WO1998041494A1 - Verfahren zur herstellung von n-butyraldehyd und/oder n-butanol - Google Patents
Verfahren zur herstellung von n-butyraldehyd und/oder n-butanol Download PDFInfo
- Publication number
- WO1998041494A1 WO1998041494A1 PCT/EP1998/001324 EP9801324W WO9841494A1 WO 1998041494 A1 WO1998041494 A1 WO 1998041494A1 EP 9801324 W EP9801324 W EP 9801324W WO 9841494 A1 WO9841494 A1 WO 9841494A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- reaction
- catalyst
- butadiene
- partial
- liquid phase
- Prior art date
Links
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 title claims abstract description 218
- ZTQSAGDEMFDKMZ-UHFFFAOYSA-N Butyraldehyde Chemical compound CCCC=O ZTQSAGDEMFDKMZ-UHFFFAOYSA-N 0.000 title claims abstract description 144
- 238000004519 manufacturing process Methods 0.000 title abstract description 17
- 238000006243 chemical reaction Methods 0.000 claims abstract description 308
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 claims abstract description 256
- 230000036961 partial effect Effects 0.000 claims abstract description 172
- 239000003054 catalyst Substances 0.000 claims abstract description 142
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 claims abstract description 75
- 239000003446 ligand Substances 0.000 claims abstract description 70
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 70
- 239000000203 mixture Substances 0.000 claims abstract description 56
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 45
- 239000001257 hydrogen Substances 0.000 claims abstract description 44
- 239000007791 liquid phase Substances 0.000 claims abstract description 41
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims abstract description 40
- 229930195733 hydrocarbon Natural products 0.000 claims abstract description 33
- 150000002430 hydrocarbons Chemical class 0.000 claims abstract description 33
- 239000004215 Carbon black (E152) Substances 0.000 claims abstract description 28
- 229910052751 metal Inorganic materials 0.000 claims abstract description 28
- 239000002184 metal Substances 0.000 claims abstract description 28
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims abstract description 20
- 230000000737 periodic effect Effects 0.000 claims abstract description 20
- 239000007848 Bronsted acid Substances 0.000 claims abstract description 14
- MQIKJSYMMJWAMP-UHFFFAOYSA-N dicobalt octacarbonyl Chemical group [Co+2].[Co+2].[O+]#[C-].[O+]#[C-].[O+]#[C-].[O+]#[C-].[O+]#[C-].[O+]#[C-].[O+]#[C-].[O+]#[C-] MQIKJSYMMJWAMP-UHFFFAOYSA-N 0.000 claims abstract description 12
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 claims abstract description 7
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims abstract description 6
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 5
- 238000000034 method Methods 0.000 claims description 178
- 230000008569 process Effects 0.000 claims description 129
- 150000001241 acetals Chemical class 0.000 claims description 78
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 69
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 claims description 64
- 239000002815 homogeneous catalyst Substances 0.000 claims description 44
- -1 platinum metals Chemical class 0.000 claims description 42
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 38
- 229910052723 transition metal Inorganic materials 0.000 claims description 38
- 229910052763 palladium Inorganic materials 0.000 claims description 35
- 229910000073 phosphorus hydride Inorganic materials 0.000 claims description 35
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 29
- 239000002638 heterogeneous catalyst Substances 0.000 claims description 28
- 238000006317 isomerization reaction Methods 0.000 claims description 23
- 239000010457 zeolite Substances 0.000 claims description 22
- 230000002378 acidificating effect Effects 0.000 claims description 21
- 150000002500 ions Chemical class 0.000 claims description 21
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 20
- 229910052759 nickel Inorganic materials 0.000 claims description 19
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 19
- 239000012429 reaction media Substances 0.000 claims description 19
- 238000002360 preparation method Methods 0.000 claims description 16
- 125000000217 alkyl group Chemical group 0.000 claims description 15
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 claims description 14
- 150000003624 transition metals Chemical class 0.000 claims description 14
- 125000003118 aryl group Chemical group 0.000 claims description 13
- 229910052697 platinum Inorganic materials 0.000 claims description 12
- 229910052703 rhodium Inorganic materials 0.000 claims description 12
- 239000010948 rhodium Substances 0.000 claims description 12
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 claims description 12
- 229910021536 Zeolite Inorganic materials 0.000 claims description 11
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 claims description 11
- 229910052741 iridium Inorganic materials 0.000 claims description 10
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 claims description 9
- 239000000377 silicon dioxide Substances 0.000 claims description 9
- 235000012239 silicon dioxide Nutrition 0.000 claims description 9
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 8
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 claims description 8
- MCMNRKCIXSYSNV-UHFFFAOYSA-N ZrO2 Inorganic materials O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 claims description 7
- 239000012876 carrier material Substances 0.000 claims description 7
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 claims description 7
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 claims description 7
- 229910052698 phosphorus Inorganic materials 0.000 claims description 6
- 239000011574 phosphorus Substances 0.000 claims description 6
- 150000003839 salts Chemical class 0.000 claims description 6
- 238000000926 separation method Methods 0.000 claims description 6
- 125000003342 alkenyl group Chemical group 0.000 claims description 4
- 239000004408 titanium dioxide Substances 0.000 claims description 4
- 125000005595 acetylacetonate group Chemical group 0.000 claims description 3
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 claims description 3
- 150000001412 amines Chemical class 0.000 claims description 3
- 229910052736 halogen Inorganic materials 0.000 claims description 3
- 150000002367 halogens Chemical class 0.000 claims description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 2
- OJMIONKXNSYLSR-UHFFFAOYSA-N phosphorous acid Chemical compound OP(O)O OJMIONKXNSYLSR-UHFFFAOYSA-N 0.000 claims 4
- VMJNTFXCTXAXTC-UHFFFAOYSA-N 2,2-difluoro-1,3-benzodioxole-5-carbonitrile Chemical group C1=C(C#N)C=C2OC(F)(F)OC2=C1 VMJNTFXCTXAXTC-UHFFFAOYSA-N 0.000 claims 1
- BPQQTUXANYXVAA-UHFFFAOYSA-N Orthosilicate Chemical compound [O-][Si]([O-])([O-])[O-] BPQQTUXANYXVAA-UHFFFAOYSA-N 0.000 claims 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 abstract description 8
- 229910052757 nitrogen Inorganic materials 0.000 abstract description 4
- 125000002777 acetyl group Chemical class [H]C([H])([H])C(*)=O 0.000 abstract 3
- 230000007062 hydrolysis Effects 0.000 description 49
- 238000006460 hydrolysis reaction Methods 0.000 description 49
- 239000011541 reaction mixture Substances 0.000 description 47
- 238000005984 hydrogenation reaction Methods 0.000 description 39
- 239000007795 chemical reaction product Substances 0.000 description 27
- 230000015572 biosynthetic process Effects 0.000 description 26
- 239000002904 solvent Substances 0.000 description 25
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 24
- 238000004821 distillation Methods 0.000 description 23
- 239000006227 byproduct Substances 0.000 description 21
- 150000002739 metals Chemical class 0.000 description 20
- 239000007788 liquid Substances 0.000 description 19
- 239000000047 product Substances 0.000 description 19
- 150000001298 alcohols Chemical class 0.000 description 18
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 17
- 150000001875 compounds Chemical class 0.000 description 15
- 241000196324 Embryophyta Species 0.000 description 14
- 238000006359 acetalization reaction Methods 0.000 description 14
- 238000004817 gas chromatography Methods 0.000 description 14
- 229910052707 ruthenium Inorganic materials 0.000 description 14
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 13
- 239000000463 material Substances 0.000 description 13
- WQRBJFZKPWPIJD-UHFFFAOYSA-N 1,1-dibutoxybutane Chemical compound CCCCOC(CCC)OCCCC WQRBJFZKPWPIJD-UHFFFAOYSA-N 0.000 description 12
- CQJQTXVQMQMNBT-UHFFFAOYSA-N 1-but-3-en-2-yloxybutane Chemical compound CCCCOC(C)C=C CQJQTXVQMQMNBT-UHFFFAOYSA-N 0.000 description 12
- 230000035484 reaction time Effects 0.000 description 12
- 239000000243 solution Substances 0.000 description 12
- ROFVEXUMMXZLPA-UHFFFAOYSA-N Bipyridyl Chemical compound N1=CC=CC=C1C1=CC=CC=N1 ROFVEXUMMXZLPA-UHFFFAOYSA-N 0.000 description 11
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical compound CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 10
- 229910017052 cobalt Inorganic materials 0.000 description 10
- 239000010941 cobalt Substances 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- 239000007789 gas Substances 0.000 description 10
- 239000002253 acid Substances 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 8
- 238000009835 boiling Methods 0.000 description 8
- 229910052799 carbon Inorganic materials 0.000 description 8
- 238000007037 hydroformylation reaction Methods 0.000 description 8
- GHVNFZFCNZKVNT-UHFFFAOYSA-N Decanoic acid Natural products CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 7
- 239000002841 Lewis acid Substances 0.000 description 7
- DGEZNRSVGBDHLK-UHFFFAOYSA-N [1,10]phenanthroline Chemical compound C1=CN=C2C3=NC=CC=C3C=CC2=C1 DGEZNRSVGBDHLK-UHFFFAOYSA-N 0.000 description 7
- 150000007942 carboxylates Chemical class 0.000 description 7
- 150000002084 enol ethers Chemical class 0.000 description 7
- 150000002170 ethers Chemical class 0.000 description 7
- 239000011521 glass Substances 0.000 description 7
- 238000007210 heterogeneous catalysis Methods 0.000 description 7
- 150000002902 organometallic compounds Chemical class 0.000 description 7
- 229940031826 phenolate Drugs 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- GYSCBCSGKXNZRH-UHFFFAOYSA-N 1-benzothiophene-2-carboxamide Chemical compound C1=CC=C2SC(C(=O)N)=CC2=C1 GYSCBCSGKXNZRH-UHFFFAOYSA-N 0.000 description 6
- MLRCQIICAYVJHD-UHFFFAOYSA-N 1-but-1-enoxybut-1-ene Chemical compound CCC=COC=CCC MLRCQIICAYVJHD-UHFFFAOYSA-N 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 6
- 150000007513 acids Chemical class 0.000 description 6
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 6
- 229920001429 chelating resin Polymers 0.000 description 6
- ORTQZVOHEJQUHG-UHFFFAOYSA-L copper(II) chloride Chemical compound Cl[Cu]Cl ORTQZVOHEJQUHG-UHFFFAOYSA-L 0.000 description 6
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Chemical group C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 6
- 238000011065 in-situ storage Methods 0.000 description 6
- 150000007517 lewis acids Chemical class 0.000 description 6
- 150000003003 phosphines Chemical class 0.000 description 6
- 150000003254 radicals Chemical class 0.000 description 6
- GCRUYRFHWGPWHJ-UHFFFAOYSA-N 1-but-1-enoxybutane Chemical class CCCCOC=CCC GCRUYRFHWGPWHJ-UHFFFAOYSA-N 0.000 description 5
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 5
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 5
- IAQRGUVFOMOMEM-UHFFFAOYSA-N but-2-ene Chemical class CC=CC IAQRGUVFOMOMEM-UHFFFAOYSA-N 0.000 description 5
- 150000002823 nitrates Chemical class 0.000 description 5
- 150000002894 organic compounds Chemical class 0.000 description 5
- 229910052762 osmium Inorganic materials 0.000 description 5
- SYQBFIAQOQZEGI-UHFFFAOYSA-N osmium atom Chemical compound [Os] SYQBFIAQOQZEGI-UHFFFAOYSA-N 0.000 description 5
- ISWSIDIOOBJBQZ-UHFFFAOYSA-M phenolate Chemical compound [O-]C1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-M 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 239000002994 raw material Substances 0.000 description 5
- 239000000376 reactant Substances 0.000 description 5
- 238000004064 recycling Methods 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 125000001424 substituent group Chemical group 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- 230000007704 transition Effects 0.000 description 5
- HOXGZVUCAYFWGR-KQQUZDAGSA-N (3e,5e)-octa-1,3,5-triene Chemical compound CC\C=C\C=C\C=C HOXGZVUCAYFWGR-KQQUZDAGSA-N 0.000 description 4
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 4
- SDRZFSPCVYEJTP-UHFFFAOYSA-N 1-ethenylcyclohexene Chemical compound C=CC1=CCCCC1 SDRZFSPCVYEJTP-UHFFFAOYSA-N 0.000 description 4
- ATVJXMYDOSMEPO-UHFFFAOYSA-N 3-prop-2-enoxyprop-1-ene Chemical compound C=CCOCC=C ATVJXMYDOSMEPO-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical group C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical group C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical group C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical group C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- QQONPFPTGQHPMA-UHFFFAOYSA-N Propene Chemical compound CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 4
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical group C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical group N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 4
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 4
- 238000006555 catalytic reaction Methods 0.000 description 4
- 239000010949 copper Substances 0.000 description 4
- 238000007172 homogeneous catalysis Methods 0.000 description 4
- 125000001145 hydrido group Chemical group *[H] 0.000 description 4
- 239000012299 nitrogen atmosphere Substances 0.000 description 4
- 150000002892 organic cations Chemical class 0.000 description 4
- 150000004707 phenolate Chemical class 0.000 description 4
- 238000001556 precipitation Methods 0.000 description 4
- 229920005989 resin Polymers 0.000 description 4
- 239000011347 resin Substances 0.000 description 4
- 229910052711 selenium Inorganic materials 0.000 description 4
- 239000011669 selenium Substances 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 125000001273 sulfonato group Chemical group [O-]S(*)(=O)=O 0.000 description 4
- RWRDLPDLKQPQOW-UHFFFAOYSA-N tetrahydropyrrole Natural products C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 4
- HGBOYTHUEUWSSQ-UHFFFAOYSA-N valeric aldehyde Natural products CCCCC=O HGBOYTHUEUWSSQ-UHFFFAOYSA-N 0.000 description 4
- 238000010626 work up procedure Methods 0.000 description 4
- DVSSQJKZHTWMOS-UHFFFAOYSA-N 1-but-2-enoxybutane Chemical compound CCCCOCC=CC DVSSQJKZHTWMOS-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 229910021592 Copper(II) chloride Inorganic materials 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 150000001299 aldehydes Chemical class 0.000 description 3
- 125000001931 aliphatic group Chemical group 0.000 description 3
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 3
- 229910052782 aluminium Inorganic materials 0.000 description 3
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 3
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 3
- KTUQUZJOVNIKNZ-UHFFFAOYSA-N butan-1-ol;hydrate Chemical compound O.CCCCO KTUQUZJOVNIKNZ-UHFFFAOYSA-N 0.000 description 3
- 150000001768 cations Chemical class 0.000 description 3
- 229910052802 copper Inorganic materials 0.000 description 3
- XTVVROIMIGLXTD-UHFFFAOYSA-N copper(II) nitrate Chemical compound [Cu+2].[O-][N+]([O-])=O.[O-][N+]([O-])=O XTVVROIMIGLXTD-UHFFFAOYSA-N 0.000 description 3
- 125000004093 cyano group Chemical group *C#N 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- BOUYBUIVMHNXQB-UHFFFAOYSA-N dicyclohexyl(2-dicyclohexylphosphanylethyl)phosphane Chemical compound C1CCCCC1P(C1CCCCC1)CCP(C1CCCCC1)C1CCCCC1 BOUYBUIVMHNXQB-UHFFFAOYSA-N 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 150000004820 halides Chemical class 0.000 description 3
- 150000002431 hydrogen Chemical class 0.000 description 3
- 238000005470 impregnation Methods 0.000 description 3
- 150000002736 metal compounds Chemical class 0.000 description 3
- 150000002825 nitriles Chemical class 0.000 description 3
- JKDRQYIYVJVOPF-FDGPNNRMSA-L palladium(ii) acetylacetonate Chemical compound [Pd+2].C\C([O-])=C\C(C)=O.C\C([O-])=C\C(C)=O JKDRQYIYVJVOPF-FDGPNNRMSA-L 0.000 description 3
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 3
- 229920001568 phenolic resin Polymers 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 3
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 3
- 150000003457 sulfones Chemical class 0.000 description 3
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 3
- 150000005045 1,10-phenanthrolines Chemical class 0.000 description 2
- CHRJZRDFSQHIFI-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;styrene Chemical compound C=CC1=CC=CC=C1.C=CC1=CC=CC=C1C=C CHRJZRDFSQHIFI-UHFFFAOYSA-N 0.000 description 2
- LVEYOSJUKRVCCF-UHFFFAOYSA-N 1,3-bis(diphenylphosphino)propane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCP(C=1C=CC=CC=1)C1=CC=CC=C1 LVEYOSJUKRVCCF-UHFFFAOYSA-N 0.000 description 2
- OCJBOOLMMGQPQU-UHFFFAOYSA-N 1,4-dichlorobenzene Chemical compound ClC1=CC=C(Cl)C=C1 OCJBOOLMMGQPQU-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N 1H-pyrrole Natural products C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- OBETXYAYXDNJHR-UHFFFAOYSA-N 2-Ethylhexanoic acid Chemical compound CCCCC(CC)C(O)=O OBETXYAYXDNJHR-UHFFFAOYSA-N 0.000 description 2
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 2
- RXGPYPPCEXISOV-UHFFFAOYSA-N 2-propylheptanoic acid Chemical compound CCCCCC(C(O)=O)CCC RXGPYPPCEXISOV-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 2
- JPVYNHNXODAKFH-UHFFFAOYSA-N Cu2+ Chemical class [Cu+2] JPVYNHNXODAKFH-UHFFFAOYSA-N 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical group C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 2
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical compound CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 description 2
- AMIMRNSIRUDHCM-UHFFFAOYSA-N Isopropylaldehyde Chemical compound CC(C)C=O AMIMRNSIRUDHCM-UHFFFAOYSA-N 0.000 description 2
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 2
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical group C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 2
- WYNCHZVNFNFDNH-UHFFFAOYSA-N Oxazolidine Chemical group C1COCN1 WYNCHZVNFNFDNH-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Chemical group C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical group C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical group C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 2
- 229910021604 Rhodium(III) chloride Inorganic materials 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 150000001242 acetic acid derivatives Chemical class 0.000 description 2
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Chemical compound CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 description 2
- 238000007259 addition reaction Methods 0.000 description 2
- 150000001340 alkali metals Chemical class 0.000 description 2
- 150000001336 alkenes Chemical class 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 125000006267 biphenyl group Chemical group 0.000 description 2
- SHZIWNPUGXLXDT-UHFFFAOYSA-N caproic acid ethyl ester Natural products CCCCCC(=O)OCC SHZIWNPUGXLXDT-UHFFFAOYSA-N 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000003638 chemical reducing agent Substances 0.000 description 2
- 150000001805 chlorine compounds Chemical class 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 230000000536 complexating effect Effects 0.000 description 2
- JGDFBJMWFLXCLJ-UHFFFAOYSA-N copper chromite Chemical compound [Cu]=O.[Cu]=O.O=[Cr]O[Cr]=O JGDFBJMWFLXCLJ-UHFFFAOYSA-N 0.000 description 2
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical class [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 2
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 2
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 229940117389 dichlorobenzene Drugs 0.000 description 2
- XNMQEEKYCVKGBD-UHFFFAOYSA-N dimethylacetylene Natural products CC#CC XNMQEEKYCVKGBD-UHFFFAOYSA-N 0.000 description 2
- 150000002028 dodecanols Chemical class 0.000 description 2
- 238000004508 fractional distillation Methods 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Chemical group CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 2
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Chemical group C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 2
- 229910021432 inorganic complex Inorganic materials 0.000 description 2
- 229910003480 inorganic solid Inorganic materials 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- NNPPMTNAJDCUHE-UHFFFAOYSA-N isobutane Chemical compound CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- 229910052750 molybdenum Inorganic materials 0.000 description 2
- 239000011733 molybdenum Substances 0.000 description 2
- 229910052901 montmorillonite Inorganic materials 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 150000002815 nickel Chemical class 0.000 description 2
- NUJGJRNETVAIRJ-UHFFFAOYSA-N octanal Chemical compound CCCCCCCC=O NUJGJRNETVAIRJ-UHFFFAOYSA-N 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 150000002989 phenols Chemical class 0.000 description 2
- 235000021317 phosphate Nutrition 0.000 description 2
- AQSJGOWTSHOLKH-UHFFFAOYSA-N phosphite(3-) Chemical class [O-]P([O-])[O-] AQSJGOWTSHOLKH-UHFFFAOYSA-N 0.000 description 2
- 239000004014 plasticizer Substances 0.000 description 2
- 229920001467 poly(styrenesulfonates) Polymers 0.000 description 2
- 229920001515 polyalkylene glycol Polymers 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 150000003138 primary alcohols Chemical class 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical group C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Chemical group COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 239000012495 reaction gas Substances 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 229910052702 rhenium Inorganic materials 0.000 description 2
- WUAPFZMCVAUBPE-UHFFFAOYSA-N rhenium atom Chemical compound [Re] WUAPFZMCVAUBPE-UHFFFAOYSA-N 0.000 description 2
- SONJTKJMTWTJCT-UHFFFAOYSA-K rhodium(iii) chloride Chemical compound [Cl-].[Cl-].[Cl-].[Rh+3] SONJTKJMTWTJCT-UHFFFAOYSA-K 0.000 description 2
- YBCAZPLXEGKKFM-UHFFFAOYSA-K ruthenium(iii) chloride Chemical compound [Cl-].[Cl-].[Cl-].[Ru+3] YBCAZPLXEGKKFM-UHFFFAOYSA-K 0.000 description 2
- 239000012266 salt solution Substances 0.000 description 2
- 150000004760 silicates Chemical class 0.000 description 2
- 150000003462 sulfoxides Chemical class 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 238000005979 thermal decomposition reaction Methods 0.000 description 2
- 125000005310 triazolidinyl group Chemical group N1(NNCC1)* 0.000 description 2
- 239000013638 trimer Substances 0.000 description 2
- 238000005829 trimerization reaction Methods 0.000 description 2
- YWWDBCBWQNCYNR-UHFFFAOYSA-N trimethylphosphine Chemical compound CP(C)C YWWDBCBWQNCYNR-UHFFFAOYSA-N 0.000 description 2
- HVLLSGMXQDNUAL-UHFFFAOYSA-N triphenyl phosphite Chemical compound C=1C=CC=CC=1OP(OC=1C=CC=CC=1)OC1=CC=CC=C1 HVLLSGMXQDNUAL-UHFFFAOYSA-N 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- MIJJHRIQVWIQGL-BQYQJAHWSA-N (6e)-8-methoxyocta-1,6-diene Chemical compound COC\C=C\CCCC=C MIJJHRIQVWIQGL-BQYQJAHWSA-N 0.000 description 1
- 125000000027 (C1-C10) alkoxy group Chemical group 0.000 description 1
- RBOZTFPIXJBLPK-HWAYABPNSA-N (NE)-N-[(2E)-1,2-bis(furan-2-yl)-2-hydroxyiminoethylidene]hydroxylamine Chemical compound O\N=C(/C(=N\O)/C1=CC=CO1)\C1=CC=CO1 RBOZTFPIXJBLPK-HWAYABPNSA-N 0.000 description 1
- JJZONEUCDUQVGR-VCFJNTAESA-N (NE)-N-[(2Z)-2-hydroxyimino-1,2-diphenylethylidene]hydroxylamine Chemical compound O\N=C(\C(=N/O)\C1=CC=CC=C1)/C1=CC=CC=C1 JJZONEUCDUQVGR-VCFJNTAESA-N 0.000 description 1
- JGUQDUKBUKFFRO-GGWOSOGESA-N (NE)-N-[(3E)-3-hydroxyiminobutan-2-ylidene]hydroxylamine Chemical compound O\N=C(/C)\C(\C)=N\O JGUQDUKBUKFFRO-GGWOSOGESA-N 0.000 description 1
- NKJOXAZJBOMXID-UHFFFAOYSA-N 1,1'-Oxybisoctane Chemical compound CCCCCCCCOCCCCCCCC NKJOXAZJBOMXID-UHFFFAOYSA-N 0.000 description 1
- BZOOCKAFKVYAOZ-UHFFFAOYSA-N 1,1-Dimethoxyoctane Chemical compound CCCCCCCC(OC)OC BZOOCKAFKVYAOZ-UHFFFAOYSA-N 0.000 description 1
- RMFRFTSSEHRKKW-UHFFFAOYSA-N 1,2-bis(diisopropylphosphino)ethane Chemical compound CC(C)P(C(C)C)CCP(C(C)C)C(C)C RMFRFTSSEHRKKW-UHFFFAOYSA-N 0.000 description 1
- ZKWQSBFSGZJNFP-UHFFFAOYSA-N 1,2-bis(dimethylphosphino)ethane Chemical compound CP(C)CCP(C)C ZKWQSBFSGZJNFP-UHFFFAOYSA-N 0.000 description 1
- QFMZQPDHXULLKC-UHFFFAOYSA-N 1,2-bis(diphenylphosphino)ethane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CCP(C=1C=CC=CC=1)C1=CC=CC=C1 QFMZQPDHXULLKC-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- VYXHVRARDIDEHS-UHFFFAOYSA-N 1,5-cyclooctadiene Chemical compound C1CC=CCCC=C1 VYXHVRARDIDEHS-UHFFFAOYSA-N 0.000 description 1
- 239000004912 1,5-cyclooctadiene Substances 0.000 description 1
- KTSVVTQTKRGWGU-UHFFFAOYSA-N 1-[2-[2-(2-butoxyethoxy)ethoxy]ethoxy]butane Chemical compound CCCCOCCOCCOCCOCCCC KTSVVTQTKRGWGU-UHFFFAOYSA-N 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical compound CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- LNETULKMXZVUST-UHFFFAOYSA-N 1-naphthoic acid Chemical compound C1=CC=C2C(C(=O)O)=CC=CC2=C1 LNETULKMXZVUST-UHFFFAOYSA-N 0.000 description 1
- KJCVRFUGPWSIIH-UHFFFAOYSA-N 1-naphthol Chemical class C1=CC=C2C(O)=CC=CC2=C1 KJCVRFUGPWSIIH-UHFFFAOYSA-N 0.000 description 1
- DRGAZIDRYFYHIJ-UHFFFAOYSA-N 2,2':6',2''-terpyridine Chemical compound N1=CC=CC=C1C1=CC=CC(C=2N=CC=CC=2)=N1 DRGAZIDRYFYHIJ-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- MIOCUERTSIJEDP-UHFFFAOYSA-N 2-diethylphosphanylethyl(diethyl)phosphane Chemical compound CCP(CC)CCP(CC)CC MIOCUERTSIJEDP-UHFFFAOYSA-N 0.000 description 1
- XJLREDNCIGVZJF-UHFFFAOYSA-N 2-dipropylphosphanylethyl(dipropyl)phosphane Chemical compound CCCP(CCC)CCP(CCC)CCC XJLREDNCIGVZJF-UHFFFAOYSA-N 0.000 description 1
- CRWNQZTZTZWPOF-UHFFFAOYSA-N 2-methyl-4-phenylpyridine Chemical compound C1=NC(C)=CC(C=2C=CC=CC=2)=C1 CRWNQZTZTZWPOF-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- WPTCSQBWLUUYDV-UHFFFAOYSA-N 2-quinolin-2-ylquinoline Chemical compound C1=CC=CC2=NC(C3=NC4=CC=CC=C4C=C3)=CC=C21 WPTCSQBWLUUYDV-UHFFFAOYSA-N 0.000 description 1
- QVNVGTVAPQGSOF-UHFFFAOYSA-L 3-[4-(9,9-dimethyl-3-aza-9-azoniabicyclo[3.3.1]nonan-3-yl)butyl]-9,9-dimethyl-3-aza-9-azoniabicyclo[3.3.1]nonane;diiodide Chemical compound [I-].[I-].C1C([N+]2(C)C)CCCC2CN1CCCCN(C1)CC2CCCC1[N+]2(C)C QVNVGTVAPQGSOF-UHFFFAOYSA-L 0.000 description 1
- YLRBBYWLBNWYGZ-UHFFFAOYSA-N 4,5-diazafluorene Chemical compound C1=CC=C2CC3=CC=CN=C3C2=N1 YLRBBYWLBNWYGZ-UHFFFAOYSA-N 0.000 description 1
- DHDHJYNTEFLIHY-UHFFFAOYSA-N 4,7-diphenyl-1,10-phenanthroline Chemical compound C1=CC=CC=C1C1=CC=NC2=C1C=CC1=C(C=3C=CC=CC=3)C=CN=C21 DHDHJYNTEFLIHY-UHFFFAOYSA-N 0.000 description 1
- BCJVBDBJSMFBRW-UHFFFAOYSA-N 4-diphenylphosphanylbutyl(diphenyl)phosphane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCCP(C=1C=CC=CC=1)C1=CC=CC=C1 BCJVBDBJSMFBRW-UHFFFAOYSA-N 0.000 description 1
- NAKGXZAOEHNQFM-UHFFFAOYSA-N 8,8-dimethoxyoct-1-ene Chemical compound COC(OC)CCCCCC=C NAKGXZAOEHNQFM-UHFFFAOYSA-N 0.000 description 1
- 239000005725 8-Hydroxyquinoline Substances 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- OMXTVXXDYZNNIH-UHFFFAOYSA-N C=1C=CC=CC=1P(C=1C=CC=CC=1)(CP)(C=1C=CC=CC=1)C1=CC=CC=C1 Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(CP)(C=1C=CC=CC=1)C1=CC=CC=C1 OMXTVXXDYZNNIH-UHFFFAOYSA-N 0.000 description 1
- UWFDFERKPLOZGU-UHFFFAOYSA-N CP(CP)(C)(C)C Chemical compound CP(CP)(C)(C)C UWFDFERKPLOZGU-UHFFFAOYSA-N 0.000 description 1
- 239000005632 Capric acid (CAS 334-48-5) Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 241000819038 Chichester Species 0.000 description 1
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 1
- 229910021590 Copper(II) bromide Inorganic materials 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- 229920000557 Nafion® Polymers 0.000 description 1
- JCXJVPUVTGWSNB-UHFFFAOYSA-N Nitrogen dioxide Chemical class O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 description 1
- 235000010678 Paulownia tomentosa Nutrition 0.000 description 1
- 240000002834 Paulownia tomentosa Species 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 239000012327 Ruthenium complex Substances 0.000 description 1
- PCSMJKASWLYICJ-UHFFFAOYSA-N Succinic aldehyde Chemical class O=CCCC=O PCSMJKASWLYICJ-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 1
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 1
- 238000002441 X-ray diffraction Methods 0.000 description 1
- AEJRNONYXDAILM-UHFFFAOYSA-N [Ba+2].[O-][Cr]([O-])=O Chemical compound [Ba+2].[O-][Cr]([O-])=O AEJRNONYXDAILM-UHFFFAOYSA-N 0.000 description 1
- WDVOMJLGVRDAGA-UHFFFAOYSA-N [O-2].[Al+3].[Si](=O)=O.[Mo](=O)(=O)=O.[Cu+2] Chemical compound [O-2].[Al+3].[Si](=O)=O.[Mo](=O)(=O)=O.[Cu+2] WDVOMJLGVRDAGA-UHFFFAOYSA-N 0.000 description 1
- ZVQOOHYFBIDMTQ-UHFFFAOYSA-N [methyl(oxido){1-[6-(trifluoromethyl)pyridin-3-yl]ethyl}-lambda(6)-sulfanylidene]cyanamide Chemical compound N#CN=S(C)(=O)C(C)C1=CC=C(C(F)(F)F)N=C1 ZVQOOHYFBIDMTQ-UHFFFAOYSA-N 0.000 description 1
- IKHGUXGNUITLKF-XPULMUKRSA-N acetaldehyde Chemical compound [14CH]([14CH3])=O IKHGUXGNUITLKF-XPULMUKRSA-N 0.000 description 1
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 238000005882 aldol condensation reaction Methods 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910000272 alkali metal oxide Inorganic materials 0.000 description 1
- 150000001447 alkali salts Chemical class 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 125000002877 alkyl aryl group Chemical group 0.000 description 1
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 239000000956 alloy Substances 0.000 description 1
- 229910045601 alloy Inorganic materials 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 1
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 1
- 235000011130 ammonium sulphate Nutrition 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 238000010719 annulation reaction Methods 0.000 description 1
- 229910052787 antimony Inorganic materials 0.000 description 1
- WATWJIUSRGPENY-UHFFFAOYSA-N antimony atom Chemical compound [Sb] WATWJIUSRGPENY-UHFFFAOYSA-N 0.000 description 1
- 239000000010 aprotic solvent Substances 0.000 description 1
- 239000003849 aromatic solvent Substances 0.000 description 1
- 229910052785 arsenic Inorganic materials 0.000 description 1
- RQNWIZPPADIBDY-UHFFFAOYSA-N arsenic atom Chemical compound [As] RQNWIZPPADIBDY-UHFFFAOYSA-N 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000005228 aryl sulfonate group Chemical group 0.000 description 1
- 235000012216 bentonite Nutrition 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- ZDZHCHYQNPQSGG-UHFFFAOYSA-N binaphthyl group Chemical group C1(=CC=CC2=CC=CC=C12)C1=CC=CC2=CC=CC=C12 ZDZHCHYQNPQSGG-UHFFFAOYSA-N 0.000 description 1
- 229910052797 bismuth Inorganic materials 0.000 description 1
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 description 1
- 238000004061 bleaching Methods 0.000 description 1
- 150000003842 bromide salts Chemical class 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- 229910052793 cadmium Inorganic materials 0.000 description 1
- BDOSMKKIYDKNTQ-UHFFFAOYSA-N cadmium atom Chemical compound [Cd] BDOSMKKIYDKNTQ-UHFFFAOYSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 150000001767 cationic compounds Chemical class 0.000 description 1
- 229910052798 chalcogen Inorganic materials 0.000 description 1
- 150000001787 chalcogens Chemical class 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 229960001701 chloroform Drugs 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 239000011651 chromium Substances 0.000 description 1
- OLFCLHDBKGQITG-UHFFFAOYSA-N chromium(3+) nickel(2+) oxygen(2-) Chemical compound [Ni+2].[O-2].[Cr+3] OLFCLHDBKGQITG-UHFFFAOYSA-N 0.000 description 1
- 238000004140 cleaning Methods 0.000 description 1
- INPLXZPZQSLHBR-UHFFFAOYSA-N cobalt(2+);sulfide Chemical compound [S-2].[Co+2] INPLXZPZQSLHBR-UHFFFAOYSA-N 0.000 description 1
- 230000009918 complex formation Effects 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 239000008139 complexing agent Substances 0.000 description 1
- 239000007859 condensation product Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 150000001879 copper Chemical class 0.000 description 1
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 1
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- MLUCVPSAIODCQM-NSCUHMNNSA-N crotonaldehyde Chemical compound C\C=C\C=O MLUCVPSAIODCQM-NSCUHMNNSA-N 0.000 description 1
- MLUCVPSAIODCQM-UHFFFAOYSA-N crotonaldehyde Natural products CC=CC=O MLUCVPSAIODCQM-UHFFFAOYSA-N 0.000 description 1
- 150000003983 crown ethers Chemical class 0.000 description 1
- 230000001186 cumulative effect Effects 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-M decanoate Chemical compound CCCCCCCCCC([O-])=O GHVNFZFCNZKVNT-UHFFFAOYSA-M 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- KHQXDNILONPNNV-UHFFFAOYSA-N dibutyl(2-dibutylphosphanylethyl)phosphane Chemical compound CCCCP(CCCC)CCP(CCCC)CCCC KHQXDNILONPNNV-UHFFFAOYSA-N 0.000 description 1
- WNZGLXFLSFWPMP-UHFFFAOYSA-N dicyclohexyl(4-dicyclohexylphosphanylbutyl)phosphane Chemical compound C1CCCCC1P(C1CCCCC1)CCCCP(C1CCCCC1)C1CCCCC1 WNZGLXFLSFWPMP-UHFFFAOYSA-N 0.000 description 1
- OWFLJHJHQKHZJR-UHFFFAOYSA-N dicyclohexyl(dicyclohexylphosphanylmethyl)phosphane Chemical compound C1CCCCC1P(C1CCCCC1)CP(C1CCCCC1)C1CCCCC1 OWFLJHJHQKHZJR-UHFFFAOYSA-N 0.000 description 1
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 1
- UVTLTCJBYXBVBA-UHFFFAOYSA-N diethylphosphanyl(diethyl)phosphane Chemical compound CCP(CC)P(CC)CC UVTLTCJBYXBVBA-UHFFFAOYSA-N 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- BVQAWSJMUYMNQN-UHFFFAOYSA-N dipyridophenazine Chemical compound C1=CC=C2C3=NC4=CC=CC=C4N=C3C3=CC=CN=C3C2=N1 BVQAWSJMUYMNQN-UHFFFAOYSA-N 0.000 description 1
- MWVXFEZPEPOQRE-UHFFFAOYSA-N ditert-butyl(2-ditert-butylphosphanylethyl)phosphane Chemical compound CC(C)(C)P(C(C)(C)C)CCP(C(C)(C)C)C(C)(C)C MWVXFEZPEPOQRE-UHFFFAOYSA-N 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- DLAHAXOYRFRPFQ-UHFFFAOYSA-N dodecyl benzoate Chemical compound CCCCCCCCCCCCOC(=O)C1=CC=CC=C1 DLAHAXOYRFRPFQ-UHFFFAOYSA-N 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 150000002191 fatty alcohols Chemical class 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 238000010285 flame spraying Methods 0.000 description 1
- SLGWESQGEUXWJQ-UHFFFAOYSA-N formaldehyde;phenol Chemical class O=C.OC1=CC=CC=C1 SLGWESQGEUXWJQ-UHFFFAOYSA-N 0.000 description 1
- 150000004675 formic acid derivatives Chemical class 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-M heptanoate Chemical compound CCCCCCC([O-])=O MNWFXJYAOYHMED-UHFFFAOYSA-M 0.000 description 1
- 238000009904 heterogeneous catalytic hydrogenation reaction Methods 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 230000003100 immobilizing effect Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 229910001411 inorganic cation Inorganic materials 0.000 description 1
- 150000004694 iodide salts Chemical class 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 239000001282 iso-butane Substances 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 229910044991 metal oxide Inorganic materials 0.000 description 1
- 150000004706 metal oxides Chemical class 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-M methanesulfonate group Chemical class CS(=O)(=O)[O-] AFVFQIVMOAPDHO-UHFFFAOYSA-M 0.000 description 1
- 230000002906 microbiologic effect Effects 0.000 description 1
- 235000013379 molasses Nutrition 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 150000002762 monocarboxylic acid derivatives Chemical class 0.000 description 1
- 150000002763 monocarboxylic acids Chemical class 0.000 description 1
- 229940105132 myristate Drugs 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical class C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- MGFYIUFZLHCRTH-UHFFFAOYSA-N nitrilotriacetic acid Chemical compound OC(=O)CN(CC(O)=O)CC(O)=O MGFYIUFZLHCRTH-UHFFFAOYSA-N 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 231100000989 no adverse effect Toxicity 0.000 description 1
- 229910052756 noble gas Inorganic materials 0.000 description 1
- 150000002835 noble gases Chemical class 0.000 description 1
- 229910000510 noble metal Inorganic materials 0.000 description 1
- SNQQPOLDUKLAAF-UHFFFAOYSA-N nonylphenol Chemical compound CCCCCCCCCC1=CC=CC=C1O SNQQPOLDUKLAAF-UHFFFAOYSA-N 0.000 description 1
- QWVGKYWNOKOFNN-UHFFFAOYSA-N o-cresol Chemical compound CC1=CC=CC=C1O QWVGKYWNOKOFNN-UHFFFAOYSA-N 0.000 description 1
- CACRRXGTWZXOAU-UHFFFAOYSA-N octadecane-1-sulfonic acid Chemical compound CCCCCCCCCCCCCCCCCCS(O)(=O)=O CACRRXGTWZXOAU-UHFFFAOYSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-M oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC([O-])=O ZQPPMHVWECSIRJ-KTKRTIGZSA-M 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- OOSUVFMPZZUMMJ-UHFFFAOYSA-N oxoplatinum oxorhodium Chemical compound O=[Rh].O=[Pt] OOSUVFMPZZUMMJ-UHFFFAOYSA-N 0.000 description 1
- 229960003540 oxyquinoline Drugs 0.000 description 1
- 239000003973 paint Substances 0.000 description 1
- 150000002940 palladium Chemical class 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 150000005041 phenanthrolines Chemical class 0.000 description 1
- OXJYINDODBGERE-UHFFFAOYSA-N phenylphosphane triphenylphosphane Chemical compound C1(=CC=CC=C1)P.C1(=CC=CC=C1)P(C1=CC=CC=C1)C1=CC=CC=C1 OXJYINDODBGERE-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 125000005547 pivalate group Chemical group 0.000 description 1
- DBJYYRBULROVQT-UHFFFAOYSA-N platinum rhenium Chemical compound [Re].[Pt] DBJYYRBULROVQT-UHFFFAOYSA-N 0.000 description 1
- 239000002574 poison Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 231100000572 poisoning Toxicity 0.000 description 1
- 230000000607 poisoning effect Effects 0.000 description 1
- 229920000867 polyelectrolyte Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 235000019353 potassium silicate Nutrition 0.000 description 1
- 238000007639 printing Methods 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- MCJGNVYPOGVAJF-UHFFFAOYSA-N quinolin-8-ol Chemical compound C1=CN=C2C(O)=CC=CC2=C1 MCJGNVYPOGVAJF-UHFFFAOYSA-N 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 229920013730 reactive polymer Polymers 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 150000003304 ruthenium compounds Chemical class 0.000 description 1
- 150000003333 secondary alcohols Chemical class 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- LIVNPJMFVYWSIS-UHFFFAOYSA-N silicon monoxide Chemical class [Si-]#[O+] LIVNPJMFVYWSIS-UHFFFAOYSA-N 0.000 description 1
- 229910052814 silicon oxide Inorganic materials 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- NTHWMYGWWRZVTN-UHFFFAOYSA-N sodium silicate Chemical compound [Na+].[Na+].[O-][Si]([O-])=O NTHWMYGWWRZVTN-UHFFFAOYSA-N 0.000 description 1
- WWNBZGLDODTKEM-UHFFFAOYSA-N sulfanylidenenickel Chemical compound [Ni]=S WWNBZGLDODTKEM-UHFFFAOYSA-N 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- 229910052714 tellurium Inorganic materials 0.000 description 1
- PORWMNRCUJJQNO-UHFFFAOYSA-N tellurium atom Chemical compound [Te] PORWMNRCUJJQNO-UHFFFAOYSA-N 0.000 description 1
- TUNFSRHWOTWDNC-UHFFFAOYSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCCC(O)=O TUNFSRHWOTWDNC-UHFFFAOYSA-N 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 238000007669 thermal treatment Methods 0.000 description 1
- 150000003585 thioureas Chemical class 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 229910052719 titanium Inorganic materials 0.000 description 1
- LBLYYCQCTBFVLH-UHFFFAOYSA-M toluenesulfonate group Chemical group C=1(C(=CC=CC1)S(=O)(=O)[O-])C LBLYYCQCTBFVLH-UHFFFAOYSA-M 0.000 description 1
- XTTGYFREQJCEML-UHFFFAOYSA-N tributyl phosphite Chemical compound CCCCOP(OCCCC)OCCCC XTTGYFREQJCEML-UHFFFAOYSA-N 0.000 description 1
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 1
- YNJBWRMUSHSURL-UHFFFAOYSA-N trichloroacetic acid Chemical class OC(=O)C(Cl)(Cl)Cl YNJBWRMUSHSURL-UHFFFAOYSA-N 0.000 description 1
- FICPQAZLPKLOLH-UHFFFAOYSA-N tricyclohexyl phosphite Chemical compound C1CCCCC1OP(OC1CCCCC1)OC1CCCCC1 FICPQAZLPKLOLH-UHFFFAOYSA-N 0.000 description 1
- WLPUWLXVBWGYMZ-UHFFFAOYSA-N tricyclohexylphosphine Chemical compound C1CCCCC1P(C1CCCCC1)C1CCCCC1 WLPUWLXVBWGYMZ-UHFFFAOYSA-N 0.000 description 1
- VTKQLVHDZYRENE-UHFFFAOYSA-N tricyclopentyl phosphite Chemical compound C1CCCC1OP(OC1CCCC1)OC1CCCC1 VTKQLVHDZYRENE-UHFFFAOYSA-N 0.000 description 1
- DHWBYAACHDUFAT-UHFFFAOYSA-N tricyclopentylphosphane Chemical compound C1CCCC1P(C1CCCC1)C1CCCC1 DHWBYAACHDUFAT-UHFFFAOYSA-N 0.000 description 1
- MOSFSEPBWRXKJZ-UHFFFAOYSA-N tridecylphosphane Chemical compound CCCCCCCCCCCCCP MOSFSEPBWRXKJZ-UHFFFAOYSA-N 0.000 description 1
- BDZBKCUKTQZUTL-UHFFFAOYSA-N triethyl phosphite Chemical compound CCOP(OCC)OCC BDZBKCUKTQZUTL-UHFFFAOYSA-N 0.000 description 1
- RXJKFRMDXUJTEX-UHFFFAOYSA-N triethylphosphine Chemical compound CCP(CC)CC RXJKFRMDXUJTEX-UHFFFAOYSA-N 0.000 description 1
- 125000002827 triflate group Chemical class FC(S(=O)(=O)O*)(F)F 0.000 description 1
- YFNKIDBQEZZDLK-UHFFFAOYSA-N triglyme Chemical compound COCCOCCOCCOC YFNKIDBQEZZDLK-UHFFFAOYSA-N 0.000 description 1
- IGNTWNVBGLNYDV-UHFFFAOYSA-N triisopropylphosphine Chemical compound CC(C)P(C(C)C)C(C)C IGNTWNVBGLNYDV-UHFFFAOYSA-N 0.000 description 1
- CYTQBVOFDCPGCX-UHFFFAOYSA-N trimethyl phosphite Chemical compound COP(OC)OC CYTQBVOFDCPGCX-UHFFFAOYSA-N 0.000 description 1
- RMZAYIKUYWXQPB-UHFFFAOYSA-N trioctylphosphane Chemical compound CCCCCCCCP(CCCCCCCC)CCCCCCCC RMZAYIKUYWXQPB-UHFFFAOYSA-N 0.000 description 1
- 150000004072 triols Chemical class 0.000 description 1
- SJHCUXCOGGKFAI-UHFFFAOYSA-N tripropan-2-yl phosphite Chemical compound CC(C)OP(OC(C)C)OC(C)C SJHCUXCOGGKFAI-UHFFFAOYSA-N 0.000 description 1
- QOPBTFMUVTXWFF-UHFFFAOYSA-N tripropyl phosphite Chemical compound CCCOP(OCCC)OCCC QOPBTFMUVTXWFF-UHFFFAOYSA-N 0.000 description 1
- KCTAHLRCZMOTKM-UHFFFAOYSA-N tripropylphosphane Chemical compound CCCP(CCC)CCC KCTAHLRCZMOTKM-UHFFFAOYSA-N 0.000 description 1
- 229960004418 trolamine Drugs 0.000 description 1
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- 239000010937 tungsten Substances 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 238000007740 vapor deposition Methods 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/132—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by reduction of an oxygen containing functional group
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C47/00—Compounds having —CHO groups
- C07C47/02—Saturated compounds having —CHO groups bound to acyclic carbon atoms or to hydrogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/01—Preparation of ethers
- C07C41/05—Preparation of ethers by addition of compounds to unsaturated compounds
- C07C41/06—Preparation of ethers by addition of compounds to unsaturated compounds by addition of organic compounds only
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/01—Preparation of ethers
- C07C41/32—Preparation of ethers by isomerisation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C41/00—Preparation of ethers; Preparation of compounds having groups, groups or groups
- C07C41/48—Preparation of compounds having groups
- C07C41/50—Preparation of compounds having groups by reactions producing groups
- C07C41/54—Preparation of compounds having groups by reactions producing groups by addition of compounds to unsaturated carbon-to-carbon bonds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/51—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition
- C07C45/511—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition involving transformation of singly bound oxygen functional groups to >C = O groups
- C07C45/515—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by pyrolysis, rearrangement or decomposition involving transformation of singly bound oxygen functional groups to >C = O groups the singly bound functional group being an acetalised, ketalised hemi-acetalised, or hemi-ketalised hydroxyl group
Abstract
Description


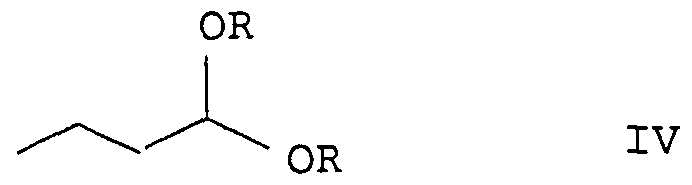













Claims


Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP98913678A EP0971868A1 (de) | 1997-03-17 | 1998-03-06 | Verfahren zur herstellung von n-butyraldehyd und/oder n-butanol |
| US09/381,452 US6166265A (en) | 1997-03-17 | 1998-03-06 | Processes for the preparation of n-butyraldehyde, n-butanol and mixtures thereof |
| JP54008198A JP2001515499A (ja) | 1997-03-17 | 1998-03-06 | n−ブチルアルデヒドおよび/またはn−ブタノールの製造方法 |
| AU68289/98A AU6828998A (en) | 1997-03-17 | 1998-03-06 | Method for producing n-butyraldehyde and/or n-butanol |
| BR9808001-6A BR9808001A (pt) | 1997-03-17 | 1998-03-06 | Processo para a preparação de n-butiraldeìdo e/ou de n-butanol |
| CA002282538A CA2282538A1 (en) | 1997-03-17 | 1998-03-06 | Method for producing n-butyraldehyde and/or n-butanol |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19710994.2 | 1997-03-17 | ||
| DE19710994A DE19710994A1 (de) | 1997-03-17 | 1997-03-17 | Verfahren zur Herstellung von n-Butyraldehyd und/oder n-Butanol |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1998041494A1 true WO1998041494A1 (de) | 1998-09-24 |
Family
ID=7823624
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1998/001324 WO1998041494A1 (de) | 1997-03-17 | 1998-03-06 | Verfahren zur herstellung von n-butyraldehyd und/oder n-butanol |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US6166265A (de) |
| EP (1) | EP0971868A1 (de) |
| JP (1) | JP2001515499A (de) |
| KR (1) | KR20000076304A (de) |
| CN (1) | CN1249738A (de) |
| AU (1) | AU6828998A (de) |
| BR (1) | BR9808001A (de) |
| CA (1) | CA2282538A1 (de) |
| DE (1) | DE19710994A1 (de) |
| ID (1) | ID22905A (de) |
| WO (1) | WO1998041494A1 (de) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000037404A1 (en) * | 1998-12-22 | 2000-06-29 | Bp Chemicals Limited | A process for making butyraldehyde from butadiene |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9823853D0 (en) | 1998-10-30 | 1998-12-23 | Bp Chem Int Ltd | A process for making n-butyl esters from butadiene |
| US9024090B2 (en) | 2012-12-19 | 2015-05-05 | Celanese International Corporation | Catalysts and processes for producing butanol |
| US8962897B2 (en) | 2012-12-19 | 2015-02-24 | Celanese International Corporation | Catalysts and processes for producing butanol |
| US9018426B1 (en) | 2013-12-19 | 2015-04-28 | Celanese International Corporation | Processes for producing multi-carbon alcohols |
| WO2017064064A1 (en) * | 2015-10-12 | 2017-04-20 | Basf Se | Hydroformylation process for producing 1,6-disubstituted hexane derivatives |
| EP3768802B1 (de) * | 2018-03-19 | 2023-03-29 | SABIC Global Technologies B.V. | Verfahren zur herstellung eines kraftstoffzusatzes |
| CN110937745B (zh) * | 2019-12-12 | 2022-07-12 | 万华化学集团股份有限公司 | 一种柠檬醛合成过程中高浓度废水的处理方法 |
| CN113877635B (zh) * | 2021-10-27 | 2024-02-06 | 南京延长反应技术研究院有限公司 | 一种铱基催化剂及其制备方法、醛化方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995019334A1 (de) * | 1994-01-14 | 1995-07-20 | Basf Aktiengesellschaft | VERFAHREN ZUR HERSTELLUNG VON n-BUTYRALDEHYD UND/ODER n-BUTANOL |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2922822A (en) * | 1957-01-08 | 1960-01-26 | Exxon Research Engineering Co | Production of methyl butenyl ethers |
| DE2751766A1 (de) * | 1977-11-19 | 1979-05-23 | Basf Ag | Verfahren zur isomerisierung von 3-buten-1-ol-verbindungen zu den entsprechenden 2-buten-1-ol-verbindungen |
-
1997
- 1997-03-17 DE DE19710994A patent/DE19710994A1/de not_active Withdrawn
-
1998
- 1998-03-06 CN CN98803008A patent/CN1249738A/zh active Pending
- 1998-03-06 ID IDW990822A patent/ID22905A/id unknown
- 1998-03-06 JP JP54008198A patent/JP2001515499A/ja active Pending
- 1998-03-06 EP EP98913678A patent/EP0971868A1/de not_active Withdrawn
- 1998-03-06 WO PCT/EP1998/001324 patent/WO1998041494A1/de not_active Application Discontinuation
- 1998-03-06 US US09/381,452 patent/US6166265A/en not_active Expired - Fee Related
- 1998-03-06 CA CA002282538A patent/CA2282538A1/en not_active Abandoned
- 1998-03-06 BR BR9808001-6A patent/BR9808001A/pt not_active IP Right Cessation
- 1998-03-06 AU AU68289/98A patent/AU6828998A/en not_active Abandoned
- 1998-03-06 KR KR1019997008405A patent/KR20000076304A/ko not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995019334A1 (de) * | 1994-01-14 | 1995-07-20 | Basf Aktiengesellschaft | VERFAHREN ZUR HERSTELLUNG VON n-BUTYRALDEHYD UND/ODER n-BUTANOL |
| DE4400837A1 (de) * | 1994-01-14 | 1995-07-20 | Basf Ag | Verfahren zur Herstellung von n-Butyraldehyd und/oder n-Butanol |
Non-Patent Citations (2)
| Title |
|---|
| B.-H. CHANG: "A facile synthesis of acetals and aldehydes from allylic ethers catalyzed by cobalt compounds", JOURNAL OF ORGANOMETALLIC CHEMISTRY, vol. 492, no. 1, 19 April 1995 (1995-04-19), LAUSANNE CH, pages 31 - 34, XP002069120 * |
| CHEMICAL ABSTRACTS, vol. 78, no. 3, 22 January 1973, Columbus, Ohio, US; abstract no. 15519k, T. YAMAHARA: "Aldehyde derivatives" page 341; column 2; XP002069121 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000037404A1 (en) * | 1998-12-22 | 2000-06-29 | Bp Chemicals Limited | A process for making butyraldehyde from butadiene |
| US6403839B1 (en) | 1998-12-22 | 2002-06-11 | Bp Chemicals Limited | Process for making butyraldehyde from butadiene |
Also Published As
| Publication number | Publication date |
|---|---|
| DE19710994A1 (de) | 1998-09-24 |
| CA2282538A1 (en) | 1998-09-24 |
| JP2001515499A (ja) | 2001-09-18 |
| KR20000076304A (ko) | 2000-12-26 |
| BR9808001A (pt) | 2000-03-08 |
| EP0971868A1 (de) | 2000-01-19 |
| US6166265A (en) | 2000-12-26 |
| ID22905A (id) | 1999-12-16 |
| AU6828998A (en) | 1998-10-12 |
| CN1249738A (zh) | 2000-04-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0739332B1 (de) | VERFAHREN ZUR HERSTELLUNG VON n-BUTYRALDEHYD UND/ODER n-BUTANOL | |
| WO2004020380A1 (de) | Verfahren zur hydroformylierung von olefinisch ungesättigten verbindungen, insbesondere olefinen in gegenwart cyclischer kohlensäureester | |
| EP0778819B1 (de) | Verfahren zur herstellung von n-butyraldehyd und/oder n-butanol | |
| WO1998041494A1 (de) | Verfahren zur herstellung von n-butyraldehyd und/oder n-butanol | |
| EP0061791B1 (de) | Verfahren zur Herstellung von Glykolaldehyd | |
| EP0761634B1 (de) | Verfahren zur Herstellung von Pentenalen | |
| DE10155520A1 (de) | Verfahren zur Herstellung von n-Butyraldehyd, n-Butanol und 2-Ethylhexanol aus 1,3-butadienhaltigen Kohlenwasserstoffströmen | |
| EP0191935A1 (de) | Verfahren zur Herstellung von 2-Alkyl-1,4-butandial | |
| DE19964298C2 (de) | Verfahren zur Herstellung von substituierten Arylhalophosphinen | |
| EP0915839B1 (de) | Verfahren zur herstellung von 6-aminocapronitril | |
| EP0931046B1 (de) | VERFAHREN ZUR HERSTELLUNG VON n-BUTYLALKYLETHERN | |
| DE2154370C3 (de) | Verfahren zur Herstellung von Äthern des 2,6-Dimethyl-2,7-octadien-1-ols und einige dieser Äther | |
| EP0150943B1 (de) | Hydroformylierung von Allylalkohol | |
| EP0966418A1 (de) | Verfahren zur herstellung von acetalen | |
| DE102005019237B4 (de) | Verfahren zur Hydroformylierung von substituierten Allylbenzolen | |
| MXPA99007576A (es) | Preparacion de n-butiraldehido y/o n-butanol | |
| CZ9903284A3 (cs) | Způsob výroby n-butyraldehydu a/nebo n-butanolu | |
| DE1793616A1 (de) | Rhodiumkomplexe | |
| DE3728981A1 (de) | Verfahren zur herstellung von ethanol im gemisch mit propanol und butanol |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 98803008.X Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AU BG BR BY CA CN CZ GE HU ID IL JP KR KZ LT LV MX NO NZ PL RO RU SG SI SK TR UA US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AM AZ BY KG KZ MD RU TJ TM AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1998913678 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/1999/007576 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2282538 Country of ref document: CA Ref document number: 2282538 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09381452 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 1998 540081 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1999-3284 Country of ref document: CZ Ref document number: 1019997008405 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 68289/98 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998913678 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1999-3284 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019997008405 Country of ref document: KR |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1998913678 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1019997008405 Country of ref document: KR |
|
| WWR | Wipo information: refused in national office |
Ref document number: PV1999-3284 Country of ref document: CZ |