JP4374249B2 - 超臨界的流体による処理方法:タンパク質微粒子の調製およびそれらの安定化 - Google Patents
超臨界的流体による処理方法:タンパク質微粒子の調製およびそれらの安定化 Download PDFInfo
- Publication number
- JP4374249B2 JP4374249B2 JP2003538186A JP2003538186A JP4374249B2 JP 4374249 B2 JP4374249 B2 JP 4374249B2 JP 2003538186 A JP2003538186 A JP 2003538186A JP 2003538186 A JP2003538186 A JP 2003538186A JP 4374249 B2 JP4374249 B2 JP 4374249B2
- Authority
- JP
- Japan
- Prior art keywords
- supercritical fluid
- solution
- protein
- stabilizer
- solvent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/30—Extraction; Separation; Purification by precipitation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/02—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length in solution
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/54—Improvements relating to the production of bulk chemicals using solvents, e.g. supercritical solvents or ionic liquids
Description
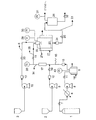
図1は、本発明の方法の実施に使用する装置の概略的なフローシートを示す。
本発明を、特に前記物質がタンパク質である場合についてさらに記述する。超臨界的流体の使用、炭水化物、アミノ酸および界面活性剤ポリマー等の種々の安定剤の使用を通じて、安定な乾燥タンパク質/安定剤微粒子の製造が可能であることが判明している。
超臨界的流体は、前記超臨界的流体の流速設定に用いるためのポンプ8により沈降容器に供給した。ライン35中における前記超臨界的流体の温度は、ノズル開口部を通じた膨張に依存して温度が低下することを考慮し、ヒーター17により、粒子形成容器内の温度よりも高い値に設定した。次に、前記調節剤を、あらかじめ決定した流速で、ポンプ9により前記超臨界的流体に添加した。タンパク質および安定剤の溶液は、定常状態(steady state conditions)が保たれている間、ポンプ10により前記粒子形成容器内に注入した。
本実施例では、本発明の方法を、アルカリホスファターゼ(ALP)およびトレハロースの共沈降混合物に用いた。
各カテゴリーにおけるいくつかのバイアルを、以下の各条件(25℃− 60% RH; 30℃− 65% RH; 40℃− 75%)で6ヶ月静置した。t = 0, 1, 2, 3, 6ヶ月時に、前記バイアル内容物のALP活性についてアッセイを行なった。表1に、この安定性調査結果をまとめる。
本実施例では、本発明の方法を、リソチームおよびトレハロースの共沈降混合物に用いた。

Claims (14)
- タンパク質またはポリペプチドとその安定剤との、ガス状アンチソルベント(anti solvent)方法による共沈降方法であり、
純粋なまたは調節剤(modifier)と混合した超臨界的流体、および、溶媒に溶解した前記タンパク質またはポリペプチドと前記安定剤とを含む溶液を、前記超臨界的流体により前記溶媒が前記溶液から抽出され、そして前記タンパク質またはポリペプチドと安定剤との共沈降が起こるように、粒子形成容器内に導入することを含み、
前記安定剤はトレハロースであり、前記溶媒は水であり、前記超臨界的流体が二酸化炭素であり、前記調節剤は前記超臨界的流体における前記溶媒の溶解度を向上させるために添加されるものであってメタノール、エタノール、およびイソプロパノールから選択される、方法。 - 前記タンパク質またはポリペプチドが、タンパク質である、請求項1に記載の方法。
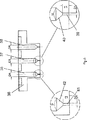
- 前記溶液および前記超臨界的流体を、別々の入口ノズルを通じて前記粒子形成容器内に導入する、請求項1または2に記載の方法。
- 前記超臨界的流体を、複数の入口ノズルを通じて前記粒子形成容器内に導入する、請求項3に記載の方法。
- 前記ノズルがディスク上に存在し、前記溶液の入口ノズルは前記ディスクの中央に存在し、その円周に沿って等間隔に配置された複数の超臨界的流体の入口ノズルにより囲まれている、請求項4に記載の方法。
- 前記溶液を調節剤と混合して前記粒子形成容器内に導入する、請求項1〜5のいずれかに記載の方法。
- 前記超臨界的流体を調節剤と混合して前記粒子形成容器内に導入する、請求項1〜6のいずれかに記載の方法。
- 前記調節剤がエタノールである、請求項1〜7のいずれかに記載の方法。
- 前記溶液中における、タンパク質またはポリペプチドと安定剤との比(重量/重量)が、1:1〜1:10である、請求項1〜8のいずれかに記載の方法。
- 前記溶液中における、タンパク質またはポリペプチドと安定剤との比(重量/重量)が1:2である、請求項9に記載の方法。
- 前記超臨界的流体を、前記流体中における音の速度と等しいかまたはそれよりも大きい速度で前記粒子形成容器中に導入する、請求項1〜10のいずれかに記載の方法。
- 調節剤を使用し、かつ、超臨界的流体の流速と調節剤の流速との比(重量/重量)が、4:1〜8:1の範囲内である、請求項1〜11のいずれかに記載の方法。
- 調節剤を使用し、かつ、調節剤の流速と溶液の流速との間の比(重量/重量)が、15:1〜25:1の範囲内である、請求項1〜12のいずれかに記載の方法。
- 請求項1〜13のいずれかに記載の方法を用いて、タンパク質またはポリペプチドと安定剤との共沈降物(coprecipitate)を製造する方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP01125048.7 | 2001-10-22 | ||
| EP01125048 | 2001-10-22 | ||
| PCT/EP2002/011761 WO2003035673A1 (en) | 2001-10-22 | 2002-10-21 | Supercritical fluids processing: preparation of protein microparticles and their stabilisation |
Publications (4)
| Publication Number | Publication Date |
|---|---|
| JP2005514341A JP2005514341A (ja) | 2005-05-19 |
| JP2005514341A6 JP2005514341A6 (ja) | 2005-08-04 |
| JP2005514341A5 JP2005514341A5 (ja) | 2005-12-22 |
| JP4374249B2 true JP4374249B2 (ja) | 2009-12-02 |
Family
ID=8179030
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003538186A Expired - Fee Related JP4374249B2 (ja) | 2001-10-22 | 2002-10-21 | 超臨界的流体による処理方法:タンパク質微粒子の調製およびそれらの安定化 |
Country Status (21)
| Country | Link |
|---|---|
| US (2) | US7250152B2 (ja) |
| EP (1) | EP1440082B1 (ja) |
| JP (1) | JP4374249B2 (ja) |
| KR (1) | KR100887429B1 (ja) |
| CN (1) | CN1575298A (ja) |
| AT (1) | ATE526340T1 (ja) |
| AU (1) | AU2002350588B2 (ja) |
| BR (1) | BR0213430A (ja) |
| CA (1) | CA2464308A1 (ja) |
| DK (1) | DK1440082T3 (ja) |
| ES (1) | ES2372032T3 (ja) |
| HU (1) | HUP0402096A3 (ja) |
| IL (2) | IL161447A0 (ja) |
| MX (1) | MXPA04003718A (ja) |
| NO (1) | NO330504B1 (ja) |
| NZ (1) | NZ532395A (ja) |
| PL (1) | PL209826B1 (ja) |
| PT (1) | PT1440082E (ja) |
| RU (1) | RU2309160C2 (ja) |
| WO (1) | WO2003035673A1 (ja) |
| ZA (1) | ZA200402899B (ja) |
Families Citing this family (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6541606B2 (en) * | 1997-12-31 | 2003-04-01 | Altus Biologics Inc. | Stabilized protein crystals formulations containing them and methods of making them |
| CA2464308A1 (en) * | 2001-10-22 | 2003-05-01 | Dompe S.P.A. | Supercritical fluids processing: preparation of protein microparticles and their stabilisation |
| GB0300338D0 (en) * | 2003-01-08 | 2003-02-05 | Bradford Particle Design Ltd | Particle formation |
| US7223445B2 (en) * | 2004-03-31 | 2007-05-29 | Eastman Kodak Company | Process for the deposition of uniform layer of particulate material |
| DE102004049850A1 (de) * | 2004-09-15 | 2006-03-02 | Cognis Ip Management Gmbh | Verfahren und Anlage zur Herstellung von Mikropartikeln oder Nanopartikeln, Verwendung der Nanopartikel und damit hergestellte Suspension oder Emulsion |
| CN1302766C (zh) * | 2005-06-23 | 2007-03-07 | 同济大学 | 超临界抗溶剂过程制备可生物降解聚合物载药微粒的方法 |
| KR100705282B1 (ko) * | 2005-06-30 | 2007-04-12 | 주식회사 케이씨아이 | 유채씨로부터 분리 단백질을 제조하는 방법 |
| EP2140003A2 (en) * | 2007-04-25 | 2010-01-06 | 3M Innovative Properties Company | Sample-processing reagent compositions, methods, and devices |
| ATE539165T1 (de) | 2007-10-19 | 2012-01-15 | Becton Dickinson Co | Verfahren zum nachweis von beta-lactamase |
| WO2010047778A1 (en) | 2008-10-20 | 2010-04-29 | Becton Dickinson And Company | Compositions for the detection of intracellular bacterial targets and other intracellular microorganism targets |
| US9138705B2 (en) | 2010-11-22 | 2015-09-22 | Hong Kong Polytechnic University | Method for precipitating a solute from a solution |
| US10821085B2 (en) | 2010-12-07 | 2020-11-03 | Kimberly-Clark Worldwide, Inc. | Wipe coated with a botanical composition having antimicrobial properties |
| US8445032B2 (en) | 2010-12-07 | 2013-05-21 | Kimberly-Clark Worldwide, Inc. | Melt-blended protein composition |
| US8524264B2 (en) | 2010-12-07 | 2013-09-03 | Kimberly-Clark Worldwide, Inc. | Protein stabilized antimicrobial composition formed by melt processing |
| US9149045B2 (en) | 2010-12-07 | 2015-10-06 | Kimberly-Clark Worldwide, Inc. | Wipe coated with a botanical emulsion having antimicrobial properties |
| US9832993B2 (en) | 2010-12-07 | 2017-12-05 | Kimberly-Clark Worldwide, Inc. | Melt processed antimicrobial composition |
| US9648874B2 (en) | 2010-12-07 | 2017-05-16 | Kimberly-Clark Worldwide, Inc. | Natural, multiple use and re-use, user saturated wipes |
| US8574628B2 (en) | 2011-12-19 | 2013-11-05 | Kimberly-Clark Worldwide, Inc. | Natural, multiple release and re-use compositions |
| US9925512B2 (en) | 2013-03-14 | 2018-03-27 | Crititech, Inc. | Equipment assembly for and method of processing particles |
| US8778181B1 (en) | 2013-03-14 | 2014-07-15 | Crititech, Inc. | Equipment assembly for and method of processing particles |
| WO2015053857A2 (en) * | 2013-10-10 | 2015-04-16 | New York University | Efficient collection of nanoparticles |
| US20180020551A1 (en) * | 2014-11-28 | 2018-01-18 | Zeon Corporation | Desmear processing method and manufacturing method for multilayer printed wiring board |
| CN105727579B (zh) * | 2016-01-28 | 2018-01-09 | 苏州鼎烯聚材纳米科技有限公司 | 浆料的低成本高效率超临界喷雾干燥方法及设备 |
| US10206873B1 (en) | 2017-08-04 | 2019-02-19 | Colorado Can Llc | Dry powder formation using a variably constrained, divided pathway for mixing fluid streams |
| CN109430514B (zh) * | 2018-11-02 | 2022-07-26 | 广东海洋大学 | 一种制备罗非鱼-豆粕共沉淀蛋白的方法 |
| KR102154923B1 (ko) * | 2019-03-05 | 2020-09-10 | 서울대학교산학협력단 | 배양액의 결정화 및 초임계 건조 장치 및 방법 |
| CN110055238A (zh) * | 2019-05-13 | 2019-07-26 | 东北农业大学 | 一种海参花碱性磷酸酶的提取方法 |
| CN114213382A (zh) * | 2021-12-10 | 2022-03-22 | 大连理工大学 | 一种基于超临界抗溶剂工艺合成微纳米级别egcg的方法 |
| CN114831930B (zh) * | 2022-03-28 | 2023-09-05 | 厦门大学 | 一种胶原蛋白组装眼药、制备方法及应用 |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2748132A1 (de) | 1977-10-27 | 1979-05-03 | Behringwerke Ag | Stabilisator fuer polysaccharid |
| JPS5874696A (ja) | 1981-10-30 | 1983-05-06 | 株式会社 ヤトロン | アデノシン三リン酸の安定化方法 |
| US4457916A (en) | 1982-08-31 | 1984-07-03 | Asahi Kasei Kogyo Kabushiki Kaisha | Method for stabilizing Tumor Necrosis Factor and a stable aqueous solution or powder containing the same |
| WO1987000196A1 (en) | 1985-07-09 | 1987-01-15 | Quadrant Bioresources Limited | Protection of proteins and the like |
| US6063910A (en) * | 1991-11-14 | 2000-05-16 | The Trustees Of Princeton University | Preparation of protein microparticles by supercritical fluid precipitation |
| US5639441A (en) | 1992-03-06 | 1997-06-17 | Board Of Regents Of University Of Colorado | Methods for fine particle formation |
| WO1994008599A1 (en) | 1992-10-14 | 1994-04-28 | The Regents Of The University Of Colorado | Ion-pairing of drugs for improved efficacy and delivery |
| GB9313642D0 (en) | 1993-07-01 | 1993-08-18 | Glaxo Group Ltd | Method and apparatus for the formation of particles |
| GB9413202D0 (en) * | 1994-06-30 | 1994-08-24 | Univ Bradford | Method and apparatus for the formation of particles |
| US5874029A (en) * | 1996-10-09 | 1999-02-23 | The University Of Kansas | Methods for particle micronization and nanonization by recrystallization from organic solutions sprayed into a compressed antisolvent |
| GB9703673D0 (en) | 1997-02-21 | 1997-04-09 | Bradford Particle Design Ltd | Method and apparatus for the formation of particles |
| GB9810559D0 (en) | 1998-05-15 | 1998-07-15 | Bradford Particle Design Ltd | Method and apparatus for particle formation |
| GB9910975D0 (en) | 1999-05-13 | 1999-07-14 | Univ Strathclyde | Rapid dehydration of proteins |
| EP1185248B1 (en) | 1999-06-09 | 2012-05-02 | Robert E. Sievers | Supercritical fluid-assisted nebulization and bubble drying |
| GB9915975D0 (en) | 1999-07-07 | 1999-09-08 | Bradford Particle Design Ltd | Method for the formation of particles |
| GB9920558D0 (en) | 1999-08-31 | 1999-11-03 | Bradford Particle Design Ltd | Methods for particle formation and their products |
| ES2223034T3 (es) * | 2001-02-26 | 2005-02-16 | Dompe S.P.A. | Aparato y metodo para la formacion de particulas micronicas y submicronicas. |
| CA2464308A1 (en) * | 2001-10-22 | 2003-05-01 | Dompe S.P.A. | Supercritical fluids processing: preparation of protein microparticles and their stabilisation |
-
2002
- 2002-10-21 CA CA002464308A patent/CA2464308A1/en not_active Abandoned
- 2002-10-21 EP EP02785262A patent/EP1440082B1/en not_active Expired - Lifetime
- 2002-10-21 JP JP2003538186A patent/JP4374249B2/ja not_active Expired - Fee Related
- 2002-10-21 HU HU0402096A patent/HUP0402096A3/hu unknown
- 2002-10-21 CN CNA028209508A patent/CN1575298A/zh active Pending
- 2002-10-21 AT AT02785262T patent/ATE526340T1/de active
- 2002-10-21 NZ NZ532395A patent/NZ532395A/xx not_active IP Right Cessation
- 2002-10-21 PT PT02785262T patent/PT1440082E/pt unknown
- 2002-10-21 IL IL16144702A patent/IL161447A0/xx unknown
- 2002-10-21 KR KR1020047005867A patent/KR100887429B1/ko not_active IP Right Cessation
- 2002-10-21 BR BR0213430-6A patent/BR0213430A/pt not_active Application Discontinuation
- 2002-10-21 WO PCT/EP2002/011761 patent/WO2003035673A1/en active Application Filing
- 2002-10-21 PL PL370303A patent/PL209826B1/pl unknown
- 2002-10-21 US US10/493,256 patent/US7250152B2/en not_active Expired - Fee Related
- 2002-10-21 DK DK02785262.3T patent/DK1440082T3/da active
- 2002-10-21 RU RU2004112424/04A patent/RU2309160C2/ru not_active IP Right Cessation
- 2002-10-21 MX MXPA04003718A patent/MXPA04003718A/es active IP Right Grant
- 2002-10-21 AU AU2002350588A patent/AU2002350588B2/en not_active Ceased
- 2002-10-21 ES ES02785262T patent/ES2372032T3/es not_active Expired - Lifetime
-
2004
- 2004-04-15 IL IL161447A patent/IL161447A/en not_active IP Right Cessation
- 2004-04-16 ZA ZA200402899A patent/ZA200402899B/en unknown
- 2004-04-21 NO NO20041634A patent/NO330504B1/no not_active IP Right Cessation
-
2007
- 2007-06-19 US US11/765,180 patent/US20070293655A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| IL161447A0 (en) | 2004-09-27 |
| WO2003035673A9 (en) | 2003-11-13 |
| NO20041634L (no) | 2004-06-17 |
| BR0213430A (pt) | 2004-11-09 |
| KR20040075856A (ko) | 2004-08-30 |
| CN1575298A (zh) | 2005-02-02 |
| RU2309160C2 (ru) | 2007-10-27 |
| AU2002350588A2 (en) | 2003-05-06 |
| ATE526340T1 (de) | 2011-10-15 |
| RU2004112424A (ru) | 2005-10-10 |
| JP2005514341A (ja) | 2005-05-19 |
| KR100887429B1 (ko) | 2009-03-09 |
| WO2003035673A1 (en) | 2003-05-01 |
| AU2002350588B2 (en) | 2008-08-28 |
| ES2372032T3 (es) | 2012-01-13 |
| NZ532395A (en) | 2006-03-31 |
| NO330504B1 (no) | 2011-05-02 |
| HUP0402096A2 (hu) | 2005-01-28 |
| US7250152B2 (en) | 2007-07-31 |
| PL370303A1 (en) | 2005-05-16 |
| ZA200402899B (en) | 2005-04-18 |
| HUP0402096A3 (en) | 2012-09-28 |
| US20070293655A1 (en) | 2007-12-20 |
| EP1440082A1 (en) | 2004-07-28 |
| PT1440082E (pt) | 2011-12-07 |
| US20050065063A1 (en) | 2005-03-24 |
| PL209826B1 (pl) | 2011-10-31 |
| CA2464308A1 (en) | 2003-05-01 |
| EP1440082B1 (en) | 2011-09-28 |
| DK1440082T3 (da) | 2012-01-16 |
| IL161447A (en) | 2010-11-30 |
| MXPA04003718A (es) | 2004-07-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4374249B2 (ja) | 超臨界的流体による処理方法:タンパク質微粒子の調製およびそれらの安定化 | |
| JP2005514341A6 (ja) | 超臨界的流体による処理方法:タンパク質微粒子の調製およびそれらの安定化 | |
| AU2002350588A1 (en) | Supercritical fluids processing: preparation of protein microparticles and their stabilisation | |
| RU2208435C2 (ru) | Внедрение активных веществ в матрицы носителей | |
| CA2462338C (en) | Powder processing with pressurized gaseous fluids | |
| US8609611B2 (en) | Synthesis of small particles | |
| US7186796B2 (en) | Method for drying water-borne materials | |
| US20030013634A1 (en) | Synthesis of small particles | |
| US7901606B2 (en) | Production of porous materials by supercritical fluid processing | |
| AU2002221320B2 (en) | Synthesis of small particles | |
| US20240082159A1 (en) | Method for preparing protein microbeads for improving stability of protein preparation | |
| AU2002365617B2 (en) | Synthesis of small particles | |
| Jovanovic et al. | Stabilization of proteins in dry powder formulations using supercritical fluid technology | |
| AU2002221320A1 (en) | Synthesis of small particles |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050927 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20060901 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20060901 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080626 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20080911 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20080919 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20081024 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090129 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090416 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090423 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090520 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090527 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090622 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090629 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090728 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090820 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090907 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120911 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130911 Year of fee payment: 4 |
|
| LAPS | Cancellation because of no payment of annual fees |