JP5600541B2 - キレート樹脂を用いるカチオンの改善された除去方法 - Google Patents
キレート樹脂を用いるカチオンの改善された除去方法 Download PDFInfo
- Publication number
- JP5600541B2 JP5600541B2 JP2010218848A JP2010218848A JP5600541B2 JP 5600541 B2 JP5600541 B2 JP 5600541B2 JP 2010218848 A JP2010218848 A JP 2010218848A JP 2010218848 A JP2010218848 A JP 2010218848A JP 5600541 B2 JP5600541 B2 JP 5600541B2
- Authority
- JP
- Japan
- Prior art keywords
- polymer beads
- beads
- futaruimi
- chelate
- calcium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 238000000034 method Methods 0.000 title claims description 37
- 150000001768 cations Chemical class 0.000 title description 29
- 229920001429 chelating resin Polymers 0.000 title description 19
- 239000011324 bead Substances 0.000 claims description 81
- 229920000642 polymer Polymers 0.000 claims description 64
- 239000013522 chelant Substances 0.000 claims description 38
- 150000002500 ions Chemical class 0.000 claims description 26
- 239000000178 monomer Substances 0.000 claims description 26
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 claims description 20
- XKJCHHZQLQNZHY-UHFFFAOYSA-N phthalimide Chemical compound C1=CC=C2C(=O)NC(=O)C2=C1 XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 claims description 20
- 150000001491 aromatic compounds Chemical class 0.000 claims description 17
- 125000000524 functional group Chemical group 0.000 claims description 15
- 239000003999 initiator Substances 0.000 claims description 13
- 239000012442 inert solvent Substances 0.000 claims description 11
- 238000006467 substitution reaction Methods 0.000 claims description 11
- 229920006037 cross link polymer Polymers 0.000 claims description 10
- 229920006216 polyvinyl aromatic Polymers 0.000 claims description 10
- 239000003361 porogen Substances 0.000 claims description 9
- 239000000725 suspension Substances 0.000 claims description 9
- 238000004519 manufacturing process Methods 0.000 claims description 8
- 239000011148 porous material Substances 0.000 claims description 8
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- 239000001257 hydrogen Substances 0.000 claims description 6
- 229910052739 hydrogen Inorganic materials 0.000 claims description 6
- 150000001875 compounds Chemical class 0.000 claims description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 58
- 239000011575 calcium Substances 0.000 description 51
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 48
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 44
- 229910052791 calcium Inorganic materials 0.000 description 44
- 239000012267 brine Substances 0.000 description 41
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 41
- 235000002639 sodium chloride Nutrition 0.000 description 36
- 229920005989 resin Polymers 0.000 description 33
- 239000011347 resin Substances 0.000 description 33
- 239000011780 sodium chloride Substances 0.000 description 30
- 238000005868 electrolysis reaction Methods 0.000 description 27
- 239000002245 particle Substances 0.000 description 26
- 239000000243 solution Substances 0.000 description 24
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 24
- MYRTYDVEIRVNKP-UHFFFAOYSA-N 1,2-Divinylbenzene Chemical compound C=CC1=CC=CC=C1C=C MYRTYDVEIRVNKP-UHFFFAOYSA-N 0.000 description 20
- 239000011777 magnesium Substances 0.000 description 17
- 239000000203 mixture Substances 0.000 description 17
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 16
- 238000010521 absorption reaction Methods 0.000 description 16
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 15
- 229910052749 magnesium Inorganic materials 0.000 description 15
- 239000012528 membrane Substances 0.000 description 15
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 14
- 229910052788 barium Inorganic materials 0.000 description 13
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 13
- 238000006116 polymerization reaction Methods 0.000 description 13
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 12
- 229910052712 strontium Inorganic materials 0.000 description 12
- CIOAGBVUUVVLOB-UHFFFAOYSA-N strontium atom Chemical compound [Sr] CIOAGBVUUVVLOB-UHFFFAOYSA-N 0.000 description 12
- 108010010803 Gelatin Proteins 0.000 description 11
- 229920000159 gelatin Polymers 0.000 description 11
- 239000008273 gelatin Substances 0.000 description 11
- 235000019322 gelatine Nutrition 0.000 description 11
- 235000011852 gelatine desserts Nutrition 0.000 description 11
- 150000001342 alkaline earth metals Chemical class 0.000 description 10
- 229910001385 heavy metal Inorganic materials 0.000 description 10
- 238000011068 loading method Methods 0.000 description 10
- 229910001514 alkali metal chloride Inorganic materials 0.000 description 9
- 239000008346 aqueous phase Substances 0.000 description 9
- 239000000126 substance Substances 0.000 description 9
- MGRVRXRGTBOSHW-UHFFFAOYSA-N (aminomethyl)phosphonic acid Chemical group NCP(O)(O)=O MGRVRXRGTBOSHW-UHFFFAOYSA-N 0.000 description 8
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 8
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 8
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 8
- 239000007864 aqueous solution Substances 0.000 description 8
- 238000009826 distribution Methods 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- 229910052783 alkali metal Inorganic materials 0.000 description 7
- 150000001340 alkali metals Chemical class 0.000 description 7
- 238000006243 chemical reaction Methods 0.000 description 7
- 239000000460 chlorine Substances 0.000 description 7
- 238000005259 measurement Methods 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- 239000003112 inhibitor Substances 0.000 description 6
- 230000008929 regeneration Effects 0.000 description 6
- 238000011069 regeneration method Methods 0.000 description 6
- 150000003839 salts Chemical class 0.000 description 6
- 238000001179 sorption measurement Methods 0.000 description 6
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- MPMBRWOOISTHJV-UHFFFAOYSA-N but-1-enylbenzene Chemical compound CCC=CC1=CC=CC=C1 MPMBRWOOISTHJV-UHFFFAOYSA-N 0.000 description 5
- 239000000084 colloidal system Substances 0.000 description 5
- 239000010949 copper Substances 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- -1 for example Substances 0.000 description 5
- 229910052757 nitrogen Inorganic materials 0.000 description 5
- 229910000510 noble metal Inorganic materials 0.000 description 5
- 230000001681 protective effect Effects 0.000 description 5
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 4
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 4
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 4
- 239000011651 chromium Substances 0.000 description 4
- 229910052802 copper Inorganic materials 0.000 description 4
- 238000011156 evaluation Methods 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 229910052742 iron Inorganic materials 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 4
- 229910052753 mercury Inorganic materials 0.000 description 4
- 229910052751 metal Inorganic materials 0.000 description 4
- 239000002184 metal Substances 0.000 description 4
- 150000002739 metals Chemical class 0.000 description 4
- 229910052759 nickel Inorganic materials 0.000 description 4
- GHMLBKRAJCXXBS-UHFFFAOYSA-N resorcinol Chemical compound OC1=CC=CC(O)=C1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 239000011701 zinc Substances 0.000 description 4
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 3
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- HRPVXLWXLXDGHG-UHFFFAOYSA-N Acrylamide Chemical group NC(=O)C=C HRPVXLWXLXDGHG-UHFFFAOYSA-N 0.000 description 3
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 3
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 3
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 3
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 3
- 229910001424 calcium ion Inorganic materials 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 229910052804 chromium Inorganic materials 0.000 description 3
- 229910017052 cobalt Inorganic materials 0.000 description 3
- 239000010941 cobalt Substances 0.000 description 3
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 description 3
- 229920001577 copolymer Polymers 0.000 description 3
- 238000009792 diffusion process Methods 0.000 description 3
- 238000004821 distillation Methods 0.000 description 3
- 238000000921 elemental analysis Methods 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 230000014509 gene expression Effects 0.000 description 3
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 229910052760 oxygen Inorganic materials 0.000 description 3
- 239000001301 oxygen Substances 0.000 description 3
- 230000002572 peristaltic effect Effects 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 238000001556 precipitation Methods 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- ISIJQEHRDSCQIU-UHFFFAOYSA-N tert-butyl 2,7-diazaspiro[4.5]decane-7-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CCCC11CNCC1 ISIJQEHRDSCQIU-UHFFFAOYSA-N 0.000 description 3
- 229910052725 zinc Inorganic materials 0.000 description 3
- XWJBRBSPAODJER-UHFFFAOYSA-N 1,7-octadiene Chemical compound C=CCCCCC=C XWJBRBSPAODJER-UHFFFAOYSA-N 0.000 description 2
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 2
- GTJOHISYCKPIMT-UHFFFAOYSA-N 2-methylundecane Chemical compound CCCCCCCCCC(C)C GTJOHISYCKPIMT-UHFFFAOYSA-N 0.000 description 2
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- VYZAMTAEIAYCRO-UHFFFAOYSA-N Chromium Chemical compound [Cr] VYZAMTAEIAYCRO-UHFFFAOYSA-N 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 2
- SGVYKUFIHHTIFL-UHFFFAOYSA-N Isobutylhexyl Natural products CCCCCCCC(C)C SGVYKUFIHHTIFL-UHFFFAOYSA-N 0.000 description 2
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 2
- JLVVSXFLKOJNIY-UHFFFAOYSA-N Magnesium ion Chemical compound [Mg+2] JLVVSXFLKOJNIY-UHFFFAOYSA-N 0.000 description 2
- YVPNKHMHNJVFAE-UHFFFAOYSA-N N=COP(OC)=O Chemical group N=COP(OC)=O YVPNKHMHNJVFAE-UHFFFAOYSA-N 0.000 description 2
- 239000004952 Polyamide Substances 0.000 description 2
- 229920002125 Sokalan® Polymers 0.000 description 2
- 238000013019 agitation Methods 0.000 description 2
- 125000005907 alkyl ester group Chemical group 0.000 description 2
- 150000001450 anions Chemical class 0.000 description 2
- 125000003118 aryl group Chemical group 0.000 description 2
- 229920005601 base polymer Polymers 0.000 description 2
- 235000019400 benzoyl peroxide Nutrition 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 229910001902 chlorine oxide Inorganic materials 0.000 description 2
- 239000007859 condensation product Substances 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- CZHYKKAKFWLGJO-UHFFFAOYSA-N dimethyl phosphite Chemical compound COP([O-])OC CZHYKKAKFWLGJO-UHFFFAOYSA-N 0.000 description 2
- 238000005538 encapsulation Methods 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 238000007306 functionalization reaction Methods 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 description 2
- 238000005342 ion exchange Methods 0.000 description 2
- VKPSKYDESGTTFR-UHFFFAOYSA-N isododecane Natural products CC(C)(C)CC(C)CC(C)(C)C VKPSKYDESGTTFR-UHFFFAOYSA-N 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 239000011572 manganese Substances 0.000 description 2
- 229910001510 metal chloride Inorganic materials 0.000 description 2
- 235000010755 mineral Nutrition 0.000 description 2
- 239000011707 mineral Substances 0.000 description 2
- QYZFTMMPKCOTAN-UHFFFAOYSA-N n-[2-(2-hydroxyethylamino)ethyl]-2-[[1-[2-(2-hydroxyethylamino)ethylamino]-2-methyl-1-oxopropan-2-yl]diazenyl]-2-methylpropanamide Chemical compound OCCNCCNC(=O)C(C)(C)N=NC(C)(C)C(=O)NCCNCCO QYZFTMMPKCOTAN-UHFFFAOYSA-N 0.000 description 2
- 229930014626 natural product Natural products 0.000 description 2
- 229910017464 nitrogen compound Inorganic materials 0.000 description 2
- 150000002830 nitrogen compounds Chemical class 0.000 description 2
- PTMHPRAIXMAOOB-UHFFFAOYSA-N phosphoramidic acid Chemical group NP(O)(O)=O PTMHPRAIXMAOOB-UHFFFAOYSA-N 0.000 description 2
- 229920002647 polyamide Polymers 0.000 description 2
- 229920000867 polyelectrolyte Polymers 0.000 description 2
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 2
- 238000002459 porosimetry Methods 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- WQGWDDDVZFFDIG-UHFFFAOYSA-N pyrogallol Chemical compound OC1=CC=CC(O)=C1O WQGWDDDVZFFDIG-UHFFFAOYSA-N 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 239000002904 solvent Substances 0.000 description 2
- AKEJUJNQAAGONA-UHFFFAOYSA-N sulfur trioxide Chemical compound O=S(=O)=O AKEJUJNQAAGONA-UHFFFAOYSA-N 0.000 description 2
- HIFJUMGIHIZEPX-UHFFFAOYSA-N sulfuric acid;sulfur trioxide Chemical group O=S(=O)=O.OS(O)(=O)=O HIFJUMGIHIZEPX-UHFFFAOYSA-N 0.000 description 2
- 230000008961 swelling Effects 0.000 description 2
- CWERGRDVMFNCDR-UHFFFAOYSA-N thioglycolic acid Chemical compound OC(=O)CS CWERGRDVMFNCDR-UHFFFAOYSA-N 0.000 description 2
- 238000004448 titration Methods 0.000 description 2
- 229920002554 vinyl polymer Polymers 0.000 description 2
- 239000002351 wastewater Substances 0.000 description 2
- OXYKVVLTXXXVRT-UHFFFAOYSA-N (4-chlorobenzoyl) 4-chlorobenzenecarboperoxoate Chemical compound C1=CC(Cl)=CC=C1C(=O)OOC(=O)C1=CC=C(Cl)C=C1 OXYKVVLTXXXVRT-UHFFFAOYSA-N 0.000 description 1
- WVAFEFUPWRPQSY-UHFFFAOYSA-N 1,2,3-tris(ethenyl)benzene Chemical compound C=CC1=CC=CC(C=C)=C1C=C WVAFEFUPWRPQSY-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- CHRJZRDFSQHIFI-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;styrene Chemical compound C=CC1=CC=CC=C1.C=CC1=CC=CC=C1C=C CHRJZRDFSQHIFI-UHFFFAOYSA-N 0.000 description 1
- QLLUAUADIMPKIH-UHFFFAOYSA-N 1,2-bis(ethenyl)naphthalene Chemical compound C1=CC=CC2=C(C=C)C(C=C)=CC=C21 QLLUAUADIMPKIH-UHFFFAOYSA-N 0.000 description 1
- PRBHEGAFLDMLAL-UHFFFAOYSA-N 1,5-Hexadiene Natural products CC=CCC=C PRBHEGAFLDMLAL-UHFFFAOYSA-N 0.000 description 1
- UXQAEOWCSOPBLF-UHFFFAOYSA-N 2,2,3,3-tetramethyloctane Chemical compound CCCCCC(C)(C)C(C)(C)C UXQAEOWCSOPBLF-UHFFFAOYSA-N 0.000 description 1
- LMLDOTHTMKGQKH-UHFFFAOYSA-N 2-(1,3-dioxoisoindol-2-yl)oxyisoindole-1,3-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1ON1C(=O)C2=CC=CC=C2C1=O LMLDOTHTMKGQKH-UHFFFAOYSA-N 0.000 description 1
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- SBYMUDUGTIKLCR-UHFFFAOYSA-N 2-chloroethenylbenzene Chemical compound ClC=CC1=CC=CC=C1 SBYMUDUGTIKLCR-UHFFFAOYSA-N 0.000 description 1
- NFEGKOIJMCGIKN-UHFFFAOYSA-N 3-(2-methylbutan-2-ylperoxymethyl)heptane Chemical compound CCCCC(CC)COOC(C)(C)CC NFEGKOIJMCGIKN-UHFFFAOYSA-N 0.000 description 1
- IWTYTFSSTWXZFU-UHFFFAOYSA-N 3-chloroprop-1-enylbenzene Chemical compound ClCC=CC1=CC=CC=C1 IWTYTFSSTWXZFU-UHFFFAOYSA-N 0.000 description 1
- JIGUICYYOYEXFS-UHFFFAOYSA-N 3-tert-butylbenzene-1,2-diol Chemical compound CC(C)(C)C1=CC=CC(O)=C1O JIGUICYYOYEXFS-UHFFFAOYSA-N 0.000 description 1
- DBCAQXHNJOFNGC-UHFFFAOYSA-N 4-bromo-1,1,1-trifluorobutane Chemical compound FC(F)(F)CCCBr DBCAQXHNJOFNGC-UHFFFAOYSA-N 0.000 description 1
- ABCGRFHYOYXEJV-UHFFFAOYSA-N 4-methylisoindole-1,3-dione Chemical compound CC1=CC=CC2=C1C(=O)NC2=O ABCGRFHYOYXEJV-UHFFFAOYSA-N 0.000 description 1
- KNIUHBNRWZGIQQ-UHFFFAOYSA-N 7-diethoxyphosphinothioyloxy-4-methylchromen-2-one Chemical compound CC1=CC(=O)OC2=CC(OP(=S)(OCC)OCC)=CC=C21 KNIUHBNRWZGIQQ-UHFFFAOYSA-N 0.000 description 1
- 0 CCc1ccc(CN(*)*)cc1 Chemical compound CCc1ccc(CN(*)*)cc1 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical compound ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- 229920001174 Diethylhydroxylamine Polymers 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920001479 Hydroxyethyl methyl cellulose Polymers 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- NHTMVDHEPJAVLT-UHFFFAOYSA-N Isooctane Chemical compound CC(C)CC(C)(C)C NHTMVDHEPJAVLT-UHFFFAOYSA-N 0.000 description 1
- 239000004201 L-cysteine Substances 0.000 description 1
- 235000013878 L-cysteine Nutrition 0.000 description 1
- YIVJZNGAASQVEM-UHFFFAOYSA-N Lauroyl peroxide Chemical compound CCCCCCCCCCCC(=O)OOC(=O)CCCCCCCCCCC YIVJZNGAASQVEM-UHFFFAOYSA-N 0.000 description 1
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 1
- 238000006683 Mannich reaction Methods 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- 229920000557 Nafion® Polymers 0.000 description 1
- VNQABZCSYCTZMS-UHFFFAOYSA-N Orthoform Chemical compound COC(=O)C1=CC=C(O)C(N)=C1 VNQABZCSYCTZMS-UHFFFAOYSA-N 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 229920002845 Poly(methacrylic acid) Polymers 0.000 description 1
- 229920002396 Polyurea Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- OKKRPWIIYQTPQF-UHFFFAOYSA-N Trimethylolpropane trimethacrylate Chemical compound CC(=C)C(=O)OCC(CC)(COC(=O)C(C)=C)COC(=O)C(C)=C OKKRPWIIYQTPQF-UHFFFAOYSA-N 0.000 description 1
- 229910052770 Uranium Inorganic materials 0.000 description 1
- AZFNGPAYDKGCRB-XCPIVNJJSA-M [(1s,2s)-2-amino-1,2-diphenylethyl]-(4-methylphenyl)sulfonylazanide;chlororuthenium(1+);1-methyl-4-propan-2-ylbenzene Chemical compound [Ru+]Cl.CC(C)C1=CC=C(C)C=C1.C1=CC(C)=CC=C1S(=O)(=O)[N-][C@@H](C=1C=CC=CC=1)[C@@H](N)C1=CC=CC=C1 AZFNGPAYDKGCRB-XCPIVNJJSA-M 0.000 description 1
- JUIBLDFFVYKUAC-UHFFFAOYSA-N [5-(2-ethylhexanoylperoxy)-2,5-dimethylhexan-2-yl] 2-ethylhexaneperoxoate Chemical compound CCCCC(CC)C(=O)OOC(C)(C)CCC(C)(C)OOC(=O)C(CC)CCCC JUIBLDFFVYKUAC-UHFFFAOYSA-N 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- XYLMUPLGERFSHI-UHFFFAOYSA-N alpha-Methylstyrene Chemical compound CC(=C)C1=CC=CC=C1 XYLMUPLGERFSHI-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 125000004103 aminoalkyl group Chemical group 0.000 description 1
- SOIFLUNRINLCBN-UHFFFAOYSA-N ammonium thiocyanate Chemical compound [NH4+].[S-]C#N SOIFLUNRINLCBN-UHFFFAOYSA-N 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- GXCSNALCLRPEAS-CFYXSCKTSA-N azane (Z)-hydroxyimino-oxido-phenylazanium Chemical compound N.O\N=[N+](/[O-])c1ccccc1 GXCSNALCLRPEAS-CFYXSCKTSA-N 0.000 description 1
- 229910001422 barium ion Inorganic materials 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 229910052793 cadmium Inorganic materials 0.000 description 1
- BDOSMKKIYDKNTQ-UHFFFAOYSA-N cadmium atom Chemical compound [Cd] BDOSMKKIYDKNTQ-UHFFFAOYSA-N 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920003086 cellulose ether Polymers 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 229910052729 chemical element Inorganic materials 0.000 description 1
- 150000008280 chlorinated hydrocarbons Chemical class 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- BLCKNMAZFRMCJJ-UHFFFAOYSA-N cyclohexyl cyclohexyloxycarbonyloxy carbonate Chemical compound C1CCCCC1OC(=O)OOC(=O)OC1CCCCC1 BLCKNMAZFRMCJJ-UHFFFAOYSA-N 0.000 description 1
- 238000010908 decantation Methods 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- FVCOIAYSJZGECG-UHFFFAOYSA-N diethylhydroxylamine Chemical compound CCN(O)CC FVCOIAYSJZGECG-UHFFFAOYSA-N 0.000 description 1
- JVSWJIKNEAIKJW-UHFFFAOYSA-N dimethyl-hexane Natural products CCCCCC(C)C JVSWJIKNEAIKJW-UHFFFAOYSA-N 0.000 description 1
- DGLRDKLJZLEJCY-UHFFFAOYSA-L disodium hydrogenphosphate dodecahydrate Chemical compound O.O.O.O.O.O.O.O.O.O.O.O.[Na+].[Na+].OP([O-])([O-])=O DGLRDKLJZLEJCY-UHFFFAOYSA-L 0.000 description 1
- ZRRLFMPOAYZELW-UHFFFAOYSA-N disodium;hydrogen phosphite Chemical compound [Na+].[Na+].OP([O-])[O-] ZRRLFMPOAYZELW-UHFFFAOYSA-N 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000009713 electroplating Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000005265 energy consumption Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 1
- STVZJERGLQHEKB-UHFFFAOYSA-N ethylene glycol dimethacrylate Substances CC(=C)C(=O)OCCOC(=O)C(C)=C STVZJERGLQHEKB-UHFFFAOYSA-N 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- 239000008098 formaldehyde solution Substances 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- PYGSKMBEVAICCR-UHFFFAOYSA-N hexa-1,5-diene Chemical compound C=CCCC=C PYGSKMBEVAICCR-UHFFFAOYSA-N 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 150000002443 hydroxylamines Chemical class 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- LRDFRRGEGBBSRN-UHFFFAOYSA-N isobutyronitrile Chemical compound CC(C)C#N LRDFRRGEGBBSRN-UHFFFAOYSA-N 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 239000011133 lead Substances 0.000 description 1
- 229910001425 magnesium ion Inorganic materials 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid group Chemical group C(\C=C/C(=O)O)(=O)O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 229910052748 manganese Inorganic materials 0.000 description 1
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- FQPSGWSUVKBHSU-UHFFFAOYSA-N methacrylamide Chemical group CC(=C)C(N)=O FQPSGWSUVKBHSU-UHFFFAOYSA-N 0.000 description 1
- STZCRXQWRGQSJD-GEEYTBSJSA-M methyl orange Chemical compound [Na+].C1=CC(N(C)C)=CC=C1\N=N\C1=CC=C(S([O-])(=O)=O)C=C1 STZCRXQWRGQSJD-GEEYTBSJSA-M 0.000 description 1
- 229940012189 methyl orange Drugs 0.000 description 1
- JYVLIDXNZAXMDK-UHFFFAOYSA-N methyl propyl carbinol Natural products CCCC(C)O JYVLIDXNZAXMDK-UHFFFAOYSA-N 0.000 description 1
- 239000012022 methylating agents Substances 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- DAHPIMYBWVSMKQ-UHFFFAOYSA-N n-hydroxy-n-phenylnitrous amide Chemical compound O=NN(O)C1=CC=CC=C1 DAHPIMYBWVSMKQ-UHFFFAOYSA-N 0.000 description 1
- ODHYIQOBTIWVRZ-UHFFFAOYSA-N n-propan-2-ylhydroxylamine Chemical compound CC(C)NO ODHYIQOBTIWVRZ-UHFFFAOYSA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 150000002832 nitroso derivatives Chemical class 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- FWFGVMYFCODZRD-UHFFFAOYSA-N oxidanium;hydrogen sulfate Chemical compound O.OS(O)(=O)=O FWFGVMYFCODZRD-UHFFFAOYSA-N 0.000 description 1
- NWVVVBRKAWDGAB-UHFFFAOYSA-N p-methoxyphenol Chemical compound COC1=CC=C(O)C=C1 NWVVVBRKAWDGAB-UHFFFAOYSA-N 0.000 description 1
- DBSDMAPJGHBWAL-UHFFFAOYSA-N penta-1,4-dien-3-ylbenzene Chemical compound C=CC(C=C)C1=CC=CC=C1 DBSDMAPJGHBWAL-UHFFFAOYSA-N 0.000 description 1
- 125000000864 peroxy group Chemical group O(O*)* 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- AQSJGOWTSHOLKH-UHFFFAOYSA-N phosphite(3-) Chemical class [O-]P([O-])[O-] AQSJGOWTSHOLKH-UHFFFAOYSA-N 0.000 description 1
- OJMIONKXNSYLSR-UHFFFAOYSA-N phosphorous acid Chemical compound OP(O)O OJMIONKXNSYLSR-UHFFFAOYSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid group Chemical group C(C=1C(C(=O)O)=CC=CC1)(=O)O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 1
- 125000005543 phthalimide group Chemical class 0.000 description 1
- JAMNHZBIQDNHMM-UHFFFAOYSA-N pivalonitrile Chemical compound CC(C)(C)C#N JAMNHZBIQDNHMM-UHFFFAOYSA-N 0.000 description 1
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical group [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 239000004848 polyfunctional curative Substances 0.000 description 1
- 239000004810 polytetrafluoroethylene Substances 0.000 description 1
- 229920002635 polyurethane Polymers 0.000 description 1
- 239000004814 polyurethane Substances 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 235000010289 potassium nitrite Nutrition 0.000 description 1
- 239000004304 potassium nitrite Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- FBCQUCJYYPMKRO-UHFFFAOYSA-N prop-2-enyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCC=C FBCQUCJYYPMKRO-UHFFFAOYSA-N 0.000 description 1
- HJWLCRVIBGQPNF-UHFFFAOYSA-N prop-2-enylbenzene Chemical compound C=CCC1=CC=CC=C1 HJWLCRVIBGQPNF-UHFFFAOYSA-N 0.000 description 1
- 229940079877 pyrogallol Drugs 0.000 description 1
- MCJGNVYPOGVAJF-UHFFFAOYSA-N quinolin-8-ol Chemical compound C1=CN=C2C(O)=CC=CC2=C1 MCJGNVYPOGVAJF-UHFFFAOYSA-N 0.000 description 1
- 239000012266 salt solution Substances 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- JVBXVOWTABLYPX-UHFFFAOYSA-L sodium dithionite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])=O JVBXVOWTABLYPX-UHFFFAOYSA-L 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 229910001415 sodium ion Inorganic materials 0.000 description 1
- 235000010288 sodium nitrite Nutrition 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- VGTPCRGMBIAPIM-UHFFFAOYSA-M sodium thiocyanate Chemical compound [Na+].[S-]C#N VGTPCRGMBIAPIM-UHFFFAOYSA-M 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 238000002798 spectrophotometry method Methods 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 150000003464 sulfur compounds Chemical class 0.000 description 1
- 238000004381 surface treatment Methods 0.000 description 1
- 238000010557 suspension polymerization reaction Methods 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- BWSZXUOMATYHHI-UHFFFAOYSA-N tert-butyl octaneperoxoate Chemical compound CCCCCCCC(=O)OOC(C)(C)C BWSZXUOMATYHHI-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- DNYWZCXLKNTFFI-UHFFFAOYSA-N uranium Chemical compound [U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U][U] DNYWZCXLKNTFFI-UHFFFAOYSA-N 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- GPPXJZIENCGNKB-UHFFFAOYSA-N vanadium Chemical compound [V]#[V] GPPXJZIENCGNKB-UHFFFAOYSA-N 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 229920003169 water-soluble polymer Polymers 0.000 description 1
Images

Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J45/00—Ion-exchange in which a complex or a chelate is formed; Use of material as complex or chelate forming ion-exchangers; Treatment of material for improving the complex or chelate forming ion-exchange properties
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F212/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring
- C08F212/02—Monomers containing only one unsaturated aliphatic radical
- C08F212/04—Monomers containing only one unsaturated aliphatic radical containing one ring
- C08F212/06—Hydrocarbons
- C08F212/08—Styrene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/30—Introducing nitrogen atoms or nitrogen-containing groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/40—Introducing phosphorus atoms or phosphorus-containing groups
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B1/00—Electrolytic production of inorganic compounds or non-metals
- C25B1/01—Products
- C25B1/34—Simultaneous production of alkali metal hydroxides and chlorine, oxyacids or salts of chlorine, e.g. by chlor-alkali electrolysis
- C25B1/46—Simultaneous production of alkali metal hydroxides and chlorine, oxyacids or salts of chlorine, e.g. by chlor-alkali electrolysis in diaphragm cells
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25B—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES FOR THE PRODUCTION OF COMPOUNDS OR NON-METALS; APPARATUS THEREFOR
- C25B15/00—Operating or servicing cells
- C25B15/08—Supplying or removing reactants or electrolytes; Regeneration of electrolytes
-
- C—CHEMISTRY; METALLURGY
- C02—TREATMENT OF WATER, WASTE WATER, SEWAGE, OR SLUDGE
- C02F—TREATMENT OF WATER, WASTE WATER, SEWAGE, OR SLUDGE
- C02F1/00—Treatment of water, waste water, or sewage
- C02F1/42—Treatment of water, waste water, or sewage by ion-exchange
- C02F2001/425—Treatment of water, waste water, or sewage by ion-exchange using cation exchangers
-
- C—CHEMISTRY; METALLURGY
- C02—TREATMENT OF WATER, WASTE WATER, SEWAGE, OR SLUDGE
- C02F—TREATMENT OF WATER, WASTE WATER, SEWAGE, OR SLUDGE
- C02F2101/00—Nature of the contaminant
- C02F2101/10—Inorganic compounds
- C02F2101/20—Heavy metals or heavy metal compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F212/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring
- C08F212/02—Monomers containing only one unsaturated aliphatic radical
- C08F212/04—Monomers containing only one unsaturated aliphatic radical containing one ring
- C08F212/06—Hydrocarbons
- C08F212/12—Monomers containing a branched unsaturated aliphatic radical or a ring substituted by an alkyl radical
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F212/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring
- C08F212/34—Monomers containing two or more unsaturated aliphatic radicals
- C08F212/36—Divinylbenzene
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Polymers & Plastics (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Electrochemistry (AREA)
- Materials Engineering (AREA)
- Metallurgy (AREA)
- Inorganic Chemistry (AREA)
- Treatment Of Water By Ion Exchange (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Polymerisation Methods In General (AREA)
- Electrolytic Production Of Non-Metals, Compounds, Apparatuses Therefor (AREA)
Description
カルシウム2+;マグネシウム2+:<20ppb
ストロンチウム2+:<100ppb
バリウム2+:<500ppb
− クロルアルカリ電解で通例起こるような、水性ブラインからの、マグネシウム、カルシウム、ストロンチウム、およびバリウムなどのアルカリ土類金属、特にカルシウムの
− 水溶液もしくは有機液体またはそれらの蒸気からの重金属および貴金属の、特にアルカリ土類金属およびアルカリ金属の水溶液からの水銀の(特にブラインまたはアルカリ金属塩化物電解からの水銀の)
著しくより良好な除去のための新規キレート形成性交換体の提供である。
a)少なくとも1種のモノビニル芳香族化合物および少なくとも1種のポリビニル芳香族化合物およびまた少なくとも1種のポロジェンおよび少なくとも1種の開始剤または開始剤の組み合わせのモノマー液滴が、噴出原理(jetting principle)またはシード供給原理(seed−feed principle)によって反応させられて単分散架橋ポリマービーズを形成し、
b)前記単分散架橋ポリマービーズの1〜15重量%のサブ部分を不活性溶媒中に存在するフタルイミド誘導体のSO3付加体へ導入し、35℃以下の温度で1〜15時間撹拌し、ここで、遊離SO3の量がフタルイミドの1モルを基準として0.28〜0.5モルであり、その後、ポリマービーズの残りの量を、不活性溶媒中に存在するフタルイミド誘導体のSO3付加体へ30〜75℃の温度で導入することによって、ポリマービーズがフタルイミド誘導体でフタルイミドメチル化され、
c)前記フタルイミドメチル化ポリマービーズがアミノメチル化ポリマービーズに転化され、そして
d)前記アミノメチル化ポリマービーズが反応させられてキレート官能基を有するイオン交換体を形成する
を特徴とする、構造要素(I)

で表されるキレート官能基を有する単分散イオン交換体の製造方法に関する。
a)少なくとも1種のモノビニル芳香族化合物および少なくとも1種のポリビニル芳香族化合物およびまた少なくとも1種のポロジェンおよび少なくとも1種の開始剤または開始剤の組み合わせのモノマー液滴を、噴出原理またはシード供給原理によって反応させて単分散架橋ポリマービーズを形成する工程と、
b)前記単分散架橋ポリマービーズの1〜15重量%のサブ部分を不活性溶媒中に存在するフタルイミド誘導体のSO3付加体へ導入し、35℃以下の温度で1〜15時間撹拌し、ここで、遊離SO3の量がフタルイミドの1モルを基準として0.28〜0.5モルであり、その後、ポリマービーズの残りの量を、不活性溶媒中に存在するフタルイミド誘導体のSO3付加体へ30〜75℃の温度で導入することによって、ポリマービーズをフタルイミド誘導体でフタルイミドメチル化する工程と、
c)前記フタルイミドメチル化ポリマービーズをアミノメチル化ポリマービーズに転化する工程と、
d)前記アミノメチル化ポリマービーズを、P−H酸化合物と組み合わせたホルマリンと懸濁液で反応させてキレート官能基を有するイオン交換体を形成する工程と
により好ましくは得られる、構造要素(I)
のキレート官能基を有し、かつ、
3.1〜3.9モル/lの総容量TCおよび70〜85nmの中央孔径を有する単分散イオン交換体に関する。
置換度は、本特許出願の場合においては、構造要素(I)においてどういう割合でアミノ基の2個の水素が、以下の基
濾過カラムに100mlの交換体を充填し、カラムを1.5時間で3重量%濃度の塩酸で溶出する。次にカラムを、溶出液が中性になるまで脱塩水で洗浄する。
X=溶出液分画の数
ΣV=溶出液の滴定での1N塩酸のml単位での総消費量
中央孔径および粒度分布は、特にDIN 66133によって明記されるように、Hg−ポロシメトリによって測定する。
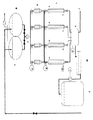
先ず、撹拌容器に5リットルの脱塩水を満たし、撹拌機を100rpmの回転速度に設定する(図2を参照されたい)。
X=脱塩水のための供給
Y=攪拌機
Z=オーバーフロー
R=測定センサー、HCB 250
A=溶出液
P=PMT 2120測定計数器
B=篩
R=パソコン
D=プリンター
B=遮断弁
T=キーボード
粒度分布のマニュアル描写は、粒度格子(例えばSelecta No.421 1/2 A4、Schleicher & Schuell)上に測定された篩画分の容積パーセントをプロットし、曲線を形成するために個々のプロット点を結ぶことによって行われる。
横座標:%単位の個々の篩での残留物合計
縦座標:mm単位の粒径
a)オートマチック評価
プログラム終了後に、オートマチック評価が、粒度分布、有効粒径および均等係数の測定を始める。
b)マニュアル評価
粒度分布のマニュアル描写は、粒度格子(例えばSelecta No.421 1/2 A4、Schleicher & Schuell)上に測定された篩画分の容積百分率をプロットし、曲線を作るために個々のプロット点を結ぶことによって進行する。
横座標:%単位の個々の篩での残留物合計
縦座標:mm単位の粒径
細孔容積を、特にDIN 66135に明記されるように、Hg−ポロシメトリによって測定する。
1a)スチレン、ジビニルベンゼンおよびエチルスチレンをベースとする単分散マクロ多孔性ポリマービーズの製造
10リットルのガラス反応器に、3000gの脱塩水を装入し、320gの脱塩水中の10gのゼラチン、16gのリン酸水素二ナトリウム12水和物および0.73gのレゾルシノールの溶液を加え、混合した。混合物を加熱し、25℃に維持した。撹拌しながら、その後、3.6重量%のジビニルベンゼンおよび0.9重量%のエチルスチレン(80%ジビニルベンゼンを含むジビニルベンゼンとエチルスチレンとの市販の異性体混合物として使用される)、0.5重量%のジベンゾイルペルオキシド、56.2重量%のスチレンおよび38.8重量%のイソドデカン(ペンタメチルヘプタンの高い割合を有する工業的異性体混合物)の、狭い粒度分布を有する3200gのマイクロカプセル化モノマー液滴であって、マイクロカプセルがゼラチンおよびアクリルアミドとアクリル酸とのコポリマーのホルムアルデヒド硬化コンプレックス・コアセルベートからなるモノマー液滴と、12のpHを有する3200gの水相との混合物を加えた。モノマー液滴の中央粒径は260μmであった。
室温で、3467gのジクロロエタン、823.9gのフタルイミドおよび579.4gの29.6重量%濃度のホルマリンを装入した。この懸濁液のpHを、水酸化ナトリウム溶液を使用して5.5〜6に設定した。その後、水を蒸留によって除去した。次に、60.4gの硫酸を加えた。生じた水を蒸留によって除去した。バッチを冷却した。30℃で、255.2gの65%濃度の発煙硫酸、その後、42.4gのプロセス工程1a)により生成した単分散ポリマービーズを加えた。懸濁液を30℃でさらに2時間撹拌した。次に、30分の間に、さらなる382gのプロセス工程1a)により生成した単分散ポリマービーズを加えた。懸濁液を70℃に加熱し、この温度でさらに6.5時間撹拌した。反応液を離水し、脱塩水を加え、残存量のジクロロエタンを蒸留によって除去した。
アミドメチル化ポリマービーズの収量:2610ml
元素分析による組成:
炭素:74.9重量%;
水素: 4.8重量%;
窒素: 5.8重量%;
残り: 酸素。
反応器で、825.2mlの50重量%濃度の水酸化ナトリウム溶液を、脱塩水で1560mlの総容積に構成した。これに、室温で2600mlの1b)からのアミドメチル化ポリマービーズを加えた。この懸濁液を180℃に2時間加熱し、この温度で8時間撹拌した。得られたポリマービーズを脱塩水で洗浄した。
収量:4200ml
元素分析による組成:
窒素:12.2重量%;
炭素:77.2重量%;
水素: 8.1重量%;
酸素: 3.1重量%
塩基性基の量の測定:樹脂の1リットル当たり2.31モル
反応器に、室温で234mlの脱塩水を装入した。これに、450mlの実施例1c)からのアミノメチル化ポリマービーズを加えた。その後、320.2gのジメチルホスファイトを15分で加えた。混合物をさらに30分間撹拌した。その後、懸濁液を60℃に加熱した。4時間の間に、815グラムの硫酸一水和物をこれに加え、その後、混合物を還流温度に加熱した。この温度で、次に、1時間の間に、367.5グラムの水性ホルムアルデヒド溶液(29.7重量%濃度)をこれに加えた。懸濁液を還流温度でさらに6時間撹拌した。その後、バッチを冷却した。得られたキレート樹脂を脱塩水で、篩上で洗浄し、次にガラスカラムへ移した。上部から、3時間で、3リットルの4重量%濃度の水性水酸化ナトリウム溶液をこれに加え、その後、カラムを3ベッド体積の脱塩水で再び洗浄した。
ナトリウム形での樹脂収量:1010ml
元素分析による組成:
窒素: 4.5重量%
リン:14.0重量%
中央ビーズ径:413μ
キレート基の量(総容量):3.39モル/l
中央孔径:75nm
元の状態での安定性:100%無傷ビーズ
ローラー試験後の安定性:97%無傷ビーズ
膨潤安定性試験後の安定性:99%無傷ビーズ
欧州特許出願公開第1 078 690 A2号明細書によるアミノメチルホスホン酸基を有するキレート樹脂(中央ビーズ径φ0.65mm±0.05mm)と比較した、アミノメチルホスホン酸基を有するキレート樹脂(中央ビーズ径0.41mm±0.05mm)を使用する30%濃度の塩化ナトリウム溶液からのカルシウム除去。
蠕動ポンプを用いて、ブラインを、測定されるべき様々な粒径のイオン交換体上に連続的にポンプ送液した。
供給濃度: 12mgCa/リットル
供給pH: 8.2
供給速度: 20BV/時
カラム温度: 60℃
破過濃度: 20μgCa/リットル
樹脂の使用量: 100ml
樹脂形: Na形
A= 300gのNaCl/リットルのブラインリザーバ容器;400リットル容量
B= 300gのNaCl/リットルのブラインリザーバ容器;400リットル容量
C= 蠕動ポンプ
D= TIC(70℃)
E= ADI(カルシウム滴定)
F= 廃水
G= QIR
H= LIC
J= ブライン混合容器600リットル
K= 加熱可能なプレフィルター(操作温度70℃)
L= 加熱可能な濾過カラム(操作温度60℃)
M= ブライン移動ビーカー/ブラインは収集容器へ蠕動ポンプを用いて移動させた
TIC=温度指示調節計
ADI=Metrohm製のADI 2040プロセス分析計の省略形
QIR=品質表示記録計(この場合Qは濾液中のCa/Mg含有率である)
LIC=調節器付きの標準指圧器
標準粒径(φ0.65mm±0.05mm)でのカルシウムの動的吸収容量:12.3gのCa/リットル
濾液中のカルシウムの量が20ppbを超えるまでの、超濾過されたブラインの数/量(ベッド体積)およびキレート樹脂の負荷継続期間:53時間で1060ベッド体積
濾液中のカルシウム量が20ppbを超えるまでの、超濾過されたブラインの数/量(ベッド体積)およびキレート樹脂の負荷継続期間:96.7時間で1935ベッド体積
アミノメチルホスホン酸基を有する実施例2に使用された2種のキレート樹脂の特性データの比較
Claims (3)
- a)少なくとも1種のモノビニル芳香族化合物および少なくとも1種のポリビニル芳香族化合物およびまた少なくとも1種のポロジェンおよび少なくとも1種の開始剤または開始剤の組み合わせのモノマー液滴が、噴出原理またはシード供給原理によって反応させられて単分散架橋ポリマービーズを形成し、
b)前記単分散架橋ポリマービーズの1〜15重量%のサブ部分を不活性溶媒中に存在するフタルイミドのSO3付加体へ導入し、35℃以下の温度で1〜15時間撹拌し、ここで、遊離SO3の量がフタルイミドの1モルを基準として0.28〜0.65モルであり、その後、ポリマービーズの残りの量を、不活性溶媒中に存在するフタルイミドのSO3付加体へ30〜75℃の温度で導入することによって、ポリマービーズがフタルイミドでフタルイミドメチル化され、
c)前記フタルイミドメチル化ポリマービーズがアミノメチル化ポリマービーズに転化され、そして
d)前記アミノメチル化ポリマービーズが反応させられてキレート官能基を有するイオン交換体を形成する
ことを特徴とする、構造要素(I)
で表されるキレート官能基を有する単分散イオン交換体の製造方法。 - 構造要素(I)
のキレート官能基を有し、かつ、3.1〜3.9モル/lの総容量TCおよび70〜85nmの中央孔径を有する単分散イオン交換体。 - a)少なくとも1種のモノビニル芳香族化合物および少なくとも1種のポリビニル芳香族化合物およびまた少なくとも1種のポロジェンおよび少なくとも1種の開始剤または開始剤の組み合わせのモノマー液滴を、噴出原理またはシード供給原理によって反応させて単分散架橋ポリマービーズを形成する工程と、
b)前記単分散架橋ポリマービーズの1〜15重量%のサブ部分を不活性溶媒中に存在するフタルイミドのSO3付加体へ導入し、35℃以下の温度で1〜15時間撹拌し、ここで、遊離SO3の量がフタルイミドの1モルを基準として0.28〜0.5モルであり、その後、ポリマービーズの残りの量を、不活性溶媒中に存在するフタルイミドのSO3付加体へ30〜75℃の温度で導入することによって、ポリマービーズをフタルイミドでフタルイミドメチル化する工程と、
c)前記フタルイミドメチル化ポリマービーズをアミノメチル化ポリマービーズに転化する工程と、
d)前記アミノメチル化ポリマービーズを、P−H酸化合物と組み合わせたホルマリンと懸濁液で反応させてキレート官能基を有するイオン交換体を形成する工程と
によって得られる、請求項2に記載のキレート官能基を有する単分散イオン交換体。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102009047848.5 | 2009-09-30 | ||
| DE102009047848A DE102009047848A1 (de) | 2009-09-30 | 2009-09-30 | Verfahren zur verbesserten Entfernung von Kationen mittels Chelatharzen |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2011116944A JP2011116944A (ja) | 2011-06-16 |
| JP2011116944A5 JP2011116944A5 (ja) | 2013-09-12 |
| JP5600541B2 true JP5600541B2 (ja) | 2014-10-01 |
Family
ID=43525178
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010218848A Expired - Fee Related JP5600541B2 (ja) | 2009-09-30 | 2010-09-29 | キレート樹脂を用いるカチオンの改善された除去方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8440730B2 (ja) |
| EP (1) | EP2305381A3 (ja) |
| JP (1) | JP5600541B2 (ja) |
| CN (1) | CN102029201A (ja) |
| DE (1) | DE102009047848A1 (ja) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102013105177A1 (de) | 2013-05-21 | 2014-11-27 | Fraunhofer-Gesellschaft zur Förderung der angewandten Forschung e.V. | Verfahren zur Gewinnung metallischer Anteile sowie von metallabgereichertem Material aus metallhaltigen Materialien |
| EP3183275B1 (de) * | 2014-08-20 | 2018-05-23 | Lanxess Deutschland GmbH | Sulfonierte, aminomethylierte chelatharze |
| DE102014221241A1 (de) * | 2014-10-20 | 2016-04-21 | Volkswagen Aktiengesellschaft | Befeuchtungseinrichtung für ein Brennstoffzellensystem sowie Brennstoffzellensystem |
| US11014832B2 (en) | 2014-12-05 | 2021-05-25 | Blue Cube Ip Llc | Brine purification process |
| JP6738145B2 (ja) * | 2015-12-22 | 2020-08-12 | 学校法人福岡大学 | 次亜塩素酸ソーダの製造方法及び次亜塩素酸ソーダの製造装置 |
| CN110003376B (zh) * | 2019-03-11 | 2021-09-28 | 西安蓝晓科技新材料股份有限公司 | 一种从含镍溶液中制备硫酸镍的螯合树脂制备方法 |
| CN110980876A (zh) * | 2019-11-21 | 2020-04-10 | 武汉百富环保工程有限公司 | 一种从钝化液中回收铜并回用铬的处理工艺 |
| CN116323693B (zh) * | 2020-09-30 | 2025-12-23 | 朗盛德国有限责任公司 | 新颖的螯合树脂 |
| JP2025510170A (ja) * | 2022-03-22 | 2025-04-14 | ランクセス・ドイチュランド・ゲーエムベーハー | アルミニウムイオン及び/又は亜鉛イオンを溶出させるための方法 |
| CN117361766A (zh) * | 2022-06-28 | 2024-01-09 | 宝山钢铁股份有限公司 | 硅钢反渗透浓缩液的零排放工艺、设备及其滤料、吸附剂及制备方法 |
| WO2025181142A1 (de) * | 2024-02-29 | 2025-09-04 | Lanxess Deutschland Gmbh | Herstellung von calciumreduziertem lithiumcarbonat |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4382124B1 (en) | 1958-07-18 | 1994-10-04 | Rohm & Haas | Process for preparing macroreticular resins, copolymers and products of said process |
| DE2418976C3 (de) * | 1974-04-19 | 1978-10-26 | Bayer Ag, 5090 Leverkusen | Verfahren zur Herstellung von wasserunlöslichen Kunstharzen mit Anionenaustauscher-Eigenschaften |
| DE3031737A1 (de) * | 1980-08-22 | 1982-04-01 | Bayer Ag, 5090 Leverkusen | Verfahren zur herstellung von perlpolymerisaten einheitlicher teilchengroesse |
| CA1166413A (en) | 1980-10-30 | 1984-05-01 | Edward E. Timm | Process and apparatus for preparing uniform size polymer beads |
| US4419245A (en) | 1982-06-30 | 1983-12-06 | Rohm And Haas Company | Copolymer process and product therefrom consisting of crosslinked seed bead swollen by styrene monomer |
| US4818773A (en) | 1983-02-24 | 1989-04-04 | Rohm And Haas Company | Alkylaminophosphonic chelating resins, their preparation and use in purifying brines |
| DE3704307A1 (de) | 1987-02-12 | 1988-08-25 | Dow Chemical Gmbh | Komplexbildendes harz des geltyps und seine verwendung zur verringerung der konzentration mehrwertiger erdalkali- und/oder schwermetallionen in loesungen |
| US5231115A (en) * | 1991-12-19 | 1993-07-27 | The Dow Chemical Company | Seeded porous copolymers and ion-exchange resins prepared therefrom |
| DE4328075A1 (de) * | 1993-08-20 | 1995-02-23 | Bayer Ag | Verfahren zur Herstellung schwachbasischer Anionenaustauscher und dafür geeigneter Reagenzien |
| US5710593A (en) | 1996-01-22 | 1998-01-20 | Tektronix, Inc. | SCH phase adjustment for digitally synthesized video test signals |
| EP1078690B1 (de) * | 1999-08-27 | 2011-10-12 | LANXESS Deutschland GmbH | Verfahren zur Herstellung von monodispersen Ionenaustauschern mit chelatisierenden Gruppen |
| DE10121163A1 (de) * | 2001-04-30 | 2002-10-31 | Bayer Ag | Verfahren zur Herstellung heterodisperser Chelatharze |
| DE10353534A1 (de) * | 2003-11-14 | 2005-06-16 | Bayer Chemicals Ag | Chelataustauscher |
| DE102006034659A1 (de) * | 2006-07-24 | 2008-02-21 | Lanxess Deutschland Gmbh | Ionenaustauscher zur Gewinnung von Wertmetallen |
| DE102007034732A1 (de) * | 2007-07-23 | 2009-01-29 | Lanxess Deutschland Gmbh | Verfahren zur Herstellung von Chelatharzen |
| DE102007034733A1 (de) * | 2007-07-23 | 2009-01-29 | Lanxess Deutschland Gmbh | Verfahren zur Herstellung monodisperser Chelatharze |
-
2009
- 2009-09-30 DE DE102009047848A patent/DE102009047848A1/de not_active Withdrawn
-
2010
- 2010-09-14 EP EP10176509A patent/EP2305381A3/de not_active Withdrawn
- 2010-09-27 US US12/890,794 patent/US8440730B2/en not_active Expired - Fee Related
- 2010-09-29 JP JP2010218848A patent/JP5600541B2/ja not_active Expired - Fee Related
- 2010-09-29 CN CN2010105021754A patent/CN102029201A/zh active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| EP2305381A3 (de) | 2011-09-14 |
| JP2011116944A (ja) | 2011-06-16 |
| US20110089116A1 (en) | 2011-04-21 |
| EP2305381A2 (de) | 2011-04-06 |
| DE102009047848A1 (de) | 2011-03-31 |
| CN102029201A (zh) | 2011-04-27 |
| US8440730B2 (en) | 2013-05-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5600540B2 (ja) | キレート樹脂を用いるカチオンの改善された除去方法 | |
| JP5600541B2 (ja) | キレート樹脂を用いるカチオンの改善された除去方法 | |
| US6649663B1 (en) | Process for preparing monodisperse ion exchangers having chelating functional groups and the use thereof | |
| CN102015107B (zh) | 氨甲基吡啶树脂 | |
| US8177982B2 (en) | Method of producing monodisperse chelate resins | |
| CN101977689B (zh) | 氨甲基吡啶树脂 | |
| JP2001089524A (ja) | チオ尿素基をもった新規単一分散型の交叉結合したビーズ状重合体を製造する方法、および金属化合物の吸着に対するその使用 | |
| RU2754099C2 (ru) | Гранулированные полимеры на основе функционализированного кватернизированным диэтилентриамином полиакрилата, способ их получения, применение этих полимеров и способ удаления оксоанионов из водных и/или органических растворов | |
| JP2007533789A (ja) | キレート交換樹脂 | |
| JP2002363216A (ja) | ヘテロ分散性キレート樹脂の製造方法 | |
| US20130245140A1 (en) | Chelate resins | |
| CN102712713B (zh) | 亚甲基氨基乙磺酸螯合树脂 | |
| RU2681852C2 (ru) | Новые, легированные алюминием, содержащие группы иминодиуксусной кислоты хелатообразующие смолы | |
| JP2007533578A (ja) | 硫酸の精製方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130725 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20130725 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20140217 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20140224 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20140520 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20140523 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140626 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140722 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20140818 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5600541 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |