JP5169727B2 - (メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂 - Google Patents
(メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂 Download PDFInfo
- Publication number
- JP5169727B2 JP5169727B2 JP2008272344A JP2008272344A JP5169727B2 JP 5169727 B2 JP5169727 B2 JP 5169727B2 JP 2008272344 A JP2008272344 A JP 2008272344A JP 2008272344 A JP2008272344 A JP 2008272344A JP 5169727 B2 JP5169727 B2 JP 5169727B2
- Authority
- JP
- Japan
- Prior art keywords
- cation exchange
- exchange resin
- meth
- weight
- resin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000003729 cation exchange resin Substances 0.000 title claims description 184
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 title claims description 181
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical class CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 title claims description 56
- 238000004519 manufacturing process Methods 0.000 title claims description 28
- 230000002378 acidificating effect Effects 0.000 title claims description 24
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 65
- 239000011347 resin Substances 0.000 claims description 65
- 229920005989 resin Polymers 0.000 claims description 65
- 238000006116 polymerization reaction Methods 0.000 claims description 51
- 230000008961 swelling Effects 0.000 claims description 41
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical group COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 claims description 38
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 38
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 19
- 238000004132 cross linking Methods 0.000 claims description 19
- 238000000034 method Methods 0.000 claims description 19
- 239000003112 inhibitor Substances 0.000 claims description 17
- 239000002253 acid Substances 0.000 claims description 16
- 238000006277 sulfonation reaction Methods 0.000 claims description 8
- 229910052757 nitrogen Inorganic materials 0.000 claims description 7
- 150000001768 cations Chemical class 0.000 claims description 5
- 238000001179 sorption measurement Methods 0.000 claims description 5
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 72
- 239000000178 monomer Substances 0.000 description 51
- 239000000243 solution Substances 0.000 description 47
- 239000012071 phase Substances 0.000 description 46
- 239000002994 raw material Substances 0.000 description 45
- 229920000642 polymer Polymers 0.000 description 44
- 238000006243 chemical reaction Methods 0.000 description 35
- 239000002245 particle Substances 0.000 description 34
- MYRTYDVEIRVNKP-UHFFFAOYSA-N 1,2-Divinylbenzene Chemical compound C=CC1=CC=CC=C1C=C MYRTYDVEIRVNKP-UHFFFAOYSA-N 0.000 description 30
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 27
- 230000000052 comparative effect Effects 0.000 description 25
- 238000005341 cation exchange Methods 0.000 description 24
- 150000003839 salts Chemical class 0.000 description 20
- 230000007935 neutral effect Effects 0.000 description 19
- 239000003921 oil Substances 0.000 description 19
- 238000000354 decomposition reaction Methods 0.000 description 18
- 239000002904 solvent Substances 0.000 description 18
- 239000000126 substance Substances 0.000 description 16
- 239000004342 Benzoyl peroxide Substances 0.000 description 14
- 235000019400 benzoyl peroxide Nutrition 0.000 description 14
- 230000007423 decrease Effects 0.000 description 14
- 238000011156 evaluation Methods 0.000 description 14
- 239000011521 glass Substances 0.000 description 14
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 13
- 239000000499 gel Substances 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- 238000004821 distillation Methods 0.000 description 11
- 239000000203 mixture Substances 0.000 description 11
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 10
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 10
- LQNUZADURLCDLV-UHFFFAOYSA-N nitrobenzene Chemical compound [O-][N+](=O)C1=CC=CC=C1 LQNUZADURLCDLV-UHFFFAOYSA-N 0.000 description 10
- 229950000688 phenothiazine Drugs 0.000 description 10
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 9
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical class CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 229940023913 cation exchange resins Drugs 0.000 description 9
- 239000003505 polymerization initiator Substances 0.000 description 9
- 239000008346 aqueous phase Substances 0.000 description 8
- 239000007864 aqueous solution Substances 0.000 description 8
- 150000001298 alcohols Chemical class 0.000 description 7
- 230000003197 catalytic effect Effects 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- NWVVVBRKAWDGAB-UHFFFAOYSA-N p-methoxyphenol Chemical compound COC1=CC=C(O)C=C1 NWVVVBRKAWDGAB-UHFFFAOYSA-N 0.000 description 7
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 6
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 6
- YIWUKEYIRIRTPP-UHFFFAOYSA-N 2-ethylhexan-1-ol Chemical compound CCCCC(CC)CO YIWUKEYIRIRTPP-UHFFFAOYSA-N 0.000 description 6
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 229910001873 dinitrogen Inorganic materials 0.000 description 6
- 239000003456 ion exchange resin Substances 0.000 description 6
- 229920003303 ion-exchange polymer Polymers 0.000 description 6
- -1 vinyl compound Chemical class 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 239000012736 aqueous medium Substances 0.000 description 5
- 230000018044 dehydration Effects 0.000 description 5
- 238000006297 dehydration reaction Methods 0.000 description 5
- 238000001035 drying Methods 0.000 description 5
- 238000001556 precipitation Methods 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- WCBPJVKVIMMEQC-UHFFFAOYSA-N 1,1-diphenyl-2-(2,4,6-trinitrophenyl)hydrazine Chemical compound [O-][N+](=O)C1=CC([N+](=O)[O-])=CC([N+]([O-])=O)=C1NN(C=1C=CC=CC=1)C1=CC=CC=C1 WCBPJVKVIMMEQC-UHFFFAOYSA-N 0.000 description 4
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 4
- XUXUHDYTLNCYQQ-UHFFFAOYSA-N 4-amino-TEMPO Chemical compound CC1(C)CC(N)CC(C)(C)N1[O] XUXUHDYTLNCYQQ-UHFFFAOYSA-N 0.000 description 4
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 4
- 230000001588 bifunctional effect Effects 0.000 description 4
- SNRUBQQJIBEYMU-UHFFFAOYSA-N dodecane Chemical compound CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 238000000691 measurement method Methods 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 239000002002 slurry Substances 0.000 description 4
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 239000008399 tap water Substances 0.000 description 4
- 235000020679 tap water Nutrition 0.000 description 4
- GTJOHISYCKPIMT-UHFFFAOYSA-N 2-methylundecane Chemical compound CCCCCCCCCC(C)C GTJOHISYCKPIMT-UHFFFAOYSA-N 0.000 description 3
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- SGVYKUFIHHTIFL-UHFFFAOYSA-N Isobutylhexyl Natural products CCCCCCCC(C)C SGVYKUFIHHTIFL-UHFFFAOYSA-N 0.000 description 3
- NHTMVDHEPJAVLT-UHFFFAOYSA-N Isooctane Chemical compound CC(C)CC(C)(C)C NHTMVDHEPJAVLT-UHFFFAOYSA-N 0.000 description 3
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 3
- 239000004372 Polyvinyl alcohol Substances 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 239000003054 catalyst Substances 0.000 description 3
- 229920001577 copolymer Polymers 0.000 description 3
- JVSWJIKNEAIKJW-UHFFFAOYSA-N dimethyl-hexane Natural products CCCCCC(C)C JVSWJIKNEAIKJW-UHFFFAOYSA-N 0.000 description 3
- 238000004817 gas chromatography Methods 0.000 description 3
- 238000005342 ion exchange Methods 0.000 description 3
- VKPSKYDESGTTFR-UHFFFAOYSA-N isododecane Natural products CC(C)(C)CC(C)CC(C)(C)C VKPSKYDESGTTFR-UHFFFAOYSA-N 0.000 description 3
- 230000007774 longterm Effects 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- GDOPTJXRTPNYNR-UHFFFAOYSA-N methyl-cyclopentane Natural products CC1CCCC1 GDOPTJXRTPNYNR-UHFFFAOYSA-N 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 230000000704 physical effect Effects 0.000 description 3
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 3
- 239000004926 polymethyl methacrylate Substances 0.000 description 3
- 229920002451 polyvinyl alcohol Polymers 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 125000001174 sulfone group Chemical group 0.000 description 3
- 230000002522 swelling effect Effects 0.000 description 3
- 238000001291 vacuum drying Methods 0.000 description 3
- 239000008096 xylene Substances 0.000 description 3
- NMRPBPVERJPACX-UHFFFAOYSA-N (3S)-octan-3-ol Natural products CCCCCC(O)CC NMRPBPVERJPACX-UHFFFAOYSA-N 0.000 description 2
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 2
- PTTPXKJBFFKCEK-UHFFFAOYSA-N 2-Methyl-4-heptanone Chemical compound CC(C)CC(=O)CC(C)C PTTPXKJBFFKCEK-UHFFFAOYSA-N 0.000 description 2
- WQBCQEDMLHHOMQ-UHFFFAOYSA-N 4-methoxycyclohexa-1,5-diene-1,4-diol Chemical compound COC1(O)CC=C(O)C=C1 WQBCQEDMLHHOMQ-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 2
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 2
- 239000004793 Polystyrene Substances 0.000 description 2
- 229920002125 Sokalan® Polymers 0.000 description 2
- 235000002597 Solanum melongena Nutrition 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 239000002826 coolant Substances 0.000 description 2
- 230000001186 cumulative effect Effects 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 230000033001 locomotion Effects 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 229960003505 mequinol Drugs 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- CXKWCBBOMKCUKX-UHFFFAOYSA-M methylene blue Chemical compound [Cl-].C1=CC(N(C)C)=CC2=[S+]C3=CC(N(C)C)=CC=C3N=C21 CXKWCBBOMKCUKX-UHFFFAOYSA-M 0.000 description 2
- 229960000907 methylthioninium chloride Drugs 0.000 description 2
- 239000011259 mixed solution Substances 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- UMFJAHHVKNCGLG-UHFFFAOYSA-N n-Nitrosodimethylamine Chemical compound CN(C)N=O UMFJAHHVKNCGLG-UHFFFAOYSA-N 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 230000000379 polymerizing effect Effects 0.000 description 2
- 229920002223 polystyrene Polymers 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 125000000542 sulfonic acid group Chemical group 0.000 description 2
- XTHPWXDJESJLNJ-UHFFFAOYSA-N sulfurochloridic acid Chemical compound OS(Cl)(=O)=O XTHPWXDJESJLNJ-UHFFFAOYSA-N 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- QEQBMZQFDDDTPN-UHFFFAOYSA-N (2-methylpropan-2-yl)oxy benzenecarboperoxoate Chemical compound CC(C)(C)OOOC(=O)C1=CC=CC=C1 QEQBMZQFDDDTPN-UHFFFAOYSA-N 0.000 description 1
- PJYUCLOYEILMHQ-UHFFFAOYSA-N 1,2-bis(1-phenylethyl)-10h-phenothiazine Chemical compound C=1C=C2SC3=CC=CC=C3NC2=C(C(C)C=2C=CC=CC=2)C=1C(C)C1=CC=CC=C1 PJYUCLOYEILMHQ-UHFFFAOYSA-N 0.000 description 1
- ZJQIXGGEADDPQB-UHFFFAOYSA-N 1,2-bis(ethenyl)-3,4-dimethylbenzene Chemical group CC1=CC=C(C=C)C(C=C)=C1C ZJQIXGGEADDPQB-UHFFFAOYSA-N 0.000 description 1
- CHRJZRDFSQHIFI-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;styrene Chemical compound C=CC1=CC=CC=C1.C=CC1=CC=CC=C1C=C CHRJZRDFSQHIFI-UHFFFAOYSA-N 0.000 description 1
- QLLUAUADIMPKIH-UHFFFAOYSA-N 1,2-bis(ethenyl)naphthalene Chemical compound C1=CC=CC2=C(C=C)C(C=C)=CC=C21 QLLUAUADIMPKIH-UHFFFAOYSA-N 0.000 description 1
- KNKRKFALVUDBJE-UHFFFAOYSA-N 1,2-dichloropropane Chemical compound CC(Cl)CCl KNKRKFALVUDBJE-UHFFFAOYSA-N 0.000 description 1
- AYMDJPGTQFHDSA-UHFFFAOYSA-N 1-(2-ethenoxyethoxy)-2-ethoxyethane Chemical compound CCOCCOCCOC=C AYMDJPGTQFHDSA-UHFFFAOYSA-N 0.000 description 1
- XMNIXWIUMCBBBL-UHFFFAOYSA-N 2-(2-phenylpropan-2-ylperoxy)propan-2-ylbenzene Chemical compound C=1C=CC=CC=1C(C)(C)OOC(C)(C)C1=CC=CC=C1 XMNIXWIUMCBBBL-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- LBQJCDLKJGOHEA-UHFFFAOYSA-N 2-ethenylbut-3-enylbenzene Chemical compound C=CC(C=C)CC1=CC=CC=C1 LBQJCDLKJGOHEA-UHFFFAOYSA-N 0.000 description 1
- TVWBTVJBDFTVOW-UHFFFAOYSA-N 2-methyl-1-(2-methylpropylperoxy)propane Chemical compound CC(C)COOCC(C)C TVWBTVJBDFTVOW-UHFFFAOYSA-N 0.000 description 1
- YAQDPWONDFRAHF-UHFFFAOYSA-N 2-methyl-2-(2-methylpentan-2-ylperoxy)pentane Chemical compound CCCC(C)(C)OOC(C)(C)CCC YAQDPWONDFRAHF-UHFFFAOYSA-N 0.000 description 1
- STGFANHLXUILNY-UHFFFAOYSA-N 3,7-dioctyl-10h-phenothiazine Chemical compound C1=C(CCCCCCCC)C=C2SC3=CC(CCCCCCCC)=CC=C3NC2=C1 STGFANHLXUILNY-UHFFFAOYSA-N 0.000 description 1
- PYSRRFNXTXNWCD-UHFFFAOYSA-N 3-(2-phenylethenyl)furan-2,5-dione Chemical compound O=C1OC(=O)C(C=CC=2C=CC=CC=2)=C1 PYSRRFNXTXNWCD-UHFFFAOYSA-N 0.000 description 1
- JIGUICYYOYEXFS-UHFFFAOYSA-N 3-tert-butylbenzene-1,2-diol Chemical compound CC(C)(C)C1=CC=CC(O)=C1O JIGUICYYOYEXFS-UHFFFAOYSA-N 0.000 description 1
- MKTOIPPVFPJEQO-UHFFFAOYSA-N 4-(3-carboxypropanoylperoxy)-4-oxobutanoic acid Chemical compound OC(=O)CCC(=O)OOC(=O)CCC(O)=O MKTOIPPVFPJEQO-UHFFFAOYSA-N 0.000 description 1
- QNRSQFWYPSFVPW-UHFFFAOYSA-N 5-(4-cyanobutyldiazenyl)pentanenitrile Chemical compound N#CCCCCN=NCCCCC#N QNRSQFWYPSFVPW-UHFFFAOYSA-N 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- YIVJZNGAASQVEM-UHFFFAOYSA-N Lauroyl peroxide Chemical compound CCCCCCCCCCCC(=O)OOC(=O)CCCCCCCCCCC YIVJZNGAASQVEM-UHFFFAOYSA-N 0.000 description 1
- UBUCNCOMADRQHX-UHFFFAOYSA-N N-Nitrosodiphenylamine Chemical compound C=1C=CC=CC=1N(N=O)C1=CC=CC=C1 UBUCNCOMADRQHX-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical group OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 1
- 240000001987 Pyrus communis Species 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 229920000147 Styrene maleic anhydride Polymers 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- 241000482268 Zea mays subsp. mays Species 0.000 description 1
- YRKCREAYFQTBPV-UHFFFAOYSA-N acetylacetone Natural products CC(=O)CC(C)=O YRKCREAYFQTBPV-UHFFFAOYSA-N 0.000 description 1
- 125000005396 acrylic acid ester group Chemical group 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 238000011001 backwashing Methods 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 239000000337 buffer salt Substances 0.000 description 1
- CDQSJQSWAWPGKG-UHFFFAOYSA-N butane-1,1-diol Chemical compound CCCC(O)O CDQSJQSWAWPGKG-UHFFFAOYSA-N 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 238000011088 calibration curve Methods 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 229920001429 chelating resin Polymers 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 238000007872 degassing Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 239000012933 diacyl peroxide Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 150000008282 halocarbons Chemical class 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- ACCCMOQWYVYDOT-UHFFFAOYSA-N hexane-1,1-diol Chemical compound CCCCCC(O)O ACCCMOQWYVYDOT-UHFFFAOYSA-N 0.000 description 1
- 229920013821 hydroxy alkyl cellulose Polymers 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- OWFXIOWLTKNBAP-UHFFFAOYSA-N isoamyl nitrite Chemical compound CC(C)CCON=O OWFXIOWLTKNBAP-UHFFFAOYSA-N 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 125000005397 methacrylic acid ester group Chemical group 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 238000003541 multi-stage reaction Methods 0.000 description 1
- DAHPIMYBWVSMKQ-UHFFFAOYSA-N n-hydroxy-n-phenylnitrous amide Chemical compound O=NN(O)C1=CC=CC=C1 DAHPIMYBWVSMKQ-UHFFFAOYSA-N 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 1
- 150000002832 nitroso derivatives Chemical class 0.000 description 1
- XNTUJOTWIMFEQS-UHFFFAOYSA-N octadecanoyl octadecaneperoxoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OOC(=O)CCCCCCCCCCCCCCCCC XNTUJOTWIMFEQS-UHFFFAOYSA-N 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- SRSFOMHQIATOFV-UHFFFAOYSA-N octanoyl octaneperoxoate Chemical compound CCCCCCCC(=O)OOC(=O)CCCCCCC SRSFOMHQIATOFV-UHFFFAOYSA-N 0.000 description 1
- 150000001451 organic peroxides Chemical class 0.000 description 1
- MMCOUVMKNAHQOY-UHFFFAOYSA-L oxido carbonate Chemical compound [O-]OC([O-])=O MMCOUVMKNAHQOY-UHFFFAOYSA-L 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- DBSDMAPJGHBWAL-UHFFFAOYSA-N penta-1,4-dien-3-ylbenzene Chemical compound C=CC(C=C)C1=CC=CC=C1 DBSDMAPJGHBWAL-UHFFFAOYSA-N 0.000 description 1
- 150000002990 phenothiazines Chemical class 0.000 description 1
- 229920000371 poly(diallyldimethylammonium chloride) polymer Polymers 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 229920000193 polymethacrylate Polymers 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 239000007870 radical polymerization initiator Substances 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 239000012488 sample solution Substances 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- HIFJUMGIHIZEPX-UHFFFAOYSA-N sulfuric acid;sulfur trioxide Chemical compound O=S(=O)=O.OS(O)(=O)=O HIFJUMGIHIZEPX-UHFFFAOYSA-N 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 238000010557 suspension polymerization reaction Methods 0.000 description 1
- 229940066767 systemic antihistamines phenothiazine derivative Drugs 0.000 description 1
- SWAXTRYEYUTSAP-UHFFFAOYSA-N tert-butyl ethaneperoxoate Chemical compound CC(=O)OOC(C)(C)C SWAXTRYEYUTSAP-UHFFFAOYSA-N 0.000 description 1
- 238000005809 transesterification reaction Methods 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
Images



Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
Landscapes
- Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
特許文献1及び2に、前記(メタ)アクリル酸エステルの製造触媒として、ポーラス型の強酸性陽イオン交換樹脂が好適に用いることができると記載されている。
また、特許文献3及び4に記載されているような従来公知のゲル型陽イオン交換樹脂では、耐久性が低いという問題があった。
(1)下記(A)、(B)、及び(C)を満たすことを特徴とする、(メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂。
(A)窒素吸着法で測定される表面積が2m2/g以下である
(B)架橋度が2重量%以上7.5%重量以下である
(C)膨潤度が2以下である
(当該膨潤度とは、下記式(I)で表される値である。
(前記強酸性陽イオン交換樹脂にメタクリル酸メチルモノマーを加え、70℃の恒温槽中で72時間静置することにより膨潤させた後の体積)/(H形に再生させた前記強酸性陽イオン交換樹脂の膨潤前の体積))・・・式(I))
(2)水分含有率が35重量%以上であることを特徴とする、(1)に記載の(メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂。
(3)重合禁止剤を含有することを特徴とする、(1)又は(2)に記載の(メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂。
(4)オリウムを0.1重量%以上含有する硫酸を用いてスルホン化する工程を有することを特徴とする、(1)〜(3)のいずれかに記載の(メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂の製造方法。
(5)(1)〜(4)のいずれかに記載の強酸性陽イオン交換樹脂を用いることを特徴とする、(メタ)アクリル酸エステルの製造方法。
さらに、本発明の強酸性陽イオン交換樹脂を用いれば、(メタ)アクリル酸エステルを高収率で製造することができる。
本発明は、(メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂、及びそれを用いた(メタ)アクリル酸エステルの製造方法に関するものである。
[陽イオン交換樹脂]
本発明の陽イオン交換樹脂は、下記(A)、(B)、及び(C)を満たすことを特徴とするものである。
(A)窒素吸着法で測定される表面積が2m2/g以下である
(B)架橋度が2重量%以上7.5%重量以下である
(C)膨潤度が2以下である
(当該膨潤度とは、下記式(I)で表される値である。
(前記強酸性陽イオン交換樹脂にメタクリル酸メチルモノマーを加え、70℃の恒温槽中で72時間静置することにより膨潤させた後の体積)/(H形に再生させた前記強酸性陽イオン交換樹脂の膨潤前の体積))・・・式(I))
(A)表面積
本発明の陽イオン交換樹脂は、表面積が比較的小さいことをその特徴とする。
は、1m2/g以下、特に好ましくは 0.2m2/g以下である。表面積が小さい陽イオン交換樹脂を用いると、(メタ)アクリル酸エステル製造の収率が向上する傾向にあるので、0.1m2/g以下であることが最も好ましい。陽イオン交換樹脂の表面積が大きす
ぎると、収率が低下する傾向にある。
(B)架橋度
本発明の陽イオン交換樹脂は、その架橋度が通常2重量%以上、好ましくは2.5重量%以上、より好ましくは3重量%以上、また、通常7.5%重量%以下、好ましくは7重量%以下、より好ましくは6重量%以下、更に好ましくは5.5重量%以下である。架橋度が高すぎると、(メタ)アクリル酸エステル製造の収率が低下する傾向にある。一方で、架橋度が低すぎると、(メタ)アクリル酸エステル製造の収率が低下する傾向にあり、さらには、通液する際に圧力損失が大きく、長期間使用するにはプロセス上の制約が多くなる場合がある。
(C)膨潤度
本発明の陽イオン交換樹脂は、膨潤性評価における膨潤度が小さいことをその特徴とする。
本発明の陽イオン交換樹脂の膨潤度は、通常2以下、好ましくは1.5以下、特に好ましくは1.3以下であり、1に近いほど好ましい。膨潤度が小さい陽イオン交換樹脂を用いると、耐久性が向上する傾向にあり、長期間の使用が可能となる。一方、膨潤度が大きい陽イオン交換樹脂を用いると、長期間使用すると樹脂が膨潤することにより体積が増加してしまうので、耐久性が低下する傾向にある。
本発明のように膨潤度の小さい陽イオン交換樹脂は、重合条件や、スルホン化条件を制御することにより得ることができる。
なお、膨潤度は、具体的には、以下の方法により測定することができる。膨潤性の評価試験には、予め以下のような処理を行ったメタクリル酸メチルモノマー(MMAモノマー)を用いる。メタクリル酸メチル(和光純薬製、特級試薬)、メトキノン(ハイドロキノンのメチルエーテルを50ppm含む。)100gを、ガラスカラムに充填した50℃で8時間真空乾燥したアルミナ(メルク製アルミナ 60メッシュ品)にSV2で通液することにより、重合禁止剤であるメトキノンを除去し、MMAモノマーを全量回収する。得られた溶液を60mmHg、36℃の条件下、減圧蒸留で精製する。具体的には、500mlのナスフラスコにクライゼンアダプターを装着し(具体的には、Y字型蒸留ヘッドを装着し、ジムロー冷却管で凝縮させる。冷却管には5℃の冷却液を循環させる。)、前記モノマー溶液を60mmHg、36℃の条件下で減圧蒸留を行なうことにより精製する。初留は仕込み重量に対し10%、釜残も10重量%残す。蒸留後は−20℃の冷蔵庫で保管し、3日以内に使用することが好ましい。
被検体である陽イオン交換樹脂に、2N−HClを10BVで通液することにより、H形に再生し、遠心分離器で付着水分を除去する。この陽イオン交換樹脂(水切り樹脂)を、メスシリンダーで20.0ml計り取り、200mlの耐圧性ガラス容器(耐圧硝子工業製)に入れ、そこに前記MMAモノマー溶液(メタノールを含む。)100gを加える。前記耐圧性ガラス容器に窒素ガスを0.5MPaまで加圧した後、大気圧まで戻す操作を3回繰り返し、圧力容器を窒素ガスで置換する。次いで、30℃で30分間加温し、その後、耐圧性ガラス容器を密栓する。この耐圧性ガラス容器を70℃の恒温槽中で静置する。72時間後、ゲル化したポリメタクリル酸メチル中の陽イオン交換樹脂の体積を測定し、試験前の20.0mlに対して、何倍に膨潤したかを計算することにより、前記膨潤度を算出することができる。
(D)水分含有率
本発明の陽イオン交換樹脂は、水分含有率が小さいことをその特徴とする。
本発明の陽イオン交換樹脂がポーラス型である場合、そのイオン形がH形であるときの水分含有率が、通常80重量%以下、好ましくは、75重量%以下、特に好ましくは、70重量%以下であり、また、通常35重量%以上、好ましくは45重量%以上、さらに好ましくは50重量%以上である。
上記水分含有率は、例えば、以下のような手順で測定することができる。
<水分含有率の測定>
被検体である陽イオン交換樹脂に塩酸水溶液を接触させて、その陽イオン交換基を再生した後、遠心分離機等の適切な機器によって脱水し、脱水した陽イオン交換樹脂の適切量を秤量する。秤量した陽イオン交換樹脂を更に真空乾燥し、真空乾燥させた陽イオン交換樹脂の質量を測定する。そして、脱水後の質量と、真空乾燥後の質量から、陽イオン交換樹脂の水分含有量を算出することができる。また、カールフィッシャー水分計で、樹脂中の水分を直接測定することもできる。
(E)粒径
本発明の陽イオン交換樹脂は、その粒径が通常100μm以上、好ましくは200μm以上、より好ましくは300μm以上、また、通常1000μm以下、好ましくは800μm以下、より好ましくは 700μm以下である。粒径が大きすぎると、陽イオン交換樹脂が破砕しやすい傾向にあり、特に、陽イオン交換樹脂を反応器に入れて原料を上向流で通液する場合においては、陽イオン交換樹脂の粒径が大きいと、陽イオン交換樹脂が流動しにくくなり、反応熱が蓄積しやすい傾向にある。粒径が小さすぎると、圧力損失が大きく原料を通液しにくい傾向にある。
陽イオン交換樹脂の粒径、即ち、粒径重量平均粒子径の測定は、例えば、以下のような手順で行われる。
篩目の径が1180μm、850μm、710μm、600μm、425μm、300μmの篩を、下方になる程、篩目の径が小さくなる様に積み重ねる。この積み重ねた篩をバットの上に置き、最上段に積み重ねられた1180μmの篩の中に陽イオン交換樹脂を約100mL入れる。
前記バットの中の小粒は次の850μmの篩の上へ戻し、また1180μmの篩の上に残った陽イオン交換樹脂はさらに別のバットに採取する。篩の目に陽イオン交換樹脂が詰まっていれば、篩をバットに逆に置き、水道水につないだゴム管に密着させ、水を強く流して篩の目に詰まった陽イオン交換樹脂を取り出す。取り出した陽イオン交換樹脂は、1180μmの篩上に残った陽イオン交換樹脂を採取したバットに移し、合計をメスシリンダーで容積を測定する。この容積をa(mL)とする。1180μmの篩を通った陽イオン交換樹脂は850μm、710μm、600μm、425μm、300μmの篩についてそれぞれ同様の操作を行い、メスシリンダーを用いて容積b(mL)、c(mL)、d(mL)、e(mL)、f(mL)を求め、最後に300μmの篩を通った樹脂の容積をメスシリンダーで測定しg(mL)とする。
前記a'〜g'より片軸に各篩の残留分累計(%)、他の軸に篩目の径(mm)をとり、これを対数確率紙上にプロットする。残留分の多い順に3点を取り、この3点を出来るだけ満足するような線を引き、この線から残留分累計が50%に相当する篩目の径(mm)を求め、これを重量平均粒子径とする。
陽イオン交換樹脂の均一係数は、懸濁重合法で製造された場合は、通常、1.5前後であるが、粒径が均一となるように、均一の液滴を発生させ、その液滴を重合する方法において製造された場合は、均一係数が1.0〜1.1程度となる。本発明においては、いずれの樹脂を使用することも可能である。バッチ反応や懸濁状態でのバッチ反応、固定床による流通反応で使用する場合、均一粒径であることが好ましい。その均一係数は1以上、1.5以下、好ましくは1.3以下、更に好ましくは1.2以下、更に好ましくは1.1以下である陽イオン交換樹脂であることが好ましい。粒径が均一な陽イオン交換樹脂を使用すると、下向流で通液する際に圧力損失が低くなる傾向にある。また、粒径が均一な陽イオン交換樹脂を上向流で通液すると、逆洗展開率が小さく、粒子の広がりが少なくなる傾向にあり、また、リフトベットで使用する場合においても、通液の圧力損失が小さくなる傾向にあり、好ましい。
(F)中性塩分解容量
本発明の陽イオン交換樹脂は、その体積当たりの中性塩分解容量(meq/ml)が、通常0.5meq/ml以上、2.5meq/ml以下である。本発明の陽イオン交換樹脂は、その体積当たりの中性塩分解容量が前記の範囲内にあることが好ましい。一般に、架橋度が高くなるほど、体積当たりの中性塩分解容量は高くなる傾向はあるが、体積当たりの中性塩分解容量が高くても、架橋度が高ければ基質である(メタ)アクリル酸やアルコール類が拡散しにくくなり、触媒活性が低下する傾向にある。
中性塩分解容量とは、強酸性陽イオン交換容量と定義され、スルホン酸基の含有率を表す。この中性塩分解容量は、例えば、以下のような手順で測定される。
本発明の陽イオン交換樹脂の中性塩分解容量は、以下の方法で分析され測定される。
先ず、陽イオン交換樹脂をカラムに詰め、これに樹脂容量の25倍量の2N−HCl水溶液を通液し、対イオンをH形に変換し、脱塩水で水洗する。この樹脂を10.0ml採り、ガラスカラムに充填し、洗浄濾液が中性になるまで十分に脱塩水で洗浄する。その後5%NaCl水溶液を25倍量通液し流出液を全て捕集する。この流出液を1N−塩酸で滴定することにより、中性塩分解容量を測定する。これを1mlの樹脂当たりに換算し、体積当たりの中性塩分解容量を算出する。一方、H形に再生した10.0mlの樹脂を取り、これを遠心分離器で水切り状態にして、水切り後の樹脂の重量を測定する。この樹脂を真空乾燥器で、50℃で8時間乾燥し、乾燥後の樹脂重量を測定する。この水切り状態の重量と乾燥後の重量から乾燥重量当たりの中性塩分解容量を算出する。
(G)化学構造
イオン交換樹脂は、イオン交換基を有する溶媒不溶性の球状又は粉末状のプラスチックと定義できる。
このプラスチックの素材としては、一般的にはスチレン系樹脂のものが多く、スチレンジビニルベンゼンの架橋共重合体で形成されており、この重合体にイオン交換基が化学結合で固定化されている。
[陽イオン交換樹脂の製造方法]
本発明の陽イオン交換樹脂は、水相と油相とを別々に調整し、水相と油相とを重合反応させることにより調整することができる。より具体的には、分散剤を溶解した水性媒体(水相)に、重合開始剤を含む架橋作用を有する二官能性不飽和単量体の混合物(油相)を加え、撹拌して懸濁液を形成し、次いで所定の重合温度に保持することにより行なわれる。
〈水相〉
水性媒体としては、水を用いることができる。
〈油相〉
架橋作用を有する二官能性不飽和単量体としては、ジビニルベンゼン、ジビニルトルエン、ジビニルキシレン、ジビニルナフタレン、ジビニルエチルベンゼン等の芳香族単量体類、エチレングリコールポリ(メタ)アクリレート等の脂肪族単量体等を用いることができる。必要に応じて、これらの溶液の2種以上を併用してもよい。
〈重合反応〉
原料モノマーの混合物と水性媒体との比率(容積比)は、一般に1:10〜1:1とし、水性媒体の方を多くすることが好ましい。
重合温度及び重合時間については、使用する重合開始剤の種類及びその使用量により異なるため一概には規定できないが、重合温度については通常30℃以上、好ましくは50℃以上、また、通常200℃以下、好ましくは150℃以下の範囲である。また、重合時間については、通常0.5時間以上、好ましくは2時間以上、また、通常24時間以下、好ましくは12時間以下の範囲である。
本発明において重合方法は特に限定されないが、球状の共重合体を生成させる為には、水または他の水性媒体中でモノマー混合物及び重合開始剤を撹拌により懸濁状態を保ちつつ重合反応を行う。
重合反応終了後は、適宜、洗浄、乾燥を行うことが好ましい。
〈陽イオン交換基の導入〉
導入する陽イオン交換基としては、スルホン基が好ましい。スルホン基の導入に用いる試薬としては、硫酸(濃硫酸であることが好ましい。)、発煙硫酸、クロロ硫酸等が挙げられ、中でも、濃硫酸が好ましい。具体的な濃硫酸の濃度としては、通常95重量%以上、好ましくは 98%重量%以上、更に好ましくは100重量%以上である。
陽イオン交換基の導入の際に用いる溶媒としては、ニトロメタン、塩化メチレン、二塩化エチレン、クロロホルム、ジクロルプロパン等のハロゲン化炭化水素類等の他、ニトロベンゼン、クロロベンゼン等も用いることができる。あるいは、無溶媒で陽イオン交換基を導入することも可能である。
陽イオン交換基の導入の際の反応温度は、通常30℃以上、好ましくは50℃以上であり、反応時間は、1時間以上、好ましくは2時間以上、また、通常24時間以下、好ましくは12時間以下、更に好ましくは8時間以下である。
〈重合禁止剤の添加〉
陽イオン交換樹脂に重合禁止剤を添加すると、樹脂表面や樹脂内部で(メタ)アクリル酸誘導体のポリマーによる有機汚染が抑制されるという効果が得られるため、好ましい。
重合禁止剤の添加量は、その種類により異なるが、陽イオン交換樹脂1Lに対し、通常10ppm以上、好ましくは50ppm以上であり、また、通常1%以下、好ましくは5000ppm以下である。
[(メタ)アクリル酸エステルの製造方法]
本発明の製造方法は、前述した本発明の陽イオン交換樹脂を用いることを必須とするものである。本発明の陽イオン交換樹脂を用いていれば、その他の製造条件については、公知の手法を用いることができる。例えば、本発明の陽イオン交換樹脂が充填された固定床あるいは攪拌機を備えたバッチ式の反応器に、原料である(メタ)アクリル酸とアルコールとを供給することにより、(メタ)アクリル酸エステルを製造することができる。
固定床で反応させる場合、反応器の容積に対して、強酸性陽イオン交換樹脂を30%から95%充填することができる。塔径に対する(樹脂を自然沈降させた)樹脂の充填層高の比率は、通常0.2以上、好ましくは0.5以上であり、また、通常10倍以下である。
(メタ)アクリル酸エステル製造反応における原料としては、(メタ)アクリル酸と、アルコール類及び/又はエステル類(以下、単に「アルコール類」と称する場合がある。)である。
エステル類としては、(メタ)アクリル酸メチル、(メタ)アクリル酸エチル等を用い
ることができる。中でも、(メタ)アクリル酸メチル等の炭素数4以下のエステルを用いることが好ましい。これらのエステル類を原料として用いれば、エステル交換反応により(メタ)アクリル酸エステルを合成することができる。
なお、製造反応の様式としては、連続的に原料を供給する方法、回分式(バッチ式)に製造する方法等が挙げられる。蒸留塔を備えたバッチ反応槽で反応を行うことも可能であり、バッチ反応槽を複数器有する場合は、一方に、(メタ)アクリル酸、もう一方にアルコール類を供給しながら、蒸留反応を行うこともできるという利点がある。
[物性値の測定方法]
後述する各実施例、及び比較例で用いた強酸性陽イオン交換樹脂の物性値、及び(メタ)アクリル酸エステルの収率は、以下の方法で測定、及び算出することができる。
<粒径>
篩目の径が1180μm、850μm、710μm、600μm、425μm、300μmの篩を、下方になる程、篩目の径が小さくなる様に積み重ねた。この積み重ねた篩をバットの上に置き、最上段に積み重ねられた1180μmの篩の中に陽イオン交換樹脂を約100mL入れた。
前記バットの中の小粒は次の850μmの篩の上へ戻し、また1180μmの篩の上に残った陽イオン交換樹脂はさらに別のバットに採取した。篩の目に陽イオン交換樹脂が詰まっていれば、篩をバットに逆に置き、水道水につないだゴム管に密着させ、水を強く流して篩の目に詰まった陽イオン交換樹脂を取り出した。取り出した陽イオン交換樹脂は、1180μmの篩上に残った陽イオン交換樹脂を採取したバットに移し、合計をメスシリンダーで容積を測定した。この容積をa(mL)とした。1180μmの篩を通った陽イオン交換樹脂は850μm、710μm、600μm、425μm、300μmの篩についてそれぞれ同様の操作を行い、メスシリンダーを用いて容積b(mL)、c(mL)、d(mL)、e(mL)、f(mL)を求め、最後に300μmの篩を通った樹脂の容積をメスシリンダーで測定しg(mL)とした。
前記a'〜g'より片軸に各篩の残留分累計(%)、他の軸に篩目の径(mm)をとり、これを対数確率紙上にプロットした。残留分の多い順に3点を取り、この3点を出来るだけ満足するような線を引き、この線から残留分累計が50%に相当する篩目の径(mm)を求め、これを重量平均粒子径(陽イオン交換樹脂の粒径)とした。
<水分含有率>
10倍量の2N−HCl水溶液を用いてH形に再生した陽イオン交換樹脂をメスシリンダーで正確に10.0mlを取り、遠心分離機によって脱水した。この樹脂を全量秤量瓶に移し、50℃で8時間真空乾燥し、真空乾燥させた陽イオン交換樹脂の質量を測定した。そして、脱水後の質量と、真空乾燥後の質量から、陽イオン交換樹脂の水分含有量を算出した。
<中性塩分解容量>
10倍量の2N−HCl水溶液を用いてH形に再生した陽イオン交換樹脂をメスシリンダーで正確に10.0mlを取った。この樹脂をカラムに充填し、25倍量の5%NaCl水溶液を通液した。食塩水の流出液を全て捕集した。この流出液を1N−NaOH又は0.1N−NaOHで滴定することにより、中性塩分解容量を測定し、これを1mlの樹脂当たりに換算し、体積当たりの中性塩分解容量を算出した。
H形に再生した10.0mlの樹脂を測り取り、これを遠心分離器で水切り状態にして、水切り後の樹脂の重量を測定した。この樹脂を真空乾燥器で、50℃で8時間乾燥し、乾燥後の樹脂重量を測定した。この水切り状態の重量と乾燥後の重量とから乾燥重量当たりの中性塩分解容量を算出した。
<表面積>
窒素吸着法により測定を行なった。具体的には、陽イオン交換樹脂を、50℃で8時間、真空乾燥した後、マイクロメトリクス社製フローソーブ2300型を用いて、表面積を測定した。
<架橋度>
ジビニルベンゼン含有量を算出し、架橋度として掲載した。
<膨潤性の評価方法>
膨潤性の評価に用いたメタクリル酸メチルモノマー(以下、「MMAモノマー」と称する場合がある。)として、予め下記のような処理を行ったものを用いた。メタクリル酸メチル(和光純薬製、特級試薬)、メトキノン(ハイドロキノンのメチルエーテルを50ppm含む。)100gを、ガラスカラムに充填した50℃で8時間真空乾燥したアルミナ(メルク製アルミナ 60メッシュ品)にSV2で通液することにより、重合禁止剤であるメトキノンを除去し、MMAモノマーを全量回収した。得られた溶液を60mmHg、36℃の条件下、減圧蒸留で精製した。具体的には、500mlのナスフラスコにクライゼンアダプターを装着し(具体的には、Y字型蒸留ヘッドを装着し、ジムロー冷却管で凝縮させた。冷却管には5℃の冷却液を循環させた。)、前記モノマー溶液を60mmHg、36℃の条件下で減圧蒸留を行なうことにより精製した。初留は仕込み重量に対し10%、釜残も10重量%残した。蒸留後は−20℃の冷蔵庫で保管し、3日以内に使用した。
被検体である陽イオン交換樹脂に、2N−HClを10BVで通液することにより、H形に再生し、遠心分離器で付着水分を除去した。この陽イオン交換樹脂(水切り樹脂)を、メスシリンダーで20.0ml計り取り、200mlの耐圧性ガラス容器(耐圧硝子工業製)に入れ、そこに前記MMAモノマー溶液(メタノールを含む。)100gを加えた。前記耐圧性ガラス容器に窒素ガスを0.5MPaまで加圧した後、大気圧まで戻す操作を3回繰り返し、圧力容器を窒素ガスで置換した。次いで、30℃で30分間加温し、その後、耐圧性ガラス容器を密栓した。この耐圧性ガラス容器を70℃の恒温槽中で静置した。72時間後、ゲル化したポリメタクリル酸メチル中の陽イオン交換樹脂の体積を測定した。膨潤前の体積(20.0ml)と、72時間後(膨潤後)の体積の値を下記式(I)に代入することにより、膨潤度を算出した。
(前記強酸性陽イオン交換樹脂にメタクリル酸メチルモノマーを加え、70℃の恒温槽中で72時間静置することにより膨潤させた後の体積)/(H形に再生させた前記強酸性陽イオン交換樹脂の膨潤前の体積))・・・式(I)
〈流通反応における評価〉
(実験装置)
液体クロマトグラフィー用ポンプ(島津製作所製、LC−6A)に、約2mのSUS製HPLC配管で、SUS316カラム(20mmφ×25cm GLサイエンス製)を接続した。前記カラムに、H形に再生した陽イオン交換樹脂75.0mlを水スラリー状態で充填した。陽イオン交換樹脂を充填したカラム出口には、反応液の冷却用に長さ1mのSUS製HPLC配管を接続し、流出液は100mlのメスシリンダーで受けられるようにした。前記メスシリンダーの下に電子天秤を設置し、流速を測定することができるようにした。前記SUSカラムを深底バス(直径24cm×深さ44cm。上下の攪拌のため、バス内にスリーワンモーターを1台設置した。)の中に入れ、SUSカラムが垂直になるように設置し、深底バス内でSUSカラムを脱塩水に浸した。水銀温度計、及びスリーワンモーターに接続した攪拌羽根により、深底バスは、内温が80.0℃となるように温度を制御した。なお、カラム出口はモノマー液を冷却するため、−5℃のブライン水を通じたシリコンチューブをトレースした。
(原料溶液の調整)
反応の原料となる試薬としては、メタクリル酸メチル、メタクリル酸、ハイドロキノン、メトキノン(4−メトキシフェノール)、フェノチアジン(以上全て、東京化成品)、メタノール(低水分50ppm以下、特級品、キシダ化学品)使用し、メタクリル酸とメタノールとが組成比でメタクリル酸1.0モルに対し、メタノール2.0モルを含む溶液となるように調製した。具体的には、メタクリル酸 860.3g、メタノール640.7gに加えて、ハイドロキノン 0.751g、メトキノン0.762g、フェノチアジン0.765gの3種類の重合禁止剤を添加し、室温で攪拌して溶解した。
(陽イオン交換樹脂の前処理)
H形に再生した陽イオン交換樹脂を、前記SUSカラムに充填した。次いで、特級メタノール(キシダ化学品 無水メタノール)150mlを、SV2の流速で、上向流で通液することにより、陽イオン交換樹脂をメタノールで置換した。水からメタノールに置換すると陽イオン交換樹脂は収縮する傾向にあるが、それもその樹脂の実力としてそのまま評価した。更に、上述のようにして調製した原料溶液(即ち、メタクリル酸とメタノールとの混合溶液)150mlを、SV2の流速で、上向流で通液することにより、陽イオン交換樹脂を原料溶液で置換した。
(MMA合成反応、及びその分析)
前記原料溶液を、樹脂量75.0mlに対し75.0g/時間の流速で(即ち、SV1の流速で)、上向流で前記SUSカラムに通液した。前記SV(空間速度)は仕込み時の体積を基準に表示したものである。
前記メスシリンダーに流出した溶液を、30分毎又は1時間毎にサンプリングした。勿論、原料溶液や反応溶液が粘調になり通液が不能になることはなかった。
〈バッチ反応(脱水系)による評価〉
(原料溶液の調整)
メタクリル酸426.4g(東京化成品)と、メタノール318.5g(キシダ化学品、無水品)とを入れ(メタノールとメタクリル酸とが2:1(モル比)となるようにした。)、さらに重合禁止剤として、ヒドロキノン(東京化成品)0.371g、メトキノン(p−メトキシヒドロキノン)(東京化成品)0.369g、フェノチアジン(東京化成品)0.364gを加えて溶解し、原料溶液を調整した。なお、フェノチアジンは10分間ほど室温で攪拌したところ、溶解した。
(陽イオン交換樹脂の前処理)
陽イオン交換樹脂に、予め、2N−HClを10BV通液し、H形に再生した。この陽イオン交換樹脂をメスシリンダーで20.0ml計り取り、これを遠心分離器で水切りした。このようにして水切りした陽イオン交換樹脂20.0mlを、300mlの三角フラスコに入れ、50℃で12時間、真空乾燥器を用いて真空乾燥を行なった。
(MMA合成反応、及びその分析)
上述のようにして得られた原料溶液を、予め50.0℃に加温していた振盪器に浸し加温した。50℃に加温した原料溶液50.0gを、上記三角フラスコ(陽イオン交換樹脂を含む。)に加え、恒温振盪器(設定温度50.0℃)を用いて120spm(ストローク長4cm)で振盪した。120spmという振盪速度は、陽イオン交換樹脂が流動状態を維持できる振盪速度である。
〈バッチ反応(水切り系)による評価〉
(原料溶液の調整)
メタクリル酸428.65g(東京化成品)と、メタノール319.11g(キシダ化学品、無水品)とを入れ(メタノールとメタクリル酸とが2:1(モル比)となるようにした。)、さらに重合禁止剤として、ヒドロキノン(東京化成品)0.388g、メトキノン(p−メトキシヒドロキノン)(東京化成品)0.357g、フェノチアジン(東京化成品)0.365gを加えて溶解し、原料溶液を調整した。
(陽イオン交換樹脂の前処理)
真空乾燥を行なわなかったこと以外は、前記〈バッチ反応(脱水系)による評価〉と同様の条件で、陽イオン交換樹脂の前処理を行なった。即ち、陽イオン交換樹脂に、予め、2N−HClを10BV通液し、H形に再生した。この陽イオン交換樹脂をメスシリンダーで20.0ml計り取り、これを遠心分離器で水切りした。このようにして水切りした陽イオン交換樹脂20.0mlを、300mlの三角フラスコに入れた。
(MMA合成反応、及びその分析)
前記〈バッチ反応(脱水系)による評価〉と同様の条件で、MMA合成反応、及びその分析を行なった。
[実施例1]
(反応器)
重合反応用の反応器として、攪拌翼、圧力計、窒素管、温度計を取り付けたガラス製重合缶を使用した。
(水相)
水相として、2%ポリビニルアルコール:以下適宜「PVA」と略す。)水溶液15mlと、0.1%メチレンブルー水溶液10mlとを含む水溶液1075mlを調製した。水相中の溶存酸素を除去するため、水相溶液を攪拌しながら窒素ガスを50ml/分で30分間流通した。
(油相(モノマー相))
原料モノマーとして、スチレン(St)249gと63%ジビニルベンゼンのモノマー溶液12.44gとを用いるとともに、重合開始剤として、75%BPO(日本油脂製 純度75%過酸化ベンゾイル)0.78gを用い、これらを混合して油相(モノマー相)を調製した。
(重合反応)
前記水相と前記油相とを、前記反応器に入れた。窒素ガスで置換する脱気操作を3回繰り返すことにより、反応器中の気相を窒素に置換した。次いで、30℃で30分間、110rpmで攪拌し、懸濁液とした。2時間かけて80℃まで昇温し、80℃で8時間保持した。得られた重合体を反応器から取り出して脱塩水で洗浄し、真空乾燥器を用いて50℃で8時間乾燥した。
(陽イオン交換基の導入)
乾燥後の重合体100gを、容量2Lの3つ口フラスコに入れ、溶媒として二塩化エチレン300g(3.0(g/重合ポリマー1g))を加え、50℃で1時間攪拌し、樹脂を膨潤させた。この中に、表1に示すように、重合ポリマー100gに対し、98%硫酸600g、及び25%オリウム350gを加えた。30℃から3時間かけて80℃まで昇温し、80℃で8時間保持した。反応終了後、反応液を室温まで冷却し、脱塩水を加えながら硫酸を除去した。その後、得られた重合体を取り出し、純水で洗浄することにより、陽イオン交換樹脂を得た。
[実施例2]
(油相(モノマー相))において、スチレンの添加量を258g、ジビニルベンゼンの含有量を18.9g、過酸化ベンゾイルの添加量を0.69gとしてモノマー相を調整したこと、及び、(カチオン交換基の導入)において、98%硫酸を4.5(g/重合ポリマー1g)、25%オリウムを2.5(g/重合ポリマー1g)の割合で添加したこと以外は、実施例1と同様の条件で陽イオン交換樹脂を製造した。
[実施例3]
(油相(モノマー相))において、スチレンの添加量を245g、ジビニルベンゼンの含有量を16.4g、過酸化ベンゾイルの添加量を0.52gとしてモノマー相を調製したこと、及び(カチオン交換基の導入)において、98%硫酸を4.5(g/重合ポリマー1g)、25%オリウムを2.0(g/重合ポリマー1g)の割合で添加し、溶媒として二塩化エチレンの代わりにニトロベンゼンを用いて、110℃で反応させたこと以外は、実施例1と同様の条件で陽イオン交換樹脂を製造した。
[実施例4]
(油相(モノマー相))において、スチレンの添加量を220g、ジビニルベンゼンの含有量を14.7g、過酸化ベンゾイルの添加量を0.75gとしてモノマー相を調製したこと、(重合反応)において、小粒径の陽イオン交換樹脂を得るために攪拌速度450rpmで重合を行なったこと(これにより、約140μmの重合ポリマーを得た。)、及び、(カチオン交換基の導入)において、98%硫酸を3.0(g/重合ポリマー1g)、25%オリウムを1.5(g/重合ポリマー1g)の割合で添加したこと以外は、実施例1と同様の条件で陽イオン交換樹脂を製造した。
[実施例5]
(油相(モノマー相))において、スチレンの添加量を224g、ジビニルベンゼンの含有量を15.2g、過酸化ベンゾイルの添加量を0.72gとしてモノマー相を調製したこと、(重合反応)において、小粒径の陽イオン交換樹脂を得るために攪拌速度550rpmで重合を行なったこと(これにより、約120μmの重合ポリマーを得た。)、(カチオン交換基の導入)において、98%硫酸を3.0(g/重合ポリマー1g)、25%オリウムを1.5(g/重合ポリマー1g)の割合で添加したこと以外は、実施例1と同様の条件で陽イオン交換樹脂を製造した。
[実施例6]
実施例3の(カチオン交換基の導入)において、98%硫酸を6.0(g/重合ポリマー1g)の割合で添加して、スルホン化を行ったこと以外は、実施例3と同様の条件で陽イオン交換樹脂を得た。
(重合禁止剤の添加)
得られた陽イオン交換樹脂(H形)を遠心分離器で水切りした。この水切りした陽イオン交換樹脂500mlを2Lのビーカーに入れ、この中へメタノールを加え、900mlの溶液とし、攪拌羽根を取り付けた攪拌機を用いて攪拌することにより、陽イオン交換樹脂をスラリー状態とした。
[実施例7]
実施例6において、フェノチアジンの代わりに、4−アミノTEMPO(4-Amino-2,2,6,6-tetramethylpiperidine 1-oxy)を用いた。
この中に、重合禁止剤としてアミノTEMPOを50mg含むメタノール溶液50mlを、滴下ロートを用いて5分間かけて滴下し、添加後、30分間攪拌した。その後、脱塩水を加えてメタノール溶液を脱塩水に置換した。
[実施例8]
4−アミノTEMPOの代わりに、DPPH(1,1−ジフェニル−2−ピクリルヒドラジル)を用いた以外は、実施例7と同様の条件で陽イオン交換樹脂を製造した。
[実施例9]
(油相(モノマー相))において、スチレンの添加量を152g、ジビニルベンゼンの
添加量を16.0g、ポリスチレン(重量平均分子量50000)の添加量を42.1g、過
酸化ベンゾイルの添加量を0.90gとしてモノマー相を調整したこと、(陽イオン交換基の導入)において、98%硫酸を1.5(g/重合ポリマー1g)、25%オリウムを3.5(g/重合ポリマー1g)、溶媒としてニトロベンゼンを3.0(g/重合ポリマー1g)の割合で使用し、105℃でスルホン化したこと以外は実施例1と同様の条件で陽イオン交換樹脂を製造した。
[実施例10]
(陽イオン交換基の導入)において、25%オリウムを7.5(g/重合ポリマー1g)の割合で添加して、105℃で5時間スルホン化したこと以外は実施例9と同様の条件で陽イオン交換樹脂を製造した。
[実施例11]
(重合反応)において、SUS製の加圧重合缶を用いて重合温度を80℃から120℃で8時間重合したこと以外は、実施例9と同様の条件で陽イオン交換樹脂を製造した。
[比較例1]
(油相(モノマー相))において、スチレンの添加量を151g、ジビニルベンゼンの含有量を5.0g、過酸化ベンゾイルの添加量を0.36gとしてモノマー相を調整したこと、及び、(カチオン交換基の導入)において、98%硫酸を8.0(g/重合ポリマー1g)、溶媒として二塩化エチレンを5.0(g/重合ポリマー1g)の割合で使用したこと以外は、実施例1と同様の条件で陽イオン交換樹脂を製造した。
[比較例2]
実施例3の(カチオン交換基の導入)において、98%硫酸とオリウムの混合溶液の代わりに、98%硫酸を6.0(g/重合ポリマー1g)の割合で用いたこと以外は、実施例3と同様の条件で陽イオン交換樹脂を製造した。
[比較例3]
(油相(モノマー相))において、スチレンの添加量を145g、ジビニルベンゼンの含有量を9.8g、2−エチルヘキサノール78.3g、過酸化ベンゾイルの添加量を0.92gとしてモノマー相を調整したこと、及び、(カチオン交換基の導入)において、98%硫酸を3.0(g/重合ポリマー1g)、25%オリウムを1.0(g/重合ポリマー1g)、溶媒として二塩化エチレンを4.0(g/重合ポリマー1g)の割合で使用したこと以外は、実施例1と同様の条件で陽イオン交換樹脂を製造した。
[比較例4]
(油相(モノマー相))において、スチレンの添加量を152g、ジビニルベンゼンの含有量を16.0g、n−ヘプタン93.6g、過酸化ベンゾイルの添加量を0.90gとしてモノマー相を調整したこと、及び、(カチオン交換基の導入)において、98%硫酸を1.5(g/重合ポリマー1g)、25%オリウムを1.0(g/重合ポリマー1g)、溶媒として二塩化エチレンを4.0(g/重合ポリマー1g)の割合で使用したこと以外は、実施例1と同様の条件で陽イオン交換樹脂を製造した。
[比較例5]
得られた陽イオン交換樹脂に重合禁止剤を添加しなかったこと以外は、実施例6と同様の条件で陽イオン交換樹脂を製造した。
[比較例6]
油相(モノマー相)において、スチレンの添加量を171.6g、ジビニルベンゼンの含有量を58.4g、過酸化ベンゾイルの添加量を0.52gとしてモノマー相を調製したこと、及び(カチオン交換基の導入)において、98%硫酸を2.0(g/重合ポリマー1g)、25%オリウムを1.5(g/重合ポリマー1g)の割合で使用したこと以外は、実施例3と同様の条件で陽イオン交換樹脂を製造した。
[比較例7]
比較例7の陽イオン交換樹脂として、ロームアンドハース社製のアンバーリストR15を用いた。
[比較例8]
比較例8の陽イオン交換樹脂として、ランクセス社製のレバチットRK2629を用いた。
[比較例9]
比較例9の陽イオン交換樹脂として、ランクセスのレバチットRSP112を用いた。[比較例10]
(油相(モノマー相))において、スチレンの添加量を157g、ジビニルベンゼンの添加量を22.8g、ポリスチレン(重量平均分子量50000)の添加量を38.6g、過
酸化ベンゾイルの添加量を0.75gとしてモノマー相を調整したこと、(陽イオン交換基の導入)において、98%硫酸を1.5(g/重合ポリマー1g)、25%オリウムを1.5(g/重合ポリマー1g)、溶媒としてニトロベンゼンを5.0(g/重合ポリマー1g)の割合で使用し、105℃でスルホン化したこと以外は実施例9と同様の条件で陽イオン交換樹脂を製造した。
[比較例11]
(油相(モノマー相))において、スチレンの添加量を78.4g、ジビニルベンゼンの含有量を51.6g、イソドデカン70.0g、過酸化ベンゾイルの添加量を1.45gとしてモノマー相を調整したこと、及び、(カチオン交換基の導入)において、98%硫酸の添加量を1.5(g/重合ポリマー1g)、25%オリウムを1.5(g/重合ポリマー1g)とし、溶媒としてニトロベンゼンを4.0(g/重合ポリマー1g)の割合で使用したこと以外は、比較例4と同様の条件で陽イオン交換樹脂を製造した。
[比較例12]
比較例12の陽イオン交換樹脂として、ロームアンドハース社製のデュオライトRC26Hを用いた。
[実施例及び比較例で得られた陽イオン交換樹脂の評価結果]
実施例及び比較例で得られた陽イオン交換樹脂を、前述の測定方法で各物性値を測定して、その結果を表1に示す。なお、表1中の「オリウム濃度(重量%)」は、98%硫酸に含まれる2%の水分を考慮して実際のオリウム濃度を算出したものである。

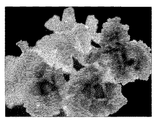
の周りにpMMAが観察されるが、陽イオン交換樹脂自体の形状の変化は観察されない。一方、図4では、陽イオン交換樹脂が膨潤し、その圧力に耐え切れずに爆裂し、ポップコーン状になっているのが観察される。
Claims (5)
- 下記(A)、(B)、及び(C)を満たす
ことを特徴とする、(メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂。
(A)窒素吸着法で測定される表面積が2m2/g以下である
(B)架橋度が2重量%以上7.5%重量以下である
(C)膨潤度が2以下である
(当該膨潤度とは、下記式(I)で表される値である。
(前記強酸性陽イオン交換樹脂にメタクリル酸メチルモノマーを加え、70℃の恒温槽中で72時間静置することにより膨潤させた後の体積)/(H形に再生させた前記強酸性陽イオン交換樹脂の膨潤前の体積))・・・式(I)) - 水分含有率が35重量%以上である
ことを特徴とする、請求項1に記載の(メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂。 - 重合禁止剤を含有する
ことを特徴とする、請求項1又は請求項2に記載の(メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂。 - オリウムを0.1重量%以上含有する硫酸を用いてスルホン化する工程を有する
ことを特徴とする、
請求項1〜3のいずれか1項に記載の(メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂の製造方法。 - 請求項1〜4のいずれか1項に記載の強酸性陽イオン交換樹脂を用いる
ことを特徴とする、(メタ)アクリル酸エステルの製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008272344A JP5169727B2 (ja) | 2008-10-22 | 2008-10-22 | (メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008272344A JP5169727B2 (ja) | 2008-10-22 | 2008-10-22 | (メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010099573A JP2010099573A (ja) | 2010-05-06 |
| JP5169727B2 true JP5169727B2 (ja) | 2013-03-27 |
Family
ID=42290694
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008272344A Active JP5169727B2 (ja) | 2008-10-22 | 2008-10-22 | (メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5169727B2 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5880376B2 (ja) * | 2012-09-27 | 2016-03-09 | 住友金属鉱山株式会社 | 複合タングステン酸化物微粒子分散液 |
| CN104845735A (zh) * | 2015-04-29 | 2015-08-19 | 四川银帆生物科技有限公司 | 利用酿酒副产物黄水制取香味液过程中的二次酯化工艺 |
| US10766848B2 (en) * | 2015-09-07 | 2020-09-08 | Rhodia Operations | Use of polymerization inhibitor compositions |
| CN113117750A (zh) * | 2021-03-17 | 2021-07-16 | 南京福昌环保有限公司 | 丙烯酸及酯类废油回收装置连续酯化复合催化剂制备应用 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH074532B2 (ja) * | 1985-04-06 | 1995-01-25 | 三菱化学株式会社 | 触媒用スルホン酸型陽イオン交換樹脂 |
| JPH0699365B2 (ja) * | 1989-07-21 | 1994-12-07 | 株式会社日本触媒 | アクリル酸エステルの製造法 |
| JP2848709B2 (ja) * | 1990-12-21 | 1999-01-20 | 徳山石油化学株式会社 | 酢酸3−エトキシブチルエステル化合物とその製造方法 |
| JP3386483B2 (ja) * | 1991-12-18 | 2003-03-17 | 三菱化学株式会社 | イオン交換樹脂の製造方法 |
| JPH07116525A (ja) * | 1993-10-21 | 1995-05-09 | Tokyo Organ Chem Ind Ltd | 陽イオン交換樹脂 |
| JP4079480B2 (ja) * | 1996-08-20 | 2008-04-23 | 三菱化学株式会社 | (メタ)アクリル酸エステルの製造方法 |
| JP2002088019A (ja) * | 2000-09-08 | 2002-03-27 | Nippon Shokubai Co Ltd | (メタ)アクリル酸エステルの製造方法 |
-
2008
- 2008-10-22 JP JP2008272344A patent/JP5169727B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2010099573A (ja) | 2010-05-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101747473B (zh) | 表面功能化的分子印迹聚合物微球及其制备方法 | |
| Svec et al. | Kinetic control of pore formation in macroporous polymers. Formation of" molded" porous materials with high flow characteristics for separations or catalysis | |
| TWI243813B (en) | Metal-doped sulfonated ion exchange resin catalysts | |
| US6646083B2 (en) | Crosslinked polymers containing tertiary amine and/or quaternary ammonium salt structures, processes for making and uses thereof | |
| Joshi et al. | Novel separation strategies based on molecularly imprinted adsorbents | |
| JPH0649139A (ja) | 架橋された球形の共重合体ビーズおよびその製造方法 | |
| EP2205672B1 (en) | Non-ionic porous, small solid resin with chemically bonded crown ether | |
| JP5169727B2 (ja) | (メタ)アクリル酸エステルの製造用強酸性陽イオン交換樹脂 | |
| WO2006132333A1 (ja) | 親水性に優れた新規充填剤、及びその製造方法 | |
| CN110479220A (zh) | 基于负载离子液体金属有机骨架的分子印迹聚合物分离富集痕量磺胺甲恶唑污染物的方法 | |
| JP2009518501A (ja) | ポリマービーズの製造方法 | |
| JP2005510609A (ja) | 多孔性支持体の後修飾 | |
| JP2005510609A5 (ja) | ||
| JPS5861463A (ja) | 液体クロマトグラフイ−用担体及び該担体を用いる脂溶性物質の分離精製方法 | |
| JP6638003B2 (ja) | 塩素化ポリ塩化ビニルを製造するためのポリ塩化ビニル粒子の提供方法 | |
| JP2003301016A (ja) | 単分散アニオン交換ゲルの製法および単分散アニオン交換ゲル | |
| JP2008208368A (ja) | 単分散弱酸性カチオン交換体 | |
| Erdem et al. | Kinetics of esterification of propionic acid with n-amyl alcohol in the presence of cation exchange resins | |
| CN108367212A (zh) | 使用聚合物大孔吸附剂色谱分离有机酸 | |
| JP5749031B2 (ja) | 液体クロマトグラフィー用カラム充填剤の製造方法、液体クロマトグラフィーによる試料の測定方法、及び、ヘモグロビン類の測定方法 | |
| CN108435146A (zh) | 一种表面带有高分子刷的核-壳型弱阳离子交换树脂微球 | |
| JP6957449B2 (ja) | 触媒担体用ビニルピリジン樹脂、その製造方法およびメタノールのカルボニル化反応用触媒 | |
| FR2568256A1 (fr) | Polymeres d'acrylate et de methacrylate de glycidyle sous forme de perles et leur procede de fabrication | |
| KR102014742B1 (ko) | 혼합염 현탁 중합방법 및 그로부터 제조된 수지 및 촉매 | |
| WO2016143673A1 (ja) | 液体クロマトグラフィー用充填剤 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110804 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20120914 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120925 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20121204 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121217 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5169727 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313111 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |