ピロリジニル基、 ピペリジニル基、 及びピペラ ジニル基から選択される 5員ないし 6員の脂肪族複素環基、
e e) 前記 d d) の 5員ないし 6員の脂肪族複素環基で置換される低級ァ ルキル基、 及び
f f ) 置換されてもよい、 ピリジル基、 ピラジュル基、 及びピリミジニル 基から選択される芳香族複素環で置換される低級アルキル基:
力 ^選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて あよい。) ;
c) 前記 b) の 4員ないし 6員の脂肪族複素環基で置換される低級アルキル 基;
d) 置換されていてもよい、 ピラゾリル基、 ピリジル基、 ビラジニル基、 ピ
リミジニル基、及ぴテトラヒドロビラニル基から選択される 5員ないし 6員の脂肪族 若しくは芳香族複素環基; 又は
e) 置換されていてもよい、 炭素数 5個ないし 6個のシクロアルキル基 (こ こで、前記シクロアルキル基の同一炭素原子に結合する水素原子 2個がォキソ基で置 換されていてもよい。) である。)、
7) 一 ORu (ここで、
R ェは、
a) く置換基群ひ >から選択される置換基で置換される低級アルキル基; b) ピロリジ -ル基、 ピペリジニル基、 ピペラジニル基、 及びモルホリノ基 から選択される 5員ないし 6員の脂肪族複素環基 (ここで、前記 5員ないし 6員の脂 肪族複素環基は、 下記 a a) ないし c c) :
a a ) 低級アルキル基、
b b) <置換基群 α>から選択される置換基、 及び
c c) <置換基群 α>から選択される置換基で置換される低級アルキル 基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて あよい。) ;
c) 前記 b) 5員ないし 6員の脂肪族複素環基で置換される低級アルキル基 である。) ;又は
d) ピロリル基、 イミダゾリル基、 ピラゾリル基、 ピリジル基、 ピラジュル 基、 及ぴピリミジ-ル基から選択される 5員ないし 6員の芳香族複素環基である。)、 及び、
8) — NHCOR13若しくは一 CH2NHCOR13 (ここで、
R13は、
a ) 低級アルキル基;
b) <置換基群 α>から選択される置換基;
c ) <置換基群 α >から選択される置換基で 1個若しくは 2個以上置換され る低級アルキル基;
d) 置換されてもよい、 炭素数 3個ないし 6個のシクロアルキル基; e) 置換されてもよい、 ピロリル基、 イミダゾリル基、 及びピラゾリル基か ら選択される 5員の芳香族複素環基で置換される低級アルキル基;又は
f )置換されてもよい、ピロリジニル基、ピペリジニル基、ピペラジニル基、 及ぴモルホリノ基から選択される 5員ないし 6員の脂肪族複素環基である。) : から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよレヽ。) である。
R9 は、 さらに好ましくは、 フエニル基 (ここで、 前記フエニル基は、 下記 1)
ないし 5) :
1) <置換基群 0^ >から選択される置換基;
2) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 及ぴピペラジニル基 から選択される 5員ないし 6員の脂肪族複素環基 (ここで、前記 5員ないし 6員の脂 肪族複素環基は、 下記 a ) ないし c ) :
a) 低級アルキル基、
b) く置換基群ひ i〉から選択される置換基、 及ぴ
c )く置換基群 αュ >から選択される置換基で置換される低級アルキル基: から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。 また、 前記 4員ないし 7員の脂肪族複素環基は、 同一炭素原子に結合する水 素原子 2個がォキソ基で置換されていてもよく、当該複素環を構成する隨接する炭素 原子間の結合が 2重結合であってもよく、当該複素環を構成する非隣接炭素原子が架 橋を形成してもよい。)、
3 )前記 2 )の 5員ないし 6員の脂肪族複素環基で置換される低級アルキル基、 4) 一 NR QR (ここで、
R i。は、 水素原子又は炭素数 1個ないし 2個の低級アルキル基であり、 R 10' は、
a) <置換基群 α 〉から選択される置換基で置換される低級アルキル基; b) ァゼチジニル基、 ピロリジニル基、 ピベリジ-ル基、 ピペラジニル基、 及ぴモルホリノ基から選択される 4員ないし 6員の脂肪族複素環基 (ここで、 前記 4 員ないし 6員の脂肪族複素環基は、 下記 a a) ないし e e) :
a a ) 低級アルキル基、
b b) く置換基群 0^ >から選択される置換基、
c c) <置換基群 α ι>から選択される置換基で置換される低級アルキル 基
d d) 置換されてもよい、 ピロリジニル基、 ピペリジニル基、 及びピペラ ジニル基から選択される 5員ないし 6員の脂肪族複素環基;及び
e e) 置換されてもよい、 ピリジル基、 ピラジ^ル基、 及ぴピリミジ-ル 基から選択される芳香族複素環で置換される低級アルキル基:
力 ^選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。) ; 及ぴ
5) 一 NHCOR1 3 (ここで、
R 1 3は、
a ) 低級アルキル基;
b) く置換基群 α ι〉から選択される置換基; 又は
c ) <置換基群 α >から選択される置換基で 1個若しくは 2個以上置換さ
れる低級アルキル基、 である。) :
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよレ、。) である。 ここで、 く置換基群 0^〉は、 ハロゲン原子、 ヒドロキシ基、 シァ ノ基、 アミノ基、 低級アルキルアミノ基、 ジ低級アルキルアミノ基、 低級アルキルス ルホ-ル基、 フッ素原子で 1個ないし 3個置換されていてもよい低級アルコキシ基、 及びフッ素原子で 1個ないし 3個置換されていてもょレ、低級アル力ノィル基である。
R 9は、 とりわけ好ましくは、 フエニル基 (ここで、 前記フエニル基は、 下記 1 ) 又は 2 ) :
1 ) 低級アルキル基及び/又は <置換基群 OLェ >から選択される置換基で 1個 若しくは 2個以上置換されもよい、 ピロリジニル基又はピペラジニル基
2 ) — N R 1 0 R 1 0, (ここで、
。は、 水素原子であり、
R 1 0 ' は、 <置換基群 0^〉から選択される置換基で 1個若しくは 2個以上置 換されもよい低級アルキル基で置換されてもよい、ァゼチジュル基又はピペリジニル 基である。) :
で、 その 3位又は 4位が置換されてもよい。) である。
また、 R 9が置換されたフエニル基であるとき、 置換基の置換位置は、 特に限定さ れないが、 2位、 3位、 4位であり、 好ましくは、 3位及ぴ Z又は 4位である。
R 9 の置換基の数は、 特に限定されないが、 1個、 2個、 又は 3個であり、 好まし くは 1個又は 2個であり、 さらに好ましくは 1個である。
また、 R 9の好ましく実施形態は、 下記に示す置換基で 3位又は 4位が置換された フエニル基と表現することもできる。
ここで、 p及ぴ qは、 同一荐しくは異なって、 1、 2又は 3であり ;
Waは、水素原子、低級アルキル基、又はく置換基群 0^〉から選択される置換基で あり ;
及ひ 2は、 同一若しくは異なって、 水素原子、 低級アルキル基、 く置換基群 α i〉から選択される置換基、又は <置換基群 αェ >から選択される置換基で置換される 低級アルキル基である。 '
R 9のより好ましい実施形態は、下記に示す置換基で 3位又は 4位が置換されたフ ェニル基と表現することもできる。
ここで、 及ぴ W2は、 前記と同じである。
R 9の特に好ましい実施形態は、 下記に示す置換基で 3位が置換されたフエ二ル基、
又は、 下記に示す置換基で 4位が置換されたフヱニル基と表現することもできる。 こ
こで、 及ぴ W2は、 前記と同じである
、Ν .
W2
く置換基群 α〉は、ハロゲン原子、 ヒドロキシ基、ニトロ基、シァノ基、アミノ基、 力ルバモイル基、 アミノスルホニル基、 低級アルキルアミノ基、 ヒ ドロキシ低級アル キルァミノ基、 ジ低級アルキルァミノ基、 イミノ基、 低級アルキルスルホニル基、 低 級アルコキシカルボ-ルァミノ基、ハロゲン原子で 1個ないし 3個置換されていても よい低級アルコキシ基、 低級アルコキシカルボニル基、 ハロゲン原子で 1個ないし 3 個置換されていてもよい低級アルカノィル基、 及ぴカルボキシル基、 であり ; 好ま しくは、ハロゲン原子、 ヒドロキシ基、シァノ基、アミノ基、低級アルキルアミノ基、 ジ低級アルキルァミノ基、 低級アルキルスルホニル基、 フッ素原子で 1個ないし 3個 置換されていてもよい低級アルコキシ基、及ぴフッ素原子で 1個ないし 3個置換され ていてもょレ、低級アル力ノィル基である。
く置換基群 ]3 >は、 ハロゲ 原子、 ヒドロキシ基、 シァノ基、 アミノ基、 ホノレミル 基、 力ルバモイル基、 アミノスルホニル基、 低級アルキルアミノ基、 ジ低級アルキル アミノ基、ヒドロキシイミノメチル基、メ トキシィミノメチル基、低級アルコキシ基、 低級アルコキシカルボニル基、低級アルコキシカルボニルァミノ基、 低級アルカノィ ル基、低級アル力ノィルォキシ基、低級アルキルチオ基、低級アルキルスルホニル基、 カルボキシル基、 及ぴテトラゾリル基、 であり ; 好ましくは、 ハロゲン原子、 シァ ノ基、 及び低級アルコキシ基である。
ηは、 1ないし 3のいずれかの整数であり、 好ましくは、 1である。
上記式 (I) の化合物は、 好ましくは、 (a) 4— (8—メチルイミダゾ [1, 2 — a] ピリジン一 3—ィル)一2— {[(1 S) —1—フエニルェチル] アミノ}一 5 —ピリ ミジンカルボ-トリル (実施例 8 )、
(b) 5—ブロモー 4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—^ fル) — N— [(1 S)一 1一フエニルェチル]一 2—ピリミジンアミン (実施例 28)、
(c) 4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— {[(1 S )— 1—フエニルェチル]アミノ}一 5—ピリミジン力ルボニトリル(実施例 30)、
(d) 5—プロモー 4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジ ンー 3—ィル]— N— [(1 S)—1—フエニルェチル]—2—ピリ ミジンアミン (実 施例 31)、
(e) 3— (5—シァノ一2— {[(1 S)一 1一フエニルェチル] ァミノ) _4ーピ リミジニル) イミダゾ [ 1, 2— a ] ピリジン一 8—カルボ-トリル(実施例 32)、
( f ) 4— [8— (ジフルォロメチル) イミダゾ [1, 2— 3 ] ピリジン一 3—ィル]
—2— {[(1 S) 一 1一フエニルェチル] アミノ}一 5—ピリミジンカルボ-トリル (実施例 33)、
(g) N— [(1 S) 一 1一フエニルェチル] 一 4— [8— (トリフルォロメチル) イミダゾ [1, 2— a]ピリジン一 3—ィル]一 2—ピリミジンアミン(実施例 34)、 (h) 5—プロモー N— [( 1 S) — 1一フエ二ルェチル] — 4一 [8— (トリフル ォロメチル) イミダゾ 2— a] ピリジン一 3—^ fル]一 2—ピリミジンアミン
(実施例 35)、
( i ) 2— {[(1 S) 一 1—フヱニルェチル] アミノ}一 4一 [8— (トリフルォロ メチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 5—ピリミジンカルボニト リル (実施例 36)、
( j ) 2— {[(I S) — 1一 (3—アミノフヱニル) ェチル] アミノ} —4— [8— (ジフルォロメチル) イミダゾ [1 , 2— a] ピリジン一 3—ィル] ピリミジン一 5 一カルボ二トリル (実施例 80)、
(k) 4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3_ィル] 一 2— [((1 S) — 1一 {3— [(1一メチルァゼチジン一 3—ィル) ァミノ] フエ 二ル} ェチル) ァミノ] ピリ ジン一 5—カルボ二トリル (実施例 88)、
(1) 4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィノレ] —2— {[(1 S) — 1— (4—ピぺラジン一 1ーィルフエニル) ェチル] ァミノ) ピ リミジン一 5—カルボ二トリル (実施例 1 10)、
(m) 4— (8—メチルイミダゾ [1, 2— 3 ] ピリジン一 3—ィル) —2— [((1 S) —1— { 3— [(1ーメチルビペリジン一 4—ィル) ァミノ] フエ二ノレ } ェチノレ) ァミノ] ピリミジン一 5—カルボ二トリル (実施例 1 1 9)、
(n) 4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) —2— [((1 S) — 1一 { 3— [ ( 1—メチルピぺリジンー 4ーィル) ァミノ]フエ二ノレ } ェチル) ァミノ] ピリミジン一 5—カルボ二トリル (実施例 123)、
(o) 4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一2— {[(1 S) 一 1一 (4ーピペラジン一 1ーィルフエニル) ェチル] アミノ} ピリミジン一 5 —カルボ二トリル (実施例 1 32)、
(p) 2— [((1 S) — 1— {4— [ (3R) 一 3—ァミノピロリジン一 1一ィル]フ ェニル } ェチル) ァミノ] —4— (8—クロロイミダゾ [1, 2i] ピリジン一 3 ーィル) ピリミジン一 5—カルボ-トリル (実施例 137)、
(q) 4— (8—ブロモイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— {[(1 S) — 1一 (4—ピぺラジン一 1—ィルフエニル) ェチル] アミノ} ピリミジン一 5 一カルボ二トリル (実施例 145)、
(r ) 4— [8— (フルォロメチル) イミダゾ [1 , 2— a] ピリジン一 3—ィノレ]一 2— {[(1 S) — 1— (4ーピペラジン一 1—ィルフヱニル) ェチル] アミノ} ピリ
ミジン一 5一カルボ二トリル (実施例 149)、
(s) 4一 [8— (フルォロメチル) イミダゾ [1, 2— 3 ] ピリジン一 3—ィノレ]— 2— {[(1 S) 一 1一 (4ーピペリジン一 4ーィルフエニル) ェチル] アミノ} ピリ ミジン一 5—カルボ二トリル (実施例 155)、
(t) 4一 [8— (1—ヒ ドロキシェチル) イミダゾ [1, 2— a] ピリジン一 3— ィル]一 2— {[( 1 S) 一' 1一 (4—ピぺラジン一 1一^ ルフ: rニル) ェチル] アミ ノ} ピリ ミジン一 5—カルボ二トリル (実施例 1 56)、 又は
(u) 4— (8—メトキシイミダゾ [1, 2— a] ピリジン一 3—ィル)一2— {[(1 S) — 1— (4ーピペラジン一 1—ィルフエニル) ェチル] アミノ} ピリ ミジン一 5 —力ルポ二トリル (実施例 158)、 或いは、 その薬学的に許容される塩若しくはェ ステ である。 また、 本願発明の好ましい形態は、 次のようにも表現することができる。 即ち、
(1) Χ 、 Χ2 、 Χ3及び Χ4が、 すべて Cであるか、 又は x2 、 x3及び x4の いずれか 1個のみが Nであり、 残りがすべて Cであり ;
R 及ぴ1^ , のいずれか一方と、 R2及ぴ R2 , のいずれか一方が、 一緒になつ て、 -X2 の結合において 2重結合を形成し;
R3及び R3 , のいずれか一方と、 R4及び R4 ' のいずれか一方が、 一緒になつ て、 X3 -X4の結合において 2重結合を形成し;
2重結合の形成に関与しない 及ぴ1^ , のいずれか一方が、 く置換基群 α>か ら選択される置換基、 く置換基群ひ >から選択される置換基で 1個若しくは 2個以上 置換されてもよい低級アルキル基、 シクロアルキル基、 ァリール基、 又はへテロァリ ール基であり ;
2重結合の形成に関与しない R2及ぴ 2 , のいずれか一方が、 水素原子、 く置換 基群 α>から選択される置換基、又は <置換基群 から選択される置換基で 1個若 しくは 2個以上置換されてもよい低級アルキル基である力、 或いは、 Νである Χ2 と 一緒になつて Νとなり ;
2重結合の形成に関与しない R 3及ぴ R 3 , のいずれか一方が、 水素原子、 <置換 基群 α >から選択される置換基、又は <置換基群ひ >から選択される置換基で 1個若 しくは 2個以上置換されてもよい低級アルキル基である力、 或いは、 Νである Χ3 と 一緒になつて Νとなり ;及ぴ、
2重結合の形成に関与しない R4及ぴ1 4 , のいずれか一方が、 水素原子である 力 或いは、 Nである X4 と一緒になって Nとなる、 上記式 (I) の化合物又はその 薬学的に許容される塩若しくはエステル;又は
(2) X 、 X2 、 X3及ぴ X4力 すべて Cであり ;
Rx及び , のいずれか一方が、 水素原子であり、 他方が、 <置換基群 α>から
選択される置換基、 <置換基群 α >から選択される置換基で 1個若しくは 2個以上置 換されてもよい低級アルキル基、 ァリール基、 又はへテロアリール基であり ;及ぴ、 R2、 R2 '、 R3、 R3 ,、 R4、 及び R4 , 、 すべて水素原子である、 上記式 ( I ) の化合物又はその薬学的に許容される塩若しくはエステル;又は
(3) R8及び R8 ' の一方が、 水素原子であり、 他方がメチル基又はェチル基であ り ; '
R9力 置換されてもよいフエ-ル基であり ;及び、
nが 1である、 上記 (1) の化合物又はその薬学的に許容される塩若しくはエステ ル;又は
(4) く置換基群 j3>が、 ハロゲン原子、 シァノ基、 及ぴ低級アルコキシ基であり ; R6 1S 水素原子、 又は <置換基群 ]3 >から選択される置換基であり、 かつ、 R7 が水素原子である、 上記 (3) の化合物又はその薬学的に許容される塩若しくはエス テル;又は
(5) Yが Nである、 上記 (4) の化合物又はその薬学的に許容される塩若しくはェ ステル;又は
(6) X , X2、 X3及ぴ X,4力 すべて Cであり ;
2重結合の形成に関与しない 及び 'のいずれか一方が、ハロゲン原子; シ ァノ基; ハロゲン原子で 1個ないし 3個置換されてもよい炭素数 1個ないし 2個の 低級アルキル基; ハロゲン原子で 1個ないし 3個置換されてもょ 、炭素数 1個ない し 2個の低級アルコキシ基; ヒドロキシ基で置換されてもよい炭素数 1個ないし 3 個の低級アルキル基; 又はシク口プロピル基であり ;
2重結合の形成に関与しない、 R2及び R2 ' のいずれか一方、 並びに R3及び R
3 , のいずれか一方が、 '同一若しくは異なって、 水素原子; ハロゲン原子; シァ ノ基; ハロゲン原子で 1個ないし 3個置換されてもよい炭素数 1個ないし 2個の低 級アルキル基; ハロゲン原子で 1個ないし 3個置換されてもよい炭素数 1個ないし
2個の低級アルコキシ基; 又は、 ヒドロキシ基で置換されてもよい炭素数 1個ない し 3個の低級アルキル基であり ;
2重結合の形成に関与しない R4及ぴ1 4 ' のいずれか一方が、水素原子である、 上記 (5) の化合物又はその薬学的に許容される塩若しくはエステル;又は
(7) R51 水素原子であり、 R6 I シァノ基であり ; R8及ぴ1 8 ' の一方 力 水素原子であり、 他方がメチル基である、 上記 (6) の化合物又はその薬学的に 許容される塩若しくはエステル;又は
(8) <置換基群 α;>が、 ハロゲン原子、 ヒドロキシ基、 シァノ基、 アミノ基、 低級 アルキルアミノ基、 ジ低級アルキルアミノ基、 低級アルキルスルホニル基、 フッ素原 子で 1個ないし 3個置換されていてもよい低級アルコキシ基、及びフッ素原子で 1個 ないし 3個置換されていてもよレ、低級アル力ノィル基であり ;
R9 ヽ フエニル基 (ここで、 前記フエニル基は、 下記 1) ないし 5) :
1) く置換基群 α>から選択される置換基;
2) ァゼチジニル基、 ピロリジニル基、 ピベリジ-ル基、 及びピペラジニル基 から選択される 5員ないし 6員の脂肪族複素環基 (ここで、前記 5員ないし 6員の脂 肪族複素環基は、 下記 a) ないし c) :
a) 低級アルキル基、
b) く置換基群 α>から選択される置換基、 及ぴ
c) く置換基群 から選択される置換基で置換される低級アルキル基: から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。 また、 前記 4員ないし 7員の脂肪族複素環基は、 同一炭素原子に結合する水 素原子 2個がォキソ基で置換されていてもよく、当該複素環を構成する隣接する炭素 原子間の結合が 2重結合であってもよく、当該複素環を構成する非隣接炭素原子が架 橋を形成してもよい。)、
3 )前記 2 )の 5員ないし 6員の脂肪族複素環基で置換される低級アルキル基、 4) 一 NR10R10 (ここで、
R i。は、 水素原子又 炭素数 1個ないし 2個の低級アルキル基であり、
IV 10 は、
a) <置換基群 α〉から選択される置換基で置換される低級アルキル基; b) ァゼチジニル基、 ピロリジニル基、 ピペリジニル基、 ピぺラジュノレ基、 及ぴモルホリノ基から選択される 4員ないし 6員の脂肪族複素環基 (ここで、 前記 4 員ないし 6員の脂肪族複素環基は、 下記 a a) ないし e e) :
a a ) 低級アルキル基、
b b) く置換基群 a;〉から選択される置換基、
c c) く置換基群 α>から選択される置換基で置換される低級アルキル基 d d) 置換されてもよい、 ピロリジニル基、 ピペリジニル基、 及びピペラ ジニル基から選択される 5員ないし 6員の脂肪族複素環基;及ぴ
e e) 置換されてもよい、 ピリジル基、 ビラジニル基、 及びピリミジニル 基から選択される芳香族複素環で置換される低級アルキル基:
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。) ; 及ぴ
5) 一 NHCOR13 (ここで、
R13は、
a ) 低級アルキル基;
b) <置換基群 a >から選択される置換基; 又は
c) <置換基群 a >から選択される置換基で 1個若しくは 2個以上置換され る低級アルキル基、 である。) :
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。) である、 上記 (7) の化合物又はその薬学的に許容される塩 しくはエス テル;又は
(9) 2重結合の形成に関与しない 及ぴ1^ , のいずれか一方が、 フッ素原子; 塩素原子; 臭素原子; シァノ基; ハロゲン原子で 1個ないし 3個置換されても よいメチル基; ハロゲン原子で 1個ないし 3個置換されてもよいメトキシ基; ヒ ドロキシ基で置換されてもょレ、炭素数 1個ないし 3個の低級アルキル基; 又はシク 口プロピル基であり ;
2重結合の形成に関与しない、 R2及ぴ R2 , のいずれか一方、 R3及ぴ R3 , の いずれか一方、並びに R4及び R4 ' のいずれか一方が、いずれも水素原子であり ; R9力 フエニル基 (ここで、 前記フエニル基は、 下記 1) 又は 2):
1 )低級アルキル基及び/又はく置換基群 α >から選択される置換基で 1個若 しくは 2個以上置換されもよい、 ピロリジニル基又はピペラジ-ル基
2) 一 NR10R10, (ここで、
。は、 水素原子であり、
R10'は、 く置換基群 >から選択される置換基で 1個若しくは 2個以上置換 されもよい低級アルキル基で置換されてもよい、ァゼチジニル基又はピペリジニル基 である。):
で、 その 3位又は 4位が置換されてもよい。) である、 上記 (8) の化合物又はその 薬学的に許容される塩若しくはエステル、 と表すことができる。
また、本願発明の別の実施形態として下記の表現も可能である。即ち、一般式 [ I。]:
ひ。)
[式中、
X1 , X2 、 X3及び X4は、 同一若しくは異なって、 C又は Nであり (但し、 X 1 , X2 、 X3及び X4のうち、 Nであるものは、 0個ないし 2個である。)、
Yは、 CH又は Nであり、
Rj 、 R1 '、 R2 、 R2 ,、 R3 、 R3 '、 R4、 及ぴ1 4 ' は、 同一若しくは異な つて、 水素原子、 く置換基群 0>から選択される置換基、 <置換基群ひ 0>から選択
される置換基で 1個若しくは 2個以上置換されてもよい低級アルキル基、了リール基、 又はへテロアリール基 (ここで、 前記ァリール基及びへテロアリール基は、 互いに独 立して、 下記 1) ないし 3) :
1) 低級アルキル基、
2) く置換基群 αο>から選択される置換基、 及ぴ
3 ) <置換基群 a 0 >から選択される置換基で置換される低級アルキル基 力 ら選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよい。) であるカ
或いは、 Raであり (ここで、 Raは、 一Z — Zs— Z 3で表され、
. Z 1 は、 O又は NHCOであり ;
Z2は、 単結合又は (CHWi) nlであり (ここで、 は、 1ないし 3のいず れかの整数であり、 iは、 1ないし η ιのいずれかの整数であり、 (CHWi) nlは、 1^= 1のとき (CHW を表し、 η ι=2のとき (CHWJ —(CHW2) を表し、 1^=3のとき (CHW ) — (CHW2) -(CHW3) を表し、 Wい W2、及び W3は、 同一若しくは異なって、 水素原子又は炭素数 1個ないし 2個の低級アルキル基であ る。); .
Z3は、低級アルコキシ基又はフエニル基であり(ここで、前記フエ-ル基は、 く置換基群 α0>から選択される置換基で 1個若しくは 2個以上置換されていてもよ い。)、
但し、 、 X2 、 X3及び X4のいずれかが Nであるとき、 Nである当 該 Xi (ここで、 iは、 1ないし 4のいずれかの整数である。) に結合する Ri及ぴ Ri , のいずれか一方は、 当該 Xi と一緒になつて Nとなり、 他方の Ri 又は Ri , は、 前記に定義したとおりであり、
また、 1^及び ' のいずれか一方と、 R2及ぴ R2 , のいずれか一方 力 一緒になつて、 — X2 の結合において 2重結合を形成してもよく、
また、 R3及ぴ R3 , のいずれか一方と、 R4及ぴ1 4 , のいずれか一方 ίΚ 一緒になつて、 Χ3 — Χ4の結合において 2重結合を形成してもよく、
R 5は、 水素原子又はメチル基であり、
R6及ぴ1 7は、 同一若しくは異なって、水素原子、 <置換基群 j3o>から選択され る置換基、 又は <置換基群) 30>から選択される置換基で 1個若しくは 2個以上置換 されてもよい低級アルキル基、 又は、 ピロリジ -ル基、 ピペリジニル基、 及びピペラ ジニル基から選択される 5員ないし 6員の脂肪族複素環基 (ここで、 前記 5員ないし 6員の脂肪族複素環基は、
1) 低級アルキル基、
2) <置換基群 0>から選択される置換基、 及び
3 ) <置換基群 0 >から選択される置換基で置換される低級アルキル基
から選択される同一若しくは異なる置換基で 1個若しくは 2個以上置換されていて もよレヽ。) であるか、
或いは、 R 6及び R 7 は一緒になって、 これらが結合する環に縮合した、 下記環:
を形成し、
R 8及ぴ1 8 , は、 同一若しくは異なって、水素原子又はく置換基群 α 0>から選択 される置換基で 1個若しくは 2個以上置換されていてもよい、低級アルキル基であり、
R 9 。 は、 ァリール基又はへテロアリール基 (ここで、 前記ァリール基及びへテロ ァリール基は、 互いに独立して、 下記 1 ) ないし 3 ) :
1 ) 低級アルキル基、
2 ) <置換基群 α: 0>から選択される置換基、 及び
3 ) <置換基群 a 0 >から選択される置換基で置換される低級アルキル基 力 ら選択される同一若しくは なる置換基で 1個若しくは 2個以上置換されていて もよい。) であり、
ηは、 1ないし 3のいずれかの整数であり、
く置換基群ひ ο >及びく置換基群 は、 下記のように定義される。
<置換基群 0! 0 > :
ハロゲン原子、 ヒ ドロキシ基、 シァノ基、 アミノ基、 力ルバモイル基、 アミノスルホ ニル基、 低級アルキルアミノ基、 ヒ ドロキシ低級アルキルアミノ基、 ジ低級アルキル アミノ基、 低級アルキルスルホニル基、 低級アルコキシ基、 低級アルコキシカルボ二 ル基、 低級アルカノィル基、 及びカルボキシル基
ぐ置換基群 jS 0>:
ハロゲン原子、 ヒドロキシ基、 シァノ基、 アミノ基、 ホノレミル基、 カルパモイル基、 ァミノスルホニル基、 低級アルキルァミノ基、 ジ低級アルキルァミノ基、 ヒドロキシ イミノメチル基、 メ トキシイミノメチル基、 低級アルコキシ基、 低級アルコキシカル ボニル基、 低級アルコキシカルボニルァミノ基、 低級アルカノィル基、 低級アルカノ ィルォキシ基、 低級アルキルチオ基、 低級アルキルスルホニル基、 カルボキシル基、 及ぴテトラゾリル基
]で示される化合物又はその薬学的に許容される塩若しくはエステル、 である。 以下に、 一般式 (I ) で示される本発明化合物の代表的な製造方法について説明す る。
スキーム 1 : 式 (I I) もしくは (I I I) の化合物から式 (I) の化合物の製 造法
スキ一ム 1
上記式 (I) の化合物 (ここで、 Xい X2、 X3、 X4、 Y、 Rい R 、 R2、 R 2 7 、 R3、 R3' 、 R4、 R 、 R5、 R6、 及ぴ R7、 R8、 R 、 及び R9は、 前 記と同義であり、 mは、 1又は 2である。) は、 下記式 (I I) もしくは (I I I) の化合物(ここで、 X X2、 X3、 X4、 Y、 Rい R 、 R2、 R2' 、 R3、 R 、 R4、 R 、 R5、 R6、 及び R7は、 前記と同義である。) と、 対応する下記式 (I V) のアルキルアミン (ここ 、 n、 R8、 R 、 及ぴ R9は前記と同義である。) と の置換反応により合成することができる。
即ち、 上記式 (I I) もしくは (I I I) の化合物と上記式 (I V) のァ/レキルァ ミンとの置換反応は、 塩基 (炭酸力リゥム等の無機塩基、 トリェチルァミン、 ジィソ プロピルェチルァミン等の有機塩基) 存在下行うことが望ましい。 溶媒はク口口ホル ム、 テトラヒ ドロフラン、 N, N—ジメチルホルムアミ ド、 ジメチルスルホキシド等 を用いることができ、 好ましくはクロ口ホルム、 テトラヒ ドロフラン、 ジメチルスル ホキシドである。 当該反応においては、 上記式 (I I) もしくは (I I I) の化合物 1モルに対して、 上記式 ( I V) のアルキルァミンを 1〜 5モル、 好ましくは 2モル 用いる。 反応温度は、 使用される原料化合物あるいは反応溶媒に応じて、 当業者が適 宜選択できるが、 通常、.室温から溶媒の沸点であり、 好ま.しくは室温から 50°Cの間 である。 また、 反応は、 通常 1時間から 120時間で完結するが、 反応時間は適宜増 減することが出来る。
なお、 上記式 (I V) の化合物は、 例えば、 3— [(1 S) _1一アミノエチル] ァニリンや 4一 [(1 S) 一 1一アミノエチル] ァニリン等であり、 市販で入手でき る力、 或いは、 市販で入手できる化合物から当業者に周知の方法またはそれに準じた 方法を用いて合成することができる。 スキーム 2 : 式 (I I) の化合物の代表的な製造法
IX
スキーム 2
まず、 上記式 (VI) の化合物 (ここで、 R6及ぴ R7は前記と同義である。) は、 テトラヒドロフラン、 1, 4一ジォキサン、 1, 2—ジメ トキシェタン等の溶媒中、 好ましくはテトラヒドロフラン中、 酢酸パラジウム、 トリフエニルホスフィン、 及び 水酸化ナトリウム水溶液等の塩基の存在下、 上記式 (V) の化合物 (ここで、 R6及 び R7は前記と同義である。) とトリス (2—エトキシビュル) ホウ素のカップリング 反応により得ることが出来る。 この場合、反応温度は使用される原料ィヒ合物あるいは 反応溶媒に応じて当業者が適宜選択することが出来る力 通常、 室温から溶媒の沸点 であり、 好ましくは室温である。 また、 反応は通常、 1時間から 24時間で完結する 力 反応時間は適宜増減できる。
次に、 上記式 (I X) の化合物 (ここで、 Xい X2、 X3、 X4、 Y、 Rい R 、 R2、 R 、 R3、 R 、 R4く R 、 R5、 R6、 及ぴ R7は前記と同義である。) は、 1, 4—ジォキサン中、 上記式 (V I I) の化合物 (ここで、 R6及ぴ R7は前記 と同義である) と上記式(VI I I) の化合物 (ここで、 Xい X2、 X3、 X4、 R 、 R2、 R 、 R3、 R 、 R4、 及ぴ R は前記と同義である。 但し、 及ぴ R のいずれか一方と、 !^及ぴ尺 のいずれか一方が、 一緒になつて、 —X2の結合において 2重結合を形成しており、 かつ、 1 3及ぴ1 のいずれか一方 と、 R4及び R のいずれか一方が、 一緒になつて、 X3— X4の結合において 2重 結合を形成している。) から合成される。 当該反応においては、 上記式 (V I I) の 化合物 1モルに対して、 上記式 (V I I I) の化合物を 1〜 3モル、 好ましくは 1モ
ル用いる。反応温度は使用される原料化合物に応じて当業者が適宜選択することがで きる力 通常、室温から溶媒の沸点であり、好ましくは室温から 50°Cである。また、 反応は 1時間から 24時間で完結するが、 反応時間は適宜増減することができる。 ここで、 上記式 (V I I) の化合物は、 上記 (V I) の化合物 (ここで、 R6及び R7は前記と同義である。) を、 1, 4_ジォキサン中、 N—プロモこはく酸イミドと 反応させて調製することができる。 当該反応においては、 上記 (V I) の化合物 1モ ルに対して、 N—ブロモこはく酸イミドを 1〜 3モル、 好ましくは 1モル用いる。 反 応显度は使用される原料ィ匕合物に応じて当業者が適宜選択することができる力 通常、 0°Cから室温であり、 好ましくは室温である。 また、 反応は、 通常、 1時間から 12 時間で完結するが、 反応時間は適宜増減することができる。 また、 得られた上記 (V I I) の化合物は単離精製することなく、 次の反応に付すことができる。
上記式 (I I) の化合物は、 上記式 ( I X) の化合物を、 ジクロロメタン、 クロ口 ホルム、 N, N—ジメチルホルムアミド等の溶媒中、 好ましくはクロ口ホルム、 N, N—ジメチルホルムアミド中、 m—クロ口過安息香酸 (m—CPBA) により酸化す ることで合成することができる。 当該反応においては、 上記式 (I X) の化合物 1モ ルに対して、 m—クロ口過安阜香酸 (m—CPBA) を 2〜5モル、 好ましくは 2モ ル用いる。 反応温度は、使用される原料化合物あるいは反応溶媒に応じて当業者が適 宜選択することができるが、 通常、 0°Cから室温であり、 好ましくは室温である。 ま た、反応は、 通常、 1時間から 24時間で完結するが、 反応時間は適宜増減すること ができる。
なお、 上記式 (V) の化合物は、 例えば、 4—クロロー 2—メチルチオピリミジン 等であり、 上記式 (V I I I) の化合物は、 例えば、 2—ァミノ一 3—ピコリン等で あり、 市販で入手できるか、 或いは、 市販で入手できる化合物から当業者に周知の方 法またはそれに準じた方法を用いて合成することができる。 スキーム 3 : 式 (I X) の化合物の別の製造法
上記式( I X)の化合物(ここで Yは Nであり、 X X
2、 X
3、 X
4、 R 、 R
2、 R
2 、 R
3、 R
3' 、 R
4、 R 、 R
5、 R
6、 及び R
7は前記と同義である。) は、 以下に示す製造法により合成することもできる。
X XI
スキーム 3
まず、上記式(X ) の化合物 (ここで Yは Nであり、 Xい X2 X3 X4 Rい
R 1 K 2 k 2 ' R,, R R R. 、 及び R 5は前記と同義である。) は、 上記式 (X) のァシル化合物 (ここで、 Xい X2 X3 X4 Rい R1' R2 R 2' R3 R R4 R 、 及ぴ 5は前記と同義である。) と N N—ジメチル ホルムアミドジメチルァセタールを反応させることにより得ることができる。 この場 合、反応温度は、 室温から N N—ジメチルホルムアミドジメチルァセタールの沸点 であり、 好ましくは沸点であ 。 反応は、 通常、 12時間から 120時間で完結する 1S 反応時間は適宜増減することができる。
次に、上記式(I X)の化合物は、メタノール、エタノール、 n—ブタノール、 N, N—ジメチルホルムアミド等の溶媒中、 好ましくは n—ブタノール中、 チォ尿素と、 ナトリウムメトキシド、 ナトリウムエトキシド等の塩基を用いて、 式 (X I) の化合 物に対して、 ピリミジン環の環化反応を行った後に、 ヨウ化メチルを用いるメチルイ匕 により合成することができる。 この場合、 反応温度は、 室温から溶媒の沸点である。 また、 反応は、 通常、 1時間から 12時間で完結するが、 反応時間は適宜増減するこ とができる。 スキーム 4 : 式 (X) の化合物の代表的な製造法
スキーム 4
上記式 (X) のァシル化合物 (ここで、 Xい X2、 X3、 X4、 Rい R 、 R2、 R2' 、 R3、 R 、 R4、 及び R は前記と同義であり、 R 5はメチル基である。 但し、 !^及ぴ のいずれか一方と、 !^及び!^' のいずれか一方が、 一緒にな つて、 — X2の結合において 2重結合を形成しており、 かつ、 1 3及ぴ1^ のい ずれか一方と、 R4及び R のいずれか一方が、 一緒になつて、 X3— X4の結合に おいて 2重結合を形成している。) は、 上記 (V I I I) の化合物 (ここで、 X 2、 X3、 X4、 Rい R 、 R2、 R 、 R3、 R 、 R4、 及び R は前記と同 義である。但し、 !^及ぴ のいずれか一方と、 2及ぴ1 ^ のいずれか一方が、 一緒になつて、 Xi_X2の結合において 2重結合を形成しており、 かつ、 R3及び R のいずれか一方と、 R4及ぴ R のいずれか一方が、 一緒になつて、 X3— X4の 結合において 2重結合を形成している。) と上記式 (X I I) のクロロケトン (ここ で、 R5は前記と同義である。) を反応させることにより合成することができる。 反応 は、公知の方法またはそれに準じた方法を用いて行うことができる (文献: B i o o r g. Me d. C h e m. L e t t. 2003, 13, 3021.、 J. Me d. C h em. 1989, 32, 2204. など)。
また、 上記式 (X) のァシル化合物 (ここで、 Xい X2、 X3、 X4、 Rい R 、 R2、 R 、 R3、 R 、 R4、 及ぴ R は前記と同義であり、 R5は水素原子又は メチル基である。 但し、 !^及び!^/ のいずれか一方と、 !^及ぴ ^ のいずれか —方が、 一緒になつて、 X — X2の結合において 2重結合を形成しており、 また、 R 3及ぴ R のいずれか一方と、 R4及ぴ R のいずれか一方が、 一緒になつて、 X3 一 X4の結合において 2重結合を形成している。) は、 上記 (X I I I) のイミダゾピ
リジン誘導体(ここで、 X X2、 X3、 χ4、 Rい R 、 R2、 R2' 、 R3、 R 、 R4、 及ぴ R は前記と同義であり、 R5は水素原子又はメチル基である。 但し、 R 及ぴ R のいずれか一方と、 !^及ぴ!^ のいずれか一方が、 一緒になって、 一 X2の結合において 2重結合を形成しており、 かつ、 !^及ぴ!^ のいずれか一方 と、 R4及び R のいずれか一方が、 一緒になつて、 X3— X4の結合において 2重 結合を形成している。) に対するフリーデル'クラフツ (F r i e d e 1— C r a f t s) アシル^ (匕反応により合成することができる。 この反応は、 常法で行うことがで きる (文献: J. Me d, Ch em. 1 970, 13, 1048.、 国際公開第 01 /014375号パンフレツトなど)。 また、 上記式 (X I I I ) のイミダゾピリジ ン誘導体は、 常法による上記式 (V I I I) の化合物 (ここで、 Xi、 X2、 X3、 X 4、 Rい R 、 R2、 R 、 R3、 R 、 R4、 及び R は前記と同義である。 伹 し、 及ぴ!^ のいずれか一方と、 1 2及ぴ1 のいずれか一方が、 一緒にな。つ て、 X — X2の結合において 2重結合を形成しており、 かつ、 1 3及ぴ1 3 / のいず れか一方と、 R4及ぴ R のいずれか一方が、 一緒になつて、 X3— X4の結合にお いて 2重結合を形成している。) と上記式 (X I V) の α—ハロゲン化カルボニル化 合物(ここで、 R5は水素原子 はメチル基である。) より、容易に調製できる (文献: J . Me d. C h e m. 1996, 39, 2856. など)。
また、 上記式 (X I I I) のイミダゾピリジン誘導体 (ここで、 Xい X
2、 X
3、 X
4、 R
x, R 、 R
2、 R 、 R
3、 R 、 R
4、 及ぴ R は前記と同義であり、 R
5は水素原子又はメチル基である。 伹し、 !^及ぴ のいずれか一方と、 R
2及 び R のいずれか一方が、 一緒になつて、 Xi— X
2の結合において 2重結合を形成 しており、また、 R
3及び R のいずれか一方と、 R
4及ぴ R のいずれか一方が、 一緒になつて、 X
3— X
4の結合において 2重結合を形成している。)を、常法により、 • ラネーニッケルを触媒とする水素添加反応に付する.ことにより、テトラヒドロイミダ ゾピリジン誘導体 (X I I I) (ここで、 Xい X
2、 X
3、 X
4、 Rい R 、 R
2、 R
2' 、 R
3、 R
3' 、 R
4、 及ぴ R は前記と同義であり.、 R
5は水素原子又はメチ ル基である。但し、 1^及ぴ R のいずれか一方と、 R
2及ぴ のいずれか一方力 一緒になつて、 Xi_X
2の結合において 2重結合を形成することはなく、 かつ、 R
3 及ぴ のいずれか一方と、 R
4及ぴ R のいずれか一;^が、 一緒になつて、 X
3 一 X の結合において 2重結合を形成することはない。) を合成することができる。 スキーム 5 : 式 (I I I) の化合物の製造法
スキーム 5
まず、上記式(XVI) の化合物(ここで、 1 6及ぴ1 7は前記と同義でぁる。) は、 テトラヒドロフラン、 1, 4—ジォキサン、 1, 2—ジメトキシェタン等の溶媒中、 好ましくはテトラヒドロフラン中、 酢酸パラジウム、 トリフエニルホスフィン、 及び 水酸化ナトリウム水溶液等の塩基の存在下、 上記式 (XV) の化合物 (ここで、 R6 及ぴ1 7は前記と同義である。) とトリス (2—エトキシビニル) ホウ素のカップリン グ反応により得ることができる。 この場合、 反応温度は、 使用される原料化合物ある いは反応溶媒に応じて当業者が適宜選択することが出来る力 通常、 室温から溶媒の 沸点であり、 好ましくは室温である。 また、 反応は通常、 1時間から 24時間で完結 するが、 反応時間は適宜増減できる。
なお、 上記式 (XV) の化合物は、 例えば、 2, 6—ジクロ口プリン等であり、 巿 販で容易に入手できる。
次に、 上記式 (I I I) の化合物 (ここで、 Xi、 X2、 X3、 X4、 R1 R 、 R2、 R2' 、 R3、 R 、 R4、 R 、 R5、 R6及ぴ R7は前記と同義である。但し、 1^及び1 のいずれか一方と、 !^及ぴ尺 のいずれか一方が、 一緒になつて、 Xi— X2の結合において 2重結合を形成しており、 また、 1 3及ぴ1 3 / のいずれか 一方と、 R4及ぴ R のいずれか一方が、 一緒になって、 X3— X4の結合において 2重結合を形成している。) は、 1, 4一ジォキサン中、 上記式 (XV I I) の化合 物 (ここで、 R6及ぴ1^7は前記と同義である。) と上記式 (VI I I) のァミノ化合 物から合成される。 当該反応は上記式 (XV I I ) の化合物 1モルに対して、 上記式 (V I I I) のァミノ化合物を 1〜 3モル、 好ましくは 1モル用いる。 反応温度は、
使用される原料ィヒ合物に応じて当業者が適宜選択することができる力 通常、 室温か ら溶媒の沸点であり、 好ましくは室温から 50°Cである。 また、 反応は 1時間から 2 4時間で完結するが、 反応時間は適宜増減することができる。
ここで、 上記式 (XV I I) の化合物は、 上記式 (XV I) の化合物 (ここで、 R 6及ぴ1 7は前記と同義である。) を 1, 4一ジォキサン中、 N—ブロモこはく酸イミ ドと反応させることにより調製できる。 当該反応は、 上記式 (XV I) の化合物 1モ ルに対して、 N—プロモこはく酸イミド 1〜3モル、 好ましくは 1モル用いる。 上記 式.(XV I) の化合物この場合において、 反応温度は、 使用される原料化合物に応じ て当業者が適宜選択することができる力 通常、 0°Cから室温であり、 好ましくは室 温である。 また、 反応は、 通常、 1時間から 1 2時間で完結するが、 反応時間は適宜 増減することができる。 また、 得られた上記式 (XV I I) の化合物は単離精製する ことなく、 次の反応に付すことができる。 スキーム 6 Yが CHである式 (I) の化合物の製造法
スキーム 6
まず、 上記式 (X I X) の化合物 (ここで、 R R7、 R8、 R 及ぴ1 9は、 前 記と同義である。) は、 クロ口ホルム、 N, N—ジメチルホルムアミド、 ジメチルス ルホキシド等の溶媒中、 好ましくはジメチルスルホキシド中、 上記式 (XV I I I ) の化合物 (ここで、 1 6及ぴ1 7は、 前記と同義である。) と上記式 (I V) のアルキ ルァミン (ここで、 R8、 R8 f 及ぴ R9は、 前記と同義である。) を反応させることに より合成できる。 当該反応は、 上記式 (XV I I I ) の化合物 1モルに対して、 上記
式 (IV) のアルキルアミンを 1〜 3モル、 好ましくは 1モル用いる。 この場合、 反 応は、 室温から溶媒の沸点であり、 好ましくは 50°Cから 100°Cである。 また、 反 応は 1時間から 24時間で完結するが、 反応時間は適宜増減できる。
次に、 上記式 (XX) の化合物 (ここで、 R6、 R7、 R8、 Rg' 及び R9は、 前記 と同義である。) は、 カップリング反応によるビエルエーテル基の導入により合成で きる。 このカップリング反応は、 上記のスキーム 2と同様に行うことができる。 また、 上記式 (I) の化合物は、 上記式 (XX I) の化合物 (ここで、 R6、 R7、 R8、 R8' 及び R9は、 前記と同義である。) と上記式 (V I I I) の化合物との環化 反応により合成することができ、 環化反応はスキーム 2と同様に行うことができる。 ここで、 上記式 (XVI I I) の化合物 (ここで、 R6及ぴ R7は、 前記と同義であ る。) は、 例えば、 2—フルオロー 4一ョードピリジン等であり、 市販で入手できる か、或いは、 市販で入手できる化合物から公知の方法またはそれに準じた方法を用い て合成することができる (文献: J. Or g. Ch e m. 1993, 58, 7832. など)。 なお、 上記のスキーム 1なレ,、し 6に記載の製造方法において、 定義した官能基が反 応条件下変化する等の場合、 当業者は、 必要に応じて、 有機合成化学で常用とされる 方法、 例えば官能基の保護、 脱保護等 [例えばプロテクティブ ·グループス 'イン · オーガニック ·シンセシス 第三版 (P r o t e c t i v e Gr oup e s i n Or g a n i c Syn t h e s i s, t h e t h i r d e d i t i o n), T. W. グリーン (T. W. Gr e en e) 著、 J o hn Wi l e y & S on s社参照] の手段を付することにより、 所望の化合物を得ることができる。
R6の導入あるいは変換は、 上述の合成中間体のいずれかの段階で行うことができ る。 以下に、 上記式 (I) で示される化合物における R6の導入あるいは変換の例に ついて説明する。 なお、 当業者は、 適宜、 公知の方法、 及び Z又は、 下記に例示する 方法もしくはこれに準ずる方法を用いることで、 R6の導入あるいは変換を行うこと ができる。
上記式(I) の化合物 (ここで、 R6はハロゲン原子であり、 Xい X2、 X3、 X4、 Y、 Rい R 、 R2、 R 、 R3、 R 、 R4、 R 、 R5、及び R7、 R8、 R 、 及び R9は、 前記と同義である。) は、 テトラヒドロフラン、 水、 酢酸、 メタノール、 エタノーノレ、 1, 4—ジォキサン、 クロロホノレム、 ジクロロメタン、 トルエン等の溶 媒中、 対応する式 (I) の化合物 (ここで、 R6は水素原子である。) を、 N—クロ口 こはく酸イミド、 N—ブロモこはく酸イミド、 N—ョードこはく酸イミド等の R6— X (ここで、 R6はハロゲン原子であり、 Xは、 例えば、 こはく酸イミドである。)、 又は塩素、 臭素、 ヨウ素等のハロゲン化剤と反応させることにより合成することがで
きる。 この場合、 反応温度は、 使用される原料化合物あるいは反応溶媒に応じて適宜 選択される力 通常、 o°cから反応に用いる溶媒の沸点である。また、反応は、通常、 1時間から 24時間で完結するが、 反応時間は適宜増減できる。
上記式 (I ) の化合物 (ここで、 R6は低級アルコキシカルボニル基であり、 X1 X2、 X3、 X4、 Y、 Rい R 、 R2、 R2' 、 R3、 R 、 R4、 R 、 R5、 及 ぴ R7、 R8、 R 、 及ぴ R9は、 前記と同義である。) は、 対応する式 (I ) の化合 物 (ここで、 R6は臭素原子である。) から合成することができる。 例えば、 式 (I) の化合物 (ここで、 R6は臭素原子である。) を、 N, N—ジメチルァセトアミド、 N 一メチルピロリ ドン、 N, N—ジメチルホルムアミ ド等の溶媒に、 メタノール、 エタ ノ一ル等のアルコ一ル類を加えた混合溶媒中で、 1, 1 '一ビス (ジフエニルホスフ イノ) フエ口セン等の配位子と酢酸パラジウム (I I) 等のパラジウム触媒と炭酸水 素ナトリウム、 トリェチルァミン等の塩基存在下、 一酸ィヒ炭素と反応させることによ り、 対応する化合物 (I) (ここで、 R6は低級アルコキシカルボニル基である。) を 合成することができる。 この場合、 反応温度は、 使用される原料化合物あるいは反応 溶媒に応じて適宜選択されるが、 通常、 5 0°Cから反応に用いる溶媒の沸点である。 また、反応は、通常、 1時間から 24時間で完結するが、反応時間は適宜増減できる。 また、上記式(I)の化合物(ここで、 R6はヒ ドロキシカルボニル基であり、 2ゝ X3ヽ 4ゝ ェヽ Rい k 1 ゝ r 2、 R 2 ゝ ν 3ゝ R3 ゝ I 4、 ゝ I 5ゝ 及 び R7、 R8、 R 、 及ぴ R9は、 前記と同義である。) は、 対応する化合物 (I) (こ こで、 R6は低級アルコキシカルボ-ル基である) の加水分解反応によって合成する ことができる。 例えば、 式 ( I ) の化合物 (ここで、 R 6は低級アルコキシカルボ二 ル基であり、 Xい X2、 X3、 X4、 Y、 Rい R 、 R2、 R 、 R3、 R3' 、 R 4、 R 、 R5、 及び R7、 R8、 R8' 、 及ぴ R9は、 前記と同義である。) を、 メタ ノール、 エタノール、 水、 テトラヒ ドロフラン等の溶媒中、 水酸化ナトリウム等を塩 基として用い、 加水分解反応に付すことにより、 式 (I) の化合物 (ここで、 R6は ヒ ドロキシカルボニル基である) を合成することができる。 この場合、 反応温度は、 使用される原料化合物に応じて、 当業者が適宜選択できる力 通常、 o°cから室温で ある。 また、 反応は、 通常、 1時間から 24時間で完結するが、 反応時間は適宜増減 できる。
また、 上記式 (I) の化合物 (ここで、 R6は力ルバモイル基であり、 X2、 X3、 X4、 Y、 Rい R 、 R2、 R 、 R3、 R3 7 、 R4、 R 、 R5、 及ぴ R7、 R8、 R8' 、 及び R9は、 前記と同義である。) は、 対応する式 (I ) の化合物 (ここ で、 R6はヒ ドロキシカルボニル基である) とァミンとの縮合反応により合成するこ とができる。 例えば、 式 (I) の化合物 (ここで、 R6はヒドロキシカルボニル基で ある) を、 テトラヒ ドロフラン、 ジメチルスルホキシド、 N, N—ジメチルホルムァ ミ ド、 1, 4—ジォキサン、 クロ口ホルム等の溶媒中、 1— (3—ジメチルアミノプ
口ピル) 一 3—ェチルカルポジィミド等の縮合剤、 および、 1ーヒドロキシベンゾト リアゾールを用いて、アンモニア等のァミンとの縮合反応に付すことにより、式(I) の化合物 (ここで、 R6はカルパモイル基である) を合成することができる。 この場 合、反応温度は、使用される原料化合物に応じて、当業者が適宜選択できるが、通常、 室温から使用される溶媒の沸点である。 また、 反応は、 通常、 1時間から 24時間で 完結するが、 反応時間は適宜増減できる。
上記式(I) の化合物 (ここで、 R6はシァノ基であり、 Xい X2、 X3、 X4、 Y、 Rい R 、 R2、 R 、 R3、 R 、 R4、 R 、 R5、 及び R7、 R8、 R8' 、 及ぴ R9は、 前記と同義である。) は、 対応する式 (I) の化合物 (ここで、 R6は力 ルパモイル基である) の脱水反応により合成することができる。 例えば、 式 (I) の 化合物 (ここで、 R6は力ルバモィル基である) を、 ピリジン中、 ォキシ塩化リンを 用いて、 脱水反応に付すことにより、 式 (I) の化合物 (ここで、 R6はシァノ基で ある) を合成することができる。 この場合、 反応温度は、 使用される原料化合物に応 じて、 当業者が適宜選択できるが、 通常、 室温から使用される溶媒の沸点である。 ま た、 反応は、 通常、 1時間から 24時間で完結するが、 反応時間は適宜増減できる。 次に、 一般式 (I) の化合物の PLK1阻害作用及ぴ細胞増殖抑制作用について以 下説明する。 P LK 1活性阻害作用の測定
(1) PLK1の調製
まず、 N末端に GST (ダルタチオン S—トランスフェラーゼ) を融合した全長の ヒト PLK1を発現するパキュロウィルスを作製した後に、スポドプテラ フルギぺ ノレダ (S p o d o p t e r a f r u g i p e r d a) (S f ) 9昆虫細胞に感染さ せた PLK1を GST—融合タンパクとして高発現させた。 その細胞を回収して、 リ シスバッファー (5 OmMトリス一塩酸バッファー (pH7. 4 ) / 150 mM 塩 ィ匕ナトリウム/ /lmM EDTA (エチレンジァミン- 4酢酸) / ImM ジチオト レイト一ノレ/ 0. 1%ポリオキシエチレンソルビタンモノラウレート) に懸濁しソニ ケーターで細胞破砕を行い、遠心後上清を回収した。 上清をダルタチオンセファロー スビーズと反応させ、 ビーズをリシスパッファーで洗浄した。 その後ビーズをプレシ ジョンプロテアーゼ入りリシスバッファーと反応させ上清を回収した。
(2) P LK 1— T 210Dの調製
ヒト PLK1の 210番目のコドンは本来スレオニンをコードするがこの部位を ァスパラギン酸に改変することによつて活性化型になることが知られている [モレキ ユラ一アンド ·セルラーバイオロジー (Mo 1. C e l l B i o l .), 17巻、 3408項 (1 997年) ]。 ヒ ト活性化型 P LK 1タンパク質を得るために、 ヒ ト
PLK 1 c DNAの 2 1 0番目のコドンに塩基置換を導入することによって、 2 1 0番目のコドンがァスパラギン酸をコードするようになった変異型 P LK 1 (P LK 1 -T 2 1 OD) の cDNAを作成した。 この PLK 1— T2 1 0D c DNAを前 述の方法と同様にバキュロウィルス発現ベクターに組み込み、昆虫細胞にて発現させ、 精製を行なった。
(3) P LK 1及び PL'Kl— T 21 ODの活性測定
1:1及ぴ卩 1:1ー丁2 1 0Dの活性測定において、基質は PLK1の基質部 位として報告されている CD C 2 5 Cのアミノ酸配列 1 9 8番目のセリン周辺配列 [ェンボ ·レポ一ト (EMBO R e p o r t)、 3卷、 34 1頁 (2002年)] を 改変した合成べプチド(ァスパラギン酸-ダルタミン酸-ロイシンーメチォニン-グルタ ミン酸ーァラニンーセリンーフエ二ルァラニンーァラニンーァスパラギン酸ーグル タミンーァスパラギン ーァラニン一リジン) を用いた。
反 、は f o y o s h l m a一 Mo r i mo t oらの方法 [ネーチヤ一 (N a t u r e)、 第 4 1 0巻、 21 5— 220頁、 (200 1年)] に準じて行った。 反応液量は 2 1. 1 Lで、 反応バッファーの組成は 2 OmM トリスー塩酸バッファー (p H 7. 4 ) / 1 0 mM 塩化マグネシウム Ζθ. 5mM ジチオトレイトールノ 1 mM EGTA (エチレングリコーノレ一ビス (ベータ一アミノエチルエーテル)一 N, N, Ν', Ν',一 4酢酸) で、 そこに精製した P LK 1と 50 Μの基質ペプチドと 50 μΜの非標識アデノシン三リン酸 (ΑΤΡ) および 1 μ C iの - 3 3 Ρ] 標識 A TP (2000— 4000 C i /mm o 1 e) を添加して、 反応温度 25 °Cで 20分 間反応させた。 その後、 10 μ Lの 35 OmMリン酸バッファーを反応系に添加して 反応を停止させ、その液を 96ウェルマルチスクリーンフォスフォセルロースフィル ターにスポットした。 7 5mMリン酸バッファーでそのフォスフォセルロースフィル ターを洗浄した後、乾燥させて放射活性を液体シンチレーシヨンカウンターで測定し た。 非標識 ΑΤΡ、 ίγ - 33 Ρ]標識 ATPはアマシャム 'バイオサイエンスから、 マルチスクリーンフォスフォセルロースフィルタ一はミリポア社からそれぞれ購入 した。
本発明化合物の反応系への添加は、予め終濃度の 20倍濃度でジメチルスルホキシ ドに溶解させた溶液を 1. 1 μ L加えることによって行った。反応系へジメチルスル ホキシドを 1. 1 μ L加えたものを対照とした。
本発明化合物の PLK 1及ぴ P LK 1— Τ 2 1 0D活性に対する I C5。値をもと めたのでその結果を下記の表 1に示す。
【表 1】
以上より、本発明化合物の PLK1及び PLK1—T210D阻害活性は著しく高 い とが明らかとなった。 細胞増殖抑制作用の測定: 細胞レベルでの P L K 1阻害活性の測定
(1) 細胞培養の方法
細胞レベルでの化合物の P LK 1阻害活性の測定にはヒト子宫頸がん細胞株 H e L a S 3細胞を用いた。 He L a S 3細胞はアメリカン タイプ カルチヤ コレク シヨン (ATTC : Ame r i c a n Ty p e Cu l t u r e Co l 1 e c t i o n) より入手し、 10%牛胎児血清添加ダルベッコ変法イーグル培地を用いて 37 で5%〇〇2存在下、 飽和水蒸気の CO 2インキュベーター内にて培養した。 (2) 本発明に係る化合物の阻害活性測定
PLK1は哺乳動物細胞の有糸分裂期 (M期) の様々な段階に重要な役割を果たし ていることが報告されている (ネイチヤー レビュー モレキュラー セル バイオ ロジー (Na t. Re v. Mo 1. Ce l l . B i o l .)、 第 5卷、 429頁、 (2 004年))。 実際、 哺乳動物細胞を PLK 1に対する s i RN Aで処理してその発現 レベルを抑制すると、 細胞周期の進行が阻害されて細胞は M期に停止する。 またこの とき、 M期における染色体凝縮に必要と考えられているヒストン H3の 10番目のセ リン残基のリン酸ィヒレベルを調べると、そのレベルが高度に亢進されていることが観 察される。 そこで、 細胞を本発明に係る化合物で処理した後にヒストン H 3のリン酸 化レベルを間接蛍光抗体法により調べ、そのレベルを指標に M期細胞を同定して M期 停止細胞の割合を解析し、 さらには各化合物の EC 50値を算出して細胞レベルでの PLK1阻害活性を評価した。
まず、 ダブルチミジン法により G1/S期に同調した H e L a S 3細胞を、 リジン 処理した 96ゥエルプレート (ファルコン社、 F a l c o n) に 1ゥエルあたり 8, 000個の割合で播種し、 前述の CO2インキュベーター内に静置した。 播種 4時間 後、終濃度が図のようになるように段階希釈した本発明に係る化合物をプレートの各 ゥェルに添カ卩し、 さらに co2インキュベータ一中に静置した。 本発明に係る化合物 を添加して 12時間後、プレートの各ゥヱル内の本発明に係る化合物を含む培地を除 去した後、 氷冷した 100 %メタノール (和光純薬) を 100 μ L添カ卩して 10分間 細胞の固定ならびに膜透過性の亢進処理を行った。 次いで、 メタノールを除去したの
ちのゥェルに、 50 Lの 1 %B S A/PB Sを添加して 30分間プロッキングを行 つた後、一次抗体反応として 50 μ Lの 2. 5mg mL 抗 p h o s p h o— H i s t o n eH3 (S e r l O)抗体(アップステート社、 Up s t a t e)を含む 1 % B S A/PB Sをゥヱルに添加し、室温で 90分間プレートを放置した。反応終了後、 PB Sで各ゥエルを 1回洗浄し、 次いで二次抗体反応として 50 μ Lの 1. 5mgZ mL Cy 5標識抗ゥサギ' I g G (H+ L) 抗体 (ケミコン社、 Ch emi c o n) ならびに核染色試薬である 10 u g/mL DAP I (シグマ社、 S I GMA) を含 む 1 % B S AZ P B Sを添カ卩し、 室温でさらに 90分間放置した。 反応終了後、 ゥェ ル内の反応液を除去して 100〃 Lの PB Sで置換した後、イン セル アナライザ —1000 (I N C e l l An a l y z e r 1000 ; GE アマシャム社 製) を用いて蛍光画像を取り込み、 各視野における M期細胞の割合 (Mi t o t i c i n d e x) を解析した。 EC 50は、 各薬剤が誘導することのできる M期停止細胞 の割合の最高値を 100 %とした時に、その 50 %を誘導することのできる薬剤濃度 として定義した。
上記の方法で求められた E C 5。値を以下の表 2に示した。
【表 2】 ,
本発明の化合物は、 強い細胞増殖抑制作用を示すことから、抗腫瘍剤として極めて 有用であると考えられる。 以上より、 本発明に係る化合物は、 優れた PLK1阻害活性を有するとともに、 さ らに、 強い細胞増殖抑制作用を示しているので、 がん細胞の増殖を強く阻害する抗が ん剤として有用であると考えられる。 即ち、本発明に係る新規置換イミダゾール誘導 体又はその医薬上許容される塩若しくはエステルを含む医薬組成物、 或いは、本発明 に係る新規置換ィミダゾール誘導体又はその医薬上許容される塩若しくはエステル を含む杭がん剤は、 がん患者の治療において有効と考えられる。 また、 該医薬組成物 及び該抗がん剤は、薬学的に許容できる担体又は希釈剤を含んでいてもよい。ここで、 「薬学的に許容できる担体又は希釈剤」 は、 賦形剤 〔例えば、 脂肪、 蜜蟎、 半固体及 び液体のポリオール、天然若しくは硬化オイルなど〕; 水 (例えば、蒸留水、特に、 注射用蒸留水など)、 生理学的食塩水、 アルコール (例えば、 エタノール)、 グリセ口 ール、 ポリオール、 プドウ糖水溶液、 マンニトール、 植物オイルなど; 添加剤 〔例 えば、増量剤、崩壌剤、結合剤、潤滑剤、湿潤剤、安定剤、乳化剤、分散剤、保存剤、 甘味料、 着色剤、 調味料若しくは芳香剤、 濃化剤、 希釈剤、 緩衝物質、 溶媒若しくは
可溶化剤、 貯蔵効果を達成するための薬剤、 浸透圧を変えるための塩、 コーティング 剤、 又は抗酸化剤〕 などを意味する。
さらに、本発明に係るイビ合物は、 エステルを含むプロドラッグとして使用すること もできる。 ここで、 「プロドラッグ」 とは、 一般に、 ある薬物分子を化学的に修飾し た誘導体で、 それ自体は生理活性を示さず、 投与後体内で、 元の薬物分子に復元し薬 効を示すものをいう。 本発'明に係る化合物のプロドラッグの例として、 例えば、 その ヒドロキシル基がリン酸基などでァシル化された上記式( I )の化合物が挙げられる。 プロドラッグ 'エステルの製造は、 当業者に周知ないし慣用の方法に従ってすること ができる。
また、本発明に係る化合物の治療効果が期待される好適な月重瘍としては、例えばヒ トの固形がん等が挙げられる。 ヒ トの固形がんとしては、 例えば、 脳がん、 頭頸部が ん、 食道がん、 甲状腺がん、 小細胞がん、 非小細胞がん、 乳がん、 胃がん、 胆のう · 胆管がん、 肝がん、 膝がん、 結腸がん、 直腸がん、 卵巣がん、 絨毛上皮がん、 子宮体 がん、 子宮頸がん、 腎盂 ·尿管がん、 膀胱がん、 前立腺がん、 陰茎がん、 睾丸がん、 胎児性がん、 ウィルムスがん、 皮膚がん、 悪性黒色腫、 神経芽細胞腫、 骨肉腫、 ユー イング腫、 軟部肉腫などが挙げられる。
次に、 上述した 「その医薬上許容される塩もしくはエステル」 について説明する。 本発明に係る化合物は、 抗がん剤などとして使用される場合には、 その薬学的に許 容しうる塩としても使用することができる。薬学的に許容しうる塩の典型例としては、 例えばナトリウム、 カリゥム等のアルカリ金属との塩、 塩酸塩、 硫酸塩、 硝酸塩、 リ ン酸塩、 炭酸塩、 炭酸水素塩、 過塩素酸塩等の無機酸塩;例えば酢酸塩、 プロピオン 酸塩、 乳酸塩、 マレイン酸塩、 フマール酸塩、 酒石酸塩、 リンゴ酸塩、 クェン酸塩、 ァスコルビン酸塩等の有機酸塩;例えばメタンスルホン酸塩、 イセチオン酸塩、 ベン ゼンスルホン酸塩、 トルエンスルホン酸塩等のスルホン酸塩;例えばァスパラギン酸 塩、 グルタミン酸塩等の酸性アミノ酸塩等を挙げることができる。
本発明に係る化合物の薬学的に許容しうる塩の製造法は、有機合成化学分野で通常 用いられる方法を適宜組み合わせて行うことができる。 具体的には、本発明に係る化 合物の遊離型の溶液をアル力リ溶液あるいは酸性溶液で中和滴定すること等が挙げ られる。
本発明に係る化合物のエステルとしては、 例えば、 メチルエステル、 ェチルエステ ルなどを挙げることができる。 これらのエステルは遊離カルボキシ基を常法に従って エステル化して製造することができる。
本発明に係る化合物を抗がん剤などとして使用する際の投与形態としては各種の 形態を選択でき、 例えば錠剤、 カプセル剤、 散剤、 顆粒剤、 液剤等の経口剤、 例えば 溶液、 懸濁液等の殺菌した液状の非経口剤等が挙げられる。
ここで、 固体の製剤は、 常法に従い、 そのまま錠剤、 カプセル剤、 顆粒剤又は粉末
の形態として製造することもできるが、適当な添加物を使用して製造することもでき る。 該添加物としては、 例えば乳糖、 ブドウ糖等の糖類; 例えばトゥモロコシ、 小 麦、 米等の澱粉類; 例えばステアリン酸等の脂肪酸; 例えばメタケイ酸ナトリウ ム、 アルミン酸マグネシゥム、 無水リン酸カルシゥム等の無機塩; 例えばポリビニ ルピロリ ドン、 ポリアルキレンダリコール等の合成高分子; 例えばステアリン酸カ ルシゥム、ステアリン酸マグネシウム等の脂肪酸塩; 例えばステアリルアルコール、 ベンジルアルコール等のアルコール類; 例えばメチルセルロース、 カルボキシメチ ノレセルロース、 ェチルセルロース、 ヒ ドロキシプロピノレメチノレセノレロース等の合成セ ルロース誘導体; その他、 水、 ゼラチン、 タルク、 植物油、 アラビアゴム等通常用 いられる添加物等が挙げられる。
これらの錠剤、 カプセル剤、 顆粒剤、 粉末等の固形製剤は、 一般的には 0 . 1〜 1 0 0重量%、 好ましくは 5〜 1 0 0重量%、 さらに好ましくは 5〜 8 5重量%、 特に 好ましくは 5〜 3 0重量%の有効成分を含むことができる。
また、 液状製剤は、 水、 アルコール類又は例えば大豆油、 ピーナツ油、 ゴマ油等の 植物由来の油等液状製剤において通常用いられる適当な添加物を使用し、懸濁液、 シ 口ップ剤、 注射剤等の形態とレて製造することができる。
特に、 非経口的に筋肉内注射、 静脈内注射、 皮下注射で投与する場合の適当な溶剤 又は希釈剤としては、例えば注射用蒸留水、塩酸リ ドカイン水溶液(筋肉内注射用)、 生理食塩水、 ブドウ糖水溶液、 エタノール、 静脈内注射用液体 (例えばクェン酸、 ク ェン酸ナトリウム等の水溶液)、 電解質溶液 (例えば点滴静注、 静脈内注射用) 等又 はこれらの混合溶液が挙げられる。
また、 これらの注射剤は予め溶解したものの他、 粉末のまま又は適当な添加物を加 えたものを要時溶解する形態もとることができる。 これらの注射液は、 通常 0 . 1〜 1 0重量%、 好ましくは 1〜 5重量%の有効成分を含むことができる。
また、 経口投与の懸濁剤又はシロップ剤等の液剤は、 0 . 5〜 1 0重量%、 好まし くは 1〜5重量%の有効成分を含むことができる。 .
本発明に係る化合物の実際に好ましい投与量は、使用される化合物の種類、配合さ れた組成物の種類、適用頻度及び治療すべき特定部位及び患者の病状によつて適宜増 減することができる。例えば、一日当りの成人一人当りの投与量は、経口投与の場合、 1 0ないし 5 0 O m g、 好ましくは 1 0ないし 2 0 O m g、 非経口投与、 好ましくは 静脈内注射の場合、 1日当り 1 0ないし 1 0 0 m g、好ましくは 1 0ないし 3 0 m g である。 なお、 投与回数は、 投与方法及ぴ症状により異なるが、 単回、 又は 2ないし 5回、 好ましくは 2ないし 3回に分けて投与することができる。
また、本発明に係る治療上有効量の上記一般式 [ I ]で示される化合物又はその薬学 的に許容し得る塩若しくはエステルを含む製剤は、 抗がん性アルキルィヒ剤、 抗がん性 代謝拮抗剤、 抗がん性抗生物質、 植物由来抗がん剤、 抗がん性白金配位化合物、 抗が
ん性カンプトテシン誘導体、抗がん性チロシンキナーゼ阻害剤、モノクローナル抗体、 インターフェロン、 生物学的応答調節剤、 及ぴその他抗がん剤からなる群から選択さ れる治療上有効量の抗がん剤又はその薬学的に許容し得る塩若しくはエステルと組 み合わせて、 同時に、 別々に、 又は順次に投与することができる。 ここで、 「製剤」 という用語は、 経口製剤及ぴ非経口製剤を含む。 経口製剤としては、 例えば、 錠剤、 カプセル剤、 散剤、 顆粒剤などであり、 一方、 非経口製剤としては、 例えば、 溶液若 しくは懸濁液等の殺菌した液状の製剤、 具体的には、 注射剤、 点滴剤などである。 上記 「抗がん性アルキル化剤」 は、 抗がん活性を有するアルキル化剤を意味し、 こ こで、 「アルキル化剤」 とは、 一般に、 有機化合物の水素原子をアルキル基で置換す るアルキル化反応において、 アルキル基を与えるものをいう。 「抗がん性アルキル化 剤」は、例えば、ナイトロジェン マスタード N—ォキシド、シクロホスフアミ ド、 ィホスフアサミ ド、 メノレファラン、 ブスノレファン、 ミ トプロニトー/レ、 力/レポコン、 チォテパ、 ラニムスチン、 二ムスチン、 テモゾロミ ド又はカルムスチンなどである。 上記 「抗がん性代謝拮抗物質」 は、 抗がん活性を有する代謝拮抗物質をいい、 ここ で、 「代鶴ォ拮抗物質」 とは、 広義には、 生体にとって重要な代 f物 (ビタミン、 補酵 素、 アミノ酸、 糖類など) と樗造上又は機能上類似しているために、 正常な物質代謝 を行わなくさせる物質や、電子伝達系を阻害することによって高エネルギー中間体を つくれなくさせる物質を包含する。 「抗がん性代謝拮抗物質」 は、 例えば、 メ トトレ キサート、 6—メルカプトプリンリボシド、 メルカプトプリン、 5—フルォロウラシ ル、 テガフール、 ドキシフルリジン、 カルモフール、 シタラビン、 シタラビンォクホ スフアート、 エノシタビン、 S— 1、 ゲムシタビン、 フルダラビン又はぺメ トレタス ド ジソディウムなどであり、 好ましくは、 5—フルォロウラシル、 S—l、 ゲムシ タビンなどである。
上記 「抗がん性抗生物質」 は、 抗がん活性を有する抗生物質をいい、 ここで、 「抗 生物質」 とは、 微生物によってつくられ、微生物その他の生物細胞の発育そ 他の機 能を阻害する物質を包含する。 「抗がん性抗生物質」 は、 例えば、 ァクチノマイシン D、 ドキソルビシン、 ダウノルビシン、 ネオカルチノスタチン、 ブレオマイシン、 ぺ プロマイシン、 マイ トマイシン C、 アクラノレビシン、 ピラノレビシン、 ェピノレビシン、 ジノスタチンスチマラマー、イダルビシン、シロリムス又はバルルビシンなどである。 上記 「植物由来抗がん剤」 は、植物を起源として見いだされた抗がん活性を有する 化合物であるか、 或いは、 その化合物を化学修飾を加えた化合物を包含する。 「植物 由来抗がん剤」 は、 例えば、 ビンクリスチン、 ビンブラスチン、 ビンデシン、 エトポ シド、 ソプゾキサン、 ドセタキセル、 パクリタキセル、 ビノレルビンなどであり、 好 ましくは、 ドセタキセル及びパクリタキセルである。
上記 「抗がん性カンプトテシン誘導体」 は、 カンプトテシン自身を含み、 構造的に カンプトテシンに関連するがん細胞増殖阻害性化合物を意味する。 「抗がん性カンプ
トテシン誘導体」 としては、 特に限定されないが、 カンプトテシン、 1 0—ヒ ドロキ シカンプトテシン、 トポテカン、 イリノテカン、 9—ァミノカンプトテシンなどが挙 げられ、好ましくは、カンプトテシン、 トポテカン、及ぴィリノテカンである。 なお、 イリノテカンは、 生体内で代謝されて S N— 3 8として抗がん作用を示す。 カンプト テシン誘導体は、 作用機構おょぴ活性はほぼカンプトテシンと同様と考えられる (新 田 他、 癌と化学療法、 1 '4, 8 5 0 - 8 5 7 ( 1 9 8 7 ) など)。
上記 「抗がん性白金配位化合物」 は、 抗がん活性を有する白金配位化合物をいい、 ここで、 「白金配位化合物」 は、 イオンの形態で白金を提供する白金配位化合物を意 味する。 好ましい白金化合物としては、 シスプラチン; シス一ジアンミンジアコ白金 (II) 一イオン; クロロ (ジエチレントリアミン) 一白金 (II) クロリ ド;ジクロロ (エチレンジァミン) —白金 (II) ; ジアンミン ( 1 , 1ーシクロブタンジカルボキ シラト) 白金 (II) (カルポプラチン) ;スピロブラチン;ィプロブラチン;ジアンミ ン ( 2—ェチルマロナト) —白金 (II) ;エチレンジァミンマロナト白金 (II) ;ァク 了 ( 1 , 2—ジアミノジシクロへキサン) スルファ ト白金 (II) ; アクア ( 1 , 2— ジアミノジシクロへキサン) マロナト白金 (II) ; ( 1, 2—ジアミノシクロへキサン) マロナト白金 (II) ; ( 4一力ノ ポキシフタラト) ( 1, 2—ジアミノシクロへキサン) 白金(II) ; ( 1 , 2—ジアミノシクロへキサン)一(イソシトラト) 白金(II) ; ( 1, 2—ジアミノシクロへキサン) 才キサラト白金 (II) ;オルマブラチン;テトラプラ チン;カルポプラチン;ネダプラチン及ぴォキザリブラチンであり、 好ましくは、 力 ルポプラチン又はォキザリブラチンである。 また、 本明細書で挙げた他の抗がん性白 金配位化合物は、 公知であり、 商業的に入手可能であり、 及び/又は、 慣用技術によ つて当業者が製造することができる。
上記の 「抗がん性チロシンキナーゼ阻害剤」 とは、 抗がん活性を有するチロシンキ ナーゼ阻害剤をいい、 ここで、 「チロシンキナーゼ阻害剤」 とは、 AT Pの τ /—リン 酸基をタンパク質の特定のチロシンのヒドロキシル基に転移する 「チロシンキ^ "一 ゼ J を阻害する化学物質をいう。 「抗がん性チロシンキナーゼ阻害剤」 としては、 ゲ フイチ二プ、 イマチニブ、 エル口チニブなどが挙げられる。
上記 「モノクローナル抗体」 は、 単クローン性抗体ともいわれ、 単一クローンの抗 体産生細胞が産生する抗体をいい、 例えば、 セツキシマプ、 ベパシズマブ、 リツキシ マブ、 ァレムッズマブ、 トラスッズマブなどが挙げられる。
上記 「インターフェロン」 とは、 抗がん活性を有するインターフェロンをいい、 一 般に、 ウィルス感染に際して、 ほとんどすべての動物細胞が生産 ·分泌する分子量約 2万の糠タンパク質であり、 ウィルス増殖抑制のみならず、 細胞 (特に腫瘍細胞) の 増殖抑制や、ナチュラルキラ一活性の増強をはじめ多様な免疫ェフエクタ一作用があ り、 サイトカインの 1種と位置づけられる。 「インタ一フエロン」 としては、例えば、 インターフェロンお、 インターフェロンひー 2 a、 インターフェロン a _ 2 b、 イン
ターフェロン 、 インターフェロン γ— 1 a、 インターフェロン γ— n 1などが挙げ られる。 '
上記 「生物学的応答調節剤」 とは、 いわゆるバイオロジカル ·レスポンス 'モディ フアイヤー (b i o l o g i c a l r e s p o n s e mo d i f i e r ; BR M) であり、 一般に、 '生体のもつ防御機構や組織細胞の生存、 増殖、 または分化など 生物学的反応を調節するこ'とによって、 腫瘍や感染あるいはその他の疾病に対して、 個体に利する方向にもっていくことを目的とする物質や薬剤の総称をいう。 「生物学 的応答調節剤」 としては、 例えば、 クレスチン、 レンチナン、 シゾフィラン、 ピシパ ニール、 ウベ二メクスなどが挙げられる。
上記 「その他抗がん剤」 とは、 抗がん活性を有する上記のいずれにも属しない抗が ん剤をいう。 「その他抗がん剤」 としては、 ミトキサントロン、 Lーァスパラギナー ゼ、 プロカルバジン、 ダカノレバジン、 ヒ ドロキシカノレパミ ド、 ペントスタチン、 トレ チノイン、 ァレファセプト、 ダルべポェチン ァノレファ、 アナスト口ゾーノレ、 ェキセ ムスタン、 ビカルタミ ド、 リュープロレリン、 フルタミ ド、 フルべストラント、 ぺガ プタニブ ォクタソディウム、 デニリューキン ジフティ トクス、 アルデスリューキ ン、 チロトロピン アルファ、,ァルセニック トリオキシド、 ボルテゾミブ、 カぺシ タビン、 ゴセレリン、 などが挙げられる。
上記「杭がん性アルキル化剤」、「杭がん性代謝拮抗物質」、 「抗がん性抗生物質」、 「植物由来抗がん剤」、「抗がん性白金配位化合物」、「抗がん性カンプトテシン誘導体」、 「抗がん性チロシンキナーゼ阻害剤」、「モノクローナル抗体」、「インターフェロン」、 「生物学的応答調節剤」、 及び 「その他杭がん剤」 は、 いずれも公知であり、 商業的 に入手可能であり、 或いは、 それ自体公知の方法ないし周知 '慣 的な方法によつ.て 当業者が製造することができる。 発明を実施するための最良の形態
実施例
以下に実施例を挙げて本発明を更に具体的に説明するが、 もとより本発明はこれら の実施例のみに限定されるものではない。 実施例において、 薄層クロマトグラフィー は、 プレートとして S i 1 i c a g e 16。F 254 (Me r c k) を、 検出法として UV検出器を用いた。 カラム用シリカゲルとしては、 Wa k o g e 1 TMC— 300又 は C— 200 (和光純薬)又は NH (FU J I S I LYS I A CHEMI CAL) を用いた。 逆相分取液体クロマトグラフィーは、 C omb i P r e p P r o C I 8 (YMC) をカラムに用い、 0. 1 %トリフルォロ酢酸水溶液、 0. 1 %トリフル ォロ酢酸ァセトニトリル溶液を移動相に用いた。 MSスペク トルは、 JMS— SX1 02 A (日本電子 (J EOL))、 QUATTRO I I (マイクロマス)、 又は LC一 MSは ZMD (マイクロマス) を用いて測定した。 NMRスペクトルは、 重ジメチル
スルホキシド溶液で測定する場合には内部基準としてジメチルスルホキシドを用い、
Gem i n i— 200 (200 MH z ; Va r i a n)、 Gemi n i— 300 (3
00 MH z; V a r i a n)、Me r c u r y 400 (400 MH z ; V a r i a n )、
1 n o v a 400 (40 OMH z ; V a r i a n)、 または J NM— AL 400 (4 00 MHz ; J EOL) 型スぺクトロメータを用いて測定し、 全 δ値を p pmで示し た。 ' 実施例で用いた略号の意味を以下に示す。
s : シングレツト
d :ダブレツト
d d :ダブノレ ダブレッ ト
d d d :ダブノレ ダブノレ ダブレッ ト
t : トリプレツト
d t :ダブノレ トリプレツト
q : クァノレテット
d q :ダブノレ クァノレテッ ト ,
m マノレチプレツ ト
b r :ブロード
b r s :プロ一ド シングレツト
b r d : ブロード ダブレツト
J :カツプリング定数
H z : ヘルツ
DMS 0— d 6:重ジメチルスルホキシド
CDC 13 :重クロ口ホンレム
CD3OD :重メタノ一ル
RT: リテンションタイム (保持時間)
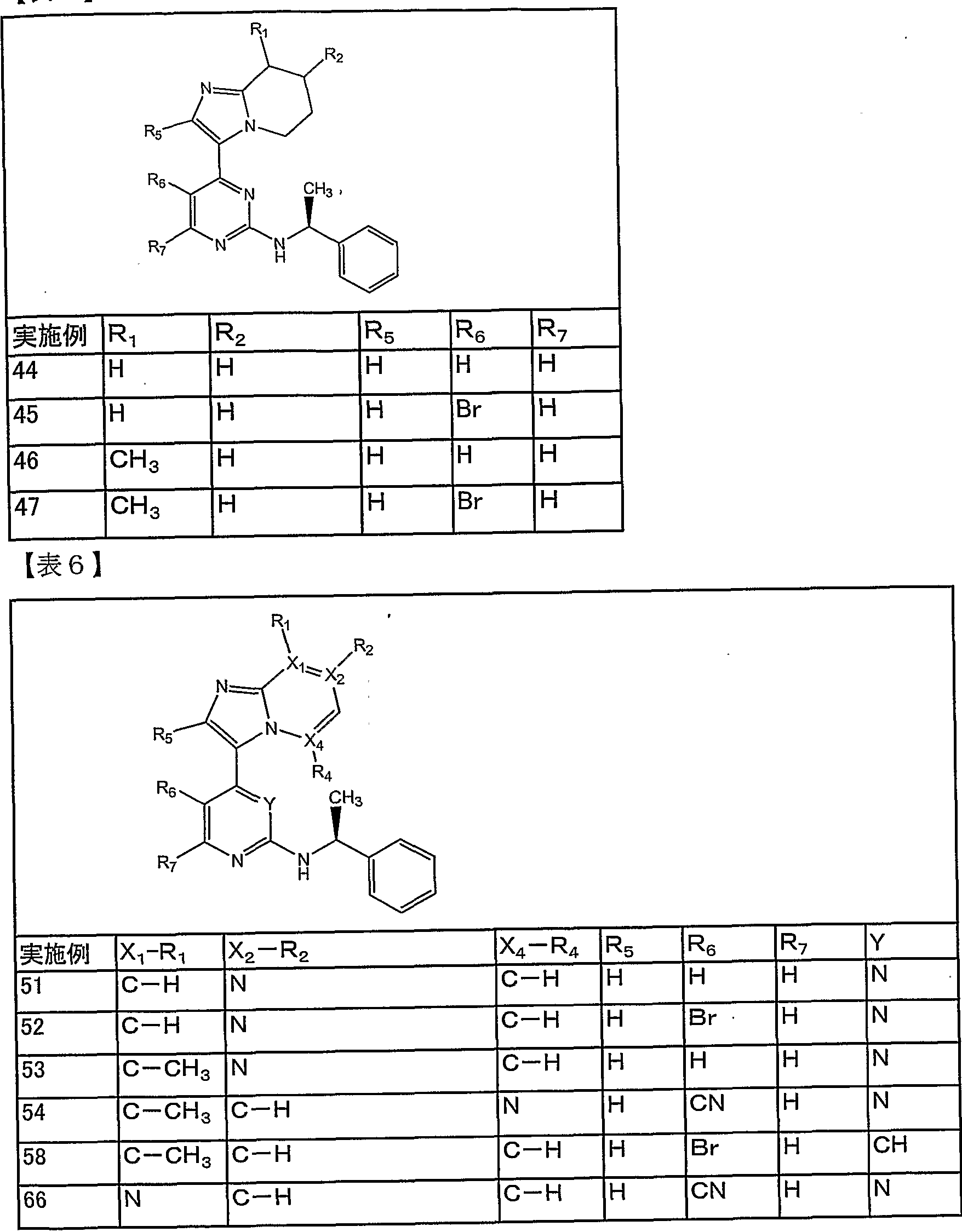
【表 4】
【表 8】
【6挲】 s
.8T9lO/SOOZdf/X3d .9SSZ0/900Z OAV
【表 1 0】
実施例 Rx *(不斉灰 )
100 CHF2 H ラセミ体 人
ヽ CH3
102 CHF2 CH2NHCOCH2NH2 H ラセミ体
103 CHF2 NHCOCH(CH3)NH2 H S体
104 CHF2 H s体
N
ヽ H
105 CHF2 H ^\ H(CH3)2 s体
ヽ H
106 CHF2 H s体
ヽ H义
107 CHF2 H S体
ヽ
ノ1^
CH3
108 CHF2 H NH(CH2)2N(CH3)2 S体
110 CHF2 H S体
NH
【表 1 2】
実施例 Ri R6 Rx *(不斉炭素)
111 CHF2 CN H ラセミ体
、。
112 CH3 CN H NH2 S体
1 13 CH3 CN H NHS02CH3 S体
114 CH3 CN H S体
ヽ H
115 CH3 CN H ラセミ体
116 CH3 CN H CH2NHCOCH2NH2 ラセミ体
117 CH3 CN H CHgSO CH3 ラセミ体
1 18 CH3 CN OCH3 H S体
119 CH3 CN H S体
^
、
120 CN H ヽ N S体
CH3
121 CN H S体
- ヽ
CH3
【表 13】
実施例 Ri R2 R3 R6 Z Ry *(不斉炭素)
133 CI H H CN C H ヽ s体
ノ ヽ CH3
135 CI H H CN C H N (CH3) (CH2)2N (CH3) 2 s体
136 CI H H CN C H s体
~ Ν: > ~ ΝΗ2
137 CI H H CN C Ή 、卜'^ \,,、、、ΝΗ2 s体
138 CI H H CN C H s体
CH3
139 CI H H CN C CI ヽ s体
ノ1 ゝ CH3
140 CI H H CN N ラセミ体
ΝΗ
141 CI H CI CN C Η s体 、
142 CI CI H CN C Η s体 、
143 F H H CN c H s体
ΝΗ
144 Br H H CN c Η s体
ノ 、 '
145 Br H H CN c H s体
ヽ Ο
【表 1 5
実施例 R2 R3 R6 Ry *(不斉 灰素)
146 Br H CH3 CN H s体 し
147 CH2F H H CN H s体 し、
148 CH2F H H CN H ヽ N ' s体 ゝ CH3
149 CH2F H H CN H s体 ヽ NH
151 CH2F H H CN H s体
152 CH2F H H CN H CH2CH2OH s体
153 CH2F H H CN H CH'2CH2N (CH3)2 s体
154 CH2F H H CN H s体
155 CH2F H H CN H s体
【表 16】
4—イミダゾ [1, 2— a] ピリジン一 3—ィルー N— [(I S) —フエニルェチル] 一 2—ピリミジンアミン [1] (以下、 化合物 [1] という) の合成。
(1) イミダゾ [1, 2— a]ピリジン 25 gをクロ口ホルム 35 OmLに溶解し、 氷冷下、 30分かけて塩ィヒアルミニウム 56. 4 gを加えた。 室温で終夜撹拌した後 に、 50°Cに加熱し、 無水酢酸 16 m Lを加え、 50 °Cにて 90分間撹拌した。 室温 に戻した後、氷水を加え、 さらに塩基性になるまで 3 N—水酸ィ匕ナトリウム水溶液を
カロえた。 不溶物をセライトにてろ別した後、 ろ液を酢酸ェチルで抽出し、 有機層を飽 和食塩水にて洗浄した。 無水硫酸ナトリウムにて乾燥後、 ろ過し、 ろ液を濃縮し、 1 一イミダゾ [ 1, 2— a] ピリジン一 3—ィルエタノン [ 1一 1] を褐色油状物とし て得た。 得られた化合物 [1— 1] は更に精製することなく、 次の反応に用いた。
(2) 未精製のイミダゾ' [ 1 , 2— a] ピリジン一 3—ィルエタノン [1ー1] を N, N—ジメチルホルムアミ ドジメチルァセタール 4 0 OmLに溶解し、 終夜過熱還 流した。 減圧下、 ジメチルホルムアミ ドジメチルァセタールを留去した後に、 残渣に ジェチルエーテルを加え、 生じた固体をろ別、 減圧乾燥し、 (E) —3— (ジメチル ァミノ) 一 1一^ (ミダゾ [ 1 , 2— a] ピリジン一 3—イノ I ^一 2—プロペン一 1—ォ ン [1— 2] 5. 0 6 gを淡褐色固体として得た。
(3) (E) —3— (ジメチルァミノ) 一1一イミダゾ [ 1 , 2— a] ピリジン一 3—イノ!^ ^一 2—プロペン一 1一オン [ 1— 2] 1 gを n—プタノ一ノレ 1 OmLに溶解 し、 チォ尿素 3 54mg、 ナトリウムメ トキシド 3 7 7m gを加え、 8 5 °Cにて 2時 間撹拌した。 室温に戻した後 、 ョウイ匕メチル 8 6 8 μ Lを加え、 室温にて 3 0分間 撹拌した。 溶媒を半分の体積になるまで留去し、 生じた固体をろ取、 ジェチルエーテ ル、 水の順で洗浄し、 減圧乾燥し、 3— [2—メチルチオ一 4一ピリミジニル] イミ ダゾ [ 1 , 2— a] ピリジン [1— 3] 7 2 8mgを灰色固体として得た。
(4) 3— [2—メチルチオ一 4一ピリミジニル] イミダゾ [1 , 2— a] ピリジ ン [1—3] 1 5 Omgをクロロホノレム 3 m Lに溶解し、 氷冷下、 m—クロ口過安息 香酸 1 6 9 m gを加え、 同温にて 3 0分間撹拌した。 飽和亜硫酸ナトリゥム水溶液、 飽和炭酸水素ナトリウム水溶液を加えた後に、 クロ口ホルムにて抽出した。 有機層を 無水硫酸ナトリゥムにて乾燥した後に、 不溶物をろ過し、 ろ液を濃縮した。 得られた 残渣をシリ^ゲルカラムクロマトグラフィーにて精製し、 .3— [2—メチルスルホニ ルー 4一ピリミジニル] イミダゾ [ 1, 2— a] ピリジン [1—4] 1 6 Omgを無 色アモルファス状物質として得た。 (5) 3— [2—メチルスルホニ Λ 4—ピリミジニル] イミダゾ [1, 2— a] ピリジン [1一 4] 7 3mgをジメチルスルホキシド l mLに溶解し、 (1 S) — 1 —フエ-ルェチルァミン 1 0 9 L、 炭酸カリゥム 1 9 6m gを加え、 8 0°Cにて 1 0 0分間撹拌した。 室温に戻した後、 反応混合物をろ過し、 逆相分取液体クロマトグ ラフィ一にて精製し、 目的化合物 [1] 1 1. l mgを淡黄色油状物として得た。 化合物 [ 1 ] のスぺク トルデータを以下に示す。
XH— NMR (CDC 1 3) 5 : 9. 24 (b r , 1 Η), 8. 2 6 (d, J = 5. 4
Η)' 8. 1 9 ( s , 1 H), 7. 6 5 (d 8 H z , 1 H), 7
4 8— 7. 24 (m, 6H), 6. 9 2 (d, J = 5. 4H z , 1 H), 6. 7 8— 6 6 6 (m, 1 H), 5. 6 8— 5. 5 2 (m, 1 H), 5. 2 3— 5. 0 8 (m, 1 H) 1. 6 3 (d, J = 6. 9H z , 3H).
ma s s : 3 1 6 (M+ 1 ) +. 実施例 2
5—プロモー 4一イミダゾ [1 , 2— a] ピリジン一 3—ィル一 N— [(1 S)一 1 一フエニルェチル]一 2—ピリミジンアミン [2] (以下、 化合物 [2] とレヽう) の 合成。 化合物 [1] 9. l mgを酢酸 0. 5mLに溶解し、 臭素 1. 8 /z Lをカロえた。 混 合物を室温にて 3 0分間撹拌した後に、 減圧下、 酢酸を留去した。 残渣を酢酸ェチル にて希釈し、飽和亜硫酸ナトリゥム水溶液、飽和炭酸水素ナトリゥム水溶液を加えた。 混合物を酢酸ェチルにて抽出し、 有機層を無水硫酸ナトリゥムにて乾燥した後、 不溶 物をろ別し、 溶媒を留去した。,得られた残渣を、 分取薄相クロマトグラフィーにて精 製し、 目的化合物 [2] 8. 5 mgを淡黄色油状物として得た。
化合物 [2] のスペクトルデータを以下に示す。
一 NMR (CDC 1 3) δ : 8. 74 ( s , 1 H), 8. 4 5 ( s, 1 H), 7. 6 7 (d, J = 1 2. 0H z , 1 H), 7. 4 1— 7. 2 6 (m, 7 H) , 6. 6 8— 6. 5 0 (m, 1 H), 5. 6 8— 5. 5 3 (m, 1 H), 5. 1 5— 5. 0 0 (m, 1 H), 1. 5 9 (d, J = 6. 9H z, 3 H).
ma s s : 3 94, 3 9 6 (M+ 1 ) +. 実施例 3
4一 (8—メチルイミダゾ [ 1 , 2— a] ピリジン一 3—ィル) 一 N— [( 1 S) 1一フエニルェチル]一 2—ピリミジンアミン [3] (以下、 化合物 [3] という) の合成。 ( 1) 2—アミノー 3—ピコリン 1 8. 2 7 gを水 1 5 OmLに溶解し、 炭酸水素 ナトリウム 1 4. 2 g、クロロアセトァノレデヒド(4 0%水溶液) 3 3. 2 gを加え、 室温にて 4時間撹拌した。 3 N—水酸化ナトリウム水溶液にて、液性を p H 1 0に調 整し、 混合物を酢酸ェチルにて抽出した。 有機層を飽和食塩水で洗浄した後に、 無水 硫酸ナトリウムにて乾燥した。 不溶物をろ別した後に、 ろ液を濃縮し、 8—メチルイ ミダゾ [1, 2— a] ピリジン [3— 1] を褐色油状物として得た。
(2) 8—メチルイミダゾ [1, 2— a] ピリジン [3— 1]から、実施例 (1) 〜 (5) の方法に準じて目的化合物 [3] を淡黄色油状物として得た。
化合物 [3] のスぺク トルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 13 (b r, 1H), 8. 24 (d, J = 5. 4 Hz, 1 H), 8. 17 ( s , 1 H), 7. 47— 7 26 (m, 5H), 7. 08 (d, J = 6. 9Hz, 1H), 6. 93 (d, J = 5. 4Hz, 1 H), 6. 72— 6. 6 0 (m, 1H), 5. 70— 5. 58 (m, 1H), 5. 22— 5. 10 (m, 1 H),
2. 62 (s, 3H), 1. 61 9H z, 3H)
ma s s : 330 (M+ 1) +. 実施例 4
5—ブロモ一4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) — N— [(1 S)一 1—フエニルェチル]一 2—ピリ ミジンアミン [4] (以下、 化合物 [4] という) の合成。 化合物 [3] 4. 5mg力 、 実施例 2の方法に準じて目的化合物 [4] 4. 0 m gを淡黄色油状物として得た。
化合物 [4] のスぺク トルデータを以下に示す。,
'H— NMR (CDC 13) δ : 8. 71 (s, 1 H), 8. 44 (s, 1 H), 7. 4 0— 7. 04 (m, 7H), 6. 58— 6. 45 (m, 1 H), 5. 63— 5. 51 (m, 1 H), 5. 14— 5. 00 (m, 1 H), 2. 63 (s, 3H), 1. 58 (d, J =6. 9H z, 3H).
ma s s : 408, 410 (M+ 1) +. , 実施例 5
メチル 4一(8—メチルイミダゾ [1, 2— a] ピリジ 一 3—ィノレ)一 2— {[(1 S) 一 1—フエニルェチル] アミノ}一 5—ピリミジンカルポキシレート [5] (以 下、 化合物 [5] という) の合成。 化合物 [4] 200mg、 酢酸パラジウム (Π) l lmg、 1, 1, 一ビスジフエ ニルホスフィノフエロセン 27. 2mg、 炭酸水素ナトリウム 123mg、 メタノー ノレ 2mL、 N, N—ジメチルホルムアミ ド 2 mLの混合物を一酸化炭素雰囲気下 (3 気圧)、 80°Cにて終夜撹拌した後、 反応混合物を水で希釈し、 酢酸ェチルにて抽出 した。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウムで乾燥した。 不溶物をろ 別し、 ろ液を減圧濃縮した後、 得られた残渣をシリ力ゲル力ラムクロマトグラフィー にて精製し、 目的化合物 [5] 186m gを淡黄色固体として得た。
化合物 [5] のスぺク トルデータを以下に示す。
NMR (CDC 1 3) δ 8. 80 (s , 1H), 8. 25 (b r, 1 H), 7. 98 (s, 1 H), 7. 43— 7. 0 2 (m, 6H), 6. 5 8— 6. 40 (m, 1H), 5. 9 3— 5. 7 8 (m, 1 H), 5. 1 8— 5. 0 5 (m, 1H), 3. 79 ( s, 3H), 2. 6 2 ( s , 3H), 1. 58 (d, J = 6. 9Hz, 3H).
ma s s : 388 (M+ 1) +. 実施例 6
4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— {[(1 S) ― 1一フエニルェチル]アミノ}一 5—ピリミジンカルボン酸 [6] (以下、化合物 [6] という) の合成。 化合物 [5] 1 0mgをメタノール 3 m Lに溶解し、 1 N—水酸ィ匕ナトリウム水溶 液を 5 1. 6 μ L力!]え、 室温にて 1時間撹拌した。 更に 1 Ν—水酸化ナトリゥム水溶 液 5 1. 6 μ Lを加え、 40 °Cにて 3時間 30分間撹拌した。 反応混合物を減圧濃縮 し、 逆相分取液体クロマトダラ.フィ一にて精製し、 目的化合物 [6] 1 0. Omgを 淡黄色固体として得た。
化合物 [6] のスぺク トルデータを以下に示す。
H— NMR (DMSO— dR) δ 9. 08— 8 73 (m, 2H+ 1 HX 1/2),
8. 5 5— 8. 34 (m, 1 Η + L ΗΧ 1/2) 7. 79— 7. 6 0 (m, 1H), 7. 48— 7. 0 3 (m, 6H), 5. 3 6— 5 20 (m, 1 H), 5. 1 0— 4. 9 7 (m, 1 H), 2. 6 2 (s, 3HX 1/2) 2 , 5 8 ( s, 3 HX 1/2), 1. 48 (t, J = 7. 5Hz, 3H).
ma s s : 3 74 (M+ 1 ) +. 実施例 Ί
4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— {[(1 S) — 1一フエニルェチル] アミノ} - -5—ピリミジンカルボキサミド [7] (以下、 化合 物 [7] という) の合成。 化合物 [5] 60. Omgをエタノール lmLに溶解し、 25%アンモニア水 0. 5mLをカロえ、 封管中 90°Cにて 7時間撹拌した。 室温に戻した後、 さらに 25%ァ ンモニァ水 lmLを加え、封管中 1 20°Cにて終夜撹拌した。 反応混合物を室温に戻 した後、 水を加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄した後、 無水硫酸ナトリウムで乾燥した。 不溶物をろ別後、 ろ液を減圧濃縮し、 得られた残渣 を分取薄層クロマトグラフィーにて精製し、 目的化合物 [7] 7. 9mgを白色固体
として得た。
化合物 [7] のスペク トルデータを以下に示す。
一 NMR (CDC 1 3) δ : 8. 6 2 ( s , 1 H), 8. 3 3 (b r, 1 H), 8. 1 4 ( s, 1 H), 7. 4 5— 7. 0 3 (m, 6 H), 6. 6 4— 6. 4 3 (m, 1 H), 5. 9 8— 5. 8 0 (m, 1 H), 5. 7 9 (b r , 2H), 5. 2 5— 5. 04 (m, 1 H), 2. 6 1 ( s, 3H), 1. 5 9 (d, J = 7. 2H z, 3H).
ma s s : 3 7 3 (M+ 1) +. 実施例 8
4一 (8—メチルイミダゾ [1 , 2— a] ピリジン一 3—ィル) 一 2— {[(1 S) ― 1一フエニルェチル] アミノ} 一 5—ピリ ミジンカルボ二トリル [8] (以下、 化合 物 [8] という) の合成。 化合物 [7] 6. Omgをピリジン l mLに溶解し、 ォキシ塩ィ匕リン 3. O ^u Lを 加え、 室温にて 3 0分間撹拌した。 減圧濃縮後、 残渣に飽和炭酸水素ナトリゥム水溶 液を加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナト リウムで乾燥した。 不溶物をろ別後、 ろ液を減圧濃縮し、 得られた残渣を分取薄層ク 口マトグラフィ一にて精製し、 目的化合物 [8] 4. Omgを白色固体として得た。 化合物 [8] のスぺク トルデータを以下に示す。
NMR (DMSO— d 6) δ : 9. 9 6 (d, J = 7. 5H z, 1 HX 1/2),
9. 1 0— 8. 84 (m, 1 H+ 1 HX 1/2), 8. 8 0 ( s , 1 HX 1/2), 8. 7 8 ( s , 1 HX 1/2), 8. 74 ( s , 1 HX 1/2), 8. 6 8 ( s , 1 HX 1 /2), 7. 5 8— 6. 9 5 (m, 7H), 5. 2 8— 5. 20 (m, 1 HX 1/2), 5. 1 7— 5. 0 2 (m, 1 HX 1 /2), 2. 6 0 ( s , 3HX 1/2), 2. 5 5 ( s, 3HX 1 /2), 1. 5 3 (d, J = 7. 5H z, 3HX 1/2), 1. 5 1 (d, J = 7. 5H z, 3HX 1/2).
ma s s : 3 5 5 (M+ 1 ) +. 実施例 9
(4一 (8—メチルイミダゾ [1 , 2— a] ピリジン一 3—ィル) 一 2— {[(1 S) 一 1—フエニルェチル] アミノ} 一 5—ピリミジニル') メタノール [9] (以下、 化 合物 [9] という) の合成。 化合物 [5] 5 8. 4mgをテトラヒドロフラン 1. 5 m Lに溶解し、 溶液を一 7 8 °Cに冷却した後、 水素化ジイソブチルアルミニウム (1. 0Mトルエン溶液) 4 5 2 Lを加え、 氷冷下、 1 B字間撹拌した。 水素化ジィソブチノレアノレミニゥム ( 1. 0
Mトルエン溶液) 452 Lをさらに加え、 同温で 30分間撹拌した。 その後、 反応 混合物に硫酸ナトリウム十水和物を加え、 終夜撹拌した。 不溶物をセライトにてろ過 し、 ろ液を減圧濃縮し、得られた残渣を分取薄層クロマトグラフィーにて精製し、 目 的化合物 [9] 34. lmgを白色固体として得た。
化合物 [9] のスペクトルデータを以下に示す。
XH— NMR (DMSO— d6) δ : 8. 40— 8. 28 (m, 2H), 7. 86 (d, J = 7. 7Hz, 1H), 7. 43— 7. 15 (m, 7H), 6. 67 (b r , 1 H), 5.. 26— 5. 23 (m, 1 H), 5. 15— 4. 96 (m, 1 H), 4. 45— 4. 30 (m, 2H), 2. 52 ( s , 3H), 1. 46 (d, J = 6. 9Hz, 3H). m a s s : 360 (M+ 1 ) +. 実施例 10
4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— {[(1 S) - 1一フエニルェチル] アミノ} 一 5—ピリミジンカルボアルデヒ ド [ 0] (以下、 化合物 [10] という) の合成。 化合物 [9] 31. 3mgをクロ口ホルム 1 OmLに加熱溶解し、 二酸化マンガン 18mgを加えた。 室温にて 5時間 30分間撹拌した後、 不溶物をセライトにてろ 過した。 ろ液を減圧濃縮し、 残渣を分取薄層クロマ 一にて精製し、 目的化 合物 [10] 20. 4 mgを白色固体として得た。
化合物 [10] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 10. 07 (s, 1 H) 9. 93 ( s , 1 H), 8. 27 (d, J = 7. 2Hz, 1 H), 7. 92 (s 1 H), 7 43— 7. 1 1 (m, 6H), 6. 54 (t, J = 7. 2H z, 1 H) , 6. 33— 6. 25 (m, 1H), 5. 21— 5. 10 (m, 1 H), 2. 64 (s, 3H), 1. 62 ( d , J = 6. 9 H z , 3H).
ma s s : 358 (M+ 1) +. 実施例 1 1
4— (8—メチルイミダゾ [1, 2_a] ピリジン一 3—ィル) 一 2— {[(1 S) — 1一フエニルェチル]アミノ}一 5—ピリミジンカルボアルデヒド ォキシム [1 1] (以下、 化合物 [1 1] という) の合成。 化合物 [10] 5. lmgをエタノールに溶解し、炭酸水素ナトリウム 2. 4m g、 塩酸ヒドロキシルァミン 1. 98m gを加え、 60°Cにて 30分間撹拌した。 反応混 合物を減圧濃縮し、 残渣を分取薄層クロマトグラフィーにて精製し、 目的化合物 [1
1] 5. 6mgを淡黄色油状物として得た。
化合物 [1 1] のスぺク トルデータを以下に示す。
XH— NMR (DMS O— d 6) δ : 1 1. 1 5 ( s , 1 Η), 8. 8 3 ( s , 1 Η), 8. 4 3— 8. 3 6 (m, 1 Η), 8. 3 1— 8. 2 2 (m, 1 Η), 8. 0 7 ( s, 1 Η), 7. 8 4— 7. 6 8 (m, 1 Η), 7. 4 2— 7. 1 8 (m, 6 Η), 6. 7 5— 6. 6 3 (m, 1 Η), '4. 5 5— 4. 4 6 (m, 1 Η), 2. 5 1 ( s, 3Η), 1. 4 7 (d, J = 6. 6H z , 3Η).
ma s s : 3 7 3 (Μ+ 1 ) +. 実施例 1 2
4一 (8—メチルイミダゾ [1 , 2— a] ピリジン一 3—ィル)一 2— {[( 1 S) ― 1一フエエルェチル] アミノ}—5—ピリミジンカルボアルデヒド O—メチルォキ シム [ 1 2] (以下、 化合物 [1 2] という) の合成。 化合物 [1 0] 5. l mgとヒドロキシルァミンメチルエーテル塩酸塩 2. 3 9m から、 実施例 1 1の方法に準じて目的化合物 [1 2] 6. 7mgを淡黄色油状物と して得た。
化合物 [1 2] のスぺクトルデータを以下に示す。
一 NMR (DMS O— d 6) δ : 8. 6 5 ( s , 1 Η), 8. 4 5— 8. 3 7 (m, 2Η), 8. 1 3 ( s, 1 H), 7. 8 5— 7. 7 0 (m, 1 H), 7. 5 3— 7. 1 8 (m, 6 H), 6. 7 5— 6. 64 (m, 1 H), 5. 0 8— 4. 9 0 (m, 1 H), 3. 8 7 ( s , 3H), 2. 54 ( s , 3 H), 1. 4 7 (d, J = 6. 3H z , 3H). ma s s : 3 8 7 (M+ 1 ) +. 実施例 1 3
5— (ジフルォロメチル) _4一 (8—メチルイミダゾ [.1, 2— a] ピリジン一 3 —ィル)— N— [(1 S) —1一フエニルェチル]—2—ピリミジンアミン [ 1 3] (以 下、 化合物 [ 1 3] という) の合成。 化合物 [ 1 0] 8. 8mgをジクロロメタン 1. 5mLに溶解し、 ジェチノレアミノ 硫黄トリフルオリ ド 9. をカロえ、 室温にて 3 0分間撹拌した。 さらにジェチル ァミノ硫黄トリフルオリ ド 1 9. 6 / Lを力!]え、 同温にて 2 0分間撹拌した。 メタノ ールを加えた後、 混合物を減圧濃縮し、残渣を分取薄層クロマトグラフィーにて精製 し、 目的化合物 [1 3] 1. 5mgを無色油状物として得た。
化合物 [ 1 3] のスぺク トルデータを以下に示す。
XH— NMR (DMS O— d 6) δ : 8. 6 4— 8. 2 9 (m, H), 8. 1 0— 7.
85 (m, 1 H), 7. 50— 6. 97 (m, 6 H), 6. 73— 6. 60 (m, 1 H), 5. 10— 4. 95 (m, 1H), 2. 54 ( s , 3H), 1. 47 ( d , J = 6. 6 Hz, 3H).
ma s s : 380 (M+ 1) +. 実施例 14 '
t一プチル 4一(8—メチルイミダゾ [1, 2— a]ピリジン一 3—ィル)一2— {[(1 S) 一 1一フエ-ルェチル] アミノ}一 5—ピリ ミジニルカルバメート [14] (以 下、 化合物 [14] という) の合成。 化合物 [6] 5 1. 8mgを、 N, N—ジメチルホルムアミ ド 3mLと 1, 4ージ ォキサン 2 m Lの混合溶媒に溶解し、混合溶液を 0 °Cに冷却した後、 トリェチルァミ ン 54. 3 μ L、 ジフエニルホスホリルアジド 38. 9 Lを力 Πえ、 室温にて 1時間 撹拌した。 t一ブチルアルコールを加え、 80°Cにて終夜撹拌した後、 水、 飽和炭酸 水素ナトリゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水で洗浄 し、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 残渣を 分取薄層クロマトグラフィーにて精製し、 目的ィ匕合物 [14] 2. 7mgを淡黄色油 状物として得た。
化合物 [14] のスぺクトルデータを以下に示す。
ΧΗ— NMR (CDC 13) δ : 8. 68 (b r , 1 Η), 8. 52 (b r , 1 Η), 8. 17 (s, 1Η), 7. 48— 7. 20 (m, 5H), 7. 08— 7. 06 (m, 1 H),
6. 57— 6. 45 (m, 1 H), 6. 10— 5. 98 (m, 1 H) , 5. 62— 5. 54 (m, 1 H), 5. 12— 5. 03 (m, 1 H), 2. 61 (s, 3H), 1. 5 7 (d, J = 6. 9Hz, 3H), 1. 49 (s, 9H).
ma s s : 445 (M+ 1 ) +. 実施例 15
4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 N2— [(1 S) ― 1一フエニルェチル]一 2, 5—ピリミジンジァミン [1 5] (以下、 化合物 [15] とレ、う) の合成。 化合物 [14] の合成過程において、 化合物 [15] も同時に生成した。 実施例 1 4の残渣の一部を、 逆相分取液体クロマトグラフィーにて精製した。 飽和炭酸水素ナ トリウム水溶液にて液性を塩基性にした後、酢酸ェチルにて抽出し、有機層を無水硫 酸ナトリゥムにて乾燥した。不溶物をろ過し、 ろ液を減圧濃縮し、 目的化合物 [15]
7. 4m gを黄色油状物として得た。
化合物 [15] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 62 (d, J = 6. 6H z , 1 H), 8. 30 (s, 1 H), 8. 03 (s, 1 H), 7. 44— 7. 25 (m, 5H), 7. 06 ( d , J =6. 6H z , 1 H), 6. 51 (t, J = 6. 6 H z , 1H), 5. 32— 5. 22 (m, 1 H), 5. 08— 4. 96 (m, 1 H), 3. 43 (b r, 2H), 2. 62 (s, 3H), 1. 55 (d, J = 6. 9Hz, 3H).
ma s s : 345 (M+ 1 ) +. 実施例 16
4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5— (4一メチル 一 1ーピぺラジュル) 一 N— [(1 S)一 1一フエニルェチル] —2—ピリミジンァ ミン [16] (以下、 化合物 [16] という) の合成。
(1) 化合物 [4] 10 Omgを無水酢酸 3mLに溶解し、 封管中 150°Cにて終 夜撹拌した。 減圧濃縮した後、 飽和炭酸水素ナトリゥム水溶液を加え、 酢酸ェチルに て抽出した。 有機層を飽和食埠水で洗浄した後、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、ろ液を減圧濃縮し、残渣を分取薄層クロマトグラフィ一にて精製し、 N— [5—プロモー 4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2—ピリミジニル]— N— [(1 S) —1一フエニルェチル] ァセトアミ ド [16 一 1] 102mgを白色固体として得た。
(2) N— [5—ブロモー 4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3 一ィル) 一 2—ピリミジニル]— N— [(1 S) —1一フエニルェチル] ァセトアミ ド [16— 1] 17mg、 ビス (ジベンジリデンァセトン)ノ ラジウム 2. 17mg、 4, 5—ビス (ジフエニルホスフイノ) 一 9, 9—ジメチルキサンテン 19mg、 炭 酸セシウム 17. 2mg、 1—メチルビペラジン 5. 0 μ L, トルエン 0. 25mL、 および 1, 4一ジォキサン 0. 5mLの混合物を、 80°Cにて 3日間撹拌した。 反応 混合物を室温に戻した後、 飽和炭酸水素ナトリウム水溶液、 水を加え、 酢酸ェチルに て抽出した。 有機層を飽和食塩水で洗浄し、 無水硫酸ナトリウムで乾燥した。 不溶物 をろ過し、 ろ液を減圧濃縮し、 残渣を逆相分取液体クロマトグラフィーにて精製し、 N— [4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一5— (4— メチルー 1ーピペラジ-ル) 一2—ピリミジェル]一 N— [(1 S) 一 1一フエニル ェチル] ァセトアミ ド [16— 2] 0. 6mgを得た。 (3) N— [4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 5 ― (4ーメチルー 1ーピペラジニル)一 2—ピリミジニル]— N— [(1 S) 一 1—
フエニルェチル] ァセトアミ ド 〔16— 2] 0. 6 mgをメタノールに溶解し、 ナト リウムメ トキシド (28%メタノール溶液) 0. lmLを加え、 60°Cにて 1時間 4 0分間撹拌した後に、 80°Cにて 2時間 20分間撹拌した。 減圧濃縮した後、 残渣を 逆相分取液体クロマトグラフィーにて精製し、 目的化合物 [16] 0. 44 m gを淡 黄色油状物として得た。
化合物 [16] のスぺクトワレデータを以下に示す。
XH— NMR (DMSO— d6) δ : 8. 90— 7. 00 (m, 1 OH), 5. 77— 5. 20 (m, 1 H), 4. 44— 4. 10 (m, 1 H), 2. 75— 2. 68 (m, 4H), 2. 57 (s, 3H), 2. 29— 2. 23 (m, 4H), 1. 48 (d, J =6. 9Hz, 3H), 1. 28 (s , 3H).
m a s s : 428 (M+ 1 ) +. 実施例 17
4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 N— [(1 S) ― 1—フエニルェチル] 一 5— (1—ピロリジニル) 一 2—ピリミジンアミン [17] (以下、 化合物 [17] という) の合成。 実施例 16— ( 1 )で得られた N— [ 5—プロモ一 4一 ( 8—メチルイミダゾ [ 1, 2— a] ピリジン一 3—ィル) 一 2—ピリミジニル] — N— [(1 S) 一 1—フエ二 ルェチル] ァセトアミ ド [16— 1] 2 Omgとピペリジン 4. 35 μ Lから、 実施 例 16— (2)、 (3) の方法に準じて目的化合物 [17] 0. 67mgを淡黄色油状 物として得た。
化合物 [17] のスぺクトルデータを以下に示す。
NMR (CDC 13) δ : 9. 00— 8. 92 (m, 1 H), 8. 78— 8. 6 5 (m, 1H), 8. 10— 7. 90 (m, 1 H), 7. 74— 7. 10 (m, 6H), 6. 73— 6. 62 (m, 1H), 5. 39— 5. 00 (m, 1H), 4. 52— 4. 15 (m, 1 H), 3. 13— 2. 75 (m, 4H), 2. 64 (s, 3H), 2. 0 0— 1. 82 (m, 4H), 1. 59 (d, J = 6. 9Hz, 3H).
ma s s : 399 (M+ 1 ) +. 実施例 18
N5, N5—ジメチルー 4一(8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) — N2— [(1 S)一 1一フエ-ルェチル]—2, 5—ピリミジンジァミン [18] (以 下、 化合物 [18] という) の合成。 実施例 16—(1)で得られた N— [5—ブロモ一4— (8—メチルイミダゾ [1,
2— a] ピリジン一 3—ィル) 一 2—ピリミジニル] — N— [(1 S) 一 1—フエ二 ルェチル] ァセトアミド [16— 1] 2 Omgとジメチルァミン (2Mテトラヒ ドロ フラン溶液) 26. から、 実施例 16— (2)、 (3) の方法に準じて目的化合 物 [ 18 ] 2. lmgを淡黄色油状物として得た。
化合物 [18] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 20 (d, J = 6. 9Hz, 1H), 8. 85 (s, 1 H), 8. 20 (s, 1 H), 7. 50— 7. 02 (m, 6H), 6. 69— 6. 5 5 ,(m, 1 H), 5. 65— 5. 40 (m, 1 H), 5. 17— 5. 03 (m, 1H), 2. 67 (s, 6H), 2. 64 (s, 3H), 1. 59 (d, J = 6. 9Hz, 3H). ma s s : 373 (M+ 1 ) +. 実施例 19
4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 5— (メチルチオ) — N— [(1 S) 一 1一フエニルェチル]—2—ピリミジンアミン [19] (以下、 化 合物 [19] という) の合成。 化合物 [4] 1001118を1^, N—ジメチルホルムアミ ド 2 mLに溶解し、 ナトリ ゥムメタンチォラート 60. 6mgをカロえ、 80°Cにて終夜撹拌した。 反応混合物を 室温に戻し、 水、 飽和炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水で洗浄し、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 残渣を分取薄層クロマトグラフィーにて精製し、 目的化合物 [1 9] 78. 5mgを淡黄色油状物として得た。
化合物 [19] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 88 (s, 1 H), 8. 45 (s, 1 H), 7. 4 8— 7. 26 (m, 6H), 7. 09 (d, J = 6. 7H z , 1H), 6. 63— 6. 50 (m, 1 H), 5. 78— 5. 62 (m, 1 H), 5. 20— 5. 08 (m, 1 H), 2. 54 (s, 3H), 2. 30 (s, 3H), 1. 59 (d, J = 6. 9Hz, 3H). m a s s : 376 (M+ 1 ) +. 実施例 20
4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一5— (メチルスル ホニル)— N— [(1 S)—1—フエニルェチル]一 2—ピリミジンアミン [20] (以 下、 化合物 [20] という) の合成。 化合物 [19] 18. 2mgを N, N—ジメチルホルムアミ ド 0. 75mLに溶解 し、 氷冷下、 m—ク口口過安息香酸 25. 1 m gを加え、 室温にて 1時間撹拌した。
水、 飽和炭酸水素ナトリウム水溶液を加え、 酢酸ェチルで抽出し、 有機層を無水硫酸 ナトリウムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 残渣を分取薄層クロマ トグラフィ一にて精製し、 目的化合物 [20] 1 1. 6 mgを黄色油状物として得た。 化合物 [20] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 04 (s, 1 H), 8. 88 ( s , 1 H), 8. 2 4 (d, J = 6. 9Hz, '1H), 7. 50— 7. 08 (m, 6 H), 6. 50— 6.
39 (m, 1 H), 6. 18— 6. 10 (m, 1 H), 5. 20— 5. 06 (m, 1 H), 2. 99 (s, 3H), 2. 59 (s, 3H), 1. 51 (d, J = 6. 9Hz, 3H). ma s s : 408 (M+ 1 ) +. 実施例 21
5—ョードー 4一 (8—メチルイミダゾ [1, 2_a] ピリジン一 3—ィル) 一 N— [(1 S)一 1一フエ-ルェチル]—2—ピリミジンアミン [21] (以下、化合物 [2 1] という) の合成。 化合物 [3] 295mgを ロロホルム 6mLに溶解し、 N—ョードこはく酸イミ ド 302m gを加え、 終夜加熱還流した。 反応混合物を室温に戻し、 飽和炭酸水素ナ トリゥム水溶液を加え、 クロロホルムにて抽出した。 有機層を飽和亜硫酸ナトリウム 水溶液、飽和食塩水で順に洗浄し、無水硫酸ナトリゥムで乾燥した。不溶物をろ過し、 ろ液を減圧濃縮し、残渣をシリカゲルカラムクロマトグラフィーにて精製し、 目的化 合物 [ 21 ] 74mgを褐色油状物として得た。
化合物 [21] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 71 ( s , 1 Η), 8. 55 (s, 1Η), 7. 4
4— 7. 00 (m, 6Η), 6. 64— 6. 39 (m, 1 H), 5. 68— 5. 54 (m, 1H), 5. 12— 4. 98 (m, 1H), 4. 83— 4. 52 (m, 1 H), 2. 6
3 (s, 3HX 1/2), 2. 62 (s, 3HX 1/2), .1. 56 (d, J = 6. 9 Hz, 3HX 1/2), 1. 41 ( d , J = 6. 9H z , 3HX 1/2).
ma s s : 456 (M+ 1 ) +. 実施例 22
5—メ トキシー 4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 N 一 [(1 S) —1一フエニルェチル] —2—ピリミジンアミン [22] (以下、 化合物
[22] とレ、う) の合成。 化合物 [21] 18. 5111§をメタノール0. 5mLに溶解し、 ヨウィ匕銅 0. 77 mg、 1, 1 0—フエナントロリン 1. 46 m g、 およぴ炭酸セシウム 26. 5 m g
を加え、 封管中 11 o°cにて終夜撹拌した。 反応混合物を減圧濃縮し、残渣を逆相分 取液体クロマトグラフィーにて精製した。飽和炭酸水素ナトリゥム水溶液にて液性を 塩基性にした後、 酢酸ェチルにて抽出し、 有機層を無水硫酸ナトリウムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 目的化合物 [22] 3. lmgを黄色ァモルフ ァスとして得た。
化合物 [22] のスぺク トルデータを以下に示す。
XH— NMR (CDC 13) δ 9. 33— 9. 24 (m, 1 H), 8. 58 ( s , 1 H), 7. 48— 7. 07 (m, 7H), 6. 67— 6. 50 (m, 1 H), 5. 61 一 5. 45 (m, 1 H), 5. 13— 5. 00 (m, 1H), 3. 92 (s, 3H), 2. 64 (s, 3H), 1. 59 (d, J = 6. 9 H z , 3H).
ma s s : 360 (M+ 1 ) +. 実施例 23
2— [(4一(8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— {[( 1 S) 一 1一フエニルェチル] アミノ}一 5—ピリ ミジニル) ァミノ] エタノール [2
3] (以下、 化合物 [23] と,いう) の合成。 化合物 [21] 18. 5mg、 ビス (ジベンジリデンアセトン) パラジウム 7. 0 mg、 4, 5—ビス (ジフエニノレホスフイノ) 一 9, 9ージメチルキサンテン 14. 1 mg、 炭酸セシウム 39. 7mg、 2—アミノエタノール 3. 7/z L、 および 1, 4一ジォキサン 0. 5mLの混合物を、 封管中 80°Cにて終夜撹拌した。 反応混合物 を減圧濃縮し、 残渣を逆相分取液体クロマトグラフィーにて精製し、 目的化合物 [2 3] 2. lmgを黄色油状物として得た。
化合物 [23] のスぺク トルデータを以下に示す。
'H— NMR (CDC 13) δ : 10. 08— 10. 01 (m, 1H), 8. 38— 8.
30 (m, 1 H), 7. 91— 7. 85 (m, 1H), 7. 5.5— 7. 20 (m, 7H), 6. 97— 6. 89 (m, 1H), 6. 78— 6. 70 (m, 1 H), 5. 12— 5. 00 (m, 1 H), 3. 86— 3. 55 (m, 2H), 3. 70 (b r, 1 H), 3.
48— 3. 40 (m, 2H), 2. 51 ( s , 3H), 1. 69 (d, J = 6. 9Hz, 3H).
ma s s : 389 (M+ 1 ) +. 実施例 24
4一(8—メチノレイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— {[(1 S) _ 1一フエニルェチル] ァミノ)一 5—ピリ ミジノール [24] (以下、 化合物 [24] という) の合成。
(1) 化合物 [21] 18. 5mg、 ヨウィ匕 §同 2. 3m g、 1, 10—フエナント 口リン 4. 5mg、 炭酸セシウム 26. 5mg、 2, 4—ジメ トキシベンジルアルコ ール 68. 3mg、 およびトルエン 0. 5mLの混合物を、 封管中 110°Cにて終夜 撹拌した。 反応混合物を減圧濃縮し、残渣を逆相分取液体ク口マトグラフィ一にて精 製し、 5— [(2, 4—ジメ トキシベンジル) ォキシ] —4一 (8—メチルイミダゾ
[1, 2— a] ピリジン一 3 fル) 一 N— [(1 S) 一 1一フエニルェチル] アミ ノ} ー5—ピリミジンァミン [24— 1] 3. 7mgを得た。 (2) 5— [(2, 4ージメ トキシベンジル) ォキシ] —4— (8—メチルイミダ ゾ [1, 2— a] ピリジン _3_ィル) 一 N— [(1 S) _1一フエニルェチル] 了 ミノ }—5—ピリミジンアミン [24— 1] 3. 7mgをクロ口ホルム 0. 5mLに 溶解し、 41[塩酸ー1, 4一ジォキサン溶液 0. 5mLを力!]え、 室温にて 3時間 40 分撹拌した。反応混合物を減圧濃縮し、逆相分取液体ク口マトグラフィ一にて精製し、 目的化合物 [24] 1. lmgを黄色油状物として得た。
化合物 [24] のスぺクトルデータを以下に示す。 .
'H— NMR (CDC 13) δ : 9. 36— 9. 23 (m, 1 H), 9. 21— 9. 1 0 (m, 1 H), 8. 12— 8. 02 (m, 1H), 7. 72— 7. 63 (m, 1H), 7. 45— 7. 00 (m, 8H), 5. 08— 4. 95 (m, 1H), 2. 76 (s, 3H), 1. 69 (d, J = 6. 9Hz, 3 H) .
ma s s : 346 (M+ 1 ) +. 実施例 25
メチル 3— (5—プロモー 2— {[(1 S) 一 1—フヱニルェチル] アミノ}一 4— ピリミジニル) イミダゾ [1, 2— a] ピリジン一 8—力ルポキシレート [25] (以 下、 化合物 [25] という) の合成。
(1) 2—ァミノニコチン酸 138m gと塩酸一メタノール溶液 6mLの混合物を、 封管中 90°Cにて終夜撹拌した。 反応混合物を減圧濃縮し、残渣に飽和炭酸水素ナト リゥム水溶液を加え、酢酸ェチルにて抽出した。 有機層を無水硫酸ナトリゥムにて乾 燥し、不溶物をろ過し、ろ液を減圧濃縮し、 2—ァミノニコチン酸メチルエステル [2 5—1] 106mgを得た。 得られた化合物 [25— 1] は更に精製することなく、 次の反応に用いた。 (2) ェチルェチュルエーテル (50%v/vへキサン溶液) 50 gをテトラヒド 口フラン 20 OmLに溶解し、 氷冷下、 ボランーテトラヒドロフラン錯体 (1. 0M
テトラヒドロフラン溶液) 1 1 9 m Lを 30分かけて滴下した後、 反応混合物を室温 まで昇温させ、さらに 4時間撹拌した。 4一クロ口一 2—メチルチオピリミジン 1 9. 1 gのテトラヒドロフラン溶液 10 OmL、 酢酸パラジウム 1. 87 g、 トリフエ二 ルホスフィン 2. 18 g、 および 3 N—水酸化ナトリゥム水溶液 1 59 m Lをカロえ、 さらに室温にて終夜撹拌した。 減圧下、 テトラヒドロフランを留去し、 残渣に水を加 え、 酢酸ェチルにて抽出し'た。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウム にて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 残渣をシリカゲルカラムクロマ トグラフィ一にて精製し、 4— [(E) 一 2—エトキシビ二ル] 一 2— (メチルチオ) ピリミジン [25— 2] 9. 7 gを褐色油状物として得た。
(3) 4一 [(E) 一 2—エトキシビニル] —2— (メチルチオ) ピリミジン [2 5— 2] 137. 2mgを 1, 4一ジォキサン 5 mLと水 2 mLの混合溶媒に溶解し、 N—ブロモこはく酸イミ ド 124. 1 m gを加えた。 室温にて 1時間撹拌した後、 2 一ァミノニコチン酸メチルエステル [25— 1] 106mgを加え、 85°Cにて 1時 間撹拌した。 反応混合物を減圧濃縮し、 残渣を分取薄層クロマトグラフィーにて精製 し、 メチル 3— [2— (メ ルチオ)一 4—ピリミジニル] イミダゾ [1, 2— a] ピリジン一 8—カルボキシレート [25— 3] 106. lmgを得た。
(4) メチル 3— [2— (メチルチオ) 一 4一ピリミジェル] イミダゾ [1, 2 —a] ピリジン一 8—カルボキシレ一ト [25— 3] 106. lmgを N, N—ジメ チルホルムアミ ド lmLに溶解し、 m—クロ口過安息香酸 183. lmgを加え、 室 温にて終夜撹拌した。 反応混合物を減圧濃縮し、 残渣を酢酸ェチルに溶解し、 飽和炭 酸水素ナトリゥム水溶液にて洗浄した。有機層を無水硫酸ナトリゥムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃縮し、 メチル 3— [2— (メチルスルホニル) 一 4 一ピリミジェル] イミダゾ [1, 2— a] ピリジン一 8—カルボキシレー卜 [25— 4] を得た。 得られた化合物 [25— 4] は、 更に精製することなく次の反応に用い た。
(5) 3— [2— (メチルスルホニル) 一4—ピリミジニル] イミダゾ [1, 2— a] ピリジン一 8—カルボキシレート [25— 4] をジメチルスルホキシド 3 m Lに 溶解し、 (1 S) — (1) 一フエニルェチルァミン 50 L、 N, N—ジイソプロピ ルェチルァミン 150 μ Lを加え、 90 °Cにて 4時間撹拌した。 反応混合物を室温に 戻した後、 水を加え、 酢酸ェチルにて抽出した。 有機層を無水硫酸ナトリウムにて乾 燥した後、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣を分取薄層クロマト グラフィ一にて精製し、 メチル 3— (2— {[(1 S) 一 1一フエニルェチル] アミ ノ} 一 4一ピリミジニル) イミダゾ [1, 2— a] ピリジン一 8—カルボキシレート
[25— 5] を得た
(6) メチル 3— (2— {[(1 S) — 1一フエ-ルェチル] アミノ} —4一ピリ ミジ -ル) イミダゾ [1, 2— a] ピリジン一 8—力ルポキシレート [25— 5] を 酢酸 2mLに溶解し、 臭素 50 を加え、 室温にて 10分間撹拌した。 反応混合物 を減圧濃縮し、得られた残渣を酢酸ェチルに溶解した。 酢酸ェチル溶液を飽和炭酸水 素ナトリゥム水溶液にて洗浄し、無水硫酸ナトリゥムにて乾燥した。不溶物をろ過し、 ろ裤を減圧濃縮し、 残渣を分取薄層クロマトグラフィーにて精製し、 目的化合物 [2 5] 106. 2m gを白色固体として得た。
化合物 [25] のスぺクトルデータを以下に示す。
NMR (CDC 13) δ : 8. 80 (s , 1 H), 8. 48 (s, 1 H), 8. 0 4 (d, J = 7. 2Hz, 1 H), 7. 47— 7. 26 (m, 5H), 6. 60 (b r , 1 H), 5. 69 (b r, 1H), 5. 07 (b r, 1 H), 4. 06 (s, 3H), 1. 58 (d, J = 7. OH z , 3H).
ma s s : 452, 454 (M+ 1 ) +. 実施例 26, 27
[3— (5_ブロモ一 2— {[(1 S)一 1ーフヱニルェチル] アミノ}—4—ピリミ ジニル) イミダゾ [1, 2— a] ピリジン一 8—ィル] メタノール [26] (以下、 化合物 [26] とレヽう)、 および [3— (2— {[(1 S) —1一フエニルェチル] ァ ミノ } —4一ピリミジニル) イミダゾ [1, 2— a] ピリジン一 8—ィル] メタノー ル [27] (以下、 化合物 [27] という) の合成。 化合物 [25] 5 Omgをテトラヒドロフラン 5mLに溶解し、 水素ィ匕リチウムァ ルミ-ゥム 10 m gを力 Bえ、 室温にて 30分間撹拌した。 反応混合物に 1 N— zk酸化 ナトリウム水溶液を加え、 有機層を分離した。 水層を酢酸 チルにて抽出し、 先の有 機層と混合し、 無水硫酸ナトリウムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し た後、 得られた残渣を分取薄層クロマトグラフィーにて精製し、 目的化合物 [26] 15. 7mgを白色固体として、 目的化合物 [27] 10. 4mgを淡黄色油状物と して得た。
化合物 [26]、 および [27] のスぺク トルデータを以下に示す。
化合物 [26]
XH— NMR (CDC 13) δ : 8. 67 (s, 1H), 8. 45 (s, 1 H), 7. 4 1— 7. 19 (m, 7H), 6. 77 (b r , 1 H), 5. 63 (b r, 1 H), 5. 08 (b r, 1H), 5. 03 (s, 2H), 1. 58 (d, J = 6. 9Hz, 3H). ma s s : 424、 426 (M+ 1) +.
化合物 [27]
XH— NMR (CDC ") δ : 8. 26 (d, J = 5. 3Hz, 1H), 8. 1 3 (s, 1H), 7. 4 6— 7. 20 (m, 7H), 6. 9 1 (d, J = 5. 3Hz, 1 H)、 6. 70 (b r、 1 H) , 5. 6 2 (b r、 1H)ゝ 5. 1 8— 5. 1 0 (m, 1 H), 5. 02 (s , 2H), 1 ·' 65 (d, J = 7. 7Hz, 3H).
ma s s : 346 (M+ 1 ) +. 実施例 28
5—ブロモ一4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル)一 N— [(1 S)一 1ーフヱニルェチル]一 2—ピリミジンアミン [28] (以下、化合物 [2 8] と ヽう) の合成。
2—アミノー 3—ク口口ピリジン 7 1 mgと 4— [(E) 一 2—エトキシビニル] 一 2—(メチルチオ) ピリミジン [25— 2] 1 0 7. 8mg力 ら、実施例 25—(3) 〜 (6) の方法に準じて、 目的化合物 [28] 2 3. 2mgを白色固体として得た。 化合物 [28] のスペクトルデータを以下に示す。
NMR (CDC 13) δ : 8. 72 (s , 1 H), 8. 4 7 (s, 1H), 7. 4 3— 7. 29 (m, 7H), 6. 47 (b r, 1 H), 5. 64 (b r , 1 H), 5. 03 (b r, 1 H), 1. 60 (d, J = 6. 9Hz, 3H).
ma s s : 428, 430 (M+ 1) +. 実施例 29
3— (5—プロモー 2— {[(1 S)一 1一フエニノレエチル] アミノ}一 4一ピリミジ -ル) イミダゾ [1 , 2— a] ピリジン一 8—カルボ二トリル [29] (以下、 化合 物 [29] という) の合成。 化合物 [25] 2 Omgをメタノール 1 mLに溶解し、 アンモニア水 (28%) 1 mLを加え、封管中 8 5°Cにて 1時間 30分撹拌した。反応混合物を室温に戻した後、 生じた固体をろ取し、 水にて洗浄した。 得られた固体をピリジン lmLに溶解し、 ォ キシ塩ィヒリン 20 μ Lを加え、 室温にて 5分間撹拌した。 反応混合物を減圧濃縮した 後、 得られた残渣を酢酸にて中性にし、 酢酸ェチルにて抽出した。 有機層を硫酸ナト リウムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃縮した。 残渣を分取薄層クロ マトグラフィ一にて精製し、 目的化合物 3. 9 8 m gを白色固体として得た。
化合物 [2 9] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 78 (s , 1 H), 8. 5 1 (s, 1 H), 7. 6
7 (d, J = 6. 9Hz, 1H)ヽ 7. 46— 7. 30 (m, 6H), 6. 56 (b r, 1 H), 5. 67 (b r, 1 H), 5. 01 (b r, 1 H), 1. 60 (d, J = 6. 9Hz, 3H).
ma s s : 419, 421 (M+ 1) +. 実施例 30 '
4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— {[(1 S) — 1一フエニルェチル] アミノ}一 5—ピリミジンカルボ二トリル [30] (以下、 ィ匕 合物 [30] という) の合成。 (1) ィ 合物 [28] 2 Omgから、 実施例 5の方法に準じてメチル 4— (8— クロロイミダゾ [1, 2— a] ピリジン一 3 fル) 一 2— {[(1 S) — 1一フエ二 ルェチル]ァミノ)一 5—ピリミジンカルボキシレート [30— l] 15mgを得た。 (2) メチル 4_ (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2
— {[(1 S)一 1—フエ-ルェチル]アミノ}一 5—ピリミジンカルボキシレート [3 0— 1] 15mgから、 実施例 7の方法に準じて 4一 (8—クロロイミダゾ [1, 2
— a] ピリジン一 3—ィル) 一 2— {[(1 S) — 1一フエ-ルェチル] アミノ}— 5 一ピリミジンカルボキサミ ド [30— 2] を得た。
(3) 4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィノレ)一 2— {[(1 S) 一 1一フエニルェチル] アミノ}一 5—ピリミジンカルポキサミド [30— 2] 力、ら、 実施例 8の方法に準じて目的化合物 [30] 1. 85 m gを白色固体として得 た。
化合物 [30] のスペクトルデータを以下に示す。
NMR (CDC 13) δ : 8. 98 ( s , 1 HX 1/3), 8. 90 ( s , 1 Η X 2/3), 8. 72 (d、 J = 5. OHz, 1H), 8. 61— 8. 55 (m, 1H), 7. 47— 7. 30 (m, 6H), 6. 59— 6. 54 (m, 1 H), 6. 14 (b r、 1H)、 5. 10 (s, 1 H), 1. 63 (d, J = 6. 9Hz, 3H).
ma s s : 375, 377 (M+ 1 ) +. , 実施例 31
5—ブロモ一4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3 一ィル]一 N— [(1 S)一 1一フエニルェチル]—2—ピリミジンアミン [31] (以 下、 化合物 [31] という) の合成。
(1) 化合物 [25] 1 22mgをテトラヒドロフラン 1 OmLに溶角早し、一 78。C にて水素化ジイソブチルアルミニウム (1. 0 1Mトルエン溶液) 0. 62mLを加 え、 同温にて 1 0分間撹拌した。 飽和塩ィ匕アンモニゥム水溶液を加え、 有機層を分離 した後、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 残渣を分取薄層クロマ トグラフィ一にて精製し、 3—(5—プロモー 2— {[(1 S)一 1—フエニルェチル] ァミノ)一 4一ピリミジ -ル) イミダゾ [ 1 , 2— a ] ピリジン一 8一カルボアルデ ヒ ド [3 1— 1] 72. 3mgを得た。
(2) 3— (5—ブロモー 2— {[(1 S) 一 1ーフヱ-ルェチル] アミノ} —4一 ピリミジ-ル) イミダゾ [ 1 , 2— a ] ピリジン一8_カルボアルデヒド [ 3 1— 1 ] 60. 3mgをジクロロメタン 2mLに溶解し、 ジェチルァミノ硫黄トリフルオリ ド 1 00 μ Lを加え、 室温にて 5分間撹拌した。 反応混合物を減圧濃縮した後、 得られ た残渣を分取薄層クロマトグラフィーにて精製し、 目的化合物 [3 1] 48. 5mg を白色固体として得た。
化合物 [3 1] のスペク トルデータを以下に示す。
ΧΗ— NMR (CDC 13) δ :, 8. 73 (s, 1 H), 8. 48 (s, 1 H), 7. 5 9— 7. 30 (m, 7H), 6. 6 1 (b r, 1H), 5. 6 8 (b r , 1 H), 5. 03 (b r, 1 H), 1. 5 9 (d, J = 7. OHz, 3H).
ma s s : 444、 446 (M+ 1) +. 実施例 32
3— (5—シァノ一2— {[(1 S) 一 1一フエニルェチル] アミノ}一 4一ピリミジ ニル) イミダゾ [1, 2— a] ピリジン一 8—カルボ二トリル [3 2] (以下、 化合 物 [3 2] という) の合成。 化合物 [2 9] 24. 9mgから、 実施例 30— ( 1 ) 〜 (3) の方法に準じて、 目的化合物 [32] 4. Omgを白色固体として得た。
化合物 [32] のスぺク トルデータを以下に示す。
XH— NMR (CDC 1 3) δ : 9. 04 ( s , 1 ΗΧ 1/3), 8. 96 (s, 1 Η
X 2X3), 8. 86 (d、 J = 7. 2Hz, 5— 8. 58 (m, 1 H), 7. 8 9— 7. 8 3 (m, 1 HX 1/3), J = 7. 2H z , 1 HX
2/3), 7. 50— 7. 3 2 (m, 6H), J = 7. OHz, 1 H), 6. 3 8 (b r、 1 H)ヽ 5. 0 9 (b r, 6 (d, J = 6. 9H z, 3H).
ma s s 3 6 6 (M+ 1 )
実施例 3 3
4一 [8— (ジフルォロメチル) イミダゾ [1 , 2— a] ピリジン一 3—ィル]一 2 一 {[(1 S) 一 1一フエニルェチル] アミノ}一 5—ピリミジンカルボ二トリル [3 3] (以下、 化合物 [3 3] という) の合成。 化合物 [3 1] 4 5. 3'mg力 ら、 実施例 3 0— (1) 〜 (3) の方法に準じて、 目的化合物 [3 3] 9. Omgを白色固体として得た。
化合物 [3 3] のスぺクトルデータを以下に示す。
XH— NMR (CDC 1 3) δ : 8. 9 8 ( s, 1 HX 1/3), 8. 9 1 ( s, 1 H X 2/3 ), 8. 8 6 (d、 J = 6. 5 H z , 1 H), 8. 6 1— 8. 5 6 (m, 1 H), 7. 7 0— 7. 7 6 (m, 1 HX 1/3), 7. 6 3 (d、 J = 6. 5H z , 1 HX 2/3), 7. 5 0— 7. 3 2 (m, 6 H), 6. 7 1 ( t, J = 7. 2H z , 1 H), 6. 1 0 (b r、 1 H)ゝ 5. 1 1 (b r, 1 H), 1. 6 6 (d, J = 6. 9H z , 3H).
ma s s : 3 9 1 (M+ 1 ) +. 実施例 34
N— [( 1 S) 一 1一フエニルェチル] —4— [8— (トリフルォロメチル) イミダ ゾ [ 1, 2— a] ピリジン一 3—ィル] 一 2—ピリミジンアミン [3 4] (以下、 化 合物 [34] という) の合成。
( 1 ) 2—クロ口一 3—トリフルォロメチルピリジン 1 0 gを 2 5%アンモニア水 1 0 OmLに溶解し、封管中 1 7 5°Cにて 3日間撹拌した。 反応混合物を室温に戻し た後、 ジェチルエーテルにて抽出した。 有機層を水、 飽和食塩水にて順に洗浄し、 無 水硫酸ナトリウムで乾燥した。 不溶物をろ過した後、 ろ液を減圧濃縮し、 3— (トリ フルォロメチル)一 2—ピリジンアミン 「34— 1」 8. .7 3 gを白色固体として得 た。
(2) 3— (トリフルォロメチル) 一 2—ピリジンアミン 「34— 1」 24 7mg と 4一 [(E) 一 2—エトキシビニル]一 2— (メチルチオ) ピリミジン 「2 5— 2 J
4 0 Omgから、 実施例 2 5— (3) 〜 (5) の方法に準じて、 目的化合物 [34]
2 3. 4 m gを淡黄色油状物として得た。
化合物 [34] のスペクトルデータを以下に示す。
XH— NMR (DMS O— d 6) δ : 1 0. 5 1 (b r , 1 HX 1/2), 9. 4 9 (b r , 1 HX 1/2), 8. 6 6— 8. 4 6 (m, 1 H), 8. 3 6— 8. 2 0 (m, 1 H), 8. 0 2— 7. 7 2 (m, 2H), 7. 4 5— 7. 1 6 (m, 7 H), 5. 2 0
一 4. 98 (πχ, 1 Η), 1. 50 (d, J = 6. 9Ηζ, 3Η).
ma s s : 384 (M+ 1 ) +. 実施例 35
5—ブロモ一N— [(I S) — 1一フエニルェチル] —4— [8— (トリフルォロメ チル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2—ピリ ミジンアミン [35] (以下、 化合物 [35] という) の合成。 化合物 [34] 15. 8mg力 ら、 実施例 2の方法に準じて目的化合物 [35] 1 5. 4m gを無色油状物として得た。
化合物 [35] のスぺク トルデータを以下に示す。
'H— NMR (CDC 13) δ : 8. 77 (s, 1 H), 8. 50 (s, 1 H), 7. 6 0 (d, J = 6. 9Hz, 1 H), 7. 42— 7. 33 (m, 6H), 6. 66— 6. 48 (m, 1 H), 5. 73— 5. 58 (m, 1 H), 5. 09— 4· 90 (m, 1H), 1. 59 (d, J = 6. 9Hz, 3H).
ma s s : 462, 464 (M+ 1 ) +. 実施例 36
2— {[(1 S)一 1一フエニルェチル] アミノ}一 4一 [8— (トリフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 5—ピリ ミジンカルボ二トリル [3 6] (以下、 化合物 [36] という) の合成。 化合物 [35] 12. lmgから、 実施例 30— (1) 〜 (3) の方法に準じて、 目的化合物 [36] 1. 3 mgを無色油状物として得た。
化合物 [36] のスペク トルデータを以下に示す。
NMR (CDC 13) δ : 8. 95 ( s , 1 H), 8. 87 (d, J = 6. 9H z , 1 H), 8. 58 (s, 1 H), 7. 66 (d, J =6. 9Hz, 1 H), 7. 4 9— 7. 30 (m, 5H), 6. 67 (t, J = 6. 9Hz, 1 H), 6. 22— 6. 15 (m, 1 H), 5. 15— 5. 02 (m, 1 H), 1. 65 (d, J = 7. 4Hz, 3H).
ma s s : 409 (M+ 1) +. 実施例 37
N— [(I S) — 1一フエ二ルェチノレ] —5— (2H—テトラゾールー 5 fル) 一 4一 [8—(トリフノレオロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ― 2—ピリミジンアミン [37] (以下、 化合物 [37] という) の合成。
ィヒ合物 [3 6] 1 Omgを N—メチルー 2—ピロリジノン 0. 5mLに溶解し、 ァ ジィ匕ナトリウム 3. 1 9mg、 トリェチルァミン塩酸塩 6. 74 m gを加え、 1 0 0 °C にて 2時間 20分撹拌した後、 1 5 0 °Cにて終夜撹拌し、 その後、 20 0 °Cにて 3時 間 3 0分撹拌した。 反応混合物を室温に戻し、 ジメチルスルホキシドで希釈した後、 逆相分取液体クロマトグラフィーにて精製し、 目的化合物 [3 7] 2. l mgを褐色 固体として得た。
化合物 [3 7] のスぺクトルデータを以下に示す。
'H— NMR (DMS O— d 6) δ : 8. 7 2— 8. 5 0 (m, 3 H), 8. 0 3— 7. 8 1 (m, 1 H), 7. 5 5— 6. 8 7 (m, 8H), 5. 3 3— 5. 0 2 (m, 1 H), 1. 54—1. 44 (m, 3H).
ma s s : 4 5 2 (M+ 1 ) +. 実施例 3 8
4— (2, 8—ジメチルイミダゾ [1 , 2— a] ピリジン一 3—ィル)一 N— [( 1 S) —1一フエニルェチル]一 2—ピリミジンアミン [3 8] (以下、 化合物 [3 8] という) の合成。
(1) 2—アミノー 3—ピコリン 2 gをテトラヒ ドロフラン 1 OmLとジェチルェ 一テル 5 mLの混合溶媒に溶解し、 3—クロロアセチルアセトン 4. 4mLを加え、 終夜加熱還流した。 反応混合物を室温まで戻し、 酢酸ェチルで希釈した後、 水、 飽和 食塩水にて順に洗浄した。 有機層を無水硫酸マグネシウムにて乾燥した後、不溶物を ろ過し、 ろ液を減圧濃縮した。 得られた残渣を少量のクロ口ホルムに溶解し、 へキサ ンを加え固ィ匕させ、 1— (2, 8—ジメチルイミダゾ [1 , 2— a] ピリジン一 3— ィル) エタノン [3 8— 1] 6 64m gを淡灰色固体として得た。
(2) 1— (2, 8—ジメチルイミダゾ [1, 2— a] ピリジン一 3—ィル) エタ ノン [3 8— 1 ] から、 実施例 1一 (2) 〜 (5) の方法に準じて、 目的化合物 [3 8] 4 9. l mgを白色固体として得た。
ィ匕合物 [3 8] のスぺクトルデータを以下に示す。
XH— NMR (CDC 1 3) δ : 8. 3 2 (d, J = 5. 1 Η ζ, 1 H), 7. 2 9— 7. 46 (m, 6H), 7. 0 1 (d, J = 6. 6H z, 1 H), 6. 7 6 (d, J = 5. 5 H z, 1 H), 6. 4 9 (b r s , 1 H), 5. 9 3 (b r s, 1 H), 5. 1 8 - 5. 1 4 (m, 1 H), 2. 6 9 ( s , 3 H), 2. 6 0 ( s, 3H), 1. 6 0 (d, J = 7. OH z , 3 H)
m a s s : 344 (M+ 1 ) +.
実施例 3 9
5—プロモー 4一 (2, 8—ジメチルイミダゾ [1, 2— a] ピリジン一 3一^ ル) 一 N— [(1 S) 一 1一フエニルェチル]一 2—ピリミジンアミン [3 9] (以下、 化 合物 [3 9] という) の合成。 化合物 [3 8] 4 9mg力 ら、実施例 2の方法に準じて、 目的化合物 [3 9] 3 8. 2m gを白色固体として得た。
化合物 [3 9] のスぺクトルデータを以下に示す。
XH— NMR (CDC 1 3) δ : 8. 45 ( s, 1 H), 7. 3 7— 7. 2 3 (m, 5 H), 7. 1 8— 7. 1 4 (m, 2H), 6. 9 8 (d, J = 6. 4H z, 1 H), 5. 7 5 (b r s, 1 H), 5. 0 3 (b r s , 1 H), 2. 6 1 ( s, 3 H), 2. 4 7 ( s, 3H), 1. 54 (d, J = 6. 8H z, 3 H)
ma s s : 4 2 2, 4 24 (M+ 1 ) +. 実施例 40 .
4一 (2—メチルイミダゾ [ 1 , 2— a] ピリジン一 3—ィル) 一 N— [( 1 S) ― 1一フエニルェチル] 一 2—ピリミジンアミン [4 0] (以下、 化合物 [4 0] とい う) の合成。
( 1) 2—アミノビリジン 2 gから、実施例 3 8_ (1)の方法に準じて、 1一(2 —メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 1一エタノン [4 0— 1] 1. 3 9 gを淡褐色固体として得た。 (2) 1— (2—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一1一エタ ノン [4 0— 1] 70 0mgから、 実施例 1— (2) 〜 (5) の方法に準じて、 目的 化合物 [40] 1 7 7mgを白色固体として得た。
化合物 [40] のスペクトルデータを以下に示す。
XH— NMR (CDC 1 3) δ : 8. 3 2 (d, J = 5. 5H z , 1 H), 7. 5 2 (d, J = 9. OH z , 1 H) 7. 4 5— 7. 2 8 (m, 6 H), 7. 2 1— 7. 1 8 (m, 1 H), 6. 7 5 (d, J = 5. 5 H z, 1 H), 6. 5 5 (b r s , 1 H), 5. 9 1 (b r s , 1 H), 5. 1 6— 5. 1 3 (m, 1 H), 2. 6 6 ( s , 3 H), 1. 5 9 (d, J = 7. 0H z , 3H)
ma s s : 3 3 0 (M+ 1) +. 実施例 4 1
5—ブロモ一 4_ (2—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 N— [(1 S)一 1ーフヱニルェチル]一 2_ピリミジンアミン [41] (以下、化合物 [4 1] という) の合成。 化合物 [40] 177mgから、 実施例 2の方法に準じて、 目的化合物 [41] 1 90. 3mgを白色固体と'して得た。
化合物 [41] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 46 ( s , 1 Η), 7. 54 (d, J = 8. 8H z, 1 H), 7. 38— 7. 23 (m, 6H), 7. 18 (m, 4H), 5. 79 (b r s, 1H), 5. 05 (b r s, 1 H), 2. 35 (s, 3H), 1. 54 (d, J =6. 8Hz, 3H)
ma s s : 408, 410 (M+ 1) +. 実施例 42
4—メチル一6— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一N— [(1 S)一 1一フエニルェチレ]一 2—ピリミジンアミン [42] (以下、化合物 [4 2] という) の合成。
(1) 4一クロ口一 2—メチルチオピリミジン 5. 93 mLをジェチルエーテル 5 OmLに溶解し、 一 78°Cにてメチルリチウム (1Mジェチルエーテル溶液) 100 mLを徐々に加えた。 0°Cにて反応液を 1時間撹拌した後、 水 2. 3mLとテトラヒ ドロフラン 15 mLを混合した溶液を加えた。同温にて反応液を 10分間撹拌した後、 2, 3—ジクロロー 5, 6—ジシァノヒドロキノン 13. 7 gのテトラヒドロフラン 溶液 50mLを加えた。 同温にて反応液を 1時間撹拌した後、 1N—水酸ィヒナトリゥ ム水溶液 100 mLを加えた。 得られた反応液をへキサンで抽出し、 有機層を 1 N— 水酸化ナトリゥム水溶液と飽和食塩水で洗浄した。 無水硫酸マグネシウムで乾燥後、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣をシリカゲルカラムクロマトグ ラフィ一で精製し、 4—クロ口一 6—メチルー 2— (メチルチオ) ピリミジン [42 —1] 5. 1 gを淡黄色固体として得た。
(2) ェチルェチェルエーテル (40%へキサン溶液) 15 gをテトラヒ ドロフラ ン 50 m Lに溶解し、 0 °Cにてボランーテトラヒ ドロフラン錯体 ( 1 Mテトラヒ ドロ フラン溶液) 29mLを加えた。 室温にて 4時間 30分撹拌した後、 4一クロロー 6 ーメチ Λ ^— 2— (メチルチオ) ピリミジン [42— 1] 4. O gのテトラヒ ドロフラ ン溶液 150mL、 3 N—水酸化ナトリウム水溶液 35 mL、 トリフエニルホスブイ ン 0. 46 g、 および酢酸パラジウム 0. 34 gを加えた。 得られた反応液を室温に
て 16時間撹拌した後、 水 20 OmLを加えた。 反応液を酢酸ェチルで抽出し、 有機 層を飽和食塩水で洗浄した。これを無水硫酸マグネシウムで乾燥後、不溶物をろ過し、 ろ液を減圧濃縮した。得られた残渣をシリカゲル力ラムクロマトグラフィ一で精製し、 4— [(E) 一 2—エトキシビュル]一 6—メチルー 2— (メチルチオ) ピリミジン [42— 2] 4. 9 gを褐色油状物として得た。 -
(3) 4一 [(E) 一 2—エトキシビニル]一 6—メチル一2— (メチルチオ) ピ リミジン [42— 2] 50 Omgと 2—アミノー 3—ピコリン 240 / Lから、 実施 例 25— (3) 〜 (5) の方法に準じて、 目的化合物 [42] 39mgを無色油状物 として得た。
化合物 [42] のスペクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 1 5 (s, 1 H), 7. 47— 7. 45 (m, 2 H), 7. 41— 7. 37 (m, 2H), 7. 29— 7. 25 (m, 2H), 7. 06 (d, J = 6. 6Hz , 1 H), 6. 84 (s, 1H), 6. 64— 6. 58 (m, 1 H), 5. 51 (b r d, J = 5. 5H z, 1H), 5. 18— 5. 12 (m, 1H), 2. 62 (s, 3H), 2. 4,0 (m, 3H), 1. 61, (d, J = 7. OHz, 3 H).
ma s s : 344 (M+ 1). 実施例 43
5—ブロモ一4—メチルー 6— (8—メチルイミダゾ [1, 2— a] ピリジン一 3— ィル)一 N— [(1 S) —1—フエ-ルェチル]一 2—ピリミジンアミン [43] (以 下、 化合物 [43] という) の合成。 化合物 [42] 37. 4mg力 ら、 実施例 2の方法に準じて、 目的化合物 [43] 37. 7mgを無色油状物として得た。
化合物 [43] のスぺクトルデータを以下に示す。
一 NMR (CDC 13) δ : 8. 55 (s, 1 H), 7. 40— 7. 36 (m, 5 H), 7. 31— 7. 25. (m, 1 H), 7. 02 (d, J = 6. 7Hz, 1 H), 6. 49— 6. 36 (m, 1 H), 5. 48 (b r d, J = 6. 7Hz, 1H), 5. 08 —5. 00 (m, 1H), 2. 61 (s, 3H), 2. 57 (m, 3H), 1. 53, (d, J = 6. 7Hz, 3H).
ma s s : 422, 424 (M+ 1) +. 実施例 44
N— [( 1 S) — 1—フエ二ルェチノレ] 一 4一 (5, 6, 7, 8—テトラヒ ドロイミ
ダゾ [ 1, 2— a] ピリジン一 3—ィル) 一 2—ピリミジンアミン [44] (以下、 化合物 [44] という) の合成。
( 1) 2—ピペリドン 1. 5 gをメシチレン 7 5mLに溶解し、 四塩ィ匕チタン 24 8 μ Lを加え、 1 40°Cに加熱した。 アミノアセトアルデヒド ジェチルァセターノレ
4. 0 2 gをメシチレン 4 5mLに溶解した溶液を調製し、 先のメシチレン溶液に 3 時間かけて滴下した。 混合物を 1 40°Cにて 7 0時間撹拌した後、 室温に戻し、 2 N 一塩酸水溶液で抽出した。得られた水層を 5 N—水酸化ナトリゥム水溶液で塩基性に した後、 クロ口ホルムにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナト リゥムで乾燥した。不溶物をろ過し、ろ液を減圧濃縮した後、減圧蒸留(3 mmHg、 1 2 0°C) により精製し、 5, 6, 7, 8—テトラヒドロイミダゾ [ 1, 2— a] ピ リジン [44一 1] 3 7 5mgを淡黄色油状物として得た。
(2) 5, 6, 7, 8—テトラヒ ドロイミダゾ [1, 2— a] ピリジン [44— 1] 3 7 5mgを四塩化炭素 1 OmLに溶解し、 N—プロモこはく酸ィミド 54 6mgを 加え、 3 0分間加熱還流した。,反応混合物を減圧濃縮し、 残渣をシリカゲル力ラムク 口マトグラフィー、 続く分取薄層クロマトグラフィーにて精製し、 3—プロモー 5, 6, 7, 8—テトラヒドロイミダゾ [1, 2— a] ピリジン [44一 2] 1 5 5 m g を淡黄色固体として得た。
(3) 3—ブロモ一5, 6, 7, 8—テトラヒドロイミダゾ [ 1, 2— a] ピリジ ン [44一 2] 1 '4 2mgをトルエン 2. 5 m Lに溶解し、 テトラキストリフエニル ホスフィンパラジウム (0) 8 1. 6mg、 トリブチル (1一エトキシビニル) すず 4 7 7 Lを加え、 1 2 0°Cにて終夜撹拌した。 反応混合物を室温に戻し、 1 0%フ ッ化カリゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を 1 0 %フッ化力リゥ ム水溶液、 飽和食塩水にて順に洗浄した後、 無水硫酸ナトリウムで乾燥した。 不溶物 をろ過し、 ろ液を減圧濃縮した後、 シリカゲルカラムクロマトグラフィーにて精製し た。 得られた化合物をクロ口ホルム 2 m Lに溶解し、 1 0 %塩^ ~メタノール溶液 2 0 0 μ Lを加え、 40分間撹拌した。 飽和炭酸水素ナトリゥム水溶液を加え塩基性に した後、 クロ口ホルムにて抽出した。 有機層を飽和食塩水で洗浄した後、 無水硫酸ナ トリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣を分取薄 層クロマトグラフィーにて精製し、 1— (5, 6, 7, 8—テトラヒドロイミダゾ [1, 2— a] ピリジン一 3—ィル) エタノン [44一 3] 6 2. 2mgを淡黄色油状物と して得た。
(4) 1— (5, 6, 7, 8—テトラヒドロイミダゾ [1 , 2— a] ピリジン一 3
一ィル) エタノン [44一 3] 62. 2mg力、ら、 実施例 1— (2) 〜 (5) の方法 に準じて目的化合物 [44] 4. 5mgを無色油状物として得た。
化合物 [44] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 17 (d, J = 6. 3Hz, 1H), 7. 49 (s, 1 H), 7. 38— 7. 20 (m, 5H), 6. 74 (d, J = 6. 3Hz, 1 H), 5. 58— 5. 45 (m, '1Η)' 5. 13— 5. 00 (m, 1H), 2. 98— 2. 80 (m, 2H), 2. 13— 1. 60 (m, 6H), 1. 57 (d, J = 6. 9H z , 3H).
ma s s : 320 (M+ 1) +. 実施例 45
5—ブロモ一N— [(1 S) — 1—フエニノレエチノレ] 一 4一 (5, 6 , 7, 8—テト ラヒ ドロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2—ピリミジンアミン [4 5] (以下、 化合物 [45] という) の合成。 化合物 [44] 2. 7mgから、 実施例 2の方法に準じて目的化合物 [45] 2. 2 mgを白色固体として得た。
化合物 [45] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 37 ( s , 1 Η), 7. 88 (s, 1Η), 7. 3 8— 7. 20 (m, 5Η), 5. 48— 5. 35 (m, 1 H), 5. 05— 4. 96 (m, 1 H), 2. 96— 2. 80 (m, 2H), 1. 92— 1. 60 (m, 6H), 1. 5 5 (d, J = 6. 9Hz, 3H).
ma s s : 398, 400 (M+ 1 ) +. 実施例 46
4一 (8—メチノ l ^一 5, 6, 7, 8—テトラヒドロイミダゾ [1, 2— a] ピリジン 一 3 ( レ) 一 N— [(1 S) 一 1ーフヱニルェチル] 一 2—ピリミジンアミン [4 6] (以下、 化合物 [46] という) の合成。 (1) 8—メチルイミダゾ [1, 2— a] ピリジン [3— 1] l gを 1ーブタノ一 ル 20mLに溶解し、 触媒量のラネーニッケルを加えた。 混合物を水素雰囲気下 (5 '気圧)、 65 °Cにて 4日間撹拌した。 反応混合物を室温に戻した後、 不溶物をセライ トにてろ過し、 メタノールで洗浄した。 ろ液を減圧濃縮し、残渣をシリカゲルカラム クロマトグラフィーにて精製し、 8—メチノレー 5, 6, 7, 8—テトラヒドロイミダ ゾ [1, 2— a] ピリジン [46— 1] 823. 3 m gを得た。
(2) 8—メチルー 5, 6, 7, 8—テトラヒドロイミダゾ [1, 2— a] ピリジ ン [4 6— 1] 8 1 Omg力 ら、実施例 44一 (2)、 (3) の方法に準じて、 1— (8 ーメチ Λ ^— 5, 6, 7, 8—テトラヒドロイミダゾ [1 , 2— a] ピリジン一 3—ィ ル) 一 1一エタノン [4 6— 2] を得た。
(3) 1— (8—メチルー 5, 6, 7, 8—テトラヒドロイミダゾ [1, 2— a] ピリジン一 3—^ fル)一 1一エタノン化合物 [4 6— 2] から、 実施例 1― (2) 〜
(5) の方法に準じて目的化合物 [46] 2 7. 3mgを白色固体として得た。 化合物 [4 6] のスぺク トルデータを以下に示す。
JH— NMR (CDC 1 3) δ : 1 0. 04 (m, 1 H), 8. 1 3 (m, 2H), 7. 3 7— 7. 2 5 (m, 5 H), 7. 0 0 (m, 1 H), 5. 0 1 (m, 1 H), 4. 3 0— 4. 2 7 (m, 1 HX 1 /2), 4. 0 1— 3. 9 8 (m, 1 HX 1/2), 3. 6 3 (m, 1 HX 1/2), 3. 5 0 (m, 1 HX 1/2), 3. 3 5— 3. 3 1 (m, 1 HX 1/2), 3. 2 1— 3. 1 9 (m, 1 HX 1/2), 2. 1 4一 1 · 8 7 (m, 3H), 1. 6 6 (d, J = 7. OH z , 3H), 1. 7 0— 1. 5 9 (m, 1 H), 1. 5 0 (d, J = 7. OH z , 3HX 1/2), 1. 4 5 (d, J = 7. OH z , 3HX 1/2).
ma s s : 3 34 (M+ 1 ) +. 実施例 4 7
5—プロモ一 4一 (8—メチル一5, 6, 7, 8—テトラヒ ドロイミダゾ [1 , 2— a] ピリジン一 3_ィル) 一 N— [(1 S) 一 1一フエ-ルェチル]一 2—ピリ ミジ ンァミン [4 7] (以下、 化合物 [4 7] という) の合成。 実施例 46で得られた化合物 [4 6] 2 5mg力ゝら、 実施例 2の方法に準じて、 目 的化合物 [4 7] 1 9. 3m gを白色固体として得た。
化合物 [4 7] のスぺクトルデータを以下に示す。
'H— NMR (CDC 1 3) δ : 8. 3 6 ( s , 1 H), 7. 8 9 ( s , 1 HX 1/2), 7. 8 8 ( s , 1 HX 1/2), 7. 3 3— 7. 2 2 (m, 5 H), 5. 5 3 (b r , 1 H), 5. 0 5— 5. 0 1 (m, 1 H), 4. 1 3— 3. 4 0 (m, 2H), 3. 0 4— 2. 9 3 (m, 1 H), 2. 04—1. 6 0 (m, 2H), 1. 5 5 (d, J = 7. OH z , 3H), 1. 5 0—1. 24 (m, 2H), 1. 4 2, (d, J = 7. OH z , 3HX 1/2), 1. 4 1 (d, J = 7. OH z , 3HX 1/2).
ma s s : 4 1 2, 4 1 4 (M+ 1 ) +. 実施例 4 8
メチノレ 3— (5—シァノ一2— {[(1 S) 一 1一フエニルェチル] アミノ} —4—ピ リミジニル) 一 8—メチルイミダゾ [ 1, 2— a] ピリジン一 7—カルボキシレート [48] (以下、 化合物 [48] という) の合成。 (1) 2—クロ口一 4—ョードピコリン 1. 0 g、 酢酸パラジウム 84mg、 1 , 1, 一ビスジフエ二ノレホス'フイノフエ口セン 218mg、炭酸水素ナトリゥム 990 mg、 N, N—ジメチルホルムアミ ド 1 OmL、 およびメタノール 1 OmLの混合物 を、 一酸化炭素雰囲気下、 80°Cにて終夜撹拌した。 反応混合物を室温に戻した後、 水、 飽和炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を飽和 食塩水で洗浄した後、 無水硫酸ナトリゥムで乾燥した。 不溶物をろ過し、 ろ液を減圧 濃縮した後、得られた残渣をシリカゲルカラムクロマトグラフィーにて精製し、 2— クロ口一 3—メチルイソニコチン酸メチルエステル [48— 1] 522mgを無色油 状物として得た。 (2) 2—クロロー 3—メチルイソニコチン酸メチルエステル [48— 1] 520 mg、 ビス (ジベンジリデンアセトン) パラジウム 16 lmg、 4, 5—ビス (ジフ ェニルホスフイノ) 一 9, 9ージメチルキサンテン 324m g、 炭酸セシウム 1· 2 8 g、 ベンゾフエノンィミン 564 L、 1, 4—ジォキサン 5mL、 およびトルェ ン 5mLの混合物を、 80°Cにて終夜撹拌した。 反応混合物を室温に戻した後、 水、 飽和炭酸水素ナトリゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩 水で洗浄した後、 無水硫酸ナトリウムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮 し、 得られた残渣をメタノール 28 mLに溶解した。 そこへ、 酢酸ナトリウム 1. 1 g、 塩酸ヒドロキシァミン 700 m gを加え、 1時間撹拌した後、 0. 1 N—水酸化 ナトリゥム水溶液を加え、 クロロホルムにて抽出した。 有機層を飽和食塩水で洗浄し た後、 無水硫酸ナトリウムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 残渣を シリカゲルカラムクロマトグラフィーにて精製し、 2—アミノー 3—メチノレイソニコ チン酸メチルエステル [48— 2] 92. 1 mgを黄色固体として得た。
(3) シス一 1—エトキシー 2—トリー n—ブチルスタニルエチレン (J. Am. Ch em. S o c. 1977, 99, 7365に記載されている方法により合成した) 2. 04 g、 4—クロロー 2— (メチルスルファニル) 一 5—ピリミジンカルボニト リル (国際公開第 2004/043936号パンフレツト、 32— 33ページに記載 されている方法により合成した) 1. 0 g、 ジクロロビス (トリフエ二ノレホスフィン) パラジウム (Π) 189mg、 およぴァセトニトリル 1 5mLの混合物を、 4時間加 熱還流した。反応混合物を室温に戻し、フッ化カリウム 1. 5 7 §と水51111^を加ぇ、 室温にて 30分間撹拌した。 不溶物をセライトにてろ過し、 ろ液に酢酸ェチルを加え
た。 得られた溶液を飽和食塩水で洗浄した後、 無水硫酸マグネシウムで乾燥した。 不 溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣を少量の酢酸ェチルに溶解した。 へ キサンを加え、 生じた固体をろ取し、 4— [(Z)一 2—エトキシビニル]一 2— (メ チルチオ) 一 5—ピリミジンカルボ二トリル [48— 3] 531. 9m gを祧色固体 として得た。
(4) 4— [(Z) —2—エトキシビュル] —2— (メチルチオ) 一 5—ピリミジ ンカルボ-ト リノレ [48— 3] 14mgと 2—アミノー 3—メチルイソニコチン酸メ チノレエステル [48— 2] 10. 5mg力 ら、 実施例 25— (3) 〜 (5) の方法に 準じて、 目的化合物 [48] 22. 7mgを淡黄色固体として得た。
化合物 [48] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 93 ( s , 1 Η), 8. 73 (d, J = 7. 4H z, 1 H), 8. 55 (s, 1H), 7. 52— 7. 10 (m, 6 H), 6. 20— 6. 1 1 (m, 1H), 5. 19— 5. 06 (m, 1 H), 3. 99 (s , 3H), 2. 9 6 (s, 3H), 1. 65 (d, J = 6. 9Hz, 3H).
ma s s : 413 (M+ 1 ) +. 実施例 49
4一 (7—メチルイミダゾ [1, 2— a] ピリジン一 3—ィノレ) 一 N— [(1 S) 1一フエニルェチル] 一 2—ピリミジンアミン [49] (以下、 化合物 [49] とい う) の合成。
4ーメチルー 2—ピリジンアミン 94. 3mgと 4— [(E) — 2_エトキシビ二 ル]一 2—(メチルチオ) ピリミジン [25— 2] 17 lmg力、ら、実施例 25—(3) 〜(5) の方法に準じて、 目的化合物 [49] 4. 2m gを淡黄色油状物として得た。 化合物 [49] のスペクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 90 (b r , 1Η), 8. 37 (b r, 1 Η), 7. 77 ( s , 1 Η), 7. 53— 7. 28 (m, 8Η), 5. 19 (q, J = 7. 7Η ζ , 1 Η), 2. 66 ( s , 3Η), 1. 67 (d, J = 7. 7Η ζ , 3Η).
ma s s : 330 (Μ+ 1 ) +. 実施例 50
5—プロモー 4一 (7—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 N— [(1 S)一 1一フエニルェチル]一 2—ピリミジンアミン [50] (以下、化合物 [5 0] とレヽう) の合成。
化合物 [49] 2. Omgから、実施例 2の方法に準じて、 目的化合物 [50] 2. lmgを白色固体として得た。
化合物 [50] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 71 (s, 1H), 8. 42 (s, 1H), 7. 5 0— 7. 28 (m, 7H), 6. 50— 6. 42 (m, 1 H), 5. 80— 5. 72 (m, 1H), 5. 12— 5. 05 (m, 1H), 2. 42 (s、 3H)、 1. 60 (d, J =6. 9H z , 3H).
ma s s : 408, 10 (M+ 1 ) +. 実施例 51
4—イミダゾ [1, 2— a] ピラジン一 3—ィルー N_ [(I S) — 1—フエ-ルェ チル] —2—ピリミジンアミン [51] (以下、 化合物 [51] という) の合成。
2—ァミノピラジン 302m gと 4一 [(E) 一 2—エトキシビュル] —2— (メ チルチオ) ピリミジン [25— 2] 623mg力 ら、 実施例 25— (3) 〜 (5) の 方法に準じて、 目的化合物 [ 1] 7 Omgを灰色固体として得た。
化合物 [51] のスぺクトルデータを以下に示す。
XH— NMR (DMSO— d6) δ : 9. 27— 9. 22 (m, 1 H), 8. 65— 8. 58 (m, 1H), 8. 31— 8. 23 (m, 1H), 8. 19— 8. 02 (m、 2H), 7. 44— 7. 34 (m, 4H), 7. 21— 7. 20 (m, 2H), 5. 05 (b r s, 1H), 1. 50 (d, J = 7. OHz, 3 H)
ma s s : 317 (M+ 1 ) +. 実施例 52
5—プロモ一 4一イミダゾ [1, 2— a] ピラジン一 3—ィル一 N— [ ( 1 S ) — 1 —フエニルェチル] 一 2—ピリミジンアミン [52] (以下、 化合物 [52] という) の合成。 化合物 [51] 3 Omgから、 実施例 2の方法に準じて、 目的化合物 [52] 28 mgを白色固体として得た。
化合物 [52] のスペクトルデータを以下に示す。
NMR (CDC 13) δ : 9. 15 ( s , 1 H), 8. 81 (s , 1H), 8. 5 1 ( s , 1 H), 7. 43— 7. 35 (m, 6H), 5. 78 (b r s, 1 H), 4. 99 (b r, 1 H), 1. 60 (d, J = 7. OHz, 3 H)
ma s s : 395, 397 (M+ 1 ) +.
実施例 53
4一 (8—メチルイミダゾ [1, 2— a] ピラジン一 3—ィル) 一 N— [(1 S) ― 1ーフヱニルェチル] 一 2—ピリ ミジンアミン [53] (以下、 化合物 [53] とい う) の合成。
( 1) 2—ク口ロー 3—メチルピラジン 5 g、ベンゾフエノンィミン 7. 44mL、 トリス (ジベンジリデンアセトン) (クロ口ホルム) ジパラジウム (0) 2 g、 2, 2, 一ビス (ジフエニルホスフイノ) 一 1, 1' ービナフチル 2. 48 g、 ナトリウ ム t一ブトキシド 5. 25 g、 およびトルエン 1 5 OmLの混合物を、 終夜加熱還流 した。 反応混合物を室温まで戻した後、 酢酸ェチルにて希釈し、 水、 飽和食塩水にて 順に洗浄し、 有機層を無水硫酸マグネシウムにて乾燥した。 不溶物をろ過し、 ろ液を 減圧濃縮した後、 残渣に 4 N塩酸一 1 , 4一ジォキサン溶液 50 m Lを加え、 1時間 静置した。 溶液を減圧濃縮し、 得られた残渣をシリカゲルカラムクロマトグラフィー にて精製した。 溶媒を留去した後、 残渣を少量のクロ口ホルムに溶解し、 へキサンを 加えた。 生じた固体をろ取し、 減圧乾燥し、 3—メチルー 2—ピラジンァミン [53 — 1] 2. 5 gを橙色固体とレて得た。
(2) 3—メチルー 2—ピラジンァミン [53— 1] 122mgと 4一 [(E) _ 2—エトキシビエル]一 2—(メチルチオ) ピリ ミジン [25— 2] 220mgから、 実施例 25— (3) 〜 (5) の方法に準じて、 目的化合物 [53] 1. 6 m gを淡黄 色油状物として得た。
化合物 [53] のスぺク トルデータを以下に示す。
ΧΗ— NMR (CD3OD) δ : 7. 86 ( s , 2Η), 7. 48— 7. 46 (m, 3 Η), 7. 39— 7. 35 (m, 4Η), 7. 26— 7. 25 (m, 1 H), 4. 85 (m, 1H), 2. 88 (s, 3H), 1. 65 (d, J = 6. 8Hz, 3 H) ma s s : 331 (M+ 1 ) +. 実施例 54
4一 (8—メチルイミダゾ [1, 2— a] ピリダジン一 3—ィル) —2— {[(1 S) —1—フエニルェチル] アミノ} 一 5—ピリミジンカルボ二トリル [54] (以下、 化合物 [54] という) の合成。
(1) (1 S) 一 1—フエニルェチルァミン 1. 84 g、 トリエチルァミン 2· 6 5mL、 1 H—ピラゾールー 1一カルボキサミジン塩酸塩 2. 23 g、 およびァセト 二トリノレ 50mLの混合物を 85 °Cにて 24時間撹拌した。反応混合物を減圧濃縮し た後、 残渣をエタノール 75 m Lに溶解した。 (エトキシメチレン) シァノ酢酸ェチ
ル 3. 52 gと 1 , 8—ジァザビシク口 [5. 4. 0]—ゥンデ力一 7—ェン 4. 5 5mLを力!]え、 6 5°Cにて 2日間撹拌した。反応混合物を室温に戻した後、水を加え、 酢酸ェチルにて洗浄した。有機層を水で抽出し、得られた水層を先の水層と混合した。 得られた水層を塩酸にて酸性にした後、 クロ口ホルムにて抽出し、得られた有機層を 無水硫酸マグネシウムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、得られた残 渣をシリカゲルカラムクロマトグラフィーにて精製し、 6—ォキソ一 2— {[(1 S) 一 1一フエニルェチル] アミノ}一 1, 6—ジヒドロー 5—ピリミジンカルボ-トリ ル [54— 1] 1. 6 9 gを黄色固体として得た。 (2) 6—ォキソ一 2— {[(1 S) — 1—フエニルェチル] ァミノ)一 1, 6—ジ ヒ ドロー 5—ピリミジンカルボ二トリル [54— 1] 1. 6 9 gをァセトニトリル 1 0 OmLに溶角军した後、 ォキシ塩化リン 5 m Lをカロえ、 8 5 °Cにて 2時間撹拌した。 反応混合物を減圧濃縮した後、得られた残渣を氷水中に入れ、酢酸ェチルにて抽出し、 有機層を水、 飽和食塩水にて順に洗浄した。 無水硫酸マグネシウムで乾燥した後、 不 溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣をシリカゲルカラムクロマトダラ フィ一にて精製し、 4一クロ口,一 2— {[(1 S)一 1—フエ-ルェチル] ァミノ)一 5—ピリミジンカルボ-トリル [54— 2] 1. 54 gを油状物として得た。
(3) 4一クロ口一 2— {[(1 S)一 1一フエニルェチル] アミノ}一 5—ピリミ ジンカルボ二トリル [54— 2] 9 7mgから、実施例 48—(3)の方法に準じて、
4— [(Z) 一 2—ェトキシビニノレ] —2— {[(1 S) 一 1—フエ-ルェチル] アミ ノ}一 5—ピリミジンカルボ二トリル [54— 3] 8 9mgを得た。
(4) 4一 [(Z) —2—エトキシビュル] —2— {[(1 S) 一 1一フエ二ルェチ ノレ] アミノ}一 5—ピリミジンカルボ二トリル [54— 3] と 4ーメチ Λ 3—ピリ ダジンァミンから、 実施例 2 5— (3) の方法に準じて、 目的化合物 [54] を黄色 固体として得た。
化合物 [54] のスぺクトルデータを以下に示す。
一 NMR (CDC 1 3) δ : 8. 63 ( s , 1HX 1/2), 8. 56 ( s , 1 Η X 1/2), 8. 4 1— 8. 30 (m, 1 Η+ 1 ΗΧ 1/2), 8. 2 5 (s, 1 ΗΧ 1/2), 7. 43— 7. 24 (m, 5Η), 7. 0 1 (d, J =4. 5Hz, 1 H), 6. 07 (m, 1 H), 5. 30 (m, 1H), 2. 7 3 ( s, 3H), 1. 70— 1. 58 (m, 3H).
ma s s : 3 56 (M+ 1 ) +. 実施例 5 5
4一 (8—アミノィミダゾ [1, 2— a] ピリダジン一 3—ィル) 一 2— {[(1 S) 一 1一フエニルェチル] アミノ} 一 5—ピリミジンカルボ二トリル [55] (以下、 化合物 [55] という) の合成。
4一 [(Z) 一 2—エトキシビニル] —2— {[(1 S) 一 1ーフヱニルェチル] ァ ミノ }一 5—ピリミジンカルボ二トリル [54— 3] 14mgと 2, 3—ピリジンジ アミンカゝら、 実施例 25— (3) の方法に準じて、 目的化合物 [55] 9m gを黄色 固体として得た。
化合物 [55] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 69 (s, 1 H), 8. 51 ( s , 1H), 8. 4 1 (m, 1 H), 7. 44— 7. 20 (m, 5H), 6. 60— 6. 50 (m, 2H), 6. 04 (m, 1H), 5. 1 7 (m, 1H), 1. 63 (d, J = 7. 2Hz, 3H). ma s s : 356 (M+ 1 ) +. 実施例 56
6— (8—メチルイミダゾ [ 2— a] ピリジン一 3—ィル) — N— [(1 S) 一 1一フエニルェチル] — 9H—プリンー 2—ァミン [56] (以下、 化合物 [56] という) の合成。 (1) 水素化ナトリウム 1. 54 gと N, N—ジメチルホルムアミ ド 10 OmLの 混合物に、氷冷下、 2, 6—ジクロロプリン 5 gを加え、同温にて 15分間撹拌した。 反応混合物に、 2— (トリメチルシリル) エトキシメチルクロリ ド 6. 81 m Lをカロ え、 同温にて 5時間撹拌した。 反応混合物に水を加え、 酢酸ェチルにて抽出した。 有 機層を水、 飽和食塩水にて順に洗浄した後、'無水硫酸マグネシウムで乾燥した。 不溶 物をろ過し、 ろ液を減圧濃縮した後、 シリカゲルカラムクロマトグラフィーにて精製 し、 2, 6—ジクロロー 9一 {[2— (トリメチノレシリル) エトキシ] メチル } —9 H—プリン [56— 1] 6. 18 gを淡褐色油状物として得た。
(2) ェチルェチュルエーテル (50%v/vへキサン溶液) 2 gをテトラヒ ドロ フラン 2 OmLに溶解し、 氷冷下、 ボランーテトラヒ ドロフラン錯体 (1. 0Mテト ラヒドロフラン溶液) 4. 7 m Lを加えた。 混合物を室温にて 3時間撹拌した後、 テ トラヒ ドロフラン 50mL、 2, 6—ジクロロー 9一 {[2— (トリメチルシリル) エトキシ] メチル } 一 9H—プリン [56— 1] 3 g、 トリフエニルホスフィン 22 2mg、酢酸パラジウム 63mg、 4 N—^酸化ナトリゥム水溶液 7. OmLを加え、 終夜加熱還流した。 反応混合物を室温まで戻し、 酢酸ェチルで希釈した後、 水、 飽和 食塩水にて順に洗浄し、 得られた有機層を無水硫酸マグネシウムにて乾燥した。 不溶
物をろ過し、 ろ液を減圧濃縮し、 残渣をシリカゲルカラムクロマトグラフィーにて精 製し、 2—クロロー 6— [(E)一 2—エトキシビュル] —9— {[2— (トリメチノレ シリル) エトキシ] メチル }一 9H一プリン [56— 2] 676mgを淡褐色油状物 として得た。
(3) 2—クロ口 _6— '[(E) —2—エトキシビュル]一 9一 {[2— (トリメチ ノレシリノレ) エトキシ] メチノレ }一 9H—プリン [56— 2] 400mg、 1, 4—ジ ォキサン 1 OmL、 およぴ水 2mLの混合物に、 氷冷下、 N—ブロモこはく酸イミド 20 Omgをカロえ、 同温にて 30分間撹拌した。 2—アミノー 3—ピコリン 122m gを加え、 50°Cにて 1時間撹拌した後、 反応混合物を室温まで戻した。 酢酸ェチル にて希釈して、 水、飽和食塩水にて順に洗浄し、 得られた有機層を無水硫酸マグネシ ゥムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮した後、 得られた残渣をシリカゲ ルカラムクロマトグラフィーにて精製し、 2—クロ口一 6—(8—メチルイミダゾ [1,
2— a] ピリジン一 3—ィル) 一 9一 {[2— (トリメチルシリル) エトキシ] メチ ル}一 9H—プリン [56— 3] 36 Omgを淡褐色固体として得た。
(4) 2—クロ口一 6— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 9一 {[2— (トリメチルシリル) ェトキシ] メチル }一 9H—プリン [56— 3]
3 Omg, トリェチルァミン 30 μ L、 ( 1 S ) — ( 1 )一フエニルェチルァミン 4 7 μ L、およぴジメチルスルホキシド 2mLの混合物を 120°Cにて 3日間撹拌した。 反応混合物を室温に戻し、酢酸ェチルにて希釈し、水、飽和食塩水にて順に洗浄した。 得られた有機層を無水硫酸マグネシウムで乾燥し、 不溶物をろ過し、 ろ液を減圧濃縮 した。残渣を分取薄層クロマトグラフィーにて精製し、 6—(8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 N— [(1 S)一 1—フエニルェチル]一 9一 {[2 一(トリメチルシリル) エトキシ] メチル }一 9H—プリンー 2—ァミン [56— 4] 25 m gを白色固体として得た。 .
(5) 6—(8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 N— [(1 S)一 1一フエニルェチル]一 9一 {[2—(トリメチルシリル) エトキシ] メチル } — 9 H—プリン一 2—ァミン [56— 4] 25 m gを 90 %トリフルォロ酢酸水溶液 1 OmLに溶解し、 室温にて 1時間静置した。 反応混合物を減圧濃縮した後、 残渣を 逆相分取液体クロマトグラフィーにて精製し、 目的化合物 [56] のトルフルォロ酢 酸塩 7. 6 mgを淡黄色固体として得た。
化合物 [56] のスぺク トルデータを以下に示す
:Η— NMR (CD3OD) δ : 9. 1 7 ( s , 1 Η), 8. 08 (s , 1 Η), 7. 8 2 (d, J = 6. 8Hz, 1 Η), 7. 47— 7. 45 (m, 2Η), 7. 34— 7.
30 (m, 4H), 7. 20— 7. 17 (m, 1H), 5. 15— 5. 10 (m, 1H), 2. 67 (s, 3H), 1. 59 (d, J = 7. OH z , 3 H)
ma s s : 370 (M+ 1 ) +. 実施例 57
6 f ミダゾ [1, 2— a'] ピリジン一 3—ィルー N— [(1 S)一 1一フエニルェ チル]— 9 H—プリン一 2—ァミン [57] (以下、化合物 [57] という) の合成。
2—ァミノピリジン 87mgと 2—ク口ロー 6— [(E)一 2—ェトキシビエル] 一 9一 {[2— (トリメチルシリル) エトキシ] メチル }一 9H—プリン [56— 2] 327. 5mg力 ら、 実施例 56— (3) 〜 (5) の方法に準じて、 目的化合物 [5 7] トルフルォロ酢酸塩 3. 4m gを淡黄色固体として得た。
化合物 [57] のスぺクトルデータを以下に示す。
ΧΗ— NMR (CDC 13) δ : 9. 26 (s, 1 H), 8. 08 (d, J = 9. 2H z , 1 H), 7. 95 (s, 1H), 7. 77— 7. 73 (m, 1 H), 7. 48— 7. 45 (m, 2H), 7. 41— 7. 38 (m, 2H), 7. 34 ( s , 2H), 7. 3 0— 7. 26 (m, 1H), 7. 1 1 (b r s, 1H), 5. 16— 5. 1 1 (m, 1 H), 3. 39— 3. 38 (m, 1 H), 1. 66 (d, J = 7. 0Hz, 3 H) ma s s : 356 (M+ 1) +. 実施例 58
5—ブロモ一4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 N— [(1 S)一 1—フエ二ルェチル]一 2—ピリジンアミン [58] (以下、 化合物 [5 8] という) の合成。
(1) 2—フルォロ一 4一ョードピリジン ( J . Or g, Ch em. 1 993, 5 8, 7832— 7838に記載されている方法により合成した) l g、 (I S) — 1 一フエ-ルェチルァミン 1. 73mL、 およびジメチルスルホキシド 2 OmLの混合 物を、 80°Cにて終夜撹拌した。反応混合物を室温に戻した後、酢酸ェチルで希釈し、 水、飽和食塩水にて順に洗浄した。 得られた有機層を無水硫酸マグネシウムで乾燥し た後、 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をシリカゲルカラムクロマ トグラフィ一にて精製し、 4—ョードー N— [(1 S) —1一フエニルェチル]一 2 一ピリジンアミン [58— 1] 744m gを淡褐色固体として得た。
(2) ェチルェチェルエーテル (50%v/vへキサン溶液) 1. 3mLをテトラ ヒ ドロフラン 1 OmLに溶解し、 氷冷下、 ボランーテトラヒ ドロフラン錯体 (1. 0
Mテトラヒドロフラン溶液) 2. 04mLを 30分かけて滴下した後、反応混合物を 室温まで昇温させ、 さらに 3時間撹拌した。 4ーョードー N— [(1 S) — 1一フエ ニルェチル]一 2—ピリジンアミン [58— 1] 33 Omgのテトラヒドロフラン溶 液 1 OmL、 酢酸パラジウム 1 1 · 5mg、 トリフエニルホスフィン 40. 2mg、 および 4 N—水酸化ナトリウム水溶液 1. 28mLを加え、 さらに室温にて 5時間撹 拌した。 反応混合物を酢酸ェチル 15 OmLにて希釈し、 水、 飽和食塩水にて順に洗 浄し、 有機層を無水硫酸マグネシウムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮 し、 残渣をシリカゲルカラムクロマトグラフィ一にて精製し、 4— [(E)一 2—ェ トキシェテュル]一 N— [(1 S)一 1一フエニルェチル]一 2—ピリジンアミン [5 8— 2] 94m gを褐色油状物として得た。
(3) 4— [(E)一 2—エトキシェテュル]一 N— [(1 S) —1一フエ二ルェチ ル] — 2—ピリジンアミン [58— 2] 94mgを 1, 4—ジォキサン 3 m Lと水 1 mLの混合溶媒に溶解し、 N—ブロモこはく酸イミド 28mgをカロえた。 室温にて 1 0分間撹拌した後、 再び、 N—プロモこはく酸イミ ド 28 m gを加え、 室温にて 30 分間撹拌した。 2—アミノー 3—ピコリン 80 Lを加え、 50°Cにて 1時間撹拌し た。 反応混合物を室温まで戻した後に、 酢酸ェチノレにて希釈し、 水、 飽和食塩水にて 順に洗浄した。 有機層を無水硫酸マグネシウムで乾燥し、 不溶物をろ過し、 ろ液を減 圧濃縮した。得られた残渣を分取薄層クロマトグラフィ一にて精製し、目的化合物 [ 5 8] 16mgを淡黄色油状物として得た。 化合物 [58] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 32 (s, 1H), 7. 72 (s, 1 H), 7. 3 7— 7. 28 (m, 6H), 7. 00 (d, J = 6. 8Hz, 1 H), 6. 56 (d d, J = 6. 8Hz, 7. OHz, 1H), 6. 23 (s, 1H), 5. 26 (d, J = 6. 4Hz, 1 H), 4. 64 (d t, J = 6. 4H z , 6. 8Hz, 1H), 1. 57 (d, J = 6. 8Hz, 3H)
ma s s : 407, 409 (M+ 1 ) +. 実施例 59
3— (5—シァノー 2— {[(I S) — 1一フエ-ルェチル] アミノ} —4一ピリミジ ニル)一8—メチルイミダゾ [1, 2— a] ピリジン一 7—力ルボン酸 [59] (以 下、 化合物 [59] という) の合成。 化合物 [48J 13. 6mg、 メタノール lmL、 テトラヒドロフラン lmL、 水 0. 5mL、 および 1 N— 7_R酸ィ匕ナトリゥム水溶液 165 μ Lの混合物を 40°Cにて
2時間撹拌した後、 2 N—塩酸水溶液 83 Lにて中和し、 減圧濃縮した。 得られた 残渣を逆相分取液体ク口マトグラフィ一にて精製し、 目的化合物 [ 59 ] 12 m gを 淡黄色固体として得た。
化合物 [59] のスぺクトルデータを以下に示す。
'H— NMR (DMSO— d6) δ : 9. 96 (d, J = 7. 4Hz, 1HX 1/2), 9. 07— 8. 66 (m, '3 H+ 1 HX 1/2), 7. 53— 7. 20 (m, 6H),
5. 30— 5. 04 (m, 1 H), 4. 02 (b r, 1H), 2. 88 (s, 3HX 1 /2), 2. 83 (s, 3HX 1/2), 1. 54 (d, J = 6. 9H z, 3HX 1/ 2), 1. 51 (d, J = 6. 9Hz, 3HX 1/2).
ma s s : 399 (M+ 1) +. 実施例 60、 61
t一ブチル N— [3— (5—シァノー 2— {[(1 S)一 1ーフヱニルェチル] アミノ} 一 4一ピリミジェル)一 8—メチルイミダゾ [1, 2— a] ピリジン一 7—ィル] 力 ルバメート [60] (以下、 化合物 [60] という)、 および 4— (7—アミノー 8— メチルイミダゾ [1, 2— a],ピリジン一 3—ィル)一2— {[(1 S)一 1—フエ二 ルェチル] アミノ }一 5—ピリミジンカルボ二トリノレ [ 61 ] (以下、 化合物 [61] という) の合成。 化合物 [59] 9mg、 トリエチルァミン 9. 2 L, Ν, Ν—ジメチルホルムァ ミ ド 0. 3mL、 および 1, 4一ジォキサン 0. 2mLの混合物に、 氷冷下、 ジフエ ニルホスホリルアジド 7. 1 Lを加え、 室温にて 1時間撹拌した。 続いて tーブチ ルアルコール 0. lmLを加え、 80°Cにて 6時間撹拌した後、 室温に戻しクロロホ ルム、 水、 飽和炭酸水素ナトリウム水溶液を加えた。 混合物をクロ口ホルムにて抽出 し、 得られた有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリゥムにて乾燥した。 不 溶物をろ過し、 ろ液を減圧濃縮した後、得られた残渣を逆相分取液体クロマトグラフ ィ一にて精製し、 目的化合物 [60] 1. 6mgを白色固体として、 目的化合物 [6 1] 4. lmgを黄色固体として得た。
化合物 [60] および [6 1] のスぺクトルデータを以下に示す。
化合物 [60]
ΧΗ— MR (CDC 13) δ 8. 98— 8. 86 (m, 1 H), 8. 54— 8. 4 5 (m, 1 H), 7. 76— 7. 62 (m, 1 H), 7. 59— 7. 10 (m, 6H),
6. 63— 6. 54 (m, 1 H), 6. 12— 6. 02 (m, 1 H), 5. 38— 5. 03 (m, 1 H), 2. 53 (s, 3H), 1. 58 (d, J = 6. 9Hz, 3H), 1. 26 (s, 9H).
ma s s : 470 (M+ 1 ) +.
化合物 [61]
ΧΗ— NMR (CDC 13) δ : 8 83 ( s , 1 Η), 8. 67 (d, J = 7. 4Η ζ, 1Η), 8. 45 ( s , 1 Η), 7. 45— 7. 20 (m, 6H), 6. 18— 5. 97 (m, 1 Η), 5. 20— 5. 02 (m, 1H), 4. 11 (b r , 2H), 2. 40 (s , 3Η), 1. 62 (d, J = 6. 9Hz, 3H).
ma s s : 370 (Μ+ 1) +. 実施例 62
4— [7— (ヒ ドロキシメチル) 一 8—メチルイミダゾ [1 — a] ピリジン一 3 一ィル) 一 2— {[(1 S) _1—フユニルェチル] アミノ} 一ピリ ミジンカルボ 二トリル [62] (以下、 化合物 [62] という) の合成。 化合物 [59] 11. 4mgをテトラヒ ドロフラン 2mLに溶解し、 1, 1, 一力 ノレポ-ノレジィミダゾール 23. 2 m gをカ卩え、 室温にて 6時間撹拌した。 水素化ほう 素ナトリウム 5. 4mgを加え、 同温にて 30分間撹拌した後、 飽和炭酸水素ナトリ ゥム水溶液を加えた。 反応混合物をクロ口ホルム一メタノール (混合比 9 : 1) の混 合溶媒にて抽出し、有機層を飽和食塩水にて洗浄した後、 無水硫酸ナトリウムで乾燥 した。 不溶物をろ過し、 ろ液を減圧濃縮し、 残渣を分取薄層クロマトグラフィーにて 精製し、 目的化合物 [62] 4. 4mgを白色固体として得た。
化合物 [62] のスペク トルデータを以下に示す。
ΧΗ— NMR (CD3OD) δ 8. 78— 8. 50 (m, 2H), 7. 98— 7. 9 2 (m, 1H), 7. 47— 6. 97 (m, 8H), 5. 34— 5. 05 (m, 1 H), 4. 83— 4. 71 (m, 2H), 2. 65 (s, 3HX 1/2), 2. 52 (s, 3 HX 1 2), 1. 59 (d, J = 6. 9Hz, 3H).
ma s s : 385 (M+ 1 ) +. 実施例 63
4一 { 8—メチノレー 7— [(メチルァミノ) メチル] イミダゾ [1, 2— a] ピリジ ンー 3—イノレ}—2— {[(1 S) —1—フエニルェチル] アミノ}一 5_ピリミジン カルボ二トリル [63] (以下、 化合物 [63] という) の合成。 化合物 [62] 3. 3mg、 N, N—ジイソプロピルェチルァミン 10. 5 μ L、 およぴクロロホルム 1. 5 mLの混合物に、 氷冷下、 塩化メタンスルホニル 3. 3 μ Lを加え、 同温にて 30分間撹拌した。 更に、 Ν, Ν—ジイソプロピルェチルァミン 3. 8 L、 および塩ィ匕メタンスルホニル 1. 7 Lを加え、 同温にて 25分間撹拌
した後、 ヨウ化ナトリウム 19. 3m g、 およぴメチルァミン (40%メタノール溶 液) 0. 5mLを加え、 60°Cにて 1時間撹拌した。 反応混合物を室温に戻した後、 飽和炭酸水素ナトリゥム水溶液と水を加え、 クロロホルムにて抽出した。 得られた有 機層を飽和食塩水にて洗浄した後、無水硫酸ナトリゥムで乾燥した。不溶物をろ過し、 ろ液を減圧濃縮し、得られた残渣を分取薄層クロマトグラフィーにて精製し、 目的化 合物 [63] 1. 8m gを'白色固体として得た。
化合物 [63] のスぺクトルデータを以下に示す。
NMR (CDC 13) δ : 8. 97— 8. 80 (m, 2H), 8. 60— 8. 4 7 (m, 1 H), 7. 44— 7. 10 (m, 6H), 6. 86— 6. 75 (m, 1 H), 6. 07— 5. 78 (m, 1 H), 5. 43— 5. 06 (m, 1 H) , 3. 91— 3. 80 (m, 2H), 2. 64 (s, 3H), 2. 52 (s, 3H), 1. 59 (d, J =6. 9Hz, 3H).
ma s s : 398 (M+ 1 ) +. 実施例 64
4一 { 7— [(4—メ トキシべ ^ジル) ォキシ]一 8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル }—2— {[(1 S)一 1一フエニルェチル] アミノ}—5—ピリ ミジンカルボ二トリル [64] (以下、 化合物 [64] という) の合成。 (1) 2—クロロー 4—ョードピコリン 234mg、 ヨウィ匕同 17. 6mg、 1, 10—フエナント口リン 33. 3m g、 炭酸セシウム 60 lmg、 4ーメ トキシベン ジルアルコール 2'30 μ L、 およびトルエン 0. 5mLの混合物を、 1 10°Cにて 4 時間撹拌した。 水と飽和炭酸水素ナトリウム水溶液を加えた後、 クロ口ホルムにて抽 出し、 有機層を飽和食塩水にて洗浄した。 得られた有機層を無水硫酸ナトリゥムで乾 燥し、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣を分取薄層クロマトダラ フィ一にて精製し、 2—クロロー 4一 [(4ーメ トキシベンジル) 才キシ]一 3—メ チルピリジン [64— 1] 207m gを白色固体として得た。
(2) 2—クロロー 4一 [(4ーメ トキシベンジル) ォキシ] —3—メチルピリジ ン [64— 1] 147mgから、 実施例 48— (2) の方法に準じて、 4一 [(4— メトキシベンジル) ォキシ]一 3—メチルー 2—ピリジンアミン [64— 2] 101 mgを黄色固体として得た。
(3) 4一 [(4ーメ トキシベンジル) ォキシ]一 3—メチルー 2—ピリジンアミ ン [64— 2] 1 19m gと 4一 [(Z) — 2—エトキシビニル]一 2— (メチルチ ォ)一 5—ピリミジンカルボ二トリル [48— 3] 108mg力 ら、実施例 25—(3)
〜 (5) の方法に準じて、 目的化合物 [64] 2 lmgを黄色固体として得た。 化合物 [64] のスぺクトルデータを以下に示す。
ΧΗ— NMR (CDC 13) δ : 8. 89 (s, 1H), 8. 82— 8. 71 (m, 1 H), 8. 46 (s, 1 H), 7. 44— 7. 26 (m, 7H), 7. 01— 6. 88 (m, 2H), 6. 48— 6. 40 (m, 1 H), 6. 06— 5. 96 (m, 1 H), 5. 16 (s, 2H), 5.' 22— 5. 00 (m, 1H), 3. 84 (s, 3H), 2. 50 (s , 3H), 1. 63 (d, J = 6. 9 H z , 3H).
ma s s : 491 (M+ 1) +. 実施例 65
4一 (7—ヒドロキシー 8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル }― 2— {[(1 S)— 1一フエニノレエチノレ] アミノ}一 5—ピリミジンカルボ二トリル [6 5] (以下、 化合物 [65] という) の合成。 化合物 [64] 2 Omgをクロ口ホルム 1. 5mLに溶解し、 氷冷下、 トリフルォ 口酢酸 1. 5mLを加え、 室 にて 30分間撹拌した。 減圧濃縮し、 得られた残渣を 逆相分取液体クロマトグラフィーにて精製し、 目的化合物 [65] 19mgを淡黄色 油状物として得た。
化合物 [65] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 63 (s, 1 H), 8. 56 (s, 1H), 8. 3 3 (d, J = 7. 6H z , 1H), 7. 47— 7. 23 (m, 6 H), 7. 08— 7. 06 (m, 1 H), 6. 92— 6. 90 (m, 1 H), 5. 02— 4. 99 (m, 1 H), 2. 14 ( s , 3H), 1. 65 (d, J = 6. 8Hz, 3H).
ma s s : 371 (M+ 1 ) +. 実施例 66
4—イミダゾ [1, 2— a] ピリミジン一 3—^ fノレ一 2— {[(1 S) — 1—フエニル ェチル] アミノ} 一 5—ピリミジンカルボ二トリル [66] (以下、 化合物 [66] という) の合成。
4— [(Z) — 2—エトキシビュル] —2— (メチルチオ) 一 5—ピリミジンカル ボニトリル [48— 3] 10 Omgと 2—ピリミジンアミン 48mg力 ら、 実施例 2 5— (3) 〜 (5) の方法に準じて、 目的化合物 [66] 15m gを淡黄色固体とし て得た。
化合物 [66] のスぺクトルデ一タを以下に示す。
'H— NMR (CDC 13) δ : 9. 07 ( s , 1 Η), 8. 94— 8. 92 (m, 1
H), 8. 62-8. 60 (m, 1 H), 8. 58 (s, 1H), 7. 45-7. 34 (m, 6H), 6. 69 -6. 66 (m, 1H), 6. 14-6. 12 (m, 1 H), 5. 08— 5. 04 (m, 1 H), 1. 66 (d, J = 7. 2Hz, 3 H)
ma s s : 342 (M+ 1) +. 実施例 67〜実施例 78 '
4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— {[(1 S) — 1一フエ-ルプロピル] ァミノ)一 5—ピリミジンカルボ-トリル [67] (以下、 化合物 [67] とレ、う)、 2— [(3—ヒドロキシ一 1一フエニルプロピル) ァミノ] —4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 5—ピリミジン カルボ二トリル [68] (以下、 化合物 [68] という)、 4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— [(3—フヱ-ルプロピル) ァミノ] — 5—ピリミジンカルボ二トリノレ [69] (以下、 化合物 [69] という)、 4— (8— メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— {[2— (2—チェニル) ェチル] アミノ} —5—ピリミジンカルボ二トリル [70] (以下、 化合物 [70] とレヽう)、 4— (8—メチルイ ^;ダゾ [1 , 2— a] ピリジン一 3—ィル)一 2— [(3 —ピリジニルメチル) ァミノ]一 5—ピリミジンカルボ二トリル [71] (以下、 化 合物 [71] という)、 2— [(2—クロ口ベンジル) ァミノ] 一 4一 (8—メチルイ ミダゾ [1, 2— a] ピリジン一 3—ィル)一 5—ピリミジンカルボ二トリノレ [72] (以下、化合物 [72] という)、 2— [(3—メ トキシベンジル) ァミノ]一 4一 (8 ーメチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 5—ピリミジンカルボ-ト リル. [73] (以下、 化合物 [73] という)、 2— [(4—メ トキシベンジル) アミ ノ] —4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 5—ピリミ ジンカルボ二トリル [74] (以下、 化合物 [74] という)、 2— [(4—ァミノべ ンジル) ァミノ] —4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) —5—ピリミジンカルボ二トリル [75] (以下、 化合物 [75] という)、 4一 (8 ーメチルイミダゾ [1, 2— 3 ] ピリジン一 3—ィル) 一 2— {[2— (トリフルォ ロメチル) ベンジル] アミノ}一 5—ピリミジンカルボ二トリル [76] (以下、 ィ匕 合物 [76] という)、 2— {[1— (4一フルオロフェニル) ェチル] アミノ}一 4 ― (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 5—ピリミジンカル ボニトリル [77] (以下、 化合物 [77] という)、 および 2— [(2, 6—ジフル ォロベンジル) ァミノ]一 4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3— ィル) 一 5—ピリミジンカルボ二トリル [78] (以下、 化合物 [78] という) の 合成。
(1) 4— [(Z) 一 2—エトキシビュル]一 2— (メチルチオ) 一 5—ピリミジ
ンカルボ二トリル [48— 3]と 2—アミノー 3—ピコリンから、実施例 25—(3)、
(4) の方法に準じて、 4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィ ル)一 2—メチルスルホニル)一 5—ピリミジンカルボ-トリル [67—1]を得た。 (2) 4_ (8—メチルイミダゾ [1, 2— a] ピリジン一 3—^ fル) 一 2—メチ ルスルホ -ル) 一 5—ピリ'ミジンカルボ二トリル [67— 1] と、 (1 S) — 1ーフ ェ-ルー 1—プロパンァミン (実施例 67の場合)、 (3 S) — 3—アミノー 3—フエ 二ルー 1一プロパノール (実施例 68の場合)、 3—フヱ-ルー 1—プロパンァミン (実施例 69の場合)、 2— (2—チェニル) 一1一エタンァミン (実施例 70の場 合)、 3—ピリジニルメタンァミン (実施例 71の場合)、 (2—クロ口フエニル) メ タンアミン (実施例 72の場合)、 (3—メ トキシフエニル) メタンァミン (実施例 7 3の場合)、 (4ーメ トキシフヱニル) メタンァミン (実施例 74の場合)、 4一 (ァ ミノメチル) ァニリン (実施例 75の場合)、 [ 2—(トリフルォロメチル) フエ-ノレ] メタンァミン (実施例 76の場合)、 1— (4一フルオロフェニル) 一 1一エタンァ ミン (実施例 77の場合)、 もしくは (2, 6—ジフルオロフェニル) メタンァミン (実施例 78の場合) 、 案施例 25— (5) の方法に準じて目的化合物 [67] 〜 [78] を得た。
化合物 [67] 〜 [78] のスぺク トルデータを以下に示す。
化合物 [67]
XH— NMR (CDC 13) δ : 9. 62 (m, 1 HX 1/3), 8. 98-8. 80 (m, 1H+ 1 HX 2/3), 8. 52 (b r , 1 H), 7. 47-7. 12 (m, 6
H), 6. 94 (m, 1 HX 1/3), 6. 66 (m, 1 HX 2/3), 6. 10 (m,
1 HX 2/3), 5. 88 (m, 1 HX 1/3), 5. 10 (m, 1 HX 1/3), 4.
92 (m, 1 HX 2/3), 2. 68 (b r, 3H), 1. 98 (m, 2H), 1. 0 3 (m, 3H).
ma s s : 369 (M+ 1 ) +. 化合物 [68]
ΧΗ— NMR (CDC 13) δ : 9. 63 (b r d, J = 7. 2H z , 1 HX 1/3), 8. 92 (b r , 1 HX 1/3), 8. 84 (b r, 1 HX 2/3), 8. 72 (b r d, J = 7. 2Hz, 1 HX 2/3), 8. 42 (b r, 1 H), 7. 48-6. 90 (m, 6H), 6. 60 (m, 1 HX 2/3), 6. 39 (m, 1 HX 1/3), 5.
42 (m, 1 HX 1/3), 5. 29 (m, 1 HX 2/3), 3. 85 (m, 2H),
2. 68 (b r , 3HX 1/3), 2. 63 (b r, 3HX 2/3), 1. 20 (m, 2H).
ma s s : 385 (M+ 1) +.
化合物 [6 9]
XH— NMR (CDC 13) δ : 9. 70 (m, 1H), 9. 0 9 (s, IHX 1/2), 8. 98 (s, IHX 1/2), 8. 59 (s, I HX l/2), 8. 5 1 ( s , 1 H X 1/2), 7. 49 (m, 1 H), 7. 36— 7. 02 (m, 5H), 6. 8 3 (m, 1 H), 6. 72 (m, 1 H), 3. 60 (m, 2H), 2. 78 (m, 2H), 2. 7 5 (s, 3HX 1/2), 2. 6 8 (s, 3HX 1 2), 2. 09 (m, 2H). ma s s : 36 9 (M+ 1) +. 化合物 [70]
ΧΗ— NMR (CDC 13) δ : 9. 70 (b r d, J = 7. 5 H z , IHX 1/2), 9. 59 (b r d, J = 6. 9Hz IHX 1/2), 9. 09 ( s, IHX 1/2), 8. 99 (s, IHX 1/2), 8 68 (s, IHX 1/2), 8. 56 ( s , 1 H X 1/2), 7. 60 (m, 1 H), 7. 3 1 -6. 50 (m, 4H): 3. 88 (m, 2H), 3. 24 (m, 2H), 2. 78 ( s , 3HX 1/2), 2. 73 ( s , 3 H X 1/2). .
m a s s : 36 1 (M+ 1 ) +. 化合物 [7 1]
'Η— NMR (CDC 13) δ : 9. 92 (b r d, J = 7. 5H z, IHX 2/3), 9. 14 (m, IHX 1/3), 9. 1 5— 8. 84 (m, 2H), 8. 75— 8. 5
7 (m, 2H), 8. 4 7 (m, 1 HX 2/3), 8. 1 7 (m, IHX 1/3), 7.
8 7 (m, I HX 2/3), 7. 6 9 (m, IHX 1/3), 7. 49 (m, 1 HX 2 /3), 7. 3 7 (m, IHX 1/3), 7. 20 (m, IHX 2/3), 6. 96 (m, I HX 1/3), 4. 90 (b r , 2H), 2. 70 ( s , 3H).
ma s s : 342 (M+ 1 ) +. 化合物 [72]
XH— NMR (CDC 13) 5 : 9. 1 0 (b r d, J = 7. 5H z, 1 H), 9. 4 (s, 1 H), 8. 6 8 ( s, 1 H), 7. 60-6. 9 7 (m, 5H), 6. 8 (m, 1 H), 4. 8 5 (b r d, J = 6. OH z , 2H), 2. 7 1 (s , 3H). ma s s : 3 75, 3 7 7 (M+ 1) +. 化合物 [73]
XH— NMR (CDC 13) δ : 9. 72 (b r d, J = 7. 2H z, I HX 1/3), 9. 05-8. 9 7 (m, 1 H+ I HX 2/3), 8. 6 6 (s, 1 HX 1/3), 8.
t
Or 5 3
0 3
X 1/3), 9. 01 ( s , 1 H), 8. 72 ( s , 1 HX 1/3), 8. 58 ( s , 1 HX 2/3), 7. 52 (m, 1 H), 7. 48-6. 56 (m, H), 4. 90 (b r d, J = 6. 3Hz, 2H), 2. 77 ( s ' 3HX 2/3), 2. 74 ( s , 3HX 1/3).
ma s s : 377 (M+ l) +. 実施例 79
2— {[1— (4—ヒドロキシフエニル) ェチル] アミノ}一 4一 (8—メチルイミ ダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—ピリミジンカルボ二トリノレ [79] (以下、 化合物 [79] という) の合成。
(1) 1— (4ーヒ ドロキシフエ二ノレ) — 1ーェタノン 5 g、 ィミダゾ一/レ 3 g、 および N, N—ジメチルホルムアミ ド 7 OmLの混合物に t一ブチルクロロジフエ二 ルシラン 1 1. 7mLを加え、 室温にて終夜撹拌した。 反応混合物に酢酸ェチル 20 OmLを加え、 水、 飽和食塩水にて順に洗浄し、 得られた有機層を無水硫酸マグネシ ゥムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をシリカゲル力 ラムクロマトグラフィーにて精製し、 1一 (4一 {[t一プチル (ジフエ二ノレ) シリ ル] ォキシ } フエニル) 一 1一エタノン [79— 1] 1 1. 7 gを白色固体として得 た。
(2) 1— (4一 {[t一ブチル (ジフエニル) シリル] ォキシ } フエニル) — 1 一エタノン [79— 1] 5 gをメタノール 5 OmLとテトラヒドロフラン 3 OmLの 混合溶媒に溶解し、 水素化ホゥ素ナトリウム 606mgを加え、 室温にて 3時間撹拌 した。 反応混合物に水 3 OmLを加え、 減圧濃縮した。 残渣に酢酸ェチル 15 OmL を加え、 水、 飽和食塩水にて順に洗浄し、 得られた有機層を無水硫酸マグネシウムで 乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をシリカゲルカラムク 口マトグラフィ一にて精製し、 1— (4— {[t—ブチル (ジフエニル) シリル] ォ キシ }フヱ-ル)一 1一エタノール [79— 2] 5. 02 gを無色油状物として得た。 (3) 1— (4一 {[t一ブチル (ジフヱニル) シリル] ォキシ } フエニル) 一 1 一エタノール [79— 2] 5. 02 gをテトラヒ ドロフラン 2 OmLに溶解し、 ジフ ェニルホスホリルアジド 3. 451111^と 1, 8—ジァザビシクロ [5. 4. 0] ゥン デカ一 7—ェン 2. 44mLを加え、 室温にて終夜撹拌した。 反応混合物に酢酸ェチ ル 1 O OmLをカロえ、 水、飽和食塩水にて順に洗浄し、 得られた有機層を無水硫酸マ グネシゥムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をシリカ ゲルカラムクロマトグラフィーにて精製し、 1一 [1— (4— {[t一ブチル (ジフ
ェニル) シリル] 才キシ } フエニル) ェチル] — 1 , 2—トリァザジェンー 2—ィゥ ム [7 9— 3] 4. 2 6 gを淡褐色油状物として得た。
(4) 1一 [1— (4一 {[ t一ブチル (ジフヱニル) シリル] ォキシ } フヱニル) ェチル]一 1, 2—トリァザジェンー 2—ィゥム [7 9— 3] 4. 2 6 gをエタノー ル 3 0mLに溶解し、 触媒量の 1 0%パラジウム炭素触媒を加え、 水素雰囲気下、 終 夜撹拌した。 不溶物をセライトにてろ過し、 エタノールにて洗浄した。 ろ液を減圧濃 縮し、 1一 (4— { [ tーブチノレ (ジフエニル) シリル] 才キシ } フエニル) ェチル ァミン [7 9— 4] 3. 94 gを淡褐色油状物として得た。
(5) 4一 (8—メチルイミダゾ [1, 2— a] ピリミジン一 3—ィル) 一 2—メ チノレスノレホニノレ)一 5—ピリミジン力ルポ二トリノレ [6 7— 1] 1 0 Omgと 1— (4 ― { [ t一ブチル (ジフエニル) シリル] 才キシ } フエニル) ェチルァミン [7 9— 4] 40 Omgから、 実施例 2 5— (5) の方法に準じて 4一 (8—メチルイミダゾ [1 , 2— a] ピリジン一 3—ィル) 一 2— {[1一 (4一 {[ t一プチル (ジフエ二 ル) シリル] ォキシ } フエ二 ェチル] アミノ} —5—ピリミジンカルボ二トリル [7 9— 5] 9 8mgを淡黄色油状物として得た。
(6) 4一( 8—メチルイミダゾ [ 1 , 2— a] ピリジン一 3—ィル)一 2— {[ 1 ― (4— { [ t一ブチル (ジフヱニル) シリル] ォキシ } フエニル) 工チル] ァミノ) 一 5—ピリミジンカルボニトリル [7 9— 5] 9 8mgをテトラヒドロフラン 3mL に溶解し、 ふつ化テトラブチルアンモニゥム (1. 0Mテトラヒドロフラン溶液) 1
6 1 μ Lを加え、室温にて 1時間撹拌した。反応混合物に酢酸ェチル 7 OmLを加え、 p H6. 8リン酸バッファー、 水、 飽和食塩水にて順に洗浄し、 得られた有機層を無 水硫酸マグネシウムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残 渣を少量のク口口ホルムとメタノールの混合溶媒 (クロ口ホルム:メタノ一ル= 9 :
1) に溶解し、 へキサンにて固化し、 生じた固体をろ取し、 目的化合物 [7 9] 4 5 m gを淡黄色固体として得た。
化合物 [7 9] のスぺク トルデータを以下に示す。
XH— NMR (DMS O— d 6) δ : 9. 9 7 (d, J = 6. 8H z , 1 HX 2/5),
9. 2 6 (d, J = 5. 9 H z , 1 H), 9. 0 1 (d, J = 6. 8H z, 1 HX 3 /5), 8. 8 8 (d, J = 7. 4H z, 1 HX 3/5), 8. 8 0 (d, J = 8. 2 H z , 1 HX 2/5), 8. 74— 8. 7 1 (m, 1 H+ 1 HX 2/5), 8. 6 0 ( s ,
1 HX 3/5), 7. 4 2— 7. 3 5 (m, 1 H), 7. 2 2— 7. 1 9 (m, 2H), 7. 1 0 ( t, J = 6. 8H z , 1 HX 2/5), 6. 9 3 ( t , J = 6. 8 H z,
1 HX 3/5), 6. 7 5— 6. 7 0 (m, 2H), 5. 1 9— 5. 1 5 (m, 1 HX
2/5), 5. 01—4. 98 (m, 1 HX 3/5), 2. 58 ( s , 3HX 2/5), 2. 54 ( s , 3HX 3/5), 1. 48 (d, J = 7. OH z , 3HX 2/5), 1. 46 (d, J = 7. OHz, 3HX 3/5)
ma s s : 371 (M+ 1) +. 実施例 80 '
2— {[(1 S) — 1— (3—ァミノフエニル) ェチル] アミノ} —4— [8— (ジフ ルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 5—力ノレ ボニトリル [80] (以下、 化合物 [80] という) の合成。
(1) 2_クロ口ニコチンァノレデヒ ド 9. 6 gをジクロロメタン 15 OmLに溶解 し、 氷冷下、 ジェチルァミノ硫黄トリフルォリ ド 22. 2mLを 10分かけて滴下し た。 0 °Cにて 40分間撹拌した後、 飽和炭酸水素ナトリウム水溶液 10 OmLを注意 深く加え、 クロ口ホルムにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナ トリウムにて乾燥した後に、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣を シリカゲルカラムクロマトグラフィーにて精製し、 2—クロロー 3— (ジフルォロメ チル) ピリジン [ 80— 1 ] 10. 1 gを淡褐色油状物として得た。
(2) 2—クロ口一 3— (ジフルォロメチル) ピリジン [80— 1] 10. l g、 ビス (ジベンジリデンアセトン) パラジウム 1. 78 g、 4, 5—ビス (ジフエ-ノレ ホスフイノ)一 9, 9ージメチルキサンテン 1 · 79 g、ベンゾフエノンィミン 12. 4 g、 炭酸セシウム 28. 2 g、 トルエン 5 OmL、 および 1, 4—ジォキサン 50 mLの混合物を 80°Cにて終夜撹拌した。 室温に戻した後、 飽和炭酸水素ナトリウム 水溶液 10 OmLと水 10 OmLを加え、 クロ口ホルムにて抽出した。 有機層を飽和 食塩水にて洗浄した後、減圧濃縮し、得られた残渣をメタノール 50 m Lに溶解した。 2N—塩酸 61. 8 mLを加え 5分間撹拌した後、 0. 5 N—塩酸 400 mLを加え 10分間撹拌した。反応溶液を 5 N—水酸化ナトリゥム水溶液にて p H= 12に調整 し、 クロロホルムにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウ ムにて乾燥した後に、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣をシリカ ゲルカラムクロマトグラフィーにて精製し、 3— (ジフルォロメチル) ピリジン一 2 ーァミン [80— 2] 2. 57 gを淡褐色油状物として得た。
(3) 3—(ジフルォロメチル) ピリジン一 2—ァミン [80— 2] 2. 57 と 4一 [(Z) —2—エトキシビエル]一 2—(メチルチオ) 一 5—ピリミジンカルボ -ル [48— 3] 4. 34 gから、 実施例 25— (3) の方法に準じて、 4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2—(メチル
チォ) ピリミジン一 5—カルボ二トリル [80— 3] 4. l l gを淡黄色ァモルファ スとして得た。
(4) 4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィ ル] 一 2— (メチルチオ) ピリミジン一 5—カルボ二トリル [80— 3] 1. 91 g をクロ口ホルム 6 OmLに溶解し、氷冷下、 m—クロ口過安息香酸 1. 77 gを加え、 0 °Cにて 30分間撹拌した。飽和炭酸水素ナトリウム水溶液 l O OmLと水 100m Lを加えた後、 クロ口ホルムとメタノールの混合溶媒 (クロ口ホルム:メタノ一ル= 9 : 1) にて抽出した。 有機層を無水硫酸ナトリウムにて乾燥した後、 不溶物をろ過 し、 ろ液を減圧濃縮した。 得られた残渣をテトラヒドロフラン 6 OmLに溶解し、 3 — [(1 S) 一 1—アミノエチル] ァニリン (国際公開第 00ノ056331号パン フレット、 67— 70ページに記載されている方法により合成した) 1. l l gとト リエチルァミン 1. 65mLのテトラヒドロフラン溶液 2 OmLに加えた。 室温にて
3時間撹拌した後、 飽和炭酸水素ナトリゥム水溶液 10。!!!しをカロぇ、 酢酸ェチルに て抽出した。有機層を飽和食塩水にて洗浄し、無水硫酸ナトリゥムにて乾燥したのち、 不溶物をろ過した。 ろ液を減 i$濃縮し、得られた残渣を逆相分取液体クロマトグラフ ィ一にて精製した。得られた溶出液を飽和炭酸水素ナトリゥム水溶液にて塩基性にし た後、 クロ口ホルムにて抽出し、 有機層を飽和食塩水で洗浄し、 無水硫酸ナトリウム にて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮した後、 残渣をエタノールに加熱溶 解し、 室温にて静置した。 析出物をろ過し、 減圧乾燥し、 目的化合物 [80] 1. 2 gを白色固体として得た。
化合物 [80] めスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 89— 9. 80 (m, 1HX 1/5), 8. 90 一 9. 02 (m, 1H+ 1 HX 4/5), 8. 62 (s, 1 HX 1/5), 8. 57 (s, 1 HX 4/5), 7. 55— 7. 10 (m, 3H), 6. 88— 6. 02 (m, 4H), 6. 06— 5. 97 (m, 1 HX 4/5), 5. 86— 5. .75 (m, 1 HX 1/5), 5. 25— 5. 18 (m, 1 HX 1/5), 5. 02— 4. 88 (m, 1HX 4/5), 3. 76 (b r s, 2H), 1. 61 (d, J = 6. 8Hz, 3 H)
ma s s : 406 (M+ 1) +. 実施例 81
N— { 3— [(1 S) 一 1一 ({5—シァノー 4— [8— (ジフルォロメチル) イミダ ゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 2—ィル } ァミノ) ェチル] フ ェニル } グリシンアミ ド [81] (以下、 化合物 [81] という) の合成。 ィ匕合物 [80] 1 Omg、 N— t一ブトキシカルボニルグリシン 1 3mg、 1ーヒド
口キシベンゾトリ了ゾール 1水和物 10 m g、 N, N—ジィソプロピルェチルァミン 12. 9 を1^, N—ジメチルホルムアミ ド 0. 5mLに溶解した後、 1ーェチノレ —3— ( 3—ジメチルァミノプロピル) カルポジィミ ド塩酸塩 14. 2mgを加え、 室温にて 5時間撹拌した。 飽和炭酸水素ナトリゥム水溶液 3 OmLを力 Pえ、 クロロホ ルムにて抽出し、 有機層を飽和食塩水にて洗浄した。 無水硫酸ナトリゥムにて乾燥し た後、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣をクロ口ホルム 0. 4m Lに溶解し、 トリフルォロ酢酸 0. 4 m Lを加え、 室温にて 1時間撹拌した。 反応液 を減圧濃縮し、 得られた残渣を逆相分取液体クロマトグラフィーにて精製した。 溶出 液を減圧濃縮した後、 メタノールに溶解し、 弱陰イオン交換樹脂 (ボンドエルート ' レギュラータイプ PSA、 ジーエルサイエンス株式会社) に通しトリフルォロ酢酸 を除いた。 溶媒を減圧留去し目的化合物 [81] 4. Omgを白色固体として得た。 化合物 [81] のスぺクトルデータを以下に示す。
ΧΗ— NMR (CDC 13) δ : 10. 07 (d, J = 7. 2H z , 1 HX 1/5), 9. 04 (d, J = 7. 2H z , 1 HX 4/5), 8. 96 ( s, 1 HX 1/5), 8. 88 (s, 1 HX 4/5), 8. 58 (s, 1 HX 1/5), 8. 57 (s , 1 HX 4 /5), 7. 87— 6. 85 (m, 9H), 5. 80— 5. 48 (m, 1 HX 1/5), 5. 14— 5. 07 (m, 1 HX 4/5), 3. 45— 3. 41 (m, 2H), 2. 5 8— 2. 57 (m, 2H), 1. 64 (d, J = 6. 8Hz, 3 H)
ma s s : 463 (M+ 1 ) +. 実施例 82
4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3一^ fル] —2 ― ({(1 S) — 1一 [3— (ピロリジン一 3—ィルァミノ) フヱニル] ェチル } アミ ノ) ピリミジン一 5—カルボ二トリル [82] (以下、 化合物 [82] という) の合 成。
(1) 化合物 [80] 23. 7mgと t一ブチル 3—ォキソピロリジン一 1一カル ボキシレート 10. 8mgをクロ口ホルム 1. 5mLに溶解し、 シァノ水素化ほう素 ナトリウム 1 lmgと塩化亜鉛 12 m gをメタノール 585 μ Lに溶解した溶液を 加え、 50°Cにて終夜撹拌した。 室温に戻した後に、 飽和炭酸水素ナトリゥム水溶液 を加え、 クロ口ホルムにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナト リウムで乾燥したのち、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣を分取 薄層クロマトグラフィーにて精製し、 t一ブチル 3— ({3— [(1 S) — 1— ({5 —シァノ一 4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3— ィル] ピリミジン一 2—ィル } ァミノ) ェチル] フエ二ル} ァミノ) ピロリジン一 1 一カルボキシレート [82— 1] 8. 7 mgを淡黄色油状物として得た。
(2) t一ブチル 3— ({ 3— [(1 S) 一 1一 ({5—シァノー 4一 [8— (ジフ ルォロメチル) イ ミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 2—イノレ} ァミノ) ェチル] フエ二ル} ァミノ) ピロリジン一 1一カルボキシレート [8 2— 1] 8. 7mgをクロ口ホルム lmLに溶解し、 氷冷下、 トリフルォロ酢酸 1 m Lを加え 30分間撹拌した。 反応液を減圧濃縮し、得られた残渣を逆相分敢液体クロマトダラ フィ一にて精製した。 溶出液を減圧濃縮した後、 メタノールに溶解し、 弱陰イオン交 換樹脂に通しトリフルォロ酢酸を除いた。 溶媒を減圧留去し目的化合物 [8 2] 6. 6mgを白色固体として得た。
化合物 [82] のスぺク トルデータを以下に示す。
ΧΗ— NMR (CDC 13) δ : 9. 1 0— 8. 96 (m, 1 H+ 1 HX 4/5), 8. 88 (s, 1 H), 8. 58 ( s, 1 HX 1/5), 8. 5 2 ( d , J = 8. OH z , 1 H), 7. 6 8— 7. 1 8 (m, 3H), 6. 88— 6. 5 3 (m, 4H), 5. 2 6— 5. 1 8 (m, 1 HX 1/5), 5. 0 1— 5. 00 (m, 1 HX 4/5), 4. 1 8— 4. 0 5 (rn, 1 H), 3. 43— 3. 14 (m, 7H), 2. 28— 1. 80 (m, 2H), 1. 62 (d, J = 6. 8Hz, 3 H)
ma s s : 4 75 (M+ 1) +. 実施例 83
4— [8— (ジフノレオロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2 一 [((1 S) —1— { 3— [(2—ヒドロキシェチル) ァミノ] フヱニル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [8 3] (以下、 化合物 [83] という) の合成。 (1) 化合物 [80] 1 0 Omgと ( tーブチルジメチルシリルォキシ) ァセトァ ルデヒド 1 5 6mLから、 実施例 82— (1) の方法に準じて 2— [((1 S) — 1一 { 3— [(2— {[ t一ブチル (ジメチル) シリル] ォキシ } ェチル) ァミノ] フエ二 ル} ェチル) ァミノ]一 4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピ リジン一 3—ィル] ピリミジン一 5—カルボ二トリル [8 3— 1] 1 22mg合成し た。
(2) 2— [((1 S) —1— { 3— [(2— {[t一ブチル (ジメチル) シリル] ォ キシ } ェチル) ァミノ] フエ二ル} ェチル) ァミノ] —4— [8— (ジフルォロメチ ル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 5—カルボ二トリル [83— 1] 1 2 2mgをテトラヒ ドロフラン 2mLに溶解し、 水冷下、 フッ化テト ラブチルアンモニゥム (1. OMテトラヒドロフラン溶液) 4 33 μ Lを加え、 室温
にて 40分間撹拌した。反応混合物に飽和炭酸水素ナトリゥム水溶液を加え、 クロ口 ホルムにて抽出し、 有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥し た。 不溶物をろ過し、 ろ液を減圧濃縮して得られた残渣を分取薄層クロマトグラフィ 一にて精製し、 目的化合物 [83] 95mgを白色固体として得た。
化合物 [83] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) 'δ : 9. 70 (d, J = 7. 2H z , 1 HX 1/5), 8. 96 (s, 1HX 1/5), 8. 92 ( d , J = 7. 2H z , 1 HX 4/5), 8. 8 7 (s, 1 HX 4/5), 8. 59 ( s , 1 HX 1/5), 8. 50 ( s, 1 HX 4, 5), 7. 64— 7. 01 (m, 3H), 6. 85— 6. 59 (m, 4H), 6. 35 一 6. 34 (m, 1 HX 4/5), 6. 08— 5. 90 (m, 1 HX 1/5), 5. 2 9— 5. 18 (m, 1 HX 1/5), 5. 08— 4. 90 (m, 1 HX 4/5), 4. 20 (b r s, 1 H), 3. 87— 3. 84 (m, 2H), 3. 49 (s , 3H), 3. 31— 3. 29 (m, 2H), 1. 61 (d, J = 6. 8H z , 3 H)
ma s s : 450 (M+ 1) +. 実施例 84 ,
4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2 ― {[(1 S) — 1— (3— {[2— (メチルスルホニル) ェチル] アミノ} フヱニル) ェチル] ァミノ) ピリミジン一 5—カルボ二トリル [84] (以下、 化合物 [84] という) の合成。 化合物 [83] '25mg、 N, N—ジイソプロピルェチルァミン 19. 4 L、 ク ロロホルム 0. 7 mLの混合物に、氷冷下、塩化メタンスルホニル 6. 5 /i Lをカロえ、 同温にて 30分間撹拌した。 飽和炭酸水素ナトリゥム水溶液を加え、 クロロホルムに て抽出した。 有機層を飽和食塩水にて洗浄した後、 無水硫酸ナトリウムで乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮して得られた残渣を、 N, N—ジメチルホルムアミ ド lmLに溶解した。 メタンスルフィン酸ナトリウム 28 mgを加え、 90°Cにて 2 時間撹拌した。反応混合物を室温に戻した後、飽和炭酸水素ナトリゥム水溶液を加え、 クロ口ホルムにて抽出した。 有機層を飽和食塩水にて洗浄した後、 無水硫酸ナトリウ ムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮して得られた残渣を分取薄層クロ マトグラフィ一にて精製し、 目的化合物 [84] 4. 7mgを白色固体として得た。 化合物 [84] のスぺク トルデータを以下に示す。
XH— NMR (CDC 13) δ 9. 72— 9. 68 (m, 1 HX 1/5), 8. 99 —8. 99 (m, 1 H+ 1 HX 4/5), 8. 61 (s, 1 HX 1/5), 8. 56 ( s , 1HX 4/5), 7. 65— 7. 19 (m, 3H), 6. 85— 6. 58 (m, 4H), 6. 15— 6. 08 (m, 1 HX 4/5), 5. 98— 5. 90 (m, 1 HX 1/5),
5. 30— 5. 18 (m, 1 HX 1/5), 5. 05— 4. 90 (m, 1 HX 4/5), 4. 37— 4. 15 (m, 3H), 3. 50— 3. 32 (m, 2H), 2. 66 (s, 3H), 1. 62 (d, J = 6. 8Hz, 3 H)
ma s s : 512 (M+ 1 ) +. 実施例 85 '
4一 [8— (ジフルォロメチル) イミダゾ [1, 2— 3 ] ピリジン一 3—ィル]一 2 一.({(1 S) — 1一 [3— (ェチルァミノ) フヱニル] ェチル } ァミノ) ピリミジン 一 5—カルボ二トリル [85] (以下、 化合物 [85] という) の合成。 化合物 [ 80 ] 15 m gと t—プチルー N— ( 2ーォキソェチル)力ーバメート 8. 8111§から実施例82— (1)、 (2) の方法に準じて、 目的化合物 [85] 8. 1 m gを白色固体として得た。
化合物 [85] のスぺク トルデータを以下に示す。
NMR (CDC 13) δ : 9. 86 (d, J = 8. OHz, 1 HX 1/5), 8.
99 (s, 1 HX 1/5), 8, 94 (d, J = 8. OHz, 1 HX 4/5), 8. 8 9 ( s , 1 HX 4/5), 8. 61 (s, 1 HX 1/5), 8. 54 ( s , 1 HX 4/ 5), 7. 74— 7. 12 (m, 3H), 6. 85— 6. 57 (m, 4H), 6. 16 —6. 14 (m, 1 HX 4/5), 8. 95— 5. 80 (m, 1 HX 1/5), 5. 3 0— 5. 17 (m, 1 HX 1/5), 5. 02— 4. 95 (m, 1 HX 4/5), 4. 23 (b r s, 1 H), 3. 19— 3. 16 (m, 2H), 2. 99— 2. 96 (m, 2H), 1. 61 (d, J = 6. 8Hz, 3H), 1. 48 (b r s, 2 H) ma s s : 449 (M+ 1 ) +. 実施例 86
2— ({(1 S) — 1一 [3— (ァゼチジン一 3—ィルァミノ) フエニル] ェチル } ァ ミノ) —4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィ ル] ピリミジン一 5—カルボ二トリル [86] (以下、 化合物 [86] という) の合 成。 化合物 [80] 15mgと tーブチルー 3ーォキソァゼチジンー 1一力ルボキシレ ート (特開 2002— 255932号公報、 6ページに記載されている方法により合 成した) 9. 5111§から実施例82— (1)、 (2) の方法に準じて、 目的化合物 [8 6] 4. 3 mgを白色固体として得た。
化合物 [86〕 のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 72— 9. 68 (m, 1 HX 1/5), 8. 99
( s , 1HX 1/5), 8. 90— 8. 88 (m, 1 HX 4/5 + 1 HX 4/5), 8. 6 1 (s, 1 HX 1/5), 8. 56 (s, 1 HX 4/5), 7. 72— 7. 19 (m, 3H), 6. 80— 6. 45 (m, 4H), 6. 09— 6. 07 (m, 1 HX 4/5), 5. 90— 5. 80 (m, 1 HX 1/5), 5. 28— 5. 20 (m, 1 HX 1/5), 5. 07— 4. 90 (m, 1 HX 4/5), 4. 42— 4. 30 (m, 1 H), 4. 2 7— 4. 15 (m, 1 H), '3. 98— 3. 85 (m, 2H), 3. 59— 3. 38 (m, 2H), 1. 62 (d, J = 6. 8Hz, 3 H)
m a s s : 461 (M+ 1) +. 実施例 87、 88
4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2 ― [((1 S) — 1— {3— [メチル (1一メチルァゼチジン一 3—ィル) ァミノ] フ ェニル } ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [87] (以下、 化合物 [87] とレヽう)、 および 4一 [8—(ジフルォロメチル) イミダゾ [1, 2— a] ピ リジン一 3—ィル]—2— [((1 S) —1— { 3— [(1一メチルァゼチジン一 3—ィ ァミノ] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [88] (以下、 化合物 [88] という) の合成。 化合物 [86] 5mgをクロ口ホルム 0. 75mLとテトラヒ ドロフラン 0. 25 mLの混合溶媒に溶解し、 ホルムアルデヒド液 (37%) 4. 05 Lを加え均一に した後、 トリァセトキシ水素化ほう素ナトリウム 1 1. 6m gを加え、 室温にて 2時 間撹拌した。 飽和炭酸水素ナトリウム水溶液を加え、 クロ口ホルムにて抽出した。 有 機層を無水硫酸ナトリウムにて乾燥した後、不溶物をろ過し、 ろ液を減圧濃縮して得 られた残渣を分取薄層クロマトグラフィーにて精製し、 目的化合物 [87] 1. Om gと目的化合物 [88] 0. 7 mgをそれぞれ白色固体として得た。
化合物 [87]、 および [88] のスぺクトルデータを以下に示す。
化合物 [87]
XH— NMR (CDC 13) δ : 9. 88— 9. 84 (m, 1 HX 1/5), 8. 99 (s, 1 HX 1/5), 8. 94— 8. 80 (m, 1 HX 4/5 + 1 HX 4/5), 8. 61 (s, 1 HX 1/5), 8. 57 (s, 1 HX 4/5), 7. 76— 7. 19 (m, 3H), 6. 83— 6. 62 (m, 4H), 6. 13— 6. 08 (m, 1 HX 4/5), 5. 98— 5. 90 (m, 1 HX 1/5), 5. 30— 5. 20 (m, 1 HX 1/5), 5. 10— 4. 90 (m, 1 HX 4/5), 4. 20— 4. 07 (m, 1 H), 3. 9 5— 3. 70 (m, 1 H), 3. 10— 2. 92 (m, 1H), 2. 84 (s, 3H), 2. 50— 2. 40 (m, 1 H), 2. 39 (s, 3H), 1. 63 (d, J = 6. 8 Hz, 3H)
ma s s : 489 (M+ 1) +. 化合物 [88]
XH— NMR (CDC 13) δ : 9. 88— 9. 84 (m, 1 HX 1/5), 8. 99 ( s , 1 HX 1/5), 8. 90— 8. 87 (m, 1 HX 4/5 + 1 HX 4/5), 8. 61 ( s, 1 HX 1/5), '8. 57 ( s, 1 HX 4/5), 7. 64— 7. 19 (m, 3H), 6. 80— 6. 45 (m, 4H), 6. 07— 5. 97 (m, 1 HX 4/5), 5. 85— 5. 77 (m, 1 HX 1/5), 5. 28— 5. 18 (m, 1 HX 1/5), 5. 05— 4. 90 (m, 1 HX 4/5), 4. 22— 4. 00 (m, 2H), 3. 7 7— 3. 65 (m, 2H), 2. 95— 2. 82 (m, 2H), 2. 36 ( s , 3H), 1. 61 (d, J = 6. 8Hz, 3H)
ma s s : 475 (M+ 1 ) +. 実施例 89
4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] 一 2 ― [((1 S) —1— {3— [(,1—イソプロピルァゼチジン一 3—ィル) ァミノ] フ ェニル } ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [89] (以下、 化合物 [89] という) の合成。 化合物 [86] 5 mgとアセトン 4 Lから、 実施例 87の方法に準じて、 目的化 合物 [89] 1. 8 mgを白色固体として得た。
化合物 [89] のスぺクトルデータを以下に示す。
ΧΗ— NMR (CDC 13) δ : 9. 88— 9. 84 (m, 1 HX 1/5), 8. 99 (s, 1 HX 1/5), 8. 90— 8. 89 (m, 1 HX 4/5 + 1 HX 4/5), 8. 61 (s, l.HX 1/5), 8. 56 (s, 1 HX 4/5), 7. 65— 7. 19 (m, 3H), 6. 82— 6. 46 (m, 4H), 6. 07— 6. 05 (m, 1HX 4/5), 5. 92— 5. 80 (m, 1 HX 1/5), 5. 28— 5. 18 (m, 1 HX 1/5), 5. 03— 4. 90 (m, 1 HX 4/5), 4. 35— 4. 00 (m, 2H), 3. 8 0— 3. 62 (m, 2H), 3. 02— 2. 85 (m, 2H), 2. 42— 2. 28 (m, 1H), 1. 61 (d, J = 6. 8Hz, 3H), 0. 96 (d, J = 6. 6Hz, 6 H)
ma s s : 503 (M+ 1 ) +. 実施例 90
4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2 一 {[(I S) —1— (3— {[1一 (2—フルォロェチル) ァゼチジン一 3—ィル]
アミノ} フエニル) ェチル] アミノ} ピリミジン一 5—カルボ二トリル [90] (以 下、 化合物 [90] という) の合成。 化合物 [86] 1 5mg、 2—フノレオ口ェチル ^一 4ーメチノレベンゼンスノレホネート (Syn t h e s i s 2004, 885に記載されている方法により合成した) 1 0. 7mg、 炭酸カリウム' 9. Omg、 およびジメチルスルホキシド lmLの混合物 を、 封管中 70°Cにて 2時間 30分撹拌した。 室温に戻した後、 逆相分取液体クロマ トグラフィ一にて精製した。 溶出液を減圧濃縮した後、 メタノールに溶解し、 弱陰ィ オン交換樹脂に通しトリフルォロ酢酸を除いた。溶媒を減圧留去し目的化合物 [90] 8. Imgを白色固体として得た。
化合物 [90] のスペクトルデータを以下に示す。
一 NMR (CDC 13) δ : 9. 86— 9. 85 (m, 1 HX 1/5), 8. 99 ( s , 1 HX 1/5), 8. 90— 8. 89 (m, 1 H X 4/ 5 + 1 H X 4/ 5), 8. 61 ( s , 1 HX 1/5), 8. 55 ( s, 1 HX 4/5), 7. 75— 7. 12 (m, 3H), 6. 80— 6. 45 (m, 4H), 6. 13— 6. 1 1 (m, 1 HX 4/5), 5. 88— 5. 86 (m, 1 H.X 1/5), 5. 30— 5. 18 (m, 1 HX 1/5), 5. 00— 4. 95 (m, 1HX 4/5), 4. 46 (t d, J =4. 8, 47. 6 Hz, 2H), 4. 22— 4. 05 (m, 2H), 3. 85— 3. 72 (m, 2H), 3. 08— 2. 93 (m, 2H), 2. 77 ( t d, J = 4. 8, 28. 8Hz, 2 H), 1. 61 (d, J = 6. 8Hz, 3 H)
m a s s : 507 (M+ 1 ) +. 実施例 91
4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] 一 2 ― [((1 S) 一 1— { 3— [(1ーメチルビペリジン一 4—ィル) ァミノ] フエ二ル} ェチル) ァミノ〕 ピリミジン一 5—カルボ-トリル [91] (以下、 化合物 [91] という) の合成。 化合物 [80] 1 1. 4mgをクロ口ホルム 0. 75mLに溶解し、 1ーメチルー 4—ピぺリ ドン 10. 4 L、 トリァセトキシ水素化ほう素ナトリウム 17. 9 m g を加え、 50°Cにて終夜撹拌した。 室温に戻した後、 飽和炭酸水素ナトリウム水溶液 を加え、 クロ口ホルムにて抽出した。 有機層を無水硫酸ナトリウムにて乾燥した後、 不溶物をろ過し、 ろ液を減圧濃縮して得られた残渣を分取薄層クロマトグラフィーに て精製し、 目的化合物 [91] 12. 5mgを淡黄色固体として得た。
化合物 [91] のスぺクトルデータを以下に示す。
XH— NMR (CDC 1 Ά) δ : 9. 86 (d, J = 7. 2H z , 1 HX 1/5), 8.
99 (s, 1 HX 1/5), 8. 9 1 (d, J = 7. 2H z , 1 HX 4/5), 8. 8 9 ( s, 1 HX 4/5), 8. 6 2 ( s ' 1 HX 1/5), 8. 55 ( s , 1 HX 4/ 5), 7. 74— 7. 1 1 (m, 3 H), 6. 8 5— 6. 5 3 (m, 4H), 6. 1 2 —6. 1 1 (m, 1 HX 4/5), 5. 96— 5. 85 (m, 1 HX 1/5), 5. 2 8— 5. 1 7 (m, 1 HX 1/5), 5. 05— 4. 8 8 (m, 1 HX 4/5), 3. 72— 3. 60 (m, 1 H), 3. 38— 3. 22 (m, 1H), 2. 82— 2. 79 (m, 2H), 2. 34 (s, 3H), 2. 1 8— 1. 90 (m, 4H), 1. 6 2 (d, J = 6. 8Hz, 3H), 1. 5 8— 1. 40 (m, 2 H)
ma s s : 503 (M+ 1 ) +. 実施例 9 2
4一 [8—(ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2 — [((1 S) — 1— { 3— [(1ーェチルビペリジン一 4一ィル) ァミノ] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [92] (以下、 化合物 [9 2] という) の合成。 化合物 [80] 1 Omgと 1ーェチルー 4ーピペリ ドン 5. 0 μ Lから、 実施例 9 1の方法に準じて、 目的化合物 [92] 5. 9mgを白色固体として得た。
化合物 [92] のスぺクトルデータを以下に示す。
ΧΗ— NMR (CDC 1 3) δ : 9. 72— 9. 6 8 (m, 1 HX 1/5), 8. 99 (s , 1 HX 1/5), 8. 92— 8. 89 (m, 1 HX 4/5 + 1 HX 4/5), 8. 62 ( s , 1 HX 1/5), 8. 5 6 ( s , 1 HX 4/5), 7. 7 3— 7. 1 2 (m, 3H), 6. 8 5— 6. 5 3 (m, 4H), 6. 06— 6. 0 5 (m, 1 HX 4/5), 5. 88— 5. 80 (m, 1 HX 1/5), 5. 25— 5. 1 7 (m, 1 HX 1/5), 5. 02— 4. 88 (m, 1 HX 4/5), 3. 66 (b r s , 1 H), 3. 4 1— 3. 28 (m, 1 H), 3. 00— 2. 85 (m, 2H), 2. 4.7 (q, J = 6. OHz, 2H), 2. 23— 2. 0 0 (m, 4H), 1. 6 2 (d, J = 6. 8H z , 3H), 1. 60— 1. 43 (m, 2H), 0. 9 1 (t, J = 6. OHz, 3 H)
ma s s : 5 1 7 (M+ 1 ) +. 実施例 93
4— [8— (ジフルォロメチル) イミダゾ [1 , 2— a] ピリジン一 3—ィル] —2 一 [((1 S) — 1— {3— [(1ーェチルビペリジン一 3—ィル) ァミノ] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ-トリル [9 3] (以下、 化合物 [9 3] という) の合成。
化合物 [80] 1 5 mgと 1—ェチルー 3—ピペリ ドン塩酸塩 1 8. 2mg力 ら、 実施例 9 1の方法に準じて、 目的化合物 [9 3] 1 3. 2mgを白色固体として得た。 化合物 [93] のスぺクトルデータを以下に示す。
XH— NMR (CDC 1 3) δ : 9. 88— 9. 84 (m, 1 HX 1/5), 8. 9 9 一 8. 9 9 (m, 1H+ 1HX 4/5), 8. 6 1 (s , 1 HX 1/5), 8. 55 ( s , 1 HX 4/5), 7. 7 3— 7. 1 5 (m, 3H), 6. 88— 6. 56 (m, 4H), 6. 1 8— 6. 0 7 (m, 1 H), 5. 3 3— 5. 20 (m, 1 HX 1/5), 5. 0 5— 4. 89 (m, 1 HX 4/5), 4. 20— 3. 9 5 (m, 1 HX 1/5), 3.
72— 3. 50 (m, 1 HX 4/5), 2. 88— 2. 1 0 (m, 4H), 1. 62 (d, J = 6. 8Hz, 3H), 1. 88— 1. 35 (m, 2H), 1. 08— 1. 03 (m,
3H)
ma s s : 5 1 7 (M+ 1) +. 実施例 94
4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2 — [((1 S) — 1— {3— [(1—イソプロピルピぺリジン一 4一ィル) ァミノ] フ ェニル } ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [94] (以下、 化合物 [94] という) の合成。 化合物 [80] 1 Omgと 1一イソプロピル一 4ーピペリ ドン 1 0. 5mg力 ら、 実施例 9 1の方法に準じて、 目的化合物 [94] 6. 8mgを白色固体として得た。 化合物 [94] めスペクトルデータを以下に示す。
NMR (CDC 1 3) δ : 9. 86 (d, J = 6. 9H z, 1 HX 1/5), 8. 9 9 (s, 1 HX 1/5), 8. 90 (d, J = 6. 9Hz, 1 HX 4/5), 8. 8 9 (s, 1 HX 4/5), 8. 6 1 (s, 1HX 1/5), 8. 5 5 (s, 1 HX 4/ 5), 7. 64— 7. 1 4 (m, 3 H), 6. 86— 6. 53 (m, 4H), 6. 1 0 一 6. 08 (m, 1 HX 4/5), 5. 9 2— 5. 80 (m, 1 HX 1/5), 5. 2
8— 5. 1 5 (m, 1 HX 1/5), 5. 02— 4. 8 9 (m, 1 HX 4/5), 3. 78— 3. 50 (m, 1H), 3. 38— 3. 20 (m, 1 H), 2. 9 7— 2. 72 (m, 3H), 2. 34 (s, 3H), 2. 39— 1. 84 (m, 4H), 1. 6 2 (d, J = 7. 0Hz, 3H), 1. 5 6— 1. 40 (m, 2H), 1. 0 8 (d, J = 6. 6H z , 6H)
m a s s : 5 3 1 (M+ 1 ) +. 実施例 95
4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2
一 {[(1 S)一 1一 (3— {[1一 (2—ヒドロキシェチル) ピぺリジン一 4一ィル] アミノ} フエニル) ェチル] アミノ} ピリミジン一 5—カルボ二トリル [9 5] (以 下、 化合物 [9 5] という) の合成。 ( 1 ) ィ匕合物 [80] 1 8 1mgと 1一 (tーブトキシカルボニル)一 4ーピぺリ ド ン 26 7mgから、 実施例 82— (1)、 (2) の方法に準じて、 4一 [8—(ジフル ォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2— ({(1 S)一 1一 [3— (ピペリジン一 4一ィルァミノ) フエニル] ェチル } ァミノ) ピリミジン一 5 一力ルポ二トリル [95一 1] 1 7 2mgを淡褐色固体として得た。
(2) 4一 [8—(ジフルォロメチル) イミダゾ [1, 2_a] ピリジン一 3—^ fル] —2— ({(1 S) —1— [3— (ピペリジン一 4ーィルアミノ) フエニル] ェチル } ァミノ) ピリミジン一 5—カルボ二トリノレ [9 5— 1] l l Omgと (tーブチルジ メチルシリルォキシ) ァセトアルデヒ ド 7 1. 5 Lから、 実施例 8 3—(1)、 (2) の方法に準じて、 目的化合物 [9 5〕 33. lmgを黄色固体として得た。
化合物 [9 5] のスぺクトルデータを以下に示す。
XH— NMR (CDC 1 3) 6 : 9. 88— 9. 84 (m, 1 HX 1/5), 8. 99 (s , 1 HX 1/5), 8. 92— 8. 8 9 (m, 1 HX 4/5 + 1 HX 4/5), 8. 6 2 ( s, 1 H X 1 / 5 ) , 8. 56 ( s , 1 HX 4/5), 7. 64— 7. 1 9 (m, 3H), 6. 8 5— 6. 53 (m, 4H), 6. 08— 6. 07 (m, 1 HX 4/5), 5. 8 8— 5. 80 (m, 1 HX 1/5), 5. 28— 5. 1 8 (m, 1 HX 1/5), 4. 9 9— 4. 96 (m, 1 HX 4/5), 3. 6 7— 3. 60 (m, 3H), 3. 4 0— 3. 25 (m, 1H), 2. 90— 2. 87 (m, 2H), 2. 80— 2. 6 5 (m, ϊ H), 2. 5 7— 2. 54 (m, 2H), 2. 25— 2. 20 (m, 2H), 2. 0 5— 2. 03 (m, 2H), 1. 6 1 (d, J = 6. 8Hz, 3H), 1. 55— 1. 35 (m, 2H)
ma s s : 5 33 (M+ 1 ) +. 実施例 96
4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2 ― ({(1 S) — 1一 [3— ({ 1— [2— (ジメチルァミノ) ェチル] ピぺリジン一 4—ィル }ァミノ) フエニル]ェチル }ァミノ) ピリミジン一 5—カルボ二トリル [9 6] (以下、 化合物 [96] という) の合成。 化合物 [9 5] 2 1. lmgとジメチルァミン (2Mテトラヒ ドロフラン溶液) 9 9 W Lから、 実施例 84の方法に準じて、 目的化合物 [96] 4. 3mgを白色固体
として得た。
化合物 [96] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 88— 9. 84 (m, 1 HX 1/5), 8. 9 9 ( s ' 1 HX 1/5), 8. 9 1— 8. 8 9 (m, 1 HX 4/5 + 1 HX 4/5), 8. 6 1 (s , 1 HX 1/5), 8. 56 (s, 1 HX 4/5), 7. 64— 7. 1 4 (m, 3H), 6. 8 5— 6. 5 2 (m, 4H), 6. 06— 6. 05 (m, 1 HX 4/5), 5. 8 9— 5. 78 (m, 1 HX 1/5), 5. 28— 5. 1 5 (m, 1 HX 1/5), 5. 02— 4. 90 (m, 1 HX 4/5), 3. 75— 3. 5 2 (m, 1 H), 3 · 3 7— 3. 22 (m, 1 H), 2. 95— 2. 82 (m, 2H), 2. 53— 2. 40 (m, 4H), 2. 2 6 ( s , 6H), 2. 23— 1. 9 5 (m, 4H), 1. 6 2 (d, J =6. 9H z, 3H), 1. 60— 1. 38 (m, 2 H)
ma s s : 5 60 (M+ 1 ) +. 実施例 9 7
4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2 ― {[(1 S) — 1— (3— {[1— (2—フルォロェチル) ピぺリジン一4—ィル] アミノ} フエニル) ェチル] アミノ} ピリミジン一 5—力ルポ二トリル [9 7] (以 下、 化合物 [9 7] という) の合成。 4— [8— (ジフルォロメチル) イミダゾ [1, 2— a」 ピリジン一 3—ィル]一 2— ({(1 S) — 1一 [3— (ピペリジン一 4一ィルァミノ) フ ニル] ェチル } ァ ミノ) ピリミジン一 5_カルボ二トリノレ [9 5— 1] 1 1. 4m gと 2—フルォロェ チルー 4—メチルベンゼンスルホネート 4. 8m から、実施例 90の方法に準じて、 目的化合物 [9 7] 4. Omgを白色固体として得た。
化合物 [97] のスぺクトルデータを以下に示す。
一 NMR (CDC 1 3) δ : 9. 88— 9. 84 (in, . 1 HX 1/5), 8. 99 ( s , 1 HX 1/5), 8. 9 2— 8. 90 (m, 1 HX 8/5), 8. 6 1 (s, 1 HX 1/5), 8. 56 ( s , 1 HX 4/5), 7. 78— 7. 1 4 (m, 3H), 6. 86— 6. 5 3 (m, 4H), 6. 08— 6. 06 (m, 1 HX 4/5), 5. 92— 5. 80 (m, 1 HX 1/5), 5. 28— 5. 1 5 (m, 1 HX 1/5), 5. 03 —4. 90 (m, 1 HX 4/5), 4. 58 ( t d, J =4. 8, 47. 5Hz, 2 H), 3. 78— 3. 44 (m, 1 H), 3. 38— 3. 22 (m, 1 H), 2. 9 9 —2. 8 3 (m, 2H), 2. 7 3 ( t d, J =4. 8, 28. 3H z , 2H), 2. 32— 2. 1 7 (m, 2H), 2. 1 2—1. 9 5 (m, 2H), 1. 62 (d, J = 6. 9Hz, 3H), 1. 60— 1. 40 (m, 2 H)
ma s s : 5 3 5 (M+ 1) +.
実施例 98
4— [8— (ジフルォロメチル) イミダゾ [1, 2— 3 ] ピリジン一 3—ィル] —2 一 [((1 S) 一 1一 { 3— [(1, 一メチルー 1, 4 ' —ビピペリジン一 4—ィノレ) ァミノ] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [98] (以 下、 化合物 [98] という) の合成。
4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ― 2— ({(1 S) 一 1一 [3— (ピペリジン一 4—ィルァミノ) フヱニル] ェチル } 了 ミノ) ピリミジン一 5_カルボ二トリル [95— 1] 20mgと 1ーメチノ I 4ーピ ペリ ドン 15. 1 μ Lから、 実施例 91の方法に準じて、 目的化合物 [98] 3. 5 m gを無色のアモ^ /ファスとして得た。
化合物 [98] のスぺク トルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 88— 9. 85 (m, 1 HX 1/5), 8. 99 (s , 1 HX 1/5), 8. 90— 8. 89 (m, 1 HX 4/5 + 1 HX 4/5), 8. 61 (s, 1 HX 1/5), 8..56 (s, 1 HX 4/5), 7. 78— 7. 10 (m, 3H), 6. 88— 6. 50 (m, 4H), 6. 10— 6. 02 (m, 1 HX 4/5), 5. 88— 5. 80 (m, 1 HX 1/5), 5. 28— 5. 18 (m, 1 HX 1/5), 5. 02— 4. 90 (m, 1 HX 4/5), 3. 75— 3. 56 (m, 1 H), 3. 3 2— 3. 20 (m, 1 H), 2. 96— 2. 82 (m, 4H), 2. 40— 2. 29 (m, 4H), 2. 28 (s, 3H), 2. 13— 1. 60 (m, 7H), 1. 62 (d, J =6. 8Hz, 3H), 1. 55— 1. 37 (m, 2 H)
ma s s : 586 (M+ 1 ) +. 実施例 99
4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2 ― {[(1 S) — 1— (3— {[1— (ピリジン一 3—ィルメチル) ピぺリジン一 4一 ィル] アミノ} フエニル) ェチル] アミノ} ピリミジン一 5—力ルポ二トリル [99] (以下、 化合物 [99] という) の合成。
4— [8— (ジフルォロメチル) イミダゾ. [1, 2— a] ピリジン一 3—^ fル] ― 2— ({(1 S) — 1— [3— (ピペリジン一 4一ィルァミノ) フヱニル] ェチル } 了 ミノ) ピリミジン一 5—カルボ二トリル [95— 1] 2 Omgと 3—ピリジンカルボ キシアルデヒ ド 1 1. 6 μ から、 実施例 91の方法に準じて、 目的化合物 [99] 9. 3m gを白色固体として得た。
化合物 [99] のスぺク トルデータを以下に示す。
XH— NMR (CDC 1 3) δ : 9. 8 6— 9. 8 5 (m, 1 HX 1/5), 8. 9 9 ( s , 1 HX 1/5), 8. 9 0— 8. 8 9 (m, 1 HX 4/5 + 1 HX 4/5), 8. 6 1 ( s , 1 HX 1/5), 8. 5 5— 8. 5 1 ( s, 1 H+ 1 HX 4/5), 7. 7 8— 7. 0 8 (m, 5H), 6. 8 5— 6. 5 3 (m, 4H), 6. 1 8— 6. 1 0 (m, 1 HX 4/5), 5. 9 3— 5. 8 5 (m, 1 HX 1/5), 5. 2 7— 5. 1 5 (m, 1 HX 1/5), 5. 0 2-^4. 8 8 (m, 1 HX 4/5), 3 · 54 ( s , 2H), 3. 3 8— 3. 2 0 (m, 2H), 2. 8 8— 2. 7 3 (m, 2H), 2. 2 5— 1. 9 3 (m, 4H), 1. 6 2 (d, J = 6. 8H z , 3H), 1. 5 8— 1. 3 8 (m, 2H)
m a s s : 5 8 0 (M+ 1 ) +. 実施例 1 0 0
4— [8— (ジフルォロメチル) イミダゾ [1, 2— 3 ] ピリジン一 3—ィル] —2 一 [((1 S) 一 1一 { 3— [(4—メチルビペラジン一 1一ィル) カルボニル] フエ 二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル [1 0 0] (以下、 化合物 [1 0 0] という) の合成。 .
(1) 3—(1ーヒ ドロキシェチル) 安息香酸メチルエステル (国際公開第 00Z 04 204 5号パンフレツト、 1 3 3ページに記載されている方法により合成した) 1. 1 6 gをクロ口ホルム 2 OmLに溶解し、 トリェチルァミン 4. 4 9mLを加え た。氷冷下、塩ィ匕メタンスルホニル 1. 4 9mLを加え、同温にて 40分間撹拌した。 飽和炭酸水素ナト 'リゥム水溶液を加え、 クロ口ホルムにて抽出した。 有機層を飽和食 塩水にて洗浄した後、 無水硫酸ナトリウムにて乾燥し、 不溶物をろ過し、 ろ液を減圧 濃縮した。 得られた残渣を N, N—ジメチルホルムアミドに溶解し、 アジィ匕ナトリウ ム 2. 0 9 gを加え、 8 0°Cにて 2時間撹拌した。 反応液を室温に戻した後、 飽和炭 酸水素ナトリゥム水溶液を加え、酢酸ェチルにて抽出した。有機層を希釈した食塩水、 飽和食塩水の順で洗浄し、無水硫酸マグネシウムにて乾燥した。不溶物をろ過した後、 ろ液を減圧濃縮し、得られた残渣をシリカゲル力ラムクロマトグラフィ一にて精製し、 メチル 3— ( 1—アジドエチル) ベンゾエート [1 0 0— 1] 1. 2 1 gを無色油 状物として得た。
(2) メチル 3— (1一アジドエチル) ベンゾエート [1 0 0— 1] 6 0 Omg をエタノール 1 5mLに溶解し、 1 0%パラジウム炭素触媒 2 0 Omgを力 Πえ、水素 雰囲気下、 終夜撹拌した。 不溶物をセライトにてろ過し、 エタノールで洗浄した後、 ろ液を減圧濃縮し、 メチル 3— (1—アミノエチル) ベンゾエート [1 0 0— 2] 3 0 Omgを無色油状物として得た。
(3) 4一 [8— (ジフノレオロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィ ル]一 2— (メチルチオ) ピリミジン一 5—カルボ二トリル [80— 3] 285mg とメチル 3— (1—アミノエチル) ベンゾエート [100— 2] 242m gから実 施例 80—(4) の方法に準じて、 メチル 3— [1— ({5—シァノ一4一 [8—(ジ フルォロメチル) イミダゾ' [1, 2— a] ピリジン一 3—ィル] ピリミジン一 2—ィ ル} ァミノ) ェチル] ベンゾエート [100— 3] 374m gを黄色油状物として得 た。 (4) メチル 3— [1— ({ 5——ンァノ一4— [8— (ジフルォロメチル) イミダ ゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 2—ィル } ァミノ) ェチル] ベ ンゾエート [100— 3] 37 Omgをメタノール 6mLとテトラヒドロフラン 6m Lの混合溶媒に懸濁させ、 1 N—水酸化ナトリゥム水溶液 4. 15mLを加え、 50°C にて 30分間撹拌した。 氷冷下、 5 N—塩酸 825 μ Lを力 11え、 反応液を中和し、 減 圧濃縮した。 得られた残渣を逆相分取液体クロマトグラフィーにて精製し、 3— [1 一 ({5—シァノー 4一 [8—.(ジフルォロメチル) イミダゾ [1, 2— a] ピリジ ンー 3—ィル] ピリミジン一 2—ィル } ァミノ) ェチル] ベンゾイツクアシッド [1 00— 4] 216mgを淡黄色固体として得た。
(5) 3— [1— ({ 5—シァノー 4一 [8— (ジフルォロメチル) イミダゾ [1 , 2— a] ピリジン一 3—ィル] ピリミジン一 2—ィル } ァミノ) ェチル] ベンゾイツ クアシッド [100— 4] 1 Omgと N—メチルビペラジン 6. lmgから、 実施例 81の方法に準じて、 目的化合物 [100] 8. Omgを白色固体として得た。 化合物 [100] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 89 (d, J = 6. 8Hz 1 HX 1/5), 9.
00-8. 94 (m, 1 H+ 1 HX 4/5), 8. 57 ( s , 1 H), 8. 02-7. 00 (m, 7H), 6. 22-6. 20 (m, 1 HX 4/5), 5 98— 5. 96 (m, 1 HX 1/5), 5. 37-5. 35 (m, 1 HX 1/5), 5. 18-5. 14 (m, 1 HX 4/5), 3. 88-3. 65 (m, 2H), 3. 48-3. 28 (m, 2H), 2. 58— 2. 15 (m, 4H), 2. 31 (s , 3H), 1. 67 (d, J = 6. 8 Hz, 3H)
ma s s : 517 (M+ 1) +. 実施例 101
4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2 ― [((1 S) 一 1一 {3— [ ( 4ーメチルピぺラジンー 1—ィル) メチル] フエ二ル}
ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル [1 0 1] (以下、 化合物 [1 0 1] とレヽう) の合成。
(1) 3— [1一({5—シァノ一4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 2—ィル } ァミノ) ェチル] ベンゾイツ クァシッド [100— 4] 1 5 lmgをテトラヒドロフラン 4 m Lに溶解し、 N, N' 一カルボニルジイミダゾール 28 21118をカ[1ぇ、 室温にて終夜撹拌した。 反応混合物 に水素化ほう素ナトリゥム 1 32m gを 2回に分けて加え、室温にて 30分間撹拌し た。 飽和炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を飽和 食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧 濃縮し、 得られた残渣を分取薄層クロマトグラフィーにて精製し、 4— [8— (ジフ ルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—^ fル] 一 2— ({ 1— [3— (ヒドロキシメチル) フエニル] ェチル } ァミノ) ピリミジン一 5—カルボ二トリル [1 0 1— 1] 5 Omgを淡黄色油状物として得た。
(2) 4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィ ル] 一 2— ({1— [3— (ヒドロキシメチル) フエニル] ェチル } ァミノ) ピリミ ジン一 5—カルボ二トリル [1 0 1— 1] 25mgと N—メチルビペラジン 1 5 m g から、 実施例 84の方法に準じて、 目的化合物 [1 01] 3. 3mgを白色固体とし て得た。
化合物 [1 0 1] のスぺクトルデータを以下に示す。
ΧΗ— NMR (CDC 1 3) δ : 9. 88 (d, J = 7. 2H z , 1 HX 1/5), 9. 05 (d, J = 7. 2Hz, 1HX 4/5), 8. 9 9 (s, 1 HX 1/5), 8. 9 4 ( s, 1 HX 4/5), 8. 60 ( s , 1 HX 1/5), 8. 57 (s, 1 HX 4/ 5), 7. 74— 6. 79 (m, 7H), 6. 1 2— 6. 1 0 (m, 1 HX 4/5), 5. 90— 5. 74 (m, 1 HX 1/5), 5. 38— 5. 32 (m, 1 HX 1/5), 5. 1 4— 5. 1 0 (m, 1 HX 4/5), 3. 5 7— 3. 47 (m, 2H), 2. 6 2— 2. 0 3 (m, 8H), 2. 26 (s, 3H), 1. 6 5 (d, J = 6. 8Hz, 3H)
ma s s : 503 (M+ 1) +. 実施例 1 02
N— { 3— [(I S) —1— ({5—シァノ一4一 [8— (ジフルォロメチル) イミダ ゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 2—ィル } ァミノ) ェチル] ベ ンジル } グリシンアミド [1 02] (以下、 化合物 [1 02] という) の合成。
(1) 4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィ ル] 一 2— ({1— [3— (ヒドロキシメチル) フエニル] ェチル } ァミノ) ピリミ ジン一 5—力ルポ二トリル [101— 1] 125mg、 N, N—ジイソプロピルェチ ルァミン 259 Lをクロ口ホルム 3m Lに溶解し、 氷冷下、 塩化メタンスルホニル 69 μ Lを加え、同温にて 30分間撹拌した。飽和炭酸水素ナトリゥム水溶液を加え、 クロ口ホルムにて抽出した。 得られた有機層を飽和食塩水にて洗浄し、無水硫酸ナト リウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮して得られた残渣を Ν, Ν— ジメチルホルムアミ ド 3 m Lに溶解した。 了ジ化ナトリウム 97mgをカ卩え、 80 °C にて 1時間撹拌した。 反応液を室温に戻した後、飽和炭酸水素ナトリゥム水溶液を加 え、 酢酸ェチルにて抽出した。 得られた有機層を飽和食塩水にて洗浄し、 無水硫酸ナ トリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮して得られた残渣を、 分取 薄層クロマトグラフィーにて精製し、 2— ({ 1— [3— (アジドメチル) フエニル] ェチル } ァミノ) —4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジ ンー 3—ィル] ピリミジン一 5—カルボ-トリル [102— 1] 87mgを淡褐色油 状物として得た。
(2) 2— ({1— [3— (アジドメチル) フエニル] ェチル } ァミノ) 一4— [8 一 (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 5—カルボ二トリノレ [102— 1] 87mgをエタノール 3mLに溶解し、 10 %パ ラジウム炭素触媒 5 Omgを加え、 水素雰囲気下、 室温にて終夜撹拌した。 不溶物を セライトにてろ過し、 ろ液を減圧濃縮し、 2— ({1一 [3— (アミノメチル) フエ ニル] ェチル } ァミノ) 一 4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 5—力ルポ二トリル [102— 2] 76m gを黄 色固体として得た。
(3) 2— ({1— [3— (アミノメチル) フエニル] ェチル } ァミノ)一4一 [8 ― (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 5—カノレボニトリル [102— 2] 10. 4mgと N— t一ブトキシカルボ二ルグリ シン 13mgから、 実施例 81の方法に準じて、 目的化合物 [102] 5. 1 1 m g を淡黄色固体として得た。
化合物 [102] のスぺクトルデータを以下に示す。
一 NMR (CDC 13) δ : 9. 99 (d, J = 6. 8Hz, 1 HX 1/5), 9. 01 (d, J =6. 8H z, 1 HX 4/5), 8. 97 (s, 1 HX 1/5), 8. 9 0 ( s , 1 HX 4/5), 8. 58 ( s , 1 HX 1/5), 8. 57 (s, 1 HX 4/ 5 ), 7. 76— 6. 51 (m, 9H), 5. 33— 5. 31 (m, 1 HX 1/5), 5. 1 3— 5. 08 (m, 1 HX 4/5), 5. 46 ( s, 2HX 1/5), 3. 37
( s, 2HX 4/5), 2. 4 2— 2. 1 8 (m, 2H), 1. 6 4 (d, J = 7. 2 H z , 3H)
ma s s : 4 7 7 (M+ 1) +. 実施例 1 0 3
N— {4— [(1 S) —1」 ({ 5—シァノー 4一 [8— (ジフルォロメチル) イミダ ゾ [ 1 , 2— a] ピリジン一 3—ィル] ピリミジン一 2—ィル } ァミノ) ェチル] フ ェニル } —Lーァラニンアミド [10 3] (以下、 化合物 [ 1 0 3] という) の合成。 (1) 4— [8—(ジフルォロメチル) イミダゾ [ 1, 2— a] ピリジン一 3— ^ f ル]— 2—(メチルチオ) ピリミジン一 5—カルボ二トリノレ [8 0— 3] 9 5mgと (S)一 α—メチノ!/ ~4— -トロベンジルァミン塩酸塩 7 3mg力 ら、 実施例 80— (4) の方法に準じて、 4一 [8—(ジフルォロメチル) イミダゾ [1, 2— a] ピ リジン一 3—ィル]一 2— {[(1 S) — 1— (4一二トロフエニル) ェチル] アミノ} ピリミジン一 5—カルボ二トリル [1 0 3— 1 ] 8 3mgを淡黄色固体として得た。
(2) 4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィ ル]—2— {[(1 S) — 1— (4一二トロフエニル) ェチル] アミノ} ピリミジン一 5—カルボ二トリル [1 0 3— 1] 8 3mgをエタノール 2mLとテトラヒ ドロフラ ン 3 m Lの混合溶媒に溶解し、 1 0%パラジウム炭素触媒 4 Omgを加え、 水素雰囲 気下、 室温にて終夜撹拌した。 不溶物をセライトにてろ過し、 ろ液を減圧濃縮して得 られた残渣を分取薄層クロマトグラフィーにて精製し、 2— {[(1 S) — 1一 (4一 アミノフエ二ノレ) ェチル] ァミノ)一4— [8— (ジフルォロメチル) イミダゾ [1 , 2— a] ピリジン一 3—ィル] ピリミジン一 5—カルボ二トリル [ 1 0 3— 2] 1 4 mgを淡黄色アモルファスとして得た。
(3) 2— {[( 1 S) 一 1一 (4一アミノフヱニル) ェチル] アミノ} —4一 [8 一(ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 5—カルボ二トリノレ [103— 2] 11 mgと N— tープトキシカルボ二ルァラニン 1 4 m gから、 実施例 8 1の方法に準じて、 目的ィ匕合物 [1 0 3] l l mgを白色固 体として得た。
化合物 [1 0 3] のスぺクトルデータを以下に示す。
XH— NMR (DMS O— d 6) δ : 1 0. 3 8—1 0. 3 6 (m, 1 H), 1 0. 2 0 (d, J = 7. 0 H z , 1 HX 1/2), 9. 1 6 (d, J = 7. 0H z , 1 HX 1Z2), 9. 0 5 (d, J = 6. 7H z , 1 HX 1 /2), 8. 9 7 ( d , J = 8. 2H z , 1 HX 1/2), 8. 7 9 ( s , 1 HX 1/2), 8. 7 8 ( s , 1 HX 1/
2), 8. 75 ( s, 1 HX 1/2), 8. 64 ( s, 1HX 1/2), 8. 1 0 (b r s , 2H), 7. 8 7 (d, J = 7. 0Hz, 1 HX 1/2), 7. 79 (d, J =
6. 7 Hz , 1 HX 1/2), 7. 6 5— 7. 3 9 (m, 5H), 7. 3 3 ( t , J =
7. 0Hz, 1 HX 1/2), 7. 1 4 ( t, J = 7. 0H z, 1 HX 1/2), 5. 23— 5. 20 (m, 1 HX 1/2), 5. 1 0— 5. 06 (m, 1 HX 1/2), 3 ·
97— 3. 9 3 (m, 1 H), 1. 53 (d, ] = 7. OH z , 3HX 1/2), 1. 49 (d, 1 = 7. OH z , 3HX 1/2), 1. 4 1 (t, J = 7. OH z , 3 H) m a s s : 4 7 7 (M+ 1 ) +. 実施例 1 04
4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2 一 [((1 S) — 1一 {4_ [(1—メチルビペリジン一 4—ィル) ァミノ] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [1 04] (以下、 化合物 [1 0 4] という) の合成。
2— {[(1 S) — 1一 (4ーァミノフエ-ル) ェチル] アミノ} 一 4一 [8— (ジフ ルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 5—カル ボニトリノレ [1 03— 2] 2 Omgと 1ーメチルー 4—ピぺリ ドン 7. 4 L力 ら、 実施例 82— (1) の方法に準じて、 目的化合物 [1 04] 7mgを白色固体として 得た。
化合物 [1 04] のスぺクトルデータを以下に示す。
'H— NMR (DMSO— d6) δ : 1 0. 20 (d, J = 6. 1 H z , 1 HX 1/2), 9. 22 (d, J = 7. 1 H z , 1 HX 1/2), 8, 9 2 (d, J = 7. 3H z, 1 HX 1/2), 8. 80 (d, J = 8. 3Hz, 1 HX 1/2), 8. 78 (s, 1 HX 1/2), 8. 76 ( s , 1 HX 1/2), 8. 7 5 ( s , 1 HX 1/2), 8. 63 (s, 1 HX 1/2), 7. 85 (d, J = 7. 6H z, 1 HX 1/2), 7. 8 〇 (d, J = 7. 3 H z , 1 HX 1/2), 7. 5 1 ( t , J = 54. 6Hz, 1 H X 1/2), 7. 4 7 ( t , J = 54. 6Hz, 1 HX 1/2), 7. 29 ( t , J = 6. 8Hz, 1 HX 1/2), 7. 1 4 (t, J = 6. 8Hz, 1 HX 1/2), 7. 1 1 (d, J = 8. 5Hz, 1 H), 7. 09 (d, J = 8. 5H z, 1H), 6. 5
5 (d, J = 8. 5H z, 1H), 6. 5 2 (d, J = 8. 5H z, 1 H), 5. 3 3 (t, J = 8. 5H z, 1H), 5. 1 5— 5. 07 (m, 1 HX 1/2), 4. 9 7 一 4. 90 (m, 1 HX 1/2), 3. 1 3— 3. 0 8 (m, 1 H), 2. 70— 2.
6 5 (m, 2H), 2. 1 3 ( s , 3 HX 1/2), 2. 1 2 ( s, 3 HX 1/2), 1. 98—1. 9 0 (m, 2H), 1. 84— 1. 7 9 (m, 2H), 1. 47 (d,
J = 6. 8H z, 3HX 1/2), 1. 44 (d, J = 6. 8H z, 3HX 1/2),
1. 3 7— 1. 29 (m, 2 H)
ma s s : 503 (M+ 1 ) +. 実施例 105
4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2 一 [((1 S) — 1一 {4」 [(1一^ f ソプロピルピぺリジン一 4—ィル) ァミノ] フ 工-ル } ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [1 0 5] (以下、 化合 物. [1 05] という) の合成。 2— {[(1 S) 一 1一 (4ーァミノフエニル) ェチル] アミノ}一 4一 [8— (ジフ ノレオロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 5—カル ポ-トリル [1 03— 2] 1 Omgと 1—イソプロピル一 4ーピペリ ドン 1 7. 4 μ L力 ら、 実施例 82— (1) の方法に準じて、 目的化合物 [1 0 5] 6mgを白色固 体として得た。
化合物 [1 05] のスぺクトルデータを以下に示す。
XH— NMR (DMSO— d6), δ : 1 0. 20 (d, J = 7. 1Hz, 1HX 1/2), 9. 22 (d, J = 6. 6Hz, 1 HX 1/2), 8, 92 (d, J = 7. 3Hz, 1 HX 1/2), 8. 80 (d, J =8. 3H z, 1HX 1/2), 8. 78 ( s , 1 HX 1/2), 8. 76 ( s , 1 HX 1/2), 8. 7 5 ( s , 1 HX 1/2), 8. 63 (s, 1 HX 1/2), 7. 8 5 (d, J = 7. 3Hz, 1 HX 1/2), 7. 8 0 (d, J = 7. 6 H z , 1 HX 1/2), 7. 5 1 ( t , J = 54. 4H z , 1 H X 1/2), 7. 47 (t, J = 54. 4H z, 1 HX 1/2), 7. 29 (t, J = 7. 1Hz, 1 HX 1/2), 7. 14 ( t , J = 7. 1 H z , 1 HX 1/2 7. 1 1— 7. 08 (m, 2H), 6. 56— 6. 5 1 (m, 2H), 5. 3 2 (t, J = 8. 4Hz, 1 H), 5. 1 5— 5. 09 (m, 1 HX 1/2), 4. 96—4. 89 (m, 1HX 1/2), 3. 1 3— 3. 07 (m, 1 H), 2. 73- -2. 6 2 (m, 3H), 2. 20— 2. 1 2 (m, 2H), 1. 8 7— 1. 8 1 (m, 2H), 1. 4 7 (d, J = 6. 8Hz, 3HX 1/2), 1 44 (d, J = 6. 8 H z , 1 HX 1/2), 1. 3 1— 1. 23 (m, 2H), 0 94 (d, J = 6. 6Hz, 3H), 0. 9 3 (d, J = 6. 6Hz, 3 H)
ma s s : 53 1 (M+ 1 ) +. 実施例 1 06
4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2 一({(1 S) — 1一 [4— (ピラジン一 2—ィルァミノ) フエニル] ェチル } ァミノ) ピリミジン一 5—カルボ二トリル [1 06] (以下、 化合物 [1 06, という) の合
成,
(1) S— (-) — 1— (4ーブロモフヱ-ル) ェチルァミン 2 gをクロ口ホルム 20mLに溶解し、 トリェチルァミン 4. 1 8mLを加えた。 氷冷下、 二炭酸ジー t 一ブチル 2. 62 gを加え、 室温にて 30分間撹拌した。 反応液を酢酸ェチル 300 mLにて希釈し、 水、 飽和食塩水で順に洗浄し、 得られた有機層を無水硫酸マグネシ ゥムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣を少量のクロ ロホノレムに溶解し、 へキサンにて固化し、 t一ブチル [(I S) —1— (4—プロモ フエニル) ェチル] カーバメート [1 06— 1] 2. 6 gを白色固体として得た。
(2) t一プチル [(1 S) — 1一 (4一ブロモフエニル) ェチル] カーバメート [1 06— 1] 1 00mg、 2—アミノビラジン 38 m g、 トリス (ジベンジリデン アセトン) (クロ口ホルム) ジパラジウム (0) 6. 8mg、 4, 5—ビス (ジフエ ニルホスフイノ)一 9, 9ージメチルキサンテン 7. 7m g、 炭酸セシウム 2 1 7 m g、 および 1, 4一ジォキサン 3 m Lの混合物を 1 00°Cにて終夜撹拌した。 反応混 合物を室温に戻した後、 不溶物をセライトにてろ過し、 ろ液を減圧濃縮して、 得られ た残渣をシリ力ゲルク口マトグラフィ一にて精製し、 t—ブチル { ( 1 S ) — 1一 [ 4 ― (ピラジン一 2—ィルァミノ) フエニル] ェチル } カーバメート [1 06— 2] 5 Omgを淡黄色固体として得た。
(3) t一プチル {(1 S) — 1一 [4一 (ピラジン一 2—ィルァミノ) フエニル] ェチル } カーバメート [1 06— 2] 5 Omgをクロ口ホルム 2mLに溶解し、 トリ フルォロ酢酸 2 m Lを加え、室温にて 1時間撹拌した。反応液を減圧濃縮し、 N— { 4 一 [(1 S)— 1—アミノエチル] フエ二ル} ピラジン一 2—ァミン [1 06— 3] のトリフルォロ酢酸塩 5 1 mgを褐色油状物として得た。
(4) 4一 [8—(ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィ ル] — 2— (メチルチオ) ピリミジン一 5—カルボ二トリノレ [80— 3] 3 7mgと N— {4— [(1 S) —1一アミノエチル] フエ二ル} ピラジン一 2—ァミン [1 0 6— 3] のトリフルォロ酢酸塩 5 1 mgから、 実施例 80— (4) の方法に準じて、 目的化合物 [1 06] 9. 2m gを淡黄色固体として得た。
化合物 [1 06] のスぺク トルデータを以下に示す。
'H— NMR (CDC 13) δ : 9. 8 3 (m, 1 HX 1/4), 9. 2 3 (m, 1 H X 3/4), 8. 9 9 (s, 1 HX 1/4), 8. 9 3 (s , 1 HX 3/4), 8. 6 2 ( s , 1 HX 1/4), 8. 5 7 ( s, 1 HX 3/4), 8. 20 (m, 1 H), 8. 1 2 (m, 1 H), 8. 00 (m, 1H), 7. 70— 7. 1 0 (m, 5H+ 1 HX 1
/4), 6. 8 8 (m, 1 HX 3/4), 6. 5 9 (m, 1 H), 6. 1 3 (m, 1 H X 3/4), 5. 8 0 (m, 1 HX 1/4), 5. 3 0 (m, 1 HX 1/4), 5. 1 0 (m, 1 HX 3/4), 1. 6 5 (d, J = 7. 6 H z , 3 H)
ma s s : 4 84 (M+ 1) +. 実施例 1 0 7 '
4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2 ― ({( 1 S) 一 1一 [4一 (4ーメチルビペラジン一 1—ィル) フエ-ル] ェチル } ァミノ) ピリミジン一 5—力ルポ二トリル [1 0 7] (以下、 化合物 [1 0 7] とい う) の合成。
(1 ) t一ブチル [( I S) —1— (4—プロモフヱニル) ェチル] カーバメート [1 0 6— 1 ] 1 0 Om gと N—メチルビペラジン 4 Om gから、 実施例 1 0 6—
(2) の方法に準じて、 t一ブチル {( 1 S) — 1一 [4— (4ーメチルビペラジン — 1—ィル) フエニル] ェチル } カーバメート [1 0 7—1] 44mgを淡褐色固体 として得た。 ,
(2) 4— [8— (ジフルォロメチル) イミダゾ [ 1 , 2— a] ピリジン一 3—ィ ル]一 2— (メチルチオ) ピリミジン一 5—カルボ二トリノレ [8 0— 3] 2 8mgと t—ブチル {( I S) —1— [4— (4—メチルビペラジン一 1一ィル) フエ-ル] ェチル } カーバメート [1 0 7— 1] 44mg力、ら、 実施例 1 0 6— (3)、 (4) の 方法に準じて、 目的化合物 [1 0 7] 1 2mgを淡黄色固体として得た。
化合物 [1 0 7] のスぺクトルデータを以下に示す。
αΗ— NMR (CDC 1 3) δ : 9. 8 3 (m, 1 HX 1/4), 9. 0 5 (d, J = 7. 2H z, 1 HX 3/4), 8. 9 9 ( s , 1 HX 1/4), 8. 9 1 ( s , 1 HX
3/4), 8. 6 2 ( s , 1 HX 1/4), 8. 5 8 ( s , . 1 HX 3/4), 7. 7 3 (m, 1 HX 1/4), 7. 6 5 (m, 1 HX 3/4), 7. 5 0— 7. 1 9 (m, 3
H), 6. 9 9— 6. 90 (m, 2H+ 1 HX 1/4), 6. 8 0 (m, 1 HX 3/4),
6. 0 0 (m, 1 HX 3/4), 5. 8 0 (m, 1 HX 1/4), 5. 3 6 (m, 1 H X 1/4), 5. 0 5 (m, 1 HX 3/4), 3. 2 3 (m, 4H), 2. 5 9 (m,
4H), 2. 3 7 ( s, 3H), 1. 6 1 (d, J = 6. 8H z , 3 H)
ma s s : 4 8 9 (M+ 1) +. 実施例 1 0 8
4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2 ― {[( 1 S) 一 1一 (4一 {[2— (ジメチルァミノ) ェチル] アミノ} フエニル)
ェチル] ァミノ) ピリミジン一 5—力ルポ二トリル [108] (以下、 化合物 [10 8] という) の合成。
(1) t一ブチル [(1 S) — 1一 (4一ブロモフエ二ル) ェチル] カーバメート [106— 1] l O Omgと N, N— メチルエチレンジァミン 35 m gから、 実施 例 106— (2) の方法に準じて、 t一ブチル [(1 S) — 1— (4— {[2— (ジメ チルァミノ) ェチル] ァミノ) フエニル) ェチル] カーバメート [108— 1] 25 mgを褐色固体として得た。 (2) 4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィ ル]一 2— (メチルチオ) ピリミジン一 5—力ルポ二トリル [80— 3] 10mgと t一プチル [(1 S) — 1一 (4— {[2— (ジメチルァミノ) ェチル] アミノ} フエ ニル)ェチル]カーバメート [108— 1] 25 mg力、ら、実施例 106—(3)、 (4) の方法に準じて、 目的化合物 [108] 1. 2m gを淡黄色固体として得た。
化合物 [108] のスぺクトルデータを以下に示す。
'H— NMR (CDC 13) δ :, 9. 85 (m, 1 HX 1/3), 9. 1 1 (d, J 7. 2Hz, 1 HX 2/3), 8. 99 ( s , 1 HX 1/3), 8. 92 (s, 1 HX 2/3), 8. 62 ( s , 1 HX 1/3), 8. 54 ( s , 1 HX 2/3), 7. 73 (m, 1 HX 1/3), 7. 65 (d, J = 7. 6Hz, 1 HX 2/3), 7. 50— 7. 10 (m, 3H), 6. 85 (m, 1 HX 2/3), 6. 74— 6. 59 (m, 2 H+ 1 HX 1/3), 6. 05 (m, 1 HX 2/3), 5. 80 (m, 1 HX 1/3), 5. 26 (m, 1 HX 1/3), 5. 01 (m, 1 HX 2/3), 3. 22 (m, 2H), 2. 69 (m, 2H), 2. 35 (s, 6H), 1. 61 (d, J = 7. 2Hz, 3 H) ma s s : 477 (M+ 1 ) +. 実施例 109
4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2 一 [((1 S)一 1一 {4— [(3R)—3— (ジメチルァミノ) ピロリジン一 1—ィル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [109] (以下、 化 合物 [109] という) の合成。
(1) t一ブチル [(1 S) —1— (4一ブロモフエニル) ェチル] カーバメート [106— 1] 10 Omgと (3R) — ( + ) — 3— (ジメチルァミノ) ピロリジン 46mgから、 実施例 106— (2) の方法に準じて、 t一ブチル ((1 S) — 1— {4— [(3 R) 一 3— (ジメチルァミノ) ピロリジン一 1—ィル] フヱニル} ェチ ル) カーバメート [109— 1] 1 16m gを淡褐色固体として得た。
(2) 4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3一^ f ノレ]— 2—(メチルチオ) ピリミジン一 5—カルボ二トリノレ [80— 3] 70mgと t一ブチル ((1 S) — 1一 {4— [(3 R) —3— (ジメチルァミノ) ピロリジン一 1一ィル] フエ-ル} ェチル) カーバメート [1 09— 1] 1 1 6mg力 ら、 実施例 1 06— (3)、 (4) の方法に準じて、 目的化合物 [1 09] 43m gを淡黄色固体 として得た。
化合物 [1 09] のスぺクトルデータを以下に示す。
XH— NMR (CDC 1 3) δ : 9. 83 (m, 1HX 1/4), 9. 23 (d, J = 6. 8H z, 1 HX 3/4), 8 · 99 ( s, 1 HX 1/4), 8. 9 3 ( s , 1 HX 3/4), 8. 6 2 (s , 1 HX 1/4), 8. 5 3 (s, 1 HX 3/4), 7. 80 —7. 1 0 (m, 4H+ 1 HX 1/4), 6. 85 (m, 1 HX 3/4), 6. 5 7 (d, J = 8. 4H z, 2H), 6. 0 7 (m, 1 HX 3/4), 5. 80 (m, 1 HX 1/ 4), 5. 26 (m, 1 HX 1/4), 5. 04 (m, 1 HX 3/4), 3. 5 3— 3. 40 (m, 2H), 3. 3 3 (m, 1H), 3. 1 7 (m, 1 H), 2. 8 9 (m, 1 H), 2. 3 3 (s, 6H), 2. 23 (m, 1 H), 1. 9 6 (m, 1 H), 1. 6 1
(d, J = 6. 8H z , 3H)
ma s s : 503 (M+ 1) +. 実施例 1 1 0
4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2— {[(1 S) —1— (4—ピぺラジン一 1ーィルフエニル) ェチル] アミノ} ピリミジ ンー 5—力ルポ二トリル [1 1 0] (以下、 化合物 [1 1 0] という) の合成。 (1) t一ブチル [(1 S) — 1— (4一ブロモフエニル) ェチル] カーバメート [1 06— 1] 1 00mg、 2, 2, 2—トリフルオロー 1ーピペラジン一 1一イノ ^ ェ タノン (T e t r a h e d o r o n L e t t . 1 9 9 5, 4 1, 735 7— 73 6 0に記載されている方法により合成した) 73mg、酢酸パラジウム 3. 7mg、 2, 2 ' 一ビス (ジフエニルホスフイノ)一 1, 1, 一ビナフチル 1 0. 7m g、 炭酸セ シゥム 2 1 7mg、 およびトルエン 3 mLの混合物を 1 00°Cにて終夜撹拌した。 反 応混合物を室温に戻した後、 不溶物をセライトにてろ過し、 ろ液を減圧濃縮して得ら れた残渣を分取薄層クロマトグラフィーにて精製し、 t一プチル((1 S)— 1一 {4 一 [4一 (トリフルォロアセチル) ピぺラジン一 1一ィル] フエ二ル} ェチル) カー バメート [1 1 0— 1] 7 3m gを白色固体として得た。
(2) 4一 [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィ
ル]一 2—(メチルチオ) ピリミジン一 5—カルボ二トリル [8 0— 3] 3 8mgと t一プチル ((1 S) — 1一 {4— [4一 (トリフルォロアセチル) ピぺラジン一 1 一ィル] フエ二ル} ェチル) カーバメート [1 1 0— 1] 7 3mg力、ら、 実施例 1 0 6— (3)、 (4) の方法に準じて、 4一 [8— (ジフルォロメチル) イミダゾ [ 1 , 2— a] ピリジン一 3—ィノレ] —2— [((1 S)一 1一 { 4一 [4一(トリフノレオ口 ァセチル) ピぺラジン _1 ィル] フエ-ル} ェチル) ァミノ] ピリミジン一 5—力 ルボニトリル [1 1 0— 2] 34mgを白色固体として得た。
(3) 4一 [8—(ジフルォロメチル) イミダゾ [1 , 2— a] ピリジン一 3—ィ ノレ]一 2— [((1 S)一 1一 {4— [4— (トリフノレオロアセチノレ) ピぺラジン一 1 一ィル] フエ二ル}ェチル) ァミノ] ピリミジン一 5—力ルポ二トリノレ [ 1 1 0— 2] 3 4mgをテトラヒドロフラン 5mLとメタノール l mLの混合溶媒に溶解し、 5 N 一水酸化ナトリゥム水溶液 2 0 Lを加え、 室温にて 3 0分間撹拌した。 反応液をク ロロホルムにて希釈し、 水、 飽和食塩水にて順に洗浄した。 得られた有機層を無水硫 酸マグネシウムにて乾燥し、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣を 少量のクロ口ホルムに溶解し、.へキサンにて固化させ、 目的ィ匕合物 [1 1 0] 2 5m gを白色固体として得た。
化合物 [1 1 0] のスぺクトルデータを以下に示す。
XH— NMR (CDC 1 3) δ 9. 84 (m, 1 HX 1/4), 9. 0 7 (d, J = 6. 8 H z, 1 HX 3/4), 8. 9 9 ( s 1 HX 1/4), 8. 9 2 ( s 1 HX
3/4), 8. 6 2 ( s , 1 HX 1/4), 8. 5 5 ( s , 1 HX 3/4), 7. 7 0 (m, 1 HX 1/4), 7. 6 5 (d, J =6. 4H z 1 HX 3/4), 7. 4 9—
7. 0 9 (m, 3H), 7. 0 1— 6. 8 8 (m 2H+ 1 HX 1/4), 6. 8 1 (m,
1 HX 3/4), 6. 0 5 (m 1 HX 3/4), 5. 8 0 (m 1 HX 1/4), 5. 2 8 (m 1 HX 1/4), 5. 0 5 (m, 1 HX 3/4), 3. 1 9 (m 4H),
3. 0 7 (m 4H), 1. 6 1 (d, J = 6. 8 H z 3 H)
ma s s : 4 7 5 (M+ 1 ) +. 実施例 1 1 1
4一 [8—.(ジフルォロメチル) イミダゾ [ 1 , 2— a] ピリジン一 3—ィル]一 2 一 [( 1— {4— [(1ーメチルビペリジン一 4一ィル) ォキシ] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ-トリノレ [1 1 1] (以下、 化合物 [1 1 1] とい う) の合成。 (1 ) 4一 [8—(ジフノレオロメチル) イミダゾ [1 , 2— a] ピリジン一 3—ィ ノレ]一 2—(メチルチオ) ピリミジン一 5—カルボ二トリル [8 0— 3] 20 Omg
と 1— (4— { [ t一ブチル (ジフエニル) シリル] 才キシ } フエニル) ェチルアミ ン [7 9— 4〕 264mgから、 実施例 8 0— (4) の方法に準じて、 2— {[1—
(4— {[ t一ブチル (ジフエニル) シリル] ォキシ } フエニル) ェチル] アミノ} 一 4一 [8— (ジフルォロメチル) イミダゾ [ 1 , 2— a] ピリジン一 3—ィル] ピ リ ミジン一 5—力ルポ二トリル [1 1 1一 1] 2 0 8m gを白色固体として得た。
(2) 2— {[1一 (4一 {[ t一ブチル (ジフヱニル) シリル] ォキシ } フエニル) ェチル] アミノ} —4— [8— (ジフルォロメチル) イミダゾ [1 , 2— a] ピリジ ン一3 ノレ] ピリ ミジン一5—力ノレボニトリノレ [1 1 1— 1] 2 0 8mgをテトラ ヒ ドロフラン 5mLに溶解し、 氷冷下、 フッ化テトラプチルアンモ -ゥム (1. 0M テトラヒドロフラン溶液) 3 2 0 μ Lを加え、 室温にて 1時間撹拌した。 反応液を酢 酸ェチル 2 0 0 m Lにて希釈し、 p H 6. 8リン酸バッファー、 飽和食塩水で順に洗 浄した。 得られた有機層を無水硫酸マグネシウムにて乾燥し、 不溶物をろ過し、 ろ液 を減圧濃縮した。 得られた残渣を少量のクロ口ホルムとメタノール混合溶媒 (クロ口 ホルム:メタノール = 9 : 1) に溶解し、 へキサンにて固化させ、 4一 [8— (ジフ ノレオロメチル) イミダゾ [1 ,. 2— a] ピリジン一 3—ィル] 一 2— {[ 1— (4— ヒ ドロキシフエニル) ェチル] アミノ} ピリ ミジン一 5—カルボ二トリル [1 1 1— 2] 1 0 7m gを白色固体として得た。 (3) 4— [8— (ジフルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィ ノレ] 一 2— {[1— (4ーヒ ドロキシフヱニル) ェチル] アミノ} ピリミジン一 5— カルボ二トリル [1 1 1— 2] 1 Omgをテトラヒドロフラン l mLに溶解し、 トリ フエ二ノレホスフィン 1 7m g、 1ーメチノレー 4ーヒ ドロキシピペリジン 5. 6mgを 加え撹拌し、 ァゾジカルボン酸ジィソプロピル 1 1 μ Lを加え、 5 0°Cにて 3時間撹 拌した。 反応混合物を室温に戻した後、 酢酸ェチル 1 O OmLにて希釈し、 水、 飽和 食塩水にて順に洗浄した。 得られた有機層を無永硫酸マグネシウムにて乾燥し、 不溶 物をろ過し、 ろ液を減圧濃縮した。 得られた残渣を分取薄層クロマトグラフィーにて 精製し、少量のクロ口ホルムに溶解し、へキサンにて固化させ、 目的化合物 [1 1 1] 5. 8 mgを白色固体として得た。
化合物 [1 1 1] のスぺク トルデータを以下に示す。
XH— NMR (DMSO— d 6) δ : 1 0. 1 9 (d, J = 7. 1 H z , 1 HX 1/2), 9. 1 6 (d, J = 6. 8 H z, 1 HX 1/2), 9. 0 1 (d, J = 7. 3 H z , 1 HX 1/2), 8. 9 1 (d, J = 8. 3H z , 1 HX 1/2), 8. 7 8 ( s , 1 HX 2/5), 8. 7 7 ( s ' 1 HX 3/5), 8. 7 6 ( s, 1 HX 2/5), 8. 6 3 ( s, 1 HX 3/5), 7. 8 5 (d, J = 7. 6H z , 1 HX 1/2), 7. 8 0 (d, J = 7. 6 H z , 1 HX 1/2), 7. 5 1 ( t , J = 5 4. 4H z, 1 H
X 1/2), 7. 46 (t, J = 54. 4Hz, 1 HX 1/2), 7. 3 1— 7. 29 (m, 2H+ 1 HX 1/2), 7. 1 3 (t, J = 7. 3H z, 1 HX 1/2), 6. 94 (d, J =8. 5H z, 2HX 1/2), 6. 9 0 (d, J =8. 8H z , 2H X 1/2), 5. 24— 5. 1 6 (m, 1 HX 1/2), 5. 08— 5. 0 1 (m, 1 HX 1/2), 4. 32— 4. 24 (m, 1H), 2. 5 9— 2. 49 (m, 2H), 2. 1 4 ( s, 3HX 1/2), 2. 1 3 ( s , 3HX 1/2), 2. 1 5— 2. 05 (m, 2H), 1. 9 1— 1. 8 1 (m, 2H), 1. 59— 1. 54 (m, 1 H), 1. 5 1 (d, J = 6. 8Hz, 3HX 1/2), 1 · 4 7 (d, J = 6. 8H z, 3 HX 1/2)
ma s s : 504 (M+ 1 ) +. 実施例 1 1 2 ·
2— {[(1 S) — 1— (4一アミノフヱニル) ェチル] アミノ} —4— (8—メチル イミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボ二トリノレ [1 1 2] (以下、 化合物 [1 1 2] という) の合成。
(1) 4一 [(Z) —2—エトキシビュル] 一 2— (メチルチオ) _5—ピリミジ ンカルボ二トリル [48— 3] 1 gと 2—アミノー 3—ピコリン 49 Omg力 ら、 実 施例 25— (3) の方法に準じて、 4一 (8—メチルイミダゾ [1, 2— a] ピリジ ンー 3—ィル)一2—(メチルチオ) ピリミジン一 5—カルボ二トリル [1 1 2— 1] 8 55 m gを褐色固体として得た。
(2) 4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一2— (メ チルチオ) ピリミジン一 5—カルボ二トリル [1 1 2— 1] 400mgと (S) — CK —メチルー 4—ニトロベンジルァミン塩酸塩 204mgから、 実施例 80— (4) の 方法に準じて、 4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—^ fル) 一2 ― {[(1 S) —1— (4一二トロフエニル) ェチル] アミノ} ピリミジン一 5—カル ボ-トリル [1 1 2— 2] 35 Omgを褐色固体として得た。 (3) 4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— {[(1 S) —1— (4一二トロフエニル) ェチル] アミノ} ピリミジン一 5—カルボ二トリ ノレ [1 1 2— 2] 35 Omgから、 実施例 1 03— (2) の方法に準じて、 目的化合 物 [1 1 2] 325mgを淡褐色固体として得た。
化合物 [1 1 2] のスぺクトルデータを以下に示す。
'H— NMR (CDC 13) δ : 9. 6 2 (m, 1 HX 1/3), 9. 03 (d, J = 7. 2H z, 1 HX 2/3), 8. 94 ( s , 1 HX 1/3), 8. 9 1 (s, 1 HX
2/3), 8. 5 8 (s, 1 HX 1/3), 8. 4 7 ( s , 1 HX 2/3), 7 · 3 0 —7. 1 2 (m, 3H), 6. 9 1 (m, 1 HX 1/3), 6. 78— 6. 6 5 (m, 2H+ 1 HX 2/3), 6. 03 (m, 1 HX 2/3), 5. 7 6 (m, 1 HX 1/3), 5. 22 (m, 1 HX 1/3), 5. 05 (m, 1 HX 2/3), 2. 66 (s, 3H), 1. 60 (d, J = 6. 8Hz, 3 H)
ma s s : 3 70 (M+ 1 ) +. 実施例 1 1 3
N— [4— ((I S) —1— {[5—シァノー 4一(8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリ ミジン一 2—ィル] ァミノ) ェチル) フエニル] メタンス ルホンアミド [1 1 3] (以下、 化合物 [1 1 3] という) の合成。 化合物 [1 1 2] 2 Omgをクロ口ホルム 5mLに溶解し、 ピリジン 25 μ Ι^を加 え、 氷冷下、 塩ィ匕メタンスルホニル 6. をカロえ、 室温にて 2時間撹拌した。 飽 和炭酸水素ナトリウム水溶液を加え、 クロ口ホルムにて抽出し、有機層を飽和食塩水 にて洗浄し、 無水硫酸マグネシウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮 し、 得られた残渣を分取薄層クロマトグラフィーにて精製し、 目的化合物 [1 1 3] 1 9 m gを白色固体として得た。
化合物 [1 1 3] のスぺク トルデータを以下に示す。
'H— NMR (CDC 13) δ : 9. 68 (m, 1 HX 1/4), 8. 9 1 (m, 1 H X 1/4), 8. 8 7 (s , 1 HX 3/4), 8. 8 1 (m, 1 HX 3/4), 8. 6 0— 8. 50 (m, 1 H), 7. 39 (m, 2H), 7. 37— 7. 1 7 (m, 3H + 1 HX 1/4), 6. 9 7 (m, 1 HX 1/4), 6. 6 9 (m, 1 HX 3/4), 6. 49 (m, 1 HX 1/4), 5. 3 1 (m, 1 HX 1/4), 5. 1 1 (m, 1 HX 3 /A), 2. 99 (s, 3HX 1/4), 2. 9 1 ( s , 3HX 3/4), 2. 6 8 ( s , 3HX 1/4), 2. 64 ( s , 3HX 3/4), 1. 6 2 (d, J = 7. 2H z, 3 H)
ma s s : 448 (M+ 1) +. 実施例 1 14
4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一2— [((1 S) — 1一 {4— [(1ーメチルビペリジン一 4一ィル) ァミノ] フエ二ル} ェチル) アミ ノ] ピリミジン一 5_カルボ二トリル [1 14] (以下、 化合物 [1 1 4] という) の合成。 化合物 [1 1 2] 24mgと 1—メチルー 4ーピペリ ドン 8 μ Lから、 実施例 8 2
― (1) の方法に準じて、 目的化合物 [1 1 4] 7mgを白色固体として得た。 化合物 [1 1 4] のスぺクトルデータを以下に示す。
XH— NMR (DMSO— d 6) δ : 9. 9 7 (d, J = 7. 3H z , 1 HX 1/2), 9. 0 7 (d, J = 6. 8 H z , 1 HX 1/2), 8, 8 3 (d, J = 7. 3 H z , 1 HX 1/2), 8. 74 (d, J = 6. 8 H z , 1 HX 1/2), 8. 7 3 ( s , 1 HX 1/2), 8. 7 1 ( 1 HX 1/2), 8. 7 0 ( s, 1 HX 1 /2), 8. 6 0 ( s , 1 HX 1/2), 7. 4 1 (d, J = 6. 8H z , 1 HX 1/2), 7. 3 5. (d, J = 7. 1 H z , 1 HX 1/2), 7. 1 1— 7. 0 8 (m, 2H+ 1 HX 1/2), 6. 9 6 ( t , J = 6. 8H z, 1 HX 1/2), 6. 5 4 (d, J = 8. 8 H z , 1 H), 6. 5 2 (d, J = 8. 8 H z , 1 H), 5. 3 4— 5. 3 0 (m, 1 H), 5. 1 5— 5. 0 7 (m, 1 HX 1/2), 4. 9 7— 4. 8 9 (m, 1 HX 1/2), 3. 1 5— 3. 0 7 (m, 1 H), 2. 7 2— 2. 6 6 (m, 2 H), 2. 5 8 ( s, 3HX 1/2), 2. 5 4 ( s, 3HX 1/2), 2. 1 5 ( s , 3 HX 1 /2), 2. 1 4 ( s , 3HX 1/2), 2. 0 1— 1. 9 7 (m, 2H), 1. 8 4 — 1 · 7 9 (m, 2H), 1. 4 7 (d, J = 7. 3H z, 3HX 1/2), 1. 44 (d, J = 7. 3 H z, 3 HX 1/2), 1. 34— 1. 3 0 (m, 2 H)
ma s s : 4 6 7 (M+ 1 ) +. 実施例 1 1 5
4— (8—メチルイミダゾ [ 1 , 2— a] ピリジン一 3—ィル) 一2— ({(1 S) 一 1— [4— (モルホリンー 4一イノレメチル) フエニル] ェチル } ァミノ) ピリミジン —5—カルボ二トリル [ 1 1 5] (以下、 化合物 [1 1 5] という) の合成。
(1) 水素化リチウムアルミニウム 1 7 7. 8mgのテトラヒ ドロフラン溶液 5m Lに、 氷冷下、 メチノレ 4— (1一アミノエチル) ベンゾエート (国際公開第 0 3/0 24 9 5 5号パンフレツト、 1 1 5ページに記載されている方法により合成した) 2 4 5mgのテトラヒドロフラン溶液 3mLを加え、 1時間撹拌した。 硫酸ナトリウム 1 0水和物を気泡が出なくなるまで加え、 室温にて終夜撹拌した。 混合物をセライト にてろ過し、 ろ液を減圧濃縮し、 [4一 (1—アミノエチル) フエ-ル] メタノール [1 1 5— 1] 1 9 5mgを無色油状物として得た。
(2) 4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一2— (メ チルチオ) ピリミジン一 5—カノレポ二トリル [1 1 2— 1] 24 2mgと [4— (1 一アミノエチル) フエニル] メタノール [1 1 5— 1] 1 9 5mg力 ら、 実施例 8 0 一 (4) の方法に準じて、 2— ({ 1— [4— (ヒドロキシメチル) フヱニル] ェチ ル} ァミノ) 一 4一 (8—メチルイミダゾ [ 1, 2— a] ピリジン一 3—ィル) ピリ
ミジン一 5—カルボ二トリル [1 1 5— 2] 1 75mgを白色固体として得た。
(3) 2— ({1— [4— (ヒ ドロキシメチル) フヱニル] ェチル } ァミノ) 一4 一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—力ノレ ボニトリル [1 15— 2] 3 Omgとモリホリン 30 Lから、 実施例 84の方法に 準じて、 目的化合物 [1 Γ5] 3. 8m gを淡黄色固体として得た。
化合物 [1 15] のスぺクトルデータを以下に示す。
2Η— NMR (DMSO— d6) δ : 9. 97 (d, J = 7. OH z, 1 HX 2/5),
8. 96 (d, J = 6. 6Hz, 1 HX 3/5), 8. 90 (d, J =8. 2Hz, 1 HX 2/5), 8. 81 (d, J = 7. 0Hz, 1 HX 3/5), 8. 73— 8. 7
2 (m, 1H), 8. 70 (s, 1 HX 2/5), 8. 58 (s, 1 HX 3/5), 7.
43— 7. 24 (m, 5H), 7. 1 2 ( t , J = 7. OH z , 1 HX 2/5), 6.
84 ( t , J = 7. OH z , 1 HX 3/5), 5. 28— 5. 20 (m, 1 HX 2/
5), 5. 10— 5. 02 (m, 1 HX 3/5), 3. 54— 3. 46 (m, 4H), 2. 58 (s, 3HX 2/5), 2. 52 (s, 3HX 3/2), 2. 30— 3. 32 (m, 6H), 1. 52 (d, J = 7. OH z , 3HX 2/5), 1. 49 (d, J =
7. 0Hz, 3HX 3/5)
ma s s : 454 (M+ 1) +. 実施例 1 16
N— [4— ((1 S) — 1— {[5—シァノー 4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 2—ィル] アミノ} ェチル) ベンジル] グリシン アミド [116] (以下、 化合物 [1 16] という) の合成。 (1) 2— ({ 1— [4— (ヒ ドロキシメチル) フヱニル] ェチル } ァミノ) 一 4 一(8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カル ボニトリル [1 15— 2] 75. 3mg力、ら、 実施例 102— (1)、 (2) の方法に 準じて、 2— ({ 1— [4— (アミノメチル) フエニル] ェチル } ァミノ) —4— (8 ーメチルイミダゾ [ 1, 2— a ] ピリジン一 3—ィル) ピリミジン一 5—カルボ二ト リル [1 16— 1] 5 lmgを白色固体として得た。
(2) 2— ({ 1一 [4— (アミノメチル) フエニル] ェチル } ァミノ) —4— (8 ーメチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボニト リル [1 16— 1] 1 5mgと N— tープトキシカルボ-ルグリシン 14mgから、 実施例 81の方法に準じて、 目的化合物 [1 16] 5. 5mgを淡黄色固体として得 た。
化合物 [ 1 1 6] のスぺク トルデータを以下に示す。
XH— NMR (DMS O— d 6) δ : 9. 9 7 (d, J = 7. OH z , 1 HX 1/2), 9. 0 4— 9. 0 0 (m, 1 H), 8. 9 4 (d, J = 8. 6 H z, 1 HX 1/2), 8. 7 9— 8. 7 3 (m, 2H), 8. 6 9 ( s , 1 HX 1/2), 8. 6 1 ( s , 1 HX 1 /2), 7. 4 5— 7. 3 7 (m, 2H+ 1 HX 1/2), 7. 2 7— 7. 2 3 (m, 1 H+ 1 HX 1/20, 7. 1 4 ( t , J = 7. OH z , 1 HX 1/2), 6. 9 7 ( t , J = 7. OH z , 1 HX 1/2), 5. 2 5— 5. 1 7 (m, 1 HX 1/ 2), 5. 1 4— 5. 0 6 (m, 1 HX 1 /2), 4. 3 0— 4. 2 9 (m, 2H), 3. 5 7— 3. 54 (m, 2H), 2. 5 8 ( s, 1 HX 1/2), 2. 5 4 ( s , 1 HX 1/2), 1. 5 1 (d, J = 7. 4H z, 3HX 1/2), 1. 4 8 (d, J = 7. 4H z, 3HX 1/2)
ma s s : 44 1 (M+ 1 ) +. 実施例 1 1 7
4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— [(( 1 S) _ 1— { 4— [(メチルスルホ二.ル) メチル] フエエル) ェチル) ァミノ] ピリ ミジン —5—カルボ二トリル [1 1 7] (以下、 化合物 [ 1 1 7] という) の合成。
2— ({ 1— [4— (ヒ ドロキシメチル) フエニル] ェチル } ァミノ) 一 4— (8 ーメチルイミダゾ [ 1 , 2— a] ピリジン一 3—ィル) ピリ ミジン一 5—力ルボニト リノレ [1 1 5— 2] 2 Omgから、実施例 84の方法に準じて、 目的化合物 [1 1 7]
1 3. 6 mgを白色固体として得た。
化合物 [1 1 7] のスぺク トルデータを以下に示す。
NMR (DMSO— d 6) δ : 9. 9 7 (d, J = 7. 1 H z , 1 HX 1/2), 9. 0 0 (d , J = 7. 3H z , 1 HX 1/2), 8. 94 (d, J = 8. OH z ,
1 HX 1/2), 8. 8 6 (d, J = 7. 1 H z , 1 HX 1/2), 8. 74 ( s , 1
HX 1/2), 8. 7 3 ( s, 1 HX 1/2), 8. 7 0 ( s , 1 HX 1/2), 8.
5 9 ( s , 1 HX 1/2), 7. 4 5— 7. 3 1 (m, 5H), 7. 1 3 ( t , J = 7.
1 H z, 1 HX 1/2), 6. 9 1 ( t , J = 7. 1 H z , 1 HX 1/2), 5. 2 9 一 5. 2 2 (m, 1 HX 1/2), 5. 1 4— 5. 0 7 (m, 1 HX 1/2), 4. 4
7— 4. 3 9 (m, 2H), 2. 8 9 ( s , 3HX 1/2), 2. 8 2 ( s, 3HX 1
/2), 2. 5 8 ( s , 3HX 1/2), 2. 5 3 ( s , 3HX 1 /2), 1. 54 (d,
J = 7. 1 H z , 3HX 1/2), 1. 5 1 (d, J = 7. 1 H z, 3HX 1/2) ma s s : 44 7 (M+ 1) +. 実施例 1 1 8
2— {[(1 S) — 1— (3—メ トキシフエニル) ェチル] アミノ}一 4一 (8—メチ ルイミダゾ [ 1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボ二トリル [ 1 1 8] (以下、 化合物 [1 1 8] という) の合成。 4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— (メチルチ ォ) ピリミジン一 5—力ルポ二トリル [1 1 2— 1] 20mgと (S)— (—) 一 1 ― (3—メ トキシフエニル) ェチルァミン 1 6mgから、 実施例 80— (4) の方法 に準じて、 化合物 [1 1 8] 1 3mgを白色固体として得た。
化合物 [1 1 8] のスぺクトルデータを以下に示す。
'H— NMR (CDC 13) δ : 9. 6 2 (m, 1 HX 1/4), 8. 94 (m, 1 H X 1/4), 8. 8 9 ( s , 1 HX 3/4), 8. 8 1 (d, J = 6. 8H z, 1 HX 3/4), 8. 5 7 (m, 1 HX 1/4), 8. 5 1 ( s , 1 HX 3/4), 7. 40 一 6. 80 (m, 5H+ 1HX 1/4), 6. 64 (m, 1 HX 3/4), 6. 0 9 (m, 1 HX 3/4), 5. 5 3 (m, 1 HX 1/4), 5. 30 (m, 1 HX 1/4), 5. 09 (m, 1 HX 3/4), 3. 8 1 (s, 3H), 2. 7 1— 2. 6 3 (m, 3H), 1. 6 2 (d, J = 7. 2H z, 3H)
ma s s : 38 5 (M+ 1 ) +. 実施例 1 1 9
4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— [((1 S)一 1一 { 3— [(1—メチルピペリジン一 4—ィル) ァミノ] フエエル) ェチル) アミ ノ] ピリミジン一 5—カルボ二トリル [1 1 9] (以下、 化合物 [1 1 9] という) の合成。 (1) 4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— (メ チルチオ) ピリミジン一 5—カルボ二トリノレ [1 1 2— 1] 50111 と 3— [(1 3) —1一アミノエチル] ァニリン 29mg力、ら、 実施例 80— (4) の方法に準じて 2 ― {[(1 S)一 1一 (3—アミノブヱニル) ェチル] アミノ}一 4一 (8—メチルイ ミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボ二トリル [1 1 9— 1] 22mgを淡黄色アモルファスとして得た。
(2) 2— {[(1 S) — 1— (3—ァミノフエニル) ェチル] アミノ} —4一 (8 ーメチルイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボニト リノレ [1 1 9一 1] 1 lmgと 1—メチルー 4—ピペリ ドン 1 1 から、 実施例 9 1の方法に準じて、 目的化合物 [1 1 9] 7. lmgを淡黄色固体として得た。 化合物 [1 1 9] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 64 (d, J = 8. OH z , 1HX 1/5), 8. 95 (s, 1HX 1/5), 8. 88 ( s , 1 HX 4/5), 8. 85 (d, J = 8. OH z, 1 HX 4/5), 8. 58 (s, 1 HX 1/5), 8. 52 ( s , 1 HX 4/ 5), 7. 30— 7. 14 (m, 2H), 6. 71— 6. 52 (m, 4H), 6. 01 — 6. 00 (m, 1 HX 4/5), 5. 88— 5. 78 (m, 1 HX 1/5), 5. 2 7— 5. 15 (m, 1 ΗΧΊ/5), 5. 03— 5. 00 (m, 1 HX 4/5), 3. 75— 3. 50 (m, 1 H), 3. 38— 3. 18 (m, 1 H), 2. 73— 2. 38 (m, 2H), 2. 69 (s, 1 HX 3/5), 2. 64 ( s ' 2H+ 1 HX 2/5), 2. 31 (s, 3H), 2. 14— 2. 01 (m, 4H), 1. 61 (d, J = 6. 8 Hz, 3H), 1. 58— 1. 40 (m, 2 H)
ma s s : 467 (M+ 1 ) +. 実施例 120
4— (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) —2— ({(1 S) ― 1— [4— (4ーメチルビペラジン一 1—ィル) フエニル] ェチル } ァミノ) ピリミ ジン一 5—カルボ二トリル [120] (以下、 化合物 [120] という) の合成。
(1) 3—ブロモー 2—二トロピリジン 327mg、 ジクロロビス (トリフエ二ノレ ホスフィン) パラジウム (Π) 256. 5mg、 トリブチル (ビニル) すず 518 μ L、 および Ν, Ν—ジメチルホルムアミ ド 6 m Lの混合物を、 80°Cにて 3時間撹拌 した。 反応混合物を室温に戻した後、 フッ化カリウム 500 m gと水 1. 5 m Lを加 え、 室温にて 30'分間撹拌した。 不溶物をセライ トにてろ過し、 飽和炭酸水素ナトリ ゥム水溶液を加え、 酢酸ェチルにて抽出した。 得られた有機層を飽和食塩水にて洗浄 し、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮した後、 得 られた残渣をシリカゲルカラムクロマトグラフィーにて精製し、 2—二トロー 3—ビ 二ルビリジン [ 120— 1 ] 236mgを淡赤色固体として得た。
(2) 2—二トロ一 3—ビュルピリジン [120— 1] 1 30m gをエタノール 5 mLに溶解し、 10%パラジウム炭素触媒 65111§をカロぇ、 水素雰囲気下、 室温にて 終夜撹拌した。 不溶物をセライトにてろ過し、 ろ液を減圧濃縮し、 3—ェチルピリジ ンー 2—ァミン [120— 2] 88. 7 mg,を黄色油状物として得た。
(3) 3—ェチルピリジン一 2—ァミン [120— 2] 85111§と4— [(2) — 2—ェトキシビュル]—2— (メチルチオ)一 5—ピリミジンカルボ-ル [48— 3] 1 54mgから、実施例 25— (3)の方法に準じて、 4一( 8—ェチルイミダゾ [ 1 , 2— a] ピリジン一 3—ィル)一 2— (メチルチオ) ピリミジン一 5—カルボ二トリ
ル [1 20— 3] 1 0 Omgを淡黄色固体として得た。
(4) 4— (8—ェチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— (メ チルチオ)ピリミジン一 5—力ルポ-トリル [1 20— 3] 2 Omgと t一プチル {(1 S) — 1— [4— (4ーメチルピペラジン一 1一ィル) フエニル] ェチル } カーバメ ート [1 07—1] 3 8m'gから、 実施例 1 06— (3)、 (4) の方法に準じて、 目 的化合物 [1 20] 5. 7mgを淡黄色固体として得た。
ィ匕合物 [1 20] のスぺクトルデータを以下に示す。
一 NMR (CDC 1 3) δ : 9. 74— 9. 6 8 (m, 1 HX 1/5), 8. 9 7 —8. 90 (m, 1 H+ 1 HX 4 5), 8. 5 7 (s, 1 HX 1/5), 8. 49 (s,
1 HX 4/5), 7. 3 5— 6. 6 5 (m, 6H), 6. 1 2— 6. 06 (m, 1 HX
4/5), 5. 82— 5. 7 3 (m, 1 HX 1/5), 5. 3 1— 5. 20 (m, 1 H
X 1/5), 5. 1 2— 5. 00 (m, 1 HX 4/5), 3. 25— 3. 1 6 (m, 4
H), 3. 30— 3. 1 5 (m, 2H), 2. 6 3— 2. 4 7 (m, 4H), 2. 3 5 (s, 3H), 1. 6 1 (d, J = 6. 6Hz, 3H), 1. 45—1. 30 (m, 3
H) ,
ma s s : 46 7 (M+ 1) +. 実施例 1 2 1
4— (8—シクロプロピルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— ({(1 S) 一 1— [4— ( 4ーメチルピぺラジン一 1—ィル) フエニル] ェチル } ァミノ) ピリミジン一 5—カルボ二トリル [1 2 1] (以下、 化合物 [1 2 1] という) の合 成。 (1) 3—シクロプロピルピリジン (T e t r a h e d o r o n L e t t. 20 02, 39, 6 98 7— 6 9 90に記載されている方法により合成した) 1. 93 g を酢酸 1 5 m Lに溶解し、 過酸化水素水 (3 1%) 2. 4 gを加え、 終夜加熱還流し た。 反応混合物を減圧濃縮し、 得られた残渣をクロ口ホルムに溶解し、 飽和炭酸水素 ナトリゥム水溶液、 水で順に洗浄し、有機層を無水硫酸ナトリゥムにて乾燥した。 不 溶物をろ過し、ろ液を減圧濃縮して、 3—シクロプロピルピリジン 1一才キシド [1 2 1— 1] 1. 47 gを褐色油状物として得た。
(2) 3—シクロプロピノレピリジン 1一ォキシド [1 2 1— 1] 1. 4 7 gをクロ 口ホルム 1 5mLに溶解し、 トリメチルシリルシアニド 1. 8 9mLを加え、 続いて ジメチルカルバモイルクロリ ド 1. 3 m Lをクロ口ホルムに 5 m Lに溶かした溶液を 30分間かけて滴下し、 室温にて終夜撹拌した。 1 0%炭酸力リゥム水溶液を 1 Om
L加え、 クロ口ホルムにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナト リウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮して得られた残渣をシリカゲ ルカラムクロマトグラフィーにて精製し、 3—シクロプロピルピリジン一 2—力ルポ 二トリノレ [121— 2] 68 Omgを白色固体として得た。
(3) 3—シクロプロピルピリジン一 2—カルボ二トリル [121— 2] 680m gと 6N—塩酸 35mLの混合物を、 終夜加熱還流した。 反応混合物を減圧濃縮し、 3—シクロプロピルピリジン一 2—力ルボン酸 [121— 3] の塩酸塩 83 Omgを 白色固体として得た。
(4) 3ーシク口プロピルピリジン一 2—力ルボン酸 [ 121— 3 ] の塩酸塩 83 0 m gを 10 %塩酸ーメタノール溶液 100 m Lに溶解し、 終夜加熱還流した。 反応 液を減圧濃縮し、 得られた残渣に飽和炭酸水素ナトリゥム水溶液を加え、 酢酸ェチル にて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮して、 メチル 3—シクロプロピルピリジン一 2—力 ルボキシレート [121— 4] , 363mgを白色固体として得た。
(5) メチル 3—シクロプロピルピリジン一 2—カルボキシレート [121— 4] 36 Omgをメタノール 4mLに溶解し、 1 N—水酸ィ匕ナトリゥム水溶液 4mLを加 え 50 °Cにて 30分間撹拌した。 反応液を減圧濃縮し、 得られた残渣をトルエンにて 2回共沸し白色固体を得た。 得られた固体を N, N—ジメチルホルムアミ ド 4mLと 1, 4一ジォキサン 3 m Lの混合溶媒に溶解し、 氷冷下、 トリェチルァミン 736 μ Lとジフエニルホスホリルアジド 569 Lを加え、 室温にて 1時間撹拌した。 その 後、 反応混合物に t—ブタノール 1. 5mLを加え、 8.0°Cにて終夜撹拌した。 反応 混合物を室温に戻した後、 飽和炭酸水素ナトリゥム水溶液を加え、 クロ口ホルムにて 抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥した。 不溶 物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をシリカゲルカラムクロマトグラフィ 一にて精製し、 3—シクロプロピルピリジン一 2—ァミン [121— 5] 41. 2m gを淡褐色油状物として得た。
(6) 3—シクロプロピルピリジン一 2—ァミン [121— 5] 41. 2mgと 4 ― [(Z) 一 2—エトキシビニル] 一 2— (メチルチオ) 一 5—ピリミジンカルボ二 トリル [48— 3] 67. 6mg力 ら、実施例 25— (3) の方法に準じて、 4— (8 ——ンク口プロピルイミダゾ [ 1 , 2— a] ピリジン一 3—ィル)一 2— (メチルチオ) ピリ ミジン一 5—カルボ二トリノレ [121— 6] 76. 8 m gを淡黄色固体として得 た。
(7) 4一 (8—シクロプロピルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2—(メチルチオ) ピリミジン一 5—カルボ二トリル [1 2 1— 6] 24mgと t一 ブチル {( 1 S) — 1— [4一 (4ーメチルピペラジン一 1一ィル) フエ-ル] ェチ ル} カーバメート [1 0 7— 1] 3 9mg力 ら、 実施例 1 0 6— (3)、 (4) の方法 に準じて、 目的化合物 [Γ2 1] 1 1. 3m gを淡黄色固体として得た。
化合物 [ 1 2 1] のスぺクトルデータを以下に示す。
XH— NMR (CDC 1 3) δ : 9. 74— 9. 6 8 (m, 1 HX 1/5), 8. 9 7 — 8. 9 2 (m, 1 H+ 1 HX 4/5), 8. 5 7 ( s , 1 HX 1/5), 8. 4 9 ( s , 1 HX 4/5), 7. 3 5— 6. 6 7 (m, 6 H), 6. 0 3— 5. 9 5 (m, 1 HX 4/5), 5. 8 0— 5. 6 8 (m, 1 HX 1/5), 5. 3 2— 5. 2 0 (m, 1 H X 1/5), 5. 1 3— 5. 0 0 (m, 1 HX 4/5), 3. 2 6— 3. 1 2 (m, 4 H), 2. 6 8— 2. 5 0 (m, 5H), 2. 3 5 ( s , 3H), 1. 6 0 (d, J = 6. 6 H z , 3H), 1. 3 0— 1. 1 2 (m, 2H), 0. 9 6— 0. 8 3 (m, 2 H)
ma s s : 4 7 9 (M+ 1 ) +,. 実施例 1 2 2
4— (8—メチルイミダゾ [1 , 2— a] ピリジン一 3—ィル)— 2— {[(1 S) — 1一 (4—ピぺラジン一 1ーィルフエニル) ェチル] アミノ} ピリミジン一 5—カル ポニトリル [ 1 22] (以下、 化合物 [1 2 2] という) の合成。
( 1) 4— (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2—(メ チノレチォ) ピリミジン一 5—カルボ二トリル [1 1 2— 1] 24. 6mgと tーブチ ル ((1 S) — 1— {4一 [4一 (トリフルォロアセチル) ピぺラジン一 1一ィル] フエ二ル} ェチル) カーバメート [ 1 1 0— 1] 5 7. 8mgから、 実施例 1 0 6— (3)、 (4) の方法に準じて、 4一 (8—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— [((1 S)一 1一 {4— [4— (トリフルォロアセチル) ピペラジ ンー 1一ィル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [1 2 2— 1] 1 2. 2m gを淡黄色固体として得た。
(2) 4一 ( 8—メチルイミダゾ [ 1 , 2— a] ピリジン一 3—ィル)一 2— [(( 1 S) — 1一 {4一 [4一(トリフルォロアセチル) ピぺラジン一 1一ィル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ノレボニトリル [ 1 2 2— 1 ] 1 2. 2mg力 ら、 実施例 1 1 0— (3) の方法に準じて、 目的化合物 [1 2 2] 5. 5mgを白色固体 として得た。
化合物 [1 22] のスぺク トルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 61 (d, J = 7. 2Hz, 1HX 1/5), 9. 00 (d, J = 7. 2H z, 1 HX 4/5), 8. 94 ( s, 1 HX 1/5), 8. 9 0 ( s , 1 HX 4/5), 8. 58 ( s ' 1 HX 1/5), 8. 49 ( s, 1 HX 4/ 5), 7. 29— 6. 67 (m, 6H), 6. 08— 6. 07 (m, 1 HX 4/5), 5. 83— 5. 72 (m, 1 HX 1/5), 5. 32— 5. 20 (m, 1 HX 1/5), 5. 14— 5. 00 (m, 1 HX 4/5), 3. 18— 3. 10 (m, 4H), 3. 0 4— 3. 01 (m, 4H), 2. 68 (b r s, 1H), 2. 65 (s, 3H), 1. 61 (d, J = 6. 8Hz, 3H)
ma s s : 439 (M+ 1 ) +. 実施例 123
4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—^ fル)一 2— [((1 S) ― 1一 { 3— [(1—メチルピペリジン一 4—ィル) ァミノ] フエエル) ェチル) アミ ノ] ピリミジン一 5—カルボ-トリル [1 23] (以下、 化合物 [1 23] という) の合成。 ,
(1) 2—ァミノ一 3—クロ口ピリジン 1 gと 4一 [(Z)一 2—エトキシビニル J 一 2— (メチルチオ)一 5—ピリミジンカルボニル [48— 3] 1. 72 gから、 実 施例 25— (3) の方法に準じて、 4— (8—クロロイミダゾ [1, 2— a] ピリジ ンー 3—ィル)一 2—(メチルチオ) ピリミジン一 5—カルボ二トリル [123—1] 846mgを黄色固体として得た。
(2) 4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2—(メ チルチオ) ピリミジン一 5—カルボ二トリノレ [123— 1] 80111§と 3— [(13)
—1—アミノエチル] ァニリン 45mgから、 実施例 80— (4) の方法に準じて、 2— {[(1 S) — 1一 (3—ァミノフエ二ノレ) ェチル] アミノ}一 4— (8—クロ口 イミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボ二トリル [1 23— 2] 60. 8mgを黄色固体として得た。
(3) 2— {[(1 S) — 1— (3—ァミノフエ-ル) ェチル] アミノ} —4— (8 一クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボニト リノレ [1 23— 2] 1 1. 7mgと 1—メチルー 4ーピぺリ ドン 1 1. 1 μ L力、ら、 実施例 91の方法に準じて、 目的化合物 [123] 10. 4mgを淡黄色固体として 得た。
化合物 [123] のスぺク トルデータを以下に示す。
XH— NMR (CDC 1 3) 6 : 9. 7 0 (d, J = 6. 9 H z , 1 HX 1/5), 8. 9 8 ( s , 1 HX 1/5), 8. 8 9 ( s , 1 HX 4/5), 8. 7 9 (d, J = 6. 9H z , 1 HX 4/5), 8. 6 2 ( s , 1 HX 1/5), 8. 5 6 ( s, 1 HX 4/ 5), 7. 54— 6. 9 0 (m, 2H), 6. 7 5— 6. 5 2 (m, 4H), 6. 0 8 —6. 00 (m, 1 HX 4/5), 5. 8 8— 5. 7 9 (m, 1 HX 1 /5), 5. 2 9— 5. 1 8 (m, 1 HX'1/5), 5. 0 3— 4. 9 0 (m, 1 HX 4/5), 3. 6 5 (d, J = 8. 2 H z , 1 H), 3. 3 5— 3. 2 9 (m, 1 H), 2. 8 8— 2. 7 0 (m, 2H), 2. 3 0 ( s, 3H), 2. 1 8— 1. 9 6 (m, 4H), 1. 6
2 (d, J = 6. 6 H z , 3H), 1. 5 8— 1. 40 (m, 2 H)
ma s s : 4 8 7, 4 8 9 (M+ 1 ) +. 実施例 1 24
4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— ({(1 S) 一 1— [3— ({ 1— [2— (ジメチルァミノ) ェチル] ピぺリジン一 4ーィル } アミ ノ) フエエル] ェチル } ァミノ) ピリミジン一 5—力ルポ二トリル [1 24] (以下、 化合物 [1 24] という) の合成。
(1) 2— {[(1 S) — 1— (3—アミノフヱニル) ェチル] アミノ}一 4— (8 一クロロイミダゾ [1 , 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボニト リル [1 2 3— 2] 2 7 Omgと 1一 (t一ブトキシカルボニル) 一 4ーピペリ ドン 4 1 4m gから、 実施例 8 2— (1)、 (2) の方法に準じて、 4一 (8—クロロイミ ダゾ [ 1 , 2— a] ピリジン一 3—ィル) 一2— ({(1 S) 一 1一 [3— (ピベリジ ン一 4—ィルァミノ) フエニル] ェチル } ァミノ) ピリミジン一 5—カルボ-トリル [1 24— 1] 1 84mgを淡褐色固体として得た。
(2) 4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— ({ ( 1 S) 一 1一 [3— (ピペリジン一 4一ィルァミノ) フエニル] ェチル } ァミノ) ピリ ミジン一 5一カルボ二トリル [1 24— 1] 8 8. l mgと (t一プチルジメチルシ リルォキシ) ァセトアルデヒド 5 9 μ から、 実施例 8 3— (1)、 (2) の方法に準 じて、 4一 (8—クロロイミダゾ [ 1 , 2— 3 ] ピリジン一 3—ィル) 一 2— {[(1 S) — 1— (3— {[1一 (2—ヒドロキシェチル) ピぺリジン一 4一ィル] アミノ} フエニル) ェチル] ァミノ) ピリミジン一 5—カルボ二トリル [ 1 24— 2] 5 6.
3 m gを黄色ァモルファスとして得た。 (3) 4一(8—クロロイミダゾ [1, 2— a]ピリジン一 3—ィル)一 2— {[(1
S) 一 1— (3— {[1— (2—ヒドロキシェチル) ピぺリジン一 4—ィル] アミノ}
フエニル) ェチル] アミノ} ピリミジン一 5—力ルポ二トリル [1 24— 2] 56. 3 m gとジメチルァミン ( 2Mテトラヒドロフラン溶液) 200 から、 実施例 8 4の方法に準じて、 目的ィ匕合物 [124] 3. 2mgを白色固体として得た。
化合物 [124] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 70 (d, J = 6. 9Hz, 1 HX 1/5), 8. 99 (s, 1HX 1/5),' 8. 88 (s, 1 HX 4/5), 8. 78 (d, J = 6. 9Hz, 1 HX 4/5), 8. 62 (s, 1HX 1/5), 8. 56 ( s , 1 H X 4 / 5), 7. 53— 6. 90 (m, 2H), 6. 70— 6. 52 (m, 4H), 6. 06 —6. 04 (m, 1 HX 4/5), 5. 88— 5. 80 (m, 1 HX 1/5), 5. 2 7— 5. 18 (m, 1 HX 1/5), 5. 02— 4. 90 (m, 1 HX 4/5), 3. 69— 3. 57 (m, 1H), 3. 33— 3. 22 (m, 1 H) 2. 93— 2. 83
(m, 2H), 2. 53— 2. 38 (m, 4H), 2. 25 (s, 6 H), 2. 20— 1. 95 (m, 4H), 1. 61 (d, J = 6. 8Hz, 3H), .. 60— 1. 40
(m, 2H)
ma s s : 544, 546 (M+ 1) +. 実施例 125
4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3_ィル) 一 5—フルオロー N ― ((1 S) — 1一 { 3— [(1ーメチルビペリジン一 4一ィル) ァミノ] フエ二ル} ェチル) ピリミジン一 2—ァミン [125] (以下、 化合物 [125] という) の合 成。
(1) 2, 4—ジクロロー 5—フルォロピリミジン 274mgから、 実施例 48— (3) の方法に準じて、 2—クロ口一 4— [(Z) 一 2—エトキシビニル] —5—フ ルォロピリミジン [125— 1] 223mgを淡褐色固体として得た。
(2) 2—クロロー 4一 [(Z) 一 2—エトキシビュル] 一 5—フルォロピリミジ ン [125—1] 10 Omgと 2—ァミノ一 3—クロ口ピリジン 63. 5mg力 ら、 実施例 25— (3) の方法に準じて、 8—クロ口一 3— (2—クロロー 5—フルォロ ピリミジン一 4 fル) イミダゾ [1, 2— a] ピリジン [125— 2] 18 Omg を黄色固体として得た。
(3) 8—クロ口一 3— (2—クロロー 5—フルォロピリミジン一 4一ィル) イミ ダゾ [1, 2— a] ピリジン [1 25— 2] 90 m gをジメチルスルホキシド 3 m L に溶解し、 3— [(1 S) — 1一アミノエチル] ァニリン 46. 6mgと炭酸力リウ ム 10 Omgを加え、 60°Cにて 5時間撹拌した。 反応混合物を室温に戻し、 逆相分
取クロマトグラフィーにて精製した。 飽和炭酸水素ナトリウム水溶液を加え、 クロ口 ホルムにて抽出し、有機層を飽和食塩水で洗浄し、無水硫酸ナトリゥムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 N— [(1 S) 一 1一 (3—アミノフヱニル) ェチル] —4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3一^ fル)一5—フ ルォロピリミジン一 2—ァミン [125— 3] 25 m gを褐色油状物として得た。
(4) N— [(1 S) — 1— (3—ァミノフエニル) ェチル] —4一 (8—クロ口 イミダゾ [1, 2— a] ピリジン一 3—ィル)一 5—フルォロピリミジン一 2_アミ ン [125— 3] 18. 5m gと 1—メチルー 4ーピペリ ドン 17. 8m gから、 実 施例 91の方法に準じて、 目的化合物 [125] 17. 5m gを白色固体として得た。 化合物 [125] のスぺクトルデータを以下に示す。
XH— NMR (CDC ") δ : 9. 20 (b r s , 1 H), 8. 41 (s, 1 H), 8. 24 ( s , 1H), 7. 39— 7. 16 (m, 2H), 6. 75— 6. 49 (m, 4H), 5. 47— 5. 46 (m, 1 H), 4. 97— 4. 85 (m, 1 H), 3. 68— 3. 45 (m, 1 H), 3. 35— 3. 20 (m, 1 H), 2. 85— 2. 70 (m, 2H), 2. 30 (s , 3H), 2. 18— 1. 95 (m, 4H), 1. 58 (d, J = 6. 6 Hz, 3H), 1. 56— 1. 40 (m, 2H)
ma s s : 480, 482 (M+ 1 ) +. 実施例 126
5—クロ口一 4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル)一 N— ((1 S) —1— { 3— [(1—メチルビペリジン一 4一ィル) ァミノ] フエ二ル} ェ チル) ピリミジン一 2—ァミン [126] (以下、化合物 [126] という) の合成。 2, 4, 5—トリクロ口ピリミジン 30 Omgから、実施例 125—(1) 〜 (4) の方法に準じて、 目的化合物 [126] 11. 2m gを淡褐色固体として得た。 化合物 [126] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 8. 72— 8. 60 (m, 2H), 8. 35 (s, 1 H), 7. 36— 7. 17 (m, 2H), 6. 72— 6. 52 (m, 4H), 5. 63 一 5. 50 (m, 1H), 5. 00— 4. 83 (m, 1H), 3. 77— 3. 47 (m, 1 H), 3. 33— 3. 20 (m, 1 H), 2. 88— 2. 72 (m, 2H), 2. 3 1 (s , 3H), 2. 20— 1. 96 (m, 4H), 1. 56 (d, J = 6. 6Hz, 3H), 1. 55— 1. 40 (m, 2 H)
ma s s : 496, 498 (M+ 1) +. 実施例 127
4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—メチル一N— ((1 S) 一 1一 { 3— [(1—メチルビペリジン一 4一ィル) ァミノ] フヱ二ル} ェ チル) ピリミジン一 2—ァミン [1 27] (以下、 化合物 [127] という) の合成。 2, 4ージクロロー 5—メチルピリミジン 50 Omgから、実施例 125—(1)
〜 (4) の方法に準じて、 '目的化合物 [1 27] 13mgを淡褐色固体として得た。 化合物 [127] のスぺクトルデータを以下に示す。
NMR (CDC 13) δ : 8. 85— 8. 68 (m, 1 H), 8. 24 (s, 1 H), 8. 07 (s, 1 H), 7. 34— 7. 12 (m, 2H), 6. 72— 6. 48 (m, 4H), 5. 45— 5. 38 (m, 1H), 5. 00— 4. 83 (m, 1H), 3. 33— 3. 25 (m, 1 H), 2. 89— 2. 78 (m, 2H), 2. 35 (s, 3H), 2. 32 (s, 3H), 2. 20— 2. 10 (m, 2H), 2. 05— 1. 9 5 (m, 4H), 1. 54 (d, J = 6. 6H z, 3 H)
ma s s : 476, 478 (M+ 1 ) +. 実施例 128 ,
4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2_ {[(1 S) ― 1— (3—ピペラジン一 1 ルフエ-ル) ェチル] アミノ} ピリミジン一 5—カル ボニトリル [128] (以下、 化合物 [128] という) の合成。
(1) 3— {(1 S) — 1一 [( t一ブトキシカルボニル) ァミノ] ェチル } フヱ- ノレトリフノレオロメタンスノレホネート (J. Me d. Ch em. 2004, 47, 28 87— 2896に記載されている方法により合成した) 306mg、 2, 2, 2—ト リフルオロー 1ーピペラジン一 1一イノ I ^一エタノン 18 lmg、 トリス (ジベンジリ デンアセトン) (クロ口ホルム) ジパラジウム (0) 47. 6mg、 2— (ジー t一 プチルホスフイノ) ビフエ二ル 49. 4mg、 リン酸三カリウム 246mg、 および トルエン 4mLの混合物を 100°Cにて終夜撹拌した。 室温に戻した後、飽和炭酸水 素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄 し、 無水硫酸ナトリウムにて乾燥し、 不溶物をろ過した。 ろ液を減圧濃縮し、 シリカ ゲルカラムクロマトグラフィーにて精製し、 t一ブチル ((1 S) — 1一 {3— [4 一 (トリフルォロアセチル) ピぺラジン一 1—ィル] フエ二ル} ェ ル) カーパメー ト [1 28— 1] 277mgを褐色油状物として得た。
(2) 4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— (メ チルチオ) ピリミジン一 5—カルボ二トリノレ [1 23— 1] 23. 6 m gと tーブチ ノレ ((1 S) — 1一 { 3— [4— (トリフルォロアセチル) ピぺラジン一 1一ィル]
フエ二ル}ェチル)カーバメート [ 1 2 8— 1] 4 7mg力 ら、実施例 1 0 6—(3)、 (4) の方法に準じて、 4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィ ル) —2— [((1 S) — 1— { 3— [4— (トリフルォロアセチル) ピぺラジン一 1 一ィル] フエ二ル}ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [1 2 8— 2] 2 7. 7mgを淡黄色固体として得た。
(3) 4一(8—クロロイミダゾ [1, 2— a] ピリジン一 3 fル)一 2— [((1 S) — 1一 { 3— [4一(トリフノレオロアセチノレ) ピぺラジン一 1一^ fル] フエ二ル} ェチル)ァミノ] ピリミジン一 5—カルボ二トリル [1 2 8— 2] 2 7. 7mg力、ら、 実施例 1 1 0— (3) の方法に準じて、 目的化合物 [1 2 8] 2 0. 7 m gを淡黄色 固体として得た。
化合物 [1 2 8] のスぺク トルデータを以下に示す。
NMR (CDC 1 3) δ : 9. 7 0 (d, J = 8. OH z , 1 HX 1/5), 8.
9 9 ( s, 1 HX 1/5), 8. 8 9 ( s, 1 HX 4/5), 8. 7 9 (d, J = 8. OH z , 1 HX 4/5), 8. 6 0 ( s ' 1 HX 1/5), 8. 5 5 ( s , 1 HX /
5), 7. 5 3— 6. 6 5 (m, 6 H), 6. 1 4— 6. 0 5 (m, 1 HX 4/5),
5. 8 8— 5. 8 0 (m, 1 HX 1/5), 5. 3 3— 5. 2 0 (m, 1 HX 1/5),
5. 1 0— 4. 9 3 (m, 1 HX 4/5), 3. 1 6— 3 L 5 (m, 4H), 3. 0
5— 3. 0 2 (m, 4H), 2. 6 5 (b r s , 1 H), ] 6 3 (d, J = 6. 8H z , 3 H)
ma s s : 4 6 0, 4 6 2 (M+ 1 ) +. 実施例 1 2 9
2— [(( I S) —1— { 3— [(3 R) —3—ァミノピロリジン一 1一ィル] フエ二 ル} ェチル) ァミノ]一 4— (8—クロロイミダゾ [ 1, 2— a] ピリジン一 3—ィ ル) ピリミジン一 5—カルボ二トリル [ 1 2 9] (以下、 化合物 [ 1 2 9] という) の合成。
( 1) 3— {(1 S) — 1一 [( t一ブトキシカルボ二ノレ) ァミノ] ェチル } フ 二 ノレトリフルォロメタンスルホネート 9 7m gと (3 R) — (+) — (トリフルォロア セトアミ ド) ピロリジン 5 7mgから、 実施例 1 2 8— (1) の方法に準じて、 t— ブチル [( 1 S) 一 1一 (3— {(3 R) 一 3— [(トリフルォロアセチル) ァミノ] ピロリジン一 1ーィル } フエニル) ェチル] カーバメート [1 2 9— 1] 2 0. 4 m gを淡黄色固体として得た。
(2) 4一 (8—クロロイミダゾ [ 1, 2— a] ピリジン一 3—ィル) 一 2— (メ
チルチオ) ピリミジン一 5—カルボ二トリル [123— 1] 12. 8mgと tーブチ ル [(1 S) — 1一 (3— {(3R) 一 3— [(トリフルォロアセチル) ァミノ] ピロ リジン一 1ーィル } フエニル) ェチル] カーバメート [129— 1] 20. 4mg力 ら、 実施例 106— (3)、 (4) の方法に準じて、 N— {.(3R) — 1一 [3— ((1 S) — 1— {[4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) —5 一シァノピリミジン一 2—'ィル]アミノ}ェチル) フエニル] ピロリジン一 3—イノレ} 一 2, 2, 2—トリフルォロアセトアミ ド [129— 2] 7. Omgを淡黄色固体と して得た。 (3) N— {(3R) — 1一 [3— ((1 S) — 1— {[4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—シァノピリミジン一 2—ィル] アミノ} ェチル) フエニル] ピロリジン一 3—ィル } ー2, 2, 2—トリフルォロアセトアミ ド [129— 2] 7. Omg力ら、 実施例 1 10— (3) の方法に準じて、 目的化合 物 [129] 4. 5mgを黄色固体として得た。
化合物 [129] のスぺク トルデータを以下に示す。
ΧΗ— NMR (CDC 13) δ :.9. 70 (d, J = 8. OH z , 1 HX 1/5), 8.
95 (s, 1HX 1/5), 8. 88 ( s , 1 HX 4/5), 8. 80 (d, J = 8.
OHz, 1 HX 4/5), 8. 60 (s, 1 HX 1/5), 8. 55 (s, 1 HX 4/
5), 7. 52— 6. 90 (m, 2H), 6. 78— 6. 45 (m, 4H), 6. 18 —6. 10 (m, 1 HX 4/5), 5. 93— 5. 80 (m, 1 HX 1/5), 5. 2
8— 5. 28 (m, 1 HX 1/5), 5. 08— 4. 92 (m, 1 HX 4/5), 3.
78— 3. 65 (m, 1 H), 3. 58— 3. 25 (m, 3H), 3. 08— 2. 93 (m, 1 H), 2. 27— 2. 12 (m, 1 H), 1. 87— 1. 73 (m, 1 H),
1. 70— 1. 50 (m, 2H), 1. 62 (d, J = 6. 8Hz, 3 H)
ma s s : 459, 461 (M+l) +. 実施例 130
4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— [((1 S) ― 1一 {4— [(1—メチルビペリジン一 4一ィル) ァミノ] フエ二ル} ェチル) アミ ノ] ピリミジン一 5—カルボ二トリル [1 30] (以下、 化合物 [1 30] という) の合成。
(1) 4— (8—クロロイミダゾ [1, 2— a」 ピリジン一 3—ィル) 一2— (メ チルチオ) ピリミジン一 5—カルボ二トリノレ [123— 1] 76mgと (S) — α— メチルー 4—ニトロベンジルァミン塩酸塩 57mg力、ら、 実施例 80— (4) の方法 に準じて、 4一(8—クロロイミダゾ [1, 2— a]ピリジン一 3—ィル)一 2— {[(1
S) — 1一 (4一二トロフエニル) ェチル] アミノ} ピリミジン一 5_カルボ-トリ ル [1 3 0— 1] 54mgを淡黄色固体として得た。
(2) 4一(8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィノレ)一 2— {[(1 S)一 1— (4—ニトロフエニル) ェチル] アミノ} ピリミジン一 5—カルボ-トリ ル [1 3 0—1] 5 4mg力 ら、 実施例 1 0 3— ( 2 ) の方法に準じて、 2— {[(1 S)— 1—(4ーァミノフエニル)ェチル]アミノ}一 4一(8—クロロイミダゾ [1 , 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボエトリノレ [ 1 3 0— 2] 3 8 mgを白色固体として得た。
(3) 2— {[(1 S) — 1— (4一アミノフヱ-ル) ェチル] アミノ} —4一 (8 一クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボニト リル [1 3 0— 2] 1 0mgと 1ーメチルー 4—ピペリ ドン 1 0 から、 実施例 9 1の方法に準じて、 目的ィ匕合物 [1 30] 6. 8 mgを白色固体として得た。
化合物 [1 3 0] のスぺクトルデータを以下に示す。
'H— NMR (DM S O— d 6) . δ : 1 0. 0 7 (d, J = 7. 1 H z , 1 HX 1/2), 9. 1 0 (d, J = 7. 1 H z, 1 HX 1/2), 8, 9 2 (d, J = 7. 6 H z, 1 HX 1/2), 8. 8 1 (d, J =8. OH z , 1 HX 1/2), 8. 7 6— 8. 7 5 (m, 1 H+ 1 HX 1/2), 8. 6 2 ( s , 1 HX 1/2), 7. 7 8 (d, J = 7. 6 H z, 1 HX 1 /2), 7. 7 3 (d, J = 7. 6H z, 1 HX 1/2), 7. 1 6 ( t , J = 7. 3H z, 1 HX 1/2), 7. 1 0 (d, J = 8. 6 H z , 2H X 1/2), 7. 0 8 (d, J = 8. 6H z , 2HX 1Z2), 7. 0 1 ( t, J = 7. 3H z , 1 HX 1/2), 6. 54 (d, J = 8. 6H z , 2HX 1/2), 6. 5 2 (d, J = 8. 6 H z , 2HX 1/2), 5. 3 3 ( t , J = 8. 4H z , 1 H), 5. 1 5— 5. 0 7 (m, 1 HX 1/2), 4. 9 6— 4. 8 8 (m, 1 HX 1/2), 3. 1 3— 3. 0 7 (m, 1 H), 2. 70— 2. 6 5 (m, 2H), 2. 1 3 ( s , 3H X 1/2), 2. 1 2 ( s, 3HX 1/2), 1. 9 8— 1. 9 3 (m, 2H), 1. 8 3— 1. 7 9 (m, 2H), 1. 4 7 (d, J = 7. 1 H z , 3 HX 1/2), 1. 4 3 (d, J = 7. 1 H z , 3HX 1/2), 1. 3 6— 1. 2 9 (m, 2 H) ma s s : 4 8 7, 4 8 9 (M+ 1 ) +. 実施例 1 3 1
4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— [((1 S) — 1— { 4— [4— (トリフルォロアセチル) ピぺラジン一 1ーィル] フエエル) ェチ ル) ァミノ] ピリミジン一 5—カルボ二トリル [1 3 1 ] (以下、 化合物 [ 1 3 1 ] という) の合成。
4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— (メチルチ ォ) ピリミジン一 5—カルボ二トリル [1 23— 1] 2 Omgと t一ブチル ((1 S) 一 1一 {4— [4一 (トリフルォロアセチル) ピぺラジン一 1一ィル] フエ二ル} ェ チル) カーバメート [1 1 0— 1] 4 Omg力、ら、 実施例 1 06— (3)、 (4) の方 法に準じて、 目的化合物 ['1 3 1] 1 7m gを白色固体として得た。
化合物 [1 3 1] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 71 (m, 1 HX 1/4), 9. 07— 8. 9 2 (m, 1 H+ 1 HX 3/4), 8. 6 2— 8. 50 (m, 1 H), 7. 55— 7. 20 (m, 3HX 3/4), 7. 4 9— 7. 0 9 (m, 3 H), 7. 04— 6. 8 9 (m, 2H+ 1 HX 1/4), 6. 6 6 (m, 1 HX 3/4), 6. 04 (m, 1 HX 3/4), 5. 80 (m, 1 HX 1/4), 5. 28 (m, 1 HX 1/4), 5. 08 (m, 1 H X 3/4), 3. 84 (m, 2H), 3. 77 (m, 2H), 3. 22 (m, 4H), 1. 6 2 (d, J = 8. OHz, 3H)
ma s s : 5 55, 5 5 7 (M+ 1 ) +. 実施例 1 32
4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) —2— {[(1 S) 一 1— (4ーピペラジン一 1ーィルフエニル) ェチル] アミノ} ピリミジン一 5—カル ボニトリル [ 1 32 ] (以下、 化合物 [1 32] という) の合成。 化合物 [1 3 1] 8mg力 ら、 実施例 1 10— (3) の方法に準じて、 目的化合物 [1 3 2] 4m gを白色固体として得た。
化合物 [1 32] のスぺクトルデータを以下に示す。
iH~NMR (CDC 13) δ : 9. 69 (d, J = 8. OHz, 1 HX 1/3), 8. 9 9— 8. 89 (m, 1 H+ 1 HX 2/3), 8. 6 2 (s, 1 HX 1/3), 8. 5 5 (s, 1 HX 3/4), 7. 50 (m, 1 HX 1/3), 7. 43 (d, J = 8. 0 H z , 1 HX 3/4), 7. 3 7— 7. 20 (m, 2H), 7. 0 1— 6. 8 9 (m, 2H+ 1 HX 1/4), 6. 66 (d d, J = 8. 0, 8. OH z, 1 HX 3/4), 6. 03 (m, 1 HX 3/4), 5. 79 (m, 1 HX 1/4), 5. 28 (m, 1 H X 1/4), 5. 05 (m, 1HX 3/4),. 3. 1 5 (m, 4H), 3. 0 3 (m, 4H), 1. 6 1 (d, J = 6. 8Hz, 3 H)
ma s s : 45 9, 46 1 (M+ 1) +. 実施例 1 33
4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— ({(1 S) —
1— [4一 (4ーメチルビペラジン一 1一ィル) フエニル] ェチル } ァミノ) ピリミ ジン一 5—力ルポ二トリル [133] (以下、 化合物 [133] という) の合成。
4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2—(メチルチオ) ピリミジン一 5—カルボ二トリル [1 23— 1] 2 Omgと t一ブチル {(1 S) — 1— [4一(4ーメチルビペラジン一 1一ィル) フエニル]ェチル }カーバメート [1 07—1] 37mg力 ら、 実施例 106— (3)、 (4) の方法に準じて、 目的化合物 [133] 8m gを白色固体として得た。
化合物 [133] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 68 (m, 1 HX 1/3), 8. 99— 8. 90 (m, 1 H+ 1 HX 2/3), 8. 62 ( s, 1 HX 1/3), 8. 56 ( s , 1 HX 3/4), 7. 50 (m, 1 HX 1/3), 7. 44 (m, 1 HX 3/4), 7. 38 一 7. 21 (m, 2H), 7. 00— 6. 90 (m, 2H+1HX 1Z4), 6. 65 (m, 1 HX 3/4), 6. 02 (m, 1 HX 3/4), 5. 79 (m, 1 HX 1/4), 5. 27 (m, 1 HX 1/4), 5. 05 (m, 1 HX 3/4), 3. 22 (m, 4H), 2. 59 (m, 4H), 2. 36 (s, 3H), 1. 61 (d, J = 6. 8H z , 3 H) ma s s : 473, 475 (M+ 1 ) +. 実施例 1 34
4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィノレ) 一 2— ({(1 S) — 1— [4— (2—ォキソピペラジン一 1一ィル) フエニル] ェチル } ァミノ) ピリミ ジン一 5—カルボ二トリル [134] (以下、 化合物 [134] という) の合成。
(1) t一ブチル [(1 S) — 1一 (4一ブロモフエ-ル) ェチル] カーバメート [106— l] 100mgと t一ブチル 3ーォキソピペラジン一 1一カルボキシレー ト 85mgから実施例 106— (2) の方法に準じて、 t一プチル 4— (4— { ( 1 S) — 1一 [( t一ブトキシカルボニル) ァミノ] ェチル } フエニル) 一 3—ォキソ ピぺラジン一 1一カルボキシレート [134— 1] 1 Omgを淡黄色固体として得た。 (2) t一プチノレ 4一(4一 {(1 S)— 1一 [(t一ブトキシカノレポ二ノレ) ァミノ] ェチル } フエニル) 一 3—ォキソピペラジ 一 1一カルボキシレート [134— 1] 1 Omgと 4一(8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル)一2—(メ チルチオ) ピリミジン一 5—カルボ二トリル [123— 1] 5mg力 ら、 実施例 10 6— (3)、 (4) の方法に準じて、 目的化合物 [134] 2mgを白色固体として得 た。
化合物 [134] のスぺクトルデータを以下に示す。
XH— NMR (CDC 1 3) δ : 9. 6 9 (d, J = 8. OH z , 1 HX 1/3), 8. 9 9 ( s, 1 HX 1/3), 8. 8 8 ( s, 1 HX 2/3), 8. 7 2 (d, J = 8. 0H z, 1 HX 1/3), 8. 6 1 ( s , 1 HX 1/3), 8. 5 8 ( s , 1 HX 2/ 3), 7. 6 0— 7. 2 0 (m, 5H+ 1 HX 1/3), 6. 8 0 (m, 1 HX 2/3), 6. 0 5 (m, 1 HX 2/3), 5. 8 9 (m, 1 HX 1/3), 5. 3 2 (m, 1 H X 1 /3), 5. 1 1 (m,' 1 HX 2/3), 3. 8 0— 3. 5 5 (m, 4H), 3. 2 3 (m, 2H), 1. 62 (d, J = 6. 8H z , 3 H)
ma s s : 4 7 3, 4 7 5 (M+ 1 ) +. 実施例 1 3 5
4一 (8—クロロイミダゾ [1 , 2— a] ピリジン一 3—^ fル) 一 2— [((1 S) ― 1— { 4— [[2— (ジメチノレアミノ) ェチル] (メチル) ァミノ] フエエル) ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [1 3 5] (以下、 化合物 [1 3 5] とい う) の合成。
(1) t一ブチル [(1 S) — 1— (4一ブロモフエ-ル) ェチル] カーバメート [1 0 6— 1] l O Omgと N, N, N,一トリメチルエチレンジァミン 4 1 m g力、 ら、 実施例 1 2 8— (1) の方法に準じて、 t一プチル ((1 S) — 1一 {4一 [[2 一 (ジメチルァミノ) ェチル] (メチル) ァミノ] フエ-ル} ェチル) カーバメート
[1 3 5— 1] 1 4m gを淡黄色固体として得た。
(2) t一ブチル ((1 S) — 1一 {4— [[2— (ジメチルァミノ) ェチル] (メ チル) ァミノ] フエ二ル} ェチル) カーバメート [1 3 5— 1] 1 4mgと 4一 (8 一クロロイミダゾ [1 , 2— a] ピリジン一 3—ィル) 一 2— (メチルチオ) ピリミ ジン一 5—カルボ二トリル [1 2 3— 1〕 9mgから、 実施例 1 0 6— (3)、 (4) の方法に準じて、 目的化合物 [1 3 5] 5. l mgを白色固体として得た。
化合物 [ 1 3 5] のスぺクトルデータを以下に示す。
XH— NMR (CDC 1 3) δ : 9. 7 0 (m, 1 HX 1/3), 9. 04 (d, J = 8. OH z , 1 HX 2/3), 8. 9 7 ( s , 1 HX 1/3), 8. 9 2 ( s , 1 HX 2/3), 8. 6 3 ( s, 1 HX 1/3), 8. 54 ( s, 1 HX 2/3), 7. 6 7 —7. 4 0 (m, 1 H), 7. 2 3 (m, 2H), 6. 9 5 (m, 1 HX 1/3), 6. 8 0— 6. 6 5 (m, 2H+ 1 HX 2/3), 6. 0 2 (m, 1 HX 2/3), 5. 8 0 (m, 1 HX 1/3), 5. 2 6 (m, 1 HX 1/3), 5. 04 (m, 1 HX 2/ 3), 3. 4 8 (m, 2H), 2. 9 6 ( s, 3 H), 2. 5 0 (m, 2H), 1. 6 1 (d, J = 6. 8H z , 3 H)
ma s s : 4 7 5, 4 7 7 (M+ 1) +.
実施例 1 36
2— ({(1 S)一 1一 [4一(3—アミノアゼチジン一 1一ィル) フヱニル] ェチル } ァミノ)一 4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジ ン一 5—カルボ-トリノレ [1 36] (以下、 化合物 [1 3 6] という) の合成。
(1) t一ブチル [(I S) — 1— (4一ブロモフエニル) ェチル] カーバメート [1 06— 1] 1 0 Omgと N—ァゼチジン一 3—イノレー 2, 2, 2—ト リ フノレオ口 ァセトアミド 6 7m gから、 実施例 1 28— (1) の方法に準じて、 t一プチル [(1 S) — 1— (4— { 3— [(トリフルォロアセチル) ァミノ] ァゼチジン一 1一^ fノレ } フエニル) ェチル] カーバメート [1 3 6— 1] 3 8mgを淡黄色固体として得た。
(2) t一ブチル [(1 S) — 1一(4一 { 3— [(トリフルォロアセチル) ァミノ] ァゼチジン一 1ーィノレ } フエニル) ェチル] カーバメート [1 36— 1] 38mgと 4一(8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2—(メチノレチォ) ピリミジン一 5—カルボ二トリル [1 23— 1] 1 9m gから、実施例 1 06— (3)、 (4) の方法に準じて、 N— { 1— [4— ((1 S) — 1— {[4— (8—クロ口イミ ダゾ [1, 2— a] ピリジン一 3—ィル)一 5—シァノピリミジン一 2—ィル] アミ ノ} ェチル) フエニル] ァゼチジン一 3 { ;V} —2, 2, 2—トリフノレオロアセト アミド [1 36— 2] 1 2mgを白色固体として得た。
(3) N— { 1— [4一 ((1 S) — 1一 {[4一 (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル)一 5—シァノピリミジン一 2—ィル] アミノ} ェチル) フ ェニル] ァゼチジン一 3—ィル } —2, 2, 2—トリフルォロアセトアミ ド [ 1 36 —2] 1 2mg力 ら、 実施例 1 1 0— (3) の方法に準じて、 目的化合物 [1 36] 6. 4m gを白色固体として得た。
化合物 [1 36] のスぺク トルデータを以下に示す。
ΧΗ— NMR (CDC 13) δ : 9. 6 9 (m, 1 HX 1/3), 9. 10 (d, J = 6. 4H z , 1 HX 2/3), 8. 97 (s , 1 HX 1/3), 8. 92 ( s , 1 HX 2 / 3), 8. 6 2 ( s, 1 HX 1/3), 8. 5 0 ( s , 1 HX 2/3), 7. 5 6 一 7. 40 (m, 1 H), 7. 2 1 (d, J = 8. 4Hz, 2H), 6. 95 (m, 1 HX 1/3), 6. 70 (m, 1HX 2/3), 6. 48 (d, J = 8. 4Hz, 2H), 6. 08 (m, 1 HX 2/3), 5. 7 9 (m, 1 HX 1/3), 5. 24 (m, 1 H X 1/3), 5. 0 3 (m, 1 HX 2/3), 4. 1 6 (m, 2H), 3. 9 5 (m, 1 H), 3. 4 5 (m, 2H), 1. 6 1 (d, J = 6. 8Hz, 3 H)
ma s s : 44 5, 447 (M+ 1) +.
実施例 1 37
2— [((1 S) — 1— {4— [(3 R) 一 3—ァミノピロリジン一 1一ィル] フエ二 ル} ェチル) ァミノ] —4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィ ル) ピリミジン一 5—カルボ二トリル [1 3 7] (以下、 化合物 [1 3 7] という) の合成。 '
(1) t—ブチル [(1 S) — 1一 (4—ブロモフエニル) ェチル] カーバメート [106— 1] l O Omgと (3R) — (+) — (トリフノレオロアセトアミ ド) ピロ リジン 7 3mgから、 実施例 1 2 8— (1) の方法に準じて、 t一プチル [(1 S) — 1— (4一 {(3 R) —3— [(トリフルォロアセチル) ァミノ] ピロリジン一 1一 ィル } フエニル) ェチル] カーバメート [1 3 7— 1] 24mgを淡黄色固体として 得た。 (2) t一ブチル [(1 S) — 1一 (4一 {(3 R) — 3— [(トリフルォロアセチ ル) ァミノ] ピロリジン一 1 ィル } フエニル) ェチル] カーバメート [1 3 7— 1] 24mgと 4一(8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル)—2—(メ チルチオ) ピリミジン一 5—カルボ二トリル [1 23— 1] 1 2mgから、 実施例 1 06— (3)、 (4) の方法に準じて、 N— {(3R) — 1— [4—((1 S)— 1一 {[4 — (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 5—シァノピリミジ ン一 2—ィル] ァミノ) ェチル) フエニル] ピロリジン一 3—ィル } —2, 2, 2— トリフルォロアセトアミド [1 3 7— 2] 1 3m gを白色固体として得た。
(3) N— {(3R) — 1— [4—((1 S) — 1一 {し4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル)一 5—シァノピリミジン一 2—ィル] アミノ} ェチル) フエニル] ピロリジン一 3—ィル } —2, 2, 2—トリフルォロアセトアミ ド [1 3 7— 2] 1 Omg力 ら、 実施例 1 1 0— (3) の方法に準じて、 目的化合物 [1 3 7] 7mgを白色固体として得た。
化合物 [1 3 7] のスぺクトルデータを以下に示す。
1 H— NMR (CDC 1 3) δ : 9. 69 (m, 1 HX 1/3), 9. 1 0 (d, J = 8. OH z, 1 HX 2/3), 8. 97 ( s ,· 1 HX 1/3), 8. 92 ( s , 1 HX 2/3), 8. 6 2 ( s , 1 HX 1/3), 8. 4 8 ( s, 1 HX 2/3), 7. 5 5 —7. 40 (m, 1 H), 7. 2 3 (d, J = 8. OH z , 2H), 6. 94 (m, 1 HX 1/3), 6. 7 3 (m, 1 HX 2/3), 6. 5 5 (d, J = 8. OH z, 2H), 6. 1 2 (m, 1 HX 2/3), 5. 80 (m, 1 HX 1/3), 5. 23 (m, 1 H X 1/3), 5. 04 (m, 1 HX 2/3), 3. 7 3 (m, 1 H), 3. 5 9— 3.
40 (m, 2H), 3. 33 (m, 1 H), 3. 02 (m, 1H), 2. 21 (m H), 1. 81 (m, 1 H), 1. 61 (d, J = 6. 8Hz, 3 H)
ma s s : 459, 461 (M+ 1 ) +. 実施例 138
4一 (8—クロ口イミダゾ' [1, 2— a] ピリジン一 3—ィル)一 2— ({(1 S) — 1一 [4— (4ーメチルー 1, 4一ジァゼパン一 1一ィル) フエ-ル] ェチル } アミ ノ) ピリミジン一 5—カルボ二トリル [1 38] (以下、 化合物 [1 38] という) の合成。
(1) t一ブチル [(1 S) — 1一 (4一ブロモフエ-ル) ェチル] カーバメート [106— 1] 10 Omgと N—メチルホモピぺラジン 46mgから実施例 106—
(2) の方法に準じて、 t一プチル {(1 S) — 1— [4— (4—メチルー 1, 4一 ジァゼパン一 1一ィル) フエニル] ェチル } カーバメート [1 38— 1] 1 5mgを 淡黄色固体として得た。
(2) t—ブチル {(1 S) — 1一 [4一 (4—メチルー 1, 4一ジァゼパン一 1 一ィル) フエニル] ェチル } カーバメート [138— 1] 15mgと 4一 (8—クロ ロイミダゾ [1, 2— a] ピリジン一 3—ィル) —2— (メチルチオ) ピリミジン一 5—カルボ二トリル [123— 1] 9mgから、 実施例 106— (3)、 (4) の方法 に準じて、 目的化合物 [138] 1. 2 mgを白色固体として得た。
化合物 [138] のスぺクトルデータを以下に示す。
ΧΗ— NMR (CDC 13) δ : 9. 69 (d, J = 8. OH z, 1 HX 1/3), 9.
07 (d, J = 8. OH z , 1 HX 2/3), 8. 97 ( s ' 1 HX 1/3), 8. 9 2 ( s , 1 HX 2/3), 8. 63 (s, 1 HX 1/3), 8. 54 ( s , 1 HX 2/
3), 7. 67— 7. 10 (m, 3H), 6. 93 (m, 1 HX 1/3), 6. 75— 6. 60 (m, 2H+ 1 HX 2/3), 6. 02 (m, 1 HX 2/3), 5. 79 (m, 1 HX 1/3), 5. 25 (m, 1 HX 1/3), 5. 04 (m, 1 HX 2/3), 3. 60 (m, 2H), 3. 48 (m, 2H), 2. 75 (m, 2H), 2. 60 (m, 2 H), 2. 40 (m, 3H), 2. 05 (m, 2H), 1. 61 (d, J = 6. 8Hz, 3H)
ma s s : 487, 489 (M+ 1 ) +. 実施例 139
4— (8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル)一2— ({(1 S) —
1一 [3—クロロー 4一 (4ーメチルビペラジン一 1一ィル) フエニル] ェチル } ァ
6187
160
ミノ) ピリミジン一 5—カルボ二トリル [1 3 9] (以下、 化合物 [1 3 9] とレヽう) の合成。
( 1 ) t—ブチル {(I S)— 1一 [4一 (4ーメチルビペラジン一 1一ィル) フ ェニル] ェチル } カーバメート [1 0 7— 1] 8 Omgをクロ口ホルム 3mLに溶解 し、 N—クロ口こはく酸イミ ド 7 Omgをカロえ、 室温にて 2時間撹拌した。 反応液を クロロホルムにて希釈し、 飽和炭酸水素ナトリゥム水溶液で洗浄し、 無水硫酸マグネ シゥムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮して、 得られた残渣を分取薄 層クロマトグラフィーにて精製し、 t—ブチル {(1 s)一 1一 [3—クロロー 4一 (4ーメチルピペラジン一 1一ィル) フエニル]ェチル }カーバメート [1 3 9— 1] 8 Omgを無色油状物として得た。
(2) t—ブチル {(I S) —1— [3—クロ口一 4— (4ーメチルビペラジン一 1一ィル) フエニル] ェチル } カーバメート [1 3 9— 1] 1 5mgと 4一 (8—ク ロロイミダゾ [1 , 2— a] ピリジン一 3—ィル)一 2—(メチルチオ) ピリミジン 一 5—力ルポ二トリル [1 2 3— 1] 1 2mg力 ら、 実施例 1 0 6— (3)、 (4) の 方法に準じて、 目的化合物 [1 3 9] 7. 5mgを白色固体として得た。
化合物 [ 1 3 9] のスぺクトルデータを以下に示す。
ΧΗ— NMR (CDC 1 3) δ : 9. 7 0 (m, 1 HX 1/3), 8. 9 8 ( s , 1 H X 1/3), 8. 9 3 ( s , 1 HX 2/3), 8. 8 6 (d, J = 8. 0H z, 1 HX
2/3), 8. 6 3 ( s , 1 HX 1/3), 8. 5 7 ( s , 1 HX 2/3), 7. 6 0
—7. 2 0 (m, 3H), 7. 0 8 (d, J = 8. OH z, 1 H), 6. 9 9 (m, 1
HX 1/3), 6. 70 (m, 1 HX 2/3), 6. 04 (m, 1 HX 2/3), 5.
8 0 (m, 1 HX 1/3), 5. 2 6 (m, 1 HX 1/3), 5. 0 3 (m, 1 HX 2 /3), 3. 1 0 (m, 4H), 2. 6 0 (m, 4H), 2. 3 7 ( s, 3H), 1. 6
1 (d, J = 6. 8H z , 3H)
m a s s : 5 0 7, 5 0 9 (M+ 1) +. 実施例 1 40
4— (8—クロロイミダゾ [1 , 2— a] ピリジン一 3—ィル)一2— {[ 1— (6 ーピペラジン一 1 fルビリジン一 3—ィル) ェチル] アミノ} ピリミジン一 5—力 ルポ二トリル [ 1 40] (以下、 化合物 [1 4 0] という) の合成。
( 1) 6—クロ口ニコチノ二トリノレ 7 0 2mg、 t一ブチルピペラジンー 1—力ノレ ボキシレー ト 9 54mg、 炭酸カリウム 1. 2 g、 および N, N—ジメチルァセトァ ミ ド 1 OmLの混合物を、 室温にて 3時間撹拌した後、 6 0°Cに昇温しさらに 4時間
7
161
撹拌した。 室温に戻した後、 水を加え、 酢酸ェチルにて抽出した。 有機層を無水硫酸 マグネシウムにて乾燥し、 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣をシ リカゲルカラムクロマトグラフィーにて精製し、 t一プチル 4— (5—シァノピリジ ンー 2—^ ル) ピぺラジン一 1一カルボキシレート [140— 1] 1. 39 gを白色 固体として得た。
(2) t一ブチル 4— ( 5—シァノビリジン一 2—^ {ル) ピぺラジン一 1一カルボ キシレート [140— 1] 90 Omgをテトラヒドロフラン 4 OmLに溶解し、 氷冷 下、 臭化メチルマグネシウム (3. 0 Mジェチルエーテル溶液) 2. 5mLを加え、 同温にて 1時間撹拌した後、 室温まで昇温し終夜撹拌した。 氷冷下、 飽和塩化アンモ ニゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄し、 無 水硫酸マグネシウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた 残渣をメタノール 50 mLに溶解し、 氷冷下、 水素化ほう素ナトリウム 850mgを 加え、 同温にて 2時間 30分撹拌した。 反応混合物に水を加え、 クロ口ホルムにて抽 出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウムにて乾燥し、 不溶物 をろ過した。 ろ液を減圧濃縮して、 得られた残渣をシリカゲルカラムクロマトグラフ ィ一にて精製し、 t—ブチル 4一 [5— (1—アミノエチル) ピリジン一 2—ィル] ピぺラジン一 1—カルボキシレート [140— 2] 95 Omgを白色固体として得た。 (3) 4一 (8—クロロイミダゾ [ 1, 2— a] ピリジン一 3—ィル) 一 2— (メ チルチオ) ピリミジン一 5—カルボ二トリノレ [123— 1] 35mgと t一ブチル 4 一 [5—(1一アミノエチル) ピリジン一 2—^ fル] ピぺラジン一 1一カルボキシレ ート [140— 2] 68mgから、 実施例 80— (4) の方法に準じて、 t一ブチル 4一 [5— (1一 {[4一(8—クロロイミダゾ [1, 2— a] ピリジン一 3—ィル) —5—シァノビリミジン一 2—ィル] ァミノ) ェチル) ピリジン一 2—ィル] ピペラ ジン一 1—カルボキシレート [140— 3] 29mgを褐色油状物として得た。
(4) t一プチノレ 4一 [5— (1一 {[4一 (8—クロロイミダ、ソ、 [1, 2— a] ピリジン一 3—ィル)一 5—シァノピリミジン一 2—ィル] アミノ} ェチル) ピリジ ンー 2—ィル] ピぺラジン一 1—カルボキシレート [140— 3] 29m gをクロ口 ホルム 3mLに溶解し、 トリフルォロ酢酸 3mLを加え、 室温にて 1時間撹拌した。 反応混合物を減圧濃縮し、 飽和炭酸水素ナトリウム水溶液を加え、 クロ口ホルムにて 抽出した。 有機層を無水硫酸マグネシウムにて乾燥し、 不溶物をろ過した。 ろ液を減 圧濃縮し、 得ちれた残渣を少量のクロ口ホルムに溶解し、 へキサンにて固化し、 目的 化合物 [140] 16. 6 mgを白色固体として得た。
化合物 [140] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 75 (m, 1HX 1/3), 9. 06 (d, J = 8. 0Hz, 1 HX 2/3), 8. 94 ( s , 1 HX 1/3), 8. 90 ( s , 1HX 2/3), 8. 60 ( s , 1 HX 1/3), 8. 55 ( s , 1 HX 2/3), 8. 22 (m, 1 H), 7. 60— 7. 40 (m, 2H), 6. 97 (m, 1 HX 1/3), 6. 85 (m, 1 HX 2/3), 6. 69 (d, J = 8. OH z , 1 H), 5. 23 (m, 1 HX 1/3), 5. 04 (hi, 1 HX 2/3), 3. 55 (m, 2H), 3. 04 (m, 2H), 1. 62 (d, J = 6. 8H z , 3 H)
ma s s : 460, 462 (M+ 1) +. 実施例 141
4一 (6, 8—ジクロロイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— [(( 1 S) — 1— { 3— [(1ーメチルビペリジン一 4_ィル) ァミノ] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [141] (以下、 化合物 [141] とい う) の合成。
(1) 2—アミノー 3, 5 ジクロ口ピリジン 10 Omgと 4一 [(Z) — 2—ェ トキシビエル]一 2—(メチルチオ)一5—ピリミジンカノレポ-ノレ [48— 3] 13 5mg力、ら、 実施例 25— (3) の方法に準じて、 4— (6, 8—ジクロロイミダゾ
[1, 2~ a] ピリジン一 3—ィル)一 2— (メチルチオ) ピリミジン一 5—カルボ 二トリル [141— 1] 205m gを淡褐色固体として得た。
(2) 4— (6, 8—ジクロロイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2 一(メチルチオ)ピリミジン一 5—カルボ二トリル [141— 1] 5 Omgと 3— [(1 S) —1—アミノエチル] ァニリン 24. 4mg力、ら、 実施例 80— (4) の方法に 準じて、 2— {[(1 S)—1— (3—ァミノフエ-ル) ェチル] アミノ}—4一(6, 8—ジクロロイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボ 二トリル [141— 2] 1 2. 6mgを淡褐色固体として得た。
(3) 2— {[(1 S)— 1— (3—アミノフヱニル) ェチル] アミノ}一 4一(6, 8—ジクロロイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボ 二トリル [141一 2] 9. 7m gと 1一メチル一4ーピペリ ドン 8. 4 から、 実施例 91の方法に準じて、 目的化合物 [141] 1 1. 7 mgを白色固体として得 た。
ィ匕合物 [141] のスぺクトルデータを以下に示す。
JH— NMR (CDC 13) δ : 9. 80 (s, 1 HX 4/5), 9. 75 (s, 1 H X 1/5), 9. 01 (s , 1 HX 4/5), 8. 95 (s, 1HX 1/5), 8. 6
2 (s , 1 HX 1/5), 8. 52 (s, 1 HX 4/5), 7. 52 ( s , 1 H), 7. 20— 7. 08 (m, 1 H), 6. 77— 6. 42 (m, 3H), 6. 1 8— 5. 88 (m, 1 H), 5. 2 8— 5. 0 6 (m, 1 H), 3. 70— 3. 4 2 (m, 1H), 3. 3 8— 3. 20 (m, 1 H), 2. 9 5— 2. 7 6 (m, 2H), 2. 3 5 ( s , 3H), 2. 2 7—1. 9 5 (m, 4H), 1. 6 6 (d, J = 6. 6Hz, 3H), 1. 60— 1. 40 (m, '2H)
ma s s : 52 1, 5 23 (M+ 1) +. 実施例 142
4一 (7, 8—ジクロロイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— [((1 S) — 1一 { 3— [(1—メチルピペリジン一 4—ィル) ァミノ] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力ルポ二トリル [142] (以下、 化合物 [1 42] とい う) の合成。 (1) 3, 4ージクロ口ピリジン一 2—ァミン (国際公開第 9 7ノ027205号 パンフレット、 22— 23ぺ ジに記載されている方法により合成した) 3 Omgと 4一 [(Z) 一 2—ェトキシビエル] —2— (メチルチオ) 一 5—ピリミジンカルボ ニル [48— 3] 40. 7mg力、ら、実施例 25—(3) の方法に準じて、 4一 (7, 8—ジクロロイミダゾ [1, 2— a] ピリジン一 3—ィル) —2— (メチルチオ) ピ リミジン一 5—力ルポ二トリル [142— 1] 2 Omgを淡褐色固体として得た。
(2) 4— (7, 8—ジクロロイミダゾ [1, 2— a」 ピリジン一 3—ィル)一 2 一(メチルチオ)ピリミジン一 5—カルボ二トリノレ [142— 1] 2 Omgと 3— [(1 S)一 1一アミノエチル] ァニリン 24. 4mg力、ら、 実施例 80— (4) の方法に 準じて、 2_ {[(1 S) — 1一(3—ァミノフエニル) ェチル] アミノ}— 4— (7, 8—ジクロロイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボ 二トリル [142— 2] 9. Omgを白色固体として得た。
(3) 2— {[(1 S) — 1— (3—アミノフヱニル) ェチル] アミノ}一 4— (7, 8—ジクロロイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボ 二トリル [1 42— 2] 8. Omgと 1一メチル一4—ピぺリ ドン 8. 力 ら、 実施例 9 1の方法に準じて、 目的化合物 [1 42] 6. 5 m gを白色固体として得た。 化合物 [142] のスぺクトルデータを以下に示す。
ΧΗ— NMR (CDC 1 3) δ : 9. 6 3 (d, J = 7. 6Hz, 1 HX 1/5), 8. 96 (s , 1 HX 1/5), 8. 8 3 (s, 1 HX 4/5), 8. 6 2 (s, 1 HX 1 5), 8. 5 6 ( s , 1 HX 4/5), 8. 5 2 (d, J = 7. 6Hz, 1 HX 4/
5), 7. 20 (d d, J = 7. 6Hz, 7. 6Hz, 1H), 7. 05 ( d , J = 7. 6Hz, 1 HX 1/5), 6. 81 (d, J = 7. 6H z , 1 HX 4/5), 6. 68 (d, J = 7. 6Hz, 1 H), 6. 60— 6. 50 (m, 2H), 6. 10— 6. 0 6 (m, 1 H X 4 / 5 ) , 5. 88— 5. 80 (m, 1 HX 1/5), 5. 23— 5. 20 (m, 1 HX 1/5), 4. 94— 4. 85 (m, 1 HX 4/5), 3. 75— 3. 70 (m, 1 H), 3. 35^3. 25 (m, 1 H), 2. 89— 2. 80 (m, 2H), 2. 32 (s, 3H), 2. 1 9— 2. 05 (m, 4H), 1. 61 (d, J = 6. 8 Hz, 3H), 1. 60— 1. 50 (m, 2 H)
ma s s : 521, 523 (M+ 1 ) +. 実施例 143
4— (8—フルォロイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— {[(1 S) — 1— (4ーピペラジン一 1—ィルフエニル) ェチル] アミノ} ピリミジン一 5—力 ルポ-トリル [143] (以下、 化合物 [143] という) の合成。
(1) 2—アミノー 3—フルォロピリジン 10 lmgと 4一 [(Z) —2—ェトキ シビュル]—2— (メチルチオ)一 5—ピリミジンカルボ-ル [48— 3] 200m から、 実施例 25— (3) の方法に準じて、 4一 (8—フルォロイミダゾ [1, 2 一 a] ピリジン一 3—ィル)一 2—(メチルチオ) ピリミジン一 5—力ルポ二トリル
[143— 1] 11 Omgを淡黄色固体として得た。
(2) 4一(8—フルォロイミダゾ [1, 2— a] ピリジン一 3—ィル)一2— (メ チルチオ)ピリミジン一 5—カルボ二トリノレ [143— 1] 25mgと t—プチル((1 S)— 1一 {4一 [4一(トリフノレ才ロアセチノレ) ピぺラジン一 1一ィル] フエ二ル} ェチル) カーバメート [1 10— 1] 52. 8mg力 ら、 実施例 106— (3)、 (4) の方法に準じて、 化合物 [143— 2] 17m gを黄色固体として得た。
(3) 化合物 [143— 2] 17mg力 ら、実施例 1 10—(3)の方法に準じて、 目的化合物 [143] 13. 7mgを淡黄色固体として得た。
化合物 [143] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 53 (d, J = 6. 9Hz, 1 HX 1/5), 8.
92 (s, 1 HX 1/5), 8. 90 (s, 1 HX 4/5), 8. 83 (d, J = 6.
9Hz, 1 HX 4/5), 8. 57 (s, 1 HX 1/5), 8. 49 ( s , 1 HX 4/
5), 7. 36— 6. 60 (m, 6 H), 6. 18— 6. 10 (m, 1 HX 4/5), 5. 87— 5. 75 (m, 1 HX 1/5), 5. 33— 5. 20 (m, 1 HX 1/5),
5. 1 7— 5. 00 (m, 1 HX 4/5), 3. 18— 3. 10 (m, 4H), 3. 0
6187
165
8— 2. 96 (m, 4H), 2. 65 (b r s , 1 H) , 62 (d, J = 6. 8H z , 3H)
ma s s : 443 (M+ 1) +. 実施例 144
4一 (8—ブロモイミダゾ' [1, 2— a] ピリジン一 3—ィル)一 2— [((1 S) ― 1— { 3— [(1ーメチルピペリジン一 4一ィル) ァミノ] フエ二ル} ェチル) アミ ノ] ピリミジン一 5—カルボ二トリル [144] (以下、 化合物 [144] という) の合成。
(1) 2—アミノー 3—プロモピリジン 55 Omgと 4一 [(Z) 一 2—エトキシ ビエル] —2—(メチルチオ)一 5—ピリミジンカルボニル [48— 3] 70 Omg から、 実施例 25—(3) の方法に準じて、 4一 (8—プロモイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2—(メチルチオ) ピリミジン一 5—力ルポ二トリル [14 4— 1] 516mgを淡褐色固体として得た。
(2) 4— (8—ブロモイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— (メ チルチオ) ピリミジン一 5—カルボ二トリル [144— 1] l O Omgと 3— [(1 S) —1—アミノエチノレ] ァニリン 59mg力 ら、 実施例 106— (3)、 (4) の方 法に準じて、 2— {[(1 S) — 1一 (3—ァミノフエニル) ェチル] ァミノ)一 4一 (8—ブロモイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5_カルボ 二トリノレ [144一 2] 61. lmgを淡黄色固体として得た。
(3) 2— {[(1 S) — 1— (3—ァミノフ ニル) ェチル] アミノ}一 4一 (8 ーブロモイミダゾ [1, 2— a] ピリジン一 3—ィル) ピリミジン一 5—カルボニト リノレ [144一 2] 21. 9mgと 1—メチルー 4ーピペリ ドン 18. 6 ^ L力、ら、 実施例 91の方法に準じて、 目的化合物 [144] 16. 4mgを白色固体として得 た。
化合物 [144] のスぺクトルデータを以下に示す。
ΧΗ— NMR (CDC 13) δ : 9. 72 (d, J =6. 9Hz, 1HX 1/5), 8.
99 (s, 1 HX 1/5), 8. 89 ( s , 1 HX 4/5), 8. 83 (d, J = 6.
9Hz, 1 HX 4/5), 8. 62 (s, 1HX 1/5), 8. 54 (s, 1 HX 4/
5), 7. 72— 6. 80 (m, 2H), 6. 70— 6. 52 (m, 4H), 6. 06 —6. 04 (m, 1 HX 4/5), 5. 88— 5. 80 (m, 1 HX 1/5), 5. 2 8— 5. 1 5 (m, 1 HX 1/5), 5. 05— 4. 89 (m, 1 HX 4/5), 3.
74— 3. 45 (m, 1 H), 3. 35— 3. 18 (m, 1H), 2. 87— 2. 68
6187
166
(m, 2H), 2. 30 (s, 3H), 2. 1 8—1. 94 (m, 4H), 1. 6 1 (d, J = 6. 6Hz, 3H), 1. 60— 1. 40 (m, 2 H)
ma s s : 5 3 1, 5 33 (M+ 1 ) +. 実施例 1 45
4一 (8—ブロモイミダゾ' [1, 2— a] ピリジン一 3—ィル)一2— {[(1 S) — 1一 (4ーピペラジン一 1一ィルフヱニル) ェチル] アミノ} ピリミジン一 5—カル ボニトリル [145] (以下、 化合物 [145] という) の合成。 (1) 4一 (8—ブロモイミダゾ [1, 2— a] ピリジン一 3—ィル)一2— (メ チルチオ) ピリミジン一 5—カルボ二トリル [144— 1] 30. 3m gと tーブチ ル ((1 S)一 1一 {4— [4— (トリフルォロアセチル) ピぺラジン一 1一ィル] フエ二ル} ェチル) カーバメート [1 1 0— 1] 5 2. 8mg力、ら、 実施例 1 06— (3)、 (4) の方法に準じて、 4一 (8—プロモイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— [((1 S) — 1— {4— [4一 (トリフルォロアセチル) ピペラジ ンー 1—ィル] フエ-ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [14 5— 1] 29. 5m gを白色固体として得た。
(3) 4— (8—プロモイミダゾ [1, 2— a] ピリジン一 3—ィル)一2— [((1 S) — 1— {4— [4— (トリフルォロアセチル) ピぺラジン一 1 fル] フエ二ル} ェチル)ァミノ] ピリミジン一 5—カルボ二トリル [1 45— 1] 29. 5mgから、 実施例 1 1 0— (3) の方法に準じて、 目的化合物 [1 45] 2 1. 2mgを淡黄色 固体として得た。
化合物 [145] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 75— 9. 73 (m, 1 HX 1/5), 8. 9 2 —8. 90 (m, 1 H+ 1 HX 4/5), 8. 6 2 (s, 1 HX 1/5), 8. 54 ( s, 1 HX 4/5), 7. 70— 6. 5 9 (m, 6 H), 6. 09— 6. 0 7 (m, 1 HX 4/5), 5. 86— 5. 72 (m, 1 HX 1/5), 5. 28— 5. 20 (m, 1 H X 1/5), 5. 07— 5. 03 (m, 1 HX 4/5), 3. 1 7— 3. 1 0 (m, 4 H), 3. 05— 3. 03 (m, 4H), 2. 6 7 (b r s , 1 H), 1. 6 1 (d, J = 6. 6Hz, 3H)
ma s s : 503, 505 (M+ 1) +. 実施例 1 46
4一 (8—プロモー 6—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— [((1 S) —1— { 3— [(1ーメチルビペリジン一 4一ィル) ァミノ] フエ二ル}
JP2005/016187
167
ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [146] (以下、 化合物 [14 6] という) の合成。 ·
(1) 2—アミノー 3—プロモー 5—メチルピリジン 169mgと 4一 [(Z) ― 2—エトキシビュル]一 2—(メチルチオ)一 5—ピリミジンカルボニル [48— 3] 2 O 0mgから、 実施例 2'5— (3) の方法に準じて、 4一 (8—プロモー 6—メチ ルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2—(メチルチオ) ピリミジン一 5—カルボ二トリル [146_1] 216mgを淡黄色固体として得た。 (2) 4一 (8—プロモー 6—メチルイミダゾ [1, 2— a」 ピリジン一 3—ィノレ) 一 2—(メチノレチォ) ピリミジン一 5—カルボ二ト リノレ [146— 1] 53. 6 m g と 3— [(I S) —1 アミノエチル] ァニリン 24. 4mg力、ら、 実施例 106— (3)、 (4) の方法に準じて、 2— {[(1 S)— 1一(3—ァミノフエニル) ェチル] アミノ}一 4一 (8—ブロモ一6—メチルイミダゾ [1, 2— a] ピリジン一 3—^ f ル) ピリミジン一 5—カルボ二トリル [146— 2] 30. 8mgを淡黄色固体とし て得た。 .
(3) 2— {[(1 S) — 1一 (3—アミノフヱニル) ェチル] アミノ}一 4一 (8 一プロモー 6—メチルイミダゾ [1, 2— a] ピリジン一 3—^ fル) ピリミジン一 5 —カルボ二トリノレ [ 146— 2] 17. 7mgと 1—メチ Λ ^— 4—ピぺリ ドン 14. から、 実施例 91の方法に準じて、 目的化合物 [146] 16. 4mgを白色 固体として得た。
化合物 [146] のスぺク トルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 47— 9. 30 (m, 1H), 8. 96— 8. 8 3 (m, 1 H), 8. 65— 8. 40 (m, 1H), 7. 60— 7. 46 (m, 1 H), 7. 30— 7. 10 (m, 1 H), 6. 77— 6. 45 (m, 2H), 6. 25— 5. 80 (m, 1 H), 5. 34— 5. 10 (m, 1 H), 3. 82— 3. 50 (m, 1 H), 3. 37— 3. 16 (m, 1H), 2. 88— 2. 78 (m, 2H), 2. 29 ( s , 3H), 2. 32— 2. 25 (m, 3H), 2. 33— 1. 92 (m, 4H), 1. 6 6 (d, J = 6. 6Hz, 3H), 1. 60— 1. 40 (m, 2H)
ma s s : 545, 547 (M+ 1 ) +. 実施例 147
4— [8— (フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2— [((1 S) — 1一 { 3— [(1—メチルビペリジン一 4一ィル) ァミノ] フユ-ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [147] (以下、 化合物 [14
7] とレヽう) の合成,
(1) 水素化リチウムアルミニウム 5. 7 gのテトラヒ ドロフラン 10 OmLの溶 液に、 2—ァミノニコチン酸 13. 8 gのテトラヒ ドロフラン溶液 2 OmLを加え、 2時間加熱還流した。 反応混合物を室温に戻した後、 硫酸ナトリウム 10水和物を気 泡が出なくなるまでゆつぐりと加え、室温にて 2時間撹拌した。 不溶物をセライトに てろ過し、 ろ液を減圧濃縮し、 (2—アミノビリジン一 3—^ fル) メタノール [14 7— 1] 10. 7 gを白色固体として得た。 (2) (2—アミノビリジン一 3_ィル) メタノール [147— 1] 4. 31 gと 4— [(Z) —2—エトキシビュル]一 2—(メチルチオ)一 5—ピリミジンカルボ 二トリノレ [48— 3] 7. 0 gから、 実施例 25— (3) の方法に準じて、 4一 [8 一(ヒドロキシメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2— (メチ ルチオ) ピリミジン一 5—カルボ二トリル [147— 2] 6. 76 gを淡褐色固体と して得た。
(3) 4— [8— (ヒ ドロキシメチル) イミダゾ [1, 2— a] ピリジン一 3—ィ ル]一 2— (メチルチオ) ピリミジン一 5—カルボ二トリノレ [147— 2] 4. 23 gをクロ口ホルム 50 OmLに溶解し、 一40°Cにてビス一 (2—メ トキシェチル) アミノサルファートリフルオリ ド 5. 2 mLを加え、 同温にて 1時間撹拌した。 さら にビス一(2—メ トキシェチル)アミノサルファートリフルオリ ド 2. 6mLを加え、 同温にて 1時間撹拌した。 飽和炭酸水素ナトリゥム水溶液をゆつく り加え、 クロロホ ルムにて抽出した。 有機層を水、 飽和食塩水にて順に洗浄し、 無水硫酸マグネシウム にて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮して得られた残渣を少量のクロロホ ルムに溶解し、 ジェチルエーテルを加え固化し、 4— [8— (フルォロメチル) イミ ダゾ [1, 2— a] ピリジン一 3—ィル]一 2—(メチルチオ) ピリミジン一 5—力 ルポ二トリル [147— 3] 3. 43 gを淡褐色固体として得た。
(4) 4一 [8— (フルォロメチル) イ ミダゾ [1, 2— a] ピリジン一 3—ィノレ] 一 2— (メチルチオ) ピリミジン一 5—カルボ二トリル [147— 3] 20 m gと 3
— [(I S) 一 1一アミノエチル] ァニリン 18mg力、ら、 実施例 80— (3) の方 法に準じて、 2— {[(1 S) 一 1一 (3—ァミノフエニル) ェチル] アミノ}一 4— [8— (フルォロメチル) イミダゾ [1, 2_a] ピリジン一 3—ィル] ピリミジン 一 5—カノレポ二 ト リノレ [147— 4] 9. Omgを淡黄色固体として得た。
(5) 2— {[(1 S) — 1一 (3—アミノフヱニル) ェチル] アミノ} —4— [8
― (フルォロメチル) イミダゾ [1 , 2— a] ピリジン一 3—ィル] ピリミジン一 5 一力ルポ二トリル [1 4 7— 4] 9. Omgと 1ーメチルー 4ーピペリ ドン 7. 4 μ から、 実施例 9 1の方法に準じて、 目的化合物 [1 4 7] 4. 3m gを白色固体と して得た。
化合物 [1 4 7] のスぺクトルデータを以下に示す。
ΧΗ— NMR (CDC 1 3) 'δ : 9. 7 5— 9. 74 (m, 1 HX 1/5), 8. 9 5 ( s, 1 HX 1/5), 8. 8 9— 8. 8 6 (m, 1 HX 4/5 + 1 HX 4/5), 8. 6 0 ( s ' 1 HX 1/5), 8. 5 5 (s , 1 HX 4/5), 7. 5 5— 7. 4 3 (m, 1 H), 7. 2 3— 7. 0 6 (m, 1 H), 6. 8 2— 6. 5 3 (m, 4H), 6. 0 4一 5. 7 6 (m, 3H), 5. 2 6— 5. 1 8 (m, 1 HX 1/5), 5. 0 3— 4. 9 6 (m, 1 HX 4/5), 3. 6 7— 3. 6 3 (m, 1 H), 3. 3 2— 3. 2 6 (m, 1 H), 2. 8 5— 2. 7 9 (m, 2H), 2. 3 2 ( s, 3 H), 2. 1 3— 2. 0 1 (m, 4H), 1. 6 1 (d, J = 6. 8H z, 3H), 1. 5 6— 1. 4 9 (m, 2H)
ma s s : 48 5 (M+ 1 ) +. 実施例 1 48
4— [8— (フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—^ fル]一 2— ({(1 S)一 1一 [4一 ( 4ーメチルピぺラジンー 1ーィル) フエニル] ェチル } ァ ミノ) ピリミジン一 5—カルボ二トリル [1 4 8] (以下、 化合物 [ 1 4 8] という) の合成。
4一 [8— (フルォロメチル) イミダゾ [1, 2— 3 ] ピリジン一 3—ィル J一 2 — (メチルチオ) ピリミジン一 5—力ルポ二 ト リノレ [1 4 7— 3] 1 5mgと tーブ チル {( 1 S) —1— [4— (4ーメチルビペラジン一 1一ィル) フエニル] ェチル } カーバメート [1 0 7— 1] 3 3. 4mg力、ら、 実施例 1 0 6— ( 3 )、 ( 4 ) の方法 に準じて、 目的化合物 [1 4 8] 1 1. 3 mgを白色固体として得た。
化合物 [1 4 8] のスぺクトルデータを以下に示す。
XH— NMR (DMSO— d 6) 6 : 1 0. 1 1 (d, J = 7. 3H z , 1 HX 1/2), 9. 1 2 (d, J = 7. 1 H z , 1 HX 1/2), 8. 9 5 (d, J = 7. 3H z , 1 HX 1/2), 8. 8 5 (d, J = 8. 3H z , 1 HX 1/2), 8. 7 5 ( s , 1 HX 1 /2), 8. 74 ( s , 1 HX 1/2), 8. 7 3 ( s, 1 HX 1/2), 8. 6 1 ( s, 1 HX 1/2), 7. 6 9 (d, J = 6. 8 H z , 1 HX 1/2), 7. 6 3 (d, J = 6. 8H z, 1 HX 1 /2), 7. 2 7— 7. 2 4 (m, 2H+ 1 HX 1/2), 7. 1 0 ( t , J = 7. 3H z, 1 HX 1/2), 6. 9 3 ( d , J = 8. 8H z , 2HX 1/2), 6. 8 9 (d, J = 8. 8H z , 2HX 1/2), 5. 8 3
TJP2005/016187
170
(d, J =4 7. 1 H z , 2HX 1/2), 5. 79 (d, J =47. 1 Η ζ, 2H X 1/2), 5. 20— 5. 1 3 (m, 1 HX 1/2), 5. 0 5— 4. 98 (m, 1 HX 1X2), 3. 08— 3. 0 5 (m, 4H), 2. 43— 2. 3 8 (m, 4H), 2. 1 9 (s , 3HX 1/2), 2. 1 8 (s , 3HX 1/2), 1. 49 (d, J = 6. 8Hz, 3HX 1/2), 1. 46 (d, J =6. 1Hz, 3HX 1/2) ma s s : 47 1 (M+ 1') +. 実施例 1 49
4一 [8—(フルォロメチル)イミダゾ [1, 2— a]ピリジン一 3—ィル]一 2— {[(1 S) — 1— (4ーピペラジン一 1ーィルフエニル) ェチル] アミノ} ピリミジン一 5 一カルボ-トリル [149] (以下、 化合物 [149] という) の合成。
(1) 4— [8— (フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2— (メチルチオ) ピリミジン一 5—カルボ二トリル [14 7— 3] 22mgと t 一プチル ((1 S) — 1一 {4— [4— (トリフルォロアセチル) ピぺラジン一 1一 ィル] フエエル) ェチル) カ バメート [1 1 0— 1] 42. 6mg力 ら、 実施例 1 0 6— (3)、 (4) の方法に準じて、 4一 [8— (フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィノレ] 一 2— [((1 S) — 1一 {4一 [4一 (トリフノレオ口 ァセチル) ピぺラジン一 1—ィル] フエエル) ェチル) ァミノ] ピリミジン一 5—力 ルボニトリル [149一 1] 1 3m gを白色固体として得た。
(2) 4— [8— (フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2— [((1 S) — 1一 {4— [4一 (トリフルォロアセチノレ) ピぺラジン一 1ーィ ノレ] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [1 4 9— 1] 1 3mg力、ら、 実施例 1 1 0— (3) の方法に準じて、 目的化合物 [1 49] 5. lm gを白色固体として得た。 .
化合物 [14 9] のスぺクトルデータを以下に示す。
XH— NMR (DMSO— d6) δ : 1 0. 1 1 (d, J = 7. 1 H z , 1 HX 1/2), 9. 1 2 (d, J = 7. 3H z, 1 HX 1/2), 8, 9 5 (d, J = 7. 3H z, 1 HX 1/2), 8. 86 (d, J = 8. 1H z, 1HX 1/2), 8. 75 (s, 1 HX 1/2), 8. 74 ( s, 1 HX 1/2), 8. 73 ( s ' 1 HX 1/2), 8. 6 1 ( s , 1 HX 1/2), 7. 6 9 (d, J = 7. 2Hz, 1 HX 1/2), 7. 6 3 (d, J = 7. 2Hz, 1 HX 1/2), 7. 2 7— 7. 2 3 (m, 2H+ 1 HX 1/2), 7. 09 ( t , J = 7. 1 H z , 1 HX 1/2), 6. 9 1 (d, J =8. 8H z , 2HX 1/2), 6. 8 7 (d, J = 8. 8Hz, 2HX 1/2), 5. 83 (d, J =4 7. 1 Hz, 2HX 1/2), 5. 7 9 (d, J =4 7. 1 Η ζ, 2H
TJP2005/016187
171
X 1/2), 5. 2 1— 5. 1 3 (m, 1 HX 1/2), 5. 04— 4. 9 6 (m, HX 1/2), 2. 9 9— 2. 9 6 (m, 4H), 2. 8 0— 2. 7 6 (m, 4 H) 1. 5 0 (d, J = 6. 8H z , 3 HX 1/2), 1. 4 6 ( d , J = 6. 8H z
3HX 1/2)
ma s s : 4 5 7 (M+ 1) +. 実施例 1 5 0
2— [(( 1 S) 一 1一 {4— [(3 R)一 3, 4一ジメチルビペラジン一 1一ィル] フエ二ル} ェチル) ァミノ]—4— [8— (フルォロメチル) ィミダゾ [1 , 2— a] ピリジン一 3—ィル] ピリミジン一 5—力ルボニトリノレ [1 5 0] (以下、化合物 [1 5 0] という) の合成。
(1) t一ブチル (3 R) — 3—メチルピペラジン一 1一カルボキシレート 5 00 mgとホルムアルデヒド液 (3 7%) 400〃 Lをクロ口ホルム 5mLに溶解し、 シ ァノトリヒドロほう酸ナトリゥム 1 5 l mgと塩化亜鉛 1 6 3 m gをメタノール 8 mLに溶解した溶液を加え、 室温にて終夜撹拌した。 反応混合物をクロ口ホルム 1 0 OmLにて希釈し、飽和炭酸水素ナトリウム水溶液にて洗浄し、 無水硫酸マグネシゥ ムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣をクロ口ホルム 4 m Lに溶解した。 トリフルォロ酢酸 4 m Lを加え、室温にて 1時間 3 0分撹拌した。 反応液を減圧濃縮し、 得られた残渣をメタノールに溶解し、 弱陰イオン交換樹脂に通 しトリフルォロ酢酸を除いた。 溶媒を減圧留去し (2 R) — 1 , 2—ジメチルビペラ ジン [1 5 0— 1] 4 6mgを褐色油状物として得た。
(2) t一プチル [(1 S) — 1一 (4一プロモフエ-ル) ェチル] カーバメート [ 1 0 6— 1] l O Omgと (2 R) — 1 , 2—ジメチルビペラジン [ 1 5 0— 1 ]
4 6mgから、 実施例 1 2 8— (1) の方法に準じて、 t—ブチル ((1 S) — 1一 {4— [(3 R)一 3, 4—ジメチルビペラジン一 1一ィル] フエ二ル} ェチル) 力 ーパメート [ 1 5 0— 2] 1 Omgを褐色油状物として得た。 (3) t一ブチル ((1 S) — 1一 {4一 [(3 R) — 3, 4—ジメチルビペラジン 一 1ーィノレ] フエ二ノレ } ェチル) カーバメート [1 5 0— 2] l Omgと 4一 [8— (フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]—2—(メチルチ ォ) ピリミジン一 5—カルボ二トリル [1 4 7— 3] 7. 51118から、 実施例1 06 一 (3)、 (4) の方法に準じて、 目的化合物 [ 1 5 0] 5. Omgを淡黄色固体とし て得た。
化合物 [1 5 0] のスぺクトルデータを以下に示す。
2005/016187
172
XH— NMR (DMS O— d 6) δ : 1 0. 1 1 (d, J = 6. 8H z, 1 HX 1/2), 9. 1 3 (d, J = 7. 3 H z , 1 HX 1/2), 8, 9 5 (d, J = 7. 3H z, 1 HX 1/2), 8. 8 5 (d, J = 8. 3H z , 1 HX 1/2), 8. 7 5 ( s , 1 HX 1 /2), 8. 7 4 ( s, 1 HX 1 /2), 8 · 7 3 ( s, 1 HX 1/2), 8. 6 1 ( s , 1 HX 1/2), 7. 6 9 (d, J = 7. 8H z , 1 HX 1/2), 7. 6 3 (d, J = 6. 3 H z , 1 HX 1/2), 7. 2 6— 7. 2 3 (m, 2H+ 1 HX 1 /2), 7. 1 0 ( t , J = 7. l H z, 1 HX 1/2), 6. 9 2 ( d , J = 8. 8 H z , 2HX 1/2), 6. 8 8 (d, J = 8. 8H z , 2HX 1/2), 5. 8 3 (d, J = 4 7. 1 H z , 2HX 1/2), 5. 7 9 (d, J =4 7. 1 H z , 2H X 1 / 2 ) , 5. 2 1— 5. 1 3 (m, 1 HX 1/2), 5. 0 5— 4. 9 7 (m, 1 HX 1 /2), 3. 4 9— 3. 44 (m, 2H), 2. 7 9— 2. 6 4 (m, 2H), 2. 34— 2. 2 6 (m, 1 H), 2. 1 9— 2. 0 7 (m, 5H), 1 · 4 9 (d, J = 7. OH z , 3 HX 1 /2), 1. 4 6 (d, J = 7. 0 H z , 3HX 1/2), 1. 34— 1. 3 0 (m, 2 H)
ma s s : 4 8 5 (M+ 1) +. 実施例 1 5 1
2— [(( I S) —1— {4— [(I S, 4 S) —2, 5—ジァザビシクロ [2. 2. 1] ヘプト一2—ィル] フエ二ル} ェチル) ァミノ]—4— [8— (フルォロメチル) イミダゾ [1 , 2— a] ピリジン一 3—ィル] ピリミジン一 5—カルボ二トリル [1 5 1] (以下、 化合物 [1 5 1] という) の合成。
( 1) t一ブチル (I S, 4 S) —2, 5—ジァザビシクロ [2. 2. 1 ] ヘプタ ンー 2—カルボキシレート 2 2 6mgをテトラヒ ドロフラン 5 mLに懸濁させ、 トリ ェチルァミン 4 7 6 μ Lを加えた。 トリフルォロ酢酸無水物 1 94 μ Lを加え、 室温 にて 1時間撹拌した。 反応混合物に水を加え、 酢酸ェチルにて抽出した。 有機層を飽 和食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減 圧濃縮して得られた残渣をシリカゲル力ラムクロマトグラフィ一にて精製し、 ( t一 プチノレ( 1 S, 4 S)— 5—(トリ フノレオロアセチノレ)一 2, 5—ジァザビシクロ [2. 2. 1] ヘプタン一 2—カルボキシレート [1 5 1— 1 ] 2 04m gを淡褐色油状物 として得た。
(2) t一ブチル (1 S, 4 S) — 5— (トリフルォロアセチル)一 2, 5—ジァ ザビシクロ [2. 2. 1] ヘプタン一 2—カルボキシレート [1 5 1— 1] 2 04m gをクロ口ホルム 3mLに溶解し、 トリフルォロ酢酸 3 mLを加え、 室温にて 1時間 撹拌した。 反応液を減圧濃縮し、 得られた残渣をメタノールに溶解し、 弱陰イオン交
6187
173
換樹脂に通しトリフルォロ酢酸を除いた。溶媒を減圧留去し(1 S, 4 S)—2— (ト リフルォロアセチル) 一 2, 5—ジァザビシク口 [2. 2. 1] ヘプタン. [1 51— 2] 156 m gを褐色油状物として得た。 (3) t—ブチル [(1 S) — 1一 (4ーブロモフヱ-ル) ェチル] カーバメート [106— 1] l O Omg'と (I S, 4 S) — 2— (トリフルォロアセチル) 一2, 5—ジァザビシクロ [2. 2. 1] ヘプタン [1 51— 2] 156mg力 ら、 実施例 1 10— (1) の方法に準じて、 t一ブチル ((I S) — 1— {4一 [(1 S, 4 S) 一 5— (トリフルォロアセチル) 一 2, 5—ジァザビシクロ [2. 2. 1] ヘプトー 2—ィル] フヱニル} ェチル) カーバメート [151— 3] 1 56mgを淡褐色固体 として得た。
(4) t—ブチノレ ((1 S) — 1— {4— [(1 S, 4 S) —5— (トリフノレオロア セチル) 一 2, 5—ジァザビシクロ [2. 2. 1] ヘプトー 2—ィル] フエ-ル} ェ チル) カーバメート [1 51— 3] 97mgと 4— [8— (フルォロメチル) ィミダ ゾ [1, 2— a] ピリジン一 3.—ィル] —2— (メチルチオ) ピリミジン一 5—カル ボニトリル [147— 3] 38mgから、 実施例 106— (3)、 (4) の方法に準じ て、 4— [8— (フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3 fル] 一. 2— [((1 S) — 1— {4— [(1 S, 4 S) —5— (トリフルォロアセチル)一 2, 5—ジァザビシクロ [2. 2. 1] ヘプトー 2—ィル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ-トリル [151— 4] 12 m gを淡褐色固体として得た。
(5) 4— [8— (フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2— [((1 S) — 1— {4— [(1 S, 4 S)一 5— (トリフルォロアセチル) ― 2, 5—ジァザビシクロ [2. 2. 1] ヘプトー 2—ィル] フエ二ル} ェチル) アミ ノ] ピリミジン一 5—カルボ二トリル [151— 4] 1 2mg力 ら、 実施例 1 10—
(3) の方法に準じて、 目的化合物 [151] 1. Omgを白色固体として得た。 化合物 [151] のスぺクトルデータを以下に示す。
ΧΗ— NMR (CDC 13) δ : 9. 75— 9. 70 (m, 1HX 1/4), 9. 25 —8. 95 (m, 1 H+ 1 HX 3/4), 8. 65— 8. 63 (s, 1 HX 1/4), 8. 51 (s, 1 HX 3/4), 8. 10— 7. 98 (m, 2H), 7. 60— 6. 4 0 (m, 5H), 6. 00— 5. 80 (m, 2H), 5. 10— 4. 40 (m, 3H), 3. 90— 3. 10 (m, 5H), 2. 30— 1. 80 (m, 2H), 1. 61 (d, J = 6. 6Hz, 3H)
ma s s : 469 (M+ 1) +.
実施例 152
4一 [8— (フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィノレ]一 2— ({(1 S) — 1— [4— (2—ヒ ドロキシェチノレ) フエニル] ェチル } ァミノ) ピリ ミジン一 5—カルボ二トリル [152] (以下、 化合物 [152] という) の合成。
(1) t一ブチル [(1 S) — 1一(4一ブロモフエ-ル) ェチル] カーバメート [106— 1] 5 Omg、 ジクロロビス (トリフエ-ルホスフィン) パラジウム (Π)
223. 4mg、 カリウムビニルトリフルォロボレート 31. 2m g、 トリェチルァ ミン 27. 9 μ L、 テトラヒ ドロフラン 0. 5mL、 およびメタノール 0. 5mLの 混合溶媒を、 100°Cにて終夜撹拌した。 反応混合物を室温まで戻し、 水を加え、 ク ロロホルムにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウムにて 乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮し、 得られた残渣を分取薄層クロマトグ ラフィ一にて精製し、 t一ブチル [(1 S) — 1一 (4一ビエルフエニル) ェチル] カーバメート [152— 1] 24. 2m gを白色固体として得た。
(2) t一ブチル [(1 S) — 1— (4一ビュルフエニル) ェチル] カーバメート [152— 1] 24. 2mgをテトラヒドロフラン lmLに溶解し、 氷冷下、 9—ポ ラビシクロ [3. 3. 1] ノナン (9—BBN、 0. 5 Mテトラヒドロフラン溶液) 783 Lを 2分間かけて滴下し、 室温にて終夜撹拌した。 3 Ν—水酸化ナトリウム 水溶液 0. 2 mLと過酸化水素水 (35%) 0. 2mLをカロえ、 室温にて 10時間撹 拌した。 反応混合物に水を加え、 クロ口ホルムにて抽出し、 有機層を飽和食塩水にて 洗浄した後、無水硫酸ナトリゥムにて乾燥した。不溶物をろ過し、ろ液を減圧濃縮し、 得られた ¾渣を分取薄層クロマトグラフィーにて精製した。 得られた化合物を N, N —ジメチルホルムァミ ド 1. 5 m Lに溶解し、 安息香酸 14. 2 m g、 4ージメチル アミノビリジン 21. 3m gを加え均一なるよう撹拌し、 氷冷下、 1ーェチルー 3— ( 3—ジメチルァミノプロピル) カルボジィミド塩酸塩 22. 3mgを加え室温にて 終夜撹拌した。 水を加え、 クロ口ホルムにて抽出し、 有機層を飽和食塩水にて洗浄し た後、 無水硫酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮して得ら れた残渣を分取薄層クロマトグラフィーにて精製し、 2— (4— {(1 S)— 1一 [(t 一ブトキシカルボニル) ァミノ] ェチル } フエニル) ェチルベンゾエート [152— 2] 15. 2mgを白色固体として得た。 .
(3) 2— (4一 {(1 S) — 1— [(t一ブトキシカルボ-ル) ァミノ] ェチル } フエニル) ェチル ベンゾエート [152— 2] 15. 2m gと 4一 [8—(フルォ ロメチル) イミダゾ [1, 2— a] ピリジンー3—ィル]一 2—(メチルチオ) ピリ ミジン一 5—力ノレボニトリノレ [147— 3] 20. lmg力 ら、実施ィ列 106—(3)、
JP2005/016187
175
(4) の方法に準じて、 2— {4— [(1 S)一 1一 ({5—シァノー 4一 [8—(フ ルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 2—ィル } ァミノ) ェチ /レ] フエ二ノレ) ェチノレべンゾエート [1 52— 3] mg 1 3. 5を白色 固体として得た。
(4) 2— {4— [(1 S)一 1一 ({ 5—シァノ一4一 [8— (フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 2—ィル } ァミノ) ェチ ル] フエュル } ェチルベンゾエート [ 1 52— 3] 1 3. 5mgをテトラヒ ドロフラ ン 2mLとメタノール 2mLの混合溶媒に溶解し、 1 N— τΚ酸化ナトリウム水溶液 1 mLを加え、 室温にて 1時間 30分撹拌した。 1 N—塩酸 1 m Lを加え、 クロ口ホル ムとメタノールの混合溶媒 (クロ口ホルム:メタノ一ル= 9 : 1) にて抽出した。 有 機層を飽和食塩水にて洗浄した後、 無水硫酸ナトリウムにて乾燥し、 不溶物をろ過し た。 ろ液を減圧濃縮し、 得られた残渣を分取薄層クロマトグラフィーにて精製し、 目 的化合物 [1 5 2] 8. 3mgを白色固体として得た。
化合物 [1 5 2] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ :.9. 7 3 (d, J = 7. 6Hz, 1 HX 1/3), 8. 94 (s, 1 HX 1/3), 8. 9 1 (d, J = 7. 6Hz, 1 HX 2/3), 8. 8 7 ( s , 1 HX 2/3), 8. 58 ( s , 1 HX 1/3), 8. 5 2 (s , 1 HX 2/ 3), 7. 52 (d, J = 7. 6H z , 1 HX 1/3), 7. 45 (d, J = 7. 6H z , 1 HX 2/3), 7. 35 (d, J = 8. OH z, 2H), 7. 2 9 (d, J = 8. 0H z, 2H), 7. 06 (d d, J = 7. 6, 7. 6H z , 1 HX 1/3), 6. 7 4 (d d, J = 7. 6, 7. 6Hz, 1 HX 2/3), 6. 1 9一 6. 1 4 (m, 1 HX 2/3), 5. 86— 5. 74 (m, 2H+ 1 HX 1/3), 5. 36— 5. 28 (m, 1 HX 1/3), 5. 1 6— 5. 08 (m, 1 HX 2/3), 3. 8 7 ( t, J = 6. 4Hz, 2H), 2. 88 ( t , J = 6. 4Hz, 2H), 1. 63 ( d , J = 6. 4Hz, 3H)
ma s s : 4 1 7 (M+ 1 ) +. 実施例 1 53
2— [((I S) — 1— {4一 [2— (ジメチルァミノ) ェチル] フエ二ル} ェチル) ァミノ] —4一 [8— (フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィ ノレ] ピリミジン一 5—カルボ二トリノレ [1 5 3] (以下、 化合物 [1 5 3] とレヽう) の合成。 化合物 [1 5 2] 7. Omgとジメチルァミン (2Mテトラヒ ドロフラン溶液) 2 mL力 ら、 実施例 84の方法に準じて、 目的化合物 [96] 5. 9mgを白色固体と
6187
176
して得た。
化合物 [1 53] のスぺクトルデータを以下に示す。
XH— NMR (CDC 13) δ : 9. 74 (d, J = 7. 6Hz, 1 HX 1/3), 8. 94 (s, 1 HX 1/3), 8. 89 (d, J = 7. 6Hz, 1 HX 2/3), 8. 8 8 ( s , 1 HX 2/3), 8. 59 (s, 1 HX 1/3), 8. 54 ( s , 1 HX 2/ 3), 7. 52 (d, J = 7. 6Hz, 1 HX 1/3), 7. 45 (d, J = 7. 6H z, 1 HX 2/3), 7. 31 (d, J = 8. 0Hz, 2H), 7. 27— 7. 25 (m, 2H), 7. 07 (d d, J = 7. 6, 7. 6Hz, 1 HX 1/3), 6. 74 ( d d, 1=1. 6, 7. 6Hz, 1 HX 2/3), 6. 10— 6. 05 (m, 1 HX 2/3), 5. 95— 5. 78 (m, 2H+ 1 HX 1/3), 5. 35— 5. 25 (m, 1 HX 1/3), 5. 12— 5. 08 (m, 1 HX 2/3), 2. 81 ( t , J =8. OHz, 2H), 2. 54 (t, J = 8. 0Hz, 2H), 2. 30 (s, 6H), 1. 62 (d, J = 6. 8Hz, 3H)
ma s s : 444 (M+ 1 ) +. 実施例 1 54 .
4一 [8— (フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2— ({(1 S) —1— [4— (1, 2, 3, 6—テトラヒ ドロピリジン一 4一ィル) フエ
-ル] ェチル } ァミノ) ピリミジン一 5—カルボ二トリル [1 54] (以下、 化合物 [1 54] という) の合成。
(1) 1一 (トリフルォロアセチル) ピぺリジン一 4一オン (特開昭 63— 227
599号公報、 24ページに記載されている方法により合成した) 2 gをテトラヒド 口フラン 2 OmLに溶解し、 _78。Cにてリチウムビス (トリメチルシリル) アミ ド (1. OMテトラヒドロフラン溶液) 1 1. 3 mLを加え、 同温にて 10分間撹拌し た。 そこへ、 N—フエニルトリフルォロメタンスルホンイミ ド 3. 7 gをテトラヒド 口フラン 9 m Lに溶解した溶液を加え、撹拌しながらー 78 °Cから室温まで 3時間か けて昇温した。 水を加え、 反応を止め、 酢酸ェチルにて抽出した。 有機層を飽和食塩 水にて洗浄した後、 無水硫酸マグネシウムにて乾燥した。 不溶物をろ過し、 ろ液を減 圧濃縮し、 化合物 [154— 1] 3. 1 2 gを淡褐色油状物として得た。 1一 (2, 2, 2—トリフルォロアセチル) 一1, 2, 3, 6—テトラヒ ドロピリジン一 4ーィ ル トリフルォロメタンスルホネート [154—1] は精製することなく、 次の反応 に付した。 (2) 1— (2, 2, 2—トリフルォロアセチル) 一 1, 2, 3, 6—テトラヒ ド 口ピリジン一 4ーィル トリフルォロメタンスルホネート [154— 1] 1 g、 を 1,
5016187
177
4一ジォキサン 3 OmLに溶解し、 1, 1 '一ビス (ジフエ-ルホスフイノ) フエ口 セン 85mg、 ジクロロビス (トリフエ-ルホスフィン) パラジウム (Π.) 125m g、 炭酸セシウム 2. 98 g、 およびビス (ピナコレート) ジボロン 775mgを加 え、 80°Cにて終夜撹拌した。 反応混合物を室温まで戻した後、 水を加え、 酢酸ェチ ルにて抽出した。 有機層を飽和食塩水にて洗浄した後、 無水硫酸マグネシウムにて乾 燥した。 不溶物をろ過し、 ろ液を減圧濃縮して得られた残渣をシリカゲルカラムクロ マトグラフィ一にて精製し、 4一 (4, 4, 5, 5—テトラメチルー 1, 3, 2—ジ ォキサボロラン一 2—ィル)一 1— (トリフルォロアセチル)一 1, 2, 3, 6—テ トラヒドロピリジン [154— 2] 40 Omgを褐色油状物として得た。
(3) 4一 (4, 4, 5, 5—テトラメチルー 1, 3, 2—ジォキサボロラン一 2 一ィル)一 1一(トリフルォロアセチル)一 1 , 2, 3, 6—テトラヒ ドロピリジン
[154— 2] 400111 を]^, N—ジメチルホルムアミ ド 10 mLに溶解し、 t一 ブチル [(1 S)— 1一(4一プロモフエニル) ェチル] カーパメート [106— 1] 196mg、 [1, 1 , 一ビス (ジフエニルホスフイノ) フエ口セン] ジクロロパラ ジゥム (Π) 54m g、 および炭酸カリウム 18 lmgを加え、 80°Cにて終夜撹拌 した。 反応混合物を室温に戻した後、 酢酸ェチル 10 OmLにて希釈し、 水、 飽和食 塩水で順に洗浄した。 得られた有機層を無水硫酸マグネシウムにて乾燥し、 不溶物を ろ過した。 ろ液を減圧濃縮し、 得られた残渣をシリ力ゲル力ラムクロマトグラフィー にて精製し、 t一ブチル ((1 S) — 1一 {4— [1一 (トリフルォロアセチル) 一 1, 2, 3, 6—テトラヒ ドロピリジン一 4一ィル] フエ二ル} ェチル) カーパメー ト [1 54— 3] 10 Omgを白色固体として得た。
(4) t一プチル ((1 S) — 1— {4一 [1一(トリフルォロアセチル) 一1, 2, 3, 6—テトラヒドロピリジン一 4—ィル] フエ-ル}ェチル) カーバメート [1
54— 3] 5 Omgと 4— [8—(フルォロメチル) イミダゾ [1, 2— a] ピリジ ンー 3—ィル]—2—(メチルチオ) ピリミジン一 5—カルボ二トリル [147— 3] 34mgから、 実施例 106— (3)、 (4) の方法に準じて、 4一 [8— (フルォロ メチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2— [((1 S) — 1— { 4 一 [1一(トリフノレオロアセチノレ)一 1, 2, 3, 6—テトラヒ ドロピリジン一 4一 ィル] フエ二ノレ) ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [154— 4] 1 Omgを白色固体として得た。
(5) 4一 [8— (フノレオロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] 一 2— [((1 S) — 1一 {4一 [1一(トリフルォロアセチル) ー1, 2, 3, 6— テトラヒ ドロピリジン一 4一ィル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力
5016187
178
ルポ二トリル [1 54— 4] l Omgから、 実施例 1 1 0— (3) の方法に準じて、 目的化合物 [1 54] 5. 6mgを淡黄色固体として得た。
化合物 [1 54] のスぺクトルデータを以下に示す。
— NMR (DMSO— d6) δ : 1 0. 1 1 (d, J = 7. 1Hz, 1 HX 1/2), 9. 1 2— 9. 02 (m, 1 H), 8. 94 (d, J = 8. OHz, 1 HX 1/2), 8. 76 (s, 1 HX 1/2), 8. 75 ( s , 1HX 1/2), 8. 7 3 ( s , 1 H X 1/2), 8. 6 1 (s , 1HX 1/2), 7. 70 (d, J = 6. 3Hz, 1 HX 1/2), 7. 6 3 (d, J = 6. 3Hz, 1 HX 1/2), 7. 43 -7. 36 (m, 4H), 7. 26 (t, J = 7. 1 Hz, 1 HX 1/2), 7. 03 (t, J = 7. 1 Hz, 1 HX 1/2), 6. 1 5— 6. 1 4 (m, 1 H), 5. 84 (d, J =4 7.
1Hz, 2HX 1/2), 5. 78 (d, J =4 7. 1Hz, 2HX 1/2), 5 7— 5. 20 (m, 1 HX 1/2), 5. 1 2— 5. 05 (m, 1 HX 1/2), 35— 3. 33 (m, 2H), 2. 9 1— 2. 86 (m, 2H), 2. 3 3— 2.
(m, 2H), 1. 5 3 (d, J = 7. 1Hz, 3HX 1/2), 1. 50 (d, 7. 1 H z , 3HX 1/2)
ma s s : 454 (M+ 1 ) +. 実施例 1 55
4一 [8—(フルォロメチル)イミダゾ [1, 2— a]ピリジン一 3—ィル]一 2— {[(1 S) 一 1一 (4ーピペリジン一 4ーィルフエニル) ェチル] アミノ} ピリミジン一 5 一カルボ二トリル [1 5 5] (以下、 化合物 [1 55] という) の合成。
(1) t—プチル ((1 S) — 1一 {4— [1— (トリフルォロアセチル)一 1, 2, 3, 6—テトラヒドロピリジン一 4一ィル] フエ二ル}ェチル) カーバメート [1 54— 3] 5 Omgをテトラヒドロフラン 3mLに溶解し、触媒量の 1 0%パラジゥ ム炭素触媒を加え、 水素雰囲気下、 室温にて 1時間撹拌した。 不溶物をセライトにて ろ過し、 ろ液を減圧濃縮して t—ブチル ((1 S) — 1一 {4一 [1— (トリフルォ ロアセチル) ピぺリジン一 4一ィル] フエ二ル}ェチル) カーバメート [1 55— 1] 53. 3m gを白色固体として得た。
(2) t—プチル ((1 S) — 1— {4一. [1一(トリフルォロアセチル) ピペリ ジン一 4—ィル] フエ二ル} ェチル) カーバメート [1 55— 1] 53. 3mgと 4 一 [8—(フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2—(メ チルチオ) ピリミジン一 5—力ルポ二トリル [1 4 7— 3] 36mg力、ら、 実施例 1 06— (3)、 (4) の方法に準じて、 4一 [8—(フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—^ fノレ]一 2_ [((1 S)一 1一 {4一 [1一(トリフノレオ口
2005/016187
179
ァセチル) ピぺリジン一 4一ィル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力 ルボニトリル [1 5 5— 2] 1 3. 5mgを淡黄色固体として得た。
(5) 4— [8— (フルォロメチル) イミダゾ [1, 2— a] ピリジン一 3—ィル] 一 2— [((1 S) 一 1一 {4— [1一 (トリフルォロアセチル) ピぺリジン一 4ーィ ル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [1 5 5— 2] 1 Omg力 ら、 実施例 1 1 0— (3) の方法に準じて、 目的化合物 [1 55] 5. 6m gを白色固体として得た。
化合物 [1 55] のスぺクトルデータを以下に示す。
ΧΗ— NMR (DMSO— d 6) δ : 1 0. 1 1 (d, J = 6. 8Hz, 1 HX 1/2), 9. 0 1— 8. 9 7 (m, 1 H), 8. 9 2 (d, J = 8. 3 H z 1 HX 1/2), 8 · 7 5 (s, 1 H), 8. 7 3 ( s , 1HX 1/2), 8. 6 1 ( s , 1 HX 1/2), 7. 70 (d, J = 6. 8H z, 1 HX 1/2), 7. 6 3 (d, J = 7. 1 Hz , 1 HX 1/2), 7. 4 3— 7. 36 (m, 4H), 7. 26 (t, J = 7. 1Hz, 1 HX 1/2), 7. 34— 7. 32 (m, 2H), 7. 27— 7. 1 7 (m, 2H + 1 HX 1/2), 7. 0 1 ( t > J = 7. 1 H z, 1 HX 1/2), 5. 84 (d, J = 47. 1 H z, 2HX 1/2), 5. 78 (d, J = 47. 1 H z, 2HX 1/2), 5. 2 6— 5. 1 9 (m, 1 HX 1/2), 5. 09— 5. 0 3 (m, 1 HX 1/2),
—2. 94 (m, 2H), 2. 5 6— 2. 5 1 (m, 2H), 6 5— 1. , 2H), 1. 5 2 (d, J = 6. 8H z, 3HX 1/2), 49 (d, lHz, 3HX 1/2)
: 45 6 (M+ 1) +. 実施例 1 56
4一 [8— (1ーヒ ドロキシェチノレ) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2— {[(1 S) 一 1一 (4—ピぺラジン一 1ーィルフエニル) ェチル] アミノ} ピリ ミジン一 5—カルボ二トリル [1 56] (以下、 化合物 [1 56] という) の合成。
(1) 2—アミノー 3—ァセチルビリジン 2 gと 4— [(Z) —2—エトキシビ二 ル]一 2—(メチルチオ)一 5—ピリミジンカルボ-ル [48— 3] 3. 2 5 gから、 実施例 25— (3) の方法に準じて、 4— (8—ァセチルイミダゾ [1, 2— a] ピ リジン一 3—ィル) —2— (メチルチオ) ピリミジン一 5—カルボ二トリル [1 56 — 1 ] 3. 2 gを淡褐色固体として得た。
(2) 4—(8—ァセチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 チルチオ) ピリミジン一 5—力ノレボニトリル [1 56— 1] 1 001118と 1
((I S) — 1一 {4— [4一 (トリフルォロアセチル) ピぺラジン一 1一ィル] フ ヱ二ル}ェチル)カーバメート [1 10— 1] 1 56mg力 ら、実施例 10.6—(3)、
(4) の方法に準じて、 4一 (8—ァセチルイミダゾ [1, 2_a] ピリジン一 3— ィル)一 2— [((1 S) — 1— {4— [4一 (トリフルォロァセチル) ピぺラジン一 1一^ fル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [156— 2] 46. 6 mgを白色固体として得た。
(3) 4一(8—ァセチルイミダゾ [1, 2— a]ピリジン一 3—ィル)ー2— [((1 S) — 1— {4— [4— (トリフノレオロアセチル) ピぺラジン一 1一ィル] フエ二ル} ェチル)ァミノ] ピリミジン一 5—カルボ-トリル [156— 2] 46. 6mg力、ら、 実施例 1 10— (3) の方法に準じて、 4— (8—ァセチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— {[(1 S)—1— (4ーピペラジン一 1ーィルフエニル) ェチノレ] アミノ} ピリミジン一 5—カノレポ二トリノレ [156— 3] 42. 2mgを黄 色固体として得た。
(4) 4一 ( 8—ァセチルイミダゾ [ 1 , 2— a]ピリジン一 3—ィノレ) 一 2— {[( 1 S)一 1一 (4ーピペラジン一 1ーィルフエニル) ェチル] アミノ} ピリミジン一 5 一カルボ二トリル [156— 3] 39 m gをテトラヒドロフラン 1 m Lとメタノール lmLの混合溶媒に溶解し、 水素化ほう素ナトリウム 2. 8mgを力 Πえ、 室温にて 1 5分間撹拌した。 飽和炭酸水素ナトリウム水溶液を加え、 クロ口ホルムとメタノール の混合溶媒 (クロ口ホルム:メタノール =9 : 1) にて抽出し、 有機層を飽和食塩水 にて洗浄した。 無水硫酸ナトリウムにて乾燥し、 不溶物をろ過した後、 ろ液を減圧濃 縮した。 得られた残渣を分取薄層クロマトグラフィーにて精製し、 4一 [8— (1— ヒドロキシェチル) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2— {[(1 S) — 1一 (4ーピペラジン一 1—ィルフエニル) ェチル] アミノ} ピリミジン一 5—力 ルポ-トリル [1 56— 4] 31. 9 mgを 2種類のジァステレオマー混合物として 得た。
(5) . 2種類のジァステレオマー混合物である 4— [8— (1—ヒドロキシェチル) イミダゾ [1, 2— a] ピリジン一 3 fル]一 2— {[(1 S) — 1— (4—ピペラ ジン一 1—ィルフエニル) ェチル] アミノ ピリミジン一 5—カルボ二トリル [1 5 6— 4] 31. 9mgを、 キラルパック AD— H (Ch i r a 1 p a k AD— H、 ダイセル化学工業) にて、 へキサンーィソプロパノールージェチルァミンを溶出液と して用い分割した。
分割条件は以下の通りである。
カラム:キラルパック AD— H (Ch i r a 1 p a k AD— H、 ダイセル化学
工業)、 直径 2 Omm、 長さ 2 5 Omm
溶出液:へキサンーィソプロパノールージェチルァミン (3 0 : 7 0 : 0.. 1) 流速: 1 5 mL/ m i n
得られた溶出液を減圧濃縮し、 ジァステレオマーのうちの一方である目的化合物 [ 1 5 6] 1 1. l mg (RT= 1 3分) を白色固体として得て、 もう一方のジァス テレオマー 4一 [8— (1 ヒ ドロキシェチル) イミダゾ [1 , 2— a] ピリジン一 3—ィル]一 2— {[(1 S) 一 1一 (4ーピペラジン一 1一ィルフヱニル) ェチル] アミノ} ピリミジン一 5—カルボ二トリル [1 5 6— 5] 1 1. 8mg (RT= 1 7 分) を白色固体として得た。
化合物 [1 5 6] のスぺクトルデータを以下に示す。
XH— NMR (CDC 1 3) δ : 9. 6 3 (d, J = 7. 2H z , 1 HX 1/5), 8. 9 8 (d, J = 7. 2H z, 1 HX 4/5), 8. 9 0 ( s , 1 HX 1/5), 8. 8 6 ( s , 1 HX 4/5), 8. 5 8 ( s , 1 HX 1/5), 8. 5 1 ( s , 1 HX 4/ 5), 7. 3 0— 6. 6 7 (m, 6H), 6. 1 7— 6. 0 5 (m, 1 HX 4/5), 5. 8 6— 5. 7 7 (m, 1 H X 1 / 5 ) , 5 · 4 0— 5. 2 2 (m, 1 H+ 1 HX 1 /5), 5. 1 2— 5. 0 0 (m, 1 HX 4/5), 3. 1 4— 3. 0 6 (m, 4H), 3. 0 4— 3. 0 1 (m, 4H), 2. 6 0 (b r s, 1 H), 2. 0 2 (b r s , 1 H), 1. 6 8 (d, J =4. 0H z , 3H), 1. 6 1 (d, J = 6. 8H z, 3 H) ma s s : 4 6 9 (M+ 1 ) +. 実施例 1 5 7
4一 [8— (1ーヒ ドロキシー 1ーメチルェチル) イミダゾ [1 , 2— a] ピリジン —3—ィル] —2— [((1 S) —1— { 3— [( 1ーメチルピペリジン一 4—ィル) ァミノ] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリノレ [1 5 7] (以 下、 化合物 [ 1 5 7] という) の合成。
( 1 ) 4一( 8—ァセチルイミダゾ [ 1 , 2— a] ピリジン一 3—ィル)一 2—(メ チルチオ) ピリミジン一 5—カルボ二トリノレ [1 5 6— 1] l O Omgをテトラヒド 口フラン 5mLに溶解し、 氷冷下、 臭化メチルマグネシウム (0. 84Mテトラヒド 口フラン溶液) 5 7 7 μ Lを加え、 室温にて終夜撹拌した。 さらに臭化メチルマグネ シゥム 2 0 0 Lを加え、 室温にて 4時間撹拌した後、 飽和塩ィ匕アンモ-ゥム水溶液 を加え反応を止めた。 クロ口ホルムにて抽出し、 有機層を無水硫酸マグネシウムにて 乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮して得られた残渣を分取薄層クロマトグ ラフィ一にて精製し、 4一 [8—(1ーヒドロキシー 1ーメチルェチル)イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2—(メチルチオ) ピリミジン一 5—カルボ-トリ ル [1 5 7— 1] 1 8m gを褐色固体として得た。
T/JP2005/016187
182
(2) 4— [8— (1ーヒドロキシー 1ーメチルェチル) イミダゾ [1., 2— a] ピリジン一 3—ィル] 一 2— (メチルチオ) ピリミジン一 5—カルボ-トリル [1 5 7— 1] 1 8mgと 3— [(1 S) — 1—アミノエチル] ァニリン 1 l mgから、 実 施例 8 0— (4) の方法に準じて、 2— {[(1 S) — 1— (3—アミノフヱ-ル) ェ ' チル] アミノ} —4— [8'— (1—ヒドロキシー 1ーメチルェチル) イミダゾ [ 1 , 2— a]ピリジン一 3—ィル]ピリミジン一 5—カルボ二トリノレ [1 5 7— 2] 1 5. 2 m gを淡褐色固体として得た。 (3) 2— {[(1 S) — 1一 (3—ァミノフエニル) ェチル] アミノ} —4— [8 一 ( 1ーヒ ドロキシー 1—メチルェチル) イミダゾ [1 , 2— a] ピリジン一 3—ィ ル] ピリミジン一 5—カルボ二トリノレ [1 5 7— 2] 1 5mgと 1ーメチノレー 4ーピ ペリ ドン 9 Lから、 実施例 9 1の方法に準じて、 目的化合物 [1 5 7] 2. 9mg を淡黄色固体として得た。
化合物 [1 5 7] のスぺクトルデータを以下に示す。
!H— NMR (DMSO— ά 6). δ : 1 0. 0 3 (d, J = 5. 8H z, 1 HX 1/3), 8. 9 3— 8. 8 9 (m, 1 HX 2/3 + 1 HX 2/3), 8. 8 2 (d, J = 8. 3H z, 1 HX 1/3), 8. 74 ( s, 1 HX 2/3), 8. 7 3 ( s , 1 HX 1/ 3), 8. 7 1 ( s, 1 HX 1/3), 8. 54 ( s , 1 HX 2/3), 7. 7 0 (d, J = 7. 3H z, 1 HX 1 /3), 7. 5 9 (d, J = 6. 3 H z , 1 HX 2/3), 7. 2 0 (d, J = 7. 1 H z , 1 HX 1/3), 7. 0 6— 6. 9 6 (m, 1 H + 1 HX 2/3), 6. 5 7— 6. 5 3 (m, 2H), 6. 4 3— 6. 3 9 (m, 1 H), 5. 5 1— 5. 4 8 (m, 1 H+ 1 HX 2/3), 5. 4 1 (d, J = 7. 6 H z, 1 HX 1/3), 5. 1 5— 5. 0 9 (m, 1 HX 1/3), 4. 9 5— 4. 8 9 (m, 1 HX 2/3), 3. 0 8 (b r s, 1 H), 2. 6 8— 2. 6 0 (m, 2H), 2. 1 4 ( s , 3 HX 2/3), 2. 0 6 ( s , 3 HX 1 /3), 1. 9 5— 1. 7 5 (m, 4H), 1. 7 2 ( s , 3HX 1/3), 1. 7 1 ( s , 3HX 1/3), 1. 6 8 ( s, 3HX 2/3), 1. 6 7 ( s , 3 HX 2/3), 1. 5 0— 1. 4 6 (m, 3H), 1. 3 8— 1. 2 3 (m, 2 H)
ma s s : 5 1 1 (M+ 1 ) +. 実施例 1 5 8
4一 (8—メ トキシイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— {[(1 S) — 1— (4ーピペラジン一 1ーィルフエニル) ェチル] アミノ} ピリミジン一 5—力 ルポ二トリノレ [ 1 5 8 ] (以下、 化合物 [1 5 8] という) の合成。
TJP2005/016187
183
(1) 2—アミノー 3—ヒドロキシピリジン 1 0 Omgと 4一 [(Z) — 2—ェト キシビエル]一 2— (メチルチオ) 一 5—ピリミジンカルボニル [4 8— 3] 2 00 mgから、実施例 2 5— (3)の方法に準じて、 4一(8—ヒ ドロキシイミダゾ [1 ,
2— a] ピリジン一 3—ィル) 一 2— (メチルチオ) ピリミジン一 5—カルボ二トリ ノレ [1 5 8— 1] 1 9 Omgを橙色固体として得た。
(2) 4一 (8—ヒドロキシイミダゾ [ 1 , 2— a] ピリジン一 3—ィル) 一 2— (メチルチオ) ピリミジン一 5—カルボ二トリル [ 1 5 8— 1 ] 1 5 Omgをジメチ ルスルホキシド 3mLに溶解し、炭酸カリゥム 2 9 3mgとヨウ化メチル 4 0 μ Lを 加え、 室温にて 3 0分間撹拌した。 クロ口ホルム 20 OmLにて希釈し、 水にて洗浄 した後、 有機層を無水硫酸マグネシウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧 濃縮して、得られた残渣を少量のクロ口ホルムとメタノールの混合溶媒(クロ口ホル ム:メタノール = 9 : 1) に溶解し、 へキサンを加えて固化し、 4一 (8—メ トキシ イミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— (メチルチオ) ピリミジン一 5 一カルボ-トリル [ 1 5 8— 2] 5 8m gを褐色固体として得た。
(3) 4— (8—メ トキシイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2—(メ チルチオ)ピリミジン一 5—カルボ二トリル [1 5 8— 2] 1 1 mgと t—ブチル(( 1 S) — 1— {4一 [4一(トリフノレオロアセチル) ピぺラジン一 1 fル] フエ二ル} ェチル) カーバメート [ 1 1 0— 1] 1 5 m gから、 実施例 1 0 6— (3)、 (4) の 方法に準じて、 4一 (8—メ トキシイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— [(( 1 S) — 1— {4— [4一(トリフルォロアセチル) ピぺラジン一 1一ィル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [ 1 5 8— 3] 1 1.
3 mgを白色固体として得た。
(3) 4— (8—メ トキシイミダゾ [1, 2— a]ピリジン一 3—ィル)一 2— [((1 S) — 1— { 4一 [4一(トリフルォロアセチル) ピぺラジン一 1—ィル] フエ二ル} ェチル)ァミノ] ピリミジン一 5—カルボ二トリル [1 5 8— 3] 1 1. 3mg力 ら、 実施例 1 1 0— (3) の方法に準じて、 目的化合物 [1 5 8] 3. 7mgを淡黄色固 体として得た。
化合物 [1 5 8] のスぺクトルデータを以 Tに示す。
'H— NMR (DMS O— d 6) δ 9. 70 (d, 1 = 6. 8H z , 1 HX 1/2), 8. 9 2 (d, J = 7. 3 H z, 1 HX 1/2), 8. 8 3 (d, J =8. 3H z , 1 HX 1/2), 8. 7 6 (d, J = 6. 6H z , 1 HX 1/2), 8. 7 2 ( s, 1 HX 1 /2), 8. 7 1 ( s, 1 HX 1/2), 8. 6 5 ( s, 1 HX 1/2), 8. 5 2 ( s , 1 HX 1/2), 7. 2 5— 7. 2 3 (m, 2H), 7. 1 0— 6. 8 6 (m,
2005/016187
184
4H), 5. 20— 5. 13 (m, 1 HX 1/2), 5. 04— 4. 96 (m, 1HX 1/2), 3. 98 ( s , 3HX 1/2), 3. 96 ( s , 3HX 1/2)., 2. 99 —2. 97 (m, 4H), 2. 83— 2. 79 (m, 4 H) ' 1. 49 (d, J = 6. 8Hz, 3HX 1/2), 1. 46 (d, J = 6. 8Hz, 3 H X 1 / 2 )
ma s s : 455 (M+ 1) +. 実施例 159
4— (8—メ トキシー 7—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2 ― {[(1 S)一 1一 (4ーピペラジン一 1一ィルフヱニル) ヱチル] アミノ} ピリミ ジン一 5—力ルポ二トリル [1 59] (以下、 化合物 [159] という) の合成。
(1) イソプロピルアミン 1. 36mLをテトラヒドロフラン 8mLに溶解し、 一 78 °Cにて n—ブチルリチウム (2. 66 Mへキサン溶液) 3. 64mLを加え、 同 温にて 10分間撹拌し、 その後 0 °Cまで昇温し 1時間撹拌した。 反応混合物を再び一 78°Cとし、 2—フエニル [1, 3] ォキサゾロ [4, 5— b] ピリジン (Sy n t h e t i c Commun. 1 992, 22 (20), 2891— 1901に記載さ れている方法により合成した) 95 Omgをテトラヒドロフラン 28mLに溶解した 溶液を加え、 同温にて 30分間撹拌した。 ョウイヒメチル 0. 36mLをテトラヒドロ フラン 16mLに溶解した溶液を加え、 同温にて 5時間撹拌した。 水を加え反応を止 め、 室温まで昇温した後、 クロ口ホルムにて抽出し、 有機層を飽和食塩水にて洗浄し た。 無水硫酸ナトリウムにて乾燥し、 不溶物をろ過し、 ろ液を減圧濃縮して得られた 残渣を、 シリカゲルカラムクロマトグラフィーにて精製し、 7—メチルー 2—フエ二 ル [1, 3] ォキサゾロ [4, 5— b] ピリジン [159— 1] 86mgを淡青色固 体として得た。
(2) 7—メチルー 2—フエ-ル [1, 3]ォキサゾロ [4, 5— b] ピリジン [1 59— 1] 86mgをテトラヒドロフラン 4 mLとエタノール 4 mLの混合溶媒に溶 解し、 33 %水酸化ナトリウム水溶液 1. 64 m L加え、 終夜加熱還流した。 室温に 戻した後、減圧濃縮した。 得られた残渣にジクロロメタン 5mLと 33%水酸ィヒナト リウム水溶液 3. 4mLを加え撹拌し、 そこにヨウ化メチル 28 ^ Lと Ad o g e n 464 (相間移動触媒、 アルドリッチ社) 30. 5 μ Ι^を力!]え、 室温にて終夜撹拌し た。 ZRを加え、クロ口ホルムにて抽出し、有機層を PH7. 0リン酸バッファー、水、 飽和食塩水にて順に洗浄した。 無水硫酸ナトリウムにて乾燥し、 不溶物をろ過し、 ろ 液を減圧濃縮し得られた残渣を分取薄層クロマトグラフィーにて精製し、 3—メトキ シー 4一メチルピリジン一 2—ァミン [159— 2] 4. 2mgを淡褐色固体として すこ。
(3) 3—メ トキシ一 4一メチルピリジン一 2—ァミン [1 59— 2] 4. 2 m g と 4— C(Z) 一 2—ェトキシビュル] —2— (メチルチオ) 一 5—ピリミジンカル ボニル [48— 3] 6. 7mg力、ら、 実施例 25— (3) の方法に準じて、 4一 (8 —メ トキシー 7—メチルイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— (メチ ルチオ) ピリ ミジン一 5—カル 二トリル [1 5 9— 3] 2. 8mgを淡黄色固体と して得た。
(4) 4一 (8—メ トキシー 7—メチルイミダゾ [1, 2— a] ピリジン一 3—ィ ル)一 2—(メチルチオ) ピリミジン一 5—カルボ二トリル [1 5 9— 3] 2. 8 m gと t一プチル ((1 S) — 1一 { 4一 [4一(トリフルォロアセチル) ピぺラジン 一 1ーィノレ] フエ二ノレ } ェチル) カーバメート [1 1 0—1] 4mg力、ら、 実施例 1 06— (3)、 (4) の方法に準じて、 4一(8—メ トキシー 7—メチノレイミダゾ [1, 2— a] ピリジン一 3—ィノレ) —2— [((1 S) 一 1一 (4一 [4一 (トリフノレオ口 ァセチル) ピぺラジン一 1—ィル] フエ二ル} ェチル) ァミノ] ピリミジン一 5—力 ルポ-トリル [1 59— 4] 3.. 6mgを淡黄色固体として得た。 '
(5) 4— (8—メ トキシー 7—メチルイミダゾ [1, 2— a] ピリジン一 3—ィ ノレ)一 2— [((1 S)一 1一 {4— [4— (トリフルォロアセチル) ピぺラジン一 1 一ィル] フエ二ル}ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [1 5 9—4] 3. 6mg力 ら、 実施例 1 1 0— (3) の方法に準じて、 目的化合物 [1 5 9] 3. Omgを淡黄色固体として得た。
化合物 [1 59] のスぺクトルデータを以下に示す。
XH— NMR (CDC 1 3) δ : 9. 40 (d, J = 6. 4Hz, 1 HX 1/5), 8. 9 0 (s , 1 HX 1/5), 8. 8 6 ( s , 1 HX 4/5), 8. 8 3 ( d , . J = 6. 4H z, 1 HX 4/5), 8. 56 (s, 1 HX 1/5), 8. 48 (s , 1 HX 4/ 5), 7. 3 5— 7. 25 (m, 2H), 6. 94 (d, J = 8. OH z , 2H), 6. 7 9 (d, J = 6. 4Hz, 1 HX 1/5), 6. 5 7 (d, J = 6. 4H z , 1 H X 4/5), 6. 05— 5. 9 7 (m, 1 HX 4/5), 5. 7 9— 5. 72 (m, 1 HX 1/5), 5. 30— 5. 2 5 (m, 1 HX 1/5), 5. 1 2— 5. 0 5 (m, 1 HX 4/5), 4. 2 1 ( s , 3H), 3. 20— 3. 1 5 (m, 4H), 3. 07 —3. 02 (m, 4H), 2. 36 (s, 3H), 1. 6 1 (d, J = 6. 8H z, 3 H)
ma s s : 46 9 (M+ 1) +. 実施例 1 60
P T/JP2005/016187
186
4一 [8— (フルォロメ トキシ) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2 ― {[(1 S)一 1一 (4ーピペラジン一 1ーィルフエ-ル) ェチル] アミノ} ピリミ ジン一 5—カルボ二トリル [160] (以下、 化合物 [160] という) の合成。 (1) 4一 (8—ヒドロキシイミダゾ [1, 2— a] ピリジン一 3—ィル) 一 2— (メチルチオ) ピリミジン ^5—カルボ二トリル [1 58— 1] l O Omgを N, N —ジメチルホルムアミド 3 mLに溶解し、炭酸カリゥム 146mgとフルォロメチル 4―メチノレべンゼンスルホネート (J. L a b e l l e d C omp d. Ra d i o p h a r m. 2003, 46, 555— 566に記載されている方法により合成した) 216m gを加え、 80°Cにて 5時間撹拌した。 反応混合物を室温に戻した後、 飽和 炭酸水素ナトリウム水溶液を加え、 クロ口ホルムとメタノールの混合溶媒 (クロロホ ルム:メタノール =9 : 1) にて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫 酸ナトリウムにて乾燥した。 不溶物をろ過し、 ろ液を減圧濃縮した。 得られた残渣を 少量のクロ口ホルムとメタノールの混合溶媒 (クロ口ホルム:メタノ一ル= 9 : 1) に溶角牟し、 ジェチルエーテルを加え固化させ、 4— [8— (フルォロメトキシ) イミ ダゾ [1, 2— a] ピリジン 3—ィル] —2— (メチルチオ) ピリミジン一 5—力 ルボニトリル [160— 1] 44. lmgを褐色固体として得た。
(2) 4一 [8—(フルォロメ トキシ) イミダゾ [1, 2— a] ピリジン一 3—ィ ル]一 2—(メチルチオ) ピリミジン一 5—カルボ二トリル [160— 1] 27. 6 mgと t一ブチル ((1 S) — 1一 {4一 [4— (トリフルォロアセチル) ピペラジ ン一 1—ィノレ] フエ二ノレ } ェチノレ) カーバメート [1 10— 1] 52. 8mg力、ら、 実施例 106— (3)、 (4) の方法に準じて、 4一 [8— (フルォロメトキシ) イミ ダゾ [1, 2— a] ピリジン一 3—ィル] —2— [((1 S)一 1一 {4一 [4— (ト リフルォロアセチル) ピぺラジン一 1一ィル] フエエル) ェチル) ァミノ] ピリミジ ンー 5—カルボ二トリノレ [160— 2] 15. 9mgを黄色アモルファスとして得た。
(3) 4一 [8— (フルォロメ トキシ) イミダゾ [1, 2— a] ピリジン一 3—ィ ル]一 2— [((1 S)一 1一 {4— [4一 (トリフルォロアセチル) ピぺラジン一 1 一ィル] フエ-ル}ェチル) ァミノ] ピリミジン一 5—カルボ二トリル [160— 2] 13. 6mgから、 実施例 1 10— (3) の.方法に準じて、 目的化合物 [160] 1 1. 3mgを淡黄色固体として得た。
化合物 [160] のスぺクトルデータを以下に示す。
'H—NMR (CDC 13) δ : 9. 52 (d, J = 7. 2Hz, 1 HX 1/5), 8. 90— 8. 82 (m, 1 H+ 1 HX 4/5), 8. 60 (s, 1 HX 1/5), 8. 5 2 (s, 1 HX 4/5), 7. 29— 6. 66 (m, 6H), 6. 13— 6. 1 1 (m,
P T/JP2005/016187
187
1 HX 4/5), 5. 97 (d, J = 52. ΟΗζ, 2Η), 5. 85— 5.
1 H X 1 / 5 ) , 5. 33— 5. 20 (m, 1 HX 1/5), 5. 12— 5.
1 HX 4/5), 3. 15— 3. 08 (m 4H), 3. 06— 3. 03 (
2. 65 (b r s, 1 H), 1. 62 (d J = 6. 8 H z , 3 H)
m a s s : 473 (M+ 1 ) +. 実施例 161
4— [8— (ジフルォロメ トキシ) イミダゾ [1, 2— a] ピリジン一 3—ィル]一 2— [((1 S) — 1一 {4— [(1ーメチルビペリジン一 4一ィル) ァミノ] フエ二 ル} ェチル) ァミノ] ピリミジン一 5—力ルポ-トリノレ [ 161 ] (以下、 化合物 [ 1 61] という) の合成。
(1) 4一 (8—ヒ ドロキシイミダゾ [1, 2— a] ピリジン一 3—ィル)一 2— (メチルチオ) ピリミジン一 5—カルボ二トリノレ [158— 1] 2 Omgをァセトニ トリル ImLに溶解し、 炭酸カリウム 2 Omg、 クロロジフルォロ酢酸ナトリウム 1 3 m gを加え、 90 °Cにて終夜撹拌した。 反応混合物を室温に戻した後、 クロ口ホル ム l O OmLにて希釈し、 水にて洗浄した。 無水硫酸マグネシウムにて乾燥し、 不溶 物をろ過した後、 ろ液を減圧濃縮し、 分取薄層クロマトグラフィーにて精製し、 4—
[8— (ジフルォロメ トキシ) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2— (メチルチオ) ピリミジン一 5一カルボ二トリル [161— 1] 1 5mgを淡褐色固 体として得た。
(2) 4— [8— (ジフルォロメ トキシ) イミダゾ [1, 2— a] ピリジン一 3— ィノレ]一 2— (メチルチオ) ピリミジン一 5—カルボ二トリル [161— 1] 1 5m gと (S) — α—メチルー 4一二トロベンジルァミン塩酸塩 14mgから、 実施例 8 0— (4) の方法に準じて、 4一 [8— (ジフルォロメ トキシ) イミダゾ [1, 2— a] ピリジン一 3—ィル] —2— {[(1 S)一 1一 (4—ニトロフヱニル) ェチル] アミノ} ピリミジン一 5—カルボ二トリル [16 1— 2] 8. 3mgを黄色固体とし て得た。
(3) 4— [8—(ジフルォロメ トキシ) .イミダゾ [1, 2— a] ピリジン一 3— ィル] 一 2— {[(1 S) 一 1一 (4一二トロフエニル) ェチル] アミノ} ピリミジン 一 5—カルボエ ト リノレ [161— 2] 8. 3mg力 ら、 実施例 103— (2) の方法 に準じて、 2— {[(1 S)— 1— (4ーァミノフエニル) ェチル]アミノ}一 4— [8 一 (ジフルォロメ トキシ) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン —5—力ノレボニトリル [161— 3] 7. 9m gを白色固体として得た。
T/JP2005/016187
188
(4) 2— {[(I S) — 1— (4—アミノフヱ-ル) ェチル] アミノ} —4一 [8 ― (ジフルォロメ トキシ) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン 一 5—力ノレボニトリノレ [1 6 1— 3] 7. 9mgと 1——メチノレー 4—ピぺリ ドン 3 μ から、 実施例 8 2— (1) の方法に準じて、 目的化合物 [ 1 6 1] 3. 2mgを白 色固体として得た。 '
化合物 [1 6 1] のスぺクトルデータを以下に示す。
NMR (DMS O— d 6) δ : 9. 9 5 (d, J = 7. 1 H z, 1 HX 1/2),
8. 9 9 (d, J = 6. 1 H z , 1 HX 1/2), 8, 9 1 (d, J = 7. 3H z, 1 HX 1/2), 8. 8 0 (d, J = 8. 3H z , 1 HX 1/2), 8. 7 6 ( s, 1
HX 1/2), 8. 7 5 ( s, 1 HX 1/2), 8. 7 3 ( s, 1 HX 1/2), 8.
5 8 ( s, 1 HX 1 /2), 7. 5 9 ( t , J = 7 3. 7H z , 1 HX 1/2), 7.
5 6 ( t , J = 7 3. 7H z , 1 HX 1/2), 7. 4 0 (d, J = 7. l H z, 1
HX 1/2), 7. 3 5 (d, J = 7. 6 H z , 1 HX 1/2), 7. 1 6 ( t , J = 7. 3H z, 1 HX 1/2), 7. 1 1— 7. 0 7 (m, 2H), 7. 0 1 ( t, J =
7. 3 H z, 1 HX 1/2), .6. 5 5 (d, J = 8. 6H z, 1 HX 1/2), 6.
5 2 (d, J = 8. 6 H z , 1 HX 1/2), 5. 3 3 ( t , J = 8. 0H z , 1 H),
5. 1 5— 5. 0 8 (m, 1 HX 1/2), 4. 9 6— 4. 8 9 (m, 1 HX 1/2),
3. 1 5— 3. 0 7 (m, 1 H), 2. 7 2— 2. 6 5 (m, 2H), 2. 1 5 ( s, 3HX 1/2), 2. 1 4 ( s , 3HX 1 /2), 2. 0 1— 1. 9 7 (m, 2H),
1. 84—1. 8 0 (m, 2H), 1. 4 7 (d, J = 6. 8 H z , 3 HX 1/2),
1. 44 (d, J = 6. 8H z , 3HX 1/2), 1. 3 8— 1. 2 9 (m, 2 H) ma s s : 5 1 9 (M+ 1) +. 実施例 1 6 2
2— [((I S) — 1— { 3— [( 1ーメチルビペリジン一 4—ィル) ァミノ] フエ二 ル} ェチル) ァミノ] 一 4一 [8— (メチルスルホニル) イミダゾ [1, 2— a] ピ リジン一 3—^ fル] ピリミジン一 5—カルボ二トリル [ 1 6 2] (以下、 化合物 [1 6 2] とレ、う) の合成。
( 1 ) 2—クロロー 3— (メチルスルホニル) ピリジン (J . O r g. C h e m. 1 9 7 9, 44, 3 0 8 0— 3 0 8 2に記載されている方法により合成した) 8 0 0 mgとアンモニア水 6 OtnLの混合物を封管中 1 7 5 °Cにて終夜撹拌した。室温に戻 した後、 ジェチルエーテルにて抽出し、 有機層を飽和食塩水にて洗浄した。 無水硫酸 ナトリウムにて乾燥し、 不溶物をろ過した。 ろ液を減圧濃縮し、 3— (メチルスルホ ニル) ピリジン一 2—ァミン [1 6 2— 1] 4 7 3m gを淡橙色固体として得た。
TJP2005/016187
189
(2) 3— (メチルスルホニル) ピリジン一 2—ァミン [162— 1] 470mgと 4— [(Z) —2—エトキシビニル]一 2— (メチルチオ) 一 5—ピリ ミジンカルボ -ル [48— 3] から、 実施例 25— (3) の方法に準じて、 4一 [8— (メチルス ルホニル) イミダゾ [1, 2— a] ピリジン一 3—/ Tル]一 2— (メチルチオ) ピリ ミジン一 5—カルボ二トリ'ル [1 62— 2] 473m gを淡黄色固体として得た。
(3) 4一 [8— (メチルスノレホニル) イミダゾ [1, 2— a] ピリジン一 3—ィ ル]一 2— (メチルチオ) ピリ ミジン一 5—カルボ二トリル [162— 2] 100m gと 3— [(1 S) 一 1一アミノエチル] ァニリン 47. 3mg力 ら、 実施例 80—
(4) の方法に準じて、 2— {[(1 S) — 1一 (3—ァミノフ ニル) ェチル] アミ ノ}一 4一 [8— (メチルスルホニル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリミジン一 5—カルボ-トリノレ [162— 3] 44. 6mgを得た。 (4) 2— {[(1 S) —1— (3—アミノフヱニル) ェチル] アミノ}—4一 [8 一 (メチルスルホニル) イミダゾ [1, 2— a] ピリジン一 3—ィル] ピリ ミジン一 5—カルボ二トリル [162— 3] 15. lmgと 1—メチノ I 4ーピペリ ドン 12. 8 から、 実施例 91の方法に準じて、 目的化合物 [162] 15. 2mgを黄色 固体として得た。
化合物 [162] のスぺク トルデータを以下に示す。
NMR (CDC 13) δ : 8. 92— 8. 89 (m, 2H), 8. 61 (s , 1 ΗΧ 1/5), 8. 59 (s, 1 ΗΧ 4/5), 8. 13 (d, J = 7. 3Ηζ, 1 Η X 1/5), 8. 04 (d, J = 7. 3Hz, 1 HX 4/5), 7. 33— 7. 12 (m, 1 H), 6. 89— 6. 56 (m, 4H), 6. 17— 6. 10 (m, 1HX4/5), 6. 02— 5. 92 (m, 1 HX 1/5), 5. 26— 5. 17 (m, 1H 1/5), 5. 00— 4. 87 (m, 1 HX 4/5), 3. 76— 3.. 60 (m, 1H), 3. 5 6 ( s, 3 HX 1/5), 3. 52 ( s, 3HX 4/5), 3. 37— 3. 20 (m, 1 H), 2. 90— 2. 75 (m, 2H), 2. 32 ( s , 3H), 2. 26— 1. 9 6 (m, 4H), 1. 62 (d, J = 6. 8Hz, 3H), 1. 60— 1. 42 (m, 2 H)
ma s s : 531 (M+ 1) +. 産業上の利用の可能性
本発明の化合物は、優れた P L K 1阻害作用及び細胞増殖阻害作用を有すること力 ら医薬の分野において有用な抗腫瘍剤として期待される。