RU2241709C2 - Производные азотсодержащих гетероциклических соединений и способ лечения расстройств, характеризующихся поражением нейронов - Google Patents
Производные азотсодержащих гетероциклических соединений и способ лечения расстройств, характеризующихся поражением нейронов Download PDFInfo
- Publication number
- RU2241709C2 RU2241709C2 RU2002103340/04A RU2002103340A RU2241709C2 RU 2241709 C2 RU2241709 C2 RU 2241709C2 RU 2002103340/04 A RU2002103340/04 A RU 2002103340/04A RU 2002103340 A RU2002103340 A RU 2002103340A RU 2241709 C2 RU2241709 C2 RU 2241709C2
- Authority
- RU
- Russia
- Prior art keywords
- compound
- mmol
- mixture
- nmr
- compounds
- Prior art date
Links
- 0 I*N1*CCCCCC1 Chemical compound I*N1*CCCCCC1 0.000 description 3
- CVPKIRXMXAEIDX-JHJVBQTASA-N CCC[C@@H](C)[C@H]1C[O](C(C)C)[C@H](C)C1 Chemical compound CCC[C@@H](C)[C@H]1C[O](C(C)C)[C@H](C)C1 CVPKIRXMXAEIDX-JHJVBQTASA-N 0.000 description 1
- RRYDMLZIKAVBRO-INIZCTEOSA-N CCOC(Cc1c[s]c([C@H](CCC2)N2C(OCc2ccccc2)=O)n1)=O Chemical compound CCOC(Cc1c[s]c([C@H](CCC2)N2C(OCc2ccccc2)=O)n1)=O RRYDMLZIKAVBRO-INIZCTEOSA-N 0.000 description 1
- KVERNDRXLDWSFC-NSHDSACASA-N NC([C@H](CCC1)N1C(OCc1ccccc1)=O)=S Chemical compound NC([C@H](CCC1)N1C(OCc1ccccc1)=O)=S KVERNDRXLDWSFC-NSHDSACASA-N 0.000 description 1
- BUASYYXQIFKXPH-SFHVURJKSA-N O=C(c1c([C@H](CCC2)N2C(OCc2ccccc2)=O)[o]cn1)OCc1cnccc1 Chemical compound O=C(c1c([C@H](CCC2)N2C(OCc2ccccc2)=O)[o]cn1)OCc1cnccc1 BUASYYXQIFKXPH-SFHVURJKSA-N 0.000 description 1
- NNNOJFNQJJZUNL-LBPRGKRZSA-N OC(c1c([C@H](CCC2)N2C(OCc2ccccc2)=O)[o]cn1)=O Chemical compound OC(c1c([C@H](CCC2)N2C(OCc2ccccc2)=O)[o]cn1)=O NNNOJFNQJJZUNL-LBPRGKRZSA-N 0.000 description 1
- TVBMSQGBIKCJQG-AWEZNQCLSA-N OCc1c[s]c([C@H](CCC2)N2C(OCc2ccccc2)=O)n1 Chemical compound OCc1c[s]c([C@H](CCC2)N2C(OCc2ccccc2)=O)n1 TVBMSQGBIKCJQG-AWEZNQCLSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Cardiology (AREA)
- Emergency Medicine (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Physical Education & Sports Medicine (AREA)
- Heart & Thoracic Surgery (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Psychology (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyrrole Compounds (AREA)
Abstract
Изобретение относится к новым производным азотсодержащих гетероциклических соединений формулы

или их фармацевтически приемлемым солям, где R1 представляет Н, COCOR2, COOR3 или SO2R3, R2 представляет С1-6алкил, С1-6алкенил, С5-7циклоалкил, 2-тиенил, 3-тиенил, фенил или замещенный фенил, R3 представляет фенилалкил,  представляет насыщенное пятичленное азотсодержащее гетероциклическое кольцо с одним атомом азота или бензоконденсированное насыщенное шестичленное азотсодержащее гетероциклическое кольцо;
представляет насыщенное пятичленное азотсодержащее гетероциклическое кольцо с одним атомом азота или бензоконденсированное насыщенное шестичленное азотсодержащее гетероциклическое кольцо;  представляет оксазол, оксадиазол или тиазол, А связан с атомом углерода пятичленного гетероароматического кольца и представляет COO(CH2)mAr,
представляет оксазол, оксадиазол или тиазол, А связан с атомом углерода пятичленного гетероароматического кольца и представляет COO(CH2)mAr,  , где R1 имеет значения, указанные выше или представляет CONR4(CH2)mAr или (CH2)mO(CH2)nAr, причем R1 не может быть COCOR2 или SO2R3, R4 представляет Н или С1-4алкил, Ar представляет 2-, 3- или 4-пиридил, m равно 1-4, n равно 0-4. Полученные соединения обладают нейротрофической активностью и могут найти применение для лечения и предупреждения нервных болезней. 4 с. и 11 з.п. ф-лы, 3 табл., 1 ил.
, где R1 имеет значения, указанные выше или представляет CONR4(CH2)mAr или (CH2)mO(CH2)nAr, причем R1 не может быть COCOR2 или SO2R3, R4 представляет Н или С1-4алкил, Ar представляет 2-, 3- или 4-пиридил, m равно 1-4, n равно 0-4. Полученные соединения обладают нейротрофической активностью и могут найти применение для лечения и предупреждения нервных болезней. 4 с. и 11 з.п. ф-лы, 3 табл., 1 ил.
Description
Настоящее изобретение относится к новым пирролидинам и пиперидинам, обладающим нейротрофической активностью. Эти соединения, а также относящиеся к ним композиции и способы полезны для лечения и предупреждения нервных болезней, таких как болезнь Паркинсона, болезнь Альцгеймера, удар, рассеянный склероз, боковой амиотрофический склероз, диабетическая невропатия и паралич Белла.
Предшествующий уровень техники
Нейродегенаративные болезни
Нейродегенеративные болезни представляют собой главную угрозу для здоровья людей во всем мире. Наиболее серьезным из таких заболеваний является болезнь Альцгеймера (БА), которая является главной причиной развития деменции у пожилых людей и занимает четвертое место среди основных причин смертности в Соединенных Штатах. По оценкам специалистов в США болезнью Альцгеймера в целом страдают от двух до трех миллионов человек и более 5% населения в возрасте свыше 65 лет. Хотя этиология БА пока еще точно не установлена, однако это заболевание характеризуется присутствием большого числа амилоидных бляшек и нейрофибриллярных клубков в области головного мозга, ответственного за познавательную функцию, и дегенерацией холинергических нейронов, восходящих от базального переднего головного мозга в области коры головного мозга и гиппокампа. В настоящее время не существует какой-либо эффективной терапии БА (Brinton, R.D. & Yamazaki R.S., Pharm. Res., 1998, 15, 386-398).
Аналогично БА болезнь Паркинсона (БП) представляет собой прогрессирующее дегенеративное заболевание центральной нервной системы (ЦНС). Частота возникновения этого заболевания среди всего населения составляет приблизительно 2%. При БП дегенерация допаминергических нейронов черной субстанции приводит к снижению уровней допамина в области головного мозга, контролирующего произвольные движения, то есть в области полосатого тела. Поэтому стандартное лечение этой болезни сосредоточено на введении агентов, таких как L-допа и бромокриптин, которые восполняют уровни допамина в пораженных областях головного мозга. Однако схемы введения допаминергических средств теряют свою эффективность по мере гибели нервных клеток и прогрессирования болезни. В это время на ранних стадиях развития БП наблюдается непроизвольный тремор, затем наступает период затруднения движения и, наконец, неподвижность. Поэтому разработка альтернативной терапии является крайне необходимой (Pahwa R. & Roller W.C. Drugs Today, 1998, 34, 95-105).
Нейродегенеративные заболевания соматосенсорной нервной системы также представляют собой класс приводящих к слабоумию и потенциально летальных состояний. Боковой амиотрофический склероз (БАС) представляет собой смертельное заболевание, характеризующееся прогрессирующей дегенерацией центральных и периферических моторных нейронов. Хотя точная этиология БАС неизвестна, однако наибольшее распространение получили теории, в которых предполагается, что стимулирующими факторами являются эксцитотоксичность и/или окислительный стресс. Рилузол является первым лекарственным средством, одобренным для лечения БАС, имеющимся в продаже. Он обладает антиэксцитотоксическими свойствами, и было показано, что он увеличивает степень выживаемости пациентов с БАС. Однако это лекарственное средство не является излечивающим, и в настоящее время проводятся клинические испытания альтернативных агентов (Louvel, E.Hugon, J. & Doble A., Trends Pharmacol. Sci., 1997, 18, 196-203).
Периферические невропатии занимают второе место среди метаболических и сосудистых состояний. В частности приблизительно 30% пациентов с сахарным диабетом страдают некоторыми формами периферической невропатии, которые могут поражать малые миелиновые волокна, вызывая потерю способности ощущать боль и температуру, либо большие волокна, вызывая дефекты двигательной и соматосенсорной функции. Фармакотерапевтическое вмешательство направлено на симптоматическое лечение, и наилучшим способом лечения и предупреждения болезни остается поддержание нормальных уровней глюкозы в крови посредством соблюдения диеты и введения инсулина (Biessels G.J. & Van Dam P.S. Neurosci. Res. Commun., 1997, 20, 1-10).
Основная масса данных, имеющихся в настоящее время, позволяет предположить, что дефицит в уровнях некоторых белковых факторов роста или нейротрофических факторов может играть ключевую роль в развитии нейродегенеративных заболеваний как периферической, так и центральной нервной системы (Tomlinson D.R., Fernyhough, P. & Diemel, L.T., Diabetes, 1997, 46 (suppl.2) S43-S-49; Hamilton, G.S., Chem. Ind., (London) 1998, 4, 127-132; Louvel, E. Hugon, J. & Doble, A. Trends Pharmacol. Sci., 1997, 18, 196-203; Ebadi, M. et al., Neurochem. Int., 1997, 30, 347-374).
Эти нейротрофические факторы могут быть разделены на два структурных класса: 1) нейтротропины, включая фактор роста нервной ткани (NGF); нейротрофический фактор роста, происходящий от глиальных клеток (GDNF); нейротрофический фактор головного мозга (BDNF); нейротропин 3 (NT-3); нейротропин 4/5 (NT-4/5); нейротропин 2 (NT-2) и цилиарный нейротрофический фактор (CNTF), который относится к цитокиновому семейству молекул. Все нейротрофические факторы промотируют отрастание нейритов, индуцируют дифференцировку и подавляют запрограммированную гибель или апоптоз клеток в конкретных субпопуляциях периферических и центральных нейронов. Так, например, NGF обладает трофическим действием на симпатические и сенсорные нейроны спинно-мозговых узлов и на холинергические нейроны медиальной перегородки в ЦНС, что позволяет предположить о его потенциальной терапевтической эффективности при БА. CNTF обладает трофическим действием на широкое поперечное сечение нейронов, включая парасимпатические нейроны, сенсорные нейроны, симпатические нейроны, двигательные нейроны, нейроны мозжечка, нейроны гиппокампа и нейроны перегородки. Особенно интересным является тот факт, что CNTF частично предупреждает атрофию скелетной мышцы после поражения нервной ткани, но он не оказывает воздействия на иннервированную мышцу, что свидетельствует о том, что CNTF действует, главным образом, при патологическом состоянии. В результате этого в настоящее время проводится исследование CNTF на его эффекты при скелетно-мышечных заболеваниях, подобных БАС.
Клиническое применение белковых нейротрофических агентов представляет серьезные трудности из-за их ограниченной биодоступности, особенно в ЦНС. Необходимость введения указанных агентов непосредственно в головной мозг для индуцирования терапевтического эффекта является относительно рискованным и трудным способом введения.
Химические агенты
Lyons W.E. и др. (Proc. Natl. Acad. Sci., 1994, 91(8), 3191-5) описывают нейротрофические эффекты иммуносуппрессорного лекарственного средства FK506, которое обладает нейротрофической активностью в культурах клеток РС12 и в чувствительных ганглиях:

Vertex Pharmaceuticals, Inc. ("Vertex") в южноафриканской заявке 964852 описывает соединения, которые, как указывается, пригодны для ингибирования ротамазной активности иммунофилина FKBP12 и стимуляции роста нейритов в клеточных культурах. Эти соединения характеризуются следующей структурой:

В заявке РСТ WO 92/19593 (Vertex) описана серия соединений, которые, как указывается, пригодны для ингибирования ротамазной активности FK506-связывающих белков (FKBP) и ингибирования активации Т-клеток. Эти соединения представлены следующей структурой:

В заявке РСТ WO 94/07858 (Vertex) описана серия соединений, которые, как указывается, пригодны в качестве сенсибилизаторов раковых клеток, резистентных к множеству лекарственных средств, для поддержания, увеличения или сохранения восприимчивости клеток к терапевтическим или профилактическим агентам. Эти соединения представлены следующей структурой:

В совместных патентах Guilford Pharmaceuticals. Inc., GPI NIL Holdings, Inc., & Johns Hopkins Uneversity School of Medicine (под общим названием "Guilford") описаны соединения, которые, как указывается, пригодны для ингибирования активности иммунофилинов типа FKBP, для промотирования роста и регенерации нейронов и для лечения неврологических расстройств.
В частности, в патенте Guilford США №5696135 и в заявке РСТ WO 96/40140 описан способ применения производных пипеколиновой кислоты, родственных FK506 и рапамицину, для лечения неврологического расстройства у животного. Описанные соединения полезны для ингибирования ротамазной активности иммунофилинов типа FKBP, для промотирования роста нейронов спинно-мозговых узлов у кур in vitro и для промотирования репарации поврежденного седалищного нерва у крыс.
В патенте США №5798355 (Guilford) описан метод применения макроциклических и ациклических производных пипеколиновой кислоты для ингибирования ферментативной активности иммунофилинов типа FKBP и для промотирования роста и регенерации нейронов.
В частности, в патентах США № 5614547 и 5795908 (Guilford) и в заявке РСТ WO 96/40633 описана серия соединений N-глиоксил-пролилового сложного эфира, которые, как указывается, пригодны для промотирования ротамазной активности иммунофилинов типа FKBP-12, для промотирования роста и регенерации нейронов и для лечения неврологических расстройств. Эти соединения представлены следующей структурой:

В патенте США №5801197 (Guilford) и в заявке РСТ WO 97/16190 описана серия не обладающих иммуносуппрессорным действием производных пипеколиновой кислоты, которые, как указывается, полезны для лечения поврежденной нервной ткани у животных. Эти соединения представляют собой аналоги серии:


В патенте США №5721256 (Guilford) описаны соединения, которые, как указывается, пригодны для ингибирования ротамазной активности FKBP, для промотирования роста и регенерации нейронов и для воздействия на нейронную активность у животных. Эта серия сульфонамидных соединений характеризуется следующей структурой:

В патенте США №5801187 (Guilford) и в заявке РСТ WO 98/13355 описана серия соединений гетероциклических сложных эфиров и амидов, которые, как указывается, пригодны для ингибирования ротамазной активности FKBP, для промотирования роста и регенерации нейронов и для воздействия на нейронную активность у животных. Эти соединения характеризуются следующей структурой:

В заявке РСТ WO 98/13343 (Guilford) описана серия соединений гетероциклических сложных тиоэфиров и кетонов, которые, как указывается, пригодны для ингибирования ротамазной активности FKBP, для промотирования роста и регенерации нейронов и для воздействия на нейронную активность у животных. Эти соединения представлены следующей структурой:

В заявке РСТ WO 98/29116 (Guilford) описана серия N-связанных сульфонамидных соединений гетероциклических сложных тиоэфиров, которые, как указывается, пригодны для ингибирования ротамазной активности FKBP, промотирования роста и регенерации нейронов и для воздействия на нейронную активность у животных. Эти соединения характеризуются следующей структурой:

В заявке РСТ WO 98/29117 (Guilford) описана серия N-связанных карбамидных и карбаматных соединений гетероциклических сложных тиоэфиров, которые, как указывается, пригодны для ингибирования ротамазной активности FKBP, для промотирования роста и регенерации нейронов и для воздействия на нейронную активность у животных. Эти соединения представлены следующей структурой:

В заявке РСТ WO 98/37882 (Guilford) описан метод применения небольших молекул карбаматных и карбамидных соединений, которые, как указывается, пригодны для ингибирования ротамазной активности иммунофилинов типа FKBP и для промотирования роста и регенерации нейронов. Эти соединения характеризуются следующей структурой:
В заявке РСТ WO 98/37885 (Guilford) описана серия N-оксидных соединений гетероциклических сложных эфиров, амидов, тиоэфиров и кетонов, которые, как указывается, пригодны для ингибирования ротамазной активности FKBP, для промотирования роста и регенерации нейронов и для лечения неврологических расстройств у животных. Эти соединения характеризуются следующей структурой:

В заявке РСТ WO 98/25950 (Guilford) описана серия тетра- и пентапептидных соединений, содержащих, по крайней мере, два пролиновых остатка, которые, как указывается, пригодны для ингибирования ротамазной активности циклофилина, для промотирования роста и регенерации нейронов и для воздействия на нейронную активность у животных.
В патентах и публикациях совместно с Ariad Gene Therapeutics, Inc. ("Ariad") описаны агенты, которые, как указывается, полезны для полимеризации иммунофилинов, для генной терапии, для активации транскрипции генов, для запуска апоптоза или для стимуляции других биологических событий в сконструированных клетках, растущих в культуре или в целых организмах.
В частности, в заявках РСТ WO 96/06097, WO 97/31898, WO 97/31899 (Ariad) и Holt, D.A. et al. (Bioorg. Med. Chem. 1998, 6(8), 1309-1335) описаны соединения, которые включают серию полимеризующих агентов, представленных следующей структурой:

В патентах совместно с Cephalon, Inc. & Kyowa Hakko Kogyo Co., Ltd. (под общим названием "Cephalon") описаны небольшие молекулы нейротрофических агентов, которые имеют потенциальное клиническое применение для лечения нейродегенеративных заболеваний.
В частности, в патентах США №5756494, 5621101 и 5461146 и в заявках РСТ WO 96/13506 и WO 94/02488 (Cephalon) описана серия индолкарбазольных ингибиторов протеинкиназы, которые, как указывается, обладают нейротрофическим действием в центральных холинергических нейронах, в спинно-мозговых узлах и в спинном мозге. Эти соединения представлены следующей структурой:

Ни один из известных агентов, обсуждаемых в данной заявке, не обладает какой-либо терапевтической или профилактической эффективностью для лечения нейродегенеративных расстройств у человека. Таким образом, в настоящее время существует крайняя необходимость в получении агентов, обладающих такой эффективностью.
Краткое описание графического материала
На чертеже показана биологическая активность in vivo соединения 30 настоящего изобретения с использованием модели сдавливания лицевого нерва крысы. В этой модели сдавливание лицевого нерва вызывает паралич мышц усов на одной стороне головы. Необработанный лицевой нерв на другой стороне головы служит в качестве внутреннего контроля. Обработка соединением 30 продемонстрировала, что подвижность усов на парализованной стороне восстанавливается более быстро по сравнению с контролем, обработанным носителем, и внутренним контролем. На этой фигуре показана степень восстановления подвижности усов на парализованной стороне по сравнению с контролем, обработанным носителем, и внутренним контролем.
Краткое описание изобретения
Настоящее изобретение относится к соединению, имеющему структуру:

или к его фармацевтически приемлемой соли,
где (a) R1 выбран из группы, состоящей из Н, COCOR2, COOR3 и SO2R3, причем
(i) R2 выбран из группы, состоящей из прямого или разветвленного O-C1-6алкила, прямого или разветвленного C1-6алкила, прямого или разветвленного C1-6алкенила, С5-7циклоалкила, 2-тиенила, 3-тиенила или фенила, где указанный фенил имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из Н, низшего алкила, низшего алкоксила, гидроксила и галогена,
(ii) R3 представляет фенилалкил, где указанный фенил имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из Н, низшего алкила, низшего алкоксила, гидроксила и галогена;
(b)  представляет четырех-шестичленное гетероциклическое кольцо, где не более чем один атом кольца представляет О или S;
представляет четырех-шестичленное гетероциклическое кольцо, где не более чем один атом кольца представляет О или S;
(c)  представляет пятичленное гетероциклическое кольцо, имеющее от двух до трех гетероатомов, выбранных из группы, состоящей из N, О и S, где, по крайней мере, одним из указанных гетероатомов является N;
представляет пятичленное гетероциклическое кольцо, имеющее от двух до трех гетероатомов, выбранных из группы, состоящей из N, О и S, где, по крайней мере, одним из указанных гетероатомов является N;
(d) А выбран из группы, состоящей из CO(CH2)mAr,  (где R1 является таким же или отличается от R1, определенного в части (а)), CONR4(CH2)mAr, (CH2)mO(СН2)nAr и (СН2)nAr, причем:
(где R1 является таким же или отличается от R1, определенного в части (а)), CONR4(CH2)mAr, (CH2)mO(СН2)nAr и (СН2)nAr, причем:
(i) R4 представляет Н или С1-4алкил;
(ii) Ar выбран из группы, состоящей из 2-пиридила, 3-пиридила, 4-пиридила и фенила, где указанный фенил имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из Н, низшего алкила, низшего алкоксила, гидроксила и галогена;
(iii) m равно 1-4;
(iv) n равно 0-4.
Настоящее изобретение также относится к соединению, имеющему структуру:

или к его фармацевтически приемлемой соли, где R" представляет прямой или разветвленный С1-4алкил.
Настоящее изобретение также относится к соединению, имеющему структуру:

или к его фармацевтически приемлемой соли,
где (a) R1 выбран из группы, состоящей из Н, COCOR2, COOR3 и SO2R3, причем
(i) R2 выбран из группы, состоящей из прямого или разветвленного O-C1-6алкила, прямого или разветвленного C1-6алкила, прямого или разветвленного C1-6алкенила, С5-7циклоалкила, 2-тиенила, 3-тиенила или фенила, где указанный фенил имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из Н, низшего алкила, низшего алкоксила, гидроксила и галогена,
(ii) R3 представляет фенилалкил, где указанный фенил имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из Н, низшего алкила, низшего алкоксила, гидроксила и галогена;
(b)  представляет четырех-шестичленное гетероциклическое кольцо, где не более чем один атом кольца представляет О или S;
представляет четырех-шестичленное гетероциклическое кольцо, где не более чем один атом кольца представляет О или S;
(c)  представляет пятичленное гетероциклическое кольцо, имеющее от двух до трех гетероатомов, выбранных из группы, состоящей из N, О и S, где, по крайней мере, одним из указанных гетероатомов является N;
представляет пятичленное гетероциклическое кольцо, имеющее от двух до трех гетероатомов, выбранных из группы, состоящей из N, О и S, где, по крайней мере, одним из указанных гетероатомов является N;
(d) В представляет (СН2)nAr или  , где n равно 0-4.
, где n равно 0-4.
Настоящее изобретение относится к способу стимуляции роста нейронов, включающему контактирование нейронов с эффективным количеством одного из соединений настоящего изобретения. Настоящее изобретение также относится к фармацевтической композиции, содержащей одно из соединений настоящего изобретения и фармацевтически приемлемый носитель.
Кроме того, настоящее изобретение относится к способу лечения индивидуума, страдающего расстройством, характеризующимся поражением нервной ткани, вызванным заболеванием или травмой, включающему введение указанному индивидууму терапевтически эффективного количества фармацевтической композиции настоящего изобретения. И наконец, настоящее изобретение относится к способу ингибирования у индивидуума возникновения расстройства, характеризующегося поражением нервной ткани, вызванным заболеванием, включающему введение указанному индивидууму профилактически эффективного количества фармацевтической композиции настоящего изобретения.
Подробное описание изобретения
Настоящее изобретение относится к новым пирролидинам и пиперидинам, обладающим неожиданно обнаруженной нейротрофической активностью. Эти соединения, а также относящиеся к ним композиции и способы полезны для лечения и предупреждения нервных болезней, таких как болезнь Паркинсона, болезнь Альцгеймера, удар, рассеянный склероз, боковой амиотрофический склероз, диабетическая невропатия и паралич Белла.
Более конкретно настоящее изобретение относится к соединению, имеющему структуру:

или к его фармацевтически приемлемой соли, где
(а) R1 выбран из группы, состоящей из Н, COCOR2, COOR3 и SO2R3, причем
(i) R2 выбран из группы, состоящей из прямого или разветвленного O-C1-6алкила, прямого или разветвленного C1-6алкила, прямого или разветвленного C1-6алкенила, С5-7циклоалкила, 2-тиенила, 3-тиенила или фенила, где указанный фенил имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из Н, низшего алкила, низшего алкоксила, гидроксила и галогена,
(ii) R3 представляет фенилалкил, где указанный фенил имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из Н, низшего алкила, низшего алкоксила, гидроксила и галогена;
(b)  представляет четырех-шестичленное гетероциклическое кольцо, где не более чем один атом кольца представляет О или S;
представляет четырех-шестичленное гетероциклическое кольцо, где не более чем один атом кольца представляет О или S;
(с)  представляет пятичленное гетероциклическое кольцо, имеющее от двух до трех гетероатомов, выбранных из группы, состоящей из N, О и S, где, по крайней мере, одним из указанных гетероатомов является N;
представляет пятичленное гетероциклическое кольцо, имеющее от двух до трех гетероатомов, выбранных из группы, состоящей из N, О и S, где, по крайней мере, одним из указанных гетероатомов является N;
(а) А выбран из группы, состоящей из COO(CH2)mAr,  (где R1 является таким же или отличается от R1, определенного в части (a)), CONR4(СН2)mAr, (СН2)mО(СН2)nAr и (СН2)nAr, причем
(где R1 является таким же или отличается от R1, определенного в части (a)), CONR4(СН2)mAr, (СН2)mО(СН2)nAr и (СН2)nAr, причем
(i) R4 представляет Н или С1-4алкил;
(ii) Ar выбран из группы, состоящей из 2-пиридила, 3-пиридила, 4-пиридила и фенила, где указанный фенил имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из Н, низшего алкила, низшего алкоксила, гидроксила и галогена;
(iii) m равно 1-4; и
(iv) n равно 0-4.
В одном из вариантов осуществления изобретения указанные соединения имеют нижеследующую структуру, где все R1 являются одинаковыми или различными.

В предпочтительном варианте осуществления изобретения указанное соединение выбрано из группы, состоящей из соединений настоящего изобретения 4, 14, 30, 31, 35, 38, 43, 44, 55, 56, 58, 60, 62 и 64.
Настоящее изобретение также относится к соединению, имеющему структуру:

или к его фармацевтически приемлемой соли, где R" представляет прямой или разветвленный С1-4алкил.
Настоящее изобретение также относится к соединению, имеющему структуру:

или к его фармацевтически приемлемой соли,
где (а) R1 выбран из группы, состоящей из Н, COCOR2, COOR3 и SO2R3, причем
(i) R2 выбран из группы, состоящей из прямого или разветвленного O-C1-6алкила, прямого или разветвленного C1-6алкила, прямого или разветвленного C1-6алкенила, С5-7циклоалкила, 2-тиенила, 3-тиенила или фенила, где указанный фенил имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из Н, низшего алкила, низшего алкоксила, гидроксила и галогена,
(ii) R3 представляет фенилалкил, где указанный фенил имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из Н, низшего алкила, низшего алкоксила, гидроксила и галогена;
(b)  представляет четырех-шестичленное гетероциклическое кольцо, где не более чем один атом кольца представляет О или S;
представляет четырех-шестичленное гетероциклическое кольцо, где не более чем один атом кольца представляет О или S;
(c)  представляет пятичленное гетероциклическое кольцо, имеющее от двух до трех гетероатомов, выбранных из группы, состоящей из N, О и S, где, по крайней мере, одним из указанных гетероатомов является N;
представляет пятичленное гетероциклическое кольцо, имеющее от двух до трех гетероатомов, выбранных из группы, состоящей из N, О и S, где, по крайней мере, одним из указанных гетероатомов является N;
(d) В представляет (CH2)nAr или  , где n равно 0-4.
, где n равно 0-4.
В предпочтительном варианте осуществления изобретения указанное соединение выбрано из группы, состоящей из соединений настоящего изобретения 24, 26, 37 и 59.
Соединения настоящего изобретения могут быть выделены и использованы в виде свободных оснований. Они могут быть также выделены и использованы в виде фармацевтически приемлемых солей. Примерами таких солей являются соли бромистоводородной, йодистоводородной, хлористоводородной, перхлорной, серной, малеиновой, фумаровой, яблочной, винной, лимонной, бензойной, миндальной, метансульфоновой, гидроэтансульфоновой, бензолсульфоновой, щавелевой, пальмовой, 2-нафталинсульфоновой, п-толуолсульфоновой, циклогексансульфамовой и сахарной кислот.
Настоящее изобретение также относится к способу стимуляции роста нейронов, включающему контактирование нейронов с эффективным количеством одного из соединений настоящего изобретения. Такое контактирование может быть осуществлено, например, in vitro, ex vivo или in vivo.
Кроме того, настоящее изобретение относится к фармацевтической композиции, включающей одно из соединений настоящего изобретения и фармацевтически приемлемый носитель.
Фармацевтически приемлемые носители хорошо известны специалистам и такими носителями являются, но не ограничиваются ими, приблизительно 0,01-0,1 М, а предпочтительно, 0,05 М фосфатный буфер или 0,8% физиологический раствор. Указанные фармацевтически приемлемые носители могут представлять собой водные или безводные растворы, суспензии и эмульсии. Примерами безводных растворителей являются пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, и инъецируемые органические сложные эфиры, такие как этилолеат. Водными носителями являются вода, этанол, спиртовые/водные растворы, глицерин, эмульсии или суспензии, включая физиологический раствор и забуференные среды. Носителями для перорального введения могут быть эликсиры, сиропы, капсулы, таблетки и т.п. Типичным твердым носителем являются инертные вещества, такие как лактоза, крахмал, глюкоза, метилцеллюлоза, стеарат магния, дифосфат кальция, маннит и т.п. Носителями для парентерального введения являются раствор хлорида натрия, декстроза Рингера, декстроза и хлорид натрия, лактированные растворы Рингера и жирные масла. Носителями для внутривенного введения являются жидкие и питательные добавки, электролитные добавки, такие как добавки на основе декстрозы Рингера и т.п. Могут присутствовать также консерванты и другие добавки, такие как противомикробные агенты, антиоксиданты, хелатобразователи, инертные газы и т.п. При необходимости все носители могут быть смешаны с дезинтегрантами, разбавителями, гранулирующими агентами, смазывающими веществами, связующими агентами и т.п., стандартными методами, известными специалистам.
Кроме того, настоящее изобретение относится к способу лечения индивидуума, страдающего расстройством, характеризующимся поражением нейронов, вызванным заболеванием или травмой, включающий введение индивидууму терапевтически эффективного количества фармацевтической композиции настоящего изобретения.
Используемый здесь термин "индивидуум" означает, но не ограничивается им, любое животное или искусственно модифицированное животное. В предпочтительном варианте осуществления настоящего изобретения указанным индивидуумом является человек.
Введение фармацевтической композиции настоящего изобретения может быть проведено или осуществлено различными методами, известными специалистам. Соединения настоящего изобретения могут быть введены, например, внутривенно, местно, внутримышечно, перорально, подкожно и непосредственно в цереброспинальную жидкость и/или в головной мозг. В предпочтительном варианте осуществления изобретения фармацевтическую композицию настоящего изобретения вводят перорально. Кроме того, такое введение может предусматривать прием индивидуумом большого количества доз в течение подходящего периода времени. Такие схемы введения могут быть определены рутинными методами.
Существует множество заболеваний, характеризующихся поражением нейронов, и такими заболеваниями являются, но не ограничиваются ими: болезнь Альцгеймера, болезнь Пика, болезнь распространенных телец Леви, прогрессирующий супернуклеарный паралич (синдром Стила-Ричардсона), мультисистемная дегенерация (синдром Шая-Дрейджера), болезни, связанные с поражением мотонейронов, включая боковой амиотрофический склероз, дегенеративная атаксия, кортикальная базальная дегенерация, комплекс БАС-паркинсонизм-деменция, обнаруженный на острове Гуам, подострый склерозирующий панэнцефалит, болезнь Гентингтона, болезнь Паркинсона, синуклеинопатии, первичная прогрессирующая афазия, стриатонигральная дегенерация, болезнь Мачадо-Джозефа/спиноцеребеллярная атаксия типа 3 и оливопонтоцеребеллярная дегенерация, расстройство Жиль де ля Туретта, бульбарный и псевдобульбарный паралич, спинальная и спинобульбарная атрофия мышц (болезнь Кеннеди), первичный боковой склероз, наследственная спастическая параплегия, болезнь Верднига-Гоффмана, болезнь Кугельберга-Веландера, болезнь Тея-Сакса, болезнь Сандхоффа, наследственное спастическое заболевание, болезнь Вольфарта-Кугельберга-Веландера, спастический парапарез, прогрессирующая многоочаговая лейкоэнцефалопатия и прионные болезни (включая болезнь Крейцфельдта-Якоба, Герцмана-Штройсслера-Шрайнкера, болезнь Куру и смертельная наследственная бессонница).
Другими расстройствами являются, но не ограничиваются ими, диффузное заболевание белого вещества (болезнь Бинсвангера), травма головы и диффузное повреждение головного мозга, повреждение спинного мозга, внутричерепные и внутрипозвоночные повреждения (включая, но не ограничиваясь ими, контузию, пенетрацию, смещение, сдавливание и разрыв ткани), удар, возникающий в результате церебральной ишемии или инфаркта, эмболическая окклюзия и тромботическая окклюзия и внутричерепная геморрагия любого типа (включая, но не ограничиваясь ими, эпидуральную, субдуральную, субарахноидальную и внутрицеребральную геморрагии).
Другими расстройствами являются, но не ограничиваются ими, демиелинизирующая болезнь, такая как рассеянный склероз; полирадикулоневрит (синдром Гийена-Барре); подострая демиелинизирующая полиневропатия; поражения головного мозга, индуцированные острым рассеянным энцефаломиелитом, острым гемморрагическим лейкоэнцефалитом или системной красной волчанкой; синдром Бехчета, ассоциированный с многоочаговыми поражениями головного мозга, невропатией и/или миелопатией; саркоидоз, ассоциированный с поражением или атрофией нервов или миелопатией; бактериальные или вирусные инфекции, приводящие к повреждению головного мозга, спинного мозга, нервной системы, менингорадикулиту, и/или миелопатии; подострая комбинированная дегенерация; поперечный миелит; наследственная невропатия Лебера; подострая некротическая энцефалопатия (болезнь Ли); митохондриальная энцефалопатия с демиелинизацией; метахромальная лейкодистрофия; болезнь Краббе; болезнь Фабри; адренолейкодистрофия; нейромиелит зрительного нерва (синдром Девика); демиелинизирующая Шваннопатия; черепная и периферическая невропатии, включая, но не ограничиваясь ими, невропатию Дежерина-Сотта и ее разновидности; болезнь Шарко-Мари-Тута и ее разновидности; наследственные полиневропатии; сенсорно-двигательные невропатии; аксонная невропатия; адреномиелоневропатия; болезнь Рефсума; невропатии, вызванные порфирией, острой или хронической интоксикацией токсинами/лекарственными средствами с аксонным, демиенилизирукздим, сенсорным, моторным и/или автономным поражением; атаксия Фридрейха; атаксия-телангиэктазия; и метахроматическая лейкодистрофия; хронические невропатии, включая, но не ограничиваясь ими, сахарный диабет и другие нарушения регуляции метаболизма и диспротеинемии (метаболические невропатии, включая невропатии, вызванные алкоголизмом); и воспалительные/иммунологические процессы (воспалительные невропатии, невропатия, ассоциированная с опоясывающим лишаем, и лепрозный неврит).
Другими расстройствами являются, но не ограничиваются ими, травматические невропатии периферических или черепных нервов, паралич Белла и другие невропатии лицевых нервов, невропатия тройничного нерва, вестибулярная невропатия, невропатия добавочного нерва, невропатия блуждающего нерва, глоссофарингеальная невропатия, невропатия зрительного нерва, невропатия глазодвигательного нерва, множественный паралич черепно-мозговых нервов, плексопатия, заболевания нервных корешков, идиопатический плечевой неврит, плексит, многоочаговая невропатия и невропатия вегетативной нервной системы.
В одном из вариантов осуществления изобретения указанное подвергаемое лечению расстройство вызвано заболеванием и выбрано из группы, состоящей из болезни Паркинсона, болезни Альцгеймера, удара, рассеянного склероза, бокового амиотрофического склероза, диабетической невропатии и паралича Белла. В другом варианте осуществления изобретения указанное подвергаемое лечению расстройство вызвано травмой головного мозга, спинного мозга или периферической нервной системы.
И наконец, настоящее изобретение относится к способу ингибирования у индивидуума возникновения расстройства, характеризующегося поражением нервной ткани, вызванным заболеванием, включающему введение указанному индивидууму профилактически эффективного количества фармацевтической композиции настоящего изобретения.
В одном из вариантов осуществления изобретения указанное ингибируемое расстройство выбрано из группы, состоящей из болезни Паркинсона, болезни Альцгеймера, удара, рассеянного склероза, бокового амиотрофического склероза, диабетической невропатии и паралича Белла.
Используемый здесь термин "терапевтически эффективная доза" фармацевтической композиции означает количество, достаточное для прекращения, реверсии или снижения прогрессирования расстройства. Используемый здесь термин "профилактически эффективная доза" фармацевтической композиции означает количество, достаточное для ингибирования возникновения заболевания, то есть предупреждения, ослабления и/или задержки начала развития заболевания. Способы определения терапевтически и профилактически эффективных доз для композиций настоящего изобретения являются известными. Эффективная доза введения фармацевтической композиции человеку может быть, например, определена математически, исходя из результатов исследовании на животных.
В одном из вариантов осуществления изобретения терапевтически и/или профилактически эффективной дозой является доза, достаточная для доставки примерно от 0,01 до 200 мг/кг соединения настоящего изобретения по массе тела. В другом варианте осуществления изобретения терапевтически и/или профилактически эффективной дозой является доза, достаточная для доставки примерно от 0,1 до 100 мг/кг соединения настоящего изобретения по массе тела. В предпочтительном варианте осуществления изобретения терапевтически и/или профилактически эффективной дозой является доза, достаточная для доставки примерно от 1 до 30 мг/кг соединения настоящего изобретения по массе тела.
Для лучшего понимания настоящего изобретения ниже приводится подробное описание экспериментальной части, которая, как очевидно для каждого специалиста, представлена лишь для иллюстрации настоящего изобретения, которое более полно описано в приведенной ниже формуле изобретения. Кроме того, в настоящем описании цитируются различные публикации. Описание этих публикаций вводится в настоящее изобретение посредством ссылки для более полного освещения научного уровня в области, к которой относится настоящее изобретение.
Подробное описание экспериментов
I. Общие методы синтеза
Репрезентативные соединения настоящего изобретения могут быть синтезированы в соответствии с общими методами синтеза, описанными ниже и проиллюстрированными на нижеследующих схемах. На этих схемах для обозначения различных соединений используется взаимозаменяемая арабская и римская нумерация. Соединения, пронумерованные арабскими цифрами в данном разделе, не следует путать с конкретными соединениями, обозначенными арабскими цифрами в таблице 1 и в других местах описания.
Схема 1
Соединение 1а общей формулы:

(где  и
и  являются такими, как они определены в настоящем описании; Z представляет прямой или разветвленный (C1-C6)алкил, прямой или разветвленный (C1-C6)алкенил, или (С5-С7)циклоалкил или фенил, где указанное фенильное кольцо имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из водорода, низшего алкила, низшего алкокси, гидрокси и галогена; Y представляет А или низший алкоксикарбонил; а А является таким, как он определен в настоящем описании) может быть получено посредством реакции соединения 1b общей формулы:
являются такими, как они определены в настоящем описании; Z представляет прямой или разветвленный (C1-C6)алкил, прямой или разветвленный (C1-C6)алкенил, или (С5-С7)циклоалкил или фенил, где указанное фенильное кольцо имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из водорода, низшего алкила, низшего алкокси, гидрокси и галогена; Y представляет А или низший алкоксикарбонил; а А является таким, как он определен в настоящем описании) может быть получено посредством реакции соединения 1b общей формулы:

(где  и
и  являются такими, как они определены в настоящем описании; R представляет прямой или разветвленный (C1-C6)алкил, Y представляет А или низший алкоксикарбонил; а А является таким, как он определен в настоящем описании) с соответствующим образом защищенным реактивом Гриньяра в инертном растворителе, таком как тетрагидрофуран или диэтиловый эфир, при температурах примерно от -78 до 0° С в течение примерно 2-6 часов в зависимости от реакционной способности оксамата.
являются такими, как они определены в настоящем описании; R представляет прямой или разветвленный (C1-C6)алкил, Y представляет А или низший алкоксикарбонил; а А является таким, как он определен в настоящем описании) с соответствующим образом защищенным реактивом Гриньяра в инертном растворителе, таком как тетрагидрофуран или диэтиловый эфир, при температурах примерно от -78 до 0° С в течение примерно 2-6 часов в зависимости от реакционной способности оксамата.
Схема 2
Альтернативно соединения 1а и 1b,
(где  и
и  являются такими, как они определены в настоящем описании; R представляет прямой или разветвленный (C1-С6)алкил, Y представляет А или низший алкоксикарбонил, А является таким, как он определен в настоящем описании; Z представляет прямой или разветвленный (C1-C6)алкил, прямой или разветвленный (С1-С6)алкенил, или (C5-C7)циклоалкил, 2-тиенил, 3-тиенил или фенил, где указанное фенильное кольцо имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из водорода, низшего алкила, низшего алкокси, гидрокси и галогена) могут быть получены посредством реакции соединения 2 общей формулы:
являются такими, как они определены в настоящем описании; R представляет прямой или разветвленный (C1-С6)алкил, Y представляет А или низший алкоксикарбонил, А является таким, как он определен в настоящем описании; Z представляет прямой или разветвленный (C1-C6)алкил, прямой или разветвленный (С1-С6)алкенил, или (C5-C7)циклоалкил, 2-тиенил, 3-тиенил или фенил, где указанное фенильное кольцо имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из водорода, низшего алкила, низшего алкокси, гидрокси и галогена) могут быть получены посредством реакции соединения 2 общей формулы:

(где  ,
,  и А являются такими, как они определены в настоящем описании) с подходящим образом защищенным хлорангидридом глиоксиловой кислоты или алкилоксалилхлоридом в инертном растворителе, таком как метиленхлорид, в течение примерно 2-6 часов. В основном реакцию проводят в присутствии органического амина, такого как диизопропилэтиламин или триэтиламин, при температуре примерно от 0 до 37° С.
и А являются такими, как они определены в настоящем описании) с подходящим образом защищенным хлорангидридом глиоксиловой кислоты или алкилоксалилхлоридом в инертном растворителе, таком как метиленхлорид, в течение примерно 2-6 часов. В основном реакцию проводят в присутствии органического амина, такого как диизопропилэтиламин или триэтиламин, при температуре примерно от 0 до 37° С.
В случае соединения 1а с определениями, указанными выше, это превращение может быть также осуществлено путем конденсации соединения 2, определенного выше, с соответствующим образом защищенной глиоксиловой кислотой в присутствии агента сочетания, такого как диизопропилкарбодиимид, дициклогексилкарбодиимид или гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония (реагент Кастро), в инертном растворителе, таком как тетрагидрофуран, диметилформамид или метиленхлорид, при температурах примерно от 0 до 37° С в течение примерно 2-24 ч.
Схема 3
Соединение 3 общей формулы:

(где  ,
,  и А являются такими, как они определены в настоящем описании; R3 представляет фенилалкил; где указанное фенильное кольцо имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из водорода, низшего алкила, низшего алкокси, гидрокси и галогена) может быть получено посредством реакции соединения 2, с определениями, описанными выше, с фенилалкилсульфонилхлоридом в инертном растворителе, таком как метиленхлорид, в течение примерно 2-24 ч. В основном эту реакцию проводят в присутствии органического амина, такого как диизопропилэтиламин или триэтиламин, при температуре примерно от 0 до 37° С.
и А являются такими, как они определены в настоящем описании; R3 представляет фенилалкил; где указанное фенильное кольцо имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из водорода, низшего алкила, низшего алкокси, гидрокси и галогена) может быть получено посредством реакции соединения 2, с определениями, описанными выше, с фенилалкилсульфонилхлоридом в инертном растворителе, таком как метиленхлорид, в течение примерно 2-24 ч. В основном эту реакцию проводят в присутствии органического амина, такого как диизопропилэтиламин или триэтиламин, при температуре примерно от 0 до 37° С.
Схема 4
Соединение 2, определенное как описано выше, может быть получено из соединения 4 общей формулы:

определенного как описано выше, стандартными методами путем удаления N-бензилоксикарбонильной группы. Такими методами являются каталитическое гидрирование в присутствии катализатора из благородного металла, такого как палладий на углероде, в спиртовом растворителе в течение примерно 4-24 ч, обычно при комнатной температуре (КТ), или реакция с трибромидом бора в инертном растворителе, таком как метиленхлорид, в течение примерно 2-6 ч, при температуре примерно от -78 до 25° С, или реакция с сильной кислотой, такой как бромистоводородная кислота в уксусной кислоте, в течение примерно 2-6 ч при температуре примерно от 20 до 100° С. В последнем методе продукт часто выделяют в виде гидробромидной соли.
Схема 5
Соединение 5а общей формулы:

(где  ,
,  , m и Ar являются такими, как они были определены в настоящем, описании) может быть получено посредством реакции соединения 5b общей формулы:
, m и Ar являются такими, как они были определены в настоящем, описании) может быть получено посредством реакции соединения 5b общей формулы:

определенного как описано выше, с ароматическим или гетероароматическим спиртом, таким как 3-гидроксипиридин. Эту реакцию обычно проводят в присутствии производного азодикарбоновой кислоты, такого как диэтилазодикарбоксилат или 1,1’-(азодикарбонил)дипиперидин, и фосфинового производного, такого как трифенилфосфин или три-н-бутилфосфин, в инертном растворителе, таком как тетрагидрофуран или толуол, в течение примерно 12-24 ч. Реакционная температура может составлять примерно от 20 до 65° С.
Схема 6
Соединения 6а и 6b общих формул:


(где  ,
,  , R4, m и Ar являются такими, как они определены в настоящем описании; R представляет COCOR2, COOR3 или SO2R3; a R2 и R3 являются такими, как они определены в настоящем описании) могут быть получены посредством реакции соединения 6с общей формулы:
, R4, m и Ar являются такими, как они определены в настоящем описании; R представляет COCOR2, COOR3 или SO2R3; a R2 и R3 являются такими, как они определены в настоящем описании) могут быть получены посредством реакции соединения 6с общей формулы:

(где  и
и  являются такими, как они определены в настоящем описании; R представляет COCOR2, COOR3 или SO2R3; а R2 и R3 являются такими, как они были определены в настоящем описании) с производным арилалкиламина или арилалканола. Реакцию осуществляют через промежуточный ацилазид или смешанный ангидрид путем добавления реагента, такого как дифенилфосфорилазид, изопропенилхлорформиат или изобутилхлорформиат, вместе с органическим аминовым основанием, таким как триэтиламин или диизопропилэтиламин, в инертном растворителе, таком как тетрагидрофуран или диметилформамид. Может быть также добавлен катализатор ацилирования, такой как диметиламинопиридин. Эту реакцию обычно проводят при температуре примерно от 0 до 25° С в течение примерно 12-24 ч.
являются такими, как они определены в настоящем описании; R представляет COCOR2, COOR3 или SO2R3; а R2 и R3 являются такими, как они были определены в настоящем описании) с производным арилалкиламина или арилалканола. Реакцию осуществляют через промежуточный ацилазид или смешанный ангидрид путем добавления реагента, такого как дифенилфосфорилазид, изопропенилхлорформиат или изобутилхлорформиат, вместе с органическим аминовым основанием, таким как триэтиламин или диизопропилэтиламин, в инертном растворителе, таком как тетрагидрофуран или диметилформамид. Может быть также добавлен катализатор ацилирования, такой как диметиламинопиридин. Эту реакцию обычно проводят при температуре примерно от 0 до 25° С в течение примерно 12-24 ч.
Схема 7
Соединение 5b может быть получено посредством реакции соединения 7 общей формулы:

(где  ,
,  и m являются такими, как они определены в настоящем описании; a R представляет низший алкил) с восстановителем, являющимся гидридом металла, таким как боргидрид лития или комбинация боргидрида натрия/хлорида лития. Эту реакцию обычно проводят в спиртовом растворителе, таком как этанол или метанол, с добавлением или без добавления тетрагидрофурана, при температуре примерно от КТ (комнатной температуры) до 65° С в течение примерно 24-72 ч.
и m являются такими, как они определены в настоящем описании; a R представляет низший алкил) с восстановителем, являющимся гидридом металла, таким как боргидрид лития или комбинация боргидрида натрия/хлорида лития. Эту реакцию обычно проводят в спиртовом растворителе, таком как этанол или метанол, с добавлением или без добавления тетрагидрофурана, при температуре примерно от КТ (комнатной температуры) до 65° С в течение примерно 24-72 ч.
Схема 8
Соединение 6с может быть получено посредством реакции соединения 8 общей формулы:

(где  и
и  являются такими, как были определены ранее; R представляет COCOR2, COOR3 или SO2R3; R2 и R3 являются такими, как они были определены в настоящем описании; а R’ представляет низший алкил) с гидроксидом щелочного металла или карбонатом щелочного металла, таким как гидроксид лития, гидроксид натрия или карбонат калия, в смешанной водной системе растворителей, такой как тетрагидрофуран/вода или этанол/вода, при температуре примерно от КТ до 80° С в течение примерно 3-24 ч.
являются такими, как были определены ранее; R представляет COCOR2, COOR3 или SO2R3; R2 и R3 являются такими, как они были определены в настоящем описании; а R’ представляет низший алкил) с гидроксидом щелочного металла или карбонатом щелочного металла, таким как гидроксид лития, гидроксид натрия или карбонат калия, в смешанной водной системе растворителей, такой как тетрагидрофуран/вода или этанол/вода, при температуре примерно от КТ до 80° С в течение примерно 3-24 ч.
Схема 9
Соединение 9а общей формулы:

(где  и m являются такими, как они были определены в настоящем описании; a R представляет низший алкил) может быть получено путем конденсации соединения 9b общей формулы:
и m являются такими, как они были определены в настоящем описании; a R представляет низший алкил) может быть получено путем конденсации соединения 9b общей формулы:

определенного, как описано выше, с α -бромкетоэфиром, таким как этилбромпируват или этил-γ -бромацетоацетат, в спиртовом растворителе, таком как этанол. Эта реакция может быть проведена при температуре примерно от 20 до 80° С в течение примерно 2-24 ч.
Схема 10
Аналогичным образом соединение 10а общей формулы:

определенное, как описано выше, может быть получено путем конденсации соединения 9b с соединением 10b общей формулы:

определенным, как описано выше, в спиртовом растворителе, таком как этанол, примерно при 80° С, в течение примерно 3-24 ч.
Схема 11
Соединение 10b, определенное, как описано выше, может быть получено посредством реакции соединения 11 общей формулы:

определенного, как описано выше, с бромистым водородом в инертном растворителе, таком как диэтиловый эфир. В основном эту реакцию проводят примерно от 0 до 25° С до тех пор, пока не завершится выделение N2.
Схема 12
Соединение 11 может быть получено из соединения 12 общей формулы:

(где  определен выше) посредством реакции хлорангидридного производного соединения 12 с диазометаном или триметилсилилдиазометаном в присутствии органического основания, такого как триэтиламин или диизопропилэтиламин. Эту реакцию в основном осуществляют в инертном растворителе, таком как тетрагидрофуран, ацетонитрил или их комбинация, при температуре примерно от 0 до 25° С в течение примерно 2-24 часа. Хлорангидрид может быть получен из соответствующей кислоты стандартными методами, описанными в литературе, такими как реакция с оксалилхлоридом в инертном растворителе, таком как метиленхлорид или тетрагидрофуран, в присутствии каталитического количества диметилформамида.
определен выше) посредством реакции хлорангидридного производного соединения 12 с диазометаном или триметилсилилдиазометаном в присутствии органического основания, такого как триэтиламин или диизопропилэтиламин. Эту реакцию в основном осуществляют в инертном растворителе, таком как тетрагидрофуран, ацетонитрил или их комбинация, при температуре примерно от 0 до 25° С в течение примерно 2-24 часа. Хлорангидрид может быть получен из соответствующей кислоты стандартными методами, описанными в литературе, такими как реакция с оксалилхлоридом в инертном растворителе, таком как метиленхлорид или тетрагидрофуран, в присутствии каталитического количества диметилформамида.
Схема 13
Соединение 13 общей формулы:

(где  является таким, как он определен в настоящем описании; a R представляет низший алкил) может быть получено посредством реакции соединения 12 с анионом, производным алкилизоцианоацетата, в полярном инертном растворителе, таком как диметилформамид, в течение примерно 12-24 ч. В основном для получения аниона используют карбонат щелочного металла, такой как карбонат калия. Для облегчения реакции карбоновую кислоту соединения 12 превращают в активную молекулу in situ, такую как ацилазид, посредством реакции с дифенилфосфорилазидом.
является таким, как он определен в настоящем описании; a R представляет низший алкил) может быть получено посредством реакции соединения 12 с анионом, производным алкилизоцианоацетата, в полярном инертном растворителе, таком как диметилформамид, в течение примерно 12-24 ч. В основном для получения аниона используют карбонат щелочного металла, такой как карбонат калия. Для облегчения реакции карбоновую кислоту соединения 12 превращают в активную молекулу in situ, такую как ацилазид, посредством реакции с дифенилфосфорилазидом.
Схема 14
Соединение 14а общей формулы:

(где  ,
,  и Ar являются такими, как они определены в настоящем описании; R представляет COCOR2, COOR3 или SO2R3; a R2 и R3 являются такими, как они были определены в настоящем описании) может быть получено путем объединения соединения 14b общей формулы:
и Ar являются такими, как они определены в настоящем описании; R представляет COCOR2, COOR3 или SO2R3; a R2 и R3 являются такими, как они были определены в настоящем описании) может быть получено путем объединения соединения 14b общей формулы:

(где Ar является таким, как он определен в настоящем описании) с соединением 14с общей формулы:

(где  является таким, как он определен в настоящем описании; R представляет COCOR2, COOR3 или SO2R3; a R2 и R3 являются такими, как они были определены в настоящем описании) в присутствии агента сочетания, такого как водорастворимый карбодиимид, диизопропилкарбодиимид или дициклогексилкарбодиимид, в инертном растворителе, таком как диглим или диоксан. В основном эту реакцию проводят при температурах примерно от 50 до 110° С в течение примерно 5-24 ч.
является таким, как он определен в настоящем описании; R представляет COCOR2, COOR3 или SO2R3; a R2 и R3 являются такими, как они были определены в настоящем описании) в присутствии агента сочетания, такого как водорастворимый карбодиимид, диизопропилкарбодиимид или дициклогексилкарбодиимид, в инертном растворителе, таком как диглим или диоксан. В основном эту реакцию проводят при температурах примерно от 50 до 110° С в течение примерно 5-24 ч.
Соединение 14b может быть получено посредством реакции аралкилнитрилов с гидрохлоридом гидроксиламина в полярном протонном растворителе, таком как этанол, в присутствии неорганического основания, такого как карбонат калия. В основном эту реакцию проводят при температурах примерно от 20 до 100° С в течение примерно 12-72 ч.
Схема 15
Соединение 15а общей формулы:

(где R представляет COCOR2, COOR3 или SO2R3; a R2 и R3 являются такими, как они были определены в настоящем описании; V представляет (СН2)nAr или  ; а Ar является таким, как он был определен в настоящем описании) может быть получено посредством реакции соединения 15b общей формулы:
; а Ar является таким, как он был определен в настоящем описании) может быть получено посредством реакции соединения 15b общей формулы:

(где R представляет COCOR2, COOR3 или SO2R3; R2 и R3 являются такими, как они были определены в настоящем описании; V представляет (CH2)nAr или  ; а Ar является таким, как он был определен в настоящем описании) с циклодегидратирующим реагентом, таким как тионилхлорид в пиридине, гексаметилдисилазан, в присутствии фторида тетра-н-бутиламмония и имидазола, Et3N+(O)2N-COOMe (реагента Бургесса) или ангидрид трифторметансульфоновой кислоты в присутствии триэтиламина. В случае использования тионилхлорида в пиридине начальную реакцию с производным бисацилгидразина проводят при температуре примерно 0° С в течение примерно 2-6 ч. Последующее замыкание кольца с образованием оксадиазола осуществляют в инертном растворителе, таком как толуол, в течение примерно 3-24 ч при температуре примерно от 80 до 150° С. Реакцию производного бисацилгидразина с гексаметилдисилазаном обычно проводят в инертном растворителе, таком как толуол или хлорбензол, при температуре примерно от 80 до 150° С в течение примерно 6-72 ч. В случае реагента Бургесса реакцию с производным бисацилгидразина обычно проводят приблизительно при КТ в течение примерно 24-72 ч в инертном растворителе, таком как тетрагидрофуран. Реакцию производного бисацилгидразина с ангидридом трифторметансульфоновой кислоты и триэтиламином обычно проводят в инертном растворителе, таком как метиленхлорид, тетрагидрофуран или диэтиловый эфир, при температуре примерно от 0 до 25° С в течение примерно 1-24 ч.
; а Ar является таким, как он был определен в настоящем описании) с циклодегидратирующим реагентом, таким как тионилхлорид в пиридине, гексаметилдисилазан, в присутствии фторида тетра-н-бутиламмония и имидазола, Et3N+(O)2N-COOMe (реагента Бургесса) или ангидрид трифторметансульфоновой кислоты в присутствии триэтиламина. В случае использования тионилхлорида в пиридине начальную реакцию с производным бисацилгидразина проводят при температуре примерно 0° С в течение примерно 2-6 ч. Последующее замыкание кольца с образованием оксадиазола осуществляют в инертном растворителе, таком как толуол, в течение примерно 3-24 ч при температуре примерно от 80 до 150° С. Реакцию производного бисацилгидразина с гексаметилдисилазаном обычно проводят в инертном растворителе, таком как толуол или хлорбензол, при температуре примерно от 80 до 150° С в течение примерно 6-72 ч. В случае реагента Бургесса реакцию с производным бисацилгидразина обычно проводят приблизительно при КТ в течение примерно 24-72 ч в инертном растворителе, таком как тетрагидрофуран. Реакцию производного бисацилгидразина с ангидридом трифторметансульфоновой кислоты и триэтиламином обычно проводят в инертном растворителе, таком как метиленхлорид, тетрагидрофуран или диэтиловый эфир, при температуре примерно от 0 до 25° С в течение примерно 1-24 ч.
Схема 16
Соединение 15b может быть получено посредством реакции смешанного ангидрида или хлор ангидридного производного соединения 14с,
(где  является таким, как он был определен в настоящем описании; R представляет COCOR2, COOR3 или SO2R3; a R2 и R3 являются такими, как они были определены в настоящем описании) с соединением 16а общей формулы:
является таким, как он был определен в настоящем описании; R представляет COCOR2, COOR3 или SO2R3; a R2 и R3 являются такими, как они были определены в настоящем описании) с соединением 16а общей формулы:

(где V представляет (СН2)nAr или  ; а Ar является таким, как он был определен в настоящем описании). В основном эту реакцию проводят в инертном растворителе, таком как тетрагидрофуран или метиленхлорид, с добавлением или без добавления третичного аминового основания, такого как триэтиламин или диизолролилэтиламин, при температуре примерно от 0 до 25° С в течение примерно 6-24 ч. Эти производные смешанного ангидрида или хлорангидрида могут быть получены из соответствующей кислоты стандартными методами, описанными в литературе, такими как реакция с изобутилхлорформиатом или этилхлорформиатом в присутствии триэтиламина или диизопропилэтиламина, или реакция с оксалилхлоридом в инертном растворителе, таком как метиленхлорид или тетрагидрофуран, в присутствии каталитического количества диметилформамида.
; а Ar является таким, как он был определен в настоящем описании). В основном эту реакцию проводят в инертном растворителе, таком как тетрагидрофуран или метиленхлорид, с добавлением или без добавления третичного аминового основания, такого как триэтиламин или диизолролилэтиламин, при температуре примерно от 0 до 25° С в течение примерно 6-24 ч. Эти производные смешанного ангидрида или хлорангидрида могут быть получены из соответствующей кислоты стандартными методами, описанными в литературе, такими как реакция с изобутилхлорформиатом или этилхлорформиатом в присутствии триэтиламина или диизопропилэтиламина, или реакция с оксалилхлоридом в инертном растворителе, таком как метиленхлорид или тетрагидрофуран, в присутствии каталитического количества диметилформамида.
В случае соединения 15b, определенного как указано выше, это превращение может быть также осуществлено путем конденсации соединения 16а, определенного как указано выше, с соединением 14с в присутствии агента сочетания, такого как диизопропилкарбодиимид, дициклогексилкарбодиимид или гексафторфосфат бензотриазол-1-илокситрис(диметиламино)-фосфония (реагент Кастро) в инертном растворителе, таком как тетрагидрофуран, диметилформамид или метиленхлорид, при температуре примерно от 0 до 37° С в течение примерно 2-24 ч.
Соединение 16а (где V представляет (CH2)nAr или  ; а Ar является таким, как он был определен в настоящем описании) может быть получено из соответствующего производного сложного низшего алкилового эфира посредством реакции с гидразином в спиртовом растворителе, таком как этанол, при кипячении с обратным холодильником в течение примерно 6-24 ч. Альтернативно соединение 16а может быть получено из соответствующего производного карбоновой кислоты через промежуточный триметилсилиловый сложный эфир посредством реакции с гидразином в инертном растворителе, таком как метиленхлорид, тетрагидрофуран или диметилформамид, при температуре примерно от 0 до 25° С в течение примерно 1-24 ч. Этот силиловый сложный эфир может быть получен in situ методами, хорошо известными специалистам, такими как реакция карбоновой кислоты с N,O-бистриметилацетамидом, при температуре примерно от 0 до 25° С в течение примерно 1-6 ч.
; а Ar является таким, как он был определен в настоящем описании) может быть получено из соответствующего производного сложного низшего алкилового эфира посредством реакции с гидразином в спиртовом растворителе, таком как этанол, при кипячении с обратным холодильником в течение примерно 6-24 ч. Альтернативно соединение 16а может быть получено из соответствующего производного карбоновой кислоты через промежуточный триметилсилиловый сложный эфир посредством реакции с гидразином в инертном растворителе, таком как метиленхлорид, тетрагидрофуран или диметилформамид, при температуре примерно от 0 до 25° С в течение примерно 1-24 ч. Этот силиловый сложный эфир может быть получен in situ методами, хорошо известными специалистам, такими как реакция карбоновой кислоты с N,O-бистриметилацетамидом, при температуре примерно от 0 до 25° С в течение примерно 1-6 ч.
Схема 17
Соединение 17 общей формулы:

(где  является таким, как он был определен в настоящем описании; R представляет COCOR2, COOR3 или SO2R3; а R2 и R3 являются такими, как они были определены в настоящем описании) может быть получено посредством реакции смешанного ангидрида или хлорангидридного производного соединения 14с,
является таким, как он был определен в настоящем описании; R представляет COCOR2, COOR3 или SO2R3; а R2 и R3 являются такими, как они были определены в настоящем описании) может быть получено посредством реакции смешанного ангидрида или хлорангидридного производного соединения 14с,
(где  является таким, как он был определен в настоящем описании, R представляет COCOR2, COOR3 или SO2R3; а R2 и R3 являются такими, как они были определены в настоящем описании) примерно с 0,5-1 эквивалентом моногидрата гидразина при температуре примерно от 0 до 25° С в течение примерно 4-24 ч.
является таким, как он был определен в настоящем описании, R представляет COCOR2, COOR3 или SO2R3; а R2 и R3 являются такими, как они были определены в настоящем описании) примерно с 0,5-1 эквивалентом моногидрата гидразина при температуре примерно от 0 до 25° С в течение примерно 4-24 ч.
Обычно реакцию проводят в инертном растворителе, таком как тетрагидрофуран или метиленхлорид, без добавления или с добавлением третичного аминового основания, такого как триэтиламин или диизопропилэтиламин. Смешанный ангидрид или производное хлорангидрида могут быть получены из соответствующей кислоты стандартными методами, описанными в литературе, такими как реакция с изобутилхлорформиатом или этилхлорформиатом в присутствии триэтиламина или диизопропилэтиламина или реакция с оксалилхлоридом в инертном растворителе, таком как метиленхлорид или тетрагидрофуран, в присутствии каталитического количества диметилформамида.
Фенилалкилсульфонилхлориды, используемые в синтезе соединения 3, производные арилалкиламинов и арилалканола, используемые в синтезе соединений 6а и 6b, соединения общей формулы соединения 12, производные низшего алкиларалкилкарбоксилата, используемые в синтезе соединений 16а, и аралкилнитрилы, используемые при получении соединения 14b, если они не являются коммерчески доступными, могут быть получены стандартными методами синтеза, ранее описанными в литературе, из легко доступных исходных материалов с использованием стандартных реагентов и реакционных условий.
Следует отметить, что если А представляет  , где
, где  и R1 являются такими, как они были определены в настоящем описании, то соединения настоящего изобретения могут содержать две группы R1. Поэтому многие реакции, описанные выше, могут быть осуществлены одновременно на обеих группах R1 путем добавления дополнительного эквивалента реагента к соответствующему субстрату. Кроме того, одна из групп R1 может быть селективно модифицирована без модификации других групп с использованием подходящей схемы защиты групп, известной специалистам.
и R1 являются такими, как они были определены в настоящем описании, то соединения настоящего изобретения могут содержать две группы R1. Поэтому многие реакции, описанные выше, могут быть осуществлены одновременно на обеих группах R1 путем добавления дополнительного эквивалента реагента к соответствующему субстрату. Кроме того, одна из групп R1 может быть селективно модифицирована без модификации других групп с использованием подходящей схемы защиты групп, известной специалистам.
В случае когда соединения настоящего изобретения имеют, по крайней мере, один хиральный центр, они могут соответственно существовать в виде энантиомеров. В случае если эти соединения имеют два или более хиральных центра, то они могут, кроме того, существовать в виде диастереомеров. Следует отметить, что все указанные изомеры и их смеси входят в объем настоящего изобретения. Кроме того, некоторые кристаллические формы данных соединений могут существовать в виде полиморфов, и эти формы входят в объем настоящего изобретения. Более того, некоторые соединения могут образовывать сольваты с водой (то есть гидраты) или с общими органическими растворителями, и такие сольваты также входят в объем настоящего изобретения.
II. Выбранные соединения настоящего изобретения
В предпочтительном варианте осуществления изобретения, соединения настоящего изобретения выбраны из группы соединений, представленных в таблице 1.
III. Конкретные методы синтеза
Конкретные соединения настоящего изобретения могут быть получены, как показано в нижеследующих примерах. Для ясности соединения настоящего изобретения, полученные в нижеследующих примерах, идентифицируются термином "Соединение", за которым следует соответствующий номер (например, Соединение 1). Промежуточные соединения в синтезе соединений настоящего изобретения обозначаются "Сравнительными примерами". Каких-либо попыток оптимизировать выходы, полученные в этих реакциях, не делалось. Каждому специалисту известны способы увеличения таких выходов, а именно путем рутинного варьирования времени реакции и температур, а также выбора растворителей и/или реагентов.
Продукты некоторых сравнительных примеров могут быть использованы в качестве промежуточных соединений для получения более чем одного соединения настоящего изобретения. В этих случаях выбор промежуточных соединений, используемых для получения последующих соединений настоящего изобретения, может быть легко осуществлен любым специалистам.
Сравнительный пример 1

К холодной (0° С) суспендированной смеси N-карбобензилокси-L-пролина (9,96 г, 40,0 ммоль) и сесквигидрата карбоната калия (26,50 г, 160,0 ммоль) в ДМФ (60 мл) добавляли дифенилфосфорилазид (12,0 мл, 55,6 ммоль) и метилизоцианоацетат (7,30 мл, 80,3 ммоль). Ледяную баню удаляли и реакционную смесь перемешивали примерно при комнатной температуре (КТ) в течение около 20 ч. Затем добавляли насыщенный раствор соли и реакционную смесь фильтровали. Фильтрат экстрагировали смесью CHCl3/СН3ОН (9:1, 150 мл). Органический раствор промывали Н2О (3× ), насыщенным раствором соли (4× ), сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенный продукт хроматографировали на силикагеле, элюируя 2% СН3ОН в CHCl3, с получением оксазола (7,47 г, выход 56%) в виде светло-коричневого масла и в виде смеси (45:55) ротамеров, на что указывал ЯМР. CIMS МН+=331 (100%). 1H-ЯМР (300 МГц, ДМСО-d6) δ 1,95-2,00 (м, 3Н), 2,30-2,45 (м, 1Н), 3,45-3,60 (м, 2Н), 3,71 (с, 0,55× 3H), 3,82 (с, 1,35Н), 4,91 (д. Jab=12,82, 1,1Н), 5,03 (д, Jab=12,82, 0,9Н), 5,45-5,48 (м, 0,45Н), 5,52-5,54 (м, 0,55Н), 7,02 (шир.с, 1Н), 7,26-7,34 (м, 4Н), 8,32-8,39 (м, 1Н).
Сравнительный пример 2

К холодному (0° С) раствору сложного метилового эфира сравнительного примера 1 (7,27 г, 22,6 ммоль) в смеси ТГФ/Н2О (2:1, 270 мл) добавляли гидроксид лития (594,7 мг, 24,8 ммоль). Полученную смесь перемешивали примерно при комнатной температуре (КТ) в течение ночи. Реакционную смесь подкисляли 4,80 г лимонной кислоты в 100 мл воды и экстрагировали CHCl3 (2× 150 мл). Объединенный органический экстракт сушили над Na2SO4, фильтровали и концентрировали с получением карбоновой кислоты (6,66 г, выход 93%) в виде белого хлопьевидного твердого вещества. CIMS М-1=315 М-45=271 (100%). 1H-ЯМР (300 МГц, ДМСО-d6), 45:55 смесь ротамеров, δ 1,95-2,05 (м, 3H), 2,25-2,40 (м, 1Н), 3,35-3,60 (м, 2Н), 4,91 (д, Jab=12,82, 0,55× 2Н), 5,03 (д, Jab=12,82, 0,45× 2Н), 5,45-5,48 (м, 0,45× 1H), 5,52-5,54 (м, 0,55× 1H), 7,00 (шир.с, 1H), 7,20-7,45 (м, 5Н), 8,25-8,35 (м, 1H).
Соединение 1

К холодному (0° С) раствору оксазол-4-карбоновой кислоты сравнительного примера 2 (632,3 мг, 2,00 ммоль) и триэтиламина (0,62 мл, 4,45 ммоль) в ДМФ (2 мл) добавляли дифенилфосфорилазид (0,48 мл, 2,22 ммоль) и 3-аминометилпиридин (0,22 мл, 2,16 ммоль). Полученную смесь перемешивали примерно при КТ в течение примерно 1 дня, разбавляли водой (25 мл) и экстрагировали EtOAc (2× 25 мл). Объединенный органический экстракт промывали водой (6× 50 мл), сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенный продукт хроматографировали на силикагеле, элюируя 100% EtOAc, в результате чего получали соединение 1 (0,49 г, выход 60%) в виде бесцветного неподвижного масла. МС (полож. петля) МН+=407 (100%); М+Na=429 (10%). 1H-ЯМР (300 МГц, CDCl3), 45:55 смесь ротамеров, δ 1,90-2,15 (м, 3H), 2,30-2,45 (м, 1Н), 3,55-3,70 (м, 2Н), 4,45-4,65 (м, 2Н), 4,85-5,20 (м, 2Н), 5,65-5,70 (м, 0,45× 1Н), 5,75-5,80 (м, 0,55× 1Н), 7,0 (шир.с, 1Н), 7,20-7,40 (м, 6Н), 8,45-8,50 (м, 1Н), 8,55-8,60 (м, 1Н).
Соединение 2

Гетерогенную смесь соединения 1 (0,39 г, 0,96 ммоль) и 10% палладия на углероде (0,05 г) в СН3ОН (25 мл) встряхивали под давлением газообразного водорода 50 фунт/кв.дюйм (3,515 кг/см3) примерно при КТ в течение примерно 3 ч. Смесь фильтровали через слой целита и осадок на фильтре промывали СН3ОН. Объединенный фильтрат концентрировали в вакууме с получением соединения 2 (0,25 г, выход 96%) в виде неподвижного масла, которое использовали без дополнительной очистки. МС (полож. петля) МН+=273 (100%). 1H-ЯМР (300 МГц, CDCl3) δ 1,8-2,2 (м, 4Н), 2,95-3,05 (м, 1Н), 3,10-3,20 (м, 1Н), 4,55-4,70 (м, 2Н), 4,90 (т, J=7,00 Гц, 1Н), 7,20-7,35 (м, 2Н), 7,60-7,75 (м, 3H), 8,53 (д, J=4,13 Гц, 1Н), 8,62 (с, 1Н).
Соединение 3

К холодному (0° С) раствору соединения 2 (0,25 г, 0,91 ммоль) и триэтиламина (140 мл, 1,00 ммоль) в метиленхлориде (10 мл) добавляли α -толуолсульфонилхлорид (184,2 мг, 0,966 ммоль). После перемешивания примерно в течение 6 ч приблизительно при КТ, реакционную смесь обрабатывали 35,6 мг сульфонилхлорида и 30 мл триэтиламина. Реакционную смесь разбавляли метиленхлоридом (40 мл), промывали водой (2× 50 мл), сушили над сульфатом натрия, фильтровали и концентрировали досуха. Неочищенный продукт подвергали перепаративной ТСХ, элюируя 100% EtOAc, с получением соединения 3 (0,17 г, выход 44%) в виде мелассообразного твердого вещества. МС (полож. петля) МН+=427 (100%). 1H-ЯМР (300 МГц, CDCl3) δ 1,72-1,80 (м, 1Н), 1,95-2,11 (м, 2Н), 2,27-2,36 (м, 1Н), 3,07-3,15 (м, 1Н), 3,44-3,52 (м, 1Н), 4,27 (д, Jab=13,97 Гц, 1Н), 4,41 (д, Jab=13,96 Гц, 1Н), 4,44-4,71 (м, 3H), 5,72-5,77 (м, 1Н), 7,28-7,45 (м, 6Н), 7,69-7,72 (м, 2Н), 8,53 (д, J=4,13 Гц, 1Н), 8,62 (с, 1Н).

К холодному (0° С) раствору оксазол-4-карбоновой кислоты сравнительного примера 2 (1,26 г, 4,00 ммоль), триэтиламина (0,62 мл, 4,445 ммоль), DMAP (49,5 мг, 0,405 ммоль) и 3-пиридилкарбинола (0,39 мл, 4,02 ммоль) в ТГФ (20 мл) добавляли изопропенилхлорформиат (0,48 мл, 4,39 ммоль). После нагревания примерно до КТ темно-желтую гетерогенную реакционную смесь перемешивали примерно при КТ в течение примерно 20 ч. Реакционную смесь разбавляли EtOAc, промывали водой и экстрагировали 1 н. водной HCl. Подкисленный водный раствор подщелачивали водным Na2CО3 и экстрагировали CHCl3 (2× 100 мл). Раствор CHCl3 сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенный продукт хроматографировали на силикагеле при элюировании 100% EtOAc, в результате чего получали соединение 4 (0,61 г, выход 37%) в виде бесцветного неподвижного масла. МС (полож. петля) МН+=408 (100%). 1H-ЯМР (300 МГц, CDCl3) δ 1,98-2,10 (м, 3H), 2,25-2,45 (м, 1Н), 3,50-3,70 (м, 2Н), 4,86-5,39 (м, 4Н), 5,59-5,61 (м, 1Н), 5,65-5,85 (м, 1Н), 7,05 (шир.с, 1Н), 7,30-7,35 (м, 5Н), 7,66-7,80 (м, 2Н), 8,58-8,71 (м, 2Н).
Сравнительный пример 3

К раствору метилового сложного эфира из сравнительного примера 1 (3,66 г, 11,0 ммоль) и хлорида лития (2,50 г, 58,9 ммоль) в смеси EtOH/ТГФ (4:3; 175 мл) двумя равными порциями добавляли боргидрид натрия (2,10 г, 55,0 ммоль). Полученную гетерогенную смесь перемешивали примерно в течение 3 дней приблизительно при КТ. Реакционную смесь гасили водным раствором NH4Cl (200 мл) и экстрагировали CHCl3 (3× 150 мл). Органический раствор сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенный продукт хроматографировали на силикагеле, элюируя 3% СН3ОН в CHCl3, в результате чего получали спирт (2,87 г, выход 86%) в виде бесцветного масла. МС (полож. петля) MH+=303 (100%). 1H-ЯМР (300 МГц, CDCl3) δ 1,90-2,10 (м, 1Н), 2,15-2,40 (м, 3H), 3,45-3,70 (м, 2Н), 4,40-4,50 (м, 1Н), 4,60-4,80 (м, 2Н), 4,90-5,15 (м, 2Н), 5,20-5,30 (м, 1Н), 7,30-7,35 (м, 5Н), 7,35 (с, 5Н), 7,75 (с, 1Н).
Соединение 5

К холодному (0° С) раствору оксазол-4-метанола сравнительного примера 3 (2,34 г, 7,74 ммоль), трифенилфосфина (3,05 г, 11,6 ммоль) и 3-гидроксипиридина (1,11 г, 11,7 ммоль) в ТГФ (60 мл) добавляли DEAD (1,86 мл, 11,7 ммоль). Полученную смесь перемешивали в течение ночи примерно при КТ и концентрировали досуха. Масло хроматографировали на силикагеле, элюируя 3% СН3ОН в CHCl3, в результате чего получали неочищенное соединение 5, содержащее побочные продукты. Полученное масло растворяли в CH2Cl2 (50 мл) и промывали 1 н. водной HCl (5× 80 мл). Подкисленный водный раствор подщелачивали NaHCO3/Na2CO3 и экстрагировали CHCl3 (3× 50 мл). Раствор CHCl3 сушили над Na2SO4, фильтровали и концентрировали с получением соединения 5 (0,47 г) в виде масла, которое отверждалось при стоянии. Указанное соединение использовали без дополнительной очистки. МС (полож. петля) MH+=380 (100%). 1H-ЯМР (300 МГц, CDCl3) 1:1 смесь ротамеров, δ 1,90-2,10 (м, 2Н), 2,15-2,30 (м, 2Н), 3,45-3,65 (м, 2Н), 4,40-4,60 (м, 1Н), 4,95-5,30 (м, 4Н), 7,10-7,25 (м, 2Н), 7,30 (с, 5Н), 7,75-8,00 (м, 1Н), 8,15 (шир.с, 0,5× 1Н), 8,25-8,30 (м, 1H), 8,40 (шир.с, 0,5× 1Н).
Соединения 6, 7 и 8
Гетерогенную смесь неочищенного соединения 5 (0,43 г) и 10% палладия на углероде (0,05 г) в СН3ОН (35 мл) встряхивали под давлением газообразного водорода 54 фунт/кв.дюйм (3,797 кг/см3) примерно при КТ в течение примерно 6,5 ч. Смесь фильтровали через слой целита и осадок на фильтре промывали СН3ОН. Фильтрат концентрировали в вакууме с получением остатка. Неочищенный продукт хроматографировали на силикагеле, элюируя смесью CHCl3:СН3ОН:NH4OH (90:9:1), с получением соединения 6 (0,10 г) в виде масла. МС (полож. петля) MH+=246 (100%).
К холодному (0° С) раствору соединения 6 (0,10 г, 0,41 ммоль) и триэтиламина (70 мл, 0,50 ммоль) в CH2Cl2 (10 мл) добавляли этилоксалилхлорид (70 мл, 0,63 ммоль). Реакционную смесь перемешивали примерно 2 ч, разбавляли CH2Cl2 (50 мл), промывали водным NaCl (3× 50 мл), сушили над Na2SO4, фильтровали и концентрировали с получением соединения 7 (0,10 г, выход 71%) в виде масла, которое использовали в последующей стадии без дополнительной очистки. МС (полож. петля) МН+=346 (100%).
К холодному (-78° С) раствору соединения 7 (0,10 г, 0,29 ммоль) в ТГФ (10 мл) добавляли избыток хлорида 1,1-диметилпропилмагния (1 М, 1,40 мл) в Et2O и полученную смесь перемешивали примерно 2 часа приблизительно при -78° С. Реакционную смесь гасили водным NH4Cl (25 мл) и экстрагировали EtOAc (2× 50 мл). EtOAc-раствор сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенный продукт хроматографировали на силикагеле, элюируя смесью EtOAc:гексан, 1:1, с получением соединения 8 (55,5 мг, выход 52%) в виде масла. МС (полож. петля) MH+=372 (100%). 1H-ЯМР (300 МГц, ДМСО-d6), смесь ротамеров (3 к 1), δ 0,66 (т, J=7 Гц, 0,25× 3H), 0,75 (т, J=7 Гц, 0,75× 3H), 0,80 (с, 0,25× 3H), 0,88 (с, 0,25× 3H), 1,07 (с, 0,75× 3H), 1,10 (с, 0,75× 3H), 1,56-1,59 (м, 2Н), 1,88-1,98 (м, 2Н), 2,08-2,29 (м, 2Н), 3,42-3,60 (м, 2Н), 4,93 (д, J=12 Гц, 0,25× 1Н), 4,99 (д, J=12 Гц, 0,25× 1Н), 5,08 (д, J=12 Гц, 0,75× 1Н), 5,18 (д, J=12 Гц, 0,75× 1Н), 5,34-5,38 (м, 1H), 7,47-7,54 (м, 1Н), 7,32-7,39 (м, 1Н), 8,18-8,20 (м, 1H), 8,34-8,35 (м, 1H), 8,35 (с, 0,75× 1H), 8,41 (с, 0,25× 1H).
Сравнительный пример 4

Гетерогенную смесь метилового сложного эфира из сравнительного примера 1 (2,00 г, 6,00 ммоль) и 10% палладия на углероде (0,20 г) в СН3ОН (40 мл) встряхивали под давлением газообразного водорода 54 фунт/кв.дюйм (3,797 кг/см3) около 20 ч примерно при КТ. Смесь фильтровали через слой целита и осадок на фильтре промывали СН3ОН (75 мл). Объединенный фильтрат концентрировали с получением пирролидина (1,20 г, выход 100%) в виде желтого твердого вещества. МС (полож. петля) MH+=197 (100%). 1H-ЯМР (300 МГц, CDCl3) δ 2,10-2,25 (м, 3H), 2,40-2,50 (м, 1H), 3,40-3,60 (м, 2Н), 3,95 (с, 3H), 5,40-5,50 (м, 1H), 7,95 (с, 1H).
Сравнительный пример 5

К холодной (0° С) смеси пирролидина из сравнительного примера 4 (1,18 г, 6,00 ммоль) и триэтиламина (0,94 мл, 6,74 ммоль) в CH2Cl2 (100 мл) добавляли этилоксалилхлорид (0,80 мл, 8,70 ммоль) в CH2Cl2 (25 мл). Реакционную смесь перемешивали примерно 2 ч приблизительно при КТ, промывали водным раствором NaCl (3× 75 мл) и сушили над Na2SO4. Затем CH2Cl2 фильтровали и концентрировали с получением остатка, который очищали с помощью хроматографии (элюирование 35% гексаном в EtOAc) с получением оксамата (1,23 г, выход 72%) в виде масла, которое затем отверждалось. МС (полож. петля) MH+=283 (5%). 1H-ЯМР (300 МГц, CDCl3), 1:1,5 смесь ротамеров, δ 1,35 (т, J=7 Гц, 0,6× 3H), 1,20 (т, J=7 Гц, 0,4× 3H), 1,95-2,20 (м, 3H), 2,40-2,50 (м, 1Н), 3,40-3,60 (м, 2Н), 3,89 (с, 0,6× 3H), 3,91 (с, 0,4× 3H), 4,00-4,15 (м, 0,4× 2Н), 4,29-4,40 (м, 0,6× 2Н), 5,70-5,75 (м, 0,6× 1Н), 5,90-5,95 (м, 0,4× 1Н), 7,75 (с, 0,6× 1Н), 7,80 (с, 0,4× 1Н).
Сравнительный пример 6

К холодному (-78° С) раствору оксамата из сравнительного примера 5 (0,87 г, 3,08 ммоль) в безводном ТГФ (10 мл) добавляли избыток хлорида 1,1-диметилпропилмагния (1 М, 4,60 мл, 4,60 ммоль) в Et2O и полученную смесь перемешивали примерно 3 часа приблизительно при -78° С. Реакционную смесь гасили водным NH4Cl (25 мл) и экстрагировали EtOAc (2× 25 мл). EtOAc-раствор сушили над Na2SO4. фильтровали и концентрировали досуха. Реакцию повторяли с 284,5 мг оксамата и 1,50 мл хлорида диметилпропилмагния. Объединенный неочищенный продукт хроматографировали на силикагеле, элюируя смесью EtOAc : гексан, 1:1, с получением оксамида (0,95 г, выход 96%) в виде бесцветного масла. МС (полож. петля) MH+=323 (90%). 1H-ЯМР (300 МГц, CDCl3), смесь ротамеров (3 к 1), δ 0,70 (т, J=7,4 Гц, 0,25× 3H), 0,80 (т, J=7,4 Гц, 0,75× 3H), 1,00 (с, 0,25× 3H), 1,05 (с, 0,25× 3H), 1,15 (с, 0,75× 6Н), 1,65-1,75 (м, 2Н), 1,90-2,20 (м, 0,75× 2Н), 2,35-2,50 (м, 0,25× 2Н), 3,55-3,70 (м, 0,75× 1Н), 3,75-3,80 (м, 0,25× 1Н), 3,90 (с, 1Н), 5,70-5,80 (м, 1Н), 7,75 (с, 0,75× 1Н), 7,80 (с, 0,25× 1H).
Сравнительный пример 7

Раствор 4-карбометоксиоксазола из сравнительного примера 6 (0,93 г, 2,88 ммоль) и гидроксида лития (76,9 мг, 3,21 ммоль) в смеси ТГФ/Н2O (2:1, 30 мл) перемешивали примерно при 0° С в течение приблизительно 1 ч и примерно при комнатной температуре в течение приблизительно 22 ч. Реакционную смесь промывали Et2O (2× 75 мл), подкисляли водной лимонной кислотой (рН 1,0) и экстрагировали CHCl3 (3× 50 мл). Раствор CHCl3 сушили над Na2SO4, фильтровали и концентрировали с получением масла. Это масло покрывали Et2O и помещали в высокий вакуум, в результате чего получали карбоновую кислоту (0,80 г, выход 90%) в виде белого твердого вещества. МС (отриц. петля) М-1=307 (100%). 1H-ЯMP (300 МГц, ТГФ-d6), смесь ротамеров (2,5 к 1), δ 0,70 (т, J=7,4 Гц, 0,30× 3H), 0,85 (т, J=7,4 Гц, 0,70× 3H), 0,92 (с, 0,30× 3H), 0,95 (с, 0,30× 3H), 1,15 (с, 0,70× 6Н), 1,45-1,65 (м, 2Н), 1,90-2,20 (м, 3H), 2,30-2,40 (м, 1Н), 3,50-3,65 (м, 2Н), 5,65-5,73 (м, 0,7× 1Н), 5,75-5,80 (м, 0,3× 1Н), 7,9 (с, 0,7× 1Н), 8,00 (с, 0,3× 1Н).
Соединение 9

К холодному (0° С) раствору оксазол-4-карбоновой кислоты из сравнительного примера 7 (0,31 г, 1,00 ммоль), триэтиламина (0,17 мл, 1,22 ммоль), DMAP (12,1 мг, 0,099 ммоль) и 3-пиридилкарбинола (0,11 мл, 1,13 ммоль) в ТГФ (15 мл) добавляли изопропенилхлорформиат (0,12 мл, 1,10 ммоль). После нагревания примерно до КТ темно-желтую гетерогенную реакционную смесь перемешивали примерно при КТ в течение примерно 20 ч. Реакционную смесь разбавляли водой и экстрагировали в CHCl3 (2× 50 мл). Раствор CHCl3 сушили над Na2SO4 фильтровали и концентрировали досуха. Неочищенный продукт хроматографировали на силикагеле при элюировании 3% СН3ОН в CHCl3, в результате чего получали соединение 9 (222,8 мг, выход 56%) в виде светло-желтого неподвижного масла. МС (полож. петля) МН+=400 (100%). 1H-ЯМР (300 МГц, ДМСО-d6), смесь ротамеров (3 к 1), δ 0,62 (т, J=7,4 Гц, 0,25× 3H), 0,76 (т, J=7,4 Гц, 0,75× 3H), 0,83 (с, 0,25× 3H), 0,86 (с, 0,25× 3H), 1,11 (с, 0,75× 6Н), 1,44-1,49 (м, 0,25× 2Н), 1,58 (кв, J=7,41, 7,42, 7,41 Гц, 0,75× 2Н), 1,88-2,04 (м, 3H), 2,25-2,34 (м, 1Н), 3,34-3,61 (м, 2Н), 5,31-5,39 (м, 2Н), 5,52-5,59 (м, 1Н), 7,42-7,46 (м, 1Н), 7,89-7,91 (м, 1Н), 8,44 (с, 0,75× 1Н), 8,50 (с, 0,25× 1Н), 8,57 (д, J=4,56 Гц, 1Н), 8,69 (с, 1Н).
Соединение 10

К холодному (0° С) раствору оксазол-4-карбоновой кислоты из сравнительного примера 7 (308,2 г, 1,00 ммоль) и триэтиламина (310 мл, 2,22 ммоль) в ДМФ (2 мл) добавляли дифенилфосфорилазид (480 мл, 1,16 ммоль) и 3-аминометилпиридин (110 мл, 2,16 ммоль). Полученную смесь перемешивали примерно при КТ в течение примерно 1 дня, разбавляли водой (25 мл) и экстрагировали EtOAc (2× 50 мл). Раствор EtOAc сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт хроматографировали на силикагеле (элюируя 5% СН3ОН в CHCl3), в результате чего получали соединение 10 (310 мг, выход 78%) в виде масла. МС (полож. петля) МН+=399 (100%). 1H-ЯМР (300 МГц, ДМСО-d6), смесь ротамеров (2 к 1), δ 0,65 (т, J=7,4 Гц, 0,33× 3H), 0,75 (т, J=7,4 Гц, 0,67× 3H), 0,85 (с, 0,33× 3H), 0,90 (с, 0,33× 3H), 1,15 (с, 0,67× 6Н), 1,45-1,65 (м, 2Н), 1,80-2,00 (м, 3H), 2,25-2,40 (м, 1Н), 3,40-3,60 (м, 2Н), 4,35-4,50 (м, 2Н), 5,60-5,65 (м, 0,67× 1Н), 5,70-5,75 (м, 0,33× 1Н), 7,20-7,30 (м, 2Н), 7,55-7,75 (м, 1Н), 8,50-8,65 (м, 2Н), 8,90-9,00 (м, 1Н).
Сравнительный пример 8

Гетерогенную смесь N-CBz-L-пропиламида (1,12 г, 4,51 ммоль) и 2,4-бис(4-метоксифенил)-1,3-дитиа-2,4-дифосфетан-2,4-дисульфида (реагент Лавессона; 911,3 мг, 2,25 ммоль) в бензоле (20 мл) перемешивали при кипячении с обратным холодильником около 1 ч. Затем добавляли еще 933,0 мг реагента Лавессона и реакцию продолжали при кипячении с обратным холодильником в течение примерно еще 1 часа. Реакционную смесь концентрировали с получением неочищенного тиоамида. Остаток растворяли в EtOH (10 мл), обрабатывали бромпируватом (300 мкл, 1,00 ммоль) и перемешивали при нагревании с обратным холодильником в течение ночи. После охлаждения до КТ, реакционную смесь обрабатывали твердым K2СО3, перемешивали примерно 10 мин и концентрировали. Остаток растворяли CHCl3 (50 мл), промывали Н2О (2× ), сушили Na2SO4, фильтровали и концентрировали. Неочищенный продукт хроматографировали на силикагеле, элюируя 2% СН3ОН в CHCl3, в результате чего получали тиазол (0,63 г, выход 39%) в виде светло-желтого масла. МС (полож. петля) МН+=361 (50%). М+Na=383 (100%). 1H-ЯМР (300 МГц, ДМСО-d6), смесь ротамеров, δ 1,23 (т, J=7,32, 7,66, 3H), 1,93-2,37 (м, 3H), 3,50-3,59 (м, 2Н), 4,30 (кв, J=7,04, 7,05, 7,08 Гц, 2Н), 4,94-5,22 (м, 3H), 7,08-7,38 (м, 5Н), 8,32 (с, 0,33Н), 8,41 (с, 0,67Н).
Сравнительный пример 9

Раствор этилового сложного эфира из сравнительного примера 8 (0,55 г, 1,53 ммоль) и гидроксида лития (37,5 мг, 1,56 ммоль) в смеси ТГФ/Н2О (2:1, 6 мл) перемешивали примерно при 0°С в течение приблизительно 1 ч и примерно при комнатной температуре в течение приблизительно 2 ч. Реакционную смесь разбавляли водным раствором NaCl, промывали CHCl3 (2× 15 мл), подкисляли лимонной кислотой (427 мг) и экстрагировали CHCl3 (2× 20 мл). Раствор CHCl3 сушили над Na2SO4, фильтровали и концентрировали с получением карбоновой кислоты (0,51 г, выход 100%) в виде масла, которое затем использовали без дополнительной очистки. МС (отриц. петля) М-1=331 (50%). 1H-ЯМР (300 МГц, ДМСО-d6) δ 1,93-2,35 (м, 3H), 2,25-2,40 (м, 1Н), 3,39-3,50 (м, 2Н), 4,94-5,21 (м, 3H), 4,94-5,22 (м, 3H), 7,04-7,12 (м, 1Н), 7,25-7,37 (м, 4Н), 8,33 (с, 1Н), 13,0 (шир.с, 1Н).
Соединение 11

К холодному раствору тиазол-4-карбоновой кислоты из сравнительного примера 9 (0,51 г, 1,53 ммоль) и триэтиламина (0,48 мл, 3,44 ммоль) в ДМФ (3 мл) добавляли дифенилфосфорилазид (376 мл, 1,72 ммоль) и 3-аминометилпиридин (180 мл, 1,77 ммоль). Полученную смесь перемешивали примерно при КТ в течение примерно 1 дня, разбавляли водой (25 мл) и экстрагировали EtOAc (2× 25 мл). Раствор EtOAc промывали водой (6× 50 мл), сушили над Na2SO4, фильтровали и концентрировали. Неочищенный продукт хроматографировали на силикагеле, элюируя 100% EtOAc, в результате чего получали соединение 11 (0,44 г, выход 68%) в виде бесцветного неподвижного масла. МС (полож. петля) MH+=423 (100%) М+Na=445 (10%). 1H-ЯМР (300 МГц, CDCl3) δ 1,90-2,05 (м, 2Н), 2,15-2,45 (м, 2Н), 3,45-3,70 (м, 2Н), 4,60-4,65 (м, 2Н), 4,95-5,25 (м, 3H), 5,65-5,85 (м, 1Н), 7,05 (шир.с, 1Н), 7,20-7,40 (м, 5Н), 7,66-7,70 (м, 1Н), 7,55-7,65 (м, 1Н), 8,00-8,05 (м, 1Н), 8,50-8,65 (м, 2Н).
Соединение 12

Смесь соединения 11 (0,38 г, 0,90 ммоль) и 30% бромистого водорода в уксусной кислоте (2 мл) перемешивали примерно при КТ в течение примерно 2 ч. Из реакционной смеси осаждалась дигидробромидная соль продукта. К реакционной смеси добавляли диэтиловый эфир и путем фильтрации собирали соединение 12 (407,8 мг, выход 100%) в виде бежевого гигроскопического твердого вещества. МС (полож. петля) MH+=289 (100%). 1H-ЯМР (300 МГц, ДМСО-d6) δ 1,95-2,25 (м, 3H), 2,40-2,50 (м, 2Н), 3,30-3,40 (м, 1Н), 3,45-3,55 (м, 1Н), 4,65-4,70 (м, 2Н), 5,15-5,25 (м, 1Н), 7,90-8,10 (м, 1Н), 8,40 (с, 1Н), 8,50-8,55 (м, 1Н), 8,80-8,85 (м, 1Н), 8,90 (с, 1Н), 9,15-9,30 (м, 1Н), 9,35-9,45 (м, 1Н), 9,75-9,90 (м, 1Н).
Соединение 13

К холодному (0°С) раствору дигидробромидной соли соединения 12 (203,0 г, 0,453 ммоль) и триэтиламина (210 мл, 1,50 ммоль) в метиленхлориде (4 мл) добавляли α -толуолсульфонилхлорид (95,0 мг, 0,498 ммоль). Через 3 ч к реакционной смеси добавляли DMAP (10 мг), триэтиламин (100 мл) и α -толуолсульфонилхлорид (44,8 мг). Реакционную смесь перемешивали в течение ночи. Растворитель и летучие вещества удаляли в вакууме. Неочищенный продукт очищали с помощью препаративной ТСХ, элюируя 5% СН3ОН в CHCl3, в результате чего получали соединение 13 (67,6 г, выход 34%) в виде бежевого твердого вещества. МС (полож. петля) МН+=443 (100%). 1H-ЯМР (300 МГц, CDCl3) δ 1,90-2,05 (м, 2Н), 2,10-2,20 (м, 2Н), 3,30-3,50 (м, 2Н), 4,3 (с, 2Н), 4,60-4,70 (м, 2Н), 4,85-4,95 (м, 1Н), 7,10-7,20 (м, 1Н), 7,30-7,45 (м, 5Н), 7,50-7,65 (м, 1Н), 7,70-7,75 (м, 1Н), 8,05 (с, 1Н), 8,50-8,55 (м, 1Н), 8,60 (с, 1Н).
Сравнительный пример 10

Раствор N-CBz-защищенного 2-пирролидино-4-карбометокситиазола из сравнительного примера 8 (3,33 г, 9,25 ммоль) в СН3СO2Н (10 мл) обрабатывали 30% HBr в СН3СO2Н (4 мл) и перемешивали примерно при КТ в течение примерно 6 ч. Осажденную дигидробромидную соль пирролидинотиазола покрывали Et2O и собирали путем фильтрации. Белое твердое вещество промывали дополнительным количеством Et2O и сушили в печи в течение ночи примерно при КТ, в результате чего получали пирролидин в виде его дигидробромидной соли (2,48 г, выход 69%). МС (полож. петля) МН+=227 (100%). Анализ для C10H14N2O2S· 2,0 HBr: Вычислено: C 30,95; H 4,15; N 7,22; S 8,26; Br 41,69. Найдено: C 31,18; H 4,07; N 7,11; S 7,91; Br 41,84. 1H-ЯМР (300 МГц, ДМСО-d6) δ 1,31 (т, J=7,12, 7,21 Гц, 3H), 2,03-2,21 (м, 3H), 2,50-2,53 (м, 1Н), 3,33-3,35 (м, 2Н), 4,33 (кв, J=7,04, 7,05, 7,08 Гц, 2Н), 5,10 (т, J=6,94, 6,94 Гц, 1Н), 8,66 (с, 1H), 9,2-9,5 (м, 2Н), 9,80 (шир.с, 1H).
Сравнительный пример 11

К холодному (0° С) раствору пирролидина из сравнительного примера 10 (1,53 г, 5,00 ммоль) в СН2Сl2 (100 мл) добавляли триэтиламин (1,70 мл, 1,21 ммоль) и этилоксалилхлорид (0,82 мл, 7,34 ммоль). Полученную реакционную смесь перемешивали приблизительно при КТ в течение примерно 2 ч, промывали водным раствором NaCl (2× 150 мл), сушили над Na2SO4, фильтровали и концентрировали, в результате чего получали оксамат (1,45 г, выход 89%) в виде масла, которое использовали без дополнительной очистки. МС (полож. петля) MH+=327 (100%). 1H-ЯМР (300 МГц, ДMCO-d6), 2:1 смесь ротамеров, δ 1,20-1,35 (м, 6Н), 1,8-2,2 (м, 3H), 2,30-2,45 (м, 1H), 3,65-3,80 (м, 2Н), 4,20-4,35 (м, 4Н), 5,35-5,40 (м, 0,67× 1H), 5,59-5,61 (м, 0,33× 1H), 8,42 (с, 0,67× 1H), 8,47 (с, 0,33× 1H).
Сравнительный пример 12

К холодному (-78° С) раствору оксамата из сравнительного примера 11 (1,39 г, 4,26 ммоль) в безводном ТГФ (25 мл) добавляли избыток хлорида 1,1-диметилпропилмагния (1 М, 7,80 мл, 7,80 ммоль) в Et2O и полученную смесь перемешивали примерно 5 ч приблизительно при -78° С. Реакционную смесь гасили водным NH4Cl (25 мл) и экстрагировали EtOAc (2× 25 мл). EtOAc-раствор сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенный продукт хроматографировали на силикагеле, элюируя 35% EtOAc в гексане, с получением оксамида (1,01 г, выход 67%) в виде белого твердого вещества. МС (полож. петля) МН+=353 (100%). 1H-ЯМР (300 МГц, ДМСО-d6), 4:1 смесь ротамеров, δ 0,65 (т, J=7,1 Гц и 1 Гц, 0,20× 3H), 0,75 (т, J=7,10 Гц, 7,10 Гц, 0,80× 3H), 0,95 (с, 0,10× 3H), 0,97 (с, 0,10× 3H), 1,15 (с, 0,80× 6Н), 1,25 (т, J=7,10 Гц, 7,10 Гц, 3H), 1,55-1,70 (м, 2Н), 1,85-2,00 (м, 2Н), 2,05-2,15 (м, 1H), 2,25-2,40 (м, 1H), 3,35-3,60 (м, 1H), 4,33 (кв, J=7,04, 7,05, 7,08 Гц, 2Н), 5,30-5,40 (м, 1H), 8,40 (с, 0,80× 1H), 8,45 (с, 0,20× 1Н).
Сравнительный пример 13

Раствор этилового сложного эфира из сравнительного примера 12 (0,95 г, 2,70 ммоль) и гидроксида лития (71,9 мг, 3,00 ммоль) в смеси ТГФ/Н2О (2,5:1, 35 мл) перемешивали примерно при 0° С в течение приблизительно 1 ч и примерно при комнатной температуре в течение приблизительно 20 ч. Реакционную смесь промывали Et2O (2× 50 мл) и подкисляли водной лимонной кислотой. Осажденную карбоновую кислоту экстрагировали CHCl3 (2× 75 мл). Раствор CHCl3 сушили над Na2SO4, фильтровали и концентрировали с получением карбоновой кислоты (0,81 г, выход 93%) в виде белого твердого вещества, которое использовали без дополнительной очистки. МС (отриц. петля) М-1=323. 1H-ЯМР (300 МГц, ДМСО-d6), 4:1 смесь ротамеров, δ 0,65 (т, J=7,1 Гц и 1 Гц, 0,20× 3H), 0,75 (т, J=7,10 Гц, 7,10 Гц, 0,80× 3H), 0,95 (с, 0,10× 3H), 0,97 (с, 0,10× 3H), 1,15 (с, 0,80× 6Н), 1,55-1,70 (м, 2Н), 1,85-2,00 (м, 2Н), 2,05-2,15 (м, 1Н), 2,25-2,40 (м, 1Н), 3,35-3,60 (м, 1Н), 5,30-5,40 (м, 1Н), 8,35 (с, 0,80× 1Н), 8,40 (с, 0,20× 1Н) 13,5 (с, 1Н).
Соединение 14

К холодному (0° С) раствору тиазол-4-карбоновой кислоты сравнительного примера 13 (322,2 мг, 1,00 ммоль) и триэтиламина (310 мл, 2,22 ммоль) в ДМФ (3 мл) добавляли дифенилфосфорилазид (250 мл, 1,16 ммоль) и 3-аминометилпиридин (110 мл, 1,08 ммоль). Полученную смесь перемешивали примерно при КТ в течение примерно 1 дня, разбавляли водным раствором NaCl (25 мл) и экстрагировали CHCl3 (3× 25 мл). Раствор CHCl3 сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенный продукт хроматографировали на силикагеле (элюируя 100% EtOAc) с получением 0,44 г соединения 14 в виде бесцветного неподвижного масла. Неочищенное масло хроматографировали на конических препаративных ТСХ-пластинах, элюируя 5% СН3ОН с CHCl3 с получением соединения 14 (271,8 мг, выход 66%) в виде бесцветного стеклообразного вещества. МС (полож. петля) MH+=415 (100%). 1H-ЯМР (300 МГц, ДМСО-d6), 4:1 смесь ротамеров, 8 0,64 (т, J=7,45 Гц, 7,45 Гц, 0,20× 3H), 0,78 (т, J=7,40 Гц, 7,40 Гц, 0,80× 3H), 0,95 (с, 0,10× 3H), 0,97 (с, 0,10× 3H), 1,15 (с, 0,80× 6Н), 1,45-1,65 (м, 2Н), 1,90-2,05 (м, 2Н), 2,10-2,20 (м, 1Н), 2,25-2,40 (м, 1Н), 3,42-3,48 (м, 1Н), 3,50-3,58 (м, 1Н), 4,47 (д, J=6,28 Гц, 2Н), 5,31-5,33 (м, 0,20× 1Н), 5,37-5,40 (м, 0,80× 1Н), 7,33-7,37 (м, 1Н), 7,70-7,73 (м, 1Н), 8,21 (с, 0,80× 1Н), 8,25 (с, 0,20× 1Н), 8,44-8,46 (м, 1Н), 8,54 (шир.с, 1Н), 9,00 (т, J = 6,17, 6,20 Гц, 1Н).
Соединение 15

К холодному (0° С) раствору тиазол-4-карбоновой кислоты из сравнительного примера 13 (322,6 мг, 1,00 ммоль), триэтиламина (0,17 мл, 1,22 ммоль), DMAP (13,0 мг, 0,100 ммоль) и 3-пиридилкарбинола (0,11 мл, 1,13 ммоль) в ТГФ (15 мл) добавляли изопропенилхлорформиат (0,12 мл, 1,10 ммоль). После нагревания до КТ гетерогенную реакционную смесь перемешивали примерно при КТ в течение примерно 20 ч. Реакционную смесь разбавляли водой и экстрагировали EtOAc (2× 25 мл). Раствор EtOAc сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенный продукт хроматографировали на 4 конических препаративных ТСХ-пластинах, элюируя 3% СН3ОН с CHCl3 с получением соединения 15 (240,0 мг, выход 59%) в виде светло-желтого твердого вещества. МС (полож. петля) MH+=416 (100%). 1H-ЯMP (300 МГц, ДМСО-d6), 4:1 смесь ротамеров, δ 0,64 (т, J=7,45 Гц, 7,45 Гц, 0,20× 3H), 0,78 (т, J=7,40 Гц, 7,40 Гц, 0,80× 3H), 0,98 (с, 0,10× 3H), 1,00 (с, 0,10× 3H), 1,14 (с, 0,80× 3H), 1,15 (с, 0,80× 3H), 1,53-1,65 (м, 2Н), 1,94-1,99 (м, 2Н), 2,05-2,15 (м, 1Н), 2,31-2,38 (м, 1Н), 3,44-3,56 (м, 2Н), 5,37 (м+с, 3H), 7,42-7,46 (м, 1Н), 7,88-7,90 (д, J=7,70 Гц, 1Н), 8,54-8,59 (м+с, 2Н), 8,69 (с, 1Н).
Сравнительный пример 14

К раствору 4-карбометокситиазола сравнительного примера 8 (3,64 г, 10,0 ммоль) и хлорида лития (2,12 г, 50,0 ммоль) в смеси EtOH (100 мл) и ТГФ (75 мл) добавляли боргидрид натрия (1,94 г, 51,3 ммоль). Через 6 часов к реакционной смеси добавляли еще 224 мг LiCl и 205 мг NaBH4 и эту смесь перемешивали в течение еще примерно 18 часов. Реакционную смесь гасили водным хлоридом аммония и экстрагировали CHCl3 (3× 100 мл). Раствор CHCl3 сушили над Na2SO4, фильтровали и концентрировали досуха. Неочищенный продукт хроматографировали на силикагеле, элюируя 5% СН3ОН в CHCl3, в результате чего получали спирт (2,39 г, выход 75%) в виде бесцветного масла. МС (полож. петля) МН+=319 (100%). 1H-ЯМР (300 МГц, CDCl3) δ 1,85-2,05 (м, 3H), 2,10-2,40 (м, 2H), 2,60-2,70 (м, 1Н), 3,40-3,55 (м, 1Н), 3,60-3,70 (м, 1Н), 4,65-4,75 (м, 2Н), 5,05-5,30 (м, 3H), 7,05 (шир.с, 1Н), 7,10 (шир.с, 1Н), 7,20-7,40 (м, 3H).
Соединение 16

Тиазол-4-метанол из сравнительного примера 14 (2,17 г, 6,83 ммоль) объединяли с трифенилфосфином (3,38 г, 12,8 ммоль), диэтилазодикарбоксилатом (1,64 мл, 10,4 ммоль) и 3-гидроксипиридином (0,97 г, 10,2 ммоль) в ТГФ (110 мл) и обрабатывали способом, описанным для получения соединения 5. В результате этой реакции получали неочищенное соединение 16 (2,08 г) и гидразиновый побочный продукт в виде масла. МС (полож. петля) MH+=396 (100%). 1H-ЯМР (300 МГц, ДМСO-d6) δ 1,85-2,05 (м, 3H), 2,10-2,40 (м, 1Н), 3,40-3,55 (м, 2Н), 4,90-5,20 (м, 5Н), 7,05 (шир.с, 1Н), 7,10 (шир.с, 1Н), 7,30-7,60 (м, 5Н), 8,12-8,15 (м, 1Н), 8,35-8,37 (м, 1Н), 9,00 (с, 1Н).
Соединение 17

Смесь неочищенного соединения 16 (1,85 г) и 30% HBr в СН3СО2Н (12 мл) перемешивали примерно при КТ в течение примерно 2 ч. Реакционную смесь разбавляли Н2O (25 мл) и экстрагировали Et2O (3× 75 мл). Подкисленный водный слой подщелачивали водным Na2CO3 и в высокой степени водорастворимое свободное основание экстрагировали CHCl3 (10× 100 мл). Водный слой концентрировали до получения влажного твердого вещества и экстрагировали CHCl3 (3× 125 мл). Объединенный раствор CHCl3 сушили (Na2SO4), фильтровали и концентрировали с получением соединения 17 (0,55 г) в виде масла. МС (полож. петля) МН+=262 (40%). 1H-ЯМР (300 МГц, ДМСО-d6) δ 1,67-1,84 (м, 2Н), 2,10-2,22 (м, 1Н), 2,82-3,00 (м, 2Н), 3,43 (шир.с, 1Н), 4,41-4,45 (м, 1Н), 5,16 (с, 2Н), 7,31-7,36 (м, 1Н), 7,47-7,51 (м, 1Н), 7,55 (с, 1Н), 8,18 (д, J=4,36 Гц, 1Н), 8,37 (д, J=2,86 Гц, 1Н).
Соединение 18

Соединение 17 (0,55 г, 2,00 ммоль), триэтиламин (0,69 мл, 4,95 ммоль) и этилоксалилхлорид (0,35 мл, 3,13 ммоль) в СН2Cl2 (100 мл) обрабатывали способом, описанным для получения соединения 7, в результате чего получали соединение 18 (0,33 г, выход 46%) в виде масла. МС (полож, петля) MH+=362 (100%). 1H-ЯМР (300 МГц, ДМСО-d6), 3:2 смесь ротамеров, δ 1,05 (т, J=7,40 Гц, 7,41 Гц, 0,40× 3H), 1,25 (т, J=7,41, 7,40 Гц, 0,60× 3H), 1,80-2,15 (м, 3H), 2,25-2,45 (м, 1Н), 3,55-3,75 (м, 2Н), 4,00 (кв, J=7,12 Гц, 7,13 Гц, 7,10 Гц, 0,4× 2Н), 4,30 (кв, J=7,12 Гц, 7,13 Гц, 7,10 Гц, 0,60× 2Н), 5,20 (с, 2Н), 5,32-5,39 (м, 0,60× 1Н), 5,40-5,55 (м, 0,40× 1Н), 7,32-7,40 (м, 1Н), 7,45-7,52 (м, 1Н), 7,69 (c, 0,60× 1Н), 7,75 (с, 0,40× 1Н), 8,19 (д, J=4,36 Гц, 1Н), 8,38-8,39 (м, 1Н).
Соединение 19

Соединение 18 (0,31 г, 0,86 ммоль) и хлорид 1,1-диметилпропилмагния (1 М, 2,25 мл, 2,25 ммоль) в ТГФ (10 мл) обрабатывали способом, описанным для получения соединения 8, в результате чего получали соединение 19 (0,17 г, выход 51%) в виде желтого масла. МС (полож. петля) МН+=388 (100%). 1H-ЯМР (300 МГц, ДМСО-d6), 4:1 смесь ротамеров, δ 0,65 (т, J=7,41 Гц, 7,42 Гц, 0,20× 3H), 0,79 (т, J=7,37, 7,40 Гц, 0,80× 3H), 0,93 (с, 0,10× 6Н), 0,97 (с, 0,10× 6Н), 1,16 (с, 0,40× 6Н), 1,17 (с, 0,40× 6Н), 1,63 (кв, J=7,40, 7,42, 7,45 Гц, 2Н), 1,95-1,97 (м, 2Н), 2,07-2,13 (м, 1Н), 2,26-2,36 (м, 1Н), 5,20 (с, 2Н), 5,34-5,40 (м, 1Н), 7,32-7,36 (м, 1Н), 7,49-7,53 (м, 1Н), 7,70 (с, 0,80× 1Н), 7,76 (с, 0,20× 1Н), 8,19 (д, J=3,96 Гц, 1Н), 8,38 (шир.с, 1Н).
Сравнительный пример 15

К холодному (0° С) раствору этилацетоацетата (65 мл, 510 ммоль) в безводном Et2O (100 мл) добавляли бром (26,40 мл, 512,4 ммоль). Реакционную смесь выдерживали примерно при КТ приблизительно в течение 1 дня, а затем выливали на лед и промывали водным Na2CO3 до тех пор, пока она не становилась щелочной. Раствор Et2O промывали насыщенным раствором соли и сушили над CaCl2 в течение приблизительно 4 дней. Затем раствор Et2O фильтровали и концентрировали, в результате чего получали 81,66 г светло-коричневого масла, которое стабилизировали твердым K2CO3. 1H-ЯМР (300 МГц, CDCl3) δ 1,50 (т, J=7,40, 7,40 Гц, 3H), 3,70 (с, 2Н), 4,00 (с, 2Н), 4,20 (кв, J=7,40, 7,40 Гц, 2Н).
Сравнительный пример 16

Раствор тиоамида N-CBz-пролина из сравнительного примера 8 (4,36 г, 16,5 ммоль) и 90%-ного этил-γ -бромацетоацетата из сравнительного примера 15 (4,80 г, 22,5 ммоль) в этаноле (170 мл) перемешивали при кипячении с обратным холодильником в течение примерно 2 ч. Реакционную смесь концентрировали досуха. Остаток хромаграфировали на силикагеле, элюируя 1% СН3ОН в CHCl3, в результате чего получали тиазол (6,25 г, выход 100%) в виде масла. МС (полож. петля) МН+=375 (40%). 1H-ЯМР (300 МГц, ДМСО-d6) δ 1,15-1,25 (м, 3H), 1,80-2,10 (м, 3H), 2,25-2,40 (м, 1Н), 3,35-3,55 (м, 2Н), 3,75 (с, 2Н), 4,05 (кв, J=7,40, 7,42, 7,45 Гц, 2Н), 4,95-5,20 (м, 3H), 7,10 (шир.с, 1Н), 7,25-7,40 (м, 5Н).
Сравнительный пример 17

Этиловый сложный эфир из сравнительного примера 16 (3,01 г, 8,00 ммоль), хлорид лития (1,72 г, 40,6 ммоль) и боргидрид натрия (1,51 г, 39,9 ммоль) в смеси EtOH/ТГФ (4:3; 175 мл) объединяли и обрабатывали способом, описанным для получения продукта сравнительного примера 14, в результате чего получали спирт (2,40 г, выход 90%) в виде светло-желтого масла. МС (полож. петля) МН+=333 (100%). 1H-ЯМР (300 МГц, ДМСО-d6) δ 1,80-2,05 (м, 3H), 2,20-2,40 (м, 1Н), 2,80 (т, J=6,52, 6,52 Гц, 2Н), 3,40-3,60 (м, 2Н), 3,65-3,70 (м, 2Н), 4,65 (т, 1Н (ОН), 4,90-5,20 (м, 3H), 7,05 (шир.с, 1Н), 7,15 (шир.с, 1Н), 7,25-7,40 (м, 4Н).
Соединение 20

Тиазол-4-этанол из сравнительного примера 17 (1,51 г, 4,55 ммоль), трифенилфосфин (2,37 г, 9,04 ммоль), диэтилазодикарбоксилат (1,00 мл, 6,35 ммоль) и 3-гидроксипиридин (0,60 г, 6,31 ммоль) в ТГФ (20 мл) обрабатывали способом, описанным для получения соединения 5, в результате чего получали неочищенное соединение 20 (0,82 г) в виде масла. МС (полож. петля) МН+=410 (100%). 1H-ЯМР (300 МГц, ДМСО-d6) δ 1,90-2,02 (м, 3H), 2,20-2,40 (м, 1Н), 3,15 (т, J=6,52, 6,52 Гц, 2Н), 3,47-3,60 (м, 2Н), 4,30-4,40 (м, 2Н), 4,93-5,20 (м, 3H), 7,05 (шир.с, 1Н), 7,20-7,70 (м, 7Н), 8,15 (д, J=4,3 Гц, 1Н), 8,28-8,32 (м, 1Н).
Соединение 21

Соединение 20 (0,71 г) и 30% HBr в СН3СО2Н (5 мл) обрабатывали способом, описанным для получения соединения 17, в результате чего получали соединение 21 (0,85 г) в виде гигроскопичной дигидробромидной соли. МС (полож. петля) MH+=276 (100%). 1H-ЯМР (300 МГц, ДМСО-d6) δ 1,90-2,20 (м, 3H), 2,40-2,50 (м, 1Н), 3,25-3,40 (м+т, J=6,52, 6,52 Гц, 4Н), 4,60 (т, J = 6,52, 6,52 Гц, 2Н), 5,00-5,10 (м, 1Н), 7,60 (с, 1Н), 7,95-8,05 (м, 1Н), 8,25-8,30 (м, 1Н), 8,65 (д, J=4,3 Гц, 1Н), 8,80 (с, 1Н), 9,10-9,25 (шир.с, 1Н), 9,75-9,90 (шир.с, 1Н).
Соединение 22

Соединение 21 (0,78 г, 1,78 ммоль), триэтиламин (0,63 мл, 4,52 ммоль) и этилоксалилхлорид (0,30 мл, 2,68 ммоль) в CH2Cl2 (25 мл) обрабатывали способом, описанным для получения соединения 7, в результате чего получали соединение 22 (0,42 г, выход 63%) в виде масла. МС (полож. петля) MH+=376 (100%). 1H-ЯМР (300 МГц, ДМСО-d6), 3:2 смесь ротамеров, δ 1,20-1,30 (м, 3H), 1,95-2,10 (м, 3H), 2,25-2,45 (м, 1Н), 3,15 (кв, J=7,40, 7,42, 7,45 Гц, 2Н), 3,50-3,75 (м, 2Н), 4,20-4,40 (м, 4Н), 5,30-5,35 (м, 0,60× 1h), 5,50-5,55 (м, 0,40× 1H), 7,25-7,33 (м, 1Н), 7,35-7,40 (м, 2Н), 8,15-8,18 (м, 1Н), 8,20-8,22 (м, 1Н), (кв, J=7,40, 7,42, 7,45 Гц, 2Н).
Соединение 23

Соединение 22 (0,41 г, 1,09 ммоль) и хлорид 1,1-диметилпропилмагния (1 М, 3,00 мл, 3,00 ммоль) в ТГФ (25 мл) обрабатывали способом, описанным для получения соединения 8, в результате чего получали соединение 23 (224,4 мг, выход 51%) в виде масла. МС (полож. петля) MH+=402 (100%). 1H-ЯМР (300 МГц, ДМСО-d6), 4:1 смесь ротамеров, δ 0,63 (т, J=7,41 Гц, 7,42 Гц, 0,20× 3H), 0,79 (т, J=7,37, 7,40 Гц, 0,80× 3H), 0,89 (с, 0,10× 6Н), 0,93 (с, 0,10× 6Н), 1,15 (с, 0,40× 6Н), 1,16 (с, 0,40× 6Н), 1,59-1,64 (м, 2Н), 1,95-2,08 (м, 3H), 2,23-2,33 (м, 1Н), 3,16 (т, J=6,30 Гц, 6,30 Гц, 2Н), 3,40-3,57 (м, 2Н), 4,36 (т, J=6,64, 6,64 Гц, 2Н), 5,30-5,37 (м, 1Н), 7,29-7,41 (м, 3H), 8,16 (д, J=4,25 Гц, 1Н), 8,26-8,29 (м, 1Н).
Соединение 24
Метод А

К перемешиваемому раствору (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты (4,557 г, 18,89 ммоль; полученной, как описано в WO 96/40633) в тетрагидрофуране (130 мл), охлажденному примерно до -15° С (МеОН/ледяная баня), добавляли триэтиламин (1,908 г, 2,63 мл, 18,89 ммоль), а затем этилхлорформиат (2,05 г, 1,806 мл, 18,86 ммоль). После перемешивания при температуре примерно от -15 до -10° С в течение примерно 30 минут осажденное твердое вещество удаляли путем фильтрации и этот фильтрат и промывки доводили до объема 170 мл путем добавления тетрагидрофурана.
При перемешивании раствора смешанного ангидрида (85 мл, 9,32 ммоль) примерно при 0° С, добавляли моногидрат гидразина (0,48 мл, 9,79 ммоль). Смесь перемешивали и оставляли на ночь для нагревания примерно до КТ. После удаления растворителя в вакууме остаток очищали колоночной хроматографией на силикагеле с использованием 50% этилацетата/дихлорметана в качестве элюента, в результате чего получали соединение 24 (0,64 г, выход 28,7%) в виде бесцветного твердого вещества, которое перекристаллизовывали из смеси эфира/пентана, т.пл. 177-178° С. CIMS 479 (MH+), 501 (М+Na+). 1H-ЯМР (300 МГц, CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 9,06 (шир.с, 2Н), 4,61 (м, 2Н), 3,50-3,46 (м, 4Н), 2,40-2,36 (м, 2Н), 2,13-1,94 (м, 6Н), 1,83-1,64 (м, 4Н), 1,25 и 1,21 (каждый с, каждый 6Н), 0,87 (т, 3H). ИК (KBr) см-1: 3261, 2970, 1706, 1684, 1636. Анализ для C24H38N4O6. Вычислено: C 60,23, Н 8,00, N 11,71. Найдено: C 60,30; H 8,03; N 11,58.
Соединение 24 также получали, как описано в Методе В.
Метод В - Соединение 24

К перемешиваемому раствору (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты (2,4229 г, 10 ммоль), полученной, как описано в WO 96/40633, в тетрагидрофуране (50 мл), примерно при КТ последовательно добавляли: триэтиламин (4,18 мл, 30 ммоль), гидрохлорид этилдиметиламинопропилкарбодиимида EDC· HCl (1,917 г, 10 ммоль) и гидрат гидроксибензотриазола HOBt· H2O (1,53 г, 10 ммоль). Примерно через 5 минут добавляли раствор гидразина в тетрагидрофуране (1 М, 5 мл, 5 ммоль) и смесь перемешивали примерно 18 ч. Тетрагидрофуран удаляли в вакууме (<35° ), а остаток растворяли в дихлорметане и последовательно промывали водой, 1% вод. HCl и водой, а затем сушили (Na2SO4). После фильтрации дихлорметан удаляли в вакууме и неочищенный продукт очищали колоночной хроматографией на силикагеле, элюируя 1,5% метанолом в дихлорметане, в результате чего получали соединение 24 (выход 18%), [α ] -95,8° (с=0,33, CHCl3), во всех отношениях идентичное соединению, полученному Методом А.
Метод А использовали для получения соединений 25-29.
Метод А - Соединение 25

С использованием (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пиперидинкарбоновой кислоты (полученной в основном, как описано для (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты в WO 96/40633) выделяли соединение 25 в виде бесцветного твердого вещества (выход 38%), т.пл. 180-181° С (эфир/пентан). CIMS 507 (МН+), 529 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 8,07 (шир.м, 2Н), 5,17 (д, 2Н), 4,29-4,08 (м, 2Н), 3,39 (д, 4Н), 2,89 (т, 1Н), 2,40-2,05 (м, 3H), 1,89-1,44 (м, 10Н), 1,23 и 1,22 (каждый с, каждый 6Н), 0,90 (т, 6Н). ИК (KBr) см-1: 3309, 2969, 1699, 1646, 1610. Анализ для C26H42N4O6: Вычислено: C 61,64, H 8,36, N 11,06. Найдено: C 61,45; H 8,58; N 10,76.
Соединение 26

С использованием (23)-1-(1,2-диоксо-3,3-диметилпентил)-2-азетидинкарбоновой кислоты (полученной в основном, как описано для (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты в WO 96/40633) выделяли соединение 26 в виде прозрачной клейкой пены (выход 35%), т.пл. <58° С. [α ] -93,4° (CHCl3), CIMS 451 (MH+), 473 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 9,70 (шир.с, 2Н), 5,02 (д, д, 2Н), 4,34-4,18 (м, 4Н), 2,85-2,73 (м, 2Н), 2,61-2,49 (м, 2Н), 1,84-1,73 (м, 6Н), 1,25 и 1,23 (каждый с, каждый 6Н), 0,84 (т, 6Н). ИК (KBr) см-1: 3498, 3247, 2972, 1703, 1636.
Соединение 27

С использованием (3S)-2-(1,2-диоксо-3,3-диметилпентил)-1,2,3,4-тетрагидроизохинолинкарбоновой кислоты (полученной в основном, как описано для (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты в WO 96/40633) выделяли соединение 27 в виде бесцветного твердого вещества (выход 41%, эфир/пентан), т.пл. 108-110° С; [α ] -55,12° (CHCl3), CIMS 603 (MH+), 625 (М+Na). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 7,22-7,03 (м, 8Н), 5,25-4,95 (м, 2Н), 4,51-4,25 (м, 4Н), 3,16-3,10 (м, 2Н), 1,72-1,66 (м, 2Н), 1,22 и 1,21 (каждый с и каждый 6Н), 0,89 (т, 6Н). ИК (KBr) см-1: 3219, 2969, 1701, 1639. Анализ для C34H42N4O6: Вычислено: C 67,75, H 7,02, N 9,30. Найдено: C 67,54; H 7,04; N 9,13.
Соединение 28

С использованием (4R)-3-(1,2-диоксо-3,3-диметилпентил)-4-тиазолидинкарбоновой кислоты (полученной в основном, как описано для (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты в WO 96/40633) выделяли соединение 28 в виде бесцветной пены (выход 39,8%), т.пл. 77-82° С (эфир/пентан); [α ] -15,6° (с=0,276, CHCl3). CIMS 515 (МН+), 537 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 8,93 (шир.с, 2Н), 5,05-4,98 (м, 2Н), 4,54-4,43 (м, 4Н), 3,59-3,54 (м, 2Н), 3,25-3,21 (9 м, 2Н), 1,78-1,74 (м, 4Н), 1,25 и 1,23 (каждый с, каждый 6Н), 0,88 (т, 3H). ИК (KBr) см-1: 3270, 2970, 1703, 1642, 1418. Анализ для С22Н34N4O6. Вычислено: C 51,34, H 6,66, N 10,89. Найдено: C 51,52; H 6,67; N 11,06.
Соединение 29

С использованием (2S)-1-бензилоксикарбонил-2-пирролидин-карбоновой кислоты выделяли соединение 29 в виде бесцветной пены с неопределенной областью плавления; [α ] -56,1° (CHCl3). CIMS 495 (MH+), 518 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 9,11 (шир.с, 2Н), 7,36 (шир.с, 10Н), 5,23-5,14 (м, 4Н), 4,41 (шир.с, 2Н), 3,55-3,44 (м, 4Н), 2,36-1,94 (м, 8Н). ИК (KBr) см-1: 3496, 1704, 1499, 1420, 1358.
Соединения 28 и 29 также получали, как описано в Методе С.
Метод С - Соединение 28

К раствору (4R)-3-(1,2-диоксо-3,3-диметилпентил)-4-тиазолидинкарбоновой кислоты (1,63 т, 6,29 ммоль) в дихлорметане (100 мл), охлажденному в ледяной бане, в течение 20 минут добавляли раствор оксалилхлорида (0,72 мл, 8,25 ммоль) в дихлорметане (5 мл). После перемешивания смеси примерно при КТ в течение примерно 2 ч растворитель удаляли путем выпаривания в вакууме при <40° С. После тщательной сушки остатка в вакууме примерно в течение 1 ч, его растворяли в сухом тетрагидрофуране (50 мл). К этому раствору при перемешивании примерно при 0° С добавляли смесь 1 М раствора гидразина в тетрагидрофуране (3,15 мл, 3,15 мМ) и триэтиламине (1,32 мл, 9,47 мМ) в течение примерно 30 минут, смесь перемешивали в течение ночи приблизительно при КТ. Примерно через 24 ч растворитель удаляли путем выпаривания в вакууме и остаток распределяли между 1 н. водной соляной кислотой и дихлорметаном. Органический слой сушили (Na2SO4) и выпаривали в вакууме. Неочищенный продукт очищали флеш-хроматографией на силикагеле, элюируя 2% метанолом в дихлорметане и получали соединение 28 (1,18 г, выход 73%), [α ] -15,6° (с=0,276, CHCl3), которое во всех отношениях идентично аутентичному образцу, полученному, как описано в Методе А.
Соединение 29

К раствору Z-пролина (10 г, 40,12 ммоль) в дихлорметане (100 мл), охлажденному примерно при 0° С, по каплям в течение примерно 20 минут в атмосфере азота добавляли оксалилхлорид (4,19 мл, 48 ммоль), а затем диметилформамид (3 капли). После перемешивания смеси примерно при КТ в течение примерно 2 ч смесь выпаривали досуха в вакууме и снова сушили в условиях высокого вакуума примерно 30 минут. Остаток растворяли в сухом тетрагидрофуране (ТГФ, 160 мл); и этот раствор добавляли к 1 М раствору гидразина в тетрагидрофуране (40 мл, 40 мМ) в течение примерно 2 минут, а затем перемешивали приблизительно при КТ в течение примерно 18 ч. Смесь выпаривали досуха в вакууме. Остаток растворяли в этилацетате (300 мл) и последовательно промывали 1% водной HCl и водой, органический слой сушили (Na2SO4), фильтровали и выпаривали в вакууме с получением бесцветного маслянистого остатка. Неочищенный продукт очищали флеш-хроматографией на силикагеле, элюируя 2% метанолом/дихлорметаном с получением соединения 29 (8,8 г, выход 88,7%), [α ] -56,1° (с=1,0, CHCl3), в виде бесцветной пены, которое идентично аутентичному образцу, полученному как описано в Методе А.
Соединение 30 получали, как описано в Методах D, Е и F.
Метод D - Соединение 30

К интенсивно перемешиваемой охлажденной льдом суспензии соединения 24 (0,567 г, 1,185 ммоль) в сухом эфире (400 мл) добавляли пиридин, (0,12 мл, 3,35 ммоль), а затем тионилхлорид (0,120 мл, 1,66 ммоль). После перемешивания смеси примерно при 0° С в течение примерно 2 ч, осажденные твердые вещества удаляли путем фильтрации, быстро промывали сухим эфиром и объединенные фильтраты выпаривали досуха в вакууме при <40° С. Остаток (пена, 0,6235 г) растворяли в сухом толуоле (25 мл) и нагревали при температуре кипения с обратным холодильником в течение примерно 3 ч. Остаток, полученный при выпаривании толуола в вакууме, очищали колоночной хроматографией на силикагеле/СН2Сl2. После элюирования смесью 1% метанол/метиленхлорид получали указанное в заголовке соединение 30 (0,354 г, выход 63,6%) в виде бесцветного твердого вещества, перекристаллизованного из эфира/пентана, т.пл. 123-124; [α ] -74,6° (с=0,8, CHCl3), CIMS 461 (MH+), 483 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 5,32 (д, д, J=3,0, 7,6), 3,59 (м, 4Н), 2,33-2,07 (м, 8Н), 1,80-1,65 (м, 4Н), 1,23 и 1,20 (каждый с, каждый 6Н), 0,86 (т, 6Н). ИК (KBr) см-1: 2972, 1704, 1641. Анализ для C24H36N4O5. Вычислено: C 62,59; H 7,88; N 12,16. Найдено: C 62,68; H 7,75; N 12,14.
Метод Е - Соединение 30

К раствору соединения 24 (0,112 г, 0,234 мМ) в хлорбензоле (10 мл) добавляли гексаметилдисилазан (0,123 мл, 0,585 мМ), имидазол (10 мг), фторид тетрабутиламмония (10 мг) и смесь нагревали при температуре кипения с обратным холодильником в течение приблизительно 72 ч. После хроматографической очистки неочищенного продукта получали соединение 30, которое было идентично аутентичному образцу, описанному в Методе D, на что указывали тонкослойная хроматография и данные масс-спектрометрии.
Метод F - Соединение 30

К раствору соединения 24 (0,2018 г, 0,42 ммоль) в тетрагидрофуране (10 мл) добавляли внутреннюю соль (метоксикарбонилсульфамоил)-триэтиламина и гидроксида (реагент Бургесса, всего 0,3014 г, 1,265 ммоль) тремя порциями, каждую из которых добавляли примерно через 30 минут. Затем смесь перемешивали примерно при КТ в течение приблизительно 72 ч. После удаления растворителя в вакууме и флэш-хроматографии реакционного остатка получали соединение 30, которое было идентично аутентичному образцу, описанному в Методе D, на что указывали тонкослойная хроматография и данные масс-спектрометрии.
Сравнительный пример 18

Смесь 3-(3-пиридил)пропионитрила (7,5 г, 56,75 мМ), гидрохлорида гидроксиламина (5,915 г, 85,12 мМ) и безводного карбоната калия (15,686 г, 113,5 мМ) в этаноле (200 мл) перемешивали и нагревали при температуре кипения с обратным холодильником в атмосфере азота примерно 64 ч. После охлаждения твердые вещества удаляли путем фильтрации и фильтраты выпаривали в вакууме досуха с получением вязкого маслянистого остатка янтарного цвета (8,95 г). После растирания в дихлорметане (300 мл) и фильтрации с последующим удалением дихлорметана в вакууме получали очень вязкий маслообразный остаток (5,84 г). Нерастворимую в дихлорметане часть растворяли в метаноле, фильтровали и выпаривали в вакууме, в результате чего получали вязкий полутвердый остаток амидоксимина (3,09 г); CIMS 166 (МН+).
Соединение 31

К раствору (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты (4,557 г, 18,89 ммоль) (полученной, как описано в WO 96/40633; 2,02 г, 8,37 ммоль) в монометиловом эфире диэтиленгликоля (диглим, 30 мл) последовательно добавляли; амидоксим из сравнительного примера 18 (1,383 г, 8,37 ммоль) и гидрохлорид 1-(диметиламинопропил)-3-этилкарбодиимида, EDC (3,21 г, 16,743 ммоль) и смесь перемешивали и нагревали в атмосфере азота в масляной бане приблизительно при 50° С в течение примерно 20 ч, а затем приблизительно при 110° С в течение примерно 5 ч. После охлаждения реакционную смесь распределяли между водой и дихлорметаном, органический слой сушили (Na2SO4) и выпаривали в вакууме с получением вязкого остатка (3,727 г). После очистки хроматографией на силикагеле/дихлорметане и элюирования смесью 1% метанол/дихлорметан получали соединение 31 в виде вязкого масла (0,39 г, выход 12,5%), CIMS 371 (MH+). 1H-ЯМР (CDCl3) смесь ротамеров) δ (для главного транс-ротамера) 8,47 (с, 2Н), 7,52 (д, 1Н, J=7,6), 7,24-7,20 (м, 1Н), 5,32 (д,д, 1Н), 3,64 (т, 2Н), 3,06 (м, 4Н), 2,42-2,33 (м, 1Н), 2,19-2,06 (м, 3H), 1,82-1,61 (м, 2Н), 1,24 и 1,22 (каждый с, каждый 3H), 0,87 (т, 3H). ИК (KBr) см-1: 2970, 1704, 1645, 1580, 1425. Анализ для C20H26N4O3: Вычислено: C 64,84, H 7,07, N 15,12. Найдено: C 64,43; H 6,95; N 14,89.
Сравнительный пример 19

Смесь этил-3-(3-пиридил)пропионата (5,92 г, 33,08 мМ) безводного гидразина (20 мл, большой избыток) и этанола (100 мл) кипятили с обратным холодильником в атмосфере азота примерно 18 ч. Растворитель удаляли путем выпаривания в вакууме и остаток растирали с эфиром и замораживали. Кристаллическое твердое вещество собирали и промывали небольшим количеством эфира с получением гидразида в виде бесцветного кристаллического твердого вещества, т.пл. 87-90° С. CIMS 166 (MH+) 1H-ЯМР (ДМСО) δ 9,04 (с, 1Н), 8,43-8,39 (м, 2Н), 7,61 (д, 1Н), 7,32-7,28 (м, 1Н), 2,83 (т, 2Н), 2,35 (т, 2Н). ИК (KBr) см-1: 3325, 3231, 3004, 1667, 1630. Анализ для С8Н11N3О: Вычислено: C 58,17, H 6,71, N 25,44. Найдено: C 57,94; H 6,49; N 25,28.
Соединение 32

С использованием процедуры Метода А, но с использованием одного эквивалента моноацилгидразина сравнительного примера 19 вместо незамещенного гидразина получали соединение 32 (выход 73%) в виде бесцветного твердого вещества, т.пл. 90-92° С (эфир/пентан). CIMS 389 (MH+). 1H-ЯМР (смесь ротамеров, CDCl3) δ (для главного транс-ротамера) 9,3 (с, 1Н), 8,83 (с, 1Н), 8,44 (с, 2Н), 7,54 (д, 1Н), 7,27-7,18 (м, 1Н), 4,59-4,56 (м, 1Н), 3,48 (т, 2Н), 2,99 (2Н), 2,57 (т, 2Н), 2,09-1,92 (м, 4Н), 1,80-1,63 (м, 2Н), 1,23 (с, 3H), 1,20 (с, 3H), 0,86 (т, 3H). ИК (KBr) см-1: 3258, 2971, 1703, 1637. Анализ для C20H28N4O4· 0,5 Н2O: Вычислено: C 60,44; H 7,35; N 14,10. Найдено: C 60,67; H 7,07; N 14,32.
Соединение 33

С использованием процедуры Метода F и соединения 32 в качестве исходного получали соединение 33 (выход 71,6%) в виде бесцветного вязкого масла. CIMS 371 (MH+). 1H-ЯMP (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 8,49 (д, 2Н), 7,54 (д, 2Н), 7,26-7,22 (м, 1Н), 5,30 (т, 2Н), 3,15 (с, 4Н), 2,35-2,04 (м, 4Н), 1,77-1,59 (м, 3H), 1,23 (с, 3H), 1,21 (с, 3H), 0,86 (т, 3H). Анализ для С20Н26N4O3: Вычислено: C 64,84; H 7,07; N 15,12. Найдено: C 64,45; H 7,07; N 15,12.
Соединение 34

С использованием процедуры Метода А, но с использованием вместо гидразина одного эквивалента 3-(3,4,5-триметоксифенил)пропионилгидразида, получали соединение 34 (выход 81,4%) в виде бесцветной стеклообразной пены CIMS 478 (МН+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 9,06 (шир.с, 1Н), 7,99 (шир.с, 1Н), 6,42 (с, 2Н), 4,61 (м, 1Н), 3,84 (с, 6Н), 3,82 (с, 3H), 3,48 (т, 2Н), 2,93 (т, 2Н), 2,54 (т, 2Н), 2,38-2,35 (м, 1Н), 2,06-1,93 (м, 3H), 1,80-1,65 (м, 2Н), 1,24 (с, 3H), 1,21 (с, 3H), 0,86 (т, 3H). ИК (KBr) см-1: 3273, 2970, 1703, 1639, 1127.
Соединение 35

С использованием процедуры Метода F и соединения 34 в качестве исходного получали соединение 35 (выход 99%) в виде бесцветного вязкого масла. CIMS 460 (МН+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 5,31 (д, д, 1Н), 3,85 (с, 6Н), 3,82 (с, 3H), 3,61-3,59 (м, 1Н), 3,25-3,00 (м, 4Н), 2,40-2,25 (м, 1Н), 2,25-1,90 (м, 4Н), 1,80-1,60 (м 2Н), 1,23 (с, 3H), 1,21 (с, 3H), 0,86 (т, 3H). ИК см-1: 2968, 1702, 1644, 1590, 1508, 1459, 1423, 1127. Анализ для C24H33N3O6·0,6 H2O: Вычислено: C 61,29; H 7,33; N 8,93. Найдено: C 61,29; H 7,17; N 8,93.
Соединение 36

В соответствии с процедурой Метода А с использованием (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пиперидинкарбоновой кислоты (полученной в основном, как описано для (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты в WO 96/40633) и с использованием гидразида сравнительного примера 19 вместо гидразина получали соединение 36 (выход 71,5%) в виде бесцветной твердой пены, т.пл. 48-52° С. CIMS 403 (МН+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 8,64 (с, 1Н), 8,47-8,43 (м, 3H), 7,56-7,53 (м, 1Н), 7,25-7,20 (м, 1Н), 5,17 (д, 1Н), 3,39 (д, 1Н), 3,03-2,98 (м, 2Н), 2,61-2,55 (м, 2Н), 2,30-2,20 (м, 1Н), 1,85-1,51 (м, 8Н), 1,23 (с, 3H), 1,22 (с, 3H), 0,89 (т, 3H). ИК (KBr) см-1: 3272, 2969, 1701, 1638, 1445. Анализ для C21H30N4O4· 0,3 Н2O: Вычислено: C 61,84; H 7,56; N 13,74. Найдено: C 61,88; H 7,40; N 13,70.
Соединение 37

В соответствии с процедурой Метода А с использованием (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пиперидинкарбоновой кислоты (полученной в основном, как описано для (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты в WO 96/40633) и с использованием 3-(3,4,5-триметоксифенил)-пропионилгидразида вместо гидразина получали соединение 37 (выход 98%) в виде прозрачного стеклообразного твердого вещества, т.пл. 52-55° С. CIMS 492 (МН+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 8,25 (шир.с, 1Н), 7,71 (шир.с, 1Н), 6,43 (д, 2Н), 5,17 (д, 1Н), 3,85 (с, 6Н), 3,83 (с, 3H), 3,38 (м, 1Н), 3,0-2,90 (м, 2Н), 2,50-2,40 (м, 2Н), 2,33 (шир.т, 1Н), 1,8-1,6 (м, 8Н), 1,24 (с, 3H), 1,23 (с, 3H). ИК (KBr) см-1: 3293, 2967, 2941, 1701, 1640, 1591, 1509, 1459, 1127. Анализ для C25H37N3O7· 0,75 H2O: Вычислено: C 59,45; H 7,68; N 8,32. Найдено: C 59,47; H 7,55; N 8,36.
Соединение 38

С использованием процедуры Метода F и соединения 36 в качестве исходного получали соединение 38 в виде бесцветного масла (выход 96%). CIMS 385 (МН+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 8,49 (д, 2Н), 7,56 (д, 1Н), 7,27-7,22 (м, 2Н), 5,93 (д, 1Н), 3,21-3,11 (м, 4Н), 2,35 (д, 1Н), 2,0-1,50 (м, 8Н), 1,24 (с, 3H), 1,21 (с, 3H), 0,90 (т, 3H). ИК (KBr) см-1: 2967, 2942, 2877, 1700, 1644, 1585, 1434. Анализ для С21Н28N4O3: Вычислено: C 65,60; H 7,34; N 14,57. Найдено: C 65,37; H 7,43; N 14,41.
Соединение 39

С использованием процедуры Метода F и соединения 37 в качестве исходного получали соединение 39 (выход 64,8%) в виде бесцветного вязкого масла. CIMS 474 (МН+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 6,42 (с, 2Н), 5,93 (д, 1Н), 3,84 (с, 6Н), 3,82 (с, 3H), 3,40 (шир.д, 1Н), 3,25-3,00 (м, 4Н), 2,36 (шир.д, 1Н), 2,0-1,50 (м, 8Н), 1,24 (3H), 1,22 (с, 3H), 0,90 (т, 3H). ИК (KBr) см-1; 3502, 2966, 2942, 1772, 1700, 1644, 1590, 1509, 1462, 1240, 1128. Анализ для С25Н35N3О6· 2 H2O: Вычислено: C 58,92; H 7,71; N 8,25. Найдено: C 59,05; H 7,45; N 8,57.
Сравнительный пример 20

К раствору N-карбобензилокси-L-пролина (2,0 г, 8,0 ммоль) в безводном метиленхлориде (20 мл) приблизительно при 0° C в атмосфере N2 по каплям добавляли оксалилхлорид (1,22 г, 9,6 ммоль), а затем 2 капли ДМФ. Раствор перемешивали примерно 3 ч, нагревали приблизительно до 25° С, а затем концентрировали. Полученный хлорангидрид растворяли в смеси ТГФ : ацетонитрил (1:1, 20 мл) и обрабатывали триэтиламином (0,87 г, 8,6 ммоль) приблизительно при 0° С в атмосфере N2. Раствор перемешивали примерно 10 мин и по каплям добавляли триметилсилилдиазометан (2,0 М раствор в гексане, 7,8 мл). Полученный раствор перемешивали примерно 2 ч приблизительно при 0° С, нагревали приблизительно до 25° С и перемешивали еще примерно 17 ч. Раствор разбавляли этилацетатом, промывали насыщенным NaHCO3 и Н2О, сушили (MgSO4) и концентрировали. Неочищенный остаток очищали колоночной хроматографией на силикагеле, элюируя 40% этилацетатом в пентане, в результате чего получали диазокетон (1,15 г, выход 53%) в виде желто-оранжевого масла. 1H-ЯМР (CDCl3, смесь цис-транс-ротамеров амида) δ 1,88-2,09, 2,17-2,38 (2 шир.м, 4Н); 3,58 (м, 2Н), 3,81, 4,03, 4,17 (с, АВ квартет, 2Н, J=4,0), 4,61 (м, 1Н), 5,13 (м, 2Н), 7,32 (м, 5Н).
Сравнительный пример 21

К раствору N-карбобензилокси-L-пролин-α -диазокетона из сравнительного примера 20 (1,0 г, 3,6 ммоль) в безводном диэтиловом эфире (10 мл) в атмосфере N2 по каплям добавляли насыщенный раствор HBr в диэтиловом эфире до тех пор, пока не прекращалось выделение N2. Раствор перемешивали примерно 1 ч приблизительно при 25° С, а затем промывали насыщенным NaHCO3, Н2O и насыщенным NaCl, сушили (MgSO4) и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле и элюировали 40% этилацетатом в пентане, в результате чего получали бромкетон (0,49 г, выход 42%) в виде прозрачного масла. 1H-ЯМР (CDCl3, смесь цис-транс-ротамеров амида) δ 1,84-2,30 (шир.м, 4Н); 3,58 (м, 2Н); 4,32 (м, 1Н); 5,17 (м, 2Н); 5,28 (т, 1Н); 7,35 (м, 5Н).
Соединение 40

К раствору тиоамида из сравнительного примера 8 (0,40 г, 1,5 ммоль) в безводном этаноле (15 мл) по каплям добавляли N-карбобензилокси-L-пролин-α -бромметилкетон из сравнительного примера 21 (0,49 г, 1,5 ммоль) в безводном этаноле (2 мл). Полученный раствор нагревали при температуре кипения с обратным холодильником в атмосфере N2 примерно 3 ч. Этот раствор охлаждали примерно до 25° С и концентрировали. Полученный остаток растворяли в смеси диэтилового эфира и насыщенного NaHCO3. Водную фазу отделяли и несколько раз экстрагировали диэтиловым эфиром. Органические слои объединяли, сушили (MgSO4) и концентрировали. Неочищенный продукт очищали колоночной хроматографией на силикагеле и элюировали 40% этилацетатом в пентане, в результате чего получали соединение 40 (0,51 г, выход 69%) в виде прозрачного масла. 1H-ЯМР (CDCl3, смесь цис-транс-ротамеров амида) δ 1,93 (м, 4Н); 2,20 (шир.м, 4Н); 3,61 (шир.м, 4Н); 5,18 (шир.м, 6Н); 6,74, 6,85 (с, с, 1Н); 7,13 (м, 2Н); 7,27 (м, 4Н); 7,39 (м, 4Н).
Соединение 41

К раствору соединения 40 (0,48 г, 0,97 ммоль) в безводном метиленхлориде (20 мл) приблизительно при 0° С по каплям добавляли 1,0 М раствор BBr3 в метиленхлориде (5 мл). Раствор перемешивали примерно 1 ч приблизительно при 0° С, а затем нагревали примерно до 25° С и перемешивали примерно 2 ч. Реакцию прекращали добавлением по каплям H2O (25 мл). Слои отделяли и органическую фазу экстрагировали Н2О. Объединенные водные фазы доводили до рН примерно 11 путем добавления по каплям 1 н. NaOH, а затем концентрировали. Полученные соли фильтровали и подвергали тщательной промывке этилацетатом. Органический фильтрат сушили (MgSO4) и концентрировали, в результате чего получали соединение 41 (0,067 г, выход 31%) в виде желтого масла. 1H-ЯМР (CDCl3) δ 1,88 (м, 6Н); 2,17 (м, 1Н); 2,31 (м, 1Н); 3,05 (м, 2Н); 3,16 (м, 2Н); 4,24 (м, 1Н); 4,57 (м, 1Н); 6,99 (с, 1Н).
Соединение 42

К раствору соединения 41 (0,067 г, 0,30 ммоль) в безводном метиленхлориде (5 мл) добавляли триэтиламин (0,13 г, 1,28 ммоль) приблизительно при 0° С. После перемешивания в течение примерно 15 мин по каплям добавляли раствор метилоксалилхлорида (0,10 г, 0,84 ммоль) в метиленхлориде (2 мл). Раствор перемешивали в течение примерно 1,5 ч приблизительно при 0° С, а затем промывали H2O, сушили (MgSO4) и концентрировали, в результате чего получали соединение 42 (0,115 г, выход 97%) в виде желтого масла. 1H-ЯМР (CDCl3, смесь 4 цис-транс-ротамеров амида) δ 1,96-2,48 (перекрывающаяся серия шир.м’с, 8Н); 3,62-4,00 (серия перекрывающихся шир.м’с, 4Н); 3,67, 3,72, 3,76, 3,91 (2 перекрывающихся с, с, с, серия перекрывающихся с, 6Н); 5,44, 5,71 (шир.м, 2Н); 6,91, 6,93, 6,98, 7,07 (с, с, с, с, 1Н).
Соединение 43
К раствору соединения 42 (0,115 г, 0,29 ммоль) в безводном ТГФ (5 мл) примерно при -78° С по каплям добавляли хлорид диметилпропилмагния (1,0 М раствор в диэтиловом эфире 0,754 мл). Раствор перемешивали в течение примерно 3 ч. приблизительно при -78° С, а затем выливали в насыщенный хлорид аммония (25 мл) и экстрагировали этилацетатом. Органические фазы объединяли, сушили (MgSO4) и концентрировали. Неочищенный остаток очищали колоночной хроматографией на силикагеле, элюируя 40% этилацетатом в пентане, в результате чего получали соединение 43 (0,075 г, выход 55%) в виде белого твердого вещества, т.пл. 127-129° С. 1H-ЯМР (CDCl3, смесь цис-транс-ротамеров амида) δ 0,60-1,02 (серия перекрывающихся с и м, 10Н); 1,12-1,21 (серия перекрывающихся с, 8Н); 1,66 (м, 4Н), 1,88-2,29 (перекрывающиеся шир.м’с, 8Н), 3,38-3,70 (шир.м, 4Н); 5,15, 5,29, 5,37, 5,42 (м, м, м, м, 2Н); 6,81, 6,84, 6,91, 6,96 (с, с, с, с, 1Н). Анализ для С25Н37N3O4S: Вычислено: C 63,13; H 7,84; N 8,83. Найдено: C 62,94; H 7,80; N 8,67.
Соединение 44

С использованием процедуры Метода F и соединения 25 в качестве исходного получали соединение 44 (выход 63%) в виде бесцветного твердого вещества, т.пл. 102-105° С (эфир/пентан). CIMS 489 (MH+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 5,98 (д, 2Н), 3,46-3,17 (м, 4Н), 2,33 (д, 2Н), 1,94-1,40 (м, 12Н), 1,25 и 1,21 (каждый с, каждый 6Н), 0,87 (т, 6Н). ИК (KBr) см-1: 1702, 1639, 1578, 1550, 1441. Анализ для C26H40N4O5· 0,6 Н2O. Вычислено: С 62,53; H 8,32; N 11,22. Найдено: C 62,52; H 8,09; N 11,14.
Соединение 45

С использованием процедуры Метода D и соединения 29 в качестве исходного получали соединение 45 (выход 68%) в виде вязкого масла; [α ]= -87,4° (CHCl3). CIMS 477 (MH+), 499 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 7,35-7,19 (м, 10Н), 5,20-5,0 (м, 6Н), 3,80-3,40 (м, 4Н), 2,40-1,85 (м, 8Н). ИК (KBr) см-1: 3584, 2956, 1705, 1584, 1498, 1446, 1410, 1355. Анализ для C26H28N4O5· 0,25 Н2О: Вычислено: C 64,92; H 5,97; N 11,65. Найдено: C 64,92; H 5,88; N 11,81.
Соединение 46
Метод G

Раствор соединения 45 (3,06 г, 6,46 ммоль) в метаноле (125 мл) гидрировали в присутствии катализатора 10% Pd/C (580 мг) примерно при 15 фунт/кв.дюйм в течение примерно 4 ч. Катализатор удаляли путем фильтрации через слой целита и фильтраты выпаривали досуха в вакууме с получением бесцветного очень вязкого масла (1,28 г, выход 95%). CIMS 209 (MH+), 231 (М+Na+). 1H-ЯМР (CDCl3) δ 4,46 (кв, 2Н), 3,17-3,01 (м, 4Н), 3,17-3,01 (м, 6Н). ИК (чистый) см-1: 3313, 2966, 2875, 1651, 1584, 1410, 1337, 1170, 1084.
Соединение 47
С использованием процедуры Метода D с большим избытком пиридина и тионилхлорида (22 и 11 эквивалентов соответственно) и соединения 27 в качестве исходного получали соединение 47 (выход 80%) в виде бесцветного пенистого вещества; [α ]=-55,12° (с=0,254, CHCl3). CIMS 585 (МН+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 7,22-6,91 (м, 8Н), 6,15-6,10 (м, 2Н), 4,41 (м, 2Н), 3,45-3,20 (м, 4Н), 1,81-1,69 (м, 4Н), 1,25 и 1,22 (каждый с, каждый 6Н), 0,90 (т, 6Н). ИК (KBr) см-1: 2969, 2928, 2879, 1702, 1621, 1586, 1556, 1499, 1429. Анализ для С34Н42Н4O5· 0,5 С2Н12: Вычислено: C 70,76; H 7,58; N 8,92. Найдено: C 70,93; H 7,38; N 8,92.
Соединение 48
Метод Н
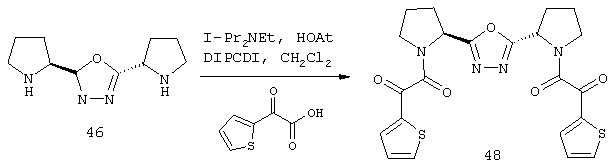
К раствору соединения 46 (0,107 г, 0,514 ммоль) в дихлорметане (10 мл) последовательно добавляли диизопропилэтиламин (2,87 мл, 1,65 ммоль), диизопропилкарбодиимид (0,323 мл), 1-гидрокси-7-азабензотриазол (0,280 г, 2,06 ммоль) и тиофен-2-глиоксиловую кислоту (0,320 г, 2,056 ммоль) в дихлорметане (5 мл). Смесь перемешивали в атмосфере аргона примерно при КТ в течение приблизительно 20 ч. Смесь выпаривали досуха в вакууме, остаток растворяли в дихлорметане (20 мл), а затем последовательно промывали 5% водной соляной кислотой, водой и насыщенным раствором бикарбоната натрия. Органический слой сушили (Na2SO4), фильтровали и выпаривали досуха с получением остатка. Неочищенный продукт очищали колоночной хроматографией на силикагеле, элюируя 1,5% метанолом в дихлорметане, в результате чего получали соединение 48 (выход 65%) в виде твердой пены, т.пл. 74-76° С. CIMS 485 (МН+), 507 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 8,05-7,96 (м, 2Н), 7,81-7,74 (м, 2Н), 7,21-7,11 (м, 2Н), 5,46-5,42 (м, 2Н), 3,87-3,66 (м, 4Н), 2,47-1,99 (м, 8Н). ИК (KBr) см-1: 3092, 2955, 2310, 1658, 1584, 1561, 1513, 1441, 1406, 1353, 1252, 1167. Анализ для C22H20N4O5S2: Вычислено: С 54,53; H 4,16; N 11,56. Найдено: C 54,49; H 4,08; N 11,34.
Соединение 51
Метод I

К охлажденному льдом перемешиваемому раствору соединения 46 (0,431 г, 2,071 ммоль) и триэтиламина (0,65 мл, 4,66 ммоль) в дихлорметане (20 мл) в атмосфере аргона в течение 30 минут добавляли метилхлороксоацетат (0,54 мл, 5,8 ммоль) в дихлорметане (9 мл). После перемешивания смеси примерно при 0° С в течение примерно еще 2 ч реакционную смесь обрабатывали путем промывки насыщенным раствором соли (3× 50 мл), сушки органического слоя (Na2SO4), фильтрации и выпаривания досуха в вакууме, в результате чего получали соединение 51 в виде пены (0,815 г, выход 95%). CIMS 381 (MH+), 403 (М+Na+).
Соединение 49

К раствору соединения 51 (0,375 г, 0,986 ммоль) в тетрагидрофуране (7 мл), перемешиваемому и охлажденному в атмосфере аргона примерно при -78° С, в течение 15 минут по каплям добавляли эфирный раствор бромида циклогексилмагния (1 мл 2 М, 2 ммоль). После дополнительного перемешивания примерно при -78° С в течение примерно 3 ч реакционную смесь обрабатывали путем выливания в насыщенный водный раствор хлорида аммония и экстракции этилацетатом, сушки органического слоя (Na2SO4), фильтрации и выпаривания досуха в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле, элюируя 0,75% метанолом в дихлорметане, в результате чего получали соединение 49 (29 мг, выход 5,8%) в виде бесцветного твердого вещества, т.пл. 132-133° С. CIMS 485 (MH+), 507 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 5,33-5,27 (м, 2Н), 3,88-3,59 (м, 4Н), 2,44-1,69 (м, 20Н), 1,38-1,00 (м, 6Н). ИК (KBr) см-1: 2880, 2927, 2852, 1706, 1641, 1582, 1560, 1445. Анализ для С26H36N4O5· 0,35 Н2O: Вычислено: C 63,61; H 7,54; N 11,41. Найдено: C 63,96; H 7,59; N 11,08.
Соединение 50

С использованием процедуры Метода А и одного эквивалента гидразида N-карбобензилоксипролина (CAS №53157-63-4) вместо гидразина получали соединение 50 (выход 56%) в виде бесцветного твердого вещества, т.пл. 145-146° С. CIMS 443 (MH+-СО), 487 (MH+), 485 (М-Н). [α ]=-96,1° (с=0,254, CHCl3). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера): 9,05 (шир.с, 1Н), 8,98 (шир.с, 1Н), 7,36 (с, 5Н), 5,23-5,11 (м, 2Н), 3,70-3,40 (м, 4Н), 2,41 (шир.с, 2Н), 2,30-1,80 (м, 6Н), 1,79-1,42 (м, 2Н), 1,25 и 1,22 (каждый с, каждый 3H), 0,87 (т, 3H). ИК (KBr) см-1: 3307, 3272, 2966, 2884, 1731, 1699, 1651, 1444. Анализ для С25Н34N4O6: Вычислено: C 61,71; H 7,04; N 11,51. Найдено: C 62,09; H 7,20; N 11,28.
Соединение 52

С использованием процедуры Метода D и соединения 26 в качестве исходного получали соединение 52 в виде бесцветного вязкого жироподобного продукта (выход 75,8%); CIMS 433 (МН+), 455 (М+Na+). 1H-ЯМР (смесь ротамеров, CDCl3) δ (для главного транс-ротамера) 5,86-5,53 (д, 2Н), 4,48-4,35 (м, 2Н), 4,27-4,14 (м, 2Н), 2,93-2,80 (м, 2Н), 2,73-2,50 (м, 2Н), 1,84-1,60 (м, 4Н), 1,21 (с, 12Н), 0,82 (т, 6Н). ИК (KBr) см-1: 2970, 2880, 1704, 1651, 1583, 1564, 1461, 1423, 1385.
Соединение 53

С использованием процедуры Метода D и соединения 50 в качестве исходного получали соединение 53 в виде бесцветного вязкого масла (выход 89%). CIMS 469 (MH+), 491 (М+Na+). [α ]=-92,7° (с=0,246, CHCl3). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 7,36-7,20 (м, 5Н), 5,20-5,03 (м, 2Н), 3,70-3,50 (м, 2Н), 2,40-1,80 (м, 4Н), 1,80-1,60 (м, 2Н). ИК (чистый) см-1: 2968, 2881, 1702, 1641, 1584, 1411, 1356. Анализ для С25H32N4O5: Вычислено: C 64,09; H 6,88; N 11,96. Найдено: C 64,20; H 6,87; N 11,83.
Соединение 54

С использованием процедуры Метода I и α -толуолсульфонилхлорида вместо метилхлороксоацетата получали соединение 54 (выход 59%) в виде бесцветного твердого вещества; т.пл. 144-145° С. CIMS 517 (MH+), 539 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 7,49-7,46 (м, 4Н), 7,40-7,37 (м, 6Н), 5,04 (кв, 2Н), 4,41 (кв, 4H), 3,39-3,31 (9 м, 2Н), 3,14-3,07 (м, 2Н), 2,33-2,12 (м, 4H), 2,09-1,94 (м, 4H). ИК (KBr) см-1: 1574, 1554, 1495, 1455, 1410, 1332, 1140. Анализ для C24H28N4O5S2: Вычислено: C 55,80; H 5,46; N 10,84. Найдено: C 55,73; H 5,42; N 10,76.
Соединение 55

С использованием процедуры Метода G и соединения 53 в качестве исходного получали соединение 55 в виде бесцветного вязкого масла (выход 90%). [α ]=-35,5° (с=0,414, CHCl3). CIMS 335 (MH+), 357 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 5,32 (дд, 2Н), 4,46 (м, 2Н), 3,61 (дт, 4H), 3,36-3,03 (м, 4H), 2,40-1,60 (м, 6Н), 1,24 и 1,21 (каждый с, каждый 3H), 0,86 (т, 3H). ИК (чистый) см-1: 3342, 2969, 2880, 1703, 1642, 1586, 1586, 1428. Анализ для C17H26N4O3: Вычислено: C 61,06; H 7,84; N 16,75. Найдено: C 60,75; H 7,64; N 16,68.
Соединение 56

С использованием процедуры Метода Н и 3,4,5-триметоксифенилглиоксиловой кислоты вместо тиофен-2-глиоксиловой кислоты получали соединение 56 в виде бесцветного твердого вещества, т.пл. 79-83° С (выход 56%). [α ]=-0,9° (с=0,260, CHCl3). CIMS 653 (MH+), 675 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 7,34 (с, 4Н), 5,41 (д, д, 2Н), 3,95 (с, 6Н), 3,93 (с, 12Н), 3,68 (т, 4Н), 3,48-2,20 (м, 8Н). ИК (KBr) см-1: 2944, 2839, 1770, 1715, 1677, 1650, 1583, 1416, 1330, 1126. Анализ для C32H36N4O11: Вычислено: C 58,89; H 5,56; N 8,58. Найдено: C 58,64; H 5,75; N 8,35.
Соединение 57

С использованием процедуры Метода I и соединения 55 в качестве исходного соединения и α -толуолсульфонилхлорида вместо метилхлороксоацетата получали соединение 57 в виде вязкого масла (выход 72%). [α ]=-28,6° (с=0,49, CHCl3). CIMS 489 (МН+), 511 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 7,49-7,46 (м, 2Н), 7,38-7,36 (м, 4Н), 5,39-5,32 (м, 1Н), 5,10-5,06 (м, 1Н), 4,39 (кв, 2Н), 3,71-3,58 (м, 2Н), 3,40-3,31 (м, 1Н), 3,11-3,00 (м, 1Н), 2,38-1,92 (м, 9Н), 1,79-1,59 (м, 2Н), 1,23 и 1,20 (каждый с, каждый 3H), 0,84 (т, 3H). ИК (чистый) см-1: 2971, 2881, 1703, 1644, 1584, 1562, 1427, 1342. Анализ для C24H32N4O5S: Вычислено: C 59,00; H 6,60; N 11,47. Найдено: C 59,24; H 6,58; N 11,39.
Соединение 58

С использованием процедуры Метода F и соединения 28 в качестве исходного получали соединение 58 (выход 44%) в виде бесцветного вязкого масла, [α ]=-12° (с=0,308, CHCl3). CIMS 497 (MH+), 519 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 5,90-5,83 (м, 2Н), 4,64-4,48 (м, 4Н), 3,53-3,34 (м, 4Н), 1,74 (м, 4Н), 1,26 и 1,23 (каждый с, каждый 6Н), 0,88 (т, 6Н). ИК см-1: 2966, 1798, 1651. Анализ для C22H32N4O5S2: Вычислено: C 53,20; H 6,49; N 11,28. Найдено: C 53,36; H 6,58; N 10,64.
Соединение 59
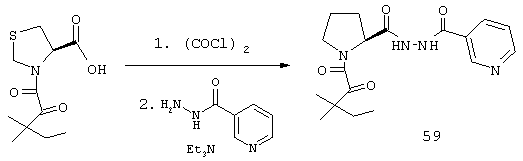
В соответствии с процедурой Метода С с использованием (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты (полученной, как описано в WO 96/40633) вместо (4R)-3-(1,2-диоксо-3,3-диметилпентил)-4-тиазолидинкарбоновой кислоты и с использованием гидразида никотиновой кислоты вместо гидразина получали соединение 59 в виде бесцветного твердого вещества, т.пл. 161-163° С (выход 42%). CIMS 361 (МН+), 383 (М+Na+). 1H-ЯМР (CDCl3) δ 9,06 (д, 1Н), 8,74 (м, 1H), 8,14 (д, 1Н), 7,37 (м, 1H), 4,68 (м, 1Н), 3,52 (т, 2Н), 2,39 (м, 1H), 2,14 (м, 2Н), 2,00 (м, 1H), 1,81-1,61 (м, 4Н). ИК (KBr) см-1: 3296, 2965, 2883, 1702, 1664, 1640, 1590, 1518. Анализ для C18H24N4O4: Вычислено: C 59,99; H 6,71; N 15,55. Найдено: C 59,88; H 6,63; N 15,38.
Соединение 60

С использованием процедуры Метода F и соединения 59 в качестве исходного получали соединение 60 в виде бесцветного твердого вещества (выход 79%), т.пл. 96-97° С. CIMS 343 (MH+), 365 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 9,25 (д, 1Н), 8,78 (м, 1Н), 8,34 (дд, 1Н), 7,47 (м, 1Н), 5,43 (д, д, 1Н), 3,67 (т, 2Н), 2,47-2,08 (м, 2Н), 1,85-1,73 (м, 2Н), 1,26 и 1,23 (каждый с, каждый 3H), 0,87 (т, 3H). ИК (KBr) см-1: 2969, 2883, 1701, 1638, 1431. Анализ для C18H22N4O3: Вычислено: C 63,14; H 6,48; N 16,36. Найдено: C 62,91; H 6,37; N 16,27.
Соединение 61

С использованием процедуры Метода А и с использованием (2S)-1-(1,2-диоксо-3,3-диметилбутил)-2-пирролидинкарбоновой кислоты (полученной в основном, как описано для (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты в WO 96/40633) в качестве исходного получали соединение 61 в виде бесцветного твердого вещества (выход 28%), т.пл. 108-110° С. CIMS 451 (МН+), 473 (М+Na+), 449 (М-Н). [α ]=-116,1° (с=0,274, CHCl3). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 9,11 (с, 2Н), 4,62 (м, 2Н), 3,47 (т, 4Н), 2,50-2,37 (м, 2Н), 2,15-1,85 (м, 4Н), 1,29 (с, 18Н). ИК (KBr) см-1: 3279, 2976, 2879, 1707, 1639, 1446. Анализ для С22H34N4O6· 0,35 Н2О: Вычислено: C 57,84; H 7,66; N 12,26. Найдено: C 58,10; H 7,75; N 11,98.
Соединение 61

В соответствии с процедурой Метода В с использованием (2S)-1-(1,2-диоксо-3,3-диметилбутил)-2-пирролидинкарбоновой кислоты (полученной в основном, как описано для (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты в WO 96/40633) в качестве исходного соединения и с использованием N-метилморфолина (NММ) в качестве основания вместо триэтиламина, Et3N, получали соединение 61 (выход 73%), которое во всех отношениях идентично соединению, полученному Методом А.
Соединение 62

С использованием процедуры Метода D и соединения 61 в качестве исходного получали соединение 62 (выход 42,5%), т.пл. 138-141° С. [α ]=-82,6° (с=0,282, CHCl3). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 5,34-5,29 (м, 2Н), 3,63-3,55 (м, 4Н), 2,40-2,00 (м, 8Н), 1,27 (с, 18Н). ИК (KBr) см-1: 2958, 1705, 1636, 1582, 1560, 1438. Анализ для C22H32N4O5: Вычислено: C 61,09; H 7,46; N 12,95. Найдено: C 61,05; H 7,44; N 12,88.
Соединение 62

С использованием процедуры Метода Н и диметилпировиноградной кислоты вместо тиофен-2-глиоксиловой кислоты получали соединение 62 (0,030 г, выход 13,5%), которое во всех отношениях идентично соединению, полученному методом D.
Соединение 63

В соответствии с процедурой Метода А с использованием (2R)-1-(1,2-диоксо-3,3-диметилпентил)-2-пиперидинкарбоновой кислоты (полученной в основном, как описано для (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты в WO 96/40633) в качестве исходного соединения вместо (2S)-1-(1,2-диоксо-3,3-диметилпентил)-2-пирролидинкарбоновой кислоты и с использованием изобутилхлорформиата вместо этилхлорформиата получали соединение 63 в виде бесцветной твердой пены (выход 37%), т.пл. 179-180° С. [α ]=+101° (с=0,474, CHCl3). CIMS 479 (MH+), 501 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 9,13 (шир.с, 2Н), 4,61 (дд, 2Н), 3,49 (м, 4Н), 2,39-2,34 (м, 2Н), 2,16-1,86 (м, 6Н), 1,84-1,61 (м, 4Н), 1,25 и 1,21 (каждый с, каждый 6Н), 0,87 (т, 6Н). ИК (KBr) см-1: 3263, 2970, 2880, 1705, 1684, 1636, 1614, 1567, 1444. Анализ для C24H38N4O5: Вычислено: C 60,23; H 8,00; N 11,71. Найдено: C 59,96; H 7,92; N 11,55.
Соединение 64

С использованием процедуры Метода D и соединения 63 в качестве исходного получали соединение 64 в виде твердого вещества цвета слоновой кости (выход 85%), т.пл. 123-124° С. [α ]=+72,2° (с=0,248, CHCl3), CIMS 461 (MH+), 483 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 5,32 (дд, 2Н), 3,68-3,53 (м, 4Н), 2,41-2,02 (м, 8Н), 1,83-1,58 (м, 4Н), 1,23 и 1,20 (каждый с, каждый 3H), 0,86 (т, 6Н). ИК (KBr) см-1: 2973, 1705, 1639, 1574, 1463, 1432, 1383, 1096. Анализ для C24H36N4O5: Вычислено: C 62,59 Н, 7,88, N 12,16. Найдено: C 62,74; H 7,81; N 12,10.
Соединение 65

С использованием процедуры Метода В и пиколилгидразида вместо гидразина получали соединение 65 в виде твердого вещества цвета слоновой кости (выход 71%), т.пл. 182-185° С. [α ]=-67,0° (с=0,26, CHCl3), CIMS 361 (MH+), 383 (М+Na+). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 10,00 (шир.с, 1Н), 9,39 (шир.с, 1Н), 8,57 (д, 1Н), 8,15 (д, 1Н), 7,86 (дт, 1Н), 7,48-7,44 (м, 1Н), 4,75-4,71 (м, 1Н), 3,53-3,47 (м, 2Н), 2,52-2,39 (м, 1Н), 2,34-1,88 (м, 4Н), 1,85-1,67 (м, 4Н), 1,28 и 1,24 (каждый с, каждый 3H), 0,89 (т, 3H). ИК (KBr) см-1: 3270, 2972, 2880, 1703, 1640. Анализ для C18H24N4O4· 0,5 Н2О: Вычислено: C 58,52, H 6,82, N 15,17. Найдено: C 58,53; H 6,44; N 14,90.
Соединение 66

С использованием процедуры Метода F и соединения 65 в качестве исходного получали соединение 66 в виде бесцветного кристаллического твердого вещества (выход 75%), т.пл. 70-72° С. CIMS 343 (MH+), 365 (М+Na+). [α ]=-36,8° (с=0,280, CHCl3). 1H-ЯМР (CDCl3, смесь ротамеров) δ (для главного транс-ротамера) 8,77 (д, 1Н), 8,23 (д, 1Н), 7,89 (д, т 1Н), 5,45 (дд, 1Н), 3,76-3,62 (м, 2Н), 2,44-2,05 (м, 4Н), 1,86-1,63 (м, 2Н), 1,27 и 1,23 (каждый с, каждый 3H), 0,87 (т, 3H). ИК (KBr) см-1: 2973, 1701, 1636, 1588, 1562, 1552, 1463, 1441. Анализ для C18H22N4O3·0,25 Н2О: Вычислено: С, 62,32; H 6,54; N 16,15. Найдено: C 62,24; H 6,38; N 16,36.
IV. Биологические анализы и активность
Результаты анализов на in vitro-активность вещества, описанного в примерах 1 и 4, приводятся в таблице 2. В примерах 2 и 3 подробно описаны методы получения клеточных культур, используемых в примере 4. Результаты анализа на in vitro-активность, описанного в примере 5, приводятся в таблице 3. Результаты анализа на in vivo-активность вещества, описанного в примере 6, представлены на чертеже.
А. Биологическая активность in vitro
Пример 1
Культура спинно-мозговых узлов (СМУ)
СМУ вырезали у новорожденных или 1-дневных крыс CD и помещали в PBS на льду. После двукратной промывки стерильной средой для культивирования, СМУ переносили с помощью изогнутого пинцета №7 в пустые лунки 6-луночного планшета, покрытого полиорнитином/аминином (Becton Dickinson Labware). Затем, очень осторожно, так чтобы не повредить СМУ, добавляли три мл/лунку среды для культивирования. Среда для культивирования представляла собой среду L-15 Лейбовица (Gibco) плюс 0,6% глюкоза, 33 мМ KCl, 10% FCS, 10 мМ Hepes и пенициллин/стрептомицин/глутамин. После инкубирования в течение ночи примерно при 37° С в 5% CO2 эту среду заменяли 3 мл/лунку аналитической среды (среда L-15 Лейбовица плюс 0,6% глюкоза, 1% FCS, 1% N-2-добавки (Gibco), 10 мкМ ara-С, 10 мМ Hepes и пенициллин/стрептомицин/глутамин), содержащей либо носитель (ДМСО, 1/200000), позитивный контроль (2-4 нг/мл NGF), либо тестируемое соединение (50-250 нМ). Все среды были свежими и приготавливались ежедневно. СМУ исследовали под микроскопом на рост нейритов в дни 1-5. В оптимальных условиях обработка носителем не индуцировала рост нейритов из СМУ. Результат эксперимента рассматривали как положительный (+), если соединение настоящего изобретения индуцировало рост нейритов диаметром ≥ 1 СМУ.
В. Анализ клеточной культуры
Пример 2
Первичные клетки гиппокампа крысы
Клетки гиппокампа иссекали из головного мозга 18-дневного эмбриона детеныша крысы и подвергали диссоциации трипсином (1 мг/мл) и растиранию. Клетки высевали при плотности 30000 клеток/лунку в 96-луночные планшеты, заполненные 100 мкл MEM и 10% FBS. На 7-й день культивирования, клетки фиксировали 4%-ным параформальдегидом и проводили иммунофлуоресцентный анализ.
Пример 3
Клетки нейробластомы M17 человека
Клетки нейробластомы M17 человека культивировали в среде ЕМЕМ и Хэмса F12 (в отношении 1:1) с 1× NEAA и 10% FBS. Эта культуральная среда содержала антибиотик 1× PSN, и ее ежедневно заменяли, а клетки пассировали в log-фазе, близкой к конфлюентности.
Пример 4
Анализ на отрастание нейрита
Культуры инкубировали с нормальной лошадиной сывороткой (1:50; Vector Labs) примерно 20 мин, промывали, а затем инкубировали с "первым" антителом, белком 1, ассоциированным с микротрубочками (антимышиный МАР-2; 1:1000; Chemicon) в течение примерно 2 ч приблизительно при комнатной температуре. После инкубирования с первым антителом культуры промывали и инкубировали с меченным флуоресцеином антителом против мышиного IgG (абсорбированного у крысы; 1:50; Vector Labs) в течение примерно 1 ч. После инкубирования с флуоресцеином культуры промывали и считывали в PBS на флуоресцентном планшет-ридере (возбуждение: 485 нм; излучение: 530 нм). Соединение рассматривалось как активное, если ответный рост нейрита превышал средний ответ ДМСО-обработанного контроля на том же самом планшете. Ответ на тестируемое соединение выражали в процентах от ДМСО-обработанного контроля. Отношение сигнал-шум было постоянным: флуоресценция от лунок с ДМСО-контролем, по крайней мере, в два раза превышала флуоресценцию в лунках с "пустышкой".
Пример 5
Анализ на связывание с никотиновым рецептором ацетилхолина
Связывание 3H-цистизина с нейронными никотиновыми рецепторами ацетилхолина осуществляли с использованием неочищенных синаптических мембранных препаратов, полученных из коры головного мозга, полосатого тела и гиппокампа крысы. Свежие или замороженные мембраны гомогенизировали в 50 объемах 10 мМ HEPES (N-2-гидроксиэтилпиперазин-N’-2-этансульфоновая кислота, рН 7,4) и центрифугировали при 42000 g. Фракцию Р2 ресуспендировали в 40 объемах 10 мМ HEPES и центрифугировали при 42000 g. Эту стадию повторяли, и P2 ресуспендировали в 25 объемах (например, 1 г ткани в 25 мл) среды, состоящей из Na+-HEPES-буфера (10 мМ, рН 7,4), 5 мМ MgCl2, 0,01% порошкообразного альбумина бычьей сыворотки (BSA) и 100 мМ NaCl. Для инициации реакции связывания соединение настоящего изобретения (100 мкл), Na-HEPES-забуференную среду для инкубации (400 мкл), 3H-цитизина (250 мкл) и суспензию биологических мембран (250 мкл) пипетировали в тест-пробирку, содержимое смешивали, а затем инкубировали примерно при 23° С приблизительно 40 мин. Реакцию связывания завершали путем фильтрации с использованием сборщика клеток Brandel Cell Harvester и количество связанного 3H-цитизина для каждого образца определяли с использованием жидкостного сцинтилляционного счетчика Wallac LKB 1205 Betaplate. Все соединения скринировали при 10 мкМ в четырех повторностях. Неспецифическое связывание определяли с использованием 10 мкМ (+)-эпибатидина для блокирования всего связывания 3H-цитизина с α -4,β -2 никотиновым рецептором ацетилхолина (α 4β 2nAChR). Активность каждого тестируемого соединения вычисляли следующим образом: после коррекции на неспецифическое связывание вычисляли процент ингибирования специфического связывания (полное связывание минус неспецифическое связывание). Затем каждое активное соединение тестировали при пяти концентрациях для построения кривой зависимости ингибирования от концентрации. Величины IC50 вычисляли путем проведения нелинейного регрессионного анализа данных с использованием стандартной регрессионной программы.

Настоящее изобретение относится к способам применения соединений 6 и 17 и фармацевтических композиций, содержащих эти соединения, для лечения болезней Паркинсона и Альцгеймера, тревожных состояний, дефицита внимания при расстройствах, характеризующихся гиперактивностью, "ADHD", синдрома Туррета, привыкания к курению, и боли.
С. Биологическая активность in vivo
Пример 6
Модель сдавливания лицевого нерва крысы
Крыс Long-Evans анестезировали кетамином (60 мг/кг)/ксилазином (6 мг/кг). Лицевой нерв открывали и механически сдавливали пинцетом возле шилососцевидного отверстия с одной стороны, при этом противоположная неповрежденная сторона служила в качестве внутреннего контроля. Сдавливание нерва приводило к параличу мышцы усов, и следовательно, к снижению подвижности усов на поврежденной стороне, которое наблюдалось сразу после восстановления от наркоза. Крысам перорально (р.о.) вводили тестируемое соединение при концентрации примерно 20 мг/кг два раза в день в течение 15 дней после хирургической операции. Контрольным крысам давали только носитель. В каждой группе тестировали от трех до восьми крыс. Восстановление подвижности усов после обработки соединениями настоящего изобретения регистрировали ежедневно в различные промежутки времени после операции в течение двух недель включительно, и полученные данные представлены на чертеже.








Claims (15)
1. Соединение, имеющее структуру

или его фармацевтически приемлемая соль, где
(a) R1 выбран из группы, состоящей из Н, COCOR2, COOR3 и SO2R3, причем
(i) R2 выбран из группы, состоящей из прямого или разветвленного C1-6алкила, прямого или разветвленного C1-6алкенила, С5-7циклоалкила, 2-тиенила, 3-тиенила или фенила, где указанный фенил имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из Н, низшего алкила, низшего алкоксила, гидроксила и галогена, и
(ii) R3 представляет фенилалкил, где указанный фенил имеет от одного до трех заместителей, независимо выбранных из группы, состоящей из Н, низшего алкила, низшего алкоксила, гидроксила и галогена;
(b)  представляет насыщенное пятичленное азотсодержащее гетероциклическое кольцо, причем указанное кольцо содержит один атом азота, или бензоконденсированное насыщенное шестичленное азотсодержащее гетероциклическое кольцо;
представляет насыщенное пятичленное азотсодержащее гетероциклическое кольцо, причем указанное кольцо содержит один атом азота, или бензоконденсированное насыщенное шестичленное азотсодержащее гетероциклическое кольцо;
(c)  представляет оксазол, оксадиазол или тиазол; и
представляет оксазол, оксадиазол или тиазол; и
(d) А связан с атомом углерода пятичленного гетероароматического кольца и выбран из группы, состоящей из COO(CH2)mAr,  ,
,
(где R1 является таким же или отличается от R1, определенного в части (a)), CONR4(CH2)mAr и (СН2)mО(СН2)nАr (где R1 не может быть COCOR2 или SO2R3),
(i) R4 представляет Н или С1-4алкил;
(ii) Ar выбран из группы, состоящей из 2-пиридила, 3-пиридила и 4-пиридила;
(iii) m равно 1-4; и
(iv) n равно 0-4.
2. Соединение по п.1, имеющее структуру

3. Соединение по п.1, имеющее структуру

4. Соединение по п.1, имеющее структуру

5. Соединение по п.1, имеющее структуру

где R1 являются одинаковыми или различными.
6. Соединение по п.5, имеющее структуру

7. Соединение по п.5, имеющее структуру

8. Соединение по п.5, имеющее структуру

9. Соединение по п.5, имеющее структуру

10. Соединение по п.5, имеющее структуру

11. Соединение по п.5, имеющее структуру:

12. Соединение по п.5, имеющее структуру

13. Соединение, имеющее структуру

или его фармацевтически приемлемая соль, где R’’ представляет прямой или разветвленный (C1-C4)алкил.
14. Способ лечения индивидуума, страдающего расстройством, характеризующимся поражением нейронов, вызванным заболеванием или травмой, включающий введение индивидууму терапевтически эффективного количества соединения по п.1.
15. Способ ингибирования у индивидуума возникновения расстройства, характеризующегося поражением нейронов, вызванным заболеванием, включающий введение указанному индивидууму профилактически эффективного количества соединения по п.1.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14300699P | 1999-07-09 | 1999-07-09 | |
| US60/143,006 | 1999-07-09 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2002103340A RU2002103340A (ru) | 2003-09-20 |
| RU2241709C2 true RU2241709C2 (ru) | 2004-12-10 |
Family
ID=22502172
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2002103340/04A RU2241709C2 (ru) | 1999-07-09 | 2000-06-14 | Производные азотсодержащих гетероциклических соединений и способ лечения расстройств, характеризующихся поражением нейронов |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US6809107B1 (ru) |
| EP (1) | EP1202990A2 (ru) |
| JP (1) | JP2003504367A (ru) |
| KR (1) | KR20020027486A (ru) |
| CN (1) | CN1382140A (ru) |
| AR (1) | AR034541A1 (ru) |
| AU (1) | AU781235B2 (ru) |
| BR (1) | BR0012327A (ru) |
| CA (1) | CA2379149A1 (ru) |
| CZ (1) | CZ200265A3 (ru) |
| HU (1) | HUP0202249A3 (ru) |
| IL (2) | IL147505A0 (ru) |
| MX (1) | MXPA02000330A (ru) |
| PL (1) | PL352432A1 (ru) |
| RU (1) | RU2241709C2 (ru) |
| TW (1) | TW589311B (ru) |
| WO (1) | WO2001004116A2 (ru) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2835254B1 (fr) * | 2002-01-25 | 2006-04-07 | Sod Conseils Rech Applic | Derives de thiazoles dans le traitement de maladies neurologiques |
| DE10240818A1 (de) * | 2002-08-30 | 2004-05-13 | Grünenthal GmbH | Substituierte 2-Pyrrolidin-2-yl-[1,3,4]-oxadiazol-Derivate |
| SI1551835T1 (sl) * | 2002-09-30 | 2007-06-30 | Neurosearch As | Novi 1,4-diazabicikloalkanski derivati, njihova priprava in uporaba |
| US20040266811A1 (en) * | 2002-11-08 | 2004-12-30 | Weinstein David E. | Methods for inducing regeneration, remyelination, and hypermyelination of nervous tissue |
| US7189746B2 (en) * | 2002-11-08 | 2007-03-13 | Gliamed, Inc. | Methods for promoting wound healing |
| WO2006116808A1 (en) * | 2005-04-29 | 2006-11-09 | Metabolic Pharmaceuticals Limited | Treating peripheral neuropathies |
| US20080015236A1 (en) * | 2006-02-08 | 2008-01-17 | Weinstein David E | Method for promoting myocardial regeneration and uses thereof |
| US7795216B2 (en) * | 2006-03-24 | 2010-09-14 | Glia Med, Inc. | Methods for promoting composite tissue regeneration and uses thereof |
| GB0701426D0 (en) * | 2007-01-25 | 2007-03-07 | Univ Sheffield | Compounds and their use |
| TWI534142B (zh) | 2011-03-15 | 2016-05-21 | 大正製藥股份有限公司 | Azole derivatives |
| CA2922607C (en) * | 2013-09-06 | 2022-08-30 | Aurigene Discovery Technologies Limited | 1,2,4-oxadiazole derivatives as immunomodulators |
| WO2016097004A1 (en) * | 2014-12-17 | 2016-06-23 | King's College London | BICYCLOHETEROARYL-HETEROARYL-BENZOIC ACID COMPOUNDS AS RETINOIC ACID RECEPTOR BETA (RARβ) AGONISTS |
| EP3795147B1 (en) * | 2015-12-04 | 2023-08-30 | The Penn State Research Foundation | Chemical reprogramming of human glial cells into neurons with small molecule cocktail |
| CN114591295B (zh) * | 2022-03-04 | 2023-11-07 | 贵州大学 | 含吡啶盐的n-(吲哚酰基)-n’-(取代的)的丙基酰肼类衍生物、其制备方法及应用 |
Family Cites Families (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4950649A (en) | 1980-09-12 | 1990-08-21 | University Of Illinois | Didemnins and nordidemnins |
| DE3721534A1 (de) * | 1986-08-02 | 1988-02-11 | Basf Ag | Thiadiazolgruppen enthaltende polymere und deren verwendung in der elektroindustrie |
| WO1992019593A1 (en) | 1991-05-09 | 1992-11-12 | Vertex Pharmaceuticals Incorporated | Novel immunosuppressive compounds |
| US5756494A (en) | 1992-07-24 | 1998-05-26 | Cephalon, Inc. | Protein kinase inhibitors for treatment of neurological disorders |
| US5461146A (en) | 1992-07-24 | 1995-10-24 | Cephalon, Inc. | Selected protein kinase inhibitors for the treatment of neurological disorders |
| US5621101A (en) | 1992-07-24 | 1997-04-15 | Cephalon, Inc. | Protein kinase inhibitors for treatment of neurological disorders |
| NZ314207A (en) | 1992-09-28 | 2000-12-22 | Vertex Pharma | 1-(2-Oxoacetyl)-piperidine-2-carboxylic acid derivatives as multi drug resistant cancer cell sensitizers |
| US5798355A (en) | 1995-06-07 | 1998-08-25 | Gpi Nil Holdings, Inc. | Inhibitors of rotamase enzyme activity |
| CA2197793C (en) | 1994-08-18 | 2007-04-24 | Dennis A. Holt | New multimerizing agents |
| US5859031A (en) | 1995-06-07 | 1999-01-12 | Gpi Nil Holdings, Inc. | Small molecule inhibitors of rotamase enzyme activity |
| US5614547A (en) | 1995-06-07 | 1997-03-25 | Guilford Pharmaceuticals Inc. | Small molecule inhibitors of rotamase enzyme |
| US5696135A (en) | 1995-06-07 | 1997-12-09 | Gpi Nil Holdings, Inc. | Inhibitors of rotamase enzyme activity effective at stimulating neuronal growth |
| US5801197A (en) | 1995-10-31 | 1998-09-01 | Gpi Nil Holdings, Inc. | Rotamase enzyme activity inhibitors |
| AU2192797A (en) | 1996-02-28 | 1997-09-16 | Ariad Gene Therapeutics, Inc. | Synthetic derivatives of rapamycin as multimerising agents for chimeric proteins with immunophilin derived domains |
| US5786378A (en) | 1996-09-25 | 1998-07-28 | Gpi Nil Holdings, Inc. | Heterocyclic thioesters |
| US5801187A (en) | 1996-09-25 | 1998-09-01 | Gpi-Nil Holdings, Inc. | Heterocyclic esters and amides |
| US5780484A (en) | 1996-11-13 | 1998-07-14 | Vertex Pharmaceuticals Incorporated | Methods for stimulating neurite growth with piperidine compounds |
| AR010744A1 (es) | 1996-12-09 | 2000-07-12 | Guildford Pharmaceuticals Inc | Inhibidores polipropilos de ciclofilina |
| US5874449A (en) | 1996-12-31 | 1999-02-23 | Gpi Nil Holdings, Inc. | N-linked sulfonamides of heterocyclic thioesters |
| US5935989A (en) | 1996-12-31 | 1999-08-10 | Gpi Nil Holdings Inc. | N-linked ureas and carbamates of heterocyclic thioesters |
| US5721256A (en) * | 1997-02-12 | 1998-02-24 | Gpi Nil Holdings, Inc. | Method of using neurotrophic sulfonamide compounds |
| ZA98825B (en) * | 1997-02-27 | 1998-10-19 | Guilford Pharm Inc | Method of using neurotrophic carbamates and ureas |
| US5846979A (en) | 1997-02-28 | 1998-12-08 | Gpi Nil Holdings, Inc. | N-oxides of heterocyclic esters, amides, thioesters, and ketones |
| JP2001516767A (ja) * | 1997-09-24 | 2001-10-02 | アムジエン・インコーポレーテツド | 感覚神経向性化合物を使用する難聴の予防及び治療法 |
| GB9804426D0 (en) | 1998-03-02 | 1998-04-29 | Pfizer Ltd | Heterocycles |
| NZ508462A (en) * | 1998-06-03 | 2004-02-27 | Guilford Pharm Inc | Aza-heterocyclic compounds used to treat neurological disorders and hair loss |
| CN1299346A (zh) * | 1998-06-03 | 2001-06-13 | Gpi;Nil控股公司 | N-杂环羧酸或羧酸等排物的脲和氨基羧酸酯 |
| US6331537B1 (en) | 1998-06-03 | 2001-12-18 | Gpi Nil Holdings, Inc. | Carboxylic acids and carboxylic acid isosteres of N-heterocyclic compounds |
| CN1306525A (zh) * | 1998-06-03 | 2001-08-01 | Gpinil控股公司 | 含有多个杂原子的杂环化合物的羧酸或其电子等排物 |
| US6284779B1 (en) | 1999-02-03 | 2001-09-04 | Schering Aktiiengesellschaft | Heteroaromatic compounds |
-
2000
- 2000-06-14 CZ CZ200265A patent/CZ200265A3/cs unknown
- 2000-06-14 JP JP2001509726A patent/JP2003504367A/ja active Pending
- 2000-06-14 EP EP00939836A patent/EP1202990A2/en not_active Withdrawn
- 2000-06-14 IL IL14750500A patent/IL147505A0/xx active IP Right Grant
- 2000-06-14 CN CN00812744A patent/CN1382140A/zh active Pending
- 2000-06-14 HU HU0202249A patent/HUP0202249A3/hu unknown
- 2000-06-14 WO PCT/US2000/016221 patent/WO2001004116A2/en not_active Application Discontinuation
- 2000-06-14 PL PL00352432A patent/PL352432A1/xx not_active Application Discontinuation
- 2000-06-14 RU RU2002103340/04A patent/RU2241709C2/ru not_active IP Right Cessation
- 2000-06-14 MX MXPA02000330A patent/MXPA02000330A/es unknown
- 2000-06-14 KR KR1020027000348A patent/KR20020027486A/ko not_active Application Discontinuation
- 2000-06-14 AU AU54855/00A patent/AU781235B2/en not_active Ceased
- 2000-06-14 CA CA002379149A patent/CA2379149A1/en not_active Abandoned
- 2000-06-14 BR BR0012327-7A patent/BR0012327A/pt not_active Application Discontinuation
- 2000-06-14 US US09/593,852 patent/US6809107B1/en not_active Expired - Lifetime
- 2000-07-07 AR ARP000103484A patent/AR034541A1/es unknown
- 2000-09-18 TW TW089113459A patent/TW589311B/zh not_active IP Right Cessation
-
2002
- 2002-01-07 IL IL147505A patent/IL147505A/en not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| IL147505A (en) | 2006-07-05 |
| KR20020027486A (ko) | 2002-04-13 |
| MXPA02000330A (es) | 2004-05-21 |
| AU5485500A (en) | 2001-01-30 |
| AR034541A1 (es) | 2004-03-03 |
| AU781235B2 (en) | 2005-05-12 |
| TW589311B (en) | 2004-06-01 |
| BR0012327A (pt) | 2002-07-02 |
| US6809107B1 (en) | 2004-10-26 |
| JP2003504367A (ja) | 2003-02-04 |
| IL147505A0 (en) | 2002-08-14 |
| WO2001004116A3 (en) | 2001-08-23 |
| EP1202990A2 (en) | 2002-05-08 |
| CZ200265A3 (cs) | 2002-07-17 |
| WO2001004116A2 (en) | 2001-01-18 |
| CA2379149A1 (en) | 2001-01-18 |
| HUP0202249A3 (en) | 2003-01-28 |
| HUP0202249A2 (hu) | 2002-12-28 |
| CN1382140A (zh) | 2002-11-27 |
| PL352432A1 (en) | 2003-08-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2241709C2 (ru) | Производные азотсодержащих гетероциклических соединений и способ лечения расстройств, характеризующихся поражением нейронов | |
| TWI655186B (zh) | S1p調節劑 | |
| US5278176A (en) | Nicotine derivatives that enhance cognitive function | |
| KR102127025B1 (ko) | S1p 조절제 및/또는 atx 조절제인 화합물들 | |
| MXPA06001558A (es) | Inhibidores de amina ciclica bace-1 que poseen un sustituyente heterociclico. | |
| US5633270A (en) | Thiazolidine derivatives, preparation thereof and drugs containing samem | |
| CA2993918A1 (en) | 1,3,4-oxadiazole amide derivative compound as histone deacetylase 6 inhibitor, and pharmaceutical composition containing same | |
| MXPA06001559A (es) | Inhibidores de amina ciclica bace-1 que poseen una benzamida como sustituyente. | |
| US20050148637A1 (en) | Heterocyclic ketone and thioester compounds and uses | |
| JP2020180129A (ja) | Rorγtのモジュレーターとしてのトリフルオロメチルアルコール | |
| US6323215B1 (en) | Neurotrophic tetrahydroisoquinolines and tetrahydrothienopyridines, and related compositions and methods | |
| US6544976B1 (en) | Neurotrophic 2-azetidinecarboxylic acid derivatives, and related compositions and methods | |
| US20050153951A1 (en) | Neurotrophic pyrrolidines and piperidines, and related compositions and methods | |
| CA3224033A1 (en) | Sulfonamide orexin receptor agonists and uses thereof | |
| US6069163A (en) | Azapeptide acids as cell adhesion inhibitors | |
| CA3221753A1 (en) | Compounds and pharmaceutical compositions for use in neurodegenerative disorders | |
| MXPA00008623A (es) | Compuestos heterociclicos como inhibidores de enzimas rotamasas | |
| MXPA06004271A (en) | Derivatives of n-[heteroaryl(piperidine-2-yl)methyl]benzamide, preparation method thereof and application of same in therapeutics |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20090615 |