JP5989299B2 - リゾホスファチジン酸結合のための組成物と方法 - Google Patents
リゾホスファチジン酸結合のための組成物と方法 Download PDFInfo
- Publication number
- JP5989299B2 JP5989299B2 JP2010510480A JP2010510480A JP5989299B2 JP 5989299 B2 JP5989299 B2 JP 5989299B2 JP 2010510480 A JP2010510480 A JP 2010510480A JP 2010510480 A JP2010510480 A JP 2010510480A JP 5989299 B2 JP5989299 B2 JP 5989299B2
- Authority
- JP
- Japan
- Prior art keywords
- antibody
- lpa
- light chain
- heavy chain
- amino acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 title claims description 280
- 230000027455 binding Effects 0.000 title claims description 113
- 238000000034 method Methods 0.000 title claims description 109
- 239000000203 mixture Substances 0.000 title claims description 80
- WRGQSWVCFNIUNZ-GDCKJWNLSA-N 1-oleoyl-sn-glycerol 3-phosphate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](O)COP(O)(O)=O WRGQSWVCFNIUNZ-GDCKJWNLSA-N 0.000 title claims 22
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 130
- 206010016654 Fibrosis Diseases 0.000 claims description 117
- 230000004761 fibrosis Effects 0.000 claims description 108
- 210000004027 cell Anatomy 0.000 claims description 102
- 201000010099 disease Diseases 0.000 claims description 100
- 239000000427 antigen Substances 0.000 claims description 85
- 108091007433 antigens Proteins 0.000 claims description 85
- 102000036639 antigens Human genes 0.000 claims description 85
- 206010028980 Neoplasm Diseases 0.000 claims description 66
- 108010021625 Immunoglobulin Fragments Proteins 0.000 claims description 63
- 102000008394 Immunoglobulin Fragments Human genes 0.000 claims description 62
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 49
- 239000003814 drug Substances 0.000 claims description 47
- 201000011510 cancer Diseases 0.000 claims description 43
- 210000001519 tissue Anatomy 0.000 claims description 39
- 208000005069 pulmonary fibrosis Diseases 0.000 claims description 32
- 208000019425 cirrhosis of liver Diseases 0.000 claims description 30
- 241001465754 Metazoa Species 0.000 claims description 29
- 208000035475 disorder Diseases 0.000 claims description 29
- 150000007523 nucleic acids Chemical class 0.000 claims description 29
- 108020004707 nucleic acids Proteins 0.000 claims description 28
- 102000039446 nucleic acids Human genes 0.000 claims description 28
- 108010047041 Complementarity Determining Regions Proteins 0.000 claims description 25
- 230000002159 abnormal effect Effects 0.000 claims description 23
- 210000002966 serum Anatomy 0.000 claims description 23
- 241000894007 species Species 0.000 claims description 23
- 230000003463 hyperproliferative effect Effects 0.000 claims description 22
- 230000033115 angiogenesis Effects 0.000 claims description 19
- 239000013598 vector Substances 0.000 claims description 17
- 238000001514 detection method Methods 0.000 claims description 16
- 238000001574 biopsy Methods 0.000 claims description 15
- 206010039710 Scleroderma Diseases 0.000 claims description 13
- 230000029663 wound healing Effects 0.000 claims description 13
- 210000004369 blood Anatomy 0.000 claims description 11
- 239000008280 blood Substances 0.000 claims description 11
- 208000008589 Obesity Diseases 0.000 claims description 7
- 230000006907 apoptotic process Effects 0.000 claims description 7
- 210000001124 body fluid Anatomy 0.000 claims description 7
- 239000010839 body fluid Substances 0.000 claims description 7
- 235000020824 obesity Nutrition 0.000 claims description 7
- 210000002381 plasma Anatomy 0.000 claims description 7
- 238000002360 preparation method Methods 0.000 claims description 7
- 208000023275 Autoimmune disease Diseases 0.000 claims description 6
- 238000008157 ELISA kit Methods 0.000 claims description 6
- 239000012530 fluid Substances 0.000 claims description 6
- 208000002780 macular degeneration Diseases 0.000 claims description 6
- 239000002773 nucleotide Substances 0.000 claims description 6
- 125000003729 nucleotide group Chemical group 0.000 claims description 6
- 208000002193 Pain Diseases 0.000 claims description 5
- 230000004770 neurodegeneration Effects 0.000 claims description 5
- 206010036790 Productive cough Diseases 0.000 claims description 4
- 230000002500 effect on skin Effects 0.000 claims description 4
- 239000003550 marker Substances 0.000 claims description 4
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 4
- 210000003802 sputum Anatomy 0.000 claims description 4
- 208000024794 sputum Diseases 0.000 claims description 4
- 230000037314 wound repair Effects 0.000 claims description 4
- 208000009329 Graft vs Host Disease Diseases 0.000 claims description 3
- 206010028594 Myocardial fibrosis Diseases 0.000 claims description 3
- 210000000941 bile Anatomy 0.000 claims description 3
- 201000002793 renal fibrosis Diseases 0.000 claims description 3
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims description 3
- 210000002700 urine Anatomy 0.000 claims description 3
- 206010052779 Transplant rejections Diseases 0.000 claims description 2
- 210000001742 aqueous humor Anatomy 0.000 claims description 2
- 210000000582 semen Anatomy 0.000 claims description 2
- 210000004127 vitreous body Anatomy 0.000 claims description 2
- 206010029113 Neovascularisation Diseases 0.000 claims 1
- 208000003589 Spider Bites Diseases 0.000 claims 1
- 230000002596 correlated effect Effects 0.000 claims 1
- 210000003097 mucus Anatomy 0.000 claims 1
- XGRLSUFHELJJAB-JGSYTFBMSA-M sodium;[(2r)-2-hydroxy-3-[(z)-octadec-9-enoyl]oxypropyl] hydrogen phosphate Chemical compound [Na+].CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](O)COP(O)([O-])=O XGRLSUFHELJJAB-JGSYTFBMSA-M 0.000 description 262
- 238000011282 treatment Methods 0.000 description 87
- 150000002632 lipids Chemical class 0.000 description 85
- 108090000623 proteins and genes Proteins 0.000 description 63
- 230000000975 bioactive effect Effects 0.000 description 60
- 239000003795 chemical substances by application Substances 0.000 description 58
- 102000004169 proteins and genes Human genes 0.000 description 49
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 48
- 235000018102 proteins Nutrition 0.000 description 48
- 150000001875 compounds Chemical class 0.000 description 47
- 108090000765 processed proteins & peptides Proteins 0.000 description 41
- 239000000243 solution Substances 0.000 description 39
- 230000014509 gene expression Effects 0.000 description 37
- 229940079593 drug Drugs 0.000 description 36
- 230000000694 effects Effects 0.000 description 36
- 238000006243 chemical reaction Methods 0.000 description 35
- 102000004196 processed proteins & peptides Human genes 0.000 description 35
- 210000004072 lung Anatomy 0.000 description 34
- 229920001184 polypeptide Polymers 0.000 description 34
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 33
- 239000012634 fragment Substances 0.000 description 32
- DUYSYHSSBDVJSM-KRWOKUGFSA-N sphingosine 1-phosphate Chemical compound CCCCCCCCCCCCC\C=C\[C@@H](O)[C@@H](N)COP(O)(O)=O DUYSYHSSBDVJSM-KRWOKUGFSA-N 0.000 description 32
- 230000001225 therapeutic effect Effects 0.000 description 32
- 230000015572 biosynthetic process Effects 0.000 description 31
- 241000699670 Mus sp. Species 0.000 description 30
- 239000000047 product Substances 0.000 description 29
- 108060003951 Immunoglobulin Proteins 0.000 description 28
- 102000018358 immunoglobulin Human genes 0.000 description 28
- -1 Lipids Lipids Chemical class 0.000 description 25
- 210000004185 liver Anatomy 0.000 description 25
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 25
- 206010061218 Inflammation Diseases 0.000 description 24
- 239000000523 sample Substances 0.000 description 24
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 23
- 210000004408 hybridoma Anatomy 0.000 description 23
- 230000004054 inflammatory process Effects 0.000 description 23
- 238000006467 substitution reaction Methods 0.000 description 23
- 102000004190 Enzymes Human genes 0.000 description 21
- 108090000790 Enzymes Proteins 0.000 description 21
- 238000005481 NMR spectroscopy Methods 0.000 description 21
- 238000003556 assay Methods 0.000 description 21
- 229940088598 enzyme Drugs 0.000 description 21
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 21
- 239000007787 solid Substances 0.000 description 21
- 238000002965 ELISA Methods 0.000 description 19
- 230000002163 immunogen Effects 0.000 description 19
- 238000004519 manufacturing process Methods 0.000 description 19
- 230000035755 proliferation Effects 0.000 description 19
- 238000003786 synthesis reaction Methods 0.000 description 19
- 229940127089 cytotoxic agent Drugs 0.000 description 18
- 102000005962 receptors Human genes 0.000 description 18
- 108020003175 receptors Proteins 0.000 description 18
- 238000012360 testing method Methods 0.000 description 18
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 17
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical group OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 17
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 17
- 235000001014 amino acid Nutrition 0.000 description 17
- 239000002246 antineoplastic agent Substances 0.000 description 17
- 238000013459 approach Methods 0.000 description 17
- 239000003153 chemical reaction reagent Substances 0.000 description 17
- 239000000463 material Substances 0.000 description 17
- 238000004809 thin layer chromatography Methods 0.000 description 17
- 235000019439 ethyl acetate Nutrition 0.000 description 16
- 150000004665 fatty acids Chemical class 0.000 description 16
- 238000005755 formation reaction Methods 0.000 description 16
- 230000028993 immune response Effects 0.000 description 16
- 230000011664 signaling Effects 0.000 description 16
- 238000003756 stirring Methods 0.000 description 16
- 235000014113 dietary fatty acids Nutrition 0.000 description 15
- 229930195729 fatty acid Natural products 0.000 description 15
- 239000000194 fatty acid Substances 0.000 description 15
- 230000004048 modification Effects 0.000 description 15
- 238000012986 modification Methods 0.000 description 15
- 239000000126 substance Substances 0.000 description 15
- 238000002560 therapeutic procedure Methods 0.000 description 15
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 14
- 241000588724 Escherichia coli Species 0.000 description 14
- 241000699666 Mus <mouse, genus> Species 0.000 description 14
- 229940024606 amino acid Drugs 0.000 description 14
- 230000004071 biological effect Effects 0.000 description 14
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 14
- 210000002950 fibroblast Anatomy 0.000 description 14
- 108020004414 DNA Proteins 0.000 description 13
- 150000001413 amino acids Chemical class 0.000 description 13
- 150000003904 phospholipids Chemical class 0.000 description 13
- 208000024891 symptom Diseases 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 description 12
- 230000000875 corresponding effect Effects 0.000 description 12
- 239000003446 ligand Substances 0.000 description 12
- 231100000241 scar Toxicity 0.000 description 12
- 102000004127 Cytokines Human genes 0.000 description 11
- 108090000695 Cytokines Proteins 0.000 description 11
- 108090000642 Lysophosphatidic Acid Receptors Proteins 0.000 description 11
- ZQQLMECVOXKFJK-NXCSZAMKSA-N N-octadecanoylsphingosine 1-phosphate Chemical compound CCCCCCCCCCCCCCCCCC(=O)N[C@@H](COP(O)(O)=O)[C@H](O)\C=C\CCCCCCCCCCCCC ZQQLMECVOXKFJK-NXCSZAMKSA-N 0.000 description 11
- 239000002253 acid Substances 0.000 description 11
- 239000000460 chlorine Substances 0.000 description 11
- 230000007882 cirrhosis Effects 0.000 description 11
- 238000003745 diagnosis Methods 0.000 description 11
- 239000000706 filtrate Substances 0.000 description 11
- 238000001727 in vivo Methods 0.000 description 11
- 238000012317 liver biopsy Methods 0.000 description 11
- 239000002609 medium Substances 0.000 description 11
- 210000000651 myofibroblast Anatomy 0.000 description 11
- 239000003921 oil Substances 0.000 description 11
- 230000008569 process Effects 0.000 description 11
- 239000011734 sodium Substances 0.000 description 11
- 238000001356 surgical procedure Methods 0.000 description 11
- 125000003396 thiol group Chemical group [H]S* 0.000 description 11
- 102000004137 Lysophosphatidic Acid Receptors Human genes 0.000 description 10
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 10
- 125000000539 amino acid group Chemical group 0.000 description 10
- 230000006378 damage Effects 0.000 description 10
- 230000003176 fibrotic effect Effects 0.000 description 10
- 230000016784 immunoglobulin production Effects 0.000 description 10
- 239000002502 liposome Substances 0.000 description 10
- 230000002265 prevention Effects 0.000 description 10
- 230000002829 reductive effect Effects 0.000 description 10
- 150000003839 salts Chemical class 0.000 description 10
- 150000003408 sphingolipids Chemical class 0.000 description 10
- 239000000758 substrate Substances 0.000 description 10
- 230000004083 survival effect Effects 0.000 description 10
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 9
- 208000000059 Dyspnea Diseases 0.000 description 9
- 206010013975 Dyspnoeas Diseases 0.000 description 9
- 208000029523 Interstitial Lung disease Diseases 0.000 description 9
- 206010035226 Plasma cell myeloma Diseases 0.000 description 9
- 239000004480 active ingredient Substances 0.000 description 9
- 239000000872 buffer Substances 0.000 description 9
- 239000000969 carrier Substances 0.000 description 9
- 238000004113 cell culture Methods 0.000 description 9
- 238000011161 development Methods 0.000 description 9
- 230000018109 developmental process Effects 0.000 description 9
- 238000000338 in vitro Methods 0.000 description 9
- 208000014674 injury Diseases 0.000 description 9
- 230000003993 interaction Effects 0.000 description 9
- 201000000050 myeloid neoplasm Diseases 0.000 description 9
- 229920000642 polymer Polymers 0.000 description 9
- 238000000746 purification Methods 0.000 description 9
- 239000002994 raw material Substances 0.000 description 9
- 230000004044 response Effects 0.000 description 9
- 239000000377 silicon dioxide Substances 0.000 description 9
- 210000003491 skin Anatomy 0.000 description 9
- WWUZIQQURGPMPG-KRWOKUGFSA-N sphingosine Chemical compound CCCCCCCCCCCCC\C=C\[C@@H](O)[C@@H](N)CO WWUZIQQURGPMPG-KRWOKUGFSA-N 0.000 description 9
- 108010035532 Collagen Proteins 0.000 description 8
- 102000008186 Collagen Human genes 0.000 description 8
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 8
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 8
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 8
- 241000282412 Homo Species 0.000 description 8
- 101001038006 Homo sapiens Lysophosphatidic acid receptor 3 Proteins 0.000 description 8
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 description 8
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 description 8
- 102000013463 Immunoglobulin Light Chains Human genes 0.000 description 8
- 108010065825 Immunoglobulin Light Chains Proteins 0.000 description 8
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 8
- 102100040388 Lysophosphatidic acid receptor 3 Human genes 0.000 description 8
- 241000124008 Mammalia Species 0.000 description 8
- 239000002202 Polyethylene glycol Substances 0.000 description 8
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 8
- 239000010425 asbestos Substances 0.000 description 8
- 229940098773 bovine serum albumin Drugs 0.000 description 8
- 229920001436 collagen Polymers 0.000 description 8
- 238000005516 engineering process Methods 0.000 description 8
- 150000002430 hydrocarbons Chemical group 0.000 description 8
- 230000001965 increasing effect Effects 0.000 description 8
- 230000005012 migration Effects 0.000 description 8
- 238000013508 migration Methods 0.000 description 8
- 230000035772 mutation Effects 0.000 description 8
- 229910052757 nitrogen Inorganic materials 0.000 description 8
- 210000000056 organ Anatomy 0.000 description 8
- 239000002245 particle Substances 0.000 description 8
- 229920001223 polyethylene glycol Polymers 0.000 description 8
- 238000001959 radiotherapy Methods 0.000 description 8
- 238000011160 research Methods 0.000 description 8
- 229910052895 riebeckite Inorganic materials 0.000 description 8
- 230000036573 scar formation Effects 0.000 description 8
- 230000037390 scarring Effects 0.000 description 8
- 208000013220 shortness of breath Diseases 0.000 description 8
- WWUZIQQURGPMPG-UHFFFAOYSA-N (-)-D-erythro-Sphingosine Natural products CCCCCCCCCCCCCC=CC(O)C(N)CO WWUZIQQURGPMPG-UHFFFAOYSA-N 0.000 description 7
- 241000894006 Bacteria Species 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 7
- 108091007491 NSP3 Papain-like protease domains Proteins 0.000 description 7
- 108090000553 Phospholipase D Proteins 0.000 description 7
- 210000000013 bile duct Anatomy 0.000 description 7
- 230000033228 biological regulation Effects 0.000 description 7
- 230000002950 deficient Effects 0.000 description 7
- 238000002405 diagnostic procedure Methods 0.000 description 7
- 239000003623 enhancer Substances 0.000 description 7
- 230000004927 fusion Effects 0.000 description 7
- 238000003780 insertion Methods 0.000 description 7
- 230000037431 insertion Effects 0.000 description 7
- 150000002500 ions Chemical class 0.000 description 7
- 239000010410 layer Substances 0.000 description 7
- 125000005647 linker group Chemical group 0.000 description 7
- 239000011159 matrix material Substances 0.000 description 7
- 210000004379 membrane Anatomy 0.000 description 7
- 239000012528 membrane Substances 0.000 description 7
- 229920006395 saturated elastomer Polymers 0.000 description 7
- 239000006228 supernatant Substances 0.000 description 7
- 230000017423 tissue regeneration Effects 0.000 description 7
- 231100000331 toxic Toxicity 0.000 description 7
- 230000002588 toxic effect Effects 0.000 description 7
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 6
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 6
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 6
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 6
- 102000009027 Albumins Human genes 0.000 description 6
- 108010088751 Albumins Proteins 0.000 description 6
- 206010003445 Ascites Diseases 0.000 description 6
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 6
- 101001038001 Homo sapiens Lysophosphatidic acid receptor 2 Proteins 0.000 description 6
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 6
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 6
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 6
- 102100040387 Lysophosphatidic acid receptor 2 Human genes 0.000 description 6
- 206010028851 Necrosis Diseases 0.000 description 6
- 102000011420 Phospholipase D Human genes 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- 239000002671 adjuvant Substances 0.000 description 6
- 239000011324 bead Substances 0.000 description 6
- 230000001588 bifunctional effect Effects 0.000 description 6
- 210000004204 blood vessel Anatomy 0.000 description 6
- 150000001720 carbohydrates Chemical class 0.000 description 6
- 238000002648 combination therapy Methods 0.000 description 6
- 229940125773 compound 10 Drugs 0.000 description 6
- 125000000151 cysteine group Chemical class N[C@@H](CS)C(=O)* 0.000 description 6
- 210000003527 eukaryotic cell Anatomy 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 230000013595 glycosylation Effects 0.000 description 6
- 238000006206 glycosylation reaction Methods 0.000 description 6
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 6
- 230000012010 growth Effects 0.000 description 6
- 210000002216 heart Anatomy 0.000 description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 6
- 229940072221 immunoglobulins Drugs 0.000 description 6
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 6
- 210000004698 lymphocyte Anatomy 0.000 description 6
- 239000002207 metabolite Substances 0.000 description 6
- 230000017074 necrotic cell death Effects 0.000 description 6
- 239000012044 organic layer Substances 0.000 description 6
- 239000008194 pharmaceutical composition Substances 0.000 description 6
- YBYRMVIVWMBXKQ-UHFFFAOYSA-N phenylmethanesulfonyl fluoride Chemical compound FS(=O)(=O)CC1=CC=CC=C1 YBYRMVIVWMBXKQ-UHFFFAOYSA-N 0.000 description 6
- 239000002243 precursor Substances 0.000 description 6
- 150000003409 sphingosine 1-phosphates Chemical class 0.000 description 6
- 210000000952 spleen Anatomy 0.000 description 6
- 230000004936 stimulating effect Effects 0.000 description 6
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 6
- 230000008733 trauma Effects 0.000 description 6
- PORPENFLTBBHSG-MGBGTMOVSA-N 1,2-dihexadecanoyl-sn-glycerol-3-phosphate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(O)=O)OC(=O)CCCCCCCCCCCCCCC PORPENFLTBBHSG-MGBGTMOVSA-N 0.000 description 5
- 241000251468 Actinopterygii Species 0.000 description 5
- OJRUSAPKCPIVBY-KQYNXXCUSA-N C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N Chemical compound C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N OJRUSAPKCPIVBY-KQYNXXCUSA-N 0.000 description 5
- 108091026890 Coding region Proteins 0.000 description 5
- 239000004971 Cross linker Substances 0.000 description 5
- 206010019280 Heart failures Diseases 0.000 description 5
- 241000711557 Hepacivirus Species 0.000 description 5
- 101000966782 Homo sapiens Lysophosphatidic acid receptor 1 Proteins 0.000 description 5
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 5
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 5
- 102100040607 Lysophosphatidic acid receptor 1 Human genes 0.000 description 5
- 239000004698 Polyethylene Substances 0.000 description 5
- 239000004793 Polystyrene Substances 0.000 description 5
- 108020004511 Recombinant DNA Proteins 0.000 description 5
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 5
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 5
- 206010046798 Uterine leiomyoma Diseases 0.000 description 5
- 241000700605 Viruses Species 0.000 description 5
- 239000012491 analyte Substances 0.000 description 5
- 230000002491 angiogenic effect Effects 0.000 description 5
- 208000035269 cancer or benign tumor Diseases 0.000 description 5
- 229930003827 cannabinoid Natural products 0.000 description 5
- 239000003557 cannabinoid Substances 0.000 description 5
- 230000004663 cell proliferation Effects 0.000 description 5
- 230000008859 change Effects 0.000 description 5
- 238000010367 cloning Methods 0.000 description 5
- 229940125758 compound 15 Drugs 0.000 description 5
- 210000002808 connective tissue Anatomy 0.000 description 5
- 238000007796 conventional method Methods 0.000 description 5
- 235000018417 cysteine Nutrition 0.000 description 5
- 238000012217 deletion Methods 0.000 description 5
- 230000037430 deletion Effects 0.000 description 5
- 230000008034 disappearance Effects 0.000 description 5
- 208000037765 diseases and disorders Diseases 0.000 description 5
- 238000000921 elemental analysis Methods 0.000 description 5
- 239000013604 expression vector Substances 0.000 description 5
- 239000000284 extract Substances 0.000 description 5
- 239000000835 fiber Substances 0.000 description 5
- 230000003352 fibrogenic effect Effects 0.000 description 5
- 235000011187 glycerol Nutrition 0.000 description 5
- 239000003102 growth factor Substances 0.000 description 5
- 208000019622 heart disease Diseases 0.000 description 5
- 210000003494 hepatocyte Anatomy 0.000 description 5
- 210000003734 kidney Anatomy 0.000 description 5
- 201000010260 leiomyoma Diseases 0.000 description 5
- 230000000670 limiting effect Effects 0.000 description 5
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 5
- 235000019341 magnesium sulphate Nutrition 0.000 description 5
- 230000002503 metabolic effect Effects 0.000 description 5
- 230000001575 pathological effect Effects 0.000 description 5
- 238000002823 phage display Methods 0.000 description 5
- 239000012071 phase Substances 0.000 description 5
- 150000003180 prostaglandins Chemical class 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- 239000007790 solid phase Substances 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- JLVSPVFPBBFMBE-HXSWCURESA-N sphingosine-1-phosphocholine Chemical compound CCCCCCCCCCCCC\C=C\[C@@H](O)[C@@H](N)COP([O-])(=O)OCC[N+](C)(C)C JLVSPVFPBBFMBE-HXSWCURESA-N 0.000 description 5
- 229940124597 therapeutic agent Drugs 0.000 description 5
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 4
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 4
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 4
- RYCNUMLMNKHWPZ-SNVBAGLBSA-N 1-acetyl-sn-glycero-3-phosphocholine Chemical compound CC(=O)OC[C@@H](O)COP([O-])(=O)OCC[N+](C)(C)C RYCNUMLMNKHWPZ-SNVBAGLBSA-N 0.000 description 4
- 238000004009 13C{1H}-NMR spectroscopy Methods 0.000 description 4
- HVAUUPRFYPCOCA-AREMUKBSSA-N 2-O-acetyl-1-O-hexadecyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCOC[C@@H](OC(C)=O)COP([O-])(=O)OCC[N+](C)(C)C HVAUUPRFYPCOCA-AREMUKBSSA-N 0.000 description 4
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- 108010006654 Bleomycin Proteins 0.000 description 4
- 108091035707 Consensus sequence Proteins 0.000 description 4
- 102100021977 Ectonucleotide pyrophosphatase/phosphodiesterase family member 2 Human genes 0.000 description 4
- 108050004000 Ectonucleotide pyrophosphatase/phosphodiesterase family member 2 Proteins 0.000 description 4
- JZNWSCPGTDBMEW-UHFFFAOYSA-N Glycerophosphorylethanolamin Natural products NCCOP(O)(=O)OCC(O)CO JZNWSCPGTDBMEW-UHFFFAOYSA-N 0.000 description 4
- 244000068988 Glycine max Species 0.000 description 4
- 235000010469 Glycine max Nutrition 0.000 description 4
- 208000005176 Hepatitis C Diseases 0.000 description 4
- 108010091358 Hypoxanthine Phosphoribosyltransferase Proteins 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- 208000002260 Keloid Diseases 0.000 description 4
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 4
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 4
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 4
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 4
- 239000004472 Lysine Substances 0.000 description 4
- 102100037611 Lysophospholipase Human genes 0.000 description 4
- 108010058864 Phospholipases A2 Proteins 0.000 description 4
- 108010003541 Platelet Activating Factor Proteins 0.000 description 4
- 206010035664 Pneumonia Diseases 0.000 description 4
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 4
- 201000010001 Silicosis Diseases 0.000 description 4
- 239000004473 Threonine Substances 0.000 description 4
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 4
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Chemical compound CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 4
- 208000027418 Wounds and injury Diseases 0.000 description 4
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 4
- ATBOMIWRCZXYSZ-XZBBILGWSA-N [1-[2,3-dihydroxypropoxy(hydroxy)phosphoryl]oxy-3-hexadecanoyloxypropan-2-yl] (9e,12e)-octadeca-9,12-dienoate Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(COP(O)(=O)OCC(O)CO)OC(=O)CCCCCCC\C=C\C\C=C\CCCCC ATBOMIWRCZXYSZ-XZBBILGWSA-N 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 238000001042 affinity chromatography Methods 0.000 description 4
- 150000001336 alkenes Chemical class 0.000 description 4
- 150000001345 alkine derivatives Chemical class 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 235000021342 arachidonic acid Nutrition 0.000 description 4
- 229940114079 arachidonic acid Drugs 0.000 description 4
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 4
- 230000009286 beneficial effect Effects 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- KGNDCEVUMONOKF-UGPLYTSKSA-N benzyl n-[(2r)-1-[(2s,4r)-2-[[(2s)-6-amino-1-(1,3-benzoxazol-2-yl)-1,1-dihydroxyhexan-2-yl]carbamoyl]-4-[(4-methylphenyl)methoxy]pyrrolidin-1-yl]-1-oxo-4-phenylbutan-2-yl]carbamate Chemical compound C1=CC(C)=CC=C1CO[C@H]1CN(C(=O)[C@@H](CCC=2C=CC=CC=2)NC(=O)OCC=2C=CC=CC=2)[C@H](C(=O)N[C@@H](CCCCN)C(O)(O)C=2OC3=CC=CC=C3N=2)C1 KGNDCEVUMONOKF-UGPLYTSKSA-N 0.000 description 4
- 229960001561 bleomycin Drugs 0.000 description 4
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 4
- 238000009534 blood test Methods 0.000 description 4
- 238000004364 calculation method Methods 0.000 description 4
- 210000000170 cell membrane Anatomy 0.000 description 4
- 230000001684 chronic effect Effects 0.000 description 4
- 230000000295 complement effect Effects 0.000 description 4
- 229940126142 compound 16 Drugs 0.000 description 4
- 229940125810 compound 20 Drugs 0.000 description 4
- 229940125833 compound 23 Drugs 0.000 description 4
- 229940125961 compound 24 Drugs 0.000 description 4
- 229940125851 compound 27 Drugs 0.000 description 4
- 239000012043 crude product Substances 0.000 description 4
- 230000007423 decrease Effects 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 206010012601 diabetes mellitus Diseases 0.000 description 4
- 230000004069 differentiation Effects 0.000 description 4
- 230000002526 effect on cardiovascular system Effects 0.000 description 4
- 150000002066 eicosanoids Chemical class 0.000 description 4
- 230000002255 enzymatic effect Effects 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 229960002989 glutamic acid Drugs 0.000 description 4
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 4
- 208000006454 hepatitis Diseases 0.000 description 4
- 230000001900 immune effect Effects 0.000 description 4
- 230000009851 immunogenic response Effects 0.000 description 4
- 230000001976 improved effect Effects 0.000 description 4
- 208000015181 infectious disease Diseases 0.000 description 4
- 208000036971 interstitial lung disease 2 Diseases 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 210000001117 keloid Anatomy 0.000 description 4
- 238000011813 knockout mouse model Methods 0.000 description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 4
- 235000005772 leucine Nutrition 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 235000018977 lysine Nutrition 0.000 description 4
- 230000003211 malignant effect Effects 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- 239000003094 microcapsule Substances 0.000 description 4
- 238000002703 mutagenesis Methods 0.000 description 4
- 231100000350 mutagenesis Toxicity 0.000 description 4
- 229940094443 oxytocics prostaglandins Drugs 0.000 description 4
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 4
- 150000008104 phosphatidylethanolamines Chemical class 0.000 description 4
- 150000003905 phosphatidylinositols Chemical class 0.000 description 4
- 239000013612 plasmid Substances 0.000 description 4
- 229920001296 polysiloxane Polymers 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 230000002206 pro-fibrotic effect Effects 0.000 description 4
- 238000007634 remodeling Methods 0.000 description 4
- 230000008439 repair process Effects 0.000 description 4
- 229960004641 rituximab Drugs 0.000 description 4
- 230000028327 secretion Effects 0.000 description 4
- 210000001626 skin fibroblast Anatomy 0.000 description 4
- 150000003384 small molecules Chemical class 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 230000008719 thickening Effects 0.000 description 4
- 230000001988 toxicity Effects 0.000 description 4
- 231100000419 toxicity Toxicity 0.000 description 4
- 238000013518 transcription Methods 0.000 description 4
- 230000035897 transcription Effects 0.000 description 4
- 210000003462 vein Anatomy 0.000 description 4
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 3
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 3
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- 229920000936 Agarose Polymers 0.000 description 3
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 3
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 3
- 239000004475 Arginine Substances 0.000 description 3
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 3
- 241000271566 Aves Species 0.000 description 3
- 108090001008 Avidin Proteins 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 241000283690 Bos taurus Species 0.000 description 3
- YDNKGFDKKRUKPY-JHOUSYSJSA-N C16 ceramide Natural products CCCCCCCCCCCCCCCC(=O)N[C@@H](CO)[C@H](O)C=CCCCCCCCCCCCCC YDNKGFDKKRUKPY-JHOUSYSJSA-N 0.000 description 3
- 102100033620 Calponin-1 Human genes 0.000 description 3
- 241000282472 Canis lupus familiaris Species 0.000 description 3
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 3
- 241000282693 Cercopithecidae Species 0.000 description 3
- 229940126657 Compound 17 Drugs 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 241000233866 Fungi Species 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 3
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 3
- 239000004471 Glycine Substances 0.000 description 3
- 206010019799 Hepatitis viral Diseases 0.000 description 3
- 101000945318 Homo sapiens Calponin-1 Proteins 0.000 description 3
- 101000652736 Homo sapiens Transgelin Proteins 0.000 description 3
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 3
- 102100029098 Hypoxanthine-guanine phosphoribosyltransferase Human genes 0.000 description 3
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 3
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 3
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 3
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 3
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 3
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 3
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical compound CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 description 3
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 3
- 208000019693 Lung disease Diseases 0.000 description 3
- 206010027476 Metastases Diseases 0.000 description 3
- CRJGESKKUOMBCT-VQTJNVASSA-N N-acetylsphinganine Chemical compound CCCCCCCCCCCCCCC[C@@H](O)[C@H](CO)NC(C)=O CRJGESKKUOMBCT-VQTJNVASSA-N 0.000 description 3
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 3
- 108091028043 Nucleic acid sequence Proteins 0.000 description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 241000288906 Primates Species 0.000 description 3
- 210000001744 T-lymphocyte Anatomy 0.000 description 3
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 229940122618 Trypsin inhibitor Drugs 0.000 description 3
- 101710162629 Trypsin inhibitor Proteins 0.000 description 3
- 208000004608 Ureteral Obstruction Diseases 0.000 description 3
- 206010064930 age-related macular degeneration Diseases 0.000 description 3
- 235000004279 alanine Nutrition 0.000 description 3
- 238000012867 alanine scanning Methods 0.000 description 3
- 230000001476 alcoholic effect Effects 0.000 description 3
- 230000009285 allergic inflammation Effects 0.000 description 3
- 239000005557 antagonist Substances 0.000 description 3
- 230000002424 anti-apoptotic effect Effects 0.000 description 3
- 230000000890 antigenic effect Effects 0.000 description 3
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 3
- 235000009697 arginine Nutrition 0.000 description 3
- 235000009582 asparagine Nutrition 0.000 description 3
- 229960001230 asparagine Drugs 0.000 description 3
- 230000002238 attenuated effect Effects 0.000 description 3
- 230000008827 biological function Effects 0.000 description 3
- 229960002685 biotin Drugs 0.000 description 3
- 235000020958 biotin Nutrition 0.000 description 3
- 239000011616 biotin Substances 0.000 description 3
- 230000017531 blood circulation Effects 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 125000002843 carboxylic acid group Chemical group 0.000 description 3
- 210000001054 cardiac fibroblast Anatomy 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 230000010261 cell growth Effects 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 230000005754 cellular signaling Effects 0.000 description 3
- 229940106189 ceramide Drugs 0.000 description 3
- ZVEQCJWYRWKARO-UHFFFAOYSA-N ceramide Natural products CCCCCCCCCCCCCCC(O)C(=O)NC(CO)C(O)C=CCCC=C(C)CCCCCCCCC ZVEQCJWYRWKARO-UHFFFAOYSA-N 0.000 description 3
- 238000002512 chemotherapy Methods 0.000 description 3
- 125000001309 chloro group Chemical group Cl* 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 239000003593 chromogenic compound Substances 0.000 description 3
- 238000003776 cleavage reaction Methods 0.000 description 3
- 229940125797 compound 12 Drugs 0.000 description 3
- 229940125782 compound 2 Drugs 0.000 description 3
- 229940126086 compound 21 Drugs 0.000 description 3
- 229940125898 compound 5 Drugs 0.000 description 3
- 239000000356 contaminant Substances 0.000 description 3
- 229920001577 copolymer Polymers 0.000 description 3
- 239000003246 corticosteroid Substances 0.000 description 3
- 125000004093 cyano group Chemical group *C#N 0.000 description 3
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 3
- 230000034994 death Effects 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 238000001212 derivatisation Methods 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 239000000539 dimer Substances 0.000 description 3
- 150000002009 diols Chemical class 0.000 description 3
- 239000000428 dust Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 210000002889 endothelial cell Anatomy 0.000 description 3
- 238000006911 enzymatic reaction Methods 0.000 description 3
- 210000002919 epithelial cell Anatomy 0.000 description 3
- 239000002024 ethyl acetate extract Substances 0.000 description 3
- 230000008622 extracellular signaling Effects 0.000 description 3
- 208000030533 eye disease Diseases 0.000 description 3
- 238000003818 flash chromatography Methods 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 210000004602 germ cell Anatomy 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 239000000833 heterodimer Substances 0.000 description 3
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 3
- 235000014304 histidine Nutrition 0.000 description 3
- 229910052739 hydrogen Inorganic materials 0.000 description 3
- 230000003053 immunization Effects 0.000 description 3
- 238000002649 immunization Methods 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 230000002757 inflammatory effect Effects 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 230000004068 intracellular signaling Effects 0.000 description 3
- 210000003292 kidney cell Anatomy 0.000 description 3
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 3
- 210000005228 liver tissue Anatomy 0.000 description 3
- 230000007774 longterm Effects 0.000 description 3
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 238000002483 medication Methods 0.000 description 3
- 230000004060 metabolic process Effects 0.000 description 3
- 230000009401 metastasis Effects 0.000 description 3
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 3
- 229930182817 methionine Natural products 0.000 description 3
- 235000006109 methionine Nutrition 0.000 description 3
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 3
- 244000005700 microbiome Species 0.000 description 3
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 3
- VVGIYYKRAMHVLU-UHFFFAOYSA-N newbouldiamide Natural products CCCCCCCCCCCCCCCCCCCC(O)C(O)C(O)C(CO)NC(=O)CCCCCCCCCCCCCCCCC VVGIYYKRAMHVLU-UHFFFAOYSA-N 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 102000013415 peroxidase activity proteins Human genes 0.000 description 3
- 108040007629 peroxidase activity proteins Proteins 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 3
- 230000004983 pleiotropic effect Effects 0.000 description 3
- 210000003240 portal vein Anatomy 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 238000010188 recombinant method Methods 0.000 description 3
- 230000008929 regeneration Effects 0.000 description 3
- 238000011069 regeneration method Methods 0.000 description 3
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 description 3
- 230000001105 regulatory effect Effects 0.000 description 3
- 230000010076 replication Effects 0.000 description 3
- 238000012827 research and development Methods 0.000 description 3
- 239000011347 resin Substances 0.000 description 3
- 229920005989 resin Polymers 0.000 description 3
- 238000012552 review Methods 0.000 description 3
- 230000007017 scission Effects 0.000 description 3
- 230000003248 secreting effect Effects 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 235000012239 silicon dioxide Nutrition 0.000 description 3
- 238000009097 single-agent therapy Methods 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- YHEDRJPUIRMZMP-ZWKOTPCHSA-N sphinganine 1-phosphate Chemical compound CCCCCCCCCCCCCCC[C@@H](O)[C@@H](N)COP(O)(O)=O YHEDRJPUIRMZMP-ZWKOTPCHSA-N 0.000 description 3
- 239000003381 stabilizer Substances 0.000 description 3
- 238000010186 staining Methods 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- JJAHTWIKCUJRDK-UHFFFAOYSA-N succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate Chemical compound C1CC(CN2C(C=CC2=O)=O)CCC1C(=O)ON1C(=O)CCC1=O JJAHTWIKCUJRDK-UHFFFAOYSA-N 0.000 description 3
- 238000013268 sustained release Methods 0.000 description 3
- 239000012730 sustained-release form Substances 0.000 description 3
- PNJXYVJNOCLJLJ-QMMMGPOBSA-N tert-butyl (4r)-4-formyl-2,2-dimethyl-1,3-oxazolidine-3-carboxylate Chemical compound CC(C)(C)OC(=O)N1[C@@H](C=O)COC1(C)C PNJXYVJNOCLJLJ-QMMMGPOBSA-N 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- WMXCDAVJEZZYLT-UHFFFAOYSA-N tert-butylthiol Chemical compound CC(C)(C)S WMXCDAVJEZZYLT-UHFFFAOYSA-N 0.000 description 3
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 3
- 229940126622 therapeutic monoclonal antibody Drugs 0.000 description 3
- 150000003568 thioethers Chemical class 0.000 description 3
- 239000003053 toxin Substances 0.000 description 3
- 231100000765 toxin Toxicity 0.000 description 3
- 108700012359 toxins Proteins 0.000 description 3
- 238000012546 transfer Methods 0.000 description 3
- 230000009261 transgenic effect Effects 0.000 description 3
- 238000002054 transplantation Methods 0.000 description 3
- 239000002753 trypsin inhibitor Substances 0.000 description 3
- 208000037999 tubulointerstitial fibrosis Diseases 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- 230000005740 tumor formation Effects 0.000 description 3
- 230000000381 tumorigenic effect Effects 0.000 description 3
- 238000002604 ultrasonography Methods 0.000 description 3
- 210000004291 uterus Anatomy 0.000 description 3
- 201000001862 viral hepatitis Diseases 0.000 description 3
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 2
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- GEYOCULIXLDCMW-UHFFFAOYSA-N 1,2-phenylenediamine Chemical compound NC1=CC=CC=C1N GEYOCULIXLDCMW-UHFFFAOYSA-N 0.000 description 2
- FALRKNHUBBKYCC-UHFFFAOYSA-N 2-(chloromethyl)pyridine-3-carbonitrile Chemical compound ClCC1=NC=CC=C1C#N FALRKNHUBBKYCC-UHFFFAOYSA-N 0.000 description 2
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 2
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 2
- GANZODCWZFAEGN-UHFFFAOYSA-N 5-mercapto-2-nitro-benzoic acid Chemical compound OC(=O)C1=CC(S)=CC=C1[N+]([O-])=O GANZODCWZFAEGN-UHFFFAOYSA-N 0.000 description 2
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 2
- 102000007469 Actins Human genes 0.000 description 2
- 108010085238 Actins Proteins 0.000 description 2
- 208000035939 Alveolitis allergic Diseases 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- 208000033116 Asbestos intoxication Diseases 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 2
- 240000007124 Brassica oleracea Species 0.000 description 2
- 235000003899 Brassica oleracea var acephala Nutrition 0.000 description 2
- 235000011301 Brassica oleracea var capitata Nutrition 0.000 description 2
- 235000001169 Brassica oleracea var oleracea Nutrition 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- 229940124638 COX inhibitor Drugs 0.000 description 2
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 2
- 208000024172 Cardiovascular disease Diseases 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- 206010011224 Cough Diseases 0.000 description 2
- 108010037462 Cyclooxygenase 2 Proteins 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 description 2
- 238000009007 Diagnostic Kit Methods 0.000 description 2
- 206010061818 Disease progression Diseases 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- 101150086780 EGD2 gene Proteins 0.000 description 2
- 238000012286 ELISA Assay Methods 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 241000283086 Equidae Species 0.000 description 2
- 208000004930 Fatty Liver Diseases 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Natural products OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 102000003886 Glycoproteins Human genes 0.000 description 2
- 108090000288 Glycoproteins Proteins 0.000 description 2
- 206010019708 Hepatic steatosis Diseases 0.000 description 2
- 241000238631 Hexapoda Species 0.000 description 2
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 2
- 101000935587 Homo sapiens Flavin reductase (NADPH) Proteins 0.000 description 2
- 101001038037 Homo sapiens Lysophosphatidic acid receptor 5 Proteins 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 2
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 2
- 208000026350 Inborn Genetic disease Diseases 0.000 description 2
- 102000004310 Ion Channels Human genes 0.000 description 2
- 108090000862 Ion Channels Proteins 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- 239000003810 Jones reagent Substances 0.000 description 2
- 206010023421 Kidney fibrosis Diseases 0.000 description 2
- 244000285963 Kluyveromyces fragilis Species 0.000 description 2
- 241001138401 Kluyveromyces lactis Species 0.000 description 2
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 2
- 108010085895 Laminin Proteins 0.000 description 2
- KFKWRHQBZQICHA-STQMWFEESA-N Leu-Phe Chemical compound CC(C)C[C@H](N)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 KFKWRHQBZQICHA-STQMWFEESA-N 0.000 description 2
- 229930184725 Lipoxin Natural products 0.000 description 2
- 208000004852 Lung Injury Diseases 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- 102100040404 Lysophosphatidic acid receptor 5 Human genes 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- 101100403820 Methanopyrus kandleri (strain AV19 / DSM 6324 / JCM 9639 / NBRC 100938) nac gene Proteins 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 2
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 2
- 206010061309 Neoplasm progression Diseases 0.000 description 2
- 101100459404 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) npc-1 gene Proteins 0.000 description 2
- 244000061176 Nicotiana tabacum Species 0.000 description 2
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 2
- 230000004989 O-glycosylation Effects 0.000 description 2
- 108091034117 Oligonucleotide Proteins 0.000 description 2
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 2
- 206010033128 Ovarian cancer Diseases 0.000 description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 description 2
- 102000004316 Oxidoreductases Human genes 0.000 description 2
- 108090000854 Oxidoreductases Proteins 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Natural products OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 2
- 102100038280 Prostaglandin G/H synthase 2 Human genes 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 239000012980 RPMI-1640 medium Substances 0.000 description 2
- 241000235347 Schizosaccharomyces pombe Species 0.000 description 2
- 229920002684 Sepharose Polymers 0.000 description 2
- 241000607720 Serratia Species 0.000 description 2
- 102000007562 Serum Albumin Human genes 0.000 description 2
- 108010071390 Serum Albumin Proteins 0.000 description 2
- 206010050207 Skin fibrosis Diseases 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- 102100025750 Sphingosine 1-phosphate receptor 1 Human genes 0.000 description 2
- 102100025747 Sphingosine 1-phosphate receptor 3 Human genes 0.000 description 2
- 102100029802 Sphingosine 1-phosphate receptor 5 Human genes 0.000 description 2
- 102000011011 Sphingosine 1-phosphate receptors Human genes 0.000 description 2
- 108050001083 Sphingosine 1-phosphate receptors Proteins 0.000 description 2
- 241000256251 Spodoptera frugiperda Species 0.000 description 2
- 241000282887 Suidae Species 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 201000009594 Systemic Scleroderma Diseases 0.000 description 2
- 206010042953 Systemic sclerosis Diseases 0.000 description 2
- 230000006052 T cell proliferation Effects 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- 108010034949 Thyroglobulin Proteins 0.000 description 2
- 102000009843 Thyroglobulin Human genes 0.000 description 2
- 206010069363 Traumatic lung injury Diseases 0.000 description 2
- 102000013127 Vimentin Human genes 0.000 description 2
- 108010065472 Vimentin Proteins 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 2
- 210000001789 adipocyte Anatomy 0.000 description 2
- 230000002776 aggregation Effects 0.000 description 2
- 238000004220 aggregation Methods 0.000 description 2
- 210000004712 air sac Anatomy 0.000 description 2
- 210000001552 airway epithelial cell Anatomy 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 125000000217 alkyl group Chemical group 0.000 description 2
- 208000026935 allergic disease Diseases 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 239000004037 angiogenesis inhibitor Substances 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 230000005875 antibody response Effects 0.000 description 2
- 206010003441 asbestosis Diseases 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 229940120638 avastin Drugs 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- 210000002469 basement membrane Anatomy 0.000 description 2
- 230000006399 behavior Effects 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- 238000002306 biochemical method Methods 0.000 description 2
- 239000012472 biological sample Substances 0.000 description 2
- 239000000090 biomarker Substances 0.000 description 2
- 229960000074 biopharmaceutical Drugs 0.000 description 2
- 238000006664 bond formation reaction Methods 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- DQXBYHZEEUGOBF-UHFFFAOYSA-N but-3-enoic acid;ethene Chemical compound C=C.OC(=O)CC=C DQXBYHZEEUGOBF-UHFFFAOYSA-N 0.000 description 2
- BTANRVKWQNVYAZ-UHFFFAOYSA-N butan-2-ol Chemical compound CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 description 2
- 210000004899 c-terminal region Anatomy 0.000 description 2
- 239000001110 calcium chloride Substances 0.000 description 2
- 229910001628 calcium chloride Inorganic materials 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 229940065144 cannabinoids Drugs 0.000 description 2
- 235000014633 carbohydrates Nutrition 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 230000000747 cardiac effect Effects 0.000 description 2
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 2
- 230000012292 cell migration Effects 0.000 description 2
- 230000033077 cellular process Effects 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 229930183167 cerebroside Natural products 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 239000007795 chemical reaction product Substances 0.000 description 2
- 210000000038 chest Anatomy 0.000 description 2
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 2
- 230000002759 chromosomal effect Effects 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 238000012875 competitive assay Methods 0.000 description 2
- 230000002860 competitive effect Effects 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940126208 compound 22 Drugs 0.000 description 2
- 229940125846 compound 25 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 238000002591 computed tomography Methods 0.000 description 2
- 230000001268 conjugating effect Effects 0.000 description 2
- 230000021615 conjugation Effects 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 229960001334 corticosteroids Drugs 0.000 description 2
- 238000004132 cross linking Methods 0.000 description 2
- 239000012228 culture supernatant Substances 0.000 description 2
- 238000012258 culturing Methods 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- 239000002254 cytotoxic agent Substances 0.000 description 2
- 231100000599 cytotoxic agent Toxicity 0.000 description 2
- 230000002354 daily effect Effects 0.000 description 2
- 230000007850 degeneration Effects 0.000 description 2
- 238000000151 deposition Methods 0.000 description 2
- 230000008021 deposition Effects 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- 239000000032 diagnostic agent Substances 0.000 description 2
- 229940039227 diagnostic agent Drugs 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- 229960005156 digoxin Drugs 0.000 description 2
- OTKJDMGTUTTYMP-UHFFFAOYSA-N dihydrosphingosine Natural products CCCCCCCCCCCCCCCC(O)C(N)CO OTKJDMGTUTTYMP-UHFFFAOYSA-N 0.000 description 2
- 230000005750 disease progression Effects 0.000 description 2
- ZGSPNIOCEDOHGS-UHFFFAOYSA-L disodium [3-[2,3-di(octadeca-9,12-dienoyloxy)propoxy-oxidophosphoryl]oxy-2-hydroxypropyl] 2,3-di(octadeca-9,12-dienoyloxy)propyl phosphate Chemical compound [Na+].[Na+].CCCCCC=CCC=CCCCCCCCC(=O)OCC(OC(=O)CCCCCCCC=CCC=CCCCCC)COP([O-])(=O)OCC(O)COP([O-])(=O)OCC(OC(=O)CCCCCCCC=CCC=CCCCCC)COC(=O)CCCCCCCC=CCC=CCCCCC ZGSPNIOCEDOHGS-UHFFFAOYSA-L 0.000 description 2
- 150000004662 dithiols Chemical class 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 238000002651 drug therapy Methods 0.000 description 2
- 208000017574 dry cough Diseases 0.000 description 2
- 239000012636 effector Substances 0.000 description 2
- 229920001971 elastomer Polymers 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- 150000002121 epoxyeicosatrienoic acids Chemical class 0.000 description 2
- 239000005038 ethylene vinyl acetate Substances 0.000 description 2
- 208000010706 fatty liver disease Diseases 0.000 description 2
- 239000007850 fluorescent dye Substances 0.000 description 2
- 238000005194 fractionation Methods 0.000 description 2
- 150000002270 gangliosides Chemical class 0.000 description 2
- 238000004817 gas chromatography Methods 0.000 description 2
- 238000001502 gel electrophoresis Methods 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 238000001415 gene therapy Methods 0.000 description 2
- 208000016361 genetic disease Diseases 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 239000004220 glutamic acid Substances 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- 208000024908 graft versus host disease Diseases 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 229940093915 gynecological organic acid Drugs 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 230000035876 healing Effects 0.000 description 2
- 201000005787 hematologic cancer Diseases 0.000 description 2
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 2
- 108060003552 hemocyanin Proteins 0.000 description 2
- 230000002440 hepatic effect Effects 0.000 description 2
- 231100000283 hepatitis Toxicity 0.000 description 2
- 208000002672 hepatitis B Diseases 0.000 description 2
- 231100000334 hepatotoxic Toxicity 0.000 description 2
- 230000003082 hepatotoxic effect Effects 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 229920002674 hyaluronan Polymers 0.000 description 2
- 229960003160 hyaluronic acid Drugs 0.000 description 2
- 239000000017 hydrogel Substances 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 238000012872 hydroxylapatite chromatography Methods 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 2
- 230000036039 immunity Effects 0.000 description 2
- 229940127121 immunoconjugate Drugs 0.000 description 2
- 230000005847 immunogenicity Effects 0.000 description 2
- 239000002955 immunomodulating agent Substances 0.000 description 2
- 229940121354 immunomodulator Drugs 0.000 description 2
- 238000001114 immunoprecipitation Methods 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 230000002458 infectious effect Effects 0.000 description 2
- 210000004969 inflammatory cell Anatomy 0.000 description 2
- 230000028709 inflammatory response Effects 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 230000009545 invasion Effects 0.000 description 2
- 230000002427 irreversible effect Effects 0.000 description 2
- 108010045069 keyhole-limpet hemocyanin Proteins 0.000 description 2
- 239000004310 lactic acid Substances 0.000 description 2
- 235000014655 lactic acid Nutrition 0.000 description 2
- 150000002617 leukotrienes Chemical class 0.000 description 2
- RGLRXNKKBLIBQS-XNHQSDQCSA-N leuprolide acetate Chemical compound CC(O)=O.CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 RGLRXNKKBLIBQS-XNHQSDQCSA-N 0.000 description 2
- 150000002639 lipoxins Chemical class 0.000 description 2
- AMXOYNBUYSYVKV-UHFFFAOYSA-M lithium bromide Chemical compound [Li+].[Br-] AMXOYNBUYSYVKV-UHFFFAOYSA-M 0.000 description 2
- 210000005229 liver cell Anatomy 0.000 description 2
- 230000005923 long-lasting effect Effects 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 231100000515 lung injury Toxicity 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 230000001394 metastastic effect Effects 0.000 description 2
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000004005 microsphere Substances 0.000 description 2
- 239000007758 minimum essential medium Substances 0.000 description 2
- 238000005065 mining Methods 0.000 description 2
- 229940126619 mouse monoclonal antibody Drugs 0.000 description 2
- 208000010125 myocardial infarction Diseases 0.000 description 2
- UMFJAHHVKNCGLG-UHFFFAOYSA-N n-Nitrosodimethylamine Chemical compound CN(C)N=O UMFJAHHVKNCGLG-UHFFFAOYSA-N 0.000 description 2
- VQSRKMNBWMHJKY-YTEVENLXSA-N n-[3-[(4ar,7as)-2-amino-6-(5-fluoropyrimidin-2-yl)-4,4a,5,7-tetrahydropyrrolo[3,4-d][1,3]thiazin-7a-yl]-4-fluorophenyl]-5-methoxypyrazine-2-carboxamide Chemical compound C1=NC(OC)=CN=C1C(=O)NC1=CC=C(F)C([C@@]23[C@@H](CN(C2)C=2N=CC(F)=CN=2)CSC(N)=N3)=C1 VQSRKMNBWMHJKY-YTEVENLXSA-N 0.000 description 2
- 201000008383 nephritis Diseases 0.000 description 2
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 2
- 239000002751 oligonucleotide probe Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 230000001590 oxidative effect Effects 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 230000001991 pathophysiological effect Effects 0.000 description 2
- AQIXEPGDORPWBJ-UHFFFAOYSA-N pentan-3-ol Chemical compound CCC(O)CC AQIXEPGDORPWBJ-UHFFFAOYSA-N 0.000 description 2
- 238000010647 peptide synthesis reaction Methods 0.000 description 2
- 210000001322 periplasm Anatomy 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 230000001766 physiological effect Effects 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 2
- 229920002451 polyvinyl alcohol Polymers 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- 230000000750 progressive effect Effects 0.000 description 2
- 210000001236 prokaryotic cell Anatomy 0.000 description 2
- 230000002062 proliferating effect Effects 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 125000006239 protecting group Chemical group 0.000 description 2
- 230000004224 protection Effects 0.000 description 2
- 230000017854 proteolysis Effects 0.000 description 2
- 230000005180 public health Effects 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 238000003127 radioimmunoassay Methods 0.000 description 2
- 230000008521 reorganization Effects 0.000 description 2
- GHMLBKRAJCXXBS-UHFFFAOYSA-N resorcinol Chemical compound OC1=CC=CC(O)=C1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 2
- 230000029058 respiratory gaseous exchange Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 238000007142 ring opening reaction Methods 0.000 description 2
- 238000002390 rotary evaporation Methods 0.000 description 2
- 239000005060 rubber Substances 0.000 description 2
- 201000000306 sarcoidosis Diseases 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 230000000391 smoking effect Effects 0.000 description 2
- OTKJDMGTUTTYMP-ZWKOTPCHSA-N sphinganine Chemical compound CCCCCCCCCCCCCCC[C@@H](O)[C@@H](N)CO OTKJDMGTUTTYMP-ZWKOTPCHSA-N 0.000 description 2
- 125000002657 sphingoid group Chemical group 0.000 description 2
- 231100000240 steatosis hepatitis Toxicity 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 229940014800 succinic anhydride Drugs 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 125000000341 threoninyl group Chemical group [H]OC([H])(C([H])([H])[H])C([H])(N([H])[H])C(*)=O 0.000 description 2
- 229940104230 thymidine Drugs 0.000 description 2
- 229960002175 thyroglobulin Drugs 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- 230000009772 tissue formation Effects 0.000 description 2
- 230000005030 transcription termination Effects 0.000 description 2
- 238000001890 transfection Methods 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 230000005751 tumor progression Effects 0.000 description 2
- 231100000588 tumorigenic Toxicity 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- 238000000108 ultra-filtration Methods 0.000 description 2
- 241000701366 unidentified nuclear polyhedrosis viruses Species 0.000 description 2
- 201000007954 uterine fibroid Diseases 0.000 description 2
- 230000008728 vascular permeability Effects 0.000 description 2
- 239000002435 venom Substances 0.000 description 2
- 210000001048 venom Anatomy 0.000 description 2
- 231100000611 venom Toxicity 0.000 description 2
- 210000005048 vimentin Anatomy 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- NTNKNFHIAFDCSJ-UHFFFAOYSA-N (2-nitrophenyl) thiohypochlorite Chemical compound [O-][N+](=O)C1=CC=CC=C1SCl NTNKNFHIAFDCSJ-UHFFFAOYSA-N 0.000 description 1
- JSPNNZKWADNWHI-PNANGNLXSA-N (2r)-2-hydroxy-n-[(2s,3r,4e,8e)-3-hydroxy-9-methyl-1-[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoctadeca-4,8-dien-2-yl]heptadecanamide Chemical compound CCCCCCCCCCCCCCC[C@@H](O)C(=O)N[C@H]([C@H](O)\C=C\CC\C=C(/C)CCCCCCCCC)CO[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O JSPNNZKWADNWHI-PNANGNLXSA-N 0.000 description 1
- KYBXNPIASYUWLN-WUCPZUCCSA-N (2s)-5-hydroxypyrrolidine-2-carboxylic acid Chemical compound OC1CC[C@@H](C(O)=O)N1 KYBXNPIASYUWLN-WUCPZUCCSA-N 0.000 description 1
- FVXDCNFHHUUREB-UHFFFAOYSA-N (7Z,10Z,13Z,16Z)-1-amino-2-hydroxydocosa-7,10,13,16-tetraen-3-one Chemical compound C(CCCC=C/CC=C/CC=C/CC=C/CCCCC)(=O)C(O)CN FVXDCNFHHUUREB-UHFFFAOYSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- TZCPCKNHXULUIY-RGULYWFUSA-N 1,2-distearoyl-sn-glycero-3-phosphoserine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCCCC TZCPCKNHXULUIY-RGULYWFUSA-N 0.000 description 1
- VILFTWLXLYIEMV-UHFFFAOYSA-N 1,5-difluoro-2,4-dinitrobenzene Chemical compound [O-][N+](=O)C1=CC([N+]([O-])=O)=C(F)C=C1F VILFTWLXLYIEMV-UHFFFAOYSA-N 0.000 description 1
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 1
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical group COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- YBBNVCVOACOHIG-UHFFFAOYSA-N 2,2-diamino-1,4-bis(4-azidophenyl)-3-butylbutane-1,4-dione Chemical compound C=1C=C(N=[N+]=[N-])C=CC=1C(=O)C(N)(N)C(CCCC)C(=O)C1=CC=C(N=[N+]=[N-])C=C1 YBBNVCVOACOHIG-UHFFFAOYSA-N 0.000 description 1
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 1
- RCRCTBLIHCHWDZ-DOFZRALJSA-N 2-arachidonoylglycerol Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(=O)OC(CO)CO RCRCTBLIHCHWDZ-DOFZRALJSA-N 0.000 description 1
- XBBVURRQGJPTHH-UHFFFAOYSA-N 2-hydroxyacetic acid;2-hydroxypropanoic acid Chemical compound OCC(O)=O.CC(O)C(O)=O XBBVURRQGJPTHH-UHFFFAOYSA-N 0.000 description 1
- LNMBCRKRCIMQLW-UHFFFAOYSA-N 2-tert-butylsulfanyl-2-methylpropane Chemical compound CC(C)(C)SC(C)(C)C LNMBCRKRCIMQLW-UHFFFAOYSA-N 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- OBWSOTREAMFOCQ-UHFFFAOYSA-N 4-(4-amino-3,5-dimethylphenyl)-2,6-dimethylaniline;hydrochloride Chemical compound Cl.CC1=C(N)C(C)=CC(C=2C=C(C)C(N)=C(C)C=2)=C1 OBWSOTREAMFOCQ-UHFFFAOYSA-N 0.000 description 1
- XZKIHKMTEMTJQX-UHFFFAOYSA-N 4-Nitrophenyl Phosphate Chemical compound OP(O)(=O)OC1=CC=C([N+]([O-])=O)C=C1 XZKIHKMTEMTJQX-UHFFFAOYSA-N 0.000 description 1
- QFVHZQCOUORWEI-UHFFFAOYSA-N 4-[(4-anilino-5-sulfonaphthalen-1-yl)diazenyl]-5-hydroxynaphthalene-2,7-disulfonic acid Chemical compound C=12C(O)=CC(S(O)(=O)=O)=CC2=CC(S(O)(=O)=O)=CC=1N=NC(C1=CC=CC(=C11)S(O)(=O)=O)=CC=C1NC1=CC=CC=C1 QFVHZQCOUORWEI-UHFFFAOYSA-N 0.000 description 1
- ZDRVLAOYDGQLFI-UHFFFAOYSA-N 4-[[4-(4-chlorophenyl)-1,3-thiazol-2-yl]amino]phenol;hydrochloride Chemical compound Cl.C1=CC(O)=CC=C1NC1=NC(C=2C=CC(Cl)=CC=2)=CS1 ZDRVLAOYDGQLFI-UHFFFAOYSA-N 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- LGZKGOGODCLQHG-CYBMUJFWSA-N 5-[(2r)-2-hydroxy-2-(3,4,5-trimethoxyphenyl)ethyl]-2-methoxyphenol Chemical class C1=C(O)C(OC)=CC=C1C[C@@H](O)C1=CC(OC)=C(OC)C(OC)=C1 LGZKGOGODCLQHG-CYBMUJFWSA-N 0.000 description 1
- 229940117976 5-hydroxylysine Drugs 0.000 description 1
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical compound OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 1
- 102100031126 6-phosphogluconolactonase Human genes 0.000 description 1
- 108010029731 6-phosphogluconolactonase Proteins 0.000 description 1
- WLCZTRVUXYALDD-IBGZPJMESA-N 7-[[(2s)-2,6-bis(2-methoxyethoxycarbonylamino)hexanoyl]amino]heptoxy-methylphosphinic acid Chemical compound COCCOC(=O)NCCCC[C@H](NC(=O)OCCOC)C(=O)NCCCCCCCOP(C)(O)=O WLCZTRVUXYALDD-IBGZPJMESA-N 0.000 description 1
- USJDOLXCPFASNV-UHFFFAOYSA-N 9-bromononan-1-ol Chemical compound OCCCCCCCCCBr USJDOLXCPFASNV-UHFFFAOYSA-N 0.000 description 1
- 208000002874 Acne Vulgaris Diseases 0.000 description 1
- 241000256118 Aedes aegypti Species 0.000 description 1
- 241000256173 Aedes albopictus Species 0.000 description 1
- 235000001674 Agaricus brunnescens Nutrition 0.000 description 1
- WQVFQXXBNHHPLX-ZKWXMUAHSA-N Ala-Ala-His Chemical compound C[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@@H](Cc1cnc[nH]1)C(O)=O WQVFQXXBNHHPLX-ZKWXMUAHSA-N 0.000 description 1
- YYSWCHMLFJLLBJ-ZLUOBGJFSA-N Ala-Ala-Ser Chemical compound C[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(O)=O YYSWCHMLFJLLBJ-ZLUOBGJFSA-N 0.000 description 1
- YYAVDNKUWLAFCV-ACZMJKKPSA-N Ala-Ser-Gln Chemical compound [H]N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(O)=O YYAVDNKUWLAFCV-ACZMJKKPSA-N 0.000 description 1
- 206010001889 Alveolitis Diseases 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- ITPDYQOUSLNIHG-UHFFFAOYSA-N Amiodarone hydrochloride Chemical compound [Cl-].CCCCC=1OC2=CC=CC=C2C=1C(=O)C1=CC(I)=C(OCC[NH+](CC)CC)C(I)=C1 ITPDYQOUSLNIHG-UHFFFAOYSA-N 0.000 description 1
- 244000046614 Annona scleroderma Species 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 101100107610 Arabidopsis thaliana ABCF4 gene Proteins 0.000 description 1
- 241000239290 Araneae Species 0.000 description 1
- BHSYMWWMVRPCPA-CYDGBPFRSA-N Arg-Arg-Ile Chemical compound CC[C@H](C)[C@@H](C(O)=O)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@@H](N)CCCN=C(N)N BHSYMWWMVRPCPA-CYDGBPFRSA-N 0.000 description 1
- 208000002109 Argyria Diseases 0.000 description 1
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 1
- PTNFNTOBUDWHNZ-GUBZILKMSA-N Asn-Arg-Met Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(O)=O PTNFNTOBUDWHNZ-GUBZILKMSA-N 0.000 description 1
- MECFLTFREHAZLH-ACZMJKKPSA-N Asn-Glu-Cys Chemical compound C(CC(=O)O)[C@@H](C(=O)N[C@@H](CS)C(=O)O)NC(=O)[C@H](CC(=O)N)N MECFLTFREHAZLH-ACZMJKKPSA-N 0.000 description 1
- KHCNTVRVAYCPQE-CIUDSAMLSA-N Asn-Lys-Asn Chemical compound [H]N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(O)=O KHCNTVRVAYCPQE-CIUDSAMLSA-N 0.000 description 1
- 241000228212 Aspergillus Species 0.000 description 1
- 241000351920 Aspergillus nidulans Species 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 241001203868 Autographa californica Species 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- 241000194108 Bacillus licheniformis Species 0.000 description 1
- 108090000363 Bacterial Luciferases Proteins 0.000 description 1
- 206010061692 Benign muscle neoplasm Diseases 0.000 description 1
- 102100026189 Beta-galactosidase Human genes 0.000 description 1
- 206010056375 Bile duct obstruction Diseases 0.000 description 1
- 206010004664 Biliary fibrosis Diseases 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 241000409811 Bombyx mori nucleopolyhedrovirus Species 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 101000800130 Bos taurus Thyroglobulin Proteins 0.000 description 1
- 241001332183 Brassica oleracea var. sabauda Species 0.000 description 1
- 235000004214 Brassica oleracea var. sabauda Nutrition 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 206010006448 Bronchiolitis Diseases 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 125000001433 C-terminal amino-acid group Chemical group 0.000 description 1
- 239000002126 C01EB10 - Adenosine Substances 0.000 description 1
- 102000009132 CB1 Cannabinoid Receptor Human genes 0.000 description 1
- 108010073366 CB1 Cannabinoid Receptor Proteins 0.000 description 1
- YKYOUMDCQGMQQO-UHFFFAOYSA-L Cadmium chloride Inorganic materials Cl[Cd]Cl YKYOUMDCQGMQQO-UHFFFAOYSA-L 0.000 description 1
- 101100242814 Caenorhabditis elegans parg-1 gene Proteins 0.000 description 1
- 241000282832 Camelidae Species 0.000 description 1
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 240000004160 Capsicum annuum Species 0.000 description 1
- 235000008534 Capsicum annuum var annuum Nutrition 0.000 description 1
- 102000005367 Carboxypeptidases Human genes 0.000 description 1
- 108010006303 Carboxypeptidases Proteins 0.000 description 1
- 208000020446 Cardiac disease Diseases 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 208000031229 Cardiomyopathies Diseases 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 206010008342 Cervix carcinoma Diseases 0.000 description 1
- 206010008469 Chest discomfort Diseases 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- 241000282552 Chlorocebus aethiops Species 0.000 description 1
- 206010008635 Cholestasis Diseases 0.000 description 1
- 206010008909 Chronic Hepatitis Diseases 0.000 description 1
- 208000000419 Chronic Hepatitis B Diseases 0.000 description 1
- 208000010471 Chronic Hepatitis D Diseases 0.000 description 1
- 208000006154 Chronic hepatitis C Diseases 0.000 description 1
- 208000032544 Cicatrix Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 240000000560 Citrus x paradisi Species 0.000 description 1
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 102000012422 Collagen Type I Human genes 0.000 description 1
- 108010022452 Collagen Type I Proteins 0.000 description 1
- 102000001187 Collagen Type III Human genes 0.000 description 1
- 108010069502 Collagen Type III Proteins 0.000 description 1
- 102000004266 Collagen Type IV Human genes 0.000 description 1
- 108010042086 Collagen Type IV Proteins 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 102000015225 Connective Tissue Growth Factor Human genes 0.000 description 1
- 108010039419 Connective Tissue Growth Factor Proteins 0.000 description 1
- 102000010970 Connexin Human genes 0.000 description 1
- 108050001175 Connexin Proteins 0.000 description 1
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 1
- 229920000742 Cotton Polymers 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- 102100037579 D-3-phosphoglycerate dehydrogenase Human genes 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- IGXWBGJHJZYPQS-SSDOTTSWSA-N D-Luciferin Chemical compound OC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1 IGXWBGJHJZYPQS-SSDOTTSWSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 1
- 108010041986 DNA Vaccines Proteins 0.000 description 1
- 230000004544 DNA amplification Effects 0.000 description 1
- 239000012623 DNA damaging agent Substances 0.000 description 1
- 229940021995 DNA vaccine Drugs 0.000 description 1
- CYCGRDQQIOGCKX-UHFFFAOYSA-N Dehydro-luciferin Natural products OC(=O)C1=CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 CYCGRDQQIOGCKX-UHFFFAOYSA-N 0.000 description 1
- 101710110818 Dermonecrotic toxin Proteins 0.000 description 1
- 101710137373 Dermonecrotic toxin LspiSicTox-betaIII1 Proteins 0.000 description 1
- 101710180073 Dermonecrotic toxin SdSicTox-betaIIB1ai Proteins 0.000 description 1
- 102100036912 Desmin Human genes 0.000 description 1
- 108010044052 Desmin Proteins 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- LTMHDMANZUZIPE-AMTYYWEZSA-N Digoxin Natural products O([C@H]1[C@H](C)O[C@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@](C)([C@H](O)C4)[C@H](C4=CC(=O)OC4)CC5)CC3)CC2)C[C@@H]1O)[C@H]1O[C@H](C)[C@@H](O[C@H]2O[C@@H](C)[C@H](O)[C@@H](O)C2)[C@@H](O)C1 LTMHDMANZUZIPE-AMTYYWEZSA-N 0.000 description 1
- BUDQDWGNQVEFAC-UHFFFAOYSA-N Dihydropyran Chemical group C1COC=CC1 BUDQDWGNQVEFAC-UHFFFAOYSA-N 0.000 description 1
- 241000255581 Drosophila <fruit fly, genus> Species 0.000 description 1
- 241000255601 Drosophila melanogaster Species 0.000 description 1
- 201000009273 Endometriosis Diseases 0.000 description 1
- 102400001047 Endostatin Human genes 0.000 description 1
- 108010079505 Endostatins Proteins 0.000 description 1
- 241000588914 Enterobacter Species 0.000 description 1
- 241000588921 Enterobacteriaceae Species 0.000 description 1
- 102400001368 Epidermal growth factor Human genes 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 241000588698 Erwinia Species 0.000 description 1
- 241001302584 Escherichia coli str. K-12 substr. W3110 Species 0.000 description 1
- 229910052693 Europium Inorganic materials 0.000 description 1
- 108091029865 Exogenous DNA Proteins 0.000 description 1
- 208000027445 Farmer Lung Diseases 0.000 description 1
- 108010067306 Fibronectins Proteins 0.000 description 1
- 102000016359 Fibronectins Human genes 0.000 description 1
- 241000724791 Filamentous phage Species 0.000 description 1
- 108090000331 Firefly luciferases Proteins 0.000 description 1
- 241000192125 Firmicutes Species 0.000 description 1
- BJGNCJDXODQBOB-UHFFFAOYSA-N Fivefly Luciferin Natural products OC(=O)C1CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 BJGNCJDXODQBOB-UHFFFAOYSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 1
- 108010015133 Galactose oxidase Proteins 0.000 description 1
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 1
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 1
- 229930182566 Gentamicin Natural products 0.000 description 1
- YNNXQZDEOCYJJL-CIUDSAMLSA-N Gln-Arg-Asp Chemical compound C(C[C@@H](C(=O)N[C@@H](CC(=O)O)C(=O)O)NC(=O)[C@H](CCC(=O)N)N)CN=C(N)N YNNXQZDEOCYJJL-CIUDSAMLSA-N 0.000 description 1
- WQWMZOIPXWSZNE-WDSKDSINSA-N Gln-Asp-Gly Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)NCC(O)=O WQWMZOIPXWSZNE-WDSKDSINSA-N 0.000 description 1
- 102000006395 Globulins Human genes 0.000 description 1
- 108010044091 Globulins Proteins 0.000 description 1
- YYOBUPFZLKQUAX-FXQIFTODSA-N Glu-Asn-Glu Chemical compound [H]N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(O)=O YYOBUPFZLKQUAX-FXQIFTODSA-N 0.000 description 1
- 108010073178 Glucan 1,4-alpha-Glucosidase Proteins 0.000 description 1
- 102100022624 Glucoamylase Human genes 0.000 description 1
- 239000004366 Glucose oxidase Substances 0.000 description 1
- 108010015776 Glucose oxidase Proteins 0.000 description 1
- 108010018962 Glucosephosphate Dehydrogenase Proteins 0.000 description 1
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 1
- 241000219146 Gossypium Species 0.000 description 1
- 206010018691 Granuloma Diseases 0.000 description 1
- 238000003747 Grignard reaction Methods 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 229910004373 HOAc Inorganic materials 0.000 description 1
- 102000002812 Heat-Shock Proteins Human genes 0.000 description 1
- 108010004889 Heat-Shock Proteins Proteins 0.000 description 1
- 101710154606 Hemagglutinin Proteins 0.000 description 1
- 208000018565 Hemochromatosis Diseases 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 206010019668 Hepatic fibrosis Diseases 0.000 description 1
- 206010019837 Hepatocellular injury Diseases 0.000 description 1
- 208000002972 Hepatolenticular Degeneration Diseases 0.000 description 1
- 101001038043 Homo sapiens Lysophosphatidic acid receptor 4 Proteins 0.000 description 1
- 101000864393 Homo sapiens Protein BUD31 homolog Proteins 0.000 description 1
- 101000693265 Homo sapiens Sphingosine 1-phosphate receptor 1 Proteins 0.000 description 1
- 101000693269 Homo sapiens Sphingosine 1-phosphate receptor 3 Proteins 0.000 description 1
- 101000653759 Homo sapiens Sphingosine 1-phosphate receptor 5 Proteins 0.000 description 1
- 101000663635 Homo sapiens Sphingosine kinase 1 Proteins 0.000 description 1
- 108090000144 Human Proteins Proteins 0.000 description 1
- 102000003839 Human Proteins Human genes 0.000 description 1
- 241000700588 Human alphaherpesvirus 1 Species 0.000 description 1
- 241000701024 Human betaherpesvirus 5 Species 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 206010020565 Hyperaemia Diseases 0.000 description 1
- 206010020880 Hypertrophy Diseases 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- DSDPLOODKXISDT-XUXIUFHCSA-N Ile-Leu-Val Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(O)=O DSDPLOODKXISDT-XUXIUFHCSA-N 0.000 description 1
- TUYOFUHICRWDGA-CIUDSAMLSA-N Ile-Met Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@H](C(O)=O)CCSC TUYOFUHICRWDGA-CIUDSAMLSA-N 0.000 description 1
- IPFKIGNDTUOFAF-CYDGBPFRSA-N Ile-Val-Arg Chemical compound CC[C@H](C)[C@H](N)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(O)=O)CCCN=C(N)N IPFKIGNDTUOFAF-CYDGBPFRSA-N 0.000 description 1
- 108010009817 Immunoglobulin Constant Regions Proteins 0.000 description 1
- 102000009786 Immunoglobulin Constant Regions Human genes 0.000 description 1
- 102000012745 Immunoglobulin Subunits Human genes 0.000 description 1
- 108010079585 Immunoglobulin Subunits Proteins 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 238000012695 Interfacial polymerization Methods 0.000 description 1
- 102100026720 Interferon beta Human genes 0.000 description 1
- 108090000467 Interferon-beta Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 208000016286 Iron metabolism disease Diseases 0.000 description 1
- 241000588748 Klebsiella Species 0.000 description 1
- 241000235058 Komagataella pastoris Species 0.000 description 1
- FADYJNXDPBKVCA-UHFFFAOYSA-N L-Phenylalanyl-L-lysin Natural products NCCCCC(C(O)=O)NC(=O)C(N)CC1=CC=CC=C1 FADYJNXDPBKVCA-UHFFFAOYSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- 241000481961 Lachancea thermotolerans Species 0.000 description 1
- 102100038609 Lactoperoxidase Human genes 0.000 description 1
- 108010023244 Lactoperoxidase Proteins 0.000 description 1
- 108091026898 Leader sequence (mRNA) Proteins 0.000 description 1
- HSQGMTRYSIHDAC-BQBZGAKWSA-N Leu-Ala Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](C)C(O)=O HSQGMTRYSIHDAC-BQBZGAKWSA-N 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- 108090001030 Lipoproteins Proteins 0.000 description 1
- 102000004895 Lipoproteins Human genes 0.000 description 1
- 206010067125 Liver injury Diseases 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 206010024971 Lower respiratory tract infections Diseases 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- DDWFXDSYGUXRAY-UHFFFAOYSA-N Luciferin Natural products CCc1c(C)c(CC2NC(=O)C(=C2C=C)C)[nH]c1Cc3[nH]c4C(=C5/NC(CC(=O)O)C(C)C5CC(=O)O)CC(=O)c4c3C DDWFXDSYGUXRAY-UHFFFAOYSA-N 0.000 description 1
- 235000007688 Lycopersicon esculentum Nutrition 0.000 description 1
- 102100040405 Lysophosphatidic acid receptor 4 Human genes 0.000 description 1
- 241000282553 Macaca Species 0.000 description 1
- 239000004907 Macro-emulsion Substances 0.000 description 1
- 108010026217 Malate Dehydrogenase Proteins 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 108010000684 Matrix Metalloproteinases Proteins 0.000 description 1
- 102000002274 Matrix Metalloproteinases Human genes 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- 102100035971 Molybdopterin molybdenumtransferase Human genes 0.000 description 1
- 101710119577 Molybdopterin molybdenumtransferase Proteins 0.000 description 1
- 241000204795 Muraena helena Species 0.000 description 1
- 102000016943 Muramidase Human genes 0.000 description 1
- 108010014251 Muramidase Proteins 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 201000004458 Myoma Diseases 0.000 description 1
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 1
- OVRNDRQMDRJTHS-CBQIKETKSA-N N-Acetyl-D-Galactosamine Chemical compound CC(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@H](O)[C@@H]1O OVRNDRQMDRJTHS-CBQIKETKSA-N 0.000 description 1
- 108010062010 N-Acetylmuramoyl-L-alanine Amidase Proteins 0.000 description 1
- MBLBDJOUHNCFQT-UHFFFAOYSA-N N-acetyl-D-galactosamine Natural products CC(=O)NC(C=O)C(O)C(O)C(O)CO MBLBDJOUHNCFQT-UHFFFAOYSA-N 0.000 description 1
- 230000004988 N-glycosylation Effects 0.000 description 1
- 101150071357 NPP2 gene Proteins 0.000 description 1
- 108091061960 Naked DNA Proteins 0.000 description 1
- 241000588652 Neisseria gonorrhoeae Species 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 208000028389 Nerve injury Diseases 0.000 description 1
- 241000221960 Neurospora Species 0.000 description 1
- 241000221961 Neurospora crassa Species 0.000 description 1
- 108091005461 Nucleic proteins Proteins 0.000 description 1
- MSHZHSPISPJWHW-UHFFFAOYSA-N O-(chloroacetylcarbamoyl)fumagillol Chemical compound O1C(CC=C(C)C)C1(C)C1C(OC)C(OC(=O)NC(=O)CCl)CCC21CO2 MSHZHSPISPJWHW-UHFFFAOYSA-N 0.000 description 1
- 206010073310 Occupational exposures Diseases 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- 108700020796 Oncogene Proteins 0.000 description 1
- 101710093908 Outer capsid protein VP4 Proteins 0.000 description 1
- 101710135467 Outer capsid protein sigma-1 Proteins 0.000 description 1
- 206010051657 Ovarian fibrosis Diseases 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 208000030852 Parasitic disease Diseases 0.000 description 1
- 208000031481 Pathologic Constriction Diseases 0.000 description 1
- 241000228168 Penicillium sp. Species 0.000 description 1
- 102000057297 Pepsin A Human genes 0.000 description 1
- 108090000284 Pepsin A Proteins 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 108700020962 Peroxidase Proteins 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- 240000007377 Petunia x hybrida Species 0.000 description 1
- FADYJNXDPBKVCA-STQMWFEESA-N Phe-Lys Chemical compound NCCCC[C@@H](C(O)=O)NC(=O)[C@@H](N)CC1=CC=CC=C1 FADYJNXDPBKVCA-STQMWFEESA-N 0.000 description 1
- KIQUCMUULDXTAZ-HJOGWXRNSA-N Phe-Tyr-Tyr Chemical compound N[C@@H](Cc1ccccc1)C(=O)N[C@@H](Cc1ccc(O)cc1)C(=O)N[C@@H](Cc1ccc(O)cc1)C(O)=O KIQUCMUULDXTAZ-HJOGWXRNSA-N 0.000 description 1
- 108010004729 Phycoerythrin Proteins 0.000 description 1
- 101100080097 Phytophthora capsici NLP2 gene Proteins 0.000 description 1
- 208000009362 Pneumococcal Pneumonia Diseases 0.000 description 1
- 206010035728 Pneumonia pneumococcal Diseases 0.000 description 1
- 229920000805 Polyaspartic acid Polymers 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 1
- 101710176177 Protein A56 Proteins 0.000 description 1
- 102100030160 Protein BUD31 homolog Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- 108010067787 Proteoglycans Proteins 0.000 description 1
- 102000016611 Proteoglycans Human genes 0.000 description 1
- 241000588769 Proteus <enterobacteria> Species 0.000 description 1
- 241000589516 Pseudomonas Species 0.000 description 1
- 206010037394 Pulmonary haemorrhage Diseases 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 208000035415 Reinfection Diseases 0.000 description 1
- 208000004756 Respiratory Insufficiency Diseases 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 238000011579 SCID mouse model Methods 0.000 description 1
- 101100068078 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) GCN4 gene Proteins 0.000 description 1
- 241000235343 Saccharomycetales Species 0.000 description 1
- 240000000111 Saccharum officinarum Species 0.000 description 1
- 235000007201 Saccharum officinarum Nutrition 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 241000293869 Salmonella enterica subsp. enterica serovar Typhimurium Species 0.000 description 1
- 241000242678 Schistosoma Species 0.000 description 1
- 238000004639 Schlenk technique Methods 0.000 description 1
- 206010070834 Sensitisation Diseases 0.000 description 1
- 229920005654 Sephadex Polymers 0.000 description 1
- 239000012507 Sephadex™ Substances 0.000 description 1
- UPLYXVPQLJVWMM-KKUMJFAQSA-N Ser-Phe-Leu Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H](CC(C)C)C(O)=O UPLYXVPQLJVWMM-KKUMJFAQSA-N 0.000 description 1
- DKGRNFUXVTYRAS-UBHSHLNASA-N Ser-Ser-Trp Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC1=CNC2=C1C=CC=C2)C(O)=O DKGRNFUXVTYRAS-UBHSHLNASA-N 0.000 description 1
- 241000607768 Shigella Species 0.000 description 1
- 229910003691 SiBr Inorganic materials 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- 240000003768 Solanum lycopersicum Species 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 101710122750 Sphingomyelinase D Proteins 0.000 description 1
- 101710155454 Sphingosine 1-phosphate receptor 1 Proteins 0.000 description 1
- 102100025749 Sphingosine 1-phosphate receptor 2 Human genes 0.000 description 1
- 101710155462 Sphingosine 1-phosphate receptor 2 Proteins 0.000 description 1
- 101710155457 Sphingosine 1-phosphate receptor 3 Proteins 0.000 description 1
- 102100029803 Sphingosine 1-phosphate receptor 4 Human genes 0.000 description 1
- 101710155458 Sphingosine 1-phosphate receptor 4 Proteins 0.000 description 1
- 101710155451 Sphingosine 1-phosphate receptor 5 Proteins 0.000 description 1
- 102100039024 Sphingosine kinase 1 Human genes 0.000 description 1
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 241000187747 Streptomyces Species 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 206010043376 Tetanus Diseases 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- GAMYVSCDDLXAQW-AOIWZFSPSA-N Thermopsosid Natural products O(C)c1c(O)ccc(C=2Oc3c(c(O)cc(O[C@H]4[C@H](O)[C@@H](O)[C@H](O)[C@H](CO)O4)c3)C(=O)C=2)c1 GAMYVSCDDLXAQW-AOIWZFSPSA-N 0.000 description 1
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 1
- COYHRQWNJDJCNA-NUJDXYNKSA-N Thr-Thr-Thr Chemical compound C[C@@H](O)[C@H](N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O COYHRQWNJDJCNA-NUJDXYNKSA-N 0.000 description 1
- 108091036066 Three prime untranslated region Proteins 0.000 description 1
- 108010022394 Threonine synthase Proteins 0.000 description 1
- 208000026062 Tissue disease Diseases 0.000 description 1
- 241001079967 Tolypocladium sp. Species 0.000 description 1
- 101710120037 Toxin CcdB Proteins 0.000 description 1
- 102000004338 Transferrin Human genes 0.000 description 1
- 108090000901 Transferrin Proteins 0.000 description 1
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 1
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 1
- 102000009618 Transforming Growth Factors Human genes 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- 241000223259 Trichoderma Species 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- KHPLUFDSWGDRHD-SLFFLAALSA-N Tyr-Tyr-Pro Chemical compound C1C[C@@H](N(C1)C(=O)[C@H](CC2=CC=C(C=C2)O)NC(=O)[C@H](CC3=CC=C(C=C3)O)N)C(=O)O KHPLUFDSWGDRHD-SLFFLAALSA-N 0.000 description 1
- 101150049278 US20 gene Proteins 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- 108010092464 Urate Oxidase Proteins 0.000 description 1
- 108010046334 Urease Proteins 0.000 description 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 1
- 206010067269 Uterine fibrosis Diseases 0.000 description 1
- 244000000188 Vaccinium ovalifolium Species 0.000 description 1
- 206010046996 Varicose vein Diseases 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 208000036141 Viral hepatitis carrier Diseases 0.000 description 1
- 244000195452 Wasabia japonica Species 0.000 description 1
- 235000000760 Wasabia japonica Nutrition 0.000 description 1
- 208000018839 Wilson disease Diseases 0.000 description 1
- 206010052428 Wound Diseases 0.000 description 1
- 108010093894 Xanthine oxidase Proteins 0.000 description 1
- 102100033220 Xanthine oxidase Human genes 0.000 description 1
- 241000235013 Yarrowia Species 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- JCABVIFDXFFRMT-DIPNUNPCSA-N [(2r)-1-[ethoxy(hydroxy)phosphoryl]oxy-3-hexadecanoyloxypropan-2-yl] octadec-9-enoate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OCC)OC(=O)CCCCCCCC=CCCCCCCCC JCABVIFDXFFRMT-DIPNUNPCSA-N 0.000 description 1
- IERHLVCPSMICTF-CCXZUQQUSA-N [(2r,3s,4s,5r)-5-(4-amino-2-oxopyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl dihydrogen phosphate Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](COP(O)(O)=O)O1 IERHLVCPSMICTF-CCXZUQQUSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- IKHGUXGNUITLKF-XPULMUKRSA-N acetaldehyde Chemical compound [14CH]([14CH3])=O IKHGUXGNUITLKF-XPULMUKRSA-N 0.000 description 1
- 239000003377 acid catalyst Substances 0.000 description 1
- 238000010306 acid treatment Methods 0.000 description 1
- 239000003929 acidic solution Substances 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 206010000496 acne Diseases 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 229960005305 adenosine Drugs 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000009824 affinity maturation Effects 0.000 description 1
- 238000001261 affinity purification Methods 0.000 description 1
- 230000004931 aggregating effect Effects 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- 108010022198 alkylglycerophosphoethanolamine phosphodiesterase Proteins 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- 229940037003 alum Drugs 0.000 description 1
- 229960003437 aminoglutethimide Drugs 0.000 description 1
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 1
- 229960003896 aminopterin Drugs 0.000 description 1
- 229960005260 amiodarone Drugs 0.000 description 1
- 238000012870 ammonium sulfate precipitation Methods 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- 239000003957 anion exchange resin Substances 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 238000011122 anti-angiogenic therapy Methods 0.000 description 1
- 229940046836 anti-estrogen Drugs 0.000 description 1
- 230000001833 anti-estrogenic effect Effects 0.000 description 1
- 230000003510 anti-fibrotic effect Effects 0.000 description 1
- 229940035635 anti-gonadotropin-releasing hormone Drugs 0.000 description 1
- 230000002529 anti-mitochondrial effect Effects 0.000 description 1
- 230000002927 anti-mitotic effect Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 230000009830 antibody antigen interaction Effects 0.000 description 1
- 238000003452 antibody preparation method Methods 0.000 description 1
- 229940124691 antibody therapeutics Drugs 0.000 description 1
- 210000000628 antibody-producing cell Anatomy 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 230000004596 appetite loss Effects 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 238000010936 aqueous wash Methods 0.000 description 1
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 1
- 239000003886 aromatase inhibitor Substances 0.000 description 1
- 229940046844 aromatase inhibitors Drugs 0.000 description 1
- 230000006793 arrhythmia Effects 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 125000000613 asparagine group Chemical group N[C@@H](CC(N)=O)C(=O)* 0.000 description 1
- 238000003149 assay kit Methods 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 208000021033 autosomal dominant polycystic liver disease Diseases 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 239000003659 bee venom Substances 0.000 description 1
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 1
- 229960002707 bendamustine Drugs 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 1
- 229960001950 benzethonium chloride Drugs 0.000 description 1
- RWCCWEUUXYIKHB-UHFFFAOYSA-N benzophenone Chemical compound C=1C=CC=CC=1C(=O)C1=CC=CC=C1 RWCCWEUUXYIKHB-UHFFFAOYSA-N 0.000 description 1
- 239000012965 benzophenone Substances 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 238000013357 binding ELISA Methods 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000012148 binding buffer Substances 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 239000013060 biological fluid Substances 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- 238000005460 biophysical method Methods 0.000 description 1
- HUTDDBSSHVOYJR-UHFFFAOYSA-H bis[(2-oxo-1,3,2$l^{5},4$l^{2}-dioxaphosphaplumbetan-2-yl)oxy]lead Chemical compound [Pb+2].[Pb+2].[Pb+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O HUTDDBSSHVOYJR-UHFFFAOYSA-H 0.000 description 1
- 210000002459 blastocyst Anatomy 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 238000010322 bone marrow transplantation Methods 0.000 description 1
- 229960001467 bortezomib Drugs 0.000 description 1
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- SXDBWCPKPHAZSM-UHFFFAOYSA-N bromic acid Chemical compound OBr(=O)=O SXDBWCPKPHAZSM-UHFFFAOYSA-N 0.000 description 1
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N butyl alcohol Substances CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940088954 camptosar Drugs 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 239000001511 capsicum annuum Substances 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 230000001269 cardiogenic effect Effects 0.000 description 1
- 230000001451 cardiotoxic effect Effects 0.000 description 1
- 239000002327 cardiovascular agent Substances 0.000 description 1
- 229940125692 cardiovascular agent Drugs 0.000 description 1
- 210000000748 cardiovascular system Anatomy 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 238000012219 cassette mutagenesis Methods 0.000 description 1
- 239000003729 cation exchange resin Substances 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 230000005779 cell damage Effects 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000006721 cell death pathway Effects 0.000 description 1
- 230000007348 cell dedifferentiation Effects 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 208000037887 cell injury Diseases 0.000 description 1
- 230000019522 cellular metabolic process Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 239000004568 cement Substances 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- RIZIAUKTHDLMQX-UHFFFAOYSA-N cerebroside D Natural products CCCCCCCCCCCCCCCCC(O)C(=O)NC(C(O)C=CCCC=C(C)CCCCCCCCC)COC1OC(CO)C(O)C(O)C1O RIZIAUKTHDLMQX-UHFFFAOYSA-N 0.000 description 1
- 150000001784 cerebrosides Chemical class 0.000 description 1
- 208000026106 cerebrovascular disease Diseases 0.000 description 1
- 201000010881 cervical cancer Diseases 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 238000009614 chemical analysis method Methods 0.000 description 1
- 238000012412 chemical coupling Methods 0.000 description 1
- 150000005829 chemical entities Chemical class 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 230000035605 chemotaxis Effects 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 235000012000 cholesterol Nutrition 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 239000012539 chromatography resin Substances 0.000 description 1
- OHNBIIZWIUBGTK-NYPSMHOZSA-N cid_23581792 Chemical compound O.Cl.C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 OHNBIIZWIUBGTK-NYPSMHOZSA-N 0.000 description 1
- 235000019504 cigarettes Nutrition 0.000 description 1
- 230000004087 circulation Effects 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- 238000003759 clinical diagnosis Methods 0.000 description 1
- 238000007374 clinical diagnostic method Methods 0.000 description 1
- 239000013599 cloning vector Substances 0.000 description 1
- 230000004186 co-expression Effects 0.000 description 1
- 238000005354 coacervation Methods 0.000 description 1
- 239000003245 coal Substances 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229910052681 coesite Inorganic materials 0.000 description 1
- 239000000084 colloidal system Substances 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- LGZKGOGODCLQHG-UHFFFAOYSA-N combretastatin Natural products C1=C(O)C(OC)=CC=C1CC(O)C1=CC(OC)=C(OC)C(OC)=C1 LGZKGOGODCLQHG-UHFFFAOYSA-N 0.000 description 1
- 230000001447 compensatory effect Effects 0.000 description 1
- 239000008139 complexing agent Substances 0.000 description 1
- 239000003636 conditioned culture medium Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 239000005289 controlled pore glass Substances 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 210000004087 cornea Anatomy 0.000 description 1
- 239000007822 coupling agent Substances 0.000 description 1
- 229910052906 cristobalite Inorganic materials 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 229910002026 crystalline silica Inorganic materials 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- UFULAYFCSOUIOV-UHFFFAOYSA-N cysteamine Chemical compound NCCS UFULAYFCSOUIOV-UHFFFAOYSA-N 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 125000001295 dansyl group Chemical group [H]C1=C([H])C(N(C([H])([H])[H])C([H])([H])[H])=C2C([H])=C([H])C([H])=C(C2=C1[H])S(*)(=O)=O 0.000 description 1
- 230000020176 deacylation Effects 0.000 description 1
- 238000005947 deacylation reaction Methods 0.000 description 1
- 230000006240 deamidation Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- YSMODUONRAFBET-UHFFFAOYSA-N delta-DL-hydroxylysine Natural products NCC(O)CCC(N)C(O)=O YSMODUONRAFBET-UHFFFAOYSA-N 0.000 description 1
- 238000006392 deoxygenation reaction Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 210000005045 desmin Anatomy 0.000 description 1
- 230000001066 destructive effect Effects 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 238000002059 diagnostic imaging Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 239000012954 diazonium Substances 0.000 description 1
- KVEAILYLMGOETO-UHFFFAOYSA-H dicalcium magnesium diphosphate Chemical compound P(=O)([O-])([O-])[O-].[Mg+2].[Ca+2].[Ca+2].P(=O)([O-])([O-])[O-] KVEAILYLMGOETO-UHFFFAOYSA-H 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- LTMHDMANZUZIPE-PUGKRICDSA-N digoxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)[C@H](O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O LTMHDMANZUZIPE-PUGKRICDSA-N 0.000 description 1
- LTMHDMANZUZIPE-UHFFFAOYSA-N digoxine Natural products C1C(O)C(O)C(C)OC1OC1C(C)OC(OC2C(OC(OC3CC4C(C5C(C6(CCC(C6(C)C(O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)CC2O)C)CC1O LTMHDMANZUZIPE-UHFFFAOYSA-N 0.000 description 1
- 102000004419 dihydrofolate reductase Human genes 0.000 description 1
- 125000005442 diisocyanate group Chemical group 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- JKNIOHXBRYZCTM-UHFFFAOYSA-N dimethyl hexanediimidate;hydrochloride Chemical compound Cl.COC(=N)CCCCC(=N)OC JKNIOHXBRYZCTM-UHFFFAOYSA-N 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- XEYBRNLFEZDVAW-ARSRFYASSA-N dinoprostone Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEYBRNLFEZDVAW-ARSRFYASSA-N 0.000 description 1
- 229960002986 dinoprostone Drugs 0.000 description 1
- 229960003983 diphtheria toxoid Drugs 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 238000002845 discoloration Methods 0.000 description 1
- VXNQMUVMEIGUJW-XNOMRPDFSA-L disodium;[2-methoxy-5-[(z)-2-(3,4,5-trimethoxyphenyl)ethenyl]phenyl] phosphate Chemical compound [Na+].[Na+].C1=C(OP([O-])([O-])=O)C(OC)=CC=C1\C=C/C1=CC(OC)=C(OC)C(OC)=C1 VXNQMUVMEIGUJW-XNOMRPDFSA-L 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 125000002228 disulfide group Chemical group 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 1
- ZWAOHEXOSAUJHY-ZIYNGMLESA-N doxifluridine Chemical compound O[C@@H]1[C@H](O)[C@@H](C)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ZWAOHEXOSAUJHY-ZIYNGMLESA-N 0.000 description 1
- 229950005454 doxifluridine Drugs 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 235000013601 eggs Nutrition 0.000 description 1
- 239000012149 elution buffer Substances 0.000 description 1
- 230000010102 embolization Effects 0.000 description 1
- 238000009261 endocrine therapy Methods 0.000 description 1
- 229940034984 endocrine therapy antineoplastic and immunomodulating agent Drugs 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 238000001839 endoscopy Methods 0.000 description 1
- 239000002158 endotoxin Substances 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 229940082789 erbitux Drugs 0.000 description 1
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 1
- 229960001433 erlotinib Drugs 0.000 description 1
- YSMODUONRAFBET-UHNVWZDZSA-N erythro-5-hydroxy-L-lysine Chemical compound NC[C@H](O)CC[C@H](N)C(O)=O YSMODUONRAFBET-UHNVWZDZSA-N 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 239000000328 estrogen antagonist Substances 0.000 description 1
- 238000012869 ethanol precipitation Methods 0.000 description 1
- 239000012259 ether extract Substances 0.000 description 1
- OGPBJKLSAFTDLK-UHFFFAOYSA-N europium atom Chemical compound [Eu] OGPBJKLSAFTDLK-UHFFFAOYSA-N 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 229960000255 exemestane Drugs 0.000 description 1
- 239000013613 expression plasmid Substances 0.000 description 1
- 210000003722 extracellular fluid Anatomy 0.000 description 1
- 201000001155 extrinsic allergic alveolitis Diseases 0.000 description 1
- 210000000416 exudates and transudate Anatomy 0.000 description 1
- 208000022195 farmer lung disease Diseases 0.000 description 1
- 229940087861 faslodex Drugs 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 229930003944 flavone Natural products 0.000 description 1
- 150000002212 flavone derivatives Chemical class 0.000 description 1
- 235000011949 flavones Nutrition 0.000 description 1
- 229930003935 flavonoid Natural products 0.000 description 1
- 150000002215 flavonoids Chemical class 0.000 description 1
- 235000017173 flavonoids Nutrition 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- 150000002222 fluorine compounds Chemical class 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 210000001733 follicular fluid Anatomy 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- 108010074605 gamma-Globulins Proteins 0.000 description 1
- 210000003976 gap junction Anatomy 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- 238000003500 gene array Methods 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 229940080856 gleevec Drugs 0.000 description 1
- 229940116332 glucose oxidase Drugs 0.000 description 1
- 235000019420 glucose oxidase Nutrition 0.000 description 1
- 125000000404 glutamine group Chemical group N[C@@H](CCC(N)=O)C(=O)* 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 150000002316 glycerophosphocholines Chemical class 0.000 description 1
- 230000034659 glycolysis Effects 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- XLXSAKCOAKORKW-AQJXLSMYSA-N gonadorelin Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 XLXSAKCOAKORKW-AQJXLSMYSA-N 0.000 description 1
- 239000010438 granite Substances 0.000 description 1
- 210000002064 heart cell Anatomy 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- XLYOFNOQVPJJNP-ZSJDYOACSA-N heavy water Substances [2H]O[2H] XLYOFNOQVPJJNP-ZSJDYOACSA-N 0.000 description 1
- 239000000185 hemagglutinin Substances 0.000 description 1
- 201000011066 hemangioma Diseases 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 231100000234 hepatic damage Toxicity 0.000 description 1
- 208000010710 hepatitis C virus infection Diseases 0.000 description 1
- 231100000437 hepatocellular injury Toxicity 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- 229960001340 histamine Drugs 0.000 description 1
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 1
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 1
- 239000000710 homodimer Substances 0.000 description 1
- 238000002744 homologous recombination Methods 0.000 description 1
- 230000006801 homologous recombination Effects 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 238000004191 hydrophobic interaction chromatography Methods 0.000 description 1
- 229920001600 hydrophobic polymer Polymers 0.000 description 1
- 125000002349 hydroxyamino group Chemical group [H]ON([H])[*] 0.000 description 1
- 229920003063 hydroxymethyl cellulose Polymers 0.000 description 1
- 229940031574 hydroxymethyl cellulose Drugs 0.000 description 1
- 230000009610 hypersensitivity Effects 0.000 description 1
- 208000022098 hypersensitivity pneumonitis Diseases 0.000 description 1
- 230000000729 hypotrophic effect Effects 0.000 description 1
- 238000009802 hysterectomy Methods 0.000 description 1
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- 230000002055 immunohistochemical effect Effects 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000006882 induction of apoptosis Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 229960003971 influenza vaccine Drugs 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 150000002484 inorganic compounds Chemical class 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 102000027411 intracellular receptors Human genes 0.000 description 1
- 108091008582 intracellular receptors Proteins 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical class II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 230000005865 ionizing radiation Effects 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 230000000302 ischemic effect Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- 235000014705 isoleucine Nutrition 0.000 description 1
- 229960003350 isoniazid Drugs 0.000 description 1
- QRXWMOHMRWLFEY-UHFFFAOYSA-N isoniazide Chemical compound NNC(=O)C1=CC=NC=C1 QRXWMOHMRWLFEY-UHFFFAOYSA-N 0.000 description 1
- 238000005304 joining Methods 0.000 description 1
- 229940057428 lactoperoxidase Drugs 0.000 description 1
- 238000002386 leaching Methods 0.000 description 1
- 239000011133 lead Substances 0.000 description 1
- 229960004942 lenalidomide Drugs 0.000 description 1
- GOTYRUGSSMKFNF-UHFFFAOYSA-N lenalidomide Chemical compound C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O GOTYRUGSSMKFNF-UHFFFAOYSA-N 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 1
- 108010071185 leucyl-alanine Proteins 0.000 description 1
- 108010044056 leucyl-phenylalanine Proteins 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- 125000003473 lipid group Chemical group 0.000 description 1
- 201000007270 liver cancer Diseases 0.000 description 1
- 230000008818 liver damage Effects 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 208000018191 liver inflammation Diseases 0.000 description 1
- 208000014018 liver neoplasm Diseases 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 208000019017 loss of appetite Diseases 0.000 description 1
- 235000021266 loss of appetite Nutrition 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 210000005265 lung cell Anatomy 0.000 description 1
- 210000004324 lymphatic system Anatomy 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 239000004325 lysozyme Substances 0.000 description 1
- 229960000274 lysozyme Drugs 0.000 description 1
- 235000010335 lysozyme Nutrition 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 208000011384 malt worker lung Diseases 0.000 description 1
- 210000001161 mammalian embryo Anatomy 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 229960003151 mercaptamine Drugs 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 230000037353 metabolic pathway Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229960004452 methionine Drugs 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 239000004530 micro-emulsion Substances 0.000 description 1
- 210000003632 microfilament Anatomy 0.000 description 1
- 108010029942 microperoxidase Proteins 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 230000004001 molecular interaction Effects 0.000 description 1
- 238000002625 monoclonal antibody therapy Methods 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- 230000004660 morphological change Effects 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 230000004899 motility Effects 0.000 description 1
- 238000000465 moulding Methods 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 230000002107 myocardial effect Effects 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- UPSFMJHZUCSEHU-JYGUBCOQSA-N n-[(2s,3r,4r,5s,6r)-2-[(2r,3s,4r,5r,6s)-5-acetamido-4-hydroxy-2-(hydroxymethyl)-6-(4-methyl-2-oxochromen-7-yl)oxyoxan-3-yl]oxy-4,5-dihydroxy-6-(hydroxymethyl)oxan-3-yl]acetamide Chemical compound CC(=O)N[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1O[C@H]1[C@H](O)[C@@H](NC(C)=O)[C@H](OC=2C=C3OC(=O)C=C(C)C3=CC=2)O[C@@H]1CO UPSFMJHZUCSEHU-JYGUBCOQSA-N 0.000 description 1
- 239000002088 nanocapsule Substances 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 229940086322 navelbine Drugs 0.000 description 1
- 230000001338 necrotic effect Effects 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 230000008764 nerve damage Effects 0.000 description 1
- 208000004296 neuralgia Diseases 0.000 description 1
- 210000002241 neurite Anatomy 0.000 description 1
- 208000021722 neuropathic pain Diseases 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229910052758 niobium Inorganic materials 0.000 description 1
- 239000010955 niobium Substances 0.000 description 1
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 1
- 229960000564 nitrofurantoin Drugs 0.000 description 1
- NXFQHRVNIOXGAQ-YCRREMRBSA-N nitrofurantoin Chemical compound O1C([N+](=O)[O-])=CC=C1\C=N\N1C(=O)NC(=O)C1 NXFQHRVNIOXGAQ-YCRREMRBSA-N 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000003865 nucleic acid synthesis inhibitor Substances 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- LUUFSCNUZAYHAT-UHFFFAOYSA-N octadecane-1,18-diol Chemical compound OCCCCCCCCCCCCCCCCCCO LUUFSCNUZAYHAT-UHFFFAOYSA-N 0.000 description 1
- IXWNTLSTOZFSCM-YVACAVLKSA-N ombrabulin Chemical compound C1=C(NC(=O)[C@@H](N)CO)C(OC)=CC=C1\C=C/C1=CC(OC)=C(OC)C(OC)=C1 IXWNTLSTOZFSCM-YVACAVLKSA-N 0.000 description 1
- 229950003600 ombrabulin Drugs 0.000 description 1
- 108091008796 oncogenic growth factors Proteins 0.000 description 1
- 229940023490 ophthalmic product Drugs 0.000 description 1
- 229940126701 oral medication Drugs 0.000 description 1
- 210000003463 organelle Anatomy 0.000 description 1
- BWKDAMBGCPRVPI-ZQRPHVBESA-N ortataxel Chemical compound O([C@@H]1[C@]23OC(=O)O[C@H]2[C@@H](C(=C([C@@H](OC(C)=O)C(=O)[C@]2(C)[C@@H](O)C[C@H]4OC[C@]4([C@H]21)OC(C)=O)C3(C)C)C)OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)CC(C)C)C(=O)C1=CC=CC=C1 BWKDAMBGCPRVPI-ZQRPHVBESA-N 0.000 description 1
- 229940127075 other antimetabolite Drugs 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000002640 oxygen therapy Methods 0.000 description 1
- LXCFILQKKLGQFO-UHFFFAOYSA-N p-hydroxybenzoic acid methyl ester Natural products COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 210000004738 parenchymal cell Anatomy 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 229960003407 pegaptanib Drugs 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- 229940111202 pepsin Drugs 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 230000003836 peripheral circulation Effects 0.000 description 1
- 239000003614 peroxisome proliferator Substances 0.000 description 1
- 230000002974 pharmacogenomic effect Effects 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- 150000008103 phosphatidic acids Chemical class 0.000 description 1
- DHRLEVQXOMLTIM-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum Chemical compound O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O DHRLEVQXOMLTIM-UHFFFAOYSA-N 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 150000003057 platinum Chemical class 0.000 description 1
- 206010035653 pneumoconiosis Diseases 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 108010054442 polyalanine Proteins 0.000 description 1
- 108010064470 polyaspartate Proteins 0.000 description 1
- 208000028589 polycystic liver disease Diseases 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920002338 polyhydroxyethylmethacrylate Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000005373 porous glass Substances 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 208000007232 portal hypertension Diseases 0.000 description 1
- 230000013155 positive regulation of cell migration Effects 0.000 description 1
- 230000004481 post-translational protein modification Effects 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 244000144977 poultry Species 0.000 description 1
- 210000000229 preadipocyte Anatomy 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 230000002250 progressing effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- XEYBRNLFEZDVAW-UHFFFAOYSA-N prostaglandin E2 Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CC=CCCCC(O)=O XEYBRNLFEZDVAW-UHFFFAOYSA-N 0.000 description 1
- 235000019419 proteases Nutrition 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 229940121649 protein inhibitor Drugs 0.000 description 1
- 239000012268 protein inhibitor Substances 0.000 description 1
- 238000001742 protein purification Methods 0.000 description 1
- 230000012743 protein tagging Effects 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 230000009325 pulmonary function Effects 0.000 description 1
- 208000008128 pulmonary tuberculosis Diseases 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- HBCQSNAFLVXVAY-UHFFFAOYSA-N pyrimidine-2-thiol Chemical compound SC1=NC=CC=N1 HBCQSNAFLVXVAY-UHFFFAOYSA-N 0.000 description 1
- 229940043131 pyroglutamate Drugs 0.000 description 1
- 239000010453 quartz Substances 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 238000002601 radiography Methods 0.000 description 1
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- 238000002708 random mutagenesis Methods 0.000 description 1
- 229960003876 ranibizumab Drugs 0.000 description 1
- 229910052761 rare earth metal Inorganic materials 0.000 description 1
- 150000002910 rare earth metals Chemical class 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 230000026267 regulation of growth Effects 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 201000004193 respiratory failure Diseases 0.000 description 1
- 210000002345 respiratory system Anatomy 0.000 description 1
- 230000008458 response to injury Effects 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 239000011435 rock Substances 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 238000005488 sandblasting Methods 0.000 description 1
- 150000004671 saturated fatty acids Chemical class 0.000 description 1
- 235000003441 saturated fatty acids Nutrition 0.000 description 1
- 230000037387 scars Effects 0.000 description 1
- 201000004409 schistosomiasis Diseases 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 230000008313 sensitization Effects 0.000 description 1
- 230000001235 sensitizing effect Effects 0.000 description 1
- 125000003607 serino group Chemical group [H]N([H])[C@]([H])(C(=O)[*])C(O[H])([H])[H] 0.000 description 1
- 230000000405 serological effect Effects 0.000 description 1
- 210000000717 sertoli cell Anatomy 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 210000002460 smooth muscle Anatomy 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- PTLRDCMBXHILCL-UHFFFAOYSA-M sodium arsenite Chemical compound [Na+].[O-][As]=O PTLRDCMBXHILCL-UHFFFAOYSA-M 0.000 description 1
- 235000017550 sodium carbonate Nutrition 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 235000011008 sodium phosphates Nutrition 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 108010035597 sphingosine kinase Proteins 0.000 description 1
- 150000003410 sphingosines Chemical class 0.000 description 1
- 238000009987 spinning Methods 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 238000007655 standard test method Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- SFVFIFLLYFPGHH-UHFFFAOYSA-M stearalkonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCCCC[N+](C)(C)CC1=CC=CC=C1 SFVFIFLLYFPGHH-UHFFFAOYSA-M 0.000 description 1
- 230000036262 stenosis Effects 0.000 description 1
- 208000037804 stenosis Diseases 0.000 description 1
- 238000011146 sterile filtration Methods 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 229910052682 stishovite Inorganic materials 0.000 description 1
- 208000022218 streptococcal pneumonia Diseases 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 210000003518 stress fiber Anatomy 0.000 description 1
- 210000002330 subarachnoid space Anatomy 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 125000001273 sulfonato group Chemical group [O-]S(*)(=O)=O 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000009469 supplementation Effects 0.000 description 1
- 230000003319 supportive effect Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000004114 suspension culture Methods 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 229950007866 tanespimycin Drugs 0.000 description 1
- AYUNIORJHRXIBJ-TXHRRWQRSA-N tanespimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(NCC=C)C(=O)C=C1C2=O AYUNIORJHRXIBJ-TXHRRWQRSA-N 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 239000003277 telomerase inhibitor Substances 0.000 description 1
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 1
- IMCGHZIGRANKHV-AJNGGQMLSA-N tert-butyl (3s,5s)-2-oxo-5-[(2s,4s)-5-oxo-4-propan-2-yloxolan-2-yl]-3-propan-2-ylpyrrolidine-1-carboxylate Chemical compound O1C(=O)[C@H](C(C)C)C[C@H]1[C@H]1N(C(=O)OC(C)(C)C)C(=O)[C@H](C(C)C)C1 IMCGHZIGRANKHV-AJNGGQMLSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229960002180 tetracycline Drugs 0.000 description 1
- 229930101283 tetracycline Natural products 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- MPLHNVLQVRSVEE-UHFFFAOYSA-N texas red Chemical compound [O-]S(=O)(=O)C1=CC(S(Cl)(=O)=O)=CC=C1C(C1=CC=2CCCN3CCCC(C=23)=C1O1)=C2C1=C(CCC1)C3=[N+]1CCCC3=C2 MPLHNVLQVRSVEE-UHFFFAOYSA-N 0.000 description 1
- 229960003433 thalidomide Drugs 0.000 description 1
- YUKQRDCYNOVPGJ-UHFFFAOYSA-N thioacetamide Chemical compound CC(N)=S YUKQRDCYNOVPGJ-UHFFFAOYSA-N 0.000 description 1
- DLFVBJFMPXGRIB-UHFFFAOYSA-N thioacetamide Natural products CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 1
- CNHYKKNIIGEXAY-UHFFFAOYSA-N thiolan-2-imine Chemical compound N=C1CCCS1 CNHYKKNIIGEXAY-UHFFFAOYSA-N 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 230000000451 tissue damage Effects 0.000 description 1
- 231100000827 tissue damage Toxicity 0.000 description 1
- 230000030968 tissue homeostasis Effects 0.000 description 1
- 208000037816 tissue injury Diseases 0.000 description 1
- 230000007838 tissue remodeling Effects 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- 229960005026 toremifene Drugs 0.000 description 1
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 1
- 239000011573 trace mineral Substances 0.000 description 1
- 235000013619 trace mineral Nutrition 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 239000012581 transferrin Substances 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 230000000472 traumatic effect Effects 0.000 description 1
- 229950001353 tretamine Drugs 0.000 description 1
- IUCJMVBFZDHPDX-UHFFFAOYSA-N tretamine Chemical compound C1CN1C1=NC(N2CC2)=NC(N2CC2)=N1 IUCJMVBFZDHPDX-UHFFFAOYSA-N 0.000 description 1
- 229910052905 tridymite Inorganic materials 0.000 description 1
- 150000005671 trienes Chemical class 0.000 description 1
- WVLBCYQITXONBZ-UHFFFAOYSA-N trimethyl phosphate Chemical compound COP(=O)(OC)OC WVLBCYQITXONBZ-UHFFFAOYSA-N 0.000 description 1
- YWWDBCBWQNCYNR-UHFFFAOYSA-N trimethylphosphine Chemical compound CP(C)C YWWDBCBWQNCYNR-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 230000005747 tumor angiogenesis Effects 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 231100000397 ulcer Toxicity 0.000 description 1
- 235000018648 unbalanced nutrition Nutrition 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 235000021122 unsaturated fatty acids Nutrition 0.000 description 1
- 150000004670 unsaturated fatty acids Chemical class 0.000 description 1
- 208000019206 urinary tract infection Diseases 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 208000010579 uterine corpus leiomyoma Diseases 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- XGOYIMQSIKSOBS-UHFFFAOYSA-N vadimezan Chemical compound C1=CC=C2C(=O)C3=CC=C(C)C(C)=C3OC2=C1CC(O)=O XGOYIMQSIKSOBS-UHFFFAOYSA-N 0.000 description 1
- 229950008737 vadimezan Drugs 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 235000014393 valine Nutrition 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 208000027185 varicose disease Diseases 0.000 description 1
- 210000004509 vascular smooth muscle cell Anatomy 0.000 description 1
- 239000004066 vascular targeting agent Substances 0.000 description 1
- 210000005166 vasculature Anatomy 0.000 description 1
- 230000009724 venous congestion Effects 0.000 description 1
- 238000009423 ventilation Methods 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- UGGWPQSBPIFKDZ-KOTLKJBCSA-N vindesine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(N)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 UGGWPQSBPIFKDZ-KOTLKJBCSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- GBABOYUKABKIAF-IELIFDKJSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-IELIFDKJSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- CILBMBUYJCWATM-PYGJLNRPSA-N vinorelbine ditartrate Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O.OC(=O)[C@H](O)[C@@H](O)C(O)=O.C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC CILBMBUYJCWATM-PYGJLNRPSA-N 0.000 description 1
- VHBFFQKBGNRLFZ-UHFFFAOYSA-N vitamin p Natural products O1C2=CC=CC=C2C(=O)C=C1C1=CC=CC=C1 VHBFFQKBGNRLFZ-UHFFFAOYSA-N 0.000 description 1
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 1
- 229960000237 vorinostat Drugs 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 208000016261 weight loss Diseases 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 229940045860 white wax Drugs 0.000 description 1
- UGBMEXLBFDAOGL-INIZCTEOSA-N zd6126 Chemical compound C1C[C@H](NC(C)=O)C2=CC(OP(O)(O)=O)=CC=C2C2=C1C=C(OC)C(OC)=C2OC UGBMEXLBFDAOGL-INIZCTEOSA-N 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Images

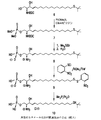





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3076—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells against structure-related tumour-associated moieties
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/92—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving lipids, e.g. cholesterol, lipoproteins, or their receptors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
- A61P29/02—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID] without antiinflammatory effect
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P41/00—Drugs used in surgical methods, e.g. surgery adjuvants for preventing adhesion or for vitreum substitution
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/44—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material not provided for elsewhere, e.g. haptens, metals, DNA, RNA, amino acids
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/577—Immunoassay; Biospecific binding assay; Materials therefor involving monoclonal antibodies binding reaction mechanisms characterised by the use of monoclonal antibodies; monoclonal antibodies per se are classified with their corresponding antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2405/00—Assays, e.g. immunoassays or enzyme assays, involving lipids
- G01N2405/04—Phospholipids, i.e. phosphoglycerides
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Hematology (AREA)
- Biochemistry (AREA)
- Urology & Nephrology (AREA)
- Biophysics (AREA)
- Cell Biology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Neurology (AREA)
- Genetics & Genomics (AREA)
- Endocrinology (AREA)
- Physics & Mathematics (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Food Science & Technology (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- Dermatology (AREA)
- Pain & Pain Management (AREA)
- Obesity (AREA)
- Reproductive Health (AREA)
Description
LPAは、成長を促す腫瘍のマイクロ環境を提供し、血管形成を促進することにより、ガンの形成と進行に関与する。さらに、LPAは、線維症、黄斑変性症のような眼病、ならびに疼痛に関連する障害にも関連している。したがって、LPAを中和するための抗体を使用するアプローチは、これらの適応症のための現行の治療手段を増やす潜在的可能性を提供する。
以下に記載の説明は、本発明を理解するために役に立ち得る情報を含む。ここに記載するいかなる情報、またはここに明示的または暗示的に引用するいかなる刊行物も、ここで請求される発明の従来技術であること、または特別に関連性があることさえも認めるものではない。
生理活性シグナル伝達脂質
脂質とそれらの誘導体は現在、単なる細胞膜中の1つの構造エレメントまたはβ酸化、解糖などの代謝プロセスの源としてではなく、医学研究のための重要な標的として認識されている。特に、ある種の生理活性脂質は、動物およびヒト疾患において重要なシグナル伝達メディエーターとして機能する。原形質膜に由来する脂質のほとんどは、構造的役割のみを果たしているにもかかわらず、それらの小部分は、細胞外刺激の細胞内への取り次ぎに関与している。これらの脂質は「生理活性脂質」、または「生理活性シグナル伝達脂質」と呼ばれている。「脂質シグナル伝達」とは、細胞膜脂質を第二のメッセンジャーとして使用する多数の細胞シグナル伝達経路のうちのいずれかを意味し、ならびに脂質シグナル伝達分子とその特異的レセプターとの直接的相互作用を意味する。脂質シグナル伝達経路は、増殖因子から炎症性サイトカインに至る様々な細胞外刺激によって活性化され、アポトーシス、分化、増殖のような細胞運命の決定を制御する。生理活性脂質シグナル伝達の研究は、精力的に科学的研究が推し進められている分野であり、ますます多くの生理活性脂質が同定され、その作用が特徴づけられている。
リゾリン脂質(LPL)は、リゾ脂質としても知られており、1つの炭化水素主鎖と1つのリン酸基 を含む1つの極性頭基を含む、 低分子量(一般的には約500ダルトン未満)の脂質である。リゾ脂質のあるものは、生理活性シグナル伝達脂質である。医学的に重要な生理活性リゾ脂質の2つの具体的な例は、LPA(グリセロール主鎖)とS1P(スフィンゴイド主鎖)である。特定のLPA、S1P、ジヒドロS1Pの構造を以下に示す。

リゾホスファチジン酸(モノ−アシルグリセロール−3−リン酸、<500ダルトン)は、1つの炭化水素主鎖と1つのリン酸基を含む1つの極性頭基からなる。LPAは、単一分子実体ではなく、異なる長さと飽和度からなる脂肪酸を有する内在性構造変異体の総称である。生物学的に関連性のあるLPAの変異体は、18:2、18:1、18:0、16:0、20:4を含む。飽和脂肪酸(16:0および18:0)と不飽和脂肪酸(16:1、18:1、18:2、20:4)の両方を有するLPA種が、血清と血漿中に検出されている。血液中に存在する主な種は16:0、18:1、18:2、20:4のLPAイソ型である。
有意な濃度(>1 μM)の生理活性LPAが、血清、唾液、卵胞液、悪性浸出液を含む様々な体液中に検出できる。
本発明は、その形態の中から、以下に詳細に説明するように、増殖過剰性障害およびその他の様々な疾患の治療または予防に有効な抗LPA薬剤を提供する。特に、本発明の特定の実施態様は、限定はされないが、LPAの18:2、18:1、18:0、16:0、20:4変異体を含む、LPAを標的とする抗体を導出する。
これらのレセプターの役割は、マウスにおけるレセプターノックアウト研究によって一部解明されている。LPA1欠損マウスは、嗅覚障害に起因する吸入異常による一部の出生後死亡率を示している。LPA1欠損マウスはまた、ブレオマイシン誘導性肺損傷による肺線維症から保護される。さらに、LPA1レセプター遺伝子を欠損するマウスは、神経損傷誘導性の神経障害性疼痛行動と現象を生じない。
LPAは、細胞増殖の誘導、細胞移動の刺激および神経突起の退縮、ギャップ結合の閉鎖、粘菌の走化性を含む、広範な生物学的応答に影響を及ぼす。LPAの生物学に関する知識体系は、より多くの細胞系がLPA応答性について試験されているため、さらに増え続けている。LPAの主要な生理学的および病態生理学的作用は、例えば、以下を含む。
細胞の成長と増殖を刺激することに加えて、LPAは、創傷の修復と再生における重要なイベントである、細胞張力と細胞表面のフィブロネクチン結合を促進することが現在公知である。
アポトーシス:最近では、抗アポトーシス活性もまたLPAに帰するとされ、ペルオキシソーム増殖レセプターγは、LPAのレセプター/標的であることが最近報告されている。
LPA産生の役割を担う分泌型リゾホスホリパーゼDであるオートタキシンは、血管形成の発達段階に必須である。さらに、不飽和LPAは、血管平滑筋細胞脱分化の誘導に主要な役割を担うことが明らかにされた。
浮腫と血管透過性:LPAは、マウスにおいて、血漿浸出とヒスタミンの遊離を誘導する。
LPAは、ヒト角膜上皮細胞において炎症メディエーターとして作用する。LPAは、角膜創傷治癒に関与し、水晶体におけるROSの放出を刺激する。LPAはまた、ウサギ角膜において、HSV−1を再活性化できる。
毒蜘蛛、Loxosceles reclusa(ブラウンレクルーススパイダー)の咬刺傷は、重篤で長期にわたり持続する組織損傷を起因し、時には死に至ることがある、壊死性潰瘍を引き起こす。この蜘蛛の咬刺傷から生じる創傷の病態は、AAとプロスタグランジンに媒介される激烈な炎症反応からなる。L. reclusa蜘蛛毒素の主成分は、しばしばスフィンゴミエリナーゼD(SMase D)と呼ばれる、スフィンゴミエリンを加水分解してC1Pを生成する、ホスホリパーゼD酵素である。ところが、様々な頭基を有するリゾリン脂質は、L. reclusa酵素によって加水分解されて、LPAを放出することが分かっている。本発明によるもののような抗LPA薬剤は、限定はされないが、L. reclusa毒物注入に起因する炎症を含む、様々なタイプの炎症の低減または治療に有効であると考えられている。
LPAは、真皮線維芽細胞において、ERK依存経路を介して、TGFが媒介するI型コラーゲンmRNAの安定性の刺激作用を抑制する。さらに、LPAは、コラーゲン遺伝子発現と線維芽細胞の増殖を刺激することによって、ある程度の直接的な線維形成効果を有する。
LPAは、S1Pのように、免疫関連細胞の調節を介して、免疫応答に役割を果たすことが示されている。これらの脂質は、免疫応答部位へのT細胞移動を促進し、T細胞の増殖ならびに様々なサイトカインの分泌を制御する。
したがって、Lpath社の抗LPAモノクローナル抗体のような、LPAの有効濃度を低下させる薬剤は、望ましくない、または過剰な、または異常なLPA濃度に関連する、創傷治癒および線維症、アポトーシス、血管形成および血管新生、血管透過性、炎症に関連する疾患と状態を治療するための方法において有効であると考えられる。
最近、本出願人は、LPAに対するいくつかのモノクローナル抗体を開発した。これらの抗LPA抗体は、様々なLPAを中和し、それらの生物学的および薬理学的作用を緩和することができる。抗LPA抗体は、したがって、過剰な、望ましくない、または異常なLPA濃度に関連する様々な疾患および状態の予防および/または治療に有効であると考えられる。
本発明を詳細に説明する前に、本発明との関連において使用されるいくつかの用語の定義を行う。これらの用語に加えて、他の用語も本明細書の他の箇所で、必要に応じて定義されている。本明細書に特に定義しない限り、本明細書に使用する技術用語は、それらの技術分野において認識されている意味を有する。
「生理活性脂質」とは、脂質シグナル伝達分子を意味する。生理活性脂質は、細胞外および/または細胞内シグナル伝達を媒介し、したがって、分化、移動、増殖、分泌、生存、その他のプロセスを調節することによって、多種の細胞の機能制御に関与するという点において、構造脂質(例えば、膜結合リン脂質)と区別される。インビボで、生理活性脂質は、細胞外液中に見出され、その他の分子、例えば、アルブミンおよびリポタンパク質のような血清タンパク質とコンジュゲートを形成するか、「遊離」型、即ち、その他の分子種とコンジュゲートを形成しない状態で存在し得る。細胞外メディエーターとして、生理活性脂質の一部は、膜結合イオンチャネルまたはGタンパク質共役レセプターまたは酵素、あるいは因子を活性化することにより細胞シグナル伝達を改変し、その結果として、複雑なシグナル伝達系を活性化し、細胞機能または生存に変化をもたらす。細胞内メディエーターとして、生理活性脂質は、酵素のような細胞内コンポーネント、またはイオンチャネル、またはアクチンのような構造エレメントと直接相互作用することによって、それらの作用を発揮することができる。
抗体または抗体フラグメントあるいは変異体との関連における「生物活性」という用語は、所望するエピトープに結合可能であり、かつ何らかの方法で生物学的作用を発現できる抗体、または抗体フラグメント、または抗体変異体を意味する。生物学的作用は、以下に限定はされないが、成長シグナルの調節、抗アポトーシスシグナルの調節、アポトーシスシグナルの調節、エフェクター機能カスケードの調節、その他のリガンド相互作用の調節を含む。
「プロモーター」という用語は、細胞内でコード配列の転写を引き起こすことが可能なすべての配列を含む。したがって、本発明によるコンストラクトで使用されるプロモーターとは、遺伝子転写の時期および/または比率の制御または調節に関与する、シス作用性転写制御エレメントおよび制御配列を含む。例えば、プロモーターは、転写制御に関与する、エンハンサー、プロモーター、転写ターミネーター、複写開始点、染色体組み込み配列、5’および3’非翻訳領域、またはイントロン配列を含む、シス作用性転写調節領域であり得る。本発明の用途に適する転写制御領域は、限定はされないが、ヒトサイトメガロウイルス(CMV)最初期エンハンサー/プロモーター、SV40初期エンハンサー/プロモーター、E.coli lac またはtrpプロモーター、原核または真核細胞またはそれらのウィルスの遺伝子発現を調節することが公知であるその他のプロモーターを含む。
「被験者」または「患者」とは、本発明による分子がもたらすことのできる処置(治療)を必要とする動物を意味する。本発明に従って治療され得る動物は、ウシ、イヌ、ウマ、ネコ、ヒツジ、ブタ、特に好ましい例である霊長類(ヒトおよび非ヒト霊長類を含む)などの哺乳類が属する、脊椎動物を含む。
「治療薬」とは、限定はされないが、以下を含む、治療効果を与えることを意図する薬剤または化合物を意味する。COX阻害薬およびその他のNSAIDSを含む抗炎症薬、抗血管新生薬、上記に定義するような化学療法剤、心血管系作用薬、免疫調節薬、神経変性疾患治療に使用される薬、眼薬など。
また、少なくとも1つのCDRペプチドをコードする、記載のヌクレオチド配列を有する規定のアイデンティティの配列を含む、単離核酸分子が供給される。この核酸分子は、免疫グロブリン重鎖または軽鎖のフラグメント、または全長免疫グロブリン重鎖または軽鎖をコードし、サカナ、トリ、哺乳類、任意に霊長類、任意にヒトに由来し得る。これらの核酸分子を含むベクターおよび宿主細胞が供給される。
特に定義しない限り、本明細書に使用されるすべての技術用語および科学用語は、本発明の属する分野の当業者によって通常理解されるのと同一の意味を有する。本発明の実施または試験には、本明細書に記載されるものと同様または同等の方法および材料を使用してよいが、適切な方法と材料については以下に記載する。本明細書に言及される全ての出版物、特許出願、特許、その他の参考文献は、その全てが参照により援用される。不一致が生じた場合、定義を含む本明細書が優先される。さらに、材料、方法、実施例は説明を目的とするもので、限定を意図してはいない。
1.組成物
本発明は、免疫原性応答(即ち、抗体産生)を促進するような方法で誘導体化されたLPAを供給する。1つの実施態様では、LPAは、LPA分子が担体分子とコンジュゲートするように、誘導体化され得る。1つの実施態様では、LPAの炭化水素鎖内の炭素原子は、ペンダント反応基によって[例えば、スルフヒドリル(チオール)基、カルボン酸基、シアノ基、エステル、ヒドロキシ基、アルケン、アルキン、酸塩素基、ハロゲン原子]で誘導体化されており、前記ペンダント反応基は保護されていても保護されていなくてもよい。この誘導体化は、分子と反応させるために(例えば、担体とコンジュゲートするために)、生理活性脂質を活性化する役割を果たす。1つの実施態様では、誘導体化LPAは、チオール化LPAである。1つの実施態様では、誘導体化LPAは、誘導体化C12またはC18LPAである。1つの実施態様では、このチオール化LPAは、クロスリンカー(例えば、IOAまたはSMCCなどの二機能性クロスリンカー)を介して、タンパク質であり得る担体にコンジュゲートされる。免疫される種において、LPAを、免疫原であるタンパク質またはその他の担体、例えば、キーホールインペットヘモシアニン(KLH)、血清アルブミン(ウシ血清アルブミンまたはBSAを含む)、ウシサイログロブリン、または大豆トリプシン阻害剤に、二機能性または誘導体化剤、例えば、マレイミドベンゾイルスクシンイミドエステル(システイン残基を介するコンジュゲート)、N−ヒドロキシスクシンイミド(リジン残基を介する)、グルタルアルデヒド、無水コハク酸、SOCl2またはR1N=C=NR(ここでRとR1は異なるアルキル基)を用いてコンジュゲートさせることは有用であり得る。非タンパク質担体(例えば、コロイド金)も、抗体産生における用途が、当該分野では公知である。
誘導体化LPAまたは誘導体化かつコンジュゲート化されたLPAは、抗LPA抗体(ポリクローナルおよび/またはモノクローナル)を産生するために使用できる。誘導体化LPAまたは誘導体化かつコンジュゲート化されたLPAはまた、本発明による方法、特に診断法において使用できる。
本発明による誘導体化LPAは、抗LPA抗体の検出および/または精製に使用することができ、前述のように担体とコンジュゲートさせることができる。この誘導体とコンジュゲートは、臨床診断法を含む診断法における用途では、固体支持体にコンジュゲートさせることが好ましい。例えば、LPA抗体の検出および/または定量は、LPAが関与する様々な医学的状態の診断に使用できる。LPA抗体の定量はまた、用量、半減期、ならびに、例えば本明細書に説明するLT3000のような抗LPA抗体による治療後の薬の量を評価するために臨床の現場において有用である。
したがって、本発明による誘導体化LPAおよび誘導体化かつコンジュゲート化されたLPAは、研究および臨床的診断のどちらにも有用である。
便宜上、本発明による誘導体化LPAは、例えば、診断アッセイ実施用の取扱説明書が添付された、規定量の試薬を組み合わせてパッケージした、キット中に供給され得る。
前述のように、1つの実施態様では、誘導体化LPAコンジュゲート(例えば、BSAまたはKLHにコンジュゲートされたチオール化LPA)は、抗LPA抗体の検出に使用されるELISAキットの(プレートをコーティングするための)レイダウン材料として使用される。かかるキットは、LPAの検出に有用である。例えば、LPA競合ELISAキットでは、(供給の)プレートは、誘導体化LPAおよび/または誘導体化かつコンジュゲート化されたLPAでコーティングされる。1つ以上のLPA標準物質と1つ以上の試料(例えば、血清または細胞培養上清)の一式を、本発明によるマウス抗LPA抗体と混合してから、誘導体化LPAでコーティングされたプレートに加える。抗体は、プレートに結合したLPA、あるいは試料または標準物質中のLPAのいずれかに対する結合を競合する。インキュベートならびに幾つかのELISA工程(取扱説明書と試薬はキットに含まれる)後、450 nmで吸光度を測定し、検量線との比較によって、試料中のLPA濃度を求める。1つの実施態様では、ELISAキットのレイダウン材料として使用されるLPAは、BSAにコンジュゲートされたチオール化C12 LPAまたはチオール化C18 LPAである。このキットで使用される抗体は、ポリクローナルまたはモノクローナル抗体であってよいが、モノクローナル抗体が好ましい。
1.はじめに
モノクローナル抗体(mAb)は、安全かつ有効な治療薬であることが示されているため、様々な疾患および障害の治療薬としてのその使用が急増している。承認済みの治療用モノクローナル抗体は、アバスチン(AvastinTM)、エルビタックス(ErbituxTM)、リツキサン(RituxanTM)を含む。その他のモノクローナル抗体は、その大部分が様々な形態の癌を標的とする様々な疾患について、臨床開発の様々なフェーズにある。一般的には、モノクローナル抗体は非ヒト哺乳類で生成される。マウスモノクローナル抗体の治療上の実用性は、非ヒト哺乳類抗体のキメラ化またはヒト化によって改善され得る。ヒト化は、投与された治療用のモノクローナル抗体に対する免疫応答の発生を大幅に減少させることによって、半減期、ならびにかかる応答の結果として生じる治療効果の低下を回避する。大部分は、ヒト化プロセスは、マウスの相補性決定領域(CDR)をヒト免疫グロブリンのフレームワーク領域(FR)中に移植することからなる。FRにある選択された残基のマウスアミノ酸残基への逆突然変異が、当初の移植コンストラクトで喪失された親和性を改善または取り戻すためにしばしば必要とされる。
LPAは、多数の疾患と障害に関連している。総説としては、Gardellら、(2006)
1264491972164_1.');
12(2):65−75、ならびにChun J. およびRosen, H.、(2006)Curr. Pharma. Design 12:161−171を参照。これらは、糖尿病・多発性硬化症・強皮症などの自己免疫疾患、癌を含む増殖過剰疾患、血管形成および血管新生に関連する疾患、肥満症、アルツハイマー病を含む神経変性疾患、統合失調症、移植拒絶および移植片対宿主病などの免疫関連疾患などを含む。
本発明の1つの形態は、増殖過剰疾患の治療方法に関する。これらの方法は、LPA関連増殖過剰疾患を罹患していることが知られているまたは疑われる哺乳類(例えば、ウシ、イヌ、ウマ、ヒツジ、ブタなどの動物、特にヒト)に、意図する適用が必要とするような、好ましくは、薬学的または獣医学的に許容される担体中にあるLPA濃度および/または活性を妨害する薬剤を含む組成物の治療有効量を投与することを含む。LPA関連の増殖過剰疾患は、腫瘍、内皮細胞増殖に関連する障害、線維形成に関連する疾患を含む。多くの場合、腫瘍は癌である。内皮細胞増殖に関連する代表的な疾患は、例えば、固形腫瘍に起因する癌、血液学的腫瘍、加齢黄斑変性のような血管形成依存性疾患である。線維形成に関連する疾患は、心不全のような、異常な心臓リモデリングを伴う疾患を含む。
癌は、おそらく最も広く認識されている増殖過剰疾患の部類の代表的なものである。癌は、破壊的な部類の疾患であると共に、心血管系疾患の次に高い致死率を有する。多くの癌は、分子レベルでは完全に理解されていない。その結果、癌は、米国政府ならびに製薬会社双方にとって、研究開発の大きな関心の的である。その結果、癌に対する闘いを支援するために、かつてないほどの研究開発の努力がなされ、多数の価値ある治療薬が製造されている。
癌は現在、主として3種類の治療法(外科手術、放射線療法、化学療法)のうちから1つまたは併用によって治療されている。外科手術は、疾患組織のバルク除去を含む。外科手術は、例えば、乳房、結腸、皮膚のような、特定の部位に局在する腫瘍の除去には往々にして有効であるが、背骨のようなその他の領域に局在する腫瘍の治療、または白血病のような播種性腫瘍状態の治療には使用できない。放射線療法は、生体組織の電離放射線への曝露を含み、曝露された細胞に死または損傷をもたらす。放射線療法の副作用は、急性かつ一過性であり得るが、不可逆的な場合もある。化学療法は、細胞複製または細胞代謝の破壊を含む。
更なる痛手は、現在の治療薬は通常、毒性および重篤な副作用の形で、患者にとって著しい難点を伴うことである。したがって、多数のグループが最近、癌と闘うための新しいアプローチに注目し始めた。これらの新しい、いわゆる「革新的療法」は、遺伝子療法およびモノクローナル抗体のような治療用タンパク質を含む。
関連する形態は、増殖過剰疾患の治療または予防のための治療手段の毒性を減少させる方法に関する。かかる方法は、増殖過剰疾患を罹患する被験者に、その増殖過剰疾患を治療または予防することを意図する治療手段の投与前、投与中、投与後に、本発明に従う薬剤(または複数の異なる薬種)の有効量を投与することを含む。細胞(例えば、癌細胞)を化学療法剤に対して感作することによって、低用量で効力が達成されるため、化学療法剤ゆえの毒性が低くなると考えられる。
さらに本発明の他の形態は、増殖過剰疾患を罹患する被験者に、被験者の生存の可能性を高めるために、その増殖過剰疾患を治療または予防することを意図する治療手段の投与前、投与中、投与後に、本発明に従う薬剤(または複数の異なる薬種)を投与することによって、増殖過剰疾患の治療を受けている被験者の生存の可能性を高める方法に関する。
線維芽細胞、特に筋線維芽細胞は、細胞損傷と炎症に対する応答において、瘢痕形成の主要な細胞エレメントである(Tomasekら(2002)、Nat Rev Mol Cell Biol, vol 3: 349−63、ならびにViragおよびMurry(2003)、Am J Pathol, vol 163: 2433−40)。筋線維芽細胞によるコラーゲン遺伝子発現は、リモデリングの特徴であり、瘢痕形成に必要である(SunおよびWeber(2000)、Cardiovasc Res, vol 46: 250−6、ならびにSunおよびWeber(1996)、J Mol Cell Cardiol, vol 28: 851−8)。
線維症は、正常な創傷治癒または発生とは対照的に、病的な修復または反応過程の一部として、臓器または組織における過剰または異常な線維性結合組織の形成または発生として説明できる。線維症の最もよくある形態は、肝臓、肺、腎臓、皮膚、子宮、卵巣線維症である。強皮症、サルコイドーシスなどの状態は、複数の臓器と組織における線維症とみなされる。
本発明による組成物と方法は、少なくとも部分的には、異常な血管新生、血管形成、線維形成、線維症、瘢痕、炎症、免疫応答によって特徴づけられる疾患および障害の治療に有効であろう。かかる疾患の1つは強皮症であり、これは全身性硬化症とも呼ばれる。
強皮症は、皮膚の瘢痕または肥厚を起因する自己免疫疾患であり、ときには、肺、心臓および/または腎臓を含む、身体の他の部分も巻き込む。強皮症は、身体の皮膚と臓器における瘢痕組織(線維症)の形成によって特徴づけられ、これは、罹患領域の肥厚と硬化を招き、その結果として機能低下に繋がる可能性がある。強皮症財団によれば、現在強皮症を罹患している米国人はおよそ30万人に上る。罹患している人々の三分の一以下は広汎性疾患を患い、残りの三分の二は主に皮膚症状を有する。この疾患が肺に影響を及ぼして瘢痕を生じると、肺はもはやこれまで通り拡張できないために、呼吸が制限される可能性がある。呼吸能力を測定するために、医師は、努力肺活量(FVC)を評価する装置を使用する。FVCが期待値の50%未満の人々では、強皮症関連肺疾患による10年死亡率はおよそ42%である。死亡率がこれほど高い1つの理由は、現在有効な治療法がないことである。
LPAは、線維芽細胞の活性化、増殖の一因となり得る線維化促進性増殖因子であり、その結果としての線維芽細胞活性の増加が、不適応な瘢痕とリモデリングに関連していたことが立証されている。さらに、皮膚の線維芽細胞活性においてLPAが果たし得る役割が実証されている。例えば、LPAがマウス皮膚線維芽細胞の移動を刺激することが示されている(Hamaら、J Biol Chem. 2004 Apr 23、279(17):17634−9)。
しばしば間質性肺疾患(ILD)と呼ばれる肺線維症は、全世界で500万人以上の人々が罹患している。米国におけるこの疾患の有病率は、実際よりも低く見積もられているが、10万人当たり3〜6症例から、10万人当たり28症例とばらつきがあるようである。欧州では、国によってこの数は異なり、明確な性差はなく、およそ10万人当たり1〜24症例と報告されている。この疾患は通常、40歳〜70歳で診断される。生存期間中央値は、3〜5年である。その有病率にも係わらず、IPFの進行を停止または逆行させるために利用可能な治療法はなく、FDAの承認を受けた治療コースもない。したがって、IPFならびに病的な組織線維症を伴うその他の疾患を治療するための新しい治療戦略について未だ満たされていないニーズがある。
職業・環境曝露:特に石綿、澱石、金属粉塵を扱う多くの仕事は、肺線維症を起因することがある。小粒子が吸入され、肺胞を損傷し、線維症を起因する。黴びた干し草のような有機物質の一部は肺線維症を惹起することもあり、これは農夫肺として公知である。
多数の異なる疾患が肺線維症を引き起こし得るが、多くの場合、その原因は不明である。そのような場合、この状態は、「特発性(原因不明の)肺線維症」と呼ばれる。特発性肺線維症では、患者の環境および職業歴を注意深く考査しても原因についての手がかりが得られない。医師と科学者の中には、この疾患を感染性またはアレルギー性の状態とする者もいるが、かかる患者の肺には、細菌およびその他の微生物が常に発見されるわけではない。他方、この状態は確かに、ウィルス様疾病を辿るように見えることもある。したがって、多くの症例における肺線維症の原因は知られているにも係わらず、特発性の変種については未だ謎のままである。
ある種の医薬品は、肺線維症を発症する望ましくない副作用を有し、その例は、ニトロフラントイン(尿路感染症治療薬としてよく使用される)、アミオダロン(不整脈の治療薬としてよく処方される)、ブレオマイシン、シクロホスファミド、メトトレキセート(癌の治療薬としてよく処方される)である。
息切れは、肺線維症の特徴である。多くの肺疾患では、息切れがその主要な症状として認められるが、それ故に診断が複雑かつ分かりにくくなる。一般的に、特発性肺線維症では、まず運動中に息切れが生じる。この状態は、労作が不可能な時点まで進行し得る。乾性咳が、よくある症状である。最後には指先が肥大して、球状の外観を呈する。これはしばしば、「ばち状指」と呼ばれる。
総合的既往歴ならびに健康診断に加えて、肺線維症の診断を絞り込み、および/または確定するには、以下の検査が必要なことがある。肺機能検査−肺の特徴と容量を測定する、肺活量測定−強制呼気量を測定する、ピークフローメーター−呼吸の変化と医薬品に対する反応を評価する、血液検査−血液中の二酸化炭素と酸素の量を分析する、X線検査、コンピューター断層撮影(CAT)スキャン、気管支鏡検査−気管支鏡と呼ばれる長く細いチューブを使って肺を検査する、気管支肺胞洗浄−炎症を特定し、特定の原因を除外するために、下気道から細胞を除去する、肺生検−病理臨床検査のために肺から組織を除去する。
肺線維症の公知の原因のうち1つがある場合、その基礎疾患の治療またはその疾患の原因となる環境から患者を排除することは、有効であり得る。これは、以下による治療を含み得る。副腎皮質ステロイドを含む経口薬物療法、インフルエンザワクチン、肺炎球菌性肺炎ワクチン、ポータブルタンクを使う酸素療法および/または肺移植。
多くの場合、治療は、肺に発生する免疫応答の治療に限られる。これは、炎症を止めることによって、肺の中に瘢痕組織や線維症が根付くことを防止することで、この疾患の進行を止めることを期待して行われる。
転帰
正確な病因は不明であるが、IPFは、肺損傷後の異常な創傷治癒に起因すると考えられている。Scotton, C.J.およびChambers, R.C.(2007)Chest, 132:1311−21。特に、肺線維芽細胞の増殖と移動の増加、ならびに瘢痕組織を形成する筋線維芽細胞の形成は、IPF発病の主要なイベントである。筋線維芽細胞は、α平滑筋アクチン(a−SMA)を発現する平滑筋様線維芽細胞であり、アクチンフィラメントと顕著なストレスファイバーに器質化する関連タンパク質から構成される収縮装置を含んでいる。組織ホメオスタシスと修復におけるこれらの通常の役割に加えて、筋線維芽細胞は数多くの線維性疾患における病態メディエーターである。Hinz, B.(2007)J Invest Dermatol. 127:526−37。肺線維症の動物モデルの線維性病巣において、筋線維芽細胞数とその密度の増加が認められている。筋線維芽細胞は組織損傷後に形成され、それによって増加した増殖因子、サイトカイン、機械的刺激の量が、常在する組織の線維芽細胞が収縮性の瘢痕組織を形成する筋線維芽細胞に変換することを促進する。肺とその他の組織において、TGFβ、CTGF、PDGF、種々の炎症性サイトカインを含む生化学的メディエーター量の持続的な上昇は、筋線維芽細胞の形成を促進し、瘢痕組織の形成を悪化させて、組織線維症に至らしめる(Scotton、2007)。したがって、IPFおよびその他の線維性疾患治療のための現在の臨床戦略は、筋線維芽細胞形成とそれに続く線維性組織の形成を促進する、生化学的因子を標的としている。
肝臓は、優れた再生能力を有しているため、再生による修復プロセスは、 restitutio ad integrum(完全回復)するように進行する。しかしながら、もし損傷が網状構造に影響を及ぼす場合、修復は瘢痕形成(線維症)によって生じ、これは血液循環の再編成と肝硬変に繋がり得る。
損傷に対する反応は、以下のように進行する。細胞の変化および組織の変化を伴う損傷(壊死)、炎症反応、修復(再生(原状回復)あるいは瘢痕(線維症)のいずれかによる)。
慢性肝疾患は、構造の撹乱である門脈高血圧症に至る線維症を誘因し、血液循環に不可逆的な再編成を生じて、肝硬変を起因し得る。線維症と肝硬変は紙一重である。線維症は、壊死、崩壊、瘢痕形成のみならず、肝臓においては、以下のカテゴリー、即ち、コラーゲン、糖タンパク質、プロテオグリカンであるマトリックスの様々な成分を合成する間葉細胞が損傷を受けたことによる、マトリックスの合成と分解の撹乱の結果、生じる。
肝線維症は、例えば、マッソントリクローム染色、鍍銀法、コラーゲン型に対する特異抗体、脂質細胞のデスミンとビメンチン染色、あるいは筋線維芽細胞のビメンチン染色のような組織学的方法、または、例えば、マトリックス中の種々の酵素あるいは良性線維症における血清ラミニンの測定による、生化学的方法によって診断し得る。
肝線維症には、先天性および後天性の2つの主要な型がある。前者は遺伝疾患であり、多嚢胞性肝疾患を起因する。後者には多数の異なるカテゴリーがあり、主として肝細胞の損傷に起因する。病理学的には、線維症は以下のように分類できる。
門脈領域から小葉に、線維芽細胞が増殖し、線維の伸展を生じる。結局は、これらの線維は連絡して橋架様隔壁を形成する。この種の線維症は、主として、ウィルス性肝炎および低栄養性肝線維症に見られる。
正常な小葉には線維芽細胞はほとんど見られない。多数の肝細胞が変性したり壊死を生じる場合、網状線維の枠組みが崩壊して、太いコラーゲン線維になる。同時に、小葉内線維性組織が増殖して、肝細胞を取り囲む。
中央線維症:増殖した線維化組織は、主として中心静脈を取り囲み、中心静脈壁の肥厚を生じる。
(微小胆管周囲の線維症)
この種の線維症は、主として、長期にわたる胆管貯留に起因し、主として、胆管周囲に発症する。顕微鏡的には、新しく形成された毛細胆管と胆管栓塞を取り囲む結合組織がある。毛細胆管の基底膜が線維化する。
免疫学的には、肝線維症は、以下のように分類できる。
肝細胞の広範な壊死および副次的な肝構造の崩壊と瘢痕形成が生じ、それが結合組織の増殖を起因する。
(能動的線維症)
リンパ細胞およびその他の炎症細胞の浸潤および再発・一貫性炎症は、結合組織の小葉への浸潤を促進する。原因により、肝線維症は以下のように分類できる。
ウィルス肝炎による線維症は、通常は、B、C、D型慢性肝炎によって発症する。全世界で、B型肝炎保菌者は3.5億人に上り、1.7億人の人々がC型肝炎に感染している。B型肝炎患者の約15%ならびにC型肝炎患者の約85%が慢性肝炎を発症し、線維症に至る。そのような場合、肝臓は、門脈周辺領域に炎症と漸次壊死ならびに線維化を呈する。そのような多数の人口集団が罹患しているため、これは肝線維症の最も重要なカテゴリーである。
この種の肝線維症は、主として、開発途上国で発症し、住血吸虫症に起因する。アジア、アフリカ、南米、中南米では2.2億人が、この感染症を罹患している。再感染と住血吸虫の卵が肝臓に蓄積して、肝線維症と肝硬変を引き起こす。
(アルコール性線維症)
これは主として、アルコールの酸化代謝物であるアセトアルデヒドに起因する。西欧諸国では、この疾患の発生率は、アルコール摂取量と正比例している。米国におけるアルコール性線維症の総症例数は、C型肝炎患者数の約3倍である。アルコール性線維症は、肝臓に以下の2つの形態学的変化を生じる。脂肪肝と細胞小器官の劣化。線維症は、まず中心静脈周辺に発症し、同時に肝実質細胞の炎症を起こす。徐々に線維症は、肝臓全体に広がる。
胆管線維症には、原発性のものと二次性のものがある。原発性胆汁性肝線維症(PBHF)は、慢性肝臓内胆汁貯留が肝線維症を起因する、自己免疫疾患である。この疾患は、40〜60歳の女性に多く発症する。血清検査では、γグロブリン値が上昇し、抗ミトコンドリア抗体に陽性を示す。病理学的研究によって、主として、微小胆管周囲の線維症と門脈周辺領域の線維症ならびに炎症があることが判明した。二次性胆管線維症は、胆管閉塞後に発症し、門脈周囲領域の炎症と進行性線維症を起因する。
このカテゴリーは一般的ではなく、稀に見られる症例である。ウィルソン病や肝レンズ核変性およびヘモクロマトーシス(血色素症)は、代謝性線維症を起因する主な疾患である。前者は遺伝病であり、銅代謝障害と肝臓沈着を起因する。後者は、鉄代謝障害であり、肝臓にヘモグロビン沈着を起因する。これらの代謝障害のどちらも、肝線維症と肝硬変を起因する。
(中毒性線維症)
四塩化炭素、有機リン、ジメチルニトロソアミン、チオアセトアミドなどの肝臓毒性物質と長期間接触したり、イソニアジド、チオ酸化ピリミジン、ウインタミン、テトラサイクリン、アセトアミノフェンなどの肝臓毒性のある医薬品を服用することは、いずれの場合も異なる度合いの肝細胞損傷、壊死、胆管貯留、またはアレルギー性炎症を生じたり、肝線維症を起因する。
この種の線維症は、主として、不十分またはバランスを欠く栄養摂取によって生じる。長期にわたる低タンパク質または高脂質食は、脂肪肝を起因し、線維症に至る。
心原性線維症:慢性鬱血性心不全は、肝細胞の虚血性変性を起因する、長期間持続する肝静脈鬱血を生じ得る。この種の肝線維症では、結合組織の肥大が、肝臓小葉の中央部から始まり、徐々に小葉の残りの部分に拡大する。
肝臓の健康を評価するための最も基準となる検査は、肝生検である。しかしながら、この方法は、皮膚に針を挿入する必要があるため、合併症の発生率は極めて低いとはいえ、合併症の可能性がある。肝生検の合併症は、内出血、ならびに肺、胃、腸、肝臓に近いその他の臓器などの他の臓器を穿刺することを含み得る。生検の正確性に関しては、正しく病期を決定し、肝生検をグレード付けするために、肝臓組織の標本サイズが重要である。その他の問題は、肝臓の一部から採取された組織は、肝臓全体を100%代表するわけではないということである。いったん肝臓組織試料が採取されると、専門家(病理学者)によって、グレード付けされ、病期が決定されるが、これは結果の解釈においてヒューマンエラーが生じる可能性に繋がる。さらに、標準化された解釈のプロトコルがないため、別の病理学者によって読解された別の生検結果と比較することが困難である。標準的な肝生検の費用は、1,500〜2,000ドルかかることがあるため、費用も1つの問題である。
肝臓学界と胃腸病学界が直面する主要な臨床上の問題の1つは、益々その数が増加している、C型肝炎ウィルス(HCV)が同定された患者に対する最良の診断・管理法である。過去10年間における、HCVについての血清学的およびウィルス学的検査の進歩ならびに治療法の向上によって、より多くの患者が同定されて、治療を求めるようになった。しかしながら、肝臓損傷、特に線維症の度合いを判定したり、個々の患者について疾患進行のリスクを予見する能力を向上する能力の進歩はほとんど達成されていない。
臨床医は、患者の生命に大きなインパクトを持ち得る、予後かつ治療法についての判断については、生検結果に依拠している。シングルパス方式の肝生検は、80%の患者において、線維症の病期または肝硬変の有無を正しく診断することができる。肝生検の診断の正確性を向上する要素は、HCVのように肝臓全体に渡る均一な疾患の存在、複数のパス、使用される生検針タイプ、長さが2cm以上のフラグメント化しない生検コアを含む。経験豊富な医師が肝生検を行い、専門の病理医が生検を解読しても、この最も基準となる検査法では、病期決定に20%以上のエラーを伴う。
LPAは、腎臓炎症と線維症に関連している。最近になって、腎臓の線維症は、LPA放出の増加とLPA1型レセプター(LPA1)を介するシグナル伝達に関連づけられている。マウスにおける一側性尿管閉塞後、LPA1ノックアウトマウスでは尿細管間質性線維症が軽減し、線維化促進性サイトカイン発現は、LPA1 アンタゴニストを投与した野生型マウスにおいて減弱した(Pradere、2007)。
子宮線維症は、子宮平滑筋腫(一般的に筋腫と呼ばれる)として公知の非悪性腫瘍である。この腫瘍は、孤立または群発し、リンゴの種のサイズから、グレープフルーツよりも大きなサイズまで、多様なサイズで生じる。子宮筋腫の診断は、一般的には、超音波検査、X線検査、CATスキャン、腹腔鏡検査および/または子宮鏡検査によって達成される。子宮筋腫の治療は、内科的(薬物治療、例えば、非ステロイドケイ抗炎症剤またはゴナドトロピン放出ホルモンアゴニスト)または外科的(例えば、筋腫切除術、子宮摘出術、子宮内膜切除または筋融解)のいずれかであり、子宮筋腫塞栓術や熱超音波焼灼術のような侵襲性の少ない方法も最近になって開発されている。
LPAはまた、コラーゲン遺伝子発現と線維芽細胞の増殖を刺激することによって、心臓の線維芽細胞において直接的な線維形成作用を有することが示されている。Chen, ら(2006)FEBS Lett. 580:4737−45。したがって、抗体のような抗LPA薬剤は、心臓細胞においても同様に抗線維化作用を有することが予測され、したがって、心筋線維症の治療に有効であることが期待される。
Lpath社の抗LPAモノクローナル抗体のような、LPAの有効濃度を低下させる薬剤は、異常な線維症によって特徴づけられる疾患と状態を治療する方法において有用であると考えられる。
LPAは、肝臓組織の線維形成と創傷治癒(Davailleら、J. Biol. Chem. 275:34268−34633、2000、 Ikedaら、Am J. Physiol. Gastrointest. Liver Physiol 279:G304−G310, 2000)、傷ついた脈管構造の治癒(Leeら、Am. J. Physiol. Cell Physiol. 278:C612−C618、2000)、その他の疾患状態、または癌のような疾患・血管形成・炎症に関連するイベント(Pyneら、 Biochem. J. 349:385−402、2000)に関与するため、本開示による組成物と方法は、これらの疾患のみならず、心臓疾患、特に組織リモデリングに関連する疾患の治療にも同様に適用し得る。LPAは、コラーゲン遺伝子発現と心臓線維芽細胞の増殖を刺激することによって、ある程度直接的な線維形成効果を有する。Chen, ら(2006)FEBS Lett. 580:4737−45。
LPA合成を担うホスホリパーゼDであるオートタキシンは、脂肪細胞によって分泌され、肥満症・糖尿病db/dbマウス由来の脂肪細胞、ならびに極度に肥満な女性被験者と2型糖尿病を罹患するヒト患者(肥満とは無関係に)においてその発現が増加していることが発見されている(Ferry ら(2003)JBC 278:18162−18169、 Boucherら(2005)Diabetologia 48:569−577、Pradereら(2007)BBA 1771:93−102に引用されている)。LPA自体が、前脂肪細胞の増殖と分化に影響を及ぼすことが示されている。Pradereら、2007. 合わせて、これは、肥満症と糖尿病の治療における抗LPA薬剤の役割を示唆しているる。
下記の実施例は、治療の観点から望ましい特性を有する、抗LPA薬剤、特に抗LPA抗体の生成を説明する。(a)LPAおよび/または18:2、18:1、18:0、16:0、12:0、20:4 LPAを含むその変異体に対する結合親和性。抗体親和性は、下記の実施例において説明するように測定される。抗体は、高親和性でLPAに結合することが好ましく、例えば、Kd値が、決して約1 x 10-7 Mを超えない、可能であれば、1 x 10-8 Mを超えない、5 x 10-9 Mを超えないこと。生理学的状況においては、抗体は、LPAレセプターに対するLPAの親和性よりもさらに高い親和性でLPAを結合することが好ましい。言うまでもなく、診断検査法のような非生理学的状況においては、必ずしもこうである必要はない。
抗LPA抗体および抗LPA抗体の変異体を生成する方法が、以下の実施例で説明される。ヒト化抗LPA抗体は、非ヒト抗LPA抗体に基づいて、調製し得る。完全ヒト抗体はまた、例えば、遺伝子改変(即ち、トランスジェニック)マウス(例えば、Medarex社から)において、免疫原を提示したときに、必ずしもCDF移植を必要としない、ヒト抗体を生成することができる。これらの抗体は、非ヒト抗体遺伝子が抑制されて、ヒト抗体遺伝子発現で置換されているマウスのような動物に由来する完全ヒト抗体(100%ヒトのタンパク質配列)である。本出願人は、該当するCDRのヒトフレームワークを生成できるようなこのような遺伝子改変マウスやその他の動物に提示したときに、生理活性脂質に対する抗体を生成できると考える。
変異体を生成する場合は、親抗体を調製する。かかる非ヒト抗体および親抗体を生成するための代表的な技術を以下の項に説明する。
抗体の生成に使用される抗原は、例えば、インタクトなLPAまたはLPAの一部(例えば、エピトープを含むLPAフラグメント)であり得る。抗体を生成するために役に立つ抗原のその他の形態は、当業者には明らかであろう。
ポリクローナル抗体は、該当する抗原とアジュバンドの複数回の皮下(sc)または腹腔内(ip)注射によって、動物(哺乳類、鳥類、軟骨魚を含む魚類を含む脊椎動物または無脊椎動物)において惹起することが好ましい。該当する抗原を、免疫される種において免疫原であるタンパク質またはその他の担体、例えば、キーホールインペットヘモシアニン(KLH)、血清アルブミン、ウシサイログロブリン、大豆トリプシン阻害剤に、二機能性または誘導体化剤、例えば、マレイミドベンゾイルスクシンイミドエステル(システイン残基を介してコンジュゲート)、N−ヒドロキシスクシンイミド(リジン残基を介する)、グルタルアルデヒド、無水コハク酸、 SOCl2または、R1N=C=NR(ここでRとR1は異なるアルキル基)、を用いてコンジュゲートさせることが有用であり得る。非タンパク質担体(例えば、コロイド金)も、抗体生成のために、当該分野に公知である。
モノクローナル抗体は、Kohlerら、Nature, 256:495(1975)が最初に説明したハイブリドーマ法を用いて作成するか、組み換えDNA法のようなその他の方法によって作成し得る(米国特許第4,816,567号)。ハイブリドーマ法では、マウスまたはその他の好適な宿主動物(ハムスターまたはマカクザルなど)を、免疫に使用したタンパク質に特異的に結合する抗体を産生する、または産生することが可能なリンパ球を誘導するために、上述のように免疫化する。あるいは、インビトロでは、リンパ球が免疫されてもよい。リンパ球を次に、ハイブリドーマ細胞を形成するために、ポリエチレングリコールのような好適な融合剤を用いて、ミエローマ細胞と融合させる(Goding、Monoclonal Antibodies: Principles and Practice(モノクローナル抗体:原理と手法)、pp.59−103(Academic Press, 1986))。
モノクローナル抗体の結合親和性は、例えば、Munsonら、Anal. Biochem., 107:220(1980)によるスキャッチャード分析によって測定することができる。
抗体のヒト化についての一般的方法は、米国特許最新5861155、US19960652558 19960606、US6479284、US20000660169 20000912、US6407213、US19930146206 19931117、US6639055、US20000705686 20001102、US6500931、US19950435516 19950504、US5530101、US5585089, US19950477728 19950607、US5693761、US19950474040 19950607、US5693762, US19950487200 19950607、US6180370、US19950484537 19950607、US2003229208、US20030389155 20030313、US5714350、US19950372262 19950113、US6350861、US19970862871 19970523、US5777085、US19950458516 19950517、US5834597、US19960656586 19960531、US5882644、US19960621751 19960322、US5932448、US19910801798 19911129、US6013256、US19970934841 19970922、US6129914、US19950397411 19950301、US6210671、v、US6329511、US19990450520 19991129、US2003166871、US20020078757 20020219、US5225539、US19910782717 19911025、US6548640、US19950452462 19950526、US5624821、US19950479752 19950607に説明されている。
抗LPA抗体のアミノ酸配列変異体は、抗LPA抗体DNA中に適切なヌクレオチドの変化を導入するか、あるいはペプチド合成によって調整される。かかる変異体は、例えば、本明細書の実施例にある抗LPA抗体のアミノ酸配列内からの残基の欠失、および/またはアミノ酸配列内への残基の挿入および/または残基の置換を含む。最終コンストラクトが所望の特性を有しているという前提で、欠失、挿入、置換の組み合わせが、最終コンストラクトに達するために使用される。また、アミノ酸の変化は、ヒト化抗LPA抗体または抗LPA抗体変異体の、グリコシル化部位の数または位置の変化といった、翻訳後プロセスを改変し得る。
Ala(A)val; leu; ile val
Arg(R)lys; gln; asn lys
Asn(N)gln; his; asp, lys; gln arg
Asp(D)glu; asn glu
Cys(C)ser; ala ser
Gln(Q)asn; glu asn
Glu(E)asp; gln asp
Gly(G)ala ala
His(H)asn; gln; lys; arg arg
Ile(I)leu; val; met; ala; leu phe; norleucine
Leu(L)norleucine; ile; val; ile met; ala; phe
Lys(K)arg; gln; asn arg
Met(M)leu; phe; ile leu
Phe(F)leu; val; ile; ala; tyr tyr
Pro(P)ala ala
Ser(S)thr thr
Thr(T)ser ser
Trp(W)tyr; phe tyr
Tyr(Y)trp; phe; thr; ser phe
Val(V)ile; leu; met; phe; leu ala; norleucine
(1)疎水性:ノルロイシン、メチオニン、アラニン、バリン、ロイシン、イソロイシン
(2)中性親水性: システイン、セリン、スレオニン
(3)酸性:アスパラギン酸、グルタミン酸
(4)塩基性:アスパラギン、グルタミン、ヒスチジン、リジン、アルギニン
(5)鎖の配向性を影響する残基:グリシン、プロリン
(6)芳香族:トリプシン、チロシン、フェニルアラニン
また、分子の酸化安定性を向上させ、異常な交差を防止するために、抗体の正しい立体構造の維持に関与しないシステイン残基はすべて置換され得る。逆に、安定性を向上させるために、(特に、抗体が、Fvフラグメントのような抗体フラグメントである場合)、システイン結合を抗体に付加してもよい。
抗体のグリコシル化は、典型的には、N結合型および/またはO結合型である。N結合型とは、アスパラギン残基の副鎖への炭水化物部分の付加を意味する。トリペプチド配列である、アスパラギン−X−セリンおよびアスパラギン−X−スレオニン(ここでXは、プロリン以外のいずれかのアミノ酸)は、アスパラギン副鎖への炭水化物部分の酵素的付加の最も一般的な認識部位である。したがって、ポリペプチド中にこれらのトリペプチド配列のいずれかが存在することによって、潜在的なグリコシル化部位が創出される。O結合型グリコシル化とは、ヒドロキシアミノ酸(5−ヒドロキシプロリンまたは5−ヒドロキシリジンもまた使用し得るが、最も一般的にはセリンまたはスレオニン)への糖類N−アセチルガラクトサミン、ガラクトース、キシロースの付加を意味する。
抗スフィンゴ脂質抗体のアミノ酸配列変異体をコードする核酸分子は、当該分野に公知の様々な方法によって調製される。これらの方法は、限定はされないが、天然源からの単離(天然アミノ酸配列変異体の場合)またはオリゴヌクレオチドを介する(または部位特異的)変異誘発、PCR変異誘発、以前に調製された変異体のカセット変異誘発または抗スフィンゴ脂質抗体の非変異体バージョンによる調製を含む。
ヒト化のほかに選択可能な方法として、ヒト抗体を生成できる。例えば、今では、免疫されると、内在性免疫グロブリンを産生することなく、ヒト抗体の完全なレパトワを生じることができるトランスジェニック動物(例えば、マウス)を作成することが可能である。例えば、キメラおよび生殖細胞系列変異マウスにおける抗体の重鎖結合領域(JH)遺伝子のホモ接合型欠失は、内在性抗体産生の完全な抑制を生じることが説明されている。かかる生殖細胞系列変異マウスへのヒト生殖細胞系列免疫グロブリン遺伝子アレイのトランスファーは、抗原攻撃に際してヒト抗体の産生を生じることになる。例えば、Jakobovitsら、Proc. Natl. Acad. Sci. USA, 90:2551(1993)、Jakobovitsら、Nature, 362:255−258(1993)、Bruggermannら、Year in Immuno., 7:33(1993)、ならびに米国特許第5,591,669、5,589,369、5,545,807号。ヒト抗体はまた、ファージディスプレイライブラリーからも生成できる(Hoogenboomら、J. Mol. Biol., 227:381(1991)、MarksらJ. Mol. Biol., 222:581−597(1991)、ならびに米国特許第5,565,332および5,573,905号)。ヒト抗体はまた、インビボで活性化されたB細胞によっても生成できる(米国特許第5,567,610および5,229,275号を参照。)
ある実施態様では、抗LPA薬剤は、抗原結合を含む、少なくとも1つの所望する活性を保持する抗体フラグメントである。抗体フラグメント産生のために、様々な技術が開発されてきている。従来より、これらのフラグメントは、インタクトな抗体のタンパク質分解性消化を介して得られた(例えば、Morimotoら、Journal of Biochemical and Biophysical Methods 24:107−117(1992)およびBrennanら、Science 229:81(1985)を参照。)しかしながら、今では、これらのフラグメントは、組み換え宿主細胞によって直接産生することができる。例えば、Fab’−SHフラグメントは、E. coliから直接回収でき、化学的に結合されて、F(ab’)2フラグメントを形成する(Carterら、Bio/Technology 10:163−167(1992))。別の実施態様では、F(ab’)2は、F(ab’)2分子のアセンブリを促進するためにロイシンジッパーGCN4を用いて形成される。別のアプローチに従って、Fv、Fab、F(ab’)2フラグメントは、組み換え宿主細胞培養から直接単離され得る。抗体フラグメント産生のためのその他の技術は、当業者には明らかであろう。
いくつかの実施態様では、抗LPA薬剤は、第一結合部分と第二結合部分からなり、ここで第一結合部分は、LPAまたはLPA代謝物である第一分子と特異的に反応し、第二結合部分は、第一分子とは異なる分子種である第二分子と特異的に反応する。かかる薬剤は、複数の第一結合部分、または複数の第二結合部分、または複数の第一結合部分と複数の第二結合部分を得る。第一結合部分と第二結合部分の比率は、約1000:1〜約1:1000の範囲に渡ってもよいが、好ましくは、約1:1であり、ここでこの比率は、原子価に換算して測定することが好ましい。
2価以上を有する抗体が検討されている。例えば、三重特異性抗体を調製することができる。Tuttら、J. Immunol. 147:60(1991)。
Fabまたはそのフラグメントにつき少なくとも2つの結合部位を含み、そのうち少なくとも1つの結合部位が第一の抗原に対し、第二の結合部位が第一の抗原と異なる第二の抗原に対する、本発明による抗体(またはポリマーまたはポリペプチド)もまた、「多重特異性」と呼ぶこととする。したがって、「二重特異性」ポリマーは、第一の抗原に対する少なくとも1つの部位および第二の抗原に対する少なくとも1つの第二の部位を含むが、「三重特異性」とは、第一の抗原に対する少なくとも1つの結合部位、第二の抗原に対する少なくとも1つの更なる結合部位、第三の抗原に対する少なくとも1つの更なる結合部位などを含む。したがって、最も単純な型では、本発明による二重特異性ポリペプチドは、本発明による二価ポリペプチド(Fabにつき)である。しかしながら、前述から明らかであるように、本発明による多重特異性ポリペプチドは、2つ以上の異なる抗原に対する結合部位をいくつでも含み得るという意味において、本発明はそれに限定されることはない。
抗LPA抗体のその他の修飾が検討されている。例えば、本発明はまた、トキシン(例えば、細菌、真菌、植物、動物由来の酵素活性を有するトキシン、またはそれらのフラグメント)のような細胞毒剤、あるいは放射性同位元素(例えば、放射性コンジュゲート)にコンジュゲートされた本明細書に記載の抗体を含む免疫コンジュゲートに関連する。コンジュゲートは、N−スクシンイミジル−3−(2−ピリジルジチオール)プロピオン酸塩(SPDP)、イミノチオラン(IT)、イミドエステルの二機能性誘導体(ジメチルアジプイミダート塩酸塩など)、活性エステル類(ジスクシイミジルスベリン酸塩など)、アルデヒド類(グルタルアルデヒドなど)、ビスアジド化合物(ビス(p−アジドベンゾイル)ヘキサンジアミンなど)、ビス−アゾニウム誘導体(ビス−(p−ジアゾニウムベンゾイル)−エチレンジアミンなど)、ジイソシアン酸塩(トリエン2,6−ジイソシアナートなど)、ビス−活性フッ素化合物(1,5−ジフルオロ−2,4−ジニトロベンゼンなど)のような様々な二機能性タンパク質カップリング剤を用いて作成される。
本発明はまた、抗LPA抗体をコードする単離核酸、核酸を含むベクターと宿主細胞、抗体産生のための組み換え技術を提供する。
抗体の組み換えによる産生には、それをコードする核酸を単離して、更なるクローニング(DNAの増幅)と発現のために複製可能なベクター中に挿入することができる。他の実施態様では、抗体は、例えば、参考文献として特に本明細書に援用されている、米国特許第5,204,244号に記載のように、相同組み換えによって産生することができる。モノクローナル抗体をコードするDNAは、従来の方法を用いて(例えば、抗体の重鎖と軽鎖をコードする遺伝子に特異的に結合可能なオリゴヌクレオチドプローブを用いて)、容易に単離および配列決定することができる。多数のベクターが使用可能である。ベクターの構成成分は、一般的に、限定はされないが、以下を1つ以上を含む。例えば、1996年7月9日に公布され、参考文献として特に本明細書に組み込まれている米国特許第5,534,615号に記載のような、シグナル配列、複製起点、1つ以上のマーカー遺伝子、エンハンサーエレメント、プロモーター、転写終止配列。
予備精製の工程後、関心対象の抗体と混入物を含む混合物は、2.5〜4.5のpHで溶出バッファーを用いて、好ましくは、低塩濃度(例えば、約0〜0.25M塩)で、低pH疎水性相互作用クロマトグラフィーにかけることができる。
本発明は、抗LPA抗体および関連する組成物と生理活性脂質LPAの血中および組織濃度を低下させるための方法を提供する。
インビボ投与用に使用される製剤は、無菌でなければならない。これは、例えば、無菌濾過膜を通して濾過することによって、容易に達成できる。
治療上の適用として、本発明による、抗LPA薬剤(例えば、抗体)は、ボーラスとして静脈内投与、または一定期間にわたって持続注入、あるいは筋肉内、腹腔内、脳脊髄内、皮下、関節内、滑液嚢内、くも膜下腔内、経口、局所、吸入ルートによってヒトに投与し得るものを含む、上記に記載のような、薬学的に許容される剤形で、哺乳類、好ましくはヒトに投与される。
疾患のタイプと重症度に基づき、例えば、1回以上の個別の投与または持続注入による、抗体約1μg・kg〜約50 mg/kg(例えば、0.1〜20 mg/kg)が、患者への投与のための初回の候補投与量である。典型的な日毎または週毎の用量は、上記に言及する理由に基づき、約1μg/kg〜約20 mg/kgまたはそれ以上の範囲である。数日間またはそれ以上にわたる反復投与については、症状によって、疾患症状の所望する抑止が起こるまで治療が反復される。しかしながら、他の投与計画も有益であり得る。この治療の進行は、例えば、X線撮影を含む、従来の技術またはアッセイによって容易にモニターされる。治療用抗体に対する患者曝露を最適化するために、抗体を用いて体液または組織中のLPA濃度を測定する検出方法を使用することができる。
かかる他の薬剤は、投与される組成物中に含まれていてもよいし、別に投与されてもよい。さらに、抗体は、適宜に連続投与、または、例えば、癌治療のための化学療法剤または放射線のような、その他の薬剤またはモダリティと併用投与される。
本発明による抗LPA薬剤(例えば、抗体)は、例えば、体液から、LPAを検出および/または精製するために使用され得る。
酵素−基質の組み合せの例は、例えば、以下を含む。
多数の他の酵素−基質の組み合せが当業者には使用可能である。これらの一般概要については、米国特許第 4,275,149号および第4,318,980号を参照。
本発明による抗体は、例えば、競合結合アッセイ、直接および間接サンドイッチアッセイ、免疫沈降アッセイなど任意の公知のアッセイ方法に用いることができる。Zola、Monoclonal Antibodies: A Manual of Techniques(モノクローナル抗体:技術マニュアル), pp.147−158(CRC Press, Inc. 1987)。
また、抗体は、インビボ診断検査法にも使用され得る。一般には、結合された標的分子を免疫シンチグラフィを用いて局在化できるように、抗体を放射性核種(例えば111In、99Tc、14C、131I、125I、3H、32P、35S)で標識する。
便宜上、本発明による抗体は、例えば、診断検査法を実施するための取扱説明書を添付する、規定量の試薬を組み合わせてパッケージしたキット中に供給され得る。抗体が酵素で標識される場合、キットには、基質と、酵素よって必要とされる補助因子(例えば、検出可能な発色団またはフルオロフォアを生じる基質前駆体)が含まれることになる。さらに、安定剤、バッファー(例えば、ブロックバッファーまたは溶解バッファー)などの他の添加物が含まれてもよい。様々な試薬の相対量は、アッセイの感受性を実質的に最適化する試薬の溶液濃度が得られるように、広範に異なってもよい。特に、試薬は、溶解することによって適切な濃度の試薬溶液が得られるように、賦形剤を含む、通常は凍結乾燥された乾燥粉末として供給され得る。
本発明の他の態様では、前述の疾患治療のために有効な物質を含む製品が供給される。この製品は、容器とラベルを具備する。好適な容器は、例えば、瓶、バイアル、シリンジ、試験管などを含む。容器は、ガラスまたはプラスチックなどの様々な材料から形成され得る。容器は、症状の治療に有効な組成物を収容し、無菌のアクセスポートを具備し得る(例えば、容器は、静脈内投与用溶液バッグ、または皮下注射針が貫通可能なストッパーを有するバイアルでありうる)。組成物中の活性薬剤は、抗スフィンゴ脂質抗体である。容器上のラベル、または容器に付随するラベルは、組成物が選択する症状の治療用であることを示す。製品はさらに、リン酸緩衝生理食塩水、リンガー液、デキストロース溶液などの薬学的に許容されるバッファーを含む第二の容器を具備してもよい。他のバッファー、希釈剤、フィルター、針、シリンジ、使用に関する説明を記載した添付文書を含む、商業上およびユーザーの観点から望ましいその他の資材をさらに含んでもよい。
本実施例で説明する合成的なアプローチでは、主に従来の有機化学を用いて構成要素を連続的に追加して抗原を調製する。本実施例で説明するアプローチのスキームを図1において提供するが、下記の合成の説明における化合物の番号は、図1において番号をふった各構造を示す。
この合成アプローチではまず、市販の15−ヒドロキシペンタデシンである1を用意し、塩化メチルスルホニルによる15−ヒドロキシ基の活性化でヒドロキシル置換を促進してそのスルホン酸塩である2を作成した。t−ブチルチオールでスルホン酸塩を置換し、保護されたチオエーテルである3を得、それをガーナーアルデヒドと縮合することによって4を作成した。アルキン部分を軽度に還元してアルケン(5)とし、次いでオキサゾリデン環の酸触媒開環によってS保護およびN保護されたスフィンゴシン・チオール置換体である6を得た。この最後の工程において、二炭酸di−t−ブチルによる再誘導体化を用いることによって、酸触媒開環中のN−BOC基の損失を緩和した。
記載した合成アプローチは、特に保護用および脱保護用の試薬の選択に関しては、例えば、実施例3に記載の二硫化トリメチル・トリフレートをチオールの脱保護に使用するなど、変更可能である。
1を(10.3 g、45.89 mmol)入れた500 mLのRBフラスコにDCM(400 mL)を添加し、その溶液を0℃まで冷却した。次に、TEA(8.34 g、82.60 mmol、9.5 mL)を一度に全部添加したのち、10分間かけてMsCl(7.88 g、68.84 mmol、5.3 mL)を滴下した。反応を室温で0.5時間または原料が消失するまで撹拌させた(Rf = 0.65、ヘキサン類/EtOAc=5:1)。反応をNH4Cl(300 mL)でクエンチし、DCM(2 X 200 mL)を抽出した。有機層をMgSO4にて乾燥し、ろ過したろ液を蒸発固化した(13.86 g、収率99.8 %)。1H NMR(CDCl3)d 4.20(t、J = 6.5 Hz、2H)、2.98(s、3H)、2.59(td、J = 7 Hz、3 Hz、2H)、1.917(t、J = 3 Hz、1H)、1.72(五重線、J = 7.5 Hz、2H)、1.505(五重線、J = 7.5 Hz、2H)、1.37(br s、4H)、1.27(br s、14H)。13C{1H} NMR(CDCl3-)d 85.45、70.90、68.72、46.69、38.04、30.22、30.15、30.14、30.07、29.81、29.76、29.69、29.42、29.17、26,09、19.06、9.31。化合物2のHRMS分析(ES−TOF)において観察された主要イオンは、m/z = 325.1804であった(C16H30O3Sとしての計算値:M+Na+325.1808)。
1Lの三つ首RBフラスコにt−ブチルチオール(4.54 g、50.40 mmol)とTHF(200 mL)を入れたのち、氷浴に入れた。30分間かけてn−BuLi(1.6 Mのヘキサン類溶液31.5 mL)を添加した。次に、化合物2(13.86 g、45.82 mmol)をTHF(100 mL)で溶解した溶液を2分間かけて添加した。反応を、1時間または原料が消失するまで撹拌する(Rf = 0.7、ヘキサン類/EtOAc=1:1)。反応を飽和NH4Cl(500 mL)でクエンチし、EtO2(2 X 250 mL)によって抽出し、MgSO4で乾燥し、ろ過したろ液を蒸発させて黄色油(11.67 g、収率86%)を得た。1H NMR(CDCl3)d 2.52(t、J = 7.5 Hz、2H)、2.18(td、J = 7 Hz、2.5 Hz、2H)、1.93(t、J = 2.5 Hz、1H)、1.55(五重線、J = 7.5 Hz、2H)、1.51(五重線、J = 7 Hz、2H)、1.38(br s、4H)、1.33(s、9H)、1.26(s、14H)。13C{1H} NMR(CDCl3-)d 85.42、68.71、68.67、54.07、42.37、31.68、30.58、30.28、30.26、30.19、30.17、29.98、29.78、29.44、29.19、29.02、19.08。
化合物3(5.0 g、16.85 mmol)を入れた250 mLのシュレンクフラスコを真空にして窒素を充填する作業を3回繰り返したのち、乾燥THF(150 mL)を添加した。得られた溶液を−78℃まで冷却した。次に、2分間かけてn−BuLi(1.6Mのヘキサン類溶液10.5 mL)を添加し、反応混合液を−78℃で18分間撹拌したのち、冷却浴を20分間取り除いた。ドライアイス浴を戻した。15分後、ガーナーアルデイド(3.36 g、14.65 mmol)の乾燥THF(10 mL)溶液を5分間かけて添加した。20分後に、冷却浴を取り除いた。2.7時間後の薄層クロマトグラフィー(TLC)では、ガーナーアルデヒドが消失していることが確認された。反応を飽和NH4Cl水溶液(300 mL)でクエンチし、Et2O(2 X 250 mL)で抽出した。合わせたEt2O相をNa2SO4にて乾燥し、ろ過したろ液を蒸発させることにより、粗化合物4およびそのシンジアステレオマー(図1に図示せず)を黄色油(9.06 g)として得た。この材料は、これ以上精製せず次の工程に使用した。
化合物4の三重結合を還元するために、上記油を窒素下で乾燥Et2O(100 mL)に溶解した。水素ガス(H2)の発生を制御するために室温でゆっくりとその溶液にRED−Al(65%のトルエン溶液20 mL)を添加した。反応を、一夜またはTLCで原料(EtOAc/ヘキサン類1:1でRf = 0.6)の消失が確認されるまで室温で撹拌し、H2の発生を制御するためにゆっくりとMeOHまたはNH4Cl水溶液でクエンチした。得られた白色懸濁液をセリットパッドでろ過し、ろ液をEtOAc(2 X 400 mL)で抽出した。合わせたEtOAc抽出液をMgSO4にて乾燥し、ろ過したろ液を蒸発させると、粗化合物5およびそのシンジアステレオマー(図1に図示せず)が黄色油(7.59 g)として残った。
化合物5が含まれている上記油をMeOH(200 mL)で溶解し、PTSA水和物(0.63 g)を添加し、その溶液を室温で1日間、その後50oCで2日間撹拌し、その時点で全原料(5)が消失していることがTLCによって示唆された。しかしながら、いくらかの極性材料が存在していたため、酸がBOC基を一部切断したことが示唆された。反応を、飽和NH4Cl水溶液(400 mL)を添加してワークアップし、エーテル(3 x 300 mL)で抽出した。混合したエーテル相をNa2SO4にて乾燥し、ろ過したろ液を乾燥するまで蒸発させると、5.14 gの油が残った。形成されたアミンを再度保護するため、上記粗産物をCH2Cl2(150 mL)に溶解し、それにBOC2O(2.44 g)およびTEA(1.7 g)を添加した。TLC(ヘキサン類/EtOAc=1:1)によって、基線に材料が残っていないことが示されたら飽和NH4Cl水溶液(200 mL)を添加し、有機相を分離したのち、混合液をCH2Cl2(3 X 200 mL)で抽出した。合わせた抽出をNa2SO4にて乾燥し、ろ液を乾燥するまで濃縮し、黄色油(7.7 g)を得、これをヘキサン類/EtOAcの(1:1までの)勾配を用いたシリカカラムクロマトグラフィーにかけることによって上記ジアステレオマーを分離した。PE/EtOAc= 1:1を用いたTLCにより、化合物6であるアンチ体のRfは0.45であった。シン体(図1に図示せず)のRfは0.40だった。化合物6の収率は2.45 g(ガーナーアルデヒドに基づいて総合的に39 %)だった。アンチ体の1H NMR(CDCl3)d 1.26(br s、20H)、1.32(s、9H)、1.45(s、9H)、1.56(五重線、2H、J = 8 Hz)、2.06(q、2H、J = 7 Hz)、2.52(t、2H、J = 7 Hz)、2.55(br s、2H)、3.60(br s、1H)、3.72(ddd、1H、J = 11.5 Hz、7.0 Hz、3.5 Hz)、3.94(dt、1H、J = 11.5 Hz、3.5 Hz)、4.32(d、1H、J = 4.5 Hz)、5.28(br s、1H)、5.54(dd、1H、J = 15.5 Hz、6.5 Hz)、5.78(dt、1H、J = 15.5 Hz、6.5 Hz)。13C {1H} NMR(CDCl3)d 156.95、134.80、129.66、80.47、75.46、63.33、56.17、42.44、32.98、31.70、30.58、30.32、30.31、30.28、30.20、30.16、30.00、29.89、29.80、29.08、29.03。
化合物6(609.5 mg、1.25 mmol)を乾燥ピリジン(2 mL)で溶解したアルコール溶液にCBr4(647.2 mg、1.95 mmol、1.56 等量)を添加した。フラスコを氷浴で冷却し、2分間かけてP(OMe)3(284.7 mg、2.29 mmol、1.84 等量)を滴下した。4分後に氷浴を取り除き12時間後に混合液をエーテル(20 mL)で希釈した。得られた混合液をHCl水溶液(10 mL、2 N)で洗浄することによりエマルションを形成し、これを水(20 mL)による希釈で分離した。水相をエーテル(2 x 10 mL)、その後EtOAc(2 x 10 mL)で抽出した。該エーテル抽出液および第1のEtOAc 抽出液を合わせ、HCl水溶液(10 mL、2 N)、水(10 mL)、および飽和炭酸水素ナトリウム水溶液(10 mL)で洗浄した。最後のEtOAc抽出液を用いて洗浄水溶液を逆抽出した。合わせた有機相をMgSO4にて乾燥し、ろ過したろ液を濃縮すると、粗産物(1.16 g)が残ったため、これをCH2Cl2、次いで CH2Cl2−EtOAc(1:20、1:6、1:3、および1:1 - 産物は、6:4、7:3で溶出し始めた)を用いたシリカ(3 x 22 cm カラム)フラッシュクロマトグラフィーにかけた。初期のフラクションは56.9 mgの油を含んでいた。後のフラクションでは、産物(化合物7、476.6 mg、64%)が透明無色の油として得られた。
元素分析値。C29H58NO7PS(595.82)としての計算値:C、58.46;H、9.81;N、2.35。実測値:C、58.09;H、9.69;N、2.41。
化合物7(333.0 mg、0.559 mmol)と撹拌棒を入れたフラスコを真空にして窒素を充填した。アセトニトリル(4 mL、CaH2から蒸留)を注射器で注入し、溶液が入ったフラスコを氷浴にて冷却した。注射器を用いて(CH3)3SiBr(438.7 mg、2.87 mmol、5.13 等量)を1分間かけて添加した。35分後、フラスコの上部を追加分のアセトニトリル(1 mL)ですすぎ、氷浴を取り除いた。さらに80分後、アリコートを取り出し、それに窒素ガスを吹き当てて溶液を乾燥し、残分をCDCl3中1H NMRで分析したが、P−OCH3部分に起因するピークはほんのわずかしかみられなかった。20分後、反応混合液に水(0.2 mL)、次いで上記アリコートの分析に使うCDCl3 溶液を添加し、混合液を回転蒸発器で約0.5 mLの体積になるまで濃縮した。アセトン(3 mL)を分割して用い、風袋重量を測った試験管に残分を移すと、薄褐色溶液が形成された。水(3 mL)を分割して添加した。0.3 mLを追加した時点で、濁りが確認された。全体で1 mLになった時点で、ゴム状の沈殿物が形成された。さらに0.6 mLの水を添加すると、さらに濁りとゴムが析出したが、最後の分の水を添加したところ、混合物の外観は変化しないようであった。この過程を終了するには全体で数時間かかった。試験管を遠心分離にかけ、ピペットで上清を除去した。もはやゴム状ではない固体をP4O10にて減圧乾燥すると、化合物8(258.2 mg、95%)が一水和物として残った。
元素分析値。C22H46NO5PS+H2O(485.66)としての計算値:C、54.40;H、9.96;N、2.88。実測値:C、54.59;H、9.84;N、2.95。
グローブボックスにおいて、撹拌棒、乾燥THF(3 mL)および氷HOAc(3 mL)が入っている試験管に化合物8(202.6 mg、0.417 mmol)を添加した。NBSCl(90 mg、0.475 mmol、1.14 等量)を添加し、0.5時間後に透明溶液を得た。全9時間後、アリコートを乾燥するまで蒸発させ、残分をCDCl3中1H NMRで分析した。CH2StBuおよびCH2SSAr と一致するピークによって反応が約75%完了していることが示唆され、スペクトルをCDCl3 中の純NBSClのものと比較したところ、反応において試薬が残っていないことが示唆された。したがって、追加分(24.7 mg、0.130 mmol、0.31 等量)を添加し、その3時間後に追加分(19.5 mg、0.103 mmol、0.25 等量)を添加した。さらに1時間後、THF(2 mL)ですすぎながら混合液を新しい試験管に移し、水(1 mL)を添加した。
上記化合物9の透明溶液にPMe3(82.4 mg、1.08 mmol、添加した塩化2−ニトロベンゼンスルフェニル総量の1.52倍)を添加した。混合液は温かくなって白濁し、経時的に沈殿物を形成した。4.5時間後、メタノールを添加し、試験管を遠心分離にかけた。沈殿物は沈殿しにくく、試験管の下方1 cmを占めた。ピペットを用いて透明黄色の上清を除去した。メタノール(5 mL、窒素で脱酸素化)を添加し、試験管を遠心分離にかけ、上清をピペットで除去した。このサイクルを3回繰り返した。濃縮すると、最後のメタノール洗浄液は4.4 mgしか残らなかった。固体残分の大半をP4O10にて減圧乾燥すると、化合物10(118.2 mg、68%)が一塩酸塩として残った。
元素分析値。C18H38NO5S+HCl(417.03)としての計算値:C、51.84;H、9.43;N、3.36。実測値:C、52.11;H、9.12;N、3.30。
化合物6(1.45 g、2.97 mmol)をAcOH(20 mL)で溶解し、NBSCl(0.56 g、2.97 mmol)を一度に全部添加した。反応を、3時間あるいは原料が消失し産物の形成がTLC [産物 Rf = 0.65、原料 Rf = 0.45、EtOAc/ヘキサン類=1:1]によって観察されるまで撹拌させた。反応を高真空管で濃縮して乾燥させ、残分をTHF/H2O(10:1を100 mL)で溶解した。
化合物11を含有する上記溶液にPh3P(0.2.33 g、8.91 mmol)を一度に全部添加し、反応を3時間または原料が消失するまで撹拌させた。粗反応混合液を高真空管で濃縮して乾燥させると、化合物12を含有する残分が残った。
化合物12を含有する上記残分をDCM(50 mL)およびTFA(10 mL)で溶解した。混合液を室温で5時間撹拌し、濃縮して乾燥させた。残分をシリカゲル入りのカラムに注入し、純DCM、次いで5% MeOH 含有DCM、その後に10% MeOHでクロマトグラフィーを実施して最終産物である化合物13(0.45 g、5からの収率46%)を粘着性の白色固体として得た。1H NMR(CDCl3)d 1.27(s)、1.33(br m,)、1.61(p、2H、J = 7.5 Hz)、2.03(br d、2H、J = 7 Hz)、2.53(q、2H、J = 7.5 Hz)、3.34(br s、1H)、3.87(br d、2H、J = 12 Hz)、4.48(br s、2H)、4.58(br s、2H)、5.42(dd、1H、J = 15 Hz、5.5 Hz)、5.82(dt、1H、J = 15 Hz、5.5 Hz)、7.91(br s、4H)。13C{1H} NMR(CDCl3)d 136.85、126.26、57。08、34.76、32.95、30.40、30.36、30.34、30.25、30.19、30.05、29.80、29.62、29.09、25.34。
本実施例に記載の合成アプローチでは、チオール化脂肪酸の調製を詳細に説明するが、これはより複雑な脂質構成に組み込まれ、任意にタンパク質または他の担体とさらに複合化され、免疫応答を誘導させるために動物に投与されるものである。本アプローチには、従来の有機化学を用いる。本実施例でとるアプローチを示すスキームを図2において提供し、下記の合成の説明における化合物番号は、図2において番号をふった各構造を示す。
(化合物15)
乾燥したシュレンクフラスコにt−ブチルチオール(12.93 g、143 mmol)を添加し、シュレンク法を用いて系を窒素下に置いた。脱気した乾燥THF(250 mL)を添加し、フラスコを氷浴において冷却した。10分間かけてn−BuLi(2.5 Mのヘキサン類溶液55 mL、137.5 mmol)を注射器でゆっくりと添加した。混合液を、0oCで一時間撹拌した。臭化酸である化合物14(10 g、36 mmol)を固体として添加し、反応を加熱して60oCで24時間撹拌した。反応を2 M HCl(250 mL)でクエンチしてエーテル(2 x 300 mL)で抽出した。合わせたエーテル層を硫酸マグネシウムで乾燥させ、ろ過したろ液を回転蒸発により濃縮し、チオエーテル酸である化合物15(10 g、収率99%)をベージュ色粉末として得た。1H NMR(CDCl3、500 MHz)δ 1.25−1.35(br s,12 H)、1.32(s、9 H)、1.35−1.40(m、2 H)、 1.50−1.60(m、2H)、1.60−1.65(m、2 H)、2.35(t、2 H、J = 7.5 Hz)、2.52(t、2 H、J = 7.5 Hz)。HRMS(ES−TOF)における主要イオンは、M+Na+ 311.2015として計算した場合、m/z 311.2020で確認された。
(化合物17)
乾燥したシュレンクフラスコに化合物16(50 g、224.2 mmol)を入れ、ナトリウム/ベンゾフェノンから蒸留した脱気した乾燥THF(250 mL)で溶解した。フラスコを氷浴において冷却したのちPTSA(0.5 g、2.6 mmol)を添加した。次に、脱気した乾燥DHP(36 g、42.8 mmol)を5分間かけてゆっくりと添加した。該混合液を室温まで温め、一夜撹拌し、TLC(PE:EtOAc= 10:1)で臭化アルコールのスポットの完全消失によって反応終了とみなされるまで監視した。次に、TEA(1 g、10 mmol)を添加してPTSAをクエンチした。その後、混合液を冷重曹溶液で洗浄し、EtOAc(3 X 250 mL)で抽出した。次いで、有機層を硫酸マグネシウムで乾燥させ、濃縮して粗産物68.2 gを得、これをカラムクロマトグラフィー(PE:EtOAc= 10:1)で精製することによって無色油60 g(収率99%)を得た。1H NMR(CDCl3、500 MHz)δ 1.31(br s、6 H)、1.41−1.44(m、2 H)、1.51−1.62(不明多胎、6 H)、1.69−1.74(m、1 H)、1.855(五重線、J = 7.6 Hz、2 H)、3.41(t、J = 7 Hz、2 H)、3.48−3.52(m、2 H)、3.73(dt、2 H、J = 6.5 Hz)、3.85−3.90(m、2 H)、4.57(t、2 H、J = 3 Hz)。
火力乾燥したシュレンクフラスコに削状マグネシウム(2.98 g、125 mmol)並びにヨウ素結晶を添加した。次いで、ナトリウムから蒸留した乾燥THF(200 mL)を添加し、シュレンク技法によって系を脱気した。その後、10分間かけて化合物17(30 g、97 mmol)をゆっくりとマグネシウムに添加し、溶液を65oCの油浴に入れ一夜撹拌した。反応は、アリコートをアセトンでクエンチして、10:1のPE:EtOAc混合液におけるRFの変化を観察することで、TLCにより完了とみなされた。その後、グリニヤール溶液をカニューレで追加の化合物17(30 g、97 mmol)が入っている窒素下の三つ首フラスコに移した。その得られた混合液が入っているフラスコを氷浴において0oCまで冷却した後、Li2CuCl4溶液(1 Mを3 mL)を注射器で添加した。反応混合液は、数分以内で非常に濃い青色になった。この混合液を一夜撹拌させた。翌朝、反応はTLC(10:1 PE:EtOAc)によって完了とみなされ、飽和NH4Cl溶液によってクエンチした後、エーテル(3 X 250 mL)に抽出した。該エーテル層を硫酸マグネシウムで乾燥し、濃縮して粗産物(40 g)を得、これをMeOHに溶解した。その後、濃HCl(0.5 mL)を添加した結果、白色のエマルションが形成され、それを3時間撹拌させた。そして前記白色のエマルションをろ過し、純ジオールである化合物18を16 g(収率58%)得た。1H NMR(CDCl3、200 MHz)δ 1.26(br s、24 H)、1.41−1.42(m、4 H)、1.51−1.68(m、4 H)、3.65(t、4 H、J = 6.5 Hz)。
対称ジオールである化合物18(11 g、38.5 mmol)を窒素下乾燥シュレンクフラスコに添加し、ナトリウムから蒸留した乾燥THF(700 mL)を添加した。系を脱気し、前記フラスコを氷浴に入れた。注射器でジイソプロピルエチルアミン(6.82 mL、42.3 mmol)を添加し、次いでMsCl(3.96 g、34.4 mmol)をゆっくりと添加、混合液を1時間攪拌させた。反応を飽和NaH2PO4 溶液(300 mL)で停止したのち、EtOAc(3 X 300 mL)で抽出した。その後、有機層を合わせ、MgSO4で乾燥させ、濃縮してジオール、モノメシレート、ジメシレートの混合物14 gを得た。NMRは、1:0.8 のCH2OH:CH2OMsプロトンの混合を示した。次いで該混合物を乾燥THF(500 mL)で溶解し、脱酸素して、それにLiBr(3.5 g、40.23 mmol)を添加した。この混合液を一夜還流させ、その時点で反応を水(150 mL)でクエンチし、EtOAc(3 X 250 mL)で抽出した。その後、有機層をMgSO4で乾燥させ、濃縮してブロム化産物の混合物を得、それをその後フラッシュクロマトグラフィー(DCM)によって精製し、化合物19(3.1 g、収率25%)を白色粉末として得た。1H NMR(CDCl3、500 MHz)δ 1.26(br s、26 H)、1.38−1.46(m、2 H)、1.55(五重線、2 H、J = 7.5 Hz)、1.85(五重線、2 H、J = 7.5 Hz)、3.403(t、2 H、J = 6.8 Hz)、3.66(t. 2 H、J = 6.8 Hz)。
化合物19(2.01 g、5.73 mmol)を丸底フラスコに入れ、固体を試薬グレードのアセトン(150 mL)に溶解した。同時に、ジョーンズ試薬を、硫酸(4 mL)にCrO3(2.25 g、22 mmol)を溶解し、冷水を10 mLゆっくりと添加し、溶液を10分間撹拌させることによって調製した。次いで、冷やしたジョーンズ試薬を5分間かけて丸底フラスコにゆっくりと添加した後、その溶液を1時間撹拌した。その結果得られたオレンジ色の溶液は数分以内で緑色になった。次に、混合液を水(150 mL)でクエンチし、エーテル(3 X 150 mL)にて2回抽出した。その後、エーテル層を硫酸マグネシウムで乾燥させ、濃縮して化合物20(2.08 g、収率98%)を白色粉末として得た。1H NMR(CDCl3、200 MHz)δ 1.27(br s、26 H)、1.58−1.71(m、2 H)、1.77−1.97(m、2H)、2.36(t、2 H、J = 7.4 Hz)、3.42(t、2 H、J = 7 Hz)。
乾燥したシュレンクフラスコにt−ブチルチオール(11.32 g、125 mmol)を添加し、ナトリウムから蒸留した乾燥THF(450 mL)に溶解した。溶液に窒素を通気することによって脱酸素したのち、フラスコを氷浴に入れた。n−BuLiのヘキサン類溶液(1.6 Mを70 mL)を10分間かけて注射器でゆっくりと添加した。この混合液を1時間撹拌させた後、化合物20(5.5 g、16.2 mmol)を添加し、溶液を60oCで一夜還流させた。翌朝アリコートをワークアップし、NMRで分析し、反応は完了したとみなされた。反応をHCl(2 Mを200 mL)でクエンチし、エーテル(3 X 250 mL)で抽出した。次に、エーテル層を硫酸マグネシウムで乾燥させ、ろ過したろ液を濃縮して、産物である化合物21を白色固体として得た(5 g、収率90%)。1H NMR(CDCl3、200 MHz)δ 1.26(br s、26 H)、1.32(br s、9 H)、1.48−1.70(m、4 H)、2.35(t、2 H、J = 7.3 Hz)、2.52(t、2 H、J = 7.3 Hz)。13C NMR(CDCl3、200 MHz)δ 24.69、28.35、29.05、29.21、29.28、29.39、29.55、29.89、31.02(3C)、33.98、41.75、179.60。
本実施例に記載の合成アプローチによってチオール化LPAが調製される。次いでこのLPA類似体は、さらに担体、例えば、タンパク質担体と複合化することができ、そしてそれを動物に投与してLPAに対する免疫原性反応を誘導することができる。このアプローチでは、有機化学および酵素反応の両方を使っており、その合成スキームは図3に示す。下記の合成の説明における化合物番号は、図3において番号をふった各構造を示す。
火力乾燥したシュレンクフラスコにチオエーテル酸、化合物15(10 g、35.8 mmol)、化合物22(グリセロホスホコリン-CdCl2 複合体、4.25 g、8.9 mmol)、DCC(7.32 g、35.8 mmol)、DMAP(2.18 g、17.8 mmol)を添加した後、フラスコを真空にして窒素を充填した。最小量の脱気した乾燥DCMを添加したところ(100 mL)、濁った混合液になった。フラスコにホイルを被せ、TLC(シリカ、DCM:MeOH:濃NH4OH=10:5:1)により完了するまで攪拌させた。化合物16は不溶性のため、その消失をTLCによりモニターすることはできなかったが、産物のスポットRf 0.1の強度が増加しないと判断された時点で反応を終了させた。これには通常3、4日間かかり、DCCおよびDMAPを更に添加する必要がある場合もある。反応が完了したら、反応混合液をろ過してろ液を濃縮し、黄色油を得、それを上記の溶媒系を用いてフラッシュクロマトグラフィーによって精製し、(CH3)3N部分、−CH2StBu部分、-CH2COO部分のピークの積分を比較した計算によると化合物23とモノアシル化産物の割合が5対1である混合物を含んでいる透明ワックスを3.6 g得た(収率50%)。HRMS(ESI−TOF)による油の分析では、M+Na+ = C40H80NNaO8PS2 + 820.4960として計算した場合、顕著なイオンがm/z 820.4972に得られた。
(化合物24)
上記化合物23とモノアセチル化産物の混合物(3.1 g、3.9 mmol)を、Et2O(400 mL)とメタノール(30 mL)に溶解した。ホウ酸塩緩衝液(100 mL、pH 7.4 0.1M、0.072 mMの塩化カルシウム)、次いでホスホリパーゼA2(ハチ毒由来、130 単位、シグマ)を添加した。得られた混合液を10時間攪拌させ、その時点で、原料(Rf = 0.7)の欠如と新しいスポット(Rf = 0.2)の発現がTLC(シリカ、MeOH:水=4:1-前記溶媒系DCM:MeOH:濃NH4OH=10:5:1は効果なし)によって確認された。有機層および水層を分別し、水層をエーテル(2 x 250 mL)で洗浄した。DCM:MeOH(2:1、2 x 50 mL)の混合液によって、水層から産物を抽出した。その後、回転蒸発により有機層を濃縮し、産物を白色ワックス(1.9 g、収率86%)として得、NMRによって純産物であることが確認された(化合物24)。1H NMR(CDCl3、500 MHz)δ 1.25−1.27(br s、12 H)、1.31(s、9 H)、1.35−1.45(m、2 H)、1.52−1.60(m、4 H)、2.31(t、2 H、J = 7.5 Hz)、2.51(t、2 H、J = 7.5 Hz)、3.28(br s、9 H)3.25−3.33(br s、2 H)、3.78−3.86(m、1 H)、3.88−3.96(m、2 H)、4.04−4.10(m、2 H)、4.26−4.34(m、2 H)。HRMS(ESI−TOF)によるワックスの分析では、顕著なイオンが m/z 550.2936に、計算ではM+Na+ 550.2943(C24H50NNaO7PS2 +)、および m/z が528.3115に、計算ではMH+ 528.3124(C24H51NO7PS2 +)が得られた。
(化合物26)
保護されたLPA試料である化合物26(0.150 g、0.34 mmol)をメタノールで洗浄し、グローブボックス内のバイアルに添加した。その後これをAcOH:THFの混合液(1:1、10 mL)に懸濁したが、1時間超音波処理した後でも完全に溶解することはなかった。その後、固体[Me2SSMe]OTf(0.114 g、0.44 mmol)を添加した。これを18時間撹拌させた。真空濃縮乾燥しつつ、アリコートを除去することによって反応をモニターし、硫黄と最も近くのCH2 ピークの1H NMR 交替を観察するために残分をCD3ODに再溶解または懸濁した。原料のピークが2.52 ppmにあるのに対し、この時点で生成した非対称の二硫化物のピークは約2.7 ppmにあった。この材料(化合物27)については、これ以上単離もキャラクテリゼーションもしなかった。
化合物27が含まれている混合物を水(100 μL)で、その直後にPMe3(0.11 g、1.4 mmol)で処理した。3時間撹拌した後、溶媒を真空によって除去し、不溶性の白色固体を得た。メタノール(5 mL)を添加し、混合液を遠心分離にかけ、母液をデカントした。減圧濃縮によってベージュ色の固体である化合物28を120 mg(収率91%)得た。化合物28はチオール化LPAハプテンであり、ジスルフィド結合形成を介して担体、例えば、アルブミンまたはKLHとコンジュゲートすることができる。化合物28のキャラクテリゼーション:1H NMR(1:1 CD3OD:CD3CO2D、500 MHz)δ 1.25−1.35(br s、12 H)、1.32−1.4(m、2 H)、1.55−1.6(m、4 H)、2.34(t、2H、J = 7)、2.47(t、2H、J = 8.5)、3.89−3.97(br s、2 H)、3.98−4.15(m、2 H)、4.21(m、1H)。メタノールに溶解した試料の負イオンESでは、m/z = 385.1にて顕著なイオンが得られた。
治療抗体の一種は、望ましくないスフィンゴ脂質と特異的に結合して、例えば、(1)心毒性効果、腫瘍形成効果、血管新生効果などの望ましくない効果を促進させるような望ましくない有毒性スフィンゴ脂質の有効濃度(および/またはそれらの代謝前駆体の濃度)を低下させ、(2)望ましくない、有毒性の、または腫瘍形成性の、または血管新生のスフィンゴ脂質と細胞受容体との結合を抑制し、および/またはかかる受容体との結合に利用可能なスフィンゴ脂質の濃度を低下させるなどの有益な効果を上げる。かかる治療効果の例として、抗S1P抗体を使用してインビボで利用可能なS1Pの血清中濃度を低下させることによって、S1Pの腫瘍形成効果や血管新生効果および心筋梗塞後の心不全、または癌、または線維形成性疾患における役割を阻害または少なくとも抑えることが挙げられるが、これに限らない。
1264491972164_2.');
、146巻(1):62−8)に従って行ったが、結果では、保持時間における差異は示されなかった。さらに、検討された各種脂質に対するモノクローナル抗体の結合特性の比較によれば、抗体が認識するエピトープには、天然S1Pの炭化水素鎖領域に含まれている二重結合が関与していないことが示されている。一方、モノクローナル抗体によって認識されるエピトープは、スフィンゴシン基本骨格のアミノアルコールを含む領域と遊離リン酸である。遊離リン酸をコリンと連鎖した場合(SPCの場合と同様)、結合はいくらか減少した。アミノ基を脂肪酸とエステル化した場合(C1Pの場合と同様)、抗体結合はみられなかった。スフィンゴシンアミノアルコール骨格をグリセリン骨格で置換した場合(LPAの場合と同様)、そこではSIP特異的モノクローナルは結合を示さなかった。これらエピトープマッピングのデータは、S1Pには、モノクローナル抗体によって認識されるエピトープが1つだけあり、このエピトープがS1Pの唯一の極性頭部基によって定義されていることを示す。
本明細書で使用する「キメラ」抗体(または「免疫グロブリン」)という用語は、特定種に由来するまたは特定の抗体クラスまたはサブクラスに属する抗体の対応する配列と同一または相同である1本の重鎖および/または軽鎖を含み、それと同時に残りの鎖は他種に由来するまたは他の抗体クラスまたはサブクラスに属する抗体の対応する配列に同一または相同であり、ならびにそれらが所望する生物活性を提示する限りにおいては、かかる抗体のフラグメントを意味する(Cabillyら、上記参照、Morrisonら、Proc. Natl. Acad. Sci. U.S.A. 81:6851(1984))。抗体 配列は、哺乳類、トリ、軟骨魚類も含むサカナを含め、脊椎動物または無脊椎動物に由来してもよい。
抗体の産生
天然のLPAに対するポリクローナル抗体は、文献(Chen JHら、Bioorg Med Chem Lett. 2000年8月7日、10(15):1691−3)で報告されているが、モノクローナル抗体は記載されていない。実施例4に記載されたアプローチと同様のアプローチを用いて、C−12チオLPA類似体(実施例3の化合物28)を、架橋薬剤としてIOAまたはSMCCを用いた標準的な化学架橋によって反応性SH基を介してタンパク質担体(KLH)と類似体を架橋して形成されるハプテンの重要な成分として、LPAに対するモノクローナル抗体を生成した。そのために、実施例4に記載の抗S1Pモノクローナル抗体の生成に関する方法を用いてマウスをチオLPA−KLHハプテン(この場合、チオール化LPA:SMCC:KLH)によって免疫化した。LPA類似体に対して免疫化した80匹のマウスのうち、LPAに対して最高の力価を示した5匹のマウス(SMCCを用いてハプテンに使用したものと同様のLPA類似体(化合物28)をBSAとコンジュゲートし、ELISAプレートに敷いたELISAを用いて判定した)をハイブリドーマの発生段階に進ませるために選んだ。
表1:LPAハイブリドーマ
6つの抗LPAモノクローナル抗体(B3、B7、B58、A63、B3A6、D22)の12:0および18:1 LPA(0.1 uM)に対する結合をELISAによって測定した。精製したモノクローナル抗体の増加する6つの濃度(0〜0.4 ug/ml)を用いて滴定曲線よりEC50値を計算した。EC50は、最大結合量50 %での抗体の有効濃度を表す。Maxは、最大結合(OD450として表される)を示す。結果を表2に示す。
表2−抗LPAモノクローナル抗体の直接結合動態
抗LPAマウスモノクローナル抗体はLPAに対して高親和性を示す
ka = M-1s-1単位の会合速度定数 kd = s-1単位の解離速度定数
表 3− LPAに対する抗LPAモノクローナル抗体の親和性
LPAは、多くのアイソフォームに生物学的に活性があることが確認されており、モノクローナル抗体がそれらの全てをある程度認識することが、治療関連上好ましい。抗LPAモノクローナル抗体の特異性は、抗体に固定化された脂質の混合物に競合体脂質を添加した競合的検定を用いて評価した。
表4. 6つの抗LPAモノクローナル抗体の特異性プロファイル
癌細胞増殖
LPAは、GPCR受容体を介するGi、Gq、G12/13の刺激および下流の情報伝達現象の活性化による細胞の残存および増殖を支援する強力な成長因子である。細胞株を、それらのLPA(0.01 mM〜10 mM)に対する増殖性応答について検討した。細胞増殖は、ケミコン社(テメクラ、カリフォルニア州)の細胞増殖分析キット(Panc−1)、ピアス社のCell−Blue力価(Caki−1)によって分析した。各データ点は、3つの独立実験の平均とした。LPAは、SKOV3およびOVCAR3(卵巣癌)、Panc−1(膵癌)、Caki−1(腎癌細胞)、DU−145(前立腺癌)、A549(肺癌)、HCT−116(結腸直腸腺癌)の細胞を含む7つのヒト由来腫瘍細胞株および1つのラット由来の腫瘍細胞株であるRBL−2H3(ラット白血病細胞)の増殖を濃度依存的に増加させた。腫瘍由来細胞は通常増殖の基礎レベルが高いが、LPAは、ほとんどの腫瘍細胞株において増殖をさらに増大させるようにみられた。選択したヒト癌細胞株においてLPA誘導性増殖を抑制する能力について、抗LPAモノクローナル抗体(B7およびB58)を検討した。LPAによる増殖誘導の増加は、抗LPAモノクローナル抗体の添加によって抑制されることが確認された。
臨床的に意義のあるレベルの化学療法剤であるパクリタキセル(タキソール)に曝露した場合の、卵巣腫瘍細胞をアポトーシスから保護するLPAの能力を研究した。SKVO3細胞を1% FBS(S)、タキソール(0.5 mM)、+/−抗LPAモノクローナル抗体によって 24時間処理した。LPAは、タキソール誘導のアポトーシスからSKVO3細胞を保護した。アポトーシスは、製造元(プロメガ)の推薦通りカスパーゼ活性の測定によって分析した。予測通り、LPAは、試験した癌細胞株のほとんどをタキソール誘導の細胞死から保護した。選択したLPA応答性細胞に抗LPA抗体LT3000を添加すると、細胞毒性化学療法剤によって誘導された死亡から細胞を保護するLPAの能力が遮断された。さらに、抗LPA抗体は、血清によってもたらされた保護力を除去することもできた。血清は約5〜20 uMのLPAを含有していると推定される。タキソールは、SKOV3細胞においてカスパーゼ−3,7活性化を誘導し、細胞へ血清を添加することにより、細胞がアポトーシスから保護された。タキソール誘導のカスパーゼ活性化は、LT3000を培地に添加することによって促進された。これは、血清中に存在するLPAの選択的な抗体による中和によって、LPAによる保護および抗アポトーシス効果が除去されたことを示唆する。
転移性癌の重要な特徴は、腫瘍細胞が接触阻害を逃れ、由来する組織から離れて移動することである。LPAは、複数の癌細胞タイプにおいて転移の可能性を促進させることが示された。従って、細胞単層スクラッチアッセイを用いて、LPA依存性細胞移動に対する抗LPAモノクローナル抗体の遮断能を複数のヒト癌細胞株において試験した。細胞を96ウェルプレートに播種し、コンフルエントになるまで増殖させた。24時間の飢餓後、ウェルの中央をピペットの先端でスクラッチした。従来から許容されているこの「スクラッチアッセイ」では、細胞がスクラッチ領域へ移動して創傷を閉鎖することによって、細胞単層に付いたスクラッチ創傷に対して画一的に反応する。移動および創傷閉鎖の進行は所望の時点で倍率10xのデジタル撮影によってモニターした。細胞は未処理(NT)であったか、Ab B7(10 μg/ml)アイソタイプが一致した非特異性抗体(NS)(10 μg/ml)の存在下または非存在下でLPA(2.5 mM)によって処理された。未処置細胞では、スクラッチ後の単層縁の間に大きい間隙が残存していた。それに対してLPA処理細胞では、同時点で小さい間隙が残存しているだけで、わずかな細胞が間隙を超えて接触していた。LPAと抗LPA抗体B7の両方で処理した細胞では、この時点で間隙がLPAのみの処理より数倍大きいものの、未処置の対照細胞ほど大きくはなかった。これは抗LPA抗体がLPA刺激による腎細胞癌(Caki−1)細胞の移動に対して抑制効果を有することを示す。同様なデータがモノクローナル抗体B3、B58でも得られた。このことは、元は転移性癌に由来する細胞株のLPAによる移動を抗LPAモノクローナル抗体が減少させられることを示す。
LPAは、増殖促進性の腫瘍微小環境を提供し、血管形成を促進することによって癌の成立および進行に関与している。特にIL−8およびVEGFなどの成長促進因子の増加が癌細胞において観察された。IL−8は、癌の進行および予後に強く関係している。IL−8は、新血管形成を促進し、好中球および内皮細胞の走化性を誘導することによって、癌において効果を発揮し得る。さらに、IL−8の過剰発現は、多数のヒト癌タイプにおいて薬抵抗性表現型の発生と相関していた。
マトリゲルプラグアッセイを用いて、抗LPAモノクローナル抗体のうち1つ(B7)を、インビボで血管形成を軽減させるその能力について試験した。このアッセイでは、マウス腫瘍に由来する基底膜を含めた腫瘍残遺物の専有混合物であるマトリゲルを使用する。マトリゲル、またはその由来物である低成長因子型(growth factor−reduced、GFR)マトリゲルを動物に皮下注射すると、固体化して「プラグ」を形成する。配置前に血管形成促進因子をマトリックスと混ぜておくと、プラグが血管内皮細胞に侵入され、最終的に血管が形成される。マトリゲルは、単独で、または組み換え成長因子(bFGF、VEGF)あるいは腫瘍細胞と混合して調製し、6週齢メスヌード(NCr Nu/Nu)マウスの脇腹に皮下注射することができる。本実施例では、Caki−1(腎癌)細胞をマトリゲル内に導入して、VEGFおよび/またはIL8および十分なLPA量を産生している。マトリゲル移植1日前から3日ごとに食塩水または10mg/kgの抗LPAモノクローナル抗体−B7で処理したマウス由来のCaki−1細胞を5x105個含有しているマトリゲルプラグを作成した。プラグを内皮CD31について染色した後、プラグ内に形成された微小脈管構造の定量化を行った。定量化データは、3つのプラグより1断面当たり少なくとも16個の視界の平均+/−SEMで行った。CD31の内皮染色による検定から、抗LPAモノクローナル抗体B7で処理したマウスのプラグは、食塩水で処理したマウスのプラグと比較して、顕著な血管形成の減少を示した。染色した血管の定量化では、食塩水で処理した動物と比較して、モノクローナル抗体B7で処理した動物のCaki−1含有プラグにおいて血管形成が50%以上低下したことが示された。これは、抗LPAモノクローナル抗体処理の結果としての腫瘍細胞血管形成における統計的に有意な低下である(食塩水に対するモノクローナル抗体B7についてスチューデントのT検定による判定はp<0.05)。
抗LPA抗体は、LPA誘導による腫瘍細胞の増殖、移動、細胞死に対する保護、サイトカイン放出を減少させる効果を持つことが多数のヒト腫瘍細胞株において(上記で)確認された。今度は、モノクローナル抗体B58、B7を腎膵癌の異種移植片モデルにおいて試験した。抗LPA抗体アプローチの潜在的な抗腫瘍形成効果を示す予備的結果を以下に記述する。
抗LPAモノクローナル抗体(B58、B7)は、循環している血管形成促進性サイトカインの量にも影響を及ぼした。抗LPAモノクローナル抗体7(Panc−1)で処理した動物では、抗体で処理したいずれの動物においてもインターロイキン8(IL−8)の血清中の量は検出できなかったのに対して、Panc−1、Caki−1の異種移植片においては、それぞれ85日後、63日後にIL−8の血清中の量が検出可能だった。より重要なことに、腫瘍の大きさとIL−8量の間には強い相関(r=0.98)があった。さらに、Caki−1腫瘍を持つ動物においてヒトIL−8の血清中の量が、食塩水による処理(r=0.55)と比較して、抗LPAモノクローナル抗体58の処理により減少した(r=0.34)。上記のごとく、循環しているサイトカインの量の低下は、腫瘍細胞自体からのサイトカイン放出が直接抑制されていることに起因しているとされている。これらのデータにより、抗LPAモノクローナル抗体には腫瘍の進行を減少させるとともに、循環している血管形成促進性化合物の量も減少させる能力があることが示されている。
腫瘍進行の重要な特徴の一つは、腫瘍は、転移して二次的な腫瘍結節を遠隔部位に形成する能力を持っているということである。本明細書に記載のインビトロ研究では、腫瘍細胞が接触阻害を逃れるように誘導して移動を促進するLPAの能力を細胞運動についてのスクラッチアッセイによって実証した。これらの研究において、抗LPAモノクローナル抗体はLPAの腫瘍増殖を促進する作動体も抑制した。インビボで腫瘍転移を抑制する抗LPAモノクローナル抗体の効果も評価された。腫瘍転移の現象を動物モデルにおいて模倣するのは困難である。多くの研究者は、腫瘍細胞を直接血流中に注射する「実験」転移モデルを用いる。
血管数が増加することは、循環に到達するまで細胞がより短い距離を移動することを意味するため、血管形成は転移において不可欠な過程である。抗LPAモノクローナル抗体が転移過程における複数の不可欠なステップを遮断できるという所見に基づくと、抗LPAモノクローナル抗体は、インビボで腫瘍細胞転移を抑制すると思われる。
高転移性マウス黒色腫(B16−F10)を用いて、3つの抗LPAモノクローナル抗体が転移に及ぼす治療効果をインビボで検討した。このモデルは、オートタキシンのcPA系阻害剤に対して高度な感受性を示した。4週齢のメス(C57BL/6)マウスに、B16−F10マウスの黒色腫腫瘍細胞(動物当たり5x 104個の細胞を100uL)を尾静脈経由で注射した。マウス(1群10匹)に25mg/kgの抗LPAモノクローナル抗体(B3またはB7のいずれか)あるいは食塩水を3日おきに腹腔内注射で投与した。18日後、肺を採取して分析した。肺器官は、黒色腫細胞にとって好ましい転移部位であるため、転移結節について精密に検討した。膨張と固定を同時に行うために、気管を経由して10%緩衝ホルマリンで肺を膨らませ、転移巣が小さくても組織学的検査によって検出可能になるようにした。肺を5つの葉に分けて、解剖顕微鏡下で腫瘍を大きさによって分類して(大 ≧5 mm、中 1−4 mm、小 <1 mm)数えた。肺を検査してみると、腫瘍数は抗体で処理した動物において明らかに減少していた。モノクローナル抗体B3で処理した動物については、大きい腫瘍が21%、中程度の腫瘍が17%、小さい腫瘍が22%減少していた。スチューデントのT検定による統計分析では、食塩水に対してモノクローナル抗体B3で処理した動物における小さい腫瘍の数についてp<0.05を得た。
ハイブリドーマ由来の3つのマウス抗体のうち一つの、活性のあるLPA結合領域を含む可変領域(Fv)とヒトIgG1免疫グロブリンのFc領域とを使用して、LPAに対するキメラ抗体を生成した。Fc領域はヒト抗体のCH1、CH2、CH3ドメインを含んだ。方法は特に限定するわけではないが、キメラ抗体は、ヒトIgG1、IgG2、IgG3、IgG4、IgA、IgMのいずれかのFc領域から産生することもできる。「ヒト化」抗体は、IgG1のフレームワーク領域などのヒト抗体フレームワーク領域(例えば、Fr1、Fr4など)とマウス抗LPAモノクローナル抗体の相補性決定領域(CDR、例えば CDR1〜4)を移植することによって生成することができることは、当業者なら理解されよう。
表5:キメラ抗LPA抗体B3、B7、B58の結合特性
ハイブリドーマ細胞株の可変領域のクローン化
抗LPAハイブリドーマ細胞株のクローンをDMEM(GlutaMAXTM I、4500mg/LのDグルコース、プルビン酸ナトリウム入りダルベッコのダルベッコ変法イーグル培地、ギブコ・インビトロジェン、カルスバッド、カリフォルニア州、111−035−003)、10% FBS(無菌胎児クローンI、Perbio Science)、1X グルタミン・ペニシリン・ストレプトマイシン(ギブコ・インビトロジェン)で増殖させた。RNeasy Mini kit(キアゲン、ドイツ・ヒルデン)に基づいた手順を用いて全RNAを107個のハイブリドーマ細胞から単離した。SMART RACE cDNA Amplification Kit(クローンテック)の製造元による手順に従ってRNAを用いて第1鎖cDNAを生成した。
インビトロジェン社のPfx DNA ポリメラーゼ・キットを10X 緩衝液、50mM MgSO4(カタログ番号11708−013)、10mM dNTP(インビトロジェン、カタログ番号18427−013)と共に使用してポリメラーゼ連鎖反応(PCR)を行った。反応混合液は、10X pfx 増幅用緩衝液を5ul、10mM dNTPを1.5ul、50mM MgSO4を1ul、オリゴヌクレオチド1を1.5ul、オリゴヌクレオチド2を1.5ul、テンプレート(〜50ng)を0.5ul、Pfx DNA ポリメラーゼを0.5ul、無菌水を38.5ul含んだ。Pfxを除いた全ての試薬を添加した後、サーマルサイクラーを開始する直前にPfxを添加した。テンプレートを95℃で3分間変性した後、95℃で30秒間、5℃の +/−勾配で58℃においてアニーリング、68℃で30秒間の伸展のサイクルを35回行った。68℃で5分間最終伸展した後、試料を4oCに保った。
ライゲーション用、または分子配列を確認する前のクローニング確認用のフラグメントを調製するためにDNAに対して制限消化を行った。制限酵素はすべてインビトロジェンまたはニュー・イングランド・バイオラボより購入したが、各酵素には必要な対応緩衝液がついてくる。DNA(通常、陽性クローンの確認には5〜10ul、連結するDNAには20〜26ul)を酵素緩衝液、制限酵素0.5〜1.0ul、無菌水と混合した(合計30ulの反応)。酵素に適した温度で反応を1時間インキュベートした。ほとんどの酵素が37℃で活性があったが、酵素よってはインキュベーション温度が室温から55℃の範囲で変動することもあった。適切に制限酵素消化した後、GeneCleanキットを用いてアガロースゲルや酵素、緩衝液などから挿入フラグメントおよびベクターを除去した。ライゲーションは、T4 DNA 2X連結緩衝液、5X DNA希釈緩衝液、T4 DNAリガーゼを含有するロシュ社のRapid Ligation Kit(カタログ番号11635379001)を用いて行った。最良の結果を出すために3:1の最終モル比でインサートとベクターを結合した。効率的なライゲーションのために挿入フラグメントを適切に希釈した。反応の5〜7ul を用いてE.coli TOP10 化学的コンピテント細胞を形質転換させた。
マイクロタイターELISAプレート(コスター、カタログ番号3361)を37oCで1時間、1M 炭酸緩衝液(pH 9.5)で希釈したウサギ抗マウスIgGのF(ab’)2フラグメント特異性抗体(ジャクソン、315−005−047)で覆った。プレートをPBSで洗浄し、PBS/BSA/Tween−20で1時間、37℃でブロッキングした。一次インキュベーションでは、非特異性のマウスIgGまたはヒトIgG、分子全体(検量線に使用)、測定する試料を希釈したものをウェルに添加した。プレートを洗浄し、ウェル当たり100 ulの1:50,000希釈のHRPコンジュゲート化抗ヒト(ジャクソン 109−035−003)を1時間、37℃でインキュベートした。洗浄後、酵素反応をテトラメチルベンジジン(シグマ、カタログ番号T0440)で検出し、1 M H2SO4を添加することにより停止した。Thermo Multiskan EXを用いて吸光度(OD)を450 nmで測定した。生データをGraphPadソフトウェアに移して分析した。
マイクロタイターELISAプレート(コスター、カタログ番号3361)を1M炭酸緩衝液(pH 9.5)に希釈したLPA−BSAで1時間、37℃でコーティングした。プレートをPBS(137 mM NaCl、2.68 mM KCl、10.1 mM Na2HPO4、1.76 mM KH2PO4、pH 7.4)で洗浄し、PBS/BSA/Tween−20により室温で1時間または4 oCで一晩ブロッキングした。試験する試料を0.4 ug/mL、0.2 ug/mL、0.1 ug/mL、0.05 ug/mL、0.0125 ug/mL、0 ug/mLに希釈して、各ウェルに100 ul 添加した。プレートを洗浄し、ウェル当たり100 ulの1:50,000希釈のHRP抗ヒト(ジャクソン 109−035−003)を1時間、37℃でインキュベートした。洗浄後、酵素反応をテトラメチルベンジジン(シグマ、カタログ番号T0440)で検出し、1 M H2SO4を添加することにより停止した。Thermo Multiskan EXを用いて吸光度(OD)を450 nmで測定した。生データをGraphPadソフトウェアに移して分析した。
293フェクチンを用い、培養に293F−FreeStyle Mediaを用いて、ヒト胎児由来腎臓細胞株293Fをベクターでトランスフェクションした。トランスフェクションは、5 μg/mLで、106細胞/mLの細胞密度で行った。トランスフェクションより3日後に、1100 rpm、25℃、5分間の遠心分離によって上清を採取した。発現量は定量的 ELISAによって定量し、結合は上記のように結合 ELISAにおいて測定した。
常法でマウスのVHおよびVLドメインを配列決定した。5つのマウス抗LPAモノクローナル抗体のクローンについて、マウスのVHおよびVLドメインの核酸およびアミノ酸配列を表8〜17に示す。各CDRH1アミノ酸配列について、Kabatにより定義されたCDRは、示された10アミノ酸の配列である。Kabat配列の太字で示された5アミノ酸の部分は、標準のCDRH1配列である。
マウス抗体クローンB7は、リード化合物として選ばれ、LpathomabTMと改名され、LT3000としても知られている。上記のように、このマウス抗LPAモノクローナル抗体は、タンパク質で誘導体化したLPA免疫原によるマウス免疫化後のハイブリドーマ細胞株に由来している。有利な特性を持つハイブリドーマ細胞株が同定されたため、これを使用して、標準的なハイブリドーマ培養法を用いてモノクローナル抗体を産生した。
- 腫瘍細胞の増殖、移動、化学療法剤に対する感受性への直接的作用
- 抗血管形成作用による腫瘍への間接的作用
- さらに、相乗的な血管形成促進成長因子の放出妨害および中和による腫瘍への間接的作用
- 線維芽細胞の増殖、移動、筋線維芽細胞表現型への形質変換および筋線維芽細胞によるコラーゲン産生への直接的作用
- 相乗的な血管形成促進性、炎症促進性、線維促進性の成長因子の発現および放出を防ぐことによる組織線維症への間接的作用
Lpathomab/LT3000は、情報伝達性脂質LPA高親和性(1〜50 pMのKD)を持ち、さらに、LT3000はLPAに対して高い特異性を示すが、100個を超える様々な生理活性脂質および生理活性タンパク質には結合親和性を示さない(その一部は構造的に類似している)。マウス抗体は、2本の同じ軽鎖と2本の同じ重鎖により構成され、合計分子量が144 kDaの全長IgG1kのアイソタイプ抗体である。生物物理学的特性を表28にまとめた。
表28:Lpathomab(LT3000)の一般的特性
LPAの多面的効果は、細胞外LPAの利用能性(有効濃度)が低くなったことにより(i)原発腫瘍の増殖、転移、血管形成を減少させること、(ii)LPAの腫瘍に対する保護的な抗アポトーシス効果の作用を中和することを示唆している。LpathomabTM/LT3000のLPAとの強力かつ特異的な結合により、癌の前臨床モデルにLT3000のインビボ処理を行うと、様々な治療上の利点が生まれるであろうと仮定した。
- インビボでの種々のヒト腫瘍異種移植モデルにおける腫瘍増殖の抑制、
- インビトロでのLPAに依存するヒト腫瘍および内皮細胞株の細胞増殖や浸潤の低下、
- インビボでの腫瘍血管形成の減少に伴う、IL6、IL8、GM−CSF、MMP2、VEGFを含む腫瘍形成・血管形成成長因子の循環量の低下、
- 転移する可能性の低下、
- 腫瘍細胞死に対するLPA誘導保護の中和。
- OVCAR3卵巣癌細胞の増殖の低下、
- インビトロでのPanc−1(膵臓)、OVCAR3、SKOV3(卵巣)腫瘍細胞によるIL−8、IL−6、VEGFのLPA誘導放出の中和、
- アポトーシスに対してSKOV3、Panc−1腫瘍細胞を保護するLPAの作用の軽減(このことは、標準の化学療法剤と併用すると効力が増大することを示唆している)、
- 腫瘍によるIL−8、IL−6、VEGFのLPA誘導放出の中和、
- LPA誘導の腫瘍細胞の移動、増殖、化学療法剤からの保護の抑制、
- 各種インビトロアッセイにおけるLPA誘導の内皮細胞管形成、移動、細胞死からの保護の中和。
- ヌードマウスに移植した、SKOV3(卵巣、下記実施例を参照)、COLO205(結腸直腸、下記実施例を参照)、DU145(前立腺)、B16 F10(マウス黒色腫、下記実施例を参照)、ルイス肺癌細胞(下記実施例を参照)を含む複数の同所性および皮下ヒト腫瘍の進行の抑制、
- 皮下SKOV3異種移植モデルおよび前立腺DU145癌細胞における腫瘍関連血管形成の劇的な減少、
- マウスマトリゲルプラグアッセイにおけるbFGF誘導およびVEGF誘導の血管形成の中和(下記実施例を参照)、
- 眼のレーザー誘導ブルーフ膜傷害モデルにおける脈絡膜の新血管形成の減少(下記実施例を参照)。
LT3000を腹腔内(IP)注射でq2d投与したところ、メスC57BL/6マウスに移植したマトリゲルプラグのFGF誘導およびVEGF誘導の血管形成が減少した。この研究は、サザン研究所(アラバマ州バーミンガム)で実施された。
メスC57/BL6マウスに移植したFGF添加およびVEGF添加のマトリゲルプラグの血管形成を遅延させるLT3000の抗血管形成効果を評価すること。
(試験デザイン)
マトリゲルのみ、あるいはbFGF、VEGFのいずれかを添加したもの(N=マウス5匹/投与群)を各マウスの脇腹に皮下(SC)注射した。マトリゲルプラグを移植する前の日に、10 mg/kgのLT3000または食塩水のIP投与による処理を開始した。処理は1日おきに(q2d)行った。殺してから、マトリゲルプラグを採取し、CD−31染色による微小血管密度(MVD)分析用に処理した。
食塩水で処理したマウスと比較してLT3000で処理したマウスでは、bFGF添加およびVEGF添加のプラグにおいて微小血管密度が約41%低下した。この低下は、統計的に有意(p<0.0001)でありCD31染色により組織学的に確認された。
(結論)
この研究は、全身投与したLT3000の抗血管形成効果によって、bFGFおよびVEGFを添加したマトリゲルプラグの新血管形成が有意に減少したことを示す。
LT3000を硝子体内注射により投与したところ、メスC57BL/6マウスにおけるレーザー誘導ブルーフ膜傷害モデルで脈絡膜の新血管形成が減少した。この研究は、ゲインズビル、フロリダ大学(Maria Grant医師の研究室)で実施された。
脈絡膜の脈管構造中の新血管形成を制限するLT3000の効力を研究すること。
(試験デザイン)
マウスをレーザー誘導のブルーフ膜破裂に付した。マウスを抗LPA抗体LT3000 0.5 μg、アイソタイプが一致した非特異性モノクローナル抗体(NSA)0.5 μg、同量の食塩水のいずれかで処理した。
処理は、レーザー破裂後、研究期間中週1回(q7d)硝子体注射して行った。ブルーフ膜の破裂から2〜4週間後にマウスを殺し、眼球を採取した。RPE・脈絡膜・強膜複合体を神経網膜から単離し、ローダミンコンジュゲート化トウゴマ凝集素Iで染色することによりCNVを評価した。判定はすべて動物当たり2〜3回の焼却について行った。
NSA処理したマウス(n=4)と比較して、LT3000で処理したマウス(n=5)においてCNV傷害の血管形成が2185014±377010(平均CNV体積±SEM)から697924±92182に減少した。これは、NSA処理したマウスと比較してLT3000処理したマウスでは68%の減少である(p≦0.05)。
(結論)
この研究は、抗LPA抗体の硝子体内投与が、傷害に応答した脈絡膜組織中での新血管形成を有意に低下させることを示す。
多数のヒト腫瘍異種移植モデルおよびげっ歯類同系モデルにおける研究により、LT3000が抗腫瘍形成活性を示すことを確認した。
LT3000をIP注射により投与した(10 mg/kg q3d)ところ、ヌードNcrマウスにおける同所性SKOV3卵巣腫瘍モデルにおける腫瘍の進行が大いに抑制された。この研究は、Lpath社によって実施された。
メス無胸腺症ヌードマウスの腹腔に移植したヒト卵巣(SKOV3)腫瘍の進行を遮断するLT3000の効果を評価すること。
(試験デザイン)
ヌードマウスの腹腔内にSKOV3腫瘍細胞を移植した。腫瘍が成立したら、IPで、10 mg/kgのLT3000をq3dで、PBS(媒体)をq3dで、毎日15mg/kgのパクリタキセル(タキソール)を4日間のいずれかでマウスを処理した。52日目の研究終了時、血清および腹水液を採取して、サイトカイン量について分析した。腫瘍を収穫して腫瘍の最終重量を量った。
この研究は、SKOV3腫瘍の進行を減少させるLT3000の能力を示す(表29)。10mg/kgのLT3000によって、腫瘍組織量は統計的に有意に減少(50%)した。予想どおり、PBSで処理した動物と比較して、パクリタキセルで処理した動物は、腫瘍組織量に大きな減少(88%)を示した。本実験において、腹腔内に腹水液が確認された動物の数は、LT3000群(6/14)の方がPBS対照群(11/13)より少なかった。さらに、PBSで処理した動物と比較して、LT3000群において腹水の蓄積容積に統計的に有意な減少が確認された。また、媒体で処理した動物と比較して、LT3000は、血管形成促進性サイトカインIL−8、IL−6、GM−CSF、VEGFの血清中濃度の低下を誘導した。最後に、LT3000は、腹水中MMP2(ヒトおよびマウス)の総量を低下させた。H&EおよびCD31染色のために選択した腫瘍切片を認定病理学者に分析させた。LT3000処理群において組織分裂(網、骨格筋、リンパ節)の低下、微小血管密度の低下が確認された。
(結論)
本研究は、抗LPA抗体の全身投与が、SKOV3腫瘍進行に有意な抑制をもたらすことを示す。
LT3000をIP注射(q3dで20 mg/kg)により投与したところ、ヌードマウスでの静脈内ルイス肺腫瘍における腫瘍進行が大きく抑制された。この研究は、Lpath社によって実施された。
LT3000は、インビトロで腫瘍細胞移動を低下させること、インビボで内皮細胞の浸潤や血管形成を有意に減少させることが確認されている。この研究の目的は、ルイス肺癌細胞を静脈内(IV)接種されたメスNcr(nu/nu)マウスの肺において腫瘍の転移を遅延させるLT3000の効果を評価することである。
肺に播種して腫瘍を発現させるためにルイス肺癌細胞をヌードマウスにIV接種した。20 mg/kgのLT3000または媒体(食塩水)による処理を同日に開始した。19日目の研究終了までマウスをq3dで処理した。研究終了時に生存動物を殺し、体重および肺重量を(腫瘍組織量の測定として)記録した。
(結果)
この研究は、抗LPA抗体の全身投与が、肺腫瘍組織量の減少、体重の増加、低いLW/BW比という結果をもたらすこと示す。
LT3000をIP注射(q3dで30 mg/kg)により投与したところ、ヌードNcrマウスにおいて皮下COLO205(結腸直腸)腫瘍異種移植片における腫瘍進行が抑制された。この研究は、サザン研究所(アラバマ州バーミンガム)によって実施された。
メスNcr(nu/nu)マウスに皮下(sc)移植して成立させたヒト結腸直腸(COLO205)癌腫瘍の進行を遅延するLT3000単独の効果を評価すること。
(試験デザイン)
COLO205腫瘍片をヌードマウスにsc移植し、腫瘍を成立させた。その後マウスを30 mg/kgのLT3000、40 mg/kgのアバスチン(商標)、15 mg/kgのパクリタキセル(商標)、媒体(食塩水)のいずれかで処理した。LT3000は3日ごとに(q3d)IP注射により投与し、アバスチン(商標)は7日ごとに(q7d)投与し、パクリタキセルは5日間毎日(q1dx5)ip注射により投与した。研究中の腫瘍の増殖は、腫瘍を3軸に沿って測定し、重量を計算することによってモニターした。
食塩水で処理した動物の腫瘍と比較して、LT3000は有意に(p<0.012)腫瘍進行を24%抑制した。研究終了時に、LT3000は腫瘍の最終重量を低下させるのにアバスチンと同様の(p<0.002)効果を示した(それぞれ32%対24%の減少)。陽性対照であるパクリタキセルは、すでに定着していた腫瘍を除去した。
(結論)
この研究は、抗LPA抗体の全身投与によってCOLO205腫瘍細胞の腫瘍進行が抑制できることを示唆した。
表31:LT3000の抗腫瘍形成活性を評価するためのマウスCOLO205異種移植モデルについての所見の数値のまとめ
このモデルは、LT3000を単独で50 mg/kg 、q3dで腹腔内注射により投与した処理に対するマウス黒色腫C56Bl/6マウスの応答を測定した。この研究は、Lpath社により実施された。
マウス黒色腫細胞株B16−F10によって誘導された肺転移の進行を減少させるLT3000の効果を評価すること。
(試験デザイン)
B16−F10腫瘍細胞の懸濁液をC57BL/6マウスに尾静脈より静脈内(IV)注射した。無作為化した後、動物を2群に分けて媒体(食塩水)または10mg/kgのLT3000をIPによってq3dで投与することによって処理し、細胞接種日を開始した。20日後、動物を殺して、心臓穿刺により血漿試料を採取し、肺を単離した。肺の最終重量を量り、体重と相関させた。また、各動物の腹腔および器官(肝、胃、卵巣、腸など)も転移病巣の存在について分析した。
表32に示すように、食塩水で処理したマウスに対し、LT3000で処理したマウスでは肺の転移容積が27%減少した。
(結論)
この研究は、抗LPA抗体の全身投与によってB16−F10肺転移の低減をもたらすことができることを示す。
細胞培養および試薬
WI−38ヒト肺線維芽細胞はATCC(バーモント州マナサス)より購入した。肺線維芽細胞は、10%ウシ胎児血清(FBS)、ペニシリン/ストレプトマイシン(100 単位/ml)を添加した最小必須培地において370C、5% CO2で保持した。a平滑筋アクチン(a−SMA)およびFAK Y397抗体はシグマ(ミズーリ州セイントルイス)より購入した。LPAはAvanti Polar Lipids社(アラバマ州アラバスター)より購入し、製造元の勧告に従って調製した。
RNA分離用に、単一のコンフルエントになったT150フラスコよりトリプシンを用いて細胞を取り出し、遠心によってペレット化して−80oCで凍結した。全RNAの単離は、Qiagen RNeasy Mini Kit(キアゲン、カリフォルニア州バレンシア)を用いて製造元の動物細胞における全RNA単離手順に従い実施した。つまり、RT−PCR用Superscript III First−Strand Synthesis System(インビトロジェン、カリフォルニア州カルスバッド)を用い、製造元の手順に従ってランダムヘキサマーを第1鎖プライマーとして用いて、2マイクログラムの全RNAを使用して第1鎖cDNAを作成した。LPA1-3受容体用のオリゴを用いて2マイクロリッターの第1鎖cDNAを増幅し、各反応においてGAPDHをコントロールとした。PCRは、Platinum Pfx DNA Polymerase(インビトロジェン、カリフォルニア州カルスバッド)を用いて設定した。その後PCR産物は1%アガロースゲル上で泳動し、エチジウムブロマイド用フィルターとUVP BioImaging Systems EpiChemi3 Darkroom(UVP Inc、カリフォルニア州アップランド)を用いて撮影した。
肺線維芽細胞を5x103細胞/ウェルの密度で96ウェルプレートに一晩播いた。播いた細胞を48時間基本培地(最小必須培地、0.1%脂肪酸フリーBSA、100単位/mLのペニシリン・ストレプトマイシン)で血清不足にした後、基本培地のみ(対照)または指示された濃度のLPAを含んでいる基本培地で72時間刺激した。細胞増殖は、製造元の手順に従ってCell Titer 96 Aqueous 細胞増殖アッセイ(プロメガ、ウィスコンシン州マディソン)を使用して測定した。吸光度は、OD450で測定し、データは対照に対する変化の倍数によって表した。吸光度の測定は4回実施した。コラーゲン産生については、I型コラーゲンのC末プロペプチド(PICP)の馴化培地での濃度を、PICP酵素免疫吸着分析(ELISA)キット(タカラバイオケミカルズ、大阪、日本)によって製造元の手順に従って測定した。α−SMA発現については、前記の{Micera、2005 #8093}のようにして細胞に基づいたELISAを実施したが、以下の変更を加えた。細胞を10%中性緩衝ホルマリンにて固定し、PBS/0.1%トリトンX−100で透過化して、内生ペルオキシダーゼを0.3% H2O2でクエンチした。細胞単層をPBS/10% FBSでブロッキングした後、α−SMAに対する一次抗体をPBS/1% BSA/0.1% Tween 20で希釈(1:1000 希釈)して細胞と4oCで一晩インキュベートした。一次抗体とのインキュベーション後、プレートをPBS/0.1% Tweenで3回洗浄し、PBS/1% BSA/0.1% Tween 20で希釈(1:1000 希釈)したHRPコンジュゲート化ヤギ抗マウス抗体と室温で1時間インキュベートした。プレートをPBSで4回洗浄し、TMB比色溶液に1〜3分間インキュベートした。同量の1M H2SO4を用いて反応を中止し、吸光度をプレートリーダーにより450 nmで読み取った。細胞増殖、コラーゲン生成、α−SMA発現のアッセイはすべて3回ずつ実施した。
肺線維芽細胞を1.5 x 104細胞/ウェルの密度で96ウェルプレートに一晩播いた。播いた細胞を24時間、基本培地(最小必須培地、0.1%脂肪酸フリーBSA、100単位/mLのペニシリン・ストレプトマイシン)で同期化させた。0時間の時点で細胞をp200ピペットチップで各ウェルの中央に沿ってスクラッチし、最少培地で洗浄し、処理する前に撮影した。その後細胞を濃度0.1〜10 μMのLPA(C18:1)または陽性対照(10% FBS)で処理した。5% CO2インキュベーターにおいて細胞を37oCで17時間刺激した。17時間処理した後、再度撮影し、写真を同サイズに調節して0時間と17時間でのスクラッチの幅を測ることによって傷の閉鎖率(%)を求めた。
LPAは肺線維芽細胞による増殖および移動を刺激する LPAは創傷治癒の制御に関与している。そのため、創傷の修復に寄与している細胞機構の2つである線維芽細胞の増殖および移動に対するLPAの用量依存的な作用を検討した。LPAは線維芽細胞の増殖を用量依存的に刺激し、10 μM LPAで最大の上昇が確認された。細胞移動に対するこのLPAの作用は、インビトロ創傷治癒アッセイでも検討した。用量依存的な細胞増殖の上昇とは異なり、LPAは低い(0.1 μM)LPA濃度で細胞移動を刺激するようであった。LPA濃度を増加させると、細胞移動が用量依存的に基礎レベルまで低下することが確認された。このデータは、肺線維芽細胞の増殖に対するLPAの濃度依存的作用と肺線維芽細胞の移動の間には反比例の関係があることを示唆している。
肺でLPAの線維化促進性の可能性を評価するために、肺線維芽細胞による筋線維芽細胞形質変換およびコラーゲンI型産生のLPAを介した刺激を、ELISAおよび免疫組織化学を用いて検討した。LPAはα−SMA(筋線維芽細胞マーカー)の発現およびプロコラーゲンI型C末ペプチド(PICP)の放出を用量依存的に促進し、10 μM LPA濃度で最大の刺激をもたらした。さらに、LPAは、α−SMAの細胞骨格張力繊維への取り込みを増加させ、接着斑キナーゼ(FAKY397)のリン酸化を刺激するが、これらは、筋線維芽細胞形質変換に必要とされる現象である。一貫して、これら形質変換した細胞は、LPA刺激後にコラーゲンI型の細胞発現の増加も示しており、このことは線維化促進性の細胞表現型へ形質変換したことを示す。
動物
20〜25 gのC57BL/6Jメスマウスをハーランより入手した。Bioquant社(カリフォルニア州サンディエゴ)の研究機関における動物の管理および使用に関する委員会(Institutional Animal Care and Use Committee、IACUC)に従って動物を処理した。マウスを空調された12時間の明暗周期の部屋に入れ、標準的な固形飼料と水道水へ自由にアクセスできるようにした。動物には通常の固形飼料(オートクレーブ済み)と水の不断給を提供した。
ブレオマイシンによる肺傷害の誘導
ケタミン(20 mg/kg)とキシラジン(2 mg/kg)の混合でマウスに麻酔をかけ、20ゲージの供給針を介して食塩水(0.9%)のみ、またはブレオマイシン(2.5 mg/kg)含有食塩水の気管内滴下(容積50 μl)を1回行った。14日間後にマウスを殺した。
マウスを無作為的に次の投与群に割り当てた。(i)食塩水。マウスに食塩水を気管内(IT)滴下して、無菌PBS(媒体)のi.p注射を打った。(ii)BLEO群。マウスにブレオマイシンをIT滴下して、無菌PBS(媒体)のi.p注射を打った。(iii)BLEO + 25 mg/kg群。マウスにブレオマイシンをIT滴下して、LT3000(25 mg/kg)のi.p注射を打った。(iv)25 mg/kg群。マウスに食塩水をIT滴下して、LT3000(25 mg/kg)のi.p注射を打った。抗体処理はすべて腹腔内(i.p.)注射により2日ごとに行った。各処理の体重およびマウス死亡率に対する効果を研究期間中記録した。
マウスを殺した後、1 mlの注射器に接続した20ゲージの血管カテーテルでマウス挿管した。気管の周辺にナイロン糸を結ぶことによってカテーテルを固定した。肺を1.0 mlの食塩水で一回洗浄した後、0.8 mlの食塩水で再度洗浄した。洗浄液を合わせ、BALFを1200 rpmで5分間遠心分離にかけた。上清を取り出し、後の分析用に−80 oCで凍結した。ペレットを0.3 mlのPBS/2%ウシ胎児血清で再懸濁し、流動細胞計測法により細胞を分類して数えた。BALF中のタンパク質量はBCAタンパク質定量試薬(ピアス、イリノイ州ロックフォード)を使用して測定した。
Trucountチューブ(BD Biosciences、カタログ番号340334)を製造元の手順に従って用いて個々の炎症細胞集団を同定し、定量化した。つまり、単細胞の懸濁液を染色緩衝液(PBS/2% FCS)で調製した。約300 μlの細胞懸濁液を12 x 75 ポリプロピレンTrucountチューブに入れた。チューブを250〜300 x g、4℃で5分間遠心分離にかけた。ピペットを用いてペレットを乱さないよう注意しながら液体を吸引した。各チューブに次のモノクローナル抗体を添加した。PEコンジュゲート化抗CD16(BD Biosciencesカタログ番号555407)、FITCコンジュゲート化抗CD14(BD Biosciencesカタログ番号555397)、PE−Cy5コンジュゲート化抗CD5(BD Biosciencesカタログ番号555354)で3色カクテルを得た。抗体の量は製造元の指示に従った。チューブをボルテックスして蓋をしたバケツ内(暗所で)で氷上に約30分間放置した。2 mlの染色緩衝液を添加することによって懸濁液を洗浄した。懸濁液をボルテックスした後、250〜300 x g、4℃で5分間遠心分離にかけて上清を削除した。工程6を2回繰り返した。その後ペレットを1 mlの染色緩衝液で再懸濁し、個々の細胞集団を蛍光補助式細胞分得(Fluorescence Assisted Cell Sorting、FACS)分析によって分析した。FACS分析については、染色した細胞を、BD FACScan(カリフォルニア州サンノゼ)を用いて1つのレーザー(488 nm アルゴン レーザー)と3つの検出器で流動細胞計測法により分析した。10,000個の細胞を収集し、データをCellQuest version 3.3ソフトウェアで分析した。
肺を切除し、個々の葉に分離し、10%緩衝ホルマリンにて室温で一晩固定した。個々の葉を3枚の水平切片に切断し、パラフィンに包埋した。葉は厚さ5 μmに切断し、ヘマトキシリン・エオシン(H&E)によって染色した。切片はすべて光学顕微鏡(倍率10x)で観察し、線維症の重症度を前記したとおり(Ashcroftら、(1988)J Clin Pathol、41:467−70)Ashcroft法を用いて半定量的に評価した。つまり、各葉の水平切片における線維症の重症度を、0〜8の尺度でスコア化した。評価基準は以下の通りだった。程度0:正常な肺、程度1:肺胞壁または細気管支壁の極小線維性肥厚、程度3:構造に明確な損傷がない中等度の壁肥厚、程度5:肺構造に明確な損傷と線維性の帯または線維性の小塊の形成を伴う線維症の増加、程度7:構造の重度の歪みおよび大きい線維性領域、程度8:線維による視界の完全閉塞。各葉の個々の切片の値を平均し、すべての葉の値を平均することにより、各動物について代表的な線維症スコアを出した。
肺切片を各5分間のキシレン(Richard Allen Scientificカタログ番号9900)洗浄3回によって脱パラフィン化した。スライドの再水和は、100%アルコール(Richard Allen Scientificカタログ番号8101)、95%アルコール(Richard Allen Scientificカタログ番号8201)、80%アルコール(Richard Allen Scientificカタログ番号8301R)の各5分間の一連のアルコール洗浄により実施した。再水和は、水道水の流水下、スライドを5分間洗浄することによって完了した。その後13分間3% H2O2(水にて30%希釈、シグマ、カタログ番号H1009)にて外来ペルオキシダーゼをクエンチした。水道水の流水下、スライドを15分間洗浄することによってH2O2を削除した。一方、クエン酸緩衝液(10mM クエン酸、pH 6.0(フィッシャー、カタログ番号A940))を蒸し器(ブラック&デッカー、Sku番号HS900)に入れて40分間95℃に予熱した。抗原の回収には、スライドを予熱したクエン酸緩衝液に移し、蒸し器で35分間95℃で加熱した。引き続いて、スライドを5分間PBS(セルグロ、カタログ番号21−040−CM)にて2回洗浄した。α平滑筋アクチン(α−SMA)あるいは結合組織成長因子(CTGF)に対してマウスIgGベクタステインABCキット(ベクター社、カタログ番号 6102)またはヤギIgGベクタステインABCキット(ベクター社、カタログ番号 PK−6105)を製造元の手順に従って用い、スライドを染色した。緩衝液を必要とする工程のすべてにPBSを使用した。製造元提案の手順に従ってアビジン・ビオチンブロッキングキット(ベクター社、カタログ番号 SP−2001)をABCキットと併用し、内生ビオチン信号を遮断した。α平滑筋アクチンに対して誘導した一次抗体(シグマ、カタログ番号 A2547)を1:5000希釈したもの、またはCTGCを再度誘導した一次抗体(サンタクルーズバイオテクノロジー、カタログ番号sc−14939)を1:50希釈したものを、指示に従って適用した。信号は、ペルオキシダーゼ基質キットDAB(ベクター社、カタログ番号 SK−4100)を用い、生産者の指示通り調製し、スライドに2分間適用することにより検出した。スライドはdiH2Oで洗浄した。対比染色はヘマトキシリン(シグマ、カタログ番号HHS32)で30秒間染色することにより実施した。スライドを水道水ですすぐことによりヘマトキシリンを除去した後、最初に水和用に使ったアルコールに対するキシレン類を逆にして脱水した。最後に、スライド当たり20 μlのパーマウント(フィッシャー、カタログ番号 SP15−100)を使用してスライドをガラス製のカバースリップでマウントした。
試験は全データが揃うまでデータ収集・分析担当者全員に完全に盲検化されていた。データはGraphPadソフトウェアを用いて分析した。実験群間の差異の統計的有意性は、独立スチューデントt検定によって計算した。
結果
LT3000はブレオマイシン傷害後の炎症および線維症を減少させる
マウスのブレオマイシンモデルを用いて、肺傷害後の肺炎症および線維症におけるLPAの役割と、これらの作用を軽減する新規のモノクローナルマウスLPA抗体(LT3000)の効果を検討した。ブレオマイシン滴下後のマウス肺の組織学的検討を行ったところ、肺胞中隔の肥厚、肺炎、肺実質の線維性閉塞など、肺組織への有意な損傷が明らかになった。LT3000で処理したマウスでは、炎症、線維症の劇的な減少と正常な肺形態の保持が確認された。これらのマウスにおける肺炎症および線維症の半定量的分析を行ったところ、LT3000処理をした場合、これらのパラメータにおいてそれぞれ56%と48%の低下が明らかになった。LT3000単独で処理した健常マウスにおいては、炎症や線維症、正常な肺形態における変化は確認されなかった。
組織の傷害度に合わせて、炎症細胞数は対照のほぼ2倍であり、ブレオマイシン滴下マウスのBAL液におけるタンパク質量は、約10倍と有意に上昇した。対照およびブレオマイシン処理マウスの両方で、LT3000の投与によりBAL液の細胞充実度が約 95%低下した。さらに、BAL液中のタンパク質量はLT3000を投与されたブレオマイシン滴下マウスにおいて40%低下した。対照マウスと比較すると、ブレオマイシン滴下マウスでは、肺傷害度と併せて、16%の体重低下が14日の時点で確認された。それに対して、LT3000処理したマウスで処理したブレオマイシン滴下マウスの体重は対照との有意差を示さなかった。
肺炎症および線維症を減少させるLT3000の能力をさらに研究するために、マウス肺組織における大食細胞の浸潤および筋線維芽細胞の密度を検討した。同様に、α−SMA染色により示される筋線維芽細胞の密度はブレオマイシン滴下マウスの線維性領域で増加していた。さらに、LT3000処理により、ブレオマイシン滴下マウスの肺における筋線維芽細胞の密度も低下した。肺線維症および炎症に対する効果は、上述の方法を用いて、肺線維症(Ashcroftスコア)、炎症(炎症スコア)の半定量的評価に従って確認した。LT3000処理によって、肺線維症および炎症はそれぞれ48%、56%低下した。
上記実施例に概説した所見では、ブレオマイシン誘導肺線維症モデルにおいてLT3000の消炎性と抗線維性の両効果が示された。したがって、ブレオマイシン傷害後に肺線維症の進行に対するLT3000の予防能力または介入能力を評価するための追加研究を行った。この実験では、マウスをランダムで次の投与群に割り当てた(表33)。(i)食塩水。マウスに食塩水を気管内(IT)滴下して、無菌PBS(媒体)のi.p注射を打った。(ii)BLEO群。マウスにブレオマイシンをIT滴下して、無菌PBS(媒体)のi.p注射を打った。(iii)予防群。マウスにブレオマイシンをIT滴下して、LT3000(50 mg/kg)のi.p注射をブレオマイシン滴下と同日から開始してq2dで6日間打った。(iv)介入群。マウスにブレオマイシンをIT滴下して、LT3000(50 mg/kg)のi.p注射をブレオマイシン滴下後から6日目に開始してq2dで打った。抗体処理はすべて腹腔内(i.p.)注射により行った。研究終了時(14日目)に、マウスを殺し、ブレオマイシン誘導の肺炎症および線維症に及ぼすLT3000の効果を下記のように評価した。(i)Aschroftらの方法(J Clin Pathol。1988 Apr、41(4):467−70)を用いて組織線維症をH&E染色した肺切片において半定量的に評価した、(ii)流動細胞計測法を用いて肺洗浄液中の炎症細胞を測定した、(iii)BCAタンパク質定量試薬(ピアス、イリノイ州ロックフォード)を用いて肺洗浄液中のタンパク質量を評価した、(iv)ブレオマイシン滴下後14日目に各マウスの体重を測定した。ブレオマイシン誘導肺炎症および線維症に及ぼすLT3000の効果の数値のまとめを表34に示す。
現在の標準治療となっている生検よりも低い侵襲性で線維症(特に肺および肝の線維症だがこれらに限定しない)をモニターできる方法が長い間望まれていた。研究者らは、疾患のマーカーのモニタリングにより、疾患の進行および/または治療効果をより低い侵襲性でモニターできるようにするために、サイトカインおよび成長因子の循環量と線維症の程度との相互の関連付けを試みた。Moraisら、(2006)Mem Inst Oswaldo Cruz、Rio de Janeiro、第101巻(付録I):353−354を参照。線維症を検出する方法は、LPA量と一つ以上の線維形成マーカー(例えば、サイトカインまたは成長因子)の量を相互に関連付けることにより、患者試料であり、臨床の場において線維症をモニタリングするのに有用であると思われる。
表35:BAL液中のサイトカイン発現量のまとめ
(ND= ブレオマイシン処置群と比較して有意差なし)
LPAは線維症や腎臓疾患に関与している多くの過程を仲介できるため、LPAとその受容体を腎線維症の動物モデルにおいて研究した。一側尿管閉塞(UUO)モデルは、炎症、線維芽細胞の活性化、細胞外基質の蓄積を含め、腎線維症の進行を加速的に模倣する。J.P. Pradere ら、(2007)J. Am. Soc. Nephrol. 18:3110-3118、J.−P. Pradereら、リゾホスファチジン酸と腎線維症[Lysophosphatidic acid and renal fibrosis]、Biochim. Biophys. Acta(2008)、doi:10.1016/j.bbalip.2008.04.001。
材料
3,3’,5,5’−テトラメチルベンジジン液体基質(TMB)はシグマ・アルドリッチ(ミズーリ州セイントルイス)から入手した。脂肪酸フリーウシ血清アルブミン(BSA)はカルバイオケム(カリフォルニア州ラ・ホーヤ)から入手した。固定化プロテインA、固定化パパイン、タンパク質脱塩スピンカラムはピアス(イリノイ州ロックフォード)から入手した。抗ヒトIgG(Fc 特異的)抗体はベチル(テキサス州モンゴメリー)より購入した。参照IgG(非特異性ヒトIgGおよびマウスIgG)、抗ヒトIgG(H+L)西洋わさびペルオキシダーゼコンジュゲートおよび抗マウスIgG(H+L)西洋わさびペルオキシダーゼコンジュゲートはジャクソン・イミュノリサーチ・ラボラトリーズ(ペンシルバニア州ウェストグローブ)より入手した。リゾホスファチジン酸(LPA)および競合ELISAで使用する他の脂質は、アバンティ・ポーラー・リピッズ(アラバマ州アラバスター)より購入した。ビオチン化LPAはエシェロン・バイオサイエンス(ユタ州ソルトレイクシティ)より購入した。
マウスCDRをヒトフレームワーク領域(FR)に移植することによってマウス抗LPAモノクローナル抗体LT3000(Lpathomab)の可変ドメインをヒト化した。Lefranc、M.P、(2003). Nucleic Acids Res、 31: 307−10、 Martin A.C. およびJ.M. Thornton、(1996)J Mol Biol、 1996. 263: 800−15、 Morea V.、 A.M. Lesk およびA. Tramontano(2000)Methods、 20: 267−79、 Foote J. およびG. Winter、(1992)J Mol Biol、 224: 487−99、 Chothia C.ら、(1985)J Mol Biol、186:651−63。
マウス抗体遺伝子をハイブリドーマよりクローニングした。ヒトフレームワーク配列およびマウスCDRを含んでいる合成遺伝子を合成オリゴヌクレオチドより構築し、平滑な制限部位を用いてpCR4Blunt−TOPOにクローニングした。配列決定して100%の配列一致を確認した後、重鎖遺伝子にはpConγベクター、軽鎖遺伝子にはpConκベクター(ロンザバイオロジックス、ニューハンプシャー州ポーツマス)を用いて、軽鎖と重鎖をクローニングし、全長IgG1キメラ抗体として発現させた。これらの各遺伝子の発現カセットには、プロモータ、コザック配列、転写終結区が含まれた。これらのプラスミドで大腸菌(One Shot Top 10化学的コンピテント大腸菌細胞、インビトロジェン、カタログ番号C4040−10)を形質変換させ、LB培地において増殖させ、グリセリンにて保存した。製造元の記載に応じて大規模なプラスミドDNAを調製した(キアゲン、エンドトキシンフリーのMAXIPREPTMキット、カタログ番号12362)。293フェクチンを用い、293F−FreeStyle Mediaを培養に用いてプラスミドをヒト胎児由来腎臓細胞株293Fにトランスフェクションした。トランスフェクションされた培養は、約2〜12 mg/Lのヒト化抗体を発現した。
プロテインAアフィニティー・クロマトグラフィーを用いて培養上清よりモノクローナル抗体を精製した。0.5 mlのProSep−vA−Ultra樹脂(ミリポア、カタログ番号115115827)を含むアリコートを自然落下法用使い捨てカラム(ピアス、カタログ番号29924)に添加し、10〜15 mlの結合用バッファー(ピアス、カタログ番号21001)で平衡化した。一過性発現したヒト化抗体を含む培養上清を結合用バッファーで1:1に希釈し、樹脂に通過させた。カラムに保持された抗体を15 mlの結合用バッファーで洗浄し、低pHの溶出用バッファー(ピアス、カタログ番号21004)で溶出し、pHを中和するために100 ulの結合用バッファーを含む1 mlのフラクションに収集した。吸光度(280 nm)>0.1のフラクションを1リットルのPBS緩衝液(セルグロ、カタログ番号021−030)で一晩透析した(Slide−A−Lyzerカセット、3500 MWCO、ピアス、カタログ番号66382)。透析した試料をセントリコンYM50(アミコン社、カタログ番号4225)濃縮器を用いて濃縮し、0.22 uMセルロースアセテート膜(コスター、カタログ番号8160)でろ過した。SDS−PAGEを用いて各産生物の純度を評価した。
各抗体試料を2−メルカプトエタノール(シグマ、カタログ番号M−3148)含有(還元型)または非含有(非還元型)のゲル添加用バッファー0.5 ug/ulで希釈した。還元試料は95 oCで5分間加熱し、非還元試料は室温でインキュベートした。1レーン当たり2 ugの抗体を4〜2%勾配ゲル(インビトロジェン、カタログ番号NP0322)に添加し、室温で1X NuPAGE MOPS SDS泳動用バッファー(インビトロジェン、カタログ番号NP0001)にて170ボルトで1時間泳動した。泳動後、ゲルを50%メタノール、10%酢酸にて10 分ほど浸漬することにより抗体を固定した。その後ゲルを200 ml 蒸留水で3回洗浄した。最後に、ゲルをGelCode(R) Blue Stain(ピアス、カタログ番号2490)にて一晩染色し、水で脱染することによりバンドを可視化した。
定量的ELISAを用いて抗体価を求めた。ヤギ抗ヒトIgG−Fc抗体(ベチル社A80−104A、1 mg/ml)を炭酸緩衝液(100mM NaHCO3、33.6 mM Na2CO3、pH 9.5)にて1:100希釈した。100 ul/ウェルのコーティング溶液を37℃で1時間インキュベートすることによりプレートをコーティングした。プレートをTBS−T(50mMトリス、0.14 M NaCl、0.05% tween−20、pH 8.0)で4回洗浄し、200 ul/ウェルTBS/BSA(50mMトリス、0.14 M NaCl、1% BSA、pH 8.0)によって37℃で1時間ブロッキングした。試料および標準を2回に検定しても十分な容量で非結合型プレートにて調製した。標準はヒト標準血清(ベチル社 RS10−110、4 mg/ml)をTBS−T/BSA(50 mMトリス、0.14 NaCl、1% BSA、0.05 % Tween−20、pH 8.0)において次の濃度に希釈することにより調製した。500 ng/ml、250 ng/ml、125 ng/ml、62.5 ng/ml、31.25 ng/ml、15.625 ng/ml、7.8125 ng/ml、0.0 ng/ml。試料の吸光度(OD)が標準の範囲内になるようにTBS−T/BSAで適切に希釈することによって試料を調製した。最も線形の範囲は125 ng/ml 15.625 ng/mlであった。プレートをTBS−Tで4回洗浄した後、100ulの標準/試料調製液を各ウェルに添加し、37℃で1時間インキュベートした。次にプレートをTBS−Tで4回洗浄し、100 ul/ウェルのTBS−T/BSAで1:150,000に希釈したHRP−ヤギ抗ヒトIgG抗体(ベチル社 A80−104P、1 mg/ml)によって37℃で1時間インキュベートした。プレートをTBS−Tで4回洗浄し、4℃に冷却した100 ul/ウェルのTMB基質を用いて展開した。7分後に1M H2SO4(100ul/ウェル)で反応を停止した。ODを450 nmで測定し、データをGraphpad Prizmソフトウェアで分析した。4つのパラメータの方程式を用いて検量線を当てはめ、これを使用して試料におけるヒトIgG含量を計算した。
ヒト化抗体のLPA結合親和性を直接結合ELISA検定により求めた。37℃で1時間0.1 M炭酸緩衝液(pH 9.5)にて希釈したImjectマリエイミド活性化ウシ血清アルブミン(BSA)(ピアス社)とコンジュゲートした1.0 ug/mlのC12:0 LPAでマイクロタイターELISAプレート(コスター)を一晩コーティングした。プレートをPBS(137mM NaCl、2.68mM KCl、10.1mM Na2HPO4、1.76mM KH2PO4、pH 7.4)で洗浄し、室温で1時間または4℃で一晩PBS/BSA/tween−20によりブロッキングした。一次インキュベーション(室温で1時間)については、抗LPA抗体(0.4ug/mL、0.2ug/mL、0.1ug/mL、0.05ug/mL、0.0125 ug/mL、0 ug/mL)の希釈系列をミクロプレート(ウェル当たり100 ml)に添加した。プレートを洗浄し、ウェル当たり100ulの1:20,000に希釈したHRPコンジュゲート化ヤギ抗ヒト(H+L)(ジャクソン、カタログ番号 109−035−003)と1時間室温でインキュベートした。洗浄後、ペルオキシダーゼをテトラメチルベンジジン基質(シグマ、カタログ番号T0440)で展開し、1 M H2SO4を添加することにより停止した。Thermo Multiskan EXを用いて450nmで吸光度(OD)を測定した。Graphpad Prismソフトウェアを用いて4つのパラメータの方程式による用量反応曲線の最小二乗を当てはめることによりEC50(半最大結合濃度)を求めた。
ヒト化抗体の特異性を競合ELISAによって求めた。C18:0 LPA コーティング材料を炭酸緩衝液(100mM NaHCO3、33.6 mM Na2CO3、pH 9.5)で0.33 ug/mlに希釈した。プレートを100 ul/ウェルのコーティング溶液でコーティングし、37℃で1時間インキュベートした。プレートをPBS(100mM Na2HPO4、20 mM KH2PO4、27 mM KCl、1.37 mM NaCl、pH 7.4)で4回洗浄し、150 ul/ウェルのPBS、1% BSA、0.1% tween−20で室温で1時間ブロッキングした。5 uM、2.5 uM、1.25 uM、0.625 uM、0.0 uMの脂質競合体(14:0 LPA(アバンティ社、カタログ番号857120)、18:1 LPA(アバンティ社、カタログ番号857130)、18:1 LPC(アバンティ社、カタログ番号845875)、cLPA(アバンティ社、カタログ番号857328)、18:1 PA(アバンティ社、カタログ番号840875)、PC(アバンティ社、カタログ番号850454)に対してヒト化抗LPA抗体を試験した。抗体をPBS、0.1 % tween−20にて0.5 ug/mlに希釈し、非結合型プレート上で、試料対抗体の比率1:3で脂質試料と合わせた。プレートをPBSで4回洗浄し、100 ul/ウェルの一次抗体・脂質の複合体と室温で1時間インキュベートした。次に、プレートをPBSで4回洗浄し、100 ul/ウェルのPBS、1% BSA、0.1% tween−20において1:20,000に希釈したHRPコンジュゲート化ヤギ抗ヒト抗体と室温で1時間インキュベートした。再度プレートをPBSで4回洗浄し、4℃でTMB基質(100 ul/ウェル)を用いて展開した。8分後に反応を100ul/ウェルの1M H2SO4によって停止した。Thermo Multiskan EXを用いて450nmで吸光度(OD)を測定した。生データをGraphPadソフトウェアに移して分析した。
加熱後のLPA結合親和性(EC50)を、直接結合ELISAを用いて測定することにより、ヒト化抗体の耐熱性を検討した。PBS(セルゴ社、カタログ番号021−040)にて溶解した抗体を25 ug/mlに希釈し、60℃、65℃、70℃、75℃、80℃で10分間インキュベートした。温度を上昇させる前に各試料を10ul取り出し、90 ulのPBSで希釈して氷上保存した。その後試料を短時間ボルテックスし、13,000 rpmで1分間遠心分離することにより不溶材料を除去した。直接LPA結合ELISAを用いて上清の結合活性を求め、加熱処理なしの同試料である対照と比較した。
結合データはすべてProteOn光学バイオセンサ(バイオラド、カリフォルニア州ハーキュリーズ)で収集した。12:0 LPAチオールおよび18:0 LPAチオールをマレイミド修飾GLCセンサー・チップ(カタログ番号176−5011)とコンジュゲートした。まず、GLCチップをスルホNHS/EDCの同量混合で7分間活性化し、次いでエチルジアミンによる7分間のブロッキング工程を実施した。次にスルホMBS(ピアス社、カタログ番号22312)をHBSランニングバッファー(10 mM HEPES、150 mM NaCl、0.005% tween−20、pH 7.4)において0.5 mMの濃度で表面上に流した。LPAチオールをHBSランニングバッファーにおいて10、1、0.1 uMの濃度に希釈して7分間注入し、3つの異なった密度のLPA表面を得た(約100、約300、約1400 RU)。次に、最高濃度として25 nMをはじめとして(元のストックをそれそれ100対1に希釈した)、3倍希釈の系列を使用してヒト化抗体の結合データを収集した。100 mM HClの10秒のパルスによって表面を復元した。データはすべて25℃で収集した。対照は、標準表面を使用し、ブランクを注入することにより処理した。各表面の応答データは、複雑な結合挙動を示すが、これはおそらく様々な程度の多価結合に起因する。結合定数の推定値を抽出するために、様々な抗体濃度のデータを、1部位および2部位のモデルを用いて全体的に当てはめた。これにより二価部位(部位1)と一価部位(部位2)の親和性の推定値を得た。
置換アッセイを用いてLPA:モノクローナル抗体のモル比を求めた。乾燥窒素気流を用いてホウケイ酸チューブ(フィッシャーブランド社、カタログ番号14−961−26)を5ナノモルのビオチン化LPA(50 ugの脂質(エシェロン・バイオサイエンヌ、カタログ番号L−012B、ロット番号F−66−136を705 ulのクロロホルム:メタノール=1:1に懸濁して100 uMの溶液を得た)でコーティングした。コーティングしたチューブを75 ul(125ピコモル)のPBS(セルグロ社、カタログ番号021−030)に溶解した抗体と室温でインキュベートした。インキュベーション3時間後、タンパク質脱塩カラム(ピアス、カタログ、番号89849)を用いてLPA:モノクローナル抗体複合体を遊離脂質から分離し、HABA/アビジン置換アッセイ(ピアス、カタログ番号28010)を製造元の指示に従って使用し、結合したビオチン化LPAのモル濃度を求めた。
抗LPA抗体はSKOV3卵巣細胞馴化培養においてLPA依存のヒトCXCL8/IL−8放出を抑制する。SKOV3細胞(ロット番号4255558、継代 14)を2 mlの1XトリプシンEDTA(メディアテック社、カタログ番号25−053−CV)で収穫し、8 mlの完全培地(10% FBS、メディアテック社カタログ番号35−011−CV)にて再懸濁した。細胞を5分間(11,000 rpm)遠心分離にかけ、5 mlの完全培地にて再懸濁した。血球計を用いて0.4%トリパンブルー(10 ul細胞に90ulトリパンブルーを添加、インビトロジェン、カタログ番号15250−061)で細胞を二回計数した。96ウェルプレートにおいて、ウェル当たり1x105個の細胞を播種した(最終容積100ul/ウェル)。一晩37°Cでインキュベートすることにより、細胞を定着させ、コンフルエントな単層を形成させた。翌日、細胞を最小培地(1mg/ml BSAのL−グルタミン含有マッコイ培地、メディアテック社、カタログ番号10−050−CV)で2回軽く洗浄した。培地を1%ペニシリン/ストレプトマイシン(メディアテック社、カタログ番号30−002 CI)2.2 g/L重炭酸ナトリウム(Mediatech社、カタログ番号25−035−CI)に調整した。次に細胞を37℃で正確に24時間血清不足にし、次いで1mg/ml BSA/PBS(カルバイオケム、カタログ番号126575)にて溶解した100 uM C18:1 LPA(アバンティ社、カタログ番号857130)によりサイトカイン刺激をLPA抗体存在下・LPA非存在下で行った。22時間の刺激後、細胞を5分間(13,500 rpm)4℃で遠心分離にかけ上清を収集した。Quantikine human CXCL8/IL−8キットを販売者の指示に従って使用し(R&Dシステムズ社、カタログ番号D8000C)、各上清のCXCL8/IL−8量を測定した。
SKOV3細胞をウェル当たり15,000細胞で96ウェルプレートに播いた。翌日、細胞を最少培地(Lグルタミン、2.2g/L重炭酸ナトリウム、1%ペニシリン/ストレプトマイシン、1mg/ml BSAを含むように調整したマッコイ5a培地)で24時間血清不足にさせた。0時間の時点で細胞をp200ピペットチップで各ウェルの中央をスクラッチし、最少培地で洗浄し、処理する前に撮影した。その後、150 ug/mlの抗体存在下・150 ug/mlの抗体非存在下で1.0 uM LPAと37oCで予めインキュベートした0.2 uM、1.0 uM、10 uMの濃度のLPA(C18:1)で細胞を処理した。陽性対照(10%FBS処理した細胞)と抗体単独も試験した。5% CO2インキュベーターにおいて細胞を37oCで17時間刺激した。17時間処理した後、再度撮影し、写真を同サイズに調節して0時間と17時間でのスクラッチの幅を定規で測ることによって傷の閉鎖率(%)を測定した。
約8〜10週齢のメスC57BL/6マウスおよびBD Biosciences((BDの)ニュージャージー州フランクリンレイクスより購入したマトリゲルマトリックス高濃度[Matrigel Matrix High Concentration]を、血管形成刺激として50 ng/ml VEGFおよび50 ng/ml bFGF、ヘパリン3 ng/mlと混合し、この研究に使った。マウス群は5つあり、0日目に10個のマトリゲルプラグを各群の5匹のマウスに接種した。一つのマウス群を対照とし、他の4つは異なった4つの用量でip注射による薬剤処理を1日おきに受けた。処理はすべて−1日目に開始し、8日目に終了した。
12日目に各群のプラグを収集した。マウスを安楽死させ、マウスの皮を引き下げてプラグを露出させた。切開してプラグを取り出し、組織学分析用に固定した。パラフィン包埋プラグの5μm切片を抗CD−31抗体で染色した。各マトリゲルプラグの断面領域における血管密度を分析した。各投与群について、少なくとも6個以上のマトリゲルプラグを定量的に分析し、微小血管密度に関して群間に統計的有意差があるかどうか評価した。
結果
LPAに対する高親和性、特異性、結合能を保持する抗体を産生する目的でマウス抗LPAモノクローナル抗体LT3000の配列をヒト化した。
LT3000のVHおよびVLのKabat CDRを受容体ヒトフレームワークに移植することによってマウス抗LPA抗体をヒト化した。7つのヒト化変種をHEK 293細胞において無血清条件下で一過性発現させ、精製した後、一連のアッセイによってキャラクテリゼーションを行った。各軽鎖および重鎖の配列を含むプラスミドを哺乳類細胞にトランスフェクションして産生した。培養5日間後に定量的ELISAを用いてモノクローナル抗体の力価を求めた。軽鎖と重鎖のすべての組み合わせで細胞培養1 ml当たり2〜12 ugの抗体を得た。
ヒト化抗LPAモノクローナル抗体変種のいずれもがキメラ抗LPA抗体(別名LT3010)およびマウス抗体LT3000と同様の低ピコモル範囲の結合親和性を示した。ヒト化変種のいずれもがLT3000と同様またはそれ以上のTMを示した。特異性に関しては、ヒト化変種はLT3000と同様の特異性プロファイルを示した。例えば、LT3000は、リゾホスファチジルコリン(LPC)、ホスファチジン酸(PA)、リゾホスファチジン酸の各種アイソフォーム(14:0および18:1 LPA、環状ホスファチジン酸(cPA)、ホスファチジルコリン(PC)に対して交差反応性を示さなかった。
インビトロ細胞アッセイにより5つのヒト化変種をさらに評価した。LPAは、癌細胞からのインターロイキン8(IL−8)放出の誘導において重要な役割を果たしていることが知られている。LT3000は、卵巣癌細胞からのIL−8放出を濃度依存的に低下させた。LT3000と比較して、ヒト化変種は同様なIL−8放出の低下を示した。
さらに、微小血管密度(MVD)に対するそれらの効果について、ヒト化変種の一部を新血管形成用マトリゲルチューブ形成アッセイにおいて試験した。両方ともMVD形成を低下させることが確認された。
本明細書において述べた特許、特許出願、文献のすべては、本発明に関連する当業者の技量を示すものである。優先権または他の利点が主張されるものも含め、特許、特許出願、文献のすべては、、それぞれの文献を個々に、参照により援用されるべく指示されている場合と同じレベルで、本明細書で参照することにより援用されている。
Claims (12)
- 生理的環境下でリゾホスファチジン酸(LPA)に結合する単離された抗体、または抗原に結合可能な抗体フラグメントであって、
前記抗体または抗体フラグメントは、下記a.に示す二つの重鎖可変ドメイン、および下記b.に示す二つの軽鎖可変ドメインを有する、ことを特徴とする抗体または抗体フラグメント:
a.各重鎖可変ドメインは、複数の重鎖フレームワーク領域に配された第1、第2および第3の重鎖相補性決定領域(CDR)を含み、ここで:
(i)前記第1の重鎖CDRは、配列番号56に示されるアミノ酸配列を有し;
(ii)前記第2の重鎖CDRは、配列番号57に示されるアミノ酸配列を有し;かつ
(iii)前記第3の重鎖CDRは、配列番号58に示されるアミノ酸配列を有する;そして、
b.前記二つの軽鎖可変ドメインは前記重鎖可変ドメインと機能的に関連しており、各軽鎖可変ドメインは、複数の軽鎖フレームワーク領域に配された第1、第2および第3の軽鎖CDRを含み、ここで:
(i)前記第1の軽鎖CDRは、配列番号59に示されるアミノ酸配列を有し;
(ii)前記第2の軽鎖CDRは、配列番号60に示されるアミノ酸配列を有し;かつ
(iii)前記第3の軽鎖CDRは、配列番号61に示されるアミノ酸配列を有する。 - 生理的環境下でリゾホスファチジン酸(LPA)に結合する単離された抗体、または抗原に結合可能な抗体フラグメントであって、
前記抗体または抗体フラグメントは、下記a.に示す二つの重鎖可変ドメイン、および下記b.に示す二つの軽鎖可変ドメインを有する、ことを特徴とする抗体または抗体フラグメント:
a.各重鎖可変ドメインは、複数の重鎖フレームワーク領域に配された第1、第2および第3の重鎖相補性決定領域(CDR)を含み、ここで:
(i)前記第1の重鎖CDRは、配列番号69に示されるアミノ酸配列を有し;
(ii)前記第2の重鎖CDRは、配列番号70に示されるアミノ酸配列を有し;かつ
(iii)前記第3の重鎖CDRは、配列番号71に示されるアミノ酸配列を有する;そして、
b.前記二つの軽鎖可変ドメインは前記重鎖可変ドメインと機能的に関連しており、各軽鎖可変ドメインは、複数の軽鎖フレームワーク領域に配された第1、第2および第3の軽鎖CDRを含み、ここで:
(i)前記第1の軽鎖CDRは、配列番号72に示されるアミノ酸配列を有し;
(ii)前記第2の軽鎖CDRは、配列番号73に示されるアミノ酸配列を有し;かつ
(iii)前記第3の軽鎖CDRは、配列番号74に示されるアミノ酸配列を有する。 - 生理的環境下でリゾホスファチジン酸(LPA)に結合する単離された抗体、または抗原に結合可能な抗体フラグメントであって、
前記抗体または抗体フラグメントは、下記a.に示す二つの重鎖可変ドメイン、および下記b.に示す二つの軽鎖可変ドメインを有する、ことを特徴とする抗体または抗体フラグメント:
a.各重鎖可変ドメインは、複数の重鎖フレームワーク領域に配された第1、第2および第3の重鎖相補性決定領域(CDR)を含み、ここで:
(i)前記第1の重鎖CDRは、配列番号81に示されるアミノ酸配列を有し;
(ii)前記第2の重鎖CDRは、配列番号82に示されるアミノ酸配列を有し;かつ
(iii)前記第3の重鎖CDRは、配列番号83に示されるアミノ酸配列を有する;そして、
b.前記二つの軽鎖可変ドメインは前記重鎖可変ドメインと機能的に関連しており、各軽鎖可変ドメインは、複数の軽鎖フレームワーク領域に配された第1、第2および第3の軽鎖CDRを含み、ここで:
(i)前記第1の軽鎖CDRは、配列番号84に示されるアミノ酸配列を有し;
(ii)前記第2の軽鎖CDRは、配列番号85に示されるアミノ酸配列を有し;かつ
(iii)前記第3の軽鎖CDRは、配列番号86に示されるアミノ酸配列を有する。 - 生理的環境下でリゾホスファチジン酸(LPA)に結合する単離された抗体、または抗原に結合可能な抗体フラグメントであって、
前記抗体または抗体フラグメントは、下記a.に示す二つの重鎖可変ドメイン、および下記b.に示す二つの軽鎖可変ドメインを有する、ことを特徴とする抗体または抗体フラグメント:
a.各重鎖可変ドメインは、複数の重鎖フレームワーク領域に配された第1、第2および第3の重鎖相補性決定領域(CDR)を含み、ここで:
(i)前記第1の重鎖CDRは、配列番号105に示されるアミノ酸配列を有し;
(ii)前記第2の重鎖CDRは、配列番号106に示されるアミノ酸配列を有し;かつ
(iii)前記第3の重鎖CDRは、配列番号107に示されるアミノ酸配列を有する;そして、
b.前記二つの軽鎖可変ドメインは前記重鎖可変ドメインと機能的に関連しており、各軽鎖可変ドメインは、複数の軽鎖フレームワーク領域に配された第1、第2および第3の軽鎖CDRを含み、ここで:
(i)前記第1の軽鎖CDRは、配列番号108に示されるアミノ酸配列を有し;
(ii)前記第2の軽鎖CDRは、配列番号109に示されるアミノ酸配列を有し;かつ
(iii)前記第3の軽鎖CDRは、配列番号110に示されるアミノ酸配列を有する。 - 前記抗体または抗体フラグメントは、抗体、キメラ抗体、ヒト化抗体、全長抗体、抗体変異体、多価抗体、および抗体フラグメントからなる群から選択される、請求項1〜4のいずれか1項に記載の抗体または抗体フラグメント。
- 前記抗体または抗体フラグメントは、1×10−8M以下の親和性でLPAに結合する、請求項1〜5のいずれか1項に記載の抗体または抗体フラグメント。
- 前記抗体または抗体フラグメントは、1×10−9M以下の親和性でLPAに結合する、請求項1〜5のいずれか1項に記載の抗体または抗体フラグメント。
- 担体、および、請求項1〜7のいずれか1項に記載の抗体または抗体フラグメントを含有する、ことを特徴とする組成物。
- 望ましくない、過剰なまたは異常なレベルのLPAに関連した疾病、疾患または状態に対する薬剤を調製するための請求項1〜7のいずれか1項に記載の抗体または抗体フラグメントの使用であって、
前記疾病または疾患は、癌を含む過剰増殖性疾患;自己免疫疾患・他移植片拒絶・移植片対宿主病を含む免疫関連疾患;神経変性疾患;肥満;2型糖尿病;黄斑変性症を含む眼疾患;疼痛;異常な血管形成または新血管新生に関連する疾患;アポトーシス;強皮症・肺線維症・腎線維症・皮膚線維症・心筋線維症・肝線維症を含む線維形成または線維症;創傷の修復および治癒;およびクモ咬傷からなる群から選択される、抗体または抗体フラグメントの使用。 - 下記a.単離核酸分子、b.ベクター、またはc.宿主細胞の少なくともいずれか一つを含有する組成物:
a.単離核酸分子は、請求項1〜4のいずれか1項に記載の抗体または抗体フラグメントの重鎖または軽鎖可変ドメインのアミノ酸配列をコードするヌクレオチド配列を含み;
b.ベクターは、上記a.に記載の核酸分子を含む核酸分子を備える;
c.宿主細胞は、(i)上記a.に記載の核酸分子を含む核酸分子、あるいは、(ii)上記b.に記載のベクターでトランスフェクションされたものである。 - 試料中のリゾホスファチジン酸(LPA)を検出する方法であって、
抗体または抗体フラグメントがLPAに結合可能な条件下で、試料中のLPAと請求項1〜7のいずれか1項に記載の抗体または抗体フラグメントとの結合を検出し、ここで:
a.前記試料は、バイオプシー試料である組織試料、または、全血、血漿、血清、尿、精液、胆汁、房水、硝子体液、粘液、気管支肺胞上皮洗浄液、および痰からなる群から選択される体液試料である;および/または
b.当該方法は、さらに下記(i)〜(iii)の少なくとも一つを含む:
(i)線維症マーカーの少なくとも1つの検出;
(ii)前記試料中のLPAレベルと同種の健常動物から得たLPAの基準レベルとの比較、ここで、基準レベルよりも相対的に高いLPAレベルが、病気の存在と相関している;
(iii)前記試料中のLPAレベルと望ましいLPAレベルとの比較。 - 試料中のリゾホスファチジン酸(LPA)を検出するために用いられるELISAキットであって、
当該キットは、請求項1〜7のいずれか1項に記載の抗体または抗体フラグメントを含む、ことを特徴とするELISAキット。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US94096407P | 2007-05-30 | 2007-05-30 | |
| US60/940,964 | 2007-05-30 | ||
| PCT/US2008/065045 WO2008150841A1 (en) | 2007-05-30 | 2008-05-29 | Compositions and methods for binding lysophosphatidic acid |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014232393A Division JP2015096507A (ja) | 2007-05-30 | 2014-11-17 | リゾホスファチジン酸結合のための組成物と方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2010530218A JP2010530218A (ja) | 2010-09-09 |
| JP2010530218A5 JP2010530218A5 (ja) | 2011-08-04 |
| JP5989299B2 true JP5989299B2 (ja) | 2016-09-07 |
Family
ID=40094082
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010510480A Active JP5989299B2 (ja) | 2007-05-30 | 2008-05-29 | リゾホスファチジン酸結合のための組成物と方法 |
| JP2014232393A Pending JP2015096507A (ja) | 2007-05-30 | 2014-11-17 | リゾホスファチジン酸結合のための組成物と方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014232393A Pending JP2015096507A (ja) | 2007-05-30 | 2014-11-17 | リゾホスファチジン酸結合のための組成物と方法 |
Country Status (9)
| Country | Link |
|---|---|
| US (3) | US8158124B2 (ja) |
| EP (1) | EP2164992B1 (ja) |
| JP (2) | JP5989299B2 (ja) |
| AU (1) | AU2008260135A1 (ja) |
| CA (1) | CA2724432A1 (ja) |
| DK (1) | DK2164992T3 (ja) |
| ES (1) | ES2585702T3 (ja) |
| LT (1) | LT2164992T (ja) |
| WO (1) | WO2008150841A1 (ja) |
Families Citing this family (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9274130B2 (en) * | 2006-05-31 | 2016-03-01 | Lpath, Inc. | Prevention and treatment of pain using antibodies to lysophosphatidic acid |
| US8796429B2 (en) * | 2006-05-31 | 2014-08-05 | Lpath, Inc. | Bioactive lipid derivatives, and methods of making and using same |
| US9274129B2 (en) * | 2006-05-31 | 2016-03-01 | Lpath, Inc. | Methods and reagents for detecting bioactive lipids |
| US9217749B2 (en) * | 2006-05-31 | 2015-12-22 | Lpath, Inc. | Immune-derived moieties reactive against lysophosphatidic acid |
| US20110064744A1 (en) * | 2007-05-30 | 2011-03-17 | Sabbadini Roger A | Prevention and treatment of pain using antibodies to lysophosphatidic acid |
| US8604172B2 (en) * | 2009-04-17 | 2013-12-10 | Lpath, Inc. | Humanized antibody compositions and methods for binding lysophosphatidic acid |
| DK2164992T3 (en) * | 2007-05-30 | 2016-08-15 | Lpath Inc | COMPOSITIONS AND METHODS FOR BONDING OF LYTHOPHOSPHATIC ACID |
| CA2722466A1 (en) | 2008-04-29 | 2009-11-05 | Tariq Ghayur | Dual variable domain immunoglobulins and uses thereof |
| US8372888B2 (en) * | 2008-04-29 | 2013-02-12 | Enzo Therapeutics, Inc. | Sphingosine kinase type 1 inhibitors, compositions and processes for using same |
| UY31861A (es) | 2008-06-03 | 2010-01-05 | Abbott Lab | Inmunoglobulina con dominio variable dual y usos de la misma |
| EP2297209A4 (en) | 2008-06-03 | 2012-08-01 | Abbott Lab | IMMUNOGLOBULINS WITH TWO VARIABLE DOMAINS AND USES THEREOF |
| WO2010006060A2 (en) | 2008-07-08 | 2010-01-14 | Abbott Laboratories | Prostaglandin e2 dual variable domain immunoglobulins and uses thereof |
| US8629247B2 (en) | 2009-04-14 | 2014-01-14 | Proscan Rx Pharma Inc. | Antibodies against prostate specific membrane antigen |
| AU2010270894A1 (en) * | 2009-06-24 | 2012-02-09 | Lpath, Inc. | Methods of increasing neuronal differntiation using antibodies to lysophoshatidic acid |
| WO2011081214A1 (ja) * | 2009-12-28 | 2011-07-07 | 国立大学法人東京大学 | オートタキシン測定による肝線維化の検査方法および検査薬 |
| WO2011106723A2 (en) * | 2010-02-26 | 2011-09-01 | Lpath, Inc. | Anti-paf antibodies |
| WO2011156242A2 (en) * | 2010-06-11 | 2011-12-15 | Lpath, Inc. | Improved anti-lysophospholipid antibody design using antibody structures |
| CA2804191C (en) * | 2010-07-14 | 2019-10-01 | Lpath, Inc. | Prevention and treatment of pain using monoclonal antibodies and antibody fragments to lysophosphatidic acid |
| JP2013537415A (ja) | 2010-08-03 | 2013-10-03 | アッヴィ・インコーポレイテッド | 二重可変ドメイン免疫グロブリンおよびその使用 |
| AU2011293253B2 (en) | 2010-08-26 | 2014-12-11 | Abbvie Inc. | Dual variable domain immunoglobulins and uses thereof |
| JP2014521723A (ja) * | 2011-08-09 | 2014-08-28 | エルパス, インコーポレーテッド | リゾホスファチジン酸の阻害剤を使用する幹細胞治療 |
| CA2861610A1 (en) | 2011-12-30 | 2013-07-04 | Abbvie Inc. | Dual specific binding proteins directed against il-13 and/or il-17 |
| US20130302831A1 (en) * | 2012-02-29 | 2013-11-14 | Roger A. Sabbadini | Methods and kits for detecting and diagnosing neurotrauma |
| DE102012021222B4 (de) * | 2012-10-27 | 2015-02-05 | Forschungszentrum Jülich GmbH | Verfahren zur Herstellung einer nanoporösen Schicht auf einem Substrat |
| NZ707641A (en) | 2012-11-01 | 2016-09-30 | Abbvie Inc | Anti-vegf/dll4 dual variable domain immunoglobulins and uses thereof |
| TW201512219A (zh) | 2013-03-15 | 2015-04-01 | Abbvie Inc | 針對IL-1β及/或IL-17之雙特異性結合蛋白 |
| WO2016025879A1 (en) * | 2014-08-15 | 2016-02-18 | Indiana University Research And Technology Corporation | Treatment of cystic diseases |
| WO2016094881A2 (en) | 2014-12-11 | 2016-06-16 | Abbvie Inc. | Lrp-8 binding proteins |
| TW201710286A (zh) | 2015-06-15 | 2017-03-16 | 艾伯維有限公司 | 抗vegf、pdgf及/或其受體之結合蛋白 |
| US9724057B2 (en) * | 2015-06-30 | 2017-08-08 | Toshiba Medical Systems Corporation | Magnetic field actuation of detectors in a computed tomography scanner |
| JP6889432B2 (ja) * | 2016-04-28 | 2021-06-18 | 学校法人甲南学園 | 樹脂固定ペプチド |
| CN112153983B (zh) | 2018-05-23 | 2024-09-13 | 杰克逊实验室 | 抗ngly-1抗体及使用方法 |
| ES2736045A1 (es) * | 2018-06-21 | 2019-12-23 | Univ Cadiz | Metodos para el diagnostico y/o tratamiento de enfermedades neurodegenerativas |
| CN113827605B (zh) * | 2021-08-27 | 2023-06-09 | 上海市肺科医院 | 甘油磷酸胆碱在制备预防和治疗结核病药物中的应用 |
| CN118541392A (zh) * | 2021-09-28 | 2024-08-23 | 准星生物医药有限公司 | 多种形式的分子复合物 |
Family Cites Families (81)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3773919A (en) * | 1969-10-23 | 1973-11-20 | Du Pont | Polylactide-drug mixtures |
| US4179337A (en) * | 1973-07-20 | 1979-12-18 | Davis Frank F | Non-immunogenic polypeptides |
| USRE30985E (en) * | 1978-01-01 | 1982-06-29 | Serum-free cell culture media | |
| US4275149A (en) * | 1978-11-24 | 1981-06-23 | Syva Company | Macromolecular environment control in specific receptor assays |
| US4318980A (en) * | 1978-04-10 | 1982-03-09 | Miles Laboratories, Inc. | Heterogenous specific binding assay employing a cycling reactant as label |
| JPS6023084B2 (ja) * | 1979-07-11 | 1985-06-05 | 味の素株式会社 | 代用血液 |
| US4376110A (en) | 1980-08-04 | 1983-03-08 | Hybritech, Incorporated | Immunometric assays using monoclonal antibodies |
| US4485045A (en) | 1981-07-06 | 1984-11-27 | Research Corporation | Synthetic phosphatidyl cholines useful in forming liposomes |
| US4640835A (en) | 1981-10-30 | 1987-02-03 | Nippon Chemiphar Company, Ltd. | Plasminogen activator derivatives |
| US4560655A (en) | 1982-12-16 | 1985-12-24 | Immunex Corporation | Serum-free cell culture medium and process for making same |
| US4657866A (en) * | 1982-12-21 | 1987-04-14 | Sudhir Kumar | Serum-free, synthetic, completely chemically defined tissue culture media |
| GB8308235D0 (en) | 1983-03-25 | 1983-05-05 | Celltech Ltd | Polypeptides |
| US4816567A (en) * | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| DD266710A3 (de) | 1983-06-06 | 1989-04-12 | Ve Forschungszentrum Biotechnologie | Verfahren zur biotechnischen Herstellung van alkalischer Phosphatase |
| US4544545A (en) | 1983-06-20 | 1985-10-01 | Trustees University Of Massachusetts | Liposomes containing modified cholesterol for organ targeting |
| US4767704A (en) | 1983-10-07 | 1988-08-30 | Columbia University In The City Of New York | Protein-free culture medium |
| US4496689A (en) * | 1983-12-27 | 1985-01-29 | Miles Laboratories, Inc. | Covalently attached complex of alpha-1-proteinase inhibitor with a water soluble polymer |
| GB8422238D0 (en) | 1984-09-03 | 1984-10-10 | Neuberger M S | Chimeric proteins |
| US4879231A (en) | 1984-10-30 | 1989-11-07 | Phillips Petroleum Company | Transformation of yeasts of the genus pichia |
| US4737456A (en) * | 1985-05-09 | 1988-04-12 | Syntex (U.S.A.) Inc. | Reducing interference in ligand-receptor binding assays |
| EP0206448B1 (en) | 1985-06-19 | 1990-11-14 | Ajinomoto Co., Inc. | Hemoglobin combined with a poly(alkylene oxide) |
| GB8516415D0 (en) | 1985-06-28 | 1985-07-31 | Celltech Ltd | Culture of animal cells |
| US4676980A (en) | 1985-09-23 | 1987-06-30 | The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Target specific cross-linked heteroantibodies |
| GB8607679D0 (en) | 1986-03-27 | 1986-04-30 | Winter G P | Recombinant dna product |
| US6548640B1 (en) | 1986-03-27 | 2003-04-15 | Btg International Limited | Altered antibodies |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| US4927762A (en) * | 1986-04-01 | 1990-05-22 | Cell Enterprises, Inc. | Cell culture medium with antioxidant |
| GB8610600D0 (en) | 1986-04-30 | 1986-06-04 | Novo Industri As | Transformation of trichoderma |
| US4791192A (en) | 1986-06-26 | 1988-12-13 | Takeda Chemical Industries, Ltd. | Chemically modified protein with polyethyleneglycol |
| US5567610A (en) | 1986-09-04 | 1996-10-22 | Bioinvent International Ab | Method of producing human monoclonal antibodies and kit therefor |
| WO1988007089A1 (en) * | 1987-03-18 | 1988-09-22 | Medical Research Council | Altered antibodies |
| US5204244A (en) * | 1987-10-27 | 1993-04-20 | Oncogen | Production of chimeric antibodies by homologous recombination |
| EP0435911B1 (en) | 1988-09-23 | 1996-03-13 | Cetus Oncology Corporation | Cell culture medium for enhanced cell growth, culture longevity and product expression |
| GB8823869D0 (en) | 1988-10-12 | 1988-11-16 | Medical Res Council | Production of antibodies |
| US5175384A (en) | 1988-12-05 | 1992-12-29 | Genpharm International | Transgenic mice depleted in mature t-cells and methods for making transgenic mice |
| US5530101A (en) * | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US20040049014A1 (en) | 1988-12-28 | 2004-03-11 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| EP0402226A1 (en) | 1989-06-06 | 1990-12-12 | Institut National De La Recherche Agronomique | Transformation vectors for yeast yarrowia |
| DE3920358A1 (de) | 1989-06-22 | 1991-01-17 | Behringwerke Ag | Bispezifische und oligospezifische, mono- und oligovalente antikoerperkonstrukte, ihre herstellung und verwendung |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| US5229275A (en) | 1990-04-26 | 1993-07-20 | Akzo N.V. | In-vitro method for producing antigen-specific human monoclonal antibodies |
| US5122469A (en) * | 1990-10-03 | 1992-06-16 | Genentech, Inc. | Method for culturing Chinese hamster ovary cells to improve production of recombinant proteins |
| DE69233482T2 (de) | 1991-05-17 | 2006-01-12 | Merck & Co., Inc. | Verfahren zur Verminderung der Immunogenität der variablen Antikörperdomänen |
| WO1992022653A1 (en) | 1991-06-14 | 1992-12-23 | Genentech, Inc. | Method for making humanized antibodies |
| ES2136092T3 (es) * | 1991-09-23 | 1999-11-16 | Medical Res Council | Procedimientos para la produccion de anticuerpos humanizados. |
| EP0617706B1 (en) | 1991-11-25 | 2001-10-17 | Enzon, Inc. | Multivalent antigen-binding proteins |
| US5932448A (en) * | 1991-11-29 | 1999-08-03 | Protein Design Labs., Inc. | Bispecific antibody heterodimers |
| US5777085A (en) | 1991-12-20 | 1998-07-07 | Protein Design Labs, Inc. | Humanized antibodies reactive with GPIIB/IIIA |
| WO1993016177A1 (en) * | 1992-02-11 | 1993-08-19 | Cell Genesys, Inc. | Homogenotization of gene-targeting events |
| US5714350A (en) | 1992-03-09 | 1998-02-03 | Protein Design Labs, Inc. | Increasing antibody affinity by altering glycosylation in the immunoglobulin variable region |
| US6129914A (en) * | 1992-03-27 | 2000-10-10 | Protein Design Labs, Inc. | Bispecific antibody effective to treat B-cell lymphoma and cell line |
| US5573905A (en) * | 1992-03-30 | 1996-11-12 | The Scripps Research Institute | Encoded combinatorial chemical libraries |
| GB9223377D0 (en) | 1992-11-04 | 1992-12-23 | Medarex Inc | Humanized antibodies to fc receptors for immunoglobulin on human mononuclear phagocytes |
| US6210671B1 (en) | 1992-12-01 | 2001-04-03 | Protein Design Labs, Inc. | Humanized antibodies reactive with L-selectin |
| GB9325182D0 (en) * | 1993-12-08 | 1994-02-09 | T Cell Sciences Inc | Humanized antibodies or binding proteins thereof specific for t cell subpopulations exhibiting select beta chain variable regions |
| US5534615A (en) | 1994-04-25 | 1996-07-09 | Genentech, Inc. | Cardiac hypertrophy factor and uses therefor |
| US5731168A (en) | 1995-03-01 | 1998-03-24 | Genentech, Inc. | Method for making heteromultimeric polypeptides |
| US6096871A (en) | 1995-04-14 | 2000-08-01 | Genentech, Inc. | Polypeptides altered to contain an epitope from the Fc region of an IgG molecule for increased half-life |
| US5882644A (en) * | 1996-03-22 | 1999-03-16 | Protein Design Labs, Inc. | Monoclonal antibodies specific for the platelet derived growth factor β receptor and methods of use thereof |
| US5834597A (en) * | 1996-05-20 | 1998-11-10 | Protein Design Labs, Inc. | Mutated nonactivating IgG2 domains and anti CD3 antibodies incorporating the same |
| US6013256A (en) | 1996-09-24 | 2000-01-11 | Protein Design Labs, Inc. | Method of preventing acute rejection following solid organ transplantation |
| WO1999029888A1 (en) | 1997-12-05 | 1999-06-17 | The Scripps Research Institute | Humanization of murine antibody |
| CA2323787A1 (en) | 1998-03-13 | 1999-09-16 | Wayne Marasco | Humanized antibody and uses thereof |
| ATE254137T1 (de) * | 1998-09-29 | 2003-11-15 | Statens Seruminstitut | ßLIGAND PRESENTING ASSEMBLYß (LPA), VERFAHREN ZU DESSEN HERSTELLUNG UND VERWENDUNGEN |
| US6248553B1 (en) * | 1998-10-22 | 2001-06-19 | Atairgin Technologies, Inc. | Enzyme method for detecting lysophospholipids and phospholipids and for detecting and correlating conditions associated with altered levels of lysophospholipids |
| AU764211C (en) | 1998-12-01 | 2006-03-30 | Abbvie Biotherapeutics Inc. | Humanized antibodies to gamma-interferon |
| JP2003530081A (ja) * | 1999-11-04 | 2003-10-14 | アボット・ラボラトリーズ | 改良後の自動化lpa検査法および癌検知法 |
| US20030003479A1 (en) * | 2001-04-19 | 2003-01-02 | Millennium Pharmaceutical, Inc. | Compositions, kits, and methods for identification, assessment, prevention, and therapy of ovarian cancer |
| WO2003100033A2 (en) * | 2002-03-13 | 2003-12-04 | Biogen Idec Ma Inc. | ANTI-αvβ6 ANTIBODIES |
| DK1517921T3 (da) * | 2002-06-28 | 2006-10-09 | Domantis Ltd | Immunglobulin-enkeltvariable antigen-bindende domæner og dobbeltspecifikke konstruktioner deraf |
| US7459285B2 (en) | 2002-12-10 | 2008-12-02 | Echelon Biosciences Incorporated | Compounds and methods of use thereof for assaying lysophospholipase D activity |
| JP4779650B2 (ja) | 2003-12-26 | 2011-09-28 | 小野薬品工業株式会社 | 高活性型リゾホスファチジン酸およびそれを用いたスクリーニング方法 |
| US7794713B2 (en) | 2004-04-07 | 2010-09-14 | Lpath, Inc. | Compositions and methods for the treatment and prevention of hyperproliferative diseases |
| DK1853631T3 (en) * | 2005-01-24 | 2016-03-29 | Univ Texas | Fc-FUSION CONSTRUCTIONS BINDING TO PHOSPHATIDYLSERINE AND THERAPEUTIC APPLICATION THEREOF |
| US20110123440A1 (en) | 2005-03-29 | 2011-05-26 | Genevieve Hansen | Altered Antibody FC Regions and Uses Thereof |
| US8796429B2 (en) * | 2006-05-31 | 2014-08-05 | Lpath, Inc. | Bioactive lipid derivatives, and methods of making and using same |
| US9217749B2 (en) * | 2006-05-31 | 2015-12-22 | Lpath, Inc. | Immune-derived moieties reactive against lysophosphatidic acid |
| US9274129B2 (en) * | 2006-05-31 | 2016-03-01 | Lpath, Inc. | Methods and reagents for detecting bioactive lipids |
| US8604172B2 (en) * | 2009-04-17 | 2013-12-10 | Lpath, Inc. | Humanized antibody compositions and methods for binding lysophosphatidic acid |
| US9163091B2 (en) * | 2007-05-30 | 2015-10-20 | Lpath, Inc. | Compositions and methods for binding lysophosphatidic acid |
| DK2164992T3 (en) * | 2007-05-30 | 2016-08-15 | Lpath Inc | COMPOSITIONS AND METHODS FOR BONDING OF LYTHOPHOSPHATIC ACID |
-
2008
- 2008-05-29 DK DK08756421.7T patent/DK2164992T3/en active
- 2008-05-29 US US12/129,109 patent/US8158124B2/en not_active Expired - Fee Related
- 2008-05-29 ES ES08756421.7T patent/ES2585702T3/es active Active
- 2008-05-29 JP JP2010510480A patent/JP5989299B2/ja active Active
- 2008-05-29 EP EP08756421.7A patent/EP2164992B1/en active Active
- 2008-05-29 LT LTEP08756421.7T patent/LT2164992T/lt unknown
- 2008-05-29 CA CA2724432A patent/CA2724432A1/en not_active Abandoned
- 2008-05-29 WO PCT/US2008/065045 patent/WO2008150841A1/en active Application Filing
- 2008-05-29 AU AU2008260135A patent/AU2008260135A1/en not_active Abandoned
-
2012
- 2012-04-16 US US13/448,269 patent/US20120195901A1/en not_active Abandoned
-
2014
- 2014-11-17 JP JP2014232393A patent/JP2015096507A/ja active Pending
-
2015
- 2015-07-29 US US14/812,325 patent/US20160146844A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| EP2164992B1 (en) | 2016-05-04 |
| US20160146844A1 (en) | 2016-05-26 |
| US8158124B2 (en) | 2012-04-17 |
| ES2585702T3 (es) | 2016-10-07 |
| JP2015096507A (ja) | 2015-05-21 |
| DK2164992T3 (en) | 2016-08-15 |
| US20120195901A1 (en) | 2012-08-02 |
| LT2164992T (lt) | 2016-09-12 |
| EP2164992A4 (en) | 2013-05-01 |
| CA2724432A1 (en) | 2008-12-11 |
| AU2008260135A1 (en) | 2008-12-11 |
| US20090136483A1 (en) | 2009-05-28 |
| EP2164992A1 (en) | 2010-03-24 |
| JP2010530218A (ja) | 2010-09-09 |
| WO2008150841A1 (en) | 2008-12-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5989299B2 (ja) | リゾホスファチジン酸結合のための組成物と方法 | |
| US9163091B2 (en) | Compositions and methods for binding lysophosphatidic acid | |
| JP6053834B2 (ja) | スフィンゴシン−1−リン酸と結合させるための組成物および方法 | |
| US8604172B2 (en) | Humanized antibody compositions and methods for binding lysophosphatidic acid | |
| JP5732182B2 (ja) | 眼疾患と症状を処置するための組成物および方法 | |
| US20110212088A1 (en) | Anti-paf antibodies | |
| US20170114125A1 (en) | Methods of treating disease using antibodies to lysophosphatidic acid | |
| US20180298111A1 (en) | Prevention and treatment of pain using antibodies to lysophosphatidic acid | |
| JP2012511026A (ja) | 抗脂抗体結晶構造を用いた抗体設計 | |
| US20170254822A1 (en) | Methods and kits for detecting and diagnosing neurotrauma | |
| JP2016102121A (ja) | リゾホスファチジン酸に対するモノクローナル抗体及び抗体断片を用いた、疼痛の予防及び治療 | |
| US8361465B2 (en) | Use of anti-sphingosine-1-phosphate antibodies in combination with chemotherapeutic agents | |
| AU2014277842A1 (en) | Compositions and Methods for Binding Lysophosphatidic Acid | |
| AU2013273727A1 (en) | Compositions and methods for treating ocular diseases and conditions | |
| AU2016204486A1 (en) | Composition and Methods for Treating Ocular Diseases and Conditions | |
| AU2015264913A1 (en) | Methods and kits for detecting and diagnosing neurotrauma |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110526 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110526 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20110603 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20110613 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110722 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120524 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130131 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130430 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130509 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130530 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130606 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130701 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130731 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130816 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130807 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130920 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20131220 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20140117 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20140121 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20140218 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20140220 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140320 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140331 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20140327 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20140716 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20141117 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20150121 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20150129 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20150226 |
|
| A912 | Re-examination (zenchi) completed and case transferred to appeal board |
Free format text: JAPANESE INTERMEDIATE CODE: A912 Effective date: 20150410 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20160421 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160520 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20160810 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5989299 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: R3D04 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |