JP4756805B2 - Dna断片の合成法 - Google Patents
Dna断片の合成法 Download PDFInfo
- Publication number
- JP4756805B2 JP4756805B2 JP2001501647A JP2001501647A JP4756805B2 JP 4756805 B2 JP4756805 B2 JP 4756805B2 JP 2001501647 A JP2001501647 A JP 2001501647A JP 2001501647 A JP2001501647 A JP 2001501647A JP 4756805 B2 JP4756805 B2 JP 4756805B2
- Authority
- JP
- Japan
- Prior art keywords
- oligonucleotide
- recognition sequence
- oligonucleotides
- sequence
- library
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Images













Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/66—General methods for inserting a gene into a vector to form a recombinant vector using cleavage and ligation; Use of non-functional linkers or adaptors, e.g. linkers containing the sequence for a restriction endonuclease
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/26—Preparation of nitrogen-containing carbohydrates
- C12P19/28—N-glycosides
- C12P19/30—Nucleotides
- C12P19/34—Polynucleotides, e.g. nucleic acids, oligoribonucleotides
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6806—Preparing nucleic acids for analysis, e.g. for polymerase chain reaction [PCR] assay
Description
当分野の現状によると、およそ2.5kbの核酸配列の合成のためには、まず、部分的に重複する約80マーのオリゴヌクレオチドを約50種、合成し精製しなければならない。次いで、これらを対又はサブセットへとハイブリダイズさせ、クレノウポリメラーゼ反応により充填するか、又はプライマーとして外部オリゴヌクレオチドを使用したポリメラーゼ連鎖反応(PCR)において構築し、一方向に連結させる(通常、取り込まれていなければならない制限部位による)。この方法は、間隙充填法として知られる。又は、酵素的もしくは化学的なライゲーションにより遺伝子断片を合成し、次いで、精製及び/又はクローニングの後、これらの断片を組み立て、より大きい遺伝子セクションを形成させることもできる(いわゆる、カセット法)。いずれの手法も、理想的な場合で少なくとも1週間、しかし通常は6〜12週間近くを要し、6ヶ月を要する場合すらある。固相と結合した逐次法は、多くの反応工程が必要とされるため収率が低く、従ってやはり信頼性が極めて低い。
【0002】
主要な問題の1つは、合成が良好に進行した場合ですら、1工程当たりのカップリング効率は99%にしか達しないために、比較的長いオリゴヌクレオチドは、回避できない終結産物の部分を必ず有するという点である。さらに、100%ではないキャッピング(capping)から生じる欠失も起こる。極めて良好な合成ですら、この部分は1カップリング工程当たり約0.25%である。合成完了後のトリチル保護基の分離も、完全には進行しない。このようにして形成された不完全なオリゴヌクレオチド産物は、非常に努力したとしても、より長いオリゴヌクレオチドから完全に分離することはできない。
【0003】
平均カップリング効率が98%であるとすると、例えば80マーの場合、所望の完全長産物の収率はわずか19.86%である。現在利用可能な精製法では、所望の最終産物は、最高でも95%の純度でしか得られない。最終的に精製されたオリゴヌクレオチドのごく一部のみに欠陥があるとしても、欠陥のある最終配列の確率は、使用されるオリゴヌクレオチドの数と共に劇的に増加する。従って、記載されたオリゴヌクレオチド50個から構成される配列は、全体の7.7%しか正確でなく、従って、通常は再作業が必要となる。これには、比較的稀な、合成中の誤カップリングによる誤塩基の取り込みは考慮されていない。
【0004】
比較的短いオリゴヌクレオチドですら、その潜在的な配列は多様であるため(30マーですら、1018越の可能性のある配列異型が存在する)、様々な遺伝子構築物のためオリゴヌクレオチドを再使用することも事実上不可能である。従って、任意の配列を生成させるために必要とされるオリゴヌクレオチドを全て利用可能にすることは、技術的に不可能である。新たな遺伝子構築物毎に、新たなオリゴヌクレオチドを合成し精製しなければならない。しかしながら、合成された材料のごく一部のみが、実際に遺伝子構成に使用され、残部は、前記の理由のため利用され得ない。遺伝子合成の過程にオリゴヌクレオチドの合成及び精製が取り込まれなければならない点が、この過程の完全な自動化に対する主要な障壁のうちの1つであり、自動化は、現在のところ、技術的に極めて困難であり、おそらく事実上達成不可能である。
【0005】
従って、本発明の目的は、任意の配列及び長さの二本鎖DNA断片の効率的な合成のための方法を提供することである。さらなる目的は、基本的な構築ブロックの限定されたライブラリーから、DNA分子を構築することを可能にする方法を提供することである。さらなる目的は、任意の遺伝子断片の平行合成及び非配列依存的連結を可能にする方法を証明することである。遺伝子合成過程を完全に自動化するためには、これら両方の先行条件が満たされねばならない。さらなる目的は、二本鎖DNA断片の自動作製のためのキットを提供することである。
【0006】
その目的は、
a)認識配列の外側を切断するIIS型制限酵素の認識配列を含有するオリゴヌクレオチドの一端を、固体マトリックスとカップリングさせる工程(ここで、カップリングは修飾により実施される)、
b)少なくとも部分的に二本鎖であり、かつ認識配列の外側を切断するIIS型制限酵素の工程a)とは異なる認識配列を含有する追加のオリゴヌクレオチドを添加する工程(ここで、このオリゴヌクレオチドはマトリックスと結合することができない)、
c)工程a)及びb)からのオリゴヌクレオチドを、ライゲートさせない末端の遮断により与えられた方向でライゲートさせる工程、
d)未消費の反応物及び酵素を除去する工程、
e)工程a)からのオリゴヌクレオチドに切断が起こるよう、認識配列の外側を切断するIIS型制限酵素により、工程c)からのライゲーション産物を切断する工程、
f)得られた核酸分子を反応混合物から分離する工程:を含む、核酸分子の作製のための方法を提供することにより達成される。
【0007】
その目的は、さらに、
前記のa)〜d)、
e)工程b)からのオリゴヌクレオチドの核酸配列に切断が起こるよう、認識配列の外側を切断するIIS型制限酵素により、工程c)からのライゲーション産物を切断する工程、
f)工程e)において得られた工程a)からの伸長オリゴヌクレオチドから反応混合物を分離する工程、
g)工程b)〜f)を少なくとも1回反復する工程:を含む、核酸分子の作製のための方法を提供することにより達成される。
【0008】
その目的は、さらに、
前記のa)〜g)、
h)工程a)からのオリゴヌクレオチドに切断が起こるよう、認識配列の外側を切断するIIS型制限酵素により、得られた核酸分子を切断する工程を含み、かつ
i)工程b)からのオリゴヌクレオチドに切断が起こるよう、認識配列の外側を切断するIIS型制限酵素により、得られた核酸分子を切断する工程を場合により含む、核酸分子の作製のための方法を提供することにより達成される。
【0009】
工程c)の後、工程c′)として、エキソヌクレアーゼ及び/又はホスファターゼ反応を実施する方法が好ましい。さらに、工程c′)の反応混合物を反応後に除去する方法が好ましい。工程b)〜f)の最後の反復において工程e)を実施しない方法がさらに好ましい。得られた核酸を、制限切断により工程a)からのオリゴヌクレオチドから分離する方法も好ましい。さらに、工程a)からのオリゴヌクレオチドを、修飾により固体マトリックスとカップリングさせる方法が好ましい。修飾がビオチン残基、ジゴキシゲニン残基、フルオレセインイソチオシアネート(FITC)、アミノ化合物、又はスクシニルエステルである方法が、特に好ましい。さらに、工程a)及び/又はb)からのオリゴヌクレオチドがループを有する方法が好ましい。工程a)からのオリゴヌクレオチドを、ループによって固体マトリックスとカップリングさせる方法が、特に好ましい。固体マトリックスがビーズ、好ましくはガラス製又はポリスチレン製のビーズ、顕微鏡スライド、DNAチップ、マイクロタイタープレートのウェル、又はテストチューブである方法が、特に好ましい。特に、固体マトリックスがストレプトアビジン残基、抗ジゴキシゲニン抗体、又は抗FITC抗体を含む方法が、好ましい。さらに、工程a)及びb)からのオリゴヌクレオチドが、ライゲートさせる末端に、相互に相補的な一本鎖突出を有する方法が、好ましい。一本鎖突出が1、2、3、4又は5ヌクレオチド長である方法が、特に好ましい。合成された核酸を、最終工程において、複製可能DNA(プラスミドベクター、ファージ又はウイルスのDNA、人工染色体、PCR産物、又はその他の人工的に作製されたDNA)と連結させる方法が、特に好ましい。コドン最適化オープンリーディングフレームを作製するため、プロモーター、エンハンサー、又はタンパク質をコードするDNAの特異的突然変異誘発のための方法が、特に好ましい。特に、本発明に係る核酸は、好ましくは、コドン最適化されたDNAワクチンとして、タンパク質ドメインの突然変異分析のため、デザイナータンパク質の鋳型として、in vitroタンパク質合成用の発現構築物として、リボザイム又はアプタマーを調製するため、病原性微生物検出用プローブとして、遺伝子発現検出用プローブとして、対立遺伝子特異的突然変異の検出のため、タンパク質/タンパク質結合、タンパク質/ペプチド結合、及び/又は低分子量物質とタンパク質との結合の検出のため、使用される。
【0010】
その目的は、
a)1〜1,048,576個の異なるオリゴヌクレオチドのライブラリー(ここで、オリゴヌクレオチドは一端で修飾により固体マトリックスとカップリングすることができ、かつオリゴヌクレオチドは認識配列の外側を切断するIIS型制限酵素の認識配列又は認識配列の一部を含有する)、
b)4〜1,048,576個の異なるオリゴヌクレオチドの付加的なライブラリー(ここで、各オリゴヌクレオチドは、a)からのIIS制限酵素とは異なる、認識配列の外側を切断するIIS型制限酵素の認識配列を含有し、かつ工程a)からの制限酵素の認識配列のその他の部分を場合により含有する)、
c)固体マトリックス、
d)核酸分子を作製するために必要とされる酵素及び/又はその他の試薬のための貯蔵器:を含む、本発明に係る方法により核酸を作製するためのキットを提供することにより、さらに達成される。
【0011】
酵素が、リガーゼ又はトポイソメラーゼ、及び/又は1個又は数個の制限酵素、及び/又はエキソヌクレアーゼ、及び/又はホスファターゼを含むキットが、好ましい。所望の塩基配列が投入された後、全ての反応工程を決定し、自動的にそれらを処理することができる自動化された機械が、特に好ましい。
【0012】
以下の図面により、本発明をさらに明確にする。
【0013】
定義
「平行」又は「平行合成」という用語は、本明細書において使用されるように、次いで、本発明に係る方法を使用してアンカー又はスプリンカーとしてライゲートさせ、伸長した核酸分子を形成させるための、異なる本発明の核酸分子が、別々の反応混合物中で同時に合成されうることを意味する。
【0014】
「スローニング(sloning)」(非配列依存的なオリゴヌクレオチドの逐次的ライゲーション)という用語は、本明細書において使用されるように、任意の配列のオリゴヌクレオチドの連続的ライゲーションのための方法をさす。
【0015】
「アンカー」又は「アンカーオリゴヌクレオチド」という用語は、本明細書において使用されるように、修飾により固体マトリックスとカップリングさせうるオリゴヌクレオチドをさす。本発明の範囲において、その二本鎖領域内のオリゴヌクレオチドは、認識配列の外側を切断するIIS型制限酵素の認識配列をさらに含有する。
【0016】
「スプリンカー(splinker)」又は「スプリンカーオリゴヌクレオチド」という用語は、本明細書において使用されるように、修飾を有しないか、又は他の型の修飾を有し、その結果、アンカーオリゴヌクレオチドをカップリングさせるマトリックスとそれ自体は結合しないオリゴヌクレオチドをさす。
【0017】
「ダンベル」という用語は、本明細書において使用されるように、2個のループに挟まれた二本鎖により特徴付けられるDNA構造をさす。
【0018】
本発明の1つの面は、
a)認識配列の外側を切断するIIS型制限酵素の認識配列を含有するオリゴヌクレオチドの一端を、固体マトリックスとカップリングさせる工程(ここで、カップリングは修飾により実施される)、
b)少なくとも部分的に二本鎖であり、かつ認識配列の外側を切断するIIS型制限酵素の工程a)とは異なる認識配列を含有する付加的なオリゴヌクレオチドを添加する工程(ここで、このオリゴヌクレオチドはマトリックスと結合することができない)、
d)工程a)及びb)からのオリゴヌクレオチドを、ライゲートさせない末端の遮断により与えられた方向でライゲートさせる工程、
h)未消費の反応物及び酵素を除去する工程、
i)工程a)からのオリゴヌクレオチドに切断が起こるよう、認識配列の外側を切断するIIS型制限酵素により、工程c)からのライゲーション産物を切断する工程、
j)得られた核酸分子を反応混合物から分離する工程:を含む核酸分子の作製のための方法に関する。
【0019】
本発明のさらなる面は、
前記のa)〜d)、
e)工程b)からのオリゴヌクレオチドの核酸配列に切断が起こるよう、認識配列の外側を切断するIIS型制限酵素により、工程c)からのライゲーション産物を切断する工程、
f)工程e)において得られた工程a)からの伸長オリゴヌクレオチドから反応混合物を分離する工程、
k)工程b)〜f)を少なくとも1回反復する工程:を含む核酸分子の作製のための方法に関する。
【0020】
本発明のさらなる面は、
前記のa)〜g)、
h)工程a)からのオリゴヌクレオチドに切断が起こるよう、認識配列の外側を切断するIIS型制限酵素により、得られた核酸分子を切断する工程を含み、かつ
i)工程b)からのオリゴヌクレオチドに切断が起こるよう、認識配列の外側を切断するIIS型制限酵素により、得られた核酸分子を切断する工程を場合により含む核酸分子の作製のための方法に関する。
【0021】
各反応工程において連結される2個のオリゴヌクレオチドのうちの1個(いわゆるアンカーオリゴヌクレオチド)は、修飾、例えばビオチン又はジゴキシゲニンのような低分子量化学化合物により固体マトリックスとカップリングしうる。好ましい実施態様において、これらは、ストレプトアビジン又は抗ジゴキシゲニンで被覆された磁性ビーズである。他方のオリゴヌクレオチド(いわゆるスプリンカーオリゴヌクレオチド)も、遮断された末端を有するが、そのような修飾は有しないか、又は他の型の修飾を有する。重要な点は、アンカーオリゴヌクレオチドが、適当なマトリックスとの結合により、スプリンカーオリゴヌクレオチドから分離されうるという点である。従って、固相との直接的又は間接的な(例えば、抗体による)結合を媒介するために適当であるならば、任意の化合物、例えばビオチン、ジゴキシゲニン、フルオレセインイソチオシアネート(FITC)、アミノ化合物、スクシニルエステル、及び当業者に周知のその他の化合物が、使用されうる。
【0022】
アンカーオリゴヌクレオチドは、好ましくはループ配列における修飾により固相とカップリングしうる1本の部分的に自己相補的なオリゴヌクレオチド、又は好ましくは一本鎖突出を有する二本鎖を形成する2本の一本鎖オリゴヌクレオチドのいずれかから構成されうる。2本の鎖のうちの1本のみをマトリックスとカップリングさせなければならないため、必要であれば、アルカリ又は熱により、他方を変性させ分離することができる(例えば、PCR反応のための鋳型として使用するため)。そのような2部アンカーオリゴヌクレオチドの場合にも、1方の末端のみがライゲートしうることを確実にするため、従ってライゲーションに必要とされない末端は遮断される。典型的なアンカーオリゴヌクレオチドの核酸配列は、以下の通りである。
【0023】
【表1】

スプリンカーオリゴヌクレオチドは、一本の部分的に自己相補的なオリゴヌクレオチド、又は好ましくは一本鎖突出を有する二本鎖を形成する2本の一本鎖オリゴヌクレオチドのいずれかから構成されうる、即ち、少なくとも部分的に相補的なオリゴヌクレオチド対を有し、2本の一本鎖のライゲートさせない各末端は遮断されていなければならない。好ましい一本鎖突出配列は、ライゲートさせる各アンカーオリゴヌクレオチドと相補的でなければならない。例示的なスプリンカーオリゴヌクレオチドの核酸配列は、以下の通りである。
【0025】
【表2】
アンカーオリゴヌクレオチド及びスプリンカーオリゴヌクレオチドは、一定の長さ、好ましい実施態様において1〜5ヌクレオチドの突出を含有しうる。ライゲートさせるオリゴヌクレオチドの場合、これらの突出は、相互に相補的であり、5′末端がリン酸化されており、1方向にのみライゲートしうる。これは、例えば、いわゆるダンベル構造を有する、ライゲートしたオリゴヌクレオチドをもたらす。全ての利用可能なアンカーヌクレオチドを完全にライゲートさせるため、ライゲートさせるスプリンカーオリゴヌクレオチドは、2〜10倍過剰で添加されうる。過剰の未反応スプリンカーは各ライゲーション工程の後、緩衝液で洗浄除去される。例えば、ストレプトアビジン被覆磁性ビーズを使用する場合、ストレプトアビジン/ビオチン結合により結合したアンカーオリゴヌクレオチドを、ライゲートしたスプリンカーと共に含有するビーズは、磁石を使用することにより反応混合物中に保持されうる。又は、例えば、ストレプトアビジンで直接被覆されたウェル、ガラスビーズ、顕微鏡スライド、DNAチップ、又は任意のその他の固相を使用することが可能である。ビーズは、比較的大きい表面を有し、従って比較的高い結合能を有するため、通常、好ましい。
【0027】
さらなるライゲーションを実施するためには、認識配列の外側で核酸配列を切断する制限エンドヌクレアーゼの認識配列が、ライゲートしたスプリンカーオリゴヌクレオチドに存在しなければならない。そのような酵素の例は、BpiI、Esp3I、Eco31I、SapI等である。本発明に係る方法にとって有用な制限酵素、及びそれらの認識配列及び切断配列は、http://rebase.neb.com/rebase/rebase.htmlのrebaseデータバンクに見出される。ライゲーション産物は、スプリンカー配列の一部がアンカーオリゴヌクレオチド上に残存するよう、スプリンカーオリゴヌクレオチド内に含まれる制限切断部位で切断される。これは、同時に、さらなるスプリンカーオリゴヌクレオチドをライゲートさせるために使用されうる配列突出を生成させる。切断された他方の部分、及びスプリンカーオリゴヌクレオチドのライゲートしなかった残余物、制限酵素、及び制限緩衝液は、さらなるサイクルが開始する際に、反応混合物から洗浄除去される。サイクルは、1回のみ実施してもよいし、又は数回反復してもよく、その後、このようにして伸長したオリゴヌクレオチドを、同時に合成された隣りの断片と連結させる。様々な制限エンドヌクレアーゼによる切断により形成された相互に相補的な突出は、合成する遺伝子に由来し、対照的に、認識配列はアンカーオリゴヌクレオチド又はスプリンカーオリゴヌクレオチドの切断除去された部分に位置するため、隣りの断片はそれらの配列と完全に無関係に連結されうる。特に、これは、大きな遺伝子ですら、わずか数回の反応工程で、多くの同時の部分的反応において組み立てることを可能にする。最適な例において、2kbの遺伝子は、例えば256回の各反応から、わずか9工程で組み立てられ得る。60マーオリゴを使用した直線的な合成(循環的ではあるが、同時ではない)の場合、同サイズの遺伝子は、30超の工程を必要とするであろう。酵素的及び化学的なライゲーション法は通常80〜90%の収率しか有しないため、全収率は、必要とされる反応工程の数と共に指数関数的に減少し、そのため、少ない反応工程を有する方法は有利である。場合により、さらなる合成から未反応アンカーオリゴヌクレオチドを排除するため、次のライゲーションに必要とされる突出又は少なくとも5′リン酸基を除去するエキソヌクレアーゼ及び/又はホスファターゼ工程を、ライゲーションの後に導入してもよい。過剰のスプリンカーオリゴヌクレオチドを使用した場合には、未反応アンカーオリゴヌクレオチドの割合は、極小さい。さらに、後続の反応は、同配列が再びライゲートした場合にのみ可能であるはずであり、そのため、未反応の、又は部分的にしか反応していないアンカーオリゴヌクレオチドの混入のリスクは比較的低いと見なすことができる。
【0028】
数回のライゲーション及び制限のサイクルの後、このようにしてライゲートした核酸配列は、続いて、最初のアンカーオリゴヌクレオチド内の核酸配列を特異的に認識する制限酵素による切断により、アンカーオリゴヌクレオチドから分離され、アンカーオリゴヌクレオチドは、マトリックスに残留する。ここで、ライゲートした核酸配列は、最後にライゲートしたスプリンカーオリゴヌクレオチドと接着している。制限酵素の不活化後、伸長したスプリンカーオリゴヌクレオチドは、最初の反応混合物から新たな反応容器へと移され、そこで、スプリンカーオリゴヌクレオチドに特異的な制限酵素で切断された、ライゲートしたアンカーオリゴヌクレオチドと連結される(第1転移)。ライゲートした核酸配列は、異なっていてもよいし同一であってもよい任意の配列であり得ることが、当業者には明らかである。第1転移から生じたライゲーション産物は、次に、アンカー特異的制限エンドヌクレアーゼにより切断され、再び、同様にして得られたライゲートしたアンカーオリゴヌクレオチドと連結される(第2転移)。結果として、ライゲートした核酸配列の長さは、各工程毎に2倍となる。毎回、DNA断片は、相補的な突出により連結されるが、これは、その他については完全に配列と無関係である。唯一の制約は、アンカー及びスプリンカーに特異的な切断部位が、合成する配列内に存在している場合、DNAが内部でも切断されてしまうであろうため、それらが合成する配列内には存在していてはならないことである。場合により、不完全にライゲートしたスプリンカーオリゴヌクレオチドの転移を防止するため、エキソヌクレアーゼ工程を、アンカー特異的制限切断部位における切断及び後続の転移の前に、毎回、導入してもよい。方法にとって必要な配列特異的切断は、原理的には、IIS制限エンドヌクレアーゼの代わりに、類似の機能を有するリボザイムによって実施されてもよい。
【0029】
2560塩基対長の二本鎖DNA配列は、わずか7回の付加的ライゲーション工程により、20塩基対の配列(4nt突出を有するスプリンカーの場合、ライブラリー由来の必要とされた最初のスプリンカーの5回の連続的なライゲーションにより得られる)から合成されうる。サイクル時間が約1hrであるとすると、この長さの任意のDNA配列が、12時間以内に合成されうる。必要とされる時間は、反応条件を最適化することにより、約6時間に半減しうる。
【0030】
4ヌクレオチド長の突出の場合、65536個の異なるスプリンカーオリゴヌクレオチドのライブラリーが、全ての可能性のある核酸配列を作製するために必要とされる。この数は、以下のような計算から得られる。可能性のある4ヌクレオチド突出は256個存在し(44=256)、次のライゲーション工程において突出を形成する直接接続ヌクレオチド4個について同数の配列異型が存在する。全体として、全ての可能性のある配列異型を表すために使用されうるスプリンカーオリゴヌクレオチドの総数は、44×44≒48≒65536個となる。必要とされるスプリンカーライブラリーの複雑度は、3ヌクレオチド突出の場合、43×43=4096、2ヌクレオチド突出の場合、42×42=256に相応じて減少し、5ヌクレオチド突出の場合には、45×45=1048576に増加するであろう。この構築ブロック系のための先行条件は、完全なスプリンカーライブラリー(2nt突出の場合256オリゴヌクレオチド、3nt突出の場合4096オリゴヌクレオチド、4nt突出の場合65536オリゴヌクレオチド、5nt突出の場合1048576ヌクレオチド)及びアンカーライブラリー(1、2、3、4又は5nt突出を有する4、16、64、256又は1024オリゴヌクレオチド)の存在である。しかしながら、適当なスプリンカーオリゴヌクレオチドを使用した予備ライゲーション工程により、同等に十分な様々な突出配列が生成しうるため、後者は絶対的には必要でない。
【0031】
原理的には、本発明に係る方法の各工程は全て、自動化が可能であり、従って、完全な遺伝子の作製が、オリゴヌクレオチドの合成と同程度に簡便である。さらに、本発明に係る方法は、コストを大きく減少させる潜在性を有する。第1に、全ての必要とされる酵素が、大規模に産生されうる。第2に、スプリンカーライブラリーのための出費は、5′突出の最後の4ヌクレオチドを除き、各スプリンカーオリゴヌクレオチドをen blocで合成することにより、大きく減少しうる。次いで、合成反応は、4つの等しい部分に分割され、次いで、4個の異なるヌクレオチドが別々の反応において次の位置(最終産物における最後から4番目の位置)に接着する。その後、4つの各反応が再び4等分され、その後、最後から3番目のヌクレオチドが接着する、等である。65536回の各合成の代わりに、相応じて大きい、従ってより好都合な規模において、256回の合成のみが必要となるであろう。さらに、256種の異なるアンカーオリゴヌクレオチドにおける平滑末端ライゲーション、それに続くエキソヌクレアーゼ処理、洗浄、及び最後のアンカー特異的制限エンドヌクレアーゼによる制限により、256種の可能性のある4ヌクレオチド突出を生成させることができる。このようにして、必要とされるスプリンカーオリゴヌクレオチド65536個は、対費用効果の高い方法で調製されうる。さらに、非反応性の欠陥のある配列はこの手法により除去されるため、これは、スプリンカーオリゴヌクレオチド65536個全ての複雑な精製を回避するであろう。使用されるオリゴヌクレオチドの極めて高い純度が、欠陥のない合成の成功にとって必須であるため、いずれにしても、これらは適切に前処理されなればならない。さらに、後続のライゲーションに必要とされる突出配列が完全なまま維持されるよう、制限及びライゲーションの工程においては、エキソヌクレアーゼがほぼ完全に存在しないことを保証する必要がある。ライゲートしなかったアンカーオリゴヌクレオチドを除去するために中間のエキソヌクレアーゼ工程を使用する場合には、これらのエキソヌクレアーゼを、徹底的に洗浄除去し、かつ/又は特異的に不活化しなければならない。
【0032】
アンカーオリゴヌクレオチド及びスプリンカーオリゴヌクレオチドは、それぞれ、自己相補的な一本鎖から構成されていてもよいし、2本の相補的なプラス鎖及びマイナス鎖から構成されていてもよい。核酸配列は、完全に相補的である必要はなく、自己相補的一本鎖オリゴヌクレオチドはループを有していてもよく、相補的なプラス鎖及びマイナス鎖は、部分的に相補的であってもよい。アンカーオリゴヌクレオチド及びスプリンカーオリゴヌクレオチドが、それぞれ、2本の相補的なプラス鎖及びマイナス鎖から構成される場合、(i)二本鎖ハイブリッドの融解温度は、集合したアンカーオリゴヌクレオチドとスプリンカーオリゴヌクレオチドの変性、及び可能性のある結果的な、固相とカップリングしていない一本鎖の意図されない転移が防止されるよう、十分に高くなければならず、(ii)伸長させない各末端は、適当な修飾により遮断されなければならない。2本の相補的なプラス鎖及びマイナス鎖からなるオリゴヌクレオチドは、自己相補的な一本鎖から構成されるオリゴヌクレオチドと比して、ある種の利点を有する。自己相補的(スナップバック)オリゴヌクレオチドは、高濃度においては網状構造を形成する傾向を有するため、しばしば、精製におけるある種の問題を引き起こす。また、一本鎖部分オリゴヌクレオチドは比較的短く、従って、より高い純度でより少ない努力で単離されうる。2本の部分オリゴヌクレオチドから構成された2部アンカーオリゴヌクレオチドは、ある種の本発明の実施態様のため使用される。
【0033】
特に好ましい実施態様において、アンカー及びスプリンカーは、以下の認識配列の組み合わせを含有する。
【0034】
【表3】
本発明のさらなる面は、本発明に係る方法による核酸の作製のためのキットである。キットは、必要なアンカーオリゴヌクレオチド及びスプリンカーオリゴヌクレオチド全てのライブラリー、さらにアンカーオリゴヌクレオチドがカップリングしうる固相、好ましくは磁性化ビーズ、適当な反応容器、リガーゼ、場合によりトポイソメラーゼ及び/又は3′−5′エキソヌクレアーゼ及び/又はホスファターゼ、認識配列の外側を切断するII型制限エンドヌクレアーゼ少なくとも2種、並びに全ての必要とされる反応緩衝液からなりうる。さらに、冷蔵試料保管コンテナを有するピペッティングステーション(pipetting station)、及び本発明に係る方法の全工程を自動的に実施する適切なソフトウェアコントロールが、好ましい。
【0036】
本発明は、再使用可能な、ある種のIIS型制限エンドヌクレアーゼ(いわゆる外側切断酵素(outside cutter))の認識配列を含有する、高純度の少なくとも部分的に二本鎖のオリゴヌクレオチドのライブラリーを提供することにより、遺伝子合成の過程全体の完全な自動化を可能にする。さらに、遺伝子断片の平行合成、及び任意の所望の部位におけるそれらの非配列依存的な連結を可能にする方法の提供により、そして固相との結合の結果としての、出発分子の方向性のある伸長により(ライゲートさせない末端は適当な修飾又はループ配列により遮断される)、そしてロボットにより処理されうる循環的な手順の明確なセットにより、自動化が可能となる。
【0037】
本発明のいくつかの面を、以下の実施例により例示するが、それらは、本発明に係る方法による遺伝子全体の完全な合成に基づいている。
【0038】
1.タンパク質配列のみが既知の場合のcDNAの作製
タンパク質のアミノ酸配列又はアミノ酸配列の一部のみが既知であり、cDNA又はゲノムの配列は不明であるという場合は多い。遺伝暗号の縮重のため、適当なcDNAライブラリーのPCRにより対応する遺伝子を直接増幅することは、通常不可能である。従って、トリプトファン、メチオニン、又はアスパラギン、アスパラギン酸、グルタミン酸、グルタミン、チロシン、フェニルアラニン、システイン、又はリジンのようなアミノ酸については、コドンが1個又は2個しか存在しないため、これらのアミノ酸が豊富に存在する領域を検索する。低縮重のプライマーを使用して予想サイズのPCR断片を得ることが可能であるならば、この断片をプローブとして使用し、cDNAバンクから各遺伝子をクローニングする。現在では、遺伝子アレイ及びクローンコレクションの利用可能性により、多くの場合、この作業は大きく単純化されているが、そのような補助は、限定された数の生物及び細胞型についてのみ利用可能であり、完全cDNAが入手可能である場合ですら、通常、適当な発現ベクターにそれを再クローニングすることがやはり必要である。プロジェクトの難度によって、必要とされる時間は、1〜2週間、極端な例においては数ヶ月から数年にまでなりうる。本発明に係る方法は、既知のタンパク質配列から出発して、1〜2日で、所望の生物のため最適化されたコドン使用を有する発現構築物を調製するために使用されうる。鋳型が利用である必要はなく、DNA配列が既知のタンパク質配列から導出され得るため、このために、タンパク質が天然に発現している生物が利用可能である必要はない。タンパク質配列決定法は改良されているため、将来は、任意の所望の生物に由来する興味深い酵素活性を有するタンパク質を配列決定し、cDNAクローニングを介した間接的な経路をとる必要なしに、本発明に係る方法により、直接的にそれらを任意の所望の発現系に移入することが可能となるであろう。
【0039】
2.デザイナー遺伝子及びデザイナータンパク質の作製
本発明のさらなる面は、デザイナー遺伝子及びデザイナータンパク質の簡便な作製、即ち、例えば新規な、又は修飾された特性を有する酵素を調製するための、様々なタンパク質の機能ドメインのカップリングである。タンパク質のX線結晶構造が既知であるならば、修飾された特異性の新規な機能をタンパク質へ導入するための、明確なリンカードメインの挿入、又は結合ポケットの再設計のような極めて特異的な修飾を施すことが可能である。例えば、標的タンパク質設計(targeted protein design)は、特異的リガンドの結合の結果としてのタンパク質のコンフォメーションの変化により活性化される制御可能な触媒中心を構築するために使用されうる。例えば、特定のウイルスタンパク質が結合した際にカスパーゼ活性を示し、次いで感染細胞のアポトーシスを誘発するデザイナータンパク質が、このようにして構築されうる。そのような高度に特異的な医薬的薬剤の最初の版は、既に記載されている。Vocero−Akbani A.M.,Heyden N.V.,Lissy N.A.,Ratner L.,Dowdy S.F.,Nat.Med.1999年1月5日:1,29−33を参照のこと。さらに、特定の位置で付加的な塩架橋を形成することができるアミノ酸を組み込むことにより、タンパク質を安定化することが可能である。これは、界面活性剤工業にとって特に有利な、高温に対する抵抗性を改良しうる。ドメイン構造が既知である場合、いくつかの機能領域の正確な発現により、所望の酵素活性を、不要な活性から分離することができる。さらに、一連の異なる反応を完全に触媒することができる多酵素複合体を構築することも可能である。これは、多くの有機化合物の合成を改良するかもしれないし、又はさらには、いくつかの合成を初めて可能にするかもしれない。将来は、そのようなデザイナー生体触媒が、現在は未だ環境的に有害な溶媒及び触媒を使用しなければならない多くの有機合成の代わりとなるかもしれないため、これは、完全に新規な展望を開くものである。
【0040】
3.ランダム突然変異誘発の代替法としての系統的突然変異誘発
生化学的に方向付けられた分子生物学においてしばしば起こる問題は、多くのタンパク質異型の中から、最も高い酵素活性、又は基質もしくは他のタンパク質との最も高い結合を有しているものを同定することである。通常の手法は、アミノ酸1個又は数個の一連のランダムな突然変異を導入し、適当なスクリーニング法において、得られた異型を分析することである。原理的には、全ての変異体を別々に調製することも可能であるが、これは時間及びコストの理由からほとんど実施されない。その過程では、ある種のアミノ酸置換が他よりも高頻度に見いだされることになるため、そして、この手法では、付加的な終止コドンの導入を回避することがほぼ不可能であるため、ランダム突然変異誘発において形成される変異体のコントロールは、本来極めて限定されている。対照的に、本発明に係る方法は、全ての所望の変異体を、特異的に、かつ過度の努力なしに調製し、タンパク質として発現させることを可能にする。
【0041】
4.特にDNAワクチンとして使用するための合成遺伝子の作製
多くの場合、異種の系におけるある種の遺伝子のタンパク質発現を最適化することが望ましい。これは、強力なプロモーターの使用によっても、部分的にしか達成されない場合が極めて多い。発現に使用される生物によって、あるアミノ酸についてのある種のコドンの使用は、達成可能な遺伝子発現に対して有利な効果を有する場合もあるし、又は不利な効果を有する場合もある。従って、例えば、多くのレトロウイルス遺伝子産物は、通常極めてATリッチであり、高等真核生物においては稀なコドンを利用しているため、真核生物においては不十分にしか翻訳されない。従って、コドン使用が哺乳動物細胞にとって最適化される場合、それは、DNAワクチンとしてのそのような遺伝子配列の適用にとって特に、大きな利点である。同様に、ある種のRNA構造は、遺伝子発現に有害な影響を与えうる転写物の不安定性を引き起こしうる。そのような要因も、コドン変化により、本発明に係る方法を用いて容易に除去されうる。
【0042】
5.欠失又は点突然変異誘発によるタンパク質ドメインの分析
変異体の分析は、タンパク質の機能的特徴決定のための好ましい方法である場合が極めて多い。欠失変異体及び点変異体を作製するための多数の確立された方法が存在するが、これらは、通常、極めて多くの時間及び労力を要する。欠失は、通常、リンカー配列の導入により、又は様々な部分配列と相補的な末端を有するプライマーを使用したPCRにより作製される。一連の明確な欠失を全て得るためには、最初に制限切断部位を導入し、次いで所望の欠失を導入するためにそれらを使用する2段階法を実施することが必要である場合が多い。適切に設計されたプライマー及び多断片ライゲーションを使用して、1段階でそのような欠失を導入することも、原理的には可能であるが、成功の確率はかなり低い。これらの全ての場合においては、野生型DNAが鋳型として存在しなければならないが、本発明に係る方法においてはそれが必要でない。さらに、まず適当な部位を見いださなければならない制限切断部位を導入する必要が全くないため、導入された突然変異がタンパク質配列の変化をもたらさないような欠失変異体を作製することができる(いわゆるサイレント部位突然変異)。本発明に係る方法は、二重又は三重の変異体の作製も可能にする。タンパク質の機能的マッピングのため、前述のサイレント部位突然変異を使用して、任意の所望の欠失の作製を補助する極めて多数の異なる制限エンドヌクレアーゼの制限切断部位を、その遺伝子配列に導入することもできる。従って、多くの例において、古典的な突然変異分析を省略し、その代わりに、より迅速かつ正確な本発明に係る方法を使用することができる。
【0043】
6.共役in vitro転写/翻訳系(「EasyPro(登録商標)」)
共役in vitro転写/翻訳系は、例えば、結合研究又は共沈アッセイのため、分析的規模でタンパク質を迅速に合成するために使用される。このためには、発現させる配列を、RNAポリメラーゼのためのプロモーターを含有するベクターへクローニングする。このポリメラーゼは、mRNAを転写するために使用され、mRNAは、RNA枯渇小麦麦芽又は網状赤血球の抽出物において所望のタンパク質へと翻訳され、タンパク質は、通常、低い収率及びより簡便な検出可能性のため、35S−メチオニン又はシステインで放射性標識される。はるかに迅速な代替法が、本発明に係る方法に基づくEasyPro(登録商標)系である。PCR産物と直接ライゲートしうる単一チミジン突出を、T7(SP6)プロモーター、内部リボソーム結合部位、及びヘキサヒスチジンタグを含有するアンカーオリゴヌクレオチドのXcmIによる制限により生成させる。様々なリーディングフレームを有する3つのEasyPro(登録商標)アンカーオリゴヌクレオチドが、正確な方向でライゲートしている全てのPCR断片を翻訳するために十分である。さらに、ターミナルトランスフェラーゼ、又は適切なスプリンカーオリゴヌクレオチドとPCR産物の3′末端とのライゲーションが、RNA転写物を安定化し、従ってより高い翻訳効率を保証する人工ポリAテールを、DNA鋳型に容易に導入するために使用されうる。さらに、所望のタンパク質をコードするDNA配列を、制限エンドヌクレアーゼによる切断の後、マッチする4nt突出を有する修飾EasyPro(登録商標)アンカーと直接ライゲートさせてもよい。
【0044】
本発明のさらなる面は、迅速なタンパク質合成のためのミニリアクタの提供である。ストレプトアビジン被覆ビーズとカップリングした発現−アンカー核酸配列の転写が、ミニリアクタの下方反応室で起こる。得られたmRNAは、3′ポリAテールを介して、やはり下方反応室に存在するオリゴdTカップリングビーズと結合する。これは、網状赤血球抽出物においてmRNAが翻訳される場所でもある。この室は、約200kDのMWCO(分子量排除)を有する限外濾過膜により、上部に位置する第2室と分離されている。この室は、目的のタンパク質と結合することができるビーズを含有する(例えば、ヘキサヒスチジンタグを有するタンパク質の場合、Ni2+−NTAビーズ)。新鮮な低分子量反応物(アミノ−アシル−tRNA、リボヌクレオチド3リン酸、CAP類似体、及びリン酸クレアチニン)を含有する緩衝溶液の継続的な供給により、作製は長期にわたり維持される。結果として、合成されたタンパク質は、同時に、下方室から上方室へと押し出され、そこで、ビーズに捕捉される。別法として、この室は、付加的な限外濾過膜により閉鎖されていてもよく、その排除サイズは、緩衝液及び比較的小さい分子にとっては透過性であるが、所望のタンパク質にとっては透過性でないよう選択される。従って、タンパク質は、上方室に集結し、精製された形態でそこから単離されうる。達成可能な収率は、大部分の分析的実験にとって適度であるのみならず、小規模なタンパク質発現実験の代わりにもなりうる。例えば、様々なタンパク質変異体の特異的酵素活性を決定することが意図される場合、従来は、これらを複雑な予備実験においてクローニングし、発現させ、精製しなければならなかった。本発明に係るEasyPro(登録商標)法には、これらの工程のほぼ全てが既に統合されているため、これは、従来の方法よりも時間的に大きく有利である。
【0045】
本発明に係る前述の方法の修飾は、抗体のエピトープマッピング、又はウイルス、細菌、もしくは真菌のタンパク質の免疫原性エピトープの同定(血清学的検出系を迅速に設定するため)に特に必要とされるペプチドライブラリーを簡便かつ安価に調製するために使用されうる。この修飾EasyPro(登録商標)のためには、アンカーオリゴヌクレオチドをスプリンカーとのライゲーションにより連続的に伸張させ、所望のペプチドをコードする配列を形成させる。最後の工程において、C末端タグ、終止コドン、及びポリAテールをコードする予め形成させた最終スプリンカーをライゲートさせる。ライゲーション産物を、記載されたミニリアクタにおいて転写し翻訳する。翻訳及び数回の洗浄工程の完了後、特異的プロテアーゼ、例えばエンテロキナーゼもしくはXa因子を使用して、EasyPro(登録商標)アンカーオリゴヌクレオチドによりコードされた切断部位において最終ペプチドを切断し、上方反応室から洗浄除去する。これらのペプチドは、後続の試験のため、C末端タグの補助により固相と結合させることができる。さらに、ペプチドは、既に精製された形態で存在しており、後続の適用に直接使用されうる。同一のアンカーオリゴヌクレオチドが毎回使用され、必要とされるスプリンカーオリゴヌクレオチドは既に製造されたセクションから2、3工程でライゲートさせることができるため、コストは従来のペプチド合成よりも低い。
【0046】
7.リボザイム又はアプタマーの作製
前記のタンパク質合成と同様に、アンカーオリゴヌクレオチドは、T7(SP6)プロモーターを使用したRNAの作製及び突然変異誘発にも使用されうる。伸張したスプリンカーオリゴヌクレオチド上のリボザイムをコードするDNA配列は、アンカーオリゴヌクレオチド上のプロモーターモジュールとライゲートしうるため、その系は、様々なリボザイムの合成に特に適している。特に、PCRにより容易に増幅されうる正確に明確なリボザイム鋳型ライブラリーを調製することが可能である。リボザイム鋳型配列は、このためのクローニング操作を実施する必要なしに、本発明に係る方法を使用して、ヌクレオチド上で正確に合成されうる。リンク配列を導入することによりリボザイムを調製することができ、これらのリボザイムを、次いで、リンク配列と相補的なDNA/RNAにより、ペプチド、核酸、脂肪族炭化水素、エステル、エーテル、又はアルコールのような任意の化学化合物とカップリングさせることができる。この化合物が固相と結合して存在する場合、この結合を切断するリボザイムが選択されうる。固相との結合から「解放」されたリボザイムは、逆転写及び後続の非対称PCRにより一本鎖DNA分子へと変換されうる。次いで、これらは、リンク配列により、適切に修飾されたアンカーオリゴヌクレオチドとハイブリダイズし、ライゲートする。アンカーオリゴヌクレオチドは、T7ポリメラーゼの補助により再びリボザイムを得ることができるよう、T7プロモーターを含有するよう構築される。不正確な逆転写酵素(例えば、HIV RT)の使用は、ランダム突然変異の導入を可能にする。選択圧は、高い活性を有するリボザイムが優先的に増幅されるよう、インキュベーションを漸進的に短くすることにより増加させることができる。固相との結合を媒介する能力を有するリボザイムは、同原理を使用して同様に選択されうる。
【0047】
8.本発明に従い作製されたssDNAの診断における使用(PathoCheck(登録商標))
感染性疾患の診断は、例えばPCRによる、病原体に関する直接的な試験を必要とする場合が多い。特に、輸血薬においては、汚染された血液試料を確実に検出し除去しうることが重要である。通常使用される血清学的アッセイは、ドナーが既にある程度の時間感染しており、抗体が既に形成されている場合にのみ、このことを保証する。例えば、HIV感染の場合、多量のウイルス複製が既に起こっていても、最大12週間の期間(極端な例ではそれ以上になることも)、血中に抗体は検出されない。全試料のルーチンのPCR検査は、コストのため多くの場所でほぼ不可能であるため、これは(実施されるとしても)各供血のプールに対して実施される。これに関する問題は、分析に使用されうる材料の量に限界があるため、極めて高いはずの感度が減少する点である。ウイルスの大部分が細胞外に存在するHIVのようなウイルス性疾患の場合、これは、遠心分離によりウイルスを濃縮することにより極めて良好に補償されうるが、主に細胞と会合しているウイルスの場合には、これは通常効率的でない。まず血液細胞からDNA又はRNAを単離することも可能であるが、非特異的PCR産物が調節不可能に増加するため、そのほんの一部のみがPCR反応に使用されうる。従って、そのような場合には、増幅する材料の予備選択を実施しなければならない。修飾アンカーオリゴヌクレオチドを使用して本発明に係る方法に従い作製された一本鎖産物が、このために使用される。この場合、2つの異なる相補鎖から構成されたアンカーオリゴヌクレオチドが使用され、その一方の鎖は5′末端で修飾、例えばビオチン化されており、他方の鎖は3′末端で遮断されている。ウイルス配列の合成後、一本鎖アンチセンスDNAが残存するよう、変性溶液による洗浄により、非ビオチン化鎖を分離する。さらなる材料が必要とされる場合には、5′ビオチン化部分的アンカーオリゴヌクレオチド、及び末端オリゴヌクレオチドを用いて、これを増幅することができる。このPCR産物の一方の鎖のみがビオチン化され、他方は変性により分離されうる。このアンチセンスDNAは、アンチセンスDNAとウイルスRNA又はDNAとを相互にハイブリダイズさせ、そのハイブリッドをストレプトアビジン被覆支持体と結合させ、ストリンジェント条件下で非ハイブリダイズ成分を洗浄除去することにより、細胞溶解物又は核酸調製物のような複雑な混合物からウイルスRNA又はDNAを濃化するために使用されうる。次いで、第2工程において、RNA又はDNAの非ハイブリダイズ部分由来のプライマーを使用した従来のPCRにより、濃縮されたRNA又はDNAを増幅し、検出することができる。これは、通常、産物のゲル電気泳動により実施され、又は、適切に修飾されたプライマーが使用されたならば、蛍光分析、又は後続ELISAにより実施される。本発明に係る方法の利点は、ほぼ任意の量の出発材料を使用することが可能であり、それが分析の感度を改良する点、異なる種類の蛍光標識を有するプライマーを使用することにより、いくつかの標的を同時に検査し、分別することも可能である点、及び細菌、真菌、又はウイルスのような任意の病原体について使用されうる点である。増幅する配列の予備濃縮も、バックグラウンド問題を大きく減少させる。適切な小型化により、1つのチップ上で多数の異なる病原体について同時に試験することが可能であり、それは、分析コストを大きく減少させる。
【0048】
9.遺伝子プロファイリング(GPro(登録商標))
分子生物学研究においては、ある種の遺伝子の発現がRNAレベルで定量的に検査されており、そして分子的診断においても、ますますそのようになっている。このための標準的な道具は、通常、放射性標識プローブと共に使用されるノーザンブロット、S1マッピング、又はリボヌクレアーゼ防御アッセイ(RPA)である。前記の一本鎖DNAは、この目的のためにも使用されうる。通常付加的なエラー源となる、分析するRNAの予備精製が、このためには必要ない。本発明に係るPathoCheck(登録商標)法と同様に、検査するmRNAを、遺伝子特異的一本鎖アンチセンスDNAを有する、過剰の、修飾、例えばビオチン化されたアンカーオリゴヌクレオチドとハイブリダイズさせ、そして例えばストレプトアビジン被覆固相上に固定化する。全タンパク質、無関係な核酸、及びその他の不純物を洗浄除去した後、これらのmRNAの別の部分と相補的な一連の直接的又は間接的な蛍光標識スプリンカー配列を用いて標的mRNAを検出する。異なる遺伝子特異的アンチセンスDNA及び異なる標識を有する検出スプリンカーオリゴヌクレオチドの使用は、数個の遺伝子の発現の同時分析を可能にする。方法全体が、単純な様式で完全に自動化されうる。分析する組織が大量のRNAseを含有していない場合、カオトロピック緩衝液中での溶解及び/又はRNasinの添加が、RNAの完全性を保証するために十分である。反応混合物中の異なるmRNAの同時検出よりも最大感度の方が重要である場合には、抗マウスIg−ペルオキシダーゼ重合体のような多価二次試薬と結合する抗体を、蛍光に基づく検出試薬の代わりに使用することができる。次いで、これらの複合体を、後続の酵素反応において、例えば適当な基質の反応により生成した化学発光により、検出する。特に頻繁に実施される研究の場合には、定量標準として合成対照mRNAを含有するGPro(登録商標)キットを、予め製剤化することができる。
【0049】
10.ハイブリッドにより媒介されるライゲーションによる対立遺伝子同定(LIMA(登録商標);ライゲーションにより媒介される変異型対立遺伝子の同定)
特に、遺伝性疾患の出生前診断のため、そして、様々な薬物に対する個体の感度を決定するためには、ある種の対立遺伝子の遺伝子型を確立しなければならない。これは、通常、検査する遺伝子座のゲノミックDNAからのPCR増幅、及びそれに続く制限分析又は配列決定により実施される。第一の場合において、これは、制限断片のゲル電気泳動による分離を必要とし、それは容易には自動化され得ない。これは、チップ配列決定法が使用されないのであれば、第2の場合にも当てはまるが、この方法は未だ完全には開発されていない。本発明に従い調製されたDNA断片は、この面にも使用されうる。先行条件は、それらが、既知の分子的に同定された対立遺伝子でなければならない点である。次いで、本発明に従い調製されるアンカーオリゴヌクレオチドを、突然変異の直前の遺伝子領域とハイブリダイズするよう、構築する。1個又は数個の蛍光標識を含有するもう1つのオリゴヌクレオチドが、本発明に従い調製されたアンカーオリゴヌクレオチドDNA及び蛍光標識オリゴヌクレオチドの2つの遊離の末端が、連続ハイブリッドが形成されるときに相互に直接隣接し、共にライゲートしうるよう、遺伝子の直接接続3′領域とハイブリダイズする。この部位の配列が異なっている場合、末端は接着せず、従ってライゲーションが起こらない。代わりに、異なる蛍光で標識されたオリゴヌクレオチドが、例えば対応する変異型配列と結合することができ、従って異なる標識がビオチン化アンカーとライゲートする。各場合に結合する蛍光色素及び従って各対立遺伝子を、レーザー励起により同定する。本発明に係る方法の感度を増加させるため、検査する遺伝子座を増幅する予備的な非対称PCRを、この場合に実施することも可能である。PCR及びハイブリダイゼーションのための反応条件が均一であるならば、1個の試料から数個の異なる対立遺伝子を同時に決定することが可能である。
【0050】
11.タンパク質アレイの直接相互作用分析(LISPA(登録商標))
ヒトゲノムプロジェクトの成功により、次の問題の1つは、約50,000個の遺伝子を分類することである。基礎研究においてのみならず、急速に発展中の分子医学の領域においても、これらの遺伝子が何をするのか、それらがどのように相互に協力するのか、いかなる状況で、どのタンパク質、ペプチド、又は低分子量物質が、どの他のタンパク質と結合するか、等を理解することが必要である。そのようなタンパク質間の協調の第1の指標は、通常、各遺伝子産物の直接的な物理的接触である。in vitroでタンパク質レベルでそのようなリンクを検査するためには、通常、精製されたタンパク質調製物が必要となる。しかしながら、50,000個のタンパク質で、これを達成することは困難である。従って、通常、潜在的な相互作用パートナーを同定するため、いわゆる酵母ツーハイブリッドスクリーニングのような遺伝学的方法が利用される。この方法は、従来極めて良好な結果をもって使用されているが、にもかかわらず、アーチファクトに対する感受性が極めて高く、面倒で、かつ現在の問題の複雑性にとっては適当でない。この目的は、本発明に係る方法の組み合わせ、バイオチップと組み合わせられた本発明に係るSloning(登録商標)法及びEasyPro(登録商標)法により達成されうる。自動化された方法は、50,000個の遺伝子を完全に合成し、発現させ、そして、それらにバイオチップの反応室における固定化のための適当なタグを供給するために使用されうる。107〜108個という量の分子が、通常、蛍光標識されたタンパク質又は低分子量化合物を用いた結合研究にとって十分である。100kDタンパク質及び5mg/mlの濃度、分子約3×1010個に相当するタンパク質溶液約1ナノリットルを、100×200×60μmのウェルに沈着させることができる。1ウェル当たりの実際の結合許容量がこの値の約1%であると仮定すると、比較的大きいタンパク質の場合ですら、未だ十分な材料が存在する。各ウェル間の間隙が約30μmであるならば、タンパク質50,000個のライブラリー全体が、わずか20cm2のチップ上に収容されうる。レーザーが、蛍光標識標的分子の結合前後の全ウェルにおける蛍光を測定し、それから、相互作用の強度が計算される。タグのみが存在するウェルが、非特異的対照として使用される。そのようなタンパク質アレイは、例えば、従来検出不可能であった薬物と細胞タンパク質との結合を検出し、複雑なシグナル伝達カスケードを理解するために使用されうる。
【0051】
12.固定化核酸アレイを使用したmRNAの平行分析(PAMINA(登録商標))
現代の薬物研究の焦点の1つは、各遺伝子の発現を選択的に操作することである。この目的のためには、可能な限り広範に、他の遺伝子の発現に対する新規活性物質の影響を検査することが必要である。シグナル伝達過程において、細胞の分化、又は疾患により誘導される代謝変化の場合には、異なる遺伝子のカスケード全体が、オン又はオフに切り替わることが多い。しかしながら、高等生物における遺伝子発現は複雑であるため、従来は、ほんの少数の遺伝子しか同時分析できなかった。しかしながら、ヒトゲノムの配列決定は、細胞の遺伝子発現全体の包括的な平行分析のための将来の基礎を提供する。利用可能な配列情報から、まず、コンピュータ化された配列比較を使用して、相互に最も低い相同性、すなわち各遺伝子のための最も高度の特異性を有する各遺伝子内の領域を同定することが可能である。遺伝子特異的一本鎖アンチセンスアンカーDNAを、これらの遺伝子セクションから導出し、次いで、それらをバイオチップ上にアレイとして固定化される。アンチセンスアンカーDNAは、全RNA/DNAハイブリッドの融解温度が狭い範囲内にあるよう、設計されうる。調査する細胞のRNA全体又はポリA+−RNAと、このアレイとの最大ストリンジェンシー条件下でのハイブリダイゼーションは、対応するmRNAが発現している場合に、バイオチップの各ウェル内にRNA/DNAハイブリッドを生成させる。第2工程において、固定化されたアンチセンスアンカーDNAを、RNaseH逆転写酵素及び好ましくは蛍光標識されている修飾オリゴヌクレオチドによる処理により、ハイブリダイズしたRNA鋳型上で伸長させる。取り込まれなかったヌクレオチドを分離するための数回の洗浄過程の後、記載されたLISPA(登録商標)技術と同様にして、cDNA反応産物を、各ウェルのレーザースキャニングにより測定することができる。
【0052】
適用例
1.アンカーオリゴヌクレオチド及びスプリンカーオリゴヌクレオチドの作製
アンカーオリゴヌクレオチド及びスプリンカーオリゴヌクレオチドは、Sinha N.D.,Biernat J.,McManus J.,Koster H.,Nucleic Acids Res,1984年1月12日:11,4539−57により記載された標準的な方法に従い、又は大規模合成の後の4分割、又はセルロース膜上での同時合成により調製した。
【0053】
2.修飾による標識
標準的な方法によりオリゴヌクレオチドを修飾した。
【0054】
3.マトリックスとのカップリング
ビオチン標識キナーゼ処理アンカーオリゴヌクレオチド20〜200pmolを、1×TE/1M NaCl、pH7.5中で50μlの総容量で、ストレプトアビジンカップリング磁性化ビーズ(MERCK)10μlに添加し、ローラー上で室温で30分間インキュベートした。続いて、未結合アンカーオリゴヌクレオチドを、毎回1×TE、pH7.5 500μlで、3回緩衝液を交換することにより洗浄除去した。
【0055】
4.第1ライゲーション工程
T4 DNAリガーゼ(Boehringer Mannheim又はNew England Biolabs)1〜5単位を含有する1×リガーゼ緩衝液(Boehringer Mannheim)中50μlの容量で、4℃、16℃、室温、又は37℃(標準16℃)で、15〜60分間ライゲーションを実施した。通常、リン酸化アンカーオリゴヌクレオチド20pmolをライゲーションに使用した。5′末端をリン酸化されたスプリンカーオリゴヌクレオチドを、1.5〜5倍モル過剰で添加した。反応後、リガーゼ及びライゲートしなかったスプリンカーオリゴヌクレオチドを、毎回1×TE、pH7.5 500μlで、3回緩衝液を交換することにより洗浄除去した。その後、1.25×制限緩衝液(Boehringer Mannheimの緩衝液A又はNew England Biolabsの緩衝液A)中にスプリンカー特異的制限酵素Eco31Iを含有する反応混合物40μlを、洗浄されたビーズに添加した。続いて、それらを前記のようにして洗浄した。
【0056】
5.第2ライゲーション工程
セクション4に記載のプロトコルに従い、他のスプリンカーオリゴヌクレオチドを用いて、さらに4回のライゲーションを実施した。
【0057】
6.転移
第5ライゲーションの後、適切な製造業者特異的緩衝液中のアンカー特異的制限酵素Esp3I又はBpiIの混合物を、洗浄後に添加し、37℃で30〜60分間インキュベートした。反応後、切断されたライゲートしたスプリンカーオリゴヌクレオチドを含む完全な混合物を取り出し、制限酵素を不活化するため、異なる反応容器で65℃で15分間、熱処理し、次いで他の反応容器で、磁性化ストレプトアビジンビーズとカップリングした適切にライゲートしたアンカーオリゴヌクレオチドとライゲートさせた。
【0058】
7.ライゲートした断片の制限調節
切断されたスプリンカーオリゴヌクレオチドの正確なサイズをモニターするため、反応混合物のうちの5μlを、18%1×TBEポリアクリルアミドゲル上で分離し、1×TBE中の0.01%SYBR−Gold(登録商標)で10分間染色し、UV光で可視化した。1〜2塩基長の差が、そのようなゲル上で検出されうる。
【図面の簡単な説明】
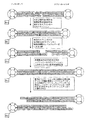
【図1】 本発明に係る方法の概略を示す図である。Bioは、アンカーオリゴヌクレオチドを固体マトリックス(例えば、ストレプトアビジン)とカップリングさせるために使用される修飾(例えば、ビオチン)を意味する。T、G、C、A及びNは、核酸塩基を意味し、Tはチミジン、Gはグアニジン、Cはシトシン、Aはアデニン、Nは4つの核酸塩基のうちのいずれかを意味する。
【図2】 PCR断片のEasyPro(登録商標)転写/翻訳系の構造を概略的に示す図である。Bioは、アンカーオリゴヌクレオチドを固体マトリックスとカップリングさせるために使用される修飾を意味する。5′UTRは、5′非翻訳領域を意味する。ATGは開始コドンを意味する。6×Hisはヒスチジンコドン6個の配列を意味する。単一T突出は、チミジン残基1個の突出を意味する。
【図3】 タンパク質合成のためのミニリアクタの概略を示す図である。
【図4】 QuickPep(登録商標)法を使用したペプチドライブラリーの作製の概略図である。Bioは、アンカーオリゴヌクレオチドを固体マトリックスとカップリングさせるために使用される修飾を意味する。T7は、T7プロモーターを意味する。rbsは内部リボソーム結合部位を意味する。ATGは開始コドンを意味する。EKは、エンテロキナーゼ切断部位を意味する。ペプチドORFは、ペプチドのオープンリーディングフレームを意味する。STOPは終止コドンを意味する。ポリAはポリAテールを意味する。
【図5】 RiboSelect(登録商標)法を使用したリボザイムの選択の概略を示す図である。
【図6】 PCRによる増幅後の病原体の検出(PathoCheck(登録商標))の概略を示す図である。Bioは、アンカーオリゴヌクレオチドを固体マトリックスとカップリングさせるために使用される修飾を意味する。
【図7】 標識スプリンカーのライゲーションによる既知の対立遺伝子の同定(LIMA(登録商標))の概略を示す図である。Bioは、アンカーオリゴヌクレオチドを固体マトリックスとカップリングさせるために使用される修飾を意味する。xは、決定された修飾が存在する部位を意味する。
【図8】 mRNAアレイの平行分析(PAMINA(登録商標))の概略を示す図である。
【図9】 アンカーオリゴヌクレオチドの概略を示す図である。Bioは、アンカーオリゴヌクレオチドを固体マトリックスとカップリングさせるために使用される修飾を意味する。T、G、C、Aは、核酸塩基を意味し、Tはチミジン、Gはグアニン、Cはシトシン、Aはアデニンを意味する。Esp3Iは制限酵素をさす。
【図10】 アンカーオリゴヌクレオチドの概略を示す図である。Bioは、アンカーオリゴヌクレオチドを固体マトリックスとカップリングさせるために使用される修飾を意味する。T、G、C、Aは、核酸塩基を意味し、Tはチミジン、Gはグアニン、Cはシトシン、Aはアデニンを意味する。BpiIは制限酵素をさす。
【図11】 2部アンカーオリゴヌクレオチドの概略を示す図である。Bioは、アンカーオリゴヌクレオチドを固体マトリックスとカップリングさせるために使用される修飾を意味する。T、G、C、Aは、核酸塩基を意味し、Tはチミジン、Gはグアニン、Cはシトシン、Aはアデニンを意味する。
【図12】 スプリンカーオリゴヌクレオチドの概略を示す図である。T、G、C、Aは、核酸塩基を意味し、Tはチミジン、Gはグアニン、Cはシトシン、Aはアデニンを意味する。BsaI及びEco31Iは制限酵素をさす。
【図13】 2部スプリンカーオリゴヌクレオチドの概略を示す図である。T、G、C、Aは、核酸塩基を意味し、Tはチミジン、Gはグアニン、Cはシトシン、Aはアデニンを意味する。BsaI及びEco31Iは制限酵素をさす。
【図14】 本発明に係る方法を使用した、長い核酸の合成経路の概略を示す図である。バーは、連続的なライゲーション/制限サイクルにより平行合成された二本鎖DNA断片を表す。ライゲートしたスプリンカーを、ライゲートしたアンカーとライゲートさせることにより、毎回、最終産物の隣接セクションを連結する。次いで、このようにして得られた大きな断片を、次の工程で、アンカー又はスプリンカーのいずれかに特異的な制限エンドヌクレアーゼで再び切断し、断片長が1工程毎に2倍となるよう、相補的突出等によって共に連結させる。使用される制限エンドヌクレアーゼの認識配列は、毎回、ライゲートした断片の切断除去される部分に位置しており、従って成長する核酸には取り込まれないため、連結は完全に配列と無関係である。バーの上の数字は、断片のサイズを塩基対で意味している。従って、20塩基対のサイズのDNA断片から出発して、これにより、最大長は、4回の転移の後に320塩基対、5回の転移の後に640塩基対、6回の転移の後に1280塩基対、7回の転移の後に2560塩基対等となる。
【配列表】
Claims (15)
- a) 下記工程:
aa) オリゴヌクレオチドの一端を固体マトリックスとカップリングさせる工程、ここで、該カップリングは修飾により実施され、該オリゴヌクレオチドは、その認識配列の外側を切断するIIS型制限酵素の認識配列を含有し、少なくとも部分的に二本鎖である、
ab) 認識配列の外側を切断するIIS型制限酵素の認識配列を含有する少なくとも部分的に二本鎖の追加のオリゴヌクレオチドを添加する工程、ここで、該認識配列は工程aa)におけるものとは異なり、このオリゴヌクレオチドはマトリックスに結合することができない、
ac) 工程aa)及びab)からのオリゴヌクレオチドを、ライゲートさせない末端の遮断により決定された方向でライゲートさせる工程、
ad) 未消費の反応物及び酵素を除去する工程、
ae) 認識配列の外側を切断するIIS型制限酵素により、工程ac)のライゲーション産物を切断する工程、ここで、該切断は、工程ab)のオリゴヌクレオチドの核酸配列に生じる、
af) 工程ae)において得られるような工程aa)の伸長オリゴヌクレオチドから反応混合物を分離する工程、
ag) 工程ab)〜af)を少なくとも1回反復する工程
により調製されるオリゴヌクレオチドを準備する工程、
b) 下記工程:
ba) オリゴヌクレオチドの一端を、固体マトリックスとカップリングさせる工程、ここで、該カップリングは、修飾により実施され、該オリゴヌクレオチドは、認識配列の外側を切断するIIS型制限酵素の認識配列を含有し、少なくとも部分的に二本鎖である、
bb) 少なくとも部分的に二本鎖であり認識配列の外側を切断するIIS型制限酵素の認識配列を有する追加のオリゴヌクレオチドを添加する工程、ここで、該認識配列は、工程ba)のものとは異なり、該オリゴヌクレオチドはマトリックスと結合することができない、
bc) 工程ba)及びbb)のオリゴヌクレオチドを、ライゲートさせない末端の遮断により決定された方向でライゲートさせる工程、
bd) 未消費の反応物及び酵素を除去する工程、
be) 認識配列の外側を切断するIIS型制限酵素により、工程bc)のライゲーション産物を切断する工程、ここで、切断は、工程bb)のオリゴヌクレオチドで生じる、
bf) このようにして伸長した核酸分子を反応混合物から分離する工程、
bg) 工程bb)〜bf)を少なくとも1回反復する工程、ここで、工程bc)における最後のライゲーション、並びに未消費の反応物及び酵素の除去の後、該ライゲーション産物をIIS型制限酵素により切断し、該切断は、工程ba)のオリゴヌクレオチドに生じる、
により調製される付加的なオリゴヌクレオチドを準備する工程、
c) 工程a)及びb)のオリゴヌクレオチドを、ライゲートさせない末端の遮断により決定された方向でライゲートさせる工程、
d) 未消費の反応物及び酵素を除去する工程、
e) 認識配列の外側を切断するIIS型制限酵素により、工程c)からのライゲーション産物を切断する工程、ここで、切断は、工程a)又はb)のオリゴヌクレオチドに生じる、
f) このようにして伸長した核酸分子を反応混合物から分離する工程
を含む、核酸分子を作製するための方法。 - 工程ab)又はbb)において使用されるオリゴヌクレオチドが、請求項1記載の方法により作製された核酸分子である、請求項1記載の方法。
- 工程ac)、bc)又はc)の後、工程ac′)、bc′)又はc′)として、エキソヌクレアーゼ及び/又はホスファターゼ反応を実施する、請求項1又は2記載の方法。
- 工程ac′)、bc′)又はc′)の反応混合物を反応後に除去する、請求項3記載の方法。
- 工程a)、aa)又はba)のオリゴヌクレオチドが、マトリックスとカップリングしていない末端で、認識配列の外側を切断するIIS型制限酵素の認識配列の一部を含有し、かつこの制限酵素の認識配列のその他の部分が工程ab)、bb)又はb)のオリゴヌクレオチドに由来する、請求項1〜3いずれか1項に記載の方法。
- 該修飾がビオチン基、ジゴキシゲニン基、フルオレセインイソチオシアネート基、アミノ化合物、又はスクシニルエステルである、請求項1〜5のいずれか1項記載の方法。
- 工程aa)、ba)若しくはa)及び/又はab)、bb)若しくはb)からのオリゴヌクレオチドがループを含む、請求項1〜6いずれか1項に記載の方法。
- 工程aa)、ba)又はa)からのオリゴヌクレオチドを、ループ領域における修飾によって固体マトリックスとカップリングさせる、請求項7記載の方法。
- 固体マトリックスがビーズ、好ましくはガラス製又はポリスチレン製のビーズ、スライド、DNAチップ、マイクロタイタープレートのウェル、又は反応チューブである、請求項1〜8いずれか1項に記載の方法。
- 固体マトリックスが、ストレプトアビジン基、抗ジゴキシゲニン抗体、又は抗フルオレセインイソチオシアネート抗体を含む、請求項1〜9いずれか1項に記載の方法。
- 工程aa)、ba)又はa)、及び、ab)、bb)又はb)のオリゴヌクレオチドが、ライゲートされる末端に、相互に相補的な一本鎖突出を有する、請求項1〜10いずれか1項に記載の方法。
- 一本鎖突出が1、2、3、4又は5ヌクレオチド長である、請求項11記載の方法。
- 様々なIIS型制限エンドヌクレアーゼが、類似の様式で切断するリボザイムと交換される、請求項1〜12いずれか1項記載の方法。
- 工程ab)、bb)又はb)におけるオリゴヌクレオチドが、PCR産物、プラスミドベクター、ファージもしくはウイルスのDNA、人工染色体、又はその他の合成DNAである、請求項1〜13いずれか1項に記載の方法。
- a)突出を有する4、16、64、256及び1024個の異なるオリゴヌクレオチドからなるライブラリー、ここで、該オリゴヌクレオチドはその突出の配列において相違し、該オリゴヌクレオチドは一端で修飾により固体マトリックスとカップリングすることができ、かつ該オリゴヌクレオチドは認識配列の外側を切断するIIS型制限酵素の認識配列又は認識配列の一部を含有する、
b)突出を有する16、256、4,096、65,536又は1,048,576個の異なるオリゴヌクレオチドからなる付加的なライブラリー、ここで、該オリゴヌクレオチドは、その突出の配列及びその後に続く配列において相違し、長さで、該突出の長さに相当し、各オリゴヌクレオチドが認識配列の外側を切断しa)の制限酵素と相違するIIS型制限酵素の認識配列を含有する、
c)固体マトリックス、
d)核酸分子を作製するために必要とされる酵素及び/又は試薬のための貯蔵器
を含む、請求項1〜14いずれか1項に記載の方法に従い、核酸配列を作製するためのキット、
ここで、該突出の長さが、1個のヌクレオチドであれば、a)のライブラリーは、4つの異なるオリゴヌクレオチドを含み、b)のライブラーは、16個の異なるオリゴヌクレオチドを含み、
該突出の長さが、2個のヌクレオチドであれば、a)のライブラリーは、16個の異なるオリゴヌクレオチドを含み、b)のライブラリーは、256個の異なるオリゴヌクレオチドを含み、
該突出の長さが、3個のヌクレオチドであれば、a)のライブラリーは、64個の異なるオリゴヌクレオチドを含み、b)のライブラリーは、4,096個の異なるオリゴヌクレオチドを含み、
該突出の長さが、4個のヌクレオチドであれば、a)のライブラリーは、256個の異なるオリゴヌクレオチドを含み、b)のライブラリーは、65,536個の異なるオリゴヌクレオチドを含み、そして
該突出の長さが、5個のヌクレオチドであれば、a)のライブラリーは、1,024個の異なるオリゴヌクレオチドを含み、b)のライブラリーは、1,048,576個の異なるオリゴヌクレオチドを含む。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19925862A DE19925862A1 (de) | 1999-06-07 | 1999-06-07 | Verfahren zur Synthese von DNA-Fragmenten |
| DE19925862.7 | 1999-06-07 | ||
| PCT/DE2000/001863 WO2000075368A2 (de) | 1999-06-07 | 2000-06-07 | Verfahren zur synthese von dna-fragmenten |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2003501069A JP2003501069A (ja) | 2003-01-14 |
| JP2003501069A5 JP2003501069A5 (ja) | 2007-07-19 |
| JP4756805B2 true JP4756805B2 (ja) | 2011-08-24 |
Family
ID=7910391
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2001501647A Expired - Lifetime JP4756805B2 (ja) | 1999-06-07 | 2000-06-07 | Dna断片の合成法 |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP1181395B1 (ja) |
| JP (1) | JP4756805B2 (ja) |
| AT (1) | ATE323179T1 (ja) |
| DE (3) | DE19925862A1 (ja) |
| DK (1) | DK1181395T3 (ja) |
| ES (1) | ES2262524T3 (ja) |
| WO (1) | WO2000075368A2 (ja) |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6670127B2 (en) * | 1997-09-16 | 2003-12-30 | Egea Biosciences, Inc. | Method for assembly of a polynucleotide encoding a target polypeptide |
| EP1205548A1 (en) * | 2000-11-09 | 2002-05-15 | Kabushiki Kaisha Toyota Chuo Kenkyusho | Methods and apparatus for successively ligating double-stranded DNA molecules on a solid phase |
| KR100758610B1 (ko) | 2001-05-07 | 2007-09-13 | (주)바이오니아 | 염기서열이 전혀 알려지지 아니한 디옥시리보핵산의선택적 증폭 방법 |
| CA2344599C (en) * | 2001-05-07 | 2011-07-12 | Bioneer Corporation | Selective polymerase chain reaction of dna of which base sequence is completely unknown |
| DE10156329A1 (de) * | 2001-07-17 | 2003-02-06 | Frieder Breitling | Verfahren und Anordnung zum Anbringen von in Transportmittel immobilisierten Substanzen sowie Monomerpartikel |
| DK1314783T3 (da) * | 2001-11-22 | 2009-03-16 | Sloning Biotechnology Gmbh | Nukleinsyrelinkere og deres anvendelse i gensyntese |
| US7563600B2 (en) | 2002-09-12 | 2009-07-21 | Combimatrix Corporation | Microarray synthesis and assembly of gene-length polynucleotides |
| ES2310197T3 (es) | 2002-10-18 | 2009-01-01 | Sloning Biotechnology Gmbh | Metodo para la preparacion de moleculas de acido nucleico. |
| FR2852605B1 (fr) * | 2003-03-18 | 2012-11-30 | Commissariat Energie Atomique | Procede de preparation de fragments d'adn et ses applications |
| EP1557464B1 (en) * | 2004-01-23 | 2010-09-29 | Sloning BioTechnology GmbH | De novo enzymatic production of nucleic acid molecules |
| US8053191B2 (en) | 2006-08-31 | 2011-11-08 | Westend Asset Clearinghouse Company, Llc | Iterative nucleic acid assembly using activation of vector-encoded traits |
| US9115352B2 (en) | 2008-03-31 | 2015-08-25 | Sloning Biotechnology Gmbh | Method for the preparation of a nucleic acid library |
| AU2010291902A1 (en) | 2009-09-14 | 2012-04-05 | Dyax Corp. | Libraries of genetic packages comprising novel HC CDR3 designs |
| US9187777B2 (en) | 2010-05-28 | 2015-11-17 | Gen9, Inc. | Methods and devices for in situ nucleic acid synthesis |
| EP4039363A1 (en) | 2010-11-12 | 2022-08-10 | Gen9, Inc. | Protein arrays and methods of using and making the same |
| CA2817697C (en) | 2010-11-12 | 2021-11-16 | Gen9, Inc. | Methods and devices for nucleic acids synthesis |
| DE102010056289A1 (de) | 2010-12-24 | 2012-06-28 | Geneart Ag | Verfahren zur Herstellung von Leseraster-korrekten Fragment-Bibliotheken |
| EP3461896B1 (en) * | 2011-07-15 | 2023-11-29 | The General Hospital Corporation | Methods of transcription activator like effector assembly |
| EP3954770A1 (en) | 2011-08-26 | 2022-02-16 | Gen9, Inc. | Compositions and methods for high fidelity assembly of nucleic acids |
| CN102584955A (zh) * | 2011-12-31 | 2012-07-18 | 北京唯尚立德生物科技有限公司 | 一种转录激活子样效应因子的固相合成方法 |
| DE102012101347B4 (de) | 2012-02-20 | 2014-01-16 | Markus Fuhrmann | Verfahren zur Herstellung einer Nukleinsäure- Bibliothek mit mindestens zwei benachbarten variablen Codon- Tripletts |
| US9150853B2 (en) | 2012-03-21 | 2015-10-06 | Gen9, Inc. | Methods for screening proteins using DNA encoded chemical libraries as templates for enzyme catalysis |
| EP2841601B1 (en) | 2012-04-24 | 2019-03-06 | Gen9, Inc. | Methods for sorting nucleic acids and multiplexed preparative in vitro cloning |
| US9890364B2 (en) | 2012-05-29 | 2018-02-13 | The General Hospital Corporation | TAL-Tet1 fusion proteins and methods of use thereof |
| JP6509727B2 (ja) | 2012-06-25 | 2019-05-15 | ギンゴー バイオワークス, インコーポレイテッド | 核酸アセンブリおよび高処理シークエンシングのための方法 |
| EP3789405A1 (en) | 2012-10-12 | 2021-03-10 | The General Hospital Corporation | Transcription activator-like effector (tale) - lysine-specific demethylase 1 (lsd1) fusion proteins |
| CA2900338A1 (en) | 2013-02-07 | 2014-08-14 | The General Hospital Corporation | Tale transcriptional activators |
| EP3143140A4 (en) * | 2014-05-16 | 2017-11-08 | The Regents Of The University Of California | Compositions and methods for single-molecule construction of dna |
| GB2559117B (en) | 2017-01-19 | 2019-11-27 | Oxford Nanopore Tech Ltd | Double stranded polynucleotide synthesis method, kit and system |
| GB2566986A (en) | 2017-09-29 | 2019-04-03 | Evonetix Ltd | Error detection during hybridisation of target double-stranded nucleic acid |
| CA3192399A1 (en) * | 2020-09-14 | 2022-03-17 | Mark S. Chee | Methods and compositions for nucleic acid assembly |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993019202A2 (en) * | 1992-03-10 | 1993-09-30 | The United States Of America, As Represented By The Secretary, Department Of Health & Human Services | Exchangeable template reaction |
| WO1995017413A1 (de) * | 1993-12-21 | 1995-06-29 | Evotec Biosystems Gmbh | Verfahren zum evolutiven design und synthese funktionaler polymere auf der basis von formenelementen und formencodes |
| WO1998010095A1 (en) * | 1996-09-05 | 1998-03-12 | Brax Genomics Limited | Characterising dna |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19633427C2 (de) * | 1996-08-20 | 2001-10-04 | Hubert S Bernauer | Verfahren zur Synthese von Nukleinsäuremolekülen mit zumindest teilweise vorbestimmter Nukleotidsequenz |
| DE19812103A1 (de) * | 1998-03-19 | 1999-09-23 | Bernauer Annette | Verfahren zur Synthese von Nucleinsäuremolekülen |
-
1999
- 1999-06-07 DE DE19925862A patent/DE19925862A1/de not_active Withdrawn
-
2000
- 2000-06-07 ES ES00945609T patent/ES2262524T3/es not_active Expired - Lifetime
- 2000-06-07 DE DE10081549T patent/DE10081549D2/de not_active Expired - Fee Related
- 2000-06-07 DE DE50012574T patent/DE50012574D1/de not_active Expired - Lifetime
- 2000-06-07 WO PCT/DE2000/001863 patent/WO2000075368A2/de active IP Right Grant
- 2000-06-07 JP JP2001501647A patent/JP4756805B2/ja not_active Expired - Lifetime
- 2000-06-07 EP EP00945609A patent/EP1181395B1/de not_active Expired - Lifetime
- 2000-06-07 AT AT00945609T patent/ATE323179T1/de active
- 2000-06-07 DK DK00945609T patent/DK1181395T3/da active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993019202A2 (en) * | 1992-03-10 | 1993-09-30 | The United States Of America, As Represented By The Secretary, Department Of Health & Human Services | Exchangeable template reaction |
| WO1995017413A1 (de) * | 1993-12-21 | 1995-06-29 | Evotec Biosystems Gmbh | Verfahren zum evolutiven design und synthese funktionaler polymere auf der basis von formenelementen und formencodes |
| WO1998010095A1 (en) * | 1996-09-05 | 1998-03-12 | Brax Genomics Limited | Characterising dna |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2000075368A3 (de) | 2001-05-17 |
| WO2000075368A2 (de) | 2000-12-14 |
| EP1181395A2 (de) | 2002-02-27 |
| JP2003501069A (ja) | 2003-01-14 |
| EP1181395B1 (de) | 2006-04-12 |
| ATE323179T1 (de) | 2006-04-15 |
| DK1181395T3 (da) | 2006-08-14 |
| DE10081549D2 (de) | 2002-07-25 |
| DE50012574D1 (de) | 2006-05-24 |
| DE19925862A1 (de) | 2000-12-14 |
| ES2262524T3 (es) | 2006-12-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4756805B2 (ja) | Dna断片の合成法 | |
| US8137906B2 (en) | Method for the synthesis of DNA fragments | |
| US11768200B2 (en) | Methods for maintaining the integrity and identification of a nucleic acid template in a multiplex sequencing reaction | |
| US10876110B2 (en) | Synthesis of sequence-verified nucleic acids | |
| JP7100680B2 (ja) | ゲノム適用および治療適用のための、核酸分子のクローン複製および増幅のためのシステムおよび方法 | |
| US6284497B1 (en) | Nucleic acid arrays and methods of synthesis | |
| JPH02501532A (ja) | 標的ポリヌクレオチド配列の選択的増幅 | |
| JPH02503054A (ja) | 核酸配列の増幅および検出 | |
| JP2002525049A (ja) | ローリングサークル増幅を用いる分子クローニング | |
| WO2007136736A2 (en) | Methods for nucleic acid sorting and synthesis | |
| JPH07509371A (ja) | 核酸配列の検出及び増幅のための方法,試薬及びキット | |
| JPH04503752A (ja) | インビトロでのdnaの増幅、ゲノムクローニングおよびマッピングの、改良された方法 | |
| US6207378B1 (en) | Method for amplifying nucleic acid molecules and method for synthesizing proteins | |
| JP5128031B2 (ja) | RecAタンパク質を用いて標識が導入された核酸を製造する方法 | |
| JP2000210082A (ja) | 標的dnaの固定化 | |
| Kimoto et al. | Unit Title | |
| JP2006132980A (ja) | タンパク質の固定化方法 | |
| JPH11178582A (ja) | 核酸分子の増幅方法及びタンパクの合成方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20070531 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20070531 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100601 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20100730 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20100819 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101201 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20110517 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20110531 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4756805 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140610 Year of fee payment: 3 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313111 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |