JP4635156B2 - シクロトリホスファゼン誘導体の製造方法 - Google Patents
シクロトリホスファゼン誘導体の製造方法 Download PDFInfo
- Publication number
- JP4635156B2 JP4635156B2 JP2005258765A JP2005258765A JP4635156B2 JP 4635156 B2 JP4635156 B2 JP 4635156B2 JP 2005258765 A JP2005258765 A JP 2005258765A JP 2005258765 A JP2005258765 A JP 2005258765A JP 4635156 B2 JP4635156 B2 JP 4635156B2
- Authority
- JP
- Japan
- Prior art keywords
- cyclotriphosphazene
- group
- general formula
- reaction
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 *P1(*)=NP(*)(*)=NP(*)(*)=NP(*)(*)=N1 Chemical compound *P1(*)=NP(*)(*)=NP(*)(*)=NP(*)(*)=N1 0.000 description 3
- PEJQKHLWXHKKGS-UHFFFAOYSA-N ClP1(Cl)=NP(Cl)(Cl)=NP(Cl)(Cl)=NP(Cl)(Cl)=N1 Chemical compound ClP1(Cl)=NP(Cl)(Cl)=NP(Cl)(Cl)=NP(Cl)(Cl)=N1 PEJQKHLWXHKKGS-UHFFFAOYSA-N 0.000 description 1
Landscapes
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
4−シアノフェノールとフェノールとの混合物をヘキサハロゲン化シクロトリホスファゼンと反応させる方法。
◎特許文献7
ヘキサハロゲン化シクロトリホスファゼンに対し、フェノールのアルカリ金属塩を反応させる方法。
◎特許文献8
アルカリ金属の水酸化物または炭酸塩を脱酸剤として用い、ヘキサハロゲン化シクロトリホスファゼンとフェノールとを反応させる方法。
◎特許文献9
フェノール類とアルカリ金属水酸化物とで調製したフェノラート類を、ヘキサハロゲン化シクロトリホスファゼンと共沸脱水で水を除きながら反応させる方法。
◎特許文献10
相間移動触媒を使用し、ヘキサハロゲン化シクロトリホスファゼンとフェノール等とを水、塩基および水と非混合性の溶媒中で反応させる方法。
◎特許文献11
無機塩類、第三級アミンおよびジメチルアミノピリジンのような有機強塩基の存在下において、ヘキサハロゲン化シクロトリホスファゼンとフェノールとを反応させる方法。
◎特許文献12
鎖状または環式の三級アミン類またはアミド化合物の存在下、アルカリ金属フェノラートとヘキサハロゲン化シクロトリホスファゼンとを反応させる方法。
一般式(5)中、Rはアルキル基を示す。
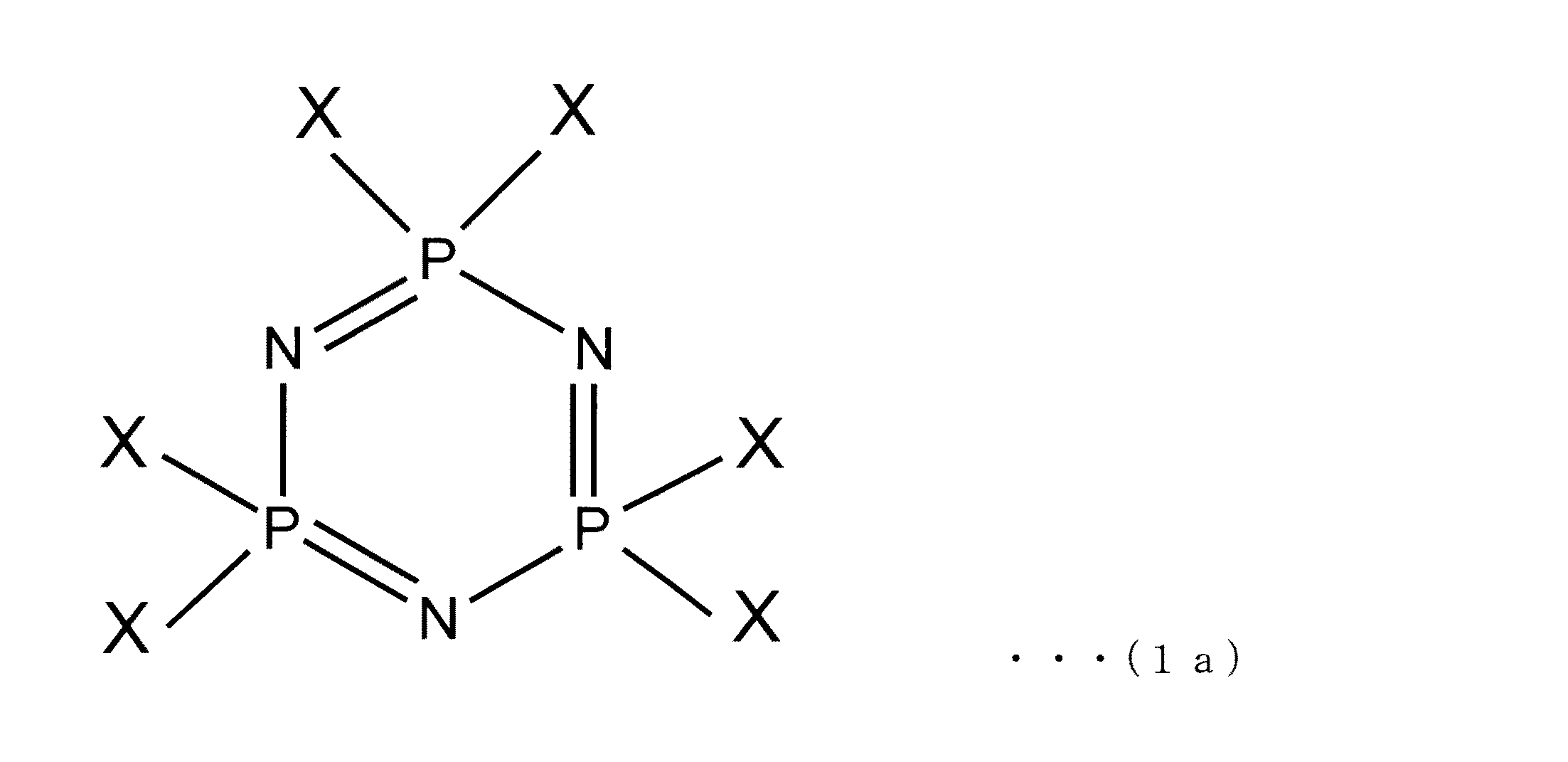
この形態に係るシクロトリホスファゼン誘導体の製造方法では、出発物質としてヘキサハロゲン化シクロトリホスファゼンを用いる。ここで用いられるヘキサハロゲン化シクロトリホスファゼンは、下記の一般式(1a)で示されるものであり、公知の物質である。
C:残留活性ハロゲン原子量(ppm)
V:測定用サンプルの滴定量(ml)
B:ブランク用サンプルの滴定量(ml)
f:0.01mol/硝酸銀水溶液のファクター
0.3545:0.01mol/硝酸銀水溶液1mlに対するハロゲンの量(mg/ml)
S:試料の採取量(mg)
この形態に係るシクロトリホスファゼン誘導体の製造方法では、出発物質として部分置換ハロゲン化シクロトリホスファゼンを用いる。ここで用いられる部分置換ハロゲン化シクロトリホスファゼンは、下記の一般式(1b)で示されるものである。
溶離液:アセトニトリル
検出器:UV 254nm
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコにモノクロロベンゼン300g、塩化アンモニウム38.6g(0.72モル)および酸化亜鉛0.81g(9.9×10−3モル)を仕込んだ。また、滴下ロートには、モノクロロベンゼン300gに五塩化リン125g(0.6モル)を溶解した溶液を仕込んだ。
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコにモノクロロベンゼン300g、塩化アンモニウム38.6g(0.72モル)、酸化亜鉛0.81g(9.9×10−3モル)およびピリジン47.4g(0.6モル)を仕込んだ。また、滴下ロートには、モノクロロベンゼン300gに五塩化リン125g(0.6モル)を溶解した溶液を仕込んだ。
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコにモノクロロベンゼン300g、塩化アンモニウム38.6g(0.72モル)および酸化亜鉛0.81g(9.9×10−3モル)を仕込んだ。また、滴下ロートには、モノクロロベンゼン300gに五塩化リン125g(0.6モル)を溶解した溶液を仕込んだ。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコにフェノール49g(0.52モル)を加え、これを200mlのシクロペンチルメチルエーテルに溶解した。一方、滴下ロートにナトリウムメトキシドを28重量%含むメタノール溶液100.3g(ナトリウムメトキシド換算で0.52モル)を仕込んだ。そして、滴下ロートの内容物を45分かけて四頚フラスコ内に滴下した。その後、四頚フラスコの内容物を106℃まで加熱してメタノールとシクロペンチルメチルエーテルの一部とを留去し、全量が250mlに調整された、フェノールナトリウム塩のシクロペンチルメチルエーテル溶液(第一溶液)を調製した。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第二溶液を仕込み、また、滴下ロートに第一溶液を仕込んだ。そして、第二溶液を0±5℃に維持しながら、第二溶液に対して第一溶液を150分かけて滴下した。滴下終了後、温度を20〜30℃に維持しながら2時間反応させた。反応終了後、反応液を濾過して食塩を分離し、この食塩を少量のシクロペンチルメチルエーテルを用いて洗浄した。これにより、塩素原子の一部がフェノキシ基により置換された部分置換ハロゲン化シクロトリホスファゼンを含む反応液を得た。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに4−シアノフェノール80g(0.67モル)を加え、これを400mlのシクロペンチルメチルエーテルに溶解した。一方、滴下ロートにナトリウムメトキシドを28重量%含むメタノール溶液129.2g(ナトリウムメトキシド換算で0.67モル)を仕込んだ。そして、滴下ロートの内容物を150分かけて四頚フラスコ内に滴下した。その後、四頚フラスコの内容物を106℃まで加熱してメタノールとシクロペンチルメチルエーテルの一部とを留去し、全量が450mlに調整された、4−シアノフェノールナトリウム塩のシクロペンチルメチルエーテル溶液(第三溶液)を調製した。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第三溶液を仕込み、これにテトラブチルアンモニウムブロマイド1gを添加して溶解した。また、滴下ロートに実施例1−aで得られた反応液の全量を仕込んだ。そして、四頚フラスコの内容物の温度を25〜50℃に維持しつつ、当該内容物に対して反応液を150分かけて滴下した。滴下終了後、四頚フラスコを加熱し、シクロペンチルメチルエーテルの還流下で2時間反応させたところ、この反応液のピリジン/アニリン呈色反応がマイナスになった。反応をさらに4時間継続し、反応を終了した。
上述の反応工程で得られた反応液を25℃まで冷却した後に濾過し、反応で生成した食塩と未反応の4−シアノフェノールナトリウム塩とを分離した。このように処理した反応液を希アルカリ水溶液と水とで数回洗浄し、洗浄後の反応液からシクロペンチルメチルエーテルを回収した。残留物に含水メタノールを加えて濾過し、得られた結晶を乾燥したところ、その重量は139gであった(収率96.9%)。この結晶の融点は103〜125℃であった。また、この結晶は、高速液体クロマトグラフィーにより分析したところ、ジフェノキシ−テトラ4−シアノフェノキシ−シクロトリホスファゼン16.10%、トリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼン80.20%およびテトラフェノキシ−ジ4−シアノフェノキシ−シクロトリホスファゼン3.08%を含むシクロトリホスファゼン誘導体混合物であり、実施例1−aにおいてヘキサクロロシクロトリホスファゼンに対して使用したフェノールナトリウム塩と、本実施例において使用した4−シアノフェノールナトリウム塩との比率に対応したシクロトリホスファゼン誘導体、すなわち、トリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼンを主に含むことが判明した。また、このシクロトリホスファゼン誘導体混合物は、既述の方法により残留活性ハロゲン原子量(残留活性塩素量)を測定したところ、50ppm未満であった。
(反応材料の調製)
攪拌装置、還流冷却器および温度計を備えた1リットルのフラスコにフェノール52.7g(0.56モル)を加え、これを200mlのシクロペンチルメチルエーテルに溶解した。そして、攪拌下のこの溶液に対し、粉末状のナトリウムメトキシド30.2g(0.56モル)を加えた。フラスコの内容物をシクロペンチルメチルエーテルの沸点まで昇温させてメタノールを留去し、全量が250mlに調整された、フェノールナトリウム塩のシクロペンチルメチルエーテル溶液(第一溶液)を得た。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第二溶液を仕込み、また、滴下ロートに第一溶液を仕込んだ。そして、第二溶液を0±5℃に維持しながら、第二溶液に対して第一溶液を150分かけて滴下した。滴下終了後、温度を20〜30℃に維持しながら2時間反応させた。反応終了後、反応液を濾過して食塩を分離し、この食塩を少量のシクロペンチルメチルエーテルを用いて洗浄した。これにより、塩素原子の一部がフェノキシ基により置換された部分置換ハロゲン化シクロトリホスファゼンを含む反応液を得た。
(反応材料の調製)
攪拌装置、還流冷却器および温度計を備えた1リットルのフラスコに4−シアノフェノール80g(0.67モル)を加え、これを800mlのアセトニトリルに溶解した。この溶液に水酸化カリウム37.5g(0.67モル)を加えて70〜80℃で1時間攪拌した。攪拌終了後、溶液にn−ヘキサン100mlを加えて加熱し、塔頂温度が55〜82℃の留分を回収して溶液から水分を除去した。さらに、当該溶液からアセトニトリルの一部を留去した後、当該溶液にシクロペンチルメチルエーテル200mlを加え、全量を300mlに調整した。これにより、4−シアノフェノールカリウム塩のシクロペンチルメチルエーテル溶液(第三溶液)を得た。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第三溶液を仕込んだ。また、滴下ロートに実施例2−aで得られた反応液の全量を仕込んだ。そして、四頚フラスコの内容物の温度を約25℃に維持しつつ、当該内容物に対して反応液を150分かけて滴下した。滴下終了後、四頚フラスコの内容物を25℃に維持しながら1時間反応させ、さらに、当該内容物を80℃まで昇温して2時間反応させたところ、この反応液のピリジン/アニリン呈色反応がマイナスになった。シクロペンチルメチルエーテルの還流下で反応をさらに4時間継続し、反応を終了した。
上述の反応工程で得られた反応液を25℃まで冷却した後に濾過し、反応で生成した塩化カリウムと未反応の4−シアノフェノールカリウム塩とを分離した。このように処理した反応液を希アルカリ水溶液と水とで数回洗浄し、洗浄後の反応液からシクロペンチルメチルエーテルを回収した。残留物に含水メタノールを加えて放置したところ、結晶が得られた。得られた結晶を乾燥したところ、その重量は137.4gであった(収率95.8%)。この結晶の融点は80〜120℃であった。また、この結晶は、高速液体クロマトグラフィーにより分析したところ、ジフェノキシ−テトラ4−シアノフェノキシ−シクロトリホスファゼン2.40%、トリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼン89.40%およびテトラフェノキシ−ジ4−シアノフェノキシ−シクロトリホスファゼン5.50%を含むシクロトリホスファゼン誘導体混合物であり、実施例2−aにおいてヘキサクロロシクロトリホスファゼンに対して使用したフェノールナトリウム塩と、本実施例において用いた4−シアノフェノールカリウム塩との比率に対応したシクロトリホスファゼン誘導体、すなわち、トリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼンを主に含むことが判明した。また、このシクロトリホスファゼン誘導体混合物は、既述の方法により残留活性ハロゲン原子量(残留活性塩素量)を測定したところ、50ppm未満であった。
(反応材料の調製)
攪拌装置、還流冷却器および温度計を備えた1リットルのフラスコにフェノール35.1g(0.373モル)を加え、これを200mlのシクロペンチルメチルエーテルに溶解した。そして、攪拌下のこの溶液に対し、粉末状のナトリウムメトキシド20.1g(0.373モル)を加えた。フラスコの内容物をシクロペンチルメチルエーテルの沸点まで昇温させてメタノールを留去し、全量が250mlに調整された、フェノールナトリウム塩のシクロペンチルメチルエーテル溶液(第一溶液)を得た。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第二溶液を仕込み、また、滴下ロートに第一溶液を仕込んだ。そして、第二溶液を0±5℃に維持しながら、第二溶液に対して第一溶液を150分かけて滴下した。滴下終了後、温度を20〜30℃に維持しながら2時間反応させた。反応終了後、反応液を濾過して食塩を分離し、この食塩を少量のシクロペンチルメチルエーテルを用いて洗浄した。これにより、塩素原子の一部がフェノキシ基により置換された部分置換ハロゲン化シクロトリホスファゼンを含む反応液を得た。
(反応材料の調製)
攪拌装置、還流冷却器および温度計を備えた1リットルのフラスコに4−シアノフェノール100g(0.85モル)を加え、これを1,000mlのアセトニトリルに溶解した。この溶液に水酸化カリウム47g(0.84モル)を加えて70〜80℃で1時間攪拌した。攪拌終了後、溶液にn−ヘキサン100mlを加えて加熱し、塔頂温度が55〜82℃の留分を回収して溶液から水分を除去した。さらに、当該溶液からアセトニトリルの一部を留去した後、当該溶液にシクロペンチルメチルエーテル200mlを加え、全量を300mlに調整した。これにより、4−シアノフェノールカリウム塩のシクロペンチルメチルエーテル溶液(第三溶液)を得た。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第三溶液を仕込んだ。また、滴下ロートに実施例3−aで得られた反応液の全量を仕込んだ。そして、四頚フラスコの内容物の温度を約25℃に維持しつつ、当該内容物に対して反応液を150分かけて滴下した。滴下終了後、四頚フラスコの内容物を25℃に維持しながら1時間反応させ、さらに、当該内容物を80℃まで昇温して2時間反応させたところ、この反応液のピリジン/アニリン呈色反応がマイナスになった。シクロペンチルメチルエーテルの還流下で反応をさらに4時間継続し、反応を終了した。
上述の反応工程で得られた反応液を25℃まで冷却した後に濾過し、反応で生成した塩化カリウムと未反応の4−シアノフェノールカリウム塩とを分離した。このように処理した反応液を希アルカリ水溶液と水とで数回洗浄し、洗浄後の反応液からシクロペンチルメチルエーテルを回収した。残留物に含水メタノールを加えて放置したところ、結晶が得られた。得られた結晶を乾燥したところ、その重量は142gであった(収率95.8%)。この結晶の融点は115〜144℃であった。また、この結晶は、高速液体クロマトグラフィーにより分析したところ、フェノキシ−ペンタ4−シアノフェノキシ−シクロトリホスファゼン10.10%、ジフェノキシ−テトラ4−シアノフェノキシ−シクロトリホスファゼン85.90%およびトリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼン3.90%を含むシクロトリホスファゼン誘導体混合物であり、実施例3−aにおいてヘキサクロロシクロトリホスファゼンに対して使用したフェノールナトリウム塩と、本実施例において用いた4−シアノフェノールカリウム塩との比率に対応したシクロトリホスファゼン誘導体、すなわち、ジフェノキシ−テトラ4−シアノフェノキシ−シクロトリホスファゼンを主に含むことが判明した。また、このシクロトリホスファゼン誘導体混合物は、既述の方法により残留活性ハロゲン原子量(残留活性塩素量)を測定したところ、50ppm未満であった。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコにフェノール47.2g(0.502モル)を加え、これを290mlのシクロペンチルメチルエーテルに溶解した。一方、滴下ロートに液状苛性ソーダ(NaOH20g/水20g)を仕込んだ。そして、シクロペンチルメチルエーテルの還流下において、四頚フラスコ内に滴下ロートの内容物を滴下して共沸脱水し、全量が250mlに調整された、フェノールナトリウム塩のシクロペンチルメチルエーテル溶液(第一溶液)を調製した。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第一溶液を仕込み、また、滴下ロートに第二溶液を仕込んだ。そして、第一溶液を0〜10℃に維持しながら、第一溶液に対して第二溶液を80分かけて滴下した。滴下終了後、温度を20〜30℃に維持しながら1時間反応させた。さらに、50℃まで1時間かけて昇温し、同温度で4時間反応した。反応終了後、反応液を濾過して食塩を分離し、この食塩を少量のシクロペンチルメチルエーテルを用いて洗浄した。これにより、塩素原子の一部がフェノキシ基により置換された部分置換ハロゲン化シクロトリホスファゼンを含む反応液を得た。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコに4−シアノフェノール84.3g(0.708モル)を加え、これを600mlのシクロペンチルメチルエーテルに溶解した。一方、滴下ロートに液状苛性カリ(KOH39g/水39g)を仕込んだ。そして、シクロペンチルメチルエーテルの還流下において、四頚フラスコ内に滴下ロートの内容物を滴下して共沸脱水し、全量が550mlに調整された、4−シアノフェノールカリウム塩のシクロペンチルメチルエーテル溶液(第三溶液)を調製した。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第三溶液を仕込み、これにテトラブチルアンモニウムブロマイド1gを添加して溶解した。また、滴下ロートに実施例4−aで得られた反応液の全量を仕込んだ。そして、四頚フラスコの内容物の温度を25〜50℃に維持しつつ、当該内容物に対して反応液を30分かけて滴下した。滴下終了後、四頚フラスコを加熱し、シクロペンチルメチルエーテルの還流下で2時間反応したところ、この反応液のピリジン/アニリン呈色反応がマイナスになった。シクロペンチルメチルエーテルの還流下で反応をさらに4時間継続し、反応を終了した。
上述の反応工程で得られた反応液を25℃まで冷却した後に濾過し、反応で生成した塩化カリウムと未反応の4−シアノフェノールカリウム塩とを分離した。このように処理した反応液を希アルカリ水溶液と水とで数回洗浄し、洗浄後の反応液からシクロペンチルメチルエーテルを完全に回収した。残留物に含水メタノールを加えて80℃に加熱し、1時間攪拌して25℃まで冷却してから濾過したところ、結晶が得られた。得られた結晶を乾燥したところ、その重量は122gであった(収率84.2%)。この結晶の融点は107〜112℃であった。また、この結晶は、高速液体クロマトグラフィーにより分析したところ、フェノキシ−ペンタ4−シアノフェノキシ−シクロトリホスファゼン3.2%、ジフェノキシ−テトラ4−シアノフェノキシ−シクロトリホスファゼン42.6%、トリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼン43.7%およびテトラフェノキシ−ジ4−シアノフェノキシ−シクロトリホスファゼン5.0%を含むシクロトリホスファゼン誘導体混合物であり、実施例4−aにおいてオクタクロロシクロテトラホスファゼン9.0%を含むヘキサクロロシクロトリホスファゼンに対して使用したフェノールナトリウム塩と、本実施例において用いた4−シアノフェノールカリウム塩との比率に対応したシクロトリホスファゼン誘導体、すなわち、ジフェノキシ−テトラ4−シアノフェノキシ−シクロトリホスファゼンとトリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼンとを主に含むことが判明した。また、このシクロトリホスファゼン誘導体混合物は、既述の方法により残留活性ハロゲン原子量(残留活性塩素量)を測定したところ、50ppm未満であった。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコにハイドロキノンモノメチルエーテル37.2g(0.3モル)およびシクロペンチルメチルエーテル300mlを仕込んだ。一方、滴下ロートに液状苛性カリ(KOH16.8g/水17g)を仕込んだ。そして、シクロペンチルメチルエーテルの還流下において、四頚フラスコ内に滴下ロートの内容物を滴下して共沸脱水し、全量が250mlに調整された、ハイドロキノンモノメチルエーテルカリウム塩のシクロペンチルメチルエーテル溶液(第一溶液)を調製した。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第一溶液を仕込み、また、滴下ロートに第二溶液を仕込んだ。そして、第一溶液を20〜25℃に維持しながら、第一溶液に対して第二溶液を60分かけて滴下した。滴下終了後、温度を20〜30℃に維持しながら1時間反応させた。さらに、60℃まで1時間かけて昇温し、同温度で10時間反応した。反応終了後、反応液を濾過して塩化カリウムを分離し、この塩化カリウムを少量のシクロペンチルメチルエーテルを用いて洗浄した。これにより、塩素原子の一部が4−メトキシフェノキシ基により置換された部分置換ハロゲン化シクロトリホスファゼンを含む反応液を得た。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコにフェノール35.3g(0.375モル)とシクロペンチルメチルエーテル250mlとを仕込んだ。一方、滴下ロートに液状苛性カリ(KOH21g/水21g)を仕込んだ。そして、シクロペンチルメチルエーテルの還流下において、四頚フラスコ内に滴下ロートの内容物を滴下して共沸脱水し、全量が200mlに調整された、フェノールカリウム塩のシクロペンチルメチルエーテル溶液(第三溶液)を調製した。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第三溶液を仕込み、また、滴下ロートに実施例5−aで得られた反応液の全量を仕込んだ。そして、四頚フラスコの内容物の温度を30〜50℃に維持しつつ、当該内容物に対して反応液を30分かけて滴下した。滴下終了後、テトラブチルアンモニウムブロマイド1gを添加して四頚フラスコを加熱し、シクロペンチルメチルエーテルの還流下で3時間反応したところ、この反応液のピリジン/アニリン呈色反応がマイナスになった。シクロペンチルメチルエーテルの還流下で反応をさらに4時間継続し、反応を終了した。
上述の反応工程で得られた反応液を25℃まで冷却した後に濾過し、反応で生成した塩化カリウムと未反応のフェノールカリウム塩とを分離した。このように処理した反応液を希アルカリ水溶液と水とで数回洗浄し、洗浄後の反応液からシクロペンチルメチルエーテルを完全に回収したところ、69.2g(収率88%)のペースト状物が得られた。このペースト状物は、高速液体クロマトグラフィーにより分析したところ、ジフェノキシ−テトラ4−メトキシフェノキシ−シクロトリホスファゼン17.8%、トリフェノキシ−トリ4−メトキシフェノキシ−シクロトリホスファゼン70.8%およびテトラフェノキシ−ジ4−メトキシフェノキシ−シクロトリホスファゼン8.8%を含むシクロトリホスファゼン誘導体混合物であり、実施例5−aにおいてヘキサクロロシクロトリホスファゼンに対して使用したハイドロキノンモノメチルエーテルカリウム塩と、本実施例において用いたフェノールカリウム塩との比率に対応したシクロトリホスファゼン誘導体、すなわち、トリフェノキシ−トリ4−メトキシフェノキシ−シクロトリホスファゼンを主に含むことが判明した。また、このシクロトリホスファゼン誘導体混合物は、既述の方法により残留活性ハロゲン原子量(残留活性塩素量)を測定したところ、50ppm未満であった。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコにフェノール35.3g(0.375モル)とシクロペンチルメチルエーテル250mlとを仕込んだ。一方、滴下ロートに液状苛性カリ(KOH21g/水21g)を仕込んだ。そして、シクロペンチルメチルエーテルの還流下において、四頚フラスコ内に滴下ロートの内容物を滴下して共沸脱水し、全量が200mlに調整された、フェノールカリウム塩のシクロペンチルメチルエーテル溶液(第四溶液)を調製した。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第四溶液を仕込み、また、滴下ロートに実施例5−aで得られた反応液の全量を仕込んだ。そして、四頚フラスコの内容物の温度を30〜50℃に維持しつつ、当該内容物に対して反応液を30分かけて滴下した。滴下終了後、四頚フラスコを加熱してシクロペンチルメチルエーテルの還流下で1時間反応した。その後、シクロペンチルメチルエーテルを反応液の温度が120〜125℃になるまで回収し、反応液を溶融状態で10時間さらに反応させた。なお、溶融状態で反応中の反応液は、溶融開始から5時間経過時にピリジン/アニリン呈色反応がマイナスになった。
上述の反応工程で得られた反応液を100℃まで冷却した後、トルエン500mlを加えて反応生成物を抽出した。抽出液を25℃に冷却して濾過し、反応で生成した塩化カリウムと未反応のフェノールカリウム塩とを分離した。このように処理した抽出液を希アルカリ水溶液と水とで数回洗浄し、洗浄後の抽出液からトルエンを完全に回収したところ、72.3g(収率92%)のペースト状物が得られた。このペースト状物は、高速液体クロマトグラフィーにより分析したところ、ジフェノキシ−テトラ4−メトキシフェノキシ−シクロトリホスファゼン15.2%、トリフェノキシ−トリ4−メトキシフェノキシ−シクロトリホスファゼン72.4%およびテトラフェノキシ−ジ4−メトキシフェノキシ−シクロトリホスファゼン10.5%を含むシクロトリホスファゼン誘導体混合物であり、実施例5−aにおいてヘキサクロロシクロトリホスファゼンに対して使用したハイドロキノンモノメチルエーテルカリウム塩と、本実施例において用いたフェノールカリウム塩との比率に対応したシクロトリホスファゼン誘導体、すなわち、トリフェノキシ−トリ4−メトキシフェノキシ−シクロトリホスファゼンを主に含むことが判明した。また、このシクロトリホスファゼン誘導体混合物は、既述の方法により残留活性ハロゲン原子量(残留活性塩素量)を測定したところ、20ppm未満であった。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコにフェノール28.2g(0.3モル)とシクロペンチルメチルエーテル300mlとを仕込んだ。一方、滴下ロートに液状苛性カリ(KOH16.8g(0.3モル)/水17g)を仕込んだ。そして、シクロペンチルメチルエーテルの還流下において、四頚フラスコ内に滴下ロートの内容物を滴下して共沸脱水し、全量が250mlに調整された、フェノールカリウム塩のシクロペンチルメチルエーテル溶液(第一溶液)を調製した。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第一溶液を仕込み、また、滴下ロートに第二溶液を仕込んだ。そして、第一溶液を20〜25℃に維持しながら、第一溶液に対して第二溶液を60分かけて滴下した。滴下終了後、温度を20〜30℃に維持しながら1時間反応させた。さらに、50℃まで1時間かけて昇温し、テトラブチルアンモニウムブロマイド0.5gを加えて同温度で2時間反応した。反応終了後、反応液を濾過して塩化カリウムを分離し、この塩化カリウムを少量のシクロペンチルメチルエーテルを用いて洗浄した。これにより、塩素原子の一部がフェノキシ基により置換された部分置換ハロゲン化シクロトリホスファゼンを含む反応液を得た。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコに4−ヒドロキシ安息香酸エチル62.3g(0.375モル)、アセトニトリル650mlおよび水酸化カリウム21g(0.375モル)を仕込んだ。そして、これを40〜50℃で攪拌し、アセトニトリルの全量を回収した。その後、シクロペンチルメチルエーテルを200ml加え、4−ヒドロキシ安息香酸エチルカリウム塩のシクロペンチルメチルエーテル溶液(第三溶液)を調製した。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第三溶液を仕込み、また、滴下ロートに実施例6−aで得られた反応液の全量を仕込んだ。そして、四頚フラスコの内容物の温度を30〜50℃に維持しつつ、当該内容物に対して反応液を30分かけて滴下した。滴下終了後、テトラブチルアンモニウムブロマイド1gを添加し、シクロペンチルメチルエーテルの還流下で3時間反応したところ、この反応液のピリジン/アニリン呈色反応がマイナスになった。シクロペンチルメチルエーテルの還流下で反応をさらに4時間継続し、反応を終了した。
上述の反応工程で得られた反応液を25℃まで冷却した後に濾過し、反応で生成した塩化カリウムと未反応の4−ヒドロキシ安息香酸エチルカリウム塩とを分離した。このように処理した反応液を希アルカリ水溶液と水とで数回洗浄し、洗浄後の反応液からシクロペンチルメチルエーテルを完全に回収したところ、82.7g(収率96%)の無色ペースト状物質が得られた。この無色ペースト状物質は、高速液体クロマトグラフィーにより分析したところ、ジフェノキシ−テトラ4−エトキシカルボニルフェノキシ−シクロトリホスファゼン3.9%、トリフェノキシ−トリ4−エトキシカルボニルフェノキシ−シクロトリホスファゼン89.9%およびテトラフェノキシ−ジ4−エトキシカルボニルフェノキシ−シクロトリホスファゼン8.8%を含むシクロトリホスファゼン誘導体混合物であり、実施例6−aにおいてヘキサクロロシクロトリホスファゼンに対して使用したフェノールカリウム塩と、本実施例において用いた4−ヒドロキシ安息香酸エチルカリウム塩との比率に対応したシクロトリホスファゼン誘導体、すなわち、トリフェノキシ−トリ4−エトキシカルボニルフェノキシ−シクロトリホスファゼンを主に含むことが判明した。また、このシクロトリホスファゼン誘導体混合物は、既述の方法により残留活性ハロゲン原子量(残留活性塩素量)を測定したところ、50ppm未満であった。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコにハイドロキノンモノメチルエーテル38.1g(0.3075モル)、フェノール28.9g(0.3075モル)およびシクロペンチルメチルエーテル550mlを仕込んだ。一方、滴下ロートに液状苛性カリ(KOH34.4g/水34g)を仕込んだ。そして、シクロペンチルメチルエーテルの還流下において、四頚フラスコ内に滴下ロートの内容物を滴下して共沸脱水し、全量が500mlに調整された、ハイドロキノンモノメチルエーテルカリウム塩とフェノールカリウム塩とのシクロペンチルメチルエーテル溶液(第一溶液)を調製した。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第一溶液を仕込み、また、滴下ロートに第二溶液を仕込んだ。そして、第一溶液を20〜25℃に維持しながら、第一溶液に対して第二溶液を120分かけて滴下した。滴下終了後、温度を20〜30℃に維持しながら1時間反応させた。さらに、60℃まで1時間かけて昇温し、テトラブチルアンモニウムブロマイド1gを加えた。その後、シクロペンチルメチルエーテルの還流下において2時間反応させたところ、この反応液のピリジン/アニリン呈色反応がマイナスになった。
得られた反応生成物のシクロペンチルメチルエーテル溶液を25℃まで冷却した後に濾過し、反応で生成した塩化カリウムと未反応のハイドロキノンモノメチルエーテルカリウム塩およびフェノールカリウム塩を分離した。このように処理したシクロペンチルメチルエーテル溶液を希アルカリ水溶液と水とで数回洗浄し、洗浄後のシクロペンチルメチルエーテル溶液からシクロペンチルメチルエーテルを完全に回収したところ、74.4g(収率95%)のペースト状物質が得られた。このペースト状物質は、高速液体クロマトグラフィーにより分析したところ、ヘキサ4−メトキシフェノキシ−シクロトリホスファゼン0.9%、フェノキシ−ペンタ4−メトキシフェノキシ−シクロトリホスファゼン4.4%、ジフェノキシ−テトラ4−メトキシフェノキシ−シクロトリホスファゼン13.6%、トリフェノキシ−トリ4−メトキシフェノキシ−シクロトリホスファゼン49.7%、テトラフェノキシ−ジ4−メトキシフェノキシ−シクロトリホスファゼン20.7%、ペンタフェノキシ−4−メトキシフェノキシ−シクロトリホスファゼン6.3%およびヘキサフェノキシ−シクロトリホスファゼン1.1%を含むシクロトリホスファゼン誘導体混合物であり、ヘキサクロロシクロトリホスファゼンに対して使用したフェノールカリウム塩とハイドロキノンモノメチルエーテルカリウム塩との比率に対応したシクロトリホスファゼン誘導体、すなわち、トリフェノキシ−トリ4−メトキシフェノキシ−シクロトリホスファゼンを主に含むことが判明した。また、このシクロトリホスファゼン誘導体混合物は、既述の方法により残留活性ハロゲン原子量(残留活性塩素量)を測定したところ、20ppm未満であった。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコにフェノール28.9g(0.3075モル)、4−シアノフェノール36.6g(0.3075モル)、シクロペンチルメチルエーテル550mlおよびn−ヘキサン100mlを仕込んだ。一方、滴下ロートに液状苛性カリ(KOH34.4g/水34g)を仕込んだ。そして、シクロペンチルメチルエーテルとn−ヘキサンとの還流下において、四頚フラスコ内に滴下ロートの内容物を滴下して共沸脱水し、全量が500mlに調整された、フェノールカリウム塩および4−シアノフェノールカリウム塩のシクロペンチルメチルエーテル溶液(第一溶液)を調製した。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第一溶液を仕込み、また、滴下ロートに第二溶液を仕込んだ。そして、第一溶液を20〜25℃に維持しつつ、第一溶液に対して第二溶液を120分かけて滴下した。滴下終了後、温度を20〜30℃に維持しながら1時間反応した。続けて、シクロペンチルメチルエーテルの還流下においてさらに20時間反応させたところ、この反応液のピリジン/アニリン呈色反応がマイナスになった。次に、反応液を加熱し、反応液の温度が120〜125℃になるまでシクロペンチルメチルエーテルを回収した。そして、反応液を溶融状態でさらに5時間反応させた。
上述の反応工程で得られた反応液を100℃まで冷却した後、シクロペンチルメチルエーテル500mlを加えて反応生成物を抽出した。抽出液を25℃に冷却して濾過し、反応で生成した塩化カリウム、未反応のフェノールカリウム塩および4−シアノフェノールカリウム塩を分離した。このように処理した抽出液を希アルカリ水溶液と水とで数回洗浄し、洗浄後の抽出液からシクロペンチルメチルエーテルを完全に回収した。この残留物に含水メタノールを加えて80℃に加熱し、1時間撹拌して25℃まで冷却したところ、結晶が得られた。得られた結晶をろ過して乾燥したところ、その重量は72.0g(収率93.0%)であった。この結晶の融点は77.4〜99.8℃であった。また、この結晶は、高速液体クロマトグラフィーにより分析したところ、ヘキサフェノキシ−シクロトリホスファゼン3.1%、ペンタフェノキシ−4−シアノフェノキシ−シクロトリホスファゼン7.6%、テトラフェノキシ−ジ4−シアノフェノキシ−シクロトリホスファゼン5.3%、トリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼン48.9%、ジフェノキシ−テトラ4−シアノフェノキシ−シクロトリホスファゼン18.5%およびフェノキシ−ペンタ4−シアノフェノキシ−シクロトリホスファゼン6.3%を含むシクロトリホスファゼン誘導体混合物であり、ヘキサクロロシクロトリホスファゼンに対して使用したフェノールカリウム塩と4−シアノフェノールカリウム塩との比率に対応したシクロトリホスファゼン誘導体、すなわち、トリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼンを主に含むことが判明した。また、このシクロトリホスファゼン誘導体混合物は、既述の方法により残留活性ハロゲン原子量(残留活性塩素量)を測定したところ、20ppm未満であった。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコにフェノール28.9g(0.3075モル)、4−シアノフェノール36.6g(0.3075モル)、アセトニトリル550ml、n−ヘキサン100mlおよび水酸化カリウム34.4gを仕込んだ。そして、アセトニトリルおよびn−ヘキサンの還流下に共沸脱水し、シクロペンチルエチルエーテル500mlを加えてシクロペンチルエチルエーテルの沸点までアセトニトリルおよびn−ヘキサンを回収した。これにより、全量が500mlに調整された、フェノールカリウム塩および4−シアノフェノールカリウム塩のシクロペンチルエチルエーテル溶液(第一溶液)を調製した。
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコに第一溶液を仕込み、また、滴下ロートに第二溶液を仕込んだ。そして、第一溶液を20〜25℃に維持しつつ、第一溶液に対して第二溶液を120分かけて滴下した。滴下終了後、温度を20〜30℃に維持しながら1時間反応した。続けて、シクロペンチルエチルエーテルの還流下においてさらに8時間反応させたところ、この反応液のピリジン/アニリン呈色反応がマイナスになった。シクロペンチルエチルエーテルの還流下で反応をさらに4時間継続し、反応を終了した。
上述の反応工程で得られた反応液を25℃まで冷却して濾過し、反応で生成した塩化カリウム、未反応のフェノールカリウム塩および4−シアノフェノールカリウム塩を分離した。このように処理した反応液を希アルカリ水溶液と水とで数回洗浄し、洗浄後の反応液からシクロペンチルエチルエーテルを完全に回収した。この残留物に含水メタノールを加えて80℃に加熱し、1時間撹拌して25℃まで冷却したところ、結晶が得られた。得られた結晶をろ過して乾燥したところ、その重量は71.3g(収率92.7%)であった。この結晶の融点は80.5〜100.2℃であった。また、この結晶は、高速液体クロマトグラフィーにより分析したところ、ヘキサフェノキシ−シクロトリホスファゼン1.4%、ペンタフェノキシ−4−シアノフェノキシ−シクロトリホスファゼン7.4%、テトラフェノキシ−ジ4−シアノフェノキシ−シクロトリホスファゼン24.2%、トリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼン49.7%、ジフェノキシ−テトラ4−シアノフェノキシ−シクロトリホスファゼン12.5%およびフェノキシ−ペンタ4−シアノフェノキシ−シクロトリホスファゼン2.8%を含むシクロトリホスファゼン誘導体混合物であり、ヘキサクロロシクロトリホスファゼンに対して使用したフェノールカリウム塩と4−シアノフェノールカリウム塩との比率に対応したシクロトリホスファゼン誘導体、すなわち、トリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼンを主に含むことが判明した。また、このシクロトリホスファゼン誘導体混合物は、既述の方法により残留活性ハロゲン原子量(残留活性塩素量)を測定したところ、50ppm未満であった。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコにフェノール59.2g(0.629モル)を加え、これを390mlのメタノールに溶解した。一方、滴下ロートにナトリウムメトキシドを28重量%含むメタノール溶液121.5g(ナトリウムメトキシド換算で0.629モル)を仕込んだ。そして、滴下ロートの内容物を45分かけて四頚フラスコ内に滴下した。四頚フラスコの内容物を30℃で1時間攪拌し、その後、メタノールの全量を留去した。これにより、73.3gのフェノールナトリウム塩を得た。得られたフェノールナトリウム塩をテトラヒドロフラン725mlに溶解し、フェノールナトリウム塩のテトラヒドロフラン溶液(第一溶液)を得た。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第二溶液を仕込み、また、滴下ロートに第一溶液を仕込んだ。そして、第一溶液を−5〜0℃に維持しながら、第二溶液に対して第一溶液を150分かけて滴下した。滴下終了後、温度を0〜5℃に維持しながら2時間反応させた。反応終了後、反応液を濾過し、少量のテトラヒドロフランを用いて洗浄した。反応液からテトラヒドロフランの全量を除去したところ、塩素原子の一部がフェノキシ基により置換された部分置換ハロゲン化シクロトリホスファゼンを含む、126.5gの白濁ペースト状物が得られた。
(反応材料の調製)
4−シアノフェノール59.6g(0.5モル)をトリエチルアミン500mlに溶解し、4−シアノフェノールのトリエチルアミン溶液(第三液)を調製した。
(反応工程)
攪拌装置、還流冷却器および温度計を備えた1リットルのフラスコに比較例1−aで得られた白濁ペースト状物の全量を仕込み、これに第三液を加えた。そして、フラスコの内容物を還流下において10時間反応させたところ、反応液のピリジン/アニリン呈色反応がマイナスになった。
上述の反応工程の反応終了後、反応液を1,500mlの冷水中に投入し、固液分離した。そして、分離された固相を希アルカリ水溶液と水とで数回洗浄し、さらにメタノール400mlで2回洗浄して乾燥したところ、65.5gの白色結晶が得られた(収率47.2%)。この結晶の融点は60〜102℃であった。また、得られた結晶を高速液体クロマトグラフィーにより分析したところ、この結晶は、ジフェノキシ−テトラ4−シアノフェノキシ−シクロトリホスファゼン1.69%、トリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼン76.08%およびテトラフェノキシ−ジ4−シアノフェノキシ−シクロトリホスファゼン18.72%を含むシクロトリホスファゼン誘導体混合物であり、比較例1−aにおいてヘキサクロロシクロトリホスファゼンに対して使用したフェノールナトリウム塩と、本比較例において用いた4−シアノフェノールとの比率に近いシクロトリホスファゼン誘導体、すなわち、トリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼンを主に含むことが判明した。但し、このシクロトリホスファゼン誘導体混合物は、既述の方法により残留活性ハロゲン原子量(残留活性塩素量)を測定したところ、2,400ppmであり、樹脂材料の改質剤としての利用は困難であった。
(反応材料の調製)
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコにフェノール52.7g(0.56モル)を加え、これを390mlのメタノールに溶解した。一方、滴下ロートにナトリウムメトキシドを28重量%含むメタノール溶液108g(ナトリウムメトキシド換算で0.56モル)を仕込んだ。そして、滴下ロートの内容物を45分かけて四頚フラスコ内に滴下した。四頚フラスコの内容物を30℃で1時間攪拌した後、メタノールの全量を留去し、残留物を725mlのテトラヒドロフランに溶解してフェノールナトリウム塩のテトラヒドロフラン溶液(第一溶液)を調製した。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに第二溶液を仕込み、また、滴下ロートに第一溶液を仕込んだ。そして、第一溶液を−5〜0℃に維持しながら、第二溶液に対して第一溶液を150分かけて滴下した。滴下終了後、温度を0〜5℃に維持しながら2時間反応させた。反応終了後、反応液を濾過し、少量のテトラヒドロフランを用いて洗浄した。反応液からテトラヒドロフランの全量を除去したところ、塩素原子の一部がフェノキシ基により置換された部分置換ハロゲン化シクロトリホスファゼンを含む白濁ペースト状物が得られた。
(反応材料の調製)
4−シアノフェノール80g(0.67モル)をトリエチルアミン800mlに溶解し、4−シアノフェノールのトリエチルアミン溶液(第三液)を調製した。
攪拌装置、還流冷却器、滴下ロートおよび温度計を備えた1リットルの四頚フラスコに比較例2−aで得られた白濁ペースト状物の全量を加え、これを200mlのトリエチルアミンに溶解した。また、滴下ロートに第三液を仕込んだ。そして、四頚フラスコの内容物に対して第三液を滴下し、さらに、1gの4−ジメチルアミノピリジンを加えて還流下で25時間反応させたところ、ピリジン/アニリン呈色反応がマイナスになった。
上述の反応工程の反応終了後、反応液を2,000mlの冷水中に投入し、固液分離した。そして、分離された固相をトルエンに溶解し、この溶液を希アルカリ水溶液と希硫酸とで数回洗浄し、さらに水洗した。洗浄後の溶液からトルエンを除去し、残留物に対してメタノールによる精製を数回繰り返して乾燥したところ、58.2gの結晶が得られた(収率40.6%)。この結晶の融点は100〜121.8℃であった。また、得られた結晶を高速液体クロマトグラフィーにより分析したところ、この結晶は、ジフェノキシ−テトラ4−シアノフェノキシ−シクロトリホスファゼン1.90%、トリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼン93.10%およびテトラフェノキシ−ジ4−シアノフェノキシ−シクロトリホスファゼン4.90%を含むシクロトリホスファゼン誘導体混合物であり、比較例2−aにおいてヘキサクロロシクロトリホスファゼンに対して使用したフェノールナトリウム塩と、本比較例において使用した4−シアノフェノールとの比率に対応したシクロトリホスファゼン誘導体、すなわち、トリフェノキシ−トリ4−シアノフェノキシ−シクロトリホスファゼンを主に含むことが判明した。但し、このシクロトリホスファゼン誘導体混合物は、既述の方法により残留活性ハロゲン原子量(残留活性塩素量)を測定したところ、800ppmであり、樹脂材料の改質剤としての利用は困難であった。
攪拌装置、還流冷却器、滴下ロート及び温度計を備えた1リットルの四頚フラスコに4−シアノフェノール20.9g(0.175モル)、フェノール16.5g(0.175モル)およびトルエン135mlを仕込んだ。そして、四頚フラスコの内容物を攪拌しながら固形水酸化ナトリウム14g(0.35モル)を添加して還流下において反応させ、6時間かけて、生成した水を共沸脱水により除去した。その後、反応液を20℃まで冷却した。
エポキシ樹脂(JER株式会社の商品名“エピコート828”)、変性ポリフェニレンエーテル樹脂(日本G.E.プラスチックス株式会社の商品名“ノリル640−111”)、硬化剤(ジアミノジフェニルメタン)並びに実施例1−b、実施例2−b、実施例3−bおよび実施例4−b(以下、これらの実施例を「評価実施例」と云う)のいずれかにおいて得られたシクロトリホスファゼン誘導体混合物を表1に示す割合で混合し、エポキシ樹脂組成物を調製した(試料1〜4)。また、比較のため、シクロトリホスファゼン誘導体混合物を添加せずに、同様のエポキシ樹脂組成物を調製した(試料5)。さらに、比較のため、評価実施例で得られたシクロトリホスファゼン誘導体混合物に代えてケミプロ化成株式会社の商品名“KD−302S”(ホスファゼン系難燃剤)を用いて同様のエポキシ樹脂組成物を調製した(試料6)。
Claims (13)
- 下記の一般式(1a)で示されるヘキサハロゲン化シクロトリホスファゼンと、下記の一般式(2)で示される有機塩とを沸点が100℃以上の非水溶性エーテル系溶媒中において反応させる工程を含み、
前記非水溶性エーテル系溶媒が下記の一般式(5)で示されるシクロペンチルアルキルエーテルである、
シクロトリホスファゼン誘導体の製造方法。
(一般式(1a)において、Xはハロゲン原子を示す。)
(一般式(2)において、M1はアルカリ金属を示し、Y1は次の一般式(3)または(4)で示される基である。
一般式(3)および(4)において、R1は、水素、アルキル基、シアノ基、アルコキシ基、アラルコキシ基またはカルボニル基含有基を示す。)
(一般式(5)中、Rはアルキル基を示す。) - 反応系から前記非水溶性エーテル系溶媒を除去し、反応生成物を溶融状態で反応させる工程をさらに含む、請求項1に記載のシクロトリホスファゼン誘導体の製造方法。
- 前記ヘキサハロゲン化シクロトリホスファゼンが15重量%未満の割合でオクタハロゲン化シクロテトラホスファゼンを含む、請求項1または2に記載のシクロトリホスファゼン誘導体の製造方法。
- 前記非水溶性エーテル系溶媒に対し、次の一般式(6)で示される四級アンモニウム塩および次の一般式(7)で示される四級アンモニウムハライドのうちの少なくとも一つを添加する、請求項1から3のいずれかに記載のシクロトリホスファゼン誘導体の製造方法。
(一般式(6)および(7)において、R3はアルキル基またはアラルキル基を、Xはハロゲン原子をそれぞれ示し、また、Y1は、前記一般式(2)のY1と同じである。) - 前記ヘキサハロゲン化シクロトリホスファゼン1.0モルに対し、前記有機塩を6.0モル未満の割合で用いる、請求項1から4のいずれかに記載のシクロトリホスファゼン誘導体の製造方法。
- 前記ヘキサハロゲン化シクロトリホスファゼン1.0モルに対し、前記有機塩を6.0モル以上の割合で用いる、請求項1から4のいずれかに記載のシクロトリホスファゼン誘導体の製造方法。
- 前記有機塩として、前記一般式(2)のY1が異なる二種類以上の有機塩の混合物を用いる、請求項6に記載のシクロトリホスファゼン誘導体の製造方法。
- 前記有機塩の混合物は、前記一般式(2)のY1がフェニル基である第一の有機塩と、前記一般式(2)のY1が4−シアノフェニル基である第二の有機塩との混合物である、請求項7に記載のシクロトリホスファゼン誘導体の製造方法。
- 前記有機塩の混合物は、前記ヘキサハロゲン化シクロトリホスファゼン1.0モルに対し、前記第一の有機塩を2.5〜3.5モル含み、前記第二の有機塩を少なくとも3.0モル含む、請求項8に記載のシクロトリホスファゼン誘導体の製造方法。
- 下記の一般式(1b)で示される部分置換ハロゲン化シクロトリホスファゼンと、下記の一般式(2)で示される有機塩とを沸点が100℃以上の非水溶性エーテル系溶媒中において反応させる工程を含み、
前記非水溶性エーテル系溶媒が下記の一般式(5)で示されるシクロペンチルアルキルエーテルである、
シクロトリホスファゼン誘導体の製造方法。
(一般式(1b)において、Zは、ハロゲン原子または下記の一般式(8a)若しくは一般式(8b)で示されるオキシアリール基を示し、少なくとも一つがハロゲン原子である。
一般式(8a)および一般式(8b)において、R4は、水素、アルキル基、シアノ基、アルコキシ基、アラルコキシ基またはカルボニル基含有基を示す。)
(一般式(2)において、M1はアルカリ金属を示し、Y1は次の一般式(3)または(4)で示される基である。
一般式(3)および(4)において、R1は、水素、アルキル基、シアノ基、アルコキシ基、アラルコキシ基またはカルボニル基含有基を示す。)
(一般式(5)中、Rはアルキル基を示す。) - 反応系から前記非水溶性エーテル系溶媒を除去し、反応生成物を溶融状態で反応させる工程をさらに含む、請求項10に記載のシクロトリホスファゼン誘導体の製造方法。
- 前記非水溶性エーテル系溶媒に対し、次の一般式(6)で示される四級アンモニウム塩および次の一般式(7)で示される四級アンモニウムハライドのうちの少なくとも一つを添加する、請求項10または11に記載のシクロトリホスファゼン誘導体の製造方法。
(一般式(6)および(7)において、R3はアルキル基またはアラルキル基を、Xはハロゲン原子をそれぞれ示し、また、Y1は、前記一般式(2)のY1と同じである。) - 前記部分置換ハロゲン化シクロトリホスファゼンに含まれる全ハロゲン原子を前記一般式(2)に由来の−OY1基で置換するために必要な量の前記有機塩を用いる、請求項10から12のいずれかに記載のシクロトリホスファゼン誘導体の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005258765A JP4635156B2 (ja) | 2004-09-27 | 2005-09-07 | シクロトリホスファゼン誘導体の製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004279025 | 2004-09-27 | ||
| JP2005258765A JP4635156B2 (ja) | 2004-09-27 | 2005-09-07 | シクロトリホスファゼン誘導体の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2006117638A JP2006117638A (ja) | 2006-05-11 |
| JP4635156B2 true JP4635156B2 (ja) | 2011-02-16 |
Family
ID=36535864
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005258765A Expired - Fee Related JP4635156B2 (ja) | 2004-09-27 | 2005-09-07 | シクロトリホスファゼン誘導体の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4635156B2 (ja) |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5578495B2 (ja) * | 2008-10-30 | 2014-08-27 | 株式会社伏見製薬所 | 難燃性樹脂組成物 |
| CN110759947A (zh) * | 2019-11-07 | 2020-02-07 | 山东省海洋化工科学研究院 | 一种六苯氧基环三磷腈的合成方法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0380616A4 (en) * | 1988-06-23 | 1990-09-05 | The Government Of The United States Of America As Represented By The Administrator Of The National Aeronautics | Process for preparing isomeric trisaryloxycyclotriphosphazene polymer precursors and intermediates |
| JPH11181429A (ja) * | 1997-02-14 | 1999-07-06 | Otsuka Chem Co Ltd | 難燃剤、難燃性樹脂組成物及び難燃性樹脂成形体 |
| JP3053617B1 (ja) * | 1999-01-05 | 2000-06-19 | 大塚化学株式会社 | ホスホニトリル酸エステルの製造法 |
| JP2001192392A (ja) * | 2000-01-11 | 2001-07-17 | Chemiprokasei Kaisha Ltd | 環状ホスファゼン化合物、環状ホスファゼン混合組成物、ホスファゼン組成物、それを有効成分とする難燃剤およびそれを含む難燃性組成物 |
| JP2002363193A (ja) * | 2001-06-05 | 2002-12-18 | Chemiprokasei Kaisha Ltd | 環状ホスファゼン類、その製造方法、それを有効成分とする難燃剤およびそれを含む樹脂組成物と成形品 |
-
2005
- 2005-09-07 JP JP2005258765A patent/JP4635156B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2006117638A (ja) | 2006-05-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2078722B1 (en) | Novel sulfonium borate complex | |
| CN1210284C (zh) | 基于双酚a双(磷酸二苯酯)的阻燃剂 | |
| US10329231B2 (en) | Epoxy compound, method for producing the same, epoxy resin composition, and cured product thereof | |
| EP1905756A1 (en) | Silver beta-ketocarboxylate, material comprising the same for forming silver metal, and use thereof | |
| JP2009024167A (ja) | ビニル化合物の製造方法 | |
| US8779162B2 (en) | Cyanate ester compounds and cured products thereof | |
| JP6138607B2 (ja) | エポキシ樹脂及び該エポキシ樹脂の製造方法 | |
| WO2014185485A1 (ja) | ビフェニル骨格含有エポキシ樹脂の製造方法 | |
| JP4635156B2 (ja) | シクロトリホスファゼン誘導体の製造方法 | |
| JP4635155B2 (ja) | シクロトリホスファゼン誘導体の製造方法 | |
| JP4635157B2 (ja) | シクロトリホスファゼン誘導体の製造方法 | |
| JP6512970B2 (ja) | 6−ヒドロキシ−2−ナフトエ酸グリシジルエーテルエステルの精製方法 | |
| JP2009107991A (ja) | 新規なヒドロキシメチル置換又はアルコキシメチル置換ビスフェノール化合物 | |
| JP6190256B2 (ja) | 新規なビス(ヒドロキシフェニル)ベンゾオキサゾール化合物 | |
| CN110002967A (zh) | 卤化物的制造方法、钾盐的制造方法、及钾盐 | |
| US7960456B2 (en) | Halogenated phosphonates, processes for their preparation and their use as flame retardants | |
| US9249169B2 (en) | Process for the preparation of phosphonium sulfonates | |
| JP4822651B2 (ja) | フェノール金属塩の製造方法 | |
| WO2003089442A1 (en) | A method of preparing phosphoric ester | |
| JP3621996B2 (ja) | エポキシ樹脂およびその製造方法 | |
| KR20230052507A (ko) | 2-시아노에틸기를 포함하는 유기화합물 및 이의 제조방법 | |
| CN102256986B (zh) | 精制而成的o-(2,6-二氯-4-甲基苯基)-o,o-二甲基硫代磷酸酯的制造方法 | |
| WO2014171358A1 (ja) | 難燃剤組成物ならびにそれを含有する難燃性樹脂組成物および成形体 | |
| JP2005272632A (ja) | シリル基含有ヨウ素化エポキシ化合物及びその製造方法 | |
| JP2007291219A (ja) | 芳香族ジメチレン−フェノール樹脂及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080731 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20100311 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100323 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100519 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100928 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20101021 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131203 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4635156 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |