WO2005113501A1 - 神経因性疼痛制御剤組成物 - Google Patents
神経因性疼痛制御剤組成物 Download PDFInfo
- Publication number
- WO2005113501A1 WO2005113501A1 PCT/JP2005/009361 JP2005009361W WO2005113501A1 WO 2005113501 A1 WO2005113501 A1 WO 2005113501A1 JP 2005009361 W JP2005009361 W JP 2005009361W WO 2005113501 A1 WO2005113501 A1 WO 2005113501A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- group
- compound
- composition according
- neuropathic pain
- Prior art date
Links
- 208000004296 neuralgia Diseases 0.000 title claims abstract description 44
- 239000000203 mixture Substances 0.000 title claims abstract description 36
- 150000001875 compounds Chemical class 0.000 claims abstract description 83
- 150000003839 salts Chemical class 0.000 claims abstract description 16
- 208000021722 neuropathic pain Diseases 0.000 claims description 42
- 125000000217 alkyl group Chemical group 0.000 claims description 15
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 claims description 14
- 239000003795 chemical substances by application Substances 0.000 claims description 12
- 238000000034 method Methods 0.000 claims description 10
- 125000003545 alkoxy group Chemical group 0.000 claims description 9
- 125000005843 halogen group Chemical group 0.000 claims description 7
- 239000000126 substance Substances 0.000 claims description 6
- 229910052760 oxygen Inorganic materials 0.000 claims description 5
- 239000001301 oxygen Substances 0.000 claims description 5
- 125000006704 (C5-C6) cycloalkyl group Chemical group 0.000 claims description 3
- 125000003342 alkenyl group Chemical group 0.000 claims description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 3
- 229910052717 sulfur Inorganic materials 0.000 claims description 3
- MYMOFIZGZYHOMD-UHFFFAOYSA-N Dioxygen Chemical compound O=O MYMOFIZGZYHOMD-UHFFFAOYSA-N 0.000 claims description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 2
- 125000004434 sulfur atom Chemical group 0.000 claims description 2
- 239000000446 fuel Substances 0.000 claims 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims 1
- 238000004519 manufacturing process Methods 0.000 claims 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 51
- 230000000202 analgesic effect Effects 0.000 description 33
- 230000000052 comparative effect Effects 0.000 description 28
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 27
- 208000002193 Pain Diseases 0.000 description 26
- 230000036407 pain Effects 0.000 description 25
- 239000000243 solution Substances 0.000 description 24
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 20
- 230000015572 biosynthetic process Effects 0.000 description 20
- 238000003786 synthesis reaction Methods 0.000 description 20
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 18
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 16
- 238000006243 chemical reaction Methods 0.000 description 15
- 238000001914 filtration Methods 0.000 description 13
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 12
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 239000013078 crystal Substances 0.000 description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 9
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 9
- -1 bromo compound Chemical class 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 8
- GWVMLCQWXVFZCN-UHFFFAOYSA-N isoindoline Chemical class C1=CC=C2CNCC2=C1 GWVMLCQWXVFZCN-UHFFFAOYSA-N 0.000 description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 7
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 238000012360 testing method Methods 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 6
- 241000699666 Mus <mouse, genus> Species 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 239000000460 chlorine Substances 0.000 description 6
- 229940079593 drug Drugs 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 238000010992 reflux Methods 0.000 description 6
- 238000005160 1H NMR spectroscopy Methods 0.000 description 5
- 241000699670 Mus sp. Species 0.000 description 5
- 239000001768 carboxy methyl cellulose Substances 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- 229930006000 Sucrose Natural products 0.000 description 4
- 229940035676 analgesics Drugs 0.000 description 4
- 239000000730 antalgic agent Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 229920000609 methyl cellulose Polymers 0.000 description 4
- 239000001923 methylcellulose Substances 0.000 description 4
- 235000010981 methylcellulose Nutrition 0.000 description 4
- 239000008194 pharmaceutical composition Substances 0.000 description 4
- 238000010898 silica gel chromatography Methods 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 229960004793 sucrose Drugs 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- ZJHZCFSWXVUBHT-UHFFFAOYSA-N 2,3-dihydro-1h-indene-5,6-dicarboxylic acid Chemical compound C1=C(C(O)=O)C(C(=O)O)=CC2=C1CCC2 ZJHZCFSWXVUBHT-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 3
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- LGRFSURHDFAFJT-UHFFFAOYSA-N Phthalic anhydride Natural products C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 150000008064 anhydrides Chemical class 0.000 description 3
- 239000001961 anticonvulsive agent Substances 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- JHIWVOJDXOSYLW-UHFFFAOYSA-N butyl 2,2-difluorocyclopropane-1-carboxylate Chemical compound CCCCOC(=O)C1CC1(F)F JHIWVOJDXOSYLW-UHFFFAOYSA-N 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 3
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 210000003141 lower extremity Anatomy 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000008018 melting Effects 0.000 description 3
- 230000004973 motor coordination Effects 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 229910000033 sodium borohydride Inorganic materials 0.000 description 3
- 239000012279 sodium borohydride Substances 0.000 description 3
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 3
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- SDJHPPZKZZWAKF-UHFFFAOYSA-N 2,3-dimethylbuta-1,3-diene Chemical compound CC(=C)C(C)=C SDJHPPZKZZWAKF-UHFFFAOYSA-N 0.000 description 2
- DVPGRIXJCJBDHP-UHFFFAOYSA-N 2-[2-(3-fluorophenyl)-5,6-dimethyl-3-oxo-1h-isoindol-1-yl]acetic acid Chemical compound O=C1C=2C=C(C)C(C)=CC=2C(CC(O)=O)N1C1=CC=CC(F)=C1 DVPGRIXJCJBDHP-UHFFFAOYSA-N 0.000 description 2
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 2
- CMQYISATYUYSAC-UHFFFAOYSA-N 5,6-dimethyl-2-benzofuran-1,3-dione Chemical compound C1=C(C)C(C)=CC2=C1C(=O)OC2=O CMQYISATYUYSAC-UHFFFAOYSA-N 0.000 description 2
- UQMLHEGKLYWPDL-UHFFFAOYSA-N 5,7-dihydrofuro[3,4-f][2]benzofuran-1,3-dione Chemical compound C1=C2C(=O)OC(=O)C2=CC2=C1COC2 UQMLHEGKLYWPDL-UHFFFAOYSA-N 0.000 description 2
- 244000215068 Acacia senegal Species 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 206010058019 Cancer Pain Diseases 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- 239000004375 Dextrin Substances 0.000 description 2
- 229920001353 Dextrin Polymers 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 229920000084 Gum arabic Polymers 0.000 description 2
- 208000007514 Herpes zoster Diseases 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 206010036376 Postherpetic Neuralgia Diseases 0.000 description 2
- 239000004373 Pullulan Substances 0.000 description 2
- 229920001218 Pullulan Polymers 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- 239000000205 acacia gum Substances 0.000 description 2
- 235000010489 acacia gum Nutrition 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 2
- 230000001773 anti-convulsant effect Effects 0.000 description 2
- 239000000935 antidepressant agent Substances 0.000 description 2
- 229940005513 antidepressants Drugs 0.000 description 2
- 229960003965 antiepileptics Drugs 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 239000002249 anxiolytic agent Substances 0.000 description 2
- 230000000949 anxiolytic effect Effects 0.000 description 2
- 229940005530 anxiolytics Drugs 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000012300 argon atmosphere Substances 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 229930002875 chlorophyll Natural products 0.000 description 2
- 235000019804 chlorophyll Nutrition 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 235000019425 dextrin Nutrition 0.000 description 2
- 206010012601 diabetes mellitus Diseases 0.000 description 2
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 208000002173 dizziness Diseases 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- VRUXDKVMRXQDKE-UHFFFAOYSA-N ethyl 2-(trifluoro-$l^{5}-phosphanylidene)acetate Chemical compound CCOC(=O)C=P(F)(F)F VRUXDKVMRXQDKE-UHFFFAOYSA-N 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- FQMQTNHJHYXQLK-UHFFFAOYSA-N furo[3,4-f][1,3]benzodioxole-5,7-dione Chemical compound C1=C2C(=O)OC(=O)C2=CC2=C1OCO2 FQMQTNHJHYXQLK-UHFFFAOYSA-N 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- RSPZSDWVQWRAEF-UHFFFAOYSA-N hepta-1,6-diyne Chemical compound C#CCCCC#C RSPZSDWVQWRAEF-UHFFFAOYSA-N 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 208000005877 painful neuropathy Diseases 0.000 description 2
- WEXRUCMBJFQVBZ-UHFFFAOYSA-N pentobarbital Chemical compound CCCC(C)C1(CC)C(=O)NC(=O)NC1=O WEXRUCMBJFQVBZ-UHFFFAOYSA-N 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 150000004885 piperazines Chemical class 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 235000019423 pullulan Nutrition 0.000 description 2
- 210000003497 sciatic nerve Anatomy 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 229960002920 sorbitol Drugs 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 230000002889 sympathetic effect Effects 0.000 description 2
- 210000002972 tibial nerve Anatomy 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- FTLYMKDSHNWQKD-UHFFFAOYSA-N (2,4,5-trichlorophenyl)boronic acid Chemical compound OB(O)C1=CC(Cl)=C(Cl)C=C1Cl FTLYMKDSHNWQKD-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- FTNJQNQLEGKTGD-UHFFFAOYSA-N 1,3-benzodioxole Chemical compound C1=CC=C2OCOC2=C1 FTNJQNQLEGKTGD-UHFFFAOYSA-N 0.000 description 1
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 1
- MBVLXJRLIIMEIF-UHFFFAOYSA-N 2-(3-fluorophenyl)-3-hydroxy-5,6-dimethyl-3h-isoindol-1-one Chemical compound O=C1C=2C=C(C)C(C)=CC=2C(O)N1C1=CC=CC(F)=C1 MBVLXJRLIIMEIF-UHFFFAOYSA-N 0.000 description 1
- USDUUSBKXDIXJB-UHFFFAOYSA-N 2-(3-fluorophenyl)-5,6-dimethyl-3-[2-(4-methylpiperazin-1-yl)-2-oxoethyl]-3h-isoindol-1-one Chemical compound C1CN(C)CCN1C(=O)CC1C2=CC(C)=C(C)C=C2C(=O)N1C1=CC=CC(F)=C1 USDUUSBKXDIXJB-UHFFFAOYSA-N 0.000 description 1
- YGZFYDFBHIDIBH-UHFFFAOYSA-N 2-[bis(2-hydroxyethyl)amino]icosan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCC(CO)N(CCO)CCO YGZFYDFBHIDIBH-UHFFFAOYSA-N 0.000 description 1
- VHMICKWLTGFITH-UHFFFAOYSA-N 2H-isoindole Chemical compound C1=CC=CC2=CNC=C21 VHMICKWLTGFITH-UHFFFAOYSA-N 0.000 description 1
- WGTASENVNYJZBK-UHFFFAOYSA-N 3,4,5-trimethoxyamphetamine Chemical compound COC1=CC(CC(C)N)=CC(OC)=C1OC WGTASENVNYJZBK-UHFFFAOYSA-N 0.000 description 1
- AEDQNOLIADXSBB-UHFFFAOYSA-N 3-(dodecylazaniumyl)propanoate Chemical compound CCCCCCCCCCCCNCCC(O)=O AEDQNOLIADXSBB-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- HRDCVMSNCBAMAM-UHFFFAOYSA-N 3-prop-2-ynoxyprop-1-yne Chemical compound C#CCOCC#C HRDCVMSNCBAMAM-UHFFFAOYSA-N 0.000 description 1
- JFHFMHPKOSJJTE-UHFFFAOYSA-N 4,5-dimethyl-2-benzofuran-1,3-dione Chemical compound CC1=CC=C2C(=O)OC(=O)C2=C1C JFHFMHPKOSJJTE-UHFFFAOYSA-N 0.000 description 1
- WPYAICCSYGUFTK-UHFFFAOYSA-N 5,6-dibromo-1,3-benzodioxole Chemical compound C1=C(Br)C(Br)=CC2=C1OCO2 WPYAICCSYGUFTK-UHFFFAOYSA-N 0.000 description 1
- UJYUDTPLHOZSGT-UHFFFAOYSA-N 5,6-dimethyl-3a,4,7,7a-tetrahydro-2-benzofuran-1,3-dione Chemical compound C1C(C)=C(C)CC2C(=O)OC(=O)C12 UJYUDTPLHOZSGT-UHFFFAOYSA-N 0.000 description 1
- NKJKYAYKHJZPFZ-UHFFFAOYSA-N 6,7-dihydro-5h-cyclopenta[f][2]benzofuran-1,3-dione Chemical compound C1=C2C(=O)OC(=O)C2=CC2=C1CCC2 NKJKYAYKHJZPFZ-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 108010011485 Aspartame Proteins 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 206010003694 Atrophy Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BOWDBWOGKSGHOX-UHFFFAOYSA-N C(=O)(OCC)CCOC=P(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound C(=O)(OCC)CCOC=P(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1 BOWDBWOGKSGHOX-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 208000000094 Chronic Pain Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 239000004287 Dehydroacetic acid Substances 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- QXNVGIXVLWOKEQ-UHFFFAOYSA-N Disodium Chemical compound [Na][Na] QXNVGIXVLWOKEQ-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- BIVBRWYINDPWKA-VLQRKCJKSA-L Glycyrrhizinate dipotassium Chemical compound [K+].[K+].O([C@@H]1[C@@H](O)[C@H](O)[C@H](O[C@@H]1O[C@H]1CC[C@]2(C)[C@H]3C(=O)C=C4[C@@H]5C[C@](C)(CC[C@@]5(CC[C@@]4(C)[C@]3(C)CC[C@H]2C1(C)C)C)C(O)=O)C([O-])=O)[C@@H]1O[C@H](C([O-])=O)[C@@H](O)[C@H](O)[C@H]1O BIVBRWYINDPWKA-VLQRKCJKSA-L 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 229930182559 Natural dye Natural products 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 229920001007 Nylon 4 Polymers 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 208000004983 Phantom Limb Diseases 0.000 description 1
- 206010056238 Phantom pain Diseases 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 208000004550 Postoperative Pain Diseases 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 244000228451 Stevia rebaudiana Species 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- QDPOOGQUCJJZAO-FCXRPNKRSA-N [(E)-3-(3,4-dihydroxyphenyl)prop-2-enoyl] (E)-3-(3,4-dihydroxyphenyl)prop-2-enoate Chemical compound C1=C(O)C(O)=CC=C1\C=C\C(=O)OC(=O)\C=C\C1=CC=C(O)C(O)=C1 QDPOOGQUCJJZAO-FCXRPNKRSA-N 0.000 description 1
- YKTSYUJCYHOUJP-UHFFFAOYSA-N [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] Chemical compound [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] YKTSYUJCYHOUJP-UHFFFAOYSA-N 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- WMGSQTMJHBYJMQ-UHFFFAOYSA-N aluminum;magnesium;silicate Chemical compound [Mg+2].[Al+3].[O-][Si]([O-])([O-])[O-] WMGSQTMJHBYJMQ-UHFFFAOYSA-N 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 230000036592 analgesia Effects 0.000 description 1
- 230000001430 anti-depressive effect Effects 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000001741 anti-phlogistic effect Effects 0.000 description 1
- 230000002921 anti-spasmodic effect Effects 0.000 description 1
- 229940125681 anticonvulsant agent Drugs 0.000 description 1
- 229940124575 antispasmodic agent Drugs 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 239000000605 aspartame Substances 0.000 description 1
- 235000010357 aspartame Nutrition 0.000 description 1
- IAOZJIPTCAWIRG-QWRGUYRKSA-N aspartame Chemical compound OC(=O)C[C@H](N)C(=O)N[C@H](C(=O)OC)CC1=CC=CC=C1 IAOZJIPTCAWIRG-QWRGUYRKSA-N 0.000 description 1
- 229960003438 aspartame Drugs 0.000 description 1
- 230000037444 atrophy Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- YTIVTFGABIZHHX-UHFFFAOYSA-N butynedioic acid Chemical compound OC(=O)C#CC(O)=O YTIVTFGABIZHHX-UHFFFAOYSA-N 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- ATNHDLDRLWWWCB-AENOIHSZSA-M chlorophyll a Chemical compound C1([C@@H](C(=O)OC)C(=O)C2=C3C)=C2N2C3=CC(C(CC)=C3C)=[N+]4C3=CC3=C(C=C)C(C)=C5N3[Mg-2]42[N+]2=C1[C@@H](CCC(=O)OC\C=C(/C)CCC[C@H](C)CCC[C@H](C)CCCC(C)C)[C@H](C)C2=C5 ATNHDLDRLWWWCB-AENOIHSZSA-M 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 235000019258 dehydroacetic acid Nutrition 0.000 description 1
- 229940061632 dehydroacetic acid Drugs 0.000 description 1
- JEQRBTDTEKWZBW-UHFFFAOYSA-N dehydroacetic acid Chemical compound CC(=O)C1=C(O)OC(C)=CC1=O JEQRBTDTEKWZBW-UHFFFAOYSA-N 0.000 description 1
- PGRHXDWITVMQBC-UHFFFAOYSA-N dehydroacetic acid Natural products CC(=O)C1C(=O)OC(C)=CC1=O PGRHXDWITVMQBC-UHFFFAOYSA-N 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 230000000994 depressogenic effect Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 150000005690 diesters Chemical class 0.000 description 1
- 235000013681 dietary sucrose Nutrition 0.000 description 1
- 229940101029 dipotassium glycyrrhizinate Drugs 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 235000013399 edible fruits Nutrition 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 1
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- JEIPFZHSYJVQDO-UHFFFAOYSA-N iron(III) oxide Inorganic materials O=[Fe]O[Fe]=O JEIPFZHSYJVQDO-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 208000013433 lightheadedness Diseases 0.000 description 1
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 description 1
- 229910052808 lithium carbonate Inorganic materials 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 229940031703 low substituted hydroxypropyl cellulose Drugs 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- WVJKHCGMRZGIJH-UHFFFAOYSA-N methanetriamine Chemical compound NC(N)N WVJKHCGMRZGIJH-UHFFFAOYSA-N 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 239000012046 mixed solvent Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 1
- 229960005181 morphine Drugs 0.000 description 1
- 230000007659 motor function Effects 0.000 description 1
- 230000003533 narcotic effect Effects 0.000 description 1
- 239000000978 natural dye Substances 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 210000000944 nerve tissue Anatomy 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 229960001412 pentobarbital Drugs 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 210000004345 peroneal nerve Anatomy 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- RFIOZSIHFNEKFF-UHFFFAOYSA-N piperazine-1-carboxylic acid Chemical compound OC(=O)N1CCNCC1 RFIOZSIHFNEKFF-UHFFFAOYSA-N 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229940068965 polysorbates Drugs 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- SSOLNOMRVKKSON-UHFFFAOYSA-N proguanil Chemical compound CC(C)\N=C(/N)N=C(N)NC1=CC=C(Cl)C=C1 SSOLNOMRVKKSON-UHFFFAOYSA-N 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- YORCIIVHUBAYBQ-UHFFFAOYSA-N propargyl bromide Chemical compound BrCC#C YORCIIVHUBAYBQ-UHFFFAOYSA-N 0.000 description 1
- UORVCLMRJXCDCP-UHFFFAOYSA-N propynoic acid Chemical group OC(=O)C#C UORVCLMRJXCDCP-UHFFFAOYSA-N 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- HELXLJCILKEWJH-NCGAPWICSA-N rebaudioside A Chemical compound O([C@H]1[C@H](O)[C@@H](CO)O[C@H]([C@@H]1O[C@H]1[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O1)O)O[C@]12C(=C)C[C@@]3(C1)CC[C@@H]1[C@@](C)(CCC[C@]1([C@@H]3CC2)C)C(=O)O[C@H]1[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O1)O)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O HELXLJCILKEWJH-NCGAPWICSA-N 0.000 description 1
- 239000006215 rectal suppository Substances 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000011514 reflex Effects 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- 229940085605 saccharin sodium Drugs 0.000 description 1
- 210000002265 sensory receptor cell Anatomy 0.000 description 1
- 102000027509 sensory receptors Human genes 0.000 description 1
- 108091008691 sensory receptors Proteins 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- OAOPAJMEZXJIAG-UHFFFAOYSA-N spiro[1,2-dihydroindene-3,3'-oxetane]-2',4'-dione Chemical compound O=C1OC(=O)C11C2=CC=CC=C2CC1 OAOPAJMEZXJIAG-UHFFFAOYSA-N 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-L sulfite Chemical class [O-]S([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-L 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 210000001590 sural nerve Anatomy 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- YKMBNBXUPNOYHE-UHFFFAOYSA-N tert-butyl 4-propan-2-ylpiperazine-1-carboxylate Chemical compound CC(C)N1CCN(C(=O)OC(C)(C)C)CC1 YKMBNBXUPNOYHE-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 239000012929 tonicity agent Substances 0.000 description 1
- 239000003204 tranquilizing agent Substances 0.000 description 1
- 230000002936 tranquilizing effect Effects 0.000 description 1
- 230000017105 transposition Effects 0.000 description 1
- IIHPVYJPDKJYOU-UHFFFAOYSA-N triphenylcarbethoxymethylenephosphorane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=CC(=O)OCC)C1=CC=CC=C1 IIHPVYJPDKJYOU-UHFFFAOYSA-N 0.000 description 1
- 210000001215 vagina Anatomy 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/44—Iso-indoles; Hydrogenated iso-indoles
- C07D209/46—Iso-indoles; Hydrogenated iso-indoles with an oxygen atom in position 1
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/58—[b]- or [c]-condensed
- C07D209/62—Naphtho [c] pyrroles; Hydrogenated naphtho [c] pyrroles
- C07D209/64—Naphtho [c] pyrroles; Hydrogenated naphtho [c] pyrroles with an oxygen atom in position 1
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
Definitions
- the present invention relates to a novel pharmaceutical use of an isoindoline derivative as a neuropathic pain control agent.
- the present invention also provides a novel isoindoline derivative.
- Neuropathic pain is pain that occurs without stimulation of peripheral sensory receptors, and is pain that is caused by direct damage or compression of nerve tissue.
- Known neuropathic pain include acute herpes zoster, postherpetic neuralgia, painful neuropathy of diabetes, and cancer pain. If the pain is severe, the patient will be depressed, and the quality of life (QOL) will be significantly reduced.
- nalgesics are generally classified into antiphlogistic analgesics represented by aspirin and narcotic analgesics represented by morphine. These analgesics have little effect on neuropathic pain. Not known. At present, neuropathic pain is prescribed with anticonvulsants, antidepressants, anxiolytics, etc. Powerful drugs have long-lasting effects even if they achieve temporary pain relief. It is often difficult to administer for a period. Thus, there is a need in the clinical setting for drugs that are effective for neuropathic pain with few side effects.
- An object of the present invention is to provide a therapeutic agent effective for suppressing neuropathic pain. Another object of the present invention is to provide a novel isoindoline derivative.
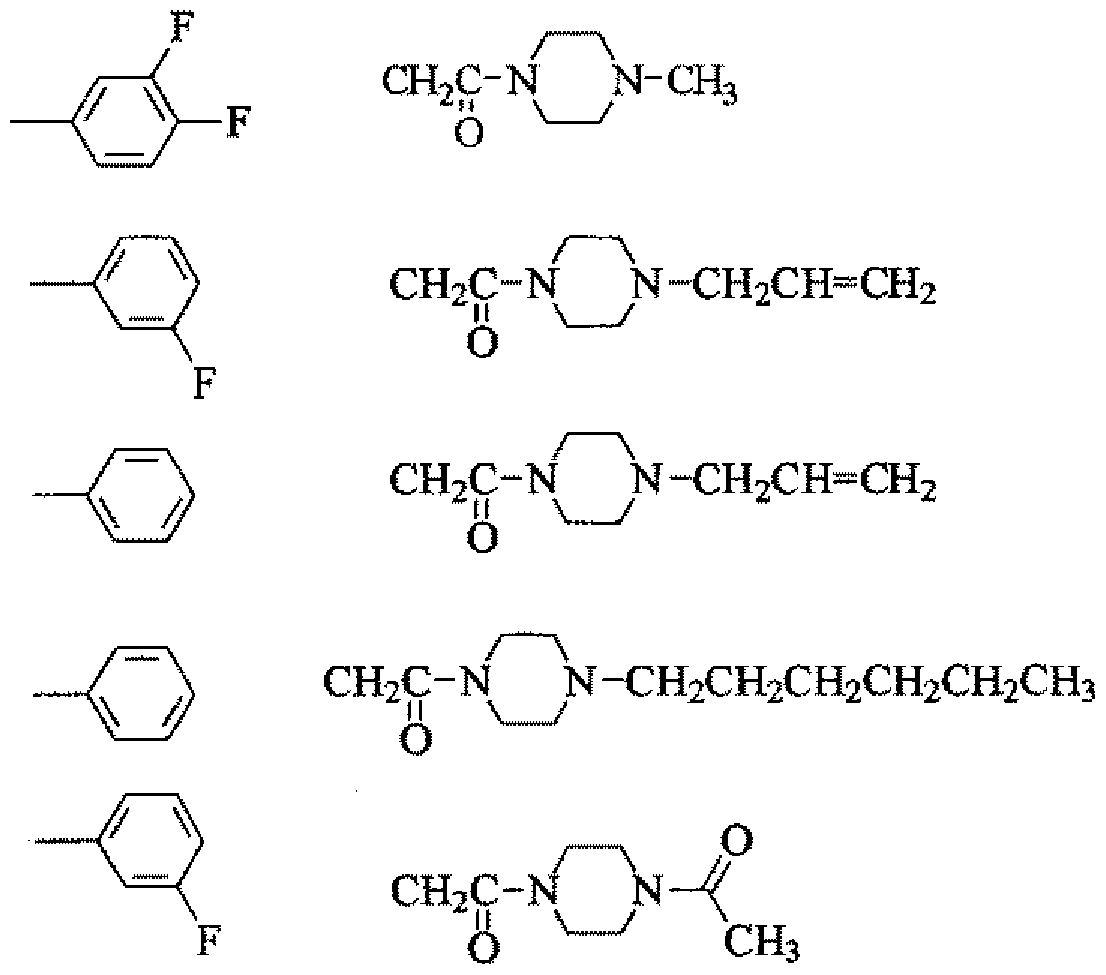
- R and R may be the same or different
- X is a halogen atom, or an alkoxy group of Cl to 6 or a phenol group to which X is bonded, with the formula: [Chemical 2]
- n is an integer of 0 to 2, provided that X is an integer of 0 or 1 when X is a Cl-6 alkoxy group;
- Y is the formula:
- R is a linear or branched alkyl group of Cl-6, a linear or branched chain of C3-6
- Alkenyl group C3-6 linear or branched alkyl group, C5-6 cycloalkyl group, formula:
- a neuropathic pain control agent composition comprising the compound or a salt thereof.
- the present invention further provides a method for controlling neuropathic pain, comprising administering an effective amount of a compound represented by the formula (I) to a subject in need of controlling neuropathic pain.
- the present invention also provides a use of the compound represented by the formula (I) for producing a neuropathic pain control agent composition.
- composition of the present invention are novel. Accordingly, the present invention also provides these novel compounds.
- novel compounds of the present invention are as follows:
- FIG. 1 is a graph showing the analgesic effect of the compound of Example 1 of the present invention and gyabapentin in a neuropathic pain model (administration of 100 mg / kg).
- FIG. 1-2 is a graph showing the analgesic effect of the compound of Example 1 of the present invention and gyabapentin in a neuropathic pain model (30 mg / kg administration).
- FIG. 2-1 is a graph showing the analgesic effect of the compounds of Examples 1 to 6 of the present invention in a neuropathic pain model.
- FIG. 2-2 is a graph showing the analgesic effect of the compounds of Examples 7 to 14 of the present invention in a neuropathic pain model.
- FIG. 2-3 is a graph showing the analgesic effect of the compounds of Examples 15 to 22 of the present invention in a neuropathic pain model.
- FIG. 2-4 is a graph showing the analgesic effect of the compounds of Examples 23 to 25 of the present invention in a neuropathic pain model.
- FIG. 3 is a graph showing the analgesic effect of the compound of Example 1 of the present invention in a (+) body and a (one) body neuropathic pain model.
- FIG. 4 is a graph showing the results of a motor coordination test of the compound of Example 1 of the present invention and giabapentin.
- R and R may be the same or different.
- a good linear or branched Cl-6 alkyl group preferably a Cl-3 alkyl group, most preferably both R and R are methyl groups.
- X forms a group shown below together with a halogen atom, an alkoxy group of Cl to 6, or a phenol group to which X is bonded.
- halogen atom examples include chlorine, fluorine, bromine and iodine, and chlorine and fluorine are preferred, and fluorine is particularly preferred.
- X is a halogen atom
- m is 1 or Or an integer of 2.
- the halogen atom is preferably bonded at the meta and Z or para positions.
- the alkoxy group is preferably an alkoxy group of Cl to 3, particularly a methoxy group. Particularly, a compound in which an alkoxy group is bonded to the para position is preferably used.
- R is a linear or branched alkyl group of Cl-6, a linear or branched C3-6
- R is an alkyl group of Cl to 6, a linear alkyl of C3 to 6,
- a cyclopentyl group and a cyclohexyl group [0022] Z is oxygen or sulfur, and oxygen is particularly preferably used.
- the compound (I) of the present invention may be produced by any known method.
- the compound (I) of the present invention is obtained by converting the compound of the formula (VII) obtained according to Scheme 1 with the corresponding aminy conjugate (VIII) and WSC [1-ethyl-3- (3 dimethylaminopropyl) carbodiimide. [Hydrochloride] and ⁇ ⁇ ⁇ ⁇ (1-hydroxybenzotriazole hydrate) in dimethylformamide or tetrahydrofuran solvent.
- Dimethyl phthalic anhydride (III 1) is obtained by heating the acid anhydride obtained by the reaction of 2,3 dimethyl-1,3-butadiene with maleic anhydride with bromine in an acetic acid solvent .
- Indandicarboxylic anhydride (III 3) is obtained by converting a diester obtained by a reaction between 1,6 heptadiyne and acetylenedicarboxylic acid getyl ester into dicarboxylic acid with hydrochloric acid, By dehydration and ring closure.
- 1,3-dihydro-2benzofuran 5,6 dicarboxylic anhydride is propargylate It can be obtained in the same manner as when (III-3) is obtained using tell.
- 1,3-benzodioxole 5,6 dicarboxylic anhydride is 1,2-dibromo
- (III-2) can be obtained from (methylenedioxy) benzene by a method similar to that for obtaining (III-2).
- 1-tert-butoxycarbylbiperazine (IX) is heated with an appropriate bromo compound or a chlorophyll compound in an acetonitrile solvent in the presence of lithium carbonate to obtain a compound (X).
- the desired piperazine derivative can be obtained by the action of oral acetic acid.
- the compound represented by the formula (I) of the present invention may be contained in the form of a salt, and such a case is also included in the scope of the present invention.
- a salt any salt with a pharmaceutically acceptable inorganic acid or organic acid is suitably used.
- a caro salt with an acid for example, a salt with an inorganic acid such as hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid, hydrobromic acid, or acetic acid or propionate
- an inorganic acid such as hydrochloric acid, sulfuric acid, nitric acid, phosphoric acid, hydrobromic acid, or acetic acid or propionate
- organic acids such as acid, fumaric acid, maleic acid, tartaric acid, citric acid, malic acid, oxalic acid, benzoic acid, methanesulfonic acid, and benzenesulfonic acid.
- the compound represented by the formula (I) has optical isomers, and the compound contained in the neuropathic pain control agent composition of the present invention may be each isomer alone or a racemic form. May be.
- the compound represented by the formula (I) is usually obtained as a racemate, but when it contains a single optical isomer, it may be separated into optically active substances by a conventional method known per se.
- the optical isomers of the compound represented by the formula (I) the (1) isomer is more preferred.
- the isoindoline derivative represented by the formula (I) has an analgesic effect on neuropathic pain in mammals, and is suitably used for controlling neuropathic pain.
- the analgesic effect of the neuropathic pain control agent composition of the present invention can be obtained at a lower dose as compared with the currently used guinea buntin, and the onset of action is rapid.
- the neuropathic pain control agent composition of the present invention is used by orally or parenterally administering to mammals including humans, for example, intravenously, epidurally, intraspinal, subcutaneously, intramuscularly, or the like. be able to.
- the dosage form is not limited and may be appropriately set by those skilled in the art.
- tablets, soft capsules, capsules such as microcapsules, granules, powders, oral preparations such as syrups, emulsions, suspensions, etc., and external preparations such as injections, ointments, creams, rectal suppositories, vagina Suppositories, etc.
- Parenteral preparations such as suppositories, pellets, drops and sustained-release preparations, and these can be administered orally or parenterally, respectively.
- the pharmaceutical composition can be produced by a method commonly used in the technical field of pharmaceutical formulation, for example, a method described in the Japanese Pharmacopoeia.
- the content of the compound of the present invention in the pharmaceutical composition can be appropriately determined depending on the dosage form, dosage and the like, and is, for example, about 0.1 to: LOO% by weight.
- the composition of the present invention can be obtained by mixing the compound represented by the formula (I) or a salt thereof with a pharmaceutically acceptable carrier or the like to prepare a pharmaceutical composition.
- a carrier or the like is used at the time of formulation, and an organic or inorganic carrier substance is used.
- Substances to be blended as an agent, an isotonic agent, a buffer, an analgesic, etc. can be used without any particular limitation. If necessary, additives such as preservatives, antioxidants, coloring agents, sweeteners and the like may be added.
- Excipients include lactose, sucrose, D-mantol, D-sorbitol, starch, starch alpha, dextrin, crystalline cellulose, low-substituted hydroxypropylcellulose, sodium carboxymethylcellulose, gum arabic, Examples include pullulan, light silicic anhydride, synthetic aluminum silicate, and magnesium aluminate metasilicate. Examples of the lubricant include magnesium stearate, calcium stearate, talc, colloidal silica and the like.
- binder examples include pregelatinized starch, sucrose, gelatin, gum arabic, methylcellulose, carboxymethylcellulose, sodium carboxymethylcellulose, crystalline cellulose, sucrose, D-man-tol, trehalose, dextrin, pullulan, Examples include hydroxypropylcellulose, hydroxypropylmethylcellulose, and polybutylpyrrolidone.
- Disintegrators include lactose, saccharose, starch, carboxymethylcellulose, carboxymethinoresenolerose kanoresum, croscanolemelose sodium, canoleboxy methinolestarch sodium, light caffeic anhydride, low substitution degree
- Examples include hydroxypropyl cellulose.
- Examples of the solvent include water for injection, physiological saline, Ringer's solution, alcohol, propylene glycol, polyethylene glycol, sesame oil, corn oil, olive oil, and cottonseed oil. I can get lost.
- Preferred examples of solubilizers include polyethylene glycol, propylene glycol, D-mantol, trehalose, benzyl benzoate, ethanol, trisaminomethane, cholesterol, triethanolamine, sodium carbonate, sodium citrate, and salici. Examples thereof include sodium luate and sodium acetate.
- suspending agent examples include surfactants such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalcodium chloride, benzethonium chloride and glyceryl monostearate; Hydrophilic alcohols such as butyl alcohol, polypyrrolidone, sodium carboxymethylcellulose, methylcellulose, hydroxymethylcellulose, hydroxyethylcellulose and hydroxypropylcellulose; polysorbates, and polyoxyethylene hydrogenated castor oil.
- surfactants such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalcodium chloride, benzethonium chloride and glyceryl monostearate
- Hydrophilic alcohols such as butyl alcohol, polypyrrolidone, sodium carboxymethylcellulose, methylcellulose, hydroxymethylcellulose, hydroxyethylcellulose and hydroxypropylcellulose
- Examples of the tonicity agent include sodium salt sodium, glycerin, D-mantol, D-sorbitol, glucose and the like.
- Examples of the buffer include buffers such as phosphate, acetate, carbonate, and citrate.
- Examples of the soothing agent include benzyl alcohol and the like.
- Examples of preservatives include noroxybenzoate esters, chlorobutanol, benzyl alcohol, phenyl alcohol, dehydroacetic acid, sorbic acid, etc.o
- antioxidants include sulfites, ascorbate, etc. Is done.
- As a coloring agent include sodium salt sodium, glycerin, D-mantol, D-sorbitol, glucose and the like.
- Examples of the buffer include buffers such as phosphate, acetate, carbonate, and citrate.
- Examples of the soothing agent include benzyl alcohol and the like.
- Examples of preservatives include noroxybenzoate esters, chlorobutanol
- Edible dyes include water-insoluble lake dyes, and natural dyes (eg, j8-potency rotin, chlorophyll, red iron oxide, etc.).
- Sweetening agents include saccharin sodium, dipotassium glycyrrhizinate, aspartame, stevia and the like.
- neuropathic pain control agent composition of the present invention may be supplemented with other medicinal ingredients unless it violates the purpose of the present invention.
- the composition of the present invention may be used for neuropathic pain, for example, acute herpes zoster, postherpetic neuralgia, painful neuropathy of diabetes, prolonged postoperative pain, reflex sympathetic atrophy, tooth extraction. It can be used for pain control for patients suffering from retroreflective sympathetic dystrophy, phantom limb pain and cancer pain.
- control of pain means not only administration when a pain attack occurs, analgesia by periodic administration for chronic pain, etc., but also administration in advance when pain is predicted to occur. It should also include preventive measures, such as preventing pain attacks by periodically administering pain if it occurs repeatedly.
- "subjects in need of controlling neuropathic pain” include patients suffering from pain as described above and patients predicted to develop pain. I do.
- the effective amount of the compound represented by the formula (I) is not particularly limited and may be appropriately selected depending on the age, sex, body weight, health condition, and severity of neuropathic pain to be administered. Just fine.
- Typical examples of the use as an oral analgesic include, but are not limited to, the isoindoline derivative of the present invention represented by the formula (I) in an amount of about 0.1 to: LOOmg / kg, preferably 3.0 to 3Og.
- Administer .OmgZkg orally The isoindoline derivative of the present invention can be orally administered at the time of pain induction since the onset of the effect is rapid. It may also be administered periodically for chronic persistent pain, for example, 'morning' noon 'and Z or three times a day before bedtime and Z or four times a day. Further, if necessary, it may be administered in combination with an analgesic aid such as an anti-inflammatory analgesic, an antidepressant, or an anticonvulsant.
- an analgesic aid such as an anti-inflammatory analgesic, an antidepressant, or an anticonvulsant.
- 1,2-Dicyano 4,5-getylbenzene (2.3 g, 12 mmol) was heated and stirred in 75% sulfuric acid (30 ml) at 150 ° C. for 3.5 hours.
- the reaction solution was poured into ice water, and the precipitated crystals were collected by filtration, washed with water, dissolved in a 10% aqueous sodium hydroxide solution, and insolubles were separated by filtration.
- the filtrate was acidified with concentrated hydrochloric acid, and the precipitated crystals were collected by filtration, washed with water and dried to obtain 1.5 g of 4,5-getylphthalic acid.
- reaction solution was concentrated under reduced pressure, the residue was dissolved in chloroform, and unreacted (carboethoxymethylene) triphenylphosphorane was adsorbed on silica gel to remove the crude product.
- the crude product was removed from methanol (40 ml), 15% The mixture was heated and stirred at 80 ° C for 4 hours in an aqueous K 2 CO solution (11 ml).
- reaction solution was concentrated under reduced pressure, water was added to the residue, extracted with getyl ether, the aqueous layer was made acidic with concentrated hydrochloric acid, and the precipitated crystals were collected by filtration, washed with water, and dried to give 2.36 g of the title compound. Obtained.
- the salt was formed with phenethylamine, and the salt was fractionally recrystallized using ethanol. The resulting salt was treated with 1N hydrochloric acid to give the title compound.
- Compound 5 is a novel compound to the applicant's knowledge.
- the method of preparing a neuropathic pain model animal was partially modified from that described in Pain, 87, 149 (2000). That is, under pentobarbital anesthesia, the right hind limb sciatic nerve of the mouse was exposed. Of the three peripheral branches of the mouse sciatic nerve, the tibial nerve (tibial nerve) was ligated all around, and the common peroneal nerve and sural nerve were not ligated. The left hind limb was used as an untreated control. One week after ligation, the feet of both hind limbs were stimulated 6 times with von Frey filament (0.6 g), and the scores of the 6 stimulations were totaled according to the following evaluation criteria to obtain a pain score. The maximum value of the pain score is 12. The escape response to the stimulus was evaluated based on the pain score. Then, after oral administration of the test substance, stimulation is performed in the same manner! ⁇ , the withdrawal response to the stimulus was evaluated.
- the response of the von Frey filament to the stimulus is determined when the mouse exerts no force to respond to the stimulus given by pressing the tip of the von Frey filament to the center of the foot so that the filament bends slightly.
- the score is 0, the score is 1 if you move your foot lightly to get away from the filament, and the score is 2 if you lick or shake vigorously.
- BALBZc male mice of 6 to 8 weeks of age were used in groups of 3 to 5 mice.
- the compound of Example 1 was orally administered at 30 or 100 mgZkg, and pain scores before and up to 3 hours after administration were measured over time.
- As a comparative drug 30 or 100 mgZkg of gyabapentin was orally administered, and the pain score was measured in the same manner. The results are shown in Figure 1.
- the analgesic effect of the compound of the present invention was expressed at a dose lower than that of giabapentin.
- the compound of the present invention showed an analgesic effect immediately after oral administration of 30 mgZkg at 5 minutes after administration.
- Test example 2 The analgesic effect on neuropathic pain of the compound produced in the above Synthesis Example was evaluated.
- the compound of the present invention has an analgesic effect on neuropathic pain.
- Example 1 The compound of Example 1 was optically resolved according to a conventional method to obtain a (+)-isomer and a (-)-isomer, respectively.
- Table 3 summarizes the analgesic effect of each compound evaluated in Test Examples 1 to 3 on neuropathic pain.
- ++ indicates that the analgesic effect was the same or more than that of the compound of Example 1
- + indicates that the compound had an analgesic effect but was lower than that of the compound of Example 1. The case where the analgesic effect was observed and helplessness was considered as one.
- Example Analgesic activity Discussion Analgesic activity Male example 1 + + Comparative example 1-Example 1 (one) + + Comparative example 2-Example Comparative example 3-
- the compound of the present invention has a strong analgesic effect at a dose (30 mg / kg) without lowering motor coordination, and it can be said that the analgesic effect occurs without affecting motor function.
- Each component is mixed with 50 mg of the compound of Example 1 per tablet such that 200 mg of lactose, 40 mg of crystalline cellulose, and 5 mg of magnesium stearate are obtained, and the mixture is press-molded by a conventional method using a tableting machine to obtain tablets.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pain & Pain Management (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Rheumatology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Indole Compounds (AREA)
Description






















Claims






















Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP05741421A EP1749817A4 (en) | 2004-05-24 | 2005-05-23 | COMPOSITION OF MEANS FOR COMBATING NEUROGENIC PAIN |
| JP2006513751A JPWO2005113501A1 (ja) | 2004-05-24 | 2005-05-23 | 神経因性疼痛制御剤組成物 |
| MXPA06013766A MXPA06013766A (es) | 2004-05-24 | 2005-05-23 | Composicion para el control del dolor neuropatico. |
| AU2005245292A AU2005245292A1 (en) | 2004-05-24 | 2005-05-23 | Neurogenic pain control agent composition |
| BRPI0511546-9A BRPI0511546A (pt) | 2004-05-24 | 2005-05-23 | composição para o controle de dor neuropática, uso e derivados de isoindolina |
| CA002563968A CA2563968A1 (en) | 2004-05-24 | 2005-05-23 | Neurogenic pain control agent composition |
| US11/587,367 US20080021042A1 (en) | 2004-05-24 | 2005-05-23 | Composition For Controlling Neuropathic Pain |
| IL178814A IL178814A0 (en) | 2004-05-24 | 2006-10-23 | Composition for controlling neuropathic pain |
| NO20064868A NO20064868L (no) | 2004-05-24 | 2006-10-25 | materiale for nevrogenisk smerteregulering |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004-153206 | 2004-05-24 | ||
| JP2004153206 | 2004-05-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005113501A1 true WO2005113501A1 (ja) | 2005-12-01 |
Family
ID=35428364
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2005/009361 WO2005113501A1 (ja) | 2004-05-24 | 2005-05-23 | 神経因性疼痛制御剤組成物 |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US20080021042A1 (ja) |
| EP (1) | EP1749817A4 (ja) |
| JP (1) | JPWO2005113501A1 (ja) |
| CN (1) | CN1956955A (ja) |
| AU (1) | AU2005245292A1 (ja) |
| BR (1) | BRPI0511546A (ja) |
| CA (1) | CA2563968A1 (ja) |
| EC (1) | ECSP067107A (ja) |
| IL (1) | IL178814A0 (ja) |
| MX (1) | MXPA06013766A (ja) |
| NO (1) | NO20064868L (ja) |
| RU (1) | RU2006145875A (ja) |
| TW (1) | TW200602318A (ja) |
| WO (1) | WO2005113501A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008008020A1 (en) * | 2006-07-12 | 2008-01-17 | Astrazeneca Ab | 3-oxoisoindoline-1-carboxamide derivatives as analgesic agents |
| US7521451B2 (en) * | 2002-11-26 | 2009-04-21 | Maruishi Pharmaceutical Co., Ltd. | Isoindoline derivative |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104803905B (zh) * | 2015-04-17 | 2017-10-10 | 复旦大学 | 一种合成异吲哚啉‑1‑酮衍生物的方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1984003089A1 (en) * | 1983-02-05 | 1984-08-16 | Takeda Chemical Industries Ltd | Condensed pyrrolinone derivatives |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2117740B1 (ja) * | 1970-12-14 | 1974-04-12 | Rhone Poulenc Sa | |
| US4590189A (en) * | 1982-04-02 | 1986-05-20 | Takeda Chemical Industries, Ltd. | Condensed pyrrolinone derivatives, their production and use |
| WO1999000121A1 (en) * | 1997-06-26 | 1999-01-07 | Eli Lilly And Company | Antithrombotic agents |
| AR042139A1 (es) * | 2002-11-26 | 2005-06-08 | Maruishi Pharma | Derivados de isoindolina, composicion farmaceutica que los contiene, y uso |
-
2005
- 2005-05-23 CN CNA2005800168374A patent/CN1956955A/zh active Pending
- 2005-05-23 WO PCT/JP2005/009361 patent/WO2005113501A1/ja active Application Filing
- 2005-05-23 US US11/587,367 patent/US20080021042A1/en not_active Abandoned
- 2005-05-23 JP JP2006513751A patent/JPWO2005113501A1/ja not_active Withdrawn
- 2005-05-23 AU AU2005245292A patent/AU2005245292A1/en not_active Abandoned
- 2005-05-23 EP EP05741421A patent/EP1749817A4/en not_active Withdrawn
- 2005-05-23 TW TW094116659A patent/TW200602318A/zh unknown
- 2005-05-23 CA CA002563968A patent/CA2563968A1/en not_active Abandoned
- 2005-05-23 MX MXPA06013766A patent/MXPA06013766A/es unknown
- 2005-05-23 BR BRPI0511546-9A patent/BRPI0511546A/pt not_active IP Right Cessation
- 2005-05-23 RU RU2006145875/04A patent/RU2006145875A/ru not_active Application Discontinuation
-
2006
- 2006-10-23 IL IL178814A patent/IL178814A0/en unknown
- 2006-10-25 NO NO20064868A patent/NO20064868L/no not_active Application Discontinuation
- 2006-12-21 EC EC2006007107A patent/ECSP067107A/es unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1984003089A1 (en) * | 1983-02-05 | 1984-08-16 | Takeda Chemical Industries Ltd | Condensed pyrrolinone derivatives |
Non-Patent Citations (2)
| Title |
|---|
| HAMAGUCHI S ET AL: "Mansei Totsu ni Taisuru Chintsu Hojoyaku.", vol. 203, no. 1, 5 October 2002 (2002-10-05), pages 59 - 63, XP008094693 * |
| See also references of EP1749817A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7521451B2 (en) * | 2002-11-26 | 2009-04-21 | Maruishi Pharmaceutical Co., Ltd. | Isoindoline derivative |
| WO2008008020A1 (en) * | 2006-07-12 | 2008-01-17 | Astrazeneca Ab | 3-oxoisoindoline-1-carboxamide derivatives as analgesic agents |
Also Published As
| Publication number | Publication date |
|---|---|
| MXPA06013766A (es) | 2007-02-08 |
| US20080021042A1 (en) | 2008-01-24 |
| TW200602318A (en) | 2006-01-16 |
| BRPI0511546A (pt) | 2008-01-02 |
| CN1956955A (zh) | 2007-05-02 |
| AU2005245292A1 (en) | 2005-12-01 |
| CA2563968A1 (en) | 2005-12-01 |
| RU2006145875A (ru) | 2008-06-27 |
| EP1749817A4 (en) | 2007-12-26 |
| IL178814A0 (en) | 2007-03-08 |
| EP1749817A1 (en) | 2007-02-07 |
| ECSP067107A (es) | 2007-02-28 |
| NO20064868L (no) | 2007-02-26 |
| JPWO2005113501A1 (ja) | 2008-03-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US4289781A (en) | Anti-psychotic phthalimidine derivatives | |
| WO1988002365A1 (en) | Cyclic amine derivatives | |
| KR20010033143A (ko) | 세로토닌 1a 수용체에 활성을 갖는 아릴피페라진 | |
| JPH08169884A (ja) | シクロプロパクロメンカルボン酸誘導体 | |
| JPH07501313A (ja) | キノロン誘導体 | |
| JP2005519073A (ja) | 睡眠標的モジュレーターを使用する睡眠障害の治療 | |
| JP4832897B2 (ja) | エステル誘導体及びその医薬用途 | |
| JPWO2005079845A1 (ja) | 片頭痛予防薬 | |
| WO2005113501A1 (ja) | 神経因性疼痛制御剤組成物 | |
| CA2345406A1 (en) | Analgesic | |
| JP2007297283A (ja) | 新規桂皮酸関連化合物 | |
| JP5004215B2 (ja) | 神経障害に伴う過活動膀胱の予防または治療用医薬組成物 | |
| JP3034604B2 (ja) | 治療用のベンズアザピン化合物 | |
| JP3158638B2 (ja) | 新規アミノフェノール誘導体及びその医薬用途 | |
| JP2006077006A (ja) | 加齢に伴う過活動膀胱治療薬 | |
| JP2009536623A (ja) | 眼圧降下剤としてのアブノーマル・カンナビジオール化合物 | |
| JPS59130868A (ja) | ピリジン誘導体 | |
| WO2006093226A1 (ja) | 止痒剤 | |
| JP4861828B2 (ja) | 酸性キノリン誘導体ならびに高血糖に関連する病状の予防および/または治療のためのその使用 | |
| JP3662566B2 (ja) | エステル化合物及びその医薬用途 | |
| WO2014063587A1 (zh) | 一类氟取代的环状胺类化合物及其制备方法、药物组合物和用途 | |
| KR102205619B1 (ko) | 신규한 인다졸 유도체 화합물 및 이를 포함하는 암의 예방, 개선 또는 치료용 약학 조성물 | |
| JP2004520348A (ja) | Ldl−受容体発現のインデューサーとしてのアリールピペリジン誘導体 | |
| KR20070018077A (ko) | 신경인성 동통제어 조성물 | |
| KR20070011551A (ko) | 알킬 옥스인돌의 피페라진 유도체 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 178814 Country of ref document: IL Ref document number: 2563968 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005245292 Country of ref document: AU Ref document number: 6250/DELNP/2006 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 550790 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005741421 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2005245292 Country of ref document: AU Date of ref document: 20050523 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12006502288 Country of ref document: PH |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005245292 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006513751 Country of ref document: JP Ref document number: 1020067024401 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2006/013766 Country of ref document: MX Ref document number: 200580016837.4 Country of ref document: CN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006/09965 Country of ref document: ZA Ref document number: 200609965 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 06124226 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200602107 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006145875 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005741421 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020067024401 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11587367 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0511546 Country of ref document: BR |
|
| WWP | Wipo information: published in national office |
Ref document number: 11587367 Country of ref document: US |