WO2004087628A1 - Ligand amino-alcool et son utilisation dans la preparation d'alcools tertiaires propargyliques et d'amines tertiaires par le biais d'une reaction d'addition eniantioselective - Google Patents
Ligand amino-alcool et son utilisation dans la preparation d'alcools tertiaires propargyliques et d'amines tertiaires par le biais d'une reaction d'addition eniantioselective Download PDFInfo
- Publication number
- WO2004087628A1 WO2004087628A1 PCT/CN2003/000462 CN0300462W WO2004087628A1 WO 2004087628 A1 WO2004087628 A1 WO 2004087628A1 CN 0300462 W CN0300462 W CN 0300462W WO 2004087628 A1 WO2004087628 A1 WO 2004087628A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- group
- benzyl
- alkyl
- fluorenyl
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/02—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions involving the formation of amino groups from compounds containing hydroxy groups or etherified or esterified hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/02—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C217/48—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being unsaturated and containing rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/78—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 2
- C07D239/80—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic System
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
Definitions
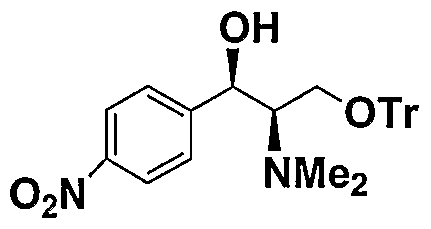
- p is hydrogen or an aryl-based protecting group
- the amino alcohol ligand is a compound having the following structure:
- R 1 , R 2 , R 4 , Z are as described above. It is particularly recommended that the chiral ligand be a compound of the following structure or its enantiomer:
- R is d ⁇ C 2 .
- Trialkylsilyl, ⁇ 2. Fluorenyl, ( 3 ⁇ ( ⁇ cycloalkyl or aryl, recommended aryl is phenyl, naphthyl, furan, thiophene, pyrrole;
- Saturated aliphatic hydrocarbon functional groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl and the like.
- the element is fluorine, chlorine, bromine or iodine.
- the alkoxycarbonyl group is preferably a carbon number of 1 to 20, and further preferably a carbon number of 1 to 4
Description




























Claims


















Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/551,770 US7439400B2 (en) | 2003-04-04 | 2003-06-16 | Amino alcohol ligand and its use in preparation of chiral proparglic tertiary alcohols and tertiary amines via enantioselective addition reaction |
| EP03816495A EP1614672B1 (en) | 2003-04-04 | 2003-06-16 | An amino alcohol ligand and its use in preparation of chiral proparglic tertiary alkohols and tertiary amines via enantioselective additon reaction |
| AU2003248217A AU2003248217A1 (en) | 2003-04-04 | 2003-06-16 | An amino alcohol ligand and its use in preparation of chiral proparglic tertiary alkohols and tertiary amines via enantioselective additon reaction |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN 03116192 CN1216036C (zh) | 2003-04-04 | 2003-04-04 | 手性氨基醇配体及其在端炔对亚氨的不对称加成中的应用 |
| CN03116192.8 | 2003-04-04 | ||
| CN03117026.9 | 2003-05-16 | ||
| CNB031170269A CN1331601C (zh) | 2003-05-16 | 2003-05-16 | 手性氨基醇配体应用于端炔对含氟烷基芳基酮的不对称加成的方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004087628A1 true WO2004087628A1 (fr) | 2004-10-14 |
Family
ID=33132421
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2003/000462 WO2004087628A1 (fr) | 2003-04-04 | 2003-06-16 | Ligand amino-alcool et son utilisation dans la preparation d'alcools tertiaires propargyliques et d'amines tertiaires par le biais d'une reaction d'addition eniantioselective |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US7439400B2 (zh) |
| EP (1) | EP1614672B1 (zh) |
| AU (1) | AU2003248217A1 (zh) |
| WO (1) | WO2004087628A1 (zh) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8115032B2 (en) | 2009-04-09 | 2012-02-14 | Lonza Ltd. | Process for the synthesis of a propargylic alcohol |
| US8283502B2 (en) | 2009-04-09 | 2012-10-09 | Lonza Ltd. | Autocatalytic process for the synthesis of chiral propargylic alcohols |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA200806473B (en) | 2008-01-31 | 2009-04-29 | Aptuit Laurus Pvt Ltd | An efficient process to induce enantioselectivity in procarbonyl compounds |
| US9073817B2 (en) * | 2008-01-31 | 2015-07-07 | Laurus Labs Private Limited | Efficient process to induce enantioselectivity in procarbonyl compounds |
| EP2448917A2 (de) | 2009-07-03 | 2012-05-09 | Archimica GmbH | Verfahren zur enantioselektiven addition von organometallischen kohlenstoffnukleophilen an trifluormethylketone und verwendung des verfahrens in der synthese von hiv reverse transcriptase inhibitoren |
| US8080655B2 (en) * | 2009-07-20 | 2011-12-20 | Apotex Pharmachem Inc. | Methods of making efavirenz and intermediates thereof |
| CN102180915B (zh) * | 2011-03-25 | 2014-08-13 | 南京理工大学 | D-果糖衍生的糖类β-氨基醇及其合成方法 |
| CN105330554B (zh) * | 2015-02-15 | 2017-07-11 | 上海迪赛诺药业股份有限公司 | 手性环丙基乙炔基叔醇类化合物的合成方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5202484A (en) * | 1989-10-20 | 1993-04-13 | Zambon Group S.P.A. | Process for the stereochemical inversion of (2s,3s)-2-amino-3-phenyl-1,3-propanediols into their (2r,3r) enantiomers |
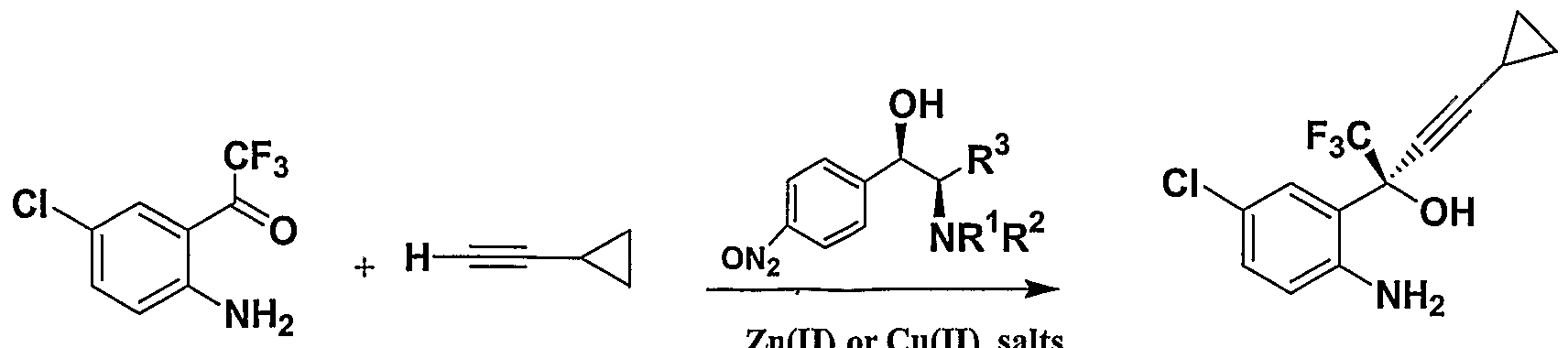
| CN1185146A (zh) * | 1995-05-25 | 1998-06-17 | 麦克公司 | (-)6-氯-4-环丙基乙炔基-4-三氟甲基-1,4-二氢-2h-3,1-苯并噁嗪-2-酮的不对称合成 |
| CN1314333A (zh) * | 2001-03-23 | 2001-09-26 | 中国科学院上海有机化学研究所 | 一种不对称合成二级丙炔醇类化合物的方法 |
| WO2001070707A2 (en) * | 2000-03-23 | 2001-09-27 | Bristol-Myers Squibb Pharma Company | Asymmetric synthesis of quinazolin-2-ones useful as hiv reverse transcriptase inhibitors |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5932726A (en) * | 1996-12-16 | 1999-08-03 | Dupont Pharmaceuticals Company | Asymmetric synthesis of benzoxazinones |
-
2003
- 2003-06-16 EP EP03816495A patent/EP1614672B1/en not_active Expired - Fee Related
- 2003-06-16 US US10/551,770 patent/US7439400B2/en not_active Expired - Fee Related
- 2003-06-16 AU AU2003248217A patent/AU2003248217A1/en not_active Abandoned
- 2003-06-16 WO PCT/CN2003/000462 patent/WO2004087628A1/zh not_active Application Discontinuation
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5202484A (en) * | 1989-10-20 | 1993-04-13 | Zambon Group S.P.A. | Process for the stereochemical inversion of (2s,3s)-2-amino-3-phenyl-1,3-propanediols into their (2r,3r) enantiomers |
| CN1185146A (zh) * | 1995-05-25 | 1998-06-17 | 麦克公司 | (-)6-氯-4-环丙基乙炔基-4-三氟甲基-1,4-二氢-2h-3,1-苯并噁嗪-2-酮的不对称合成 |
| WO2001070707A2 (en) * | 2000-03-23 | 2001-09-27 | Bristol-Myers Squibb Pharma Company | Asymmetric synthesis of quinazolin-2-ones useful as hiv reverse transcriptase inhibitors |
| CN1314333A (zh) * | 2001-03-23 | 2001-09-26 | 中国科学院上海有机化学研究所 | 一种不对称合成二级丙炔醇类化合物的方法 |
Non-Patent Citations (3)
| Title |
|---|
| J. W. CORBETT ET AL., ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, 1999, pages 2893 - 2897 |
| JIANG B. ET AL., CHEM. COMMUN., 2002, pages 1524 - 1525 |
| See also references of EP1614672A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8115032B2 (en) | 2009-04-09 | 2012-02-14 | Lonza Ltd. | Process for the synthesis of a propargylic alcohol |
| US8283502B2 (en) | 2009-04-09 | 2012-10-09 | Lonza Ltd. | Autocatalytic process for the synthesis of chiral propargylic alcohols |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2003248217A1 (en) | 2004-10-25 |
| EP1614672A4 (en) | 2007-04-04 |
| EP1614672A1 (en) | 2006-01-11 |
| EP1614672B1 (en) | 2010-09-22 |
| US20060217552A1 (en) | 2006-09-28 |
| US7439400B2 (en) | 2008-10-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA1043349A (en) | Therapeutic 1-hydroxyaryl-2-amidoalkylaminoethanol derivatives | |
| CZ404699A3 (cs) | Způsob výroby klíčového meziproduktu | |
| JP2024509797A (ja) | オメカムチブメカルビルの合成 | |
| JP5514218B2 (ja) | シナカルセトを調製するためのプロセス | |
| WO2004087628A1 (fr) | Ligand amino-alcool et son utilisation dans la preparation d'alcools tertiaires propargyliques et d'amines tertiaires par le biais d'une reaction d'addition eniantioselective | |
| JP2001519810A (ja) | 新規中間体を経由するベンゾオキサジノン類の不斉合成 | |
| CN103664816B (zh) | Hiv逆转录酶抑制剂依法韦伦类化合物的一锅法不对称合成工艺 | |
| WO2008035687A1 (fr) | PROCÉDÉ DE FABRICATION D'UN ACIDE α-AMINÉ CONTENANT DU PHOSPHORE ET INTERMÉDIAIRE DE FABRICATION | |
| CZ293246B6 (cs) | Způsob výroby 2-aryl-3-aryl-5-halogenpyridinů | |
| JP2001510174A (ja) | アミノアリールアセチレンを調製するためのプロセス | |
| CN110128345A (zh) | 一种吡唑酮衍生物的制备方法 | |
| EP0091851A1 (fr) | Procédé de préparation d'éthers d'aryle portant des substituants différents sur les deux noyaux aromatiques | |
| CN1216036C (zh) | 手性氨基醇配体及其在端炔对亚氨的不对称加成中的应用 | |
| CN1449865A (zh) | 手性氨基醇配体应用于端炔对含氟烷基芳基酮的不对称加成的方法 | |
| KR20140011383A (ko) | 치환된 디페닐 유도체 | |
| JP2001514648A (ja) | アリールスルホニルクロリドの製造のためのプロセス | |
| CN104774183A (zh) | 一种甲酰基瑞舒伐汀钙中间体的制备方法 | |
| Galons et al. | Organic reactions without solvent: Michael additions on an unsaturated sulfone and sulfoxide | |
| CN110713466B (zh) | 一种四氮唑导向的间位硝化的c-h活化新方法 | |
| JP2002030050A (ja) | アミン化合物、中間体、製造法および光学分割剤 | |
| JPH0841047A (ja) | 新規なフルオル化されたo−ジアミノベンゾ−1,4−ジオキセン | |
| CN112321451B (zh) | 一种用于制备盐酸西那卡塞药物中间体的方法 | |
| JP4120239B2 (ja) | 9,10−ジブロモアントラセン類の製造方法 | |
| EP1314727B1 (fr) | Synthèse directe de sels quaternaires de phenanthridinium | |
| JPS6343382B2 (zh) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SC SD SE SG SK SL TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006217552 Country of ref document: US Ref document number: 10551770 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1060/MUMNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003816495 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003816495 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10551770 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: JP |