JP6882981B2 - Rpe細胞集団およびそれを作製する方法 - Google Patents
Rpe細胞集団およびそれを作製する方法 Download PDFInfo
- Publication number
- JP6882981B2 JP6882981B2 JP2017535834A JP2017535834A JP6882981B2 JP 6882981 B2 JP6882981 B2 JP 6882981B2 JP 2017535834 A JP2017535834 A JP 2017535834A JP 2017535834 A JP2017535834 A JP 2017535834A JP 6882981 B2 JP6882981 B2 JP 6882981B2
- Authority
- JP
- Japan
- Prior art keywords
- cells
- rpe
- cell
- human
- differentiated
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 210000004027 cell Anatomy 0.000 claims description 705
- 238000000034 method Methods 0.000 claims description 120
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 claims description 96
- 241000282414 Homo sapiens Species 0.000 claims description 95
- 102000004887 Transforming Growth Factor beta Human genes 0.000 claims description 56
- 108090001012 Transforming Growth Factor beta Proteins 0.000 claims description 56
- 235000005152 nicotinamide Nutrition 0.000 claims description 51
- 210000001671 embryonic stem cell Anatomy 0.000 claims description 46
- 239000011570 nicotinamide Substances 0.000 claims description 45
- 229960003966 nicotinamide Drugs 0.000 claims description 45
- 108010023082 activin A Proteins 0.000 claims description 39
- 238000012258 culturing Methods 0.000 claims description 39
- 239000000126 substance Substances 0.000 claims description 23
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 20
- 239000001301 oxygen Substances 0.000 claims description 20
- 229910052760 oxygen Inorganic materials 0.000 claims description 20
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 claims description 19
- 210000001778 pluripotent stem cell Anatomy 0.000 claims description 17
- 102100024785 Fibroblast growth factor 2 Human genes 0.000 claims description 14
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 claims description 14
- 239000000463 material Substances 0.000 claims description 14
- 230000001464 adherent effect Effects 0.000 claims description 13
- 210000001316 polygonal cell Anatomy 0.000 claims description 9
- 239000000790 retinal pigment Substances 0.000 claims description 4
- 210000003583 retinal pigment epithelium Anatomy 0.000 description 287
- 230000028327 secretion Effects 0.000 description 103
- 102100028001 Retinaldehyde-binding protein 1 Human genes 0.000 description 86
- 101710101931 Retinaldehyde-binding protein 1 Proteins 0.000 description 86
- 230000004069 differentiation Effects 0.000 description 74
- 238000012369 In process control Methods 0.000 description 66
- 238000004519 manufacturing process Methods 0.000 description 65
- 238000010965 in-process control Methods 0.000 description 63
- 238000012360 testing method Methods 0.000 description 63
- 210000001508 eye Anatomy 0.000 description 57
- 239000002609 medium Substances 0.000 description 55
- 241001465754 Metazoa Species 0.000 description 47
- 102000005754 Cytokine Receptor gp130 Human genes 0.000 description 44
- 108010006197 Cytokine Receptor gp130 Proteins 0.000 description 44
- 102100026262 Metalloproteinase inhibitor 2 Human genes 0.000 description 41
- 108010031372 Tissue Inhibitor of Metalloproteinase-2 Proteins 0.000 description 40
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 39
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 39
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 39
- 210000004694 pigment cell Anatomy 0.000 description 38
- 102100022987 Angiogenin Human genes 0.000 description 37
- 108010072788 angiogenin Proteins 0.000 description 37
- 230000014509 gene expression Effects 0.000 description 30
- 238000002347 injection Methods 0.000 description 28
- 239000007924 injection Substances 0.000 description 28
- 229920001184 polypeptide Polymers 0.000 description 28
- 102000004196 processed proteins & peptides Human genes 0.000 description 28
- 108090000765 processed proteins & peptides Proteins 0.000 description 28
- 108091008695 photoreceptors Proteins 0.000 description 27
- 108090000623 proteins and genes Proteins 0.000 description 23
- 238000003556 assay Methods 0.000 description 22
- 210000000130 stem cell Anatomy 0.000 description 22
- 238000005138 cryopreservation Methods 0.000 description 20
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 20
- 210000001519 tissue Anatomy 0.000 description 20
- 238000002054 transplantation Methods 0.000 description 20
- 238000002965 ELISA Methods 0.000 description 19
- 102000007354 PAX6 Transcription Factor Human genes 0.000 description 19
- 230000001965 increasing effect Effects 0.000 description 19
- 102000004169 proteins and genes Human genes 0.000 description 19
- 210000002950 fibroblast Anatomy 0.000 description 18
- 102100022430 Melanocyte protein PMEL Human genes 0.000 description 17
- 108010032788 PAX6 Transcription Factor Proteins 0.000 description 17
- 241000700159 Rattus Species 0.000 description 17
- 238000012744 immunostaining Methods 0.000 description 17
- 239000006228 supernatant Substances 0.000 description 17
- 102100035423 POU domain, class 5, transcription factor 1 Human genes 0.000 description 16
- 101710126211 POU domain, class 5, transcription factor 1 Proteins 0.000 description 16
- 239000013642 negative control Substances 0.000 description 16
- 210000001525 retina Anatomy 0.000 description 16
- 239000000523 sample Substances 0.000 description 16
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- 238000004113 cell culture Methods 0.000 description 15
- 210000002966 serum Anatomy 0.000 description 15
- 210000002459 blastocyst Anatomy 0.000 description 14
- 239000003550 marker Substances 0.000 description 14
- 230000002207 retinal effect Effects 0.000 description 14
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 13
- 239000000853 adhesive Substances 0.000 description 13
- 230000001070 adhesive effect Effects 0.000 description 13
- 238000004458 analytical method Methods 0.000 description 13
- 238000011534 incubation Methods 0.000 description 13
- 239000000203 mixture Substances 0.000 description 13
- 239000013641 positive control Substances 0.000 description 13
- 235000020945 retinal Nutrition 0.000 description 13
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 12
- 230000004888 barrier function Effects 0.000 description 12
- 238000011156 evaluation Methods 0.000 description 12
- 238000011194 good manufacturing practice Methods 0.000 description 12
- 239000010410 layer Substances 0.000 description 12
- 239000002356 single layer Substances 0.000 description 12
- 101100517196 Arabidopsis thaliana NRPE1 gene Proteins 0.000 description 11
- 101100190825 Bos taurus PMEL gene Proteins 0.000 description 11
- 102100033356 Lecithin retinol acyltransferase Human genes 0.000 description 11
- 102100030173 Muellerian-inhibiting factor Human genes 0.000 description 11
- 101100073341 Oryza sativa subsp. japonica KAO gene Proteins 0.000 description 11
- 239000003814 drug Substances 0.000 description 11
- 230000012010 growth Effects 0.000 description 11
- 239000003102 growth factor Substances 0.000 description 11
- 108010084957 lecithin-retinol acyltransferase Proteins 0.000 description 11
- 208000002780 macular degeneration Diseases 0.000 description 11
- 239000000049 pigment Substances 0.000 description 11
- 239000011604 retinal Substances 0.000 description 11
- 101150005492 rpe1 gene Proteins 0.000 description 11
- 239000000758 substrate Substances 0.000 description 11
- 230000004083 survival effect Effects 0.000 description 11
- NCYCYZXNIZJOKI-UHFFFAOYSA-N vitamin A aldehyde Natural products O=CC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C NCYCYZXNIZJOKI-UHFFFAOYSA-N 0.000 description 11
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- 101000620359 Homo sapiens Melanocyte protein PMEL Proteins 0.000 description 10
- 101000670189 Homo sapiens Ribulose-phosphate 3-epimerase Proteins 0.000 description 10
- 241000699666 Mus <mouse, genus> Species 0.000 description 10
- 239000008186 active pharmaceutical agent Substances 0.000 description 10
- 229940088679 drug related substance Drugs 0.000 description 10
- 230000006870 function Effects 0.000 description 10
- 238000010166 immunofluorescence Methods 0.000 description 10
- 239000012535 impurity Substances 0.000 description 10
- 150000005480 nicotinamides Chemical class 0.000 description 10
- 239000012071 phase Substances 0.000 description 10
- NCYCYZXNIZJOKI-IOUUIBBYSA-N 11-cis-retinal Chemical compound O=C/C=C(\C)/C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C NCYCYZXNIZJOKI-IOUUIBBYSA-N 0.000 description 9
- 108050002823 Bestrophin Proteins 0.000 description 9
- 108050003623 Bestrophin-1 Proteins 0.000 description 9
- 101001023986 Homo sapiens Growth/differentiation factor 3 Proteins 0.000 description 9
- 108060004872 MIF Proteins 0.000 description 9
- 201000007737 Retinal degeneration Diseases 0.000 description 9
- 102000004330 Rhodopsin Human genes 0.000 description 9
- 108090000820 Rhodopsin Proteins 0.000 description 9
- 230000001605 fetal effect Effects 0.000 description 9
- 238000009472 formulation Methods 0.000 description 9
- 230000002062 proliferating effect Effects 0.000 description 9
- 238000003908 quality control method Methods 0.000 description 9
- 238000010186 staining Methods 0.000 description 9
- 230000001225 therapeutic effect Effects 0.000 description 9
- 102000012304 Bestrophin Human genes 0.000 description 8
- 108020004414 DNA Proteins 0.000 description 8
- 230000005856 abnormality Effects 0.000 description 8
- 206010064930 age-related macular degeneration Diseases 0.000 description 8
- 239000000090 biomarker Substances 0.000 description 8
- 239000006285 cell suspension Substances 0.000 description 8
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 8
- 230000036512 infertility Effects 0.000 description 8
- 230000035755 proliferation Effects 0.000 description 8
- 229940124597 therapeutic agent Drugs 0.000 description 8
- 102100035364 Growth/differentiation factor 3 Human genes 0.000 description 7
- 108010085895 Laminin Proteins 0.000 description 7
- 102000007547 Laminin Human genes 0.000 description 7
- 101710130208 Melanocyte protein PMEL Proteins 0.000 description 7
- 102100039364 Metalloproteinase inhibitor 1 Human genes 0.000 description 7
- 241000699670 Mus sp. Species 0.000 description 7
- 108010031374 Tissue Inhibitor of Metalloproteinase-1 Proteins 0.000 description 7
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 7
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 7
- 239000003855 balanced salt solution Substances 0.000 description 7
- 210000004556 brain Anatomy 0.000 description 7
- 150000001875 compounds Chemical class 0.000 description 7
- 238000012137 double-staining Methods 0.000 description 7
- 108010014606 glutathione-bicarbonate-Ringer solution Proteins 0.000 description 7
- 210000005260 human cell Anatomy 0.000 description 7
- 230000003902 lesion Effects 0.000 description 7
- 238000013411 master cell bank Methods 0.000 description 7
- 238000005259 measurement Methods 0.000 description 7
- 230000008569 process Effects 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- 238000003498 protein array Methods 0.000 description 7
- 230000004258 retinal degeneration Effects 0.000 description 7
- 210000000844 retinal pigment epithelial cell Anatomy 0.000 description 7
- 230000035899 viability Effects 0.000 description 7
- 230000000007 visual effect Effects 0.000 description 7
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 6
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- ZHAFUINZIZIXFC-UHFFFAOYSA-N [9-(dimethylamino)-10-methylbenzo[a]phenoxazin-5-ylidene]azanium;chloride Chemical compound [Cl-].O1C2=CC(=[NH2+])C3=CC=CC=C3C2=NC2=C1C=C(N(C)C)C(C)=C2 ZHAFUINZIZIXFC-UHFFFAOYSA-N 0.000 description 6
- 239000000488 activin Substances 0.000 description 6
- 230000006378 damage Effects 0.000 description 6
- 230000007423 decrease Effects 0.000 description 6
- 201000010099 disease Diseases 0.000 description 6
- 210000002744 extracellular matrix Anatomy 0.000 description 6
- 239000001963 growth medium Substances 0.000 description 6
- 238000000338 in vitro Methods 0.000 description 6
- 208000014674 injury Diseases 0.000 description 6
- 230000007774 longterm Effects 0.000 description 6
- 230000008929 regeneration Effects 0.000 description 6
- 238000011069 regeneration method Methods 0.000 description 6
- 230000003248 secreting effect Effects 0.000 description 6
- 210000000278 spinal cord Anatomy 0.000 description 6
- 239000003981 vehicle Substances 0.000 description 6
- 108010059616 Activins Proteins 0.000 description 5
- 108010010803 Gelatin Proteins 0.000 description 5
- 102100026818 Inhibin beta E chain Human genes 0.000 description 5
- 241000283973 Oryctolagus cuniculus Species 0.000 description 5
- 208000007014 Retinitis pigmentosa Diseases 0.000 description 5
- 208000027418 Wounds and injury Diseases 0.000 description 5
- 238000004115 adherent culture Methods 0.000 description 5
- 239000000427 antigen Substances 0.000 description 5
- 102000036639 antigens Human genes 0.000 description 5
- 108091007433 antigens Proteins 0.000 description 5
- 230000004663 cell proliferation Effects 0.000 description 5
- 230000004438 eyesight Effects 0.000 description 5
- 239000012634 fragment Substances 0.000 description 5
- 229920000159 gelatin Polymers 0.000 description 5
- 239000008273 gelatin Substances 0.000 description 5
- 235000019322 gelatine Nutrition 0.000 description 5
- 235000011852 gelatine desserts Nutrition 0.000 description 5
- 210000004263 induced pluripotent stem cell Anatomy 0.000 description 5
- 210000003734 kidney Anatomy 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- 230000000877 morphologic effect Effects 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- 210000000056 organ Anatomy 0.000 description 5
- 108091005709 peripheral membrane receptors Proteins 0.000 description 5
- 238000010257 thawing Methods 0.000 description 5
- 230000003442 weekly effect Effects 0.000 description 5
- 102000029816 Collagenase Human genes 0.000 description 4
- 108060005980 Collagenase Proteins 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- 206010021143 Hypoxia Diseases 0.000 description 4
- 239000012480 LAL reagent Substances 0.000 description 4
- 238000011529 RT qPCR Methods 0.000 description 4
- 208000017442 Retinal disease Diseases 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 230000003833 cell viability Effects 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- 229960002424 collagenase Drugs 0.000 description 4
- 239000000356 contaminant Substances 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 210000002257 embryonic structure Anatomy 0.000 description 4
- 229940088598 enzyme Drugs 0.000 description 4
- 210000002919 epithelial cell Anatomy 0.000 description 4
- 238000000684 flow cytometry Methods 0.000 description 4
- 239000012530 fluid Substances 0.000 description 4
- 210000002216 heart Anatomy 0.000 description 4
- 230000001146 hypoxic effect Effects 0.000 description 4
- 238000002513 implantation Methods 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 210000004185 liver Anatomy 0.000 description 4
- 210000004072 lung Anatomy 0.000 description 4
- 210000001165 lymph node Anatomy 0.000 description 4
- 210000002752 melanocyte Anatomy 0.000 description 4
- 230000001537 neural effect Effects 0.000 description 4
- 230000007170 pathology Effects 0.000 description 4
- 239000002243 precursor Substances 0.000 description 4
- 230000020509 sex determination Effects 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 210000000952 spleen Anatomy 0.000 description 4
- 230000008093 supporting effect Effects 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- GSCPDZHWVNUUFI-UHFFFAOYSA-N 3-aminobenzamide Chemical compound NC(=O)C1=CC=CC(N)=C1 GSCPDZHWVNUUFI-UHFFFAOYSA-N 0.000 description 3
- 102000003916 Arrestin Human genes 0.000 description 3
- 108090000328 Arrestin Proteins 0.000 description 3
- 241000283707 Capra Species 0.000 description 3
- 108010035532 Collagen Proteins 0.000 description 3
- 102000008186 Collagen Human genes 0.000 description 3
- 102000004127 Cytokines Human genes 0.000 description 3
- 108090000695 Cytokines Proteins 0.000 description 3
- 206010011906 Death Diseases 0.000 description 3
- 101000899111 Homo sapiens Hemoglobin subunit beta Proteins 0.000 description 3
- 108090001005 Interleukin-6 Proteins 0.000 description 3
- 102000004889 Interleukin-6 Human genes 0.000 description 3
- 241000204031 Mycoplasma Species 0.000 description 3
- 208000012641 Pigmentation disease Diseases 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 102000040945 Transcription factor Human genes 0.000 description 3
- 108091023040 Transcription factor Proteins 0.000 description 3
- 230000002159 abnormal effect Effects 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 239000006143 cell culture medium Substances 0.000 description 3
- 230000010261 cell growth Effects 0.000 description 3
- 239000002771 cell marker Substances 0.000 description 3
- 229920001436 collagen Polymers 0.000 description 3
- 230000002596 correlated effect Effects 0.000 description 3
- 210000004544 dc2 Anatomy 0.000 description 3
- 230000034994 death Effects 0.000 description 3
- 231100000517 death Toxicity 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 230000002950 deficient Effects 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 230000004720 fertilization Effects 0.000 description 3
- 238000003384 imaging method Methods 0.000 description 3
- 238000003125 immunofluorescent labeling Methods 0.000 description 3
- 238000011065 in-situ storage Methods 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- 238000004190 ion pair chromatography Methods 0.000 description 3
- -1 lecithin aldehyde Chemical class 0.000 description 3
- 238000012423 maintenance Methods 0.000 description 3
- 210000001161 mammalian embryo Anatomy 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 210000002780 melanosome Anatomy 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 238000010369 molecular cloning Methods 0.000 description 3
- 210000005157 neural retina Anatomy 0.000 description 3
- 208000015122 neurodegenerative disease Diseases 0.000 description 3
- 239000008194 pharmaceutical composition Substances 0.000 description 3
- 230000019612 pigmentation Effects 0.000 description 3
- 230000010287 polarization Effects 0.000 description 3
- 230000035935 pregnancy Effects 0.000 description 3
- 238000004886 process control Methods 0.000 description 3
- 238000011002 quantification Methods 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 230000004304 visual acuity Effects 0.000 description 3
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- 108010005853 Anti-Mullerian Hormone Proteins 0.000 description 2
- 208000037663 Best vitelliform macular dystrophy Diseases 0.000 description 2
- 108010007726 Bone Morphogenetic Proteins Proteins 0.000 description 2
- 102000007350 Bone Morphogenetic Proteins Human genes 0.000 description 2
- 102100021943 C-C motif chemokine 2 Human genes 0.000 description 2
- 101710155857 C-C motif chemokine 2 Proteins 0.000 description 2
- 102000014914 Carrier Proteins Human genes 0.000 description 2
- 208000002111 Eye Abnormalities Diseases 0.000 description 2
- 108010067306 Fibronectins Proteins 0.000 description 2
- 102000016359 Fibronectins Human genes 0.000 description 2
- XUMBMVFBXHLACL-UHFFFAOYSA-N Melanin Chemical compound O=C1C(=O)C(C2=CNC3=C(C(C(=O)C4=C32)=O)C)=C2C4=CNC2=C1C XUMBMVFBXHLACL-UHFFFAOYSA-N 0.000 description 2
- 108010050345 Microphthalmia-Associated Transcription Factor Proteins 0.000 description 2
- 238000011789 NOD SCID mouse Methods 0.000 description 2
- 108010035042 Osteoprotegerin Proteins 0.000 description 2
- 102000008108 Osteoprotegerin Human genes 0.000 description 2
- 101150081664 PAX6 gene Proteins 0.000 description 2
- 102000012338 Poly(ADP-ribose) Polymerases Human genes 0.000 description 2
- 108010061844 Poly(ADP-ribose) Polymerases Proteins 0.000 description 2
- 229920000776 Poly(Adenosine diphosphate-ribose) polymerase Polymers 0.000 description 2
- 229920001213 Polysorbate 20 Polymers 0.000 description 2
- 238000010240 RT-PCR analysis Methods 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- 206010038848 Retinal detachment Diseases 0.000 description 2
- 206010057430 Retinal injury Diseases 0.000 description 2
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 2
- DFPAKSUCGFBDDF-ZQBYOMGUSA-N [14c]-nicotinamide Chemical compound N[14C](=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-ZQBYOMGUSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 230000003698 anagen phase Effects 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 239000000868 anti-mullerian hormone Substances 0.000 description 2
- 230000003460 anti-nuclear Effects 0.000 description 2
- 238000003491 array Methods 0.000 description 2
- 230000037444 atrophy Effects 0.000 description 2
- 230000003416 augmentation Effects 0.000 description 2
- 238000011888 autopsy Methods 0.000 description 2
- 239000007640 basal medium Substances 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 108091008324 binding proteins Proteins 0.000 description 2
- 210000004155 blood-retinal barrier Anatomy 0.000 description 2
- 230000004378 blood-retinal barrier Effects 0.000 description 2
- 210000001185 bone marrow Anatomy 0.000 description 2
- 229940112869 bone morphogenetic protein Drugs 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 230000024245 cell differentiation Effects 0.000 description 2
- 238000007385 chemical modification Methods 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 210000003161 choroid Anatomy 0.000 description 2
- 230000004186 co-expression Effects 0.000 description 2
- 230000001427 coherent effect Effects 0.000 description 2
- 239000003636 conditioned culture medium Substances 0.000 description 2
- 239000012228 culture supernatant Substances 0.000 description 2
- 230000007850 degeneration Effects 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 239000003599 detergent Substances 0.000 description 2
- 230000001627 detrimental effect Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 239000000975 dye Substances 0.000 description 2
- 230000004064 dysfunction Effects 0.000 description 2
- 238000007667 floating Methods 0.000 description 2
- 238000007710 freezing Methods 0.000 description 2
- 230000008014 freezing Effects 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 230000007045 gastrulation Effects 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 238000013537 high throughput screening Methods 0.000 description 2
- 230000002519 immonomodulatory effect Effects 0.000 description 2
- 238000003018 immunoassay Methods 0.000 description 2
- 238000003364 immunohistochemistry Methods 0.000 description 2
- 238000010874 in vitro model Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 230000033001 locomotion Effects 0.000 description 2
- 108010082117 matrigel Proteins 0.000 description 2
- 230000035800 maturation Effects 0.000 description 2
- 102000006240 membrane receptors Human genes 0.000 description 2
- 108020004084 membrane receptors Proteins 0.000 description 2
- 238000010232 migration assay Methods 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 239000002547 new drug Substances 0.000 description 2
- 235000015097 nutrients Nutrition 0.000 description 2
- XXUPLYBCNPLTIW-UHFFFAOYSA-N octadec-7-ynoic acid Chemical compound CCCCCCCCCCC#CCCCCCC(O)=O XXUPLYBCNPLTIW-UHFFFAOYSA-N 0.000 description 2
- 210000001328 optic nerve Anatomy 0.000 description 2
- 210000001672 ovary Anatomy 0.000 description 2
- 230000003076 paracrine Effects 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 2
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 238000011552 rat model Methods 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000001850 reproductive effect Effects 0.000 description 2
- 230000004264 retinal detachment Effects 0.000 description 2
- 201000003849 retinal lattice degeneration Diseases 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 210000004927 skin cell Anatomy 0.000 description 2
- 238000012421 spiking Methods 0.000 description 2
- 230000002269 spontaneous effect Effects 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 210000000498 stratum granulosum Anatomy 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 238000001356 surgical procedure Methods 0.000 description 2
- 238000004114 suspension culture Methods 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 230000002123 temporal effect Effects 0.000 description 2
- 210000001550 testis Anatomy 0.000 description 2
- 210000001541 thymus gland Anatomy 0.000 description 2
- 210000001578 tight junction Anatomy 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 230000002110 toxicologic effect Effects 0.000 description 2
- 231100000027 toxicology Toxicity 0.000 description 2
- 210000003954 umbilical cord Anatomy 0.000 description 2
- 230000004382 visual function Effects 0.000 description 2
- 201000007790 vitelliform macular dystrophy Diseases 0.000 description 2
- 208000020938 vitelliform macular dystrophy 2 Diseases 0.000 description 2
- 210000004340 zona pellucida Anatomy 0.000 description 2
- CTLOSZHDGZLOQE-UHFFFAOYSA-N 14-methoxy-9-[(4-methylpiperazin-1-yl)methyl]-9,19-diazapentacyclo[10.7.0.02,6.07,11.013,18]nonadeca-1(12),2(6),7(11),13(18),14,16-hexaene-8,10-dione Chemical compound O=C1C2=C3C=4C(OC)=CC=CC=4NC3=C3CCCC3=C2C(=O)N1CN1CCN(C)CC1 CTLOSZHDGZLOQE-UHFFFAOYSA-N 0.000 description 1
- AJHPGXZOIAYYDW-UHFFFAOYSA-N 3-(2-cyanophenyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]propanoic acid Chemical compound CC(C)(C)OC(=O)NC(C(O)=O)CC1=CC=CC=C1C#N AJHPGXZOIAYYDW-UHFFFAOYSA-N 0.000 description 1
- MDOJTZQKHMAPBK-UHFFFAOYSA-N 4-iodo-3-nitrobenzamide Chemical compound NC(=O)C1=CC=C(I)C([N+]([O-])=O)=C1 MDOJTZQKHMAPBK-UHFFFAOYSA-N 0.000 description 1
- ZLWYEPMDOUQDBW-UHFFFAOYSA-N 6-aminonicotinamide Chemical compound NC(=O)C1=CC=C(N)N=C1 ZLWYEPMDOUQDBW-UHFFFAOYSA-N 0.000 description 1
- 102100027211 Albumin Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 239000012114 Alexa Fluor 647 Substances 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 102100023635 Alpha-fetoprotein Human genes 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 206010003694 Atrophy Diseases 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 101100454433 Biomphalaria glabrata BG01 gene Proteins 0.000 description 1
- 101100454434 Biomphalaria glabrata BG04 gene Proteins 0.000 description 1
- 201000004569 Blindness Diseases 0.000 description 1
- 102100024506 Bone morphogenetic protein 2 Human genes 0.000 description 1
- 102100024504 Bone morphogenetic protein 3 Human genes 0.000 description 1
- 102100024505 Bone morphogenetic protein 4 Human genes 0.000 description 1
- 102100022526 Bone morphogenetic protein 5 Human genes 0.000 description 1
- 102100022525 Bone morphogenetic protein 6 Human genes 0.000 description 1
- 102100022544 Bone morphogenetic protein 7 Human genes 0.000 description 1
- 108090000715 Brain-derived neurotrophic factor Proteins 0.000 description 1
- 102000004219 Brain-derived neurotrophic factor Human genes 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 102000019034 Chemokines Human genes 0.000 description 1
- 108010012236 Chemokines Proteins 0.000 description 1
- 108010005939 Ciliary Neurotrophic Factor Proteins 0.000 description 1
- 102100031614 Ciliary neurotrophic factor Human genes 0.000 description 1
- 206010010541 Congenital melanosis Diseases 0.000 description 1
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine Chemical compound NCCCC[C@@H](N)C(O)=O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 1
- 206010012689 Diabetic retinopathy Diseases 0.000 description 1
- 101100296720 Dictyostelium discoideum Pde4 gene Proteins 0.000 description 1
- QRLVDLBMBULFAL-UHFFFAOYSA-N Digitonin Natural products CC1CCC2(OC1)OC3C(O)C4C5CCC6CC(OC7OC(CO)C(OC8OC(CO)C(O)C(OC9OCC(O)C(O)C9OC%10OC(CO)C(O)C(OC%11OC(CO)C(O)C(O)C%11O)C%10O)C8O)C(O)C7O)C(O)CC6(C)C5CCC4(C)C3C2C QRLVDLBMBULFAL-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 238000008157 ELISA kit Methods 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- 208000003098 Ganglion Cysts Diseases 0.000 description 1
- 208000031448 Genomic Instability Diseases 0.000 description 1
- 102100024208 Homeobox protein MIXL1 Human genes 0.000 description 1
- 102100030634 Homeobox protein OTX2 Human genes 0.000 description 1
- 102100027345 Homeobox protein SIX3 Human genes 0.000 description 1
- 102100025448 Homeobox protein SIX6 Human genes 0.000 description 1
- 101000762366 Homo sapiens Bone morphogenetic protein 2 Proteins 0.000 description 1
- 101000762375 Homo sapiens Bone morphogenetic protein 3 Proteins 0.000 description 1
- 101000762379 Homo sapiens Bone morphogenetic protein 4 Proteins 0.000 description 1
- 101000899388 Homo sapiens Bone morphogenetic protein 5 Proteins 0.000 description 1
- 101000899390 Homo sapiens Bone morphogenetic protein 6 Proteins 0.000 description 1
- 101000899361 Homo sapiens Bone morphogenetic protein 7 Proteins 0.000 description 1
- 101001052462 Homo sapiens Homeobox protein MIXL1 Proteins 0.000 description 1
- 101000584400 Homo sapiens Homeobox protein OTX2 Proteins 0.000 description 1
- 101000651928 Homo sapiens Homeobox protein SIX3 Proteins 0.000 description 1
- 101000835956 Homo sapiens Homeobox protein SIX6 Proteins 0.000 description 1
- 101001020544 Homo sapiens LIM/homeobox protein Lhx2 Proteins 0.000 description 1
- 102000010781 Interleukin-6 Receptors Human genes 0.000 description 1
- 108010038501 Interleukin-6 Receptors Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 102100036132 LIM/homeobox protein Lhx2 Human genes 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241000699673 Mesocricetus auratus Species 0.000 description 1
- 108050006602 Metalloproteinase inhibitor 2 Proteins 0.000 description 1
- 102000010750 Metalloproteins Human genes 0.000 description 1
- 108010063312 Metalloproteins Proteins 0.000 description 1
- 102000013760 Microphthalmia-Associated Transcription Factor Human genes 0.000 description 1
- 102100030157 Microphthalmia-associated transcription factor Human genes 0.000 description 1
- 208000009795 Microphthalmos Diseases 0.000 description 1
- 101100096242 Mus musculus Sox9 gene Proteins 0.000 description 1
- BAWFJGJZGIEFAR-NNYOXOHSSA-N NAD zwitterion Chemical compound NC(=O)C1=CC=C[N+]([C@H]2[C@@H]([C@H](O)[C@@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 BAWFJGJZGIEFAR-NNYOXOHSSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 108010025020 Nerve Growth Factor Proteins 0.000 description 1
- 102000007072 Nerve Growth Factors Human genes 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- 238000000636 Northern blotting Methods 0.000 description 1
- 239000012661 PARP inhibitor Substances 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 229930040373 Paraformaldehyde Natural products 0.000 description 1
- 208000034247 Pattern dystrophy Diseases 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 206010057249 Phagocytosis Diseases 0.000 description 1
- 101100082610 Plasmodium falciparum (isolate 3D7) PDEdelta gene Proteins 0.000 description 1
- 229940121906 Poly ADP ribose polymerase inhibitor Drugs 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 101710115195 Protease inhibitor 2 Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical group C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 1
- 208000019155 Radiation injury Diseases 0.000 description 1
- 229940127361 Receptor Tyrosine Kinase Inhibitors Drugs 0.000 description 1
- 206010038933 Retinopathy of prematurity Diseases 0.000 description 1
- 208000005400 Synovial Cyst Diseases 0.000 description 1
- 206010043276 Teratoma Diseases 0.000 description 1
- QIOZLISABUUKJY-UHFFFAOYSA-N Thiobenzamide Chemical class NC(=S)C1=CC=CC=C1 QIOZLISABUUKJY-UHFFFAOYSA-N 0.000 description 1
- 102100023935 Transmembrane glycoprotein NMB Human genes 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 102100039094 Tyrosinase Human genes 0.000 description 1
- 108060008724 Tyrosinase Proteins 0.000 description 1
- 206010046798 Uterine leiomyoma Diseases 0.000 description 1
- 108091008605 VEGF receptors Proteins 0.000 description 1
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 1
- 206010047531 Visual acuity reduced Diseases 0.000 description 1
- 206010047571 Visual impairment Diseases 0.000 description 1
- 101000998548 Yersinia ruckeri Alkaline proteinase inhibitor Proteins 0.000 description 1
- 210000002718 aborted fetus Anatomy 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 108010023079 activin B Proteins 0.000 description 1
- 210000004504 adult stem cell Anatomy 0.000 description 1
- 238000011316 allogeneic transplantation Methods 0.000 description 1
- 108010026331 alpha-Fetoproteins Proteins 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 238000000540 analysis of variance Methods 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 244000037640 animal pathogen Species 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001772 anti-angiogenic effect Effects 0.000 description 1
- 230000003127 anti-melanomic effect Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 238000000149 argon plasma sintering Methods 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 238000002306 biochemical method Methods 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 238000004820 blood count Methods 0.000 description 1
- 210000004271 bone marrow stromal cell Anatomy 0.000 description 1
- 239000012888 bovine serum Substances 0.000 description 1
- 210000005252 bulbus oculi Anatomy 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 230000011712 cell development Effects 0.000 description 1
- 239000012560 cell impurity Substances 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 238000002659 cell therapy Methods 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- HWGQMRYQVZSGDQ-HZPDHXFCSA-N chembl3137320 Chemical compound CN1N=CN=C1[C@H]([C@H](N1)C=2C=CC(F)=CC=2)C2=NNC(=O)C3=C2C1=CC(F)=C3 HWGQMRYQVZSGDQ-HZPDHXFCSA-N 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 210000002987 choroid plexus Anatomy 0.000 description 1
- 210000004240 ciliary body Anatomy 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 238000010226 confocal imaging Methods 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 210000000795 conjunctiva Anatomy 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 210000004087 cornea Anatomy 0.000 description 1
- 230000000875 corresponding effect Effects 0.000 description 1
- 238000012136 culture method Methods 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- UVYVLBIGDKGWPX-KUAJCENISA-N digitonin Chemical compound O([C@@H]1[C@@H]([C@]2(CC[C@@H]3[C@@]4(C)C[C@@H](O)[C@H](O[C@H]5[C@@H]([C@@H](O)[C@@H](O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)CO7)O)[C@H](O)[C@@H](CO)O6)O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O7)O)[C@@H](O)[C@@H](CO)O6)O)[C@@H](CO)O5)O)C[C@@H]4CC[C@H]3[C@@H]2[C@@H]1O)C)[C@@H]1C)[C@]11CC[C@@H](C)CO1 UVYVLBIGDKGWPX-KUAJCENISA-N 0.000 description 1
- UVYVLBIGDKGWPX-UHFFFAOYSA-N digitonine Natural products CC1C(C2(CCC3C4(C)CC(O)C(OC5C(C(O)C(OC6C(C(OC7C(C(O)C(O)CO7)O)C(O)C(CO)O6)OC6C(C(OC7C(C(O)C(O)C(CO)O7)O)C(O)C(CO)O6)O)C(CO)O5)O)CC4CCC3C2C2O)C)C2OC11CCC(C)CO1 UVYVLBIGDKGWPX-UHFFFAOYSA-N 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 238000002845 discoloration Methods 0.000 description 1
- 108010007093 dispase Proteins 0.000 description 1
- 238000002224 dissection Methods 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 210000003981 ectoderm Anatomy 0.000 description 1
- 210000001705 ectoderm cell Anatomy 0.000 description 1
- 238000002571 electroretinography Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 210000001900 endoderm Anatomy 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003344 environmental pollutant Substances 0.000 description 1
- 230000007515 enzymatic degradation Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- YQGOJNYOYNNSMM-UHFFFAOYSA-N eosin Chemical compound [Na+].OC(=O)C1=CC=CC=C1C1=C2C=C(Br)C(=O)C(Br)=C2OC2=C(Br)C(O)=C(Br)C=C21 YQGOJNYOYNNSMM-UHFFFAOYSA-N 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 239000013020 final formulation Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 210000003953 foreskin Anatomy 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 210000004602 germ cell Anatomy 0.000 description 1
- 210000001654 germ layer Anatomy 0.000 description 1
- 210000004907 gland Anatomy 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 230000007236 host immunity Effects 0.000 description 1
- 102000053076 human PMEL Human genes 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 210000001822 immobilized cell Anatomy 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 238000003365 immunocytochemistry Methods 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 230000004410 intraocular pressure Effects 0.000 description 1
- 230000001788 irregular Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 244000144972 livestock Species 0.000 description 1
- 210000004874 lower jaw Anatomy 0.000 description 1
- 230000000938 luteal effect Effects 0.000 description 1
- 241001515942 marmosets Species 0.000 description 1
- 238000010297 mechanical methods and process Methods 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 210000002901 mesenchymal stem cell Anatomy 0.000 description 1
- 210000003716 mesoderm Anatomy 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 239000003475 metalloproteinase inhibitor Substances 0.000 description 1
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 1
- 230000002906 microbiologic effect Effects 0.000 description 1
- 201000010478 microphthalmia Diseases 0.000 description 1
- 238000000386 microscopy Methods 0.000 description 1
- 230000001617 migratory effect Effects 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 238000004264 monolayer culture Methods 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- 229950006238 nadide Drugs 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 210000001020 neural plate Anatomy 0.000 description 1
- 230000000508 neurotrophic effect Effects 0.000 description 1
- 229960002715 nicotine Drugs 0.000 description 1
- SNICXCGAKADSCV-UHFFFAOYSA-N nicotine Natural products CN1CCCC1C1=CC=CN=C1 SNICXCGAKADSCV-UHFFFAOYSA-N 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- PCHKPVIQAHNQLW-CQSZACIVSA-N niraparib Chemical compound N1=C2C(C(=O)N)=CC=CC2=CN1C(C=C1)=CC=C1[C@@H]1CCCNC1 PCHKPVIQAHNQLW-CQSZACIVSA-N 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- 238000007899 nucleic acid hybridization Methods 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 208000013441 ocular lesion Diseases 0.000 description 1
- FDLYAMZZIXQODN-UHFFFAOYSA-N olaparib Chemical compound FC1=CC=C(CC=2C3=CC=CC=C3C(=O)NN=2)C=C1C(=O)N(CC1)CCN1C(=O)C1CC1 FDLYAMZZIXQODN-UHFFFAOYSA-N 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 238000001543 one-way ANOVA Methods 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 230000000242 pagocytic effect Effects 0.000 description 1
- 210000002741 palatine tonsil Anatomy 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 229920002866 paraformaldehyde Polymers 0.000 description 1
- 230000008186 parthenogenesis Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 230000008823 permeabilization Effects 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- 230000008782 phagocytosis Effects 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 210000000608 photoreceptor cell Anatomy 0.000 description 1
- 210000002826 placenta Anatomy 0.000 description 1
- 231100000719 pollutant Toxicity 0.000 description 1
- 238000013105 post hoc analysis Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 238000012514 protein characterization Methods 0.000 description 1
- 238000001742 protein purification Methods 0.000 description 1
- 238000013094 purity test Methods 0.000 description 1
- 238000012797 qualification Methods 0.000 description 1
- 239000001397 quillaja saponaria molina bark Substances 0.000 description 1
- 238000003127 radioimmunoassay Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000009256 replacement therapy Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000004283 retinal dysfunction Effects 0.000 description 1
- 230000009201 retinal pigment epithelium development Effects 0.000 description 1
- 210000001164 retinal progenitor cell Anatomy 0.000 description 1
- 238000003757 reverse transcription PCR Methods 0.000 description 1
- FCCGJTKEKXUBFZ-UHFFFAOYSA-N rucaparib phosphate Chemical compound OP(O)(O)=O.C1=CC(CNC)=CC=C1C(N1)=C2CCNC(=O)C3=C2C1=CC(F)=C3 FCCGJTKEKXUBFZ-UHFFFAOYSA-N 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 238000003118 sandwich ELISA Methods 0.000 description 1
- 229930182490 saponin Natural products 0.000 description 1
- 150000007949 saponins Chemical class 0.000 description 1
- 210000003786 sclera Anatomy 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 208000022417 sinus histiocytosis with massive lymphadenopathy Diseases 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 238000003307 slaughter Methods 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 238000009331 sowing Methods 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000012430 stability testing Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 210000001562 sternum Anatomy 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000003319 supportive effect Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 210000000115 thoracic cavity Anatomy 0.000 description 1
- 208000008732 thymoma Diseases 0.000 description 1
- 239000003104 tissue culture media Substances 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 108091007466 transmembrane glycoproteins Proteins 0.000 description 1
- 230000008736 traumatic injury Effects 0.000 description 1
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 1
- 230000001228 trophic effect Effects 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 230000000381 tumorigenic effect Effects 0.000 description 1
- 210000000689 upper leg Anatomy 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 239000012808 vapor phase Substances 0.000 description 1
- 239000013598 vector Substances 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- JNAHVYVRKWKWKQ-CYBMUJFWSA-N veliparib Chemical compound N=1C2=CC=CC(C(N)=O)=C2NC=1[C@@]1(C)CCCN1 JNAHVYVRKWKWKQ-CYBMUJFWSA-N 0.000 description 1
- 229950011257 veliparib Drugs 0.000 description 1
- 230000007332 vesicle formation Effects 0.000 description 1
- 208000029257 vision disease Diseases 0.000 description 1
- 230000004393 visual impairment Effects 0.000 description 1
- 230000009012 visual motion Effects 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
Images


















































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0618—Cells of the nervous system
- C12N5/0621—Eye cells, e.g. cornea, iris pigmented cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/30—Nerves; Brain; Eyes; Corneal cells; Cerebrospinal fluid; Neuronal stem cells; Neuronal precursor cells; Glial cells; Oligodendrocytes; Schwann cells; Astroglia; Astrocytes; Choroid plexus; Spinal cord tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/02—Atmosphere, e.g. low oxygen conditions
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/30—Organic components
- C12N2500/38—Vitamins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/115—Basic fibroblast growth factor (bFGF, FGF-2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/15—Transforming growth factor beta (TGF-β)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/16—Activin; Inhibin; Mullerian inhibiting substance
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2506/00—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells
- C12N2506/02—Differentiation of animal cells from one lineage to another; Differentiation of pluripotent cells from embryonic cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2533/00—Supports or coatings for cell culture, characterised by material
- C12N2533/50—Proteins
- C12N2533/52—Fibronectin; Laminin
Description
(a)分化細胞を生成するように、ヒト胚性幹細胞を、ニコチンアミドを含みアクチビンAを欠如している培地中で培養する段階;
(b)RPE系列にさらに分化している細胞を生成するように、分化細胞を、ニコチンアミドおよびアクチビンAを含む培地中で培養する段階;ならびに
(c)RPE系列にさらに分化している細胞を、ニコチンアミドを含みアクチビンAを欠如している培地中で培養する段階
によって作製される。
(a)分化細胞を生成するように、多能性幹細胞を、分化物質を含みトランスフォーミング成長因子β(TGFβ)スーパーファミリーメンバーを欠如している培地中で培養する段階;
(b)RPE系列にさらに分化している細胞を生成するように、分化細胞を、トランスフォーミング成長因子β(TGFβ)スーパーファミリーのメンバーおよび分化物質を含む培地中で培養する段階;
(c)RPE細胞を生成するように、RPE系列にさらに分化している細胞を、分化物質を含みトランスフォーミング成長因子β(TGFβ)スーパーファミリーのメンバーを欠如している培地中で培養する段階
を含む、RPE細胞を作製する方法であって、段階(a)〜(c)が、大気酸素レベルが約10%未満の条件下において行われる、方法が提供される。
(i)分化細胞を含む細胞のクラスターを生成するように、培養されるヒト多能性幹細胞の集団を、ニコチンアミドを含む培地中で、アクチビンAの非存在下で非付着条件下において培養する段階、および、その後に、
(ii)(i)の分化細胞を、ニコチンアミドを含む培地中で、アクチビンAの非存在下で付着条件下において培養する段階
を含む。
ヒト多角RPE細胞の集団であって、それらの細胞の少なくとも95%がプレメラノソームタンパク質(PMEL17)と細胞レチンアルデヒド結合タンパク質(CRALBP)とを同時発現し、該細胞の集団の経上皮電気抵抗が100オームより大きい、ヒト多角RPE細胞の集団。
[本発明1002]
ヒトRPE細胞の集団であって、それらの細胞の少なくとも80%がプレメラノソームタンパク質(PMEL17)と細胞レチンアルデヒド結合タンパク質(CRALBP)とを同時発現し、かつ該集団内の細胞が、アンジオゲニン、組織メタロプロテアーゼ阻害物質2(TIMP2)、可溶性糖タンパク質130(sgp130)、および可溶型腫瘍壊死因子α遍在性膜受容体1(sTNF-R1)のそれぞれを分泌する、ヒトRPE細胞の集団。
[本発明1003]
前記集団内の細胞が、アンジオゲニン、組織メタロプロテアーゼ阻害物質2(TIMP2)、可溶性糖タンパク質130(sgp130)、および可溶型腫瘍壊死因子α遍在性膜受容体1(sTNF-R1)のそれぞれを分泌する、本発明1001の細胞集団。
[本発明1004]
前記細胞が、前記アンジオゲニン、前記TIMP2、前記sgp130、または前記sTNF-R1を極性化様式で分泌する、本発明1002または1003の細胞集団。
[本発明1005]
前記細胞が、前記アンジオゲニン、前記TIMP2、前記sgp130、および前記sTNF-R1のそれぞれを極性化様式で分泌する、本発明1002または1003の細胞集団。
[本発明1006]
sgp130の基底側分泌に対するsgp130の頂端側分泌の比が1より大きい、本発明1004または1005の細胞集団。
[本発明1007]
sTNF-R1の基底側分泌に対するsTNF-R1の頂端側分泌の比が1より大きい、本発明1004または1005の細胞集団。
[本発明1008]
アンジオゲニンの頂端側分泌に対するアンジオゲニンの基底側分泌の比が1より大きい、本発明1004または1005の細胞集団。
[本発明1009]
TIMP2の基底側分泌に対するTIMP2の頂端側分泌の比が1より大きい、本発明1004または1005の細胞集団。
[本発明1010]
前記集団中のOct4 + TRA-1-60 + 細胞の数が1:250,000を下回る、本発明1001または1002の細胞集団。
[本発明1011]
免疫染色によって測定された場合に、前記細胞の少なくとも80%がベストロフィン1(Bestrophin 1)を発現する、本発明1001〜1010のいずれかの細胞集団。
[本発明1012]
免疫染色によって測定された場合に、前記細胞の少なくとも80%が小眼球症関連転写因子(MITF)を発現する、本発明1001〜1011のいずれかの細胞集団。
[本発明1013]
FACSによって測定された場合に、前記細胞の50%超がペアードボックス遺伝子6(PAX-6)を発現する、本発明1001〜1012のいずれかの細胞集団。
[本発明1014]
前記細胞が、1日につき1mlあたり750ng超の色素上皮由来因子(PEDF)を分泌する、本発明1001〜1013のいずれかの細胞集団。
[本発明1015]
前記細胞がPEDFおよび血管内皮増殖因子(VEGF)を極性化様式で分泌する、本発明1001〜1014のいずれかの細胞集団。
[本発明1016]
PEDFの基底側分泌に対するPEDFの頂端側分泌の比が1より大きい、本発明1015の細胞集団。
[本発明1017]
2〜8℃における8時間のインキュベーション後に、前記比が依然として1より大きい、本発明1016の細胞集団。
[本発明1018]
前記細胞の集団の経上皮電気抵抗が100オームより大きい、本発明1002の細胞集団。
[本発明1019]
2〜8℃における8時間のインキュベーション後に、前記細胞の前記経上皮電気抵抗が依然として100オームより大きい、本発明1001または1018の細胞集団。
[本発明1020]
VEGFの頂端側分泌に対するVEGFの基底側分泌の比が1より大きい、本発明1015または1016の細胞集団。
[本発明1021]
2〜8℃における8時間のインキュベーション後に、前記比が依然として1より大きい、本発明1020の細胞集団。
[本発明1022]
網膜下投与後にRCSラットにおける視力をレスキューすることができる、本発明1001〜1021のいずれかの細胞集団。
[本発明1023]
RCSラットにおいて網膜下投与後少なくとも180日間にわたって光受容体をレスキューすることができる、本発明1001〜1021のいずれかの細胞集団。
[本発明1024]
ヒト胚性幹細胞のエクスビボ分化によって作製された、本発明1001〜1023のいずれかの細胞集団。
[本発明1025]
(a)分化細胞を生成するように、ヒト胚性幹細胞を、ニコチンアミドを含みアクチビンAを欠如している培地中で培養する段階;
(b)RPE系列にさらに分化している細胞を生成するように、該分化細胞を、ニコチンアミドおよびアクチビンAを含む培地中で培養する段階;ならびに
(c)該RPE系列にさらに分化している細胞を、ニコチンアミドを含みアクチビンAを欠如している培地中で培養する段階
によって作製された、本発明1001〜1024のいずれかの細胞集団。
[本発明1026]
前記胚性幹細胞が、bFGFおよびTGFβを含む培地中で増殖される、本発明1025の細胞集団。
[本発明1027]
前記胚性幹細胞がヒト帯線維芽細胞上で培養される、本発明1025の細胞集団。
[本発明1028]
段階(a)〜(c)が、大気酸素レベルが約10%未満の条件下において行われる、本発明1025〜1027のいずれかの細胞集団。
[本発明1029]
段階(c)の後に、分化した前記細胞を、ニコチンアミドの存在下で大気酸素レベルが約10%を超える条件下において培地中で培養する段階をさらに含む、本発明1028の細胞集団。
[本発明1030]
活性物質としての本発明1001〜1029のいずれかの細胞集団と、薬学的に許容される担体とを含む、薬学的組成物。
[本発明1031]
網膜変性を処置するための、本発明1001〜1030のいずれかの細胞集団の使用。
[本発明1032]
(a)分化細胞を生成するように、多能性幹細胞を、分化物質を含みトランスフォーミング成長因子β(TGFβ)スーパーファミリーのメンバーを欠如している培地中で培養する段階;
(b)RPE系列にさらに分化している細胞を生成するように、該分化細胞を、該トランスフォーミング成長因子β(TGFβ)スーパーファミリーのメンバーおよび該分化物質を含む培地中で培養する段階;
(c)RPE細胞を生成するように、該RPE系列にさらに分化している細胞を、分化物質を含みトランスフォーミング成長因子β(TGFβ)スーパーファミリーのメンバーを欠如している培地中で培養する段階
を含む、RPE細胞を作製する方法であって、
段階(a)〜(c)が、大気酸素レベルが約10%未満の条件下において行われる、方法。
[本発明1033]
段階(a)が非付着条件下において行われる、本発明1032の方法。
[本発明1034]
前記非付着条件が非付着性培養プレートを含む、本発明1033の方法。
[本発明1035]
段階(a)が、
(i)分化細胞を含む細胞のクラスターを生成するように、培養されるヒト多能性幹細胞の集団を、ニコチンアミドを含む培地中で、アクチビンAの非存在下で非付着条件下において培養する段階、および、その後に、
(ii)(i)の分化細胞を、ニコチンアミドを含む培地中で、アクチビンAの非存在下で付着条件下において培養する段階
を含む、本発明1032の方法。
[本発明1036]
段階(ii)の前に前記細胞のクラスターを解離して、細胞の凝集塊または細胞の単一細胞懸濁液を作製する段階をさらに含む、本発明1035の方法。
[本発明1037]
段階(c)の後に、分化した前記細胞を、分化物質の存在下で大気酸素レベルが約10%を超える条件下において培地中で培養する段階をさらに含む、本発明1032の方法。
[本発明1038]
前記トランスフォーミング成長因子β(TGFβ)スーパーファミリーのメンバーが、TGFβ1、TGFβ3、およびアクチビンAからなる群より選択される、本発明1032の方法。
[本発明1039]
段階(a)の分化物質および段階(c)の分化物質が同一である、本発明1032の方法。
[本発明1040]
段階(a)の分化物質がニコチンアミド(NA)または3-アミノベンズアミドである、本発明1032の方法。
[本発明1041]
段階(c)の後に、多角細胞を選択する段階をさらに含む、本発明1032の方法。
[本発明1042]
前記多角細胞を増殖させる段階をさらに含む、本発明1041の方法。
[本発明1043]
前記増殖させる段階が付着性表面上で行われる、本発明1042の方法。
[本発明1044]
前記多能性幹細胞が胚性幹細胞を含む、本発明1032の方法。
[本発明1045]
前記胚性幹細胞が、bFGFおよびTGFβを含む培地中で増殖される、本発明1044の方法。
[本発明1046]
前記胚性幹細胞がヒト帯線維芽細胞上で培養される、本発明1044の方法。
特に定義のない限り、本明細書で使用する技術用語および科学用語は全て、本発明が属する当業者に一般的に理解されているものと同じ意味を有する。本明細書に記載のものと同様のまたは等価な方法および材料を本発明の態様の実施または試験において使用することができるが、例示的な方法および/または材料を下記で説明する。矛盾する場合は、定義を含む本明細書が優先される。さらに、材料、方法、および実施例は例示にすぎず、必ず限定することが意図されない。
本発明は、その一部の態様では、網膜色素上皮細胞に関し、さらに詳細には、治療用物質として、このような細胞を評価することに関するが、これに限定されない。本発明はまた、ヒト胚性幹細胞からの網膜色素上皮細胞の作製に関する。
1.ホルムアルデヒド、続いて、界面活性剤: ホルムアルデヒドによる固定(例えば、4.5%以下で10〜15分間(これによりタンパク質が安定化される)、続いて、界面活性剤、例えば、TritonまたはNP-40(PBS中で0.1〜1%)、Tween20(PBS中で0.1〜1%)、サポニン、ジギトニン、およびLeucoperm(例えば、PBS中で0.5%v/v)による膜の破壊;
2.ホルムアルデヒド(例えば、4.5%以下)、続いてメタノール;
3.メタノール、続いて、界面活性剤(例えば、80%メタノール、次いで、0.1%Tween20);
4.アセトン固定および透過処理
を含む。
-分化の間に、培養細胞はTGFβシグナル伝達に応答すること;
-RPE細胞は、最終分化を示すマーカー、例えば、ベストロフィン1、CRALBP、および/またはRPE65を発現すること;
-移植後に(すなわち、インサイチューで)、RPE細胞は、RPE細胞に隣接する光受容体を支持する栄養作用を示すこと;
-さらに、インサイチューで、RPE細胞は、これらの光受容体の正常な再生プロセスの一環として、脱落した光受容体外節の食作用を伴って機能することができること;
-さらに、インサイチューで、RPE細胞は網膜関門を作製し、視サイクルにおいて機能することができること。

(a)ESCを第1の分化物質(例えば、ニコチンアミド)を含む培地中で培養する段階;ならびに
(b)段階(a)から得られた細胞を、TGFβスーパーファミリーのメンバー(例えば、アクチビンA)および第1の分化物質(例えば、ニコチンアミド)を含む培地中で培養する段階。
-ノックアウト血清代用品(knock out serum replacement)(KOSR)、Nutridoma-CS、TCH(商標)、N2、N2誘導体、もしくはB27、または組み合わせなどがあるが、これに限定されない、血清または血清代用品を含有する培地。
-フィブロネクチン、ラミニン、コラーゲン、およびゼラチンなどがあるが、これに限定されない、細胞外マトリックス(ECM)成分。ECMは、増殖因子の1つまたは複数のTGFβスーパーファミリーのメンバーを運ぶのに用いられてもよい。
-ペニシリンおよびストレプトマイシンなどがあるが、これに限定されない、抗菌剤。
-非必須アミノ酸(NEAA)。BDNF、NT3、NT4などがあるが、これに限定されない、培養中のSCの生存の促進において役割を果たすことが公知であるニューロトロフィン。
(a)分化細胞を生成するように、多能性幹細胞を、分化物質を含みトランスフォーミング成長因子β(TGFβ)スーパーファミリーのメンバーを欠如している培地中で培養する段階;
(b)RPE系列にさらに分化している細胞を生成するように、分化細胞を、トランスフォーミング成長因子β(TGFβ)スーパーファミリーのメンバーおよび分化物質を含む培地中で培養する段階;
(c)RPE系列にさらに分化している細胞からの色素上皮由来因子(PEDF)の分泌を分析する段階;ならびに
(d)RPE細胞を生成するように、RPE系列にさらに分化している細胞を、分化物質を含みトランスフォーミング成長因子β(TGFβ)スーパーファミリーのメンバーを欠如している培地中で培養する段階
を含み、
PEDF量が所定のレベルを上回る場合に段階(d)が行われる、
網膜上皮細胞を作製する方法が提供される。
CRALBP/PMEL17二重染色FACS法の適格性判定
この研究の目的は、少なくとも3日の試験日にわたる少なくとも6回の独立したスパイキング(spiking)アッセイ法において、CRALBP/PMEL17二重染色FACS法の正確度および精度を証明することによって、CRALBP/PMEL17二重染色FACS法に適格性を判定することであった。陽性対照細胞としてOpRegen(登録商標)バッチ5Cを使用し、陰性対照細胞としてHAD-C 102-hESCを使用して、このアッセイ法の適格性判定を行った。異なるスパイキングポイントの正確度および精度を試験するために、hESCにスパイクした既知量のRPE(OpRegen(登録商標)5C)の較正曲線を使用した。予想された正確度および精度は、全ポイントで25%までであった。
正確度:アッセイ法の正確度を4つのレベルのスパイク済RPE(25%、50%、75%、および95%)の試験結果から求めた。潜在的に100%のRPE細胞であることに対して、RPEストック(OpRegen(登録商標)5C)の正確度を求めた。それぞれのレベル値を6回の独立したラン/測定によって分析した。
本明細書に示した結果から、開示された方法は適格であると判定され、OpRegen(登録商標)最終産物におけるRPEの純度およびOpRegen(登録商標)の生産工程に沿った異なる段階でのRPEの純度をインビトロで求める所期の用途に適していることが分かる。50%〜99.5%の範囲のRPE細胞において、相対バイアスの正確度は<25%、%CVの精度は<20%であった。
OpRegen(登録商標)純度のレベルの評価
ヒト網膜色素上皮細胞(RPE)の純度ならびにRPE細胞中の非RPE細胞不純物のレベルを評価するためのFACSに基づく方法を開発した。視サイクル成分の1つである細胞レチンアルデヒド結合タンパク質(CRALBP)が、成熟RPE細胞のユニークなマーカーとしてバイオインフォマティクスを用いて同定された。CRALBP特異的モノクローナル抗体を用いた予備研究から、本明細書に記載の方法に従って作製されたRPE細胞において98%を超える純度が示されている。さらに、これらの結果は、RPEに認められるメラノソームマーカーであるPMEL17の免疫染色によって裏付けられた。さらに、いくつかのRPE特異的マーカーとは異なり、CRALBPは、可能性のある神経冠細胞汚染物質であるメラノサイトにおいて発現していない。
抗CRALBPモノクローナル抗体を用いた初期FACSデータから、OpRegen(登録商標)の純度レベルは98%を上回ることが分かった。可能性のある神経冠細胞汚染物質であるメラノサイトは、ユニークなRPE特異的マーカーであるCRALBPが陰性であることが見出された。(1.7%)。親株HADC102-hESCは予想した通りCRALBP陰性であった(0.2%)。
製造工程および工程管理の説明
OpRegen(登録商標)は、放射線照射された、ゼノフリーGMPグレードのヒト臍帯線維芽細胞フィーダー上で増殖させた、ゼノフリーGMPグレードのHAD-C 102 hESC株から製造される。臨床グレードヒト線維芽細胞フィーダー細胞株(CRD008; MCB)およびワーキング細胞バンク(WCB)を医薬品及び医薬部外品の製造管理及び品質管理規則(Good Manufacturing Practice)(GMP)の下で、かつゼノフリー条件下において生産し、適切に試験し、特徴決定し、バンク化(bank)した。次いで、GMPの下で、かつゼノフリー条件下において、余ったヒト胚盤胞から臨床グレードのhESC HAD-C 102株を得る際に、これらを使用した。
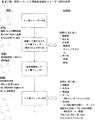
工程管理ポイント
IPCポイントを図16に図示した。生産工程に沿ってhESC不純物およびRPE純度を評価するために選択したサンプリングポイントを以下で説明する。
TRA-1-60+Oct4+hESCの定量:生産工程に沿って収集した様々な試料中のhESCのレベルを、高感度のロバストなOct4/TRA-1-60二重染色FACS法を用いて決定した。初期の神経/眼野(eye field)分化を支持する増殖条件下において、多能性細胞増殖を支持するフィーダーならびに増殖因子(TGFβおよびbFGF)を除去して1週間後に、TRA-1-60+Oct4+細胞は0.0106〜2.7%しかなかった(IPCポイント3、球状体)。RPE分化を促進するアクチビンAを添加した後に、TRA-1-60+Oct4+細胞のレベルは0.00048〜0.0168%までさらに減少した(IPCポイント4、アクチビンの終了)。分化終了時に、非色素細胞を切除した後に、TRA-1-60+Oct4+細胞のレベルは0.00033〜0.03754%であった(IPCポイント7、色素細胞)。生産工程が終了する2段階前であるP0では、0.00009〜0.00108%(LODを下回る-LLOQに近い)のレベルのTRA-1-60+Oct4+細胞が検出された(IPCポイント8)。P1(IPCポイント9)、凍結保存前のP2(原体;IPCポイント10)、および凍結保存後のP2(DP;IPCポイント11)におけるTRA-1-60+Oct4+細胞のレベルはアッセイLLOQを下回る(すなわち、それぞれ、0.00004〜0.00047%、0.00000〜0.00016%、および0.00000〜0.00020%)。
3つの模擬試験生産ラン(模擬試験ラン2、4、および5)を、臨床バッチのGMP生産で使用した同じGMP生産方法、ゼノフリーGMPグレード細胞(放射線照射済みCRD008フィーダー上で増殖させたHAD-C 102 hESC)、ゼノフリーGMPグレード試薬、およびGMPグレード実験器具を用いて研究グレード条件下において行った。模擬試験生産2、4、および5は、生産に沿ってhESC不純物のレベルを評価することを目的とし、模擬試験生産4および5はまた、工程品質管理において重要なものを同定することも目的とした。
有効度評価
実験の構成:本発明者らは、実施例4に記載のように作製したRPE細胞の網膜下移植によって、Royal College of Surgeons(RCS)ラットモデルにおけるRDD進行を遅らせることができるかどうか調べた。
細胞数:細胞を計数した後に、適切な投与濃度にアリコートした。全注射時点について注射前の細胞生存率の平均は94.0%±0.03であった。注射後の細胞生存率の平均は92.4%±0.02であった。
眼底画像化:細胞で処置した眼の剖検時に収集した眼底画像から、外科手術中に網膜下小疱が形成された場所; 細胞が網膜下空間に沈着した場所に対応する網膜の色素過剰区域および色素不足区域が明らかになった(図19A〜C)。これらのまだら模様の区域は、BSS+を注射した眼でも注射をしていない眼でも、はっきりと分からなかった。
RCSラットの網膜下空間に移植した場合に、RPE細胞は、試験された全齢で、RCSラットの視力を対照の視力を上回ってレスキューした。移植片が、評価するのに十分に大きいか、または評価のために到達可能な網膜区域にあった場合に、ERG応答は保護された。移植して180日後までに移植区域において桿体光受容体および錐体光受容体はレスキューされた。まとめると、このデータから、OpRegen(登録商標)は宿主網膜の機能完全性および構造完全性を長期間にわたって維持することが証明される。従って、OpRegen(登録商標)は、RPおよびAMDなどのヒトRPE細胞障害の処置に大きな可能性を秘めている。
RPE細胞の安定性
短期安定性
BSS plus中の製剤化されたRPE細胞(実施例4に記載のように作製した)をバイアル1個あたり600〜1000μlの最終体積で調製した。短期安定性を0時間、4時間、8時間、および24時間の時点で試験した。細胞は全ての時点で安定なことが見出された。
・低濃度(BSS plus 100μlにつき70×103個)は時点0時間の93%±5から時点8時間の91%±1に変化し、有意な減少はなかった。
・高濃度(BSS plus 100μlにつき70×103個)は時点0時間の92%±3から時点8時間の91%±2に変化し、有意な減少はなかった。
生存率、総細胞数/バイアル、およびRPE同一性は3年間にわたってずっと維持された。さらに、示したように、データから、保存前に収集したものと同様のレベルで有効性および純度が証明された。
安全性および生体内分布
本研究の目的は、雄NOD-SCIDマウスおよび雌NOD-SCIDマウスに6ヶ月の研究期間にわたって網膜下投与した後に、(実施例4に記載のように作製した)RPE細胞の生存、生体内分布、および安全性を評価することであった。
・臨床観察;
・体重;
・眼科検査(肉眼検査および生体顕微鏡検査(biomicroscopic examination)を含む) ;
・LEICA M80 Stereo顕微鏡を用いた、網膜下注射の質の外科的顕微鏡検査(眼底検査);
・全血球数および血液化学;
・剖検および肉眼的病理学;
・臓器重量(絶対的ならびに体重および脳の重量に対して相対的);
・視神経を含む対側性の処置および無処置の眼の収集、固定、およびパラフィンブロッキング;
・眼ならびに組織(骨髄のある胸骨、脳、心臓、腎臓、肝臓、肺、下顎リンパ節、脊髄、脾臓、胸腺、腫瘤、および肉眼的病変)の盲検式ヘマトキシリン-エオジン組織病理学;
・ヘマトキシリン-エオジン染色スライドにおける色素細胞の盲検式半定量;
・ヒトマーカー(ヒト核)+RPEマーカー(ヒトPMEL17)を対象にした、眼における色素細胞移植片を証明する、代表的なヘマトキシリン-エオジンスライドに隣接する選択されたスライドの盲検式免疫染色、ならびにヒトRPE細胞および非RPE細胞、ヒトマーカー(ヒト核)+増殖マーカー(ヒトKi67)の評価、ならびにヒト増殖細胞および非ヒト増殖細胞、RPEマーカー(RPE65)+増殖マーカー(ヒトKi67)の評価、ならびにRPEヒト増殖細胞および非RPEヒト増殖細胞の評価;
・ヒト起源を排除するためのヒトマーカー(ヒト核)を対象にした、テラトーマ、腫瘍、異常細胞、および病変部を証明する、代表的なヘマトキシリン-エオジンスライドに隣接する選択されたスライドの盲検式免疫染色;
・血液、骨髄(大腿から収集した)、脳、視神経のある左眼および右眼、心臓、左腎臓および右腎臓、肝臓、肺、下顎リンパ節、卵巣、骨格大腿二頭筋、脊髄、脾臓、精巣、ならびに胸腺からのゲノムDNAの収集および抽出、ならびにヒトβグロビンのqPCR分析;
・同じ群および時点からの動物においてヒトβグロビン陽性であることが見出された(上記以外の)組織に対するヘマトキシリン-エオジン組織病理学。
詳細な臨床観察、体重、眼科検査、ならびに血液学および血清臨床化学で構成される臨床病理学を含む生存中の検査において、RPEに関連する毒物学的所見はなかった。詳細な臨床観察および眼科検査において、両用量レベルのRPE色素細胞で処置したマウスでは、アルビノバックグラウンドを有する左眼に「眼が黒っぽく変色した」と観察されたことが見出された。生き残っている動物の眼科検査から、この観察は、硝子体中央の(mid-vitreal)黒っぽい色素沈着のある病巣(foci)で構成されることが分かった。色素沈着のある病巣は、側頭部後側(temporal posterior)水晶体包から鼻網膜表面に及ぶ線に沿ってランダムに分布していた。これらの病巣は、注射中に見られる硝子体の逆流により裏付けられるように、注射後に眼から注射カニューレを取り外す際に注射カニューレから漏れたRPE細胞、または網膜下移植後に硝子体液内に漏れたRPE細胞であると解釈された。
RPEを100,000細胞/μl/眼までの用量レベルで1回注射した後、6ヶ月の研究期間の間に、NOD/SCID動物モデルにおいて局所または全身の毒物学的作用、致死作用、または腫瘍形成作用は観察されなかった。RPE細胞の生体内分布は、処置された左眼に限定され、網膜下細胞は時間の関数として網膜下注射部位から局所的に広がった。2週間、2ヶ月、および6ヶ月の間隔で、高用量群において検査された動物のほとんどにおいてRPE細胞は主に網膜下空間に存在し、硝子体がこれに続き、ヒト核特異的バイオマーカーおよび/またはヒトRPE特異的バイオマーカーに対する抗体による免疫染色の陽性は一定でなかった。眼におけるRPE細胞の持続性は少なくとも6ヶ月と見積もられ、細胞増殖は非常に限られていた。限られた増殖は主に硝子体で起こり、有害な作用を及ぼさなかった。処置された眼においてRPE細胞数が経時的に増加するという証拠があったが、これには、検査された網膜下集団における増殖発生の低下が伴った。RPE特異的マーカーであるRPE65およびPMEL17は、硝子体内とは対照的に、主に網膜下空間内のRPE細胞で発現した。硝子体内にKi67陽性細胞発生の大部分が見出された。後者は、RPE細胞の経時的な増加が硝子体空間に限定され、特異的なRPE65およびPMEL17 RPEマーカーの発現が微小環境によって調節される可能性があることを示唆している。結論として、上記で示されたデータに基づいて、ビヒクル対照群と比較した場合に、本明細書で説明されたRPE細胞の注射に関連する重大な安全問題はなかった。
RPE細胞におけるPax-6発現
目的:ヒト網膜色素上皮(RPE)細胞においてPAX-6レベルを評価するためのFACSに基づく方法の開発。
(実施例4に記載のように作製した)凍結RPE細胞を融解し、遠心沈殿し、1mlのPBS minusに再懸濁し、35μMセルストレーナーで濾過し、NC-200細胞カウンターを用いて計数した。細胞濃度を、PBS minus中1mlにつき約1×106個の細胞まで調整した。細胞懸濁液1mlにつき1μl/mlのFVS450を添加した後に、ボルテックスし、37℃で6分間インキュベートした。FVS450を0.1%BSA(-Ig)-PBS minusでクエンチし、0.1%BSA(-Ig)-Fc-block(RTで5分間)に再懸濁して、細胞上の全Fcエピトープをブロックした。次いで、細胞を固定し、抗Pax-6抗体(AF647カタログ番号562249)で染色した。
図29から分かるように、P0およびP2における細胞はPAX6陽性である(P0では81.5%〜82.5%、P2では91.3%〜96.1%)。P2は生産工程の終了時に継代したものであり、P0は2増加段階前のものである。図29および図30に示したように、データはバッチ全体にわたって首尾一貫していることが示された。さらに、本発明者らは、FACS分析によって、RPE細胞がPAX-6およびCRALBPに対して二重染色されることを示した(図31)。
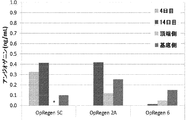
RPE細胞によって分泌されたタンパク質の同定
目的:バッチリリース有効性アッセイ法ならびに工程管理アッセイ法として使用することができる、OpRegen(登録商標)(RPE細胞)によって分泌されたタンパク質(既知および新規)のシグネチャーを同定すること。
1. 12ウェルプレート上で4日間および14日間培養した、融解後のRPE製剤細胞(継代3代目で0.5×106細胞/ウェル)(本明細書中ではOpRegen(登録商標)と呼ぶ)。
2. 12ウェルプレート上で14日間培養し、次いで、(AM-RPE-15に従って)トランズウェル上で3週間培養し、500Ωを超えるTEERを示した、融解後のRPE製剤細胞。頂端側チャンバーおよび基底側チャンバーから上清を採取した。
3. アクチビンA処理前(QC3)およびアクチビンA処理後(QC4)の、実施例3に記載のプロトコールに従って作製した細胞。
4. TGFβおよびFGFが添加されていないNutristem培地(Nut-)。
1. それぞれ、12ウェルプレート上で14日間培養し、次いで、(AM-RPE-15に従って)トランズウェル上で3週間培養し、それぞれ355Ωおよび505ΩのTEERを示した、融解後のOpRegen(登録商標)製剤細胞。上清を、14日目(継代3代目)から、ならびに頂端側チャンバーおよび基底側チャンバーから採取した。
2. 12ウェルプレート上で14日間培養した、融解後のRPE7細胞(継代3代目で0.5×106細胞/ウェル)。
3. (実施例3に記載のように)酵素または機械によって単離した後にラミニン521上で増殖させた、生産工程の継代1代目の終了時の模擬試験VI細胞。これらの細胞をAM-RPE-15に従って有効性について試験し、12ウェルプレート上で14日目の細胞(継代2代目)から、ならびにトランズウェル上で3週間後の頂端側チャンバーおよび基底側チャンバーから採取した細胞から、上清を収集した。
4. 4日目および14日目の継代3代目の胎児HuRPE細胞(0.5×106細胞/ウェル)。
G7アレイ結果を本明細書中の以下の表9に示した。
OpRegen(登録商標)とRPE1およびRPE7との比較
目的:OpRegen(登録商標)(RPE細胞)と、Idelsonら、2009のプロトコールに従って作製したRPE細胞を比較すること。
OpRegen(登録商標)(RPE細胞)を実施例3に記載のように作製した。
Claims (6)
- (a)分化細胞を生成するように、多能性幹細胞を、分化物質を含みトランスフォーミング成長因子β(TGFβ)スーパーファミリーのメンバーを欠如している培地中で培養する段階;
(b)網膜色素上皮(RPE)系列にさらに分化している細胞を生成するように、該分化細胞を、該トランスフォーミング成長因子β(TGFβ)スーパーファミリーのメンバーおよび該分化物質を含む培地中で培養する段階;および
(c)RPE細胞を生成するように、該RPE系列にさらに分化している細胞を、分化物質を含みトランスフォーミング成長因子β(TGFβ)スーパーファミリーのメンバーを欠如している培地中で培養する段階
を含む、RPE細胞を作製する方法であって、
段階(a)〜(c)が、大気酸素レベルが約10%未満の条件下において行われる、方法。 - 段階(a)が、
(i)分化細胞を含む細胞のクラスターを生成するように、培養されるヒト多能性幹細胞の集団を、ニコチンアミドを含む培地中で、アクチビンAの非存在下で非付着条件下において培養する段階、および、その後に、
(ii)(i)の分化細胞を、ニコチンアミドを含む培地中で、アクチビンAの非存在下で付着条件下において培養する段階
を含む、請求項1に記載の方法。 - 段階(c)の後に、分化した前記細胞を、分化物質の存在下で大気酸素レベルが約10%を超える条件下において培地中で培養する段階をさらに含む、請求項1または2に記載の方法。
- 前記トランスフォーミング成長因子β(TGFβ)スーパーファミリーのメンバーが、TGFβ1、TGFβ3、およびアクチビンAからなる群より選択される、請求項1〜3のいずれか一項に記載の方法。
- 段階(c)の後に、多角細胞を選択する段階をさらに含む、請求項1または2に記載の方法。
- 前記多能性幹細胞が胚性幹細胞を含み、かつ該胚性幹細胞が、bFGFおよびTGFβを含む培地中で増殖される、請求項2に記載の方法。
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462097753P | 2014-12-30 | 2014-12-30 | |
| US62/097,753 | 2014-12-30 | ||
| US201562116972P | 2015-02-17 | 2015-02-17 | |
| US62/116,972 | 2015-02-17 | ||
| US201562195309P | 2015-07-22 | 2015-07-22 | |
| US62/195,309 | 2015-07-22 | ||
| PCT/IL2015/051269 WO2016108239A1 (en) | 2014-12-30 | 2015-12-30 | Rpe cell populations and methods of generating same |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021012746A Division JP2021072832A (ja) | 2014-12-30 | 2021-01-29 | Rpe細胞集団およびそれを作製する方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2018502576A JP2018502576A (ja) | 2018-02-01 |
| JP2018502576A5 JP2018502576A5 (ja) | 2019-02-14 |
| JP6882981B2 true JP6882981B2 (ja) | 2021-06-02 |
Family
ID=55315463
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017535834A Active JP6882981B2 (ja) | 2014-12-30 | 2015-12-30 | Rpe細胞集団およびそれを作製する方法 |
| JP2021012746A Pending JP2021072832A (ja) | 2014-12-30 | 2021-01-29 | Rpe細胞集団およびそれを作製する方法 |
| JP2023010056A Pending JP2023052646A (ja) | 2014-12-30 | 2023-01-26 | Rpe細胞集団およびそれを作製する方法 |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021012746A Pending JP2021072832A (ja) | 2014-12-30 | 2021-01-29 | Rpe細胞集団およびそれを作製する方法 |
| JP2023010056A Pending JP2023052646A (ja) | 2014-12-30 | 2023-01-26 | Rpe細胞集団およびそれを作製する方法 |
Country Status (17)
| Country | Link |
|---|---|
| US (5) | US11891622B2 (ja) |
| EP (2) | EP3674397A1 (ja) |
| JP (3) | JP6882981B2 (ja) |
| KR (1) | KR20170115514A (ja) |
| CN (1) | CN107406830A (ja) |
| AU (2) | AU2015373050B2 (ja) |
| BR (1) | BR112017014345A2 (ja) |
| CA (1) | CA2972575A1 (ja) |
| DK (1) | DK3240892T5 (ja) |
| EA (1) | EA201791416A1 (ja) |
| ES (1) | ES2774456T3 (ja) |
| HK (1) | HK1246348A1 (ja) |
| IL (3) | IL305070A (ja) |
| MX (1) | MX2017008737A (ja) |
| PH (1) | PH12017501218A1 (ja) |
| SG (1) | SG11201705379VA (ja) |
| WO (1) | WO2016108239A1 (ja) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IL301479A (en) | 2009-11-17 | 2023-05-01 | Astellas Inst For Regenerative Medicine | Methods for preparing human RPE cells and pharmaceutical preparations of human RPE cells |
| CN107427534A (zh) | 2014-12-30 | 2017-12-01 | 细胞治疗神经科学有限公司 | 治疗视网膜疾病的方法 |
| JP2018502575A (ja) | 2014-12-30 | 2018-02-01 | セル キュア ニューロサイエンシズ リミテッド | 網膜色素上皮細胞集団の評価法 |
| MX2017008737A (es) | 2014-12-30 | 2018-01-25 | Cell Cure Neurosciences Ltd | Poblaciones de celulas rpe y metodos para generar las mismas. |
| EP3331995A1 (en) * | 2015-08-05 | 2018-06-13 | Cell Cure Neurosciences Ltd. | Preparation of photoreceptors for the treatment of retinal diseases |
| AU2016303632B2 (en) | 2015-08-05 | 2022-09-29 | Cell Cure Neurosciences Ltd. | Preparation of retinal pigment epithelium cells |
| US20200085882A1 (en) * | 2017-03-16 | 2020-03-19 | Lineage Cell Therapeutics, Inc. | Methods for measuring therapeutic effects of retinal disease therapies |
| CN109957545A (zh) | 2017-12-26 | 2019-07-02 | 江苏艾尔康生物医药科技有限公司 | 一种消化rpe细胞的方法 |
| WO2020229628A1 (en) * | 2019-05-15 | 2020-11-19 | Novo Nordisk A/S | Methods for obtaining eye field progenitor cells from human pluripotent stem cells |
| CN111972399B (zh) * | 2020-08-06 | 2021-11-30 | 温州医科大学 | 维持肝细胞活性的保存液 |
| AU2022319764A1 (en) | 2021-07-28 | 2024-01-25 | Lineage Cell Therapeutics, Inc. | Expansion of retinal pigment epithelium cells |
| CN115840045B (zh) * | 2023-02-23 | 2023-04-28 | 中国农业大学 | 一种猪胚胎期毛囊基板前体细胞的鉴定方法及其应用 |
Family Cites Families (79)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL154600B (nl) | 1971-02-10 | 1977-09-15 | Organon Nv | Werkwijze voor het aantonen en bepalen van specifiek bindende eiwitten en hun corresponderende bindbare stoffen. |
| NL154598B (nl) | 1970-11-10 | 1977-09-15 | Organon Nv | Werkwijze voor het aantonen en bepalen van laagmoleculire verbindingen en van eiwitten die deze verbindingen specifiek kunnen binden, alsmede testverpakking. |
| NL154599B (nl) | 1970-12-28 | 1977-09-15 | Organon Nv | Werkwijze voor het aantonen en bepalen van specifiek bindende eiwitten en hun corresponderende bindbare stoffen, alsmede testverpakking. |
| US3901654A (en) | 1971-06-21 | 1975-08-26 | Biological Developments | Receptor assays of biologically active compounds employing biologically specific receptors |
| US3853987A (en) | 1971-09-01 | 1974-12-10 | W Dreyer | Immunological reagent and radioimmuno assay |
| US3867517A (en) | 1971-12-21 | 1975-02-18 | Abbott Lab | Direct radioimmunoassay for antigens and their antibodies |
| NL171930C (nl) | 1972-05-11 | 1983-06-01 | Akzo Nv | Werkwijze voor het aantonen en bepalen van haptenen, alsmede testverpakkingen. |
| US3850578A (en) | 1973-03-12 | 1974-11-26 | H Mcconnell | Process for assaying for biologically active molecules |
| US3935074A (en) | 1973-12-17 | 1976-01-27 | Syva Company | Antibody steric hindrance immunoassay with two antibodies |
| US3996345A (en) | 1974-08-12 | 1976-12-07 | Syva Company | Fluorescence quenching with immunological pairs in immunoassays |
| US4034074A (en) | 1974-09-19 | 1977-07-05 | The Board Of Trustees Of Leland Stanford Junior University | Universal reagent 2-site immunoradiometric assay using labelled anti (IgG) |
| US3984533A (en) | 1975-11-13 | 1976-10-05 | General Electric Company | Electrophoretic method of detecting antigen-antibody reaction |
| US4098876A (en) | 1976-10-26 | 1978-07-04 | Corning Glass Works | Reverse sandwich immunoassay |
| US4879219A (en) | 1980-09-19 | 1989-11-07 | General Hospital Corporation | Immunoassay utilizing monoclonal high affinity IgM antibodies |
| US5011771A (en) | 1984-04-12 | 1991-04-30 | The General Hospital Corporation | Multiepitopic immunometric assay |
| US4666828A (en) | 1984-08-15 | 1987-05-19 | The General Hospital Corporation | Test for Huntington's disease |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4801531A (en) | 1985-04-17 | 1989-01-31 | Biotechnology Research Partners, Ltd. | Apo AI/CIII genomic polymorphisms predictive of atherosclerosis |
| US5272057A (en) | 1988-10-14 | 1993-12-21 | Georgetown University | Method of detecting a predisposition to cancer by the use of restriction fragment length polymorphism of the gene for human poly (ADP-ribose) polymerase |
| SG49267A1 (en) | 1989-08-14 | 1998-05-18 | Photogenesis Inc | Surgical instrument and cell isolation and transplantation |
| US5192659A (en) | 1989-08-25 | 1993-03-09 | Genetype Ag | Intron sequence analysis method for detection of adjacent and remote locus alleles as haplotypes |
| US6045791A (en) | 1992-03-06 | 2000-04-04 | Photogenesis, Inc. | Retinal pigment epithelium transplantation |
| US5281521A (en) | 1992-07-20 | 1994-01-25 | The Trustees Of The University Of Pennsylvania | Modified avidin-biotin technique |
| US5405742A (en) | 1993-07-16 | 1995-04-11 | Cyromedical Sciences, Inc. | Solutions for tissue preservation and bloodless surgery and methods using same |
| US5755785A (en) | 1994-08-12 | 1998-05-26 | The University Of South Florida | Sutureless corneal transplantation method |
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| US5854015A (en) | 1995-10-31 | 1998-12-29 | Applied Food Biotechnology, Inc. | Method of making pure 3R-3'R stereoisomer of zeaxanthin for human ingestion |
| US5941250A (en) | 1996-11-21 | 1999-08-24 | University Of Louisville Research Foundation Inc. | Retinal tissue implantation method |
| US6090622A (en) | 1997-03-31 | 2000-07-18 | The Johns Hopkins School Of Medicine | Human embryonic pluripotent germ cells |
| GB9715584D0 (en) | 1997-07-23 | 1997-10-01 | Eisai Co Ltd | Compounds |
| GB0001930D0 (en) | 2000-01-27 | 2000-03-22 | Novartis Ag | Organic compounds |
| EE200300360A (et) | 2001-01-31 | 2003-12-15 | Pfizer Products Inc. | PDE4 isosüümide inhibiitoritena kasutatavad nikotiinamiidi biarüülderivaadid |
| AU2003201745A1 (en) | 2002-02-11 | 2003-09-04 | Pfizer Limited | Nicotinamide derivatives and a tiotropium salt in combination for the treatment of e.g. inflammatory, allergic and respiratory diseases |
| US7288658B2 (en) | 2003-07-15 | 2007-10-30 | Hoffmann-La Roche Inc. | Process for preparation of pyridine derivatives |
| US7794704B2 (en) | 2004-01-23 | 2010-09-14 | Advanced Cell Technology, Inc. | Methods for producing enriched populations of human retinal pigment epithelium cells for treatment of retinal degeneration |
| EP1799810A2 (en) | 2004-10-12 | 2007-06-27 | Technion Research & Development Foundation Ltd. | Isolated primate embryonic cells and methods of generating and using same |
| ES2525684T3 (es) | 2004-12-29 | 2014-12-29 | Hadasit Medical Research Services And Development Ltd. | Sistemas de cultivo de células madre |
| US20080003676A1 (en) | 2006-06-26 | 2008-01-03 | Millipore Corporation | Growth of embryonic stem cells |
| EP2128244A4 (en) | 2007-01-18 | 2012-05-09 | Riken | INDUCTION / DIFFERENTIATION METHOD IN PHOTORECEPTOR CELL |
| US8956866B2 (en) | 2007-04-18 | 2015-02-17 | Hadasit Medical Research Services And Development Ltd. | Retinal pigment epithelial cells differentiated from embryonic stem cells with nicotinamide and activin A |
| HUE055815T2 (hu) | 2007-10-05 | 2021-12-28 | Univ Wayne State | Vegyületek nyújtott felszabadítására alkalmas dendrimerek |
| JP2011500024A (ja) | 2007-10-12 | 2011-01-06 | アドバンスド セル テクノロジー, インコーポレイテッド | Rpe細胞を生成する改良された方法およびrpe細胞の組成物 |
| US20090226955A1 (en) | 2007-12-21 | 2009-09-10 | University Of Miami | Immortalized retinal pigmented epithelial cells |
| JP2009180811A (ja) | 2008-01-29 | 2009-08-13 | Canon Inc | レンズ鏡筒および撮像装置 |
| KR101025880B1 (ko) | 2008-10-23 | 2011-03-30 | 공희숙 | 성체 줄기세포를 이용한 인슐린 분비세포로의 분화 유도방법 |
| CA2762584A1 (en) | 2009-05-20 | 2010-11-25 | Cardio3 Biosciences S.A. | Pharmaceutical composition for the treatment of heart diseases |
| IL301479A (en) | 2009-11-17 | 2023-05-01 | Astellas Inst For Regenerative Medicine | Methods for preparing human RPE cells and pharmaceutical preparations of human RPE cells |
| WO2012092420A2 (en) | 2010-12-30 | 2012-07-05 | Anthrogenesis Corporation | Methods for cryopreserving and encapsulating cells |
| US20120258451A1 (en) | 2011-04-08 | 2012-10-11 | Advanced Cell Technology, Inc. | Laser isolation of viable cells |
| EP2702135B1 (en) | 2011-04-29 | 2019-04-17 | University of Southern California | Method of cryopreservation of stem cell-derived retinal pigment epithelial cells on polymeric substrate |
| US9107897B2 (en) | 2011-05-18 | 2015-08-18 | The Regents Of The University Of California | Methods for isolating mammalian retinal progenitor cells |
| FI2780022T4 (fi) | 2011-11-14 | 2023-07-06 | Pharmaceutical preparations of human rpe cells and uses thereof | |
| US9658216B2 (en) | 2012-01-31 | 2017-05-23 | Cell Cure Neurosciences Ltd. | Methods of selecting retinal pigmented epithelial cells |
| US9850463B2 (en) | 2012-02-01 | 2017-12-26 | The Regents Of The University Of California | Methods of culturing retinal pigmented epithelium cells, including xeno-free production, RPE enrichment, and cryopreservation |
| CN102618497B (zh) * | 2012-03-15 | 2013-10-02 | 中国人民解放军第三军医大学第一附属医院 | 利用人骨髓间充质干细胞制备视网膜色素上皮细胞的方法 |
| AU2013245406B2 (en) | 2012-04-03 | 2017-07-20 | Reneuron Limited | Stem cell microparticles |
| JP2015519370A (ja) | 2012-05-30 | 2015-07-09 | ニューロテック ユーエスエー, インコーポレイテッド | 低温保存される植込み型細胞培養デバイスおよびその使用 |
| EP2882846B1 (en) | 2012-06-05 | 2018-09-05 | The Regents of the University of California | Methods and compositions for the rapid production of retinal pigmented epithelial cells from pluripotent cells |
| KR20150032552A (ko) | 2012-06-11 | 2015-03-26 | 매큐클리어 인코포레이션 | 치료 제제 및 치료 방법 |
| DK2925856T3 (en) | 2012-12-03 | 2018-05-07 | Biolamina Ab | Process for the preparation of RPE cells |
| EP2951290B1 (en) | 2013-02-01 | 2017-11-29 | The United States of America, as represented by The Secretary, Department of Health and Human Services | Method for generating retinal pigment epithelium (rpe) cells from induced pluripotent stem cells (ipscs) |
| US9943545B2 (en) | 2013-03-15 | 2018-04-17 | Fate Therapeutics, Inc. | Stem cell culture media and methods of enhancing cell survival |
| US20150159134A1 (en) | 2013-12-11 | 2015-06-11 | Pfizer Limited | Method for producing retinal pigment epithelial cells |
| AU2014363032A1 (en) * | 2013-12-11 | 2016-06-09 | Pfizer Limited | Method for producing retinal pigment epithelial cells |
| SG11201604994UA (en) | 2013-12-20 | 2016-07-28 | Essential Pharmaceuticals Llc | Media for cell culture |
| WO2015175504A1 (en) * | 2014-05-12 | 2015-11-19 | The Johns Hopkins University | Differentiation of human pluripotent stem cells into retinal pigment epithelium using hif1 inhibitors |
| ES2880948T3 (es) | 2014-05-12 | 2021-11-26 | Roosterbio Inc | Células listas para imprimir y dispositivos integrados |
| CN107427534A (zh) | 2014-12-30 | 2017-12-01 | 细胞治疗神经科学有限公司 | 治疗视网膜疾病的方法 |
| MX2017008737A (es) | 2014-12-30 | 2018-01-25 | Cell Cure Neurosciences Ltd | Poblaciones de celulas rpe y metodos para generar las mismas. |
| JP2018502575A (ja) | 2014-12-30 | 2018-02-01 | セル キュア ニューロサイエンシズ リミテッド | 網膜色素上皮細胞集団の評価法 |
| US11230696B2 (en) | 2015-07-29 | 2022-01-25 | Hadasit Medical Research Services And Development Ltd. | Large scale production of retinal pigment epithelial cells |
| EP3331995A1 (en) | 2015-08-05 | 2018-06-13 | Cell Cure Neurosciences Ltd. | Preparation of photoreceptors for the treatment of retinal diseases |
| AU2016303632B2 (en) | 2015-08-05 | 2022-09-29 | Cell Cure Neurosciences Ltd. | Preparation of retinal pigment epithelium cells |
| IL299326A (en) | 2015-08-18 | 2023-02-01 | Astellas Inst For Regenerative Medicine | Clinical formulations |
| CN105284787A (zh) | 2015-10-23 | 2016-02-03 | 深圳爱生再生医学科技有限公司 | 骨髓间充质干细胞的保存试剂及其应用 |
| US20180312805A1 (en) | 2015-10-26 | 2018-11-01 | Cell Cure Neurosciences Ltd. | Preparation of Retinal Pigment Epithelium Cells |
| KR101982801B1 (ko) | 2015-10-28 | 2019-05-30 | 서울대학교 산학협력단 | 망막색소상피 분화 유도용 조성물 |
| US20200085882A1 (en) | 2017-03-16 | 2020-03-19 | Lineage Cell Therapeutics, Inc. | Methods for measuring therapeutic effects of retinal disease therapies |
| EP3731789A4 (en) | 2017-12-29 | 2021-09-08 | Cell Cure Neurosciences Ltd. | RETINAL PIGMENTARY EPITHELIUM CELL COMPOSITIONS |
-
2015
- 2015-12-30 MX MX2017008737A patent/MX2017008737A/es unknown
- 2015-12-30 WO PCT/IL2015/051269 patent/WO2016108239A1/en active Application Filing
- 2015-12-30 EA EA201791416A patent/EA201791416A1/ru unknown
- 2015-12-30 AU AU2015373050A patent/AU2015373050B2/en active Active
- 2015-12-30 CN CN201580076740.6A patent/CN107406830A/zh active Pending
- 2015-12-30 EP EP19207191.8A patent/EP3674397A1/en active Pending
- 2015-12-30 KR KR1020177021264A patent/KR20170115514A/ko unknown
- 2015-12-30 EP EP15832827.8A patent/EP3240892B9/en active Active
- 2015-12-30 CA CA2972575A patent/CA2972575A1/en active Pending
- 2015-12-30 BR BR112017014345-3A patent/BR112017014345A2/pt not_active Application Discontinuation
- 2015-12-30 US US15/539,473 patent/US11891622B2/en active Active
- 2015-12-30 IL IL305070A patent/IL305070A/en unknown
- 2015-12-30 ES ES15832827T patent/ES2774456T3/es active Active
- 2015-12-30 JP JP2017535834A patent/JP6882981B2/ja active Active
- 2015-12-30 SG SG11201705379VA patent/SG11201705379VA/en unknown
- 2015-12-30 IL IL273405A patent/IL273405B2/en unknown
- 2015-12-30 DK DK15832827.8T patent/DK3240892T5/da active
-
2017
- 2017-06-29 IL IL253238A patent/IL253238B/en active IP Right Grant
- 2017-06-29 PH PH12017501218A patent/PH12017501218A1/en unknown
-
2018
- 2018-05-03 HK HK18105713.5A patent/HK1246348A1/zh unknown
-
2021
- 2021-01-29 JP JP2021012746A patent/JP2021072832A/ja active Pending
- 2021-05-18 US US17/323,441 patent/US20210332325A1/en not_active Abandoned
- 2021-12-08 US US17/546,011 patent/US20220169981A1/en not_active Abandoned
- 2021-12-08 US US17/546,013 patent/US20220154141A1/en active Pending
- 2021-12-16 US US17/552,743 patent/US20220169982A1/en not_active Abandoned
-
2022
- 2022-12-22 AU AU2022291539A patent/AU2022291539A1/en active Pending
-
2023
- 2023-01-26 JP JP2023010056A patent/JP2023052646A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6882981B2 (ja) | Rpe細胞集団およびそれを作製する方法 | |
| US20230028133A1 (en) | Methods of treating retinal diseases | |
| JP2021078513A (ja) | 網膜色素上皮細胞集団の評価法 | |
| NZ733006A (en) | Rpe cell populations and methods of generating same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171004 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190104 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20190104 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20191023 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20191114 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20200212 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20201014 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210129 |
|
| C60 | Trial request (containing other claim documents, opposition documents) |
Free format text: JAPANESE INTERMEDIATE CODE: C60 Effective date: 20210129 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20210208 |
|
| C21 | Notice of transfer of a case for reconsideration by examiners before appeal proceedings |
Free format text: JAPANESE INTERMEDIATE CODE: C21 Effective date: 20210210 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20210412 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20210507 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6882981 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |