JP4969002B2 - rAAV形質導入を増加するための化合物および方法 - Google Patents
rAAV形質導入を増加するための化合物および方法 Download PDFInfo
- Publication number
- JP4969002B2 JP4969002B2 JP2001501645A JP2001501645A JP4969002B2 JP 4969002 B2 JP4969002 B2 JP 4969002B2 JP 2001501645 A JP2001501645 A JP 2001501645A JP 2001501645 A JP2001501645 A JP 2001501645A JP 4969002 B2 JP4969002 B2 JP 4969002B2
- Authority
- JP
- Japan
- Prior art keywords
- raav
- virus
- infection
- aav
- use according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 238000010361 transduction Methods 0.000 title claims description 145
- 230000026683 transduction Effects 0.000 title claims description 145
- 150000001875 compounds Chemical class 0.000 title claims description 13
- 238000000034 method Methods 0.000 title description 79
- 230000001965 increasing effect Effects 0.000 title description 42
- 241000700605 Viruses Species 0.000 claims description 184
- 239000003795 chemical substances by application Substances 0.000 claims description 72
- 230000003612 virological effect Effects 0.000 claims description 71
- 229940079156 Proteasome inhibitor Drugs 0.000 claims description 55
- 239000003207 proteasome inhibitor Substances 0.000 claims description 54
- 239000012528 membrane Substances 0.000 claims description 51
- 125000000217 alkyl group Chemical group 0.000 claims description 42
- 238000012546 transfer Methods 0.000 claims description 42
- 230000000694 effects Effects 0.000 claims description 40
- 238000012545 processing Methods 0.000 claims description 30
- 239000003814 drug Substances 0.000 claims description 29
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical group OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 claims description 26
- 229940024606 amino acid Drugs 0.000 claims description 24
- 150000001413 amino acids Chemical class 0.000 claims description 24
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 24
- 235000001014 amino acid Nutrition 0.000 claims description 23
- 229910052739 hydrogen Inorganic materials 0.000 claims description 19
- 239000001257 hydrogen Substances 0.000 claims description 19
- -1 benzyl ester Chemical class 0.000 claims description 16
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 claims description 16
- 238000004519 manufacturing process Methods 0.000 claims description 16
- 108090000848 Ubiquitin Proteins 0.000 claims description 15
- 102000044159 Ubiquitin Human genes 0.000 claims description 15
- 125000001589 carboacyl group Chemical group 0.000 claims description 15
- 125000003118 aryl group Chemical group 0.000 claims description 13
- 150000003839 salts Chemical class 0.000 claims description 13
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 claims description 11
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 claims description 11
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical group NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 claims description 10
- 102000006275 Ubiquitin-Protein Ligases Human genes 0.000 claims description 9
- 108010083111 Ubiquitin-Protein Ligases Proteins 0.000 claims description 9
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 claims description 9
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 9
- 210000004962 mammalian cell Anatomy 0.000 claims description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 8
- SNDPXSYFESPGGJ-BYPYZUCNSA-N L-2-aminopentanoic acid Chemical group CCC[C@H](N)C(O)=O SNDPXSYFESPGGJ-BYPYZUCNSA-N 0.000 claims description 8
- SNDPXSYFESPGGJ-UHFFFAOYSA-N L-norVal-OH Chemical group CCCC(N)C(O)=O SNDPXSYFESPGGJ-UHFFFAOYSA-N 0.000 claims description 8
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical group CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 claims description 8
- 125000001429 N-terminal alpha-amino-acid group Chemical group 0.000 claims description 8
- 230000015572 biosynthetic process Effects 0.000 claims description 8
- 230000000903 blocking effect Effects 0.000 claims description 8
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 8
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Chemical group CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 claims description 8
- 229960000310 isoleucine Drugs 0.000 claims description 8
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical group CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 claims description 7
- HSQGMTRYSIHDAC-BQBZGAKWSA-N Leu-Ala Chemical group CC(C)C[C@H](N)C(=O)N[C@@H](C)C(O)=O HSQGMTRYSIHDAC-BQBZGAKWSA-N 0.000 claims description 7
- FRJIAZKQGSCKPQ-FSPLSTOPSA-N His-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@@H](N)CC1=CN=CN1 FRJIAZKQGSCKPQ-FSPLSTOPSA-N 0.000 claims description 6
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 claims description 6
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical group CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 claims description 5
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Chemical group CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 claims description 5
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 5
- 239000004475 Arginine Chemical group 0.000 claims description 4
- 239000004471 Glycine Chemical group 0.000 claims description 4
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical group NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 claims description 4
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical group CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 claims description 4
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Chemical group CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 claims description 4
- 235000004279 alanine Nutrition 0.000 claims description 4
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 claims description 4
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Chemical group OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 claims description 4
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 4
- 239000004474 valine Chemical group 0.000 claims description 4
- 102100020696 Ubiquitin-conjugating enzyme E2 K Human genes 0.000 claims description 3
- 230000004913 activation Effects 0.000 claims description 3
- 125000003277 amino group Chemical group 0.000 claims description 3
- 150000002431 hydrogen Chemical class 0.000 claims description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 3
- 238000003786 synthesis reaction Methods 0.000 claims description 3
- 108010084736 ubiquitin carrier proteins Proteins 0.000 claims description 3
- 125000001433 C-terminal amino-acid group Chemical group 0.000 claims description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 claims description 2
- 125000002252 acyl group Chemical group 0.000 claims description 2
- 150000001408 amides Chemical class 0.000 claims description 2
- 125000006239 protecting group Chemical group 0.000 claims description 2
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 claims description 2
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 claims 5
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims 2
- 210000004899 c-terminal region Anatomy 0.000 claims 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims 1
- UUEVFMOUBSLVJW-UHFFFAOYSA-N oxo-[[1-[2-[2-[2-[4-(oxoazaniumylmethylidene)pyridin-1-yl]ethoxy]ethoxy]ethyl]pyridin-4-ylidene]methyl]azanium;dibromide Chemical compound [Br-].[Br-].C1=CC(=C[NH+]=O)C=CN1CCOCCOCCN1C=CC(=C[NH+]=O)C=C1 UUEVFMOUBSLVJW-UHFFFAOYSA-N 0.000 claims 1
- 210000004027 cell Anatomy 0.000 description 242
- 208000015181 infectious disease Diseases 0.000 description 179
- 241000702421 Dependoparvovirus Species 0.000 description 149
- 108090000623 proteins and genes Proteins 0.000 description 144
- 208000002267 Anti-neutrophil cytoplasmic antibody-associated vasculitis Diseases 0.000 description 131
- 230000014509 gene expression Effects 0.000 description 96
- 239000002245 particle Substances 0.000 description 94
- 108091033319 polynucleotide Proteins 0.000 description 90
- 102000040430 polynucleotide Human genes 0.000 description 90
- 239000002157 polynucleotide Substances 0.000 description 90
- 210000000981 epithelium Anatomy 0.000 description 53
- 108700019146 Transgenes Proteins 0.000 description 52
- 239000013598 vector Substances 0.000 description 49
- 210000004379 membrane Anatomy 0.000 description 48
- 230000012202 endocytosis Effects 0.000 description 44
- 238000011282 treatment Methods 0.000 description 44
- 241000282414 Homo sapiens Species 0.000 description 41
- 108020004414 DNA Proteins 0.000 description 40
- 239000000203 mixture Substances 0.000 description 37
- 238000004806 packaging method and process Methods 0.000 description 35
- 102000004169 proteins and genes Human genes 0.000 description 32
- 239000013608 rAAV vector Substances 0.000 description 32
- 230000006870 function Effects 0.000 description 30
- 210000004072 lung Anatomy 0.000 description 30
- 241000701161 unidentified adenovirus Species 0.000 description 30
- 235000018102 proteins Nutrition 0.000 description 29
- 210000004940 nucleus Anatomy 0.000 description 27
- 230000010076 replication Effects 0.000 description 27
- 229940079593 drug Drugs 0.000 description 25
- 238000001415 gene therapy Methods 0.000 description 24
- 239000002953 phosphate buffered saline Substances 0.000 description 24
- 239000013612 plasmid Substances 0.000 description 24
- 238000012360 testing method Methods 0.000 description 23
- 239000013607 AAV vector Substances 0.000 description 22
- TZYWCYJVHRLUCT-VABKMULXSA-N N-benzyloxycarbonyl-L-leucyl-L-leucyl-L-leucinal Chemical compound CC(C)C[C@@H](C=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)OCC1=CC=CC=C1 TZYWCYJVHRLUCT-VABKMULXSA-N 0.000 description 22
- 108010002974 benzyloxycarbonylleucyl-leucyl-leucine aldehyde Proteins 0.000 description 22
- 239000002609 medium Substances 0.000 description 22
- 239000000243 solution Substances 0.000 description 22
- 238000001727 in vivo Methods 0.000 description 21
- 230000001404 mediated effect Effects 0.000 description 21
- 229920000642 polymer Polymers 0.000 description 20
- 238000013518 transcription Methods 0.000 description 20
- 230000035897 transcription Effects 0.000 description 20
- 238000009281 ultraviolet germicidal irradiation Methods 0.000 description 20
- 239000007788 liquid Substances 0.000 description 19
- 238000002360 preparation method Methods 0.000 description 19
- 239000000047 product Substances 0.000 description 19
- 210000001519 tissue Anatomy 0.000 description 19
- 108090000565 Capsid Proteins Proteins 0.000 description 18
- 108091026890 Coding region Proteins 0.000 description 18
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 18
- 125000003545 alkoxy group Chemical group 0.000 description 18
- 210000002950 fibroblast Anatomy 0.000 description 18
- 108020003175 receptors Proteins 0.000 description 18
- 241000699666 Mus <mouse, genus> Species 0.000 description 17
- 210000005058 airway cell Anatomy 0.000 description 17
- 230000001939 inductive effect Effects 0.000 description 17
- 102000005962 receptors Human genes 0.000 description 17
- 102100023321 Ceruloplasmin Human genes 0.000 description 16
- 125000003342 alkenyl group Chemical group 0.000 description 16
- 125000000304 alkynyl group Chemical group 0.000 description 16
- 239000000543 intermediate Substances 0.000 description 16
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 15
- 229910052736 halogen Inorganic materials 0.000 description 15
- 150000002367 halogens Chemical class 0.000 description 15
- 102000004196 processed proteins & peptides Human genes 0.000 description 15
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 15
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 14
- 108090000708 Proteasome Endopeptidase Complex Proteins 0.000 description 14
- 102000004245 Proteasome Endopeptidase Complex Human genes 0.000 description 14
- 238000002105 Southern blotting Methods 0.000 description 14
- 239000004480 active ingredient Substances 0.000 description 14
- 238000009472 formulation Methods 0.000 description 14
- 239000000463 material Substances 0.000 description 14
- 230000037361 pathway Effects 0.000 description 14
- 238000006243 chemical reaction Methods 0.000 description 13
- 238000011534 incubation Methods 0.000 description 13
- 239000003112 inhibitor Substances 0.000 description 13
- 230000003834 intracellular effect Effects 0.000 description 13
- 239000011159 matrix material Substances 0.000 description 13
- 210000002345 respiratory system Anatomy 0.000 description 13
- 238000002474 experimental method Methods 0.000 description 12
- 230000007774 longterm Effects 0.000 description 12
- 229920001184 polypeptide Polymers 0.000 description 12
- 230000008569 process Effects 0.000 description 12
- 238000009790 rate-determining step (RDS) Methods 0.000 description 12
- 230000001105 regulatory effect Effects 0.000 description 12
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 11
- 241001465754 Metazoa Species 0.000 description 11
- 238000003556 assay Methods 0.000 description 11
- 239000002552 dosage form Substances 0.000 description 11
- 210000001163 endosome Anatomy 0.000 description 11
- 210000004185 liver Anatomy 0.000 description 11
- 230000004807 localization Effects 0.000 description 11
- 230000007246 mechanism Effects 0.000 description 11
- 239000000843 powder Substances 0.000 description 11
- 238000010798 ubiquitination Methods 0.000 description 11
- 201000003883 Cystic fibrosis Diseases 0.000 description 10
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 10
- 241000699670 Mus sp. Species 0.000 description 10
- 208000036142 Viral infection Diseases 0.000 description 10
- 230000001225 therapeutic effect Effects 0.000 description 10
- 238000001890 transfection Methods 0.000 description 10
- 238000013519 translation Methods 0.000 description 10
- 230000034512 ubiquitination Effects 0.000 description 10
- 230000009385 viral infection Effects 0.000 description 10
- 108020005202 Viral DNA Proteins 0.000 description 9
- 210000001552 airway epithelial cell Anatomy 0.000 description 9
- 210000000234 capsid Anatomy 0.000 description 9
- 230000015556 catabolic process Effects 0.000 description 9
- 238000006731 degradation reaction Methods 0.000 description 9
- 239000000839 emulsion Substances 0.000 description 9
- 239000003623 enhancer Substances 0.000 description 9
- 238000009396 hybridization Methods 0.000 description 9
- 239000000178 monomer Substances 0.000 description 9
- 230000002688 persistence Effects 0.000 description 9
- 238000010186 staining Methods 0.000 description 9
- 239000013603 viral vector Substances 0.000 description 9
- 102000053602 DNA Human genes 0.000 description 8
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 8
- 102000008055 Heparan Sulfate Proteoglycans Human genes 0.000 description 8
- 229920002971 Heparan sulfate Polymers 0.000 description 8
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 8
- 108090000054 Syndecan-2 Proteins 0.000 description 8
- 239000011324 bead Substances 0.000 description 8
- KQNZDYYTLMIZCT-KQPMLPITSA-N brefeldin A Chemical compound O[C@@H]1\C=C\C(=O)O[C@@H](C)CCC\C=C\[C@@H]2C[C@H](O)C[C@H]21 KQNZDYYTLMIZCT-KQPMLPITSA-N 0.000 description 8
- 230000001413 cellular effect Effects 0.000 description 8
- 210000000349 chromosome Anatomy 0.000 description 8
- 229920001577 copolymer Polymers 0.000 description 8
- 125000000753 cycloalkyl group Chemical group 0.000 description 8
- 239000002852 cysteine proteinase inhibitor Substances 0.000 description 8
- 238000012217 deletion Methods 0.000 description 8
- 230000037430 deletion Effects 0.000 description 8
- 210000002919 epithelial cell Anatomy 0.000 description 8
- 229960004452 methionine Drugs 0.000 description 8
- 230000004048 modification Effects 0.000 description 8
- 238000012986 modification Methods 0.000 description 8
- 239000000523 sample Substances 0.000 description 8
- 230000032258 transport Effects 0.000 description 8
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 7
- 102000012605 Cystic Fibrosis Transmembrane Conductance Regulator Human genes 0.000 description 7
- 108010079245 Cystic Fibrosis Transmembrane Conductance Regulator Proteins 0.000 description 7
- 239000002998 adhesive polymer Substances 0.000 description 7
- 210000000424 bronchial epithelial cell Anatomy 0.000 description 7
- 230000000295 complement effect Effects 0.000 description 7
- 230000007423 decrease Effects 0.000 description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 7
- 239000006185 dispersion Substances 0.000 description 7
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 7
- 230000002068 genetic effect Effects 0.000 description 7
- 238000000338 in vitro Methods 0.000 description 7
- 125000003729 nucleotide group Chemical group 0.000 description 7
- 101150066583 rep gene Proteins 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 239000003981 vehicle Substances 0.000 description 7
- 210000002845 virion Anatomy 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 6
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 6
- 241000701022 Cytomegalovirus Species 0.000 description 6
- 241000829100 Macaca mulatta polyomavirus 1 Species 0.000 description 6
- 108091028043 Nucleic acid sequence Proteins 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 102000016611 Proteoglycans Human genes 0.000 description 6
- 108010067787 Proteoglycans Proteins 0.000 description 6
- 230000009471 action Effects 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 229920002678 cellulose Polymers 0.000 description 6
- 238000000576 coating method Methods 0.000 description 6
- 201000010099 disease Diseases 0.000 description 6
- 210000003527 eukaryotic cell Anatomy 0.000 description 6
- 239000012091 fetal bovine serum Substances 0.000 description 6
- 238000000799 fluorescence microscopy Methods 0.000 description 6
- 238000001476 gene delivery Methods 0.000 description 6
- 238000010166 immunofluorescence Methods 0.000 description 6
- 238000011065 in-situ storage Methods 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 230000010354 integration Effects 0.000 description 6
- 238000002372 labelling Methods 0.000 description 6
- 230000000670 limiting effect Effects 0.000 description 6
- 230000002132 lysosomal effect Effects 0.000 description 6
- 239000008194 pharmaceutical composition Substances 0.000 description 6
- 229920001223 polyethylene glycol Polymers 0.000 description 6
- 210000001533 respiratory mucosa Anatomy 0.000 description 6
- 210000003491 skin Anatomy 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 230000002103 transcriptional effect Effects 0.000 description 6
- 241001529453 unidentified herpesvirus Species 0.000 description 6
- 241000271566 Aves Species 0.000 description 5
- 241000701931 Canine parvovirus Species 0.000 description 5
- 108091006146 Channels Proteins 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 108010010803 Gelatin Proteins 0.000 description 5
- VSNHCAURESNICA-UHFFFAOYSA-N Hydroxyurea Chemical compound NC(=O)NO VSNHCAURESNICA-UHFFFAOYSA-N 0.000 description 5
- 241000124008 Mammalia Species 0.000 description 5
- 229930040373 Paraformaldehyde Natural products 0.000 description 5
- 241000700584 Simplexvirus Species 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- 229930006000 Sucrose Natural products 0.000 description 5
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 5
- 102000004338 Transferrin Human genes 0.000 description 5
- 108090000901 Transferrin Proteins 0.000 description 5
- 102000004142 Trypsin Human genes 0.000 description 5
- 108090000631 Trypsin Proteins 0.000 description 5
- GBOGMAARMMDZGR-UHFFFAOYSA-N UNPD149280 Natural products N1C(=O)C23OC(=O)C=CC(O)CCCC(C)CC=CC3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 GBOGMAARMMDZGR-UHFFFAOYSA-N 0.000 description 5
- 239000000443 aerosol Substances 0.000 description 5
- 150000001299 aldehydes Chemical class 0.000 description 5
- 230000000692 anti-sense effect Effects 0.000 description 5
- 230000001580 bacterial effect Effects 0.000 description 5
- 108010005774 beta-Galactosidase Proteins 0.000 description 5
- AIYUHDOJVYHVIT-UHFFFAOYSA-M caesium chloride Chemical compound [Cl-].[Cs+] AIYUHDOJVYHVIT-UHFFFAOYSA-M 0.000 description 5
- 239000000969 carrier Substances 0.000 description 5
- 235000010980 cellulose Nutrition 0.000 description 5
- 239000001913 cellulose Substances 0.000 description 5
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Natural products ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 5
- 125000004122 cyclic group Chemical group 0.000 description 5
- GBOGMAARMMDZGR-JREHFAHYSA-N cytochalasin B Natural products C[C@H]1CCC[C@@H](O)C=CC(=O)O[C@@]23[C@H](C=CC1)[C@H](O)C(=C)[C@@H](C)[C@@H]2[C@H](Cc4ccccc4)NC3=O GBOGMAARMMDZGR-JREHFAHYSA-N 0.000 description 5
- GBOGMAARMMDZGR-TYHYBEHESA-N cytochalasin B Chemical compound C([C@H]1[C@@H]2[C@@H](C([C@@H](O)[C@@H]3/C=C/C[C@H](C)CCC[C@@H](O)/C=C/C(=O)O[C@@]23C(=O)N1)=C)C)C1=CC=CC=C1 GBOGMAARMMDZGR-TYHYBEHESA-N 0.000 description 5
- 230000001419 dependent effect Effects 0.000 description 5
- 238000001514 detection method Methods 0.000 description 5
- 239000003085 diluting agent Substances 0.000 description 5
- 238000009826 distribution Methods 0.000 description 5
- 229940088598 enzyme Drugs 0.000 description 5
- 238000011156 evaluation Methods 0.000 description 5
- 239000012634 fragment Substances 0.000 description 5
- 239000008273 gelatin Substances 0.000 description 5
- 229920000159 gelatin Polymers 0.000 description 5
- 235000019322 gelatine Nutrition 0.000 description 5
- 235000011852 gelatine desserts Nutrition 0.000 description 5
- 238000012239 gene modification Methods 0.000 description 5
- 230000005017 genetic modification Effects 0.000 description 5
- 235000013617 genetically modified food Nutrition 0.000 description 5
- 229960001330 hydroxycarbamide Drugs 0.000 description 5
- 238000010348 incorporation Methods 0.000 description 5
- 230000002458 infectious effect Effects 0.000 description 5
- 238000001802 infusion Methods 0.000 description 5
- 239000004615 ingredient Substances 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- 238000012423 maintenance Methods 0.000 description 5
- 229930182817 methionine Natural products 0.000 description 5
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 5
- 239000002773 nucleotide Substances 0.000 description 5
- 229920002866 paraformaldehyde Polymers 0.000 description 5
- 230000006798 recombination Effects 0.000 description 5
- 238000005215 recombination Methods 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- 210000002966 serum Anatomy 0.000 description 5
- 239000002904 solvent Substances 0.000 description 5
- 239000005720 sucrose Substances 0.000 description 5
- 238000011200 topical administration Methods 0.000 description 5
- 230000001988 toxicity Effects 0.000 description 5
- 231100000419 toxicity Toxicity 0.000 description 5
- 230000037317 transdermal delivery Effects 0.000 description 5
- 239000012581 transferrin Substances 0.000 description 5
- 239000012588 trypsin Substances 0.000 description 5
- WHTVZRBIWZFKQO-AWEZNQCLSA-N (S)-chloroquine Chemical compound ClC1=CC=C2C(N[C@@H](C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-AWEZNQCLSA-N 0.000 description 4
- 102000007469 Actins Human genes 0.000 description 4
- 108010085238 Actins Proteins 0.000 description 4
- 102100026189 Beta-galactosidase Human genes 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- 102000029749 Microtubule Human genes 0.000 description 4
- 108091022875 Microtubule Proteins 0.000 description 4
- 206010028980 Neoplasm Diseases 0.000 description 4
- 239000002202 Polyethylene glycol Substances 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 4
- 239000003242 anti bacterial agent Substances 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 230000003115 biocidal effect Effects 0.000 description 4
- 230000033228 biological regulation Effects 0.000 description 4
- JUMGSHROWPPKFX-UHFFFAOYSA-N brefeldin-A Natural products CC1CCCC=CC2(C)CC(O)CC2(C)C(O)C=CC(=O)O1 JUMGSHROWPPKFX-UHFFFAOYSA-N 0.000 description 4
- 210000003123 bronchiole Anatomy 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 239000001506 calcium phosphate Substances 0.000 description 4
- 229910000389 calcium phosphate Inorganic materials 0.000 description 4
- 235000011010 calcium phosphates Nutrition 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 229960003677 chloroquine Drugs 0.000 description 4
- 230000001886 ciliary effect Effects 0.000 description 4
- 238000011260 co-administration Methods 0.000 description 4
- 239000011248 coating agent Substances 0.000 description 4
- 239000002270 dispersing agent Substances 0.000 description 4
- 238000004520 electroporation Methods 0.000 description 4
- 229960005420 etoposide Drugs 0.000 description 4
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 4
- 239000011888 foil Substances 0.000 description 4
- 230000004927 fusion Effects 0.000 description 4
- 229940045109 genistein Drugs 0.000 description 4
- TZBJGXHYKVUXJN-UHFFFAOYSA-N genistein Natural products C1=CC(O)=CC=C1C1=COC2=CC(O)=CC(O)=C2C1=O TZBJGXHYKVUXJN-UHFFFAOYSA-N 0.000 description 4
- 235000006539 genistein Nutrition 0.000 description 4
- ZCOLJUOHXJRHDI-CMWLGVBASA-N genistein 7-O-beta-D-glucoside Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=C2C(=O)C(C=3C=CC(O)=CC=3)=COC2=C1 ZCOLJUOHXJRHDI-CMWLGVBASA-N 0.000 description 4
- 235000011187 glycerol Nutrition 0.000 description 4
- 239000001963 growth medium Substances 0.000 description 4
- 210000005260 human cell Anatomy 0.000 description 4
- 238000003780 insertion Methods 0.000 description 4
- 230000037431 insertion Effects 0.000 description 4
- 238000007918 intramuscular administration Methods 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 239000003550 marker Substances 0.000 description 4
- 238000001000 micrograph Methods 0.000 description 4
- 210000004688 microtubule Anatomy 0.000 description 4
- 210000003205 muscle Anatomy 0.000 description 4
- 210000000056 organ Anatomy 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- 239000000546 pharmaceutical excipient Substances 0.000 description 4
- 229920000058 polyacrylate Polymers 0.000 description 4
- 230000008488 polyadenylation Effects 0.000 description 4
- 239000003755 preservative agent Substances 0.000 description 4
- 230000002265 prevention Effects 0.000 description 4
- 230000001566 pro-viral effect Effects 0.000 description 4
- 238000011002 quantification Methods 0.000 description 4
- 230000002285 radioactive effect Effects 0.000 description 4
- 230000000717 retained effect Effects 0.000 description 4
- 230000002441 reversible effect Effects 0.000 description 4
- 239000007921 spray Substances 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 239000008107 starch Substances 0.000 description 4
- 229910052717 sulfur Inorganic materials 0.000 description 4
- 239000004094 surface-active agent Substances 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- 230000008685 targeting Effects 0.000 description 4
- 230000001052 transient effect Effects 0.000 description 4
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 4
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 3
- OPIFSICVWOWJMJ-AEOCFKNESA-N 5-bromo-4-chloro-3-indolyl beta-D-galactoside Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1OC1=CNC2=CC=C(Br)C(Cl)=C12 OPIFSICVWOWJMJ-AEOCFKNESA-N 0.000 description 3
- 244000215068 Acacia senegal Species 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- 241000702423 Adeno-associated virus - 2 Species 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- 108020004638 Circular DNA Proteins 0.000 description 3
- 101000749287 Clitocybe nebularis Clitocypin Proteins 0.000 description 3
- 101000767029 Clitocybe nebularis Clitocypin-1 Proteins 0.000 description 3
- 102000008186 Collagen Human genes 0.000 description 3
- 108010035532 Collagen Proteins 0.000 description 3
- 229940094664 Cysteine protease inhibitor Drugs 0.000 description 3
- 102000004127 Cytokines Human genes 0.000 description 3
- 108090000695 Cytokines Proteins 0.000 description 3
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 3
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 3
- 108010053770 Deoxyribonucleases Proteins 0.000 description 3
- 102000016911 Deoxyribonucleases Human genes 0.000 description 3
- 108010016626 Dipeptides Proteins 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- 229920000084 Gum arabic Polymers 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 238000000636 Northern blotting Methods 0.000 description 3
- 108700026244 Open Reading Frames Proteins 0.000 description 3
- 108700008625 Reporter Genes Proteins 0.000 description 3
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 3
- 108020004440 Thymidine kinase Proteins 0.000 description 3
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 3
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 3
- 229940127507 Ubiquitin Ligase Inhibitors Drugs 0.000 description 3
- 206010046865 Vaccinia virus infection Diseases 0.000 description 3
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 3
- 235000010489 acacia gum Nutrition 0.000 description 3
- 239000000205 acacia gum Substances 0.000 description 3
- 108700019030 adenovirus E4orf6 Proteins 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 210000000270 basal cell Anatomy 0.000 description 3
- 230000001588 bifunctional effect Effects 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 239000007894 caplet Substances 0.000 description 3
- 239000013592 cell lysate Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 229920001436 collagen Polymers 0.000 description 3
- 238000011109 contamination Methods 0.000 description 3
- 239000006071 cream Substances 0.000 description 3
- 210000000805 cytoplasm Anatomy 0.000 description 3
- 230000001086 cytosolic effect Effects 0.000 description 3
- 230000007547 defect Effects 0.000 description 3
- 238000000502 dialysis Methods 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 108010048367 enhanced green fluorescent protein Proteins 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 238000003306 harvesting Methods 0.000 description 3
- 229920000669 heparin Polymers 0.000 description 3
- 229960002897 heparin Drugs 0.000 description 3
- 108010038082 heparin proteoglycan Proteins 0.000 description 3
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 3
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 3
- 239000000411 inducer Substances 0.000 description 3
- 230000009545 invasion Effects 0.000 description 3
- 239000002502 liposome Substances 0.000 description 3
- 230000033001 locomotion Effects 0.000 description 3
- 239000006210 lotion Substances 0.000 description 3
- 108020004999 messenger RNA Proteins 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- 244000005700 microbiome Species 0.000 description 3
- 210000003632 microfilament Anatomy 0.000 description 3
- 238000007479 molecular analysis Methods 0.000 description 3
- 238000007491 morphometric analysis Methods 0.000 description 3
- VOFUROIFQGPCGE-UHFFFAOYSA-N nile red Chemical compound C1=CC=C2C3=NC4=CC=C(N(CC)CC)C=C4OC3=CC(=O)C2=C1 VOFUROIFQGPCGE-UHFFFAOYSA-N 0.000 description 3
- 239000002674 ointment Substances 0.000 description 3
- 230000001575 pathological effect Effects 0.000 description 3
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 3
- 229920006254 polymer film Polymers 0.000 description 3
- 229920005862 polyol Polymers 0.000 description 3
- 150000003077 polyols Chemical class 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 3
- 230000001681 protective effect Effects 0.000 description 3
- 238000000159 protein binding assay Methods 0.000 description 3
- 230000001177 retroviral effect Effects 0.000 description 3
- 210000002027 skeletal muscle Anatomy 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 239000000375 suspending agent Substances 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 235000012222 talc Nutrition 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- 239000002562 thickening agent Substances 0.000 description 3
- 210000003437 trachea Anatomy 0.000 description 3
- 230000002463 transducing effect Effects 0.000 description 3
- 230000006663 ubiquitin-proteasome pathway Effects 0.000 description 3
- 238000005199 ultracentrifugation Methods 0.000 description 3
- 208000007089 vaccinia Diseases 0.000 description 3
- 235000015112 vegetable and seed oil Nutrition 0.000 description 3
- 239000008158 vegetable oil Substances 0.000 description 3
- 229960003048 vinblastine Drugs 0.000 description 3
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 3
- 238000001262 western blot Methods 0.000 description 3
- 239000000080 wetting agent Substances 0.000 description 3
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 2
- IPJDHSYCSQAODE-UHFFFAOYSA-N 5-chloromethylfluorescein diacetate Chemical compound O1C(=O)C2=CC(CCl)=CC=C2C21C1=CC=C(OC(C)=O)C=C1OC1=CC(OC(=O)C)=CC=C21 IPJDHSYCSQAODE-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- 229920000936 Agarose Polymers 0.000 description 2
- 208000035143 Bacterial infection Diseases 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 2
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 101150044789 Cap gene Proteins 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 108090000994 Catalytic RNA Proteins 0.000 description 2
- 102000053642 Catalytic RNA Human genes 0.000 description 2
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 2
- 208000003322 Coinfection Diseases 0.000 description 2
- 206010053138 Congenital aplastic anaemia Diseases 0.000 description 2
- 241000938605 Crocodylia Species 0.000 description 2
- 102000005927 Cysteine Proteases Human genes 0.000 description 2
- 108010005843 Cysteine Proteases Proteins 0.000 description 2
- 102000000311 Cytosine Deaminase Human genes 0.000 description 2
- 108010080611 Cytosine Deaminase Proteins 0.000 description 2
- 239000012623 DNA damaging agent Substances 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 241000283086 Equidae Species 0.000 description 2
- 201000004939 Fanconi anemia Diseases 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 2
- 241000287828 Gallus gallus Species 0.000 description 2
- 108700007698 Genetic Terminator Regions Proteins 0.000 description 2
- 108010051696 Growth Hormone Proteins 0.000 description 2
- 241000700588 Human alphaherpesvirus 1 Species 0.000 description 2
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 2
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 2
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 description 2
- 208000026350 Inborn Genetic disease Diseases 0.000 description 2
- FFOPEPMHKILNIT-UHFFFAOYSA-N Isopropyl butyrate Chemical compound CCCC(=O)OC(C)C FFOPEPMHKILNIT-UHFFFAOYSA-N 0.000 description 2
- FFEARJCKVFRZRR-UHFFFAOYSA-N L-Methionine Natural products CSCCC(N)C(O)=O FFEARJCKVFRZRR-UHFFFAOYSA-N 0.000 description 2
- 229930195722 L-methionine Natural products 0.000 description 2
- 108010007622 LDL Lipoproteins Proteins 0.000 description 2
- 102000007330 LDL Lipoproteins Human genes 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 108090000157 Metallothionein Proteins 0.000 description 2
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 2
- 241000713333 Mouse mammary tumor virus Species 0.000 description 2
- 206010065764 Mucosal infection Diseases 0.000 description 2
- QIAFMBKCNZACKA-UHFFFAOYSA-N N-benzoylglycine Chemical compound OC(=O)CNC(=O)C1=CC=CC=C1 QIAFMBKCNZACKA-UHFFFAOYSA-N 0.000 description 2
- KYRVNWMVYQXFEU-UHFFFAOYSA-N Nocodazole Chemical compound C1=C2NC(NC(=O)OC)=NC2=CC=C1C(=O)C1=CC=CS1 KYRVNWMVYQXFEU-UHFFFAOYSA-N 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 208000030852 Parasitic disease Diseases 0.000 description 2
- 208000018737 Parkinson disease Diseases 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- 241000286209 Phasianidae Species 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 239000004743 Polypropylene Substances 0.000 description 2
- 241000125945 Protoparvovirus Species 0.000 description 2
- 239000012083 RIPA buffer Substances 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 208000035415 Reinfection Diseases 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- BQCADISMDOOEFD-UHFFFAOYSA-N Silver Chemical compound [Ag] BQCADISMDOOEFD-UHFFFAOYSA-N 0.000 description 2
- 108020004682 Single-Stranded DNA Proteins 0.000 description 2
- 102100038803 Somatotropin Human genes 0.000 description 2
- 101710172711 Structural protein Proteins 0.000 description 2
- 241000701093 Suid alphaherpesvirus 1 Species 0.000 description 2
- 241000282887 Suidae Species 0.000 description 2
- 101710137500 T7 RNA polymerase Proteins 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- 108700005077 Viral Genes Proteins 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000011543 agarose gel Substances 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- NOFOAYPPHIUXJR-APNQCZIXSA-N aphidicolin Chemical compound C1[C@@]23[C@@]4(C)CC[C@@H](O)[C@@](C)(CO)[C@@H]4CC[C@H]3C[C@H]1[C@](CO)(O)CC2 NOFOAYPPHIUXJR-APNQCZIXSA-N 0.000 description 2
- SEKZNWAQALMJNH-YZUCACDQSA-N aphidicolin Natural products C[C@]1(CO)CC[C@]23C[C@H]1C[C@@H]2CC[C@H]4[C@](C)(CO)[C@H](O)CC[C@]34C SEKZNWAQALMJNH-YZUCACDQSA-N 0.000 description 2
- 208000022362 bacterial infectious disease Diseases 0.000 description 2
- 210000002469 basement membrane Anatomy 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 238000009395 breeding Methods 0.000 description 2
- 230000001488 breeding effect Effects 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 229940127093 camptothecin Drugs 0.000 description 2
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- 235000013330 chicken meat Nutrition 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 210000000254 ciliated cell Anatomy 0.000 description 2
- 238000010367 cloning Methods 0.000 description 2
- 238000012761 co-transfection Methods 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 230000002860 competitive effect Effects 0.000 description 2
- 238000012937 correction Methods 0.000 description 2
- 238000004132 cross linking Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 2
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 230000006862 enzymatic digestion Effects 0.000 description 2
- 239000013604 expression vector Substances 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 230000009395 genetic defect Effects 0.000 description 2
- 208000016361 genetic disease Diseases 0.000 description 2
- 238000009650 gentamicin protection assay Methods 0.000 description 2
- 239000000122 growth hormone Substances 0.000 description 2
- 229910001385 heavy metal Inorganic materials 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- 230000001744 histochemical effect Effects 0.000 description 2
- 230000006801 homologous recombination Effects 0.000 description 2
- 238000002744 homologous recombination Methods 0.000 description 2
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 2
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 2
- 238000012760 immunocytochemical staining Methods 0.000 description 2
- 238000001114 immunoprecipitation Methods 0.000 description 2
- 238000012744 immunostaining Methods 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 230000006698 induction Effects 0.000 description 2
- 229940060367 inert ingredients Drugs 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- 239000007972 injectable composition Substances 0.000 description 2
- 239000002054 inoculum Substances 0.000 description 2
- 102000006495 integrins Human genes 0.000 description 2
- 108010044426 integrins Proteins 0.000 description 2
- 239000007927 intramuscular injection Substances 0.000 description 2
- 238000010255 intramuscular injection Methods 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 2
- 101150066555 lacZ gene Proteins 0.000 description 2
- 229960003136 leucine Drugs 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 238000001638 lipofection Methods 0.000 description 2
- 244000144972 livestock Species 0.000 description 2
- 210000003712 lysosome Anatomy 0.000 description 2
- 230000001868 lysosomic effect Effects 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 102000006240 membrane receptors Human genes 0.000 description 2
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 2
- 239000004530 micro-emulsion Substances 0.000 description 2
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 2
- 239000008108 microcrystalline cellulose Substances 0.000 description 2
- 229940016286 microcrystalline cellulose Drugs 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 230000002438 mitochondrial effect Effects 0.000 description 2
- 230000003562 morphometric effect Effects 0.000 description 2
- 238000013425 morphometry Methods 0.000 description 2
- 210000004165 myocardium Anatomy 0.000 description 2
- 239000007923 nasal drop Substances 0.000 description 2
- 229940100662 nasal drops Drugs 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 229950006344 nocodazole Drugs 0.000 description 2
- 230000025308 nuclear transport Effects 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 238000011275 oncology therapy Methods 0.000 description 2
- 238000005457 optimization Methods 0.000 description 2
- 230000008520 organization Effects 0.000 description 2
- 239000012188 paraffin wax Substances 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000007170 pathology Effects 0.000 description 2
- 230000002085 persistent effect Effects 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 229920000515 polycarbonate Polymers 0.000 description 2
- 239000004417 polycarbonate Substances 0.000 description 2
- 239000005020 polyethylene terephthalate Substances 0.000 description 2
- 229920000193 polymethacrylate Polymers 0.000 description 2
- 229920001155 polypropylene Polymers 0.000 description 2
- 229920001296 polysiloxane Polymers 0.000 description 2
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 2
- 239000004810 polytetrafluoroethylene Substances 0.000 description 2
- 239000004814 polyurethane Substances 0.000 description 2
- 229920002635 polyurethane Polymers 0.000 description 2
- 229920000915 polyvinyl chloride Polymers 0.000 description 2
- 239000004800 polyvinyl chloride Substances 0.000 description 2
- 210000003240 portal vein Anatomy 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 239000003380 propellant Substances 0.000 description 2
- 230000026938 proteasomal ubiquitin-dependent protein catabolic process Effects 0.000 description 2
- 230000017854 proteolysis Effects 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000003757 reverse transcription PCR Methods 0.000 description 2
- 108091092562 ribozyme Proteins 0.000 description 2
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical compound C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 208000007056 sickle cell anemia Diseases 0.000 description 2
- 229910052709 silver Inorganic materials 0.000 description 2
- 239000004332 silver Substances 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 230000008093 supporting effect Effects 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- 238000013268 sustained release Methods 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 230000002195 synergetic effect Effects 0.000 description 2
- 238000007910 systemic administration Methods 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 235000010215 titanium dioxide Nutrition 0.000 description 2
- 239000004408 titanium dioxide Substances 0.000 description 2
- 238000004448 titration Methods 0.000 description 2
- 239000012049 topical pharmaceutical composition Substances 0.000 description 2
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 241001430294 unidentified retrovirus Species 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 230000028973 vesicle-mediated transport Effects 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 2
- DIGQNXIGRZPYDK-WKSCXVIASA-N (2R)-6-amino-2-[[2-[[(2S)-2-[[2-[[(2R)-2-[[(2S)-2-[[(2R,3S)-2-[[2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2S,3S)-2-[[(2R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2R)-2-[[2-[[2-[[2-[(2-amino-1-hydroxyethylidene)amino]-3-carboxy-1-hydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxypropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1,5-dihydroxy-5-iminopentylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxybutylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1,3-dihydroxypropylidene]amino]-1-hydroxyethylidene]amino]-1-hydroxy-3-sulfanylpropylidene]amino]-1-hydroxyethylidene]amino]hexanoic acid Chemical compound C[C@@H]([C@@H](C(=N[C@@H](CS)C(=N[C@@H](C)C(=N[C@@H](CO)C(=NCC(=N[C@@H](CCC(=N)O)C(=NC(CS)C(=N[C@H]([C@H](C)O)C(=N[C@H](CS)C(=N[C@H](CO)C(=NCC(=N[C@H](CS)C(=NCC(=N[C@H](CCCCN)C(=O)O)O)O)O)O)O)O)O)O)O)O)O)O)O)N=C([C@H](CS)N=C([C@H](CO)N=C([C@H](CO)N=C([C@H](C)N=C(CN=C([C@H](CO)N=C([C@H](CS)N=C(CN=C(C(CS)N=C(C(CC(=O)O)N=C(CN)O)O)O)O)O)O)O)O)O)O)O)O DIGQNXIGRZPYDK-WKSCXVIASA-N 0.000 description 1
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- NPDBDJFLKKQMCM-SCSAIBSYSA-N (2s)-2-amino-3,3-dimethylbutanoic acid Chemical group CC(C)(C)[C@H](N)C(O)=O NPDBDJFLKKQMCM-SCSAIBSYSA-N 0.000 description 1
- CQYBNXGHMBNGCG-FXQIFTODSA-N (2s,3as,7as)-2,3,3a,4,5,6,7,7a-octahydro-1h-indol-1-ium-2-carboxylate Chemical compound C1CCC[C@@H]2[NH2+][C@H](C(=O)[O-])C[C@@H]21 CQYBNXGHMBNGCG-FXQIFTODSA-N 0.000 description 1
- LJRDOKAZOAKLDU-UDXJMMFXSA-N (2s,3s,4r,5r,6r)-5-amino-2-(aminomethyl)-6-[(2r,3s,4r,5s)-5-[(1r,2r,3s,5r,6s)-3,5-diamino-2-[(2s,3r,4r,5s,6r)-3-amino-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-hydroxycyclohexyl]oxy-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl]oxyoxane-3,4-diol;sulfuric ac Chemical compound OS(O)(=O)=O.N[C@@H]1[C@@H](O)[C@H](O)[C@H](CN)O[C@@H]1O[C@H]1[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](N)C[C@@H](N)[C@@H]2O)O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)N)O[C@@H]1CO LJRDOKAZOAKLDU-UDXJMMFXSA-N 0.000 description 1
- BWKMGYQJPOAASG-UHFFFAOYSA-N 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid Chemical compound C1=CC=C2CNC(C(=O)O)CC2=C1 BWKMGYQJPOAASG-UHFFFAOYSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- HNAGHMKIPMKKBB-UHFFFAOYSA-N 1-benzylpyrrolidine-3-carboxamide Chemical compound C1C(C(=O)N)CCN1CC1=CC=CC=C1 HNAGHMKIPMKKBB-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- FUOOLUPWFVMBKG-UHFFFAOYSA-N 2-Aminoisobutyric acid Chemical compound CC(C)(N)C(O)=O FUOOLUPWFVMBKG-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- SENMPMXZMGNQAG-UHFFFAOYSA-N 3,4-dihydro-2,5-benzodioxocine-1,6-dione Chemical compound O=C1OCCOC(=O)C2=CC=CC=C12 SENMPMXZMGNQAG-UHFFFAOYSA-N 0.000 description 1
- BXRLWGXPSRYJDZ-UHFFFAOYSA-N 3-cyanoalanine Chemical compound OC(=O)C(N)CC#N BXRLWGXPSRYJDZ-UHFFFAOYSA-N 0.000 description 1
- XOJWAAUYNWGQAU-UHFFFAOYSA-N 4-(2-methylprop-2-enoyloxy)butyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCCCCOC(=O)C(C)=C XOJWAAUYNWGQAU-UHFFFAOYSA-N 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- JHWGFJBTMHEZME-UHFFFAOYSA-N 4-prop-2-enoyloxybutyl prop-2-enoate Chemical compound C=CC(=O)OCCCCOC(=O)C=C JHWGFJBTMHEZME-UHFFFAOYSA-N 0.000 description 1
- YSMODUONRAFBET-UHFFFAOYSA-N 5-hydroxylysine Chemical group NCC(O)CCC(N)C(O)=O YSMODUONRAFBET-UHFFFAOYSA-N 0.000 description 1
- GJCOSYZMQJWQCA-UHFFFAOYSA-N 9H-xanthene Chemical compound C1=CC=C2CC3=CC=CC=C3OC2=C1 GJCOSYZMQJWQCA-UHFFFAOYSA-N 0.000 description 1
- 108010022579 ATP dependent 26S protease Proteins 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 102100029457 Adenine phosphoribosyltransferase Human genes 0.000 description 1
- 108010024223 Adenine phosphoribosyltransferase Proteins 0.000 description 1
- 241000701242 Adenoviridae Species 0.000 description 1
- 108010024878 Adenovirus E1A Proteins Proteins 0.000 description 1
- 108010057856 Adenovirus E2 Proteins Proteins 0.000 description 1
- 108700026758 Adenovirus hexon capsid Proteins 0.000 description 1
- 102100027211 Albumin Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 108010087765 Antipain Proteins 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- 108010087504 Beta-Globulins Proteins 0.000 description 1
- 208000019838 Blood disease Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 239000004255 Butylated hydroxyanisole Substances 0.000 description 1
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- AFWTZXXDGQBIKW-UHFFFAOYSA-N C14 surfactin Natural products CCCCCCCCCCCC1CC(=O)NC(CCC(O)=O)C(=O)NC(CC(C)C)C(=O)NC(CC(C)C)C(=O)NC(C(C)C)C(=O)NC(CC(O)=O)C(=O)NC(CC(C)C)C(=O)NC(CC(C)C)C(=O)O1 AFWTZXXDGQBIKW-UHFFFAOYSA-N 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- 101710132601 Capsid protein Proteins 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 102000004225 Cathepsin B Human genes 0.000 description 1
- 108090000712 Cathepsin B Proteins 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 108010036867 Cerebroside-Sulfatase Proteins 0.000 description 1
- 239000004709 Chlorinated polyethylene Substances 0.000 description 1
- 108010077544 Chromatin Proteins 0.000 description 1
- 102000005853 Clathrin Human genes 0.000 description 1
- 108010019874 Clathrin Proteins 0.000 description 1
- 208000032170 Congenital Abnormalities Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 244000303965 Cyamopsis psoralioides Species 0.000 description 1
- 102000015833 Cystatin Human genes 0.000 description 1
- 108010061635 Cystatin B Proteins 0.000 description 1
- 108010061642 Cystatin C Proteins 0.000 description 1
- 102100026891 Cystatin-B Human genes 0.000 description 1
- 102100026897 Cystatin-C Human genes 0.000 description 1
- 238000007400 DNA extraction Methods 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 241000450599 DNA viruses Species 0.000 description 1
- 229920004934 Dacron® Polymers 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- SCMSYZJDIQPSDI-SRVKXCTJSA-N E-64c Chemical compound CC(C)CCNC(=O)[C@H](CC(C)C)NC(=O)[C@H]1O[C@@H]1C(O)=O SCMSYZJDIQPSDI-SRVKXCTJSA-N 0.000 description 1
- 101150029662 E1 gene Proteins 0.000 description 1
- 101150005585 E3 gene Proteins 0.000 description 1
- LTLYEAJONXGNFG-DCAQKATOSA-N E64 Chemical compound NC(=N)NCCCCNC(=O)[C@H](CC(C)C)NC(=O)[C@H]1O[C@@H]1C(O)=O LTLYEAJONXGNFG-DCAQKATOSA-N 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 229920000219 Ethylene vinyl alcohol Polymers 0.000 description 1
- 108010076282 Factor IX Proteins 0.000 description 1
- 108010054218 Factor VIII Proteins 0.000 description 1
- 102100023593 Fibroblast growth factor receptor 1 Human genes 0.000 description 1
- 101710182386 Fibroblast growth factor receptor 1 Proteins 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 208000015872 Gaucher disease Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 108010017544 Glucosylceramidase Proteins 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 208000037357 HIV infectious disease Diseases 0.000 description 1
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 1
- 208000031220 Hemophilia Diseases 0.000 description 1
- 208000009292 Hemophilia A Diseases 0.000 description 1
- 108010068250 Herpes Simplex Virus Protein Vmw65 Proteins 0.000 description 1
- 108091027305 Heteroduplex Proteins 0.000 description 1
- 241001135569 Human adenovirus 5 Species 0.000 description 1
- 241000598171 Human adenovirus sp. Species 0.000 description 1
- 241000701027 Human herpesvirus 6 Species 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 108010091358 Hypoxanthine Phosphoribosyltransferase Proteins 0.000 description 1
- 102100029098 Hypoxanthine-guanine phosphoribosyltransferase Human genes 0.000 description 1
- 206010062016 Immunosuppression Diseases 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 108091092195 Intron Proteins 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- ZGUNAGUHMKGQNY-ZETCQYMHSA-N L-alpha-phenylglycine zwitterion Chemical compound OC(=O)[C@@H](N)C1=CC=CC=C1 ZGUNAGUHMKGQNY-ZETCQYMHSA-N 0.000 description 1
- RHGKLRLOHDJJDR-BYPYZUCNSA-N L-citrulline Chemical compound NC(=O)NCCC[C@H]([NH3+])C([O-])=O RHGKLRLOHDJJDR-BYPYZUCNSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- 125000000174 L-prolyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])[C@@]1([H])C(*)=O 0.000 description 1
- DGYHPLMPMRKMPD-UHFFFAOYSA-N L-propargyl glycine Natural products OC(=O)C(N)CC#C DGYHPLMPMRKMPD-UHFFFAOYSA-N 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- 125000000510 L-tryptophano group Chemical group [H]C1=C([H])C([H])=C2N([H])C([H])=C(C([H])([H])[C@@]([H])(C(O[H])=O)N([H])[*])C2=C1[H] 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 101710192606 Latent membrane protein 2 Proteins 0.000 description 1
- 241000713666 Lentivirus Species 0.000 description 1
- LESXFEZIFXFIQR-LURJTMIESA-N Leu-Gly Chemical compound CC(C)C[C@H](N)C(=O)NCC(O)=O LESXFEZIFXFIQR-LURJTMIESA-N 0.000 description 1
- DNDWZFHLZVYOGF-KKUMJFAQSA-N Leu-Leu-Leu Chemical compound CC(C)C[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O DNDWZFHLZVYOGF-KKUMJFAQSA-N 0.000 description 1
- GDBQQVLCIARPGH-UHFFFAOYSA-N Leupeptin Natural products CC(C)CC(NC(C)=O)C(=O)NC(CC(C)C)C(=O)NC(C=O)CCCN=C(N)N GDBQQVLCIARPGH-UHFFFAOYSA-N 0.000 description 1
- 229940113306 Ligase inhibitor Drugs 0.000 description 1
- 208000019693 Lung disease Diseases 0.000 description 1
- 241000282560 Macaca mulatta Species 0.000 description 1
- 241000712079 Measles morbillivirus Species 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 102000003792 Metallothionein Human genes 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 208000021642 Muscular disease Diseases 0.000 description 1
- 102000003505 Myosin Human genes 0.000 description 1
- 108060008487 Myosin Proteins 0.000 description 1
- RHGKLRLOHDJJDR-UHFFFAOYSA-N Ndelta-carbamoyl-DL-ornithine Natural products OC(=O)C(N)CCCNC(N)=O RHGKLRLOHDJJDR-UHFFFAOYSA-N 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 108091092724 Noncoding DNA Proteins 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- BZQFBWGGLXLEPQ-UHFFFAOYSA-N O-phosphoryl-L-serine Natural products OC(=O)C(N)COP(O)(O)=O BZQFBWGGLXLEPQ-UHFFFAOYSA-N 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 229920002367 Polyisobutene Polymers 0.000 description 1
- 229920001328 Polyvinylidene chloride Polymers 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 206010036790 Productive cough Diseases 0.000 description 1
- 102000017143 RNA Polymerase I Human genes 0.000 description 1
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 241000220317 Rosa Species 0.000 description 1
- 230000018199 S phase Effects 0.000 description 1
- 108010077895 Sarcosine Proteins 0.000 description 1
- 241000710961 Semliki Forest virus Species 0.000 description 1
- 206010040880 Skin irritation Diseases 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 238000000692 Student's t-test Methods 0.000 description 1
- 101150003725 TK gene Proteins 0.000 description 1
- 101710109576 Terminal protein Proteins 0.000 description 1
- BGNXCDMCOKJUMV-UHFFFAOYSA-N Tert-Butylhydroquinone Chemical compound CC(C)(C)C1=CC(O)=CC=C1O BGNXCDMCOKJUMV-UHFFFAOYSA-N 0.000 description 1
- 208000002903 Thalassemia Diseases 0.000 description 1
- 102000006601 Thymidine Kinase Human genes 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 108700029229 Transcriptional Regulatory Elements Proteins 0.000 description 1
- OKKRPWIIYQTPQF-UHFFFAOYSA-N Trimethylolpropane trimethacrylate Chemical compound CC(=C)C(=O)OCC(CC)(COC(=O)C(C)=C)COC(=O)C(C)=C OKKRPWIIYQTPQF-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 108091000117 Tyrosine 3-Monooxygenase Proteins 0.000 description 1
- 108010005705 Ubiquitinated Proteins Proteins 0.000 description 1
- 241000700618 Vaccinia virus Species 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 241000711975 Vesicular stomatitis virus Species 0.000 description 1
- 229920002433 Vinyl chloride-vinyl acetate copolymer Polymers 0.000 description 1
- 108070000030 Viral receptors Proteins 0.000 description 1
- NGBKFLTYGSREKK-PMACEKPBSA-N Z-Val-Phe-H Chemical compound N([C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C=O)C(=O)OCC1=CC=CC=C1 NGBKFLTYGSREKK-PMACEKPBSA-N 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 108010091545 acetylleucyl-leucyl-norleucinal Proteins 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 108700010877 adenoviridae proteins Proteins 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 230000002009 allergenic effect Effects 0.000 description 1
- SRVFFFJZQVENJC-IHRRRGAJSA-N aloxistatin Chemical compound CCOC(=O)[C@H]1O[C@@H]1C(=O)N[C@@H](CC(C)C)C(=O)NCCC(C)C SRVFFFJZQVENJC-IHRRRGAJSA-N 0.000 description 1
- 229940087168 alpha tocopherol Drugs 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 239000010775 animal oil Substances 0.000 description 1
- 238000000137 annealing Methods 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000002668 anti-heparin effect Effects 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- SDNYTAYICBFYFH-TUFLPTIASA-N antipain Chemical compound NC(N)=NCCC[C@@H](C=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 SDNYTAYICBFYFH-TUFLPTIASA-N 0.000 description 1
- 101150067977 ap gene Proteins 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000012736 aqueous medium Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 239000002473 artificial blood Substances 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 230000003190 augmentative effect Effects 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- 238000000211 autoradiogram Methods 0.000 description 1
- 239000003855 balanced salt solution Substances 0.000 description 1
- 239000007640 basal medium Substances 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- 125000002527 bicyclic carbocyclic group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 238000010170 biological method Methods 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- ZPOLOEWJWXZUSP-WAYWQWQTSA-N bis(prop-2-enyl) (z)-but-2-enedioate Chemical compound C=CCOC(=O)\C=C/C(=O)OCC=C ZPOLOEWJWXZUSP-WAYWQWQTSA-N 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 229940124630 bronchodilator Drugs 0.000 description 1
- 239000000168 bronchodilator agent Substances 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 239000004067 bulking agent Substances 0.000 description 1
- OBNCKNCVKJNDBV-UHFFFAOYSA-N butanoic acid ethyl ester Natural products CCCC(=O)OCC OBNCKNCVKJNDBV-UHFFFAOYSA-N 0.000 description 1
- 229920005549 butyl rubber Polymers 0.000 description 1
- 235000019282 butylated hydroxyanisole Nutrition 0.000 description 1
- 229940043253 butylated hydroxyanisole Drugs 0.000 description 1
- CZBZUDVBLSSABA-UHFFFAOYSA-N butylated hydroxyanisole Chemical compound COC1=CC=C(O)C(C(C)(C)C)=C1.COC1=CC=C(O)C=C1C(C)(C)C CZBZUDVBLSSABA-UHFFFAOYSA-N 0.000 description 1
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 1
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 108010007877 calpain inhibitor III Proteins 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 239000012830 cancer therapeutic Substances 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 229960004424 carbon dioxide Drugs 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 230000006652 catabolic pathway Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 229940112822 chewing gum Drugs 0.000 description 1
- 235000015218 chewing gum Nutrition 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- KXKPYJOVDUMHGS-OSRGNVMNSA-N chondroitin sulfate Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](OS(O)(=O)=O)[C@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](C(O)=O)O1 KXKPYJOVDUMHGS-OSRGNVMNSA-N 0.000 description 1
- 210000003483 chromatin Anatomy 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 210000004081 cilia Anatomy 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 229960002173 citrulline Drugs 0.000 description 1
- 235000013477 citrulline Nutrition 0.000 description 1
- 229930193282 clathrin Natural products 0.000 description 1
- 210000002777 columnar cell Anatomy 0.000 description 1
- 230000002301 combined effect Effects 0.000 description 1
- 230000002995 comedolytic effect Effects 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 230000001143 conditioned effect Effects 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 238000004624 confocal microscopy Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 230000037029 cross reaction Effects 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 108050004038 cystatin Proteins 0.000 description 1
- 230000006743 cytoplasmic accumulation Effects 0.000 description 1
- 230000003436 cytoskeletal effect Effects 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 238000005034 decoration Methods 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 239000005547 deoxyribonucleotide Substances 0.000 description 1
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 229950006137 dexfosfoserine Drugs 0.000 description 1
- GUJOJGAPFQRJSV-UHFFFAOYSA-N dialuminum;dioxosilane;oxygen(2-);hydrate Chemical compound O.[O-2].[O-2].[O-2].[Al+3].[Al+3].O=[Si]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O GUJOJGAPFQRJSV-UHFFFAOYSA-N 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940042935 dichlorodifluoromethane Drugs 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- UGMCXQCYOVCMTB-UHFFFAOYSA-K dihydroxy(stearato)aluminium Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[Al](O)O UGMCXQCYOVCMTB-UHFFFAOYSA-K 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 239000004205 dimethyl polysiloxane Substances 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 1
- 230000003828 downregulation Effects 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 241001493065 dsRNA viruses Species 0.000 description 1
- 210000001198 duodenum Anatomy 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000007515 enzymatic degradation Effects 0.000 description 1
- 229920005558 epichlorohydrin rubber Polymers 0.000 description 1
- 210000004783 epithelial tight junction Anatomy 0.000 description 1
- 230000003628 erosive effect Effects 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000013305 flexible fiber Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 238000002073 fluorescence micrograph Methods 0.000 description 1
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000011194 food seasoning agent Nutrition 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 239000012737 fresh medium Substances 0.000 description 1
- 108010074605 gamma-Globulins Proteins 0.000 description 1
- UHBYWPGGCSDKFX-VKHMYHEASA-N gamma-carboxy-L-glutamic acid Chemical compound OC(=O)[C@@H](N)CC(C(O)=O)C(O)=O UHBYWPGGCSDKFX-VKHMYHEASA-N 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 239000003349 gelling agent Substances 0.000 description 1
- 238000012224 gene deletion Methods 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 230000009477 glass transition Effects 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000007773 growth pattern Effects 0.000 description 1
- 229920000591 gum Polymers 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- 210000003709 heart valve Anatomy 0.000 description 1
- 208000014951 hematologic disease Diseases 0.000 description 1
- 208000018706 hematopoietic system disease Diseases 0.000 description 1
- 108010057863 heparin receptor Proteins 0.000 description 1
- 210000005161 hepatic lobe Anatomy 0.000 description 1
- 229920001903 high density polyethylene Polymers 0.000 description 1
- 239000004700 high-density polyethylene Substances 0.000 description 1
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- 229960002591 hydroxyproline Drugs 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000001506 immunosuppresive effect Effects 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000007901 in situ hybridization Methods 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 238000013101 initial test Methods 0.000 description 1
- 230000000266 injurious effect Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 230000006662 intracellular pathway Effects 0.000 description 1
- 230000006525 intracellular process Effects 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000000741 isoleucyl group Chemical group [H]N([H])C(C(C([H])([H])[H])C([H])([H])C([H])([H])[H])C(=O)O* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 235000015110 jellies Nutrition 0.000 description 1
- 239000003410 keratolytic agent Substances 0.000 description 1
- 238000012933 kinetic analysis Methods 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- GDBQQVLCIARPGH-ULQDDVLXSA-N leupeptin Chemical compound CC(C)C[C@H](NC(C)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C=O)CCCN=C(N)N GDBQQVLCIARPGH-ULQDDVLXSA-N 0.000 description 1
- 108010052968 leupeptin Proteins 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 239000000436 ligase inhibitor Substances 0.000 description 1
- 230000029226 lipidation Effects 0.000 description 1
- 239000008263 liquid aerosol Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 210000005229 liver cell Anatomy 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 229920001684 low density polyethylene Polymers 0.000 description 1
- 239000004702 low-density polyethylene Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000005265 lung cell Anatomy 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 239000006166 lysate Substances 0.000 description 1
- 230000006674 lysosomal degradation Effects 0.000 description 1
- 230000002101 lytic effect Effects 0.000 description 1
- 238000002803 maceration Methods 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 239000000395 magnesium oxide Substances 0.000 description 1
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 230000021121 meiosis Effects 0.000 description 1
- 108020004084 membrane receptors Proteins 0.000 description 1
- 230000003458 metachromatic effect Effects 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 229940071648 metered dose inhaler Drugs 0.000 description 1
- RFKMCNOHBTXSMU-UHFFFAOYSA-N methoxyflurane Chemical compound COC(F)(F)C(Cl)Cl RFKMCNOHBTXSMU-UHFFFAOYSA-N 0.000 description 1
- 229960002455 methoxyflurane Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 210000001724 microfibril Anatomy 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 230000025090 microtubule depolymerization Effects 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 230000011278 mitosis Effects 0.000 description 1
- 230000009456 molecular mechanism Effects 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- 229910052901 montmorillonite Inorganic materials 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 229940051866 mouthwash Drugs 0.000 description 1
- 210000004877 mucosa Anatomy 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 229940097496 nasal spray Drugs 0.000 description 1
- 239000007922 nasal spray Substances 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- PGSADBUBUOPOJS-UHFFFAOYSA-N neutral red Chemical compound Cl.C1=C(C)C(N)=CC2=NC3=CC(N(C)C)=CC=C3N=C21 PGSADBUBUOPOJS-UHFFFAOYSA-N 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 230000004942 nuclear accumulation Effects 0.000 description 1
- 210000000633 nuclear envelope Anatomy 0.000 description 1
- 239000003865 nucleic acid synthesis inhibitor Substances 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 210000003463 organelle Anatomy 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 108700025694 p53 Genes Proteins 0.000 description 1
- 230000036281 parasite infection Effects 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 229960001639 penicillamine Drugs 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- BZQFBWGGLXLEPQ-REOHCLBHSA-N phosphoserine Chemical compound OC(=O)[C@@H](N)COP(O)(O)=O BZQFBWGGLXLEPQ-REOHCLBHSA-N 0.000 description 1
- USRGIUJOYOXOQJ-GBXIJSLDSA-N phosphothreonine Chemical compound OP(=O)(O)O[C@H](C)[C@H](N)C(O)=O USRGIUJOYOXOQJ-GBXIJSLDSA-N 0.000 description 1
- DCWXELXMIBXGTH-UHFFFAOYSA-N phosphotyrosine Chemical compound OC(=O)C(N)CC1=CC=C(OP(O)(O)=O)C=C1 DCWXELXMIBXGTH-UHFFFAOYSA-N 0.000 description 1
- 238000000053 physical method Methods 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 230000003169 placental effect Effects 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229920001084 poly(chloroprene) Polymers 0.000 description 1
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920000139 polyethylene terephthalate Polymers 0.000 description 1
- 229920000151 polyglycol Polymers 0.000 description 1
- 239000010695 polyglycol Substances 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 239000005033 polyvinylidene chloride Substances 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 230000001323 posttranslational effect Effects 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- FBCQUCJYYPMKRO-UHFFFAOYSA-N prop-2-enyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OCC=C FBCQUCJYYPMKRO-UHFFFAOYSA-N 0.000 description 1
- QTECDUFMBMSHKR-UHFFFAOYSA-N prop-2-enyl prop-2-enoate Chemical compound C=CCOC(=O)C=C QTECDUFMBMSHKR-UHFFFAOYSA-N 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 229940024999 proteolytic enzymes for treatment of wounds and ulcers Drugs 0.000 description 1
- 230000004063 proteosomal degradation Effects 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 238000009613 pulmonary function test Methods 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 239000013646 rAAV2 vector Substances 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 230000008521 reorganization Effects 0.000 description 1
- 230000003362 replicative effect Effects 0.000 description 1
- 238000002271 resection Methods 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 208000023504 respiratory system disease Diseases 0.000 description 1
- 230000006965 reversible inhibition Effects 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 210000005241 right ventricle Anatomy 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 229940043230 sarcosine Drugs 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000012764 semi-quantitative analysis Methods 0.000 description 1
- 239000012679 serum free medium Substances 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229920002379 silicone rubber Polymers 0.000 description 1
- 230000036556 skin irritation Effects 0.000 description 1
- 231100000475 skin irritation Toxicity 0.000 description 1
- 239000000344 soap Substances 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 239000008275 solid aerosol Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 210000003802 sputum Anatomy 0.000 description 1
- 208000024794 sputum Diseases 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000012128 staining reagent Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 238000011146 sterile filtration Methods 0.000 description 1
- 239000008174 sterile solution Substances 0.000 description 1
- 230000001954 sterilising effect Effects 0.000 description 1
- 238000004659 sterilization and disinfection Methods 0.000 description 1
- 239000003270 steroid hormone Substances 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 210000000434 stratum corneum Anatomy 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 230000004960 subcellular localization Effects 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- NJGWOFRZMQRKHT-UHFFFAOYSA-N surfactin Natural products CC(C)CCCCCCCCCC1CC(=O)NC(CCC(O)=O)C(=O)NC(CC(C)C)C(=O)NC(CC(C)C)C(=O)NC(C(C)C)C(=O)NC(CC(O)=O)C(=O)NC(CC(C)C)C(=O)NC(CC(C)C)C(=O)O1 NJGWOFRZMQRKHT-UHFFFAOYSA-N 0.000 description 1
- NJGWOFRZMQRKHT-WGVNQGGSSA-N surfactin C Chemical compound CC(C)CCCCCCCCC[C@@H]1CC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)O1 NJGWOFRZMQRKHT-WGVNQGGSSA-N 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 229920003002 synthetic resin Polymers 0.000 description 1
- 239000000057 synthetic resin Substances 0.000 description 1
- 231100000057 systemic toxicity Toxicity 0.000 description 1
- 210000004876 tela submucosa Anatomy 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000008719 thickening Effects 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229940033663 thimerosal Drugs 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 229960000984 tocofersolan Drugs 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- FGMPLJWBKKVCDB-UHFFFAOYSA-N trans-L-hydroxy-proline Natural products ON1CCCC1C(O)=O FGMPLJWBKKVCDB-UHFFFAOYSA-N 0.000 description 1
- 230000005030 transcription termination Effects 0.000 description 1
- 238000013271 transdermal drug delivery Methods 0.000 description 1
- 230000014723 transformation of host cell by virus Effects 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 230000014616 translation Effects 0.000 description 1
- 230000005945 translocation Effects 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 241000712461 unidentified influenza virus Species 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- JXLYSJRDGCGARV-CUGARIAGSA-N vinblastine Chemical compound C([C@@](C1)(O)CC)C(C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 JXLYSJRDGCGARV-CUGARIAGSA-N 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 210000004885 white matter Anatomy 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- XOOUIPVCVHRTMJ-UHFFFAOYSA-L zinc stearate Chemical compound [Zn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O XOOUIPVCVHRTMJ-UHFFFAOYSA-L 0.000 description 1
- 239000002076 α-tocopherol Substances 0.000 description 1
- 235000004835 α-tocopherol Nutrition 0.000 description 1
Images
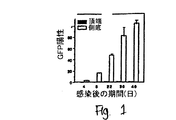

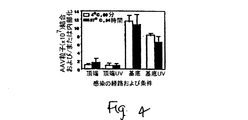

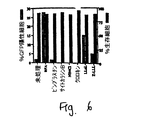















Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
- G01N33/5067—Liver cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/197—Carboxylic acids, e.g. valproic acid having an amino group the amino and the carboxyl groups being attached to the same acyclic carbon chain, e.g. gamma-aminobutyric acid [GABA], beta-alanine, epsilon-aminocaproic acid or pantothenic acid
- A61K31/198—Alpha-amino acids, e.g. alanine or edetic acid [EDTA]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/21—Esters, e.g. nitroglycerine, selenocyanates
- A61K31/27—Esters, e.g. nitroglycerine, selenocyanates of carbamic or thiocarbamic acids, meprobamate, carbachol, neostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/365—Lactones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4706—4-Aminoquinolines; 8-Aminoquinolines, e.g. chloroquine, primaquine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/02—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving viable microorganisms
- C12Q1/025—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving viable microorganisms for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/502—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics for testing non-proliferative effects
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
- G01N33/56983—Viruses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2799/00—Uses of viruses
- C12N2799/02—Uses of viruses as vector
- C12N2799/021—Uses of viruses as vector for the expression of a heterologous nucleic acid
- C12N2799/025—Uses of viruses as vector for the expression of a heterologous nucleic acid where the vector is derived from a parvovirus
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/005—Assays involving biological materials from specific organisms or of a specific nature from viruses
- G01N2333/01—DNA viruses
- G01N2333/015—Parvoviridae, e.g. feline panleukopenia virus, human Parvovirus
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/005—Assays involving biological materials from specific organisms or of a specific nature from viruses
- G01N2333/01—DNA viruses
- G01N2333/075—Adenoviridae
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/81—Protease inhibitors
- G01N2333/8107—Endopeptidase (E.C. 3.4.21-99) inhibitors
- G01N2333/8139—Cysteine protease (E.C. 3.4.22) inhibitors, e.g. cystatin
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/90—Enzymes; Proenzymes
- G01N2333/914—Hydrolases (3)
- G01N2333/948—Hydrolases (3) acting on peptide bonds (3.4)
- G01N2333/95—Proteinases, i.e. endopeptidases (3.4.21-3.4.99)
- G01N2333/964—Proteinases, i.e. endopeptidases (3.4.21-3.4.99) derived from animal tissue
- G01N2333/96425—Proteinases, i.e. endopeptidases (3.4.21-3.4.99) derived from animal tissue from mammals
- G01N2333/96427—Proteinases, i.e. endopeptidases (3.4.21-3.4.99) derived from animal tissue from mammals in general
- G01N2333/9643—Proteinases, i.e. endopeptidases (3.4.21-3.4.99) derived from animal tissue from mammals in general with EC number
- G01N2333/96466—Cysteine endopeptidases (3.4.22)
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Organic Chemistry (AREA)
- Cell Biology (AREA)
- Epidemiology (AREA)
- Biotechnology (AREA)
- Analytical Chemistry (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Physics & Mathematics (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Food Science & Technology (AREA)
- Toxicology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Zoology (AREA)
- Virology (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Wood Science & Technology (AREA)
- Gastroenterology & Hepatology (AREA)
Description
米国政府権利の言明
本発明は、少なくともその一部分が、アメリカ合衆国政府からの補助金(米国国立衛生研究所からの契約第HL51887号)により行われた。米国政府が本発明において一定の権利を有する可能性がある。
発明の背景
組換えアデノ随伴ウイルス(rAAV)は、多数の標的臓器および遺伝疾患に対する遺伝子治療ベクターとしてその可能性が強調されている種々の特性を有する。rAAVベクター系は、アデノウイルスおよびレトロウイルスに対して大きい利益を提供する可能性がある。これらには、非−分裂細胞のゲノム内への組込みに対するrAAVの能力、すべてのウイルス遺伝子を欠損できることによる可能な免疫反応の欠失、およびrAAVが高力価まで濃縮できるという事実を含む。
【0002】
アデノ随伴ウイルスタイプ−2(AAV−2)は、嚢胞性繊維症の遺伝子治療のために非常に有望なベクターであると示唆されている(Conrad et al., 1996;Flotte et al., 1993; Halbert et al., 1997)。ラビットおよびアカゲザル(rhesus macaques) の気道へのAAVベクターの生体内投与は、ウイルスDNAの6カ月間までの持続という長期残存を証明した(Conrad et al., 1996; Halbert et al., 1997) 。このベクター系は種々のヒト、サル、およびネズミ細胞株内の非常に広い宿主親和性を有することが主張されている事実にもかかわらず(Lebkowski et al., 1998; Muzyczka, 1992)、ヒト気道上皮内の総合形質導入効率はかなり低いように思われる。外的刺激、例えばDNA損傷剤、トポイソメラーゼ阻害剤、またはアデノウイルス初期遺伝子産物が存在しないと(Alexander et al., 1997; Alexander et al., 1996; Ferrari et al., 1996; Fisher et al., 1996;Halbert et al., 1997; Russel et al., 1996)、rAAVを用いる一次細胞およびその他の非−分裂細胞の生体内形質導入は著しく非効率である。反対に、S−期において活発に分裂する細胞のrAAV形質導入は、著しく効率的である(Russel et al., 1994) 。しかし、rAAVは、骨格筋およびCNSニューロンのin situ 形質導入において著しい効率を示し、rAAVベクターが非−分裂細胞の一定の集団を効果的に形質導入でき、そして細胞特異性特性がウイルスプロセシングに対して大きい影響を有することを示す(Duan et al., 1998; Kaplitt et al., 1994: Xiao et al., 1996)。
【0003】
2件の研究は、ウイルスゲノムの一本鎖から二本鎖への転換が、AAV媒介遺伝子導入に対する律速段階であろうと示唆している(Ferrari et al., 1996; Fisher et al., 1996) 。これらの研究は、アデノウイルスE4orf6が、一本鎖DNAゲノムの線型、二本鎖複製形ダイマー(Rfd)およびモノマー(Rfm)へのrAAV複製の溶解期の特性的な経路を通じる転換を増加させることを示した。これらの複製形の構造は、1個の共有結合末端を有する頭−頭および尾−尾方向線型コンカテマー(concatamer)から成る(Ferrari et al., 1996; Ficheret al., 1996) 。反対に最近の研究は、頭−尾モノマーおよびコンカテマー構造を有する二本鎖環状中間体へのrAAVゲノムの転換のための別の経路を解明した(Duan et al., 1999; Duan et al., 1998; Sanlioglu et a l., 1999) 。環状AAVゲノムまたはRf中間体のいずれかの形成に導く明確な経路は、種々の細胞因子により調節されるように見える。例えば、アデノウイルスE4orf6発現は、環状ゲノム形成を低下させ、一方アデノウイルスE2aはその形成を増加させる(Duan et al., 1999) 。同様に、UV照射もAAV環状中間体形成を増加させるが、しかしRf中間体は増加しない(Sanlioglu et a l., 1999) 。環状AAV中間体が、筋肉中での長期エピソーム持続および導入遺伝子発現にに関連するという知見(Duan et al., 1998) およびUV照射が環状中間体および組込みの範囲の両方を増加するという知見に基づいて、AAV環状中間体は潜在的な相の前組込み構造であろう。
【0004】
ヒト気道は、特別な繊毛性(ciliated)または非繊毛性上皮細胞により覆われており、これは外的環境から保護するだけでなく、気道内腔と下の粘膜下組織との間の分子の交換の制御に関係する機能も行っている。これらの細胞機能は、膜タンパク質および細胞レベルより下の細胞小器官の分布に関して高度に極性の構成により支持されている(Rodriguez-Boulan et al., 1993; Willis et al., 1996)。従来の研究は、この非対称な空間構成が、遺伝子導入の効率に対して著しい影響を有することを示唆している。例えば、頂端面上のインテグリンおよびアデノウイルス繊維受容体の欠失が、分化した繊毛性気道上皮のこのウイルス系による非効率的な感染を説明するであろう(Goldman et al., 1995; Zabner et al., 1997) 。極性化気道上皮細胞に関する最近の研究は、レトロウイルス感染性に対する同様の面依存性も明らかにし、これは部分的には側底膜に対するレトロウイルス受容体の分配により説明されるであろう(Wang et al., 1998) 。さらに、極性化気道上皮のrAAV感染における極性の同様な知見が報告されている(Duan et al., 1998) 。これらの研究は、気道上皮の側底側からのrAAVの200倍大きい感染性を証明する。
【0005】
上皮細胞内への極性侵入も、種々の他のウイルスに対して周知の現象である。例えば、ワクシニアウイルス、水疱性口内炎ウイルス、サイトメガロウイルス、イヌパルボウイルス(CPV)、およびセムリキ森林ウイルスは、極性上皮を主として側底膜を通して形質導入する(Basak et al., 1989; Fuller et al., 1984; Rodriguez et al., 1991; Tugizov et al., 1996) 。反対に、サルウイルス40および麻疹ウイルスは、優先的に頂端膜を介して感染する(Blau et al., 1995; Clayspri et al., 1988)。一般に、細胞膜受容体の非対称分布は、これらのウイルスにより示される感染の極性が関係すると信じられる。
【0006】
しかし、他の律速段階がウイルス形質導入の総合効率に関係することも妥当と思われる。これらの段階は、ウイルス結合、エンドサイトーシス、エンドソームプロセシング、核輸送、脱外被、遺伝子変換、転写および翻訳を含む。これに関して、極性化MDCK細胞における従来の研究は、頂端面からの被覆ピットの遅い成熟を証明し、これは 頂端膜と側底膜の間のエンドサイトーシスの速度の差を示す(Naim et al., 1995) 。
【0007】
AAVに対して、ウイルスベクター形質導入における律速段階を増強させるために、2種の方法が使用されている。これらは、細胞表面受容体(Qing et al.,1997) および/またはウイルス外被タンパク質内の受容体リガンド(Wickham etal., 1996a; Wickham et al., 1996b; Bartlett et al., 1999) の操作を含む。別の方法は、rAAVの場合の発現可能な形への非機能性ウイルスゲノムの分子変換を増加するか(Fisher et al., 1996; Sanlioglu et al., 1999) 、または導入遺伝子発現カセットを改変して転写および翻訳効率を増加させる(Zabner et al., 1996; Xiao et al., 1998)ことにより導入遺伝子発現を増加しようと試みている。
【0008】
嚢胞性繊維症は、白色人種で最も一般的な遺伝病であり、この疾患に対する遺伝子治療は肺気道上皮を標的とする。嚢胞性繊維症処置のための遺伝子治療ビヒクルとしてのAAVの開発は、そのウイルス生物学に基づいて種々の独自の利点を有する。例えば、野生型AAV感染は、呼吸器上皮に起きることは知られているが、しかし関連する病理は知られていない。しかし、上記のように、完全に分化した気道上皮は、rAAV−2だけでなく他の現在使用されているアデノウイルス、レンチウイルス、テトロウイルスおよびAAVを含むウイルスベクターのすべての他のタイプの頂端面からの感染に極めて耐性である。
【0009】
従って、要求されているのは、rAAV形質導入を生体内で改変、例えば増加または増強できる薬剤の同定である。同様に要求されているのは、非−分裂細胞、例えば肝臓および気道内のものにおけるrAAV内の導入遺伝子の発現を増加または増強する薬剤の同定である。
発明の概要
本発明は、真核細胞、例えば哺乳類細胞、例えば哺乳類の肺または肝細胞、または真核細胞の集団のアデノ随伴ウイルス形質導入を改変する薬剤を同定する方法を提供する。この方法は、細胞または細胞集団を薬剤およびウイルスと接触させることを含んでなる。次いで、ウイルス形質導入が改変されたかどうかを決定する。好ましい細胞は、哺乳類、鳥類、爬虫類、特には家畜化された哺乳類および鳥類、例えばヒト、非ヒト霊長類、畜牛、ヒツジ、ブタ、ウマ、イヌ、ネコ、マウス、ラット、ラビット、ニワトリ、およびシチメンチョウのものを含む。例えば、空気−液体界面またはヒト気管支異種移植片で増殖した極性化ヒト気道上皮細胞は、ウイルス形質導入を改変する薬剤を同定するために有用である。好ましい薬剤は、例えばウイルスエンドサイトーシスの増強、エンドソーム内のウイルス核酸またはタンパク質分解の低下、および/または核へのウイルス輸送を増加することによりウイルス形質導入を増加させるものである。従って、ウイルス形質導入を増加させる薬剤は、治療ペプチドまたはポリペプチドを導入および/または発現するためにrAAVを用いる遺伝子治療に特に有用である。
【0010】
以下に記載するように、ウイルス結合、例えばウイルス受容体の制限された分布、および気道上皮の頂端膜におけるAAV−2のエンドサイトーシスは、この組織タイプの形質導入における主要な律速段階ではない。実際に、分化したヒト気道上皮は、rAAV−2を著しく効率的に頂端面から内部化(internalize) する。どちらかというば、核への内部化したウイルスのエンドソームプロセシングおよび転送(trafficking) が、気道の頂端膜からの感染に続いてAAV−2が遭遇する主要な障害である。一本鎖AAV DNAの環状ゲノムへの効率的な変換に導く側底側感染とは異なり、頂端側感染は、転写的に不活性な状態の細胞内一本鎖ウイルスDNAの50日にも達する持続を起こさせた。AAV−2のエンドソームプロセシングおよび核への細胞内経路の効率を増加させるプロテアソーム阻害剤を用いて、頂端面から200倍以上の著しく増加した形質導入が観察され、ほとんど側底面からの形質導入のものに近い。AAVキャプシドタンパク質がエンドサイトーシスの後にユビキチン化され、そして侵入するウイルスのユビキチン媒介プロテアソーム分解が、プロテアソームまたはユビキチンリガーゼ阻害剤のいずれかを用いる処理によりブロックできることも見いだされた。
【0011】
従って、ウイルスキャプシドのユビキチン化は、一本鎖DNAゲノムを転写的に活性な状態に変換するための核および/または核プロセシングイベントへのウイルスの転送の効率を改変するために主要な障害であると考えられる。さらに重要なことは、マウス肺内のプロテアソーム阻害剤の生体内適用は、rAAV遺伝子導入を大きい細気管支内の上皮細胞の検出不能レベルから平均10.4±1.6%まで上昇させた。従って、気道内の形質導入に対する主要なエンドソームプロセシング障害を回避するためのプロテアソーム阻害剤の使用は、呼吸器疾患、例えば嚢胞性繊維症の生体内AAV媒介遺伝子治療に対して、ならびにウイルスプロセシングが律速イベントと思われる他の組織に対する臨床的に有用な戦略を提供するであろう。
【0012】
細胞、例えばヒト細胞のrAAVによる形質導入を増加させる好ましい薬剤は、ペプチドシステインプロテアーゼ阻害剤、例えばLLnLを含む。従って、本発明は、さらに真核細胞をウイルスおよび、式(I):R1 −A−(B)n −C〔式中、R1 はN−末端アミノ酸ブロック基であり、それぞれAおよびBは独立してアミノ酸であり、Cはアミノ酸であり、ここで末端カルボキシ基はホルミル(CHO)基により置換されており、そしてnは0、1、2または3である〕の化合物またはその薬剤的に許容できる塩を含んでなる薬剤と接触させる方法を提供する。
【0013】
【化4】

本発明の別の好ましい薬剤は、式(II)
〔式中、
R2 は、N−末端アミノ酸ブロック基であり、
R3 、R4 およびR5 が、それぞれ独立して水素、(C1 〜C10)アルキル、アリールまたはアリール(C1 〜C10)アルキルであり、そして
R6 、R7 およびR8 が、それぞれ独立して水素、(C1 〜C10)アルキル、アリールまたはアリール(C1 〜C10)アルキルである〕の化合物またはこの薬剤的に許容できるこの塩である。
【0015】
本発明の方法に有用な他の好ましい薬剤は、式(III)
【0016】
【化5】
〔式中、
R1 は、H、ハロゲン、(C1 〜C10)アルキル、(C1 〜C10)アルケニル、(C1 〜C10)アルキニル、(C1 〜C10)アルコキシ、(C1 〜C10)アルカノイル、(=O)、(=S)、OH、SR、CN、NO2 、トリフルオロメチルまたは(C1 〜C10)アルコキシであり、ここであらゆるアルキル、アルケニル、アルキニル、アルコキシまたはアルカノイルは場合により1個またはそれ以上のハロゲン、OH、SH、CN、NO2 、トリフルオロメチル、NRRまたはSRで置換されてもよく、ここで、それぞれのRは独立してHまたは(C1 〜C10)アルキルであり、
R2 は、(=O)または(=S)であり、
R3 は、H、(C1 〜C10)アルキル、(C1 〜C10)アルケニル、(C1 〜C10)アルキニル、(C1 〜C10)アルコキシまたは(C3 〜C8 )シクロアルキルであり、ここであらゆるアルキル、アルケニル、アルキニル、アルコキシまたはシクロアルキルは場合により1個またはそれ以上のハロゲン、OH、CN、NO2 、トリフルオロメチル、SR、またはNRRで置換されてもよく、ここで、それぞれのRは独立してHまたは(C1 〜C10)アルキルであり、
R4 は、H、(C1 〜C10)アルキル、(C1 〜C10)アルケニル、(C1 〜C10)アルキニル、(C1 〜C10)アルコキシまたは(C3 〜C8 )シクロアルキルであり、ここであらゆるアルキル、アルケニル、アルキニル、アルコキシまたはシクロアルキルは場合により1個またはそれ以上のハロゲン、OH、CN、NO2 、トリフルオロメチル、SR、またはNRRで置換されてもよく、ここで、それぞれのRは独立してHまたは(C1 〜C10)アルキルであり、
R5 は、H、ハロゲン、(C1 〜C10)アルキル、(C1 〜C10)アルケニル、(C1 〜C10)アルキニル、(C1 〜C10)アルコキシ、(C1 〜C10)アルカノイル、(=O)、(=S)、OH、SR、CN、NO2 、またはトリフルオロメチルであり、ここであらゆるアルキル、アルケニル、アルキニル、アルコキシまたはアルカノイルは場合により1個またはそれ以上のハロゲン、OH、SH、CN、NO2 、トリフルオロメチル、NRRまたはSRで置換されてもよく、ここでそれぞれのRは独立してHまたは(C1 〜C10)アルキルであり、そして
Xは、O、SまたはNR(式中、RはHまたは(C1 〜C10)アルキルである)である〕
の化合物またはこの薬剤的に許容できる塩である。
【0018】
好ましくは、R1 がOHである。R2 が(=O)であり、R3 がHまたは(C1 〜C10)アルキルであっても好ましく、そして最も好ましくはR3 がメチルである。その他の好ましい態様は、R4 がHまたは(C1 〜C10)アルキルであるものを含み、そしてさらに好ましくはR4 がHであり、R5 がハロゲン、CN、NO2 、トリフルオロメチル、またはOHであり、そしてさらに好ましくはR5 がOHである。式(III)の化合物は、XがOまたはS、好ましくはOであるものを含み、ここで両方の-----が単結合であるか、一方の-----が二重結合であるか、または両方の-----が二重結合である。さらに好ましい態様では、R1 がOHであり、R2 が(=O)であり、R3 がメチルであり、R4がHであり、R5 がOHであり、XがOであり、そして両方の-----が二重結合である。
【0019】
さらに本発明の方法に有用な好ましい薬剤は、式(III)
【0020】
【化6】
〔式中、R1 はハロゲン、CN、NO2 、トリフルオロメチルまたはOHである〕の化合物である。好ましくはR1 がOHである。R2 が(=O)であり、R3 がHまたは(C1 〜C10)アルキルであっても好ましく、そして最も好ましくはR3 がメチルである。その他の好ましい態様は、R4 がHまたは(C1 〜C10)アルキルであるものを含み、そしてさらに好ましくはR4 がHであり、R5 がハロゲン、CN、NO2 、トリフルオロメチル、またはOHであり、さらに好ましくはR5 がOHである。式(IV)の化合物は、XがOまたはS、好ましくはOであるものを含み、ここで両方の-----が単結合であるか、一方の-----)が二重結合であるか、または両方の-----が二重結合である。さらに好ましい態様では、R1 がOHであり、R2 が(=O)であり、R3 がメチルであり、R4 がHであり、R5 がOHであり、XがOであり、そして両方の-----が二重結合である。
【0022】
本発明の方法に有用な他の好ましい薬剤は、ユビキチンの活性化、ユビキチンキャリヤータンパク質へのユビキチンの転移、ユビキチンリガーゼ、またはこれらの組み合わせを阻害する薬剤を含む好ましいユビキチンリガーゼ阻害剤は、式(IV)
R−A−A1 −R1
〔式中、Rは、水素、アミノ酸、またはペプチドであり、ここでN末端アミノ酸は場合によりアセチル、アシル、トルフルオロアセチル、またはベンジルオキシカルボニルを用いてアミノ基において保護されることができ、
Aはアミノ酸または直接結合であり、
A1 はアミノ酸であり、そして
R1 は、ヒドロキシまたはアミノ酸であり、ここでC−末端アミノ酸は、場合により(C1 〜C6 )アルキル、フェニル、ベンジルエステルまたはアミド(例えばC(=O)NR2 (ここで、それぞれのRは独立して水素または(C1 〜C6 )アルキルである)を用いてカルボキシ基において保護されている〕
の化合物またはこの薬剤的に許容できる塩を含む.
Rの特定の値は水素である。
【0023】
Aの特定の値はアミノ酸である。Aの他の特定の値はIle、LeuまたはHisである。Aの他の特定の値はLeuまたはHisである。
【0024】
A1 の特定の値はAlaまたはGlyである。A1 の他の特定の値はAlaである。
【0025】
R1 の特定の値はヒドロキシである。
【0026】
特定すると、ペプチドはジペプチド(すなわちアミノ酸2個を含んでなることができる)であることができる。
【0027】
特定すると、ペプチドはH−Leu−Ala−OH、H−His−Ala−OH、H−Leu−Gly−OH、H−His−Gly−OH、H−Ile−Ala−OH、H−Ile−Gly−OHであることができる。さらに特定すると、ペプチドはH−Leu−Ala−OHまたはH−His−Ala−OHであることができる。
【0028】
さらに、本明細書中に記載のように、ウイルスのエンドソームプロセシングを阻害する薬剤の活性は、薬剤、例えばEDTAまたはEGTAの添加により増加してもよく、これらはエンドソームプロセシングに関連する経路において分子を改変するものであって、例えばカルシウムキレート化剤または細胞内カルシウムレベルの調節剤のような薬剤である。従って、本発明は、1)エンドソームプロセシングの阻害剤、および2)阻害剤の活性を増加する薬剤を含んでなる組成物またはキットも提供する。阻害剤、またはこれらの組み合わせは、本発明の方法で使用してもよく、または阻害剤の活性を増加させる薬剤と一緒に使用してもよい。
【0029】
従って、本発明は、真核細胞または細胞集団のアデノ随伴ウイルス形質導入を改変するための方法も提供する、この方法は、細胞を、ウイルス形質導入を改変するために有効な一定量の本発明の薬剤および一定量のウイルスと接触することを含んでなる。薬剤は、細胞とウイルスの接触の前または細胞とウイルスの接触の後に、ウイルスとを同時に細胞に接触させてもよい。1種または複数の薬剤および/またはウイルスは、所望の効果、すなわちrAAV形質導入を増加させるために、それぞれ一回投与、または反復投与してもよい。アデノ随伴ウイルスは、広い宿主範囲(肺発現に対する)を有し、そして筋肉内で持続することが証明されているので、rAAVは、あらゆる動物、および特には哺乳類および鳥類、魚類、および爬虫類、ことには家畜化された哺乳類、鳥類、例えば畜牛、ヒツジ、ブタ、ウマ、イヌ、ネコ、ニワトリ、およびシチメンチョウ内での遺伝子発現のためにに使用してもよい。ヒトおよび家畜の両者への使用が特に好ましい。
【0030】
発現される遺伝子は、あらゆる所望の制御要素(例えばプロモーター、オペレーター)を有するポリペプチドをコードするDNA断片、または転写がある種のRNA含有分子(例えば転写制御要素、+RNA、またはアンチセンス分子)のすべてまたは一部分を産生する非−コーディングDNA断片のいずれであることもできる。
【0031】
ベクターが有用な治療または予防治療は、血液疾患(例えば鎌状赤血球貧血、サラセミア、血友病、およびファンコーニ貧血)、神経疾患、例えばアルツハイマー病およびパーキンソン病、および骨格筋、心筋および平滑菌に関係する筋肉疾患、ならびに肺の疾患、例えば嚢胞性繊維症および喘息を含む。特には、本発明のベクターが有用な治療遺伝子は、β−グロブリン遺伝子、γ−グロブリン遺伝子、第8因子遺伝子、第9因子遺伝子、嚢胞性繊維症膜貫通コンダクタンス受容体遺伝子(CFTR)、ファンコーニ貧血補体グループ、リボザイムをコードする遺伝子、アンチセンス遺伝子、低密度リポタンパク質(LDL)遺伝子、チロシンヒドロキシラーゼ遺伝子(パーキンソン病)、グルコセレブロシダーゼ遺伝子(ゴーシェ病)、アリールスルファターゼa遺伝子(異染性白質ジストロフィー)または他のポリペプチドまたはタンパク質をコードする遺伝子を含む。本発明の範囲内には、組換えアデノ随伴ウイルスベクター内の1個またはそれ以上のオープンリーディングフレームも包含し、すなわち複数の遺伝子が一個のベクターに存在してもよい。さらに、環状中間体として、コンカテマーであってもよく、そのコンカテマーのそれぞれのモノマーは異なる遺伝子を含んでなってもよい。
【0032】
組換えアデノ随伴ウイルスの環状化中間体は、Hela細胞、繊維芽細胞および筋肉細胞に連結した配列にエピソーム持続性を与える。従って、組換えアデノ随伴ウイルスの生体内での持続性は、上昇したエピソーム安定性を有する組込み前中間体を表すエピソーム環状化ゲノムを通じて起きることができる。従って、組換えアデノ随伴ウイルスは、アデノ随伴ウイルスの環状中間体の1種またはそれ以上の最初のDNAセグメント、すなわちその生物的活性セグメントまたはその変種を含んでなるベクターから好ましくは調製され、そのDNAセグメントは宿主細胞内のベクターのエピソーム的安定性または持続性の増加を与え、そして第二DNAセグメントは遺伝子を含んでなる。好ましくは第二DNAセグメントは、治療的に有効なポリペプチドをコードする。
【0033】
[発明の詳細な説明]
定義
本明細書中に使用する「ベクター」は、ポリヌクレオチドを含んでなるかまたはこれと会合する巨大分子または巨大分子の会合物を呼び、そしてこれは生体内および生体外での細胞へのポリヌクレオチドの送達を媒介するために使用できる。例示としてのベクターは、例えばプラスミド、ウイルスベクター、リポソームおよびその他の遺伝子送達媒体を含む。送達されるポリヌクレオチド、場合により「標的ポリヌクレオチド」または「導入遺伝子」と呼ばれるものは、遺伝子治療に重要なコーディング配列(例えば治療的に重要なタンパク質をコードする遺伝子)および/または選択可能または検出可能な標識を含んでなってもよい。
【0034】
「AAV」は、アデノ随伴ウイルスであり、そしてウイルス自体またはその誘導体を呼ぶために使用してもよい。この用語は、すべてのサブタイプおよび天然に存在の形および組換え形の両方を含み、除外を要求される場合を除く。略字「rAAV」は、組換えアデノ随伴ウイルスを呼び、組換えAAVベクター(または「rAAV」ベクター)とも呼ばれる。
【0035】
本明細書中に使用する「形質導入(transduction)」または「形質導入(transducing) 」は、外在ポリヌクレオチド、例えばrAAVベクター内の導入遺伝子を宿主細胞内へ導入してポリヌクレオチド、例えば細胞内の導入遺伝子発現に導く過程を呼ぶ用語である。この過程は、1)細胞膜へのウイルスの結合、2)エンドサイトーシス、3)エンドソームからの脱出および核への転送、4)ウイルス粒子の外被を外し、環状中間体を含む発現可能な二本鎖形を形成するための第二のDNA鎖の合成、および5)宿主ゲノム内への組込みを含み、例えば本発明の薬剤によるこれらのいずれか、またはこれらまたはこれらの組み合わせの改変は、宿主細胞または細胞集団内に導入されたポリヌクレオチドの改変された発現または持続をもたらす。rAAVを介して導入されたポリヌクレオチドの改変された発現または持続は、タンパク質、発現、およびDNAおよびRNAハイブリダイゼーションを含み、これらに限定されない当該技術分野では周知の方法により測定できる。本発明の薬剤は、好ましくは、ウイルスエンドサイトーシス、エンドソームからの脱出および核への転送、および/または核内でのウイルス粒子の脱外被を増加し、導入されたポリヌクレオチド、例えばrAAVベクター内の導入遺伝子の発現を生体外でも生体内でも改変する。外因性ポリヌクレオチドの導入のために使用される方法は、周知の技術、例えばトランスフェクション、リポフェクション、ウイルス感染、形質転換、および電気さく孔法、ならびに非−ウイルス遺伝子送達形質技術を含む。導入されたポリヌクレオチドは、安定または一時的に宿主細胞内に維持されてもよい。安定な維持は、代表的には、導入されたポリヌクレオチドが宿主細胞と許容性の複製始点を含むか、または宿主細胞のレプリコン、例えば染色体外レプリコン(例えばプラスミド)または核またはミトコンドリア染色体内に組み込むことを必要とする。
【0036】
「遺伝子送達」は、遺伝子導入のために外在ポリヌクレオチドを細胞内に導入することを呼び、そしてターゲティング、結合、取り込み、輸送、局在化、レプリコン組込みおよび発現を包含してもよい。
【0037】
「遺伝子導入」は、外在ポリヌクレオチドの細胞内への導入を呼び、これはターゲティング、結合、取り込み、輸送、局在化およびレプリコン組込みを包含してもよいが、しかしその後の遺伝子の発現とは異なり、これを含まない。
【0038】
「遺伝子発現」または「発現」は、遺伝子転写、翻訳、および翻訳後の装飾の過程を呼ぶ。
【0039】
「検出可能な標識(marker)遺伝子」は、遺伝子を有する細胞を特異的に検出させる遺伝子である(例えば標識遺伝子を持っていない細胞から区別する)。多様なこのような遺伝子が当該技術分野では公知である。
【0040】
「選択可能な標識遺伝子」は、相当する選択剤の存在において、遺伝子を有する細胞に特異的に存在または不在を選択させる遺伝子である。例としては、抗生物質耐性遺伝子は、相当する抗生物質の存在において、宿主細胞を陽性に選択させるための陽性選択可能標識遺伝子として使用できる。種々の陽性および陰性の選択可能標識が当該技術分野では公知であり、その一部は以下に記載する。
【0041】
本明細書中に使用する「rAAVベクター」は、AAV起源ではないポリヌクレオチド配列(すなわちAAVに非相同のポリヌクレオチド)を含んでなるAAVベクターを呼び、代表的には、細胞の遺伝的形質転換のために重要な配列である。本発明の好ましいベクター構築物中では、非相同ポリヌクレオチドは、少なくとも1個、好ましくは2個のAAV逆方向末端反復配列(ITR)が近接している。用語rAAVベクターは、rAAVベクター粒子およびrAAVベクタープラスミドの両方を含む。
【0042】
「AAVウイルス」または「AAVウイルス粒子」は、少なくとも1個のAAVキャプシドタンパク質(好ましくは野生型AAVのすべてのキャプシドタンパク質)およびキャプシドに入ったポリヌクレオチドから成るウイルス粒子を呼ぶ。粒子が非相同ポリヌクレオチド(すなわち野生型AAVゲノム以外のポリヌクレオチド、例えば哺乳類細胞に送達されるべき導入遺伝子)を含んでなる場合には、これは代表的には「rAAV」と呼ばれる。
【0043】
AAVに対する「ヘルパーウイルス」は、AAV(例えば野生型AAV)を哺乳類細胞により複製およびパッケージングさせるウイルスを呼ぶ。AAVに対して種々のこのようなヘルパーウイルスが当該技術分野では公知であり、アデノウイルス、ヘルペスウイルスおよびポックスウイルス、例えばワクシニアが含まれる。アデノウイルスは、多数の異なるサブグループを包含するが、サブグループCのアデノウイルスタイプ5が最も一般的に使用される。ヒト、ヒト以外の哺乳類および鳥類起源の多数のアデノウイルスが既知であり、そして寄託機関、例えばATCCから入手できる。ヘルペスファミリーのウイルスは、例えば単純ヘルペスウイルス(HSV)およびエプスタイン−バー−ウイルス(EBV)、ならびにサイトメガロウイルス(CMV)および仮性狂犬病ウイルス(PRV)を含み、これらもATCCなどの寄託機関から入手できる。
【0044】
「感染性」ウイルスまたウイルス粒子は、ウイルス粒子が栄養性である細胞内に送達できるポリヌクレオチド成分を含んでなるものである。この用語は、ウイルスのいかなる複製能力を必ずしも意味しない。
【0045】
「複製可能」ウイルス(例えば複製可能AAV、ときには「RCA」と省略される)は、感染性で、そして感染した細胞内で複製可能な(すなわち、ヘルパーウイルスまたはヘルパーウイルス機能の存在下で)表現型的に野生型のウイルスを呼ぶ。AAVの場合に、複製可能性は、一般に機能性AAVパッケージング遺伝子の存在を必要とする。本明細書中に記載する好ましいrAAVベクターは、1個またはそれ以上のAAVパッケージング遺伝子の欠失により哺乳類細胞(特にはヒト細胞)中で複製不可能である。好ましくは、このようなrAAVベクターは、AAVパッケージング遺伝子と侵入したrAAVベクターとの間の組換えによりRCAが産生される可能性を最小化するために、あらゆるAAVパッケージング遺伝子配列を欠失している。本明細書中に記載する好ましいrAAVベクター調製物は、あったとしても少数のRCAを含むものである(好ましくは102 rAAV粒子あたりに約1RCA以下、より好ましくは104 rAAV粒子あたりに約1RCA以下、さらに好ましくは108 rAAV粒子あたりに約1RCA以下、それ以上に好ましくは1012rAAV粒子あたりに約1RCA以下、最も好ましくはRCAが含まれない)。
【0046】
用語「ポリヌクレオチド」は、あらゆる長さのヌクレオチドのポリマー形を呼び、デオキシリボヌクレオチドまたはリボヌクレオチドまたはこれらの類似体を含む。ポリヌクレオチドは、修飾ヌクレオチド、例えばメチル化またはキャップしたヌクレオチドおよびヌクレオチド類似体を含んでなってもよく、そして非−ヌクレオチド成分で中断されていてもよい。存在する場合に、ヌクレオチド構造に対する修飾は、ポリマー構築の前または後に与えられてもよい。本明細書中有に使用する用語ポリヌクレオチドは、二本鎖および一本鎖分子に互換可能に使用される。特に規定または要求しない限り、ポリヌクレオチドであると記載された本明細書中に本発明のあらゆる態様は、二本鎖形および二本鎖形を形成していることが既知または予想される2本の相補一本鎖形それぞれの両方を包含する。
【0047】
本明細書中に使用する「転写調節配列」または「TRS」は、遺伝子またはこれが操作的に連結するコーディング配列の転写を制御するゲノム領域を呼ぶ。本発明内で使用する転写調節配列は、一般に少なくとも1個の転写プロモーターを含み、そして1個またはそれ以上の転写のエンハンサーおよび/またはターミネーターを含んでもよい。
【0048】
「操作的に連結」は、このように記載された成分が協同した様式で機能できるような関係にある2個またはそれ以上の成分の配置を呼ぶ。例としては、転写調節配列またはプロモーターは、TRSまたプロモーターがコーディング配列の転写を促進する場合に、コーディング配列に操作的に連結している。操作的に連結したTRSは、一般にコーディング配列とシスで結合するが、しかしかならずしも直接これに隣接する必要はない。
【0049】
「非相同」は、比較される全体の他の部分と遺伝子型的に異なる部分から誘導されることを意味する。例えば、異なる細胞タイプ内に遺伝子操作技術により導入されたポリヌクレオチドは、非相同ポリヌクレオチドである(そして、発現された場合には、非相同ポリペプチドをコードできる)。同様に、本来のコーディング配列から取り去られそして異なるコーディング配列に操作的に連結されたTRSまたはプロモーターは、非相同TRSまたはプロモーターである。
【0050】
「レプリコン」は、ポリヌクレオチドの複製を適当な宿主細胞内で可能とする機転または複製を含んでなるポリヌクレオチドを呼ぶ。レプリコンの例は、エピソーム(プラスミドを含む)、ならびに染色体(例えば核またはミトコンドリア染色体)を含む。細胞内へのポリヌクレオチドの「安定な組込み」は、細胞内で安定に維持されるようになるレプリコン内へポリヌクレオチドが組み込まれることを意味する。プラスミドのようなエピソームは、時には数世代も維持できるが、エピソーム的に保持される遺伝物質は、一般に染色体に組み込まれた物質よりも欠失する可能性が高い。しかし、ポリヌクレオチドの維持は、しばしばポリヌクレオチド内またはこれに隣接する選択可能な標識の取り込みにより行うことができ、次いでポリヌクレオチドを有する細胞を選択的圧力下で維持する。ある場合には、配列が染色体内に組み込まれない限り、これらは有効に安定的に維持できない。従って、選択可能な標識を含んでなる配列の保持のための選択は、その中で標識が染色体内に安定に組み込まれた細胞の選択をもたらすことができる。抗生物質耐性遺伝子は、この場合に便利に利用でき、これは当該技術分野では周知である。代表的には、安定に組み込まれたポリヌクレオチドは、平均して少なくとも約20世代、好ましくは少なくとも約100世代、さらに好ましくは永久に維持されるように予想される。真核生物染色体のクロマチン構造は、組み込まれたポリヌクレオチドの発現レベルに影響できる。エピソーム上に保持された遺伝子を有すると、特定の遺伝子の複数の安定維持コピーを有するよう希望する場合に特に有用であることができる。本発明の範囲内で特に望ましい性質を有する安定細胞株の選択は、以下に記載して説明する。
【0051】
本明細書中に使用する「パッケージング」は、ウイルスベクター、特にはAAVベクターの構築およびキャプシド化をもたらす一連の細胞スケール以下のイベントを呼ぶ。従って、適当なベクターが適当な条件下でパッケージング細胞株内に導入される場合に、これはウイルス粒子内に集積できる。ウイルスベクター、特にはAAVベクターのパッケージングを伴う機能は、本明細書中および当該技術分野に記載されている。
【0052】
「ターミネーター」は、リード・スルー転写が低減または防止するようになるポリヌクレオチド配列を呼ぶ(すなわち、これはターミネーターの一方側からの転写のターミネーターの他方側への継続を現象または防止する)。転写が中断される程度は、代表的には、ターミネーター配列の塩基配列および/または長さの関数である。特に、多数の分子生物学的系で周知のように、特定のDNA配列、一般に「転写終止配列」と呼ばれるものは、RNAポリメラーゼによるリード・スルー・転写を中断する特定の配列であり、RNAポリメラーゼ分子を転写されているDNAから停止および/または外すことによると推測される。このような配列特異性ターミネーターの代表的な例は、ポリアデニル化(「ポリA」)配列、例えばSV40ポリAを含む。このような配列特異性ターミネーターに加えてまたはこれに代えて、プロモーターとコーディング領域との間への比較的長いDNA配列の挿入もコーディング領域の転写を中断する傾向があり、これは一般に中間挿入配列の長さに比例する。この効果は、RNAポリメラーゼ分子が常に転写中のDNA分子から離れる傾向があるために起きると推測され、そしてコーディング領域に到着する前に通過しなければならない配列の長さが長くなると、一般に、コーディング領域の転写が完了または場合によれば開始する前でも離脱が起きる可能性が高くなる。従って、ターミネーターは、転写を一方向のみから(「一方向ターミネーター」)または両方向から(「二方向ターミネーター」)防止してもよく、そして配列特異性終止配列または配列非特異性終止配列または両方を含んでなってもよい。種々のこのようなターミネーター配列が当該技術分野では公知であり、そして本発明の範囲内でのこのような配列の例示的な使用は、以下に記載される。
【0053】
「宿主細胞」、「細胞株」、「細胞カルチャー」および「パッケージング細胞株」およびその他のこのような用語は、本発明に有用な高級真核細胞、好ましくは哺乳類細胞、最も好ましくはヒト細胞を表す。これらの細胞は、組換えベクター、ウイルスまたはその他の転移ポリヌクレオチドのレシピエントとして使用でき、そして形質導入された最初の細胞の後代を含む。単一細胞の後代は、最初の親細胞と完全に一致(形態的またはゲノム集合内で)する必要はない。
【0054】
「治療遺伝子」「標的ポリヌクレオチド」「導入遺伝子」、「関係する遺伝子」などは、一般にベクターを用いて導入される遺伝子または複数の遺伝子を呼ぶ。代表的には、本発明の範囲内で、このような遺伝子はrAAVベクター内に位置する(このベクターは逆方向末端反復(ITR)領域に近接し、従って複製およびrAAV粒子内にキャプシド化できる)。標的ポリヌクレオチドは、本発明内で、多数の異なる用途のためのrAAVベクターの作製のために使用できる。このようなポリヌクレオチドは、下記を含み、これらの限定はされない。(i)構造タンパク質または酵素の欠失、欠損または最適状態に及ばないレベルにより起きる欠陥を救済するための遺伝子治療の他の形で有用なタンパク質をコードするポリヌクレオチド、(ii)アンチセンス分子内に転写されるポリヌクレオチド、(iii)転写または翻訳因子を結合するデコイ内に転写されるポリヌクレオチド、(iv)細胞調節体、例えばサイトカインをコードするポリヌクレオチド、(v)特定の薬剤に対して可能なレシピエント細胞を産生できるポリヌクレオチド、例えばヘルペスウイルスチミジンキナーゼ遺伝子、および(vi)ガン治療のためのポリヌクレオチド、例えば種々のガン治療のためのE1Aガンサプレッサー遺伝子またはp53ガンサプレッサー遺伝子。レシピエント宿主細胞内の導入遺伝子の発現を起こすために、これは好ましくはプロモーターそれ自体または非相同的プロモーターに操作的に連結される。多数の適合するプロモーターが当該技術分野では公知であり、その選択は、標的ポリヌクレオチドの発現の所望のレベルに依存し、構成的発現、誘導的発現、細胞特異性または組織特異性発現などを希望するかどうかによる。rAAVベクターは、選択可能な標識を含んでもよい。
【0055】
「遺伝子」は、転写および翻訳された後に特定のタンパク質をコード可能な少なくとも1個のオープンリーディングフレームを含むポリヌクレオチドを呼ぶ。
【0056】
ポリヌクレオチドに適用された場合の「組換え」は、ポリヌクレオチドがクローニング、制限および/または結合ステップ、および天然に見られるポリヌクレオチドとは異なる構築物をもたらす他の操作の種々の組み合わせの産物であることを意味する。組換えウイルスは、組換えポリヌクレオチドを含んでなるウイルス粒子である。この用語は、それぞれ最初のポリヌクレオチド構築物の複製物および最初のウイルス構築物の後代を含む。
【0057】
「制御要素」または「制御配列」は、ポリヌクレオチドの複製、重複、転写、スプライシング、翻訳、または分解を含むポリヌクレオチドの機能性調節に寄与する分子の相互作用に含まれるヌクレオチド配列である。調節は、過程の頻度、速度、または特異性に影響してもよく、そして本質的に増強または阻害性であってもよい。当該技術分野で公知の制御要素は、例えば転写調節配列、例えばプロモーターまたはエンハンサーである。プロモーターは、一定の条件下でRNAポリメラーゼを結合しそして通常はプロモーターから下流(3’方向)に位置するコーディング領域の転写を開始する能力があるDNA領域である。プロモーターはAAVプロモーター、例えばP5、P19、P40およびAAV ITRプロモーター、ならびに非相同プロモーターを含む。
【0058】
「発現ベクター」は、関係するポリペプチドをコードし、そして意図する標的細胞内にタンパク質の発現を起こすために使用される領域を含んでなるベクターである。発現ベクターは、標的内のタンパク質の促進を容易にするためにコード化領域に操作的に連結する制御要素も含んでなる。発現のために操作的に連結する制御要素と遺伝子または複数の遺伝子との組み合わせは、時に「発現カセット」と呼ばれ、その多数が既知であり、そして当該技術分野で利用できるかまたは当該技術分野で利用できる成分から容易に構築できる。
【0059】
「遺伝的改変」は、遺伝要素が有糸分裂または減数分裂以外により細胞内に導入される過程を呼ぶ。要素は細胞に非相同性であってもよく、または細胞内にすでに存在する要素の追加のコピーまたは改善されたものであってもよい。遺伝的改変は、例えば、組換えプラスミドまたは他のポリヌクレオチドを用いて当該技術分野では公知のあらゆる過程、例えば電気さく孔、リン酸カルシウム沈降、またはポリヌクレオチド−リポソーム複合体との接触を通じて細胞をトランスフェクションして行ってもよい。遺伝的改変は、例えばDNAまたはRNAウイルスまたはウイルスベクターを用いる形質導入または感染により行ってもよい。好ましくは、遺伝要素は、細胞内の染色体またはミニクロモソーム内に導入される。しかし、細胞およびその後代の表現型および/または遺伝型を変化させるあらゆる改変は、この用語内に含まれる。
【0060】
細胞は、生体外で細胞の長期間の培養の間にその機能を行うために配列が利用できる場合には、遺伝子配列を「安定して」改変、形質導入または形質転換されるといわれる。好ましい例では、このような細胞は、遺伝子改変が改変された細胞の後代によっても遺伝可能なように導入されると「遺伝的」に改変される。
【0061】
用語「ポリペプチド」および「タンパク質」は、本明細書内であらゆる長さのアミノ酸のポリマーを互換可能に呼ぶように用いられる。この用語は、変性、例えばジスルフィド結合形成、グリコシル化、アセチル化、リン酸化、脂質化または標識化合物と複合されたアミノ酸ポリマーも包含する。例えば「CFTR」および類似のポリペプチドは、遺伝子治療およびそのための組成物の範囲内で考察する場合には、それぞれ不変のポリペプチド、またはそのあらゆる断片または遺伝子的に操作された誘導体であって、当初のタンパク質の所望の生化学的機能を保持しているものを呼ぶ。同様に、CFTR、およびその他の遺伝子治療に使用するためのこのような遺伝子(代表的には、レシピエント細胞への送達される「導入遺伝子」と呼ばれる)への言及は、所望の生化学的機能を有する不変のポリペプチドまたはあらゆる断片または遺伝子的に操作された誘導体をコードするポリヌクレオチドを含む。
【0062】
「単離された」プラスミド、ウイルスまたはその他の物質は、物質または類似の物質が天然に存在または最初に調製された形で存在するであろう他の成分の少なくとも一部が存在しない物質の調製を呼ぶ。従って、例えば、単離された物質は、起源の混合物からそれを濃縮技術を用いて調製されてもよい。濃縮は、絶対的基準、例えば溶液の体積あたりの重量で測定でき、またはこれは期限混合物中に存在する第二のあらゆる介在物質に関連して測定できる。本発明の態様の濃縮が上昇すると好ましさも上昇する。従って、例えば、2倍の濃縮は好ましく、10倍の濃縮は一層好ましく、100倍の濃縮はさらに好ましく、1000倍の濃縮はそれ以上さらに好ましい。
【0063】
AAVの調製は、感染性AAV粒子と感染性ヘルパーウイルスとの比が少なくとも102 :1、好ましくは少なくとも104 :1、さらに好ましくは少なくとも106 :1、さらに好ましくは少なくとも108 :1であるとヘルパーウイルスを「本質的に含まない」を言われる。調製物は、好ましくは相当する量のヘルパーウイルスタンパク質(すなわち、上記のヘルパーウイルス粒子不純物が破壊された形で存在する場合に、このようなヘルパーウイルスレベルの結果として存在するタンパク質)も含まない。ウイルスおよび/または細胞タンパク質汚染物は、一般にSDSゲル上のクーマシー染色バンドの存在として観察できる(例えばAAVキャプシドタンパク質VP1、VP2およびVP3に相当するものとは異なるバンドの出現)。
【0064】
ウイルス産生、複製またはパッケージングの記載に使用される場合の「効率」は、方法の有用な性質を呼ぶ。特には、細胞あたりに産生されたウイルス粒子の増殖速度および数である。「高効率」産生は、細胞あたりに少なくとも100ウイルス粒子の産生、好ましくは少なくとも約10,000個およびさらに好ましくは少なくとも約100,000個の粒子が、規定の生育期間中で産生されることを示す。
【0065】
本発明に従って処理される「個体」または「患者」は、脊椎動物、特には哺乳類の構成員を呼び、そして家畜動物、スポーツ用動物、およびヒトを含む霊長類を呼び、これらには限定されない。
【0066】
個体または細胞の「処理」は、処理を開始した時点での個体または細胞の本来の経過を改変、例えば個体内の予防、治療またはその他の有益な効果を引き出すための試みにおける介入のあらゆる形式である。例えば個体の処理は、あらゆる病理学的状態、遺伝的または誘発的遺伝子欠失、ウイルス、細菌または寄生生物の感染、新生物または形成不全性状態、または免疫系機能不全例えば自己免疫または免疫抑制を含む(これらに限定はされない)状態により起きた病理を低下または制限するために採用しもよい。処理は、組成物、例えば薬剤組成物の投与、および組成物で処理された適合する細胞の投与を含む(これらに限定はされない)。処理は、予防的にも治療的にも行ってもよい。すなわち、病理学的イベントの開始または病因的薬剤への接触の前またはその後である。
【0067】
本発明の実施は、別途断らない限り、分子生物学、ウイルス学、微生物学、組換えDNA、および免疫学の慣用の技術を用い、これらは当該技術分野の範囲内である。このような技術は、文献に十分に説明されている。例えば下記を参照のこと。
【0068】
【表1】
1.rAAVベクター
組換えAAVベクターは、ヒト遺伝子治療、特に疾患、例えば嚢胞性繊維症および鎌状細胞貧血に対して可能な強力な方法である。遺伝子治療の方法において他の方法に対するrAAVベクターの主要な利点は、これが一般に宿主細胞内に安定に組み込まれるようになるために標的細胞の継続した複製を要求しないことにある。
【0070】
rAAVベクターおよび/またはウイルスは1種またはそれ以上の検出可能標識を含んでもよい。種々のこのような標識は公知であり、例示として、細菌性β−ガラクトシダーゼ(lacZ)遺伝子、ヒト胎盤アルカリホスホターゼ(AP)遺伝子および生体外および生体内の両方でのレポーター分子として使用されている種々の細胞表面標識をコードする遺伝子を含む。rAAVベクターおよび/またはウイルスは、1個またはそれ以上の選択可能な標識を含んでもよい。
【0071】
組換えAAVベクターおよび/またはウイルスは、タンパク質をコードしないポリヌクレオチド、例えば二倍体を形成して正常mRNAの翻訳を遮断するために使用できるアンチセンスmRNA(mRNAの補体)をコードするポリヌクレオチドおよびリボザイムをコードするポリヌクレオチド(RNA触媒)を含むポリヌクレオチドを含んでなることもできる。
II.AAVベクターの選択および調製
種々の血清型は遺伝子レベルでも機能的および構造的に関連しているので、あらゆる血清型のアデノ随伴ウイルスが遺伝レベルでもrAAVの調製に使用できる(例えばBlacklow, Parvoviruses and Human Disease, J.R. Pattison ed. (1988), pp165-174;およびRose, Comprehensive Virology, 3,1, 1974 参照)。すべてのAAV血清型は、相同性rep遺伝子により媒介される同様の複製性を明らかに示し、そしてすべてが一般的にAAV2で発現されるものと同様の3種の関連キャプシドタンパク質を有する。関連性の程度は、さらに、ゲノムの長さに沿った血清型の間の広範な交差ハイブリダイゼーション、およびITRに相当する末端における類似した自己アニーリングセグメントの存在を明らかにするヘテロ二本鎖分析により示唆される。同様の感染パターンも、それぞれの血清型内の複製機能は、同様の調節制御下にあることを示唆する。種々のAAV血清型の中で、AAV2が最も一般的に使用される。
【0072】
本発明のAAVベクターは、代表的にはAAVに非相同性のポリヌクレオチドを含んでなる。ポリヌクレオチドは、遺伝子治療の範囲内で標的細胞に機能、例えばある種の表現型の発現の上昇および下降調節を提供する能力のために代表的には関連がある。このような非相同性ポリヌクレオチドまたは「導入遺伝子」は、一般に所望の機能またはコード化遺伝子を提供するために十分な長さである。
【0073】
非相同性ポリヌクレオチドの転写が意図する標的細胞内に所望される場合には、例えば標的細胞内の転写の所望のレベルおよび/または特異性に関連して、それ自体または非相同性プロモーターに操作的に連結していてもよく、これは当該技術分野では公知である。種々のプロモーターおよびエンハンサーがこの目的に適合する。構成的プロモーターは、遺伝子転写の継続するレベルを提供し、そして治療的ポリヌクレオチドが継続ベースで発現されることが望まれる場合には好ましい。誘導可能なプロモーターは、一般に、誘発要因が存在しないと低い活性を示し、そして誘導要因が存在すると上昇制御される。一定の時点または一定の場所でのみの発現が望まれる場合、または誘発剤を用いる発現レベルの滴定が望まれる場合には、これらが好ましいであろう。プロモーターおよびエンハンサーは、組織特異性であってもよい。すなわち、これらの活性は一定の細胞タイプ内でのみ発現し、これは遺伝子調節要素がこれらの細胞内のみで見いだされると推測される。
【0074】
プロモーターの例示的な例は、サルウイルス40からのSV40後期プロモーター、バキュロウイルスポリヘドロンエンハンサー/プロモーター要素、単純ヘルペスウイルスチミジンキナーゼ(HSV tk)、サイトメガロウイルス(CMV)からの極初期プロモーターおよびLTR要素を含む種々のレトロウイルスプロモーターである。誘導可能プロモーターは、重金属イオン誘導可能プロモーター(例えばマウス乳ガンウイルス(mMTV)プロモーターまたは種々の成長ホルモンプロモーター)、およびT7 RNAポリメラーゼの存在下で活性であるT7ファージからのプロモーターを含む。例示として、組織特異性プロモーターの例は、種々のサーファクチン(surfactin) プロモーター(肺内での発現のため)、ミオシンプロモーター(例えば筋肉内)、およびアルブミンプロモーター(肝臓内発現のため)を含む。各種の他のプロモーターが既知であり、そして一般に当該技術分野で既知で入手可能であり、そしてこのようなプロモーターの多数の配列が配列データベース、例えばジーンバンク・データベース(GeneBank database) から利用できる。
【0075】
意図する標的細胞内で翻訳を希望する場合には、非相同ポリヌクレオチドは、好ましくは翻訳を促進する制御要素(例えばリボソーム結合部位すなわち「RBS」およびポリアデニル化シグナル)も含んでなる。従って、非相同ポリヌクレオチドは、一般に、適切なプロモーターに操作的に連結する少なくとも1個のコーディング領域を含んでなり、そして例えば、操作的に連結するエンハンサー、リボソーム結合部位およびポリ−Aシグナルも含んでなってもよい。非相同性ポリヌクレオチドは、一個のコード化領域、または1個を越えるコード化領域を同じかまたは異なるプロモーターの制御下で含んでなってもよい。制御要素およびコード化領域の組み合わせを含む単位全体は、しばしば発現カセットと呼ばれる。
【0076】
非相同性ポリヌクレオチドは、組換え技術によりAAVゲノムコーディング領域内または好ましくはその代わり(すなわちAAV repおよびcap遺伝子の代わりに)に組み込まれるが、しかし、一般に両側でAAV逆方向末端反復領域(ITR)がどちらかの側で近接している。これは、ITRがコーディング配列から上流側および下流側の両方に現れ、いずれも直接近位にあり、好ましくは(必ずしも必要ではないが)複製−可能なAAVゲノムを再生するかもしれない組換えの可能性を低下させるためにAAV起源のいかなる介在配列もないことを意味する。しかし、単独のITRでも、2個のITRを含んでなる配置と通常は関連する機能を実行するために十分であろうし(例えばWO94/13788参照)、従ってITR1個のみを有するベクター構築物は、本発明のパッケージングおよび調製方法と関連して使用できる。
【0077】
repに対する本来のプロモーターは自己制御性であり、そして産生されるAVの量を制限できる。rep遺伝子も、repがベクター構築物の一部分として提供されてもまたは別々のいずれかであっても、非相同プロモーターに操作的に連結できる。rep遺伝子発現により強くは下降制御されないあらゆる非相同プロモーターが適合する。しかし、rep遺伝子の構成的発現が宿主細胞に負の影響を有するために、誘導可能なプロモーターが好ましい。各種の誘導可能なプロモーターが当該技術分野では既知であり、例示として、重金属イオン誘導可能プロモーター(例えばメタロチオネインプロモーター)、ステロイドホルモン誘導可能プロモーター(例えばMMTVプロモーターまたは成長ホルモンプロモーター)、およびT7 RNAポリメラーゼの存在下で活性であるT7ファージからのプロモーターが含まれる。誘導可能プロモーターの特に好ましいサブクラスは、rAAVベクターの複製およびパッケージングを補足するために使用されるヘルパーウイルスにより誘導されるものである。多数のヘルパーウイルス誘導可能プロモーターが記述されており、これには、アデノウイルスE1Aタンパク質により誘導可能なアデノウイルス初期遺伝子プロモーター、アデノウイルス主要後期プロモーター、ヘルペスウイルスタンパク質、例えばVP16または1CP4により誘導可能なヘルペスウイルスプロモーター、ならびにワクシニアまたはポックスウイルス誘導可能プロモーターを含む。
【0078】
ヘルパーウイルス誘導可能プロモーターの同定および試験のための方法は、記載されている(例えばWO96/17947号参照)。従って、候補プロモーターがヘルパーウイルス誘導可能であるかどうかおよびこれらが高効率パッケージング細胞の産生に有用であるかどうかを決定する方法は当該技術分野で公知である。要約すると、一つのこのような方法は、AAVrep遺伝子のp5プロモーターを推定ヘルパーウイルス誘導可能プロモーターと置換することを含む(当該技術分野で既知であるかまたは周知の技術、例えばプロモーターを含まない「レポーター」遺伝子への連結を用いて同定)。AAVrep−cap遺伝子(p5置換を有する)、好ましくは陽性選択可能標識に連結したもの、例えば抗生物質耐性遺伝子を、次いで適当な宿主細胞(例えばHeLaまたはA549細胞、以下に例示)中に安定に組み込む。選択条件(例えば抗生物質の存在下)で比較的良く生育する細胞を、次いでヘルパーウイルスを加えてrepおよびcap遺伝子を発現するためのこれらの能力に関して試験する。repおよび/またはcap発現のための最初の試験として、細胞はRepおよび/またはCapタンパク質を検出する免疫蛍光を用いて容易にスクリーニングできる。次いで、パッケージング能力および効率の確認は、侵入するrAAVベクターの複製およびパッケージングに対する機能試験により決定できる。この方法を用いて、マウスのメタロチオネイン遺伝子から誘導したヘルパー−ウイルス−誘導可能プロモーターが、p5プロモーターに対する適当な置換として同定され、そしてrAAV粒子の高い力価を生成するために使用される(WO96/17947に記載のように)。
【0079】
種々のAAVゲノムの相対キャプシド化サイズ限界を考慮すると、ゲノム内への大きい非相同ポリヌクレオチドの挿入が、AAV配列の一部分の除去を必要とする。1個またはそれ以上の遺伝子の除去は、複製可能なAAV(”RCA”)産生の見込みを低下するためにいかなる場合にも望ましい。従って、rep、capまたは双方のコード化またはプロモーター配列は、好ましくは除去されるが、それはこれらの遺伝子により与えられる機能は、トランス状態で与えられることができるからである。
【0080】
得られたベクターは、これらの機能に関して「欠失」していると呼ばれる。ベクターを複製およびパッケージするために、欠失した機能をパッケージング遺伝子または複数のこれら遺伝子で補足し、これらは一緒になって種々の欠失したrepおよび/またはcap遺伝子産物に必要な機能をコードする。パッケージング遺伝子または遺伝子カセットは、好ましくはAAV ITRで近接されず、そして好ましくはrAAVゲノムといかなる本質的な相同性も共有しない。従って、ベクター配列と別途に供給されたパッケージング遺伝子との間の複製の間の相同組換えを最小にするために、2個のポリヌクレオチド配列の重複を回避することが望ましい。組み替えの相同性のレベルおよび相当する頻度は、相同配列の長さおよびこれらの共有する部分のレベルが増加するとと増加する。与えられた系で関心を呼び起こす相同のレベルは、理論的に決定しそして実験的に確認でき、これは当該技術分野では公知である。しかし、代表的には、全長の少なくとも80%一致の際に重複配列が約25ヌクレオチド配列以下の場合、または全長の少なくとも70%一致の際に重複配列が約50ヌクレオチド配列以下の場合に、組換えは本質的に低下または排除される。当然ながら、一層低いレベルの相同性が望ましく、それはこれが組み替えの見込みをさらに低下させるからである。いかなる重複がなくても、RCA産生のいくらかの残留頻度はあるように見える。RCA産生の頻度のこれ以上の低下(例えば非相同性組換えにより)が、AAVの複製およびキャプシド化機能の「切除」により得ることがきで、これはアレンら(Allen et al., WO98/27204)に記載されている。
【0081】
AAVベクター構築物、および相補パッケージング遺伝子構築物は、本発明中で多数の異なる形で実行できる。ウイルス粒子、プラスミド、および安定に形質転換された宿主細胞は、すべてこのような構築物をパッケージング細胞内に、一時的または永久に導入するために使用できる。
【0082】
本発明のある態様では、AAVベクターおよび存在する場合に相補パッケージング遺伝子は、細菌プラスミド、AAV粒子、またはこれらのあらゆる組み合わせの形で提供される。他の態様では、AAVベクター配列、パッケージング遺伝子、または双方のいずれかが、遺伝子的に改変(好ましくは遺伝的に改変)された真核細胞の形で提供される。AAVベクター配列、AAVパッケージング遺伝子、または双方を発現するように遺伝的に改変された宿主細胞の開発は、信頼できるレベルで発現される物質の確立された供給源を提供する。
【0083】
従って、種々の異なる遺伝子的に改変された細胞は、本発明の範囲内で使用できる。例示として、哺乳類宿主細胞は、安定に組み込まれたrAAVベクターの少なくとも1個の不変のコピーと一緒に使用してもよい。プロモーターに操作的に連結した少なくとも1個のAAV rep遺伝子を含んでなるAAVパッケージングプラスミドは、複製機能を提供するために使用できる(米国特許(US)第5,658,776号に記載の通り)。あるいは、プロモーターに操作的に連結したAAV rep遺伝子を有する安定な哺乳類細胞株は、複製機能を提供するために使用できる(例えばTrempe et al., WO95/13392 号; Burstein et al(WO98/23018 号);およびJohnson et al(米国特許(US)第5,656,785 号) 参照) 。上記のようにキャプシド化タンパク質を提供するAAV cap遺伝子は、AAV rep遺伝子と一緒または別々に提供できる(例えば以上に引用した出願および特許ならびにAllen et al. (WO98/27204号) 参照)。その他の組み合わせも可能であり、本発明の範囲内に含まれる。
III.rAAV作製
遺伝子治療などの目的に有用な組換えAAV粒子を作製するために、パッケージング細胞株に、好ましくは、1個またはそれ以上の関係するポリヌクレオチド(すなわち標的ポリヌクレオチド)の周囲のAAV逆方向末端反復(ITR)領域を含んでなる組換えAAVベクターが供給される。
【0084】
標的ポリヌクレオチドは、一般にそれ自体または非相同プロモーターのいずれかのプロモーターに操作的に連結している。多数の適合するプロモーターが当該技術分野では公知であり、その選択は標的ポリヌクレオチドの発現の希望するレベル(すなわち、構成性発現、誘導性発現、細胞特異性または組織特異性発現などのいずれを希望するか)に依存する。
【0085】
好ましくは、rAAVベクターは、rAAVベクターによりすでに感染している細胞の選択を可能とするために陽性選択可能標識も含む。必要または希望されるようになった場合にこれらの同じ細胞に対する選択の手段として、陰性選択可能標識が含まれてもよい。好ましい態様では、S.D. Lupton が記載した「二官能性選択可能融合遺伝子」が使用できる。例えばPCT/US91/08442号およびPCT/US94/05601号参照。要約すると、これらの構築物は、優勢な陽性選択可能標識と陰性選択可能標識の間の直接翻訳融合を含む、好ましい陽性選択可能標識は、hph、neo、およびgptから成る群より選ばれる遺伝子から誘導され、そして好ましい陰性選択可能遺伝子は、シトシンデアミナーゼ、HSV−1 TK、VZV TK、HPRT、APRTおよびgptから成る群より選ばれる遺伝子から誘導される。特に好ましい標識は、二官能性選択可能融合遺伝子であり、ここで陽性選択可能標識はhphまたはneoから誘導され、そして陰性選択可能標識はシトシンデアミナーゼまたはTK遺伝子から誘導される。
【0086】
有用な標的ポチヌクレオチドは、多数の異なる応用のためにrAAVベクター中に使用できる。このようなポリヌクレオチドは、下記を含むが、これに限定はされない。(i)構造タンパク質または酵素の欠失、欠陥または最適でないレベルにより起きる欠失を救済するための遺伝子治療の他の形に有用なタンパク質をコードするポリヌクレオチド、(ii)アンチセンス分子内に転写されるポリヌクレオチド、(iii)転写または翻訳因子を結合するデコイ内に転写されるポリヌクレオチド、(iv)細胞調節因子、例えばサイトカインをコードするポリヌクレオチド、(v)特異性薬剤、例えばレシピエント細胞をヘルペスウイルスチミジンキナーゼ遺伝子を可能とするレシピエント産生ができるポリヌクレオチド、および(VI)ガン治療のためのポリヌクレオチド、例えば、ある種の肺ガンおよび乳ガンに関連する欠損または破壊されたp53遺伝子の置換のための野生型p53ガンサプレッサーcDNA、または乳ガンおよび卵巣ガンを含む各種の異なるガンの腫瘍発生および/または転移を阻害する能力があるE1Aガンサプレッサー遺伝子。
【0087】
得られた組換えAAV粒子の治療特異性は、導入された特定のベクターまたはプロ−ベクターにより決定されるので、同じ基本パッケージング細胞株がこれらのいずれの応用に対しても変性できる。例えば、特異性標的ポリヌクレオチドを含んでなるベクターは、数種の可能な方法のいずれかによりAAVベクターの産生のためのパッケージング細胞内に導入でき、これには、例えばrAAVプロ−ベクターを含んでなるプラスミドの電気さく孔またはトランスフェクション、またはrAAVベクターまたはプロベクターを含んでなるrAAVまたはヘルパーウイルスを用いる感染が含まれる。
【0088】
ヘルペスウイルスは、rAAVベクターの導入の前、その間またはその後に導入できる。例えば、プラスミドは、ヘルパーウイルスと一緒に培地内に同時感染でき、次いで、細胞を十分な期間、代表的には2〜5日間、当該技術分野では公知の複製およびパッケージングのために適する条件下で培養できる(引用文献は上記、そして実施例は下記参照)。溶解物を調製し、そして組換えAAVベクター粒子を当該技術分野では公知の方法を用いて精製する。
【0089】
下記の実施例でも説明する好ましい態様では、組換えAAVベクターは、自体が、パッケージングのために使用される哺乳類細胞内に安定に組み込まれる。次いで、このようなrAAV「産生細胞」を培養し、使用するまで保管する。このような産生細胞からrAAV粒子の産生を誘導するために、使用者は細胞をヘルパーウイルスに感染させそしてAAVの複製およびパッケージングに適する条件下で細胞を培養するだけでよい(下記の通り)。
【0090】
あるいは、1種またはそれ以上のAAVスプリット−パッケージング遺伝子またはrAAVベクターは組換えヘルパーウイルスの一部分として導入できる。例えば、アデノウイルスのE1、E3および/またはE4遺伝子は、1種またはそれ以上のスプリット−パッケージング遺伝子またはrAAVベクターと置換できる。アデノウイルスベクター、例えばE1および/またはE3領域内へのクローニングを容易にするための技術は、当該技術分野では公知である(例えばBett,A.J. et al., Proc. Natl. Acad. Sci. USA, 91, 8802-8806 (1994) 参照)。従って、組換えアデノウイルスのようなヘルパーウイルスは、ヘルパーウイルス機能を与えるために使用でき、ならびにAAVパッケージング遺伝子および/またはrAAVプロ−ベクターも同様であり、これは(当該技術分野では公知のように)このようなヘルパーウイルス内の多数の遺伝子(例えばアデノウイルスのE3遺伝子)が、ヘルパーウイルス活性を失うことなく置換できるからである。トランス状態であらゆる必要なヘルパーウイルス機能を提供することにより、追加の遺伝子がのようなヘルパーウイルス内に挿入できる。例えば、ヒト293細胞は、アデノウイルスE1変異体を補足できるアデノウイルス遺伝子を含む。従って、細胞がこのようなアデノウイルス機能をトランス状態で有効に提供できる細胞内で使用するために、非相同遺伝子もE1遺伝子が欠失しているアデノウイルス内にクローニングできる。あるいは、ヘルパーウイルスの使用は、パッケージング細胞内にすべての必要なヘルパーウイルス機能を提供することで不要となる。
IV.細胞内への遺伝物質の導入
当該技術分野に記載されそして本明細書中および上記の引用文献中に例示されているように、遺伝物質はこのような細胞を形質転換または形質導入する各種の手段のいずれかを用いて細胞(例えばAAV産生のための哺乳類「産生細胞)内に導入できる。例示として、このような技術は、例えば細菌プラスミドを用いるトランスフェクション、ウイルスベクターを用いる感染、電気さく孔、リン酸カルシウム沈降、および種々の脂質に基づく組成物のいずれかを用いる導入(例えばしばしば「リポフェクション」と呼ばれる方法)を含む。これらの技術を実行するための方法および組成物は、当該技術分野で記載され、広く入手可能である。
【0091】
適当に改変された細胞の選択は、当該技術分野のいかなる技術によっても実行できる、例えば、細胞を改変するために使用されるポリヌクレオチド配列は、当該技術分野では公知の1種またはそれ以上の検出可能または選択的に検出可能な標識に同時に導入または操作的に連結できる。例示として、選択可能な標識として薬剤耐性遺伝子を使用できる。次いで、薬剤耐性細胞を取り出して培養し、次いで所望の配列、すなわちパッケージング遺伝子産物、または非相同ポリヌクレオチドの産物の発現を適宜試験する。導入したポリヌクレオチドの取得、定位および/または維持に対する試験は、DNAハイブリダゼーションに基づく技術(例えばサザンブロッティングおよびその他の当該技術分野で公知の方法)を用いて行うことができる。発現のための試験は、遺伝子的に改変された細胞から抽出したRNAのノーザン分析により、または相当する遺伝子産物に対する間接免疫蛍光により容易に行うことができる。パッケージング能力および効率の試験および確認は、AAVおよびヘルパーウイルスの残った機能成分を細胞に導入してAAV粒子の産生を試験できる。細胞が複数のポリヌクレオチド構築物を用いて遺伝的に改変されている場合には、これらを細胞に分離して導入し、そして各段階を順次実証すると一般的にさらに便利である(本質的ではないが)。このような技術を記載した引用文献は、本明細書中の引用文献に含まれる。
V.ヘルパーウイルスの選択および調製
以上に考察したように、AAVは自己複製を欠失したパルボウイルスであり、そして一般にある種の複製機能を提供するためにヘルパーウイルスに依存しなければならない。多数のこのようなヘルパーウイルスが同定され、これにはアデノウイルス、ヘルペスウイルス(HSV1、サイトメガリウイルスおよびHHV−6を含み、これらには限定されない)、およびポックスウイルス(とくにはワクシニア)を含む。このようなあらゆるウイルスを本発明に使用してもよい。
【0092】
しばしば、ヘルパーウイルスは目的とする宿主細胞を感染できるタイプおよびサブグループのアデノウイルスである。サブグループC、特には血清型1、2、4、6および7のヒトアデノウイルスが一般に使用される。血清型5が一般に好ましい。
【0093】
アデノウイルスの特徴および生育パターンは、当該技術分野では公知である。読者は、例えばHorowitz "Adenoviridae and their replication", pp771-816、Fundamental Virology, Fields et al., eds、が参照できる。パッケージされたアデノウイルスゲノムは、線状DNA分子であり、左手および右手末端において末端タンパク質複合体を通じてアデノウイルスITRを通じて連結し、環を形成する。初期、中間および後期成分に対する制御およびコード化領域はゲノム内で重複している。初期領域遺伝子は、アデノウイルスゲノムの複製に関係し、そしてE1、E2、E3およびE4領域内へのこれらの位置に依存して分類される。
【0094】
本質的ではないけれども、原理的に、ヘルパーウイルス株が、最終的に遺伝子治療を受けるために最終的に患者内での複製に対して欠失していることが望ましい。従って、rAAV調製物内に存在するあらゆる残留ヘルパーウイルスは、複製不能である。E1AまたはE1AとE3領域の両方が欠失しているアデノウイルスは殆どのヒト細胞に感染性ではない。これらは欠失した活性を補足できる許容細胞株(例えばヒト293細胞株)内で複製できる。ヘルパー機能と関連するように見えるアデノウイルスの領域、ならびに関連しない領域が同定され、当該技術分野で記載されている(例えばP. Colosi et al., WO97/17458号、およびこの中に引用されている文献参照)。
VI.遺伝子治療のためのrAAVの使用
AAVベクターは、遺伝子治療の目的で個体に投与するために使用できる。遺伝子治療に適する疾患は、ウイルス、細菌、または寄生虫感染、種々の悪性疾患および高度増殖性状態(hyperproliferation)、自己免疫状態、および先天性欠陥により誘導されるものを含み、これらの限定はされない。
【0095】
遺伝子治療は、細胞内または細胞により分泌される特定のタンパク質の発現のレベルを増加して行うことができる。本発明のベクターは、遺伝子標識、欠失または欠陥遺伝子の置換、または治療遺伝子の挿入のいずれかのために細胞と遺伝的に改変するために使用してもよい。あるいは、発現のレベルを低下させる細胞にポリヌクレオチドを供給してもよい。これは、望ましくない表現型、例えば悪性疾患の経過の間での増幅または過剰発現される遺伝子、または微生物感染の経過の間で導入または過剰発現された遺伝子の産物の抑制のために使用してもよい。発現レベルは、例えば標的遺伝子またはRNA転写物(アンチセンス治療)のいずれかとの安定なハイブリッドを形成する能力、関連するmRNAを切断するためのリボザイムとして作用する能力、または標的遺伝子の産物に対するデコイとして作用する能力がある配列を含んでなる治療ポリヌクレオチドの供給により低下させてもよい。
【0096】
本発明の方法によるrAAVベクターの導入は、当該技術分野で利用できて周知であるあらゆる数の送達技術(外科的または非外科的)の使用を含む。このような送達技術は、例えば血管カテーテル挿入、カニューレ挿入、注射、吸入、塗擦、局所、経口、経皮、動脈内、静脈内、および/または腹腔投与を含む。ベクターは、例えば例示として、人工血管(PTFEまたはダクロン)、心臓弁、血管内ステント、血管内ペーヴィング(intravascular paving)ならびにその他の非血管補てつ物を含むバイオプロステーゼにより導入されることができる。ベクター溶液の送達、頻度、組成および投与範囲に関する一般的技術は、当該技術分野の技術の範囲内である。
【0097】
特に、本発明のベクターの組織への送達のために、ベクターを宿主動物に導入するあらゆる物理的および生物学的方法が使用できる。ベクターは、裸の組換えベクターおよびウイルス外被タンパク質内にパッケージされたベクターDNAの両方を意味し、これはAAV投与のために周知である。AAVベクターをリン酸塩緩衝生理食塩水内に単に溶解することは、筋肉組織発現のために有用な媒体を供給するために十分であることが確証されており、そしてベクターと同時投与できるキャリヤーまたは他の成分に既知の制限はない(しかしDNAを分解する成分はベクターの正常の方法では回避される)。薬剤組成物は、注射用製剤としてまたは経皮輸送による筋肉への送達するために局所用製剤として調製できる。筋肉内注射および経皮輸送の両者のための多数の製剤がこれまで開発されそして本発明の実施のために使用できる、ベクターは、投与および取り扱いを容易にするためにあらゆる薬剤的に許容できるキャリヤーと一緒に使用できる。
【0098】
筋肉内注射の目的で、アジュバント、例えばゴマ油またはラッカセイ油またはプロピレングリコール内の溶液が使用でき、ならびに滅菌した水溶液も使用できる。このような水溶液は、所望の場合には緩衝してもよく、そして液体希釈剤は、最初に生理食塩水またはグルコースを用いて等張性を与えられる。遊離酸(酸性リン酸基を含むDNA)または薬剤的に許容できる塩としてのAAVベクターの溶液は、界面活性剤、例えばヒドロキシプロピルセルロースと適当に混合された水中で調製できる。AAVウイルス粒子の分散は、グリセロール、液状ポリエチレングリコールおよびこれらの混合物中および油中でも調製できる。保管および使用の通常の条件下で、これらの調製剤は、微生物の生育を防止するために保存剤を含む。これに関して、使用される滅菌水性媒体は、当該技術分野の熟練者には周知の標準技術により容易に得られる。
【0099】
注射用に適する剤形は、滅菌液剤または分散剤および滅菌注射液剤または分散剤の即時調合のための滅菌散剤を含む。すべての場合に、剤形は滅菌されていなければならず、そして容易に注射器で使用できるように液状でなければならない。これは製造よび貯蔵の条件下で安定でなければならず、そして微生物、例えば細菌および真菌の汚染作用に対抗して保存されなければならない。キャリヤーは、例えば水、エタノール、ポリオール(例えばグリセロール、プロピレングリコール、液状ポリエチレングリコールなど)を含む溶剤または分散剤、これらの適当な混合物、および植物油であることができる。例えば被覆、例えばレシチンの使用により、分散液の場合に所要の粒径の維持により、または界面活性剤の使用により適当な流動性を維持できる。微生物の作用の防止は、種々の抗細菌および抗真菌剤、例えばパラベン、クロロブタノール、フェノール、ソルビン酸、チメロサールなどにより得ることができる。多くの場合に、等張剤、例えば砂糖または塩化ナトリウムを含むことも好ましい。注射用組成物の長期の吸収は、吸収遅延のための薬剤、例えばモノステアリン酸アルミニウムおよびゼラチンを使用して得ることができる。
【0100】
滅菌注射用液剤は、必要量のAAVベクターを種々の上記の他の成分を必要に応じて含む適当な溶剤中に入れ、次いで濾過滅菌を行って調製される。一般に、分散液は、滅菌した有効成分を、塩基性分散媒体よび所要の上記の他の成分を含む滅菌したビヒクル中に入れて調製される。滅菌した注射用液剤の調製のための滅菌粉末の場合には、調製の好ましい方法は、有効成分の粉末に加えて上記のこれらの滅菌濾過溶液からのあらゆる追加の所望の成分を生成する真空乾燥および凍結乾燥技術である。
【0101】
局所投与の目的には、それ以外は上記の非蛍光溶液に同じ希薄滅菌、水溶液(通常濃度約0.1〜5%)が、経皮貼付剤内への混和に適する容器内で調製され、そして公知のキャリヤー、例えば薬品グレードのジメチルスルホキシド(DMSO)を含むこともできる。
【0102】
特に重要なことは、適切に作用する嚢胞性繊維症膜貫通コンダクタンス調節剤(CFTR)の気道上皮への供給による嚢胞性繊維症の遺伝子欠陥の修正である。従って、本来のCFTRタンパク質をコードするrAAVベクター、およびこれらの変異体および断片は、すべて本発明の好ましい態様である。
【0103】
本発明の組成物は、生体内でも生体外でも使用してよい。生体内遺伝子治療は、本発明のベクターを直接患者に投与することを含んでなる。薬剤組成物は液状の溶液または懸濁液として、エマルションとして、または使用前に液体内に溶解または懸濁するために適する固体形で供給することができる。気道内に投与するために、投与の好ましい方法は、適当な噴霧装置を用いて固体または液体のいずれかのエーロゾルを供給する組成物を用いるエーロゾルによる。気道へ好ましい他の投与方法では、ベクターを点滴注入するための可とう性光ファイバー気管支鏡を用いる。代表的には、ウイルスベクターは、薬剤的に許容でき、発熱物質を含まない緩衝液、例えばリンゲル平衡塩溶液(pH7.4)内に入れる。要求はされないけれども、薬剤組成物は、場合により正確な量の投与に適する単位用量形として供給してもよい。
【0104】
処置する患者に応じて、有効量のウイルスを投与する。有効量は、一回または分割した投与で与えてもよい。低いパーセントの形質導入で遺伝的欠陥を治療できる場合には、処置の目的は、一般に形質導入のこのレベルに達するかまたは越えることである。 ある場合には、この形質導入レベルは、標的細胞の約1〜5%の形質導入だけで達成されるが、しかしさらに代表的には希望する組織タイプの細胞の20%、通常は少なくとも約50%、好ましくは少なくとも約80%、さらに好ましくは少なくとも約95%、その上さらに好ましくは希望する組成タイプの細胞の少なくとも約99%である。指針として、気管支鏡により投与される一回投与中に存在するベクター粒子の数は、一般に少なくとも約1x108 、さらに代表的には5x108 、1x1010、およびある場合には1x1011粒子であり、これらはDNアーゼ耐性およびDNアーゼ感受性粒子の両方を含む。DNアーゼ耐性粒子として、用量は一般に1x106 〜1x1014粒子の間、さらに一般的には1x108 〜1x1012粒子の間である。処置は、2または3週間毎に必要に応じて反復ができるが、しかし180日の間に1回の処置で十分であろう。
【0105】
宿主細胞中の所望のDNA配列の存在を確認するために、種々のアッセイを行ってもよい。このようなアッセイには、例えば当該技術分野の熟練者には周知の「分子生物学的」アッセイ、例えばサザンおよびノーザンブロット法、RT−PCRおよびPCR、「生物化学的」アッセイ、例えば免疫学的手段(免疫沈降、免疫アフィニティーカラム、ELISAおよびウエスタンブロット法)によるベクター内に存在する遺伝子から発現されたポリペプチドの存在を検出する方法、または本発明の範囲内に入る特定の核酸分子の存在および/または発現を同定するために有用な他のアッセイを含む。
【0106】
導入されたDNAセグメントから産生されたRNAを検出および定量するために、RT−PCRが使用される。PCRのこの応用において、最初に逆転写酵素などの酵素を用いてRNAをDNAに逆転写することが必要であり、次いで慣用のPCR技術を用いてDNAを増幅する。多くの場合に、有用なPCR技術は、RNA産物の完全性を証明しない。RNA産物の性質に関するこれ以上の情報は、ノーザンブロッティングにより得てもよい。この技術は、RNA種の存在を証明しそしてそのRNAの完全性に関する情報を与える。RNA種の存在または不在は、ドット(dot) またはスロットブロット(slot blot) ノーザンハイブリダイゼーションを用いて決定もできる。これらの技術は、ノーザンブロット法の変形であり、RNA種の存在または不在を証明するだけである。
【0107】
サザンブロット法およびPCRは問題のDNAセグメントを検出するために使用できるが、これらはDNAセグメントが発現されているかどうかに関する情報は提供しない。発現は、導入されたDNA配列のポリペプチド産物を特異的に同定するかまたは宿主細胞内に導入されたDNAセグメントの発現によりもたらされる表現型変化を評価して評価してもよい。
【0108】
従って、遺伝子改変の有効性は、種々の基準により測定できる。生検または外科摘出により取り出された試料は、in situ ハイブリダゼーション、ベクター特異性プローブを用いるPCR増幅、RNアーゼプロテクション、免疫組織学、または免疫蛍光細胞計数により分析してもよい。ベクターが気管支鏡により投与される場合には、肺機能試験を行ってもよく、そして炎症性サイトカインの存在に関して気管支洗浄を行ってもよい。処置された患者は臨床徴候に関して検査してもよく、そして治療用ポリヌクレオチドにより運搬された筈の機能を細胞が発現するかどうかを決定する。
【0109】
生体内および生体外治療を使用するかどうか、および特定の組成物、用量、および投与経路の選択の決定は、処置する状態および患者の特徴を含みこれに限定はされない多数の異なる因子に依存する。このような特徴および適当な治療方式の設計の評価は、最終的には処方する医師の責任である。
【0110】
上記は、なかでもヘルパーウイルス(例えばアデノウイルス)および細胞タンパク質を本質的に含まない組換えAAVベクターの高力価調剤を作製するための方法を提供する。本発明の精神から離れることなく、当該技術分野の熟練者によりこれらの方法に対して変更を与えることが可能と理解される。
VII.本発明の実施に有用な薬剤
本発明の実施に有用な薬剤は、rAAV形質導入効率を改変する薬剤を含む。好ましい薬剤は、rAAV形質導入を増強または増加する薬剤を含む。このような薬剤は、ウイルスエンドサイトーシスを増加する薬剤、例えばブレフェルジンA、エンドソームプロセシングおよび/または核への転送では例えばシステインプロテアーゼ阻害剤を含む。好ましくは、阻害剤は、エンドソーム、例えばリソソーム性システインプロテアーゼ阻害剤である。さらに好ましくは、本発明の薬剤は可逆性システインプロテアーゼ阻害剤である。本発明の範囲内のシステインプロテアーゼ阻害剤は、シスタチン、例えばシスタチンBまたはシスタチンC、アンチパイン、ロイペプチン、E−64、E−64c、E−64d、KO2(Wacher et al., J. Pharma. Sci. 87. 1322(1988) )、LLnL、Z−LLL、CBZ−Val−Phe−H、システインプロテアーゼ阻害剤、例えば米国特許(US)第5,607,831 号、第5,374,623 号、第5,639,732 号、第5,658,906 号、第5,714,484 号、第5,560,937 号、第5,374,623 号、第5,607,831 号、第5,723,580 号、第5,744,339 号、第5,827,877 号、第5,852,007 号、および第5,776,718号、特開平10-77276号、特開平8-198870号、特開平8-81431 号、特開平7-126294号、特開平4-202170号、WO96/21006号およびWO96/40737号に開示されたものを含む。
【0111】
好ましいシステインプロテアーゼ阻害剤は、ペプチドおよびこれらの類似体である。本発明の範囲内の好ましいペプチドシステインプロテアーゼ阻害剤は、アミノ酸残基2〜20個、好ましくは3〜10個、さらに好ましくは3〜8個を含んでなる。「アミノ酸」は、DまたはL形の天然アミノ酸(例えばAla、Arg、Asn、Asp、Cys、Glu、Gln、Gly、His、Hyl、Hyp、Ile、Leu、Lys、Met、Phe、Pro、Ser、Thr、Trp、Tyr、およびVal)、ならびに非天然アミノ酸(例えばホスホセリン、ホスホトレオニン、ホスホチロシン、ヒドロキシプロリン、γ−カルボキシグルタミン酸、馬尿酸、オクタヒドロインドール−2−カルボン酸、スタチン、1,2,3,4−テトラヒドロイソキノリン−3−カルボン酸、ペニシラミン、オルニチン、シトルリン、α−メチル−アラニン、パラ−ベンゾイルフェニルアラニン、フェニルグリシン、プロパルギルグリシン、サルコシン、ノル−ロイシン、ノル−バリン、およびt−ブチルグリシン)の残基を含んでなる。ペプチド類似体は、少なくとも1種のD形のアミノ酸および/または非天然アミノ酸、またはその他の非天然アミノ酸部分を含んでなる分子である。
【0112】
好ましいペプチドシステインプロテアーゼ阻害剤は、式(I):R1 −A−(B)n −C
〔式中、R1 はN−末端アミノ酸ブロック基であり、AおよびBそれぞれは独立してアミノ酸であり、Cはアミノ酸であり、ここで末端カルボキシ基はホルミル(CHO)基により置換されており、そしてnは0、1、2または3である〕の化合物またはその薬剤的に許容できる塩を含む。一つの好ましい態様では、R1 が(C1 〜C10)アルカノイル、アセチルまたはベンジルオキシカルボニルである。別の好ましい態様では、AおよびBそれぞれが、独立してアラニン、アルギニン、グリシン、イソロイシン、ロイシン、バリン、ノル−ロイシンまたはノル−バリンであり、さらに好ましいくは、AおよびBそれぞれがイソロイシンである。さらに別の好ましい態様では、Cがアラニン、アルギニン、グリシン、イソロイシン、ロイシン、バリン、ノル−ロイシンまたはノル−バリン〔式中、末端カルボキシ基がホルミル(CHO)基により置換されている〕であり、そしてさらに好ましくは、Cがノル−ロイシンまたはノル−バリン〔式中、末端カルボキシ基がホルミル(CHO)基により置換されている〕である。
【0113】
さらに好ましい態様では、R1 が(C1 〜C10)アルカノイルまたはベンジルオキシカルボニルであり、AおよびBがそれぞれイソロイシンであり、Cがノル−ロイシンまたはノル−バリンであり〔式中、末端カルボキシ基がホルミル(CHO)基により置換されている〕、そしてNが1である。
【0114】
本発明の範囲内に含まれるのは、式(II)
【0115】
【化7】
〔式中、
R2 は、N−末端アミノ酸ブロック基であり、
R3 、R4 およびR5 は、それぞれ独立して水素、(C1 〜C10)アルキル、アリールまたはアリール(C1 〜C10)アルキルであり、そして
R6 、R7 およびR8 は、それぞれ独立して水素、(C1 〜C10)アルキル、アリールまたはアリール(C1 〜C10)アルキルである〕の化合物またはこの薬剤的に許容できる塩である。好ましくは、R2 が(C1 〜C10)アルカノイル、アセリルまたはベンジルオキシカルボニルである。また好ましくはR3 が水素または(C1 〜C10)アルキル、例えば2−メチルプロピルである。R4 が水素または(C1 〜C10)アルキル、例えば2−メチルプロピルが好ましい。
【0117】
別の好ましい態様では、R5 が水素または(C1 〜C10)アルキル、例えばブチルまたはプロピルである。
【0118】
さらに別の好ましい態様では、R2 がアセチルまたはベンジルオキシカルボニルであり、R3 およびR4 がそれぞれ2−メチルプロピルであり、R5 がブチルまたはプロピルであり、そしてR6 、R7 およびR8 は、それぞれ独立して水素である。
【0119】
本発明の方法に有用な別の好ましい薬剤は、式(III)
【0120】
【化8】
〔式中、
R1 は、H、ハロゲン、(C1 〜C10)アルキル、(C1 〜C10)アルケニル、(C1 〜C10)アルキニル、(C1 〜C10)アルコキシ、(C1 〜C10)アルカノイル、(=O)、(=S)、OH、SR、CN、NO2 、トリフルオロメチルまたは(C1 〜C10)アルコキシであり、ここであらゆるアルキル、アルケニル、アルキニル、アルコキシまたはアルカノイルは場合により1個またはそれ以上のハロゲン、OH、SH、CN、NO2 、トリフルオロメチル、NRRまたはSRで置換されてもよく、ここで、それぞれのRは独立してHまたは(C1 〜C10)アルキルであり、
R2 は、(=O)または(=S)であり、
R3 は、H、(C1 〜C10)アルキル、(C1 〜C10)アルケニル、(C1 〜C10)アルキニル、(C1 〜C10)アルコキシまたは(C3 〜C8 )シクロアルキルであり、ここであらゆるアルキル、アルケニル、アルキニル、アルコキシまたはシクロアルキルは場合により1個またはそれ以上のハロゲン、OH、CN、NO2 、トリフルオロメチル、SR、またはNRRで置換されてもよく、ここで、それぞれのRは独立してHまたは(C1 〜C10)アルキルであり、
R4 は、H、(C1 〜C10)アルキル、(C1 〜C10)アルケニル、(C1 〜C10)アルキニル、(C1 〜C10)アルコキシまたは(C3 〜C8 )シクロアルキルであり、ここであらゆるアルキル、アルケニル、アルキニル、アルコキシまたはシクロアルキルは場合により1個またはそれ以上のハロゲン、OH、CN、NO2 、トリフルオロメチル、SR、またはNRRで置換されてもよく、ここで、それぞれのRは独立してHまたは(C1 〜C10)アルキルであり、
R5 は、H、ハロゲン、(C1 〜C10)アルキル、(C1 〜C10)アルケニル、(C1 〜C10)アルキニル、(C1 〜C10)アルコキシ、(C1 〜C10)アルカノイル、(=O)、(=S)、OH、SR、CN、NO2 、またはトリフルオロメチルであり、ここであらゆるアルキル、アルケニル、アルキニル、アルコキシまたはアルカノイルは場合により1個またはそれ以上のハロゲン、OH、SH、CN、NO2 、トリフルオロメチル、NRRまたはSRで置換されてもよく、ここでそれぞれのRは独立してHまたは(C1 〜C10)アルキルであり、そして
Xは、O、SまたはNR(RはHまたは(C1 〜C10)アルキルである)である〕
の化合物またはこの薬剤的に許容できる塩である。
【0122】
下記の定義は、特に別途断らない限り適用される。アルキルは、直鎖または分枝上鎖の基を表すが、しかし個別の基への言及、例えば「プロピル」は直鎖基のみを含み、分枝状異性体、例えば「イソプロピル」は特定して言及される。アリールはフェニル基または環原子約9〜10個を有するオルト融合二環式炭素環基でその中の少なくとも1個の環は芳香族環であるものを表す。
【0123】
適するN−アミノ酸ブロック基は、当該技術分野の熟練者には公知である(例えば、T.W. Greene, Protecting Groups In Organic Synthesis; Wiley: New York, 1981、および該明細書に引用された参考文献を参照)。R1 の好ましい値は、(C1 〜C10)アルカノイル(例えばアセチル)およびベンジルオキシカルボニルである。
VIII.本発明の薬剤の投与の用量、製剤よび経路
本発明に従って同定された薬剤の投与は、連続または断続であってもよく、これは例えばレシピエントの生理学的条件、投与の目的が治療または予防であるかどうか、および熟練した医師には公知のその他の因子に依存する。本発明の薬剤の投与は、あらかじめ選定された期間にわたり本質的に連続であるか、または間隔をおいた一連の期間で行ってもよい。局所または全身投与の両者が考慮される。本発明の薬剤が予防目的で使用される場合には、本発明の薬剤は、慢性的使用に従うべきであり、好ましくは全身投与による。
【0124】
式(I)、(II)または(III)の化合物のこれらの塩を含む本発明の薬剤は、好ましくは約0.01μM〜約1mM、さらに好ましくは約0.1μM〜約40μM、その上にさらに好ましくは約1μM〜約40μMの用量で投与されるが、しかし他の用量も有益な効果を生じる。例えば、好ましいLLnLの用量は、約1μM〜約40μMを含むが、ZLLの好ましい用量は、約0.1μM〜約4μMを含む。
【0125】
以下に記載のように場合により徐放のために処方してもよい本発明の薬剤を含んでなる1種またはそれ以上の適当な単位用量形は、経口、または直腸、経皮、皮下、静脈内、筋肉内、腹腔内、胸郭内、肺内および鼻経由を含む非経口を含む種々の経路で投与できる。例えば、肝臓への投与には、静脈内投与が好ましい。肺への投与は、気道投与が好ましい。製剤は、適合する場合には、区分された単位用量形で提供してもよくそして薬剤学では周知のあらゆる方法で調製してもよい。このような方法は、薬剤と液状キャリヤー、固体マトリックス、半固体キャリヤー、微細に粉砕した固体キャリヤーまたはこれらの組み合わせと一緒にし、次いで、必要な場合には製品を所望の投与形に賦形または成形する工程を含んでもよい。
【0126】
本発明の薬剤が経口投与のために調製される場合には、これらは好ましくは薬剤的に許容できるキャリヤー、希釈剤または賦形剤と組み合わせて、薬剤製剤または単位用量形に形成する。このような製剤内の全有効成分は、製剤の0.1〜99.9重量%を含んでなる。「薬剤的に許容できる」としては、キャリヤー、希釈剤、賦形剤および/または塩が、製剤の他の成分と許容性であり、そしてこれらのレシピエントに対して無害でなければならないことを意味する。経口投与のための有効成分は、粉末として、または顆粒として、溶液、懸濁液またはエマルションとして、または到達できる基材、例えばチューインガムから有効成分の摂取のための合成樹脂として存在してもよい。有効成分は、大型丸薬、舐薬またはペーストとして提供してもよい。
【0127】
本発明の薬剤を含む製剤は、当該技術分野では周知の方法により、周知で容易に使用できる成分を用いて調製できる。例えば、薬剤は、通常の賦形剤、希釈剤、またはキャリヤーを用いて処方でき、そして錠剤、カプセル、懸濁剤、散剤、などに成形してもよい。このような薬剤に適する賦形剤、希釈剤、またはキャリヤーの例は、下記を含む。充填剤および増量剤、例えばデンプン、糖類、ナンニトール、およびケイ素誘導体、結合剤、例えばカルボキシメチルセルロース、HPMCおよびその他のセルロース誘導体、アルギン酸化合物、ゼラチン、およびポリビニル−ピロリドン、湿潤剤、例えばグリセロール、崩壊剤、例えば炭酸カルシウムおよび炭酸水素ナトリウム、溶解遅延のための薬剤、例えばパラフィン、再吸収促進剤、例えば第四級アンモニウム化合物、界面活性剤、例えばセチルアルコール、モノステアリン酸グリセロール、吸収性キャリヤー、例えばカオリンおよびベントナイト、および滑剤、例えばタルク、ステアリン酸カルシウムおよびステアリン酸マグネシウム、および固体ポリエチルグリコール。
【0128】
例えば、本発明の薬剤を含む錠剤およびカプレットは、緩衝剤、例えば炭酸カルシウム、酸化マグネシウムおよび炭酸マグネシウムを含むことができる。カプレットおよび錠剤は、不活性成分、例えばセルロース、プレゼラチン化したデンプン、二酸化ケイ素、ヒドロキシプロピルメチルセルロース、ステアリン酸マグネシウム、マイクロクリスタリンセルロース、デンプン、タルク、二酸化チタン、安息香酸、クエン酸、トウモロコシデンプン、鉱油、ポリプロピレングリコール、リン酸ナトリウム、およびステアリン酸亜鉛などを含むこともできる。本発明の薬剤を含む硬質および軟質ゼラチンカプセルは、不活性成分、例えばゼラチン、マイクロクリスタリンセルロース、ラウリル硫酸ナトリウム、デンプン、タルクおよび二酸化チタン等ならびに液体ビヒクル、例えばポリエチレングリコール(PEG)および植物油を含むことができる。さらに本発明の薬剤の腸溶被覆カプレットまたは錠剤は、胃内での消化に耐え、そして十二指腸のさらに中性からアルカリ性環境内で溶解するように設計される。
【0129】
本発明の薬剤は、経口投与のためのエリキシル剤または液剤として、または非経口投与、例えば筋肉内、皮下または静脈内経路のために適する液剤として製剤されてもよい。
【0130】
本発明の薬剤の製剤は、水性または非水性の溶液または分散液の形、あるいはエマルションまたは懸濁液の形をとることができる。
【0131】
従って、治療剤は、非経口投与(例えば注射、例えばボーラス注射または連続輸液)のために製剤してもよく、そしてアンプル、充填済注射器、小容量輸液容器の単独用量形または保存剤を加えた複数用量容器内で提供してもよい。有効成分は、油性または水性ビヒクル内の懸濁液、溶液またはエマルションの形を取ってもよく、そして製剤助剤、例えば懸濁剤、安定剤および/または分散剤などを含んでもよい。あるいは、有効成分は、適当なビヒクル、例えば滅菌して発熱物質を含まない水を用いる調剤のために使用前に、滅菌固体の無菌単離により、または溶液からの凍結乾燥により得られる粉末形であってもよい。
【0132】
これらの調剤は、従来技術から周知の薬剤的に許容できるビヒクルおよびアジュバントを含むことができる。例えば、生理学的観点から、水に加えて、溶剤、例えばアセトン、エタノール、イソプロピルアルコール、グリコールエーテル、例えば名称「ダウアノール(Dowanol) 」として販売されている製品、ポリグリコールおよびポリエチレングリコール、短鎖の酸のC1 〜C4 −アルキルエステル、好ましくは酪酸エチルまたは酪酸イソプロピル、脂肪酸トリグリセリド、例えば名称「ミグリオール(Miglyol) 」として販売されている製品、ミリスチン酸イソプロピル、動物油、鉱油および植物油およびポリシロキサンから選択される1種またはそれ以上の有機溶剤を用いて溶液を用いる液剤を調製することが可能である。
【0133】
本発明による組成物は、増粘剤、例えばセルロースおよび/またはセルロース誘導体を含むこともできる。これらは、ガム類、例えばキサンタン、グアールおまたはカルボガムまたはアラビアゴム、またはポリエチレングリコール、ベントン(bentone) およびモンモリロン石等も含むことができる。
【0134】
必要な場合には、抗酸化剤、界面活性剤、その他の保存剤、皮膜形成剤、角質溶解剤、コメド溶解剤(comedolytic agent) 、香料および着色剤から選ばれるアジュバントを加えることも可能である。また、上記状態またはその他の状態のいずれに対するものであっても、他の活性成分を加えてもよい。
【0135】
例えば、抗酸化剤の中では、t−ブチルヒドロキノン、ブチル化ヒドロキシアニソール、ブチル化ヒドロキシトルエンおよびα−トコフェロールおよびその誘導体が挙げられる。主として局所投与のために条件を整える剤形は、クリーム、乳剤、ゲル、分散液またはミクロエマルションの形、高度または低度に濃縮したローション剤、含浸パッド、膏薬または貼付剤、または噴霧または発泡形のエーロゾル製剤の形または石鹸ケークの形を取る。
【0136】
さらに、薬剤は徐放投与形などとしての製剤にも良く適合する。製剤は、有効成分を消化管または気道にのみまたは好ましくはこれらの特定の部分にのみ、可能なら一定の期間にわたって放出するように構成できる。被覆、エンベロープ、および保護マトリックスは、例えばポリマー状物質、例えばポリラクチド−グリコレート、リポソーム、ミクロエマルション、ミクロ粒子、ナノ粒子またはワックスから作製してもよい。これらの被覆、エンベロープ、および保護マトリックスは、留置装置、例えばステント、カテーテル、腹膜透析などを被覆するために有用である。
【0137】
本発明の薬剤は、経皮投与のために貼付剤を介して送達できる。薬剤の経皮送達に適する貼付剤の例については、米国特許(US)第5,560,922 号を参照のこと。経皮送達のための貼付剤は、バッキング層と、薬剤と同時に1種またはそれ以上の皮膚透過助剤を中に分散または溶解したポリマーマトリックスを含んでなることができる。バッキング層は、薬剤に非透過性のあらゆる適当な材料からなることができる。バッキング層はマトリックス層の保護カバーとして役立ち、そして支持機能も与える。バッキングは、ポリマーマトリックスと本質的に同じ大きさであるかまたはポリマーマトリックスの側部分を越えて延びてもよい大きい寸法またはポリマーマトリックスの側を覆い、次いでバッキング層の表面が接着手段のための基部となることができるような形でもよい。あるいは、ポリマーマトリックスは、接着性ポリマー、例えばポリアクリレートまたはアクリレート/酢酸ビニルコポリマーを含むかまたは配合していてもよい。長期の適用のために、皮膚の含水または浸軟を最小にできるように、微細孔および/または通気性バッキングラミネートを用いることも望ましいであろう。
【0138】
バッキング層作製のために適する材料の例は、高密度および低密度ポリエチレン、ポリプロピレン、ポリウレタン、ポリ塩化ビニル、ポリエステル、例えばポリ(エチレンフタレート)、金属フォイル、これらの適当なポリマーフィルムの金属フォイル積層物などである。好ましくは、バッキング層に使用される材料は、このようなポリマーフィルムと金属フォイル、例えばアルミニウムフォイルとの積層物である。このような積層物中で、積層物のポリマーフィルムが、通常接着性ポリマーマトリックスと接触する。
【0139】
バッキング層は、所望の保護および支持機能を与えるために適するあらゆる適当な厚さであることができる。適当な厚さは、約10〜約200μmである。
【0140】
一般に、生物学的に受容できる接着性ポリマー層を形成するために使用されるこれらのポリマーは、薬剤が制御された速度で通過できる成形された本体、薄い壁またはコーティングを形成できるものである。適当なポリマーは、生物学的および薬学的に許容性、非アレルギー性およびこれらが接触する体液または組織中に不溶性で許容性の液体である。可溶性ポリマーの使用は、皮膚水分によるマトリックスの溶解または浸食が、薬剤の放出速度ならびに取り外しに便利なようにその位置にとどまる投与単位の能力に影響するので、避けるべきである。
【0141】
接着性ポリマー層を作製するための材料の例は、ポリエチレン、ポリプロピレン、ポリウレタン、エチレン/プロピレンコポリマー、エチレン/アクリル酸エチルコポリマー、エチレン/酢酸ビニルコポリマー、シリコーンエラストマー、特には薬品グレードポリジメチルシロキサン、ネオプレンゴム、ポリイソブチレン、ポリアクリレート、塩素化ポリエチレン、ポリ塩化ビニル、塩化ビニル−酢酸ビニルコポリマー、架橋ポリメタクリレートポリマー(ヒドロゲル)、ポリ塩化ビニリデン、ポリ(エチレンテレフタレート)、ブチルゴム、エピクロロヒドリンゴム、エチレンビニルアルコールコポリマー、エチレンビニルオキシエタノールコポリマー、シリコーンコポリマー、例えばポリシロキサン−ポリカーボネートコポリマー、ポリシロキサンポリエチレンオキシドコポリマー、ポリシロキサン−ポリメタクリレートコポリマー、ポリシロキサン−アルキレンコポリマー(例えばポリシロキサン−エチレンコポリマー)、ポリシロキサン−アルキレンシランコポリマー(ポリシロキサン−エチレンシランコポリマー)など、セルロースポリマー、例えばメチルまたはエチルセルロース、ヒドロキシプロピルメチルセルロース、およびセルロースエステル、ポリカーボネート、ポリテトラフルオロエチレンなどを含む。
【0142】
好ましくは、生物学的に受容できる接着性ポリマーマトリックスは、室温以下のガラス転移点を有するポリマーから選択しなければならない。ポリマーは室温である程度の結晶度を持ってもよいが、しかし必要ではない。架橋性モノマー単位または部位をこのようなポリマー内に組み込むことができる。例えば、架橋性モノマーは、ポリアクリレートポリマー内に組み込むことができ、これらは薬剤をポリマー内に分散した後にマトリックスを架橋するための部位を提供する。ポリアクリレートポリマーのための既知の架橋性モノマーは、ポリオールのポリメタクリルエステル、例えばブチレンジアクリレートおよびジメタクリレート、トリメチロールプロパントリメタクリレートなどを含む。このような部位を提供するその他のモノマーには、アクリル酸アリル、メタクリル酸アリル、マレイン酸ジアリルなどが含まれる。
【0143】
好ましくは、可塑剤および/または湿潤剤が接着性ポリマーマトリックス中に分散される。水溶性ポリオールが一般にこの目的に適する。調剤中に湿潤剤の組込みは、皮膚表面で投与単位に水分を吸収させ、これはさらに皮膚刺激を低下し、そして送達系の接着性ポリマー層の剥離を防止することに役立つ。
【0144】
経皮送達系から放出された薬剤は、それぞれの皮膚の層を透過できなければならない。薬剤の透過速度を上昇するために、経皮薬剤投与系は、分子透過に最大の抵抗をする皮膚の最外層、角質層の透過性を特に上昇できなければならない。薬剤の経皮投与のための貼付剤の調製は、当該技術分野に周知である。
【0145】
吸入による上部(鼻)または下部気道への投与のために、本発明の薬剤は、吹送器、ネブライザーまたは加圧容器またはその他のエーロゾル噴霧投与に便利な手段から便利に投与される。加圧容器は、適当な噴霧剤、例えばジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、二酸化炭素またはその他の適合する気体を含んでなってもよい。加圧エーロゾルの場合に、投与単位は、一定量を供給する弁を備えて決定されてもよい。
【0146】
あるいは、吸入または吹送による投与のために、組成物は乾燥散剤、例えば薬剤および適当な粉末基剤、例えばラクトースまたはデンプンとの粉末混合物の形を取ってもよい。粉末組成物は、粉末が吸入器、吹送器又は定量吸入器を用いて投与できる例えばカプセルまたはカートリッジ、または例えばゼラチンまたはブリスター包装内の単位投与形内で提供してもよい。
【0147】
鼻内投与のために、薬剤は、点鼻剤、液体噴霧剤、例えばプラスチックビンの噴霧器または定量投与吸入器を介して投与してもよい。代表的な噴霧器は、Mistometer(Wintrop) およびMedihaler(Riker)である。
【0148】
本発明の薬剤の局所投与は、薬剤を疾患の部位またはその近くに送達する各種の技術により行うことができる。部位特異性または標的局部送達技術の例は、利用できる技術を限定する意図ではなく、その例示である。例には、局所送達カテーテル、例えば輸液または留置カテーテル、例えば針輸液カテーテル、シャントおよびステントまたはその他の移植可能な器具、部位指定キャリヤー、直接注射、または直接適用を含む。
【0149】
局所投与のために、薬剤は当該技術分野では公知のようにして標的領域に直接適用してもよい。この目的のための便利な形には、創傷包帯、被覆包帯またはその他のポリマー被覆、膏薬、クリーム、ローション、ペースト、ジェリー、噴霧、およびエーロゾルが含まれる。膏薬およびクリームは、例えば水性または油性基剤に適当な増粘および/またはゲル化剤を加えて製剤してもよい。ローションは、水性または油性基剤を用いて製剤されてもよくそして一般に1種またはそれ以上の乳化剤、安定剤、分散剤、懸濁剤、増粘剤または着色剤を含む。有効成分はイオン導入によっても送達でき、例えば米国特許(US)第4,140,122 号、第4,383,529 号または第4,051,842 号に開示されている。局所製剤における本発明の薬剤の重量百分率は、種々の因子に関係するが、しかし調剤の全重量の0.01%〜95%、そして代表的には0.1〜25重量%である。
【0150】
滴下剤、例えば点眼剤または点鼻剤は、水性または非水性基剤を用いて処方され、同時に1種またはそれ以上の分散剤、可溶化剤または懸濁剤を含んでなってもよい。液状噴霧剤は、便利には加圧容器から送達される。滴下剤は、簡単な点眼用フタ付きビン、または液状内容物を特殊な形状の容器を介して滴下するために適したプラスチックビンを介して送達することができる。
【0151】
薬剤は、さらに口または喉の局所投与のために製剤してもよい。例えば、有効成分を、さらに調味基剤、通常はスクロースまたはアラビアゴムまたはトラガントを含んでなるトローチ剤として、不活性基剤例えばゼラチンおよびグリセリンまたはスクロースおよびアラビアゴム中の組成物を含んでなる芳香錠剤、および適当な液状キャリヤー内に本発明の組成物を含んでなる口洗浄剤として製剤してもよい。
【0152】
本明細書中に記載した製剤および組成物は、その他の成分、例えば抗微生物剤、または保存剤を含んでもよい。さらに、有効成分は、他の薬剤、例えば気管支拡張剤と組み合わせて使用してもよい。
【0153】
本発明の薬剤は、哺乳類に、単独または薬剤的に許容できるキャリヤーと組み合わせて投与してもよい。上記のように、有効成分とキャリヤーとの相対比率は、化合物の溶解度および化合物性質、投与の選択した経路および標準的な調剤方法により決定される。
【0154】
本薬剤の用量は、投与の形、選定した特定の化合物および治療する特定の患者の生理学的性質により変化する。一般に、最初は少量を使用し、必要に応じて、場合により最適量に達するまで少しずつ増量する。
【0155】
本発明は、以下の実施例によりさらに説明するが、限定するものではない。
実施例1
気管支上皮細胞法におけるrAAV形質導入の極性および時間経過
極性ヒト気管支上皮の一次カルチャー。一次ヒト気道上皮細胞を従来の記載のようにして肺移植体からの気管支試料の酵素消化により捕集した(Kondo et al., 1991; Zhang et al., 1995)。単離した気道一次細胞を、コラーゲン被覆ミリセル−HAカルチャーインサート(Millicell-HA culture insert, Millipore Corp., Bedford, MA) 上に密度5x105 細胞/cm2 で接種した。一次カルチャーを空気−液体界面で2週間以上培養し、その時点で粘膜微繊毛上皮への分化が起きた。細胞の側底面側へのみの供給に使用したカルチャー培地は、49%DMEM、49%ハム(Ham) のF12および2%ウルトラサー(Ultraser)G(BioSpectra, Cedex,France) を含んでいた。
【0156】
rAAVの産生。組換えAAVウイルスをCaPO4 同時−トランスフェクションプロトコールにより産生し、そして以前に記載されたように(Duan et al.,1997) 定密度塩化セシウム超遠心分離3回で精製した。プロウイルスプラスミドpCisAV.GFP3oriを、CMVエンハンサー/プロモーターおよびSV40ポリアデニル化シグナルの転写制御下で、GFPレポーター遺伝子をコードするrAAV(AV.GFP3ori)産生に使用した(Duan et al., 1998)。組み合えウイルスストックを58℃で60分間加熱して汚染ヘルパーアデノウイルスを不活性化した。代表的な収量は、プラスミド標準に対するDNAスロットブロットハイブリダゼーションアッセイに基づいて2〜5x1012粒子/mlであった。繁殖に使用した組換えアデノウイルス(Ad. CMVAlkphos;Duan et al., 1997) に対する第二レポターアッセイに基づいて、アデノウイルス汚染のレベルは、rAAVの1x1010DNA粒子当たりに1機能性粒子以下であった(感度限界)。ウイルス調製物は、以前の記載のように(Duan et al., 1998) 、抗Rep抗体(American Research Products, Inc., Belmont, MA) を用いて293細胞に同時感染させたAV.GFP3ori/Ad.CMVLacZの免疫細胞化学染色によりwtAAVの汚染を評価した。感染に1x1010rAAV粒子(感度限界)を使用した場合に、すべてのrAAVストックが、Rep免疫反応性の存在しないことを示した。Rep/Capコーディングプラスミドを用いるトランスフェクションは、Repタンパク質の抗体染色に対する対照として役立った。
【0157】
極性化気道上皮の感染。一次気道カルチャー上に適用する前に、rAAVの精製したストックをPBS中で透析した。気道細胞の感染のために、5μl AV.GFP3ori(5x109 粒子、近似MOI=5000)を培地100ulと混合し、そしてミリセルインサートの頂端側または側底側コンパートメント内に直接適用した。頂端側および側底側の両方の感染に対して、rAAVを含む培地を24時間後に除去し、そして新しい培地で置換(基底側に対し)または空気に暴露(頂端側に対し)した。
【0158】
ヘパリン硫酸プロテオグリカンの免疫蛍光定位。極性化気道上皮内のAAVタイプ−2受容体(膜−会合ヘパリン硫酸プロテオグリカン)の定位を、15分間の4%パラホルムアルデヒド固定、スクロース中の凍結保護、およびOCT中の固定に続いて凍結切片について行った。8μm切片を20%ヤギ血清/PBS中で20分間ブロックし、次いでラット抗ヘパリン硫酸プロテオグリカンモノクローナル抗体(Chemico International Inc., Temecula, CA)の1:200希釈中でインキュベーションした。抗原は、FITC−接合ヤギ抗−ラットIgG(Jackson ImmunoResearch Laboratories, Inc., West Grove,PA)の1:250希釈を用いる間接免疫蛍光法により検出した。核はヨウ化プロピジウム(5μg/ml)を用いて対比染色した。免疫細胞化学染色の特異性は、特異競争性ヘパリン硫酸(Sigma, St. Louis, MO)、または非特異競争性コンドロイチン硫酸C(Sigma, St. Louis, MO)のいずれかを用い、最終濃度5μg/mlで8時間、4℃、次いで切片に抗体を適用して、一次抗体の前染色により行った競争実験で確認した。
結果
気道上皮細胞の極性化カルチャーの感染に著しい極性が証明され、側底膜は頂端膜より200倍形質導入可能であった(図1)。さらに、これらの試験は、レポーターGFP導入遺伝子の発現は、最高レベルに達するために著しい時間(40日間)を要したことを示した。同様の知見は、他のrAAVレポター、例えばAV.RSV Alkphosを用いても得られ、形質導入に要する時間遅れは、導入遺伝子カセットのためではないことを示唆している。免疫蛍光定位法を用いて、既知AAV−2受容体の一つ(膜会合ヘパリン硫酸プロテオグリカン、HSPG)が、分化した気道細胞の基底面に極限された限定された発現のパターンを有することが見いだされた(図2)。
【0159】
従って、rAAVによる極性化気道上皮の形質導入は、頂端面と比較して側底面からの方がかなり大きい。この差は、基底面上のHSPGの制限された定位に少なくとも一部分は起因すると考えられる。HSPGの同じ定位パターンが、独立した抗−HSPG抗体2個を用いる本来のヒト気管支組織で観察された(データは記載しない)。等張EGTA溶液を用いる頂端面の前処理による上皮密着結合の一時的な破壊は、頂端面に適用されたrAAVの形質導入を8倍増加させた(Duan et al., 1998) 。従って、側底膜への限定された接近は、rAAV形質導入に対する障害であろう。気道内でのrAAV形質導入生物学の他の興味ある特徴は、有効な導入遺伝子発現に要する長い時間である。この知見には2種の可能な説明がある。第一は、核へのrAAVの輸送が遅い律速段階である。あるいは、発現可能な二本鎖形への一本鎖rAAVゲノムの転換が律速段階である。
実施例2
側底膜におけるAAV結合
受容体の存在量が、rAAV結合が頂端面で律速であるという意見を支持し、最終的にこの事実はウイルス結合の直接評価を必要とすることを示す。この目的で、エンドサイトーシスが存在しない4℃における結合を研究するために放射能標識ウイルスを使用した。さらに、上記の遺伝子発現試験で記載したと同じ条件で全結合および侵入を研究した。他の系でrAAVを増加することが知られている環境刺激(すなわちUV照射)も評価した。
方法
ウイルス結合および取り込みアッセイ。トリチウム標識AV.GFP3oriを以前記載されたプロトコール(Summerford et al., 1998) に従い、いくつかの変更を加えて調製した。要約すると、pCisAV.GFP3oriおよびpRepCapプラスミドを用いるトランスフェクションおよびAd.CMVLacZを用いる感染の7時間後に、メチル− 3Hチミジン(比放射能3159GBq/ミリモル、NET-027Z, NEN Life Science Products, Inc. MA) を加えて最終濃度1μCi/mlの細胞培地とした。 3H−AV.GFP3oriを精製した。代表的な収量は、3.6x108 粒子/μl、比放射能4x10-7cpm/ビリオンであった。極性化気管支上皮細胞へのrAAVの結合を評価するために、100μl 3H−AV.GFP3ori(MOI=60,000粒子/細胞、全体で1.2x104 cpm、3x1010粒子)を頂端面または側底面のいずれかに上記のように加え、そして4℃で90分間インキュベーションした。分化した気道上皮内へのrAAVの複合結合/取り込みを同じ設定で測定したが、ただし、採取する前にカルチャーを37℃で24時間インキュベーションした。これらの複合ウイルス結合/取り込みアッセイを、終点として導入遺伝子発現を用いるrAAV形質導入の機能性試験に使用しと同じ感染条件で行った。PBS中で3回洗浄した後、in situ でレディーセイフ(Ready Safe)液体シンチレーションカクテル(Beckman Instruments, Inc., Fullerton, CA)5mlを用い、室温で5分間細胞を溶解し、そしてシンチレーションカウンターで放射能を定量した。結合および内部化されたrAAV粒子の量の計算は、 3H−標識ビリオンの既知の比放射能に基づいて行った。
【0160】
UV照射。UV照射実験のためにミリセルインサートを一時的に空の100mm組織カルチャープレート(頂端面が上)内に置き、そして25j/m2 のUV光(254nm)に暴露した。照射の後、ミリセルインサートを、側底面上にウルトラサー(Ultraser)−G培地を含むプレートに迅速に返還した。上記のように支持膜の頂端面または側底面のいずれかに100μl中の5x109 rAAV粒子の適用によるUV照射の直後に、rAAVの感染を行った。
結果
rAAV形質導入効率は、不死化した細胞株および非分裂一次細胞の両者において、細胞の生存率または増殖能力を著しくは改変しない用量においてUV照射により改善できることが従来知られていた(Alexander et al., 1994; Ferrari et al., 1996)。一次カルチャーの頂端側または側底側のいずれかへのrAAVの適用の前のUV照射(25j/m2 )の効果を評価した。図3Aに示すように、ウイルスをUV刺激の後に頂端面に加えた場合に、導入遺伝子発現における30倍の増加が、処理の40日後に観察された。この結果は、UVを用いてAAV形質導入を増加した以前の成功を確認した(Alexander et al., 1994; Ferrari etal., 1996)。興味あることに、AAV感染をUV照射カルチャー室の側底側で行った場合に、効率は、やはり側底側から感染された非照射対照と比較して、感染40日後に2倍低下した(図3B)。これらの結果は、UV照射が、極性化気道一次カルチャー内でrAAV形質導入を調節する能力があることを示唆する。しかし、この調節の大きさおよび方向は感染の細胞面に依存して異なる。UV照射後の頂端面側からの増加した形質導入は、基底および頂端コンパートメント内のAAVの非対称侵入および/またはプロセシング経路によるようである。UV照射後のHSPGの定位研究は、頂端面での検出できる増加は証明しなかったが、しかし基底膜での免疫反応性のほほ完全な低下を証明した(Duan et al., 1988)。これらの知見は、UV照射後のrAAV形質導入における2種類の可能な変動の説明を示唆する。第一は、UV後の基底感染性の低下は、部分的にはHSPG受容体の減少によるものであろう。第二は、検出可能なHSPG受容体が存在しない場合の頂端膜でのrAAV形質導入をUVが増加できることは、AAV結合および取り込みのための別の経路が頂端膜で起きているに違いないことを示唆する。
【0161】
UV増強の機構およびHSPG受容体存在量とrAAV形質導入との相関をさらに研究するために、放射能標識rAAVを結合およびウイルス取り込みを試験するために使用した。これらの研究からの知見は、4℃における側底膜へのより高い結合を確認し、従ってHSPG定位研究を支持する(図4)。しかし、結合の差が6〜7倍に過ぎないことは、結合の他にrAAV形質導入より他の現象が、頂端膜および側底膜からの形質導入における200倍の変動を起こしているに違いないことを示唆する。第二のユニークな知見は、37℃で24時間でのウイルス結合および取り込みが、4℃における頂端膜および側底膜の両方からのものと同等であったことである(図4)。この観察は、ウイルス結合が比較的効率的であり、そしてウイルスの取り込み(またはエンドサイトーシス)がrAAV形質導入に関係する限定因子であることを示唆する。さらに、頂端面形質導入を30倍増加するUV照射に続いて、4℃または37℃のいずれでも頂端面からのウイルス結合/取り込みに検出可能な変化をもたらさない。この知見は、rAAV結合単独ではrAAV形質導入の効率と相関しないことを強調する。
【0162】
頂端膜および側底膜からのrAAV形質導入における差を起こすその他の因子をさらに評価するために、ナイル・レッドビーズ(Nile Red beads)(AAV粒子と類似した大きさ、20μm、を有する)を頂端膜および側底膜からのエンドサイトーシス速度の評価に利用した。これらの研究は、側底膜が頂端膜よりも著しく高い受容体非依存性エンドサイトーシスの速度を有することを証明した(図5)。さらに、UV照射の後に、ナイル・レッドビーズの取り込みが著しく増加した(図5)。これらの知見は、増加したベクター結合が存在しない場合に、UV照射により誘導される律速段階としてのウイルスのエンドサイトーシスを意味する。
【0163】
これらの知見は、極性化気道上皮における頂端膜と側底膜との間の機能的な相違を強調し、そして基底膜における一層高い結合レベルおよび一層高いエンドサイトーシス速度が、増加した形質導入を起こしているらしいことを示唆する。さらに、微繊毛を含む細胞骨格構造の著しい再構成を起こす(Duan et al., 1988)頂端膜へのUV照射が、ウイルス結合における変化がない場合に頂端膜エンドサイトーシスで著しい増加をもたらし、側底膜エンドサイトーシスではもたらさない。これらの研究は、頂端膜からのrAAV形質導入における律速段階としてのエンドサイトーシスの速度および/または核へのウイルスの転送を示唆する。
実施例3
Cy3標識rAAVを用いるウイルスエンドサイトーシスおよび核への転送の評価
AAVのエンドサイトーシスおよび核転送をこれらの過程を調節する種々の薬剤を用いる処理の後で試験するために、蛍光標識ウイルスを調製した。ウイルスを下記のプロトコールを用いて標識した。3回CsClバンド精製した(banded)rAAV(AV.GFP3ori)をAmersham(Piscataway, NJ)からの変更した方法を利用して2官能性NHS−エステル カルボシアニン−Cy3と接合した。この方法は、Cy3をウイルスキャプシド内のアミン基をエステル結合として接合する。要約すると、ウイルス5x1011粒子を30分間、4℃で、NHS−エステル カルボシアニン−Cy3の上昇する濃度を用いて丁度1mlの反応容積中でインキュベーションした。架橋反応の数種の反応条件で、染料:rAAV粒子比0.2、1、5、25、100、および200を用いて評価した。溶液を透析室(10,000MWカットオフ、Gibco BRL, Gaithersburg, MD )に移送し、そして24時間、20mM HEPES pH7.5、150mM NaClを含む透析緩衝液を2回交換して透析した。最後に、試料を血清を含まないDMEM中で一晩透析し、そしてセントリコン(Centricon, Amicon) 中で濃縮した。この溶液を、ガラススライド上のHeLa細胞のウイルス感染のために4℃で90分間、血清を用いないで直接使用した。ウイルスの4℃結合の後、スライドを血清を含まない媒体中で2回洗浄し、そして分析のために固定するかまたは10%血清含有媒体の存在下での連続感染のために37℃シフトした。100の染料−対−粒子比を用いた条件が最善の結果を与えた。
【0164】
これらの標識実験からの結果を図18に示し、そして標識ウイルスがエンドサイトーシスがない場合の4℃において細胞表面へ有効に結合することを、予想どおりに証明した。60分間、37℃において、細胞質内のウイルスの存在量の目に見える上昇が認められ、これは120分間、37℃で核に有効に輸送されたものである(図18AおよびB)。さらに、標識ウイルスは、同様の擬標識したウイルスストックと比較して、HeLa細胞上の感染24時間後のGFP発現の95%以上の機能性形質導入を保持した。ウイルス結合は、感染の時点でヘパリンにより濃度依存性で阻害された(図18C)。ヘパリンは、ヘパリン硫酸プロテオグリカン受容体との結合をブロックすることにより、HeLa細胞内の大部分のrAAV形質導入を阻害することがすでに知られている。Cy3−標識rAAVは、GFP導入遺伝子発現により測定して、その官能性形質導入活性の95%以上を保持した。従って、大部分の標識ビリオンは、エンドサイトーシスに対して機能的に活性である。極性化気道上皮細胞モデルにおけるrAAVのエンドサイトーシスに関して、Cy−3標識rAAVとFITC標識トランスフェリンとの同時定位は、大部分のAAV粒子がトランスフェリン含有ベシクル内にエンドサイトーシスされていることを証明する(図18D)。従って、rAAVはクラスリン被覆ピットを通じてエンドサイトーシスされるようである。
実施例4
エンドソーム処理がAAV形質導入を制限する
側底膜がより高いエンドサイトーシス速度を有しそしてUV照射が頂端膜からのエンドソーム取り込みおよびrAAV形質導入を増加するという知見に基づいて、ウイルス取り込みおよび核への輸送に影響するエンドソーム経路が頂端膜からでは制約であることが可能である。反対に、これらの経路は、気道上皮細胞の側底膜からでは最大レベルで活性であろう。エンドソームプロセシングの重要性をさらに研究するために、エンドソームプロセシングを改変することが知られている数種の化合物の効果を評価した。
方法
最初の研究は、用量滴定および毒性が迅速にアッセイできるので、集密一次ヒト繊維芽細胞内で行った。選択した化合物をrAAV感染の前に繊維芽細胞単層を処理するために使用した。rAAV形質導入は、感染96時間後にFACS分析により評価し、そして死滅細胞の割合をヨウ化プロピジウムの組込みにより同時に評価した。
【0165】
これらの化合物は、ノコダゾール(nocodazole, Sigma, St. Louis, Mo;解重合した微小管で、リソソーム散乱を起こす)、硫酸ビンブラスチン(vinblastin sulfate, Sigma, St. Louis, Mo;解重合した微小管で、細胞内エンソドームおよびリソソームの運動をブロックしてエンドサイトーシスを阻害する)、サイトカラシンB(cytochalasin B, Sigma, St. Louis, Mo; 解重合した微小繊維、すなわちアクチンで、エンドソームとリソソームとの融合をブロックする。細胞内のエンドソームとリソソームの運動をブロックしてエンドサイトーシスを阻害する)、ブレフェルジンA(brefeldin A, BFA, Sigma, St. Louis, Mo; ERからゴルジへのタンパク質輸送を可逆的にブロックする。BFAは頂端側からのエンドサイトーシスを増加するが側底側からは増加しないことが知られている、Prydz et al., (1992)参照)、NH4 Cl(Sigma, St. Louis, Mo; エンドソームpHを上昇させる向リソソーム試薬であって、そしてイヌパルボウイルス脱外被を阻害することが示されている。Basak et al.(1992)参照)、クロロキン(Sigma, St. Louis, Mo; エンドソームpHを上昇させそしてリソソームシステインプロテアーゼのカテプシンBを阻害する向リソソーム試薬であって、そしてイヌパルボウイルス脱外被を阻害することが示されている。Basak et al.(1992)参照)、およびLLnL(N−アセチル−L−ロイシニル−L−ロイシナル−L−ノルロイシナル、Calbiochem-Novabiochem Corp. LaJolla, CA) およびZ−LLL(N−カルボベンゾキシル−L−ロイシニル−L−ロイシニル−L−ノルバリナル、Calbiochem-Novabiochem Corp. LaJolla, CA) 、これらはトリペプチト性のアルデヒドシステインプロテアーゼ阻害剤である。これらのトリペプチドは、構造的にクロロキンに類似しているが、異なる脂質溶解性およびシステインプロテアーゼに対する特異性を有する(Seglen, 1983)。これらの分子は、pH変化とは異なる機構で分子のエンドソーム分解を低下する。これらは26Sユビキチンおよびプロテアソーム依存性タンパク質分解経路を阻害することも知られている(Rock et al., 1994) 。
結果
イヌカルボウイルスに関して以前に報告されたように(Basake et al., 1992)、エンドソームpHを上昇させるNH4 Clおよびクロロキンの両者は、rAAV形質導入を著しく阻害した(図6)。これらの結果は、ウイルス放出の促進および/または感染後の脱外被におけるエンドソームpHの重要性を支持する。さらに、微小フィラメントの形成を中断するサイトカラシンBのような試薬は、rAAV形質導入に著しい低下をもたらし、アクチン微小フィラメントがrAAV形質導入にある役割を有することを示唆する。さらに、微小管解重合を促進しそしてMDCK内でのエンドサイトーシスを低下の両方するビンブラスチンは、rAAV形質導入にほとんど作用しない。
【0166】
しかし、最も興味があるのは、ERからゴルジへの小胞輸送を中断しそしてMDCK細胞内の頂端膜エンドサイトーシスを増加することが知られているBFAを用いる処理(Prydz et al., 1992)が、著しいrAAV形質導入増加に導くことである。ERからゴルジへの小胞輸送の重要性は明らかではないが、しかし、UV照射が膜エンドサイトーシスも増加しそしてBFAも同じことを行うことが示唆されている知見を考慮すると、これらの知見は、受容体結合rAAVの膜エンドサイトーシスの速度が形質導入における律速段階ではないかと示唆する。BFAに対すると同様に、2種のエンドソームプロテアーゼ阻害剤(トリペプチドのLLnLおよびZ−LLL)は、両者ともrAAV形質導入を著しく増加する。これらのトリペプチドは、これまでプラスミドDNAのトランスフェクション効率の上昇のために使用され、そしてDNAのリソソーム分解を阻害すると考えられていた(Coonrod etal., 1997) 。
【0167】
データは、エンドサイトーシスおよびエンドソームプロセシングがrAAV形質導入において鍵となる律速段階であるという仮説を支持する。微小管ではなくて、アクチン微小フィラメントがrAAV形質導入に重要であり、核へのrAAV輸送を促進することにより作用するであろうと考えられる。さらに、サイトカラシンBは、極性化MDCK細胞のインフルエンザウイルスによる頂端側感染を有効にブロックし、側底側はしない(Gottlieb et al., 1993) 。これらの知見は、エンドサイト小胞は極性化上皮細胞の2面で形成される過程に基本的な差があり、そしてアクチン繊維の完全性および/または重合が頂端面で要求されることを示す。しかし、微小管解重合剤、例えばビンブラスチンがrAAV−2形質導入を阻害しないという知見は、イヌパルボウイルスのノコダゾール阻害に関する以前の報告(Vihinen-Tanta et al., 1998)とは異なる。最後に、トリペプチドプロテアーゼ阻害剤を用いた研究は、rAAV形質導入における著しい増加を証明した。この知見は、ウイルスのエンドソーム分解および/またはエンドソーム放出が、rAAV形質導入における重要な律速段階であることを示唆する。
実施例5
放射能標識rAAVを用いるエンドサイトーシスおよび核への転送の評価
放射能標識ウイルスは、エンドサイトーシスおよび核転送に関する試験でCy3−標識ウイルスに対していくつかの独特の利点を提供する。第一に、Cy3−標識ウイルスとは異なり、35S−および 3H標識ウイルスを用いると、ビリオンに対する共有結合修飾を必要としない。従って、共有結合修飾した分子による転送に対する凝集またはその他の未知の作用に遭遇しない。第二に、35S−および 3H標識ウイルスと比較して、ウイルスキャプシドタンパク質およびDNAの運命がそれぞれ評価できる。rAV.GFP3oriは、下記のプロトコールで標識された。準集密(subconfluent)293細胞の20枚の150mmプレートに、Ad.LacZ(5pfu/細胞)を用いて1時間感染させ、次いでpCisAV.GFG1ori(250μg)およびpRepCap(750μg)のリン酸カルシウムトランスフェクション。細胞をさらに10時間インキュベーションし、その時点で培地を2%FBSメチオニン不含DMEMに45〜60分間交換した。次いで培地を、2%FBSメチオニン不含DMEM中400mlあたりの35S−メチオニンの15mCiを含む標識培地に再交換し(最終=1.49MBq/ml)そして細胞を1.5時間、37で標識適用(pulse) した。標識適用の後、L−メチオニンを最終濃度30mg/Lまで再度加え、細胞をさらに30時間、37℃でインキュベーションした。細胞溶解物を調製しそしてウイルスを精製した。標識したウイルスの代表的な比放射能は、5x10-6cpm/粒子であり、これは5.5x10-7cpm/粒子の比放射能のこの分野での他の報告よりもいくらか高かった(Baetlett et al., 1999) 。
【0168】
LLnLの存在または不在でのrAAVが核へ転送する効果を比較した結果を図19に示す。LLnLの存在または不在での頂端側と側底側からの35S標識AV.GFP3ori(MOI=50,000粒子/細胞)で感染した極性気道上皮細胞の凍結切片を写真エマルションの上に置きそして5週間暴露し、次いで現像した。図19AおよびBの顕微鏡写真は、LLnLの存在下での側底面感染の後の銀粒子により示されるウイルスの核および細胞質蓄積を証明する。銀粒子に関連する細胞質および核の形態測定分析を図19Cに示し、そしてこれは感染2時間後のLLnLの存在下での核転送の著しい増加を証明する。ウイルスの核蓄積の増加は、頂端側と側底側感染の後のLLnLの存在下で認められる。従って、ウイルスのエンドサイトーシス速度および核転送は、気道内での律速であるようであり、そしてこれらの過程はトリペプチドプロテアーゼ阻害剤により増加した。
実施例6
エンドソームプロセシング阻害剤は、極性化気道細胞内のrAAV形質導入を増加する
材料および方法
ヒト気管支上皮の一次カルチャーおよび使用した試薬
一次ヒト気道上皮細胞は、以前の記載のように、肺移植体からの気管支試料の酵素消化により捕集した(Kondo et al., 1991; Zabner et al., 1996) 。単離した一次気道細胞を密度5x105 細胞/cm2 でコラーゲン被覆ミリセル−HAカルチャーインサート(Millicell-HA culture inserts, Millicell Corp., Bedford, MA)上に接種した。一次カルチャーを空気−液体界面で2週間以上培養し、その時点で粘膜微繊毛上皮細胞への分化が起きた。細胞の側底側への供給のみに使用した培地は、49%DMEM、49%Ham’sF12および2%ウルトラサーG(BioSepra, Cedex, France) を含んでいた。ジメチルスルホキシド(DMSO)、カンプトテシン(Camp)、エトポシド(Etop)、アフィジコリン(Aphi)、ヒドロキシ尿素(HU)およびゲニステイン(Geni)は、Sigma(St. Louis, MO)から購入した。トリペプチドアルデヒドプロテアソーム阻害剤N−アセチル−L−ロイシル−L−ロイシル−ノルロイシン(LLnL)およびベンゾキシカルボニル−L−ロイシル−L−ロイシル−L−ロイシナル(Z−LLL)は、Calbiochem-Novabiochem Corp.(LaJolla, CA) から購入した。ユビキチンリガーゼ(E3)阻害剤は、Bachem Bioscience (King of Prussia, PA) から入手した。抗−AVVキャプシドモノクローナル抗体(抗−VP−1、2および3)は、American Research Products (Belmont, MA)から購入し、そして抗ユビキチン抗体はSanta Cruz Biotechnology Inc.(Santa Cruz, CA) から購入した。
【0169】
組換えAAVウイルスストックの調製。組換えAAVは、CaPO4 同時トランスフェクションプロトコールにより作製し、そして上記の実施例1記載のようにして、等密度の塩化セシウム超遠心分離3回により精製した。プロウイルスプラスミドpCisAV.GFP3oriは、Duan et al.(1998) により記載されている。RSVプロモーターおよびSV40ポリ−アデニル化シグナルの転写制御下でアルカリホスホターゼレポーター遺伝子をコードするプロウイルスプラスミドpCisRSV.AlkphosをAV.Alkphos作製に使用した(Yang et al., 1999) 。AV.LacZ作製に使用したプロウイルスプラスミドpCisRSV.LacZは、最初に3474bp NotI消化β−ガラクトシダーゼ遺伝子(pCMVβ、Clontechから)をpRep4(Invitrogen)のNotI側に挿入して作製した。RSVプロモーター、β−ガラクトシダーゼレポター遺伝子およびSV40polyAシグナルを含む全β−ガラクトシダーゼ発現カセットをSalIにより切除し、次いでpSub201骨格中に平滑末端連結によりクローンした(Samulski et al., 1987) 。組換えウイルスストックを58℃に60分間加熱して汚染しているヘルパーアデノウイルスを不活性化した。代表的な収量は、プラスミド標準に対するDNAスロットブロットハイブリダゼーションアッセイに基づいて5x105 〜5x109 細胞/μlであった。繁殖に対して使用した組換えアデノウイルス(AV.GFP3oriに対してはAd.CMVAlkphos、AV.Alkphosに対してはAd.CMVLacZ、AV.LacZに対してはAd.CMVGFP)に対する第二レポーターアッセイ(Duan et al., 1997) に基づいて、アデノウイルス汚染のレベルは、アデノウイルスの存在下での293細胞の感染に使用したrAAV粒子1x1010あたりに機能性粒子1個以下であった。Rep/Capコード化プラスミドを用いるトランスフェクションは、Repタンパク質の抗体染色に対する対照として役立った。生体外または生体内感染の前に、ウイルスをPBS中で透析した。
【0170】
極性化気道上皮細胞および一次ヒト繊維芽細胞の形質導入。完全に分化した気管支細胞のrAAV感染は、Duan et al.(1998) の記載のようにして行った。気道細胞の頂端面からの感染のために、5μlrAAVを培地50μlと混合しそしてミリセルインサートの頂端コンパートメントに直接適用した(MOI=10,000粒子/細胞)。頂端面感染の間に、ミリセルの側底側は連続的に培地に浸した。基底側への遺伝子転移は、ミリセルインサートを反転しそして支持しているフィルター膜の底にウイルスベクター体積50μlで2時間適用して行った。次いで、当初のウイルス接種物の連続した存在と追加の培地450μl中でミリセルインサートを正しい位置に戻した。頂端側および側底側の両方の感染のために、rAAV含有培地を24間後に取り去り、そして新しいカルチャー培地に交換(基底側)または空気に暴露(頂端側)した。極性化気道細胞内のAAV形質導入の効力に対する種々の薬剤の効果を試験するために、気道上皮感染の前にそれぞれの溶液1μlをAAVと混合した。薬剤は、別途断らない限り、通常24時間のAAV感染期間中存在した。大部分の薬剤はDMSO中に溶かしたが、ただし、ヒドロキシ尿素(リン酸塩緩衝生理食塩水中に溶解)、H−Leu−Ala−OH(0.9%氷酢酸中に溶解)およびH−His−Ala−OH(50%メタノール中に溶解)を除く。薬剤の作用濃度は下記である。0.1μMカンプトテチン、10μMエトポシド、5μg/mlアフィジコリン、40mMヒドロキシ尿素、50μMゲニステイン、40μM LLnLおよび4μM Z−LLL。ユビキチンリガーゼ(E3)阻害剤(H−Leu−Ala−OHおよびH−His−Ala−OH)を使用する場合には、気道細胞を、感染の前に最終濃度2mMの両方の阻害剤の混合物を60分間前処理し、次いで側底面からの24時間感染の全期間に阻害剤(0.2mM)を連続した存在させた。EGTA処理を含む研究は、水中の3mM EGTAを用いる10分間の極性化気道上皮の頂端膜の一時的処理により行った(Duan et al., 1998) 。等張EGTA処理の後、カルチャーを培地を用いて2回洗浄し、そして40μM LLnLの存在または不在においてrAAVを用いて感染させた。ヒト一次繊維芽細胞(P4)を10%ウシ胎児血清(FBS)、1%ペニシリンおよびストレプトマイシン、および89%DMEM中に維持した。AV.GEP3oriを用いる感染は、2%FBS DMEM中の1000DNA粒子/細胞のMOIにおける80%集密繊維芽細胞を用いて24時間行った。
【0171】
rAAVのS 35 標識。rAV.GEP3oriのキャプシドタンパク質中のメチオニン残留物を、変更を加えた以前に公開したプロトコール(Mizukami et al., 1996) に従って放射性ウイルスストックの作製の間に標識した。要約すると、準集密293細胞の20枚の150mmプレートを、Ad.LacZ(5pfu/細胞)を用いて1時間感染させ、次いでpCisAV.GFP3ori(250μg)およびpRepCap(750μg)のリン酸カルシウムトランスフェクションを行った。細胞をさらに10時間インキュベーションし、その時点で培地を2%FBSメチオニン不含DMEMに45〜60分間交換した。培地を2%PBSメチオニン不含DEDMの400mlあたりにS35メチオニン15mlを含む標識培地(最終=1.49MBq/ml)に再交換し、そして細胞を1.5時間、37℃で標識適用した。標識の後、L−メチオニンを最終濃度30mg/Lまで加えて戻し、そして細胞をさらに30時間、37℃でインキュベーションした。細胞溶解物を調製し、そしてウイルスを上記のようにして等密度塩化セシウム超遠心分離により精製した。標識ウイルス調製物の代表的な比放射能は、5x10-6cpm/粒子であり、これは他の研究者により報告された5.5x10-7cpm/粒子より少し高い(Bartlet et al., 1999)。
【0172】
ウイルス結合/侵入アッセイおよびウイルス粒子の in situ 定位。極性化気管支上皮細胞へのrAAVの結合を評価するために、S35標識AV.GFP3oriを頂端面または側底面のいずれかに適用し(MOI=50,000粒子/細胞)、次いで4℃で60分間培養した。分化した気道上皮内へのrAAVの複合結合/侵入を同じ条件下で測定したが、ただし、採取の前に培地を37℃でさらに2〜24時間インキュベーションした。これらの複合ウイルス結合/侵入アッセイは、同じ条件下で、導入遺伝子発現を終点とするrAAV形質導入の機能性研究のためのものに使用して行った。PBSを用いた洗浄3回の後に、細胞をin situ で液体シンチレーションカクテル5mlを加えて室温で5分間溶解させ、そして放射能をシンチレーションカウンターで定量した。
【0173】
極性化ヒト気管支上皮細胞内のrAAV粒子の細胞下レベル定位を分析するために、S35標識ウイルス(MOI=50,000粒子/細胞)を粘膜または漿膜面に適用して感染を行った。感染の2時間後に、トランスウエル(transwell) を培地を用いて3回洗浄しそして4%パラホルムアルデヒド内で一晩固定し、次いで凍結保護および凍結割断のために埋め込んだ。10μm凍結切片を写真エマルション上に置き、前に公開したプロトコールに従って5週間現像した(Duan et al., 1998) 。
【0174】
極性化気道上皮カルチャーの感染の後のrAAVウイルスゲノムの分子分析。結合してエンドサイトーシスしたウイルスの分子状態を、rAAV感染の後に種々の時点でアッセイした。細胞表面に付着したウイルスの量を試験するために、rAAV感染を4℃で1時間行った。結合の後、ウイルスの内部化の程度を、ウイルスの存在下、37℃で4〜24時間の連続インキュベーションにより評価した。ウイルスDNAを変更したヒルト(Hirt)プロトコールに従って抽出し、そしてハイボンド(Hybond)N+ナイロン膜(Amersham)を用いてサザンブロットを行った(Duan et al., 1997) 。ウイルスゲノムからの1.6kb一本鎖ウイルスDNA、2.7kb二本鎖環状中間体、および4.7kb二本鎖複製物を、5x106 cpm/mlでの導入遺伝子EGFP特異性プローブを用いて検出した。ブロットを0.2xSSC/0.1%SDS、55℃で20分間のストリンジェンシーで2回洗浄した。ウイルス内部化を評価する目的の研究において、細胞表面に付着したウイルスを、0.5%トリプシン、および5.3mM EDTAを含む緩衝液1mlを用い37℃で10分間でトリプシン処理して除去し(ミリセルインサートの頂端側および側底側コンパートメントに500μl緩衝液を加えた)、次いで氷冷PBSを用いて2回洗浄した。外部に結合したAAVウイルスを、4℃で60分間感染した細胞から抽出したHirt DNA中の1.6kbウイルスゲノムバンドの強度により決定した。内部化したウイルスは、感染後に37℃で4および24時間トリプシン処理した細胞から抽出したHirt DNA中の1.6kbウイルスゲノムバンドの強度により決定した。内部化ウイルスの分子構造内の動的変化は、ウイルスを培地から取り出した後、2、10、20、30および50日でアッセイした。
【0175】
免疫沈降によるユビキチン化AAVキャプシドタンパク質の検出。AAVユビキチン化に対するプロテアソーム阻害剤の効果を分析するために、ヒト一次繊維芽細胞をウイルス感染の6時間後に、1X RIPA緩衝液中で溶解させた。次いで、細胞溶解物を30μlタンパク質A−アガロースを用いて透明化(cleared)した。上清をモノクローナル抗−VP1、2、および3抗体(クローンB1、ARP)の10μlと一緒にインキュベーションし、次いで30μlタンパク質A−アガロースを加えた。ペレットをIX RIPA緩衝液を用いて4回洗浄し、そして10%SDS−PAGEで分離した。ニトロセルロースフィルター上に転移した後、ブロットを、抗−ユビキチンモノクローナル抗体(クローンP4D1、Santa Cruzカタログ#SC8017)の1:1000希釈物、次いで1:500HRP−接合二次抗体(BMB)を用いてプローブした。最終洗浄の後、免疫反応性をECLシステム(Amersham)を用いて可視化した。
【0176】
マウス内の生体内試験。動物実験は、アイオワ大学の制度化した指針に従って行った。マウス肺内のAAV媒介遺伝子転移に対するプロテアソーム阻害剤の影響を決定するために、6週齢BALB/cマウスをメトキシフルラン室を用いて軽く麻酔した。AV.LacZ(5x1010粒子)を単独または10μl点滴注入液中の400μM Z−LLを用いて、Walters et al.(2000)が記載したようにして鼻噴霧により投与した。DMSO溶剤の予期しない毒性を防止するために、プロテアソーム阻害剤Z−LLLをエタノール中に40mMストック溶液として溶かし、そして1%最終濃度でウイルス接種物中に含ませた。Z−LLLを含まないウイルス感染対照も1%最終濃度のエタノールを含んでいた。一次培養したヒト気道細胞および繊維芽細胞の両方の研究が40μM LLnLおよび4μM Z−LLLの間で近い増加効率であることを証明し、そしてエタノール中のLLnLの低い溶解度のために(実施例7では、気管に投与されたDMSO中のLLnLの低い用量を使用した)、この特定のマウス肺研究にはZ−LLLのみを試験した。感染の2、10および150日後に動物を安楽死させそしてPBS(10ml)を右心室中に点滴注入し、次いで肺および心臓を不変のカセットとして取り出した。気道に管を挿入し、水圧力10cmで下記の溶液を順番に点滴注入した。PBS、0.5%グルタルアルデヒド、1mM MgCl2 /PBS、および最後に室温で一晩のインキュベーションのためのX−gal染色試薬。次いで、X−gal染色したマウス肺を10%中性緩衝ホルマリン中、48時間、室温で後固定し、そして順番に、10%、20%および30%スクロース/PBS溶液中で凍結保存した。肺(それぞれの条件でN=3)をOCT(最適切断温度、Baxter, Warrendale, PA) 内に埋め込みそして15μmの順次切片を、気道上皮内の陽性細胞の割合を計算して遺伝子導入に関して分析した。気道の直径を形態測定分析の結果の分類のために記録した(>360μm、260〜350μm、160〜250μm、<150μm)。150より大きい気道断面をそれぞれの実験条件に対して定量した。
結果
極性化気道上皮内のrAAVゲノムの分子分析
最近の研究は、分化した気道上皮の頂端面において、AAV−2受容体、ヘパリン硫酸プロテオグリカンおよび共受容体、FGFR−1およびαVβ5インテグリンの欠失を明らかにした(Duan et al., 1998; Duan et al., 1999; Hugheset al., 1993; Goldman et al., 1999) 。しかし、頂端膜および側底膜における放射性ウイルスの結合の差は、4〜7倍に過ぎない(側底>頂端)(Duan et al., 1998) 。結合におけるこれらの差は、rAAV−2を用いる感染の極性で観察された200倍の変動(側底>>頂端)を説明するには不十分である(Duan et al., 1998) 。これらの知見は、ウイルス結合および/または取り込みが、気道上皮内の不十分な粘膜導入に寄与する単独の限定因子ではないことを示唆する。この目的で、空気−液体界面でカルチャーされたヒト気管支上皮の頂端側および側底側感染の50日後におけるrAAV DNAの分子状態を評価した。この時点で、EGPFレポーターから測定された遺伝子発現は、側底側に感染されたカルチャー内が200倍以上高かった(データは記載しない)(Duan et al., 1998)。カルチャーからのHirt DNAを、32P標識EGPFプローブを用いるサザンブロットハイブリダゼーションにより評価した。図8に示すように、頂端側適用rAAVの顕著な量が気道細胞を感染することができた。しかし、この時点(50日後)では、一本鎖ウイルスゲノム(ssDNA)のみが検出された。この結果は、rAAVが粘膜表面からエンドサイトーシスすることができ、そしてエンドサイトーシスされたウイルスssDNAはある未知の細胞内コンパートメント内に安全に隔離されていることを明らかに示唆する。反対に、大部分の側底側適用rAAVは二本鎖形に変換され、これらは1%非−変性アガロースゲル中で2.8kbおよび>12kbで移動したことを明らかに示唆する(図8)。従来の報告と一致して(Sanlioglu et al., 1999; Duan et al., 1999) 、その後のHirt DNAの制限酵素マッピングおよびサザンブロットは、2.8kbバンドがスーパーコイル化した環状エピソーム分子であることを確認した(データは記載しない)。側底側感染の後に著しく強くなる>12kbバンドの実体は、現在では未知であるがしかしAAVゲノムのエピソーム環状コンカテマーを表すであろう。総合すると、これらの結果は、AAVウイルスDNAの環状ゲノムへの不十分な分子転換が、気道の頂端面からのrAAV媒介遺伝子導入に対する著しい障害となっていることを示唆する。さらに、自己開始による線型複製ではなくて環状化が、極性化気道上皮内のrAAV変換に対する主要な経路である。
【0177】
プロテアソーム阻害剤は、極性化気道上皮内のrAAV感染を劇的に増加する。rAAVが気道内で頂端感染の後に、ある細胞コンパートメント内に潜在しているように見えること、およびウイルスゲノムの分子転換を改変する薬剤が気道上皮内のrAAV形質導入を増加させるのではないかとの事実を考慮して、数種の薬剤をこの観点から試験し、これには、DNA損傷剤(Alexander et al., 1994)、DNA合成およびトポイソメラーゼ阻害剤(Russel et al., 1995) 、および細胞チロシンキナーゼ阻害剤(Qing et al., 1997; Man et al., 1998) が含まれる。カンプトテチン、エトポシド、ヒドロキシ尿素、およびゲニステインの適用は、側底面からのrAAV形質導入に10〜60倍の増加をもたらした(図9)。しかし、興味あることには、これらの薬剤のいずれも頂端面からのrAAV形質導入を促進しなかった(データは記載しない)。他の細胞タイプ内でrAAV形質導入に関与する核内イベントに影響することが知られている化学薬品(Sanlioglu et al., 1999)は、気道内でのrAAV頂端側感染を促進しないことが知られているので、細胞内過程、例えばユビキチン−プロテアソーム経路に影響する他の薬品を試験した。プロテアソーム系は、HIVなどのウイルスを含む多数の外来および内在性分子の細胞内プロセシングを調節することが知られている(Schwartz et al., 1998) 。プロテアソーム経路の数種の特異性、細胞透過性のペプチドアルデヒド阻害剤が最近発見されている(Rock et al., (1994); Fenteany et al., 1995)。これらの阻害剤は、プロテアソームコア内のタンパク質分解性酵素の活性部位に結合し、そしてこれらの機能を可逆的にブロックする(Rubin etal., 1995)。プロテアソームが感染後にrAAVを封鎖する細胞内コンパートメントであるかどうかを試験するために、トリペプチド性アルデヒドプロテアソーム阻害剤(システインプロテアーゼ阻害剤)N−アセチル−L−ロイシニル−L−ロイシナル−L−ノルロイシナル(LLnL、カルパイン阻害剤Iとも呼ばれる)を、rAAV感染の時点でヒト気管支上皮細胞の極性化カルチャーに適用した。意外にも、40μM LLnLをrAAVと同時に漿膜面に適用すると、導入遺伝子発現に200倍以上の増加が感染後2日で得られた(図9)。同様の結果は、他のユビキチン−プロテアソーム経路阻害剤のN−カルボベンゾキシル−L−ロイシニル−L−ロイシニル−L−ロイシナル(Z−LLL、MG132とも呼ばれる)(Jansen et al., 1995) を試験しても得られた(データは記載しない)。しかし、最も重要な知見は、これらのプロテアソーム阻害剤も本質的に粘膜面からのrAAV形質導入を増加することである(下記参照)。他の薬剤と比較して、プロテアソーム阻害剤は気道上皮におけるrAAV形質導入の最も強力なエンハンサーであることが見いだされた。
【0178】
プロテアソーム阻害剤は極性化された様式で気道上皮内のrAAV形質導入を増加させる。プロテアソーム阻害剤は漿膜面からのrAAV形質導入の効果を著しく増加するように見えるけれども、気道内の遺伝子送達の臨床適用に最も適切な経路は粘膜面である。頂端膜からのrAAV形質導入に対するプロテアソーム阻害剤の効果を試験するために、LLnL処理後の気道上皮の粘膜面および漿膜面の両方からのrAAV形質導入の対比速度比較を行った。図7Aに示すように、粘膜面へのLLnLとrAAVの同時投与は、AAV形質導入の持続的な増加をもたらし、これは感染の22日後にピークに達した。粘膜感染とは対照的に、LLnLの存在下での漿膜面からのrAAV感染は、感染の72時間後の遺伝子発現に一過性のピークをもたらしただけであり、これは22日までに非処理試料のものと同等のレベルに戻った(図7B)。これらの結果は、AAVウイルスが頂端膜または側底膜のいずれかに適用された場合に、プロテアソーム阻害剤LLnLが、異なる増加プロファイルを生成することを示唆する。気道上皮によるLLnLの極性化取り込みにより起きる可能な作用を除外するために、粘膜面および漿膜面の両方からのrAAVおよびLLnL投与の種々の組み合わせを試験した。感染の間にLLnLがrAAVと同じかまたは異なる面に適用されることに関係なく、AAV形質導入に対する同様の増加パターンが得られた(データは記載しない)。
【0179】
LLnL投与が特定の気道細胞タイプのrAAV形質導入を増加させるかどうかを試験するために、アルカリホスホターゼ遺伝子(Alkphos)をコードするrAAVベクターを使用した。形質導入された細胞タイプは、この問題に対応するために、Alkphosに対する標準組織化学的染色により評価した。LLnLが存在しないと、rAAVは、ウイルスの漿膜適用の3日後に優先的に基底細胞を形質導入した(図10AおよびC)。AV.GFP3oriウイルスを用いた従来の知見と一致して、LLnLの同時投与は、AV.Alkphos形質導入の劇的な増加をもたらした(図10BおよびD)。興味あることに、繊毛細胞形質導入は、rAAV感染の時点におけるLLnLを用いる処理により最も著しく増加した(図10E)。反対に、基底細胞は、LLnL処置にほとんど反応しなかった。これらの知見は、LLnL作用の機構が、ある種の細胞特異性成分を有し、これは極性化(すなわち繊毛性)とも非極性化(すなわち基底性)細胞タイプとも異なる。
【0180】
LLnL強化rAAV形質導入の最適化。
【0181】
LLnLの存在下で到達されるrAAV形質導入の増加をさらに改善する目的で、rAAV感染後のLLnL投与の時期および回数を変化させる数種の詳細な速度論的研究を行った(図11)。これらの研究が数件の重要な結論が得られた。第一は、側基底投与後3日に1回のLLnL投与が、30日後のレベルが感染の時点での1回の投与のものと同じであったにもかかわらず、ピーク導入遺伝子発現の長さを延長した。第二は、LLnLの連続投与が細胞に毒性でありそして10日目には導入遺伝子発現がなくなった。第三は、LLnLの存在下での7、10、15日後のrAAVを用いるカルチャーの再感染が、同様の増加パターンをもたらし、そして、予想どおりに、30日目において観察された導入遺伝子発現の最終レベルを上昇させた(15日目の第二回感染からのデータのみを記載する)。しかし最も注目すべきは、側底側から感染されたすべてのカルチャーは、LLnLが投与されたかどうかには関係なく、たがいに2〜3倍の範囲内の同様な長期導入遺伝子発現レベルを生成したことである。
【0182】
ウイルス感染の時点でのLLnL投与が頂端面および側底面の両方からのrAAV形質導入を増加した事実にもかかわらず、この形質導入の速度は、著しく異なっていた。側底側感染の後の増加は一時的であり、一方頂端側感染の後の増加は長期であった(図7)。さらに、頂端膜からのLLnLを用いる誘導は長期に継続したけれども、30日で導入遺伝子発現の最高レベルは基底感染からもたらされたものの8分の1に過ぎなかった(図7A)。等張EGTA溶液の適用は、頂端面からのAAV形質導入を7〜10倍増加した(Duan et al., 1998; Walters et al., 2000) 。従って、EGTAとLLnLとの複合投与は、頂端面からのrAAV形質導入におけるこれ以上の増加をもたらすであろう。興味あることに、LLnLの存在下においてrAAVを用いる感染前のEGTAを用いる気道カルチャーの処理は、最初の3日間の一時的な形質導入のピークを与え、そして著しく増加した(200倍)継続的な導入遺伝子発現のレベルを30日まで与えた(図11B)。この導入遺伝子発現の長期継続的レベルは、基底面からのrAAV感染と同程度であるが、EGTA単独で頂端側感染した上皮に観察されたレベルよりはるかに高い(図11B)。要約すると、これらの結果は、EGTAおよびLLnLがrAAV形質導入に相乗効果を有し、頂端面からの形質導入に対して側底側感染後にのみ通常観察されるようなレベルで形質導入されることを証明している。
【0183】
ウイルス結合および内部化は、LLnL処理により影響されない。LLnLの作用は、代表的にはプロテアソーム系の選択的および可逆的阻害に帰せられる。しかし、ウイルス結合および/またはエンドサイトーシスに対するあらゆる可能な効果を除外することが重要である。タイプ1単純ヘルペスウイルスに見られるように(Everett et al., 1998)、LLnL処理は4℃でのrAAV結合に著しい効果を有していない(図12)。同様に、37℃における2時間感染期間におけるS35標識rAAVの取り込みは、LLnL処理により変化されない(図12)。この結果を考慮すると、LLnLは、ウイルス結合および侵入の点から遠位の点で作用する。興味あることには、感染の24時間後に、細胞内放射能の量の著しい低下が、LLnLを用いて処理された上皮内で観察され、これはどの表面が感染されたかには関係なかった(図12)。この時点における導入遺伝子発現の一致した増加(図12)を考慮すると、LLnLは核へのウイルスのプロセシングおよび移動を加速し、その際、S35標識キャプシドタンパク質の脱外被および除去が起きる。この機構で、S35異性体は、培地内に薄められそして細胞関連カウントの低下を説明できるであろう。
【0184】
LLnLは、rAAVのエンドソームプロセシングおよび核転送を増加する。LLnLがrAAVの核への転送を増加するという仮説を試験すすために、頂端面および側底面からの感染に続くS35標識rAAV粒子のin situ 定位を、LLnLの存在および不在で行った。不変の放射能標識キャプシドタンパク質の損失が感染の24時間後に起こったので、2時間の時点をこの分析のために選定した。写真エマルションオーバーレイを用いて、S35標識rAAV粒子の細胞レベルより下の分布をブラインド(blinded) 形態測定分析により評価した。図13に示すように、ウイルス粒子の大部分は、LLnLが存在しない場合には細胞質に位置していた。これは、感染が頂端面または側底面からのいずれから行われても成り立った。反対に、LLnL処理は、放射能標識rAAV粒子の細胞内分布を本質的に変化し、核関連粒子の著しい移動をもたらした。これらの結果は、LLnLがウイルス脱外被およびその後のS35異性体の媒体内への損失を増加すると示唆する、感染の24時間後での全細胞カウントからの知見を確証した。
【0185】
LLnLは、感染後の特定時間枠内でrAAV形質導入を増加する。これまでの証拠は、LLnLが核へのrAAVの細胞内移動を増加するように作用するであろうと示唆していた。さらに、LLnL作用は投与される上皮面とは無関係である(すなわち、LLnLの漿膜投与は、粘膜感染を増加しそして逆も起きる)。これは、形質導入を増加するためにLLnLが必ずしもAAV粒子と一緒にエンドサイトーシスされる必要がないことを示す。従って、LLnLは、rAAVエンドサイトーシスの後の特定時点で作用するがしかしエンドソームプロセシングの間ではないのであろう。この仮説に機能的な支持を与えるために、側底面からの感染後の種々の時点でのLLnL作用の速度論的分析を行った。これらの実験で、LLnLを、AAV感染の時点または感染後の種々の時点のいずれかで培地に加えた。ウイルス媒介導入遺伝子発現は、感染の後、24時間間隔で定量した。増加は、ウイルス感染の1、24、48、および72時間後のLLnLの投与に関係なく起きた。しかし、24または48時間でのLLnLの添加は、増加の最強のレベルを与えた。AAV発現を低下させるLLnLの能力は、感染の27時間後には低下しているように見え(図14)、そして最初のAAV感染から15日後には完全に失われた(データは記載しない)。総合すると、rAAVが細胞に侵入した後、これは、プロテアソーム阻害剤−促進遊離に感受性である細胞内コンパートメントを標的とするであろうと考えられる。さらに、感染の15日後のLLnL増加効果の消滅は、導入遺伝子産物の増加した転写、翻訳、および/または安定性が、この観察に関係する機構に寄与しないようことを示唆する。
【0186】
LLnLとEGTAとの組み合わせ処理は、内部化rAAVの分解を防止する。LLnLによるrAAV形質導入の増加に関係する分子機構をさらに解明するために、感染した細胞内のrAAVゲノムをHirt DNAをサザンブロッティングして分析した。S35標識ウイルスを用いた研究と一致して(図12)、極性化気道上皮の両方の面に対するrAAV結合はLLnL投与により影響されなかった(図15A、頂端側感染に対してはレーン8および9、基底側感染に対してはレーン11および12)。サザンブロッティングは、さらに、基底面からの2〜7倍高いウイルス結合を証明し、これは種々の組織試料間では変動した(データは記載しない)。ウイルス内部化の程度を、表面に結合したウイルスをストリッピングした後にトリプシンで比較した。以前の結果を確認して(図8、12および13)、rAAVの著しい量が、感染期間に頂端面からエンドサイトーシスされ(図15B、レーン2および3、レーン8および9)、一方、ウイルス取り込みは側底面からの方が活性である(図18B、レーン5および6、レーン11および12)。LLnL単独は内部化したAAVウイルスDNAの酵素分解を本質的に防止せず(図15B、図11も参照)、これは核内への増加に関してウイルス転送がLLnLによる観察された増加より重要であることを示す。しかし、等張EGTAおよびLLnLの両方を用いる処理は、頂端面から内部化したウイルスの量を本質的に増加した(図15、レーン2、4とレーン8、10との比較、図18Cレーン1、2および5、6および10、12の比較)。等張EGTA処理のみは、頂端側感染の後、AAV DNAの持続性(4倍以下、図18C、レーン11)、またはAAV媒介遺伝子発現(10倍以下、図5B)をわずかに増加するだけであるので(Duan et al., 1998; Walters et al., 2000) 、EGTAとLLnLとの複合効果が関係する主要な機構は、内部化したウイルスの低下した分解およびエンドサイトーシスの増加した速度によるものであろう。これらのEGTAとLLnLとの相乗効果は、頂端膜からのrAAV形質導入を200倍以上増加する。さらに、一本鎖ウイルスゲノムの線状複製物または環状形への転換は、アデノウイルス初期遺伝子産物またはUV照射によりそれぞれ増加されるAAV形質導入と関連する(Fisher et al., 1996; Sanlioglu et al., 1999; Duan et al., 1999)。Ad.d1802およびrAAVを用いて同時感染されたカルチャーからのHirt DNAのサザンブロットに示されるように(図15C、レーン9)、LLnLが増加したAAV形質導入は、明らかに線状複製中間体(4.7kbバンド)の形成を通じては媒介されず、これはAd.d1802同時感染により産生されたアデノウイルスE4orf6タンパク質の存在から分かる。
【0187】
気道内のrAAV感染の後のウイルスキャプシドタンパク質のユビキチン化は、導入の効率を改変する。ユビキチン化分子のプロテアソーム依存性分解は、内在性および外来タンパク質の両方の処理のための主要な経路である(Schwartz et al., 1999) 。この経路の制御における数種の異なる段階が同定され、これには、活性化酵素(E1)によるユビキチンの活性化、ユビキチンキャリヤータンパク質(E2)への活性化ユビキチンの転移、および引き続いてのユビキチンリガーゼ(E3)によるタンパク質物質への活性化ユビキチンの送達を含む。最終的に、ユビキチン化したタンパク質はATP−依存性過程を通って26Sプロテアソームにより分解される。プロテアソーム阻害剤によるrAAV形質導入の増加がエンドソームコンパートメントからのユビキチン化したウイルスの遊離を含むかどうかを試験するために、感染後のAAVキャプシドタンパク質上のユビキチン側鎖の程度を試験し、プロテアソーム阻害剤を用いる処理がユビキチン化の程度を改変するかどうかも同時に試験した。AAVキャプシドタンパク質を、ウイルス感染6時間後のrAAV感染ヒト極性化気道細胞および集密ヒト繊維芽細胞から抗−VP1、2、3抗体を用いて免疫沈降した。抗−ユビキチン特異性抗体を用いるその後のウエスタン分析は、プロテアソーム処理後に繊維芽細胞内のユビキチン化AAVキャプシドの細胞レベルでの著しい増加を証明した(図16B)。ユビキチン化は、キャプシドタンパク質の分子量(63kd、73kdおよび87kd)を220〜250kdまで著しく増加しそしてこれは他の分子に対するユビキチン化後の大きさ変化と一致する(Bregman et al., 1996)。不幸なことには、空気−液体界面培養ヒト気道細胞から回収できるウイルスの限定された量は、この系内でのユビキチン化キャプシドを検出する能力をなくした(データは記載しない)。それにもかかわらず、集密繊維芽細胞も、プロテアソーム阻害剤処理後の導入遺伝子発現の増加(10倍)を証明した。従って、プロテアソーム阻害剤は、プロテアソーム内のユビキチン化AAVのターゲティングおよび/または分解を低下させてrAAV形質導入を増加する。このようなプロテアソーム阻害剤の真の結果は、ユビキチン化ウイルスキャプシドの存在量を増加すると期待される。
【0188】
極性化気道モデルにおける技術的な限界がユビキチン化ウイルスキャプシドの直接検出を妨げるために、ユビキチン−プロテアソーム経路内の他の段階の調節が、プロテアソーム阻害剤LLnLおよびZ−LLLで観察されたと同様にrAAV形質導入を増加するかどうかを決定した。数種のジペプチド、例えばH−Leu−Ala−OHおよびH−His−Ala−OHは、ユビキチンリガーゼ(E3)を阻害することが知られている(Obin et al., 1999) 。これらのユビキチンリガーゼ阻害剤の適用は、実際に、ヒト気道細胞の側底面からのrAAV形質導入を増加した(図16、パネルC)。総合すると、繊維芽細胞および極性化気道上皮における両方のデータは、AAVキャプシドがエンドサイトーシスの後にユビキチン化され、そしてこの過程がrAAV形質導入への障害となることを示唆する。トリペプチドプロテアソーム阻害剤によるrAAV形質導入の増加に関係する最も説得力がある機構は、ユビキチン化されたウイルス分解および/またはプロテアソームに対するターゲティングの防止を含む。
【0189】
生体内のプロテアソーム阻害剤によるrAAV形質導入の長期的増加。生体内遺伝子治療に対するプロテアソーム阻害剤の潜在的な有用性を評価するために、マウス肺内の生体内rAAV媒介遺伝子導入に対するこれらの薬剤の毒性および効力を試験した。マウス内でのこれらのプロテアソーム阻害剤の毒性を評価するために、極性化気道細胞内の遺伝子導入を誘導するために使用されるよりも10、100、および1000倍高いLLnLまたはZ−LLLの有効用量を、気管内および全身(IV)投与の両方で投与した。肺および肝臓の組織学的評価により、毒性は示されず(データは記載しない)、動物の死亡によっても証明されなかった。これらのプロテアソーム阻害剤が生体内のrAAV形質導入を改善できるかどうかを研究するために、AV.LacZ(5x1010粒子)を単独または400μM Z−LLLの存在下で、鼻内投与により送達した。マウス肺を感染の3、10および150日後に採取し、短期および長期作用を評価した。基底面(図7B)からまたは頂端面からのEGTAとの組み合わせた(図11B)プロテアソーム阻害剤処理は、明白で迅速なrAAV形質導入の増加をもたらしたが、しかし感染の3および10日後の肺組織のX−gal染色は、プロテアソーム阻害剤処理または非処理群内で検出可能な導入遺伝子発現は示さなかった(データは記載しない)。反対に、Z−LLL処理試料において150日目に著しい形質導入が達成された(図17E参照)。ターゲティングした導入遺伝子発現は、実質組織内ではなくて、主として導通する気道に局限される。肺胞壁細胞はほとんど形質導入されない。平均して気道細胞の約5.88%がAV.LacZにより形質導入され、そしてLacZ陽性細胞は全導通気道で観察され、特性的な導入プロフィールが明らかであった。大きい細気管支(>350mm)内の形質導入効率は、気道上皮の平均して10.36±1.63%に達し、一方、小さい細気管支(<150mm)では気道細胞の1.37±0.41%で、β−ガラクトシダーゼ導入遺伝子を発現した(図17F)。遠位および近位の気道内の導入遺伝子発現の範囲は、それぞれ0〜4%および5〜18%であった。大きい気道内の高くそして一致する形質導入を示す形質導入プロフィールは、小さい気道を包含する肺の領域へのウイルスの一層不均衡な送達を反映する。AV.LacZ単独により感染された肺からの凍結切片の検査は、全部で315の気道切片中で(n=3動物)ただ2個のlacZ陽性細胞を明らかにした。
考察
気道の頂端面からの不十分な遺伝子導入は、組換えアデノウイルス(Walters et al., 1999; Pickles et al., 1998)、アデノ随伴ウイルス(Duan et al., 1998) 、レトロウイルス(Wang et al., 1998) 、および非ウイルスのリポソームベクター(Chu et al., 1999)を用いる嚢胞性繊維症のための多数の遺伝子治療法における主要な障害であった。一般に、気道上皮の頂端膜を通る不十分なウイルス媒介遺伝子送達は、主としてこの面上の受容体または共受容体の不足によると考えられていた。
【0190】
極性化気道上皮内のrAAV感染の分子分析は、数点の独自の知見を明らかにした。第一に、気道上皮の頂端膜における既知のAAV−2受容体および共受容体の従来報告されていた不足(Duan et al., 1999) は、AAV感染を排除しない決定的な証拠がある。側底面からの形質導入(導入遺伝子発現により決定されるもの)は、頂端膜からのものより200倍効率的であり、S35標識ウイウルスを用いるウイルスエンドサイトーシスの定量的および半定量的分析またはサザンブロッティングは、頂端面からのウイルス取り込みが、側底膜からのものより2〜7倍効率が低いのみであることを証明した。従って、以前に同定されなかった別の受容体/共受容体および/または受容体に非依存性の機構が、気道の粘膜面からのAAV取り込みに含まれているであろうと推定することが号知的である。
【0191】
極性が多くのタンパク質のエンドソームプロセシングに著しく影響すると広く認められており、そして明確な選別機構が頂端側および側底側コンパートメントについて記載されている(Odorizzi et al., 1996; Rodriguez-Boulan et al., 1993)。側底側および頂端側感染の後のウイルス取り込み効率と導入遺伝子発現との間に直接相関がないことは、追加的なエンドサイトーシス後の障害がrAAV媒介遺伝子導入に存在することを示唆する。側底面に対する頂端面経路感染に続くAAV核転送の程度の差は、基底および頂端細胞コンパートメントがAAV形質導入の極性に影響する明確な生物学的性質を有することを示唆する。rAAV形質導入に対するエンドソームプロセシング障害は、極性化上皮細胞には限られないであろう。この意見の支持として、核へのウイルス粒子の阻害された細胞内転送がNIH 3T3細胞内で観察された。さらに、rAAVはUV照射により機能的活性状態に回復されるまで、集密化一次繊維芽細胞内に7日間も不活性状態で止まることができる。従って、感染に対するエンドサイトーシス後の障害は、複数の細胞タイプ内に存在する。
【0192】
気道内で、粘膜面からのrAAV形質導入における主要な律速段階は、内部化したウイルスの不十分なエンドソームプロセシングを含むと考えられる。プロテアソムを通じる調節された細胞内タンパク質分解は、多くの生理学および病理学的状態において重要な役割を演じる(Schwartz et al., 1999; Kato, 1999) 。多くの特異性プロテアソーム阻害剤の最近の同定は、新規の治療方法としてのこの細胞酵素系内の薬理学的介入のための基礎を設定した。例えば、数種の細胞透過性合成トリペプチドアルデヒド(例えば本発明に使用したLLnLおよびZ−LLL)は、有望なガン治療剤または抗炎症薬剤であると証明されている(Goldberg et al., 1995; Klotzel, 1998; Wojcik, 1999)。さらに、プロテアソームはHIV感染において抗ウイルス機能を有すると示唆されており(Schwartz et al.,1998) 、プロテアソーム機能の阻害は、組換えウイルスを用いる形質導入の促進に有利なことを意味する。気道内のrAAVの頂端側感染が侵入後のイベントにより著しく限定されるという分子的証拠に基づいて、ユビキチン/プロテアソーム経路は、この遮断に関して有力と考えられる。プロテアソームは、一般に内因性および外来タンパク質の排除のためのコンパートメントとして知られている。しかし、最近の研究は、プロテアソーム系がエンドサイトーシスを調節することに関与することも示唆している(Bonifacino et al., 1998; Strous et al., 1999)。遺伝子送達の観点から、プロテアソーム阻害剤は、トランスフェクションされたプラスミドDNAを分解から保護することが示された(Coonrod et al., 1997)。本明細書中に記載した結果は、気道上皮へのrAAV媒介遺伝子導入も、プロテアソーム阻害剤により著しく増加されることを証明している。さらに、この増加は、プロテアソーム阻害剤で刺激された核へのウイルス転送と相関する。プロテアソーム阻害剤は頂端面からのAAV形質導入の長期レベルを増加するが、側底側感染へのこれらの影響は形質導入の長期レベルではなくて、主としてその速度を改変すと考えられる。これらの差は、頂端面および側底面からのrAAV形質導入に関係する基本的に異なる経路を強調する。
【0193】
数種の知見は、エンドサイトーシス後のウイルスのユビキチン化が、侵入AAVを選別するための重要な機構であるという意見を支持する。第一に、タンパク質のユビキチン依存性分解を遮断するとして知られているプロテアソーム阻害剤を用いる気道上皮の処理は、rAAV遺伝子導入を増加する。第二に、気道上皮内のユビキチンE3リガーゼ活性の阻害も形質導入を増加する。最後に、rAAVキャプシドタンパク質は、集密化ヒト繊維芽細胞内に感染後にユビキチン化され、そしてこのユビキチン化の程度は、ユビキチン−プロテアソーム分解経路の阻害により増加する(図16)。
【0194】
適用した観点から、本研究の最も重要な知見の一つは、マウス肺内でプロテアソーム阻害剤により誘導されるrAAV形質導入の永続的な高いレベルである。Z−LLLとrAAVとの同時投与は、感染の150日後で、近位気管支上皮細胞で検出不可能レベルから10.36±1.63%まで導入遺伝子発現を増加する。この遺伝子発現のレベルは、CFTR欠失の治療修正のために十分と考えられる(Crystal, 1999) 。臨床適用のためのこの戦略の実行可能性は、さらにマウスへのプロテアソーム阻害剤投与の後で、検出できるほどの局所または全身毒性がないことでも支持される。さらに、数種の別の臓器、例えば心臓骨格筋および肝臓における予備研究は、rAAV2のユビキチン化が臓器特異性様式で起きるであろうことを示唆した。骨格筋および心筋内へのプロテアソーム阻害剤の適用は、短期または長期rAAV媒介遺伝子導入のいずれにも影響しなかった。しかし肝臓内へのZ−LLLの適用(実施例7参照)は、感染1カ月後でrAAV形質導入に7倍の増加をもたらした。これらの知見は、トリペプチドプロテアソーム阻害剤がウイルスプロセシングの細胞生物学に応じて、肺以外の臓器内の遺伝子導入を増加させるために使用できることを示唆する。
【0195】
結論として、rAAV−2を用いる気道内の頂端側感染に対する主要な障害は、エンドソームプロセシングおよびユビキチン化のレベルにある。ユビキチン−プロテアソーム系の調節は、嚢胞性繊維症の遺伝子治療に対する気道の粘膜面からのrAAV形質導入を増加する革新的な戦略を明らかにした。
実施例7
生体内での肺気道上皮および肝臓内のアルカリホスファターゼ遺伝子の発現
肺または肝臓内の本発明の薬剤の存在または不在におけるrAAVの生体内活性は、アルカリホスファターゼ(AP)遺伝子を用いて試験することができる。AP遺伝子、組換えAV.Alkphosを含むrAAVベクター(5x1010粒子)を、PBS中のウイルス単独またはPBS中で40μM LLnLと一緒にしたウイルスのいずれかとして、マウス肺に投与した。ウイルスは、全体で体積30μlを、30G針を用いてC57Balb/cマウス気管内に直接点滴注入した。マウス肺内でのウイルスの拡散を確かめるために、ウイルス投与の直後に同じ注射薬を通じて空気50μlを肺内にポンプ送入した。感染の90日後に、肺をそのまま採取し、4パパラホルムアルデヒド中に固定し、次いで凍結割断した。AAV媒介導入遺伝子発現を、耐熱性アルカリホスファターゼに対して染色した10μm組織切片により評価した(図17A〜C)。
【0196】
組換えAV.Alkphos(5x1010粒子)もPBS中のウイルス単独またはPBS中で40μm LLnL、またはPBS中の40μm Z−LLLと一緒にしたウイルスのいずれかとしてマウス肝臓に投与した。ウイルスをC57B6マウスの門脈内に直接点滴注入した。AAV媒介アルカリホスファターゼ導入遺伝子発現を組織学染色により、感染の2および4週間後に冷凍組織切片内で評価した(図12)。
【0197】
【表2】
【表3】
【表4】
【表5】
【表6】
【表7】
【表8】
【表9】
【表10】
【表11】
【表12】
【表13】
【表14】
【表15】
【表16】
すべての出版物、特許および特許出願は、引用することにより本明細書中に含まれる。以上の明細書中で、本発明は一部の好ましい態様と関係して記載され、そして多くの詳細は説明の目的で記載されているが、本発明は別の態様が可能であり、そして本明細書中の一部は、本発明の基本原理から離れることなくかなりの変化が可能であることは、当該技術分野の熟練者には明らかであろう。
【図面の簡単な説明】
【図1】 分化した気管支上皮細胞内のrAAV形質導入の極性および時間経過。極性化気道上皮を、頂端側および側底側を経由して、AV.GFP3oriウイルス(MOI=10,000粒子/細胞)の5x109 粒子を用いて24時間感染した。GFP導入遺伝子発現細胞の存在量は、4、8、22、30、および40日目に間接蛍光顕微鏡により定量した。パネルAの棒グラフは、それぞれの条件における4回の独立した実験の平均(±SEM)を表す。
【図2】 極性化気管支上皮カルチャー中のAAV−2受容体、ヘパリン硫酸プロテオグリカンの免疫細胞化学定位。完全に分化したヒト気管支カルチャーの切片をヘパリン硫酸プロテオグリカンに対するモノクローナル抗体を用いてインキュベーションした。免疫反応性は、FITC標識二次抗体を用いて間接免疫蛍光法により検出した。パネルAは、極性化気管支上皮の基底面に主として位置するヘパリン硫酸プロテオグリカンに対する基底免疫染色を描いたものである。免疫反応性の特異性は、免疫染色前の遊離ヘパリン硫酸を用いる一次抗体の予備インキュベーションにより確認した(パネルB)。
【図3】 極性化気道上皮を、AV.GFP3oriウイルス(MOI=10,000粒子/細胞)による感染の前に一次カルチャーの頂端側(パネルA)または側底側(パネルB)からUV(25j/m2 )を用いて処理した。GFP導入遺伝子発現細胞の存在量は、4、8、22、30、および40日目に間接蛍光顕微鏡により定量した。結果は、それぞれの条件における4回の独立した実験の平均(±SEM)を表す。
【図4】 頂端面または側底面のいずれかに適用したウイルスを結合および内部化するための極性化気管支上皮カルチャーの能力を、放射能標識したrAAV( 3H−AV.GFP3ori)を用いて定量した。二つの実験条件で、ウイルス結合(ウイルスを用いる4℃で90分間のインキュベーション)または長期インキュベーション期間での結合および内部化ウイルスの全量(ウイルスを用いる37℃で24時間のインキュベーション)のいずれかで評価した。結果は、それぞれの条件における5回の独立した実験の平均(±SEM)を表す。
【図5】 極性化気道上皮内で200nmナイルレッド蛍光ビーズ(AAVビリオンの大きさ)を用いてエンドサイトーシスを評価した。ウイルス感染実験と同じ条件下でビースを頂端面または側底面に適用した。パネルA、B、D、E、G、およびH中の2−D共焦点再構成像は、ビーズの頂端側(A〜C)および側底側(D〜F)適用の後の細胞質の頂端面下コンパートメント内に内部化された蛍光ビーズの断面定位を描いている。パネルG〜Iは、ビーズの頂端側適用の前にUV(25j/m2 )を用いて処理したカルチャーを表す。数点の独立したインキュベーション時点も評価し、その中で5分(A、DおよびG)および6時間(B、EおよびH)を示す。低倍率(2.5x)間接蛍光顕微鏡により撮影した正面(en face) 顕微鏡写真も、インキュベーション6時間の時点のものを示す(C、FおよびI)。明らかでない理由で、適用の上皮側とは関係なくビーズは頂端面下コンパートメントに蓄積した。
【図6】 一次集密繊維芽細胞を、ブレフェルジンA(10μg/ml)、ビンブラスチン(22μM)、サイトカラシンB(1μM)、NH4 Cl(2mM)、クロロキン(20μM)、LLnL(400μM)、およびZ−LLL(4μM)(最終濃度を括弧内に示す)を用いて処理した後に、AV.GFP3oriウイルスをMOI=10,000粒子/細胞で感染した。感染の96時間後に細胞を採取し、そして形質導入効率をFACS分析によりGFP発現に関して未処理rAAV感染細胞と比較した。化合物の毒性により起きる非−特異性効果を制御するために、生存細胞の割合もヨウ化プロピジウム組込みのないことにより評価した。非毒性の投与のみを示した。
【図7】 プロテアソーム阻害剤は、気道上皮の頂端面または側底面からのrAAV形質導入を違って増加する。rAAV形質導入の効率および時間経過は、40μM LLnLの存在または不在におけるrAV.GFPori3(MOI=10,000粒子/細胞)を用いる感染の後に、極性化気道上皮カルチャー内で評価した。導入遺伝子発現は、間接蛍光顕微鏡により、記載の時点において、10x視野でのGFP陽性細胞の平均数を定量して測定した(各時点に対して3個の独立した試料の平均±SEM)。LLnL処理の効果は、頂端面(パネルA)または側底面(パネルB)からの感染の後のそれぞれの時点における組織試料の相当する組の間で比較した。それぞれのパネルの右側の顕微鏡写真は、感染時点から3および22日後の代表的な20x視野を示す。
【図8】 極性化気道上皮内のAAVゲノムのサザンブロット分析。分化したヒト気管支上皮カルチャーをAV.GFP3oriを用いて10,000粒子/細胞のMOIで頂端面(レーン1および3)または側底面(レーン2および4)から感染した。感染の50日後に、Hirt DNAを採取し、そして1%アガロースゲル中で電気泳動した。それぞれのレーンは、2個のトランスウエルカルチャーからの一緒にしたHirt DNAを表しそして2個のゲルは2個の独立した組織試料に由来した。サザンブロットをP32標識EGFPにハイブリダゼーションし、そしてフィルムに48時間暴露した。分子量標準をオートラジオグラムの左に記載した。AAVウイルスDNAの一本鎖および環状モノマー形は、それぞれ1.6kbおよび2.8kbで移動した。ssDNAは一本鎖ウイルスDNA、CMは環状モノマー、CCは環状コンカテマー、極性化気道上皮の環状中間体である。
【図9】 極性化気道上皮の基底面からのAAV形質導入に対する種々の薬剤の効果。完全に分化したヒト気管支気道上皮を、指定の薬剤を用いて、AAV感染(MOI=10,000粒子/細胞)の時点で側底面から処理した。Camp:0.1μMのカンプトテシン、Etop:10μMのエトポシド、Ahpi:5μg/mlのアフィドコリン、HU:50mMのヒドロキシ尿素、Geni:50μMのゲニステイン、LLnLは40μMで使用した。大部分の試験薬剤(ヒドロキシ尿素を除く)はDMSO中に溶かし、1%(体積/体積)DMSOのビヒクル対照も非−特異性効果を除外するために行った。GFP発現は、感染の48時間後に、カルチャー皿内でランダムに10x視野10個あたりのGFP陽性細胞の数を計数して測定した。データは、それぞれの試験薬品に対する3個の独立した感染の平均±SEMを表す。
【図10】 プロテアソーム阻害剤LLnLは、繊毛性細胞内にAAV形質導入を優先的に誘導する。AAVにより形質導入された細胞タイプを、40μM LLnLの存在または不在におけるAV.Alkphos感染(MOI=10,000粒子/細胞)を用いる極性化上皮カルチャーの側底側感染の3日後に、アルカリホスファターゼ染色により試験した。パネルA(LLnLなし)およびパネルB(LLnLあり)の正面顕微鏡写真は、LLnLの存在下における増加したrAAV形質導入を描いている。8μmパラフィン切片を、導入された細胞のタイプを組織学的定量のために使用した。下記3種を使用した。繊毛性細胞(頂端面および微繊毛に定位したalkphos発現)、内腔と接触しない上皮の下半分に存在する基底細胞、および非繊毛性、円柱状細胞。パネルC(LLnLなし)およびパネルD(LLnLあり)は、それぞれの条件における代表的な中性レッドで対比染色した断面を示す。AAVにより形質導入された種々の細胞タイプに対するalkphos染色細胞/2000上皮細胞の数をパネルEに示す(3個の独立したミリセルインサート試料からの平均±SEM)。
【図11】 極性化気管支上皮内のLLnL増加形質導入の最適化。分化したトランスウエルカルチャーを、AV.GFP3ori(10,000粒子/細胞)を用いて側底面(パネルA)または頂端面(パネルB)のどちらかから、LLnLおよび/またはEGTAを含む指定の処理を行って感染させた。すべの感染は24時間行いそしてGFP導入遺伝子発現を指定の時点において間接蛍光顕微鏡により測定した。データは、それぞれの実験条件に対する平均(±SEM、N=6)を示す。実験は、2人の異なる患者から得た試料から誘導したトランスウエル上で3回行った。下記の条件をパネルAの側底側感染に対して評価した。1)AV.GFP3oriのみを用いる単回感染(黒線)、2)40μM LLnL存在下でのAV.GFP3oriを用いる単回感染(紫実線)、3)40μM LLnL存在下でのAV.GFP3oriを用いる単回感染、その後基底コンパートメントカルチャー媒体内に40μM LLnLの5時間暴露を3日毎に反復(赤実線)、4)40μM LLnL存在下でのAV.GFP3oriを用いる単回感染、その後rAAVを除去した後に基底媒体内に40μM LLnLの連続暴露(緑実線)、および5)感染の時点で24時間にわたり40μM LLnLの存在下で、1日目および15日目にAV.GFP3oriを用いる反復感染(青破線)。下記の条件をパネルBの頂端側感染に対して評価した。1)AV.GFP3oriのみを用いる単回感染(黒実線)、2)ウイルス感染の前に3mM等張EGTAを用いる前処理の後にAV.GFP3oriを用いる単回感染(紫実線)、3)40μM LLnL存在下でのAV.GFP3oriを用いる単回感染(緑実線)、4)ウイルス感染前の3mM等張EGTAを用いる前処理の後に40μM LLnL存在下でのAV.GFP3oriを用いる単回感染(赤実線)。
【図12】 完全に分化したヒト気管支上皮内のS35標識AV.GFP3oriの結合および取り込み。頂端面または側底面からウイルスを結合または内部化する極性化気管支上皮の能力をS35標識rAAVを用いて定量した。結合アッセイは、ウイルスを用いて4℃で1時間インキュベーションし、PBS内で反復洗浄した後に行った。一緒にした結合および内部化したウイルスを、4℃で1時間以下のインキュベーション、そして引き続いて37℃での2時間および24時間インキュベーションの後に定量した。放射能標識ウイルスの非特異性背景結合は、平行した実験で、気管支細胞を接種しなかったコラーゲン被覆の空のチェンバーで決定した。背景カウント(平均15.67±5.17cpm/ウエル)を分析前の実験試料カウントから差し引いた。それぞれのパネルの右側のデータは、結合/内部化ウイルスのネットcpmとして示す(原カウントから空のトランスウエルの背景カウントを差し引く)。結果は、それぞれの条件に対する6個の独立したトランスウエルの平均(±SEM)を示す。実験は、2種の独立した組織試料に対して3回行った。それぞれの試料の対(LLnL使用または不使用)の間の差の有意性は、スチューデント(Student) のt−検定を用いて評価し、そしてp−値はそれぞれの条件のデータの上に括弧で示した。放射性ウイルスの取り込みとrAAVコード化導入遺伝子の機能的発現とを相関させるために、試料の同じ組からのGFP発現を感染の24時間後に間接蛍光顕微鏡により定量した。結果(平均±SEM、N=6)は、それぞれのパネルの右側に棒グラフで示した。
【図13】 S35標識ウイルスを用いる極性化気道上皮内のrAAVのin situ 定位。極性化ヒト気道上皮をS35標識ウイルス(MOI=50,000粒子/細胞)を用いて頂端側または側底側のいずれかから感染させた(±LLnL)。感染の2時間後に、トランスウエルを培地を用いて3回洗浄しそして4%パラホルムアルデヒド内に一晩固定し、次いでスクロースおよびOCT埋め込みをして凍結保護した。凍結切片10μmに写真エマルションを重ね、5週間暴露した後現像した。パネルAは、LLnLの存在下での側底側感染の後の代表的な定位パターンである。矢印は核会合ウイルスを示す。パネルBは、パネルA中の四角で囲った部分の拡大である。ブラインド形態測定定量を、パネルAの10個のランダムに選定した視野の核関連および細胞質放射性粒子の数を数えて行った。全体で細胞60個を視野毎に定量して、全体で試料当たりに細胞600個および条件あたりに細胞2400個を得た。パネルCの結果は、それぞれの条件あたりに独立した試料4個の平均±SEMである。
【図14】 LLnLは、感染後の一定期間にrAAV形質導入を誘導する。AAV形質導入に対するLLnL投与の速度論的効果を研究するために、プロテアソーム阻害剤LLnLを、気道上皮の側底面からのrAAV感染の後、種々の時点で培地に加えた。すべてのカルチャーをAV.GFP3ori(10,000粒子/細胞)で0時間目に感染させた。40μM LLnLを、感染時点(0時間目)または感染後24時間間隔で加えた(24、48、および72時間)。LLnL処理を行わなかった場合のAAV形質導入の基線も示した。GFP発現は、間接蛍光顕微鏡法を用いて感染の1、2、3、および4日後に定量しそして10x視野あたりのGFP陽性細胞として表す。それぞれのパネルのデータは、3個の独立して試料からの平均(±SEM)を表す。LLnL添加後の導入遺伝子媒介のrAAV中の即時増加は、細胞質からウイルス粒子が発現できる核へのウイルス粒子のエンドソームプロセシングを増加することによりLLnLが作用するという仮説に一致する。さらに、感染の2〜3日後のLLnLの添加は、感染時点でのLLnLの添加よりも高い導入遺伝子発現レベルを与える。これらの結果は、ウイルス結合および内部化は、LLnLにより増強される段階ではないであろうという意見を支持する。
【図15】 ウイルスDNAのサザンブロット分析によるrAAVエンドサイトーシスの検討。AV.GFP3ori感染または模擬感染したヒト気管支上皮からのHirt DNA(パネルAおよびBの両方でレーン1および7)を、P32標識EGFPプローブに対するサザンブロッティングによるウイルスゲノムの直接試験のために抽出した。パネルAは、頂端側または側底側感染前1時間、4℃でのLLnLの存在下または不在、EGTAを用いるか用いないウイルス結合試験を描く。細胞表面結合ウイルスは、トリプシンにより完全に除去された(パネルA、レーン2〜6)。表面結合rAAVの量を決定するために、細胞をAV.GFP3oriを用いて1時間、4℃でで感染させそしてHirt DNA抽出の前にはトリプシン処理しなかった。パネルA レーン8:頂端側AAV感染、レーン9:LLnLが存在する場合の頂端側AAV感染、レーン10:LLnLが存在する場合の頂端側感染の前に、細胞を等張EGTAで前処理した、レーン11:側底側感染、レーン12:LLnLが存在する場合の側底側感染。パネルBは、LLnLの存在下または不在における頂端面または側底面のいずれかからのrAAV内部化、および等張EGTAおよびLLnLで複合処理した後の頂端面からの内部化を評価する研究の結果を示す。内部化したウイルスゲノムのネットの量を検出するために、パネルB内のすべての試料を、Hirt DNA採取の直前にトリプシンを用いて処理した。感染後4時間(パネルB、レーン2〜6)および24時間(パネルB、レーン8〜12)、37℃でのインキュベーションにおける内部化ウイルスの程度は、1.6kb一本鎖ウイルスゲノムバンドの強度により表される。パネルB レーン2:4時間の頂端側AAV感染、レーン3:LLnLの存在下での4時間の頂端側AAV感染、レーン4:LLnLの存在下での4時間の頂端側感染の前に等張EGTAを用いて細胞を前処理した。レーン5:4時間の基底感染、レーン6:LLnLの存在下での4時間の側底側感染、レーン8:24時間の頂端側AAV感染、レーン9:LLnLの存在下での24時間の頂端側感染、レーン10:LLnLの存在下での24時間の頂端側感染の前に等張EGTAを用いて細胞を前処理した、レーン11:24時間の側底側感染、レーン12:LLnLの存在下での24時間の側底側感染。パネルCは、頂端膜(レーン1、2、5、6、10、11および12)および側底膜(レーン3、4、7、8、9、13および14)からの24時間感染の2、10、30日後におけるrAAVゲノムに対するLLnL/EGTAの効果を比較する。処理条件は、それぞれのレーン上に記載した。トランスウエルは、Hirt DNAを採取する前にトリプシンを用いて処理しなかった。別の対照は、Ad.d1802(MOI=500部/細胞)を用いた同時感染を含み、モノマーからの複製を証明した(レーン9、4.7kb)。異なる暴露時間をC内の異なるパネルで使用したことに注意すること(レーン1〜4は3時間、レーン5〜8は15時間、レーン10〜14は12時間)。未感染カルチャーからの相当するDNA試料は、検出可能はハイブリダゼーションを証明しなかった(データは示さない)。
【図16】 ウイルスユビキチン化状態の変性がrAAV形質導入を促進する。極性化ヒト気道細胞と同様に、ヒト一次集密繊維芽細胞中のrAAV形質導入もトリペプチドプロテアソーム阻害剤により増加する。80%集密ヒト一次繊維芽細胞を1000DNAのMOIのAV.GFP3oriで感染させた。パネルAは、感染の96時間後における40μM LLnLの不在(左の写真)および存在(右の写真)でのGFP導入遺伝子発現を描いている。同様の効果が4μM Z−LLLを用いても得られる(データは記載しない)。上および下パネルは、明視野およびFITC−チャンネル蛍光顕微鏡写真をそれぞれ示す。GFP発現細胞のFACS選別により測定したrAAVを形質導入された細胞の平均(±SEM,N=3)百分率をパネルBの棒グラフに表す。パネルCは、一次集密繊維芽細胞の感染の6時間後のユビキチン化AAVキャプシドタンパク質(矢じり印で標識)の同定を示す。本研究において、感染された細胞からのrAAVを最初に抗−VP1,2,3(AAV−キャプシド)モノクローナル抗体を用いて免疫沈降し、次いで抗ユビキチンモノクローナル抗体を用いてユビキチン側鎖のウエスタンブロット検出を行った。約65および25kdで移動する二つの大きい背景バンドは、二次抗体と交差反応する重鎖および軽鎖抗体サブユニットを表す。さらに、低分子量交差反応バンド(30〜40kd)の等しい強度は、タンパク質の等しい負荷の内部対照として役立つ。パネルDは、ユビキチンE3リガーゼの阻害による極性化気道上皮内のrAAV形質導入の増加を表す。ユビキチンリガーゼ阻害剤ジペプチド(0.2mM H−Leu−Ala−OHおよび0.2mM H−His−Ala−OH)を用いる処理の後に、上皮をAV.GFP3oriウイルス(MOI=10,000粒子/細胞)を用いて側底面から感染させた。結果は、感染の1および15日後における10x視野あたりのGFP発現細胞の平均(±SEM,N=3)数を示す。
【図17】 プロテアソーム阻害剤によるマウス導通気道内のrAAV媒介遺伝子導入の持続性誘導。パネルA〜C)組み合えAV.Alkphos(5x1010粒子)をPBS中のウイルス単独またはPBS中の40μM LLnLと一緒のウイルスとしてのいずれかでマウス肺に投与した。全体で30μlの体積で30G針を用いてC57Balb/cマウス気管内に直接点滴注入した。マウス肺内でのウイルスの拡散を確かめるために、ウイルスを投与した後に直ちに同じ注射器を通して空気50μlを肺内に送った。感染の90日後に、肺をそのまま採取しそして4%パラホルムアルデヒド中に固定し、次いで冷凍割断した。AAV媒介導入遺伝子発現は、耐熱性アルカリホスファターゼに対する10μm組織切片染色により評価した。パネルA:AAV単独を用いる感染、パネルBおよびC:40m LLnLを補足したAAVを用いる感染,パネルD〜F)6週齢BALB/cマウス(各グループ中の動物N=3)を、400μM Z−LLLの存在または不在でAV.LacZの5x1010DNA粒子を用いて鼻点滴注入により感染させた。感染の150日後の大きい細気管支内のLacZ発現に対する組織化学的染色の代表的な例をパネルDおよびEに示す。それぞれのパネルの右および左側は、ノマルスキーおよび明視野顕微鏡写真である。100μmのスケールバーは、すべての顕微鏡写真に適用される。気道の種々のレベルにおけるLacZ発現上皮細胞の平均(±SEM)百分率は、方法の項に概略を記載した形態測定法を用いて定量し、そして分析は、それぞれのグループに対する3匹の独立した動物からの結果を示す(パネルC)。
【図18】 HeLa細胞内のCy3標識rAAV。HeLa細胞をCy3標識AV.GFP3oriの500粒子/細胞のMOIで90分間、4℃において血清の不在で感染させた。次いで、細胞を洗浄し、そして2%パラホルムアルデヒド中で固定するかまたは固定の前に37℃でさらに60または120分間インキュベーションのいずれかを行った。ウイルス感染の前に、細胞を30分間、1μM 5−クロロメチルフルオレセイン二酢酸(Cell TrackerTM Green CMFDA, Molecular Probe)内でインキュベーションして二重蛍光チャンネル像内の細胞および標識ウイルスを可視化した。90分間、4℃で感染し、次いで60および120分間、37℃でインキュベーションした細胞の代表的な共焦点像を、Cy3および二重Cy3/FITCチャンネルの両方で示す(パネルA)。二重チャンネル像内の核をNuで標示する。図示した共焦点像は、細胞の中心領域内で取った0.5μm3層から併合したものである。パネルBは、90分間、4℃で感染し(パネルBの左側)次いで120分間、37℃でインキュベーションした細胞に対するノマルスキーおよびCy3チャンネルを用いた非−共焦点像を示す(パネルBの右側)。4℃で結合したウイルスは、細胞の表面膜に位置する。37℃のインキュベーション時間が長くなると、ウイルスは核膜に転位する。4℃でのウイルス結合は、パネルCに記入した指定濃度における遊離ヘパリンの添加により完了する(共焦点像は3件からの併合)。FITC標識トランスフェリンおよびCy3標識rAAVのエンドサイトーシスは、HeLa細胞内で観察された(パネルD)。90分間4℃においてFITC標識トランスフェリンの存在下でCy3rAAVを用いてHeLa細胞を感染し、次いで洗浄した。次いで、細胞を固定の前に37℃で30分間静置し、そして共焦点顕微鏡法により分析した。パネルD内の像は、Cy3、FITCおよび組み合わせたチャンネル(合併)の単独の0.5μm断面であった。結果は、rAAVおよびトランスフェリンの大部分のエンドサイト小胞内でのコロニー化を証明する。
【図19】 プロテアソーム阻害剤Z−LLLの同時投与は、生体内でマウス肝臓のAAV媒介遺伝子導入を増加する。組換えAV.Alkphos(5x1010粒子)をPBS中のウイルス単独またはPBS中の40μM LLnLと一緒のウイルスとして、またはPBS中の200μM LLnLと一緒のウイルスとしてのいずれかでマウスの肝臓内に投与した。ウイルスは、C57B6マウスの門脈内に直接点滴注入した。AAV媒介アルカリホスファターゼ導入遺伝子発現は、組織学的染色により感染2〜4週間後に冷凍組織切片で評価した。それぞれの動物個体のすべての6個の肝臓葉内のalkphos陽性細胞の平均百分率を、NIHイメージ分析ソフトウエアにより定量した。データは、それぞれのグループ内のそれぞれの時点に対する3匹の独立したマウスの平均(±SEM)を表す。
Claims (24)
- 医薬の製造における、哺乳類細胞のAAV形質導入を増加させる量のプロテアソーム阻害剤の使用であって、該プロテアソーム阻害剤が、哺乳類細胞の膜へのウイルスの結合の後であってウイルスゲノムの発現可能な形態をもたらす第二の鎖の合成の前に、AAV形質導入を増加させ、
ここで、該プロテアソーム阻害剤が、場合によりN−及び/又はC−末端保護基を有するジ−またはトリ−ペプチドである、上記使用。 - 組換えAAVのエンドソームプロセシングが改変される、請求項1記載の使用。
- 該プロテアソーム阻害剤が式(I):R1 −A−(B)n −C〔式中、R1 はN−末端アミノ酸ブロック基であり、それぞれAおよびBは独立してアミノ酸であり、Cはアミノ酸であり、ここで末端カルボキシ基はホルミル(CHO)基により置換されており、そしてnは0、1、2または3である〕の化合物またはその薬剤的に許容できる塩である、請求項1記載の使用。
- R1 が(C1 〜C10)アルカノイルである、請求項3記載の使用。
- R1 がアセチルまたはベンジルオキシカルボニルである、請求項3記載の使用。
- それぞれAおよびBが、独立してアラニン、アルギニン、グリシン、イソロイシン、ロイシン、バリン、ノル−ロイシンまたはノル−バリンである、請求項3記載の使用。
- それぞれAおよびBが、イソロイシンである、請求項3記載の使用。
- 末端カルボキシ基がホルミル(CHO)基により置換されている、Cがアラニン、アルギニン、グリシン、イソロイシン、ロイシン、バリン、ノル−ロイシンまたはノル−バリンである請求項3記載の使用。
- R1 が(C1 〜C10)アルカノイルまたはベンジルオキシカルボニルであり、AおよびBがそれぞれイソロイシンであり、Cがノル−ロイシンまたはノル−バリンである請求項3記載の使用であって、末端カルボキシ基がホルミル(CHO)基により置換されており、そしてNが1である上記使用。
- 該プロテアソーム阻害剤が式(II)
R2 は、N−末端アミノ酸ブロック基であり、
R3 、R4 およびR5 は、それぞれ独立して水素、(C1 〜C10)アルキル、アリールまたはアリール(C1 〜C10)アルキルであり、そして
R6 、R7 およびR8 は、それぞれ独立して水素、(C1 〜C10)アルキル、アリールまたはアリール(C1 〜C10)アルキルである〕の化合物またはこの薬剤的に許容できる塩である、請求項1記載の使用。 - R2 が(C1 〜C10)アルカノイルである、請求項10記載の使用。
- R2 がアセチルまたはベンジルオキシカルボニルである、請求項10記載の使用。
- R3 が水素または(C1 〜C10)アルキルである、請求項10記載の使用。
- R3 が2−メチルプロピルである、請求項10記載の使用。
- R4 が水素または(C1 〜C10)アルキルである、請求項10記載の使用。
- R4 が2−メチルプロピルである、請求項10記載の使用。
- R5 が水素または(C1 〜C10)アルキルである、請求項10記載の使用。
- R5 がブチルまたはプロピルである、請求項10記載の使用。
- R2 がアセチルまたはベンジルオキシカルボニルであり、R3 およびR4 がそれぞれ2−メチルプロピルであり、R5 がブチルまたはプロピルであり、そしてR6 、R7 およびR8 が、それぞれ独立して水素である、請求項10記載の使用。
- 該プロテアソーム阻害剤がユビキチンの活性化、ユビキチンキャリヤータンパク質、ユビキチンリガーゼ、およびこれらの組み合わせへのユビキチンの転移を阻害する、請求項1記載の使用。
- 該プロテアソーム阻害剤が式(V)
R−A−A1 −R1
〔式中、Rは、水素、アミノ酸、またはペプチドであり、ここでN末端アミノ酸は場合によりアセチル、アシル、トルフルオロアセチル、またはベンジルオキシカルボニルを用いてアミノ基において保護されることができ、Aはアミノ酸または直接結合であり、A1 はアミノ酸であり、そして
R1 は、ヒドロキシまたはアミノ酸であり、ここでC−末端アミノ酸は、場合により(C1 〜C6 )アルキル、フェニル、ベンジルエステルまたはアミド(例えばC(=O)NR2 、ここで、それぞれのRは独立して水素または(C1 〜C6 )アルキルである)を用いてカルボキシ基において保護されていてもよい〕
の化合物またはこの薬剤的に許容できる塩である、請求項1記載の使用。 - 該プロテアソーム阻害剤がH−Leu−Ala−OH、H−His−Ala−OH、またはこれらの組み合わせである、請求項21記載の使用。
- 該プロテアソーム阻害剤の活性を増加させる第二の薬剤をさらに含んでなる、請求項1記載の使用。
- 第二の薬剤がEGTAである、請求項23記載の使用。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13818899P | 1999-06-08 | 1999-06-08 | |
| US60/138,188 | 1999-06-08 | ||
| US20108900P | 2000-05-02 | 2000-05-02 | |
| US60/201,089 | 2000-05-02 | ||
| PCT/US2000/015700 WO2000075365A2 (en) | 1999-06-08 | 2000-06-08 | Compounds and methods to enhance raav transduction |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2003501068A JP2003501068A (ja) | 2003-01-14 |
| JP2003501068A5 JP2003501068A5 (ja) | 2007-08-09 |
| JP4969002B2 true JP4969002B2 (ja) | 2012-07-04 |
Family
ID=26835948
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2001501645A Expired - Lifetime JP4969002B2 (ja) | 1999-06-08 | 2000-06-08 | rAAV形質導入を増加するための化合物および方法 |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US8846030B2 (ja) |
| EP (1) | EP1190249A2 (ja) |
| JP (1) | JP4969002B2 (ja) |
| AU (1) | AU782966B2 (ja) |
| CA (1) | CA2376400A1 (ja) |
| WO (1) | WO2000075365A2 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11684679B2 (en) | 2016-03-07 | 2023-06-27 | University Of Iowa Research Foundation | AAV-mediated expression using a synthetic promoter and enhancer |
| US11702672B2 (en) | 2016-02-08 | 2023-07-18 | University Of Iowa Research Foundation | Methods to produce chimeric adeno-associated virus/bocavirus parvovirus |
| US11999965B2 (en) | 2017-01-13 | 2024-06-04 | University Of Iowa Research Foundation | Bocaparvovirus small noncoding RNA and uses thereof |
| US12173305B2 (en) | 2016-05-26 | 2024-12-24 | University Of Iowa Research Foundation | cis and trans requirements for terminal resolution of human bocavirus 1 |
Families Citing this family (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6924128B2 (en) | 1994-12-06 | 2005-08-02 | Targeted Genetics Corporation | Packaging cell lines for generation of high titers of recombinant AAV vectors |
| US6995006B2 (en) | 1997-09-05 | 2006-02-07 | Targeted Genetics Corporation | Methods for generating high titer helper-free preparations of released recombinant AAV vectors |
| US6346415B1 (en) | 1997-10-21 | 2002-02-12 | Targeted Genetics Corporation | Transcriptionally-activated AAV inverted terminal repeats (ITRS) for use with recombinant AAV vectors |
| US6436392B1 (en) | 1998-05-20 | 2002-08-20 | University Of Iowa Research Foundation | Adeno-associated virus vectors |
| US6893865B1 (en) | 1999-04-28 | 2005-05-17 | Targeted Genetics Corporation | Methods, compositions, and cells for encapsidating recombinant vectors in AAV particles |
| JP4969002B2 (ja) | 1999-06-08 | 2012-07-04 | ユニバーシテイ・オブ・アイオワ・リサーチ・フアウンデーシヨン | rAAV形質導入を増加するための化合物および方法 |
| US7241447B1 (en) * | 1999-10-07 | 2007-07-10 | University Of Iowa Research Foundation | Adeno-associated virus vectors and uses thereof |
| US8241622B2 (en) | 2001-07-13 | 2012-08-14 | University Of Iowa Research Foundation | Adeno-associated virus vectors with intravector heterologous terminal palindromic sequences |
| JP2007524379A (ja) * | 2003-03-31 | 2007-08-30 | ユニバーシテイ・オブ・アイオワ・リサーチ・フアウンデーシヨン | 上皮ナトリウムチャンネル関連障害のPharmico−遺伝子治療 |
| AU2007243184A1 (en) * | 2006-04-28 | 2007-11-08 | University Of Iowa Research Foundation | Methods and compounds to modulate parvovirus transduction of mammalian cells or to alter virus infection, method to identify a viral receptor or co-receptor |
| CA2788682C (en) * | 2010-02-05 | 2019-03-05 | The University Of North Carolina At Chapel Hill | Compositions and methods for enhanced parvovirus transduction |
| WO2011117258A2 (en) * | 2010-03-22 | 2011-09-29 | Association Institut De Myologie | Methods of increasing efficiency of vector penetration of target tissue |
| WO2015191508A1 (en) | 2014-06-09 | 2015-12-17 | Voyager Therapeutics, Inc. | Chimeric capsids |
| CN119876138A (zh) | 2014-11-14 | 2025-04-25 | 沃雅戈治疗公司 | 调节性多核苷酸 |
| CA3193811A1 (en) | 2014-11-14 | 2016-05-19 | Voyager Therapeutics, Inc. | Compositions and methods of treating amyotrophic lateral sclerosis (als) |
| HK1245326A1 (zh) | 2014-12-12 | 2018-08-24 | Voyager Therapeutics, Inc. | 用於生产scaav的组合物和方法 |
| EP3448874A4 (en) | 2016-04-29 | 2020-04-22 | Voyager Therapeutics, Inc. | COMPOSITIONS FOR TREATING A DISEASE |
| EP3448987A4 (en) | 2016-04-29 | 2020-05-27 | Voyager Therapeutics, Inc. | COMPOSITIONS FOR TREATING A DISEASE |
| CA3024448C (en) | 2016-05-18 | 2025-09-09 | Voyager Therapeutics, Inc. | MODULATING POLYNUCLEOTIDES |
| CA3024449A1 (en) | 2016-05-18 | 2017-11-23 | Voyager Therapeutics, Inc. | Compositions and methods of treating huntington's disease |
| WO2018044933A1 (en) | 2016-08-30 | 2018-03-08 | The Regents Of The University Of California | Methods for biomedical targeting and delivery and devices and systems for practicing the same |
| JP2020518259A (ja) | 2017-05-05 | 2020-06-25 | ボイジャー セラピューティクス インコーポレイテッドVoyager Therapeutics,Inc. | ハンチントン病治療組成物および方法 |
| EP3618839A4 (en) | 2017-05-05 | 2021-06-09 | Voyager Therapeutics, Inc. | COMPOSITIONS AND METHODS OF TREATMENT OF AMYOTROPHIC LATERAL SCLEROSIS (ALS) |
| JP7229989B2 (ja) | 2017-07-17 | 2023-02-28 | ボイジャー セラピューティクス インコーポレイテッド | 軌道アレイガイドシステム |
| JP7221275B2 (ja) | 2017-08-03 | 2023-02-13 | ボイジャー セラピューティクス インコーポレイテッド | Aavを送達するための組成物および方法 |
| MX2020002148A (es) * | 2017-08-25 | 2020-07-20 | Ovid Therapeutics Inc | Vectores adenoasociados recombinantes. |
| US20200263199A1 (en) | 2017-09-29 | 2020-08-20 | Voyager Therapeutics, Inc. | Rescue of central and peripheral neurological phenotype of friedreich's ataxia by intravenous delivery |
| TW202413649A (zh) | 2017-10-16 | 2024-04-01 | 美商航海家醫療公司 | 肌萎縮性脊髓側索硬化症(als)之治療 |
| WO2019079242A1 (en) | 2017-10-16 | 2019-04-25 | Voyager Therapeutics, Inc. | TREATMENT OF AMYOTROPHIC LATERAL SCLEROSIS (ALS) |
| US12281321B2 (en) | 2018-09-28 | 2025-04-22 | Voyager Therapeutics, Inc. | Frataxin expression constructs having engineered promoters and methods of use thereof |
| AU2021265103A1 (en) | 2020-04-29 | 2023-01-19 | Bristol-Myers Squibb Company | Miniaturized dystrophins having spectrin fusion domains and uses thereof |
| WO2025222013A1 (en) * | 2024-04-17 | 2025-10-23 | Solid Biosciences Inc. | Recombinant aav production process |
Family Cites Families (74)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4487934A (en) | 1980-06-06 | 1984-12-11 | Hoechst-Roussel Pharmaceuticals Inc. | Method of preparing poly-substituted acylbenzenes |
| US4501729A (en) | 1982-12-13 | 1985-02-26 | Research Corporation | Aerosolized amiloride treatment of retained pulmonary secretions |
| DE3326472A1 (de) | 1983-07-22 | 1985-02-14 | Hoechst Ag, 6230 Frankfurt | Neue insulin-derivate, verfahren zu deren herstellung und deren verwendung sowie pharmazeutische mittel zur behandlung des diabetes mellitus |
| DE3567209D1 (en) | 1984-04-06 | 1989-02-09 | Zambon Spa | Optically active ketals, processes for their preparation and their use in the synthesis of apha-arylakanoic acids |
| AU654979B2 (en) | 1990-10-05 | 1994-12-01 | University Of North Carolina At Chapel Hill, The | Method of administering amiloride |
| US5173414A (en) | 1990-10-30 | 1992-12-22 | Applied Immune Sciences, Inc. | Production of recombinant adeno-associated virus vectors |
| US5292498A (en) | 1991-06-19 | 1994-03-08 | The University Of North Carolina At Chapel Hill | Method of treating lung disease with uridine triphosphates |
| US5478745A (en) | 1992-12-04 | 1995-12-26 | University Of Pittsburgh | Recombinant viral vector system |
| US5686436A (en) | 1993-05-13 | 1997-11-11 | Hiv Diagnostics, Inc. | Multi-faceted method to repress reproduction of latent viruses in humans and animals |
| US5512269A (en) | 1993-06-09 | 1996-04-30 | Burroughs Wellcome, Co. | Method of treating retained pulmonary secretions |
| US5498531A (en) | 1993-09-10 | 1996-03-12 | President And Fellows Of Harvard College | Intron-mediated recombinant techniques and reagents |
| US5750398A (en) * | 1993-11-30 | 1998-05-12 | David C. Johnson | Vector, element and method for inhibiting immune recognition |
| US5604090A (en) | 1994-06-06 | 1997-02-18 | Fred Hutchinson Cancer Research Center | Method for increasing transduction of cells by adeno-associated virus vectors |
| US5656256A (en) | 1994-12-14 | 1997-08-12 | The University Of North Carolina At Chapel Hill | Methods of treating lung disease by an aerosol containing benzamil or phenamil |
| US5843742A (en) | 1994-12-16 | 1998-12-01 | Avigen Incorporated | Adeno-associated derived vector systems for gene delivery and integration into target cells |
| US6281010B1 (en) | 1995-06-05 | 2001-08-28 | The Trustees Of The University Of Pennsylvania | Adenovirus gene therapy vehicle and cell line |
| US5635160A (en) | 1995-06-07 | 1997-06-03 | The University Of North Carolina At Chapel Hill | Dinucleotides useful for the treatment of cystic fibrosis and for hydrating mucus secretions |
| US5628984A (en) | 1995-07-31 | 1997-05-13 | University Of North Carolina At Chapel Hill | Method of detecting lung disease |
| JPH11511968A (ja) | 1995-08-21 | 1999-10-19 | デューク ユニバーシティ | 抗原提示細胞上にて抗原密度を増加させる方法 |
| US5801030A (en) | 1995-09-01 | 1998-09-01 | Genvec, Inc. | Methods and vectors for site-specific recombination |
| WO1997009997A1 (en) | 1995-09-12 | 1997-03-20 | Genentech, Inc. | Cystic fibrosis therapy |
| DE69636937T3 (de) | 1995-12-15 | 2011-01-05 | Virxsys Corp. | Durch trans-spaltung erhaltene therapeutische molekule |
| US6083702A (en) | 1995-12-15 | 2000-07-04 | Intronn Holdings Llc | Methods and compositions for use in spliceosome mediated RNA trans-splicing |
| US6420347B1 (en) | 1997-03-27 | 2002-07-16 | Inspire Pharmaceuticals, Inc. | Method of treating ciliary dyskinesia with uridine triphosphates and related compounds |
| EP0932418B1 (en) | 1996-09-06 | 2007-12-05 | The Trustees Of The University Of Pennsylvania | Method for recombinant adeno-associated virus-directed gene therapy |
| AU5588298A (en) | 1996-12-02 | 1998-06-29 | Cell Genesys, Inc. | Adeno-associated viral vector-mediated delivery of dna to cells of the liver |
| US6287569B1 (en) | 1997-04-10 | 2001-09-11 | The Regents Of The University Of California | Vaccines with enhanced intracellular processing |
| FR2763845B1 (fr) | 1997-05-30 | 2003-04-18 | Centre Nat Rech Scient | Produits anti-cancereux pour le traitement de la mucoviscidose |
| US6156303A (en) | 1997-06-11 | 2000-12-05 | University Of Washington | Adeno-associated virus (AAV) isolates and AAV vectors derived therefrom |
| US6037177A (en) | 1997-08-08 | 2000-03-14 | Cell Genesys, Inc. | Method for increasing the efficiency of recombinant AAV production |
| ID29198A (id) | 1997-08-29 | 2001-08-09 | Univ North Carolina | Pemakaian uridin 5'-difosfat dan persamaannya untuk mengobati penyakit paru-paru |
| US6436392B1 (en) | 1998-05-20 | 2002-08-20 | University Of Iowa Research Foundation | Adeno-associated virus vectors |
| CA2332540A1 (en) | 1998-05-22 | 1999-12-02 | Benjamin R. Yerxa | Therapeutic dinucleotide and derivatives |
| US6290951B1 (en) | 1998-08-01 | 2001-09-18 | Alfacell Corporation | Alteration of the cell cycle in vivo, and particularly for inducing apoptosis of tumor cells |
| US6200560B1 (en) | 1998-10-20 | 2001-03-13 | Avigen, Inc. | Adeno-associated virus vectors for expression of factor VIII by target cells |
| EP1143896B1 (en) | 1998-10-20 | 2006-12-13 | The University of North Carolina at Chapel Hill | Methods of hydrating the nasal mucosal surface |
| US6221349B1 (en) | 1998-10-20 | 2001-04-24 | Avigen, Inc. | Adeno-associated vectors for expression of factor VIII by target cells |
| US6855549B1 (en) | 1998-11-23 | 2005-02-15 | The University Of Iowa Research Foundation | Methods and compositions for increasing the infectivity of gene transfer vectors |
| US6926911B1 (en) | 1998-12-22 | 2005-08-09 | The University Of North Carolina At Chapel Hill | Compounds and methods for the treatment of airway diseases and for the delivery of airway drugs |
| CN1339970A (zh) | 1999-02-10 | 2002-03-13 | 鹤尾隆 | 抗癌药物增强剂 |
| US6306663B1 (en) * | 1999-02-12 | 2001-10-23 | Proteinex, Inc. | Controlling protein levels in eucaryotic organisms |
| JP4969002B2 (ja) | 1999-06-08 | 2012-07-04 | ユニバーシテイ・オブ・アイオワ・リサーチ・フアウンデーシヨン | rAAV形質導入を増加するための化合物および方法 |
| US7122335B1 (en) | 1999-06-08 | 2006-10-17 | University Of Iowa Research Foundation | Compounds and methods to enhance rAAV transduction |
| KR20020038694A (ko) | 1999-07-19 | 2002-05-23 | 프란시스 제이 메이어 | 소듐 채널 차단제의 콘쥬게이트 및 그 이용방법 |
| US6475537B1 (en) | 2000-07-27 | 2002-11-05 | Rhodia Inc. | Hops acid antibacterial compositions |
| US6416759B1 (en) | 1999-09-30 | 2002-07-09 | The Regents Of The University Of California | Antiproliferative Sgk reagents and methods |
| EP1224313A1 (en) | 1999-10-07 | 2002-07-24 | University of Iowa Research Foundation | Adeno-associated viruses and uses thereof |
| AU7879000A (en) | 1999-10-12 | 2001-04-23 | University Of North Carolina At Chapel Hill, The | Adeno-associated virus vectors encoding factor viii and methods of using the same |
| WO2001029243A1 (en) | 1999-10-15 | 2001-04-26 | Dalhousie University | Method and vector for producing and transferring trans-spliced peptides |
| US20020132770A1 (en) | 1999-10-27 | 2002-09-19 | Caplan Michael T. | Conductance of improperly folded proteins through the secretory pathway and related methods for treating disease |
| WO2001066782A1 (en) | 2000-03-03 | 2001-09-13 | The Board Of Trustees Of The Leland Stanford Junior University | Adeno-associated viral compositions and methods |
| CA2302627A1 (en) | 2000-03-23 | 2001-09-23 | Rashmi Kothary | Constructs comprising the enac promoter sequence and a reporter gene for screening pharmaceuticals |
| US20020076754A1 (en) | 2000-04-20 | 2002-06-20 | Liangwu Sun | Overcoming AAV vector size limitation through viral DNA hetero-dimerization |
| CA2406743A1 (en) | 2000-04-28 | 2001-11-08 | The Trustees Of The University Of Pennsylvania | Recombinant aav vectors with aav5 capsids and aav5 vectors pseudotyped in heterologous capsids |
| US20020128203A1 (en) | 2000-09-20 | 2002-09-12 | Laurent Schild | Methods of identifying inhibitory compounds and uses thereof |
| CA2423170A1 (en) | 2000-09-22 | 2002-03-28 | Galephar M/F | Pharmaceutical semi-solid composition of isotretinoin |
| WO2002028348A2 (en) | 2000-10-04 | 2002-04-11 | The Children's Hospital Of Philadelphia | Compositions and methods for treatment of cystic fibrosis |
| AU2002251844A1 (en) | 2001-02-02 | 2002-10-03 | Corixa Corporation | Compositions and methods for the therapy and diagnosis of colon cancer |
| US20030013137A1 (en) | 2001-03-13 | 2003-01-16 | Barak Larry S. | Automated methods of detecting receptor activity |
| US6586416B2 (en) | 2001-04-19 | 2003-07-01 | James K. Bubien | Methods of treatment using an epithelial sodium channel blocker and an inhibitor of the mineralocorticoid receptor |
| WO2002087306A2 (en) | 2001-05-01 | 2002-11-07 | Senomyx, Inc. | High throughput cell-based assay for monitoring sodium channel activity and discovery of salty taste modulating compounds |
| US20030003583A1 (en) | 2001-06-08 | 2003-01-02 | Raphael Hirsch | Regulation of transgene expression following AAV transduction |
| EP1419245A4 (en) | 2001-07-13 | 2006-04-05 | Univ Iowa Res Found | ADENO-ASSOCIATED PSEUDOTYPE VIRUSES AND USES THEREOF |
| US8241622B2 (en) | 2001-07-13 | 2012-08-14 | University Of Iowa Research Foundation | Adeno-associated virus vectors with intravector heterologous terminal palindromic sequences |
| AU2002364612A1 (en) | 2001-12-31 | 2003-07-24 | Algos Therapeutics, Inc. | METHODS AND MATERIALS FOR MODULATING ENaC-BETA |
| CA2483105C (en) | 2002-05-13 | 2011-05-10 | Arexis Ab | Autoimmune conditions and nadph oxidase defects |
| JP2007524379A (ja) | 2003-03-31 | 2007-08-30 | ユニバーシテイ・オブ・アイオワ・リサーチ・フアウンデーシヨン | 上皮ナトリウムチャンネル関連障害のPharmico−遺伝子治療 |
| EP1486567A1 (en) | 2003-06-11 | 2004-12-15 | Deutsches Krebsforschungszentrum Stiftung des öffentlichen Rechts | Improved adeno-associated virus (AAV) vector for gene therapy |
| SI3235827T1 (sl) | 2003-06-19 | 2021-05-31 | Genzyme Corporation | AAV virioni z zmanjšano imunoreaktivnostjo in uporaba za ta namen |
| WO2005049850A2 (en) | 2003-11-14 | 2005-06-02 | University Of Washington | Compositions and methods for systemic nucleic acid sequence delivery |
| WO2005116224A2 (en) | 2004-05-18 | 2005-12-08 | Children's Memorial Hospital | Tetracycline-regulated adeno-associated viral (aav) vectors for gene delivery to the nervous system |
| EP1602926A1 (en) | 2004-06-04 | 2005-12-07 | University of Geneva | Novel means and methods for the treatment of hearing loss and phantom hearing |
| AU2007243184A1 (en) | 2006-04-28 | 2007-11-08 | University Of Iowa Research Foundation | Methods and compounds to modulate parvovirus transduction of mammalian cells or to alter virus infection, method to identify a viral receptor or co-receptor |
| WO2009028387A1 (ja) * | 2007-08-24 | 2009-03-05 | Kyowa Hakko Kirin Co., Ltd. | プロテアーゼ阻害剤耐性を有する癌の治療薬 |
-
2000
- 2000-06-08 JP JP2001501645A patent/JP4969002B2/ja not_active Expired - Lifetime
- 2000-06-08 EP EP00944624A patent/EP1190249A2/en not_active Withdrawn
- 2000-06-08 CA CA002376400A patent/CA2376400A1/en not_active Abandoned
- 2000-06-08 WO PCT/US2000/015700 patent/WO2000075365A2/en not_active Ceased
- 2000-06-08 AU AU58694/00A patent/AU782966B2/en not_active Expired
-
2005
- 2005-12-13 US US11/301,601 patent/US8846030B2/en not_active Expired - Fee Related
-
2007
- 2007-08-07 US US11/890,777 patent/US20080213221A1/en not_active Abandoned
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11702672B2 (en) | 2016-02-08 | 2023-07-18 | University Of Iowa Research Foundation | Methods to produce chimeric adeno-associated virus/bocavirus parvovirus |
| US11684679B2 (en) | 2016-03-07 | 2023-06-27 | University Of Iowa Research Foundation | AAV-mediated expression using a synthetic promoter and enhancer |
| US12173305B2 (en) | 2016-05-26 | 2024-12-24 | University Of Iowa Research Foundation | cis and trans requirements for terminal resolution of human bocavirus 1 |
| US11999965B2 (en) | 2017-01-13 | 2024-06-04 | University Of Iowa Research Foundation | Bocaparvovirus small noncoding RNA and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US20060093585A1 (en) | 2006-05-04 |
| WO2000075365A2 (en) | 2000-12-14 |
| AU5869400A (en) | 2000-12-28 |
| US20080213221A1 (en) | 2008-09-04 |
| WO2000075365A3 (en) | 2001-03-01 |
| WO2000075365A9 (en) | 2002-05-02 |
| US8846030B2 (en) | 2014-09-30 |
| AU782966B2 (en) | 2005-09-15 |
| EP1190249A2 (en) | 2002-03-27 |
| CA2376400A1 (en) | 2000-12-14 |
| JP2003501068A (ja) | 2003-01-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4969002B2 (ja) | rAAV形質導入を増加するための化合物および方法 | |
| US20080206198A1 (en) | Compounds and methods for pharmico-gene therapy of epithelial sodium channel associated disorders | |
| JP2004534543A (ja) | シュードタイプ化アデノ関連ウイルスおよびその使用 | |
| JP6516725B2 (ja) | キメラアデノ随伴ウイルス/ボカウイルスパルボウイルスベクター | |
| KR102268883B1 (ko) | 누트린-3a 및 펩티드에 의한 폐 섬유증의 저해 | |
| US20050095270A1 (en) | Drug delivery to the inner ear and methods of using same | |
| JP2009535034A (ja) | 哺乳動物細胞のパルボウイルス形質導入を調節若しくはウイルス感染を変えるための方法および化合物、ウイルス受容体若しくは共受容体の同定方法 | |
| US7122335B1 (en) | Compounds and methods to enhance rAAV transduction | |
| HUP0104298A2 (hu) | Parvovíruskapszid-részecskék alkalmazása a sejtproliferáció és -migráció gátlására | |
| US6818612B2 (en) | Use of parvovirus capsid particles in the inhibition of cell proliferation and migration | |
| AU2018210990A1 (en) | Compositions for reducing sarcolipin expression and preventing and treating muscular dystrophy and cardiomyopathy and methods of use | |
| WO2023225619A2 (en) | Delivery of focal adhesion kinase for treating vascular and/or capillary tissue injury |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20070531 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20070531 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20090212 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100608 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100908 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20101005 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20101005 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101207 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20110906 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120110 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20120125 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120327 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120403 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150413 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4969002 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |