JP4989891B2 - 金属酸化物系蛍光体微粒子を利用した蛍光体及びその製造方法並びに蛍光変換体 - Google Patents
金属酸化物系蛍光体微粒子を利用した蛍光体及びその製造方法並びに蛍光変換体 Download PDFInfo
- Publication number
- JP4989891B2 JP4989891B2 JP2005517448A JP2005517448A JP4989891B2 JP 4989891 B2 JP4989891 B2 JP 4989891B2 JP 2005517448 A JP2005517448 A JP 2005517448A JP 2005517448 A JP2005517448 A JP 2005517448A JP 4989891 B2 JP4989891 B2 JP 4989891B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- metal oxide
- phosphor
- fine particles
- fluorescent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 title claims description 116
- 229910044991 metal oxide Inorganic materials 0.000 title claims description 99
- 150000004706 metal oxides Chemical class 0.000 title claims description 99
- 239000010419 fine particle Substances 0.000 title claims description 83
- 238000004519 manufacturing process Methods 0.000 title claims description 26
- 238000000034 method Methods 0.000 claims description 55
- 229920005989 resin Polymers 0.000 claims description 48
- 239000011347 resin Substances 0.000 claims description 48
- 229910052751 metal Inorganic materials 0.000 claims description 41
- 239000002245 particle Substances 0.000 claims description 41
- 239000002184 metal Substances 0.000 claims description 38
- 239000002904 solvent Substances 0.000 claims description 37
- 239000002612 dispersion medium Substances 0.000 claims description 33
- 239000006185 dispersion Substances 0.000 claims description 31
- 239000002244 precipitate Substances 0.000 claims description 24
- 125000000524 functional group Chemical group 0.000 claims description 21
- 150000002894 organic compounds Chemical class 0.000 claims description 21
- 239000013078 crystal Substances 0.000 claims description 20
- 125000000962 organic group Chemical group 0.000 claims description 18
- 238000010438 heat treatment Methods 0.000 claims description 16
- 150000001875 compounds Chemical class 0.000 claims description 14
- 238000009835 boiling Methods 0.000 claims description 10
- 229910052684 Cerium Inorganic materials 0.000 claims description 9
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 9
- 238000000926 separation method Methods 0.000 claims description 9
- 229910052765 Lutetium Inorganic materials 0.000 claims description 7
- GWXLDORMOJMVQZ-UHFFFAOYSA-N cerium Chemical compound [Ce] GWXLDORMOJMVQZ-UHFFFAOYSA-N 0.000 claims description 7
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical group [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 claims description 7
- 239000011159 matrix material Substances 0.000 claims description 6
- 239000002609 medium Substances 0.000 claims description 6
- 238000004062 sedimentation Methods 0.000 claims description 6
- 229910052782 aluminium Inorganic materials 0.000 claims description 5
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 claims description 5
- 239000011362 coarse particle Substances 0.000 claims description 5
- 238000010304 firing Methods 0.000 claims description 5
- 229910052727 yttrium Inorganic materials 0.000 claims description 5
- OHSVLFRHMCKCQY-UHFFFAOYSA-N lutetium atom Chemical compound [Lu] OHSVLFRHMCKCQY-UHFFFAOYSA-N 0.000 claims description 4
- 235000021384 green leafy vegetables Nutrition 0.000 claims description 3
- 230000003287 optical effect Effects 0.000 claims description 3
- 238000007789 sealing Methods 0.000 claims description 3
- 238000005245 sintering Methods 0.000 claims 1
- -1 B 4 O 12 Inorganic materials 0.000 description 239
- 238000006243 chemical reaction Methods 0.000 description 49
- 239000010408 film Substances 0.000 description 48
- 239000000843 powder Substances 0.000 description 37
- 239000002994 raw material Substances 0.000 description 29
- WERYXYBDKMZEQL-UHFFFAOYSA-N butane-1,4-diol Chemical compound OCCCCO WERYXYBDKMZEQL-UHFFFAOYSA-N 0.000 description 26
- 239000011521 glass Substances 0.000 description 25
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 18
- 239000000047 product Substances 0.000 description 18
- 239000000243 solution Substances 0.000 description 17
- 239000010409 thin film Substances 0.000 description 17
- 230000005284 excitation Effects 0.000 description 15
- 238000005259 measurement Methods 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 14
- 238000002441 X-ray diffraction Methods 0.000 description 13
- 238000001816 cooling Methods 0.000 description 13
- 239000007787 solid Substances 0.000 description 13
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 12
- 239000007788 liquid Substances 0.000 description 12
- 239000000203 mixture Substances 0.000 description 12
- 239000000126 substance Substances 0.000 description 10
- 239000000758 substrate Substances 0.000 description 10
- 238000002189 fluorescence spectrum Methods 0.000 description 9
- 239000000463 material Substances 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- 125000004432 carbon atom Chemical group C* 0.000 description 8
- 239000007850 fluorescent dye Substances 0.000 description 8
- 229920001223 polyethylene glycol Polymers 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- 238000000861 blow drying Methods 0.000 description 7
- 238000005119 centrifugation Methods 0.000 description 7
- AERUOEZHIAYQQL-UHFFFAOYSA-K cerium(3+);triacetate;hydrate Chemical compound O.[Ce+3].CC([O-])=O.CC([O-])=O.CC([O-])=O AERUOEZHIAYQQL-UHFFFAOYSA-K 0.000 description 7
- 238000005401 electroluminescence Methods 0.000 description 7
- 238000006862 quantum yield reaction Methods 0.000 description 7
- 238000002834 transmittance Methods 0.000 description 7
- AIQRTHPXPDTMBQ-UHFFFAOYSA-K yttrium(3+);triacetate;tetrahydrate Chemical compound O.O.O.O.[Y+3].CC([O-])=O.CC([O-])=O.CC([O-])=O AIQRTHPXPDTMBQ-UHFFFAOYSA-K 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- 239000002202 Polyethylene glycol Substances 0.000 description 6
- 230000000052 comparative effect Effects 0.000 description 6
- 238000000295 emission spectrum Methods 0.000 description 6
- 230000004580 weight loss Effects 0.000 description 6
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 5
- 238000000695 excitation spectrum Methods 0.000 description 5
- 238000001914 filtration Methods 0.000 description 5
- 239000003960 organic solvent Substances 0.000 description 5
- 125000006850 spacer group Chemical group 0.000 description 5
- 150000001298 alcohols Chemical class 0.000 description 4
- 125000000217 alkyl group Chemical group 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 239000000975 dye Substances 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- 239000000178 monomer Substances 0.000 description 4
- 125000004430 oxygen atom Chemical group O* 0.000 description 4
- 239000000049 pigment Substances 0.000 description 4
- LLHKCFNBLRBOGN-UHFFFAOYSA-N propylene glycol methyl ether acetate Chemical compound COCC(C)OC(C)=O LLHKCFNBLRBOGN-UHFFFAOYSA-N 0.000 description 4
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- 229910015999 BaAl Inorganic materials 0.000 description 3
- 229910052693 Europium Inorganic materials 0.000 description 3
- 229910052688 Gadolinium Inorganic materials 0.000 description 3
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 3
- 229910004283 SiO 4 Inorganic materials 0.000 description 3
- 229910052771 Terbium Inorganic materials 0.000 description 3
- 238000002835 absorbance Methods 0.000 description 3
- 238000000862 absorption spectrum Methods 0.000 description 3
- 238000004220 aggregation Methods 0.000 description 3
- 230000002776 aggregation Effects 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- 239000010953 base metal Substances 0.000 description 3
- VBVAVBCYMYWNOU-UHFFFAOYSA-N coumarin 6 Chemical compound C1=CC=C2SC(C3=CC4=CC=C(C=C4OC3=O)N(CC)CC)=NC2=C1 VBVAVBCYMYWNOU-UHFFFAOYSA-N 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 238000010586 diagram Methods 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- OGPBJKLSAFTDLK-UHFFFAOYSA-N europium atom Chemical compound [Eu] OGPBJKLSAFTDLK-UHFFFAOYSA-N 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 238000000634 powder X-ray diffraction Methods 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 239000010453 quartz Substances 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N silicon dioxide Inorganic materials O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- 229910019655 synthetic inorganic crystalline material Inorganic materials 0.000 description 3
- GZCRRIHWUXGPOV-UHFFFAOYSA-N terbium atom Chemical compound [Tb] GZCRRIHWUXGPOV-UHFFFAOYSA-N 0.000 description 3
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 2
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 2
- YEJRWHAVMIAJKC-UHFFFAOYSA-N 4-Butyrolactone Chemical compound O=C1CCCO1 YEJRWHAVMIAJKC-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- KFSLWBXXFJQRDL-UHFFFAOYSA-N Peracetic acid Chemical compound CC(=O)OO KFSLWBXXFJQRDL-UHFFFAOYSA-N 0.000 description 2
- 229910003668 SrAl Inorganic materials 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 150000001335 aliphatic alkanes Chemical class 0.000 description 2
- 125000003342 alkenyl group Chemical group 0.000 description 2
- 150000004703 alkoxides Chemical class 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- 150000004645 aluminates Chemical class 0.000 description 2
- 239000003849 aromatic solvent Substances 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 125000004104 aryloxy group Chemical group 0.000 description 2
- 239000012965 benzophenone Substances 0.000 description 2
- 150000008366 benzophenones Chemical class 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 150000007942 carboxylates Chemical class 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- VYXSBFYARXAAKO-WTKGSRSZSA-N chembl402140 Chemical compound Cl.C1=2C=C(C)C(NCC)=CC=2OC2=C\C(=N/CC)C(C)=CC2=C1C1=CC=CC=C1C(=O)OCC VYXSBFYARXAAKO-WTKGSRSZSA-N 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 229920001577 copolymer Polymers 0.000 description 2
- JHIVVAPYMSGYDF-UHFFFAOYSA-N cyclohexanone Chemical compound O=C1CCCCC1 JHIVVAPYMSGYDF-UHFFFAOYSA-N 0.000 description 2
- DOIRQSBPFJWKBE-UHFFFAOYSA-N dibutyl phthalate Chemical compound CCCCOC(=O)C1=CC=CC=C1C(=O)OCCCC DOIRQSBPFJWKBE-UHFFFAOYSA-N 0.000 description 2
- 238000002296 dynamic light scattering Methods 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- 230000005281 excited state Effects 0.000 description 2
- UIWYJDYFSGRHKR-UHFFFAOYSA-N gadolinium atom Chemical compound [Gd] UIWYJDYFSGRHKR-UHFFFAOYSA-N 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 239000003999 initiator Substances 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- ZXEKIIBDNHEJCQ-UHFFFAOYSA-N isobutanol Chemical compound CC(C)CO ZXEKIIBDNHEJCQ-UHFFFAOYSA-N 0.000 description 2
- 239000011572 manganese Substances 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical group [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 2
- 229910052753 mercury Inorganic materials 0.000 description 2
- 150000002736 metal compounds Chemical class 0.000 description 2
- 229920003145 methacrylic acid copolymer Polymers 0.000 description 2
- 229940117841 methacrylic acid copolymer Drugs 0.000 description 2
- CKFGINPQOCXMAZ-UHFFFAOYSA-N methanediol Chemical compound OCO CKFGINPQOCXMAZ-UHFFFAOYSA-N 0.000 description 2
- 230000000737 periodic effect Effects 0.000 description 2
- 229960003742 phenol Drugs 0.000 description 2
- 235000021317 phosphate Nutrition 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- WQGWDDDVZFFDIG-UHFFFAOYSA-N pyrogallol Chemical compound OC1=CC=CC(O)=C1O WQGWDDDVZFFDIG-UHFFFAOYSA-N 0.000 description 2
- 229910052761 rare earth metal Inorganic materials 0.000 description 2
- 150000002910 rare earth metals Chemical class 0.000 description 2
- PYWVYCXTNDRMGF-UHFFFAOYSA-N rhodamine B Chemical compound [Cl-].C=12C=CC(=[N+](CC)CC)C=C2OC2=CC(N(CC)CC)=CC=C2C=1C1=CC=CC=C1C(O)=O PYWVYCXTNDRMGF-UHFFFAOYSA-N 0.000 description 2
- 238000007650 screen-printing Methods 0.000 description 2
- 239000000565 sealant Substances 0.000 description 2
- 150000004760 silicates Chemical class 0.000 description 2
- 238000003980 solgel method Methods 0.000 description 2
- 229910001220 stainless steel Inorganic materials 0.000 description 2
- 239000010935 stainless steel Substances 0.000 description 2
- 125000005504 styryl group Chemical group 0.000 description 2
- 150000005846 sugar alcohols Polymers 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 238000012719 thermal polymerization Methods 0.000 description 2
- 229920001187 thermosetting polymer Polymers 0.000 description 2
- 238000000411 transmission spectrum Methods 0.000 description 2
- 238000001132 ultrasonic dispersion Methods 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- DNIAPMSPPWPWGF-VKHMYHEASA-N (+)-propylene glycol Chemical compound C[C@H](O)CO DNIAPMSPPWPWGF-VKHMYHEASA-N 0.000 description 1
- YPFDHNVEDLHUCE-UHFFFAOYSA-N 1,3-propanediol Substances OCCCO YPFDHNVEDLHUCE-UHFFFAOYSA-N 0.000 description 1
- 125000006083 1-bromoethyl group Chemical group 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000001478 1-chloroethyl group Chemical group [H]C([H])([H])C([H])(Cl)* 0.000 description 1
- 125000004066 1-hydroxyethyl group Chemical group [H]OC([H])([*])C([H])([H])[H] 0.000 description 1
- 125000001637 1-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C(*)=C([H])C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000004134 1-norbornyl group Chemical group [H]C1([H])C([H])([H])C2(*)C([H])([H])C([H])([H])C1([H])C2([H])[H] 0.000 description 1
- 125000004343 1-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- WJFKNYWRSNBZNX-UHFFFAOYSA-N 10H-phenothiazine Chemical compound C1=CC=C2NC3=CC=CC=C3SC2=C1 WJFKNYWRSNBZNX-UHFFFAOYSA-N 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- XNWFRZJHXBZDAG-UHFFFAOYSA-N 2-METHOXYETHANOL Chemical compound COCCO XNWFRZJHXBZDAG-UHFFFAOYSA-N 0.000 description 1
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 description 1
- 125000006280 2-bromobenzyl group Chemical group [H]C1=C([H])C(Br)=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000005999 2-bromoethyl group Chemical group 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- POAOYUHQDCAZBD-UHFFFAOYSA-N 2-butoxyethanol Chemical compound CCCCOCCO POAOYUHQDCAZBD-UHFFFAOYSA-N 0.000 description 1
- 125000006282 2-chlorobenzyl group Chemical group [H]C1=C([H])C(Cl)=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000001340 2-chloroethyl group Chemical group [H]C([H])(Cl)C([H])([H])* 0.000 description 1
- 125000001731 2-cyanoethyl group Chemical group [H]C([H])(*)C([H])([H])C#N 0.000 description 1
- ZNQVEEAIQZEUHB-UHFFFAOYSA-N 2-ethoxyethanol Chemical compound CCOCCO ZNQVEEAIQZEUHB-UHFFFAOYSA-N 0.000 description 1
- SVONRAPFKPVNKG-UHFFFAOYSA-N 2-ethoxyethyl acetate Chemical compound CCOCCOC(C)=O SVONRAPFKPVNKG-UHFFFAOYSA-N 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000006290 2-hydroxybenzyl group Chemical group [H]OC1=C(C([H])=C([H])C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000006481 2-iodobenzyl group Chemical group [H]C1=C([H])C(I)=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- LWRBVKNFOYUCNP-UHFFFAOYSA-N 2-methyl-1-(4-methylsulfanylphenyl)-2-morpholin-4-ylpropan-1-one Chemical compound C1=CC(SC)=CC=C1C(=O)C(C)(C)N1CCOCC1 LWRBVKNFOYUCNP-UHFFFAOYSA-N 0.000 description 1
- 125000001622 2-naphthyl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C(*)C([H])=C([H])C2=C1[H] 0.000 description 1
- 125000004135 2-norbornyl group Chemical group [H]C1([H])C([H])([H])C2([H])C([H])([H])C1([H])C([H])([H])C2([H])* 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- 125000003852 3-chlorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C(Cl)=C1[H])C([H])([H])* 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000006291 3-hydroxybenzyl group Chemical group [H]OC1=C([H])C([H])=C([H])C(=C1[H])C([H])([H])* 0.000 description 1
- 125000006482 3-iodobenzyl group Chemical group [H]C1=C([H])C(=C([H])C(I)=C1[H])C([H])([H])* 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- JIGUICYYOYEXFS-UHFFFAOYSA-N 3-tert-butylbenzene-1,2-diol Chemical compound CC(C)(C)C1=CC=CC(O)=C1O JIGUICYYOYEXFS-UHFFFAOYSA-N 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000006281 4-bromobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1Br)C([H])([H])* 0.000 description 1
- 125000006283 4-chlorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1Cl)C([H])([H])* 0.000 description 1
- 125000003143 4-hydroxybenzyl group Chemical group [H]C([*])([H])C1=C([H])C([H])=C(O[H])C([H])=C1[H] 0.000 description 1
- 125000006483 4-iodobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1I)C([H])([H])* 0.000 description 1
- GZVHEAJQGPRDLQ-UHFFFAOYSA-N 6-phenyl-1,3,5-triazine-2,4-diamine Chemical compound NC1=NC(N)=NC(C=2C=CC=CC=2)=N1 GZVHEAJQGPRDLQ-UHFFFAOYSA-N 0.000 description 1
- 229910016066 BaSi Inorganic materials 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- FIPWRIJSWJWJAI-UHFFFAOYSA-N Butyl carbitol 6-propylpiperonyl ether Chemical group C1=C(CCC)C(COCCOCCOCCCC)=CC2=C1OCO2 FIPWRIJSWJWJAI-UHFFFAOYSA-N 0.000 description 1
- 229910004762 CaSiO Inorganic materials 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- MQIUGAXCHLFZKX-UHFFFAOYSA-N Di-n-octyl phthalate Natural products CCCCCCCCOC(=O)C1=CC=CC=C1C(=O)OCCCCCCCC MQIUGAXCHLFZKX-UHFFFAOYSA-N 0.000 description 1
- 229910052692 Dysprosium Inorganic materials 0.000 description 1
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- 239000004354 Hydroxyethyl cellulose Substances 0.000 description 1
- 229920000663 Hydroxyethyl cellulose Polymers 0.000 description 1
- 229910002420 LaOCl Inorganic materials 0.000 description 1
- 239000004640 Melamine resin Substances 0.000 description 1
- 229920000877 Melamine resin Polymers 0.000 description 1
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 1
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 1
- 229910017625 MgSiO Inorganic materials 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- ALQSHHUCVQOPAS-UHFFFAOYSA-N Pentane-1,5-diol Chemical compound OCCCCCO ALQSHHUCVQOPAS-UHFFFAOYSA-N 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 229910052777 Praseodymium Inorganic materials 0.000 description 1
- 229910052772 Samarium Inorganic materials 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- 229910006404 SnO 2 Inorganic materials 0.000 description 1
- 244000028419 Styrax benzoin Species 0.000 description 1
- 235000000126 Styrax benzoin Nutrition 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 235000008411 Sumatra benzointree Nutrition 0.000 description 1
- 229910052775 Thulium Inorganic materials 0.000 description 1
- DAKWPKUUDNSNPN-UHFFFAOYSA-N Trimethylolpropane triacrylate Chemical compound C=CC(=O)OCC(CC)(COC(=O)C=C)COC(=O)C=C DAKWPKUUDNSNPN-UHFFFAOYSA-N 0.000 description 1
- 230000009102 absorption Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- 150000008062 acetophenones Chemical class 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 125000003670 adamantan-2-yl group Chemical group [H]C1([H])C(C2([H])[H])([H])C([H])([H])C3([H])C([*])([H])C1([H])C([H])([H])C2([H])C3([H])[H] 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 229920000180 alkyd Polymers 0.000 description 1
- SMZOGRDCAXLAAR-UHFFFAOYSA-N aluminium isopropoxide Chemical compound [Al+3].CC(C)[O-].CC(C)[O-].CC(C)[O-] SMZOGRDCAXLAAR-UHFFFAOYSA-N 0.000 description 1
- JNDMLEXHDPKVFC-UHFFFAOYSA-N aluminum;oxygen(2-);yttrium(3+) Chemical compound [O-2].[O-2].[O-2].[Al+3].[Y+3] JNDMLEXHDPKVFC-UHFFFAOYSA-N 0.000 description 1
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 1
- 125000002078 anthracen-1-yl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C([*])=C([H])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 1
- 125000000748 anthracen-2-yl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C3C([H])=C([*])C([H])=C([H])C3=C([H])C2=C1[H] 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- 239000002518 antifoaming agent Substances 0.000 description 1
- 229910001439 antimony ion Inorganic materials 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 229910052788 barium Inorganic materials 0.000 description 1
- DSAJWYNOEDNPEQ-UHFFFAOYSA-N barium atom Chemical compound [Ba] DSAJWYNOEDNPEQ-UHFFFAOYSA-N 0.000 description 1
- 229960002130 benzoin Drugs 0.000 description 1
- BJQHLKABXJIVAM-UHFFFAOYSA-N bis(2-ethylhexyl) phthalate Chemical compound CCCCC(CC)COC(=O)C1=CC=CC=C1C(=O)OCC(CC)CCCC BJQHLKABXJIVAM-UHFFFAOYSA-N 0.000 description 1
- 150000001642 boronic acid derivatives Chemical class 0.000 description 1
- 125000005997 bromomethyl group Chemical group 0.000 description 1
- CDQSJQSWAWPGKG-UHFFFAOYSA-N butane-1,1-diol Chemical compound CCCC(O)O CDQSJQSWAWPGKG-UHFFFAOYSA-N 0.000 description 1
- AQCDIIAORKRFCD-UHFFFAOYSA-N cadmium selenide Chemical compound [Cd]=[Se] AQCDIIAORKRFCD-UHFFFAOYSA-N 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000001721 carbon Chemical class 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 238000003763 carbonization Methods 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 229910052798 chalcogen Inorganic materials 0.000 description 1
- 150000004770 chalcogenides Chemical class 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 239000011651 chromium Substances 0.000 description 1
- 229910001430 chromium ion Inorganic materials 0.000 description 1
- 238000000975 co-precipitation Methods 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 150000004696 coordination complex Chemical class 0.000 description 1
- 238000002447 crystallographic data Methods 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 238000010908 decantation Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- ISAOCJYIOMOJEB-UHFFFAOYSA-N desyl alcohol Natural products C=1C=CC=CC=1C(O)C(=O)C1=CC=CC=C1 ISAOCJYIOMOJEB-UHFFFAOYSA-N 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- KBQHZAAAGSGFKK-UHFFFAOYSA-N dysprosium atom Chemical compound [Dy] KBQHZAAAGSGFKK-UHFFFAOYSA-N 0.000 description 1
- 229920006332 epoxy adhesive Polymers 0.000 description 1
- 239000003822 epoxy resin Substances 0.000 description 1
- WGXGKXTZIQFQFO-CMDGGOBGSA-N ethenyl (e)-3-phenylprop-2-enoate Chemical compound C=COC(=O)\C=C\C1=CC=CC=C1 WGXGKXTZIQFQFO-CMDGGOBGSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 235000019256 formaldehyde Nutrition 0.000 description 1
- 229910052733 gallium Inorganic materials 0.000 description 1
- 230000005283 ground state Effects 0.000 description 1
- 235000019382 gum benzoic Nutrition 0.000 description 1
- XXMIOPMDWAUFGU-UHFFFAOYSA-N hexane-1,6-diol Chemical compound OCCCCCCO XXMIOPMDWAUFGU-UHFFFAOYSA-N 0.000 description 1
- 239000012510 hollow fiber Substances 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 235000019447 hydroxyethyl cellulose Nutrition 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 229940035429 isobutyl alcohol Drugs 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000002596 lactones Chemical class 0.000 description 1
- 229910052746 lanthanum Inorganic materials 0.000 description 1
- FZLIPJUXYLNCLC-UHFFFAOYSA-N lanthanum atom Chemical compound [La] FZLIPJUXYLNCLC-UHFFFAOYSA-N 0.000 description 1
- RVPVRDXYQKGNMQ-UHFFFAOYSA-N lead(2+) Chemical compound [Pb+2] RVPVRDXYQKGNMQ-UHFFFAOYSA-N 0.000 description 1
- ZMTOUBHBKDQYAB-UHFFFAOYSA-K lutetium(3+);triacetate;hydrate Chemical compound O.[Lu+3].CC([O-])=O.CC([O-])=O.CC([O-])=O ZMTOUBHBKDQYAB-UHFFFAOYSA-K 0.000 description 1
- 125000003564 m-cyanobenzyl group Chemical group [H]C1=C([H])C(=C([H])C(C#N)=C1[H])C([H])([H])* 0.000 description 1
- 125000000040 m-tolyl group Chemical group [H]C1=C([H])C(*)=C([H])C(=C1[H])C([H])([H])[H] 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- 229910001437 manganese ion Inorganic materials 0.000 description 1
- 125000005394 methallyl group Chemical group 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000002159 nanocrystal Substances 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- 125000006504 o-cyanobenzyl group Chemical group [H]C1=C([H])C(C#N)=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000003261 o-tolyl group Chemical group [H]C1=C([H])C(*)=C(C([H])=C1[H])C([H])([H])[H] 0.000 description 1
- 239000011368 organic material Substances 0.000 description 1
- 150000001451 organic peroxides Chemical class 0.000 description 1
- 125000006505 p-cyanobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C#N)C([H])([H])* 0.000 description 1
- NWVVVBRKAWDGAB-UHFFFAOYSA-N p-methoxyphenol Chemical compound COC1=CC=C(O)C=C1 NWVVVBRKAWDGAB-UHFFFAOYSA-N 0.000 description 1
- 125000006503 p-nitrobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1[N+]([O-])=O)C([H])([H])* 0.000 description 1
- 125000001037 p-tolyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C([H])([H])[H] 0.000 description 1
- 238000000059 patterning Methods 0.000 description 1
- XCRBXWCUXJNEFX-UHFFFAOYSA-N peroxybenzoic acid Chemical class OOC(=O)C1=CC=CC=C1 XCRBXWCUXJNEFX-UHFFFAOYSA-N 0.000 description 1
- 239000005011 phenolic resin Substances 0.000 description 1
- 229950000688 phenothiazine Drugs 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 239000003504 photosensitizing agent Substances 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229910052696 pnictogen Inorganic materials 0.000 description 1
- 229920003229 poly(methyl methacrylate) Polymers 0.000 description 1
- 229920006122 polyamide resin Polymers 0.000 description 1
- 229920001230 polyarylate Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 229920000647 polyepoxide Polymers 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 239000004926 polymethyl methacrylate Substances 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920000166 polytrimethylene carbonate Polymers 0.000 description 1
- 229920005749 polyurethane resin Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 235000019422 polyvinyl alcohol Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- PUDIUYLPXJFUGB-UHFFFAOYSA-N praseodymium atom Chemical compound [Pr] PUDIUYLPXJFUGB-UHFFFAOYSA-N 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 229940079877 pyrogallol Drugs 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 230000009103 reabsorption Effects 0.000 description 1
- 239000011342 resin composition Substances 0.000 description 1
- KZUNJOHGWZRPMI-UHFFFAOYSA-N samarium atom Chemical compound [Sm] KZUNJOHGWZRPMI-UHFFFAOYSA-N 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000004065 semiconductor Substances 0.000 description 1
- 229920002545 silicone oil Polymers 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 239000006104 solid solution Substances 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 238000004528 spin coating Methods 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 150000003464 sulfur compounds Chemical class 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- ZLUSCZLCHQSJRU-UHFFFAOYSA-N thallium(1+) Chemical compound [Tl+] ZLUSCZLCHQSJRU-UHFFFAOYSA-N 0.000 description 1
- 238000005979 thermal decomposition reaction Methods 0.000 description 1
- 238000002411 thermogravimetry Methods 0.000 description 1
- 229920005992 thermoplastic resin Polymers 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- FRNOGLGSGLTDKL-UHFFFAOYSA-N thulium atom Chemical compound [Tm] FRNOGLGSGLTDKL-UHFFFAOYSA-N 0.000 description 1
- 229910001432 tin ion Inorganic materials 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 229910021642 ultra pure water Inorganic materials 0.000 description 1
- 239000012498 ultrapure water Substances 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000013585 weight reducing agent Substances 0.000 description 1
- 229910019901 yttrium aluminum garnet Inorganic materials 0.000 description 1
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 1
Images















Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K11/00—Luminescent, e.g. electroluminescent, chemiluminescent materials
- C09K11/08—Luminescent, e.g. electroluminescent, chemiluminescent materials containing inorganic luminescent materials
- C09K11/77—Luminescent, e.g. electroluminescent, chemiluminescent materials containing inorganic luminescent materials containing rare earth metals
- C09K11/7766—Luminescent, e.g. electroluminescent, chemiluminescent materials containing inorganic luminescent materials containing rare earth metals containing two or more rare earth metals
- C09K11/7774—Aluminates
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K11/00—Luminescent, e.g. electroluminescent, chemiluminescent materials
- C09K11/02—Use of particular materials as binders, particle coatings or suspension media therefor
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K11/00—Luminescent, e.g. electroluminescent, chemiluminescent materials
- C09K11/08—Luminescent, e.g. electroluminescent, chemiluminescent materials containing inorganic luminescent materials
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K11/00—Luminescent, e.g. electroluminescent, chemiluminescent materials
- C09K11/08—Luminescent, e.g. electroluminescent, chemiluminescent materials containing inorganic luminescent materials
- C09K11/77—Luminescent, e.g. electroluminescent, chemiluminescent materials containing inorganic luminescent materials containing rare earth metals
-
- H—ELECTRICITY
- H05—ELECTRIC TECHNIQUES NOT OTHERWISE PROVIDED FOR
- H05B—ELECTRIC HEATING; ELECTRIC LIGHT SOURCES NOT OTHERWISE PROVIDED FOR; CIRCUIT ARRANGEMENTS FOR ELECTRIC LIGHT SOURCES, IN GENERAL
- H05B33/00—Electroluminescent light sources
- H05B33/12—Light sources with substantially two-dimensional radiating surfaces
- H05B33/14—Light sources with substantially two-dimensional radiating surfaces characterised by the chemical or physical composition or the arrangement of the electroluminescent material, or by the simultaneous addition of the electroluminescent material in or onto the light source
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K59/00—Integrated devices, or assemblies of multiple devices, comprising at least one organic light-emitting element covered by group H10K50/00
- H10K59/30—Devices specially adapted for multicolour light emission
- H10K59/38—Devices specially adapted for multicolour light emission comprising colour filters or colour changing media [CCM]
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/70—Nanostructure
- Y10S977/778—Nanostructure within specified host or matrix material, e.g. nanocomposite films
- Y10S977/783—Organic host/matrix, e.g. lipid
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S977/00—Nanotechnology
- Y10S977/70—Nanostructure
- Y10S977/778—Nanostructure within specified host or matrix material, e.g. nanocomposite films
- Y10S977/786—Fluidic host/matrix containing nanomaterials
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/25—Web or sheet containing structurally defined element or component and including a second component containing structurally defined particles
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/25—Web or sheet containing structurally defined element or component and including a second component containing structurally defined particles
- Y10T428/256—Heavy metal or aluminum or compound thereof
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/29—Coated or structually defined flake, particle, cell, strand, strand portion, rod, filament, macroscopic fiber or mass thereof
- Y10T428/2982—Particulate matter [e.g., sphere, flake, etc.]
- Y10T428/2991—Coated
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Luminescent Compositions (AREA)
Description
(1)素子から発せられる光の色純度を高めるには、励起光源から発せされた光を、蛍光変換膜において効率よく他の波長の光に変換する必要がある。励起光源からの光が変換されずに膜を通過すると、色純度の低下の原因となる。蛍光変換膜の変換効率を高め、変換された光の強度(蛍光強度)を大きくするためには、励起光源から発せられる光を、膜に十分吸収させる必要がある。そのために、蛍光変換膜中の有機蛍光色素濃度を大きくしていくと、膜中で有機蛍光色素同士が会合するため、光源から吸収したエネルギーが隣接した色素に奪われ、いわゆる濃度消光という現象が避けられず、高い蛍光量子収率を得ることができなかった。
(2)光透過性媒体としては、膜の耐熱性向上や生産性向上などの理由により、光硬化性樹脂や熱硬化性樹脂などの反応硬化性樹脂が主に用いられている。この場合、樹脂中の反応成分と有機蛍光色素が反応し、色素が分解したり構造が変化して蛍光性がさらに低下するという問題があった。
この問題について、実験例をもとに具体的に説明する。図16は、蛍光変換膜中の色素濃度を変えたときの吸光度と蛍光量子収率の関係を示す図である。図中、丸は、ベンゾグアナミン樹脂に有機蛍光色素としてローダミン6Gを分散した蛍光変換膜を、白抜き三角は、同樹脂に有機蛍光色素としてクマリン6を分散した蛍光変換膜を、黒塗り三角は、光硬化性樹脂にクマリン6を分散した蛍光変換膜を示している。ローダミン6G分散膜には534nm、クマリン6分散膜には、456nmをピークとする光源からの光を照射した。図16において、横軸は、その波長における吸光度、縦軸は、蛍光量子収率を示している。
図16から明らかなように、低濃度では80%以上の高い蛍光量子収率を示す色素を用いても、励起光に対する吸光度が1を越える領域では、蛍光量子収率は50%未満となる。特に、反応性樹脂である光硬化性樹脂中に分散した場合には、30%程度の低い蛍光量子収率となることがわかる。
特許文献4では、無機蛍光体として、セリウムで付活されたイットリウム・アルミニウム・ガーネット系蛍光体(通称:YAG:Ce蛍光体)を用い、これを熱可塑性樹脂シートに分散させたシートを蛍光変換膜としている。ここでは、焼成したYAG:Ce成形体を粉砕して微粒子を製造しているため、微粒子の粒径がマイクロメートルのオーダーとなる。そのため、励起光源を散乱させることなく、十分吸収させるためには、樹脂シートの厚みを、例えば120μmといったような大きな厚みとし、かつ低濃度で微粒子を分散させる必要があり、有機エレクトロルミネッセンス素子用の蛍光変換膜としては適用が困難であった。
特許文献5では、金属酸化物蛍光体を製造する方法として、蛍光体の母体及び付活剤を構成する金属元素の炭酸塩、硝酸塩、水酸化物、硫酸塩、リン酸塩、ホウ酸塩、ケイ酸塩、アルミン酸塩、カルボン酸塩、ハロゲン化物及びアルコキシドの中から選ばれた1種以上の化合物と、オキシカルボン酸又はポリアミノキレート剤とを反応させて得た金属錯体を、溶媒中でポリオールと重合反応させて錯体重合体を形成し、これを焼成する製造方法が開示されている。しかしながら、この方法では、800℃以上の高温で焼成するため、生成に含まれる有機成分が熱分解する。そのため粒子が二次凝集を起こし、粒径としては100nm程度であり、未だ不十分であった。また、同じ理由により、有機溶媒や樹脂への分散性が悪いという問題があった。
また、非特許文献1では、ゾル−ゲル法で得た微粒子を800℃以上の高温で焼成し、35nm程度のYAG:Ge蛍光体を得ている。しかしながら、特許文献5と同様に、微粒子に有機成分が含まれないため、有機溶媒や樹脂への分散性が悪いという問題があった。
特許文献6では、カドミウムセレナイド(CdSe)のようなII−VI属半導体のナノクリスタルを樹脂に分散した膜を蛍光変換膜として用いている。しかしながら、一般に、金属カルコゲナイド化物は、耐水性、耐薬品性、耐熱性など耐久性に劣るという課題があった。
前記蛍光性ペーストを250〜300℃の温度で焼成処理する蛍光体の製造方法、
前記蛍光体を単独、もしくは該蛍光体に樹脂又は溶媒を添加し、固化してなる蛍光変換体、及び前記蛍光性を樹脂又は溶媒に分散してなる蛍光変換体
を提供するものである。
前記有機基は、末端又は側鎖に官能基を一つ以上有する有機化合物から少なくとも一つの該官能基が解離したものであり、前記有機基は、母体結晶の金属酸化物における金属原子又は酸素原子に配位しており、酸素原子に配位している場合には、例えば、図2に示すように、有機化合物(X−R)から官能基Xが解離して、酸素原子に有機基Rが配位している。なお、図2中、Meは金属原子、Oは酸素原子である。
なお、例えば、(YxGd1-x)3Al5O12という記載は、Y3Al5O12の結晶格子内で、イットリウム(Y)のうちの(1−x)at%がガドリニウム(Gd)に置換されていることを意味する。
(A)周期表第II族、第III族、第IV族、第V族、第VI族の金属元素
アンチモンイオン(Sb3+)、スズイオン(Sn2+)、鉛イオン(Pb2+)、タリウムイオン(Tl+ )、水銀原子(Hg)
(B)遷移金属元素
マンガンイオン(Mn2+,Mn4+)、クロムイオン(Cr3+)
(C)希土類元素金属元素
La3+、Lu3+、Pr3+、Nd3+,Pm3+、Sm3+、Eu3+、Gd3+、Tb3+、Dy3+、Ho3+、Er3+、Tm3+、Yb3+、Sm2+、Eu2+、Yb2+、Ce3+
以上の元素の中で、希土類金属元素イオンが好ましく、発光効率が高いという点で、ユーロピウム(Eu)、テルビウム(Tb)、プラセオジム(Pr)、セリウム(Ce)、サマリウム(Sm)、ツリウム(Tm)、ジスプロシウム(Dy)及びルテチウム(Lu)の中から選ばれる一種以上であると特に好ましい。
組合せて得られる化合物の具体例としては、ユーロピウム(Eu)を発光中心とした例として、BaAl8O13:Eu2+、BaMgAl10O17:Eu2+、BaMgAl14O23:Eu2+、Y2O3:Eu2+、Ba2GdNbO5 :Eu3+、BaMgAl10O17:Eu3+、BaMg2Al16O27:Eu3+、GdBO3:Eu3+、LuBO3:Eu3+、Y3Al5O12:Eu3+、YBO3:Eu3+、(YxGd1-x)2O3 :Eu3+、Y2O2S:Eu3+、Y2O3:Eu3+等が挙げられ、セリウム(Ce)を発光中心とした例として、Gd2SiO5:Ce3+、LaBO3:Ce3+、LaPO4:Ce3+、YAl3B4O12:Ce3+、Y3Al3Ga2O12:Ce3+、Y3(AlxGa1-x)5O12:Ce3+、(YxGd1-x)3Al5O12:Ce3+、Y2SiO5:Ce3+、LuAlO3:Ce3+、Lu2SiO5:Ce3+等が挙げられ、テルビウム(Tb)を発光中心とした例として、CeMgAl11O19:Tb3+、Gd2O2S:Tb3+、GdBO3:Tb3+、GdMgB5O10:Tb3+、(GdxY1-x)2O2S:Tb3+、LaPO4:Ce3+、LuBO3:Tb3+、Y3(AlxGa1-x)5O12:Tb3+、Y3Al3Ga2O12:Tb3+、Y3Al5O12:Tb3+、Y2SiO5:Tb3+等が挙げられる。
前記有機基としては、例えば、置換もしくは無置換の炭素数1〜50のアルキル基、置換もしくは無置換の炭素数1〜50のアルケニル基、置換もしくは無置換の炭素数1〜50のアルコキシ基、置換もしくは無置換の炭素数1〜50のシクロアルキル基、置換もしくは無置換の炭素数6〜50のアリール基、置換もしくは無置換の炭素数5〜50のヘテロアリール基、置換もしくは無置換の炭素数7〜50のアラルキル基、置換もしくは無置換の炭素数5〜50のアリールオキシ基等が挙げられる。
置換もしくは無置換のアルコキシ基は、−OYで表される基であり、Yの例としては、前記アルキル基で例示した具体的等が挙げられる。
置換もしくは無置換のシクロアルキル基の例としては、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、4−メチルシクロヘキシル基、1−アダマンチル基、2−アダマンチル基、1−ノルボルニル基、2−ノルボルニル基等が挙げられる。
置換もしくは無置換のアリール基の例としては、フェニル基、1−ナフチル基、2−ナフチル基、1−アントリル基、2−アントリル基、9−アントリル基、1−フェナンスリル基、2−フェナンスリル基、3−フェナンスリル基、4−フェナンスリル基、9−フェナンスリル基、1−ナフタセニル基、2−ナフタセニル基、9−ナフタセニル基、1−ピレニル基、2−ピレニル基、4−ピレニル基、2−ビフェニルイル基、3−ビフェニルイル基、4−ビフェニルイル基、p−ターフェニル−4−イル基、p−ターフェニル−3−イル基、p−ターフェニル−2−イル基、m−ターフェニル−4−イル基、m−ターフェニル−3−イル基、m−ターフェニル−2−イル基、o−トリル基、m−トリル基、p−トリル基、p−t−ブチルフェニル基、p−(2−フェニルプロピル)フェニル基、3−メチル−2−ナフチル基、4−メチル−1−ナフチル基、4−メチル−1−アントリル基、4’−メチルビフェニルイル基、4”−t−ブチル−p−ターフェニル−4−イル基等が挙げられる。
置換もしくは無置換のアリールオキシ基は、−OY’で表される基であり、Y’の例としては、前記アリール基で例示した具体的等が挙げられる。
以上の各基の具体例は、1価の基であるが、さらに水素が解離した2価以上の基であってもよく、さらに水素が下記官能基で置換されたものであってもよい。
本発明の金属酸化物系蛍光体微粒子の平均粒径は、1〜100nmと小さく、1〜60nmであると好ましい。
前記末端又は側鎖に官能基を一つ以上有する有機化合物の例としては、イソブチルアルコール、1,4−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、グリセロール、エチレングリコール、トリメチレングリコール、1,3−プロパンジオール、1,4−ヒドロキシベンゼン、1,3−ヒドロキシベンゼン、1,2−ヒドロキシベンゼン、2−ヒドロキシエチルメルカプタン、2−ヒドロキシエチルアミン等が挙げられ、1,4−ブタンジオール、グリセロール、エチレングリコールが好ましい。
本発明の金属酸化物系蛍光体微粒子の表面に前記有機基が配位しているかどうか確認する手段としては、TG−DTA(示差熱−熱重量分析)による方法が挙げられる。例えば、1,4−ブタンジオールOH(CH2)4OHの末端OHが解離した有機基(CH2)4OHが微粒子表面に配位しているかどうかは、1,4−ブタンジオールの沸点229℃を超えて昇温し続けた場合でも重量減少が継続して見られるかどうかで判定することができる。昇温し続けると、表面に配位した有機基が熱分解することにより重量減少が継続する。
このように、本発明の金属酸化物系蛍光体微粒子分散液は、(b)成分として前記した本発明の金属酸化物系蛍光体微粒子を用い、(a)成分として(b)成分で用いたものと同種の有機化合物を含む分散媒を用いることにより、極めて分散性が良い。
また、前記(a)成分の分散媒は、前記有機化合物の他に、公知の他の成分を含んでいてもよく、例えば、メチルエチルケトン、メチルイソブチルケトン、シクロヘキサノン等のケトン類、メチルセロソルブ、エチルセロソルブ、ブチルセロソルブ、セロソルブアセテート等のセロソルブ類、γ−ブチロラクトン等のラクトン類、ポリエチレングリコールなどが挙げられる。
樹脂成分としては、非硬化型樹脂、熱硬化型樹脂、光硬化型樹脂等が挙げられ、具体的には、オリゴマーもしくはポリマー形態のメラミン樹脂、フェノール樹脂、アルキド樹脂、エポキシ樹脂、ポリウレタン樹脂、マレイン酸樹脂、ポリアミド樹脂、又はポリメチルメタクリレート、ポリアリレート、ポリカーボネート、ポリビニルアルコール、ポリビニルピロリドン、ヒドロキシエチルセルロース、カルボキシメチルセルロース等、及びこれらを形成するモノマーを構成成分とする共重合体が挙げられる。
これらの樹脂のうち、特に、蛍光変換膜を任意の形状にパターニングする目的がある場合には、光硬化型樹脂成分を用いる。光硬化型樹脂としては、通常感光剤を含む反応性ビニル基を有するアクリル酸、メタクリル酸系、メタクリル酸エステル−メタクリル酸共重合体の光重合型や、ポリケイ皮酸ビニルなどの光架橋型が用いられる。また、必要に応じ、光重合可能なエチレン性不飽和基を有するモノマー及び/又はオリゴマーや、光重合開始剤又は増感剤を加えることができる。
前記モノマー、オリゴマー成分の具体例としては、水酸基を有するモノマーとして2−ヒドロキシエチル(メタ)アクリレート,2−ヒドロキシプロピル(メタ)アクリレート,2−ヒドロキシヘキシル(メタ)アクリレート等が挙げられ、(メタ)アクリル酸エステル類としてエチレングリコールジ(メタ)アクリレート,ジエチレングリコールジ(メタ)アクリレートなどを挙げることができる。
前記光重合開始剤又は増感剤としては、例えば、アセトフェノン類、ベンゾフェノン類、ベンゾインエーテル類、イオウ化合物、アントラキノン類、有機過酸化物又はチオール類等が好適に使用される。
さらに、本発明の金属酸化物系蛍光体微粒子分散液には、必要に応じて硬化促進剤、熱重合禁止剤、可塑剤、消泡剤、レベリング剤などの添加剤を配合することができる。硬化促進剤としては、例えば、過安息香酸誘導体、過酢酸、ベンゾフェノン類等があり、熱重合禁止剤としては、例えば、ハイドロキノン、ハイドロキノンモノメチルエーテル、ピロガロール、t−ブチルカテコール、フェノチアジン等があり、可塑剤としては、例えば、ジブチルフタレート、ジオクチルフタレート、トリクレジル等を挙げることができる。
光透過性樹脂としては、前述したものと同じである。
蛍光変換膜の作製は、特に限定されず、本発明の金属酸化物系蛍光体微粒子、分散媒及び光透過性樹脂の混合された分散液を、公知の成膜方法、例えば、スピンコート法、スクリーン印刷法、ディップ法、インクジェット法などの方法によって、支持基板上に成膜する。成膜後、分散媒の沸点、蒸気圧、膜の厚みに応じて適宜加熱して膜中から分散媒を蒸発させ、光透過性樹脂中に金属酸化物系蛍光体微粒子の分散された蛍光変換膜を得る。
蛍光変換膜の厚さは、通常0.1μm〜1mm、好ましくは1μm〜100μmである。
蛍光変換膜中の金属酸化物系蛍光体微粒子の量は、通常0.1〜90質量%であり、1〜70質量%であると好ましい。該微粒子の量が0.1質量%以上であると、励起光源から発せられる光を十分に吸収することができ、結果として得られる蛍光強度も大きい。また、微粒子の量が90質量%以下であれば、膜の平滑性が良好で、機械的強度も高いため好ましい。
本発明の蛍光変換膜、蛍光性液体、蛍光性ペースト、蛍光体及び蛍光変換体は、例えば、蛍光変換膜を例とすると図4に示すように、励起光源の上に配置され、励起光源から発せられた励起光が通過する際に、その波長を、例えば、より波長の長い光に変換(例えば青色を緑色又は赤色に変換)した蛍光を発するものである。励起光源としては、例えば、有機エレクトロルミネッセンス素子、無機エレクトロルミネッセンス素子、発光ダイオード、冷陰極管、蛍光管、レーザー等が挙げられ、特に有機エレクトロルミネッセンス素子、発光ダイオードに適している。
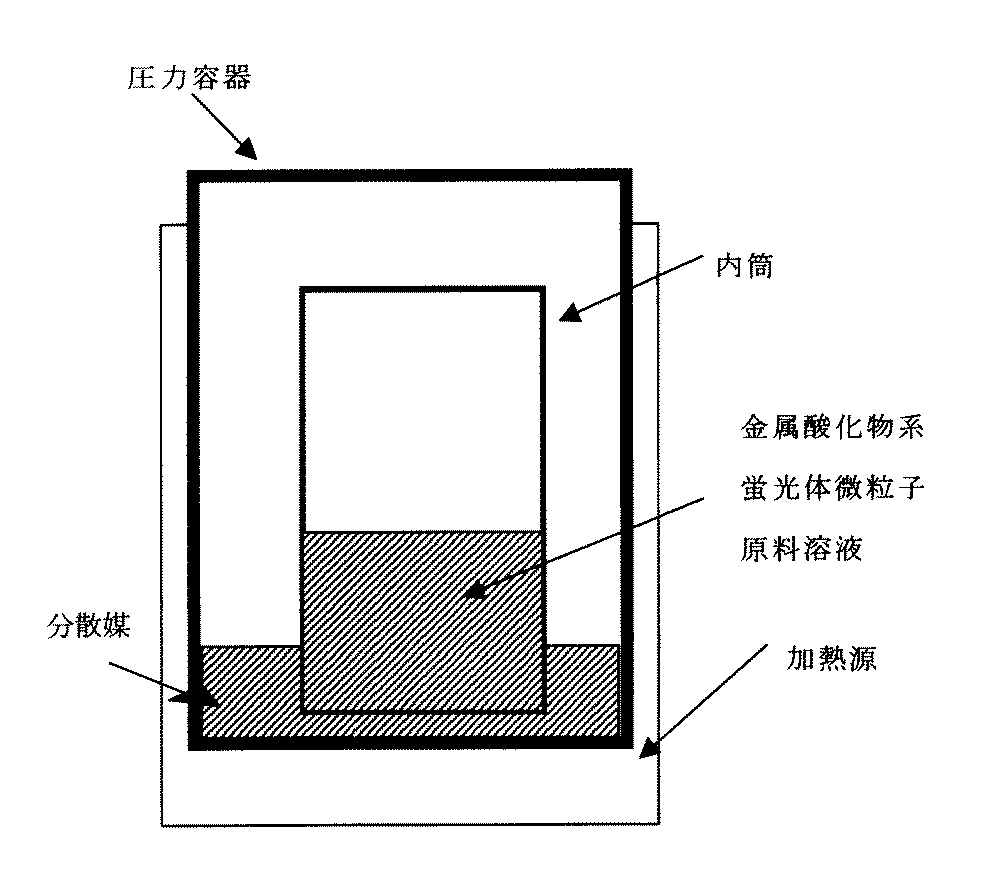
本発明の金属酸化物系蛍光体微粒子の製造方法は、図5に示すように、金属酸化物からなる母体を形成する金属元素の化合物と、発光中心である金属元素の化合物とを、末端又は側鎖に官能基を一つ以上有する有機化合物を含む分散媒に溶解又は分散させた原料溶液を圧力容器内に封入し、前記有機化合物の沸点以上の温度で加熱する。
具体的な製造方法としては、大きく分けて(1)原料(母体金属酸化物源、発光中心の金属元素源、前記有機化合物及び分散媒)の選定・調合・仕込み、(2)加熱、(3)精製の三つの工程からなる。以下、順に説明する。
本発明の金属酸化物系蛍光体微粒子を構成する母体金属酸化物を形成する金属元素、及び発光中心の金属元素の供給源の具体例としては、前記母体の金属酸化物及び発光中心である金属元素の、炭酸塩、酢酸塩、硝酸塩、水酸化物、硫酸塩、リン酸塩、ホウ酸塩、ケイ酸塩、アルミン酸塩、カルボン酸塩、ハロゲン化物、アルコキシド等の化合物及びこれらの水和物などを挙げることができる。これらを、常法により目的の化合物が得られる配合比で調合し、前記分散媒に溶解又は分散させ、原料溶液を得る。調合した原料溶液を、オートクレーブに代表される圧力容器に封入する。
前記末端又は側鎖に官能基を一つ以上有する有機化合物を含む分散媒は、前記金属酸化物系蛍光体微粒子分散液で説明したものと同様である。
前記母体の金属酸化物を形成する金属元素の化合物及び発光中心である金属元素の化合物の合計量(金属化合物)と分散媒との配合比は、最終的に得られる分散液の所望の固形分濃度、及び原料の均一性を考慮して決めることができるが、通常、金属化合物:分散媒=0.1:100〜50:50(質量比)の範囲であり、1:99〜50:50であると好ましい。分散媒の比率を上げることができず、結果として、後述する加熱工程にて、圧力容器内の圧力が上がらない場合には、図6に示すように、所定の配合比にて調合した原料溶液を入れた内筒容器を、分散媒のみ入れた圧力容器内に入れるという方法が有効である。
また、圧力容器の内容積に対する原料溶液の体積比率(容積率)は、後述する加熱時間を減らすことができるためできるだけ大きいことが好ましく、具体的には、40%以上であると好ましく、60%以上であるとさらに好ましい。
前記原料溶液の反応を進行させるため、圧力容器の外部に設置された加熱源から熱を供給する。一般に、金属酸化物系蛍光体微粒子を製造するためには、通常800℃以上の高温で焼成する必要がある。しかし、本発明による金属酸化物系蛍光体微粒子の製造方法においては、高圧下にて反応を進行させるため、例えば200℃〜500℃といった低温での反応が可能であり、そのため、通常得られる粒径よりも小さな微粒子を得ることができる。すなわち、圧力容器内で圧力を上げる必要があり、加熱温度としては、用いる分散媒の沸点以上の温度で加熱する。本発明において、反応時の圧力としては、通常、0.5〜10MPaであり、1〜8MPaであると好ましい。
加熱時間は、目的とする金属酸化物蛍光体微粒子の種類、原料化合物、分散媒の種類により到達圧力が異なるため、適宜選択することができるが、通常、1時間〜10時間の範囲である。1時間より時間が短いと、微粒子の結晶性が上がらず、必要な蛍光強度が得られない。また、10時間より時間が長くなると、分散媒の炭化による不純物の混入や、微粒子の二次凝集進行などが生じ易い。
反応終了後、室温まで冷却する。必要に応じ、生成物を遠心分離し、残った沈殿物に有機溶媒を添加し、再度遠心分離を行う。共沈法、ゾル−ゲル法などの公知の製造方法では、反応終了後、結晶性を上げるために1000℃以上の高温で焼成する必要があった。すると、二次凝集が進行し結果として100nm以上の大きな微粒子しか得ることができなかった。本発明の製造方法によれば、反応後の焼成の必要がないため、粒径の小さな微粒子を得ることができる。さらに、精製の必要があれば、有機溶媒を添加し、遠心分離を複数回繰り返してもよい。
また、金属酸化物系蛍光体微粒子と溶媒との混合物に、該微粒子の分散状態を変化させる溶媒を添加た後、遠心分離、フィルター処理又は自然沈降を単独もしくは組み合わせて分級処理し、溶媒を含んだ透明な金属酸化物系蛍光体微粒子として分離すると好ましい。 前記微粒子の分散状態を変化させる溶媒としては、例えば、下記金属酸化物系蛍光体微粒子の合成に使用する溶媒や水なども好適に使用でき、該微粒子の分散状態を変化させる働きを有する。例えば、蛍光体合成に使用する溶媒(例えば、1,4ブタンジオール)を加えると、分散している微粒子と溶媒の比率が変化し、吸着平衡が変化し、分散性が変化する。また、アセトンのような有機溶媒では、微粒子分散が不安定になり、沈降を加速的に促進させる。代表的な溶媒としては、直鎖もしくは分岐鎖アルコール、単価アルコール、多価アルコール、アルカン、ケトン、エーテル、エステル、芳香族溶媒、水等が挙げられる。溶媒の添加量は、状態を見ながら適宜最適値に設定すればよい。
本発明の蛍光性ペーストは、溶媒に本発明の金属酸化物系蛍光体微粒子を50重量%以上含み、該金属酸化物系蛍光体微粒子中の金属酸化物に起因する発光波長の光を光路長150μm換算で50%以上透過する透明な蛍光性ペーストである。なお、ここで言う蛍光性ペーストとは、一般には1,000cp以上の粘性を持ったものを示し、狭義には50,000cp以上の粘性をもったものを示す。
前記蛍光性液体及び蛍光性ペーストにおける溶媒としては、1,4−ブタンジオールの他、直鎖もしくは分岐アルコール、単価アルコール、多価アルコール、アルカン、ケトン、エーテル、エステル、芳香族溶媒、水などが挙げられる。
前記蛍光性液体及び前記蛍光性ペーストを、それぞれ500℃以下の温度で焼成処理することで透明固体である蛍光体を得ることができる。焼成の雰囲気は空気中、不活性ガス中など適宜選ぶことができる。
本発明の蛍光変換体に用いる樹脂としては、特に限定されず公知のものを用いればよく、前記(c)樹脂成分と同様の例が挙げられる。
本発明の蛍光変換体に用いる溶媒としては、特に限定されず公知のものを用いればよい。
前記(2)の反応終了後、室温まで冷却する。この生成物から金属酸化物系蛍光体微粒子及び金属酸化物系蛍光体微粒子表面に配位する有機基及び溶媒の混合物として蛍光体を取り出す。このとき、適宜条件を選ぶと透明な蛍光体として分離が可能となる。分離手順は、まず、後処理の分離を助けるため、前述した分散状態を変化させる溶媒を1種類〜数種類添加する。この溶媒の添加量は、生成物の容量に対して1/20〜2/1、好ましく1/5〜1/1である。添加後、超音波ホモジナイザーや機械式ホモジナイザー等により混合を促進させてもよい。また、生成物の状態により、後処理の分離が容易に可能ならば、溶媒を添加しなくてもよい。
分離した蛍光体は、ペースト状態である。さらにこのペーストを、例えば窒素気流下、通常500℃以下の温度、好ましくは400℃以下、さらに好ましくは250〜300℃の温度で焼成すると透明な蛍光体(固体)が得られる。焼成時間は処理するペースト総量によるが、10分〜1時間程度が好適である。
また、各測定は以下のようにして行った。
(1)X線回折による同定
得られた生成物粉体をガラス基板に固定したものを試料とし、X線回折測定装置(Rint2200、理学電機製)を用いてX線回折角度と回折強度の関係を求めた。粉体の同定は、JCPDSカード(ICDD(International Center for Diffraction Data:国際回折データセンター) のJCPDS(Joint Committee on Powder Diffraction Standards)で編集された粉末X線回折データベース)との比較により行った。
(2)ICPによる定量分析
得られた生成物粉体0.02gを白金皿に取り、水/硫酸/塩酸を加えて溶解した試料とし、ICP発光分光分析を行い、試料中の金属元素の質量%を求めた。
(3)動的光散乱による平均粒径測定
得られた生成物粉体0.02gに1,4−ブタンジオール 1ミリリットルを加え、超音波分散を10分間行い分散液を得た。超純水30ミリリットル中に得られた分散液数滴を加え、さらに超音波分散を10分間行ったものを試料とし、動的光散乱測定装置(機種名:HPPS、Malvern Instruments社製)を用いて数平均の粒径分布を求めた。
なお、上記のように粉体から分散液を作るのではなく、加熱保持、室温まで冷却して得られた生成物溶液を直接超純水溶液に加え、以下同様な操作によって得たものを試料とすることもできる。
(4)蛍光強度測定
粉体を石英セルに充填し、蛍光分光光度計(FP−6500、日本分光社製)を用いて、励起スペクトルのピーク波長λEXを求めた。次に、λEXを励起波長としたときの蛍光スペクトルのピーク波長λEM及びピーク強度Iを求めた。
同様に、薄膜サンプルについては、薄膜を前記石英セルの替わりに配置し、薄膜サンプルの背面には、拡散反射率の高い酸化マグネシウム板を置いて後方への蛍光も効率的に測定できるようにした。薄膜に対して45°の角度で励起光を入射し、45°方向の蛍光を測定しピーク強度を測定した。
(5)分光光度計による透過率測定
蛍光体サンプルの透過率の測定は、紫外〜可視光分光光度計(島津製作所UV3100)により行った。薄膜サンプルは、薄膜フォルダーで挟み基板に垂直に入射し透過した透過率を測定した。溶液サンプルは、1cm角の溶液セルに入れて測定した。
(i)酢酸イットリウム・4水和物(関東化学社製)2.51g、酢酸セリウム・1水和物(関東化学社製)0.025g、アルミニウムトリイソプロポキシド(以下AIP、関東化学社製)2.55gを、分散媒1,4−ブタンジオール(以下1,4−BD、関東化学製、沸点:229℃)52.8ミリリットルを入れたガラス内筒に投入した。
(ii)ガラス内筒と圧力容器との隙間に1,4−BD 10.8ミリリットルを添加した。なお、圧力容器の内容積は120ミリリットルであり、原料仕込み体積は63.6ミリリットル、圧力容器内容積に対する原料溶液の仕込み容積率は53.0%であった。
(iii) 攪拌機にて300rpmの速度にて攪拌しながら、昇温速度3.1℃/分にて300℃まで昇温した。
(iv)その後300℃で2時間保持し反応を進行させた。2時間後の到達圧力は5.1MPaであった。
(v) 室温まで冷却後、生成物を遠心分離し、残った沈殿物にエタノールを添加した。この操作を4回繰返したのち、50℃の送風乾燥を行い、粉体を得た。
これらの原料及び製造条件を表1に示す。
得られた粉体について、前記(1)〜(4)の方法にて、各測定を行った結果を以下に示す。
前記(1)の方法でX線回折による同定を行った。図7に、得られたX線回折パターン(上段)、及びY3Al5O12に対応するJCPDSカード(下段)を示す。図7から分かるように、最も回折強度の強い(420)面のピークを始めとして、全てのピークがY3Al5O12に対応するJCPDSカードと一致し、母体結晶がY3Al5O12からなることを確認した。
前記(2)の方法で粉体中のCeの質量%を測定したところ0.53質量%であり、Ceがドープされていることを確認した。
前記(3)の方法で粉体の平均粒径を測定したところ46nmであった。
前記(4)の方法で波長λEX、λEM及びピーク強度Iを測定したところ、λEX=452nm、λEM=528nm、I=25.9(任意単位)であった。図8に測定で得られた励起スペクトル及び蛍光スペクトルを示す。
(i)酢酸イットリウム・4水和物(関東化学社製)2.51g、酢酸セリウム・1水和物(関東化学社製)0.025g、AIP(関東化学社製)2.55gを、分散媒1,4−BD 52.8ミリリットルを入れたガラス内筒に投入した。
(ii)ガラス内筒と圧力容器との隙間に1,4−BD 10.8ミリリットルを添加した。なお、圧力容器の内容積は120ミリリットルであり、原料仕込み体積は63.6ミリリットル、圧力容器内容積に対する原料溶液の仕込み容積率は53.0%であった。
(iii) 攪拌機にて300rpmの速度にて攪拌しながら、昇温速度3.1℃/分にて300℃まで昇温した。
(iv)その後300℃で4時間保持し反応を進行させた。4時間後の到達圧力は5.5MPaであった。
(v) 室温まで冷却後、生成物を遠心分離し、残った沈殿物にエタノールを添加した。この操作を4回繰返したのち、50℃の送風乾燥を行い、粉体を得た。
これらの原料及び製造条件を表1に示す。
得られた粉体について、前記(1)〜(4)の方法にて、各測定を行った結果を以下に示す。
前記(1)の方法でX線回折による同定を行ったところ、全てのピークがY3Al5O12に対応するJCPDSカードと一致し、母体結晶がY3Al5O12からなることを確認した。
前記(2)の方法で粉体中のCeの質量%を測定したところ0.54質量%であり、Ceがドープされていることを確認した。
前記(3)の方法で粉体の平均粒径を測定したところ52nmであった。
前記(4)の方法で波長λEX、λEM及びピーク強度Iを測定したところ、λEX=453nm、λEM=528nm、I=38.3(任意単位)であった。
(i)酢酸イットリウム・4水和物(関東化学社製)2.26g、酢酸ルテチウム・1水和物(関東化学社製)0.13g、酢酸セリウム・1水和物(関東化学社製)0.025g、AIP(関東化学社製)2.55gを、分散媒1,4−BD 52.8ミリリットルを入れたガラス内筒に投入した。
(ii)ガラス内筒と圧力容器との隙間に1,4−BD 10.8ミリリットルを添加した。なお、圧力容器の内容積は120ミリリットルであり、原料仕込み体積は63.6ミリリットル、圧力容器内容積に対する原料溶液の仕込み容積率は53.0%であった。
(iii) 攪拌機にて300rpmの速度にて攪拌しながら、昇温速度3.1℃/分にて300℃まで昇温した。
(iv)その後300℃で2時間保持し反応を進行させた。2時間後の到達圧力は4.5MPaであった。
(v) 室温まで冷却後、生成物を遠心分離し、残った沈殿物にエタノールを添加した。この操作を4回繰返したのち、50℃の送風乾燥を行い、粉体を得た。
これらの原料及び製造条件を表1に示す。
得られた粉体について、前記(1)〜(4)の方法にて、各測定を行った結果を以下に示す。
前記(1)の方法でX線回折による同定を行ったところ、全てのピークがY3Al5O12に対応するJCPDSカードと一致し、母体結晶がY3Al5O12からなることを確認した。
前記(2)の方法で粉体中のCe及びLuの質量%を測定したところ、Ceが0.52質量%、Luが3.51質量%であり、Ce及びLuがドープされていることを確認した。
前記(3)の方法で粉体の平均粒径を測定したところ56nmであった。
前記(4)の方法で波長λEX、λEM及びピーク強度Iを測定したところ、λEX=452nm、λEM=526nm、I=18.8(任意単位)であった。
(i)酢酸イットリウム・4水和物(関東化学社製)2.51g、酢酸セリウム・1水和物(関東化学社製)0.025g、アルミニウムトリ−セカンダリーブトキシド(以下ASB、関東化学社製)3.07gを、分散媒1,4−BD 52.8ミリリットルを入れたガラス内筒に投入した。
(ii)ガラス内筒と圧力容器との隙間に1,4−BD 10.8ミリリットルを添加した。なお、圧力容器の内容積は120ミリリットルであり、原料仕込み体積は63.6ミリリットル、圧力容器内容積に対する原料溶液の仕込み容積率は53.0%であった。
(iii) 攪拌機にて300rpmの速度にて攪拌しながら、昇温速度3.1℃/分にて300℃まで昇温した。
(iv)その後300℃で2時間保持し反応を進行させた。2時間後の到達圧力は4.5MPaであった。
(v) 室温まで冷却後、生成物を遠心分離し、残った沈殿物にエタノールを添加した。この操作を4回繰返したのち、50℃の送風乾燥を行い、粉体を得た。
これらの原料及び製造条件を表1に示す。
得られた粉体について、前記(1)〜(4)の方法にて、各測定を行った結果を以下に示す。
前記(1)の方法でX線回折による同定を行ったところ、全てのピークがY3Al5O12に対応するJCPDSカードと一致し、母体結晶がY3Al5O12からなることを確認した。
前記(2)の方法で粉体中のCeの質量%を測定したところ0.46質量%であり、Ceがドープされていることを確認した。
前記(3)の方法で粉体の平均粒径を測定したところ82nmであった。
前記(4)の方法で波長λEX、λEM及びピーク強度Iを測定したところ、λEX=452nm、λEM=530nm、I=15.8(任意単位)であった。
(i)酢酸イットリウム・4水和物(関東化学社製)2.51g、酢酸セリウム・1水和物(関東化学社製)0.025g、AIP(関東化学社製)2.55gを、分散媒エチレングリコール(以下EG、関東化学社製、沸点:198℃)52.8ミリリットルを入れたガラス内筒に投入した。
(ii)ガラス内筒と圧力容器との隙間にEG 10.8ミリリットルを添加した。なお、圧力容器の内容積は120ミリリットルであり、原料仕込み体積は63.6ミリリットル、圧力容器内容積に対する原料溶液の仕込み容積率は53.0%であった。
(iii) 攪拌機にて300rpmの速度にて攪拌しながら、昇温速度3.1℃/分にて300℃まで昇温した。
(iv)その後300℃で2時間保持し反応を進行させた。2時間後の到達圧力は2.0MPaであった。
(v) 室温まで冷却後、生成物を遠心分離し、残った沈殿物にエタノールを添加した。この操作を4回繰返したのち、50℃の送風乾燥を行い、粉体を得た。
これらの原料及び製造条件を表1に示す。
得られた粉体について、前記(1)〜(4)の方法にて、各測定を行った結果を以下に示す。
前記(1)の方法でX線回折による同定を行ったところ、全てのピークがY3Al5O12に対応するJCPDSカードと一致し、母体結晶がY3Al5O12からなることを確認した。
前記(2)の方法で粉体中のCeの質量%を測定したところ0.52質量%であり、Ceがドープされていることを確認した。
前記(3)の方法で粉体の平均粒径を測定したところ51nmであった。
前記(4)の方法で波長λEX、λEM及びピーク強度Iを測定したところ、λEX=452nm、λEM=530nm、I=15.8(任意単位)であった。
(i)酢酸イットリウム・4水和物(関東化学社製)2.51g、酢酸セリウム・1水和物(関東化学社製)0.025g、AIP(関東化学社製)2.55gを、分散媒1,4−BD 26.4ミリリットルを入れたガラス内筒に投入し、さらに分散媒ポリエチレングリコール#200(以下PEG、関東化学社製)26.4ミリリットルをガラス内筒に投入した。
(ii)ガラス内筒と圧力容器との隙間に、1,4−ブタンジオールとポリエチレングリコール♯200(関東化学社製)を体積比1:1で混合した溶液10.8ミリリットルを添加した。なお、圧力容器の内容積は120ミリリットルであり、原料仕込み体積は63.6ミリリットル、圧力容器内容積に対する原料溶液の仕込み容積率は53.0%であった。
(iii) 攪拌機にて300rpmの速度にて攪拌しながら、昇温速度3.1℃/分にて300℃まで昇温した。
(iv)その後300℃で4時間保持し反応を進行させた。2時間後の到達圧力は1.5MPaであった。
(v) 室温まで冷却後、生成物を遠心分離し、残った沈殿物にエタノールを添加した。この操作を4回繰返したのち、50℃の送風乾燥を行い、粉体を得た。
これらの原料及び製造条件を表1に示す。
得られた粉体について、前記(1)〜(4)の方法にて、各測定を行った結果を以下に示す。
前記(1)の方法でX線回折による同定を行ったところ、全てのピークがY3Al5O12に対応するJCPDSカードと一致し、母体結晶がY3Al5O12からなることを確認した。
前記(2)の方法で粉体中のCeの質量%を測定したところ0.48質量%であり、Ceがドープされていることを確認した。
前記(3)の方法で粉体の平均粒径を測定したところ52nmであった。
前記(4)の方法で波長λEX、λEM及びピーク強度Iを測定したところ、λEX=452nm、λEM=528nm、I=48.1(任意単位)であった。
(i)実施例1で得られた粉体1.0gに、分散媒として1,4−BDを1.0g、1−メトキシ−2−アセトキシプロパンを4.0g加え、超音波分散を10分間行った。さらに、光透過性樹脂として、以下のA〜C成分及び組成比からなる樹脂組成物を1g加え、攪拌処理を行い、分散液を調製した。
A:メタクリル酸−メタクリル酸メチル共重合体(メタクリル酸共重合比15%、重量平均分子量Mw=2万)
B:トリメチロールプロパントリアクリレート
C:光重合開始剤(商品名:イルガキュア907、チバスペシャリティケミカル社製)
A:B:C(重量比)=55:42:3
(ii)得られた分散液を用い、スクリーン印刷法によりガラス基板上に成膜し、150℃で20分間の乾燥処理を行った。続いて、300mJ/cm2 の高圧水銀灯の光を空気中で照射し、その後に空気中で200℃で60分間の熱処理を行い、膜厚10μmの蛍光変換膜を得た。
(iii) 青色光を発する有機EL素子(輝度100nit、CIE色度座標(0.16,0.10)、ELピーク波長445nm)の発光面上に、シリコーンオイルを介して得られた蛍光変換膜を貼り付け、発光スペクトルを測定した。その結果、CIE色度(0.22、0.45)、輝度139nitの緑色光を得た。図9に、有機EL素子の発光スペクトルと、蛍光変換膜を通して得られた発光スペクトルを示す。
(i)酢酸イットリウム・4水和物(関東化学社製)2.51g、酢酸セリウム・1水和物(関東化学社製)0.025g、AIP(関東化学社製)2.55gを、解離する官能基を末端又は側鎖に持たない分散媒1−メトキシ−2−アセトキシプロパン(以下PGMEA、和光純薬社製、沸点:146℃)52.8ミリリットルを入れたガラス内筒に投入した。
(ii)ガラス内筒と圧力容器との隙間にPGMEA 10.8ミリリットルを添加した。
(iii) 攪拌機にて300rpmの速度にて攪拌しながら、昇温速度3.1℃/分にて300℃まで昇温した。
(iv)その後265℃で2時間保持し反応を進行させた。2時間後の到達圧力は、1.5MPaであった。
(v) 室温まで冷却後、生成物を遠心分離し、残った沈殿物にエタノールを添加した。この操作を4回繰返したのち、50℃の送風乾燥を行い、粉体を得た。
これらの原料及び製造条件を表1に示す。
得られた粉体について、前記(1)の方法でX線回折による同定を行った。得られたX線回折パターン、及びY3Al5O12に対応するJCPDSカードを図10に示す。図10から分かるように、得られたX線回折パターンは、Y3Al5O12に対応するJCPDSカードと全く一致せず、生成物を同定することができなかった。
また、前記(4)の方法で波長λEX、λEM及びピーク強度Iを測定したが、有意な蛍光を検出することができなかった。
実施例1の(i)〜(iv)と同様に操作した。
(v) 室温まで冷却後、そのままで、2週間静置して上澄みを取り出し、遠心分離機(日立製作所製高速冷却遠心機:himacCR22)にて遠心力2000gで60分処理して沈殿を分離した。沈殿を少量取り前記(3)の方法にて平均粒径を測定した。このときの粒径は55nmであった。
(vi)沈殿と溶媒の固液界面付近より、透明な分散液を1.0g得た。この操作を複数回繰り返して集めた透明な分散液を、(4)の方法で石英セルに入れ蛍光を測定した。このときの蛍光ピーク波長は520nmであった。(5)の吸収スペクトル測定より波長520nmの透過率は88%であった。この透明な分散液を300℃まで加熱した時の重量減より、分散液に含まれている溶媒は85重量%であった。すなわち固形分濃度は15%であった。
(vii) 沈殿部分より得られた蛍光体は黄色透明のペーストであった。そのペーストを窒素中300℃まで加熱した時の重量減によりペーストに含まれている溶媒は約20重量%であった。すなわち固形分濃度は80重量%であった。加熱後の蛍光体は、透明な塊となった。
(viii)スペーサにより、2 枚のガラス基板に150μmの隙間を作り、その中に前記ペーストを挟んでエポキシ接着剤からなる封止剤で封止し、透明な薄膜サンプルが得られた。その模式図及び上面から見た写真を図11及び図12に示す。この薄膜サンプルにより得られた透過スペクトルを図13、蛍光スペクトル及び励起スペクトルを図14に示す。このとき蛍光スペクトルのピーク波長は520nmであった。また、(5)の吸収スペクトル測定より、波長520nmの透過率89%であった。図12の写真に示したように、ペーストの薄膜を通した文字がはっきり読み取れ、透明性の高さが確認できた。
実施例1で得られた粉末とポリエチレングリコール(分子量600)を75:25の重量比率で混合し、黄白色に濁ったペーストを作製した。スペーサにより2枚のガラス板に150μmの隙間を作り、その中に前記ペーストを挟んで封止剤で封止し、透明な薄膜サンプルを得た。前記(4)の方法で測定したところ、蛍光スペクトルのピーク波長は525nmであり、(5)の吸収スペクトル測定より、波長525nmの透過率は45%であった。
実施例1の(i) 〜(iv)と同様に操作した。
(v) 室温まで冷却後、遠心分離機にて遠心力500gで30分処理して沈殿を取り除き、さらに遠心分離機にて遠心力2000gで60分処理して沈殿を分離した。沈殿を少量取り前記(3)の方法で平均粒径を測定した。このときの粒径は65nmであった。
(vi)沈殿部分より得られた蛍光体は黄色透明のペーストであった。そのペーストを窒素中300℃まで加熱した時の重量減によりペーストに含まれている溶媒は約25重量%であることが分かった。すなわち固形分濃度は75重量%であった。加熱後の蛍光体は、体積が減少し、透明な塊となった。ペーストより溶媒を取り除き透明な固形物を得た。
(vii) スペーサにより、2枚のガラス板に150μmの隙間を作り、その中に前記ペーストを挟んで封止し、透明な薄膜サンプルを得た。このサンプルに波長365nmの紫外光を照射すると、緑色の発光が確認でき、蛍光体であることが確認できた。
実施例1の(i) 〜(iv)と同様に操作した。
(v) 室温まで冷却後、得られた生成物の体積約80ccと同容積のメタノールを添加してよく攪拌した。さらに遠心分離機にて遠心力2000gで60分処理して沈殿を分離した。沈殿を少量取り(3)の方法で平均粒径を測定した。このときの粒径は45nmであった。沈殿は窒素気流下50℃で1日放置しメタノールを除去した。
(vi)沈殿部分より得られた蛍光体は黄色透明のペーストであった。そのペーストを窒素中300℃まで加熱した時の重量減によりペーストに含まれている溶媒は約45重量%であることが分かった。すなわち固形分濃度は55重量%であった。加熱後の蛍光体は、体積が減少し、透明な塊となった。ペーストより溶媒を取り除き透明な固形物を得た。
(vii) スペーサにより、2枚のガラス板に150μmの隙間を作り、その中に前記ペーストを挟んで封止し、透明な薄膜サンプルを得た。このサンプルに波長365nmの紫外光を照射すると、緑色の発光が確認でき、蛍光体であることが確認できた。
実施例1の(i) 〜(iv)と同様に操作した。
(v) 室温まで冷却後、得られた生成物の体積約80ccと同容積のアセトンを添加してよく攪拌した。さらに遠心分離機にて遠心力2000gで60分処理して沈殿を分離した。沈殿を少量取り(3)の方法で平均粒径を測定した。このときの粒径は45nmであった。沈殿は窒素気流下50℃で1日放置しアセトンを除去した。
(vi)沈殿部分より得られた蛍光体は黄色透明のペーストであった。そのペーストを窒素中300℃まで加熱した時の重量減によりペーストに含まれている溶媒は約45重量%であることが分かった。すなわち固形分濃度は55重量%であった。加熱後の蛍光体は、体積が減少し、透明な塊となった。ペーストより溶媒を取り除き透明な固形物を得た。
(vii) スペーサにより、2枚のガラス板に150μmの隙間を作り、その中に前記ペーストを挟んで封止し、透明な薄膜サンプルを得た。このサンプルに波長365nmの紫外光を照射すると、緑色の発光が確認でき、蛍光体であることが確認できた。
実施例9で得られた黄色透明ペーストを窒素置換したオーブンで300℃まで加熱し、30分保持した。冷却後取り出したところ、透明な固形物が得られた。この固形物に波長365nmの紫外線を当てると緑色に光り、蛍光変換体であることが確認できた。
実施例12
実施例9で得られた黄色透明ペーストとPEG(平均分子量2000)を重量比で1:10になるようにビーカーに計り取り、ホットプレート上約150℃で混合した混合物を、ガラス基板上に塗り広げ、黄色の薄膜を得た。同様の混合物を加熱状態で耐熱性の箱(ステンレス製)に流し込み、厚み2mmの樹脂基板を得た。薄膜、樹脂基板ともに波長365nmの紫外線を当てると緑色に光り、蛍光変換体であることが確認できた。
実施例7の(viii)において、得られた黄色透明ペーストを2枚のガラス板で挟む際、その隙間(膜厚)を100μm、200μm、300μmと変化させ、得られる蛍光スペクトルのピーク強度依存性を測定した。
横軸に膜厚、縦軸にピーク強度を蛍光強度としてプロットした結果を図15に示す。同図に示すように、膜厚と蛍光強度は、ほぼ正比例関係にあり、ペーストが透明であるため蛍光が効率よく取り出せることが分かった。
比較例2において、得られた黄白色ペーストを2枚のガラス板で挟む際、その隙間(膜厚)を100μm、200μm、300μmと変化させ、得られる蛍光スペクトルのピーク強度依存性を測定した。
横軸に膜厚、縦軸にピーク強度を蛍光強度としてプロットした結果を図15に示す。同図に示すように、膜厚と蛍光強度は、依存の少ない関係が得られ、ペーストが不透明であるために膜厚を増やしても、蛍光が効率よく取り出せないことが分かった。
実施例7で得られた黄色透明ペーストをポリエチレングリコール(分子量6000)と75:25の重量比率で150℃に昇温しながら混合した。この混合物をステンレスバットの中に流し込み、室温まで冷却することで膜厚2mmの基板を得た。365nmの光を当てると緑色に光り、蛍光変換体として機能する蛍光変換板であることが確認できた。
実施例15
実施例7で得られた黄色透明ペーストをポリエチレングリコール(分子量6000)と75:25の重量比率で150℃に昇温しながら混合した。青色発光ダイオード(日亜化学社製NSPE520S)の樹脂を一部分丁寧に取り外し、取り除いた発光部分に前記混合物を滴下し、冷却した。発光ダイオードを点灯させると黄色〜緑白色の発光が確認できた。
参考例1の(i)において、粉体の代わりに実施例7で得られた黄色透明ペーストを用いた以外は同様にして、(i)〜(iii) の操作を行い、(iii) において、同じ光源で励起したときの発光スペクトルを測定した。その結果、CIE色度座標(0.25、0.44)、輝度130nitの緑色光を得た。
Claims (4)
- 金属酸化物からなる母体を形成する金属元素の化合物と、発光中心である金属元素の化合物とを、末端又は側鎖にOH基の官能基を一つ以上有する有機化合物を含む分散媒に溶解又は分散させた溶液を圧力容器内に封入し、前記有機化合物の沸点以上の温度で加熱し、圧力0.5〜10Mpa、温度200〜500℃で反応して得られた金属酸化物系蛍光体微粒子と前記分散媒との混合物を、遠心分離又は自然沈降を単独もしくは組み合わせて分級処理して粗大粒子を除き、さらに遠心分離した後、前記分散媒を含んだ透明な金属酸化物系蛍光体微粒子の沈殿物として得られる蛍光性ペーストであって、
前記金属酸化物からなる母体結晶中に、前記発光中心である金属元素がドープされた前記金属酸化物系蛍光体微粒子において、該蛍光体微粒子表面に有機基が配位してなり、前記母体結晶の金属酸化物における金属元素が、イットリウム(Y)及びアルミニウム(Al)であり、かつ発光中心である金属元素が、セリウム(Ce)及びルテチウム(Lu)から選ばれる少なくとも一種であり、前記有機基が、末端又は側鎖にOH基の官能基を一つ以上有する有機化合物から少なくとも一つの該官能基が解離したものであり、平均粒径1〜100nmの前記金属酸化物系蛍光体微粒子を、前記分散媒に50重量%以上含み、該金属酸化物系蛍光体微粒子中の金属酸化物に起因する発光波長の光を光路長150μm換算で50%以上透過する透明な前記蛍光性ペーストを250〜300℃の温度で焼成処理してなる蛍光体。 - 金属酸化物からなる母体を形成する金属元素の化合物と、発光中心である金属元素の化合物とを、末端又は側鎖にOH基の官能基を一つ以上有する有機化合物を含む分散媒に溶解又は分散させた溶液を圧力容器内に封入し、前記有機化合物の沸点以上の温度で加熱し、圧力0.5〜10Mpa、温度200〜500℃で反応して得られた金属酸化物系蛍光体微粒子と前記分散媒との混合物を、遠心分離又は自然沈降を単独もしくは組み合わせて分級処理して粗大粒子を除き、さらに遠心分離した後、前記分散媒を含んだ透明な金属酸化物系蛍光体微粒子の沈殿物として得られる蛍光性ペーストであって、
前記金属酸化物からなる母体結晶中に、前記発光中心である金属元素がドープされた前記金属酸化物系蛍光体微粒子において、該蛍光体微粒子表面に有機基が配位してなり、前記母体結晶の金属酸化物における金属元素が、イットリウム(Y)及びアルミニウム(Al)であり、かつ発光中心である金属元素が、セリウム(Ce)及びルテチウム(Lu)から選ばれる少なくとも一種であり、前記有機基が、末端又は側鎖にOH基の官能基を一つ以上有する有機化合物から少なくとも一つの該官能基が解離したものであり、平均粒径1〜100nmの前記金属酸化物系蛍光体微粒子を、前記分散媒に50重量%以上含み、該金属酸化物系蛍光体微粒子中の金属酸化物に起因する発光波長の光を光路長150μm換算で50%以上透過する透明な前記蛍光性ペーストを250〜300℃の温度で焼成処理する蛍光体の製造方法。 - 請求項1に記載の蛍光体を単独、もしくは該蛍光体に樹脂及び/又は溶媒を添加し、固化してなる蛍光変換体。
- 請求項1に記載の蛍光体を樹脂及び/又は溶媒に分散してなる蛍光変換体。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005517448A JP4989891B2 (ja) | 2004-01-29 | 2005-01-26 | 金属酸化物系蛍光体微粒子を利用した蛍光体及びその製造方法並びに蛍光変換体 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004021488 | 2004-01-29 | ||
| JP2004021488 | 2004-01-29 | ||
| PCT/JP2005/000976 WO2005073342A1 (ja) | 2004-01-29 | 2005-01-26 | 金属酸化物系蛍光体微粒子及びその製造方法、それを利用した分散液、蛍光変換膜、金属酸化物系蛍光体微粒子の分離方法、蛍光性液体、蛍光性ペースト、蛍光体及びその製造方法並びに蛍光変換体 |
| JP2005517448A JP4989891B2 (ja) | 2004-01-29 | 2005-01-26 | 金属酸化物系蛍光体微粒子を利用した蛍光体及びその製造方法並びに蛍光変換体 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2005073342A1 JPWO2005073342A1 (ja) | 2007-09-13 |
| JP4989891B2 true JP4989891B2 (ja) | 2012-08-01 |
Family
ID=34823795
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005517448A Expired - Fee Related JP4989891B2 (ja) | 2004-01-29 | 2005-01-26 | 金属酸化物系蛍光体微粒子を利用した蛍光体及びその製造方法並びに蛍光変換体 |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US20070166543A1 (ja) |
| EP (1) | EP1715022A4 (ja) |
| JP (1) | JP4989891B2 (ja) |
| KR (1) | KR20060123537A (ja) |
| CN (1) | CN1934217A (ja) |
| TW (1) | TW200528538A (ja) |
| WO (1) | WO2005073342A1 (ja) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006222403A (ja) * | 2005-02-14 | 2006-08-24 | Kri Inc | 光増幅素器 |
| WO2007080555A1 (en) * | 2006-01-16 | 2007-07-19 | Koninklijke Philips Electronics N.V. | Phosphor converted light emitting device |
| FR2896791B1 (fr) * | 2006-01-30 | 2008-10-10 | Rhodia Recherches & Tech | Dispersion colloidale d'un borate de terre rare, son procede de preparation et son utilisation comme luminophore |
| RU2315135C2 (ru) | 2006-02-06 | 2008-01-20 | Владимир Семенович Абрамов | Метод выращивания неполярных эпитаксиальных гетероструктур на основе нитридов элементов iii группы |
| KR100950418B1 (ko) * | 2008-02-29 | 2010-03-29 | 부경대학교 산학협력단 | 백색 엘이디용 가넷계 결정 형광체 및 이의 제조방법 |
| US8152586B2 (en) | 2008-08-11 | 2012-04-10 | Shat-R-Shield, Inc. | Shatterproof light tube having after-glow |
| EP2161763A1 (de) * | 2008-09-04 | 2010-03-10 | Bayer MaterialScience AG | Konversionsfolie und ein Verfahren zu deren Herstellung |
| TWI478373B (zh) * | 2009-04-24 | 2015-03-21 | Kunshan Nano New Material Technology Co Ltd | A centrifugal separation apparatus, a method for manufacturing a solid particle by centrifugal separation, and a solid particle for a light emitting diode device |
| KR20140015763A (ko) | 2012-07-24 | 2014-02-07 | 삼성디스플레이 주식회사 | Led 패키지 및 이를 갖는 표시 장치 |
| US10510914B2 (en) * | 2013-03-21 | 2019-12-17 | Board Of Trustees Of Michigan State University | Transparent energy-harvesting devices |
| CN107180908B (zh) * | 2017-06-27 | 2019-12-27 | 深圳Tcl新技术有限公司 | 一种led、背光模组及液晶显示装置 |
| KR102318332B1 (ko) * | 2019-05-08 | 2021-10-28 | 신동현 | 벌브형 레이저 발광램프 |
| CN114851657A (zh) * | 2022-05-12 | 2022-08-05 | 深圳大学 | 可编辑的动态磷光柔性薄膜及其应用方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09127686A (ja) * | 1995-11-02 | 1997-05-16 | Hitachi Chem Co Ltd | 感光性樹脂組成物、これを用いた感光性フィルム及び蛍光パターンの製造法 |
| JPH10168442A (ja) * | 1996-12-06 | 1998-06-23 | Philips Electron Nv | ヒドロキシカルボン酸コーティングを有する蛍光体組成物 |
| JPH11181419A (ja) * | 1997-12-25 | 1999-07-06 | Kasei Optonix Co Ltd | 金属酸化物系蛍光体の製造方法 |
| JP2002121549A (ja) * | 2000-06-26 | 2002-04-26 | Mitsubishi Chemicals Corp | 半導体超微粒子 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5985173A (en) * | 1997-11-18 | 1999-11-16 | Gray; Henry F. | Phosphors having a semiconductor host surrounded by a shell |
| US6576155B1 (en) * | 1998-11-10 | 2003-06-10 | Biocrystal, Ltd. | Fluorescent ink compositions comprising functionalized fluorescent nanocrystals |
| JP3797812B2 (ja) * | 1999-01-11 | 2006-07-19 | 日立マクセル株式会社 | 無機蛍光体およびその製造方法 |
| US7241399B2 (en) * | 2000-09-08 | 2007-07-10 | Centrum Fuer Angewandte Nanotechnologie (Can) Gmbh | Synthesis of nanoparticles |
| JP2002156498A (ja) * | 2000-11-20 | 2002-05-31 | Fuji Photo Film Co Ltd | 放射線像変換パネル |
| DE10153829A1 (de) * | 2001-11-05 | 2003-05-28 | Bayer Ag | Assay basierend auf dotierten Nanoteilchen |
| US6783699B2 (en) * | 2002-10-17 | 2004-08-31 | Medgene, Inc. | Europium-containing fluorescent nanoparticles and methods of manufacture thereof |
| JP2006028354A (ja) * | 2004-07-16 | 2006-02-02 | Keio Gijuku | 蛍光体及び蛍光体の製造方法 |
| US7393618B2 (en) * | 2006-09-15 | 2008-07-01 | Idemitsu Kosan Co., Ltd. | Composition for color converting member and production method of color conversion substrate using the same |
-
2005
- 2005-01-26 KR KR1020067016260A patent/KR20060123537A/ko not_active Application Discontinuation
- 2005-01-26 WO PCT/JP2005/000976 patent/WO2005073342A1/ja active Application Filing
- 2005-01-26 JP JP2005517448A patent/JP4989891B2/ja not_active Expired - Fee Related
- 2005-01-26 US US10/587,631 patent/US20070166543A1/en not_active Abandoned
- 2005-01-26 EP EP05704113A patent/EP1715022A4/en not_active Withdrawn
- 2005-01-26 CN CNA2005800087377A patent/CN1934217A/zh active Pending
- 2005-01-28 TW TW094102787A patent/TW200528538A/zh unknown
-
2009
- 2009-08-24 US US12/546,162 patent/US7883641B2/en not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09127686A (ja) * | 1995-11-02 | 1997-05-16 | Hitachi Chem Co Ltd | 感光性樹脂組成物、これを用いた感光性フィルム及び蛍光パターンの製造法 |
| JPH10168442A (ja) * | 1996-12-06 | 1998-06-23 | Philips Electron Nv | ヒドロキシカルボン酸コーティングを有する蛍光体組成物 |
| JPH11181419A (ja) * | 1997-12-25 | 1999-07-06 | Kasei Optonix Co Ltd | 金属酸化物系蛍光体の製造方法 |
| JP2002121549A (ja) * | 2000-06-26 | 2002-04-26 | Mitsubishi Chemicals Corp | 半導体超微粒子 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2005073342A1 (ja) | 2005-08-11 |
| US20070166543A1 (en) | 2007-07-19 |
| US20100047561A1 (en) | 2010-02-25 |
| TW200528538A (en) | 2005-09-01 |
| US7883641B2 (en) | 2011-02-08 |
| KR20060123537A (ko) | 2006-12-01 |
| CN1934217A (zh) | 2007-03-21 |
| EP1715022A4 (en) | 2009-04-15 |
| EP1715022A1 (en) | 2006-10-25 |
| JPWO2005073342A1 (ja) | 2007-09-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4989891B2 (ja) | 金属酸化物系蛍光体微粒子を利用した蛍光体及びその製造方法並びに蛍光変換体 | |
| JP4739185B2 (ja) | 有機エレクトロルミネッセンス表示装置 | |
| EP1990844B1 (en) | Organic electroluminescent device | |
| EP1860097B1 (en) | Aromatic amine derivative and organic electroluminescence device utilizing the same | |
| EP2261302B2 (en) | Organic electroluminescence device and anthracene derivative | |
| EP2239259B1 (en) | Aromatic amine derivative and use of the same in an organic electroluminescent device | |
| EP2008992A1 (en) | Aromatic amine derivative and organic electroluminescent device using the same | |
| JPWO2006009039A1 (ja) | カラー発光装置 | |
| JPWO2006070712A1 (ja) | 有機エレクトロルミネッセンス素子用発光性インク組成物 | |
| JPWO2008023623A1 (ja) | 有機エレクトロルミネッセンス素子 | |
| JPWO2005117500A1 (ja) | 白色系有機エレクトロルミネッセンス素子 | |
| EP1724323A1 (en) | Material for organic electroluminescence device and organic electroluminescence device utilizing the same | |
| KR20100121489A (ko) | 유기 발광 매체 및 유기 el 소자 | |
| JPWO2005109964A1 (ja) | 有機エレクトロルミネッセンス表示装置 | |
| JPWO2006085434A1 (ja) | ビスアントラセン誘導体及びそれを利用した有機エレクトロルミネッセンス素子 | |
| JP4652516B2 (ja) | ピラン誘導体 | |
| EP1404160A1 (en) | Organic electroluminescence device | |
| JP2008115093A (ja) | アミノジベンゾフルオレン誘導体及びそれを用いた有機エレクトロルミネッセンス素子 | |
| JPWO2007097178A1 (ja) | 有機エレクトロルミネッセンス素子用材料、その製造方法及び有機エレクトロルミネッセンス素子 | |
| JP2002124385A (ja) | 有機エレクトロルミネッセンス素子 | |
| US20020041978A1 (en) | Organic electroluminescent device | |
| JP2008133241A (ja) | ジベンゾ[c,g]トリフェニレン誘導体及びそれを用いた有機エレクトロルミネッセンス素子 | |
| JP2008069301A (ja) | シリコンナノシート、そのコロイド溶液、およびシリコンナノシートの製造方法。 | |
| US20010006741A1 (en) | Organic electroluminescent device | |
| EP4417670A1 (en) | Organic solid up-conversion material |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20070921 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110308 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110427 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120117 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120312 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120403 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120501 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150511 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |