JP4580234B2 - 表面活性炭水化物脂肪酸エステルを製造するためのトランスアシドリシスプロセス - Google Patents
表面活性炭水化物脂肪酸エステルを製造するためのトランスアシドリシスプロセス Download PDFInfo
- Publication number
- JP4580234B2 JP4580234B2 JP2004517462A JP2004517462A JP4580234B2 JP 4580234 B2 JP4580234 B2 JP 4580234B2 JP 2004517462 A JP2004517462 A JP 2004517462A JP 2004517462 A JP2004517462 A JP 2004517462A JP 4580234 B2 JP4580234 B2 JP 4580234B2
- Authority
- JP
- Japan
- Prior art keywords
- fatty acid
- carbohydrate
- acid ester
- producing
- unreacted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- -1 carbohydrate fatty acid esters Chemical class 0.000 title claims description 89
- 239000000194 fatty acid Substances 0.000 title claims description 78
- 235000014113 dietary fatty acids Nutrition 0.000 title claims description 77
- 229930195729 fatty acid Natural products 0.000 title claims description 77
- 238000000034 method Methods 0.000 title claims description 36
- 230000008569 process Effects 0.000 title claims description 28
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical group CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 claims description 27
- 239000002904 solvent Substances 0.000 claims description 24
- 238000004519 manufacturing process Methods 0.000 claims description 21
- 238000006243 chemical reaction Methods 0.000 claims description 20
- 239000011541 reaction mixture Substances 0.000 claims description 20
- 235000021588 free fatty acids Nutrition 0.000 claims description 18
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 claims description 16
- 150000001720 carbohydrates Chemical class 0.000 claims description 16
- 239000003377 acid catalyst Substances 0.000 claims description 13
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 claims description 13
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 claims description 12
- 238000001816 cooling Methods 0.000 claims description 10
- 150000002772 monosaccharides Chemical class 0.000 claims description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 6
- 230000007062 hydrolysis Effects 0.000 claims description 6
- 238000006460 hydrolysis reaction Methods 0.000 claims description 6
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 6
- 229920001296 polysiloxane Polymers 0.000 claims description 6
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 5
- 150000002016 disaccharides Chemical class 0.000 claims description 5
- 238000001556 precipitation Methods 0.000 claims description 5
- 238000000746 purification Methods 0.000 claims description 5
- 150000004043 trisaccharides Chemical class 0.000 claims description 5
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 claims description 4
- 238000000605 extraction Methods 0.000 claims description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 4
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 claims description 3
- 239000002253 acid Substances 0.000 claims description 3
- 239000003960 organic solvent Substances 0.000 claims description 3
- 230000001376 precipitating effect Effects 0.000 claims description 3
- 125000003843 furanosyl group Chemical group 0.000 claims description 2
- 125000003132 pyranosyl group Chemical group 0.000 claims description 2
- 230000010933 acylation Effects 0.000 claims 4
- 238000005917 acylation reaction Methods 0.000 claims 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical group OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 claims 2
- 125000002252 acyl group Chemical group 0.000 claims 2
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 claims 2
- 229940126062 Compound A Drugs 0.000 claims 1
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 claims 1
- 125000004122 cyclic group Chemical group 0.000 claims 1
- 238000010791 quenching Methods 0.000 claims 1
- 230000000171 quenching effect Effects 0.000 claims 1
- 238000000638 solvent extraction Methods 0.000 claims 1
- 239000011877 solvent mixture Substances 0.000 claims 1
- 235000014633 carbohydrates Nutrition 0.000 description 44
- 239000000047 product Substances 0.000 description 32
- 150000004665 fatty acids Chemical class 0.000 description 14
- 229930006000 Sucrose Natural products 0.000 description 13
- 239000005720 sucrose Substances 0.000 description 13
- 239000000376 reactant Substances 0.000 description 12
- MUPFEKGTMRGPLJ-RMMQSMQOSA-N Raffinose Natural products O(C[C@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O[C@@]2(CO)[C@H](O)[C@@H](O)[C@@H](CO)O2)O1)[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 MUPFEKGTMRGPLJ-RMMQSMQOSA-N 0.000 description 11
- MUPFEKGTMRGPLJ-UHFFFAOYSA-N UNPD196149 Natural products OC1C(O)C(CO)OC1(CO)OC1C(O)C(O)C(O)C(COC2C(C(O)C(O)C(CO)O2)O)O1 MUPFEKGTMRGPLJ-UHFFFAOYSA-N 0.000 description 11
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 10
- MUPFEKGTMRGPLJ-ZQSKZDJDSA-N raffinose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO[C@@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)O)O1 MUPFEKGTMRGPLJ-ZQSKZDJDSA-N 0.000 description 10
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 9
- 230000020176 deacylation Effects 0.000 description 8
- 238000005947 deacylation reaction Methods 0.000 description 8
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 8
- 229960004592 isopropanol Drugs 0.000 description 7
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 7
- 239000002244 precipitate Substances 0.000 description 7
- 238000005292 vacuum distillation Methods 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 229940070765 laurate Drugs 0.000 description 6
- 229940049964 oleate Drugs 0.000 description 6
- 239000003054 catalyst Substances 0.000 description 5
- NAYZMSNLBRPCBX-SSPAHAAFSA-N dodecanoic acid;(2r,3s,4r,5r)-2,3,4,5,6-pentahydroxyhexanal Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O.CCCCCCCCCCCC(O)=O NAYZMSNLBRPCBX-SSPAHAAFSA-N 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- 239000000203 mixture Substances 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 238000006467 substitution reaction Methods 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- 239000005639 Lauric acid Substances 0.000 description 4
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 4
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 4
- 150000002190 fatty acyls Chemical group 0.000 description 4
- 239000012456 homogeneous solution Substances 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 238000004064 recycling Methods 0.000 description 4
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 150000001735 carboxylic acids Chemical class 0.000 description 3
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- ZIJKGAXBCRWEOL-UHFFFAOYSA-N sucrose octaacetate Chemical compound CC(=O)OC1C(OC(C)=O)C(COC(=O)C)OC1(COC(C)=O)OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(COC(C)=O)O1 ZIJKGAXBCRWEOL-UHFFFAOYSA-N 0.000 description 3
- 230000002194 synthesizing effect Effects 0.000 description 3
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 2
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 2
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- 239000005642 Oleic acid Substances 0.000 description 2
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 239000003814 drug Substances 0.000 description 2
- 238000006317 isomerization reaction Methods 0.000 description 2
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 2
- 229920000728 polyester Polymers 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 238000005809 transesterification reaction Methods 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- 208000019399 Colonic disease Diseases 0.000 description 1
- 239000004129 EU approved improving agent Substances 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 208000035150 Hypercholesterolemia Diseases 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- ZIJKGAXBCRWEOL-SAXBRCJISA-N Sucrose octaacetate Chemical compound CC(=O)O[C@H]1[C@H](OC(C)=O)[C@@H](COC(=O)C)O[C@@]1(COC(C)=O)O[C@@H]1[C@H](OC(C)=O)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O1 ZIJKGAXBCRWEOL-SAXBRCJISA-N 0.000 description 1
- 239000001083 [(2R,3R,4S,5R)-1,2,4,5-tetraacetyloxy-6-oxohexan-3-yl] acetate Substances 0.000 description 1
- 239000001344 [(2S,3S,4R,5R)-4-acetyloxy-2,5-bis(acetyloxymethyl)-2-[(2R,3R,4S,5R,6R)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxyoxolan-3-yl] acetate Substances 0.000 description 1
- UAOKXEHOENRFMP-ZJIFWQFVSA-N [(2r,3r,4s,5r)-2,3,4,5-tetraacetyloxy-6-oxohexyl] acetate Chemical compound CC(=O)OC[C@@H](OC(C)=O)[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](OC(C)=O)C=O UAOKXEHOENRFMP-ZJIFWQFVSA-N 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 229940040526 anhydrous sodium acetate Drugs 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 201000001883 cholelithiasis Diseases 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- JZWRVBFHEOUCDY-KNRYUCGFSA-N dodecanoic acid (2R,3S,4R,5R)-2,3,4,5,6-pentahydroxyhexanal Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O.CCCCCCCCCCCC(O)=O.CCCCCCCCCCCC(O)=O JZWRVBFHEOUCDY-KNRYUCGFSA-N 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000011067 equilibration Methods 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 235000019387 fatty acid methyl ester Nutrition 0.000 description 1
- 125000001924 fatty-acyl group Chemical group 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 150000002303 glucose derivatives Chemical class 0.000 description 1
- 229910001385 heavy metal Inorganic materials 0.000 description 1
- 230000000749 insecticidal effect Effects 0.000 description 1
- 229910052745 lead Inorganic materials 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000000197 pyrolysis Methods 0.000 description 1
- 238000000275 quality assurance Methods 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- 238000001223 reverse osmosis Methods 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 239000000344 soap Substances 0.000 description 1
- 238000000859 sublimation Methods 0.000 description 1
- 230000008022 sublimation Effects 0.000 description 1
- 229940013883 sucrose octaacetate Drugs 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- 238000000108 ultra-filtration Methods 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Images

Landscapes
- Saccharide Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
【0001】
発明の背景
(a)本発明の分野
本発明は、表面活性炭水化物脂肪酸エステル、特に、減圧下および酸触媒の存在下での、低分子量カルボン酸と遊離脂肪酸との低温での無溶媒トランスアシドリシス反応に関する。
【背景技術】
【0002】
(b)従来技術の説明
炭水化物脂肪酸エステルは、優れた生分解性を有する非刺激性、非イオン界面活性剤である。それは、膜タンパク質を可溶化し、多くのグレードの洗剤、薬剤、食品および化粧品を作るために使用される。炭水化物脂肪酸エステルは、治療薬としても使用される。1998年4月発行のヨコヤマおよびヨネダによる米国特許第5,739,117号には、脳代謝改善薬としてのグルコースエステルの使用が記載されている。炭水化物エステルは、胆石症(米国特許第4,264,583号)、結腸疾患(米国特許第5,840,860号)および高コレステロール血症(米国特許第4,241,054号)の治療にも使用することができる。それらは、抗菌活性および殺虫活性を示すことも知られている。しかしながら、炭水化物脂肪酸エステルの合成方法は多くの制限に直面している。
【0003】
溶媒および非溶媒環境におけるショ糖脂肪酸エステルの合成法;以下のバッチ式または連続反応方式が現在存在する。従来技術は、米国特許第4,614,718号、同第4,927,920号;同第4,966,966号;同第4,968,791号;同第4,996,309号;同第5,043,438号;および同第5,908,922号に記述されている。しかしながら、炭水化物脂肪エステルを製造するための、従来技術で記載の従来の方法は、以下の不利点および制限に直面している:
【0004】
(1)炭水化物脂肪エステルの大部分の工業生産者によって現在支持されているエステル交換反応法は、基質の糖および脂肪酸部位が、極性の差異のために不混和性であることから、不十分な転化平衡を有する可逆過程である。したがって、そのプロセスは、低い生成物収率によって特徴付けられる;
(2)優れた安全な保証を有する相互溶媒(mutual solvent)を見つけること。ピリジン、N,N−ジメチルホルムアミド(DMF)、ジメチルアセトアミド(DMA)、ジメチルスルホキシド(DMSO)、クロロホルム、ベンゼンおよびトルエンなどの相互溶媒が基質を可溶化するのに必要である。これらの相互溶媒は毒性があり、現在の規則に準ずるレベルまで除去することができず、このため、生成物の用途が制限される。
(3)複雑かつ費用のかかる生成物精製手順。
(4)公知の大部分のプロセスに通常必要とされる、100℃を超える温度での反応物炭水化物の熱分解および異性化。
(5)特に酵素的プロセスにおける、高い触媒添加。
【0005】
米国特許第5,945,519号には、ショ糖脂肪酸エステルの無溶媒製造プロセスならびに非糖ポリオール脂肪酸エステとのそれらの混合物が記載されている。このプロセスでは、炭素原子6〜20個の鎖長を有する1種または複数種の脂肪酸アルキルエステルとショ糖を温度120〜160℃で反応させ、次いで、その反応混合物を脂肪酸アルキルエステルと減圧で反応させ、次いで溶媒を添加することなく濾過する。
【0006】
毒性相互溶媒の使用に付随する問題に取り組むため、米国特許第4,996,309号では、水性反応系においてアルカリ触媒の存在下にて、ショ糖と脂肪酸アルキルエステルとを反応させることによってショ糖脂肪酸エステルを製造する方法が開示されている。触媒によって、生成物において大量の石鹸が生じる。したがって、逆浸透および限外濾過ステップを要し、分離および精製が面倒になり、費用がかかる。その場合でさえ、通常得られる生成物純度は、わずか70%である。
【0007】
米国特許第5,872,245号には、固定(stationary)エステル交換反応触媒の存在下にて、「担体」溶媒としての限定量のメタノール中で、ショ糖を脂肪酸メチルエステルと反応させることによってショ糖脂肪酸エステルを合成し、機械的乳化するための連続プロセスが記載されている。このプロセスでは、触媒量の水酸化ナトリウムの他に、Zn、Cu、SnおよびPbなどの重金属触媒が用いられる。生成物の分離は、密度の区別(density differentiation)によって達成される。
【0008】
しかしながら、このプロセスは、水酸化ナトリウムの存在および熱によって誘導される色および反事実(antifact)を示さない。さらに、6種類を超える同様な生成物の生成物混合物と反応物との有効な密度の区別によって、プロセスサイクルにおけるホールドアップ時間が長くなる。モノ、ジ、トリおよびポリエステルの形成は選択的に制御することができず、したがって、プロセスで形成される「過剰エステル化」ポリオールが蓄積する。
【0009】
本発明の主な目的は、表面活性脂肪酸エステルを製造するための無溶媒トランスアシドリシスプロセスであって、反応中に溶媒を添加せず、エステル化を迅速に進め100℃未満の温度でポリエステルを形成し、必要であれば、一部脱アシル化して親水性親油性比(HLB)値が1〜16の目的の生成物を形成することによって、上記の欠点および/または制限を無くしたプロセスを提供することである。
【発明の開示】
【課題を解決するための手段】
【0010】
本発明の概要
したがって、本発明は、表面活性炭水化物脂肪酸エステルを製造するためのトランスアシドリシスプロセスであって、減圧下および酸触媒存在下での、低分子量カルボン酸(C2またはC3)および遊離脂肪酸(C6〜C22)の炭水化物エステルの間の低温でのトランスアシドリシス反応を含むプロセスを提供する。
【0011】
一態様において、本発明は:
(a)酸触媒の存在下にて、減圧下でアシル化炭水化物を遊離脂肪酸と反応させるステップと、
(b)再循環のために、ステップ(a)で得られた反応混合物から、未反応脂肪酸を脱色し、抽出または結晶化するステップと、
(c)再循環のために、ステップ(a)で得られた反応混合物から、未反応アシル化炭水化物を沈殿させるステップと、
(d)炭水化物脂肪酸エステル層を回収するステップと、
を含む、表面活性炭水化物脂肪酸エステルを製造するための無溶媒トランスアシドリシスプロセスを提供する。
【0012】
ステップ(a)での反応は、反応混合物に溶媒を添加することなく行われる。その反応は、60〜95℃の温度範囲で行われることが好ましい。ステップ(c)での未反応アシル化炭水化物の沈殿は、炭水化物脂肪酸エステル層を−4〜10℃の温度に冷却することによって達成される。
【0013】
表面活性炭水化物脂肪酸エステルを製造するトランスアシドリシスを提供することによって、本発明に従って、親水性親油性比(HLB値)1〜10を有する、C2またはC3−アシル化炭水化物のモノ、ジおよびポリ脂肪酸エステルが、脂肪酸部位の置換度および鎖長に応じて得ることができる。
【0014】
本発明のその他の態様において、HLB値8〜16の遊離ヒドロキシル基を有する炭水化物脂肪酸エステルが、酸触媒の存在下での部分加水分解の更なるステップによって得ることができる。
【0015】
本発明の他の態様では、未反応遊離脂肪酸および低分子量カルボン酸の未反応炭水化物エステルを精製中に除去し、開始反応物混合物に再循環する。
【0016】
従来技術の上記の制限のうちの少なくとも1つを避ける、本発明によるプロセスの利点は:
(1)反応媒体としての溶媒を避けることである。反応物炭水化物に結合されたC2−またはC3−アシル基は、優れた保護基および脱離基であり、それと同時に、脂肪酸において炭水化物部位の溶解性を高める;
(2)低いエネルギー要求量で反応平衡に前進させるために低い圧力を用いること。この結果、高い収率(>90%)が得られ、生成物の異性化および分解が少なくなる。
(3)供給原料は、再生可能天然資源であり、容易に入手可能であり、かつ安価である。未反応基質は再循環される。したがって、このプロセスは工業的に存立可能である。
(4)本発明は、バッチ式および連続式炭水化物脂肪酸エステルプロセスのどちらにも適用可能である。
(5)高いグレード(純度98%)の炭水化物脂肪酸エステルが得られる。
(6)このプロセスは、多くの炭水化物脂肪酸エステルの製造に対応可能である。
【0017】
本発明の範囲を制限することなく、本発明の他の目的および利点は、以下の詳細な説明および添付の図面を参照すれば明らかになるだろう。
【発明を実施するための最良の形態】
【0018】
詳細な説明
本発明の炭水化物脂肪酸エステルを製造するためのプロセスに従って、好ましい反応物C2−またはC3−アシル化炭水化物としては、単糖ないし三糖炭水化物が挙げられ、好ましい遊離脂肪酸は、ゼロ、一価または二価不飽和を有するC6−C22の鎖長を含む。
【0019】
本発明のプロセスにおいて、酸触媒としては、単糖の場合には、硫酸およびカンファースルホン酸;二糖および三糖の場合には、三フッ化ホウ素ジエチルエーテラート、アルキルスルホン酸ポリシロキサンおよびポリシロキサンおよびトシル酸が挙げられる。
【0020】
本発明のプロセスの炭水化物脂肪エステルは以下の化学構造:
【化1】
【0021】
ステップ1:
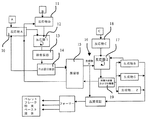
図1を参照すると、反応器1(12)において、C2−またはC3−アシル化炭水化物、反応物A、10、(好ましくは、単糖ないし三糖)をC6〜C22遊離脂肪酸(反応物B、11)、11と反応させる。反応は、酸触媒の存在下で、温度範囲60〜95℃(好ましくは、単糖の場合には80〜90℃、二糖および三糖の場合には60〜75℃)で、範囲4〜20トル(好ましくは、5〜10トル)の減圧下にて、いずれの溶媒も添加することなく、絶え間なく攪拌しながら行われる。
【0022】
その反応は3〜6時間続けられ、その後にそれを止め、次いで、イソプロパノール、n−プロパノール、酢酸エチルまたはエタノールなどの有機溶媒中で、反応混合物を取り上げる。
【0023】
トランスアシドリシスの副生成物である、低分子量C2−またはC3−カルボン酸は、凝縮によって捕集され、他の用途のために保管しておく。
ステップ2:
次いで、反応混合物を脱色装置(decoloriser)13に通し、そこで、活性炭などの吸着剤と接触することによって、反応混合物は脱色され、次いで濾過される。
次いで、未反応遊離脂肪酸(反応物B、11)および未反応アシル化炭水化物(反応物A、10)を回収し、以下に記述するようにステップ3またはステップ4のいずれかによって再循環される:
ステップ3:
【0024】
ステップ2の溶媒は、蒸留器15において、温度30〜50℃で真空蒸留することによって除去し、次いで未反応脂肪酸をヘキサンで抽出する。次いで、温度30〜50℃でヘキサンを真空蒸留し、未反応脂肪酸を回収する。残りの反応混合物をステップ2で使用した溶媒中に再溶解して30〜40%(w/v)溶液が形成され、冷却器分離器(chiller separator)14において−4〜10℃に冷却し、未反応アシル化炭水化物沈殿物が生じる。次いで、濾液溶媒を真空蒸留により除去し、生成物のアシル化炭水化物脂肪酸エステル(生成物A、16)が得られる。回収された未反応脂肪酸(反応物B、11)およびアシル化炭水化物(反応物A、10)は、反応器1に再循環するために保管しておく。
【0025】
ステップ4:
ステップ3の代替方法としては、ステップ2の溶媒を水10〜20%(V/V)でスパイキング(spiking)し、冷却器分離器14でその溶媒を8〜12℃に冷却することによって、未反応脂肪酸を結晶化する。次いで、その溶液をさらに−4〜0℃に冷却することによって、未反応アシル化炭水化物を沈殿物として除去する。次いで、濾液溶媒を真空蒸留により除去し、アシル化炭水化物脂肪酸エステル生成物(生成物A、16)が得られる。回収された未反応脂肪酸およびアシル化炭水化物は、反応器1(12)に再循環するために保管しておく。
【0026】
ステップ5
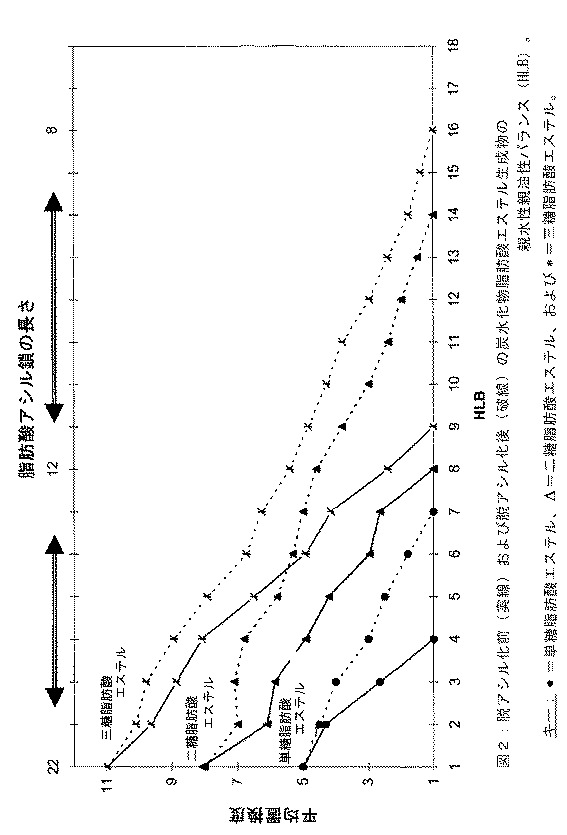
生成物の親水性親油性比(HLB)プロファイルを望ましいレベルに改変するために、ステップ3または4で得られたアシル化炭水化物脂肪酸エステル(生成物A、16)を第2反応器(反応器2(17))に通し、そこで、脂肪酸アシル基の置換、脱アシル化および鎖長の程度に応じて、酸触媒(反応物C、18)の存在下で部分加水分解することによって遊離ヒドロキシル基を釣り合わせ、必要なHLBプロファイルが得られる。1〜16にわたる生成物のHLBプロファイルの詳細は図2に示される。
【0027】
加水分解中に釣り合わすことができる遊離脂肪酸は、冷却器−分離器19のステップ(Stage Cooler-Separator)、で結晶化することによって除去され、反応器1(12)に再循環される。
炭水化物脂肪酸エステルにおける脱アシル化の程度および脂肪酸アシル鎖の長さを変えることによって、図2で詳述されるように、異なるHLB値1〜16を有する生成物(生成物B、C、D...)を製造することができる。これらの生成物は様々な溶解性および昇華温度を有し、したがって、存在するエステルに応じて、10〜−15℃の温度に段階的に冷却することによって、炭水化物脂肪酸エステルに分離される。
【0028】
ステップ6:
段階的な冷却によって生成物を分離した後、次いで、品質保証に通し、下流ユーザーに供給するために、フォーマーに通してペレット、フレーク、ペーストまたは液体などの所望の形状にすることができる。
【0029】
以下の実施例によって、本発明がさらに説明されるだろう。
【実施例】
実施例1:アセチル化グルコースラウレートおよび部分脱アシル化グルコースラウレートの製造
a)アセチル化グルコースラウレートの製造
電磁スターラー、活栓、液体窒素冷トラップに通じる真空引取り(vacuum take-off)ラインおよび真空ポンプを備えた三つ口丸底フラスコに、ラウリン酸(Fluka Chemika社,スイス)0.03mol(6.00g)を添加した。グルコースペンタアセテート、GPA(Fluka Chemika社,スイス)0.01mol(3.90g)を添加し、均一な溶液が形成するまで、その混合物を絶え間なく攪拌しながら油浴において80〜100℃に加熱した。次いで、濃硫酸(または0.2%カンファースルホン酸)0.1%(w/w)(0.01g)を添加し、圧力5〜10トルをかけた。反応を3〜6時間続けた。次いで、その反応を止め、n−プロパノール100mlで採取した。次いで、溶液を1M NaHCO3水溶液で中和し、活性炭1.0gで脱色し、温かいまま濾過した。次いで、N−プロパノールを真空蒸留によって除去した。
【0030】
未反応ラウリン酸を50×4mlヘキサンで抽出した。ヘキサンを真空蒸留して、未反応のラウリン酸を回収し、再循環のために保管しておいた。グルコース脂肪酸エステルを温かいn−プロパノール中に再溶解し、30〜40%(w/v)溶液が形成され、−4〜0℃に冷却し、濾過して、沈殿物として未反応GPAを得た。次いで、濾液であるn−プロパノールを真空蒸留により除去し、純度70〜85%のアセチル化グルコースラウレートを得た。脂肪酸の抽出およびGPAの沈殿を繰り返し行うことによって、純度90〜95%の生成物(収率90.2%)が得られた。生成物のHLBプロファイルの詳細を図2に示し、その図で、脱アシル化前(実線)および脱アシル化後(破線)の炭水化物脂肪酸エステル生成物の親水性親油性バランス(HLB)が示される。
キー(KEY):・=単糖脂肪酸エステル、△=二糖脂肪酸エステル、および*=三糖脂肪酸エステル。
【0031】
代替方法としては、図1に示されていない反応において、n−プロパノール溶液を水10〜20%(V/V)でスパイキングし、その溶液を8〜12℃に冷却することによって、未反応脂肪酸を結晶化し、次いでその溶液をさらに−4〜0℃に冷却することによって、未反応GPAを沈殿物として除去する。
都合および設備の選択に応じて、脂肪酸を抽出するためのいずれの方法を用いてもよい。
【0032】
b)アセチル化グルコースラウレートの部分脱アシル化
上記で得られたアセチル化グルコースラウレートの一部をCF3COOH:H20(7:3)に添加し、20〜50%(w/v)溶液を形成し、室温(22〜33℃)で15分から2時間の間、攪拌した。処理の継続時間、脂肪酸アシル鎖の長さおよび置換の程度に応じて、図2に詳述される、様々なHLB値の部分脱アシル化グルコース脂肪酸エステルが得られた。溶媒を減圧下で蒸留除去した。生成物を温度10〜−15℃に段階的に冷却することによって、様々な程度の脱アシル化および置換の炭水化物脂肪酸エステルに分離する。
【0033】
実施例2:アセチル化オレイン酸スクロースおよび部分脱アシル化オレイン酸スクロースの製造
a)アセチル化オレイン酸スクロースの製造
電磁スターラー、活栓、液体窒素冷トラップに通じる真空引取りラインおよび真空ポンプを備えた三つ口丸底フラスコに、オレイン酸(Fluka Chemika社,スイス)0.06mol(16.93g)を計り入れた。スクロースオクタアセテート、SOA(Fluka Chemika社,スイス)0.01mol(6.79g)を添加し、均一な溶液が形成するまで、その混合物を絶え間なく攪拌しながら、油浴で80〜95℃に加熱した。次いで、トシル酸0.01%(w/w)(またはアルキルスルホン酸ポリシロキサン、またはBF3.OEt2)を添加した。次いで反応温度を60〜75℃に下げ、圧力5〜10トルをかけた。反応を3〜6時間続けた。生成物をイソ−プロパノール250mlで採取し、1M NaHCO3水溶液で中和し、活性炭2.0gで脱色し、濾過した。次いで、イソ−プロパノールを真空蒸留によって除去した。未反応オレイン酸を50×4mlヘキサンで抽出し、再利用のために保管しておいた。ショ糖脂肪酸エステルを温かいイソプロパノールに再溶解して30〜40%(w/v)溶液が形成され、−4〜0℃に冷却し、濾過して、未反応SOAを沈殿物として得た。次いで、濾液であるイソプロパノールを蒸留除去し、純度70〜85%のアセチル化オレイン酸スクロースを得た。ヘキサンによる抽出およびSOAの沈殿を繰り返し行うことによって、純度90〜95%の生成物(収率87%)が得られた。
【0034】
b)アセチル化オレイン酸スクロースの部分脱アシル化
上記で得られたアセチル化オレイン酸スクロースの一部をCF3COOH:H20(7:3)に添加し、20〜50%(w/v)溶液を形成し、室温(22〜33℃)で15分から2時間の間、攪拌した。処理の継続時間、脂肪酸アシル鎖の長さおよび置換の程度に応じて、図2に詳述される、様々なHLB値の部分脱アシル化ショ糖脂肪酸エステルが得られた。溶媒を減圧下で蒸留除去した。所望の場合には、生成物を温度10〜−15℃に段階的に冷却することによって、様々な程度の脱アシル化および/または置換の炭水化物脂肪酸エステルに分離する。
【0035】
実施例3:アセチル化ラウリン酸ラフィノースおよび部分脱アシル化ラウリン酸ラフィノースの製造
a)過酢酸ラフィノース(RA)の製造
無水ラフィノース(252g、0.50mol)を無水酢酸ナトリウム60gに添加し、予め100℃に平衡化された無水酢酸800mL中で攪拌した。完全に均一な溶液が30〜50分で得られる。反応を1〜2時間続けたままにし、その後、反応混合物を氷4リットルに注ぎ、2時間静置しておいた。形成された過酢酸ラフィノース沈殿物を上澄み液から分離し、ジクロロメタンに再溶解し、中性のpHの飽和重炭酸ナトリウム溶液で3回洗浄した。次いで、ジクロロメタン溶液を活性炭2.0gで脱色し、真空乾燥させて、白い粉末の過酢酸ラフィノース492g(収率90%)を得た。
【0036】
b)アセチル化ラウリン酸ラフィノースの製造
電磁スターラー、活栓、液体窒素冷トラップに通じる真空引取りラインおよび真空ポンプを備えた三つ口丸底フラスコに、ラウリン酸(Fluka Chemika社,スイス)0.06mol(12.0g)を計り入れた。過酢酸ラフィノース、RA(上記で製造された)0.01mol(11.54g)を添加し、均一な溶液が形成するまで、その混合物を絶え間なく攪拌しながら、油浴で80〜100℃に加熱した。次いで、BF3.OEt20.01%(w/w)(またはトシル酸またはアルキルスルホン酸ポリシロキサン)を添加した。反応温度を60〜75℃に下げ、圧力5〜10トルをかけた。反応を2〜6時間続けた。生成物を酢酸エチル250ml中で採取し、1M NaHCO3水溶液でクエンチし、活性炭2.0gで脱色し、濾過した。次いで酢酸エチルを真空蒸留によって除去した。
【0037】
未反応ラウリン酸を50×4mlヘキサンで抽出し、再利用のために保管しておいた。ラフィノース脂肪酸エステルを温かいイソプロパノールに再溶解して30〜40%(w/v)溶液が形成され、−4〜0℃に冷却し、濾過して、未反応RAを沈殿物として得た。次いで、濾液であるイソプロパノールを蒸留除去し、純度70〜85%のアセチル化ラウリン酸ラフィノースを得た。ヘキサンによる抽出およびRAの沈殿を繰り返し行うことによって、純度90〜95%の生成物(収率85%)が得られた。
【0038】
c)アセチル化ラウリン酸ラフィノースの部分脱アシル化
上記で得られたアセチル化ラウリン酸ラフィノースの一部をCF3COOH:H20(7:3)に添加し、20〜50%(w/v)溶液を形成し、室温(22〜33℃)で15分〜2時間の間、攪拌した。処理の継続時間、脂肪酸アシル鎖の長さおよび置換の程度に応じて、図2に詳述される、様々なHLB値の部分脱アシル化ラウリン酸ラフィノースが得られた。溶媒を減圧下で蒸留除去した。生成物を温度10〜−15℃に段階的に冷却することによって、様々な程度の脱アシル化および/または置換の炭水化物脂肪酸エステルに分離する。
【0039】
本発明は好ましい実施形態に関して説明されているが、本発明の精神および範囲から逸脱することなく、本発明に修正および改良を加えることができることは、当業者には明らかであるだろう。したがって、本発明は、特定の説明的な実施形態によって制限されるものではなく、添付の特許請求の範囲によってのみ制限される。
【図面の簡単な説明】
【0040】
本発明の実施形態は、図面を参照して以下に記述される。
【図1】本発明の好ましい実施形態のプロセスフローチャートである。
【図2】脱アシル化前および後の生成物炭水化物脂肪酸エステルの親水性親油性比(HLB)プロファイルのグラフである。
Claims (21)
- (a)無溶媒トランスアシドリシスにより、酸触媒の存在下にて、減圧下で、C2又はC3のアシル化炭水化物エステルを遊離脂肪酸と反応させるステップと、
(b)ステップ(a)で得られた反応混合物から、未反応遊離脂肪酸を脱色し、分離するステップと、
(c)ステップ(b)で得られた反応混合物から、未反応C2又はC3のアシル化炭水化物エステルを沈殿させるステップと、
(d)ステップ(c)で得られた反応混合物から、炭水化物脂肪酸エステルを回収するステップとを含む炭水化物脂肪酸エステルの製造方法。 - ステップ(a)において、溶媒が反応混合物に添加されない請求項1に記載の炭水化物脂肪酸エステルの製造方法。
- ステップ(b)における反応混合物中の前記未反応遊離脂肪酸が、制御温度で溶媒混合物から沈殿によって除去される請求項1に記載の炭水化物脂肪酸エステルの製造方法。
- ステップ(b)における反応混合物中の前記未反応遊離脂肪酸が、溶媒抽出によって反応混合物から除去される請求項1に記載の炭水化物脂肪酸エステルの製造方法。
- ステップ(b)における反応混合物を範囲−4〜10℃の温度に冷却することによって、前記未反応C2又はC3のアシル化炭水化物エステルをステップ(c)で沈殿させる請求項1に記載の炭水化物脂肪酸エステルの製造方法。
- 精製ステップ(b)および(c)中に除去される、前記未反応遊離脂肪酸および未反応C2又はC3のアシル化炭水化物エステルが、反応混合物に再循環される請求項1に記載の炭水化物脂肪酸エステルの製造方法。
- ステップ(a)が、4〜20トルの圧力範囲で行われる請求項1に記載の炭水化物脂肪酸エステルの製造方法。
- ステップ(a)が、5〜10トルの圧力範囲で行われる請求項1に記載の炭水化物脂肪酸エステルの製造方法。
- 様々な親水性親油性比(HLB)値のC2又はC3のアシル化炭水化物エステルのモノ、ジおよびポリ脂肪酸エステルが得られる請求項1に記載の炭水化物脂肪酸エステルの製造方法。
- 炭水化物脂肪酸エステル生成物の前記親水性親油性比(HLB)値が、1〜10の範囲である請求項1に記載の炭水化物脂肪酸エステルの製造方法。
- (e)酸触媒の存在下で、C2又はC3のアシル化炭水化物脂肪酸エステルを所定の時間、部分加水分解することによって遊離ヒドロキシル基を遊離させ、所定のHLB値の遊離ヒドロキシル基を有する炭水化物脂肪酸エステルを得るステップをさらに含む請求項1に記載の炭水化物脂肪酸エステルの製造方法。
- 炭水化物脂肪酸エステル生成物の前記HLB値が、8〜16の範囲である請求項11に記載の炭水化物脂肪酸エステルの製造方法。
- ステップ(a)が、60〜95℃の温度範囲で行われる請求項1に記載の炭水化物脂肪酸エステルの製造方法。
- (a)無溶媒トランスアシドリシスにより、酸触媒の存在下にて、減圧下で、アシル基がアセチル基又はプロピオニル基であるアシル化炭水化物エステルを遊離脂肪酸と反応させるステップと、
(b)ステップ(a)で得られた反応混合物から、未反応遊離脂肪酸を脱色し、分離するステップと、
(c)ステップ(b)で得られた反応混合物から、未反応アシル化炭水化物エステルを沈殿させるステップと、
(d)未反応遊離脂肪酸および未反応アシル基がアセチル基又はプロピオニル基であるアシル化炭水化物エステルを精製中に除去し、除去された未反応遊離脂肪酸およびアシル化炭水化物エステルを開始反応物混合物に再循環させるステップと、
(e)酸触媒の存在下で、アシル化炭水化物脂肪酸エステルを、部分加水分解することによって、遊離ヒドロキシル基を遊離させ、親水性親油性比(HLB)値1〜16の遊離ヒドロキシル基を有する炭水化物脂肪酸エステルを得るステップとを含む炭水化物脂肪酸エステルの製造方法。 - (a)ステップのアシル化炭水化物エステルが、一部または完全アシル化の単糖、二糖および三糖からなる群を含み、その1又は複数の単糖単位がフラノシル、ピラノシルまたはC2〜C6非環式構造である請求項1または14に記載の製造方法。
- 前記酸触媒が、単糖の場合には、硫酸またはカンファースルホン酸を含むとともに、二糖および三糖の場合には、三フッ化ホウ素ジエチルエーテラート、アルキルスルホン酸ポリシロキサンおよびポリシロキサンまたはトシル酸を含む請求項1または14に記載の製造方法。
- さらに、反応を停止させるステップ及び反応混合物を有機溶媒中に採取するステップを含み、これらのステップが、エタノール、イソプロパノール、n−プロパノール及び酢酸エチルからなる群から選択される有機溶媒によって行われる請求項1または14に記載の製造方法。
- 前記抽出溶媒がヘキサンである請求項4に記載の製造方法。
- 前記遊離脂肪酸が、ゼロ、一価、または二価不飽和を有するC6〜C22鎖長を有する請求項1または14に記載の製造方法。
- 前記加水分解酸触媒がトリフルオロ酢酸である請求項11または14に記載の製造方法。
- 前記の部分加水分解された炭水化物脂肪酸エステルが、アシル化の程度に応じて、−15〜10℃の制御温度範囲で段階的に冷却することによってさらに分離される請求項11または14に記載の製造方法。
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/SG2002/000133 WO2004002997A1 (en) | 2001-02-24 | 2002-06-27 | Trans-acidolysis process for the preparation of surface-active carbohydrate fatty-acid esters |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2005538970A JP2005538970A (ja) | 2005-12-22 |
| JP4580234B2 true JP4580234B2 (ja) | 2010-11-10 |
Family
ID=34215084
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2004517462A Expired - Fee Related JP4580234B2 (ja) | 2002-06-27 | 2002-06-27 | 表面活性炭水化物脂肪酸エステルを製造するためのトランスアシドリシスプロセス |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP1515980A1 (ja) |
| JP (1) | JP4580234B2 (ja) |
| AU (1) | AU2002305964A1 (ja) |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE227137C (ja) * | ||||
| AT333781B (de) * | 1972-11-06 | 1976-12-10 | Krems Chemie Gmbh | Verfahren zur herstellung neuer gemischter partialester |
| FR2503167A1 (fr) * | 1981-04-06 | 1982-10-08 | Blohorn Sa | Procede de fabrication d'esthers de sucre et notamment de saccharose |
| US5440027A (en) * | 1993-10-05 | 1995-08-08 | Kraft General Foods, Inc. | Method for preparing saccharide fatty acid polyesters by transesterification |
| US5424420A (en) * | 1993-10-05 | 1995-06-13 | Kraft Foods, Inc. | Method for preparing saccharide polyesters by transesterification |
-
2002
- 2002-06-27 JP JP2004517462A patent/JP4580234B2/ja not_active Expired - Fee Related
- 2002-06-27 AU AU2002305964A patent/AU2002305964A1/en not_active Abandoned
- 2002-06-27 EP EP02733770A patent/EP1515980A1/en not_active Withdrawn
Also Published As
| Publication number | Publication date |
|---|---|
| AU2002305964A1 (en) | 2004-01-19 |
| EP1515980A1 (en) | 2005-03-23 |
| JP2005538970A (ja) | 2005-12-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US4334061A (en) | Process for recovery of polyol fatty acid polyesters | |
| EP2828275B1 (en) | Synthesis of the trisaccharide 3-o-fucosyllactose and intermediates thereof | |
| CN108997520B (zh) | 一种降冰片烯衍生物含异类糖单元均聚物及其合成方法 | |
| US8283464B2 (en) | Process for synthesizing and purifying sucralose | |
| CN103703012A (zh) | 乳糖-n-四糖的制造 | |
| KR20120004504A (ko) | 6˝-시알릴락토오스 염 및 그의 합성 및 기타 α-시알릴올리고사카라이드의 합성 방법 | |
| CA2131785A1 (en) | Method for preparing saccharide fatty acid polyesters by transesterification | |
| JP3231765B2 (ja) | デメチルエピポドフィロトキシンの製造方法 | |
| JP4580234B2 (ja) | 表面活性炭水化物脂肪酸エステルを製造するためのトランスアシドリシスプロセス | |
| EP0570056A1 (en) | Process for preparing alkyl polyglucosides | |
| US6846916B2 (en) | Trans-acidolysis process for the preparation of carbohydrate fatty-acid esters | |
| EP0647652A2 (en) | Method for preparing saccharide polyesters by transesterification | |
| WO2002018400A2 (en) | Methods of preparing disaccharide and trisaccharide c6-c12fatty acid esters with high alpha content and materials therefrom | |
| KR20080020570A (ko) | 수크로오스-6-에스테르를 제조하는 방법 | |
| WO2004002997A1 (en) | Trans-acidolysis process for the preparation of surface-active carbohydrate fatty-acid esters | |
| CN113683650A (zh) | β-D-(1,4)-甘露糖醛酸寡糖及其中间体的制备方法 | |
| US20140349347A1 (en) | Synthesis of n-acetyl-d-neuraminic acid | |
| JPH093088A (ja) | アミノ二糖及びキチン又はその類似多糖類の製造方法 | |
| EP1537134B1 (en) | Process for producing a ribofuranose | |
| CN118894895A (zh) | 一种新型疫苗佐剂mpla中间体的合成方法 | |
| JP4698775B2 (ja) | エトポシドの製造方法 | |
| JP3884493B2 (ja) | フッ化β−キシロビオシル及びキシロオリゴ糖の製造方法 | |
| KR100550160B1 (ko) | 알부틴 및 알파형 알부틴 제조용 중간체의 제조방법 | |
| KR850000078B1 (ko) | 4,1',6'-트리클로로-4,1',6'-트리데옥시-갈락토슈크로오스의 제법 | |
| KR100543639B1 (ko) | 2-분지쇄-3-히드록시지방산 및 그 염의 제조방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090317 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090615 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090622 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090716 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090722 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090724 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100803 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100827 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130903 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |