JP7254792B2 - グルコース感受性アルブミン結合誘導体 - Google Patents
グルコース感受性アルブミン結合誘導体 Download PDFInfo
- Publication number
- JP7254792B2 JP7254792B2 JP2020525911A JP2020525911A JP7254792B2 JP 7254792 B2 JP7254792 B2 JP 7254792B2 JP 2020525911 A JP2020525911 A JP 2020525911A JP 2020525911 A JP2020525911 A JP 2020525911A JP 7254792 B2 JP7254792 B2 JP 7254792B2
- Authority
- JP
- Japan
- Prior art keywords
- nhch
- cooh
- formula
- linker
- covalent bond
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 title description 35
- 239000008103 glucose Substances 0.000 title description 35
- 102000009027 Albumins Human genes 0.000 title description 12
- 108010088751 Albumins Proteins 0.000 title description 12
- -1 diboron compound Chemical class 0.000 claims description 234
- ZOCHARZZJNPSEU-UHFFFAOYSA-N diboron Chemical compound B#B ZOCHARZZJNPSEU-UHFFFAOYSA-N 0.000 claims description 68
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 62
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 claims description 58
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 50
- 229910052731 fluorine Inorganic materials 0.000 claims description 46
- 150000001875 compounds Chemical class 0.000 claims description 36
- HCMJWOGOISXSDL-UHFFFAOYSA-N (2-isothiocyanato-1-phenylethyl)benzene Chemical compound C=1C=CC=CC=1C(CN=C=S)C1=CC=CC=C1 HCMJWOGOISXSDL-UHFFFAOYSA-N 0.000 claims description 35
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 35
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 claims description 35
- 229910052757 nitrogen Inorganic materials 0.000 claims description 32
- 239000004471 Glycine Substances 0.000 claims description 31
- KYXOSSZIPTXYDE-UHFFFAOYSA-N 2-[(5-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)-[2-[(5-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)amino]ethyl]amino]acetic acid Chemical compound C12=C(C=C(C(=C2)C(=O)NCCN(C(=O)C2=C(C=C3COB(C3=C2)O)F)CC(=O)O)F)COB1O KYXOSSZIPTXYDE-UHFFFAOYSA-N 0.000 claims description 29
- 239000008186 active pharmaceutical agent Substances 0.000 claims description 25
- 229940088679 drug related substance Drugs 0.000 claims description 25
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 24
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 claims description 22
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 22
- KMTNEBVTDNRXGF-UHFFFAOYSA-N 2-[(1-hydroxy-4-methylsulfonyl-3H-2,1-benzoxaborole-6-carbonyl)-[2-[(1-hydroxy-4-methylsulfonyl-3H-2,1-benzoxaborole-6-carbonyl)amino]ethyl]amino]acetic acid Chemical compound CS(=O)(=O)C1=CC(=CC2=C1COB2O)C(=O)NCCN(CC(O)=O)C(=O)C1=CC2=C(COB2O)C(=C1)S(C)(=O)=O KMTNEBVTDNRXGF-UHFFFAOYSA-N 0.000 claims description 13
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 13
- 150000008575 L-amino acids Chemical group 0.000 claims description 11
- 239000004472 Lysine Substances 0.000 claims description 11
- WCOQAGPRGRKKLM-UHFFFAOYSA-N pyrrolidine-3,4-diamine Chemical compound NC1CNCC1N WCOQAGPRGRKKLM-UHFFFAOYSA-N 0.000 claims description 11
- 229910052717 sulfur Inorganic materials 0.000 claims description 11
- 238000002360 preparation method Methods 0.000 claims description 9
- BSTWEDGDAUAJGS-INIZCTEOSA-N (2S)-2,3-bis[[1-hydroxy-4-(trifluoromethyl)-3H-2,1-benzoxaborole-6-carbonyl]amino]propanoic acid Chemical compound OB1OCC2=C1C=C(C=C2C(F)(F)F)C(=O)NC[C@H](NC(=O)C1=CC2=C(COB2O)C(=C1)C(F)(F)F)C(O)=O BSTWEDGDAUAJGS-INIZCTEOSA-N 0.000 claims description 7
- IFPSNUVIKOCJJS-UHFFFAOYSA-N 2-[[4-(difluoromethyl)-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl]-[2-[[4-(difluoromethyl)-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl]amino]ethyl]amino]acetic acid Chemical compound B1(C2=CC(C(=O)N(CCNC(=O)C3=CC4=C(C(C(F)F)=C3)COB4O)CC(=O)O)=CC(C(F)F)=C2CO1)O IFPSNUVIKOCJJS-UHFFFAOYSA-N 0.000 claims description 7
- MRGFTTLPAZFITK-UHFFFAOYSA-N 3-borono-5-(3-borono-5-fluorobenzoyl)benzoic acid Chemical compound B(O)(O)C=1C=C(C(=O)O)C=C(C=1)C(C1=CC(=CC(=C1)F)B(O)O)=O MRGFTTLPAZFITK-UHFFFAOYSA-N 0.000 claims description 7
- AGGOZXDCVADXOG-PMACEKPBSA-N 4-[(3S,4S)-3,4-bis[[1-hydroxy-4-(trifluoromethyl)-3H-2,1-benzoxaborole-6-carbonyl]amino]pyrrolidin-1-yl]-4-oxobutanoic acid Chemical compound OB1OCC2=C1C=C(C=C2C(F)(F)F)C(=O)N[C@H]1CN(C[C@@H]1NC(=O)C=1C=C(C2=C(B(OC2)O)C=1)C(F)(F)F)C(CCC(=O)O)=O AGGOZXDCVADXOG-PMACEKPBSA-N 0.000 claims description 7
- AVXAWRYJOASUAO-UHFFFAOYSA-N 2-[(6-fluoro-1-hydroxy-3H-2,1-benzoxaborole-5-carbonyl)-[2-[(6-fluoro-1-hydroxy-3H-2,1-benzoxaborole-5-carbonyl)amino]ethyl]amino]acetic acid Chemical compound B1(O)C2=CC(F)=C(C(=O)NCCN(C(=O)C3=CC4=C(B(OC4)O)C=C3F)CC(=O)O)C=C2CO1 AVXAWRYJOASUAO-UHFFFAOYSA-N 0.000 claims description 6
- YYEDEFNVBGUOAE-UHFFFAOYSA-N 2-[(7-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)-[2-[(7-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)amino]ethyl]amino]acetic acid Chemical compound B1(OCC2=CC=C(C(=O)N(CCNC(=O)C3=C(C4=C(COB4O)C=C3)F)CC(=O)O)C(F)=C12)O YYEDEFNVBGUOAE-UHFFFAOYSA-N 0.000 claims description 6
- PNJCUNQJHAZVNW-UHFFFAOYSA-N 2-[[1-hydroxy-4-(trifluoromethyl)-3H-2,1-benzoxaborole-6-carbonyl]-[2-[[1-hydroxy-4-(trifluoromethyl)-3H-2,1-benzoxaborole-6-carbonyl]amino]ethyl]amino]acetic acid Chemical compound FC(C1=C2COB(C2=CC(C(=O)N(CCNC(=O)C2=CC(C(F)(F)F)=C3COB(C3=C2)O)CC(=O)O)=C1)O)(F)F PNJCUNQJHAZVNW-UHFFFAOYSA-N 0.000 claims description 6
- FYDSFWPETZKCEZ-INIZCTEOSA-N (2S)-2,3-bis[(4-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)amino]propanoic acid Chemical compound OB1OCC2=C1C=C(C=C2F)C(=O)NC[C@H](NC(=O)C1=CC2=C(COB2O)C(F)=C1)C(O)=O FYDSFWPETZKCEZ-INIZCTEOSA-N 0.000 claims description 5
- WKRRJDCYDXEALH-LBPRGKRZSA-N (2S)-2,3-bis[(7-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)amino]propanoic acid Chemical compound OB1OCC2=C1C(F)=C(C=C2)C(=O)NC[C@H](NC(=O)C1=C(F)C2=C(COB2O)C=C1)C(O)=O WKRRJDCYDXEALH-LBPRGKRZSA-N 0.000 claims description 5
- WBNFCQHAVHZZNR-UHFFFAOYSA-N 2-[(4-chloro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)-[2-[(4-chloro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)amino]ethyl]amino]acetic acid Chemical compound OB1OCC2=C1C=C(C=C2Cl)C(=O)NCCN(CC(O)=O)C(=O)C1=CC2=C(COB2O)C(Cl)=C1 WBNFCQHAVHZZNR-UHFFFAOYSA-N 0.000 claims description 5
- GYHHUFZIOCQQPE-UHFFFAOYSA-N 2-[(4-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)-[2-[(4-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)amino]ethyl]amino]acetic acid Chemical compound C12=C(C(=CC(=C2)C(=O)NCCN(C(=O)C2=CC3=C(COB3O)C(F)=C2)CC(=O)O)F)COB1O GYHHUFZIOCQQPE-UHFFFAOYSA-N 0.000 claims description 5
- YHAQWGCPUZAMBH-UHFFFAOYSA-N 3-borono-5-(5-borono-2,4-difluorobenzoyl)benzoic acid Chemical compound B(O)(O)C=1C=C(C(=O)O)C=C(C=1)C(C1=C(C=C(C(=C1)B(O)O)F)F)=O YHAQWGCPUZAMBH-UHFFFAOYSA-N 0.000 claims description 5
- RGILTGWUMCGOFP-INIZCTEOSA-N (2S)-2,3-bis[(5-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)amino]propanoic acid Chemical compound OB1OCC2=C1C=C(C(=O)NC[C@H](NC(=O)C1=CC3=C(COB3O)C=C1F)C(O)=O)C(F)=C2 RGILTGWUMCGOFP-INIZCTEOSA-N 0.000 claims description 4
- PHMHAPKORFPYJN-UHFFFAOYSA-N 2-[[3-borono-5-(3-borono-5-fluorobenzoyl)benzoyl]amino]acetic acid Chemical compound B(O)(O)C=1C=C(C(=O)NCC(=O)O)C=C(C=1)C(C1=CC(=CC(=C1)F)B(O)O)=O PHMHAPKORFPYJN-UHFFFAOYSA-N 0.000 claims description 4
- NDBOJEVGWQXYHB-UHFFFAOYSA-N 2-[[3-borono-5-[[3-borono-5-(trifluoromethyl)phenyl]-difluoromethyl]benzoyl]amino]acetic acid Chemical compound B(O)(O)C=1C=C(C(=O)NCC(=O)O)C=C(C=1)C(F)(F)C1=CC(=CC(=C1)C(F)(F)F)B(O)O NDBOJEVGWQXYHB-UHFFFAOYSA-N 0.000 claims description 4
- XDBBUIGDFHGGBH-UHFFFAOYSA-N 2-[[bis[3-borono-5-(trifluoromethyl)phenyl]-oxo-lambda6-sulfanylidene]amino]acetic acid Chemical compound B(O)(O)C=1C=C(C=C(C=1)C(F)(F)F)S(=O)(C1=CC(=CC(=C1)C(F)(F)F)B(O)O)=NCC(=O)O XDBBUIGDFHGGBH-UHFFFAOYSA-N 0.000 claims description 4
- ZKLFZDRMVOOGEA-UHFFFAOYSA-N 3,5-bis[[(4-borono-2-fluorobenzoyl)amino]methyl]benzoic acid Chemical compound B(O)(O)C1=CC(=C(C(=O)NCC=2C=C(C(=O)O)C=C(C=2)CNC(C2=C(C=C(C=C2)B(O)O)F)=O)C=C1)F ZKLFZDRMVOOGEA-UHFFFAOYSA-N 0.000 claims description 4
- SNUJJPVZCDPFAI-UHFFFAOYSA-N 3,5-bis[[(4-borono-3-fluorobenzoyl)amino]methyl]benzoic acid Chemical compound B(O)(O)C1=C(C=C(C(=O)NCC=2C=C(C(=O)O)C=C(C=2)CNC(C2=CC(=C(C=C2)B(O)O)F)=O)C=C1)F SNUJJPVZCDPFAI-UHFFFAOYSA-N 0.000 claims description 4
- GBJWMUIRJPSXDI-UHFFFAOYSA-N 3,5-bis[[(7-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)amino]methyl]benzoic acid Chemical compound OB1OCC2=C1C(F)=C(C=C2)C(=O)NCC1=CC(=CC(CNC(=O)C2=C(F)C3=C(COB3O)C=C2)=C1)C(O)=O GBJWMUIRJPSXDI-UHFFFAOYSA-N 0.000 claims description 4
- DMHQMKRFJWBZLV-UHFFFAOYSA-N 3-borono-5-(3-borono-4-fluorophenyl)sulfonyl-4-fluorobenzoic acid Chemical compound B(O)(O)C=1C=C(C(=O)O)C=C(C=1F)S(=O)(=O)C1=CC(=C(C=C1)F)B(O)O DMHQMKRFJWBZLV-UHFFFAOYSA-N 0.000 claims description 4
- WHBKQAURLKIMOC-HOTGVXAUSA-N 4-[(3S,4S)-3,4-bis[(7-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)amino]pyrrolidin-1-yl]-4-oxobutanoic acid Chemical compound OB1OCC2=C1C(F)=C(C=C2)C(=O)N[C@H]1CN(C[C@@H]1NC(=O)C1=C(F)C2=C(COB2O)C=C1)C(=O)CCC(O)=O WHBKQAURLKIMOC-HOTGVXAUSA-N 0.000 claims description 4
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 claims description 4
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- SNAPCMGWKWKWID-HNNXBMFYSA-N (2S)-2,4-bis[(4-borono-3-fluorobenzoyl)amino]butanoic acid Chemical compound B(O)(O)C1=C(C=C(C(=O)N[C@H](C(=O)O)CCNC(C2=CC(=C(C=C2)B(O)O)F)=O)C=C1)F SNAPCMGWKWKWID-HNNXBMFYSA-N 0.000 claims description 3
- MDVFAWOHRRNTKZ-UHFFFAOYSA-N 2-[[1-hydroxy-4-(trifluoromethylsulfonyl)-3H-2,1-benzoxaborole-6-carbonyl]-[2-[[1-hydroxy-4-(trifluoromethylsulfonyl)-3H-2,1-benzoxaborole-6-carbonyl]amino]ethyl]amino]acetic acid Chemical compound C(F)(S(=O)(=O)C1=C2COB(C2=CC(C(=O)N(CCNC(=O)C2=CC(S(=O)(=O)C(F)(F)F)=C3COB(C3=C2)O)CC(=O)O)=C1)O)(F)F MDVFAWOHRRNTKZ-UHFFFAOYSA-N 0.000 claims description 3
- AYUFKECOJZWSET-UHFFFAOYSA-N 2-[[3-borono-5-(3-borono-5-fluorophenyl)sulfonylbenzoyl]amino]acetic acid Chemical compound B(O)(O)C=1C=C(C(=O)NCC(=O)O)C=C(C=1)S(=O)(=O)C1=CC(=CC(=C1)F)B(O)O AYUFKECOJZWSET-UHFFFAOYSA-N 0.000 claims description 3
- VSRBGIOHJQEUQH-UHFFFAOYSA-N 2-[[bis(3-borono-5-chlorophenyl)-oxo-lambda6-sulfanylidene]amino]acetic acid Chemical compound B(O)(O)C=1C=C(C=C(C=1)Cl)S(=O)(C1=CC(=CC(=C1)Cl)B(O)O)=NCC(=O)O VSRBGIOHJQEUQH-UHFFFAOYSA-N 0.000 claims description 3
- TZHIKJDMMOQQOE-UHFFFAOYSA-N 2-[[bis(3-boronophenyl)-oxo-lambda6-sulfanylidene]amino]acetic acid Chemical compound B(O)(O)C=1C=C(C=CC=1)S(=O)(C1=CC(=CC=C1)B(O)O)=NCC(=O)O TZHIKJDMMOQQOE-UHFFFAOYSA-N 0.000 claims description 3
- IWSSNZPGZWGHCX-UHFFFAOYSA-N 2-[[bis[1-hydroxy-4-(trifluoromethyl)-3H-2,1-benzoxaborol-6-yl]-oxo-lambda6-sulfanylidene]amino]acetic acid Chemical compound OB1OCC2=C1C=C(C=C2C(F)(F)F)S(=O)(=NCC(O)=O)C1=CC2=C(COB2O)C(=C1)C(F)(F)F IWSSNZPGZWGHCX-UHFFFAOYSA-N 0.000 claims description 3
- OMCMNRVUXKYUSM-UHFFFAOYSA-N 2-[[bis[3-borono-5-(difluoromethyl)phenyl]-oxo-lambda6-sulfanylidene]amino]acetic acid Chemical compound OB(O)C1=CC(=CC(=C1)S(=O)(=NCC(O)=O)C1=CC(=CC(=C1)C(F)F)B(O)O)C(F)F OMCMNRVUXKYUSM-UHFFFAOYSA-N 0.000 claims description 3
- KVEOIDBXMCFVPO-UHFFFAOYSA-N 3-[2,3-bis[(4-chloro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)amino]propanoylamino]propanoic acid Chemical compound OB1OCC2=C1C=C(C=C2Cl)C(=O)NCC(NC(=O)C1=CC2=C(COB2O)C(Cl)=C1)C(=O)NCCC(O)=O KVEOIDBXMCFVPO-UHFFFAOYSA-N 0.000 claims description 3
- YSNDNJMKHKLTNK-INIZCTEOSA-N (2S)-2,3-bis[(1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)amino]propanoic acid Chemical compound OB1OCC2=C1C=C(C=C2)C(=O)NC[C@H](NC(=O)C1=CC2=C(COB2O)C=C1)C(O)=O YSNDNJMKHKLTNK-INIZCTEOSA-N 0.000 claims description 2
- DDVKTHUXRVZKTQ-UHFFFAOYSA-N 2-[[3-borono-5-(3-boronophenyl)sulfonylbenzoyl]amino]acetic acid Chemical compound B(O)(O)C=1C=C(C(=O)NCC(=O)O)C=C(C=1)S(=O)(=O)C1=CC(=CC=C1)B(O)O DDVKTHUXRVZKTQ-UHFFFAOYSA-N 0.000 claims description 2
- NFDMEVUWFFHGTD-UHFFFAOYSA-N 2-[[3-borono-5-[3-borono-5-(trifluoromethyl)benzoyl]benzoyl]amino]acetic acid Chemical compound B(O)(O)C=1C=C(C(=O)NCC(=O)O)C=C(C=1)C(C1=CC(=CC(=C1)C(F)(F)F)B(O)O)=O NFDMEVUWFFHGTD-UHFFFAOYSA-N 0.000 claims description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 2
- 125000000896 monocarboxylic acid group Chemical group 0.000 claims 65
- FSBSTRDJNASFCP-UHFFFAOYSA-N 4-borono-2-[3-borono-5-(trifluoromethyl)phenyl]sulfonyl-6-(trifluoromethyl)benzoic acid Chemical compound OB(O)C1=CC(=CC(=C1)S(=O)(=O)C1=C(C(O)=O)C(=CC(=C1)B(O)O)C(F)(F)F)C(F)(F)F FSBSTRDJNASFCP-UHFFFAOYSA-N 0.000 claims 1
- 150000001732 carboxylic acid derivatives Chemical group 0.000 claims 1
- 238000001212 derivatisation Methods 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 588
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 534
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 307
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 299
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 289
- 239000000203 mixture Substances 0.000 description 180
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 175
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 166
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 164
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 139
- 239000011541 reaction mixture Substances 0.000 description 137
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 132
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 132
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 129
- 239000007787 solid Substances 0.000 description 127
- 239000000243 solution Substances 0.000 description 126
- 238000006243 chemical reaction Methods 0.000 description 125
- 125000005647 linker group Chemical group 0.000 description 123
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 118
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 108
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 104
- 238000000105 evaporative light scattering detection Methods 0.000 description 91
- 239000012267 brine Substances 0.000 description 80
- 239000000741 silica gel Substances 0.000 description 80
- 229910002027 silica gel Inorganic materials 0.000 description 80
- 229960001866 silicon dioxide Drugs 0.000 description 80
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 80
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 78
- 239000012044 organic layer Substances 0.000 description 75
- 239000003480 eluent Substances 0.000 description 66
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 63
- 238000003756 stirring Methods 0.000 description 63
- 238000000034 method Methods 0.000 description 56
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 54
- 230000002829 reductive effect Effects 0.000 description 54
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 54
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 51
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 50
- 238000003818 flash chromatography Methods 0.000 description 48
- 239000012043 crude product Substances 0.000 description 47
- LDOKWNQBODDHOT-UHFFFAOYSA-N tert-butyl 2-[[oxo-bis[3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-lambda6-sulfanylidene]amino]acetate Chemical compound O=S(C1=CC(=CC=C1)B1OC(C(O1)(C)C)(C)C)(C1=CC(=CC=C1)B1OC(C(O1)(C)C)(C)C)=NCC(=O)OC(C)(C)C LDOKWNQBODDHOT-UHFFFAOYSA-N 0.000 description 47
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 45
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 45
- 229910052938 sodium sulfate Inorganic materials 0.000 description 36
- 235000011152 sodium sulphate Nutrition 0.000 description 36
- 239000000377 silicon dioxide Substances 0.000 description 33
- 239000002904 solvent Substances 0.000 description 33
- 239000002244 precipitate Substances 0.000 description 32
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 30
- 239000000706 filtrate Substances 0.000 description 30
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 29
- 229960002449 glycine Drugs 0.000 description 28
- 238000001914 filtration Methods 0.000 description 27
- 235000011056 potassium acetate Nutrition 0.000 description 27
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 26
- 238000000806 fluorine-19 nuclear magnetic resonance spectrum Methods 0.000 description 26
- 239000003921 oil Substances 0.000 description 26
- 235000019198 oils Nutrition 0.000 description 26
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 26
- 239000000725 suspension Substances 0.000 description 25
- 239000003039 volatile agent Substances 0.000 description 25
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 24
- 239000012074 organic phase Substances 0.000 description 23
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 22
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 22
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 21
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 21
- 229910052681 coesite Inorganic materials 0.000 description 21
- 229910052906 cristobalite Inorganic materials 0.000 description 21
- 239000000047 product Substances 0.000 description 21
- 235000012239 silicon dioxide Nutrition 0.000 description 21
- 229910052682 stishovite Inorganic materials 0.000 description 21
- 229910052905 tridymite Inorganic materials 0.000 description 21
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 20
- 238000001816 cooling Methods 0.000 description 20
- 239000000843 powder Substances 0.000 description 20
- PDSMXCBNXMITIY-UHFFFAOYSA-N tert-butyl 2-(trifluoromethyl)-6-[3-(trifluoromethyl)phenyl]sulfanylbenzoate Chemical compound FC(C1=C(C(=O)OC(C)(C)C)C(=CC=C1)SC1=CC(=CC=C1)C(F)(F)F)(F)F PDSMXCBNXMITIY-UHFFFAOYSA-N 0.000 description 20
- CSCPPACGZOOCGX-WFGJKAKNSA-N acetone d6 Chemical compound [2H]C([2H])([2H])C(=O)C([2H])([2H])[2H] CSCPPACGZOOCGX-WFGJKAKNSA-N 0.000 description 19
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 18
- GCOVMLYKRBOYFK-UHFFFAOYSA-N tert-butyl 2-[[3-[3-fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfonyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoyl]amino]acetate Chemical compound CC(C)(C)OC(=O)CNC(=O)C1=CC(=CC(=C1)B1OC(C)(C)C(C)(C)O1)S(=O)(=O)C1=CC(F)=CC(=C1)B1OC(C)(C)C(C)(C)O1 GCOVMLYKRBOYFK-UHFFFAOYSA-N 0.000 description 17
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 16
- 239000006260 foam Substances 0.000 description 16
- JMCOYJWUCDGIPI-UHFFFAOYSA-N methyl 3-fluoro-4-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate Chemical compound COC(=O)C1=CC(F)=C(C)C(B2OC(C)(C)C(C)(C)O2)=C1 JMCOYJWUCDGIPI-UHFFFAOYSA-N 0.000 description 16
- KHIWWQKSHDUIBK-UHFFFAOYSA-N periodic acid Chemical compound OI(=O)(=O)=O KHIWWQKSHDUIBK-UHFFFAOYSA-N 0.000 description 16
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 15
- NHZJZGSLUCGXFL-UHFFFAOYSA-N 2-[[3-[3-fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]sulfonyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoyl]amino]acetic acid Chemical compound CC1(C)OB(OC1(C)C)C1=CC(=CC(F)=C1)S(=O)(=O)C1=CC(=CC(=C1)C(=O)NCC(O)=O)B1OC(C)(C)C(C)(C)O1 NHZJZGSLUCGXFL-UHFFFAOYSA-N 0.000 description 15
- 239000003814 drug Substances 0.000 description 14
- 239000012071 phase Substances 0.000 description 14
- IVDFJHOHABJVEH-UHFFFAOYSA-N pinacol Chemical compound CC(C)(O)C(C)(C)O IVDFJHOHABJVEH-UHFFFAOYSA-N 0.000 description 14
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 13
- 229940022663 acetate Drugs 0.000 description 13
- 239000002253 acid Substances 0.000 description 13
- 238000004440 column chromatography Methods 0.000 description 13
- 229910000027 potassium carbonate Inorganic materials 0.000 description 13
- 235000011181 potassium carbonates Nutrition 0.000 description 13
- 238000002953 preparative HPLC Methods 0.000 description 13
- LEHBURLTIWGHEM-UHFFFAOYSA-N pyridinium chlorochromate Substances [O-][Cr](Cl)(=O)=O.C1=CC=[NH+]C=C1 LEHBURLTIWGHEM-UHFFFAOYSA-N 0.000 description 13
- 229940095102 methyl benzoate Drugs 0.000 description 12
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical class [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 12
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 11
- XVSAONVFHVFAJI-UHFFFAOYSA-N 4-(difluoromethyl)-1-hydroxy-3H-2,1-benzoxaborole-6-carboxylic acid Chemical compound OB1OCC2=C1C=C(C=C2C(F)F)C(O)=O XVSAONVFHVFAJI-UHFFFAOYSA-N 0.000 description 11
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 11
- DHMQDGOQFOQNFH-UHFFFAOYSA-M Aminoacetate Chemical compound NCC([O-])=O DHMQDGOQFOQNFH-UHFFFAOYSA-M 0.000 description 11
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 11
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Substances IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 11
- 238000005481 NMR spectroscopy Methods 0.000 description 11
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 11
- 108090000765 processed proteins & peptides Proteins 0.000 description 11
- 238000000746 purification Methods 0.000 description 11
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 11
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 10
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 10
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- 229960003646 lysine Drugs 0.000 description 10
- WUSQONUPNHFBOU-UHFFFAOYSA-N methyl 3-bromo-5-iodobenzoate Chemical compound COC(=O)C1=CC(Br)=CC(I)=C1 WUSQONUPNHFBOU-UHFFFAOYSA-N 0.000 description 10
- 235000015497 potassium bicarbonate Nutrition 0.000 description 10
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 10
- 239000011736 potassium bicarbonate Substances 0.000 description 10
- 150000003839 salts Chemical class 0.000 description 10
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 10
- 235000019345 sodium thiosulphate Nutrition 0.000 description 10
- 239000007821 HATU Substances 0.000 description 9
- 229910004298 SiO 2 Inorganic materials 0.000 description 9
- 229940079593 drug Drugs 0.000 description 9
- 238000010438 heat treatment Methods 0.000 description 9
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 9
- 235000018102 proteins Nutrition 0.000 description 9
- 102000004169 proteins and genes Human genes 0.000 description 9
- 108090000623 proteins and genes Proteins 0.000 description 9
- 239000011347 resin Substances 0.000 description 9
- 229920005989 resin Polymers 0.000 description 9
- NEACBPHJNVVONN-UHFFFAOYSA-N 6,6-dichlorohexyl-[2-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane Chemical group CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1PCCCCCC(Cl)Cl NEACBPHJNVVONN-UHFFFAOYSA-N 0.000 description 8
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 8
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 8
- 239000008346 aqueous phase Substances 0.000 description 8
- 229910052786 argon Inorganic materials 0.000 description 8
- 239000012300 argon atmosphere Substances 0.000 description 8
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 8
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 8
- 239000010410 layer Substances 0.000 description 8
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 8
- XBNGYFFABRKICK-UHFFFAOYSA-N 2,3,4,5,6-pentafluorophenol Chemical compound OC1=C(F)C(F)=C(F)C(F)=C1F XBNGYFFABRKICK-UHFFFAOYSA-N 0.000 description 7
- 102000008100 Human Serum Albumin Human genes 0.000 description 7
- 108091006905 Human Serum Albumin Proteins 0.000 description 7
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 7
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical class OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 7
- 239000012230 colorless oil Substances 0.000 description 7
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 7
- 239000000543 intermediate Substances 0.000 description 7
- 208000030159 metabolic disease Diseases 0.000 description 7
- 239000012299 nitrogen atmosphere Substances 0.000 description 7
- 125000005062 perfluorophenyl group Chemical group FC1=C(C(=C(C(=C1F)F)F)F)* 0.000 description 7
- OSWULUXZFOQIRU-UHFFFAOYSA-N tert-butyl 2-aminoacetate;hydrochloride Chemical compound Cl.CC(C)(C)OC(=O)CN OSWULUXZFOQIRU-UHFFFAOYSA-N 0.000 description 7
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 6
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- 210000004369 blood Anatomy 0.000 description 6
- 239000008280 blood Substances 0.000 description 6
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 6
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 6
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 6
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 6
- DBTNVRCCIDISMV-UHFFFAOYSA-L lithium;magnesium;propane;dichloride Chemical compound [Li+].[Mg+2].[Cl-].[Cl-].C[CH-]C DBTNVRCCIDISMV-UHFFFAOYSA-L 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 6
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 6
- SKWCZPYWFRTSDD-DKWTVANSSA-N (2s)-2,3-diaminopropanoic acid;hydrochloride Chemical compound Cl.NC[C@H](N)C(O)=O SKWCZPYWFRTSDD-DKWTVANSSA-N 0.000 description 5
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 5
- PIINGYXNCHTJTF-UHFFFAOYSA-N 2-(2-azaniumylethylamino)acetate Chemical compound NCCNCC(O)=O PIINGYXNCHTJTF-UHFFFAOYSA-N 0.000 description 5
- JTNCEQNHURODLX-UHFFFAOYSA-N 2-phenylethanimidamide Chemical compound NC(=N)CC1=CC=CC=C1 JTNCEQNHURODLX-UHFFFAOYSA-N 0.000 description 5
- 239000005711 Benzoic acid Substances 0.000 description 5
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 5
- 239000012425 OXONE® Substances 0.000 description 5
- 241000209094 Oryza Species 0.000 description 5
- 235000007164 Oryza sativa Nutrition 0.000 description 5
- BVCZEBOGSOYJJT-UHFFFAOYSA-N ammonium carbamate Chemical compound [NH4+].NC([O-])=O BVCZEBOGSOYJJT-UHFFFAOYSA-N 0.000 description 5
- 235000019270 ammonium chloride Nutrition 0.000 description 5
- 235000010233 benzoic acid Nutrition 0.000 description 5
- KXDHJXZQYSOELW-UHFFFAOYSA-N carbonic acid monoamide Natural products NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 5
- 206010012601 diabetes mellitus Diseases 0.000 description 5
- 239000005457 ice water Substances 0.000 description 5
- 239000004033 plastic Substances 0.000 description 5
- OKBMCNHOEMXPTM-UHFFFAOYSA-M potassium peroxymonosulfate Chemical compound [K+].OOS([O-])(=O)=O OKBMCNHOEMXPTM-UHFFFAOYSA-M 0.000 description 5
- 235000009566 rice Nutrition 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 229910000029 sodium carbonate Inorganic materials 0.000 description 5
- BNWCETAHAJSBFG-UHFFFAOYSA-N tert-butyl 2-bromoacetate Chemical compound CC(C)(C)OC(=O)CBr BNWCETAHAJSBFG-UHFFFAOYSA-N 0.000 description 5
- PCRLZGCXLNNMFL-UHFFFAOYSA-N 3-bromo-5-(trifluoromethyl)benzaldehyde Chemical compound FC(F)(F)C1=CC(Br)=CC(C=O)=C1 PCRLZGCXLNNMFL-UHFFFAOYSA-N 0.000 description 4
- MKJBJYCBKXPQSY-UHFFFAOYSA-N 3-bromo-5-iodobenzoic acid Chemical compound OC(=O)C1=CC(Br)=CC(I)=C1 MKJBJYCBKXPQSY-UHFFFAOYSA-N 0.000 description 4
- 125000004179 3-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(Cl)=C1[H] 0.000 description 4
- AGGOZXDCVADXOG-WOJBJXKFSA-N 4-[(3R,4R)-3,4-bis[[1-hydroxy-4-(trifluoromethyl)-3H-2,1-benzoxaborole-6-carbonyl]amino]pyrrolidin-1-yl]-4-oxobutanoic acid Chemical compound OB1OCC2=C1C=C(C=C2C(F)(F)F)C(=O)N[C@@H]1CN(C[C@H]1NC(=O)C1=CC2=C(COB2O)C(=C1)C(F)(F)F)C(=O)CCC(O)=O AGGOZXDCVADXOG-WOJBJXKFSA-N 0.000 description 4
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 4
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 4
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- 208000013016 Hypoglycemia Diseases 0.000 description 4
- 102000004877 Insulin Human genes 0.000 description 4
- 108090001061 Insulin Proteins 0.000 description 4
- 102000036770 Islet Amyloid Polypeptide Human genes 0.000 description 4
- 108010041872 Islet Amyloid Polypeptide Proteins 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 4
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 4
- 229920001971 elastomer Polymers 0.000 description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 230000008020 evaporation Effects 0.000 description 4
- 125000001841 imino group Chemical group [H]N=* 0.000 description 4
- 229940125396 insulin Drugs 0.000 description 4
- 229910052740 iodine Inorganic materials 0.000 description 4
- 229940098779 methanesulfonic acid Drugs 0.000 description 4
- DJGNICGQUHOEAO-UHFFFAOYSA-N methyl 3-bromo-5-[3-bromo-5-(trifluoromethyl)benzoyl]benzoate Chemical compound BrC=1C=C(C(=O)OC)C=C(C=1)C(C1=CC(=CC(=C1)C(F)(F)F)Br)=O DJGNICGQUHOEAO-UHFFFAOYSA-N 0.000 description 4
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 239000012312 sodium hydride Substances 0.000 description 4
- 229910000104 sodium hydride Inorganic materials 0.000 description 4
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 4
- MNKCGUKVRJZKEQ-MIXQCLKLSA-N (1z,5z)-cycloocta-1,5-diene;iridium;methanol Chemical compound [Ir].[Ir].OC.OC.C\1C\C=C/CC\C=C/1.C\1C\C=C/CC\C=C/1 MNKCGUKVRJZKEQ-MIXQCLKLSA-N 0.000 description 3
- LCHKCXJFJWILII-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate Chemical compound CC1(C)OB(OC1(C)C)C1=CC(F)=C(C=C1)C(=O)ON1C(=O)CCC1=O LCHKCXJFJWILII-UHFFFAOYSA-N 0.000 description 3
- UFDULEKOJAEIRI-UHFFFAOYSA-N (2-acetyloxy-3-iodophenyl) acetate Chemical compound CC(=O)OC1=CC=CC(I)=C1OC(C)=O UFDULEKOJAEIRI-UHFFFAOYSA-N 0.000 description 3
- OFNXSUANJLHGQN-UHFFFAOYSA-N 1,3-dibromo-5-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC(Br)=CC(Br)=C1 OFNXSUANJLHGQN-UHFFFAOYSA-N 0.000 description 3
- PAAKRIQUPXJQIW-UHFFFAOYSA-N 2-fluoro-3-iodo-4-methylbenzoic acid Chemical compound CC1=CC=C(C(O)=O)C(F)=C1I PAAKRIQUPXJQIW-UHFFFAOYSA-N 0.000 description 3
- BKJBHQXEYLPXTQ-UHFFFAOYSA-N 3,5-bis(aminomethyl)benzoic acid;dihydrochloride Chemical compound Cl.Cl.NCC1=CC(CN)=CC(C(O)=O)=C1 BKJBHQXEYLPXTQ-UHFFFAOYSA-N 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 229920000742 Cotton Polymers 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 229930091371 Fructose Natural products 0.000 description 3
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 3
- 239000005715 Fructose Substances 0.000 description 3
- 101710198884 GATA-type zinc finger protein 1 Proteins 0.000 description 3
- 102400000322 Glucagon-like peptide 1 Human genes 0.000 description 3
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000021615 conjugation Effects 0.000 description 3
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 3
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 3
- 208000016097 disease of metabolism Diseases 0.000 description 3
- 238000010828 elution Methods 0.000 description 3
- IIEWJVIFRVWJOD-UHFFFAOYSA-N ethyl cyclohexane Natural products CCC1CCCCC1 IIEWJVIFRVWJOD-UHFFFAOYSA-N 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 230000002218 hypoglycaemic effect Effects 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 239000002198 insoluble material Substances 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 235000018977 lysine Nutrition 0.000 description 3
- KTFQDZCNPGFKAH-UHFFFAOYSA-N methyl 3-chloro-4-methylbenzoate Chemical compound COC(=O)C1=CC=C(C)C(Cl)=C1 KTFQDZCNPGFKAH-UHFFFAOYSA-N 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 238000001953 recrystallisation Methods 0.000 description 3
- KAYNEFHCQUKTAV-UHFFFAOYSA-N (2,3,4,5,6-pentafluorophenyl) 1-hydroxy-3H-2,1-benzoxaborole-6-carboxylate Chemical compound OB1OCC2=C1C=C(C=C2)C(=O)OC1=C(C(=C(C(=C1F)F)F)F)F KAYNEFHCQUKTAV-UHFFFAOYSA-N 0.000 description 2
- LTMPMNIGELAWSL-KBPBESRZSA-N (2,5-dioxopyrrolidin-1-yl) 4-[(3S,4S)-3,4-bis[(2-methylpropan-2-yl)oxycarbonylamino]pyrrolidin-1-yl]-4-oxobutanoate Chemical compound C(C)(C)(C)OC(=O)N[C@H]1CN(C[C@@H]1NC(=O)OC(C)(C)C)C(CCC(=O)ON1C(CCC1=O)=O)=O LTMPMNIGELAWSL-KBPBESRZSA-N 0.000 description 2
- FNBSHKZKRBBDLS-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 6-fluoro-1-hydroxy-3H-2,1-benzoxaborole-5-carboxylate Chemical compound OB1OCC2=C1C=C(F)C(=C2)C(=O)ON1C(=O)CCC1=O FNBSHKZKRBBDLS-UHFFFAOYSA-N 0.000 description 2
- QYCQNXJLWFPDLH-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 7-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carboxylate Chemical compound OB1OCC2=C1C(F)=C(C=C2)C(=O)ON1C(=O)CCC1=O QYCQNXJLWFPDLH-UHFFFAOYSA-N 0.000 description 2
- SLDOOASMMAMZQK-UHFFFAOYSA-N (3-bromo-4-fluoro-5-iodophenyl)methanol Chemical compound OCc1cc(Br)c(F)c(I)c1 SLDOOASMMAMZQK-UHFFFAOYSA-N 0.000 description 2
- NRDBEGMTZGNVTH-UHFFFAOYSA-N (3-bromo-5-nitrophenyl)-pentafluoro-$l^{6}-sulfane Chemical compound [O-][N+](=O)C1=CC(Br)=CC(S(F)(F)(F)(F)F)=C1 NRDBEGMTZGNVTH-UHFFFAOYSA-N 0.000 description 2
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 2
- GETTZEONDQJALK-UHFFFAOYSA-N (trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC=CC=C1 GETTZEONDQJALK-UHFFFAOYSA-N 0.000 description 2
- RLMQWRRODRPLSJ-UHFFFAOYSA-N 1-(trifluoromethyl)-3-[3-(trifluoromethyl)phenyl]sulfanylbenzene Chemical compound FC(F)(F)C1=CC=CC(SC=2C=C(C=CC=2)C(F)(F)F)=C1 RLMQWRRODRPLSJ-UHFFFAOYSA-N 0.000 description 2
- UDNHRKCCVZPOAK-UHFFFAOYSA-N 1-bromo-5-iodo-2-methyl-3-(trifluoromethyl)benzene Chemical compound CC1=C(Br)C=C(I)C=C1C(F)(F)F UDNHRKCCVZPOAK-UHFFFAOYSA-N 0.000 description 2
- VECFIRWWXYYZIG-UHFFFAOYSA-N 1-chloro-3-(3-chlorophenyl)sulfanylbenzene Chemical compound ClC1=CC=CC(SC=2C=C(Cl)C=CC=2)=C1 VECFIRWWXYYZIG-UHFFFAOYSA-N 0.000 description 2
- VXDGEAYKTHEYPI-UHFFFAOYSA-N 1-hydroxy-3h-2,1-benzoxaborole-6-carboxylic acid Chemical compound C1=C(C(O)=O)C=C2B(O)OCC2=C1 VXDGEAYKTHEYPI-UHFFFAOYSA-N 0.000 description 2
- IJBFCJIIWBYEHT-UHFFFAOYSA-N 1-hydroxy-4-(trifluoromethyl)-3H-2,1-benzoxaborole-6-carboxylic acid Chemical compound OB1OCC2=C1C=C(C=C2C(F)(F)F)C(O)=O IJBFCJIIWBYEHT-UHFFFAOYSA-N 0.000 description 2
- JYRJERNGMHBYSQ-UHFFFAOYSA-N 1-hydroxy-5-(trifluoromethyl)-3H-2,1-benzoxaborole-6-carboxylic acid Chemical compound OB1OCC2=C1C=C(C(O)=O)C(=C2)C(F)(F)F JYRJERNGMHBYSQ-UHFFFAOYSA-N 0.000 description 2
- RKMGAJGJIURJSJ-UHFFFAOYSA-N 2,2,6,6-tetramethylpiperidine Chemical compound CC1(C)CCCC(C)(C)N1 RKMGAJGJIURJSJ-UHFFFAOYSA-N 0.000 description 2
- QITBKIGOBMSJBN-UHFFFAOYSA-N 2-(9h-fluoren-9-ylmethoxycarbonylamino)-5,5,5-trifluoro-4-methylpentanoic acid Chemical compound C1=CC=C2C(COC(=O)NC(CC(C)C(F)(F)F)C(O)=O)C3=CC=CC=C3C2=C1 QITBKIGOBMSJBN-UHFFFAOYSA-N 0.000 description 2
- WCJSBZQQLKPJSB-UHFFFAOYSA-N 2-bromo-5-iodo-3-methylbenzoic acid Chemical compound CC1=CC(I)=CC(C(O)=O)=C1Br WCJSBZQQLKPJSB-UHFFFAOYSA-N 0.000 description 2
- HYKCLWQMKWGJDD-UHFFFAOYSA-N 2-fluoro-3-iodo-4-methylbenzonitrile Chemical compound CC1=CC=C(C#N)C(F)=C1I HYKCLWQMKWGJDD-UHFFFAOYSA-N 0.000 description 2
- APOYTRAZFJURPB-UHFFFAOYSA-N 2-methoxy-n-(2-methoxyethyl)-n-(trifluoro-$l^{4}-sulfanyl)ethanamine Chemical compound COCCN(S(F)(F)F)CCOC APOYTRAZFJURPB-UHFFFAOYSA-N 0.000 description 2
- LINBWYYLPWJQHE-UHFFFAOYSA-N 3-(9h-fluoren-9-ylmethoxycarbonylamino)propanoic acid Chemical compound C1=CC=C2C(COC(=O)NCCC(=O)O)C3=CC=CC=C3C2=C1 LINBWYYLPWJQHE-UHFFFAOYSA-N 0.000 description 2
- KVEOIDBXMCFVPO-SFHVURJKSA-N 3-[[(2S)-2,3-bis[(4-chloro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)amino]propanoyl]amino]propanoic acid Chemical compound ClC1=CC(=CC=2B(OCC=21)O)C(=O)N[C@H](C(=O)NCCC(=O)O)CNC(=O)C=1C=C(C2=C(B(OC2)O)C=1)Cl KVEOIDBXMCFVPO-SFHVURJKSA-N 0.000 description 2
- UFNYFGZMALWWQR-UHFFFAOYSA-N 3-bromo-4-fluoro-5-iodobenzoic acid Chemical compound OC(=O)C1=CC(Br)=C(F)C(I)=C1 UFNYFGZMALWWQR-UHFFFAOYSA-N 0.000 description 2
- DKEKDYDTVBYPGJ-UHFFFAOYSA-N 3-bromo-4-methyl-5-(trifluoromethyl)aniline Chemical compound CC1=C(Br)C=C(N)C=C1C(F)(F)F DKEKDYDTVBYPGJ-UHFFFAOYSA-N 0.000 description 2
- FUKITJMBFMUDGQ-UHFFFAOYSA-N 3-bromo-4-methyl-5-(trifluoromethyl)benzoic acid Chemical compound BrC=1C=C(C(=O)O)C=C(C=1C)C(F)(F)F FUKITJMBFMUDGQ-UHFFFAOYSA-N 0.000 description 2
- ZFJOMUKPDWNRFI-UHFFFAOYSA-N 3-bromo-4-methylbenzoic acid Chemical compound CC1=CC=C(C(O)=O)C=C1Br ZFJOMUKPDWNRFI-UHFFFAOYSA-N 0.000 description 2
- NUGDEICXEZSLCZ-UHFFFAOYSA-N 3-bromo-5-(3-bromo-4-fluorophenyl)sulfonyl-4-fluorobenzoic acid Chemical compound BrC=1C=C(C(=O)O)C=C(C=1F)S(=O)(=O)C1=CC(=C(C=C1)F)Br NUGDEICXEZSLCZ-UHFFFAOYSA-N 0.000 description 2
- QCQHJTZTZKIKQJ-UHFFFAOYSA-N 3-bromo-5-(3-bromophenyl)sulfonylbenzoic acid Chemical compound BrC=1C=C(C(=O)O)C=C(C=1)S(=O)(=O)C1=CC(=CC=C1)Br QCQHJTZTZKIKQJ-UHFFFAOYSA-N 0.000 description 2
- HCROKBUNMALNHZ-UHFFFAOYSA-N 3-bromo-5-(pentafluoro-$l^{6}-sulfanyl)aniline Chemical compound NC1=CC(Br)=CC(S(F)(F)(F)(F)F)=C1 HCROKBUNMALNHZ-UHFFFAOYSA-N 0.000 description 2
- BIOUKJLBCCXLHW-UHFFFAOYSA-N 3-bromo-5-(trifluoromethyl)benzenethiol Chemical compound FC(F)(F)C1=CC(S)=CC(Br)=C1 BIOUKJLBCCXLHW-UHFFFAOYSA-N 0.000 description 2
- MEFQRXHVMJPOKZ-UHFFFAOYSA-N 3-bromo-5-fluorobenzaldehyde Chemical compound FC1=CC(Br)=CC(C=O)=C1 MEFQRXHVMJPOKZ-UHFFFAOYSA-N 0.000 description 2
- LYVFIZDTOOBDPB-UHFFFAOYSA-N 3-bromo-5-iodo-4-methylbenzoic acid Chemical compound CC1=C(Br)C=C(C(O)=O)C=C1I LYVFIZDTOOBDPB-UHFFFAOYSA-N 0.000 description 2
- LDAXWVOHZOKFEL-USPAICOZSA-N 4-[(3S,4S)-3,4-diaminopyrrolidin-1-yl]-4-oxobutanoic acid dihydrochloride Chemical compound Cl.Cl.N[C@H]1CN(C[C@@H]1N)C(CCC(=O)O)=O LDAXWVOHZOKFEL-USPAICOZSA-N 0.000 description 2
- TVNVDRMGOOLVOX-UHFFFAOYSA-N 4-borono-3-fluorobenzoic acid Chemical compound OB(O)C1=CC=C(C(O)=O)C=C1F TVNVDRMGOOLVOX-UHFFFAOYSA-N 0.000 description 2
- PHJOGJTZXVBKJO-UHFFFAOYSA-N 4-methyl-2-(trifluoromethyl)benzoic acid Chemical compound CC1=CC=C(C(O)=O)C(C(F)(F)F)=C1 PHJOGJTZXVBKJO-UHFFFAOYSA-N 0.000 description 2
- UFNBTKFAPDUBCY-UHFFFAOYSA-N 5-bromo-2,4-difluorobenzaldehyde Chemical compound FC1=CC(F)=C(C=O)C=C1Br UFNBTKFAPDUBCY-UHFFFAOYSA-N 0.000 description 2
- BGBAXZILVNOSAT-UHFFFAOYSA-N 5-bromo-2-fluoro-4-methylbenzoic acid Chemical compound CC1=CC(F)=C(C(O)=O)C=C1Br BGBAXZILVNOSAT-UHFFFAOYSA-N 0.000 description 2
- XNTKXXOTDXHIKP-UHFFFAOYSA-N 5-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carboxylic acid Chemical compound FC=1C(=CC2=C(COB2O)C=1)C(=O)O XNTKXXOTDXHIKP-UHFFFAOYSA-N 0.000 description 2
- CHKAVLMEPLZHHC-UHFFFAOYSA-N 5-iodo-4-methyl-2-(trifluoromethyl)benzoic acid Chemical compound IC=1C(=CC(=C(C(=O)O)C=1)C(F)(F)F)C CHKAVLMEPLZHHC-UHFFFAOYSA-N 0.000 description 2
- ATFMSPPPQNUERA-UHFFFAOYSA-N 7-fluoro-1-hydroxy-3h-2,1-benzoxaborole-6-carboxylic acid Chemical compound C1=C(C(O)=O)C(F)=C2B(O)OCC2=C1 ATFMSPPPQNUERA-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- 241000270322 Lepidosauria Species 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- 235000019502 Orange oil Nutrition 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 2
- XEMGYXULHWMCCC-UHFFFAOYSA-N [3-bromo-5-(3-bromo-4-fluorophenyl)sulfonyl-4-fluorophenyl]methanol Chemical compound BrC=1C=C(C=C(C=1F)S(=O)(=O)C1=CC(=C(C=C1)F)Br)CO XEMGYXULHWMCCC-UHFFFAOYSA-N 0.000 description 2
- ZBIKORITPGTTGI-UHFFFAOYSA-N [acetyloxy(phenyl)-$l^{3}-iodanyl] acetate Chemical compound CC(=O)OI(OC(C)=O)C1=CC=CC=C1 ZBIKORITPGTTGI-UHFFFAOYSA-N 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- YQUSJUJNDKUWAM-UHFFFAOYSA-N benzenesulfonylsulfanylsulfonylbenzene Chemical compound C=1C=CC=CC=1S(=O)(=O)SS(=O)(=O)C1=CC=CC=C1 YQUSJUJNDKUWAM-UHFFFAOYSA-N 0.000 description 2
- QUKGYYKBILRGFE-UHFFFAOYSA-N benzyl acetate Chemical compound CC(=O)OCC1=CC=CC=C1 QUKGYYKBILRGFE-UHFFFAOYSA-N 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 125000005620 boronic acid group Chemical class 0.000 description 2
- 235000011089 carbon dioxide Nutrition 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 239000002024 ethyl acetate extract Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- NNPPMTNAJDCUHE-UHFFFAOYSA-N isobutane Chemical compound CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- JBJGSVBGUBATNH-UHFFFAOYSA-N methyl 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate Chemical compound COC(=O)C1=CC=CC(B2OC(C)(C)C(C)(C)O2)=C1 JBJGSVBGUBATNH-UHFFFAOYSA-N 0.000 description 2
- VVACSDNKQJXQHR-UHFFFAOYSA-N methyl 3-bromo-4-(bromomethyl)-5-methylsulfonylbenzoate Chemical compound BrC=1C=C(C(=O)OC)C=C(C=1CBr)S(=O)(=O)C VVACSDNKQJXQHR-UHFFFAOYSA-N 0.000 description 2
- CFJQBNFFHMBNKQ-UHFFFAOYSA-N methyl 3-bromo-4-(bromomethyl)benzoate Chemical compound COC(=O)C1=CC=C(CBr)C(Br)=C1 CFJQBNFFHMBNKQ-UHFFFAOYSA-N 0.000 description 2
- XJLBJHXGEARPTH-UHFFFAOYSA-N methyl 3-bromo-4-methyl-5-(trifluoromethyl)benzoate Chemical compound BrC=1C=C(C(=O)OC)C=C(C=1C)C(F)(F)F XJLBJHXGEARPTH-UHFFFAOYSA-N 0.000 description 2
- KIEZGTKEYSTCMS-UHFFFAOYSA-N methyl 3-bromo-5-(difluoromethyl)-4-methylbenzoate Chemical compound BrC=1C=C(C(=O)OC)C=C(C=1C)C(F)F KIEZGTKEYSTCMS-UHFFFAOYSA-N 0.000 description 2
- MBOAEPYPUBWIRI-UHFFFAOYSA-N methyl 3-bromo-5-fluoro-4-methylbenzoate Chemical compound COC(=O)C1=CC(F)=C(C)C(Br)=C1 MBOAEPYPUBWIRI-UHFFFAOYSA-N 0.000 description 2
- CWYZDZOQRNCQNW-UHFFFAOYSA-N methyl 3-bromo-5-iodo-4-methylbenzoate Chemical compound COC(=O)C1=CC(Br)=C(C)C(I)=C1 CWYZDZOQRNCQNW-UHFFFAOYSA-N 0.000 description 2
- AEZMUBHCIUTXLG-UHFFFAOYSA-N methyl 4-(bromomethyl)-3-chloro-5-iodobenzoate Chemical compound COC(C1=CC(=C(C(=C1)I)CBr)Cl)=O AEZMUBHCIUTXLG-UHFFFAOYSA-N 0.000 description 2
- XETVWRMKXDTMNL-UHFFFAOYSA-N methyl 5-bromo-2-fluoro-4-(2-oxopropyl)benzoate Chemical compound BrC=1C(=CC(=C(C(=O)OC)C=1)F)CC(C)=O XETVWRMKXDTMNL-UHFFFAOYSA-N 0.000 description 2
- OEZJLPRXYNPFHE-UHFFFAOYSA-N methyl 5-bromo-4-(bromomethyl)-2-fluorobenzoate Chemical compound COC(C1=C(C=C(C(=C1)Br)CBr)F)=O OEZJLPRXYNPFHE-UHFFFAOYSA-N 0.000 description 2
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 239000010502 orange oil Substances 0.000 description 2
- 229910052763 palladium Inorganic materials 0.000 description 2
- IZUPBVBPLAPZRR-UHFFFAOYSA-N pentachloro-phenol Natural products OC1=C(Cl)C(Cl)=C(Cl)C(Cl)=C1Cl IZUPBVBPLAPZRR-UHFFFAOYSA-N 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 235000019260 propionic acid Nutrition 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 2
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 2
- 235000010288 sodium nitrite Nutrition 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical compound O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 2
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- SZZZXFKLFTXERQ-UHFFFAOYSA-N tert-butyl 2-(2-aminoethylamino)acetate hydrochloride Chemical compound Cl.NCCNCC(=O)OC(C)(C)C SZZZXFKLFTXERQ-UHFFFAOYSA-N 0.000 description 2
- STTPWCAWYGNDQF-UHFFFAOYSA-N tert-butyl 2-[[3-bromo-5-(3-bromo-5-fluorophenyl)sulfonylbenzoyl]amino]acetate Chemical compound BrC=1C=C(C(=O)NCC(=O)OC(C)(C)C)C=C(C=1)S(=O)(=O)C1=CC(=CC(=C1)F)Br STTPWCAWYGNDQF-UHFFFAOYSA-N 0.000 description 2
- ALSQQVSJPVVTFY-UHFFFAOYSA-N tert-butyl 2-[[3-bromo-5-[3-bromo-5-(trifluoromethyl)benzoyl]benzoyl]amino]acetate Chemical compound BrC=1C=C(C(=O)NCC(=O)OC(C)(C)C)C=C(C=1)C(C1=CC(=CC(=C1)C(F)(F)F)Br)=O ALSQQVSJPVVTFY-UHFFFAOYSA-N 0.000 description 2
- OHHCGMHFFHGOOV-UHFFFAOYSA-N tert-butyl 2-[[3-bromo-5-[3-bromo-5-(trifluoromethyl)phenyl]sulfonylbenzoyl]amino]acetate Chemical compound BrC=1C=C(C(=O)NCC(=O)OC(C)(C)C)C=C(C=1)S(=O)(=O)C1=CC(=CC(=C1)C(F)(F)F)Br OHHCGMHFFHGOOV-UHFFFAOYSA-N 0.000 description 2
- FEKPJTKLMHEVJH-UHFFFAOYSA-N tert-butyl N-[3-bromo-4-methyl-5-(trifluoromethyl)phenyl]carbamate Chemical compound BrC=1C=C(C=C(C=1C)C(F)(F)F)NC(OC(C)(C)C)=O FEKPJTKLMHEVJH-UHFFFAOYSA-N 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- SPWGLKRZUCQLGL-UHFFFAOYSA-N (2,3,4,5,6-pentafluorophenyl) 1-hydroxy-4-(trifluoromethyl)-3H-2,1-benzoxaborole-6-carboxylate Chemical compound OB1OCC2=C1C=C(C=C2C(F)(F)F)C(=O)OC1=C(C(=C(C(=C1F)F)F)F)F SPWGLKRZUCQLGL-UHFFFAOYSA-N 0.000 description 1
- MOCLUDZKYQWTMY-UHFFFAOYSA-N (2,3,4,5,6-pentafluorophenyl) 1-hydroxy-4-methylsulfonyl-3H-2,1-benzoxaborole-6-carboxylate Chemical compound OB1OCC2=C1C=C(C=C2S(=O)(=O)C)C(=O)OC1=C(C(=C(C(=C1F)F)F)F)F MOCLUDZKYQWTMY-UHFFFAOYSA-N 0.000 description 1
- IVECMDOMTWLCLU-UHFFFAOYSA-N (2,3,4,5,6-pentafluorophenyl) 1-hydroxy-5-(trifluoromethyl)-3H-2,1-benzoxaborole-6-carboxylate Chemical compound OB1OCC2=C1C=C(C(=C2)C(F)(F)F)C(=O)OC1=C(C(=C(C(=C1F)F)F)F)F IVECMDOMTWLCLU-UHFFFAOYSA-N 0.000 description 1
- OPLMXOVJWUFAAH-UHFFFAOYSA-N (2,3,4,5,6-pentafluorophenyl) 4-(difluoromethyl)-1-hydroxy-3H-2,1-benzoxaborole-6-carboxylate Chemical compound FC(C1=CC(=CC=2B(OCC=21)O)C(=O)OC1=C(C(=C(C(=C1F)F)F)F)F)F OPLMXOVJWUFAAH-UHFFFAOYSA-N 0.000 description 1
- MJXZNLFJGVWKNR-UHFFFAOYSA-N (2,3,4,5,6-pentafluorophenyl) 4-chloro-1-hydroxy-3H-2,1-benzoxaborole-6-carboxylate Chemical compound ClC1=CC(=CC=2B(OCC=21)O)C(=O)OC1=C(C(=C(C(=C1F)F)F)F)F MJXZNLFJGVWKNR-UHFFFAOYSA-N 0.000 description 1
- KGXZNKQWFNCEPO-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 1-hydroxy-4-(trifluoromethyl)-3H-2,1-benzoxaborole-6-carboxylate Chemical compound OB1OCC2=C1C=C(C=C2C(F)(F)F)C(=O)ON1C(CCC1=O)=O KGXZNKQWFNCEPO-UHFFFAOYSA-N 0.000 description 1
- MGACDMGJHWUYBH-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 3-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate Chemical compound CC1(C)OB(OC1(C)C)C1=C(F)C=C(C=C1)C(=O)ON1C(=O)CCC1=O MGACDMGJHWUYBH-UHFFFAOYSA-N 0.000 description 1
- JORVCDBLGWIVAI-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 4-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carboxylate Chemical compound OB1OCC2=C1C=C(C=C2F)C(=O)ON1C(=O)CCC1=O JORVCDBLGWIVAI-UHFFFAOYSA-N 0.000 description 1
- CEEGOSWFFHSPHM-PMERELPUSA-N (2s)-2,3-bis(9h-fluoren-9-ylmethoxycarbonylamino)propanoic acid Chemical compound C12=CC=CC=C2C2=CC=CC=C2C1COC(=O)N[C@H](C(=O)O)CNC(=O)OCC1C2=CC=CC=C2C2=CC=CC=C21 CEEGOSWFFHSPHM-PMERELPUSA-N 0.000 description 1
- CKAAWCHIBBNLOJ-QTNFYWBSSA-N (2s)-2,4-diaminobutanoic acid;dihydrochloride Chemical compound Cl.Cl.NCC[C@H](N)C(O)=O CKAAWCHIBBNLOJ-QTNFYWBSSA-N 0.000 description 1
- DQUHYEDEGRNAFO-QMMMGPOBSA-N (2s)-6-amino-2-[(2-methylpropan-2-yl)oxycarbonylamino]hexanoic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)CCCCN DQUHYEDEGRNAFO-QMMMGPOBSA-N 0.000 description 1
- BYEAHWXPCBROCE-UHFFFAOYSA-N 1,1,1,3,3,3-hexafluoropropan-2-ol Chemical compound FC(F)(F)C(O)C(F)(F)F BYEAHWXPCBROCE-UHFFFAOYSA-N 0.000 description 1
- ASWYHZXKFSLNLN-UHFFFAOYSA-N 1,3-dibromo-5-fluorobenzene Chemical compound FC1=CC(Br)=CC(Br)=C1 ASWYHZXKFSLNLN-UHFFFAOYSA-N 0.000 description 1
- PPUZKAPOPPRMFE-UHFFFAOYSA-N 1,5-dibromo-2,4-difluorobenzene Chemical compound FC1=CC(F)=C(Br)C=C1Br PPUZKAPOPPRMFE-UHFFFAOYSA-N 0.000 description 1
- DBHKCZVHGMSFKH-UHFFFAOYSA-N 1-(difluoromethyl)-3-[3-(difluoromethyl)phenyl]sulfanylbenzene Chemical compound FC(C=1C=C(C=CC=1)SC1=CC(=CC=C1)C(F)F)F DBHKCZVHGMSFKH-UHFFFAOYSA-N 0.000 description 1
- HROHDLSABVNCEZ-UHFFFAOYSA-N 1-(difluoromethyl)-3-iodobenzene Chemical compound FC(F)C1=CC=CC(I)=C1 HROHDLSABVNCEZ-UHFFFAOYSA-N 0.000 description 1
- GKZISXVNJDHSSG-UHFFFAOYSA-N 1-bromo-2-(ethoxymethoxymethyl)-5-iodo-3-(trifluoromethyl)benzene Chemical compound CCOCOCC1=C(Br)C=C(I)C=C1C(F)(F)F GKZISXVNJDHSSG-UHFFFAOYSA-N 0.000 description 1
- MICMHFIQSAMEJG-UHFFFAOYSA-N 1-bromopyrrolidine-2,5-dione Chemical compound BrN1C(=O)CCC1=O.BrN1C(=O)CCC1=O MICMHFIQSAMEJG-UHFFFAOYSA-N 0.000 description 1
- JFLSOKIMYBSASW-UHFFFAOYSA-N 1-chloro-2-[chloro(diphenyl)methyl]benzene Chemical compound ClC1=CC=CC=C1C(Cl)(C=1C=CC=CC=1)C1=CC=CC=C1 JFLSOKIMYBSASW-UHFFFAOYSA-N 0.000 description 1
- JMLWXCJXOYDXRN-UHFFFAOYSA-N 1-chloro-3-iodobenzene Chemical compound ClC1=CC=CC(I)=C1 JMLWXCJXOYDXRN-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- TWINDMKDQZVATN-UHFFFAOYSA-N 1-hydroxy-4-methylsulfonyl-3H-2,1-benzoxaborole-6-carboxylic acid Chemical compound CS(=O)(=O)C1=CC(=CC2=C1COB2O)C(O)=O TWINDMKDQZVATN-UHFFFAOYSA-N 0.000 description 1
- IGISPMBUGPHLBY-UHFFFAOYSA-N 1-iodo-3-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC=CC(I)=C1 IGISPMBUGPHLBY-UHFFFAOYSA-N 0.000 description 1
- SKWCZPYWFRTSDD-UHFFFAOYSA-N 2,3-bis(azaniumyl)propanoate;chloride Chemical compound Cl.NCC(N)C(O)=O SKWCZPYWFRTSDD-UHFFFAOYSA-N 0.000 description 1
- AOQYIRCFPBPZMA-UHFFFAOYSA-N 2-(2-aminoethylamino)acetic acid;hydrochloride Chemical compound Cl.NCCNCC(O)=O AOQYIRCFPBPZMA-UHFFFAOYSA-N 0.000 description 1
- BXWGNHMLCCNMPQ-UHFFFAOYSA-N 2-[[3-borono-5-[3-borono-5-(trifluoromethyl)phenyl]sulfonylbenzoyl]amino]acetic acid Chemical compound B(O)(C1=CC(=CC(S(=O)(=O)C2=CC(C(F)(F)F)=CC(B(O)O)=C2)=C1)C(=O)NCC(=O)O)O BXWGNHMLCCNMPQ-UHFFFAOYSA-N 0.000 description 1
- ALFWHEYHCZRVLO-UHFFFAOYSA-N 2-fluoro-4-methylbenzoic acid Chemical compound CC1=CC=C(C(O)=O)C(F)=C1 ALFWHEYHCZRVLO-UHFFFAOYSA-N 0.000 description 1
- WCGNLBCJPBKXCN-UHFFFAOYSA-N 2-fluoro-4-methylbenzonitrile Chemical compound CC1=CC=C(C#N)C(F)=C1 WCGNLBCJPBKXCN-UHFFFAOYSA-N 0.000 description 1
- BMIBJCFFZPYJHF-UHFFFAOYSA-N 2-methoxy-5-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical compound COC1=NC=C(C)C=C1B1OC(C)(C)C(C)(C)O1 BMIBJCFFZPYJHF-UHFFFAOYSA-N 0.000 description 1
- STJUSBSLJXOSDU-UHFFFAOYSA-N 2-methyl-5-(trifluoromethyl)benzoic acid Chemical compound CC1=CC=C(C(F)(F)F)C=C1C(O)=O STJUSBSLJXOSDU-UHFFFAOYSA-N 0.000 description 1
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 1
- UMVOQQDNEYOJOK-UHFFFAOYSA-N 3,5-dimethylbenzoic acid Chemical compound CC1=CC(C)=CC(C(O)=O)=C1 UMVOQQDNEYOJOK-UHFFFAOYSA-N 0.000 description 1
- JHUUPUMBZGWODW-UHFFFAOYSA-N 3,6-dihydro-1,2-dioxine Chemical compound C1OOCC=C1 JHUUPUMBZGWODW-UHFFFAOYSA-N 0.000 description 1
- SCURCOWZQJIUGR-UHFFFAOYSA-N 3-(trifluoromethyl)benzenethiol Chemical compound FC(F)(F)C1=CC=CC(S)=C1 SCURCOWZQJIUGR-UHFFFAOYSA-N 0.000 description 1
- YHEYITLLMWYECH-UHFFFAOYSA-N 3-[3-fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoyl]-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoic acid Chemical compound CC1(C)OB(OC1(C)C)C1=CC(=CC(F)=C1)C(=O)C1=CC(=CC(=C1)C(O)=O)B1OC(C)(C)C(C)(C)O1 YHEYITLLMWYECH-UHFFFAOYSA-N 0.000 description 1
- GZWACXGVBBELRA-UHFFFAOYSA-N 3-bromo-2-fluoro-4-methylbenzoic acid Chemical compound CC1=CC=C(C(O)=O)C(F)=C1Br GZWACXGVBBELRA-UHFFFAOYSA-N 0.000 description 1
- LXWFFEUPSMBYBS-UHFFFAOYSA-N 3-bromo-4-(bromomethyl)-5-(trifluoromethyl)benzoic acid Chemical compound C1=C(C=C(C(=C1C(F)(F)F)CBr)Br)C(=O)O LXWFFEUPSMBYBS-UHFFFAOYSA-N 0.000 description 1
- FAHZIKXYYRGSHF-UHFFFAOYSA-N 3-bromo-4-fluorobenzaldehyde Chemical compound FC1=CC=C(C=O)C=C1Br FAHZIKXYYRGSHF-UHFFFAOYSA-N 0.000 description 1
- HXQRAQKAGXRFEM-UHFFFAOYSA-N 3-bromo-4-fluorobenzenethiol Chemical compound FC1=CC=C(S)C=C1Br HXQRAQKAGXRFEM-UHFFFAOYSA-N 0.000 description 1
- ONELILMJNOWXSA-UHFFFAOYSA-M 3-bromo-4-fluorobenzoate Chemical compound [O-]C(=O)C1=CC=C(F)C(Br)=C1 ONELILMJNOWXSA-UHFFFAOYSA-M 0.000 description 1
- ONELILMJNOWXSA-UHFFFAOYSA-N 3-bromo-4-fluorobenzoic acid Chemical compound OC(=O)C1=CC=C(F)C(Br)=C1 ONELILMJNOWXSA-UHFFFAOYSA-N 0.000 description 1
- BGQIBTIPNDMTNO-UHFFFAOYSA-N 3-bromo-4-methyl-5-methylsulfonylbenzoic acid Chemical compound CC1=C(Br)C=C(C(O)=O)C=C1S(C)(=O)=O BGQIBTIPNDMTNO-UHFFFAOYSA-N 0.000 description 1
- ZFJOMUKPDWNRFI-UHFFFAOYSA-M 3-bromo-4-methylbenzoate Chemical compound CC1=CC=C(C([O-])=O)C=C1Br ZFJOMUKPDWNRFI-UHFFFAOYSA-M 0.000 description 1
- VJHFBALSYNJCED-UHFFFAOYSA-N 3-bromo-5-[3-bromo-5-(trifluoromethyl)phenyl]sulfonylbenzoic acid Chemical compound BrC=1C=C(C(=O)O)C=C(C=1)S(=O)(=O)C1=CC(=CC(=C1)C(F)(F)F)Br VJHFBALSYNJCED-UHFFFAOYSA-N 0.000 description 1
- XRECGLNXYCDFBH-UHFFFAOYSA-N 3-bromo-5-[[3-bromo-5-(trifluoromethyl)phenyl]-difluoromethyl]benzoic acid Chemical compound BrC=1C=C(C(=O)O)C=C(C=1)C(F)(F)C1=CC(=CC(=C1)C(F)(F)F)Br XRECGLNXYCDFBH-UHFFFAOYSA-N 0.000 description 1
- GGVUNSZTZDPBEY-UHFFFAOYSA-N 3-bromo-5-fluorobenzenethiol Chemical compound FC1=CC(S)=CC(Br)=C1 GGVUNSZTZDPBEY-UHFFFAOYSA-N 0.000 description 1
- TXJRHFOYXQUVKW-UHFFFAOYSA-N 3-bromo-5-formyl-4-methylbenzoic acid Chemical compound BrC=1C=C(C(=O)O)C=C(C=1C)C=O TXJRHFOYXQUVKW-UHFFFAOYSA-N 0.000 description 1
- HNGQQUDFJDROPY-UHFFFAOYSA-N 3-bromobenzenethiol Chemical compound SC1=CC=CC(Br)=C1 HNGQQUDFJDROPY-UHFFFAOYSA-N 0.000 description 1
- SDKUOEOJAXGCLU-UHFFFAOYSA-N 3-chloro-4-methylbenzoic acid Chemical compound CC1=CC=C(C(O)=O)C=C1Cl SDKUOEOJAXGCLU-UHFFFAOYSA-N 0.000 description 1
- XUQCONCMPCVUDM-UHFFFAOYSA-N 3-fluoro-4-methylbenzoic acid Chemical compound CC1=CC=C(C(O)=O)C=C1F XUQCONCMPCVUDM-UHFFFAOYSA-N 0.000 description 1
- RZODAQZAFOBFLS-UHFFFAOYSA-N 3-iodobenzaldehyde Chemical compound IC1=CC=CC(C=O)=C1 RZODAQZAFOBFLS-UHFFFAOYSA-N 0.000 description 1
- BNFRSBIMCFBJHK-UHFFFAOYSA-N 3-methyl-2-methylsulfonylbenzoic acid Chemical compound CC1=CC=CC(C(O)=O)=C1S(C)(=O)=O BNFRSBIMCFBJHK-UHFFFAOYSA-N 0.000 description 1
- CZDWJVSOQOMYGC-UHFFFAOYSA-N 4-borono-2-fluorobenzoic acid Chemical compound OB(O)C1=CC=C(C(O)=O)C(F)=C1 CZDWJVSOQOMYGC-UHFFFAOYSA-N 0.000 description 1
- RROAIZSLTZINPA-UHFFFAOYSA-N 4-chloro-1-hydroxy-3H-2,1-benzoxaborole-6-carboxylic acid Chemical compound OB1OCC2=C1C=C(C=C2Cl)C(O)=O RROAIZSLTZINPA-UHFFFAOYSA-N 0.000 description 1
- UWFGVERUNYQOIE-UHFFFAOYSA-N 4-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carboxylic acid Chemical compound OB1OCC2=C1C=C(C=C2F)C(O)=O UWFGVERUNYQOIE-UHFFFAOYSA-N 0.000 description 1
- WBKLSDSNRDYOJN-UHFFFAOYSA-N 4-fluoro-1-hydroxy-3h-2,1-benzoxaborole Chemical compound C1=CC=C(F)C2=C1B(O)OC2 WBKLSDSNRDYOJN-UHFFFAOYSA-N 0.000 description 1
- CAPKAYDTKWGFQB-UHFFFAOYSA-N 4-methyl-3-(trifluoromethyl)benzoic acid Chemical compound CC1=CC=C(C(O)=O)C=C1C(F)(F)F CAPKAYDTKWGFQB-UHFFFAOYSA-N 0.000 description 1
- TXNLQUKVUJITMX-UHFFFAOYSA-N 4-tert-butyl-2-(4-tert-butylpyridin-2-yl)pyridine Chemical group CC(C)(C)C1=CC=NC(C=2N=CC=C(C=2)C(C)(C)C)=C1 TXNLQUKVUJITMX-UHFFFAOYSA-N 0.000 description 1
- GIPODNSBTNDSML-UHFFFAOYSA-N 6-fluoro-1-hydroxy-3H-2,1-benzoxaborole-5-carboxylic acid Chemical compound OB1OCC2=C1C=C(F)C(=C2)C(O)=O GIPODNSBTNDSML-UHFFFAOYSA-N 0.000 description 1
- LIGKOZHDSGAHME-UHFFFAOYSA-N 7-fluoro-1-hydroxy-3h-2,1-benzoxaborole Chemical compound C1=CC(F)=C2B(O)OCC2=C1 LIGKOZHDSGAHME-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- LUGFXTJIJHEVSS-UHFFFAOYSA-N C(C)(=O)OIOC(C)=O Chemical compound C(C)(=O)OIOC(C)=O LUGFXTJIJHEVSS-UHFFFAOYSA-N 0.000 description 1
- PSXLCTPHDAEPLK-UHFFFAOYSA-N CC(C)[Mg] Chemical compound CC(C)[Mg] PSXLCTPHDAEPLK-UHFFFAOYSA-N 0.000 description 1
- XWTGJEVGKWHPER-UHFFFAOYSA-N ClC=1C=C(C=CC=1)S(=O)(C1=CC(=CC=C1)Cl)=NCC(=O)OC(C)(C)C Chemical compound ClC=1C=C(C=CC=1)S(=O)(C1=CC(=CC=C1)Cl)=NCC(=O)OC(C)(C)C XWTGJEVGKWHPER-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- RWSOTUBLDIXVET-UHFFFAOYSA-N Dihydrogen sulfide Chemical compound S RWSOTUBLDIXVET-UHFFFAOYSA-N 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- YBTTUPKAVLJCGU-UHFFFAOYSA-N IN1C(C(CC1=O)Br)=O.IN1C(CCC1=O)=O Chemical compound IN1C(C(CC1=O)Br)=O.IN1C(CCC1=O)=O YBTTUPKAVLJCGU-UHFFFAOYSA-N 0.000 description 1
- 235000019766 L-Lysine Nutrition 0.000 description 1
- BVHLGVCQOALMSV-JEDNCBNOSA-N L-lysine hydrochloride Chemical compound Cl.NCCCC[C@H](N)C(O)=O BVHLGVCQOALMSV-JEDNCBNOSA-N 0.000 description 1
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- TWUWSMPTAVGVMJ-UHFFFAOYSA-N [2-bromo-4-iodo-6-(trifluoromethyl)phenyl]methyl acetate Chemical compound C(C)(=O)OCC1=C(C=C(C=C1C(F)(F)F)I)Br TWUWSMPTAVGVMJ-UHFFFAOYSA-N 0.000 description 1
- WZXLJRQIBCKECK-UHFFFAOYSA-N [3-(5-borono-2,4-difluorobenzoyl)-5-methoxycarbonylphenyl]boronic acid Chemical compound B(O)(O)C=1C=C(C(=O)C=2C(=CC(=C(C=2)B(O)O)F)F)C=C(C=1)C(=O)OC WZXLJRQIBCKECK-UHFFFAOYSA-N 0.000 description 1
- SJZAPSHKTOTRBQ-UHFFFAOYSA-N [dimethylamino(triazolo[4,5-b]pyridin-1-yl)methylidene]-dimethylazanium Chemical compound C1=CC=C2[N+](=C(N(C)C)N(C)C)N=NC2=N1 SJZAPSHKTOTRBQ-UHFFFAOYSA-N 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- AVCOQPBBUQWIEX-UHFFFAOYSA-N acetic acid 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N(=NC(C#N)(C)C)C(C#N)(C)C.C(C)(=O)O AVCOQPBBUQWIEX-UHFFFAOYSA-N 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- RGCKGOZRHPZPFP-UHFFFAOYSA-N alizarin Chemical compound C1=CC=C2C(=O)C3=C(O)C(O)=CC=C3C(=O)C2=C1 RGCKGOZRHPZPFP-UHFFFAOYSA-N 0.000 description 1
- 239000003472 antidiabetic agent Substances 0.000 description 1
- 239000006286 aqueous extract Substances 0.000 description 1
- 125000005605 benzo group Chemical group 0.000 description 1
- 229940007550 benzyl acetate Drugs 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 239000012455 biphasic mixture Substances 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 1
- 125000005621 boronate group Chemical group 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- FCYRSDMGOLYDHL-UHFFFAOYSA-N chloromethoxyethane Chemical compound CCOCCl FCYRSDMGOLYDHL-UHFFFAOYSA-N 0.000 description 1
- 238000010549 co-Evaporation Methods 0.000 description 1
- 230000009918 complex formation Effects 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- JZCCFEFSEZPSOG-UHFFFAOYSA-L copper(II) sulfate pentahydrate Chemical compound O.O.O.O.O.[Cu+2].[O-]S([O-])(=O)=O JZCCFEFSEZPSOG-UHFFFAOYSA-L 0.000 description 1
- 239000002178 crystalline material Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- JCWIWBWXCVGEAN-UHFFFAOYSA-L cyclopentyl(diphenyl)phosphane;dichloropalladium;iron Chemical compound [Fe].Cl[Pd]Cl.[CH]1[CH][CH][CH][C]1P(C=1C=CC=CC=1)C1=CC=CC=C1.[CH]1[CH][CH][CH][C]1P(C=1C=CC=CC=1)C1=CC=CC=C1 JCWIWBWXCVGEAN-UHFFFAOYSA-L 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- VBXDEEVJTYBRJJ-UHFFFAOYSA-N diboronic acid Chemical compound OBOBO VBXDEEVJTYBRJJ-UHFFFAOYSA-N 0.000 description 1
- LXCYSACZTOKNNS-UHFFFAOYSA-N diethoxy(oxo)phosphanium Chemical compound CCO[P+](=O)OCC LXCYSACZTOKNNS-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- LFQRKUIOSYPVFY-UHFFFAOYSA-L dipotassium diacetate Chemical compound [K+].[K+].CC([O-])=O.CC([O-])=O LFQRKUIOSYPVFY-UHFFFAOYSA-L 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 125000006575 electron-withdrawing group Chemical group 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 230000007515 enzymatic degradation Effects 0.000 description 1
- 238000010931 ester hydrolysis Methods 0.000 description 1
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 150000002303 glucose derivatives Chemical class 0.000 description 1
- 230000010030 glucose lowering effect Effects 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229910000037 hydrogen sulfide Inorganic materials 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 239000004026 insulin derivative Substances 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- MPOOBUXJJDKJOG-UHFFFAOYSA-M lithium;chloride;hydrochloride Chemical compound [Li+].Cl.[Cl-] MPOOBUXJJDKJOG-UHFFFAOYSA-M 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 235000012054 meals Nutrition 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- VONMYVUXQPKGOT-UHFFFAOYSA-N methyl 2,3-dibromo-5-fluoro-4-methylbenzoate Chemical compound BrC1=C(C(=O)OC)C=C(C(=C1Br)C)F VONMYVUXQPKGOT-UHFFFAOYSA-N 0.000 description 1
- AUQMLSDKYGMQFZ-UHFFFAOYSA-N methyl 2-fluoro-3-iodo-4-methylbenzoate Chemical compound COC(=O)C1=CC=C(C)C(I)=C1F AUQMLSDKYGMQFZ-UHFFFAOYSA-N 0.000 description 1
- ZRXGXOINRRURRP-UHFFFAOYSA-N methyl 2-fluoro-4-(2-oxopropyl)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate Chemical compound COC(=O)C1=C(F)C=C(CC(C)=O)C(=C1)B1OC(C)(C)C(C)(C)O1 ZRXGXOINRRURRP-UHFFFAOYSA-N 0.000 description 1
- OSWYZVITFZFCMM-UHFFFAOYSA-N methyl 2-fluoro-4-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate Chemical compound COC(=O)C1=CC=C(C)C(B2OC(C)(C)C(C)(C)O2)=C1F OSWYZVITFZFCMM-UHFFFAOYSA-N 0.000 description 1
- VKDYWLKLAIJZPL-UHFFFAOYSA-N methyl 3,5-bis(bromomethyl)benzoate Chemical compound COC(=O)C1=CC(CBr)=CC(CBr)=C1 VKDYWLKLAIJZPL-UHFFFAOYSA-N 0.000 description 1
- JITYFJMCTZXHAX-UHFFFAOYSA-N methyl 3,5-bis[(diformylamino)methyl]benzoate Chemical compound COC(=O)C1=CC(CN(C=O)C=O)=CC(CN(C=O)C=O)=C1 JITYFJMCTZXHAX-UHFFFAOYSA-N 0.000 description 1
- OWGAFGUXZHWZHK-UHFFFAOYSA-N methyl 3-benzoylsulfanyl-5-bromo-4-methylbenzoate Chemical compound C(C1=CC=CC=C1)(=O)SC=1C=C(C(=O)OC)C=C(C=1C)Br OWGAFGUXZHWZHK-UHFFFAOYSA-N 0.000 description 1
- BYRUHRFBYDFJCT-UHFFFAOYSA-N methyl 3-bromo-4-(2-oxopropyl)-5-(trifluoromethyl)benzoate Chemical compound BrC=1C=C(C(=O)OC)C=C(C=1CC(C)=O)C(F)(F)F BYRUHRFBYDFJCT-UHFFFAOYSA-N 0.000 description 1
- IRPLVPJDEJYBGM-UHFFFAOYSA-N methyl 3-bromo-4-(bromomethyl)-5-(trifluoromethyl)benzoate Chemical compound BrC=1C=C(C(=O)OC)C=C(C=1CBr)C(F)(F)F IRPLVPJDEJYBGM-UHFFFAOYSA-N 0.000 description 1
- LPQWURVUVUJPKX-UHFFFAOYSA-N methyl 3-bromo-4-methyl-5-methylsulfanylbenzoate Chemical compound BrC=1C=C(C(=O)OC)C=C(C=1C)SC LPQWURVUVUJPKX-UHFFFAOYSA-N 0.000 description 1
- ZRGBIDIAIHONSF-UHFFFAOYSA-N methyl 3-bromo-4-methyl-5-methylsulfonylbenzoate Chemical compound BrC=1C=C(C(=O)OC)C=C(C=1C)S(=O)(=O)C ZRGBIDIAIHONSF-UHFFFAOYSA-N 0.000 description 1
- MASRAGFWFYHMFI-UHFFFAOYSA-N methyl 3-bromo-4-methylbenzoate Chemical compound COC(=O)C1=CC=C(C)C(Br)=C1 MASRAGFWFYHMFI-UHFFFAOYSA-N 0.000 description 1
- OIJDSGFFXDCCGY-UHFFFAOYSA-N methyl 3-bromo-5-(3-bromo-5-fluorobenzoyl)benzoate Chemical compound BrC=1C=C(C(=O)OC)C=C(C=1)C(C1=CC(=CC(=C1)F)Br)=O OIJDSGFFXDCCGY-UHFFFAOYSA-N 0.000 description 1
- SFIXIWISXWJMQO-UHFFFAOYSA-N methyl 3-bromo-5-(3-bromo-5-fluorophenyl)sulfanylbenzoate Chemical compound BrC=1C=C(C(=O)OC)C=C(C=1)SC1=CC(=CC(=C1)F)Br SFIXIWISXWJMQO-UHFFFAOYSA-N 0.000 description 1
- SSSHVIYLUKEBCZ-UHFFFAOYSA-N methyl 3-bromo-5-(5-bromo-2,4-difluorobenzoyl)benzoate Chemical compound BrC=1C=C(C(=O)OC)C=C(C=1)C(C1=C(C=C(C(=C1)Br)F)F)=O SSSHVIYLUKEBCZ-UHFFFAOYSA-N 0.000 description 1
- BSWXJWAJBGXCFD-UHFFFAOYSA-N methyl 3-bromo-5-[3-bromo-5-(trifluoromethyl)phenyl]sulfanylbenzoate Chemical compound BrC=1C=C(C(=O)OC)C=C(C=1)SC1=CC(=CC(=C1)C(F)(F)F)Br BSWXJWAJBGXCFD-UHFFFAOYSA-N 0.000 description 1
- YIEMWQJHQKHJOW-UHFFFAOYSA-N methyl 3-bromo-5-formyl-4-methylbenzoate Chemical compound COC(=O)C1=CC(Br)=C(C)C(C=O)=C1 YIEMWQJHQKHJOW-UHFFFAOYSA-N 0.000 description 1
- YLJUMFCAQGZAIZ-UHFFFAOYSA-N methyl 3-chloro-5-iodo-4-methylbenzoate Chemical compound COC(=O)C1=CC(Cl)=C(C)C(I)=C1 YLJUMFCAQGZAIZ-UHFFFAOYSA-N 0.000 description 1
- NPXOIGSBRLCOSD-UHFFFAOYSA-N methyl 3-iodobenzoate Chemical compound COC(=O)C1=CC=CC(I)=C1 NPXOIGSBRLCOSD-UHFFFAOYSA-N 0.000 description 1
- WSWBRLCFFPJOTG-UHFFFAOYSA-N methyl 4-(acetyloxymethyl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5-(trifluoromethyl)benzoate Chemical compound COC(=O)C1=CC(B2OC(C)(C)C(C)(C)O2)=C(COC(C)=O)C(=C1)C(F)(F)F WSWBRLCFFPJOTG-UHFFFAOYSA-N 0.000 description 1
- CPDBPEBBNOXSKY-UHFFFAOYSA-N methyl 4-(acetyloxymethyl)-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate Chemical compound COC(=O)C1=CC=C(COC(C)=O)C(B2OC(C)(C)C(C)(C)O2)=C1 CPDBPEBBNOXSKY-UHFFFAOYSA-N 0.000 description 1
- UYVZNCGLCUCRSO-UHFFFAOYSA-N methyl 4-(acetyloxymethyl)-3-(difluoromethyl)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate Chemical compound COC(=O)C1=CC(C(F)F)=C(COC(C)=O)C(=C1)B1OC(C)(C)C(C)(C)O1 UYVZNCGLCUCRSO-UHFFFAOYSA-N 0.000 description 1
- KLLALTMJKAMULG-UHFFFAOYSA-N methyl 4-(acetyloxymethyl)-3-bromo-5-(difluoromethyl)benzoate Chemical compound C(C)(=O)OCC1=C(C=C(C(=O)OC)C=C1C(F)F)Br KLLALTMJKAMULG-UHFFFAOYSA-N 0.000 description 1
- WCJCZQASQPXLBZ-UHFFFAOYSA-N methyl 4-(acetyloxymethyl)-3-bromo-5-(trifluoromethyl)benzoate Chemical compound BrC=1C=C(C(=O)OC)C=C(C=1COC(C)=O)C(F)(F)F WCJCZQASQPXLBZ-UHFFFAOYSA-N 0.000 description 1
- XBKBBLSWYAIMFL-UHFFFAOYSA-N methyl 4-(acetyloxymethyl)-3-bromobenzoate Chemical compound COC(=O)C1=CC=C(COC(C)=O)C(Br)=C1 XBKBBLSWYAIMFL-UHFFFAOYSA-N 0.000 description 1
- GNMNZWYXAHHPKF-UHFFFAOYSA-N methyl 4-(acetyloxymethyl)-3-chloro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate Chemical compound COC(=O)C1=CC(Cl)=C(COC(C)=O)C(=C1)B1OC(C)(C)C(C)(C)O1 GNMNZWYXAHHPKF-UHFFFAOYSA-N 0.000 description 1
- JFHWRMFVPLFVSW-UHFFFAOYSA-N methyl 4-(acetyloxymethyl)-5-iodo-2-(trifluoromethyl)benzoate Chemical compound C(C)(=O)OCC1=CC(=C(C(=O)OC)C=C1I)C(F)(F)F JFHWRMFVPLFVSW-UHFFFAOYSA-N 0.000 description 1
- GXBIEXNBANHJRX-UHFFFAOYSA-N methyl 4-(bromomethyl)-3-fluoro-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate Chemical compound COC(=O)C1=CC(F)=C(CBr)C(=C1)B1OC(C)(C)C(C)(C)O1 GXBIEXNBANHJRX-UHFFFAOYSA-N 0.000 description 1
- HXRZJJNEXLYOBD-UHFFFAOYSA-N methyl 5-bromo-2-fluoro-4-methylbenzoate Chemical compound COC(=O)C1=CC(Br)=C(C)C=C1F HXRZJJNEXLYOBD-UHFFFAOYSA-N 0.000 description 1
- CQSMCYHUVCAXEJ-UHFFFAOYSA-N methyl 5-iodo-4-methyl-2-(trifluoromethyl)benzoate Chemical compound FC(C1=C(C(=O)OC)C=C(C(=C1)C)I)(F)F CQSMCYHUVCAXEJ-UHFFFAOYSA-N 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- CMWYAOXYQATXSI-UHFFFAOYSA-N n,n-dimethylformamide;piperidine Chemical compound CN(C)C=O.C1CCNCC1 CMWYAOXYQATXSI-UHFFFAOYSA-N 0.000 description 1
- DUWWHGPELOTTOE-UHFFFAOYSA-N n-(5-chloro-2,4-dimethoxyphenyl)-3-oxobutanamide Chemical compound COC1=CC(OC)=C(NC(=O)CC(C)=O)C=C1Cl DUWWHGPELOTTOE-UHFFFAOYSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 1
- FSTNQYCPXJMFMT-UHFFFAOYSA-N pentafluoro-(3-nitrophenyl)-$l^{6}-sulfane Chemical compound [O-][N+](=O)C1=CC=CC(S(F)(F)(F)(F)F)=C1 FSTNQYCPXJMFMT-UHFFFAOYSA-N 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- VYMDGNCVAMGZFE-UHFFFAOYSA-N phenylbutazonum Chemical compound O=C1C(CCCC)C(=O)N(C=2C=CC=CC=2)N1C1=CC=CC=C1 VYMDGNCVAMGZFE-UHFFFAOYSA-N 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 1
- JCBJVAJGLKENNC-UHFFFAOYSA-M potassium ethyl xanthate Chemical compound [K+].CCOC([S-])=S JCBJVAJGLKENNC-UHFFFAOYSA-M 0.000 description 1
- 150000003214 pyranose derivatives Chemical group 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 239000012047 saturated solution Substances 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229940001584 sodium metabisulfite Drugs 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- 229940048181 sodium sulfide nonahydrate Drugs 0.000 description 1
- QJXDSDLNUKLDBP-UHFFFAOYSA-M sodium;n-formylmethanimidate Chemical compound [Na+].O=C[N-]C=O QJXDSDLNUKLDBP-UHFFFAOYSA-M 0.000 description 1
- WMDLZMCDBSJMTM-UHFFFAOYSA-M sodium;sulfanide;nonahydrate Chemical compound O.O.O.O.O.O.O.O.O.[Na+].[SH-] WMDLZMCDBSJMTM-UHFFFAOYSA-M 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 229960002317 succinimide Drugs 0.000 description 1
- LFQDNHWZDQTITF-UHFFFAOYSA-N tavaborole Chemical compound FC1=CC=C2B(O)OCC2=C1 LFQDNHWZDQTITF-UHFFFAOYSA-N 0.000 description 1
- UMCLNRCYCLQFTJ-UHFFFAOYSA-N tert-butyl 2-[(6-fluoro-1-hydroxy-3H-2,1-benzoxaborole-5-carbonyl)-[2-[(6-fluoro-1-hydroxy-3H-2,1-benzoxaborole-5-carbonyl)amino]ethyl]amino]acetate Chemical compound FC=1C(=CC2=C(B(OC2)O)C=1)C(=O)N(CC(=O)OC(C)(C)C)CCNC(=O)C1=CC2=C(B(OC2)O)C=C1F UMCLNRCYCLQFTJ-UHFFFAOYSA-N 0.000 description 1
- YRNJKGXSVFJWJC-UHFFFAOYSA-N tert-butyl 2-[(7-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)-[2-[(7-fluoro-1-hydroxy-3H-2,1-benzoxaborole-6-carbonyl)amino]ethyl]amino]acetate Chemical compound FC1=C(C=CC2=C1B(OC2)O)C(=O)N(CC(=O)OC(C)(C)C)CCNC(=O)C=1C=CC2=C(B(OC2)O)C=1F YRNJKGXSVFJWJC-UHFFFAOYSA-N 0.000 description 1
- JPHOVMGSUKPVEB-UHFFFAOYSA-N tert-butyl 2-[[3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5-[3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-5-(trifluoromethyl)benzoyl]benzoyl]amino]acetate Chemical compound CC(C)(C)OC(=O)CNC(=O)C1=CC(=CC(=C1)C(=O)C1=CC(=CC(=C1)C(F)(F)F)B1OC(C)(C)C(C)(C)O1)B1OC(C)(C)C(C)(C)O1 JPHOVMGSUKPVEB-UHFFFAOYSA-N 0.000 description 1
- LSGHLVYZLMYVAI-UHFFFAOYSA-N tert-butyl 2-[[3-bromo-5-(3-bromophenyl)sulfonylbenzoyl]amino]acetate Chemical compound BrC=1C=C(C(=O)NCC(=O)OC(C)(C)C)C=C(C=1)S(=O)(=O)C1=CC(=CC=C1)Br LSGHLVYZLMYVAI-UHFFFAOYSA-N 0.000 description 1
- FCGLAXMHBIGMBF-UHFFFAOYSA-N tert-butyl 2-[[3-bromo-5-[[3-bromo-5-(trifluoromethyl)phenyl]-difluoromethyl]benzoyl]amino]acetate Chemical compound BrC=1C=C(C(=O)NCC(=O)OC(C)(C)C)C=C(C=1)C(F)(F)C1=CC(=CC(=C1)C(F)(F)F)Br FCGLAXMHBIGMBF-UHFFFAOYSA-N 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- GKASDNZWUGIAMG-UHFFFAOYSA-N triethyl orthoformate Chemical compound CCOC(OCC)OCC GKASDNZWUGIAMG-UHFFFAOYSA-N 0.000 description 1
- 239000007966 viscous suspension Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Images






Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/025—Boronic and borinic acid compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/69—Boron compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K33/00—Medicinal preparations containing inorganic active ingredients
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q3/00—Condition responsive control processes
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B30/00—Methods of screening libraries
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B30/00—Methods of screening libraries
- C40B30/04—Methods of screening libraries by measuring the ability to specifically bind a target molecule, e.g. antibody-antigen binding, receptor-ligand binding
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B30/00—Methods of screening libraries
- C40B30/10—Methods of screening libraries by measuring physical properties, e.g. mass
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N24/00—Investigating or analyzing materials by the use of nuclear magnetic resonance, electron paramagnetic resonance or other spin effects
- G01N24/08—Investigating or analyzing materials by the use of nuclear magnetic resonance, electron paramagnetic resonance or other spin effects by using nuclear magnetic resonance
- G01N24/088—Assessment or manipulation of a chemical or biochemical reaction, e.g. verification whether a chemical reaction occurred or whether a ligand binds to a receptor in drug screening or assessing reaction kinetics
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/536—Immunoassay; Biospecific binding assay; Materials therefor with immune complex formed in liquid phase
- G01N33/542—Immunoassay; Biospecific binding assay; Materials therefor with immune complex formed in liquid phase with steric inhibition or signal modification, e.g. fluorescent quenching
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/66—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving blood sugars, e.g. galactose
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
- G01N33/6803—General methods of protein analysis not limited to specific proteins or families of proteins
- G01N33/6827—Total protein determination, e.g. albumin in urine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/435—Assays involving biological materials from specific organisms or of a specific nature from animals; from humans
- G01N2333/76—Assays involving albumins other than in routine use for blocking surfaces or for anchoring haptens during immunisation
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2800/00—Detection or diagnosis of diseases
- G01N2800/04—Endocrine or metabolic disorders
- G01N2800/042—Disorders of carbohydrate metabolism, e.g. diabetes, glucose metabolism
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Physics & Mathematics (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Biomedical Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Analytical Chemistry (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- General Physics & Mathematics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Pathology (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Food Science & Technology (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Cell Biology (AREA)
- High Energy & Nuclear Physics (AREA)
- Biophysics (AREA)
- General Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Inorganic Chemistry (AREA)
- Diabetes (AREA)
- Bioinformatics & Computational Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
一態様において、本発明は、一般式I’によって表されるジボロンコンジュゲートを提供し、
R1’-X’-R2’
式I’中、
X’が、式Ia’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表し、
Dが、原薬を表し、
W’が、共有結合、もしくは
-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表すか、または
X’が、式Ib’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表し、
R3’が、 -(CH2)m’(C=O)-W’-Dを表し、式中、
m’が、1~4の範囲の整数を表し、
W’が、共有結合、もしくは-N-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、
Dが、原薬を表すか、または
X’が、式Ic’のリンカーを表し、

式中、
----が、R1’もしくはR2’に対する共有結合を表し、
n’が、1~4の範囲の整数を表し、
W’が、共有結合、もしくは
-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、
Dが、原薬を表すか、または
X’が、式Id’のリンカーを表し、

式中、
----が、R1’もしくはR2’に対する共有結合を表し、
R4’が、 -(C=O)(CH2)p’(C=O)-W’-Dを表し、式中、
W’が、共有結合、もしくは-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および
-NHCH(COOH)CH2CH2(C=O)-(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、
式中、p’が、1~4の範囲の整数を表し、
Dが、原薬を表すか、または
X’が、式Ie’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表すか、または
X’が、式If’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表すか、または
X’が、式Ig’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表すか、または
X’が、式Ih’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表すか、または
X’が、式Ii’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表し、
W’が、共有結合、もしくは-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、もしくは-NHCH(COOH)CH2CH2(C=O)-(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、
Dが、原薬を表し、
R1’およびR2’が、同一であっても異なっていてもよく、各々が式IIa’または式IIb’の基を表し、

1~4つのY’が、Hを表し、
0、1、または2つのY’が、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/またはSO2CF3を表し、
1つのY’が、式I’のXに対する付着点(を表す共有結合)を表し、
「X」が式Ie’、If’、Ig’またはIh’であるとき、R1’またはR2’のいずれかのY’が、-(C=O)-W’-Dを表し、式中、W’が、共有結合、または-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、
式中、Dが、原薬を表す。
別の態様において、本発明は、式Iによって表されるジボロン化合物、具体的にはジボロネートまたはジボロキソール誘導体を提供し、
R1-X’-R2
式I中、
Xが、式Iaのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表し、
Wが、OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表すか、または
Xが、式Ibのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表し、
R3が、-(CH2)m(C=O)-Wを表し、
式中、mが、1~4の範囲の整数を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表すか、または
Xが、式Icのリンカーを表し、

式中、
----が、R1またはR2に対する共有結合を表し、
nは、1~4の範囲の整数を表し、
Wが、OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表すか、または
式中、Xが、式Idのリンカーを表し、

式中、
----が、R1またはR2に対する共有結合を表し、
R4が、-(C=O)(CH2)m(C=O)-Wを表し、
式中、mが、1~4の範囲の整数を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表すか、または
Xが、式Ieのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
Xが、式Ifのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
Xが、式Igのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
Xが、式Ihのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
Xが、式Iiのリンカーを表し、

----が、R1またはR2に対する共有結合を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1~4つのYが、Hを表し、
0、1、または2つのYが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/またはSO2CF3を表し、
1つのYが、式IのXに対する付着点(を表す共有結合)を表し、
Xが式Ie、If、Ig、またはIhであるとき、R1またはR2のいずれかの1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表す。

----が、R1またはR2に対する共有結合を表し、
Wが、OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IaのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/またはSO2CF3を表し、
残りのYが、Hを表す。
Wが、-OHを表す。
Wが、-OHを表し、
Yのうちの1つが、FまたはCF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。
Wが、-OHを表し、
Yのうちの1つが、Fを表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。
Wが、-OHを表し、
R1およびR2が、同一であり、式IIaの基を表し、
Yのうちの1つが、Fを表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。
Wが、-OHを表し、
R1およびR2が、同一であり、式IIbの基を表し、
Yのうちの1つが、Fを表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R3が、-(CH2)m(C=O)-Wを表し、
式中、mが、1~4の範囲の整数を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IbのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/またはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。
mが、1であり、Wが、-OHである。

式中、
----が、R1またはR2に対する共有結合を表し、
nは、1~4の範囲の整数を表し、
Wが、OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOHを表し(後者は、L-ガンマ-GluまたはD-ガンマ-Glu残基を表す)、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IcのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/またはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。
nが、1~3の範囲の整数であり、Wが、-OHである。
nが、1、2または3であり、
Wが、-OHを表し、
R1およびR2が、同一であり、式IIaまたはIIbの基を表し、
Yのうちの1つが、FまたはCF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。
nが、1、2または3であり、
Wが、-OHを表し、
R1およびR2が、同一であり、式IIaの基を表し、
Yのうちの1つが、Fを表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。
さらなる実施形態において、本発明のジボロン化合物は、式Iによって表され、式中、Xが、上記に定義される式Icのリンカーを表し、式中、
nが、1または2であり、
Wが、-OHを表し、
R1およびR2が、同一であり、式IIbの基を表し、
Yのうちの1つが、FまたはCF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。

式中、
----が、R1またはR2に対する共有結合を表し、
R4が、-(C=O)(CH2)p(C=O)-Wを表し、
式中、pが、1~4の範囲の整数を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOHを表し(後者は、L-ガンマ-GluまたはD-ガンマ-Glu残基を表す)、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IdのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/またはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。
pが、2であり、Wが、-OHである。
pが、2を表し、
Wが、-OHを表し、
R1およびR2が、同一であり、式IIbの基を表し、
Yのうちの1つが、FまたはCF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Ieのリンカー-CO-に対する付着点(を表す共有結合)を表し、
R1またはR2のいずれかの1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/またはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。
Wが、-OHである。
R1およびR2が、同一であり、式IIaの基を表し、
Wが、-OHを表し、
1つのYが、-COOHまたは-CONHCH2COOHを表し、
1つのYが、FまたはCF3を表し、
Yの残りが、Hを表す。

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Ifのリンカー-SO-に対する付着点(を表す共有結合)を表し、
R1またはR2のいずれかの1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/またはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Igのリンカー-(SO2)-に対する付着点(を表す共有結合)を表し、
R1またはR2のいずれかの1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/またはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。
R1およびR2が、同一であり、式IIaの基を表し、
R1またはR2のいずれかの1つのYが、-(C=O)-Wを表し、式中、Wが、-NHCH2COOHを表し、
1つのYが、F、CF3、またはSF5を表し、
Yの残りが、Hを表す、請求項1に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Ihのリンカー-(CF2)-に対する付着点(を表す共有結合)を表し、
R1またはR2のいずれかの1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/またはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。
R1およびR2が、同一であり、式IIaの基を表し、
R1またはR2のいずれかの1つのYが、-(C=O)-Wを表し、式中、Wが、-NHCH2COOHを表し、
1つのYが、CF3を表し、
Yの残りが、Hを表す、請求項1に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または
-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IiのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/またはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。
Wが、-OHである。

1~4つのYが、Hを表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/またはSO2CF3を表し、
1つのYが、式IのXに対する付着点(を表す共有結合)を表し、
Xが式Ie、If、Ig、またはIhであるとき、R1またはR2のいずれかの1つのYが、 -(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOHを表す(後者は、L-ガンマ-GluまたはD-ガンマ-Glu残基を表す)。

1~4つのYが、Hを表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/またはSO2CF3を表し、
1つのYが、式IのXに対する付着点(を表す共有結合)を表し、
Xが式Ie、If、Ig、またはIhであるとき、R1またはR2のいずれかの1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOHを表す(後者は、L-ガンマ-GluまたはD-ガンマ-Glu残基を表す)。
3,5-ビス((4-ボロノ-2-フルオロベンズアミド)メチル)安息香酸、
3,5-ビス((4-ボロノ-3-フルオロベンズアミド)メチル)安息香酸、
N,N’-ビス(4-ボロノ-3-フルオロベンズアミド)-N-エチル-グリシンアミド、
(S)-2,4-ビス(4-ボロノ-3-フルオロベンズアミド)ブタン酸、
N-(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、
3,5-ビス((7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)メチル)安息香酸、
N-(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、
N-(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、
N-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボニル)-N-(2-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボキサミド)エチル)グリシン、
N2,N6-ビス(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボニル)-L-リジン、
3-ボロノ-5-((3-ボロノ-4-フルオロフェニル)スルホニル)-4-フルオロ安息香酸、
3-ボロノ-5-(3-ボロノ-5-フルオロベンゾイル)安息香酸、
3-ボロノ-5-(5-ボロノ-2,4-ジフルオロベンゾイル)安息香酸、
N6-(4-ボロノ-2-フルオロベンゾイル)-N2-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]-オキサボロール-5-カルボニル)-L-リジン、
(S)-2,3-ビス(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-プロパン酸、
(S)-2,3-ビス(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-プロパン酸、
(S)-2,3-ビス(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-プロパン酸、
(3-ボロノ-5-((3-ボロノ-5-(トリフルオロメチル)フェニル)ジフルオロメチル)ベンゾイル)グリシン、
(3-ボロノ-5-(3-ボロノ-5-(トリフルオロメチル)ベンゾイル)ベンゾイル)グリシン、
(3-ボロノ-5-((3-ボロノ-5-(トリフルオロメチル)フェニル)スルホニル)グリシン、
4-((3S,4S)-3,4-ビス(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)ピロリジン-1-イル)-4-オキソブタン酸、
(3-ボロノ-5-(3-ボロノ-5-フルオロベンゾイル)ベンゾイル)グリシン、
(3-(6-ボロノ-2-(エトキシカルボニル)-8-フルオロ-1,1-ジオキシド-4H-ベンゾ[b][1,4]チアジン-4-イル)-5-フルオロフェニル)ボロン酸、
(3-ボロノ-5-(3-ボロノ-5-フルオロフェニル)スルホニル)ベンゾイル)グリシン、
N-(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-エチル)グリシン、
(S)-2,3-ビス(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)プロパン酸、
4-((3S,4S)-3,4-ビス(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)ピロリジン-1-イル)-4-オキソブタン酸、
(3-ボロノ-5-((3-ボロノ-5-(ペンタフルオロ-λ6-スルファニル)フェニル)スルホニル)ベンゾイル)グリシン、
4-[(3R,4R)-3,4-ビス[[1-ヒドロキシ-4-(トリフルオロメチル)-3H-2,1-ベンゾオキサボロール-6-カルボニル]-アミノ]ピロリジン-1-イル]-4-オキソブタン酸、
2-((ビス(3-ボロノ-5-(トリフルオロメチル)フェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸、
N-(4-(ジフルオロメチル)-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(4-(ジフルオロメチル)-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-エチル)グリシン、
N-(4-クロロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(4-クロロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、
3-(2,3-ビス(4-クロロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-プロパンアミド)プロパン酸、
2-((ビス(3-ボロノ-5-(ジフルオロメチル)フェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸、
2-((ビス(3-ボロノ-5-クロロフェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸、および
2-((ビス(3-ボロノフェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸、からなる群から選択される。
別の態様から見ると、本発明は、薬剤として、具体的には代謝障害または症状の治療のための薬剤として使用するための新規のジボロンコンジュゲートを提供する。
別の態様から見ると、本発明は、治療有効量の本発明のジボロンコンジュゲートを含む新規の医薬組成物を提供する。一実施形態において、本発明は、本発明のジボロンコンジュゲートと、1つ以上の賦形剤とを含む医薬組成物を提供する。
別の態様から見ると、本発明は、本発明の新規のジボロンコンジュゲートの製造において中間化合物として使用するための新規のジボロン化合物を提供する。
本発明のジボロン化合物は、例えば、実施例に記載されるような化学合成のための従来の方法によって調製されてもよい。
別の態様から見ると、本発明は、生体動物体の代謝疾患または障害または症状を治療、予防、または緩和する方法を提供し、その方法は、治療有効量の本発明のジボロンコンジュゲートを、それを必要としているそのような生体動物体に投与するステップを含む。
本発明は、以下の非限定的実施形態によってさらに説明される:
1.一般式I’によって表される、ジボロンコンジュゲートであって、


Xが、式Iaのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表し、
Wが、-OHもしくは-NHCH2COOHを表すか、または
Xが、式Ibのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表し、
R3が、-(CH2)mCOOHを表し、
式中、mが、1~4の範囲の整数を表すか、または
Xが、式Icのリンカーを表し、

式中、
----が、R1もしくはR2に対する共有結合を表し、
nは、1~4の範囲の整数を表し、
Wが、-OHもしくは-NHCH2COOHを表すか、または
式中、Xが、式Idのリンカーを表し、

式中、
----が、R1もしくはR2に対する共有結合を表し、
R4が、-(C=O)(CH2)m(COOH)を表し、
式中、mが、1~4の範囲の整数を表すか、または
Xが、式Ieのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
Xが、式Ifのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
Xが、式Igのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
Xが、式Ihのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表し、
式Ia、Ib、Ic、Id、Ie、If、Ig、およびIh中、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1~4つのYが、Hを表し、
Yのうちの0、1、または2つが、F、CF3、および/またはSF5を表し、
1つのYが、式IのXに対する付着点(を表す共有結合)を表し、
式Ie、If、Ig、およびIh中、Yが、-COOHまたはCONHCH2COOHを表す、ジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
Wが、-OHまたは-NHCH2COOHを表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IaのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態2に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R3が、 -(CH2)mCOOHを表し、
式中、mが、1~4の範囲の整数を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IbのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態2に記載のジボロン化合物。

式中、
----が、R1またはR2に対する共有結合を表し、
nは、1~4の範囲の整数を表し、
Wが、-OHまたは-NHCH2COOHを表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IcのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態2に記載のジボロン化合物。

式中、
----が、R1またはR2に対する共有結合を表し、
R4が、-(C=O)(CH2)m(COOH)を表し、
式中、mが、1~4の範囲の整数を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IdのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態2に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Ieのリンカー-CO-に対する付着点(を表す共有結合)を表し、
1つのYが、-COOHまたは-CONHCH2COOHを表し、
Yのうちの0、1、または2つが、F、CF3、および/またはSF5を表し、
Yの残りが、Hを表す、実施形態2に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Ifのリンカー-SO-に対する付着点(を表す共有結合)を表し、
1つのYが、-COOHまたは-CONHCH2COOHを表し、
Yのうちの0、1、または2つが、F、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態2に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Igのリンカー-(SO2)-に対する付着点(を表す共有結合)を表し、
1つのYが、-COOHまたは-CONHCH2COOHを表し、
Yのうちの0、1、または2つが、F、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態2に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Ihのリンカー-(CF2)-Xに対する付着点(を表す共有結合)を表し、
1つのYが、-COOHまたは-CONHCH2COOHを表し、
Yのうちの0、1、または2つが、F、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態2に記載のジボロン化合物。
3,5-ビス((4-ボロノ-2-フルオロベンズアミド)メチル)安息香酸、3,5-ビス((4-ボロノ-3-フルオロベンズアミド)メチル)安息香酸、N,N’-ビス(4-ボロノ-3-フルオロベンズアミド)-N-エチル-グリシンアミド、(S)-2,4-ビス(4-ボロノ-3-フルオロベンズアミド)ブタン酸、N-(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、3,5-ビス((7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)メチル)安息香酸、N-(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、N-(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、N-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボニル)-N-(2-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボキサミド)エチル)グリシン、N2,N6-ビス(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボニル)-L-リジン、3-ボロノ-5-((3-ボロノ-4-フルオロフェニル)スルホニル)-4-フルオロ安息香酸]、3-ボロノ-5-(3-ボロノ-5-フルオロベンゾイル)安息香酸、3-ボロノ-5-(5-ボロノ-2,4-ジフルオロベンゾイル)安息香酸、
N6-(4-ボロノ-2-フルオロベンゾイル)-N2-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボニル)-L-リジン、(S)-2,3-ビス(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ-[c][1,2]オキサボロール-6-カルボキサミド)-プロパン酸、(S)-2,3-ビス(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-プロパン酸、(S)-2,3-ビス(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-プロパン酸、
(3-ボロノ-5-((3-ボロノ-5-(トリフルオロメチル)フェニル)ジフルオロメチル)ベンゾイル)グリシン、
3-ボロノ-5-(3-ボロノ-5-(トリフルオロメチル)ベンゾイル)グリシン)、(3-ボロノ-5-((3-ボロノ-5-(トリフルオロメチル)フェニル)スルホニル)グリシン、4-((3S,4S)-3,4-ビス(7-フルオロ-1-ヒドロキシ-1,3-ヒドロベンゾ-[c][1,2]オキサボロール-6-カルボキサミド)ピロリジン-1-イル)-4-オキソブタン酸、(3-ボロノ-5-(3-ボロノ-5-フルオロベンゾイル)ベンゾイル)グリシン、(3-(6-ボロノ-2-(エトキシカルボニル)-8-フルオロ-1,1-ジオキシド-4H-ベンゾ[b][1,4]チアジン-4-イル)-5-フルオロフェニル)ボロン酸、(3-ボロノ-5-((3-ボロノ-5-フルオロフェニル)スルホニル)ベンゾイル)-グリシン、N-(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-エチル)グリシン、(S)-2,3-ビス(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)プロパン酸、4-((3S,4S)-3,4-ビス(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-ピロリジン-1-イル)-4-オキソブタン酸、(3-ボロノ-5-((3-ボロノ-5-(ペンタフルオロ-λ6-スルファニル)-フェニル)スルホニル)ベンゾイル)グリシン、および4-[(3R,4R)-3,4-ビス[[1-ヒドロキシ-4-(トリフルオロ-メチル)-3H-2,1-ベンゾオキサボロール-6-カルボニル]-アミノ]ピロリジン-1-イル]-4-オキソブタン酸、からなる群から選択される、ジボロン化合物。
R1’-X’-R2’
式I’中、
X’が、式Ia’のリンカーを表し、

----が、R1 ’もしくはR2 ’に対する共有結合を表し、
Dが、原薬を表し、
W’が、共有結合、もしくは
-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表すか、または
X’が、式Ib’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表し、
R3’が、-(CH2)m’(C=O)-W’-Dを表し、式中、
m’が、1~4の範囲の整数を表し、
W’が、共有結合、もしくは-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、
Dが、原薬を表すか、または
X’が、式Ic’のリンカーを表し、

式中、
----が、R1’もしくはR2’に対する共有結合を表し、
n’が、1~4の範囲の整数を表し、
W’が、共有結合、もしくは
-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、
Dが、原薬を表すか、または
X’が、式Id’のリンカーを表し、

式中、
----が、R1’もしくはR2’に対する共有結合を表し、
R4’が、-(C=O)(CH2)p’(C=O)-W’-Dを表し、式中、
W’が、共有結合、もしくは-N-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-
(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、
式中、p’が、1~4の範囲の整数を表し、
および
Dが、原薬を表すか、または
X’が、式Ie’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表すか、または
X’が、式If’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表すか、または
X’が、式Ig’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表すか、または
X’が、式Ih’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表すか、または
X’が、式Ii’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表し、
W’が、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1’およびR2’が、同一であっても異なっていてもよく、各々が式IIa’または式IIb’の基を表し、

1~4つのY’が、Hを表し、
0、1、または2つのY’が、F、Cl、CF2、CF3、および/またはSF5を表し、
1つのY’が、式I’のXに対する付着点(を表す共有結合)を表し、
X’が式Ie’、If’、Ig’またはIh’であるとき、R1’またはR2’のいずれかのYが、-(C=O)-W’-Dを表し、式中、W’が、共有結合、または-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、
式中、Dが、原薬を表す、ジボロンコンジュゲート。

Xが、式Iaのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表すか、または
Xが、式Ibのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表し、
R3が、-(CH2)m(C=O)-Wを表し、
式中、mが、1~4の範囲の整数を表し、
Wが、OH、NHCH2COOH、NHCH2CH2COOH、NHCH2CH2CH2COOH、もしくはNHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表すか、または
Xが、式Icのリンカーを表し、

式中、
----が、R1またはR2に対する共有結合を表し、
nは、1~4の範囲の整数を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表すか、または
式中、Xが、式Idのリンカーを表し、

式中、
----が、R1またはR2に対する共有結合を表し、
R4が、-(C=O)(CH2)p(C=O)-Wを表し、
式中、pが、1~4の範囲の整数を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表すか、または
Xが、式Ieのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
Xが、式Ifのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
Xが、式Igのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
Xが、式Ihのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
Xが、式Iiのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1~4つのYが、Hを表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、および/またはSF5を表し、
1つのYが、式IのXに対する付着点(を表す共有結合)を表し、
Xが式Ie、If、Ig、またはIhであるとき、R1またはR2のいずれかの1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOHを表す(後者は、L-ガンマ-GluまたはD-ガンマ-Glu残基を表す)、ジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IaのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態16に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R3が、-(CH2)m(C=O)-Wを表し、
式中、mが、1~4の範囲の整数を表し、
Wが、OH、NHCH2COOH、NHCH2CH2COOH、NHCH2CH2CH2COOH、またはNHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IbのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態16に記載のジボロン化合物。

式中、
----が、R1またはR2に対する共有結合を表し、
nは、1~4の範囲の整数を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IcのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態16に記載のジボロン化合物。

式中、
----が、R1またはR2に対する共有結合を表し、
R4が、-(C=O)(CH2)p(C=O)-Wを表し、
式中、pが、1~4の範囲の整数を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IdのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態16に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Ieのリンカー-CO-に対する付着点(を表す共有結合)を表し、
R1またはR2のいずれかの1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、および/またはSF5を表し、
Yの残りが、Hを表す、実施形態16に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Ifのリンカー-SO-に対する付着点(を表す共有結合)を表し、
R1またはR2のいずれかの1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態16に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Igのリンカー-(SO2)-に対する付着点(を表す共有結合)を表し、
R1またはR2のいずれかの1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態16に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Ihのリンカー-(CF2)-に対する付着点(を表す共有結合)を表し、
R1またはR2のいずれかの1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態16に記載のジボロン化合物。

----が、R1またはR2に対する共有結合を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(後者は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IiのリンカーXに対する付着点(を表す共有結合)を表し、
Yのうちの0、1、または2つが、F、Cl、CF2、CF3、および/またはSF5を表し、
残りのYが、Hを表す、実施形態16に記載のジボロン化合物。
3,5-ビス((4-ボロノ-2-フルオロベンズアミド)メチル)安息香酸、3,5-ビス((4-ボロノ-3-フルオロ-ベンズアミド)メチル)安息香酸、N,N’-ビス(4-ボロノ-3-フルオロベンズアミド)-N-エチル-グリシンアミド、(S)-2,4-ビス(4-ボロノ-3-フルオロベンズアミド)ブタン酸、N-(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、3,5-ビス((7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)メチル)安息香酸、N-(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、N-(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、N-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボニル)-N-(2-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボキサミド)エチル)グリシン、N2,N6-ビス(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボニル)-L-リジン、3-ボロノ-5-((3-ボロノ-4-フルオロフェニル)スルホニル)-4-フルオロ安息香酸、3-ボロノ-5-(3-ボロノ-5-フルオロベンゾイル)安息香酸、3-ボロノ-5-(5-ボロノ-2,4-ジフルオロベンゾイル)安息香酸、
N6-(4-ボロノ-2-フルオロベンゾイル)-N2-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]-オキサボロール-5-カルボニル)-L-リジン、(S)-2,3-ビス(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ-[c][1,2]オキサボロール-6-カルボキサミド)プロパン酸、(S)-2,3-ビス(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)プロパン酸、(S)-2,3-ビス(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)プロパン酸、(3-ボロノ-5-((3-ボロノ-5-(トリフルオロメチル)フェニル)ジフルオロメチル)ベンゾイル)-グリシン、(3-ボロノ-5-(3-ボロノ-5-(リフルオロメチル)ベンゾイル)ベンゾイル)グリシン、(3-ボロノ-5-((3-ボロノ-5-(トリフルオロメチル)フェニル)スルホニル)グリシン、4-((3S,4S)-3,4-ビス(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)ピロリジン-1-イル)-4-オキソブタン酸、
(3-ボロノ-5-(3-ボロノ-5-フルオロベンゾイル)ベンゾイル)グリシン、(3-(6-ボロノ-2-(エトキシ-カルボニル)-8-フルオロ-1,1-ジオキシド-4H-ベンゾ[b][1,4]チアジン-4-イル)-5-フルオロフェニル)ボロン酸、(3-ボロノ-5-((3-ボロノ-5-フルオロフェニル)スルホニル)ベンゾイル)グリシン、N-(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、(S)-2,3-ビス(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)プロパン酸、4-((3S,4S)-3,4-ビス(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)ピロリジン-1-イル)-4-オキソブタン酸、(3-ボロノ-5-((3-ボロノ-5-(ペンタフルオロ-λ6-スルファニル)フェニル)スルホニル)ベンゾイル)グリシン、2-((ビス(3-ボロノ-5-(トリフルオロメチル)フェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸、N-(4-(ジフルオロメチル)-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(4-(ジフルオロメチル)-1-ヒドロキシ-1,3-ジヒドロメチル[c][1,2]オキサボロール-6-カルボキサミド)エチル)-グリシン、N-(4-クロロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(4-クロロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、
3-(2,3-ビス(4-クロロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-プロパンアミド)プロパン酸、2-((ビス(3-ボロノ-5-(ジフルオロメチル)フェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸;2-((ビス(3-ボロノ-5-クロロフェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸、および2-((ビス(3-ボロノフェニル)(オキソ)-λ6-スルファニリデン)-アミノ)酢酸、からなる群から選択される、ジボロン化合物。
AA 酢酸
AIBN 2,2’-アゾビス(2-メチルプロピオニトリル)
ARS アリザリンレッドナトリウム
DAST (ジエチルアミノ)硫黄トリフルオリド
DCC N,N’-ジシクリオヘキシルカルボジイミド
DMSO ジメチルスルホキシド
EDC N-(3-ジメチルアミノプロピル)-N’-エチルカルボジイミド
ELSD エレクトロスプレー検出
F-NMR フッ素-19核磁気共鳴分光法
FA ギ酸
HATU 1-((ジメチルアミノ)(ジメチルイミニオ)メチル)-1H-[1,2,3]トリアゾロ[4,5-b]ピリジン3-オキシドヘキサフルオロホスフェート
HSA ヒト血清アルブミン
LCMS 液体クロマトグラフィ質量分析
NBS N-ブロモスクシンイミド(1-ブロモピロリジン-2,5-ジオン)
NIS N-ヨードスクシンイミド(1-ヨードブロモピロリジン-2,5-ジオン)
NMR 核磁気共鳴分光法
HOSu、NOHSu N-ヒドロキシスクシンイミド
PCC クロロクロム酸ピリジニウム
XPhos 2-ジクロヘキシルホスフィノ-2’,4’,6’-トリイソプロピルビフェニル
実施例1
3,5-ビス((4-ボロノ-2-フルオロベンズアミド)メチル)安息香酸



3,5-ビス((4-ボロノ-3-フルオロベンズアミド)メチル)安息香酸

N,N’-ビス(4-ボロノ-3-フルオロベンズアミド)-N-エチル-グリシンアミド

(S)-2,4-ビス(4-ボロノ-3-フルオロベンズアミド)ブタン酸


N-(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン



3,5-ビス((7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)メチル)安息香酸


収量:13.0g(70%)。
N-(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン


N-(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン



N-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボニル)-N-(2-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボキサミド)エチル)グリシン


N 2 ,N 6 -ビス(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボニル)-L-リジン


3-ボロノ-5-((3-ボロノ-4-フルオロフェニル)スルホニル)-4-フルオロ安息香酸


LC-MS m/z:443.2(M+H)+。
3-ボロノ-5-(3-ボロノ-5-フルオロベンゾイル)安息香酸


3-ボロノ-5-(5-ボロノ-2,4-ジフルオロベンゾイル)安息香酸



N 6 -(4-ボロノ-2-フルオロベンゾイル)-N 2 -(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボニル)-L-リジン


(S)-2,3-ビス(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)プロパン酸


(S)-2,3-ビス(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)プロパン酸



(S)-2,3-ビス(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)プロパン酸


(3-ボロノ-5-((3-ボロノ-5-(トリフルオロメチル)フェニル)ジフルオロメチル)ベンゾイル)グリシン



(3-ボロノ-5-(3-ボロノ-5-(トリフルオロメチル)ベンゾイル)ベンゾイル)グリシン


(3-ボロノ-5-((3-ボロノ-5-(トリフルオロメチル)フェニル)スルホニル)グリシン


4-((3S,4S)-3,4-ビス(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)ピロリジン-1-イル)-4-オキソブタン酸



(3-ボロノ-5-(3-ボロノ-5-フルオロベンゾイル)ベンゾイル)グリシン


(3-ボロノ-5-((3-ボロノ-5-フルオロフェニル)スルホニル)ベンゾイル)グリシン


N-(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン


(S)-2,3-ビス(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)プロパン酸

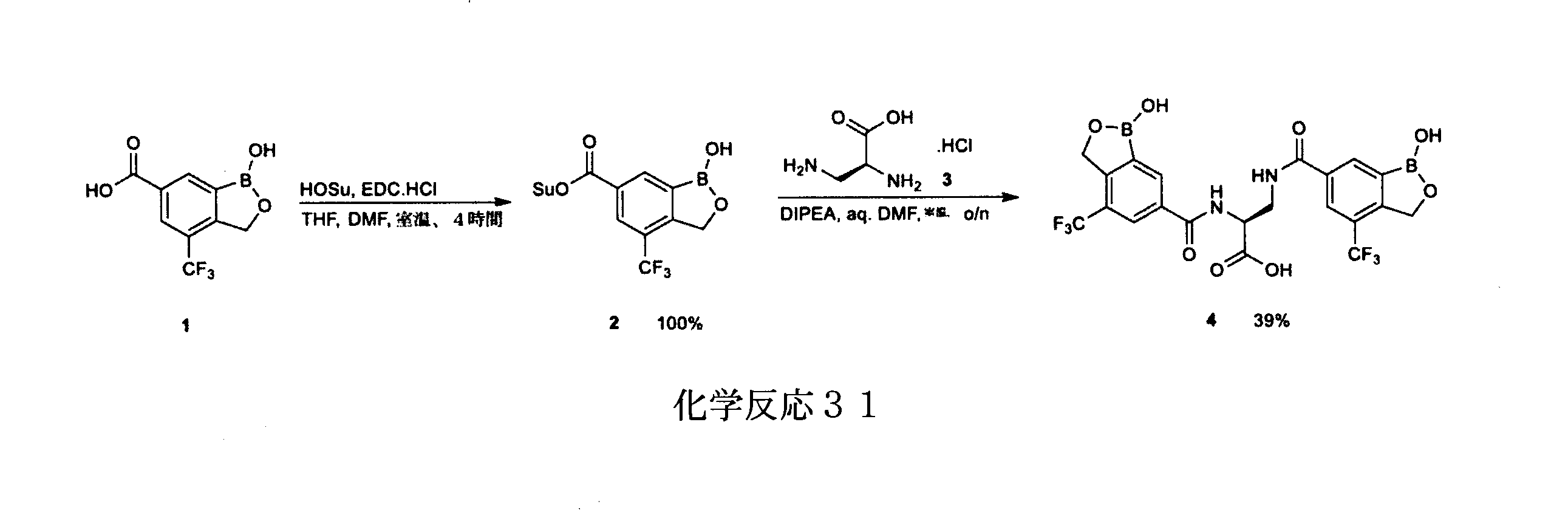
4-((3S,4S)-3,4-ビス(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)ピロリジン-1-イル)-4-オキソブタン酸


(3-ボロノ-5-((3-ボロノ-5-(ペンタフルオロ-λ 6 -スルファニル)フェニル)スルホニル)ベンゾイル)グリシン


4-[(3R,4R)-3,4-ビス[[1-ヒドロキシ-4-(トリフルオロメチル)-3H-2,1-ベンゾオキサボロール-6-カルボニル]アミノ]ピロリジン-1-イル]-4-オキソブタン酸

2-((ビス(3-ボロノ-5-(トリフルオロメチル)フェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸


収量:4.01g(62%)。
N-(4-(ジフルオロメチル)-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(4-(ジフルオロメチル)-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン


収量:87.8g(99%)。
1H NMRスペクトル(300 MHz,DMSO-d6,δH):8.30(d, J=1.7 Hz,1 H);8.06(d,J=1.7 Hz,1 H);2.64(s,3 H)。
収量:6.24g(96%)。
収量:2.82g(53%)。
N-(4-クロロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(4-クロロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン


収量:32.0g(73%)。
(S)-3-(2,3-ビス(4-クロロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-プロパンアミド)プロパン酸


0mL、60.0mmol)の溶液を樹脂に添加し、混合物を5時間振盪した。樹脂をジクロロメタン(3×100mL)、2-プロパノール(3×100mL)、N,N-ジメチルホルムアミド(3×100mL)、およびジクロロメタン(5×100mL)で洗浄した。
2-((ビス(3-ボロノ-5-(ジフルオロメチル)フェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸

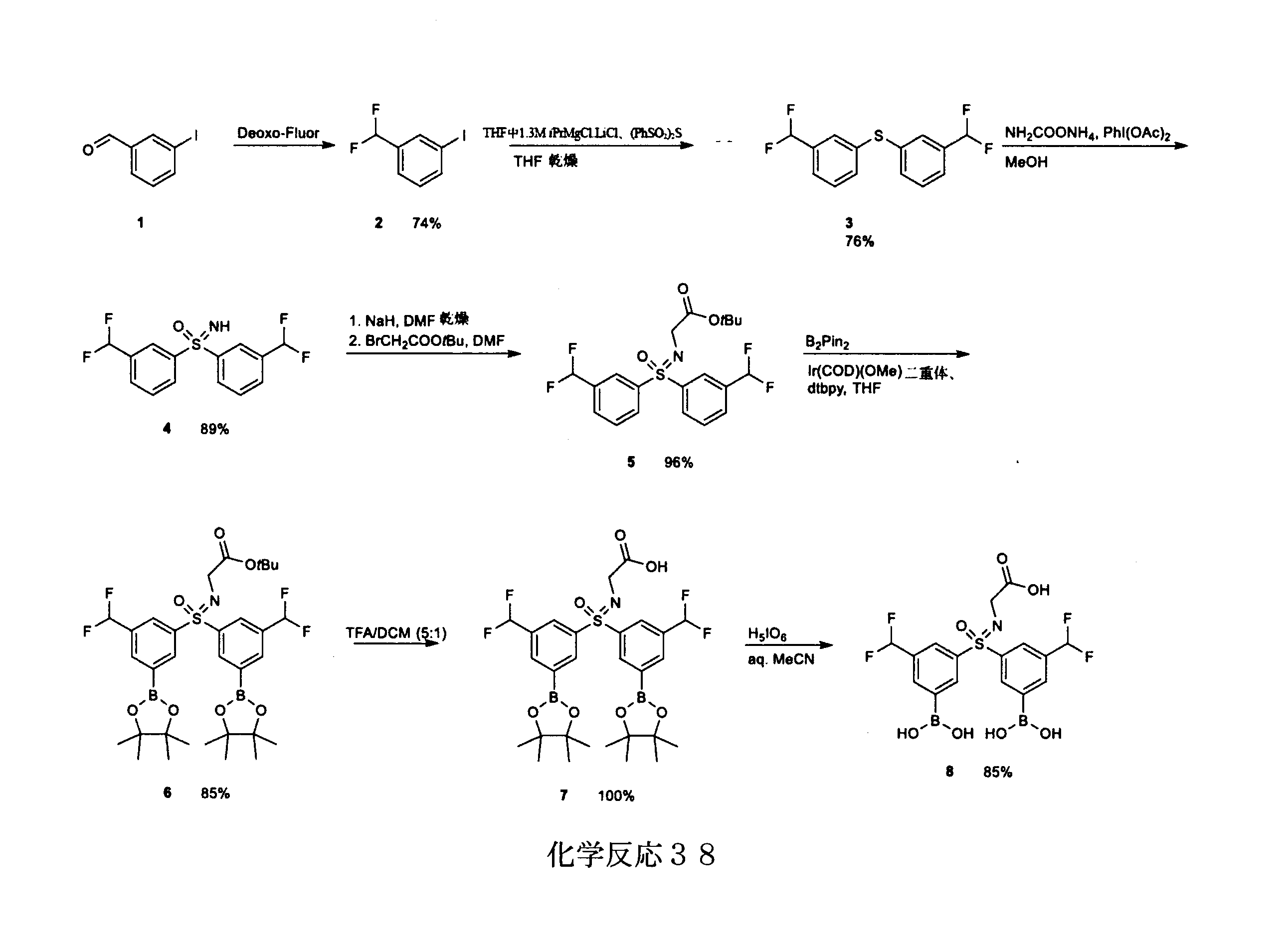
2-((ビス(3-ボロノ-5-クロロフェニル)(オキソ)- λ6-スルファニリデン)アミノ)酢酸


1H NMRスペクトル(300 MHz,DMSO-d6,δH) :7.45-7.38(m,6 H);7.35-7.29(m,2 H)。
2-((ビス(3-ボロノフェニル)(オキソ)- λ6-スルファニリデン)アミノ)酢酸


N-(1-ヒドロキシ-5-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(1-ヒドロキシ-5-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン


2-((ビス(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-イル)(オキソ)- λ6-スルファニリデン)アミノ)酢酸


収量:4.64g(定量)。
1H NMRスペクトル(300 MHz,CDCl3,δH):8.18(s,1 H);7.98(s,1 H);4.87(d,J=6.9 Hz,2 H),2.08(t,J=6.9 Hz,1 H)。
1H NMRスペクトル(300 MHz,CDCl3,δH):7.76(s,2 H);7.65(s,2 H);4.84(s,4 H);4.79(s,4 H);3.71(q,J=7.1 Hz,4 H);1.26(t,J=7.1 Hz,6 H)。

チルエーテル中に溶解し、ヘキサン(3体積)を添加した。沈殿生成物を濾過によって収集し、低温ヘキサン(2×5mL)で洗浄し、空気乾燥させて、オフホワイト色固体として2-((ビス(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ-[c][1,2]オキサボロール-6-イル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸(5)を得た。
(3-ボロノ-5-((3-ボロノフェニル)スルホニル)ベンゾイル)グリシン


収量:26.0g(76%)。
乾燥させ、濾過し、蒸発させた。粗生成物を最小量の湿潤(水と十分に振盪された)酢酸エチル中に溶解し、その溶液を氷冷n-ヘキサン(5mL)に撹拌しながら滴下し、生成物の沈殿物を得た。固体を濾過によって収集し、ヘキサン(2×5mL)で洗浄して、無色固体として表題の(3-ボロノ-5-((3-ボロノフェニル)スルホニル)ベンゾイル)グリシン(9)を得た。収量:221.0mg(54%)。
(S)-2,3-ビス(1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)プロパン
酸


RF(SiO2、シクロヘキサン/酢酸エチル9:1):0.45。
N-(1-ヒドロキシ-4-(メチルスルホニル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(1-ヒドロキシ-4-(メチルスルホニル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-エチル)グリシン


1H NMRスペクトル(300 MHz,DMSO-d6,δH):8.46(d,J=1.7 Hz,1 H);8.40(d,J=1.8 Hz,1 H);3.91(s,3 H);3.34(s,3 H);2.79(s,3 H)。
1H NMRスペクトル(300 MHz,DMSO-d6,δH):8.47(d,J=1.1 Hz,2 H);5.14(bs,2 H);3.92(s,3 H);3.44(s,3 H)。
3-ブロモ-4-(ブロモメチル)-5-(メチルスルホニル)安息香酸メチル(4、2.70g、7.00mmol)を、アセトニトリル(70mL)中のカリウム酢酸カリウム(1.37g、14.0mmol)と75Cで一晩撹拌した。懸濁液をセライトパッドを通して濾過し、真空下で蒸発させた。粗生成物をジクロロメタン中に溶解し、再び濾過した。濾液を蒸発させ、カラムクロマトグラフィ(Silicagel 60、0.040~0.063mm、溶離液:シクロヘキサン/酢酸エチル7:1)によって精製して、白色固体として4-(アセトキシメチル)-3-ブロモ-5-(メチルスルホニル)安息香酸メチル(5)を得た。収量:4.24g(54%)。
1H NMRスペクトル(300 MHz,アセトン-d6/D2O,δΗ):8.67(s,1 H);8.54(s,1 H);5.40(s,2 H);3.23(s,3 H)。
19F NMRスペクトル(282 MHz,アセトン-d6/D2O,δF):-154.63(d,J=17.0 Hz);-159.62(t,J=21.3 Hz);164.43(t,J=19.1 Hz)。
LC-MS m/z:421.0(M-H)-。
N-(1-ヒドロキシ-4-((トリフルオロメチル)スルホニル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(1-ヒドロキシ-4-((トリフルオロメチル)スルホニル)-1,3-ジヒドロベンゾ-[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン


1H NMRスペクトル(300 MHz,DMSO-d6,δH):8.30(d, J=1.7 Hz,1 H);8.06(d,J=1.7 Hz,1 H);2.64(s,3 H)。
RF(SiO2、シクロヘキサン/ジクロロメタン1:1):0.30。
1H NMRスペクトル(300 MHz,CDCl3,δΗ):8.33(d,J=1.7 Hz,1 H);8.14(d,J=1.8 Hz,1 H);8.08-8.00(m,2 H);7.69-7.61(m,1 H);7.56-7.49(m,2 H);3.93(s,3 H);2.59(s,3 H)。
19F NMRスペクトル(282 MHz,CDCl3,δF):-77.39(s)。
1H NMRスペクトル(300 MHz,DMSO-d6,δΗ):9.93(s,1 H);9.09(s,1 H);8.65(s,1 H);5.41(s,2 H)。
4-ボロノ-2-((3-ボロノ-5-(トリフルオロメチル)フェニル)スルホニル)-6-(トリフルオロメチル)安息香酸


1H NMRスペクトル(300 MHz,CDCl3,δH):8.78(s,1 H);8.60(s,1 H);8.34(s,2 H);8.25(s,1 H);1.39(s,12 H);1.36(s,12 H)。
アセトニトリル(4mL)中の上記の酸(7、210mg、0.33mmol)の溶液を水(1mL)で希釈し、続いて過ヨウ素酸(301mg、1.32mmol)を添加した。得られる混合物を1時間撹拌し、次いで、これを酢酸エチル(30mL)と水(30mL)との間で分配した。相を分離し、有機相を水(1×30mL)およびブライン(1×30mL)で洗浄し、無水硫酸ナトリウム上で乾燥させ、濾過し、蒸発乾固させた。残渣を、20%水酸化ナトリウム水溶液(10mL)中に溶解し、ジクロロメタン(2×40mL)で洗浄した。水層を1M塩酸水溶液(30mL)で酸性化し、酢酸エチル(2×50mL)で抽出した。酢酸エチル抽出物を混合し、無水硫酸ナトリウム上で乾燥させ、濾過し、生成物が沈殿し始めるまで真空下で濃縮した。懸濁液をn-ヘキサン(50mL)で希釈した。沈殿物を濾過によって収集し、n-ヘキサンで洗浄し、アセトニトリル(30mL)中に溶解し、凍結乾燥させて、ベージュ色粉末として表題化合物(8)を得た。
2-((ビス(3-ボロノ-5-(トリフルオロメトキシ)フェニル)(オキソ)- λ6-スルファニリデン)アミノ)酢酸


アリザリンレッド結合アッセイは、ボロネート化合物のグルコースへの阻害親和性を決定するために使用される比色アッセイである。アッセイは、ボロネートへの結合時のアリザリンレッドの色のシフトに基づき、このシフトの後には、330~340nm領域の吸光度の変化があり得る。
アリザリンレッド(ARS)とボロネート化合物との間の解離定数(Kd)の決定については、200μMのARSを20mMのリン酸緩衝液pH7.4中に溶解し、1、0.5、0.25、0.125、62.5、31.25、15.625、7.812、3.906、1.953、0.9767、0.488、および0.244mMのボロン酸を用いて、96ウェルプレートに三つ組で滴定する。4000rpmで5分間遠心分離した後、プレートを吸収検出のためにマルチウェル分光計(SpectraMax,Molecular Devices)内に置く。
ボロネートと炭水化物との間の阻害定数(Ki)の決定については、400 μMのボロン酸を、穏やかに撹拌しながら20mMのリン酸緩衝液pH7.4中に溶解する。化合物が完全に溶解すると、200μMのアリザリンレッド(ARS)を溶液に添加する。次いで、ARS-ボロネート溶液を、適切な炭水化物を用いて96マルチウェルプレート(黒、平坦、かつ透明な底部)1:1にアリコートする。具体的には、D-グルコースおよびL-乳酸溶液を、これらの濃度:1000、500、250、100、50、25、10、5、2.5、1、0.25、0.1mM、および2500、1000、500、100、50、10、5、1、0.5、0.1、0.05、0.01mMのそれぞれにおいて、20mMリン酸緩衝液pH7.4中で調製する。ARS-ボロネートを炭水化物と混合したプレートを室温で20分間インキュベートする。4000rpmで5分間遠心分離した後、プレートを吸収検出のためにマルチウェル分光計(SpectraMax,Molecular Devices)内に置く。
13Cグルコースアッセイは、未結合グルコース(A状態)とジボロネートに結合したグルコース(B状態)との間のゆっくりとした化学シフト交換を利用するNMRベースのアッセイである。


グルコースとボロネート化合物との間の解離定数(Kd)の決定については、10mMのリン酸緩衝液および5%D2O中のpH7.4の1mMのボロネートを有する、および有しない1mMのグルコースC13の試料を調製する。次いで、試料をBruker NMR機器(RT、32スキャン)における標準炭素HSQCを用いて調査する。遊離グルコースおよびボロネートに結合したグルコースのピークの強度を、TOPSPINプログラム(Bruker)の使用によって決定する。遊離グルコースピークとグルコース結合ピークとの強度の差は、平衡における生成物ABの濃度を示し、これにより、Kdは、方程式1を使用して計算され得る。

このアッセイは、19F NMRスペクトルの決定方法を説明し、これは、アルブミンへのジボロン化合物結合、およびジボロン化合物のグルコース感受性アルブミン結合を示す。
Claims (16)
- 式I:

・式I中、
・Xが、式Iaのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表すか、または
・Xが、式Ibのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表し、
R3が、-(CH2)m(C=O)-Wを表し、
式中、mが、1~4の範囲の整数を表し、
Wが、OH、NHCH2COOH、NHCH2CH2COOH、NHCH2CH2CH2COOH、もしくはNHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表すか、または
・Xが、式Icのリンカーを表し、
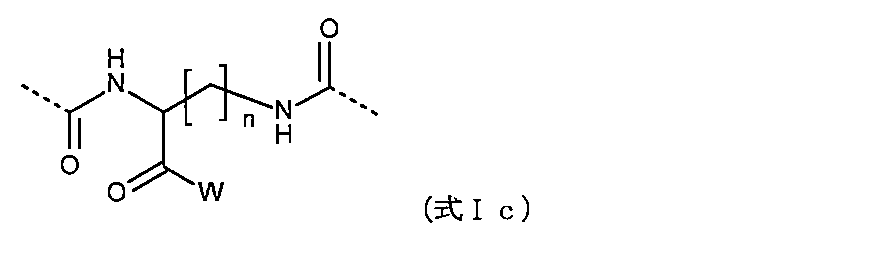
式中、
----が、R1もしくはR2に対する共有結合を表し、
nは、1~4の範囲の整数を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表すか、または
・式中、Xが、式Idのリンカーを表し、

式中、
----が、R1もしくはR2に対する共有結合を表し、
R4が、-(C=O)(CH2)p(C=O)-Wを表し、
式中、pが、1~4の範囲の整数を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表すか、または
・Xが、式Ieのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
・Xが、式Ifのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
・Xが、式Igのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
・Xが、式Ihのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表すか、または
・Xが、式Iiのリンカーを表し、

----が、R1もしくはR2に対する共有結合を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、もしくは-NHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1~4つのYが、Hを表し、
Yが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3ではないか、または、Yのうちの1もしくは2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3を表し、
1つのYが、式IのXに対する付着点(を表す共有結合)を表し、
Xが式Ie、If、Ig、またはIhであるとき、R1またはR2のいずれかにおける1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表す、ジボロン化合物。 - 式Iによって表される、請求項1に記載のジボロン化合物であって、式中、Xが、式Iaのリンカーを表し、

----が、R1またはR2に対する共有結合を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IaのリンカーXに対する付着点(を表す共有結合)を表し、
Yが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3ではないか、または、Yのうちの1もしくは2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。 - 式Iによって表される、請求項1に記載のジボロン化合物であって、式中、Xが、式Ibのリンカーを表し、

----が、R1またはR2に対する共有結合を表し、
R3が、-(CH2)m(C=O)-Wを表し、
式中、mが、1~4の範囲の整数を表し、
Wが、OH、NHCH2COOH、NHCH2CH2COOH、NHCH2CH2CH2COOH、またはNHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IbのリンカーXに対する付着点(を表す共有結合)を表し、
Yが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3ではないか、または、Yのうちの1もしくは2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。 - 式Iによって表される、請求項1に記載のジボロン化合物であって、式中、Xが、式Icのリンカーを表し、

式中、
----が、R1またはR2に対する共有結合を表し、
nは、1~4の範囲の整数を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IcのリンカーXに対する付着点(を表す共有結合)を表し、
Yが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3ではないか、または、Yのうちの1もしくは2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。 - 式Iによって表される、請求項1に記載のジボロン化合物であって、式中、Xが、式Idのリンカーを表し、

式中、
----が、R1またはR2に対する共有結合を表し、
R4が、-(C=O)(CH2)p(C=O)-Wを表し、
式中、pが、1~4の範囲の整数を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IdのリンカーXに対する付着点(を表す共有結合)を表し、
Yが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3ではないか、または、Yのうちの1もしくは2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。 - 式Iによって表される、請求項1に記載のジボロン化合物であって、式中、Xが、式Ieのリンカーを表し、

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Ieのリンカー-CO-に対する付着点(を表す共有結合)を表し、
R1またはR2のいずれかにおける1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
Yが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3ではないか、または、Yのうちの1もしくは2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3を表し、
Yの残りが、Hを表す、請求項1に記載のジボロン化合物。 - 式Iによって表される、請求項1に記載のジボロン化合物であって、式中、Xが、式Ifのリンカーを表し、

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Ifのリンカー-SO-に対する付着点(を表す共有結合)を表し、
R1またはR2のいずれかにおける1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
Yが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3ではないか、または、Yのうちの1もしくは2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。 - 式Iによって表される、請求項1に記載のジボロン化合物であって、式中、Xが、式Igのリンカーを表し、

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、
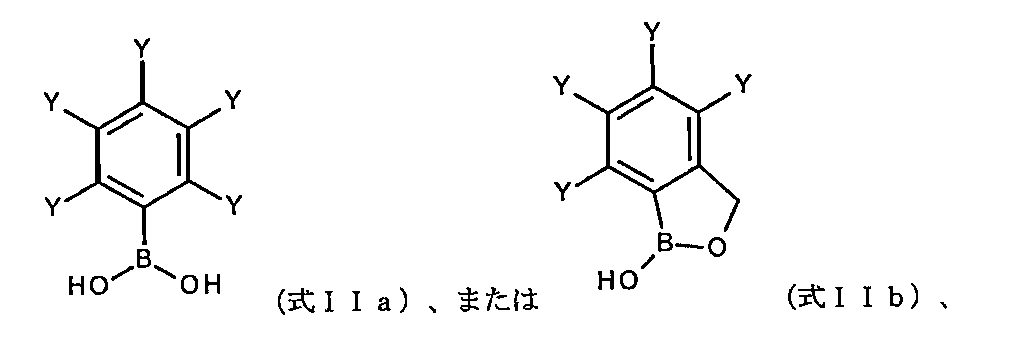
1つのYが、式Igのリンカー-(SO2)-に対する付着点(を表す共有結合)を表し、
R1またはR2のいずれかにおける1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
Yが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3ではないか、または、Yのうちの1もしくは2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。 - 式Iによって表される、請求項1に記載のジボロン化合物であって、式中、Xが、式Ihのリンカーを表し、

----が、R1またはR2に対する共有結合を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式Ihのリンカー-(CF2)-に対する付着点(を表す共有結合)を表し、
R1またはR2のいずれかにおける1つのYが、-(C=O)-Wを表し、式中、Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
Yが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3ではないか、または、Yのうちの1もしくは2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。 - 式Iによって表される、請求項1に記載のジボロン化合物であって、式中、
Xが、式Iiのリンカーを表し、

----が、R1またはR2に対する共有結合を表し、
Wが、-OH、-NHCH2COOH、-NHCH2CH2COOH、-NHCH2CH2CH2COOH、または-NHCH(COOH)CH2CH2COOH(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)を表し、
R1およびR2が、同一であっても異なっていてもよく、各々が式IIaまたはIIbの基を表し、

1つのYが、式IiのリンカーXに対する付着点(を表す共有結合)を表し、
Yが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3ではないか、または、Yのうちの1もしくは2つが、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3を表し、
残りのYが、Hを表す、請求項1に記載のジボロン化合物。 - 3,5-ビス((4-ボロノ-2-フルオロベンズアミド)メチル)安息香酸、
3,5-ビス((4-ボロノ-3-フルオロベンズアミド)メチル)安息香酸、
N,N’-ビス(4-ボロノ-3-フルオロベンズアミド)-N-エチル-グリシンアミド、
(S)-2,4-ビス(4-ボロノ-3-フルオロベンズアミド)ブタン酸、
N-(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、
3,5-ビス((7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)メチル)安息香酸、
N-(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、
N-(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、
N-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボニル)-N-(2-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボキサミド)エチル)グリシン、
N2,N6-ビス(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-5-カルボニル)-L-リジン、
3-ボロノ-5-((3-ボロノ-4-フルオロフェニル)スルホニル)-4-フルオロ安息香酸、
3-ボロノ-5-(3-ボロノ-5-フルオロベンゾイル)安息香酸、
3-ボロノ-5-(5-ボロノ-2,4-ジフルオロベンゾイル)安息香酸、
N6-(4-ボロノ-2-フルオロベンゾイル)-N2-(6-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]-オキサボロール-5-カルボニル)-L-リジン、
(S)-2,3-ビス(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-プロパン酸、
(S)-2,3-ビス(5-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-プロパン酸、
(S)-2,3-ビス(4-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-プロパン酸、
(3-ボロノ-5-((3-ボロノ-5-(トリフルオロメチル)フェニル)ジフルオロメチル)ベンゾイル)グリシン、
(3-ボロノ-5-(3-ボロノ-5-(トリフルオロメチル)ベンゾイル)ベンゾイル)グリシン、
(3-ボロノ-5-((3-ボロノ-5-(トリフルオロメチル)フェニル)スルホニル)グリシン、
4-((3S,4S)-3,4-ビス(7-フルオロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)ピロリジン-1-イル)-4-オキソブタン酸、
(3-ボロノ-5-(3-ボロノ-5-フルオロベンゾイル)ベンゾイル)グリシン、
(3-ボロノ-5-((3-ボロノ-5-フルオロフェニル)スルホニル)ベンゾイル)グリシン、
N-(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-エチル)グリシン、
(S)-2,3-ビス(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)プロパン酸、
4-((3S,4S)-3,4-ビス(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)ピロリジン-1-イル)-4-オキソブタン酸、
(3-ボロノ-5-((3-ボロノ-5-(ペンタフルオロ-λ6-スルファニル)フェニル)スルホニル)ベンゾイル)グリシン、
2-((ビス(3-ボロノ-5-(トリフルオロメチル)フェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸、
N-(4-(ジフルオロメチル)-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(4-(ジフルオロメチル)-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-エチル)グリシン、
N-(4-クロロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(4-クロロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、
3-(2,3-ビス(4-クロロ-1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)プロパンアミド)プロパン酸、
2-((ビス(3-ボロノ-5-(ジフルオロメチル)フェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸、
2-((ビス(3-ボロノ-5-クロロフェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸、
2-((ビス(3-ボロノフェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸、
N-(1-ヒドロキシ-5-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(1-ヒドロキシ-5-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-エチル)グリシン、
2-((ビス(1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-イル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸、
(3-ボロノ-5-((3-ボロノフェニル)スルホニル)ベンゾイル)グリシン、
(S)-2,3-ビス(1-ヒドロキシ-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)プロパン酸、
N-(1-ヒドロキシ-4-(メチルスルホニル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(1-ヒドロキシ-4-(メチルスルホニル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボキサミド)-エチル)グリシン、
N-(1-ヒドロキシ-4-((トリフルオロメチル)スルホニル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボニル)-N-(2-(1-ヒドロキシ-4-((トリフルオロメチル)スルホニル)-1,3-ジヒドロベンゾ-[c][1,2]オキサボロール-6-カルボキサミド)エチル)グリシン、
4-ボロノ-2-((3-ボロノ-5-(トリフルオロメチル)フェニル)スルホニル)-6-(トリフルオロメチル)安息香酸、および
2-((ビス(3-ボロノ-5-(トリフルオロメトキシ)フェニル)(オキソ)-λ6-スルファニリデン)アミノ)酢酸からなる群から選択される、請求項1に記載のジボロン化合物。 - 請求項1~11のいずれか一項に記載のジボロン化合物が、任意のリンカーを介して原薬にコンジュゲートしている、ジボロンコンジュゲート。
- 前記ジボロン化合物が、前記ジボロン化合物のカルボン酸部分の誘導体化によって、任意のリンカーを介して前記原薬にコンジュゲートしている、請求項12に記載のジボロンコンジュゲート。
- 一般式I’:
R1’-X’-R2’
によって表される、ジボロンコンジュゲートであって、
・式I’中、
・X’が、式Ia’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表し、
Dが、原薬を表し、
W’が、共有結合、もしくは-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表すか、または
・X’が、式Ib’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表し、
R3’が、-(CH2)m’(C=O)-W’-Dを表し、式中、
m’が、1~4の範囲の整数を表し、
W’が、共有結合、もしくは-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、
Dが、原薬を表すか、または
・X’が、式Ic’のリンカーを表し、

式中、
----が、R1’もしくはR2’に対する共有結合を表し、
nが、1~4の範囲の整数を表し、
W’が、共有結合、もしくは-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、
Dが、原薬を表すか、または
・X’が、式Id’のリンカーを表し、

式中、
----が、R1’もしくはR2’に対する共有結合を表し、
R4’が、-(C=O)(CH2)p’(C=O)-W’-Dを表し、式中、
W’が、共有結合、もしくは-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、
式中、p’が、1~4の範囲の整数を表し、
Dが、原薬を表すか、または
・X’が、式Ie’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表すか、または
・X’が、式If’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表すか、または
・X’が、式Ig’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表すか、または
・X’が、式Ih’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表すか、または
・X’が、式Ii’のリンカーを表し、

----が、R1’もしくはR2’に対する共有結合を表し、
Dが、原薬を表し、
W’が、共有結合、もしくは-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、
R1’およびR2’が、同一であっても異なっていてもよく、各々が式IIa’または式IIb’の基を表し、

1~4つのY’が、Hを表し、
Y’が、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3ではないか、または、1もしくは2つのY’が、F、Cl、CF2、CF3、SF5、OCF3、SO2CH3、および/もしくはSO2CF3を表し、
1つのY’が、式I’のX’に対する付着点(を表す共有結合)を表し、
・X’が、式Ie’、If’、Ig’、またはIh’であるとき、R1’またはR2’のいずれかにおける1つのYが、-(C=O)-W’-Dを表し、式中、W’が、共有結合、もしくは-NHCH2(C=O)-、-NHCH2CH2(C=O)-、-NHCH2CH2CH2(C=O)-、および-NHCH(COOH)CH2CH2(C=O)-(前記基は、L-ガンマ-GluもしくはD-ガンマ-Glu残基を表す)からなる群から選択されるリンカーを表し、Dが、原薬を表す、
ジボロンコンジュゲート。 - 請求項12~14のいずれか一項に記載のジボロンコンジュゲートの製造のための中間化合物として使用するための、請求項1~11のいずれか一項に記載のジボロン化合物。
- 請求項12~14のいずれか一項に記載のジボロンコンジュゲートを含む、医薬組成物。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023051919A JP7622123B2 (ja) | 2017-11-09 | 2023-03-28 | ジボロン化合物および糖尿病関連ペプチドまたはタンパク質を含むジボロンコンジュゲートならびに1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボン酸 |
| JP2025005534A JP2025061325A (ja) | 2017-11-09 | 2025-01-15 | グルコース感受性アルブミン結合誘導体 |
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17200734.6 | 2017-11-09 | ||
| EP17200734 | 2017-11-09 | ||
| EP18178294.7 | 2018-06-18 | ||
| EP18178294 | 2018-06-18 | ||
| PCT/EP2018/080650 WO2019092125A1 (en) | 2017-11-09 | 2018-11-08 | Glucose-sensitive albumin-binding derivatives |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023051919A Division JP7622123B2 (ja) | 2017-11-09 | 2023-03-28 | ジボロン化合物および糖尿病関連ペプチドまたはタンパク質を含むジボロンコンジュゲートならびに1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボン酸 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2021502372A JP2021502372A (ja) | 2021-01-28 |
| JP2021502372A5 JP2021502372A5 (ja) | 2021-11-18 |
| JP7254792B2 true JP7254792B2 (ja) | 2023-04-10 |
Family
ID=64049277
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020525911A Active JP7254792B2 (ja) | 2017-11-09 | 2018-11-08 | グルコース感受性アルブミン結合誘導体 |
| JP2023051919A Active JP7622123B2 (ja) | 2017-11-09 | 2023-03-28 | ジボロン化合物および糖尿病関連ペプチドまたはタンパク質を含むジボロンコンジュゲートならびに1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボン酸 |
| JP2025005534A Pending JP2025061325A (ja) | 2017-11-09 | 2025-01-15 | グルコース感受性アルブミン結合誘導体 |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023051919A Active JP7622123B2 (ja) | 2017-11-09 | 2023-03-28 | ジボロン化合物および糖尿病関連ペプチドまたはタンパク質を含むジボロンコンジュゲートならびに1-ヒドロキシ-4-(トリフルオロメチル)-1,3-ジヒドロベンゾ[c][1,2]オキサボロール-6-カルボン酸 |
| JP2025005534A Pending JP2025061325A (ja) | 2017-11-09 | 2025-01-15 | グルコース感受性アルブミン結合誘導体 |
Country Status (7)
| Country | Link |
|---|---|
| US (4) | US11186595B2 (ja) |
| EP (2) | EP4227313B1 (ja) |
| JP (3) | JP7254792B2 (ja) |
| CN (2) | CN111315751B (ja) |
| ES (1) | ES2956032T3 (ja) |
| MA (2) | MA71363A (ja) |
| WO (1) | WO2019092125A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2025061325A (ja) * | 2017-11-09 | 2025-04-10 | ノヴォ ノルディスク アー/エス | グルコース感受性アルブミン結合誘導体 |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016179568A1 (en) | 2015-05-06 | 2016-11-10 | Protomer Technologies, Inc. | Glucose responsive insulins |
| BR112021016782A2 (pt) * | 2019-03-29 | 2021-11-30 | Novo Nordisk As | Composto, composição, e, método para o tratamento ou prevenção de diabetes, diabetes do tipo 1, diabetes do tipo 2, tolerância à glicose comprometida, hiperglicemia e síndrome metabólica |
| CN112174989B (zh) * | 2019-07-02 | 2023-06-20 | 江西同和药业股份有限公司 | 一种克立硼罗的制备方法 |
| WO2021022116A1 (en) * | 2019-07-31 | 2021-02-04 | Thermalin Inc. | Insulin analogues with glucose regulated conformational switch |
| BR112022019687A2 (pt) * | 2020-03-31 | 2022-12-20 | Protomer Tech Inc | Conjugados para responsividade seletiva a dióis vicinais |
| CA3198757A1 (en) * | 2020-11-19 | 2022-05-27 | Protomer Technologies Inc. | Aromatic boron-containing compounds and insulin analogs |
| US20240000897A1 (en) * | 2020-12-02 | 2024-01-04 | The Regents Of The University Of California | Injectable biodegradable polymeric complex for glucose-responsive insulin delivery |
| WO2022235691A1 (en) * | 2021-05-03 | 2022-11-10 | The Trustees Of Indiana University | Molecular design of glucose sensors in glucose-responsive insulin analogues |
| CN114478597B (zh) * | 2021-12-29 | 2023-08-29 | 宁波大学 | 一种快速识别葡萄糖手性的试剂及其制备方法和应用 |
| PE20251287A1 (es) | 2022-05-18 | 2025-05-14 | Protomer Tech Inc | Compuestos aromaticos que contienen boro y analogos de insulina relacionados |
| PE20252681A1 (es) * | 2023-04-11 | 2025-11-24 | Protomer Tech Inc | Compuestos que contienen uno o mas diboronatos y analogos de insulina relacionados |
| AR133270A1 (es) | 2023-07-18 | 2025-09-10 | Novo Nordisk As | Composiciones farmacéuticas de derivados de la insulina |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003535106A (ja) | 2000-06-02 | 2003-11-25 | ノボ ノルディスク アクティーゼルスカブ | グルコース検知性インスリン誘導体からの、インスリンのグルコース依存性放出 |
| US20150320837A1 (en) | 2012-12-12 | 2015-11-12 | Massachusetts Institute Of Technology | Insulin derivatives for diabetes treatment |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU628674B2 (en) | 1989-10-19 | 1992-09-17 | Nippon Oil And Fats Company, Limited | Polymer complexes of a sugar response type |
| US7316999B2 (en) | 2000-06-02 | 2008-01-08 | Novo Nordisk A/S | Glucose dependent release of insulin from glucose sensing insulin derivatives |
| US7317000B2 (en) | 2001-12-02 | 2008-01-08 | Novo Nordisk A/S | Glucose-dependent insulins |
| AU2002349295A1 (en) | 2001-12-02 | 2003-06-17 | Novo Nordisk A/S | Glucose dependant release of insulin from glucose sensing insulin derivatives |
| AU2003223930A1 (en) | 2002-06-14 | 2003-12-31 | Novo Nordisk A/S | Pharmaceutical use of boronic acids and esters thereof |
| WO2009112583A2 (en) | 2008-03-14 | 2009-09-17 | Novo Nordisk A/S | Protease-stabilized insulin analogues |
| DE102008052314A1 (de) * | 2008-10-15 | 2010-04-22 | Syntatec Chemicals Gmbh | Aromatische und heteroaromatische Poly-trifluoroborate und Verfahren zur Herstellung |
| ES2611040T3 (es) | 2009-06-30 | 2017-05-04 | Novo Nordisk A/S | Derivados de insulina |
| TWI458732B (zh) * | 2012-06-27 | 2014-11-01 | 國立交通大學 | 具硼酸基團連接子及含有其之生物感測元件 |
| US20170000133A1 (en) | 2013-12-23 | 2017-01-05 | Syngenta Participations Ag | Benzoxaborole fungicides |
| WO2015106292A1 (en) * | 2014-01-13 | 2015-07-16 | Coferon, Inc. | Bcr-abl tyrosine-kinase ligands capable of dimerizing in an aqueous solution, and methods of using same |
| AR103397A1 (es) | 2015-01-13 | 2017-05-10 | Syngenta Participations Ag | Oxoborazoles microbicidas |
| BR112017019515A2 (pt) | 2015-03-13 | 2018-04-24 | Case Western Reserve University | análogo de insulina, composição farmacêutica e método de tratamento de um paciente |
| WO2016179568A1 (en) | 2015-05-06 | 2016-11-10 | Protomer Technologies, Inc. | Glucose responsive insulins |
| PE20190119A1 (es) | 2016-05-12 | 2019-01-17 | Anacor Pharmaceuticals Inc | Esteres de oxaborol y sus usos |
| US10690675B2 (en) * | 2017-04-14 | 2020-06-23 | Georgia Tech Research Corporation | Methods for enriching glycopeptides for global analysis of glycoproteins |
| CN111315751B (zh) * | 2017-11-09 | 2023-12-12 | 诺和诺德股份有限公司 | 葡萄糖敏感性白蛋白结合衍生物 |
| US20210214412A1 (en) | 2018-04-16 | 2021-07-15 | University Of Utah Research Foundation | Glucose-responsive insulin |
-
2018
- 2018-11-08 CN CN201880072571.2A patent/CN111315751B/zh active Active
- 2018-11-08 JP JP2020525911A patent/JP7254792B2/ja active Active
- 2018-11-08 MA MA71363A patent/MA71363A/fr unknown
- 2018-11-08 US US16/759,378 patent/US11186595B2/en active Active
- 2018-11-08 EP EP23172873.4A patent/EP4227313B1/en active Active
- 2018-11-08 ES ES18795692T patent/ES2956032T3/es active Active
- 2018-11-08 CN CN202311566982.6A patent/CN117624207A/zh active Pending
- 2018-11-08 EP EP18795692.5A patent/EP3707145B1/en active Active
- 2018-11-08 MA MA050552A patent/MA50552A/fr unknown
- 2018-11-08 WO PCT/EP2018/080650 patent/WO2019092125A1/en not_active Ceased
-
2021
- 2021-10-13 US US17/500,242 patent/US11767332B2/en active Active
-
2023
- 2023-03-28 JP JP2023051919A patent/JP7622123B2/ja active Active
- 2023-06-16 US US18/210,763 patent/US12227528B2/en active Active
-
2024
- 2024-12-18 US US18/985,160 patent/US20250122226A1/en active Pending
-
2025
- 2025-01-15 JP JP2025005534A patent/JP2025061325A/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003535106A (ja) | 2000-06-02 | 2003-11-25 | ノボ ノルディスク アクティーゼルスカブ | グルコース検知性インスリン誘導体からの、インスリンのグルコース依存性放出 |
| US20150320837A1 (en) | 2012-12-12 | 2015-11-12 | Massachusetts Institute Of Technology | Insulin derivatives for diabetes treatment |
Non-Patent Citations (1)
| Title |
|---|
| Hoeg-Jensen, Thomas,Preparationand screening of diboronate arrays for identification of carbohydrate binders,QSAR &Combinatorial Science,2004年,Vol.23, No.5,pp.344-351 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2025061325A (ja) * | 2017-11-09 | 2025-04-10 | ノヴォ ノルディスク アー/エス | グルコース感受性アルブミン結合誘導体 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3707145A1 (en) | 2020-09-16 |
| JP2023080126A (ja) | 2023-06-08 |
| JP7622123B2 (ja) | 2025-01-27 |
| CN111315751A (zh) | 2020-06-19 |
| EP4227313C0 (en) | 2025-10-22 |
| JP2021502372A (ja) | 2021-01-28 |
| JP2025061325A (ja) | 2025-04-10 |
| CN111315751B (zh) | 2023-12-12 |
| WO2019092125A1 (en) | 2019-05-16 |
| US11767332B2 (en) | 2023-09-26 |
| US20220081451A1 (en) | 2022-03-17 |
| MA50552A (fr) | 2020-09-16 |
| CN117624207A (zh) | 2024-03-01 |
| ES2956032T3 (es) | 2023-12-12 |
| US20250122226A1 (en) | 2025-04-17 |
| EP3707145B1 (en) | 2023-06-21 |
| EP4227313A1 (en) | 2023-08-16 |
| US11186595B2 (en) | 2021-11-30 |
| US12227528B2 (en) | 2025-02-18 |
| EP4227313B1 (en) | 2025-10-22 |
| US20230331745A1 (en) | 2023-10-19 |
| MA71363A (fr) | 2025-04-30 |
| US20200325160A1 (en) | 2020-10-15 |
| EP3707145C0 (en) | 2023-06-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7254792B2 (ja) | グルコース感受性アルブミン結合誘導体 | |
| JP7640462B2 (ja) | グルコース感受性インスリン誘導体 | |
| CN113164763A (zh) | Nlrp3抑制剂 | |
| CN113474338A (zh) | 吡嗪类衍生物及其在抑制shp2中的应用 | |
| EA027120B1 (ru) | Противофиброзные пиридиноны | |
| US20200095217A1 (en) | Hydroxy formamide derivatives and their use | |
| CN113423691A (zh) | 肽结合剂 | |
| JP2005504040A (ja) | カテプシンK阻害剤としてのα−ケトアミド誘導体 | |
| AU2020360342A1 (en) | Sulfo-substituted biaryl compound or salt thereof, preparation method therefor, and use thereof | |
| CN102131802A (zh) | 用作凝血酶抑制剂的新型杂环甲酰胺 | |
| AU2014354488B2 (en) | Amide derivatives for GPR119 agonist | |
| EP2470514B1 (fr) | Pseudo-dipeptides comme inhibiteurs de mmp | |
| EP4545517A1 (en) | Nav1.8 inhibitor | |
| HK40057173A (en) | Nlrp3 inhibitors | |
| HK40061204A (en) | Pyrazine derivative and application thereof in inhibiting shp2 | |
| HK1246280A1 (en) | Pyridine derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20211004 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20211004 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20220915 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220920 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20221215 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20230227 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20230329 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7254792 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |