JP5555872B2 - 金属(iv)テトラ−アミジネート化合物ならびに蒸着においての使用 - Google Patents
金属(iv)テトラ−アミジネート化合物ならびに蒸着においての使用 Download PDFInfo
- Publication number
- JP5555872B2 JP5555872B2 JP2009518210A JP2009518210A JP5555872B2 JP 5555872 B2 JP5555872 B2 JP 5555872B2 JP 2009518210 A JP2009518210 A JP 2009518210A JP 2009518210 A JP2009518210 A JP 2009518210A JP 5555872 B2 JP5555872 B2 JP 5555872B2
- Authority
- JP
- Japan
- Prior art keywords
- metal
- hafnium
- compound
- zirconium
- vapor
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images



Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C257/00—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines
- C07C257/10—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. amidines
- C07C257/14—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. amidines having carbon atoms of amidino groups bound to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic System
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F11/00—Compounds containing elements of Groups 6 or 16 of the Periodic System
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
-
- C—CHEMISTRY; METALLURGY
- C23—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; CHEMICAL SURFACE TREATMENT; DIFFUSION TREATMENT OF METALLIC MATERIAL; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL; INHIBITING CORROSION OF METALLIC MATERIAL OR INCRUSTATION IN GENERAL
- C23C—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; SURFACE TREATMENT OF METALLIC MATERIAL BY DIFFUSION INTO THE SURFACE, BY CHEMICAL CONVERSION OR SUBSTITUTION; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL
- C23C16/00—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes
- C23C16/22—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes characterised by the deposition of inorganic material, other than metallic material
- C23C16/30—Deposition of compounds, mixtures or solid solutions, e.g. borides, carbides, nitrides
- C23C16/34—Nitrides
-
- C—CHEMISTRY; METALLURGY
- C23—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; CHEMICAL SURFACE TREATMENT; DIFFUSION TREATMENT OF METALLIC MATERIAL; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL; INHIBITING CORROSION OF METALLIC MATERIAL OR INCRUSTATION IN GENERAL
- C23C—COATING METALLIC MATERIAL; COATING MATERIAL WITH METALLIC MATERIAL; SURFACE TREATMENT OF METALLIC MATERIAL BY DIFFUSION INTO THE SURFACE, BY CHEMICAL CONVERSION OR SUBSTITUTION; COATING BY VACUUM EVAPORATION, BY SPUTTERING, BY ION IMPLANTATION OR BY CHEMICAL VAPOUR DEPOSITION, IN GENERAL
- C23C16/00—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes
- C23C16/22—Chemical coating by decomposition of gaseous compounds, without leaving reaction products of surface material in the coating, i.e. chemical vapour deposition [CVD] processes characterised by the deposition of inorganic material, other than metallic material
- C23C16/30—Deposition of compounds, mixtures or solid solutions, e.g. borides, carbides, nitrides
- C23C16/40—Oxides
- C23C16/405—Oxides of refractory metals or yttrium
Description

イソブチルアミン(7.3 グラム、0.1 mol)、アセトニトリル(4.1 グラム、0.1 mol)及びランタントリフレート、La(CF3SO3)3、(1.2 グラム、0.002 mol)溶液は窒素雰囲気下で30時間還流された。未反応出発原料と副生物2,4,6−トリメチル−1,3,5−トリアジンは40℃でおよそ0.2 Torrにおいて分別蒸留により分離された。次に無色N,N’−ジイソブチルアセトアミジンは95℃及び0.06 torrにおいて留出された。さらなる精製は第二蒸留により行なわれた。収量:6.4 グラム(75%イソブチルアミンに基づく)。1H NMR(ベンゼン−d6+少量のCD3OD、ppm):3.1(d, 4、NCH2)、1.9(m, 2、CH(CH3)2)、1.7(s, 3、CCH3)、1.0(d, 12、CH(CH3)2)。
0.8グラム(3 mmol)のテトラキス(ジメチルアミド)ジルコニウム(IV)、Zr(NMe2)4は10 mLのトルエンに溶解され、次に−30℃まで30分間冷却された。この溶液に2.3グラム(13.5 mmol)N,N’−ジイソブチルアセトアミジン、iBu−AMDが加えられ、混合物は一夜中90℃に加熱された(配位交換反応)。−30℃に冷却した後に無色結晶質は沈殿され、濾過分離された。収量:1.85グラム(80%)。1HNMR(ベンゼン−d6, ppm):3.10(d, 16,J= 6.9 Hz、NCH2)、1.89(m, 8、CH(CH3)2)、1.71(s, 12、CCH3)1.00(d, 48,J= 6.6 Hz, d、CH(CH3)2)。13C NMR(ベンゼン−d6, ppm):174(CCH3)、55.76(NCH2)、31.74(CH(CH3)2)、21.134(CH(CH3)2、12.10(CCH3)。C40H84N8Zrについて分析計算:C62.53、H11.02、N14.58。実測:C62.76、H11.25、N14.50。
この化合物は実施例2においてZr(iBu−AMD)4について記載されたものと同様の方法で、3mmolのテトラキス(ジメチルアミド)ハフニウム(IV)、Hf(NMe2)4から開始して調製された。生成物は白色粉末として単離された。収量:2.17グラム(85%)。1H NMR(ベンゼン−d6, ppm):3.15(d, 16,J= 7.2 Hz、NCH2)、1.87(m, 8、CH(CH3)2),1.70(s, 12、CCH3)、0.99(d, 48,J= 6.8 Hz、CH(CH3)2)。C40H84HfN8について分析計算:C56.15、H9.90、N13.10。実測:55.85、H9.97、N13.30。
無水物ランタントリフレート(3.00グラム、5.12 mmol)は圧力容器に配置された。乾燥アセトニトリル(23.3グラム、0.568 mol)は常温容器に凝縮された。容器は液体窒素で冷却され、無水メチルアミン(53.1グラム、1.71 mol)が加えられた。容器は密封され、室温に温めることを許容される。副生物アンモニアは日々に解放された。反応は大部分3日後に完全であった。次に無色N,N’−ジメチルアセトアミジンは20℃及び0.04 torrにおいての昇華による副生物N−メチルアセトアミジンを除去することにより単離された。1H NMR(ベンゼン−d6):2.60(s, 6、NCH3)、1.40(s, 3、CCH3)。
この化合物はZr(iBu−AMD)4について実施例2に記載されるものと同様にN,N’−ジイソブチルアセトアミジンの代わりにN,N’−ジメチルアセトアミジンを用いて配位交換により調製された。代わりにZrCl4はジオキサンに溶解されたN,N’−ジメチルアセトアミジンリチウム塩と反応された(塩複分解反応)。この反応混合物は加熱されて8時間還流された。ジオキサンの蒸発後に固体残留物はペンタンに加えて抽出された。沈殿塩化リチウムを取り除く懸濁液のデカンテーション後にペンタンは減圧下に蒸発され、粗生成物を生じ、それが次に60℃及び40ミリバールの圧力において昇華により精製された。大気圧において160℃で昇華されることもできる。収率は合成が小規模で行なわれたときには昇華後に24%であった。1H NMR(ベンゼン−d6, ppm):3.03(s, 24、NCH3)、1.57(s, 12、CCH3).13C NMR(ベンゼン−d6, ppm):175.99(s、CCH3)、34.55(s、NCH3),9.84(s、CCH3)。C16H36N8Zrについて分析計算:C44.51、H8.40、N25.95。実測:C45.31、H7.92、N25.60または第二分析においてC43.30、H8.76、N24.87。この生成物はTG曲線からそのT1/2値は0.6%残存量で216℃であるためにテトラキス(N,N’−ジイソブチルアセトアミジナート)ジルコニウム(IV)、実施例2の生成物よりも揮発性である。その融点は約168℃である。
この化合物は実施例5において記載される塩複分解反応によりHfCl4から調製された。1H NMR(ベンゼン−d6, ppm) :3.07 (s, 24、NCH3) , 1.55 (s, 12, CCH3)。13C NMR(ベンゼン−d6, ppm) :175.69 (s、CCH3) 、34.31 (s、NCH3) 、10.09 (s、CCH3) 。C16H36HfN8について分析計算:C37.03、H6.99、N21.59。実測:37.00、H6.89、N21.34。この生成物はTG曲線からそのT1/2値は221℃であるためにテトラキス (N, N' −ジイソブチルアセトアミジナート) ハフニウム(IV)、実施例3の生成物よりも揮発性である。蒸発後のその残量はごくわずか、1%未満であり、その融点は約171℃である。
リチウムN,N’−ジメチルプロピオンアミジネートのジオキサン溶液は実施例4の記載方法によりアセトニトリルの代わりにプロピオンニトリルを用いて調製された。この溶液は次に、テトラキス(N,N’−ジメチルプロピオンアミジナート)ジルコニウム(IV)を調製するために実施例5に記載される塩複分解方法でZrCl4と使用された。この化合物は実施例2に記載されるものと類似する配位交換反応によっても調製されることが可能である。1H NMR(ベンゼン−d6, ppm):3.07(s, 24、NCH3)、2.10(q, 8,J= 7.6 Hz、CH2CH3)、0.96(t, 12,J= 7.6 Hz、CH2CH3)。13C NMR(ベンゼン−d6, ppm):180.12(s、CCH2CH3),33.92(s、NCH3)、17.31(s、CCH2CH3)、10.41(s、CCH2CH3)。C20H44N8Zrについて分析計算:C49.24、H9.09、N22.97。実測:49.42、H9.04、N22.43。その融点は109℃であり十分に低いので、バブラーで気化させるのに十分な高さの温度において液体である。TG曲線からそのT1/2値は0.6%のごくわずかの残量で245℃である。
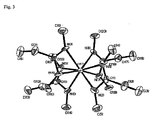
この化合物は実施例7において記載される塩複分解反応によりHfCl4から調製された。実施例1に記載されるものに類似する配位交換反応によっても調製されることが可能である。1H NMR(ベンゼン−d6, ppm):3.10(s, 24、NCH3)、2.08(q, 8,J= 7.6 Hz、CH2CH3)、0.95(t, 12,J= 7.6 Hz、CH2CH3)。13C NMR(ベンゼン−d6, ppm):179.75(s、CCH2CH3)、33.71(s、NCH3)、17.51(s、CCH2CH3)、10.40(s、CCH2CH3)。C20H44HfN8について分析計算:C41.77、H7.71、N19.48。実測:42.32、H8.11、N19.18。その融点は114℃であり十分に低いので、バブラーで気化させるのに十分な高さの温度において液体である。TG曲線からそのT1/2値はごくわずかの不揮発残留物で252℃である。X線結晶学は図3において示されるテトラキス(N,N’−ジメチルプロピオンアミジナート)ハフニウム(IV)の分子構造を決定するために使用され、ただし、原子は50%熱長円体により示され、水素原子は明瞭のために省略される。各分子は8窒素原子により四アミジネート配位子から取り囲まれた一つのハフニウム原子を含む。
リチウムN,N’−ジメチルブチルアミジネートのジオキサン溶液はアセトニトリルの代わりにブチロニトリルを用いて、実施例4に記載される方法により調製された。この溶液は次に、テトラキス(N,N’−ジメチルブチルアミジナート)ジルコニウム(IV)を調製するために実施例5に記載される塩複分解方法でZrCl4と使用された。この化合物は実施例1に記載されるものと類似する配位交換反応によっても調製されることが可能である。化合物は室温で液体であるので、昇華ではなく蒸留により精製された。1H NMR(ベンゼン−d6, ppm):3.11(s, 24、NCH3)、2.15(t, 8,J= 8.0 Hz、CCH2CH2CH3)、1.49(m, 8、CCH2CH2CH3)、0.90(t, 12,J= 6.8 Hz、CCH2CH2CH3)。13C NMR(ベンゼン−d6, ppm):179.27(s、CCH2CH2CH3)、34.28(s、NCH3)、26.14(s、CCH2CH2CH3)、19.82(s、CCH2CH2CH3)、14.47(s、CCH2CH2CH3)。C24H52N8Zrについて分析計算:C52.99、H9.63、N20.60。実測:53.63、H9.87、N20.89。そのT1/2値は246℃であり、蒸発してごくわずか残留物が残る。
この化合物は実施例9に記載される塩複分解反応によりHfCl4から調製された。実施例1に記載されるものと類似する配位交換反応によっても調製されることが可能である。化合物は室温において液体であるので、昇華ではなく蒸留により精製された。1H NMR(ベンゼン−d6, ppm):3.15(s, 24、NCH3)、2.13(t, 8,J= 8.0 Hz、CCH2CH2CH3)、1.49(m, 8、CCH2CH2CH3)、0.89(t, 12,J= 6.8 Hz、CCH2CH2CH3)。13C NMR(ベンゼン−d6, ppm):178.87(s、CCH2CH2CH3)、34.08(s、NCH3)、26.29(s、CCH2CH2CH3)、19.82(s、CCH2CH2CH3)、14.41(s、CCH2CH2CH3)。C24H52HfN8について分析計算:C45.67、H8.30、N17.75。実測:45.31、H8.81、N17.61。そのT1/2値は252℃であり、蒸発してごくわずか残留物が残る。
リチウムN,N’−ジエチルアセトアミジナートのジオキサン溶液はメチルアミンの代わりにエチルアミンを用いて、実施例4に記載される方法により調製された。この溶液はテトラキス(N,N’−ジエチルアセトアミジナート)ジルコニウム(IV)を調製するために実施例5に記載される塩複分解方法でZrCl4と使用された。この化合物は実施例1に記載されるものと類似する配位交換反応によっても調製されることが可能である。化合物は室温において液体であるので、昇華の代わりに蒸留により精製された。1H NMR(ベンゼン−d6, ppm):3.32(q, 16,J= 7.2 Hz、NCH2CH3)、1.63(s, 12、CCH3)、1.10(t, 24,J= 7.2 Hz、NCH2CH3)。13C NMR(ベンゼン−d6, ppm):173.59(s、CCH3)、41.38(s、NCH2CH3)、18.00(s、NCH2CH3)、10.20(s、CCH3)。C24H52N8Zrについて分析計算:C52.99、H9.63、N20.60。実測:52.86、H9.40、N20.99。そのT1/2値は242℃であり、蒸発してごくわずか残留物が残る。
この化合物は実施例11に記載される塩複分解方法によりHfCl4から調製された。実施例1に記載されるものと類似する配位交換反応によっても調製されることが可能である。1H NMR(ベンゼン−d6, ppm):3.32(q, 16,J= 7.2 Hz、NCH2CH3)、1.63(s, 12、CCH3)、1.10(t, 24,J= 7.2 Hz、NCH2CH3)。13C NMR(ベンゼン−d6, ppm):173.07(s、CCH3)、41.08(s、NCH2CH3)、18.00(s、NCH2CH3)、10.59(s、CCH3)。C24H52N8Hfについて分析計算:C45.67、H8.30、N17.75。実測:46.17、H7.93、N17.27。そのT1/2値は264℃であり、蒸発してごくわずか残留物が残る。
この化合物は二段階で五塩化タンタルから調製された。第一段階は五塩化タンタルエーテル溶液(20 mLエーテルに0.95 mmol、331 mg)をリチウムN,N’−ジエチルアセトアミジネート溶液(20 mLエーテルに2 mmol、アミジンとn−ブチルリチウムヘキサン溶液(2.6 M)から現場で調製される)について−78℃において窒素下の添加を含む。トリシュロロ−ビス(N,N’−ジエチルアセトアミジナート)タンタル(V)と暫定的に記載される中間体はエーテルに部分的可溶であり、橙色溶液を与える。多少のジオキサンは中間体の溶解を促進するために加えられ、そしてもう2当量のリチウムN,N’−ジエチルアセトアミジネート(20 mLエーテルで2 mmol、アミジンとn−ブチルリチウムヘキサン溶液(2.6 M)から現場で調製される)が室温で添加された。1当量のナトリウムアマルガムはさらに加えられた(22.8 mg、0.645%ナトリウムを有する重量で3.53 g水銀アマルガムについて)。溶液は一夜中室温において撹拌された。溶液は濃紫色に12時間内に変わった。エーテルは減圧下にストリップ除去され、ペンタン(20 mL)が加えられた。紫色溶液は沈殿を分離され、水銀および不溶物質から分離される。真空下に乾燥され、生成物、紫色固体を生じ、それはテトラキス(N,N’−ジエチルアセトアミジナート)タンタル(IV)と暫定的に記載されることができる。生成物は真空下に昇華されることができた、そして昇華画分は分解なく150℃で0.01 mmHgの圧力において再昇華されることができた。生成物は不純物を含むが;その色と真空下に分解なく昇華されることができたことは揮発性タンタル(IV)アミジネートであるらしいことを示す。
他の金属中心を含有する化合物はZrCl4に代わる好適な金属源を用いて実施例3に記載されるものと同様に調製されることができる。例えば、テトラキス(N,N’−ジメチルアセトアミジナート)タングステン(IV)は塩化タングステン(IV)、WCl4を用いて調製される;テトラキス(N,N’−ジメチルアセトアミジナート)錫(IV)は塩化錫(IV)、SnCl4を用いて調製される;テトラキス(ジメチルアセトアミジナート)テルル(IV)はTeCl4から調製される;及び、テトラキス(N,N’−ジメチルアセトアミジナート)ウラン(IV)は塩化ウラン(IV)、UCl4を用いることで調製される。
10 nmol cm-2投与量のテトラキス(N,N’−ジメチルプロピオンアミジナート)ハフニウム(IV)蒸気は200℃において直接液体注入設備から10 Torr−secの暴露で400℃のALD反応器内に、20 nmol cm -2 投与量のオゾンの10 Torr−sec暴露と交互に投入される。酸化ハフニウム膜は80:1の高縦横比をもつ狭孔内部に相似沈着される。
10 nmol cm-2投与量のテトラキス(N,N’−ジメチルプロピオンアミジナート)ハフニウム(IV)蒸気は200℃において直接液体注入設備から10 Torr−secの暴露で400℃の反応器内に、20 nmol cm -2 投与量の水蒸気の10 Torr−sec暴露と交互に投入される。酸化ハフニウム膜は80:1の高縦横比の細孔内部に相似沈着される。
10 nmol cm-2投与量のテトラキス(N,N’−ジメチルブチルアミジナート)ハフニウム(IV)蒸気は200℃において直接液体注入設備から10 Torr−secの暴露で400℃の反応器内に、20 nmol cm -2 投与量のアンモニアの10 Torr−sec暴露と交互に投入される。窒化ハフニウム膜は80:1の高縦横比の細孔内部に相似沈着される。
過剰の無水メチルアミン(39 g、1.26 mol)を、無水シアン化ナトリウム(30.2 g、0.617 mol)、無水塩酸メチルアミン(41.6 g、0.617 mol)、及びランタントリフレート(30.2 g、8.88 mmol)の混合物上に、−196℃で密閉圧力容器中で凝縮させた。混合物は室温に温めることを許容され、次に72時間40〜50℃において超音波をあてて分解された。揮発ガスは室温で反応容器から逃げることを許容され、無水メチルアミン(10 g、0.322 mol)を、冷却反応混合物上に凝縮させた。蒸発、メチルアミン添加及び超音波処理のこの作業周期はもう二回反復された。全揮発物は、反応容器を室温で緩やかに排気することにより最終的に放出された。ジエチルエーテル(50 mL)が加えられ、反応混合物は窒素下に大型フラスコに注入された。全揮発物は室温の反応混合物から−196℃で真空下の大型Schlenkフラスコに蒸気の球体から球体への移動を通じて移動された。N,N’−ジメチルホルムアミジンはエーテル無色溶液として得られた。1H NMR(ベンゼン−d6 +少量のCD3OD、ppm):7.29(s, 1、CH)、2.65(br, 6、NCH3)。
メチルアミンのナトリウム塩は分解して例18と同生成物N,N’−ジメチルホルムアミジンを生じるまで約100℃に加熱された。
1.16 g(3.29 mmol)のテトラキス(ジメチルアミド)ハフニウム(IV)、Hf(NMe2)4は20 mLのジエチルエーテルに溶解され、次に−30℃に30分間冷却された。この溶液に20 mLジエチルエーテルについて0.968 g(13.4 mmol)N,N’−ジメチルホルムアミジンが加えられ、混合物は室温で一夜中撹拌された。少量の白色副生物は沈殿した(ジメチルホルムアミジン中のモノメチルホルムアミジン不純物のために起こりそうな)。上澄みエーテル溶液は沈殿と分離され、減圧下に蒸発されて白色結晶として生成物を生じる。その融点は140〜142℃である。1H NMR(ベンゼン−d6, ppm):3.03(s, 24、NCH3)、8.00(s, 4、CH)。13C NMR(ベンゼン−d6, ppm):171.60(CH)、38.67(NCH3)。そのT1/2値は187℃であり、蒸発してごくわずか残留物が残る。
蒸発速度についての恒温熱重量分析は図4において4つの異なるハフニウムアミジネート(実施例6、実施例8、実施例10及び実施例19の)について比較される。これらの測定が各温度でテトラキス(N,N’−ジメチルホルミジナート)ハフニウム(IV)は最も高い蒸発速度を有することを示す。142℃(その融点)を越えるバブラー温度で液体であるのでその高蒸発速度は極再現性がある。したがって、多用途においてテトラキス(N,N’−ジメチルホルミジナート)ハフニウム(IV)はハフニウム含有物質の蒸着に好ましい先駆物質である。室温において液体が必要とされる用途では同様の沸点の溶媒中溶液として供給されることが可能である。好適な溶媒はトリグリムまたはテトラグリムのようなポリエーテルを含む。代わりに室温で液体のテトラキス(N,N’−ジメチルブチルアミジナート)ハフニウム(IV)は気化設備への混り気のない液体の供給に使用可能である。
例20の工程はテトラキス(ジメチルアミド)ハフニウム(IV)、Hf(NMe2)4の代わりにテトラキス(ジメチルアミド)ジルコニウム(IV)で繰返される。テトラキス(N,N’−ジメチルホルミジナート)ハフニウム(IV)類似性質を有するテトラキス(N,N’−ジメチルホルムアミジナート)ジルコニウム(IV)は得られる。
Claims (23)
- 構造式
を有する化合物。 - 金属Mはジルコニウム、ハフニウム、錫、タンタル、ニオブ、タングステン、モリブデン、ウラン、レニウム、白金、オスミウム、イリジウム、ルテニウム、パラジウム、チタン、ロジウム、バナジウム、セリウム及び鉛からなる群から選択される、請求項1に記載の化合物。
- 該金属Mはハフニウム、ジルコニウム、タンタル、ニオブ、タングステン、モリブデン、錫、テルルおよびウランからなる群から選択される、請求項1に記載の化合物。
- 該金属Mはジルコニウムである、請求項1に記載の化合物。
- 該金属Mはハフニウムである、請求項1に記載の化合物。
- R1、R3、R4、R6、R7、R9、R10およびR12はα位において枝分れしていないアルキル基である、請求項1に記載の化合物。
- 支持体上に金属を含む薄膜を形成する方法であって、熱せられた該支持体の表面を一以上の揮発性金属テトラアミジネート化合物の蒸気に暴露することを含み、該金属テトラアミジネート化合物は構造式
方法。 - 金属Mはジルコニウム、ハフニウム、錫、タンタル、ニオブ、タングステン、モリブデン、ウラン、レニウム、白金、オスミウム、イリジウム、ルテニウム、パラジウム、チタン、ロジウム、バナジウム、セリウム、テルルおよび鉛からなる群から選択される、請求項7に記載の方法。
- 該金属Mはハフニウム、ジルコニウム、タンタル、ニオブ、タングステン、モリブデン、錫、テルルおよびウランからなる群から選択される、請求項7に記載の方法。
- 該金属Mはジルコニウムである、請求項7に記載の方法。
- 該金属Mはハフニウムである、請求項7に記載の方法。
- 該支持体は酸素源にもさらされ、該薄膜は金属酸化物を含む、請求項7に記載の方法。
- 該酸素源は水蒸気を含む、請求項12に記載の方法。
- 該酸素源は酸素分子を含む、請求項12に記載の方法。
- 該酸素源はオゾンを含む、請求項12に記載の方法。
- 該支持体は窒素源にもさらされ、該薄膜は金属窒化物を含む、請求項7に記載の方法。
- 該窒素源はアンモニアを含む、請求項16に記載の方法。
- 該薄膜はCVD工程で付着される、請求項7に記載の方法。
- 該薄膜はALD工程で沈積される、請求項7に記載の方法。
- 該蒸気は固体金属テトラアミジネート化合物を気化することにより得られる、請求項7に記載の方法。
- 該蒸気は液体金属テトラアミジネート化合物を気化することにより得られる、請求項7に記載の方法。
- 該蒸気は金属テトラアミジネートを100〜250℃の範囲の温度で揮発することにより得られる、請求項7に記載の方法。
- 該表面は200〜500℃の範囲の温度である、請求項7に記載の方法。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US81720906P | 2006-06-28 | 2006-06-28 | |
| US60/817,209 | 2006-06-28 | ||
| US11/581,986 US7264486B2 (en) | 2005-10-17 | 2006-10-17 | Electrical connector |
| US11/581,986 | 2006-10-17 | ||
| PCT/US2007/014768 WO2008002546A1 (en) | 2006-06-28 | 2007-06-26 | Metal(iv) tetra-amidinate compounds and their use in vapor deposition |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013081411A Division JP2013209375A (ja) | 2006-06-28 | 2013-04-09 | 金属(iv)テトラ−アミジネート化合物ならびに蒸着においての使用 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2009542654A JP2009542654A (ja) | 2009-12-03 |
| JP2009542654A5 JP2009542654A5 (ja) | 2010-08-12 |
| JP5555872B2 true JP5555872B2 (ja) | 2014-07-23 |
Family
ID=40328880
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009518210A Active JP5555872B2 (ja) | 2006-06-28 | 2007-06-26 | 金属(iv)テトラ−アミジネート化合物ならびに蒸着においての使用 |
| JP2013081411A Pending JP2013209375A (ja) | 2006-06-28 | 2013-04-09 | 金属(iv)テトラ−アミジネート化合物ならびに蒸着においての使用 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013081411A Pending JP2013209375A (ja) | 2006-06-28 | 2013-04-09 | 金属(iv)テトラ−アミジネート化合物ならびに蒸着においての使用 |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP2032529B1 (ja) |
| JP (2) | JP5555872B2 (ja) |
| KR (1) | KR101467587B1 (ja) |
| CN (2) | CN102993050A (ja) |
| WO (1) | WO2008002546A1 (ja) |
Families Citing this family (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG171683A1 (en) | 2006-05-12 | 2011-06-29 | Advanced Tech Materials | Low temperature deposition of phase change memory materials |
| KR101467587B1 (ko) * | 2006-06-28 | 2014-12-01 | 프레지던트 앤드 펠로우즈 오브 하바드 칼리지 | 금속(ⅳ) 테트라-아미디네이트 화합물 및 기상증착에서의 그의 용도 |
| CN101495672B (zh) | 2006-11-02 | 2011-12-07 | 高级技术材料公司 | 对于金属薄膜的cvd/ald有用的锑及锗复合物 |
| JP5571547B2 (ja) * | 2007-04-09 | 2014-08-13 | プレジデント アンド フェローズ オブ ハーバード カレッジ | 銅の相互接続体のための窒化コバルト層及びそれらを形成する方法 |
| JP5437594B2 (ja) | 2007-06-05 | 2014-03-12 | ローム・アンド・ハース・エレクトロニック・マテリアルズ,エル.エル.シー. | 有機金属化合物 |
| KR20100084157A (ko) | 2007-09-17 | 2010-07-23 | 레르 리키드 쏘시에떼 아노님 뿌르 레드 에렉스뿔라따시옹 데 프로세데 조르즈 클로드 | Gst 필름 증착용 텔루륨 전구체 |
| US8834968B2 (en) | 2007-10-11 | 2014-09-16 | Samsung Electronics Co., Ltd. | Method of forming phase change material layer using Ge(II) source, and method of fabricating phase change memory device |
| KR101458953B1 (ko) | 2007-10-11 | 2014-11-07 | 삼성전자주식회사 | Ge(Ⅱ)소오스를 사용한 상변화 물질막 형성 방법 및상변화 메모리 소자 제조 방법 |
| SG178736A1 (en) | 2007-10-31 | 2012-03-29 | Advanced Tech Materials | Amorphous ge/te deposition process |
| US20090215225A1 (en) | 2008-02-24 | 2009-08-27 | Advanced Technology Materials, Inc. | Tellurium compounds useful for deposition of tellurium containing materials |
| US8802194B2 (en) | 2008-05-29 | 2014-08-12 | L'Air Liquide, Société Anonyme pour l'Etude et l'Exploitation des Procédés Georges Claude | Tellurium precursors for film deposition |
| CN102046838A (zh) | 2008-05-29 | 2011-05-04 | 乔治洛德方法研究和开发液化空气有限公司 | 用于膜沉积的碲前体 |
| US8636845B2 (en) | 2008-06-25 | 2014-01-28 | L'Air Liquide, Société Anonyme pour l'Etude et l'Exploitation des Procédés Georges Claude | Metal heterocyclic compounds for deposition of thin films |
| US8236381B2 (en) | 2008-08-08 | 2012-08-07 | L'air Liquide Societe Anonyme Pour L'etude Et L'exploitation Des Procedes Georges Claude | Metal piperidinate and metal pyridinate precursors for thin film deposition |
| US8330136B2 (en) | 2008-12-05 | 2012-12-11 | Advanced Technology Materials, Inc. | High concentration nitrogen-containing germanium telluride based memory devices and processes of making |
| KR101805211B1 (ko) | 2009-09-02 | 2017-12-05 | 레르 리키드 쏘시에떼 아노님 뿌르 레드 에렉스뿔라따시옹 데 프로세데 조르즈 클로드 | 게르마늄 함유 막 침착을 위한 디할라이드 게르마늄(ⅱ) 전구체 |
| SE1000070A1 (sv) | 2010-01-26 | 2011-06-07 | Formox Ab | Katalysator för framställning av aldehyd |
| KR20120123126A (ko) | 2010-02-03 | 2012-11-07 | 레르 리키드 쏘시에떼 아노님 뿌르 레?드 에렉스뿔라따시옹 데 프로세데 조르즈 클로드 | 박막 증착용 칼코게나이드-함유 전구체, 그의 제조 방법 및 사용 방법 |
| KR101706809B1 (ko) | 2010-03-26 | 2017-02-15 | 엔테그리스, 아이엔씨. | 게르마늄 안티몬 텔루라이드 물질 및 이를 포함하는 장치 |
| WO2011146913A2 (en) | 2010-05-21 | 2011-11-24 | Advanced Technology Materials, Inc. | Germanium antimony telluride materials and devices incorporating same |
| EP2609102B1 (en) | 2010-08-27 | 2014-12-31 | Sigma-Aldrich Co. LLC | Molybdenum (iv) amide precursors and use thereof in atomic layer deposition |
| CN102418084A (zh) * | 2011-12-14 | 2012-04-18 | 无锡迈纳德微纳技术有限公司 | 一种源气隔离的固态源原子层沉积装置和方法 |
| WO2014070682A1 (en) | 2012-10-30 | 2014-05-08 | Advaned Technology Materials, Inc. | Double self-aligned phase change memory device structure |
| CN104307572B (zh) * | 2014-10-16 | 2016-06-22 | 山西大学 | 一种脒基铝金属催化剂及其制备方法 |
| CN113652672B (zh) * | 2015-05-27 | 2023-12-22 | Asm Ip 控股有限公司 | 用于含钼或钨薄膜的ald的前体的合成和用途 |
| KR101581314B1 (ko) * | 2015-07-20 | 2015-12-31 | (주)마이크로켐 | 텅스텐 전구체 및 이를 포함하는 텅스텐 함유 필름 증착방법 |
| US10662527B2 (en) | 2016-06-01 | 2020-05-26 | Asm Ip Holding B.V. | Manifolds for uniform vapor deposition |
| US10358407B2 (en) | 2016-10-12 | 2019-07-23 | Asm Ip Holding B.V. | Synthesis and use of precursors for vapor deposition of tungsten containing thin films |
| US10643838B2 (en) * | 2017-06-20 | 2020-05-05 | Applied Materials, Inc. | In-situ formation of non-volatile lanthanide thin film precursors and use in ALD and CVD |
| CN108641073B (zh) * | 2018-04-17 | 2020-09-29 | 山西大学 | 一种三核有机亚锡金属催化剂及其制备方法和应用 |
| US11492701B2 (en) | 2019-03-19 | 2022-11-08 | Asm Ip Holding B.V. | Reactor manifolds |
| CN110128373B (zh) * | 2019-06-12 | 2023-03-24 | 鸿翌科技有限公司 | 哌嗪基锡配合物及其制备方法、薄膜、太阳能电池 |
| KR20210048408A (ko) | 2019-10-22 | 2021-05-03 | 에이에스엠 아이피 홀딩 비.브이. | 반도체 증착 반응기 매니폴드 |
| TW202136571A (zh) | 2020-02-10 | 2021-10-01 | 荷蘭商Asm Ip 控股公司 | 高深寬比孔內的氧化鉿之沉積 |
| KR20220167299A (ko) * | 2020-04-10 | 2022-12-20 | 가부시키가이샤 아데카 | 아미디네이트 화합물, 그 2 량체 화합물, 박막 형성용 원료 및 박막의 제조 방법 |
| KR20230157999A (ko) | 2021-03-18 | 2023-11-17 | 가부시키가이샤 아데카 | 주석 화합물, 박막 형성용 원료, 박막, 박막의 제조 방법 및 할로겐 화합물 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5139825A (en) * | 1989-11-30 | 1992-08-18 | President And Fellows Of Harvard College | Process for chemical vapor deposition of transition metal nitrides |
| US6255414B1 (en) * | 1998-09-01 | 2001-07-03 | E. I. Du Pont De Nemours And Company | Polymerization of olefins |
| AU2003248850A1 (en) * | 2002-07-12 | 2004-02-02 | President And Fellows Of Harvard College | Vapor deposition of tungsten nitride |
| KR102220703B1 (ko) * | 2002-11-15 | 2021-02-26 | 프레지던트 앤드 펠로우즈 오브 하바드 칼리지 | 금속 아미디네이트를 이용한 원자층 증착법 |
| US7396949B2 (en) * | 2003-08-19 | 2008-07-08 | Denk Michael K | Class of volatile compounds for the deposition of thin films of metals and metal compounds |
| JP4639686B2 (ja) * | 2004-07-27 | 2011-02-23 | Jsr株式会社 | 化学気相成長材料及び化学気相成長方法 |
| KR101467587B1 (ko) * | 2006-06-28 | 2014-12-01 | 프레지던트 앤드 펠로우즈 오브 하바드 칼리지 | 금속(ⅳ) 테트라-아미디네이트 화합물 및 기상증착에서의 그의 용도 |
-
2007
- 2007-06-26 KR KR1020097001242A patent/KR101467587B1/ko active IP Right Grant
- 2007-06-26 CN CN2012104614215A patent/CN102993050A/zh active Pending
- 2007-06-26 WO PCT/US2007/014768 patent/WO2008002546A1/en active Application Filing
- 2007-06-26 EP EP07796442A patent/EP2032529B1/en active Active
- 2007-06-26 CN CN2007800298108A patent/CN101500989B/zh active Active
- 2007-06-26 JP JP2009518210A patent/JP5555872B2/ja active Active
-
2013
- 2013-04-09 JP JP2013081411A patent/JP2013209375A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| KR101467587B1 (ko) | 2014-12-01 |
| EP2032529A1 (en) | 2009-03-11 |
| KR20090024803A (ko) | 2009-03-09 |
| CN102993050A (zh) | 2013-03-27 |
| CN101500989B (zh) | 2012-12-19 |
| WO2008002546A1 (en) | 2008-01-03 |
| CN101500989A (zh) | 2009-08-05 |
| JP2009542654A (ja) | 2009-12-03 |
| EP2032529B1 (en) | 2012-10-17 |
| JP2013209375A (ja) | 2013-10-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5555872B2 (ja) | 金属(iv)テトラ−アミジネート化合物ならびに蒸着においての使用 | |
| JP2009542654A5 (ja) | ||
| US7638645B2 (en) | Metal (IV) tetra-amidinate compounds and their use in vapor deposition | |
| TWI359804B (en) | Metal-containing compound, method for producing th | |
| US20190292659A1 (en) | Volatile dihydropyrazinly and dihydropyrazine metal complexes | |
| US9453036B2 (en) | Group 11 mono-metallic precursor compounds and use thereof in metal deposition | |
| EP1563117B1 (en) | Atomic layer deposition using metal amidinates | |
| US7816550B2 (en) | Processes for the production of organometallic compounds | |
| EP2065364A1 (en) | Metal complexes of tridentate beta-ketoiminates | |
| EP1849789A1 (en) | Metal complexes of polydentate beta-ketoiminates | |
| WO2020116364A1 (ja) | ルテニウム錯体からなる化学蒸着用原料及び該化学蒸着用原料を用いた化学蒸着法 | |
| US7956168B2 (en) | Organometallic compounds having sterically hindered amides | |
| US20180273550A1 (en) | Metal bicyclic amidinates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20100622 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20100622 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20121002 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20121009 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130108 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130116 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130409 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130716 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20131015 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20131022 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140108 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20140408 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20140508 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5555872 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |