JP5060751B2 - 酢酸ビニルの単離法 - Google Patents
酢酸ビニルの単離法 Download PDFInfo
- Publication number
- JP5060751B2 JP5060751B2 JP2006211544A JP2006211544A JP5060751B2 JP 5060751 B2 JP5060751 B2 JP 5060751B2 JP 2006211544 A JP2006211544 A JP 2006211544A JP 2006211544 A JP2006211544 A JP 2006211544A JP 5060751 B2 JP5060751 B2 JP 5060751B2
- Authority
- JP
- Japan
- Prior art keywords
- distillation column
- organic phase
- gas
- vinyl acetate
- column
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- XTXRWKRVRITETP-UHFFFAOYSA-N Vinyl acetate Chemical compound CC(=O)OC=C XTXRWKRVRITETP-UHFFFAOYSA-N 0.000 title claims abstract description 88
- 238000002955 isolation Methods 0.000 title 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims abstract description 208
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims abstract description 120
- 239000007789 gas Substances 0.000 claims abstract description 117
- 238000004821 distillation Methods 0.000 claims abstract description 87
- 239000012074 organic phase Substances 0.000 claims abstract description 67
- 239000007788 liquid Substances 0.000 claims abstract description 47
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims abstract description 40
- 239000000203 mixture Substances 0.000 claims abstract description 39
- 229910001868 water Inorganic materials 0.000 claims abstract description 39
- 238000005406 washing Methods 0.000 claims abstract description 31
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 claims abstract description 22
- 239000005977 Ethylene Substances 0.000 claims abstract description 22
- 239000012071 phase Substances 0.000 claims abstract description 20
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims abstract description 18
- 238000006243 chemical reaction Methods 0.000 claims abstract description 17
- 238000009835 boiling Methods 0.000 claims abstract description 12
- 238000001816 cooling Methods 0.000 claims abstract description 12
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims abstract description 11
- 239000003054 catalyst Substances 0.000 claims abstract description 11
- 239000001301 oxygen Substances 0.000 claims abstract description 11
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 11
- 229910052763 palladium Inorganic materials 0.000 claims abstract description 9
- 150000002941 palladium compounds Chemical class 0.000 claims abstract description 4
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 claims abstract 2
- 238000000034 method Methods 0.000 claims description 52
- 239000008346 aqueous phase Substances 0.000 claims description 15
- FGUUSXIOTUKUDN-IBGZPJMESA-N C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 Chemical compound C1(=CC=CC=C1)N1C2=C(NC([C@H](C1)NC=1OC(=NN=1)C1=CC=CC=C1)=O)C=CC=C2 FGUUSXIOTUKUDN-IBGZPJMESA-N 0.000 claims description 5
- 230000003134 recirculating effect Effects 0.000 claims description 4
- GNFTZDOKVXKIBK-UHFFFAOYSA-N 3-(2-methoxyethoxy)benzohydrazide Chemical compound COCCOC1=CC=CC(C(=O)NN)=C1 GNFTZDOKVXKIBK-UHFFFAOYSA-N 0.000 claims description 2
- 238000005201 scrubbing Methods 0.000 claims 1
- 238000000926 separation method Methods 0.000 abstract description 6
- 238000005086 pumping Methods 0.000 abstract description 2
- 239000002253 acid Substances 0.000 abstract 1
- 230000018044 dehydration Effects 0.000 description 37
- 238000006297 dehydration reaction Methods 0.000 description 37
- 239000000047 product Substances 0.000 description 29
- 238000010533 azeotropic distillation Methods 0.000 description 15
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 12
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 12
- 239000000463 material Substances 0.000 description 8
- 229910052786 argon Inorganic materials 0.000 description 6
- 239000001569 carbon dioxide Substances 0.000 description 6
- 229910002092 carbon dioxide Inorganic materials 0.000 description 6
- 150000001875 compounds Chemical class 0.000 description 6
- 239000011261 inert gas Substances 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 229920000642 polymer Polymers 0.000 description 5
- 239000006227 byproduct Substances 0.000 description 4
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 4
- 239000010931 gold Substances 0.000 description 4
- 229910052737 gold Inorganic materials 0.000 description 4
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 4
- 238000010992 reflux Methods 0.000 description 4
- JTXMVXSTHSMVQF-UHFFFAOYSA-N 2-acetyloxyethyl acetate Chemical compound CC(=O)OCCOC(C)=O JTXMVXSTHSMVQF-UHFFFAOYSA-N 0.000 description 3
- 239000012190 activator Substances 0.000 description 3
- 229910052783 alkali metal Inorganic materials 0.000 description 3
- 238000004140 cleaning Methods 0.000 description 3
- 238000005260 corrosion Methods 0.000 description 3
- 230000007797 corrosion Effects 0.000 description 3
- 238000005265 energy consumption Methods 0.000 description 3
- 229940117927 ethylene oxide Drugs 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 238000010626 work up procedure Methods 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 229910052793 cadmium Inorganic materials 0.000 description 2
- BDOSMKKIYDKNTQ-UHFFFAOYSA-N cadmium atom Chemical compound [Cd] BDOSMKKIYDKNTQ-UHFFFAOYSA-N 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 235000011056 potassium acetate Nutrition 0.000 description 2
- 229910052761 rare earth metal Inorganic materials 0.000 description 2
- 150000002910 rare earth metals Chemical class 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052788 barium Inorganic materials 0.000 description 1
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 1
- PVEOYINWKBTPIZ-UHFFFAOYSA-N but-3-enoic acid Chemical compound OC(=O)CC=C PVEOYINWKBTPIZ-UHFFFAOYSA-N 0.000 description 1
- LHQLJMJLROMYRN-UHFFFAOYSA-L cadmium acetate Chemical compound [Cd+2].CC([O-])=O.CC([O-])=O LHQLJMJLROMYRN-UHFFFAOYSA-L 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 238000011278 co-treatment Methods 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000007599 discharging Methods 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- -1 nitrogen and argon Chemical compound 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/04—Preparation of carboxylic acid esters by reacting carboxylic acids or symmetrical anhydrides onto unsaturated carbon-to-carbon bonds
- C07C67/05—Preparation of carboxylic acid esters by reacting carboxylic acids or symmetrical anhydrides onto unsaturated carbon-to-carbon bonds with oxidation
- C07C67/055—Preparation of carboxylic acid esters by reacting carboxylic acids or symmetrical anhydrides onto unsaturated carbon-to-carbon bonds with oxidation in the presence of platinum group metals or their compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/48—Separation; Purification; Stabilisation; Use of additives
- C07C67/52—Separation; Purification; Stabilisation; Use of additives by change in the physical state, e.g. crystallisation
- C07C67/54—Separation; Purification; Stabilisation; Use of additives by change in the physical state, e.g. crystallisation by distillation
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
H2C=CH2+CH3−COOH+0.5H2O → H2C=CH−O−CO−CH3+H2O
完全には避けることができないエチレンの完全酸化でCO2と水が生じる:
H2C=CH2+3O2 → 2CO2+2H20
それ故に1モルの酢酸ビニル当たり1モルより多い水が生じる。一般には水の重量は生じる酢酸ビニルの重量の約1/4に成る。
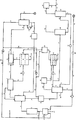
a)反応域から出るガス混合物を第一の蒸留塔に導入し、
b)第一の蒸留塔の塔頂部から出るガス混合物を−20〜+50℃ に冷却し、その際に生じる凝縮物を水性相と有機相とに分離し、
c)段階b)で生じた水性相を除去し、
d)段階b)で生じる有機相の全部または一部を段階a)で利用した第一蒸留塔の塔頂部に再循環液として戻しそして有機相の、再循環液として利用しない部分を取り出し、
e)段階b)で凝縮されない酢酸ビニル含有ガスを洗浄塔において少なくとも90%濃度の水性酢酸で洗浄しそしてその際に塔底部の所で酢酸ビニル含有の酢酸溶液を得、
f)酢酸ビニル、酢酸エチル、酢酸および水を含有する、段階a)の塔底部生成物を収集容器に供給しそして加圧下にある該液体を圧力開放し、その際にガスが生じ、
g)段階f)での圧力開放の際に生じる液体を第二の蒸留塔に供給しそしてそれの塔底部の上方の富化域から酢酸エチル含有側流を引き出し、
h)酢酸および水を含有する、段階g)の塔底部生成物の全部または一部を段階e)でガス洗浄のために利用し、
i)段階g)の塔頂部の蒸気を冷却し、その際に生じる凝縮液を水性相と有機相とに分離し、
j)段階i)で生じた水性相を除去し、
k)段階i)で生じる有機相の一部を再還流液として段階g)で利用する第二の蒸留塔の塔頂部に戻しそして残りの部分を引き出す
の各段階を行う酢酸ビニルの分離方法において、
l)段階e)で利用する洗浄塔の塔底部生成物の一部をポンプ循環下に最初に冷却しそして段階e)で利用する洗浄塔の塔底部に戻し、残りの部分を引出しそしてその引き出された部分を少なくとも30℃の温度に加熱しそして段階a)で利用した第一の蒸留塔の下方部分に供給し、
m)段階d)で引き出された残りの有機相を圧力開放し、その圧力開放の際に生じるガスを段階f)で生じるガスと一緒にしそしてその一緒にしたガスを再び方法系に戻し、
n)段階m)で得られる有機相を段階i)で得られる有機相と一緒にしそして段階k)で引き出されそして再循環液として使用されない残りの有機相を第三の蒸留塔に導入し、
o)段階n)の第三の蒸留塔の塔頂留出物を冷却しそしてその際に得られる低沸点留分とその際に生じる水とに分離し、
p)段階n)の第三の蒸留塔の塔底部生成物を第四の蒸留塔に導入し、
q)段階p)で利用した第四の蒸留塔の塔頂から純粋酢酸ビニルを引き出す
ことを特徴とする、上記方法である。
5・・・酢酸ビニル反応器
7・・・予備脱水器
21・・・洗浄塔(器)
34・・・再循環ガス圧縮器
37・・・第二の蒸留塔
41・・・相分離器
48・・・第三の蒸留器
61・・・第四の蒸留塔
Claims (11)
- 気相においてパラジウムまたはパラジウム化合物含有触媒に接してエチレンと酢酸および酸素とを反応させる際に生じるガス混合物から酢酸ビニルを分離するに際して、
a)反応域から出るガス混合物を第一の蒸留塔に導入し、
b)第一の蒸留塔の塔頂部から出るガス混合物を−20〜+50℃に冷却し、その際に生じる凝縮物を水性相と有機相とに分離し、
c)段階b)で生じた水性相を除去し、
d)段階b)で生じる有機相の全部または一部を段階a)で利用した第一蒸留塔の塔頂部に再循環液として戻しそして有機相の、再循環液として利用しない部分を取り出し、
e)段階b)で凝縮されない酢酸ビニル含有ガスを洗浄塔において少なくとも90%濃度の水性酢酸で洗浄しそしてその際に塔底部の所で酢酸ビニル含有の酢酸溶液を得、
f)酢酸ビニル、酢酸エチル、酢酸および水を含有する、段階a)の塔底部生成物を収集容器に供給しそして加圧下にある該液体を圧力開放し、その際にガスが生じ、
g)段階f)での圧力開放の際に生じる液体を第二の蒸留塔に供給しそしてそれの塔底部の上方の富化域から酢酸エチル含有側流を引き出し、
h)酢酸および水を含有する、段階g)の塔底部生成物の全部または一部を段階e)でガス洗浄のために利用し、
i)段階g)の塔頂部の蒸気を冷却し、その際に生じる凝縮液を水性相と有機相とに分離し、
j)段階i)で生じた水性相を除去し、
k)段階i)で生じる有機相の一部を再還流液として段階g)で利用する第二の蒸留塔の塔頂部に戻しそして残りの部分を引き出す
の各段階を行う酢酸ビニルの分離方法において、
l)段階e)で利用する洗浄塔の塔底部生成物の一部をポンプ循環下に最初に冷却しそして段階e)で利用する洗浄塔の塔底部に戻し、残りの部分を引出しそしてその引き出された部分を少なくとも30℃の温度に加熱しそして段階a)で利用した第一の蒸留塔の下方部分に供給し、
m)段階d)で引き出された残りの有機相を圧力開放し、その圧力開放の際に生じるガスを段階f)で生じるガスと一緒にしそしてその一緒にしたガスを再び方法系に戻し、
n)段階m)で得られる有機相を段階i)で得られる有機相と一緒にしそして段階k)で引き出されそして再循環液として使用されない残りの有機相を第三の蒸留塔に導入し、
o)段階n)の第三の蒸留塔の塔頂留出物を冷却しそしてその際に得られる低沸点留分とその際に生じる水とに分離し、
p)段階n)の第三の蒸留塔の塔底部生成物を第四の蒸留塔に導入し、
q)段階p)で利用した第四の蒸留塔の塔頂から純粋酢酸ビニルを引き出す
ことを特徴とする、上記方法。 - 段階l)において洗浄塔から引き出される塔底部生成物を60℃〜120℃に加熱する、請求項1に記載の方法。
- 段階l)において引き出される塔底部生成物を段階a)において利用する第一の蒸留塔の、該塔の塔底部から数えて2〜15段に供給する、請求項1または2に記載の方法。
- 段階l)で引き出される塔底部生成物の、段階a)で利用する第一の蒸留塔への還流を、第一の蒸留塔の塔底部留出物が80℃〜150℃の温度を有する様に行う、請求項1〜3のいずれか一つに記載の方法。
- 段階f)において段階a)からの塔底部生成物を収集容器で0.02〜0.2MPaの圧力に圧力開放する、請求項1〜4のいずれか一つに記載の方法。
- 段階m)において段階d)で引き出される残留有機相を圧力開放容器に供給しそして0.02〜0.2MPaの圧力に圧力開放する、請求項1〜5のいずれか一つに記載の方法。
- 段階b)での冷却温度、および段階b)で生じる有機相の、段階d)で再循環液として利用する部分を、全ての酢酸エチルが段階a)の第一の蒸留塔の塔底部生成物中にできるだけ含まれる様に選択する、請求項1〜6のいずれか一つに記載の方法。
- 有機相の、段階d)で再循環液として使用しない部分を増やし、その際に段階n)において相分離器で一緒にした有機相ができるだけ少ない酢酸エチル含有量である請求項1〜7のいずれか一つに記載の方法。
- 段階n)において、第二の蒸留塔の塔頂部蒸気ができるだけ少ない酢酸および酢酸エチルしか含まない様な量でしか、一緒にされた有機相を再循環液として戻さない、請求項1〜8のいずれか一つに記載の方法。
- 段階o)において、低沸点留分および水の十分な部分を分離するために、冷却された塔頂生成物を再循環液としてそれだけ第三の蒸留塔に供給する、請求項1〜9のいずれか一つに記載の方法。
- 段階a)で、反応領域から流出するガス混合物を最初に向流式熱交換器において、より冷たい循環ガスで115〜150℃に冷却しそして次いで始めて第一の蒸留塔に導入する、請求項1〜10のいずれか一つに記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102005036930.8 | 2005-08-05 | ||
| DE102005036930.8A DE102005036930B4 (de) | 2005-08-05 | 2005-08-05 | Verfahren zur Isolierung von Vinylacetat |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2007045821A JP2007045821A (ja) | 2007-02-22 |
| JP2007045821A5 JP2007045821A5 (ja) | 2009-09-17 |
| JP5060751B2 true JP5060751B2 (ja) | 2012-10-31 |
Family
ID=37603063
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2006211544A Active JP5060751B2 (ja) | 2005-08-05 | 2006-08-03 | 酢酸ビニルの単離法 |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US7301048B2 (ja) |
| EP (1) | EP1760065B1 (ja) |
| JP (1) | JP5060751B2 (ja) |
| CN (1) | CN1907943B (ja) |
| AT (1) | ATE425954T1 (ja) |
| BR (1) | BRPI0603175B1 (ja) |
| CA (1) | CA2555391C (ja) |
| DE (2) | DE102005036930B4 (ja) |
| ES (1) | ES2323541T3 (ja) |
| MX (1) | MXPA06008721A (ja) |
| MY (1) | MY138283A (ja) |
| PL (1) | PL1760065T3 (ja) |
| SG (1) | SG130116A1 (ja) |
| TW (1) | TWI308146B (ja) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102006038689B4 (de) * | 2006-08-17 | 2015-01-22 | Celanese Chemicals Europe Gmbh | Verfahren zur Aufarbeitung von Vinylacetat |
| DE102008001366A1 (de) * | 2008-04-24 | 2009-10-29 | Wacker Chemie Ag | Verfahren zur Herstellung von ungesättigten Carbonsäureestern |
| RU2477268C1 (ru) * | 2008-12-13 | 2013-03-10 | Силаниз Кемикалз Юроп Гмбх | Способ получения винилацетата |
| KR20110099313A (ko) * | 2008-12-13 | 2011-09-07 | 셀라네제 쉐미칼스 오이로페 게엠베하 | 비닐 아세테이트의 제조 방법 |
| DE102009002666A1 (de) * | 2009-04-27 | 2010-10-28 | Wacker Chemie Ag | Verfahren zur Herstellung von Vinylacetat |
| DE102010001097A1 (de) | 2010-01-21 | 2011-07-28 | Wacker Chemie AG, 81737 | Verfahren zur Herstellung von Vinylacetat |
| DE102012214064A1 (de) | 2012-08-08 | 2014-02-13 | Wacker Chemie Ag | Verfahren zur Aufreinigung von Kohlendioxid enthaltenden Prozessgasen aus der Herstellung von Vinylacetat |
| US9045413B2 (en) * | 2012-08-30 | 2015-06-02 | Celanese International Corporation | Process for vinyl acetate production having sidecar reactor for predehydrating column |
| DE102014206450A1 (de) | 2014-04-03 | 2015-10-08 | Wacker Chemie Ag | Verfahren zur Aufreinigung von Kohlendioxid enthaltenden Prozessgasen aus der Herstellung von Vinylacetat |
| DE102016205487A1 (de) * | 2016-04-04 | 2017-10-05 | Wacker Chemie Ag | Verfahren zur Herstellung von Vinylacetat |
| BR112022000672B1 (pt) * | 2019-08-05 | 2024-02-20 | Lyondellbasell Acetyls, Llc | Método para purificar uma corrente de alimentação de acetato de vinila líquido bruto |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1278430C2 (de) * | 1967-02-25 | 1974-05-30 | Verfahren zur abtrennung der mischung eines vinylesters im molaren ueberschuss mit der entsprechenden carbonsaeure | |
| US3591463A (en) * | 1968-10-25 | 1971-07-06 | Du Pont | Separation of chloroform and/or ethyl acetate and/or methylethyl ketone from vinyl acetate by extractive distillation |
| DE2610624C2 (de) * | 1976-03-13 | 1985-02-14 | Hoechst Ag, 6230 Frankfurt | Verfahren zur Gewinnung von Vinylacetat aus den Gasgemischen, die bei dessen Herstellung anfallen |
| DE2943985A1 (de) * | 1979-10-31 | 1981-05-14 | Hoechst Ag, 6000 Frankfurt | Verfahren zur abtrennung von wasser aus gemischen mit vinylacetat und essigsaeure |
| DE3422575A1 (de) * | 1984-06-18 | 1985-12-19 | Hoechst Ag, 6230 Frankfurt | Verfahren zur abtrennung von vinylacetat |
| JPS6219554A (ja) * | 1985-07-17 | 1987-01-28 | Nippon Synthetic Chem Ind Co Ltd:The | 酢酸ビニル反応生成物の精製方法 |
| DE3934614A1 (de) * | 1989-10-17 | 1991-04-18 | Hoechst Ag | Verfahren zur isolierung von vinylacetat |
| DE10042695A1 (de) * | 2000-08-31 | 2002-03-28 | Wacker Chemie Gmbh | Verfahren zur Reduzierung von Polymerisationsinhibitoren bei der Herstellung von Vinylacetatmonomer (VAM) |
-
2005
- 2005-08-05 DE DE102005036930.8A patent/DE102005036930B4/de not_active Expired - Fee Related
-
2006
- 2006-07-11 US US11/484,419 patent/US7301048B2/en active Active
- 2006-07-26 AT AT06015532T patent/ATE425954T1/de active
- 2006-07-26 DE DE502006003159T patent/DE502006003159D1/de active Active
- 2006-07-26 ES ES06015532T patent/ES2323541T3/es active Active
- 2006-07-26 PL PL06015532T patent/PL1760065T3/pl unknown
- 2006-07-26 EP EP06015532A patent/EP1760065B1/de active Active
- 2006-08-01 MX MXPA06008721A patent/MXPA06008721A/es active IP Right Grant
- 2006-08-02 CN CN2006101083543A patent/CN1907943B/zh active Active
- 2006-08-03 TW TW095128412A patent/TWI308146B/zh not_active IP Right Cessation
- 2006-08-03 CA CA2555391A patent/CA2555391C/en not_active Expired - Fee Related
- 2006-08-03 SG SG200605232-8A patent/SG130116A1/en unknown
- 2006-08-03 JP JP2006211544A patent/JP5060751B2/ja active Active
- 2006-08-04 BR BRPI0603175A patent/BRPI0603175B1/pt active IP Right Grant
- 2006-08-04 MY MYPI20063773A patent/MY138283A/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| BRPI0603175B1 (pt) | 2015-11-03 |
| PL1760065T3 (pl) | 2009-06-30 |
| DE502006003159D1 (de) | 2009-04-30 |
| DE102005036930B4 (de) | 2014-01-23 |
| MXPA06008721A (es) | 2007-02-05 |
| US20070032678A1 (en) | 2007-02-08 |
| BRPI0603175A (pt) | 2007-05-15 |
| DE102005036930A1 (de) | 2007-02-08 |
| TW200706533A (en) | 2007-02-16 |
| SG130116A1 (en) | 2007-03-20 |
| CA2555391C (en) | 2013-09-24 |
| TWI308146B (en) | 2009-04-01 |
| ATE425954T1 (de) | 2009-04-15 |
| MY138283A (en) | 2009-05-29 |
| EP1760065B1 (de) | 2009-03-18 |
| CN1907943A (zh) | 2007-02-07 |
| CA2555391A1 (en) | 2007-02-05 |
| ES2323541T3 (es) | 2009-07-20 |
| JP2007045821A (ja) | 2007-02-22 |
| CN1907943B (zh) | 2011-09-21 |
| US7301048B2 (en) | 2007-11-27 |
| EP1760065A1 (de) | 2007-03-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5060751B2 (ja) | 酢酸ビニルの単離法 | |
| JP5411288B2 (ja) | 酢酸ビニルの製造方法 | |
| JP2859419B2 (ja) | 酢酸ビニルの分離法 | |
| JP2007045821A5 (ja) | ||
| CN1102530C (zh) | 处理将HCl氧化成氯时的反应气体的方法 | |
| JPS6121603B2 (ja) | ||
| EP2089352B1 (en) | Process for working up vinyl acetate | |
| KR102641477B1 (ko) | 분리벽을 갖는 증류 컬럼을 포함하는 (메트)아크릴산의 정제 방법 | |
| RU2494092C2 (ru) | Улучшенный способ совместного получения акрилонитрила и циановодорода | |
| JPS6112646A (ja) | 酢酸ビニルの分離方法 | |
| TW201125846A (en) | Process for preparing vinyl acetate | |
| CN106715377B (zh) | 用于处理乙酸生产单元的废气的方法和装置 | |
| JP2012524757A (ja) | 酢酸ビニルの製造方法 | |
| JP4677066B2 (ja) | 液状の粗酢酸ビニルの後処理法 | |
| CN111372909A (zh) | 用于生产乙酸乙烯酯的方法 | |
| JPH0138775B2 (ja) | ||
| KR20070017031A (ko) | 비닐 아세테이트의 분리방법 | |
| RU2477268C1 (ru) | Способ получения винилацетата | |
| KR100744753B1 (ko) | 부탄올을 이용한 공비증류에 의한 초산의 회수방법 | |
| JPS5826895B2 (ja) | メタクロレイン含有ガス混合物から水蒸気を除去する方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090731 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090804 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20100526 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20120307 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120410 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120615 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120731 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120806 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150810 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5060751 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313111 |
|
| R370 | Written measure of declining of transfer procedure |
Free format text: JAPANESE INTERMEDIATE CODE: R370 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313111 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |