JP6123097B2 - チエノピリミジン化合物を製造するための方法 - Google Patents
チエノピリミジン化合物を製造するための方法 Download PDFInfo
- Publication number
- JP6123097B2 JP6123097B2 JP2015536109A JP2015536109A JP6123097B2 JP 6123097 B2 JP6123097 B2 JP 6123097B2 JP 2015536109 A JP2015536109 A JP 2015536109A JP 2015536109 A JP2015536109 A JP 2015536109A JP 6123097 B2 JP6123097 B2 JP 6123097B2
- Authority
- JP
- Japan
- Prior art keywords
- acid
- give
- piperazin
- reacting
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- VEFJGLZHHCKBPO-PKPIPKONSA-N C[C@@H](C(N1C=CNCC1)O)O Chemical compound C[C@@H](C(N1C=CNCC1)O)O VEFJGLZHHCKBPO-PKPIPKONSA-N 0.000 description 1
- YOVVNQKCSKSHKT-HNNXBMFYSA-N C[C@@H](C(N1CCN(Cc2c(C)c(nc(-c3cnc(N)nc3)nc3N4CCOCC4)c3[s]2)CC1)=O)O Chemical compound C[C@@H](C(N1CCN(Cc2c(C)c(nc(-c3cnc(N)nc3)nc3N4CCOCC4)c3[s]2)CC1)=O)O YOVVNQKCSKSHKT-HNNXBMFYSA-N 0.000 description 1
- VRPXEWRMAZIPSK-AWEZNQCLSA-N C[C@@H](C(N1CCN(Cc2c(C)c(nc(C)nc3N4CCOCC4)c3[s]2)CC1)=O)O Chemical compound C[C@@H](C(N1CCN(Cc2c(C)c(nc(C)nc3N4CCOCC4)c3[s]2)CC1)=O)O VRPXEWRMAZIPSK-AWEZNQCLSA-N 0.000 description 1
- YVPBSLIVIGORID-ZDUSSCGKSA-N C[C@@H](C(N1CCN(Cc2c(C)c(nc(nc3N4CCOCC4)Cl)c3[s]2)CC1)=O)O Chemical compound C[C@@H](C(N1CCN(Cc2c(C)c(nc(nc3N4CCOCC4)Cl)c3[s]2)CC1)=O)O YVPBSLIVIGORID-ZDUSSCGKSA-N 0.000 description 1
- JQRBBKFUEZDIRG-LURJTMIESA-N C[C@@H](C(N1CCNCC1)=O)O Chemical compound C[C@@H](C(N1CCNCC1)=O)O JQRBBKFUEZDIRG-LURJTMIESA-N 0.000 description 1
- HAHDZQASMZYULP-UHFFFAOYSA-N Cc1c(C=O)[s]c(C(N2CCOCC2)=N2)c1NC2Cl Chemical compound Cc1c(C=O)[s]c(C(N2CCOCC2)=N2)c1NC2Cl HAHDZQASMZYULP-UHFFFAOYSA-N 0.000 description 1
- YICRPERKKBDRSP-UHFFFAOYSA-N Cc1c[s]c(C(OC)=O)c1N Chemical compound Cc1c[s]c(C(OC)=O)c1N YICRPERKKBDRSP-UHFFFAOYSA-N 0.000 description 1
- CGHYQZASLKERLV-UHFFFAOYSA-N Nc1ncc(B(O)O)cn1 Chemical compound Nc1ncc(B(O)O)cn1 CGHYQZASLKERLV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/185—Radicals derived from carboxylic acids from aliphatic carboxylic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Health & Medical Sciences (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
Description
本非仮出願は、37 CFR§1.53(b)に基づき出願されたものであるが、全体を参考することにより援用される2012年10月10日に出願された米国仮出願番号61/711900の35USC§119(e)に基づく利益を主張する。
用語「キラル」は、鏡像パートナーの重ね合わせができない特性を有する分子を意味し、一方で、「アキラル」という用語は、それらの鏡像パートナーに重ね合わせることができる分子を指す。
本発明は、GDC−0980合成のためのプロセス、方法、試薬、及び中間体を含み、GDC−0980とは、以下の構造
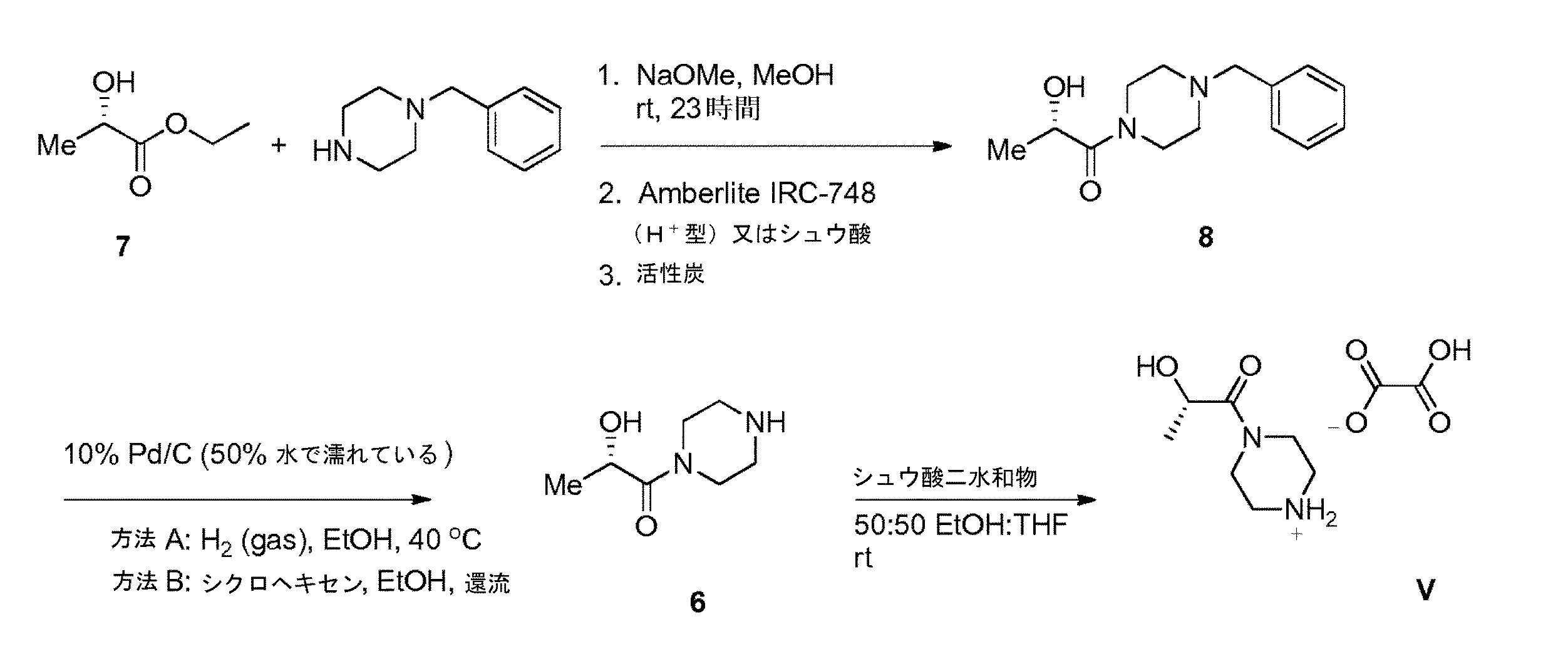
スキーム3は、(S)−2−ヒドロキシプロパン酸 (L−乳酸) 1からの中間体、(S)−2−ヒドロキシ−1−(ピペラジン−1−イル)プロパン−1−オンのシュウ酸塩Vの合成を示す。1のアセチル化によって(S)−2−アセトキシプロパン酸 2が与えられ、続いて、塩化オキサリルのような塩素化試薬での処理が行われ、酸塩化物、(S)−1−クロロ−1−オキソプロパン−2−イル アセテート 3(実施例6)が与えられた。トリエチルアミンの存在下でのジクロロメタン中の1−ベンジルピペラジンのジヒドロクロリド 塩との3の反応により、(S)−1−(4−ベンジルピペラジン−1−イル)−1−オキソプロパン−2−イル アセテート 4(実施例7)が与えられた。水酸化リチウムでの4のアセテートの加水分解により、(S)−1−(4−ベンジルピペラジン−1−イル)−2−ヒドロキシプロパン−1−オン 5(実施例8)が与えられ、続いてN−ベンジル基を除去するために水素添加が行われ、((S)−2−ヒドロキシ−1−(ピペラジン−1−イル)プロパン−1−オン 6(実施例9)が与えられた。シュウ酸塩は、エタノール及びテトラヒドロフラン中のシュウ酸を用いて6から形成され、V(実施例9)が与えられた。
GDC−0980は、ヒトを含む哺乳動物における過剰増殖性疾患の治療的処置(予防的治療を含む)のための治療的組み合わせにおいて使用するための標準的な薬務に従って処方され得る。本発明は、1又は複数の薬学的に許容される担体、流動促進剤、希釈剤、又は賦形剤と関連してGDC−0980を含む医薬組成物を提供する。
メチル 3−アミノ−4−メチルチオフェン−2−カルボンキシレート IX(100g、0.584mol)及び酢酸(750ml、13.1mol)を、透明な溶液を得るために5分間撹拌した。水(120mL)中のシアン酸カリウム(56.8g、0.70mol)の溶液を20分にわたりゆっくりと加え、混合物を1.5時間撹拌した。水(120mL)中の追加のシアン酸カリウム(56.8g、0.70mol)を20分にわたりゆっくりと加え、混合物を2時間撹拌した。水(600mL)を加え、混合物を10℃に冷却し、2時間攪拌した。固体を濾過により回収し、冷水(250mL)で洗浄した。次に、固体を水(1.4L)中の水酸化ナトリウム溶液(79.4g、1.99mol)中で12時間攪拌した。pHは、水性濃塩酸溶液(35wt%、110mL)をゆっくりと加えることにより6−7に調整され、次いで、5分間撹拌した。得られた固体を濾過により回収し、水(2×250mL)で洗浄し、24時間50℃で減圧乾燥させ、7−メチルチエノ[3,2−d]ピリミジン−2,4(1H,3H)−ジオン VIIIがオフホワイトの固体(89.6g、84%の収率)として得られた。1H NMR(500MHz、DMSO−d6)δ7.68(s、1H)、2.20(s、3H);LCMS(ESI pos)m/z[M+H]183。
アセトニトリル(450mL)中の7−メチルチエノ[3,2−d]ピリミジン−2,4(1H,3H)−ジオン VIII(89.4g、0.491mol)とN,N−ジメチルアニリン(44.6g、0.368mol)との混合物に、10分にわたってオキシ塩化リン(312g、2.04mol)を加えた。反応混合物を85℃に加熱し、24時間撹拌した。室温に冷却した後、10℃未満の温度を維持しながら、混合物をゆっくりと氷(900g)と水(300mL)との混合物に移した。混合物を30分間その温度で撹拌した。固体を濾過により回収し、水(450mL)で洗浄し、24時間50℃で減圧乾燥させ、2,4−ジクロロ−7−メチルチエノ[3,2−d]ピリミジン VIIがオフホワイトの固体(97.0g、90%の収率)として得られた。1H NMR(400MHz、CDCl3)δ7.75(s、1H)、2.50(s、3H);LCMS(ESI pos)m/z[M+H]220。
2,4−ジクロロ−7−メチルチエノ[3,2−d]ピリミジン VII(90g、0.411mol)とメタノール(900mL)との混合物を10℃に冷却した。15℃未満の温度を維持しながら、モルホリン(89.5g、1.03mol)を加えた。反応混合物を2時間撹拌し、次いで5℃まで冷却し、さらに1時間撹拌した。固体を濾過により回収し、水(450mL)で洗浄し、24時間50℃で減圧乾燥し、白色固体(105g、95%の収率)として4−(2−クロロ−7−メチルチエノ[3,2−d]ピリミジン−4−イル)モルホリノ VIが得られた。1H NMR(400MHz、CDCl3)δ7.94(s、1H)、3.95−3.86(m、4H)、3.80−3.71(m、4H)、2.9(s、3H);LCMS(ESI pos)m/z [M+H]270。
4−(2 クロロ−7−メチルチエノ[3,2 d]ピリミジン 4 yl)モルホリン VI(27.0g、100mmol)を適切な大きさの反応器に充填し、テトラヒドロフラン(無水、270mL)が加えられた。反応混合物を−10℃未満に冷却し、テトラヒドロフラン(25.7g、50.0mmol)中のi−PrMgClの20wt%溶液をゆっくりと加え、続いて、−10℃未満の内部温度を維持しながら、ヘプタン(30.0g、117mmol)中のn−BuLiの25wt%溶液をゆっくりと加えた。混合物は、−10℃未満で2時間攪拌することが許された。−10℃未満の内部温度を維持しながら、無水N,N−ジメチルホルムアミド(14.6g、200mmol)をゆっくりと加えた。反応混合物を1−2時間撹拌し、80%酢酸、37%塩酸水溶液、イソプロパノール及び水の混合物に移した。得られたスラリーを50−55℃に加熱し、1−3時間撹拌した。懸濁液を減圧濃縮し、テトラヒドロフランを除去した。次に、懸濁液を室温に冷却し、濾過し、水ですすいだ。ケーキを40−60℃で減圧乾燥し、2−クロロ−7−メチル−4−モルホリノチエノ[3,2−d]ピリミジン−6−カルボアルデヒド IVが黄色固体(29.2g、98%の収率)として得られた。1H NMR(400MHz、CDCl3)δ10.38(s、1H)、4.03−4.05(m、4H)、3.85−3.87(m、4H)、2.76(s、3H)。
ジクロロメタン(50.0kg)中の(S)−2−ヒドロキシプロパン酸(L−乳酸)1(35.0kg、388mol)の溶液を5−10℃に冷却し、反応温度を10−20℃に維持しながら、塩化アセチル(75.0kg、955mol)を加えた。反応混合物を10−20℃で4時間撹拌した。ジクロロメタン(240kg)が加えられ、続いて、反応温度を0−15℃で維持する速度で塩化オキサリル(139kg、1095mol)が加えられた。反応混合物を10−20℃で10時間熟成させ、その混合物を減圧濃縮し、(S)−1−クロロ−1−オキソプロパン−2−イル アセテート 3を含む残留物が与えられた。
容器にジクロロメタン(260kg)を充填し、続いて、トリエチルアミン(65.0kg、642mol)を加えた。混合物を0−10℃に冷却し、4−ベンジルピペラジン ジヒドロクロリド(31.8kg、128mol)を加えた。反応温度を5−15℃に維持しながら、混合物に、実施例6からの(S)−1−クロロ−1−オキソプロパン−2−イル アセテート 3を加えた。反応混合物を10−20℃で10時間撹拌した。氷水(50kg)を加え、層を分離した。水層をジクロロメタン(2×50kg)で抽出した。有機層を合わせ、5−10℃まで冷却した。HCl水溶液(4N)溶液をゆっくりと加え、pHを6−7に調整した。層を分離し、水層をジクロロメタン(2×50kg)で抽出した。有機層を組み合わせ、無水硫酸ナトリウム上で乾燥させ、濾過し、減圧濃縮して、(S)−1−(4−ベンジルピペラジン−1−イル)−1−オキソプロパン−2−イル アセテート 4を含む残留物が与えられた。
メタノール(300kg)を(S)−1−(4−ベンジルピペラジン−1−イル)−1−オキソプロパン−2−イル アセテート 4を含む実施例7からの残留物に加え、混合物を0−10℃に冷却した。反応温度を0−15℃に維持する速度で、水(100kg)中の水酸化リチウム一水和物の溶液(13.6kg、324mol)を加えた。2時間熟成させた後、5−15℃で酢酸(4.5kg、75mol)により、pHを7に調整した。混合物を減圧濃縮した。残留物にジクロロメタン(150kg)を加え、層を分離した。水層をジクロロメタン(2×150kg)で抽出した。有機層を組み合わせ、無水硫酸ナトリウム上で乾燥させ、濾過し、減圧濃縮した。エチルアセテート(31kg)を残留物に加え、続いて、シクロヘキサン(183kg)をゆっくりと加えた。混合物を40−50℃に加熱し1時間撹拌した。混合物を0−10℃に冷却し、8時間熟成させた。固体を濾過により回収し、冷シクロヘキサンで洗浄し、50℃で12時間、減圧乾燥させ、(S)−1−(4−ベンジルピペラジン−1−イル)−2−ヒドロキシプロパン−1−オン 5(46kg、71%の収率、HPLCによる98%の純度)が得られた。1H NMR(300MHz、CDCl3)δ7.42−7.20(m、5H)、4.43(q、J=6.4Hz、1H)、3.84(一般的な(broad) s、1H)、3.76−3.55(m,2H)、3.53(s、2H)、3.48−3.32(m、2H)、2.46(s、4H)、1.32(dd、J=6.6、3.9Hz、3H)。
エタノール(350kg)、パラジウム(活性炭に対して10%)(8.40kg、7.89mol)及び(S)−1−(4−ベンジルピペラジン−1−イル)−2−ヒドロキシプロパン−1−オン 5(70.0kg、282mol)を反応器に充填し、混合物を窒素でパージし、加熱還流した。シクロヘキセン(70.0kg、852mol)をゆっくりと加えた。反応混合物を加熱還流し、24時間撹拌した。周囲温度に冷却した後、混合物をCELITE(登録商標)のパッドを通して濾過し、ケーキをエタノール(20kg)で洗浄した。濾液を10−15℃に冷却し、テトラヒドロフラン(156kg)中のシュウ酸二水和物の溶液(36.0kg、286mol)を、反応温度を10−20℃で維持する速度で、ゆっくりと加えた。2時間熟成させた後、固体を濾過により回収し、エタノール(100kg)で洗浄し、50−55℃で減圧乾燥させ、(S)−2−ヒドロキシ−1−(ピペラジン−1−イル)プロパン−1−オン シュウ酸塩 V(58.2kg、83%)が与えられた。1H NMR(300MHz、D2O)δ4.73−4.51(m、1H)、3.93−3.59(m、4H)、3.26(dd、J=8.8,4.0Hz、4H)、1.27(d、J=6.7Hz、3H)。
1−ベンジルピペラジン(5.0g,28.40mmol,1.00当量)を充填したフラスコを10℃に冷却した。エチル (2S)−2−ヒドロキシプロパノエート 7(10.1g,85.1mmol,3.00当量)を、20℃未満の温度を維持する速度で加えた。20℃未満の温度を維持しながら、ナトリウムメトキシド(MeOH中25wt%)(4.9mL、21.3mmol、0.75当量)を加えた。冷水浴を除去し、次いで、反応混合物を周囲温度まで温め、16時間熟成させた。混合物をエタノール(25mL)で希釈し、AMBERLITE(登録商標)IRC−748樹脂(Dow Chemical Co.,Na+型31.6g、1.8meq/g、2当量、5%HCl水溶液を使用してH+型に予め調節されている)で処理した。懸濁液を周囲温度で2時間撹拌した。樹脂をCELITE(登録商標)(3.5g)のパッドを通した濾過により除去し、パッドをエタノール(2×33.8mL)で洗浄した。濾液及び洗浄液を合わせ、50mLまで減圧濃縮した。溶液に活性炭(DARCO(登録商標)KB−WJ、Norit Inc.,生成物3.52gの100%の理論上の収率に基づく50wt%)を加えた。懸濁液を周囲温度で18時間撹拌した。懸濁液をCELITE(登録商標)(7g)のパッドを通して濾過し、パッドをEtOH(2×33.6mL)で洗浄した。濾液及び洗浄液を合わせ、真空濃縮を行い、(S)−1−(4−ベンジルピペラジン−1−イル)−2−ヒドロキシプロパン−1−オン 8を含む残留物が得られた。
ピペラジン(10.0g、116mmol)を充填したフラスコ及び(S)−エチル 2−ヒドロキシプロパノエート 7(17.84g、151mmol、1.30当量)を10℃に冷却した。20℃未満の温度を維持しながら、ナトリウムメトキシド(MeOH中25wt%)(12.55g、58.1mmol、0.50当量)をゆっくりと加えた。冷水浴を除去し、反応混合物を周囲温度に温め、19時間熟成させた。水(6.23g、346mmol、3.0当量)を加え、混合物を16時間熟成させた。混合物をエタノール(40mL)で希釈し、減圧濃縮した。残留物をエタノール(40mL)で希釈し、エタノール(30mL)中のシュウ酸二水和物の溶液(6.58g、52.2mmol)で処理し、pHを7.5に調整した。懸濁液を10℃未満に冷却し、CELITE(登録商標)のパッドを通して濾過し、エタノール(2×60mL)で洗浄した。濾液及び洗浄液を合わせ、50mLまで濃縮した。溶液を10℃に冷却し、エタノール(60mL)中のシュウ酸二水和物の溶液(16.1g、128mmol)をゆっくりと加えた。懸濁液を周囲温度に温め、1時間攪拌した。懸濁液は、10℃に冷却され、固体を濾過により回収し、冷エタノール(2×18mL)で洗浄し、50℃で24時間減圧乾燥させ、(S)−2−ヒドロキシ−1−(ピペラジン−1−イル)プロパン−1−オンのシュウ酸塩Vが白色固体(15.2g、53%)として得られた。1H NMR(300MHz、D2O)δ4.73−4.51(m、1H)、3.93−3.59(m、4H)、3.26(dd、J=8.8、4.0Hz、4H)、1.27(d、J=6.7Hz、3H)。
ジメチルホルムアミド(DMF)(319L)中の、5−ブロモピリミジン−2−アミン 9(80.0kg、460mol)と、ジ−tert−ブチル ジカーボネート(Boc2O)(250kg、1140mol)と、トリエチルアミン(139kg,1370mol)との混合物に、4−ジメチルアミノピリジン(DMAP)(5.70kg、46.6mol)がゆっくりと加えられた。反応混合物を70−90℃に加熱し、3時間撹拌した。15−40℃まで冷却した後、混合物を氷水(6000kg)でゆっくりとクエンチし、懸濁液を1時間撹拌した。固体を濾過により回収し、水(200kg)で1時間、攪拌した。得られた固体を濾過により回収し、50℃で10時間、真空乾燥して、ビス−tert−ブチル 5−ブロモピリミジン−2−イル−ジカルバメート 10(216kg、HPLCで97A%を上回る、定量収率)が得られた。1H NMR(500MHz、CDCl3)δ8.71(s、2H)、1.40(s、18H);LCMS(ESI)m/z[M−H]373。
無水エタノール(1692L)中の2−[ビス(tert−ブトキシカルボニル)アミノ]−5−ブロモピリミジン10の溶液(216kg粗、460mol、前工程で定量収率と仮定)に、温度を0−20℃で維持しながら、水(344L)中の水酸化ナトリウムの溶液(55.2kg、1380mol)をゆっくりと加えた。2−[ビス(tert−ブトキシカルボニル)−アミノ]−5−ブロモピリミジン (10)の含有量がHPLCにより0.5%以下になるまで、混合物をその温度で撹拌した。反応混合物を0−5℃に冷却し、温度を5℃未満に維持しながら、シュウ酸(86.0kg,955mol)を加えることによって、pHを7に調整した。次に、温度を50℃未満に制御しながら、混合物を500−600Lの容積まで真空蒸留した。水(800kg)を加え、混合物を1時間撹拌した。固体を濾過により回収し、水(2×500L)で撹拌した。得られた固体を濾過により回収し、50℃で減圧乾燥し、tert−ブチル 5−ブロモピリミジン−2−イルカルバメート 11(107kg、二工程にわたって85%の収率)が得られた。1H NMR(500MHz、CDCl3)δ8.63(s、2H)、8.12(s、1H)、1.55(s、9H)。LCMS(ESI)m/z[M+H−Boc]176。
テトラヒドロフラン(910L)中のtert−ブチル(5−ブロモピリミジン−2−イル)カルバメート 11(45.0kg、164mol)にトリイソプロピル ホウ酸塩(Sigma−Aldrich社、CAS番号5419−55−6、77.4kg,412mol)をゆっくりと加え、混合物を−70℃(摂氏マイナス70度)に冷却した。温度を−65℃未満に維持しながら、n−ブチルリチウム(ヘキサン中の2.5M溶液、264L、660mol)を加え、tert−ブチル(5−ブロモピリミジン−2−イル)カルバメート 11の含有量がHPLCにより0.5%以下になるまで、反応混合物を撹拌した。温度を40℃未満に維持しながら、精製水(5kg)を加えた。混合物を5℃に冷却し、25%含水硫酸水素ナトリウム(aqueous sodium hydrogen sulfate)(270kg)を加えることによって、pHを7に調節した。混合物を50℃に加熱し、有機溶媒を減圧下で除去した。水(600kg)を加え、混合物を5℃未満に冷却し、25%含水硫酸水素ナトリウム(aqueous sodium hydrogen sulfate)(60kg)を加えることによって、pHを3.5に調整した。固体を濾過により回収し、水(240kg)で30分間、攪拌した。得られた固体を濾過により回収し、水(550kg)で再スラリー化し、この混合物を0−5℃に冷却した。温度を10℃未満に維持しながら、10% 水酸化ナトリウム水溶液(aqueous sodium hydroxide solution)を加え、混合物を2時間撹拌した。水相を石油エーテル(2x40kg)で抽出した。次いで、温度を0−10℃に維持しながら、水相のpHを、25% 硫酸水素ナトリウム水溶液(aqueous sodium hydrogen sulfate solution)を加えることによって、3.5に調節した。スラリーを濾過し、固体を水(400kg)で1時間、再スラリー化した。固体を濾過により回収し、フィルター上で乾燥させ、Boc保護された、2−(tert−ブトキシカルボニルアミノ)ピリミジン−5−イルボロン酸 12(40kg湿性、HPLCで49wt%、50%の収率)が得られた。1H NMR(500MHz、DMSO−d6)δ10.08(s、1H)、8.82(s、2H)、8.42(s、2H)、1.46(s、9H)。
水(245kg)中の[2−[(tert−ブトキシカルボニル)アミノ]ピリミジン−5−イル]ボロン酸 12(40.0kg、HPLCにより49wt%、82.0mol)の混合物に、温度を30℃未満に維持しながら濃塩酸(39.6L)を加えた。反応混合物を12時間攪拌し、次いで10℃に冷却した。温度を15℃未満に維持しながら、混合物のpHを、50%水酸化ナトリウム水溶液を加えることにより6.5に調整し、次いで、混合物を1時間撹拌した。水(69.0kg)を加え、混合物を30分間熟成させた。得られたスラリーを濾過し、ケーキを50℃で真空乾燥させ、2−アミノピリミジン−5−イルボロン酸 III(10.2kg、90%の収率)が得られた。1H NMR(300MHz、DMSO−d6)δ8.50(s、2H)、7.97(s、2H)、6.74(s、2H)。
2−[ビス(tert−ブトキシカルボニル)アミノ]−5−ブロモピリミジン 10(10g、27mmol)、クロロ(2−ジシクロヘキシルホスフィノ−2′,4′,6′−トリイソプロピル−1,1′−ビフェニル)[2−(2′−アミノ−1,1′−ビフェニル)]パラジウム(II)(101mg,0.128mmol),4,4,4’,4’,5,5,5’,5’−オクタメチル−2,2’−ビ(1,3,2−ジオキサボロラン)又はビス(ピナコラート)ジボロンとして知られているもの、B2Pin2、ピナコールジボラン(13.6g、53.4mmol)及びナトリウムアセテート(7.9g、80mmol)の溶液に、50mLのトルエンが加えられた。混合物を85℃で7時間加熱した。20℃まで冷却された後、水(90mL)中の1NのNaOHを加えた。二相混合物をセライトで濾過し、有機層を廃棄した。有機層を80℃に加熱し、水(21.2g)中の37%HClを加えた。溶液を2時間撹拌し、0℃に冷却した。水(23.7g)中の28%NaOHの溶液を、pHが7になるまで加えた。得られた懸濁液を濾過し、水ですすいだ。灰白色の固体を50℃で16時間真空乾燥させた。
Claims (15)
- 以下の構造
を有する(S)−1−(4−((2−(2−アミノピリミジン−5−イル)−7−メチル−4−モルホリノチエノ[3,2−d]ピリミジン−6−イル)メチル)ピペラジン−1−イル)−2−ヒドロキシプロパン−1−オン(I)及びその立体異性体、幾何異性体、互変異性体、並びに薬学的に許容される塩を調製するための方法であって、
(a) 以下の(S)−1−(4−((2−クロロ−7−メチル−4−モルホリノチエノ[3,2−d]ピリミジン−6−イル)メチル)ピペラジン−1−イル)−2−ヒドロキシプロパン−1−オン(II)を与えるために、以下の(S)−2−ヒドロキシ−1−(ピペラジン−1−イル)プロパン−1−オン(6)のシュウ酸塩及び以下の2−クロロ−7−メチル−4−モルホリノチエノ[3,2−d]ピリミジン−6−カルボアルデヒド(IV)を還元剤と反応させ、
;及び
(b) 上記(I)を与えるために、上記(II)、パラジウム触媒、及び以下の構造を有する2−アミノピリミジン−5−イルボロン酸(III)を反応させる
ことを含む方法。 - (S)−2−ヒドロキシ−1−(ピペラジン−1−イル)プロパン−1−オンのシュウ酸塩は、(S)−エチル 2−ヒドロキシプロパノエートを、ピペラジン、続いてシュウ酸と反応させることを含む方法によって調製される、請求項1に記載の方法。
- 以下の構造
を有する(S)−2−ヒドロキシ−1−(ピペラジン−1−イル)プロパン−1−オンのシュウ酸塩(V)は:
(a) 以下の構造を有する(S)−1−(4−ベンジルピペラジン−1−イル)−2−ヒドロキシプロパン−1−オン(8)を与えるために(S)−エチル 2−ヒドロキシプロパノエート(7)を1−ベンジルピペラジンと反応させ
;
(b) 以下の構造を有する(S)−2−ヒドロキシ−1−(ピペラジン−1−イル)プロパン−1−オン(6)を与えるために上記(8)をパラジウム触媒で還元し
;及び
(c) 上記(V)を与えるために(S)−2−ヒドロキシ−1−(ピペラジン−1−イル)プロパン−1−オンをシュウ酸と反応させる
ことを含む方法によって調製される、請求項1に記載の方法。 - (S)−2−ヒドロキシ−1−(ピペラジン−1−イル)プロパン−1−オンのシュウ酸塩(V)は:
(a) (S)−2−アセトキシプロパン酸を与えるために(S)−2−ヒドロキシプロパン酸(L−乳酸)(1)をアセチル化し;
(b) (S)−1−クロロ−1−オキソプロパン−2−イル アセテートを与えるために(S)−2−アセトキシプロパン酸を塩素化試薬と反応させ;
(c) (S)−1−(4−ベンジルピペラジン−1−イル)−1−オキソプロパン−2−イル アセテートを与えるためにS)−1−クロロ−1−オキソプロパン−2−イル アセテートを1−ベンジルピペラジンと反応させ;
(d) (S)−1−(4−ベンジルピペラジン−1−イル)−2−ヒドロキシプロパン−1−オンを与えるために(S)−1−(4−ベンジルピペラジン−1−イル)−1−オキソプロパン−2−イル アセテートのアセテートを加水分解し;
(e) (S)−2−ヒドロキシ−1−(ピペラジン−1−イル)プロパン−1−オンを与えるために(S)−1−(4−ベンジルピペラジン−1−イル)−2−ヒドロキシプロパン−1−オンのベンジル基をパラジウム触媒で還元除去し;及び
(f) 上記(V)を与えるために(S)−2−ヒドロキシ−1−(ピペラジン−1−イル)プロパン−1−オンをシュウ酸と反応させる
ことを含む方法によって調製される、請求項1に記載の方法。 - 2−アミノピリミジン−5−イルボロン酸(III)は、
(a) 以下の構造を有するビス−tert−ブチル 5−ブロモピリミジン−2−イル−ジカルバメート(10)を与えるために5−ブロモピリミジン−2−アミンをBoc保護試薬と反応させ
;
(b) 以下の構造を有するtert−ブチル 5−ブロモピリミジン−2−イルカルバメート(11)を与えるために1つのBoc基を塩基性加水分解し
;
(c) 2−(tert−ブトキシカルボニルアミノ)ピリミジン−5−イルボロン酸(12)を与えるために上記(11)をアルキルリチウム試薬でメタレーションして、ホウ酸トリアルキル試薬でボリレーションし;及び
(d) 上記(III)を与えるために(12)のBoc基を酸性脱保護する
ことを含む方法によって調製される、請求項1から4の何れか一項に記載の方法。 - 2−アミノピリミジン−5−イルボロン酸(III)は、
(a) 5−ブロモピリミジン−2−アミンを、リチウムビス(トリメチルシリル)アミド、次にn−ブチルリチウム、続いてホウ酸トリアルキル試薬と反応させ;及び
(b) 上記(III)を与えるために上記混合物を水性の酸で処理すること
を含む方法によって調製される、請求項1から4の何れか一項に記載の方法。 - 2−クロロ−7−メチル−4−モルホリノチエノ[3,2−d]ピリミジン−6−カルボアルデヒド(IV)が、以下の構造を有する4−(2−クロロ−7−メチルチエノ[3,2−d]ピリミジン−4−イル)モルホリノ(VI)を、グリニャール試薬、アルキルリチウム試薬、及びジメチルホルムアミドと反応させることを含む
方法によって調製される、請求項1から6の何れか一項に記載の方法。 - 2−アミノピリミジン−5−イルボロン酸(III)が、
(a) 以下の構造を有するビス−tert−ブチル 5−(4,4,5,5−テトラメチル−1,3,2−ジオキサボロラン−2−イル)ピリミジン−2−イルジカルバメートを与えるために4,4,4’,4’,5,5,5’,5’−オクタメチル−2,2’−ビ(1,3,2−ジオキサボロラン及びビス−tert−ブチル5−ブロモピリジン−2−イル−ジカルバメート(10)を、以下:
から選択される触媒の存在条件下で反応させ
;及び
(b) 上記(III)を与えるために両方のBoc基及びピナコール基を酸加水分解する
ことを含む方法によって調製される、請求項1から7の何れか一項に記載の方法。 - 4−(2−クロロ−7−メチルチエノ[3,2−d]ピリミジン−4−イル)モルホリノ(VI)が、以下の構造を有する2,4−ジクロロ−7−メチルチエノ[3,2−d]ピリミジン(VII)をモルホリンと反応させることを含む
方法によって調製される、請求項7に記載の方法。 - 2,4−ジクロロ−7−メチルチエノ[3,2−d]ピリミジン(VII)が、以下の構造を有する7−メチルチエノ[3,2−d]ピリミジン−2,4(1H、3H)−ジオン(VIII)をオキシ塩化リンと反応させることを含む
方法によって調製される、請求項9に記載の方法。 - 7−メチルチエノ[3,2−d]ピリミジン−2,4(1H、3H)−ジオン(VIII)が、以下の構造を有するメチル 3−アミノ−4−メチルチオフェン−2−カルボンキシレート(IX)をシアン酸カリウムと反応させることを含む
方法によって調製される、請求項10に記載の方法。 - パラジウム触媒が、PdCl2(PPh3)2、Pd(t−Bu)3、PdCl2dppfCH2Cl2、Pd(PPh3)4、Pd(OAc)/PPh3、Cl2Pd[(Pet3)]2、Pd(DIPHOS)2、Cl2Pd(Bipy)、[PdCl(Ph2PCH2PPh2)]2、Cl2Pd[P(o−tol)3]2、Pd2(dba)3/P(o−tol)3、Pd2(dba)/P(furyl)3、Cl2Pd[P(furyl)3]2、Cl2Pd(PMePh2)2、Cl2Pd[P(4−F−Ph)3]2、Cl2Pd[P(C6F6)3]2、Cl2Pd[P(2−COOH−Ph)(Ph)2]2、及びCl2Pd[P(4−COOH−Ph)(Ph)2]2から選択される、請求項1から11の何れか一項に記載の方法。
- 還元剤が、トリアセトキシ水素化ホウ素ナトリウム、2−ピコリンボラン、又は5−エチル−2−メチルピリジンボランである、請求項1から12の何れか一項に記載の方法。
- (II)、パラジウム触媒、及び2−アミノピリミジン−5−イルボロン酸(III)を反応させた後に、活性炭を通して反応混合物を濾過することをさらに含む、請求項1から13の何れか一項に記載の方法。
- 以下の構造を有する(S)−2−ヒドロキシ−1−(ピペラジン−1−イル)プロパン−1−オンのシュウ酸塩
。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261711900P | 2012-10-10 | 2012-10-10 | |
| US61/711,900 | 2012-10-10 | ||
| PCT/EP2013/070994 WO2014056955A1 (en) | 2012-10-10 | 2013-10-09 | Process for making thienopyrimidine compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2015534572A JP2015534572A (ja) | 2015-12-03 |
| JP6123097B2 true JP6123097B2 (ja) | 2017-05-10 |
Family
ID=49322377
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015536109A Active JP6123097B2 (ja) | 2012-10-10 | 2013-10-09 | チエノピリミジン化合物を製造するための方法 |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US8895729B2 (ja) |
| EP (1) | EP2906566B1 (ja) |
| JP (1) | JP6123097B2 (ja) |
| KR (1) | KR101698283B1 (ja) |
| CN (2) | CN104718212B (ja) |
| AR (1) | AR092960A1 (ja) |
| BR (1) | BR112015007970A8 (ja) |
| CA (1) | CA2883513A1 (ja) |
| ES (1) | ES2594078T3 (ja) |
| MX (1) | MX349551B (ja) |
| RU (1) | RU2637309C2 (ja) |
| WO (1) | WO2014056955A1 (ja) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8895729B2 (en) * | 2012-10-10 | 2014-11-25 | Genentech, Inc. | Process for making thienopyrimidine compounds |
| JP6456925B2 (ja) * | 2013-05-06 | 2019-01-23 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | ボロン酸中間体の調製のための方法 |
| US20170216286A1 (en) | 2014-01-28 | 2017-08-03 | Mayo Foundation For Medical Education And Research | Killing senescent cells and treating senescence-associated conditions using a src inhibitor and a flavonoid |
| IL286427B2 (en) | 2014-01-28 | 2024-08-01 | Mayo Found Medical Education & Res | Inhibitors bcl-2 anti-apoptotic protein family members for treatment of non cancer pulmonary disease or an opthalmic disease |
| TW201813963A (zh) | 2016-09-23 | 2018-04-16 | 美商基利科學股份有限公司 | 磷脂醯肌醇3-激酶抑制劑 |
| TW201825465A (zh) | 2016-09-23 | 2018-07-16 | 美商基利科學股份有限公司 | 磷脂醯肌醇3-激酶抑制劑 |
| TW201815787A (zh) | 2016-09-23 | 2018-05-01 | 美商基利科學股份有限公司 | 磷脂醯肌醇3-激酶抑制劑 |
| EA201991069A1 (ru) * | 2016-11-02 | 2019-10-31 | Комбинированная терапия ингибитором фосфоинозитид-3-киназы и связывающим цинк агентом | |
| WO2019180141A1 (en) | 2018-03-23 | 2019-09-26 | Bayer Aktiengesellschaft | Combinations of rogaratinib |
| CN113717049A (zh) * | 2021-08-05 | 2021-11-30 | 江西兄弟医药有限公司 | 一种制备(s)-乙酰氧基丙酰氯的半连续方法 |
Family Cites Families (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9208135D0 (en) | 1992-04-13 | 1992-05-27 | Ludwig Inst Cancer Res | Polypeptides having kinase activity,their preparation and use |
| US5846824A (en) | 1994-02-07 | 1998-12-08 | Ludwig Institute For Cancer Research | Polypeptides having kinase activity, their preparation and use |
| US6274327B1 (en) | 1992-04-13 | 2001-08-14 | Ludwig Institute For Cancer Research | Polypeptides having kinase activity, their preparation and use |
| US6395916B1 (en) | 1998-07-10 | 2002-05-28 | Massachusetts Institute Of Technology | Ligands for metals and improved metal-catalyzed processes based thereon |
| US6307087B1 (en) | 1998-07-10 | 2001-10-23 | Massachusetts Institute Of Technology | Ligands for metals and improved metal-catalyzed processes based thereon |
| US7223879B2 (en) | 1998-07-10 | 2007-05-29 | Massachusetts Institute Of Technology | Ligands for metals and improved metal-catalyzed processes based thereon |
| KR20040024564A (ko) | 2001-07-12 | 2004-03-20 | 아베시아 리미티드 | 마이크로캡슐화된 촉매, 이의 제조 방법 및 이의 사용 방법 |
| GB0423653D0 (en) | 2004-10-25 | 2004-11-24 | Piramed Ltd | Pharmaceutical compounds |
| TWI498332B (zh) | 2006-04-26 | 2015-09-01 | Hoffmann La Roche | 作為pi3k抑制劑之嘧啶衍生物及相關製備方法、醫藥組合物、用途、套組及產物 |
| GB0608264D0 (en) * | 2006-04-26 | 2006-06-07 | Piramed Ltd | Pharmaceutical compounds |
| ZA200904534B (en) * | 2006-12-07 | 2010-09-29 | Hoffmann La Roche | Phosphoinositide 3-kinase inhibitor compounds and methods of use |
| RU2470936C2 (ru) * | 2006-12-07 | 2012-12-27 | Дженентек, Инк. | Соединения-ингибиторы фосфоинозитид 3-киназы и способы применения |
| MX2009005950A (es) * | 2006-12-07 | 2009-10-12 | Genentech Inc | Compuestos inhibidores de fosfoinositido 3-quinasas y metodos de uso. |
| EP2205242B1 (en) * | 2007-09-12 | 2015-04-15 | Genentech, Inc. | Combinations of phosphoinositide 3-kinase inhibitor compounds and chemotherapeutic agents, and methods of use |
| JP5348725B2 (ja) | 2007-10-25 | 2013-11-20 | ジェネンテック, インコーポレイテッド | チエノピリミジン化合物の製造方法 |
| CN101952298B (zh) | 2007-12-12 | 2015-06-17 | 麻省理工学院 | 用于过渡金属催化的交联偶合反应的配体及其使用方法 |
| JP5709766B2 (ja) | 2009-03-12 | 2015-04-30 | ジェネンテック, インコーポレイテッド | 造血器腫瘍の治療のためのホスホイノシチド3キナーゼ阻害剤化合物と化学療法剤の併用 |
| US20120308562A1 (en) | 2011-06-03 | 2012-12-06 | Derynck Mika K | Methods of treating mesothelioma with a pi3k inhibitor compound |
| CN102399235A (zh) * | 2011-10-25 | 2012-04-04 | 江苏弘和药物研发有限公司 | 一种2-氨基-5-嘧啶硼酸频哪醇酯的合成方法 |
| CN102367260A (zh) * | 2011-12-12 | 2012-03-07 | 南京药石药物研发有限公司 | 2-氨基嘧啶-5-硼酸的合成方法 |
| CN102675323B (zh) * | 2012-06-01 | 2014-04-09 | 南京药石药物研发有限公司 | 吡咯并[2,1-f][1,2,4]三嗪衍生物及其抗肿瘤用途 |
| US8895729B2 (en) * | 2012-10-10 | 2014-11-25 | Genentech, Inc. | Process for making thienopyrimidine compounds |
| CN102993163A (zh) * | 2012-12-07 | 2013-03-27 | 青岛前线生物工程有限公司 | 3-甲基噻吩-2-甲醛的合成方法 |
-
2013
- 2013-10-09 US US14/049,477 patent/US8895729B2/en active Active
- 2013-10-09 MX MX2015004467A patent/MX349551B/es active IP Right Grant
- 2013-10-09 BR BR112015007970A patent/BR112015007970A8/pt not_active Application Discontinuation
- 2013-10-09 CN CN201380052049.5A patent/CN104718212B/zh active Active
- 2013-10-09 KR KR1020157009108A patent/KR101698283B1/ko not_active Expired - Fee Related
- 2013-10-09 WO PCT/EP2013/070994 patent/WO2014056955A1/en not_active Ceased
- 2013-10-09 CA CA2883513A patent/CA2883513A1/en not_active Abandoned
- 2013-10-09 CN CN201710374289.7A patent/CN107188898B/zh active Active
- 2013-10-09 AR ARP130103666A patent/AR092960A1/es unknown
- 2013-10-09 JP JP2015536109A patent/JP6123097B2/ja active Active
- 2013-10-09 EP EP13774174.0A patent/EP2906566B1/en active Active
- 2013-10-09 RU RU2015113747A patent/RU2637309C2/ru not_active IP Right Cessation
- 2013-10-09 ES ES13774174.0T patent/ES2594078T3/es active Active
Also Published As
| Publication number | Publication date |
|---|---|
| RU2015113747A (ru) | 2016-12-10 |
| CA2883513A1 (en) | 2014-04-17 |
| BR112015007970A2 (pt) | 2017-07-04 |
| KR101698283B1 (ko) | 2017-01-19 |
| CN104718212B (zh) | 2017-06-16 |
| ES2594078T3 (es) | 2016-12-15 |
| MX2015004467A (es) | 2015-07-14 |
| HK1244272A1 (zh) | 2018-08-03 |
| JP2015534572A (ja) | 2015-12-03 |
| HK1211026A1 (en) | 2016-05-13 |
| US8895729B2 (en) | 2014-11-25 |
| MX349551B (es) | 2017-08-02 |
| BR112015007970A8 (pt) | 2019-08-27 |
| EP2906566A1 (en) | 2015-08-19 |
| US20140100366A1 (en) | 2014-04-10 |
| KR20150054945A (ko) | 2015-05-20 |
| WO2014056955A1 (en) | 2014-04-17 |
| EP2906566B1 (en) | 2016-08-24 |
| CN104718212A (zh) | 2015-06-17 |
| AR092960A1 (es) | 2015-05-06 |
| CN107188898A (zh) | 2017-09-22 |
| CN107188898B (zh) | 2019-12-03 |
| RU2637309C2 (ru) | 2017-12-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6123097B2 (ja) | チエノピリミジン化合物を製造するための方法 | |
| KR101725696B1 (ko) | 신규한 이환형 피리딘온 | |
| JP6505023B2 (ja) | Cnsおよび他の障害を治療するための、pde4アイソザイムの阻害薬としてのアザベンゾイミダゾール化合物 | |
| JP6663459B2 (ja) | ベンゾオキサゼピン化合物の作製方法 | |
| JP6506833B2 (ja) | イミダゾピリダジン化合物 | |
| US9975856B2 (en) | Process for the preparation of (E)-3-(4-((E)-2-(2-chloro-4-fluorophenyl)-1-(1H-indazol-5-yl)but-1-en-1-yl)phenyl)acrylic acid | |
| CA3015166C (en) | 6,7-dihydro-5h-pyrazolo[5,1-b][1,3]oxazine-2-carboxamide compounds | |
| CA2953004C (en) | Aromatic heterocyclic derivatives and pharmaceutical applications thereof | |
| CN104725249A (zh) | 苄胺类衍生物及其在药物上的应用 | |
| JP2019501930A (ja) | 6,7,8,9−テトラヒドロ−5H−ピリド[2,3−d]アゼピンドーパミンD3リガンド | |
| JP2018520167A (ja) | PI3K/mTOR阻害剤としての溶融キノリン化合物 | |
| GB2583606A (en) | Indoleamine 2,3-Dioxygenase inhibitors and use of same in medicine | |
| HK1244272B (zh) | 制备噻吩并嘧啶化合物的方法 | |
| HK1211026B (en) | Process for making thienopyrimidine compounds | |
| HK40058692A (en) | 6,7-dihydro-5h-pyrazolo[5,1-b][1,3]oxazine-2-carboxamide compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20150603 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20160607 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20160831 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20161207 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20170228 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20170309 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6123097 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |