WO2002006197A2 - Verfahren zur herstellung von bicyclischen 1,3-diketonen - Google Patents
Verfahren zur herstellung von bicyclischen 1,3-diketonen Download PDFInfo
- Publication number
- WO2002006197A2 WO2002006197A2 PCT/EP2001/007639 EP0107639W WO0206197A2 WO 2002006197 A2 WO2002006197 A2 WO 2002006197A2 EP 0107639 W EP0107639 W EP 0107639W WO 0206197 A2 WO0206197 A2 WO 0206197A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- ketone
- alkyl
- halogen
- bicyclic
- Prior art date
Links
- 0 CC(*)(C1*2)C(C)(*)C2C=C(*)C1O Chemical compound CC(*)(C1*2)C(C)(*)C2C=C(*)C1O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/42—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by hydrolysis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/45—Carboxylic acid nitriles having cyano groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C255/47—Carboxylic acid nitriles having cyano groups bound to carbon atoms of rings other than six-membered aromatic rings to carbon atoms of rings being part of condensed ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/27—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by oxidation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/27—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by oxidation
- C07C45/29—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by oxidation of hydroxy groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/27—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by oxidation
- C07C45/29—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by oxidation of hydroxy groups
- C07C45/298—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by oxidation of hydroxy groups with manganese derivatives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/673—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by change of size of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C49/00—Ketones; Ketenes; Dimeric ketenes; Ketonic chelates
- C07C49/385—Saturated compounds containing a keto group being part of a ring
- C07C49/417—Saturated compounds containing a keto group being part of a ring polycyclic
- C07C49/423—Saturated compounds containing a keto group being part of a ring polycyclic a keto group being part of a condensed ring system
- C07C49/427—Saturated compounds containing a keto group being part of a ring polycyclic a keto group being part of a condensed ring system having two rings
- C07C49/433—Saturated compounds containing a keto group being part of a ring polycyclic a keto group being part of a condensed ring system having two rings the condensed ring system containing seven carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/36—Systems containing two condensed rings the rings having more than two atoms in common
- C07C2602/44—Systems containing two condensed rings the rings having more than two atoms in common the bicyclo ring system containing eight carbon atoms
Definitions
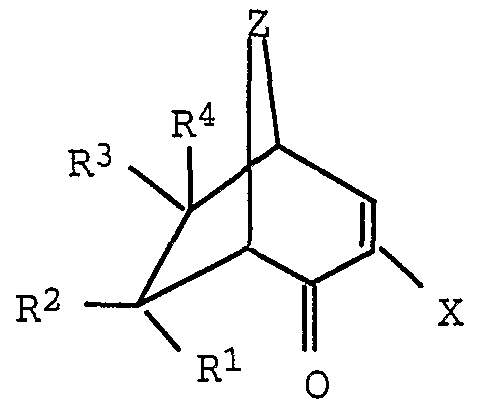
- the present invention relates to a process for the preparation of bicyclic 1,3-diketones of the formula I,
- R 1, R 2, R 3 and R 4 are hydrogen, C 4 -alkyl, C 4 alkoxycarbonyl, halogen, cyano, nitro, C 1 -C 4 -Alkylth.io, C ⁇ -C4 alkylsulfenyl or -CC 4 alkylsulfonyl and " 'j
- R 5 C 1 -C 4 alkyl and C 1 -C 4 alkylcarbonyl
- Bicyclic 1,3-diketones are valuable compounds that are considered
- JP 10 265 441 and JP 10 256 415 are based on very expensive norbornanone. The high feedstock costs do not make this process appear economical.
- R 1 , R 2 , R 3 and R 4 are hydrogen, C 1 -C 4 alkyl, C 1 -C 4 alkoxycarbonyl, halogen, cyano, nitro, C 1 -C 4 alkylthio, C 1 -C 4 alkylsulfenyl and C ⁇ -C 4 alkylsulfonyl and
- R 1 -R 4 and Z have the meaning given above and
- the allylic halogen of the compound of formula III can be oxidized to the unsaturated ketone of formula V.
- R l, R 2, R 3 and R 4 is hydrogen, Ci -C4 alkyl, Ci -C 4 alkoxy carbonyl, halogen, cyano, nitro, C 1 -C 4 alkylthio, C ⁇ -C 4 -alkylsulfenyl and -C-C 4 alkylsulfonyl and
- Bicyclic 1,3-diketones of the formula I can be present in keto-enol tautomers la and Ib.
- This invention also relates to a process for the preparation of tautomers of the formulas Ia and Ib.
- the process according to the invention for the preparation of compounds I essentially comprises one or more of process steps a) - e). Such reaction processes can also be considered which one or more of process steps a) e) are combined into one step (one-pot synthesis).
- inventive method also includes the synthesis of the other enantiomer.
- the implementation takes place, for example, under the following conditions:
- This stage runs over a dihalocarbene, preferably dichlorocarbene, which is produced from haloform and a base.
- Haloform preferably chloroform
- a base such as e.g. Alkali metal hydroxides, alkaline earth metal hydroxides, alkali metal alcoholates or alkali metal amides, preferably NaOH, KOH, sodium methylate and optionally a phase transfer catalyst such as e.g. Tetrabutylammonium chloride, trimethyl-benzyl-ammonium chloride or Aliquat 336 without solvent or in an inert hydrocarbon or halogenated hydrocarbon such as e.g. Hexane, heptane, petroleum ether, dichloromethane, carbon tetrachloride, dichloroethane or chlorobenzene and optionally water.
- a base such as e.g. Alkali metal hydroxides, alkaline earth metal hydroxides, alkali metal alcoholates or alkali metal amides, preferably NaOH, KOH, sodium methylate and optionally a phase transfer catalyst such as e.g. Tetrabuty
- the stoichiometric ratios are, for example, as follows: 1 equivalent of compound II is 1-4 equivalents. Haloform, if necessary .0.0001-0.10 equiv. Phase transfer catalyst and 1-4 equivalents of base used.
- the addition takes place, for example, in the following sequence: Compound II and haloform are optionally mixed with phase transfer catalyst in the inert solvent and the base is preferably added at 30 ° -60 ° C. at 0 ° C.-100 ° C.
- the processing takes place e.g. by stirring the product mixture into water and subsequent extraction and optionally distillation of the residue obtained under reduced pressure.
- the processing can also be carried out without purification by distilling off the solvent and direct use of the crude product in stage b).
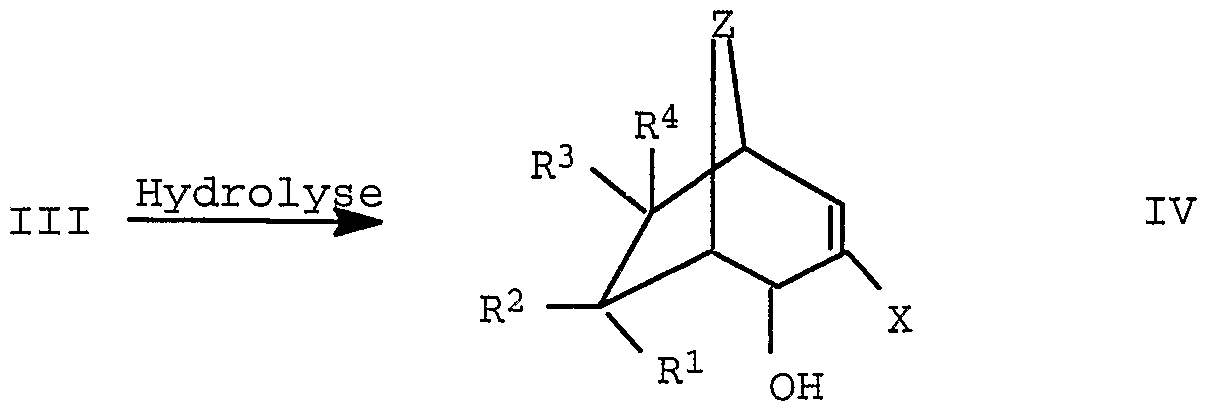
- the hydrolysis takes place e.g. under the following conditions: Water may be used as solvent, optionally with the addition of a phase transfer catalyst, tetrahydrofuran, dimethylformamide or dimethyl sulfoxide.
- the hydrolysis is e.g. with alkali metal hydroxides such as sodium hydroxide or potassium hydroxide or alkaline earth metal hydroxides such as e.g. Magnesium hydroxide or calcium hydroxide carried out, NaOH and KOH are preferred.
- reaction takes place at temperatures from 0 ° C. to the boiling point of the solvent, preferably at room temperature to the reflux temperature of the particular solvent.
- the stoichiometric ratios are as follows: 1 equivalent of compound III is 1-5 equiv. Base, preferably 1-1.5 equiv. Base used.
- the processing takes place e.g. by stirring into water and extraction with an organic solvent and subsequent fractional distillation. If water is used as the solvent, the extraction can take place directly.
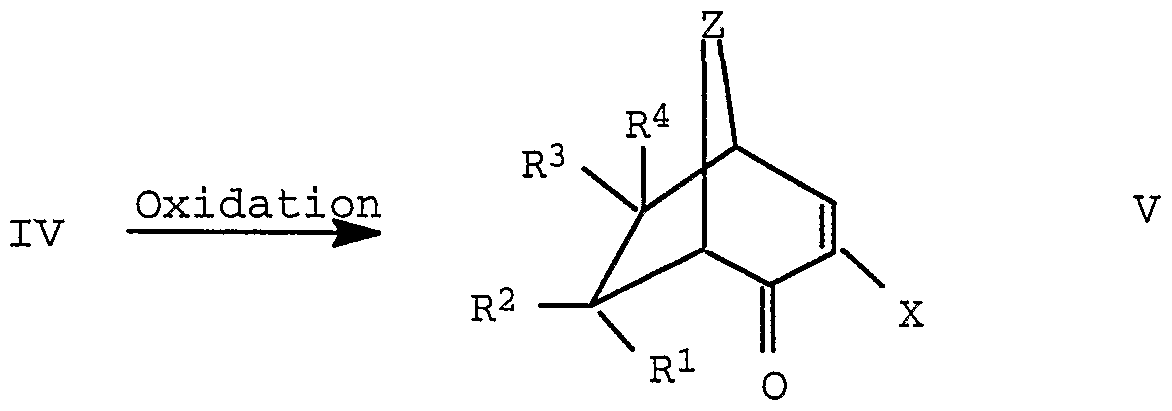
- the oxidation can be carried out, for example, with the following oxidizing agents: air, manganese dioxide, potassium permanganate, Jones reagent (chromic acid / sulfuric acid), dimethyl sulfoxide, optionally with additives such as NaHC0 3 , potassium hydrogen phosphate or potassium dihydrogen phosphate or activators such as oxalyl chloride, phosphorus trichloride, phosphorus oxychloride, phosphorus oxychloride , Thionyl chloride, acetyl chloride, acetic anhydride, sulfur trioxide-pyridine complex, tertiary amine oxides such as trimethylamine oxide or N-methylmorpholine-N-oxide, hydrogen peroxide, optionally with a catalyst such as sodium wolfmat, sodium hypochlorite, peracids such as perbenzoic acids , Peracetic acid or pertrifluoroacetic acid, bromine, chlorine, ruthenium tetraoxide, optionally cata
- a catalyst such as e.g. Sodium tungstate, air
- additives such as e.g. Potassium hydrogen phosphate / potassium dihydrogen phosphate or activators
- Oxalyl chloride, thionyl chloride, acetic anhydride or phosphorus trichloride such as e.g. Oxalyl chloride, thionyl
- inert hydrocarbons such as hexane, heptane or petroleum ether
- inert chlorinated hydrocarbons such as dichloromethane or chlorobenzene are suitable as solvents. If the oxidizing agent is a liquid, there is no need for additional solvents.
- the oxidation is e.g. carried out at a temperature of -60 ° C to the boiling point of the respective solvent.
- nucleophilic, negative charge s abil isdes ion are, for example cyanides, sulfites, C ⁇ -Cg-Alkylsulfinate or optionally substituted by C 1 -C 3 -alkyl, C 3 alkoxy, C 1 -C 3 alkylthio, C ⁇ -C 3 -Alkylsulfonyl, halogen, cyano, nitro or sulfonate substituted phenylsulfinate and mixtures thereof.
- Sources of cyanide can be, for example, hydrocyanic acid, alkali metal cyanides such as lithium cyanide, sodium cyanide or potassium cyanide or organic compounds such as trimethylsilyl cyanide or acetone cyanohydrin.
- Suitable sulfite sources are, for example, sulphurous acid, alkali metal sulfites such as sodium sulfite or potassium sulfite or alkali metal hydrogen sulfites such as sodium hydrogen sulfite.
- Suitable sulfinates are alkyl sulfinates such as sodium methyl sulfinate or aryl sulfinates such as sodium tolyl sulfinate.
- the base comes e.g. Nitrogen bases such as triethylamine, pyridine, diazabicycloundecane (DBU) or dimethylaminopyridine (DMAP) or alkali metal hydroxides such as lithium hydroxide, sodium hydroxide or potassium hydroxide, alkaline earth metal hydroxides such as barium hydroxide or calcium hydroxide, alkali metal carbonates such as sodium carbonate or potassium carbonate, alkali metal acetate or potassium hydrogen carbonate such as potassium hydrogen carbonate or sodium hydrogen carbonate, consideration.
- Nitrogen bases such as triethylamine, pyridine, diazabicycloundecane (DBU) or dimethylaminopyridine (DMAP) or alkali metal hydroxides such as lithium hydroxide, sodium hydroxide or potassium hydroxide, alkaline earth metal hydroxides such as barium hydroxide or calcium hydroxide, alkali metal carbonates such as sodium carbonate or potassium carbonate, alkali metal acetate or potassium hydrogen carbonate
- This reaction is a process for converting a 2-halo-alk-2-en-1-one into, for example, 3-cyano-alk-2-en-1-one, if stabilizing for the nucleophilic negative charge Ion Y "is the cyano group.
- Y can also stand for alkyl sulfinate, aryl sulfinate or sulfite.
- Reactions of 2-bromo-cycloalk-2-en-l-ones with NaCN or KCN are from Tetrahedron Lett. 1987, 28, 6485-6488; Tetrahedron 1987, 43, 5593-5604 known. Stage e)
- the reaction takes place, for example, under the following conditions: Alcohols such as methanol, ethanol, propanol or isopropanol, water, acetonitrile, dioxane, tetrahydrofuran, preferably methanol, ethanol and water.
- Alcohols such as methanol, ethanol, propanol or isopropanol, water, acetonitrile, dioxane, tetrahydrofuran, preferably methanol, ethanol and water.
- the hydrolysis can be carried out, for example, by alkali metal hydroxides such as lithium hydroxide, sodium hydroxide or potassium hydroxide, alkaline earth metal hydroxides such as calcium hydroxide or barium hydroxide, aluminum hydroxide, alkali metal carbonates such as sodium carbonate or potassium carbonate, alkali metal hydrogen carbonates such as sodium hydrogen carbonate or potassium hydrogen carbonate, acetates such as sodium acetate or nitrogen acetate or sodium acetate or sodium acetate or ammonia dissolved in water.
- alkali metal hydroxides such as lithium hydroxide, sodium hydroxide or potassium hydroxide
- alkaline earth metal hydroxides such as calcium hydroxide or barium hydroxide
- aluminum hydroxide alkali metal carbonates such as sodium carbonate or potassium carbonate
- alkali metal hydrogen carbonates such as sodium hydrogen carbonate or potassium hydrogen carbonate
- acetates such as sodium acetate or nitrogen acetate or sodium acetate or sodium acetate or ammonia dissolved in water
- Hydrochloric acid sulfuric acid, phosphoric acid, nitric acid, perchloric acid, chloric acid, hydrobromic acid and / or hydroiodic acid, organic acids such as e.g. Formic acid, acetic acid, propionic acid, butyric acid, stearic acid, oleic acid, benzoic acids and phenols.
- the reaction can be carried out at temperatures from -40 ° C. to 150 ° C., preferably at room temperature to reflux temperature of the particular solvent.
- the stoichiometric ratios are, for example, 1-5 equivalents, preferably 1-2 equivalents, of acid or base, per equivalent of compound VI.
- Steps d) and e) can also be carried out as a one-pot reaction with the stated amounts of reagents.
- the -H-NMR shows a mixture of about 60% product and 40% starting material.
- Variant F Phosphorus trichloride (1.91 g, 0.0139 mol) in dichloromethane (25 ml) was cooled to -30 ° C. and a solution of dimethyl sulfoxide (3.4 g, 0.044 mol) in dichloromethane (5 ml) was added dropwise. After 10 min, exo-3-chloro-bicyclo [3.2.1] oct-3-en-2-ol (2.0 g, 0.0126 mol) in dichloromethane (10 ml) was added and the mixture was stirred for a further 15 min. The mixture was allowed to warm up slowly to ambient temperature and the pH was adjusted to 1 using hydrochloric acid. The organic phase was separated, dried with sodium sulfate and concentrated.
- Triethlyamine (201 g, 1.99 mol) was stirred into cold hydrochloric acid, the organic phase was washed with water, dried over sodium sulfate and concentrated.
- Potassium hydroxide solution (0.5%, 20 mol) was added to 4-cyano-bicyclo [3.2.1] oct-3-en-2-one (0.02 g, 0.14 mmol) and the mixture was stirred at room temperature for 2 h. The mixture was acidified with hydrochloric acid and extracted with ethyl acetate. The organic phase was dried over sodium sulfate and concentrated.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description













Claims




Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR0112214-2A BR0112214A (pt) | 2000-07-06 | 2001-07-04 | Processo para preparar 1,3-dicetonas bicìclicas, e, cetona bicìclica |
| IL15341301A IL153413A0 (en) | 2000-07-06 | 2001-07-04 | Method for producing bicyclic 1,3-diketones |
| SK7-2003A SK72003A3 (en) | 2000-07-06 | 2001-07-04 | Method for producing bicyclic 1,3-diketones |
| AU2001281951A AU2001281951A1 (en) | 2000-07-06 | 2001-07-04 | Method for producing bicyclic 1,3-diketones |
| JP2002512104A JP2004504287A (ja) | 2000-07-06 | 2001-07-04 | 二環式1,3−ジケトンの製造方法 |
| NZ523835A NZ523835A (en) | 2000-07-06 | 2001-07-04 | Method for producing bicyclic 1,3-diketones |
| EA200300101A EA200300101A1 (ru) | 2000-07-06 | 2001-07-04 | Способ получения бициклических 1,3-дикетонов |
| MXPA02012323A MXPA02012323A (es) | 2000-07-06 | 2001-07-04 | Procedimiento para la obtencion de 1,3-dicetonas biciclicas. |
| CA002414895A CA2414895A1 (en) | 2000-07-06 | 2001-07-04 | Method for producing bicyclic 1,3-diketones |
| KR10-2003-7000120A KR20030013524A (ko) | 2000-07-06 | 2001-07-04 | 비시클릭 1,3-디케톤의 제조방법 |
| EP01960460A EP1296920A2 (de) | 2000-07-06 | 2001-07-04 | Verfahren zur herstellung von bicyclischen 1,3-diketonen |
| PL01360846A PL360846A1 (en) | 2000-07-06 | 2001-07-04 | Method for producing bicyclic 1,3-diketones |
| HU0300902A HUP0300902A3 (en) | 2000-07-06 | 2001-07-04 | Method for producing bicyclic 1,3-diketones |
| US10/332,034 US6815563B2 (en) | 2000-07-06 | 2001-07-04 | Method for producing bicyclic 1,3-diketones |
| BG107470A BG107470A (en) | 2000-07-06 | 2003-01-16 | Method for producing bicyclic 1,3-diketones |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10032396 | 2000-07-06 | ||
| DE10032396.0 | 2000-07-06 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2002006197A2 true WO2002006197A2 (de) | 2002-01-24 |
| WO2002006197A3 WO2002006197A3 (de) | 2002-05-02 |
Family
ID=7647695
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2001/007639 WO2002006197A2 (de) | 2000-07-06 | 2001-07-04 | Verfahren zur herstellung von bicyclischen 1,3-diketonen |
Country Status (20)
| Country | Link |
|---|---|
| US (1) | US6815563B2 (de) |
| EP (1) | EP1296920A2 (de) |
| JP (1) | JP2004504287A (de) |
| KR (1) | KR20030013524A (de) |
| CN (1) | CN1440376A (de) |
| AR (1) | AR028786A1 (de) |
| AU (1) | AU2001281951A1 (de) |
| BG (1) | BG107470A (de) |
| BR (1) | BR0112214A (de) |
| CA (1) | CA2414895A1 (de) |
| CZ (1) | CZ200314A3 (de) |
| EA (1) | EA200300101A1 (de) |
| HU (1) | HUP0300902A3 (de) |
| IL (1) | IL153413A0 (de) |
| MX (1) | MXPA02012323A (de) |
| NZ (1) | NZ523835A (de) |
| PL (1) | PL360846A1 (de) |
| SK (1) | SK72003A3 (de) |
| WO (1) | WO2002006197A2 (de) |
| ZA (1) | ZA200300988B (de) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009523330A (ja) * | 2005-10-31 | 2009-06-18 | マイクロソフト コーポレーション | モバイル装置とコンピューティング装置との間のボイスインスタントメッセージング |
| AU2005321407B2 (en) * | 2004-12-30 | 2011-04-07 | Enrico Garaci | Retrotransposon inhibition in therapy |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012033548A2 (en) | 2010-09-07 | 2012-03-15 | E. I. Du Pont De Nemours And Company | Herbicidal bis-nitrogen-containing oxo and sulfono heterocycles |
| MX2012002973A (es) | 2009-09-09 | 2012-04-19 | Du Pont | Derivados de pirimidona herbicidas. |
| CN105693569A (zh) * | 2016-01-06 | 2016-06-22 | 江苏理工学院 | 3-[4-(甲基磺酰基)-2-氯苯甲酰基]二环[3.2.1]-2,4-辛二酮的合成方法 |
| CN116514635A (zh) * | 2023-01-19 | 2023-08-01 | 山东潍坊润丰化工股份有限公司 | 3-氯双环[3.2.1]-3-辛烯-2-醇的制备方法 |
| CN116041167B (zh) * | 2023-01-28 | 2023-07-07 | 山东潍坊润丰化工股份有限公司 | 双环酮类化合物及其制备方法、双环[3.2.1]-3-辛烷-2,4-二酮的制备方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0338992A2 (de) * | 1988-04-18 | 1989-10-25 | Sandoz Ag | Substituierte Aryl- und Heteroaryl- Bicyclodionen |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1038093A (zh) | 1988-04-18 | 1989-12-20 | 山道士有限公司 | 对有机化合物和关于有机化合物的改进 |
| US5536703A (en) | 1995-01-13 | 1996-07-16 | Sandoz Ltd. | Herbicidal substituted benzoyl bicycloalkanediones |
| JP3712137B2 (ja) | 1995-08-08 | 2005-11-02 | 株式会社エス・ディー・エス バイオテック | 水田用除草剤組成物 |
| JP4159001B2 (ja) | 1997-03-25 | 2008-10-01 | 株式会社エス・ディー・エス バイオテック | メチレンノルカンファーの製造方法 |
| JP3929545B2 (ja) | 1997-03-25 | 2007-06-13 | 株式会社エス・ディー・エス バイオテック | 3−アセチル−シクロペンタンカルボン酸エステルの製造方法 |
-
2001
- 2001-07-04 MX MXPA02012323A patent/MXPA02012323A/es not_active Application Discontinuation
- 2001-07-04 NZ NZ523835A patent/NZ523835A/en unknown
- 2001-07-04 HU HU0300902A patent/HUP0300902A3/hu unknown
- 2001-07-04 EA EA200300101A patent/EA200300101A1/ru unknown
- 2001-07-04 IL IL15341301A patent/IL153413A0/xx unknown
- 2001-07-04 JP JP2002512104A patent/JP2004504287A/ja not_active Withdrawn
- 2001-07-04 PL PL01360846A patent/PL360846A1/xx unknown
- 2001-07-04 EP EP01960460A patent/EP1296920A2/de not_active Withdrawn
- 2001-07-04 WO PCT/EP2001/007639 patent/WO2002006197A2/de not_active Application Discontinuation
- 2001-07-04 US US10/332,034 patent/US6815563B2/en not_active Expired - Fee Related
- 2001-07-04 BR BR0112214-2A patent/BR0112214A/pt not_active IP Right Cessation
- 2001-07-04 AU AU2001281951A patent/AU2001281951A1/en not_active Abandoned
- 2001-07-04 CA CA002414895A patent/CA2414895A1/en not_active Abandoned
- 2001-07-04 CZ CZ200314A patent/CZ200314A3/cs unknown
- 2001-07-04 SK SK7-2003A patent/SK72003A3/sk unknown
- 2001-07-04 KR KR10-2003-7000120A patent/KR20030013524A/ko not_active Application Discontinuation
- 2001-07-04 CN CN01812350A patent/CN1440376A/zh active Pending
- 2001-07-05 AR ARP010103202A patent/AR028786A1/es unknown
-
2003
- 2003-01-16 BG BG107470A patent/BG107470A/xx unknown
- 2003-02-05 ZA ZA200300988A patent/ZA200300988B/en unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0338992A2 (de) * | 1988-04-18 | 1989-10-25 | Sandoz Ag | Substituierte Aryl- und Heteroaryl- Bicyclodionen |
Non-Patent Citations (2)
| Title |
|---|
| DUROCHIER J G ET AL: "L'EFFET REFLEXE DANS LE BICYCLO[3.2.1ÜOCTANEDIOL-2BETA,4BETA" CANADIAN JOURNAL OF CHEMISTRY - JOURNAL CANADIEN DE CHIMIE, XX, XX, Bd. 42, Nr. 2, 1964, Seiten 260-267, XP001022681 ISSN: 0008-4042 * |
| GLEITER R ET AL: "THE ORBITAL SEQUENCE IN CYCLIC 1,3-DIKETONES" TETRAHEDRON, ELSEVIER SCIENCE PUBLISHERS, AMSTERDAM, NL, Bd. 36, Nr. 5, 1980, Seiten 655-659, XP001026048 ISSN: 0040-4020 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2005321407B2 (en) * | 2004-12-30 | 2011-04-07 | Enrico Garaci | Retrotransposon inhibition in therapy |
| JP2009523330A (ja) * | 2005-10-31 | 2009-06-18 | マイクロソフト コーポレーション | モバイル装置とコンピューティング装置との間のボイスインスタントメッセージング |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20030013524A (ko) | 2003-02-14 |
| US6815563B2 (en) | 2004-11-09 |
| PL360846A1 (en) | 2004-09-20 |
| US20030187290A1 (en) | 2003-10-02 |
| IL153413A0 (en) | 2003-07-06 |
| WO2002006197A3 (de) | 2002-05-02 |
| ZA200300988B (en) | 2004-03-08 |
| EP1296920A2 (de) | 2003-04-02 |
| AU2001281951A1 (en) | 2002-01-30 |
| CN1440376A (zh) | 2003-09-03 |
| BR0112214A (pt) | 2003-05-06 |
| CZ200314A3 (cs) | 2003-08-13 |
| HUP0300902A2 (hu) | 2003-08-28 |
| SK72003A3 (en) | 2003-07-01 |
| JP2004504287A (ja) | 2004-02-12 |
| NZ523835A (en) | 2004-03-26 |
| MXPA02012323A (es) | 2003-04-25 |
| EA200300101A1 (ru) | 2003-06-26 |
| CA2414895A1 (en) | 2003-01-03 |
| AR028786A1 (es) | 2003-05-21 |
| HUP0300902A3 (en) | 2005-02-28 |
| BG107470A (en) | 2003-09-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0014409A1 (de) | Verfahren zur Herstellung von Alpha-Hydroxycarbonsäureamiden | |
| DE69627166T2 (de) | Verfahren zur herstellung von 2-chlor-5-chlomethyl-thiazol-derivaten | |
| DE19544800A1 (de) | Verfahren zur Herstellung von 1,3-Dimethyl-5-fluor-pyrazol-4-carboxaniliden | |
| EP0463464A1 (de) | Verfahren zur Herstellung von 2-Chlor-5-methyl-pyridin | |
| EP0556683B1 (de) | Verfahren zur Herstellung von 2-Chlor-5-methyl-pyridin | |
| WO2002006197A2 (de) | Verfahren zur herstellung von bicyclischen 1,3-diketonen | |
| EP0001980B1 (de) | Verfahren zur Herstellung von Halogenvinyl-gamma-butyrolactonen, sowie Zwischenprodukte und ihre Herstellung | |
| EP0132733A2 (de) | Neue Fluorpivalsäurefluoride und Verfahren zu ihrer Herstellung | |
| EP0454623B1 (de) | Verfahren zur Herstellung von linearen 1,3-Diketonen | |
| EP0439745B1 (de) | Verfahren zur Herstellung von 2-Chlor-5-methyl-pyridin | |
| EP0089485B1 (de) | Verfahren zur Herstellung von 5-Chlor-1H-tetrazol-1-carbonsäureestern sowie Verfahren zur Herstellung der erforderlichen Dichlorisonitril-carbonsäureester | |
| CH620666A5 (de) | ||
| EP0632025B1 (de) | Verfahren zur Herstellung von 2-Chlor-5-chlormethylpyridin | |
| EP0048370A1 (de) | Verfahren zur Herstellung von trans-3-(Z-2-Chlor-2-aryl-vinyl)-2,2-dimethyl-cyclopropan-1-carbon-säure-derivaten, neue Zwischenprodukte hierfür, Verfahren zu deren Herstellung und Verwendung von Zwischenprodukten in Schädlingsbekämpfungsmitteln | |
| EP0290903B1 (de) | Beta-Fluoracyl-beta-halogenvinylalkylether | |
| EP0103749B1 (de) | Dialkoxymethyl-butyrolactone, Verfahren zu ihrer Herstellung, Zwischenprodukte dafür und ihre Verwendung | |
| EP0002477B1 (de) | Verfahren zur Herstellung von 1,1-Dihalogen-4-methyl-1,3-pentadienen | |
| EP0224256A1 (de) | 4-Alkoxy-2-oxo-pyrrolidin-1-yl-essigsäurealkylester | |
| EP0035754B1 (de) | Verfahren zur Herstellung von 1,1-Dichlor-alkenen | |
| DD209443A5 (de) | Verfahren zur herstellung von acylaminoderivaten von 1-(aryl- oder subst.-aryl)amino-1-thioalkancarboxysaeuren | |
| DE1963061C3 (de) | Verfahren zur Herstellung von Carbamoylformhydroxamoylchloridverbindungen | |
| EP1207147A1 (de) | Verfahren zur Herstellung von Orthokohlensäureester | |
| WO1998040363A1 (de) | Verfahren zur herstellung von chinazol-4-onen | |
| DE1468624B2 (de) | Verfahren zur herstellung von beta-cyanketonen | |
| DE2443742A1 (de) | Cyclopenten-derivate und zwischenprodukte und verfahren zu ihrer herstellung |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 153413 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2002/012323 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001960460 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2002 512104 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001281951 Country of ref document: AU Ref document number: 2414895 Country of ref document: CA Ref document number: 72003 Country of ref document: SK Ref document number: PV2003-14 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020037000120 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 018123503 Country of ref document: CN Ref document number: 10332034 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 10747001 Country of ref document: BG Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200300069 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 164/CHENP/2003 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 523835 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200300101 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003/00988 Country of ref document: ZA Ref document number: 200300988 Country of ref document: ZA |
|
| ENP | Entry into the national phase |
Ref document number: 20030127 Country of ref document: UZ Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020037000120 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001960460 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2003-14 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 523835 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 523835 Country of ref document: NZ |
|
| WWR | Wipo information: refused in national office |
Ref document number: PV2003-14 Country of ref document: CZ |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2001960460 Country of ref document: EP |