WO1997006177A1 - Procede d'elaboration de derives d'erythromycine - Google Patents
Procede d'elaboration de derives d'erythromycine Download PDFInfo
- Publication number
- WO1997006177A1 WO1997006177A1 PCT/JP1996/002191 JP9602191W WO9706177A1 WO 1997006177 A1 WO1997006177 A1 WO 1997006177A1 JP 9602191 W JP9602191 W JP 9602191W WO 9706177 A1 WO9706177 A1 WO 9706177A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- lower alkyl
- reaction
- hydroxyl group
- formula
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
Definitions
- the present invention relates to a method for producing an erythromycin derivative and a fumarate crystal of the erythromycin derivative obtained by the production method.
- a compound represented by O 97/06177 was obtained, the hydroxyl group at the 12-position of this compound was alkylated, and the acetyl group at the 2'-position and the formyl group at the 4 "-position were removed.
- R 2 represents a lower alkyl group
- the compound is reacted with benzyloxycarbonyl chloride under basic conditions. After removing the hydroxyl group, alkylating the nitrogen atom at the 3'-position, converting it to a fumarate, then recrystallizing the crude crystal with an alcohol-based solvent and then recrystallizing with hydrous ethyl acetate.
- R 2 represents a lower alkyl group
- the present invention also relates to erythromycin A (formula (I)) P
- R 2 represents a lower alkyl group
- the compound is reacted with benzyloxycarbonyl chloride under basic conditions.
- the formula (II) is characterized in that after removing the hydroxyl group and then alkylating the nitrogen atom at the 3′-position, a fumarate is obtained.
- R 2 represents a lower alkyl group
- the acetylation of the hydroxyl group at the 2′-position of erythromycin A, the formylation of the hydroxyl group at the 4 ′′ -position, and the hemi-ketalization reaction are preferably carried out in one pot.
- the term “one-pot” refers to This means that the reaction is performed in one step without isolating and purifying the reaction product of each step.
- the alkylation reaction of the hydroxyl group at the 12-position and the removal reaction of the acetyl group at the 2′-position and the formyl group at the 4 ′′ -position are preferably performed in one pot.
- the acetylation of the 2'-hydroxyl group, the formylation and the hemi-ketalization reaction of the 4'-hydroxyl group of erythromycin A were performed in one pot, and the alkylation reaction of the 12-hydroxyl group and the 2 ' It is particularly preferred to carry out the reaction for removing the acetyl group and the formyl group at the 4 "position in one pot.
- the present invention relates to erythromycin A
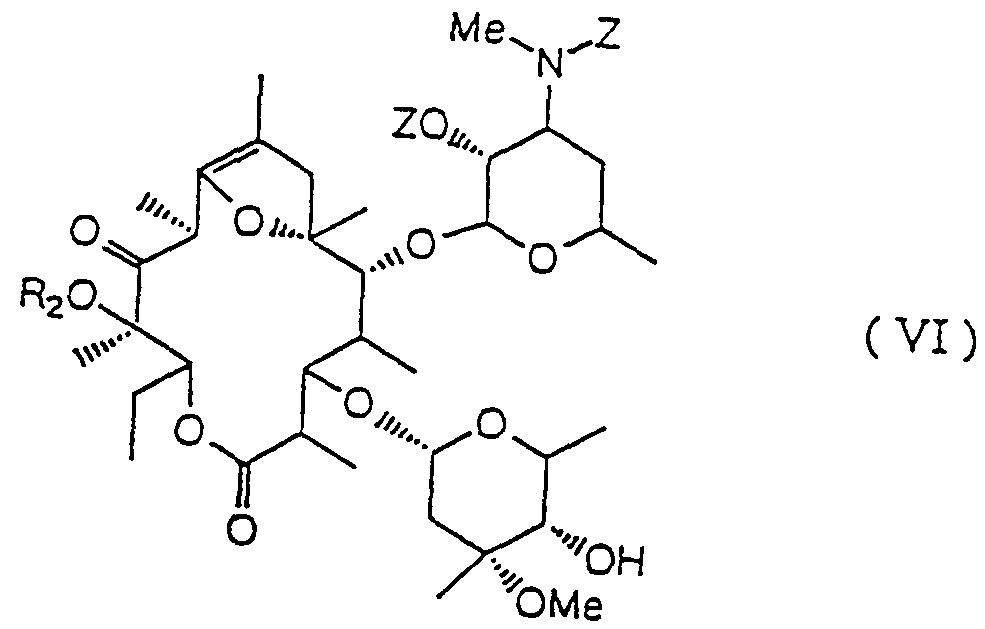
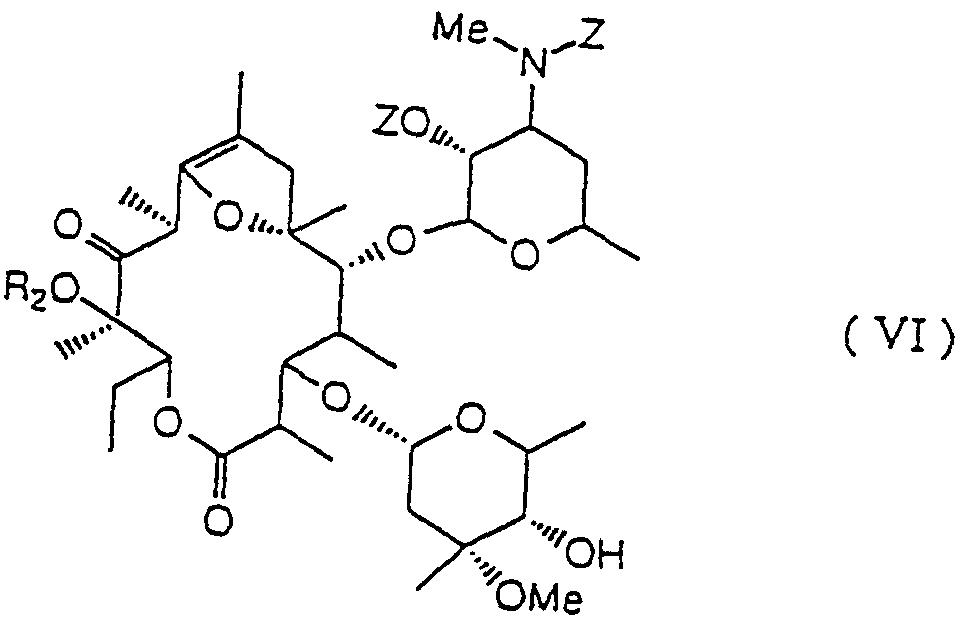
- R 2 represents a lower alkyl group and Z represents a benzyloxycarbonyl group).
- R 2 represents a lower alkyl group
- Figure 1 shows the X-ray powder X-ray spectrum of a ⁇ -shaped crystal.
- Figure 2 shows a powder X-ray spectrum of the C-type crystal.
- Figure 3 shows the powder X-ray spectrum of the D-type crystal.
- FIG. 4 shows a DSC curve in the thermal analysis of Form A crystal.
- FIG. 5 shows a DSC curve in the thermal analysis of Form C crystal.
- FIG. 6 shows the DSC curve in the thermal analysis of Form D crystal.
- FIG. 7 is a view showing the residual ratio of each crystal in a heating stability test.
- FIG. 8 is a diagram showing the residual ratio of each crystal in a humidification stability test. BEST MODE FOR CARRYING OUT THE INVENTION
- the lower alkyl group means, for example, a linear or branched alkyl group having 1 to 6 carbon atoms, specifically, for example, a methyl group, an ethyl group, an n-propyl group, an isopropyl group And n-butyl group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group, hexyl group and the like, and preferably methyl group, ethyl group, n-propyl group and isopropyl group.
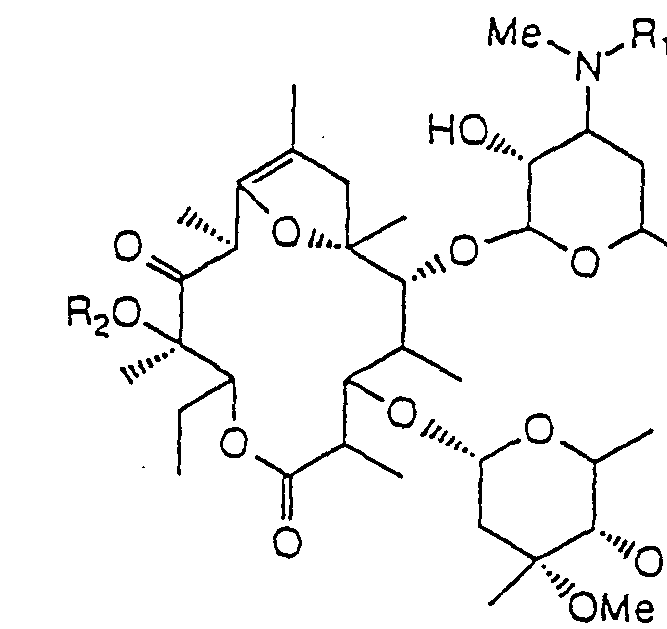
- R i is an isopropyl group
- a particularly preferred example of R 2 is a methyl group.
- reaction route D An example of the production method of the present invention is shown below (reaction route D.
- erythromycin A the compound represented by the formula (I)
- the compound represented by the formula (m) is obtained:
- the three-stage reaction of acetylation, formylation, and hemi-ketalization is preferably performed in one pot.
- Examples include organic bases such as, for example, the inorganic base diamine, preferably organic bases such as pyridine, triethylamine, disopropylethylamine, pyrrolidine, piperidine, morpholine, getylamine, diisopropylamine.
- Solvents used include acetylation, formylation, and hemi-catalyst.
- Those which are inert in the three-step reaction of the oxidation are preferably, for example, ethyl acetate, acetone, dichloromethane, chloroform, and the like, more preferably, ethyl acetate and acetone, and most preferably, acetic acid.
- acetylating agent examples include acetic anhydride, acetyl chloride, sodium acetate, and the like, preferably acetic anhydride and acetyl chloride, and most preferably acetic anhydride.
- the reaction temperature is preferably about 0 ° C. to 50 ° C., more preferably about room temperature, and the reaction time is about 30 minutes to 3 hours, preferably 1 hour to 2 hours.
- Preferred examples of the formylating agent used in the second-stage formylation reaction include, for example, formic acid-acetic anhydride and sodium formate acetyl monochloride, and more preferably, formic acid monoacetic anhydride and the like.
- Examples of the base used include, for example, organic bases such as inorganic base pyridine, and preferably pyridine, triethylamine, diisopropylethylamine, pyrrolidine, piperidine, morpholine, getylamine, diisopropylamine. And more preferably pyridine.
- the reaction temperature is preferably about 40 ° C to 5 ° C. 20 ° C to 0 ° C is preferred.
- the reaction time is about 1 to 1 hour, preferably about 5 to 12 hours.
- the third stage hemicetalization reaction is carried out under acidic conditions.
- the acidic condition means that an acid is present in the reaction system.
- Examples of the acid used here include organic acids, preferably carboxylic acids such as acetic acid and formic acid, and preferably acetic acid.
- the reaction temperature is preferably from about room temperature to about 60 ° C, more preferably from 40 ° C to 50 ° C.
- the reaction time is about 1 to 1 hour, preferably about 2 to 12 hours.
- the resulting compound represented by the formula (II) is subjected to an oxidation reaction to oxidize the hydroxyl group at the 11-position.
- the oxidizing agent include organic oxidizing agents such as dimethyl sulfoxide and Dess-Martin Periodinane reagent, and metal oxides such as ruthenium tetroxide.
- organic oxidizing agents such as dimethyl sulfoxide and Dess-Martin Periodinane reagent
- metal oxides such as ruthenium tetroxide.
- dimethyl sulfoxide doicyclohexyl carbonyl is used.
- dimethylsulfoxide trifluoroacetic anhydride is particularly preferred is dimethylsulfoxide trifluoroacetic anhydride.
- the solvent used may be any solvent that is inert to the reaction, but when dimethylsulfoxide trifluoroacetic anhydride is used as the oxidizing agent, halogen-based solvents such as chloroform, dichloromethane and the like are preferable. Are preferred.
- the reaction temperature is preferably about 160 ° C to 0 ° C, more preferably about 120 ° C to 110 ° C.
- the reaction time is about 30 minutes to 5 hours, preferably 1 hour to 2 hours.
- An alkylating agent is allowed to act on the obtained compound represented by the formula (IV) under basic conditions to alkylate the hydroxyl group at the 12-position.
- the protecting groups at the 2'-position and the 4 "-position are removed.
- the alkylation and the removal of the protecting group are preferably performed in one pot.
- alkylating agent used in the first-stage alkylation reaction examples include, for example, alkyl halides, alkyl tosylates, and alkyl imidates. Gerare, preferably wherein c, alkyl tosylate, such as alkyl halide and the like, preferably a methyl group as the alkyl moiety.
- alkyl tosylate such as alkyl halide and the like
- methyl group as the alkyl moiety methylating agent
- the base used include metal hydrides, metal hydroxides, metal alkoxides and the like, preferably metal hydrides and the like, and particularly preferably sodium hydride.
- the solvent used may be any solvent that is inert to the reaction, but an aprotic polar solvent is preferred, and dimethylimidazolidinone, dimethylformamide, dimethylacetamide, tetrahydrofuran, acetonitrile, etc. And particularly preferably dimethylimidazolidinone and dimethylformamide.
- the reaction temperature is preferably about 0 ° C to 60 ° C, more preferably 0 ° C to 30 ° C.
- the reaction time is about 1 hour to 12 hours, preferably 2 hours to 8 hours.
- the second step of removing the protective group is carried out by a usual method of removing an acetyl group or a formyl group, and is preferably removed under basic conditions.
- the base to be used include inorganic bases such as sodium hydrogen carbonate and potassium carbonate, and more preferable is sodium hydrogen carbonate.
- the solvent used may be any solvent that is inert to the reaction, but is preferably an alcohol-based solvent, and more preferably methanol, ethanol, or the like.
- the reaction temperature is preferably about 40 ° C to 80 ° C, more preferably 50 ° C to 60 ° C.
- the reaction time is about 1 hour to 12 hours, preferably 3 hours to 8 hours.
- the base is used in an excessive amount, for example, at least 2 equivalents, preferably about 2 equivalents, in the first-stage alkylation reaction.
- the solvent can be exchanged as necessary at each stage, such as adding the second stage reaction solvent to the first stage reaction solvent.
- benzyloxycarbonylating agent used in the first step for example, benzyloxycarbonyl chloride is preferable.
- the base used include inorganic bases such as sodium bicarbonate and potassium carbonate, and preferably sodium bicarbonate and the like.
- the solvent used may be any solvent that is inert to the reaction, but is preferably an aromatic hydrocarbon solvent, and more preferably toluene.
- the reaction temperature is preferably about 30 ° C. to 80 ° C., more preferably 45 ° C. to 70 ° C., and particularly preferably about 60 ° C.
- the reaction time is about 2 hours to 12 hours, preferably 4 hours to 8 hours.
- the benzyloxycarbonylating agent is required in an excess amount relative to the compound represented by the general formula (V), preferably from 9 to 15 equivalents, and more preferably from 10 to 12 equivalents.
- the removal reaction of the benzyloxycarbonyl group in the second step is carried out by a usual deprotection method.
- the deprotection method include catalytic hydrogenation, and preferably catalytic hydrogenation using a palladium-carbon catalyst.
- the hydrogen source in addition to hydrogen, ammonium formate and the like can be used.
- the catalytic hydrogenation may be performed under pressure, and the pressure under pressurization is preferably about 2 to 5 atm, more preferably 3 to 5 atm. 4 atm.
- the solvent used may be any solvent that is inert to the reaction, but is preferably an alcohol-based solvent, and more preferably methanol, ethanol, or the like.
- the reaction temperature is about 0 ° C. to 50 ° C., preferably about 10 ° C. to 30 ° C., and more preferably around room temperature.
- the reaction time is about 30 minutes to 3 hours, preferably 1 hour to 2 hours.
- the solvent used may be any solvent that is inert to the reaction, but may be an alcohol-based solvent. And more preferably, methanol and ethanol.
- the reaction temperature is preferably about 50 ° C. to 100 ° C., and more preferably 60 ° C. to 90 ° C.
- the reaction time is about 30 minutes to 3 hours, preferably 1 hour to 2 hours.
- alkylating agent used in the third-stage alkylation reaction examples include, for example, an alkyl halide, an alkyl tosylate and the like, preferably an alkyl halide and the like.
- an isopropyl group is particularly preferred as the alkyl moiety.
- Preferred examples of the isopropylylating agent include isopropylyl iodide.
- the base to be used includes, for example, an inorganic base in addition to an organic base such as amine.
- Preferred examples include diisopropylethylamine, triethylamine, morpholine, piperidine, pyrrolidine, pyridine and the like, and particularly preferred. Is triethylamine.
- the solvent used may be any solvent that is inert to the reaction, but is preferably an aprotic polar solvent, an alcoholic solvent, or the like, and more preferably dimethylimidazolidinone, dimethylformamide, or dimethylacetamide. And acetonitrile, tetrahydrofuran, methanol, ethanol and the like, and more preferably dimethylimidazolidinone, dimethylformamide and acetonitrile.
- the reaction temperature is preferably about 50 ° C to 100 ° C, more preferably 60 ° C to 80 ° C.
- the reaction time is about 3 to 10 hours, preferably 5 to 10 hours.
- the conversion reaction to the fumarate in the fourth step is carried out by a usual method for forming a salt.
- the solvent used is preferably an alcohol solvent, an ether solvent such as tetrahydrofuran, acetone, and the like, and more preferably, methanol, ethanol, isopropanol, and the like.
- the reaction temperature is preferably from about 120 ° C to 50 ° C, and more preferably from about 15 ° C to room temperature.
- the reaction time is about 1 to 6 hours, preferably 3 to 4 hours.
- the obtained fumarate of the compound represented by the general formula (II) is purified if necessary. Recrystallization is preferred as a purification method.
- the recrystallization solvent for example, an ester solvent, an alcohol solvent, an ether solvent, or a mixed solvent thereof, which may contain water, is preferably used.
- the ratio of methanol to isopropanol is, for example, about 10:90 to 50:50, preferably about 20:80 to 30:70.
- the ratio of the mixed solvent of ethyl acetate and water is, for example, 99.5: 0.5 to 97: 3, preferably 99: 1 to 98: 2, and more preferably 98.5: 1. It is about 5.
- recrystallization is performed using ethyl acetate or a mixed solvent of ethyl acetate and water
- recrystallization is performed using a mixed solvent of methanol and isopropanol described in JP-A-6-56873 (form A). Crystals of different crystal forms (C-type and D-type) were obtained. X-ray powder diffraction and thermal analysis (DSC) data of these crystals are shown in the figures ( Figures 1, 2, 3, 4, 5, and 6).
- the molar ratio of the compound represented by the formula (VE) and the fumaric acid in the crystal (form A crystal) obtained by recrystallization with a mixed solvent of methanol and isopropanol was 2: 1.
- the crystals obtained by recrystallization using ethyl acetate or a mixed solvent of ethyl acetate and water have a molar ratio of the compound represented by the formula (VH) to fumaric acid of 1: 1 ( (C-type crystal) and 2: 1 (D-type crystal).
- the fumarate salt of the compound represented by the formula (VE) (for example, A-form crystal), which is roughly purified by a method such as recrystallization with a mixed solvent of methanol and isopropanol, is added to ethyl acetate.
- Crystals obtained by recrystallization with a mixed solvent of water and water (D-type crystals) have superior stability as compared with crystals of other crystal forms, such as excellent quality as a drug or a pharmaceutical raw material. It is clear that you have
- Erythromycin A (20.0 g, 0.027 mol 1) dissolved in acetic anhydride (3.34 g, 0.033 mol 1), pyridine (3.45 g, 0.044 mol 1) and ethyl acetate (80 ml) The mixture was stirred at room temperature for 1 hour. Thereafter, formic acid (11.29 g, 0.245 mol 1) and acetic anhydride (12.52 g, 0.123 mol 1) were added dropwise under ice cooling (0 ° C.), and the mixture was stirred for 3 hours while cooling with ice. Thereafter, the temperature was gradually returned to room temperature, and left overnight.
- Example 3 Synthesis of Deprotected Compound (Compound (R 2 : Methyl) Represented by Formula (V)) 60% sodium hydride (1.02 g, 0.026 mol 1) was added to dimethylformamide (30 ml). Was added, and the compound (10.0 g, 0.013mo1) obtained in Example 2 was further added under ice-cooling (0 ° C), followed by stirring for 30 minutes. After dropwise addition of methyl tosylate (2.38 g, 0.013mo1), the mixture was stirred at 0 to 5 ° C for 1 hour and at 15 to 20 ° C for 1.5 hours. Methanol (60 ml) was added, and the mixture was heated at 60 ° C for 5 hours. After heating, it was left as it was for 10 minutes.
- Example 3 To the toluene (55 ml), the compound obtained in Example 3 (5.5 g, 0.0076 mol) and solid sodium hydrogen carbonate (9.5 g, 0.113 mol) were added. Then, under stirring, benzyloxycarbonyl chloride (18.0 g, 0.106 mol) was added dropwise at 70 to 80 ° C, and the mixture was heated at the same temperature for 4 hours. The reaction mixture was left at room temperature overnight. .
- Example 6 Fumarate Form (Formula ( ⁇ ) Synthesis of the fumarate of the compound to be prepared) To the dimethylimidazolidinone (35 ml) was added the compound obtained in Example 5 (10 g, 0.014 mol 1), isopropyl iodide (23.8 g. 0.14 mol), Triethylamine (16.95 g, 0.17mo1) was added and dissolved, heated at 70-75 ° C for 7-8 hours, and left overnight.
- Example 7 Crude Purification of Fumarate Form (Fumarate of Compound Represented by Formula ( ⁇ ))
- the compound (10.0 g, 0, 0123mo 1) obtained in Example 6 was dissolved in methanol (25 ml). Then, isopropanol (75 ml) was gradually added dropwise to this solution. The reaction mixture was stirred at room temperature for 1 hour, at 0 ° C for 1 hour, and at 15 ° C for 1 hour for crystallization and filtered under reduced pressure. A crude product of the title compound as white crystals (9.25 g, 92.5%) was obtained.
- Example 8 Purification of Fumarate Form (Fumarate of Compound Represented by Formula ( ⁇ ))
- the crude product (10 g, 0.0123 mol) obtained in Example 7 was converted to ethyl acetate (100 m 2). 1), and water (1.5 ml) was added dropwise.
- the mixture was stirred at 110 ° C for 1 hour and at 110 ° C for 4 hours, and filtered under reduced pressure to obtain a purified crystal of the title compound as white crystals (9.04 g, 90.4%).
- Example 9 Synthesis of Fumarate Form (Fumarate Salt of Compound Represented by Formula (VK))
- the compound obtained in Example 3 (9.5 g, 0.013mo1) in toluene (55 ml) was solid-form.
- Sodium bicarbonate (16.4 g, 0.195 mol) was added.
- benzyloxycarbonyl chloride (31.1 g, 0.183 mol 1) was added dropwise, and the mixture was heated at that temperature for 4 hours, and then left overnight at room temperature.
- Pyridine (6.94 g, 0.086 mol) was added to the reaction mixture, and the mixture was stirred for 30 minutes.
- the crystals are obtained by recrystallizing a fumarate of the compound represented by the formula (II) from methanol-isopropanol and recrystallizing it from ethyl acetate or a mixed solvent of ethyl acetate and water. Obtained crystal (C-type crystal,
- HP LC measurement conditions 100 ml was injected into a HPLC under the following conditions as 10 ml, and the residual ratio was determined from the peak area ratio of the sample used and the internal standard. HP LC measurement conditions
- Fig. 7 shows the results. Under these conditions, the A-type crystal had a residual rate of about 60% at 70%, whereas the C-type and D-type crystals had about 80% remaining at 70%.
- Example 11 Stability test of fumarate salt crystals of a compound represented by formula ( ⁇ ) under humidified conditions
- the stability of the compound represented by the formula ( ⁇ ) under humidification conditions due to the difference in the crystal form of the fumarate crystals was tested.
- the test was performed in the same manner as in Example 10 except that the severe test was performed in a desiccator at 80 ° C adjusted to a relative humidity of 75% with a saturated sodium chloride aqueous solution.
- Fig. 8 shows the results. As is clear from the graph, the humidification stability of Form A and Form D crystals was much higher than that of Form C crystals.
- the production method of the present invention comprises the following steps: (1) each reaction step required to obtain a purified final product; (2) acetylation of hydroxyl group at 2'-position of erythromycin A, formylation and hydroxylation of hydroxyl group at 4 "-position of erythromycin A in one pot
- the number of steps can be reduced compared to the conventional production method. It can be said that this is an excellent industrial production method, such as the possibility of reduction.
- the fumarate salt crystals of the compound represented by the formula (VH) obtained according to the present invention are superior in stability, as compared with conventionally obtained crystals, and are excellent as pharmaceuticals or pharmaceutical raw materials. Have quality.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Saccharide Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description

























Claims


















Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU66306/96A AU710368C (en) | 1995-08-03 | 1996-08-05 | Process for producing erythromycin derivatives |
| US09/011,142 US5959088A (en) | 1995-08-03 | 1996-08-05 | Process for producing erythromycin derivatives |
| EP96925989A EP0846697A4 (en) | 1995-08-03 | 1996-08-05 | PROCESS FOR THE PREPARATION OF ERYTHROMYCIN DERIVATIVES |
| CA002228254A CA2228254C (en) | 1995-08-03 | 1996-08-05 | Process for producing erythromycin derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP22959895 | 1995-08-03 | ||
| JP7/229598 | 1995-08-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1997006177A1 true WO1997006177A1 (fr) | 1997-02-20 |
Family
ID=16894695
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1996/002191 WO1997006177A1 (fr) | 1995-08-03 | 1996-08-05 | Procede d'elaboration de derives d'erythromycine |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US5959088A (ja) |
| EP (1) | EP0846697A4 (ja) |
| KR (1) | KR100354690B1 (ja) |
| CN (4) | CN1070864C (ja) |
| AU (1) | AU710368C (ja) |
| CA (1) | CA2228254C (ja) |
| IL (1) | IL119002A (ja) |
| MX (1) | MX9800904A (ja) |
| RU (1) | RU2266295C2 (ja) |
| TW (1) | TW486484B (ja) |
| WO (1) | WO1997006177A1 (ja) |
| ZA (1) | ZA966601B (ja) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PT102202B (pt) * | 1998-09-10 | 2001-04-30 | Hovione Sociedade Quimica S A | Processo para a purificacao de roxitromicina |
| CA2420847C (en) * | 2000-09-01 | 2008-02-05 | Chugai Seiyaku Kabushiki Kaisha | Process for producing erythromycin derivative |
| CA2425913C (en) * | 2000-10-12 | 2008-07-15 | Chugai Seiyaku Kabushiki Kaisha | Erythromycin derivative having novel crystal structures and processes for their production |
| DE60206921T2 (de) * | 2001-06-13 | 2006-07-20 | Ube Industries, Ltd., Ube | Verfahren zur herstellung von erythromycinverbindungen |
| US6916792B2 (en) * | 2001-06-13 | 2005-07-12 | Ube Industries, Ltd. | Process for preparing erythromycin compound |
| TW200302830A (en) * | 2002-01-11 | 2003-08-16 | Chugai Pharmaceutical Co Ltd | Anhydrate/hydrate of an erythromycin derivative and processes for preparing said anhydrate/hydrate |
| US7407941B2 (en) * | 2003-08-26 | 2008-08-05 | Pfizer, Inc. | N-desmethyl-N-substituted-11-deoxyerythromycin compounds |
| US20050113319A1 (en) * | 2003-08-26 | 2005-05-26 | Christopher Carreras | 11-Deoxy-6,9-ether erythromycin compounds |
| US7211568B2 (en) * | 2003-12-18 | 2007-05-01 | Kosan Biosciences Incorporated | 9-Desoxoerythromycin compounds as prokinetic agents |
| US7582611B2 (en) * | 2005-05-24 | 2009-09-01 | Pfizer Inc. | Motilide compounds |
| NZ568763A (en) * | 2005-12-08 | 2010-04-30 | Pfizer | Method for demethylating the 3'-dimethylamino group of erythromycin compounds |
| RU2422453C2 (ru) * | 2006-12-05 | 2011-06-27 | Пфайзер Инк. | Полиморфы мотилида |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4849980A (ja) * | 1971-10-30 | 1973-07-14 | ||
| JPS6399092A (ja) * | 1985-08-31 | 1988-04-30 | Kitasato Inst:The | エリスロマイシン誘導体およびその製造法 |
| JPH0656873A (ja) * | 1992-05-26 | 1994-03-01 | Chugai Pharmaceut Co Ltd | エリスロマイシン誘導体 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6056873A (ja) * | 1983-09-06 | 1985-04-02 | 東京貿易株式会社 | ボルト・ナットの締付方法 |
| US5008249A (en) * | 1985-08-31 | 1991-04-16 | Kitasato Kenkyusho | Therapeutic method of stimulating digestive tract contractile motion in mammals |
| US4672056A (en) * | 1985-11-12 | 1987-06-09 | Abbott Laboratories | Erythromycin A derivatives and method of use |
-
1996
- 1996-08-02 ZA ZA9606601A patent/ZA966601B/xx unknown
- 1996-08-03 TW TW085109381A patent/TW486484B/zh not_active IP Right Cessation
- 1996-08-04 IL IL11900296A patent/IL119002A/xx not_active IP Right Cessation
- 1996-08-05 CN CN96196915A patent/CN1070864C/zh not_active Expired - Fee Related
- 1996-08-05 KR KR10-1998-0700802A patent/KR100354690B1/ko not_active IP Right Cessation
- 1996-08-05 CA CA002228254A patent/CA2228254C/en not_active Expired - Fee Related
- 1996-08-05 CN CNB011012188A patent/CN1156489C/zh not_active Expired - Fee Related
- 1996-08-05 US US09/011,142 patent/US5959088A/en not_active Expired - Lifetime
- 1996-08-05 AU AU66306/96A patent/AU710368C/en not_active Ceased
- 1996-08-05 EP EP96925989A patent/EP0846697A4/en not_active Withdrawn
- 1996-08-05 CN CNB01101220XA patent/CN1141313C/zh not_active Expired - Fee Related
- 1996-08-05 RU RU98103451/04A patent/RU2266295C2/ru not_active IP Right Cessation
- 1996-08-05 WO PCT/JP1996/002191 patent/WO1997006177A1/ja active IP Right Grant
-
1998
- 1998-02-02 MX MX9800904A patent/MX9800904A/es not_active IP Right Cessation
-
2001
- 2001-01-11 CN CN01101219A patent/CN1316429A/zh active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4849980A (ja) * | 1971-10-30 | 1973-07-14 | ||
| JPS6399092A (ja) * | 1985-08-31 | 1988-04-30 | Kitasato Inst:The | エリスロマイシン誘導体およびその製造法 |
| JPH0656873A (ja) * | 1992-05-26 | 1994-03-01 | Chugai Pharmaceut Co Ltd | エリスロマイシン誘導体 |
Non-Patent Citations (4)
| Title |
|---|
| JONES P.H. ET AL: "Chemical modifications of erythromycin antibiotics. 3. Synthesis of 4'' and 11 esters of erythromycin A and B", JOURNAL OF MEDICINAL CHEMISTRY, vol. 15, no. 6, 1972, WASHINGTON, US, pages 631 - 634, XP002178020 * |
| KOGA H. ET AL: "POTENT, ACID-STABLE AND ORALLY ACTIVE MACROLIDE-TYPE MOTILIN RECEPTOR AGONISTS, GM-611 AND THE DERIVATIVES", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 4, no. 11, 1994, OXFORD, GB, pages 1347 - 1352, XP001037172 * |
| See also references of EP0846697A4 * |
| TADANIER J. ET AL: "SOME CHEMICAL AND STEREOCHEMICAL MODIFICATIONS OF THE ERYTHROMYCIN LACTONE RINGS", JOURNAL OF ORGANIC CHEMISTRY, vol. 39, no. 17, 1974, EASTON, US, pages 2495 - 2501, XP000670002 * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0846697A1 (en) | 1998-06-10 |
| MX9800904A (es) | 1998-11-30 |
| CA2228254C (en) | 2003-10-28 |
| CN1314358A (zh) | 2001-09-26 |
| AU710368C (en) | 2002-11-07 |
| IL119002A0 (en) | 1996-11-14 |
| IL119002A (en) | 2000-09-28 |
| CN1320606A (zh) | 2001-11-07 |
| CN1316429A (zh) | 2001-10-10 |
| TW486484B (en) | 2002-05-11 |
| KR100354690B1 (ko) | 2003-01-24 |
| ZA966601B (en) | 1997-02-19 |
| EP0846697A4 (en) | 2002-04-17 |
| RU2266295C2 (ru) | 2005-12-20 |
| CA2228254A1 (en) | 1997-02-20 |
| CN1141313C (zh) | 2004-03-10 |
| CN1196058A (zh) | 1998-10-14 |
| KR19990036134A (ko) | 1999-05-25 |
| AU710368B2 (en) | 1999-09-16 |
| CN1156489C (zh) | 2004-07-07 |
| US5959088A (en) | 1999-09-28 |
| AU6630696A (en) | 1997-03-05 |
| CN1070864C (zh) | 2001-09-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| FI80708C (fi) | Foerfarande foer selektiv metylation av derivat av erytromycin a. | |
| EP0619319B1 (en) | 5-0-desosaminylerythronolide derivative | |
| WO1997006177A1 (fr) | Procede d'elaboration de derives d'erythromycine | |
| JPH08239374A (ja) | 2,2−ジフルオロ−3−カルバモイルリボーススルホネート化合物およびβ−ヌクレオシドの製造方法 | |
| US6555677B2 (en) | Phase transfer catalyzed glycosidation of an indolocarbazole | |
| KR100317907B1 (ko) | 신규한 중간체, 이를 이용한 마크로라이드계 항생제의제조방법 | |
| WO2009053259A1 (en) | Process for the production of telithromycin | |
| EP0245013B1 (en) | Erythromycin derivatives | |
| JPS6360031B2 (ja) | ||
| JP3258914B2 (ja) | エリスロマイシン誘導体の製造方法 | |
| US4933439A (en) | Tylosin derivatives and processes for producing the same | |
| JP3978006B2 (ja) | エリスロマイシン誘導体の製造方法 | |
| JP2007501268A (ja) | 新規な9−デオキソ−9−ジヒドロ−9a−アザ−9a−ホモエリスロマイシンAの3−デクラジノシル9a−N−カルバモイル及び9a−N−チオカルバモイル誘導体 | |
| EP0511799A1 (en) | Pharmaceutical compounds | |
| JP2007204490A (ja) | エリスロマイシン誘導体の製造方法 | |
| WO2005044832A1 (en) | Process for the preparation of 1-chloro-3,5-di-o-acyl-2-deoxy-l-ribofuranoside derivatives | |
| AU2003261708A1 (en) | Process for producing indolopyrrolocarbazole derivative | |
| WO2004007518A1 (en) | Erythromycin a 9-o-pseudosaccharinyloxime derivatives and process for the preparation of clarithromycin using the same | |
| US6504035B1 (en) | 3-deoxy-desmycosin derivatives and process for their preparation | |
| US20050159371A1 (en) | Process for producing erythromycin a derivative | |
| JP2843695B2 (ja) | 10,11,12,13−テトラヒドロ−デスマイコシン誘導体、その製造法及びその医薬としての用途 | |
| JP2004217540A (ja) | 没食子酸配糖体の製造方法 | |
| JPH0529038B2 (ja) | ||
| JPH07224067A (ja) | 7−アルコキシまたはヒドロキシスタウロスポリン系物質の製造法 | |
| JPS59222500A (ja) | マクロライド抗生物質誘導体の製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 96196915.6 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AU AZ BB BG BR BY CA CN CU CZ EE GE HU IL IS KE KG KR KZ LK LR LS LT LV MD MG MK MN MW MX NO NZ PL RO RU SD SG SI SK TJ TM TR TT UA UG US UZ VN AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): KE LS MW SD SZ UG AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2228254 Country of ref document: CA Ref document number: 2228254 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1996925989 Country of ref document: EP Ref document number: PA/A/1998/000904 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1019980700802 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09011142 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1996925989 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019980700802 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1019980700802 Country of ref document: KR |