JP6798673B2 - 保護膜前駆体液、保護膜形成方法、および複合体 - Google Patents
保護膜前駆体液、保護膜形成方法、および複合体 Download PDFInfo
- Publication number
- JP6798673B2 JP6798673B2 JP2015236767A JP2015236767A JP6798673B2 JP 6798673 B2 JP6798673 B2 JP 6798673B2 JP 2015236767 A JP2015236767 A JP 2015236767A JP 2015236767 A JP2015236767 A JP 2015236767A JP 6798673 B2 JP6798673 B2 JP 6798673B2
- Authority
- JP
- Japan
- Prior art keywords
- protective film
- sample
- paramylon
- acetylparamylon
- complex
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images







Landscapes
- Paints Or Removers (AREA)
- Other Surface Treatments For Metallic Materials (AREA)
- Application Of Or Painting With Fluid Materials (AREA)
- Polyethers (AREA)
Description
図1は、パラミロンからアセチルパラミロンへの合成スキームである。以下の手順で、パラミロンからアセチルパラミロンを合成した。まず、非特許文献1に記載された方法で、ミドリムシからパラミロンを抽出した。つぎに、このパラミロン10.00g(グルコース部が61.70mmol)と、N,N−ジメチルアセトアミド(DMAc)500mLと、塩化リチウム(LiCl)7.85g(185.19mmol)を、温度120℃の窒素雰囲気下で0.5時間撹拌し、均一な溶液を得た。そして、この溶液の温度を70℃に下げた後、DMAc1000mLと、ピリジン168mL(2.09mol)と、無水酢酸240mL(2.54mol)をこの溶液に加えて撹拌し、反応混合液を得た。
1H NMR(CD2Cl2):δ=5.03-4.48 (m), 4.51-4.12 (m), 4.11-3.91 (m), 3.88-3.36 (m), 2.12 (s), 2.06 (s), 2.00 (s). DSace 2.01.
FT-IR(cm-1):1737, 1631, 1434, 1367, 1213, 1077, 1038, 1026, 892.
1H NMR(CD2Cl2):δ=4.97-4.70 (m), 4.41-4.21 (m), 4.05-3.91 (m), 3.74-3.47 (m), 2.11 (s), 2.06 (s), 2.00 (s). DSace 2.37.
FT-IR(cm-1):1736, 1629, 1434, 1368, 1211, 1029, 891, 834, 669.
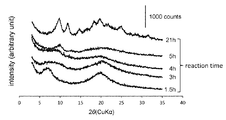
複合体の原料となるアセチルパラミロンの合成に適した反応時間を決定するために、各反応時間にサンプリングした反応中間体の粉末X線回折を測定した。その結果を図2に示す。反応時間1.5時間の試料は6.8°と19.6°にブロードピークが見られた。出発原料であるパラミロン粒子では6.8°、19.3°、20.5°、24.0°に鋭いピークが見られることから、反応時間1.5時間のサンプルにはパラミロン粒子の結晶性がある程度残っていると考えられる。反応時間3時間および4時間のサンプルについては、ピークのブロード化が見られた。反応時間5時間のサンプル(試料A)については9.8°にピークが見られ、さらに反応時間21時間のサンプル(試料B)では9.8°、11.8°、14.8°、18.2°、19.9°、21.3°、21.8°、25.1°、27.3°、および31.7°に新たなピークが出現した。
図3は、セルロースからアセチルセルロースへの合成スキームである。以下の手順で、主成分がセルロースである溶解パルプからアセチルセルロースを合成した。まず、溶解パルプ2.00g(グルコース部が12.33mmol)とDMAc75mLを、温度150℃の窒素雰囲気下で1時間撹拌し、不均一な混合物を得た。つぎに、LiCl3.50g(82.6mmol)をこの混合物に加え、さらに1時間撹拌した。そして、この混合物を室温で22時間放置して溶液を得た。つぎに、DMAc75mLと、ピリジン34mL(422mmol)と、無水酢酸48mL(508mmol)をこの溶液に加え、70℃で6時間撹拌した後、室温で15時間撹拌した。そして、この溶液にメタノール300mLを加えて白色固形分を沈殿させた。つぎに、遠心分離で上澄液を除去し、残った固形物をメタノール100mLで15分間撹拌洗浄した。
1H NMR(CD2Cl2):δ=5.12-4.97 (m), 4.79-4.68 (m), 4.47-4.26 (m), 4.13-3.93 (m), 3.78-3.65 (m), 3.57-3.46 (m), 2.07 (s), 1.98 (s), 1.92 (s). DSace 2.75.
FT-IR(cm-1):1733, 1362, 1208, 1030, 895.
図3に示す反応スキームに従って、微結晶性セルロースからアセチルセルロースを合成した。まず、微結晶性セルロース2.00g(グルコース部が12.33mmol)とDMAc75mLを、温度150℃の窒素雰囲気下で2時間撹拌して混合物を得た。つぎに、LiCl3.51g(82.8mmol)をこの混合物に加え、さらに1時間撹拌した。そして、この混合物を室温で22時間放置して溶液を得た。つぎに、DMAc75mLと、ピリジン34mL(422mmol)と、無水酢酸48mL(508mmol)をこの溶液に加え、70℃で6時間撹拌した後、室温で15時間撹拌した。そして、この溶液にメタノール300mLを加えて白色固形分を沈殿させた。つぎに、遠心分離で上澄液を除去し、残った固形物をメタノール100mLで15分間撹拌洗浄した。
1H NMR(CD2Cl2):δ=5.06-4.97 (m), 4.76-4.70 (m), 4.44-4.26 (m), 4.12-4.00 (m), 3.77-3.67 (m), 3.57-3.47 (m), 2.07 (s), 1.98 (s), 1.92 (s). DSace 2.82.
FT-IR(cm-1):1733, 1623, 1420, 1363, 1209, 1076, 1038, 1024, 967, 894.
溶媒キャスト法により自立透明薄膜を調製した。すなわち、75mm×100mmのフッ素樹脂皿に、試料A250mgを30mLのクロロホルムに溶かした溶液を注ぎ、この皿をアルミ箔で覆い、アルミ箔に直径約1mmの穴を5箇所設けた。試料B、試料C、および試料D250mgをそれぞれ30mLのジクロロメタンに溶かした溶液も同様に調製した。室温で8時間以上かけて溶媒を徐々に蒸発させて、試料A、試料B、試料C、および試料Dの透明薄膜を得た。
上記膜の調製方法と同様の方法で、試料A、試料B、試料C、および試料Dからそれぞれ構成される保護膜(以下、試料Aから構成される保護膜を「保護膜A」と、試料Bから構成される保護膜を「保護膜B」と、試料Cから構成される保護膜を「保護膜C」と、試料Dから構成される保護膜を「保護膜D」とそれぞれいうことがある)を基材上に形成して、複合体(以下、保護膜Aを備える複合体を「複合体A」と、保護膜Bを備える複合体を「複合体B」と、保護膜Cを備える複合体を「複合体C」と、保護膜Dを備える複合体を「複合体D」とそれぞれいうことがある)を得た。
図6は、保護膜A、保護膜B、保護膜C、および保護膜DのX線回折の分析結果を示している。なお、回折チャートの明確化のため、保護膜CのX線回折強度を1/10にした。保護膜Aでは、9.5°付近に幅広い散乱が観察された。ブランク測定では、この角度付近に散乱と回折が見られないことから、この散乱は低結晶領域に由来するアモルファスのハローパターンと推測される。また、弱いハローパターンが16°〜18°付近に見られたが、これもアモルファス領域に由来するものと考えられる。これに対して保護膜Bでは、ハローパターンに加えて、鋭いピークが9.3°、16.2°、16.5°、18.7°、および25.2°に見られた。以上の結果は、保護膜Aと保護膜Bが結晶領域とアモルファス領域から構成されていること、保護膜Bは保護膜Aより結晶化度が高いことを示している。
各試料の数平均分子量(Mn)と重量平均分子量(Mw)は、Wyatt Technology社製の多角度光散乱検出器miminiDAWN、動的光散乱モジュールQELS、および高性能示差屈折率検出器Optilab rEX一式に、昭和電工社製カラムKD-805を接続したSEC-MALLSシステム(移動相:クロロホルム、カラム温度:40℃、流速:1.0mL/min)により決定した。
1H NMR(CDCl3):δ=5.23-4.97 (m), 4.83-4.21 (m), 4.13-3.92 (m), 3.89-3.23 (m), 2.41-2.21 (m), 2.12 (s), 2.00 (s), 1.93 (s), 1.61 (m), 1.25 (s), 0.88 (t, J = 6.0). DSace 2.15, DSlac 0.50.
FT-IR(cm-1):2923, 2851, 1748, 1467, 1427, 1365, 1231, 1156, 1082, 1038, 900, 627, 602.
示差走査熱量測定(DSC)は、理学電機社製Thermo PlusEVO II DSC8230(昇温速度:10.0℃/min、25℃〜230℃)を用いて行い、ガラス転移温度(Tg)は2回目のスキャンで得られたサーモグラムから決定した。熱重量分析(TG)は、理学電機社製Thermo plus EVO II TG8120(昇温速度:10.0℃/min、25℃〜500℃、窒素気流:100mL/min)を用いて行い、5%重量減少温度(Td5)を決定した。

溶媒キャスト法による膜の調製工程は、ハロゲン系溶媒に溶けているアセチルパラミロンやアセチルセルロースが固化する過程を含んでいる。膜の調製工程でのアセチルパラミロンの分子配列能の効果について知見を得ることを目的として、試料Bと試料Cのジクロロメタン溶液の粘度を測定した。その結果を表1に示す。試料Bの溶液の粘度は、試料Cの溶液の粘度より大きいことが明らかとなった。この結果は、試料Bの溶液は試料Cの溶液よりも規則性の高い高分子、すなわち、らせんを基本構造とする分子集合体を含んでいることを示している。この結果より、試料Bから構成される膜の高結晶性は、粘度測定から示唆される溶液中での試料Bの分子が本来持っている分子配列能の高さに起因するものと考えられた。
テンシロン万能材料試験機(RTG-1225、エー・アンド・デイ社)を用いて、試料A、試料B、および試料Cからそれぞれ構成される膜の引張試験を室温で行った。縦50mm×横10mm×厚さ約0.05mmの試験片で30mmの間隔を設け、引張速度3mm/分で引っ張った。表1にこの結果を示している。試料Bの最大応力は、試料Cの最大応力と同等であった。破断伸びは試料Aが最大であった。これは、試料Aが非晶質であることに起因すると考えられる。ヤング率は試料Cが最大、試料Aが最小で、試料Bのヤング率は試料Cのヤング率の約85%であった。この結果から、アセチルパラミロンはアセチルセルロースより変形しやすい、すなわち柔軟性が高いことがわかった。
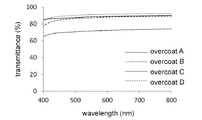
分光光度計(UV-2500、島津製作所)を用いて、保護膜A、保護膜B、保護膜C、および保護膜Dのそれぞれの薄膜(約10mm×10mmの大きさ)の光透過率を測定した。測定に用いた膜の厚さは、保護膜Aが21.8±2.1μm、保護膜Bが42.3±11.6μm、保護膜Cが25.3±5.3μm、保護膜Dが8.6±1.1μmであった。保護膜A、保護膜B、保護膜C、および保護膜Dの透過率スペクトルを図9に示す。図9に示すように、保護膜Aまたは保護膜Bでは、可視光域での光透過率が約90%であった。
波長632nmの光を用いて、保護膜A、保護膜B、および保護膜Dの屈折率をプリズムカプラー(2010/M、Metricon社)で測定した。なお、保護膜Cはその透明性の低さのため測定対象から除外した。測定に用いた膜の大きさは全て40mm×40mmで、膜の厚さは、保護膜Aが21.8±2.1μm、保護膜Bが42.3±11.6μm、保護膜Dが8.6±1.1μmであった。表2に屈折率(nx/ny)と複屈折(Δn = |nx-ny|)を示す。
Claims (7)
- 基材上に塗布した後に乾燥させて、前記基材上に保護膜を形成するための保護膜前駆体液であって、
パラミロンを構成するグルコースの一部のヒドロキシル基のHがアセチル基のみで置換されているとともに、前記グルコース1分子当たりのアセチル基置換度が2.01以上2.37以下であるアセチルパラミロンと、
有機溶剤と、
を含有する保護膜前駆体液。 - 請求項1において、
前記アセチルパラミロンが、ミドリムシ由来のパラミロンをアセチル化したものである保護膜前駆体液。 - 請求項1または2において、
前記有機溶剤が、クロロホルムおよびジクロロメタンの少なくとも一方である保護膜前駆体液。 - 基材上に保護膜を形成する保護膜形成方法であって、
請求項1から3のいずれかの保護膜前駆体液を、前記基材上に塗布して塗布層を形成する塗布工程と、
前記塗布層から前記有機溶剤を除去する乾燥工程と、
を有する保護膜形成方法。 - 基材と、前記基材上に形成された保護膜とを有する複合体であって、
前記保護膜の主成分がアセチルパラミロンであり、
前記アセチルパラミロンは、パラミロンを構成するグルコースの一部のヒドロキシル基のHがアセチル基のみで置換されており、
前記アセチルパラミロンのグルコース1分子当たりのアセチル基置換度が2.01以上2.37以下である複合体。 - 請求項5において、
前記アセチルパラミロンが、ミドリムシ由来のパラミロンをアセチル化したものである複合体。 - 請求項5または6において、
前記基材が液晶基板である複合体。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015236767A JP6798673B2 (ja) | 2015-12-03 | 2015-12-03 | 保護膜前駆体液、保護膜形成方法、および複合体 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015236767A JP6798673B2 (ja) | 2015-12-03 | 2015-12-03 | 保護膜前駆体液、保護膜形成方法、および複合体 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2017101177A JP2017101177A (ja) | 2017-06-08 |
| JP2017101177A5 JP2017101177A5 (ja) | 2018-10-11 |
| JP6798673B2 true JP6798673B2 (ja) | 2020-12-09 |
Family
ID=59016295
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015236767A Active JP6798673B2 (ja) | 2015-12-03 | 2015-12-03 | 保護膜前駆体液、保護膜形成方法、および複合体 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6798673B2 (ja) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017218566A (ja) * | 2016-06-03 | 2017-12-14 | 株式会社ユーグレナ | パラミロンエステル誘導体及び繊維 |
| US12570769B2 (en) | 2020-05-08 | 2026-03-10 | Nec Corporation | Paramylon-based resin, molding material, molded body, and production method for paramylon-based resin |
| JP7640069B2 (ja) * | 2021-01-07 | 2025-03-05 | 国立研究開発法人産業技術総合研究所 | 疎水性ナノファイバー及びその製造方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3069206B2 (ja) * | 1992-12-02 | 2000-07-24 | 富士写真フイルム株式会社 | 液晶表示装置 |
| JP4501333B2 (ja) * | 2002-09-04 | 2010-07-14 | 日本製紙株式会社 | 塗料組成物及びハードコートフィルム |
| JP2008107659A (ja) * | 2006-10-26 | 2008-05-08 | Kaneka Corp | 光学補償用塗工膜、該光学補償用塗工膜形成用塗工液、偏光板、液晶表示装置、更にはその形成方法 |
| TWI425258B (zh) * | 2009-02-03 | 2014-02-01 | Jiro Corporate Plan Inc | A polarizing element outer protective film, a polarizing plate, and a liquid crystal display element |
| JP5720492B2 (ja) * | 2011-08-23 | 2015-05-20 | コニカミノルタ株式会社 | 位相差フィルム、偏光板及び液晶表示装置 |
| EP2921504B1 (en) * | 2012-11-14 | 2017-07-12 | National Institute of Advanced Industrial Science and Technology | Beta-1,3-glucan derivative and method for producing beta-1,3-glucan derivative |
| JP2014098095A (ja) * | 2012-11-14 | 2014-05-29 | National Institute Of Advanced Industrial & Technology | β−1,3−グルカン誘導体、及びβ−1,3−グルカン誘導体の製造方法 |
| JP2015011236A (ja) * | 2013-06-28 | 2015-01-19 | 株式会社ケンシュー | 光学用樹脂材料、光学用部品、及び光学用樹脂材料の非晶性の判定方法 |
-
2015
- 2015-12-03 JP JP2015236767A patent/JP6798673B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2017101177A (ja) | 2017-06-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2811340T3 (es) | Preparación de poli(ésteres de alfa-1,3-glucano) usando anhídridos orgánicos cíclicos | |
| KR101118879B1 (ko) | 위상차 필름용 셀룰로오스 디아세테이트 | |
| CN110358099B (zh) | Pva基复合材料及其前驱体、重塑产品、复合水凝胶、复合薄膜及制备和应用 | |
| BR112015015609B1 (pt) | Filmes e método para a preparação de um filme de éster de poli alfa-1,3-glucano | |
| JP6798673B2 (ja) | 保護膜前駆体液、保護膜形成方法、および複合体 | |
| Wang et al. | Rapid microwave-assisted ionothermal dissolution of cellulose and its regeneration properties | |
| Shibakami et al. | Preparation of transparent self-standing thin films made from acetylated euglenoid β-1, 3-glucans | |
| EP3663328A1 (en) | Hydrophilic polyamide or polyimide | |
| Zhang et al. | Preparation and characterization of regenerated cellulose blend films containing high amount of poly (vinyl alcohol)(PVA) in ionic liquid | |
| Liu et al. | Efficient synthesis of cellulose acetate through one-step homogeneous acetylation of cotton cellulose in binary ionic liquids | |
| JP6093947B2 (ja) | β−1,3−グルカンナノファイバー及びその製造方法 | |
| Xiao et al. | Synthesis and characterization of cellulose-graft-poly (L-lactide) via ring-opening polymerization. | |
| Ding et al. | Favorable compatibility efficiency and thermal stability of PLA/P4HB/PGMA blends contributed by phase interface-located chain expansion reaction | |
| Gardella et al. | On stereocomplexed polylactide materials as support for PAMAM dendrimers: synthesis and properties | |
| Li et al. | Study on the structure–property relationship of starch/PLA composite films modified by the synergistic effect of aromatic rings and aliphatic chains | |
| Lee et al. | Synthesis and characterization of dextrin derivatives by heterogeneous esterification | |
| WO2019004212A1 (ja) | d14圧電定数を有する圧電フィルム製造用多糖類組成物およびd14圧電定数を有する圧電フィルムの製造方法 | |
| Számel et al. | Thermal analysis of cellulose acetate modified with caprolactone | |
| Liu et al. | Chiral, pH responsive hydrogels constructed by N‐Acryloyl‐alanine and PEGDA/α‐CD inclusion complex: preparation and chiral release ability | |
| CN108463473B (zh) | 乙酸纤维素及乙酸纤维素的制造方法 | |
| Liebert et al. | Microscopic visualization of nanostructures of cellulose derivatives | |
| Hesse et al. | Studies on the Film Formation of Polysaccharide Based Furan‐2‐Carboxylic Acid Esters | |
| Pan et al. | Design and fabrication of xylan-graft-poly (methyl methacrylate) thermoplastic via SARA ATRP | |
| Ifuku et al. | Preparation of 6-O-(4-alkoxytrityl) celluloses and their properties | |
| Batra et al. | Enhancement of functional properties by blending cocoon extracted Antheraea mylitta silk fibroin with polyvinyl alcohol for applications in biomedical field |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A80 | Written request to apply exceptions to lack of novelty of invention |
Free format text: JAPANESE INTERMEDIATE CODE: A80 Effective date: 20151210 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20180828 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180829 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20190724 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20190821 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20191018 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20191220 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200520 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200717 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20201104 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20201112 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6798673 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |