JP4367687B2 - オレフィン重合用の架橋メタロセン化合物およびそれを用いたオレフィンの重合方法 - Google Patents
オレフィン重合用の架橋メタロセン化合物およびそれを用いたオレフィンの重合方法 Download PDFInfo
- Publication number
- JP4367687B2 JP4367687B2 JP2002342815A JP2002342815A JP4367687B2 JP 4367687 B2 JP4367687 B2 JP 4367687B2 JP 2002342815 A JP2002342815 A JP 2002342815A JP 2002342815 A JP2002342815 A JP 2002342815A JP 4367687 B2 JP4367687 B2 JP 4367687B2
- Authority
- JP
- Japan
- Prior art keywords
- added
- mmol
- solution
- polymerization
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Description
【発明が属する技術分野】
本発明は、特定の構造を有するオレフィン重合用の触媒または触媒成分として有用なメタロセン化合物、および該メタロセン化合物を含む触媒の存在下で、オレフィンを重合する方法に関する。
【0002】
【従来の技術】
オレフィン重合用の均一系触媒としては、いわゆるメタロセン化合物がよく知られている。メタロセン化合物を用いてオレフィンを重合する方法、特にα−オレフィンを立体規則的に重合する方法は、W. Kaminskyらによってアイソタクテイック重合が報告(Angew. Chem. Int. Ed. Engl., 24, 507(1985))されて以来、重合活性の更なる向上や立体規則性改善の視点から多くの改良研究が行なわれている。このような研究の一環としてシクロペンタジエニル配位子とフルオレニル配位子を炭素一原子で架橋したメタロセン化合物を用いた重合結果がJ. A. Ewenによって報告されている(J. Am. Chem. Soc.,110, 6255(1988))。さらにシクロペンタジエニル配位子とフルオレニル配位子を炭素ニ原子で架橋(エチレン架橋)したメタロセン化合物を用いたポリプロピレンの重合結果がM. H. Leeによって報告されている(J. Organomet. Chem., 561, 37(1998))。
しかしながら、これらの重合活性は未だ十分でなく、さらに重合活性の優れたメタロセン化合物およびこのようなメタロセン化合物を含むオレフィン重合用触媒の出現が望まれていた。
【0003】
【発明が解決しようとする課題】
本発明は、上記の課題を解決するために行なわれたものであり、すなわち公知のメタロセン化合物を用いた場合に比して高い重合活性を有するオレフィン重合用の架橋メタロセン化合物を提供すること、および該架橋メタロセン化合物を含む触媒の存在下で重合する方法を提供することを目的としている。
【0004】
【課題を解決するための手段】
本発明の架橋メタロセン化合物は、下記一般式[1]で表される架橋メタロセン化合物である。
【0005】
【化2】
このような架橋メタロセン化合物を含む触媒の存在下、エチレンおよびα-オレフィンから選ばれる1種以上のモノマーであり、モノマーの少なくとも1種がエチレンまたはプロピレンであるモノマーを重合することにより効率良くポリオレフィンを製造することが可能となる。
【0006】
以下、前記一般式[1]で表わされる架橋メタロセン化合物、好ましい架橋メタロセン化合物の例示、本発明の架橋メタロセン化合物の製造方法、本発明の架橋メタロセン化合物をオレフィン重合用触媒に供する際の好ましい形態、そして最後にこのオレフィン重合用触媒の存在下でオレフォンを重合する方法について順次説明する。
【0007】
架橋メタロセン化合物
本発明の架橋メタロセン化合物は、前記一般式[1]で表わされる。一般式[1]において、R1、R2、R3、R4、R5、R6、R7、R8、R9、R10、R11、R12は水素原子、炭化水素基、ケイ素含有基から選ばれ、それぞれ同一でも異なっていても よく、R5からR12までの隣接した置換基は互いに結合して環を形成してもよい。
【0008】
炭化水素基としては、炭素数1から20のアルキル基、炭素原子数7〜20のアリールアルキル基、炭素原子数6〜20のアリール基などが挙げられる。例えば、メチル基、エチル基、n-プロピル基、イソプロピル基、アリル(allyl)基、n-ブチル基、t-ブチル基、アミル基、n-ペンチル基、n-ヘキシル基、n-ヘプチル基、n-オクチル基、n-ノニル基、n-デカニル基、3-メチルペンチル基、1,1-ジエチルプロピル基、1,1-ジメチルブチル基、1-メチル-1-プロピルブチル基、1,1-プロピルブチル基、1,1-ジメチル-2-メチルプロピル基、1-メチル-1-イソプロピル-2-メチルプロピル基、シクロペンチル基、シクロヘキシル基、シクロヘプチル基、シクロオクチル基、ノルボルニル基、アダマンチル基、フェニル基、ナフチル基、ビフェニル基、フェナントリル基、アントラセニル基、ベンジル基、クミル基、メトキシ基、エトキシ基、フェノキシ基、N-メチルアミノ基、N,N−ジメチルアミノ基、N-フェニルアミノ基などが挙げられる。
ケイ素含有基としては、トリメチルシリル基、トリエチルシリル基、ジフェニルメチルシリル基、ジメチルフェニルシリル基などを挙げることができる。
【0009】
フルオレン環に置換するR5からR12までの隣接した置換基は互いに結合して環を形成していてもよい。このような置換フルオレニル基としては、ベンゾフルオレニル基、ジベンゾフルオレニル基、オクタメチルオクタヒドロジベンゾフルオレニル基、オクタメチルテトラヒドロジシクロペンタフルオレニル基を例示することができる。
【0010】
本発明の架橋メタロセン化合物において重要なのはn=1のとき前記R1からR16が全て水素原子である架橋メタロセン化合物が除外されていることである。n=1のときはR1からR16のいずれか一つ以上の基が炭化水素基またはケイ素含有基で置換されることによって高い重合活性が発現する。R1〜R16で表わされる基は、それぞれ同一でも異なっていてもよい。R1からR16の好ましい態様、すなわち重合活性の視点から好ましい架橋メタロセン化合物としては、R6、R7、R10、R11の任意の二つ以上の置換基が炭素数1〜20の炭化水素基である化合物である架橋メタロセン化合物が挙げられる。ここで炭素数1〜20の炭化水素基としては、メチル基、エチル基、n-プロピル基、イソプロピル基、アリル(allyl)基、n-ブチル基、tert-ブチル基、アミル基、n-ペンチル基等である。これらの中では配位子の合成上の容易さから左右対称、すなわちR6とR11、R7とR10は同一の基であることが好ましい。このような好ましい態様の中には、R6とR7が脂肪族環(AR-1)を形成し、且つR10とR11が脂肪族環(AR-1)と同一な脂肪族環(AR-2)を形成している架橋メタロセン化合物も含まれる。
【0011】
Y1およびY2は第14族原子であり相互に同一でも異なっていてもよい。第14族炭化水素原子としては、炭素原子、ケイ素原子、ゲルマニウム原子およびスズ原子を例示できるが、この中では炭素原子またはケイ素原子が好ましく、特にY1とY2 が同一原子であることが更に好ましい。R13、R14、R15、R16は水素原子または炭化水素基であり、炭化水素基としては前記炭化水素基と同様な基を例示することができる。nは1〜3の整数であり、好ましくはn=1である。R13とR15は互いに結合して環を形成してもよく、R13とR15は互いに結合して環を形成すると同時にR14とR16は互いに結合して環を形成してもよい。R13とR15が環を形成する場合、n=1の場合は隣接する基が環形成することであり、n=2の場合はR13とY1の隣接位(α位)またはβ位のR15と環形成してもよく、n=3の場合はR13とY1の隣接位(α位)、β位またはγ位のR15と環形成していてもよい。R14とR16が環を形成する場合も同じように、n=1の場合は隣接する基が環形成することであり、n=2の場合はR14とY1の隣接位(α位)またはβ位のR16と環形成してもよく、n=3の場合はR14とY1の隣接位(α位)、β位またはγ位のR16と環形成していてもよい。MはTi、ZrまたはHfであり、Qはハロゲン、炭化水素基、アニオン配位子または孤立電子対で配位可能な中性配位子から同一または異なる組合せで選んでもよく、jは1〜4の整数である。
【0012】
Qはハロゲン、炭化水素基、アニオン配位子または孤立電子対で配位可能な中性配位子から同一または異なる組合せで選ばれる。jは1〜4の整数であり、jが2以上の時は、Qは互いに同一でも異なっていてもよい。
ハロゲンの具体例としては、フッ素、塩素、臭素、ヨウ素であり、炭化水素基の具体例としては前記と同様のものが挙げられる。
アニオン配位子の具体例としては、メトキシ、tert−ブトキシ、フェノキシなどのアルコキシ基、アセテート、ベンゾエートなどのカルボキシレート基、メシレート、トシレートなどのスルホネート基が挙げられる。
孤立電子対で配位可能な中性配位子の具体例としては、トリメチルホスフィン、トリエチルホスフィン、トリフェニルホスフィン、ジフェニルメチルホスフィンなどの有機リン化合物、テトラヒドロフラン、ジエチルエーテル、ジオキサン、1,2−ジメトキシエタンなどのエーテル類が挙げられる。Qは、少なくても一つがハロゲンまたはアルキル基であることが好ましい。
【0013】
架橋メタロセン化合物およびその例示
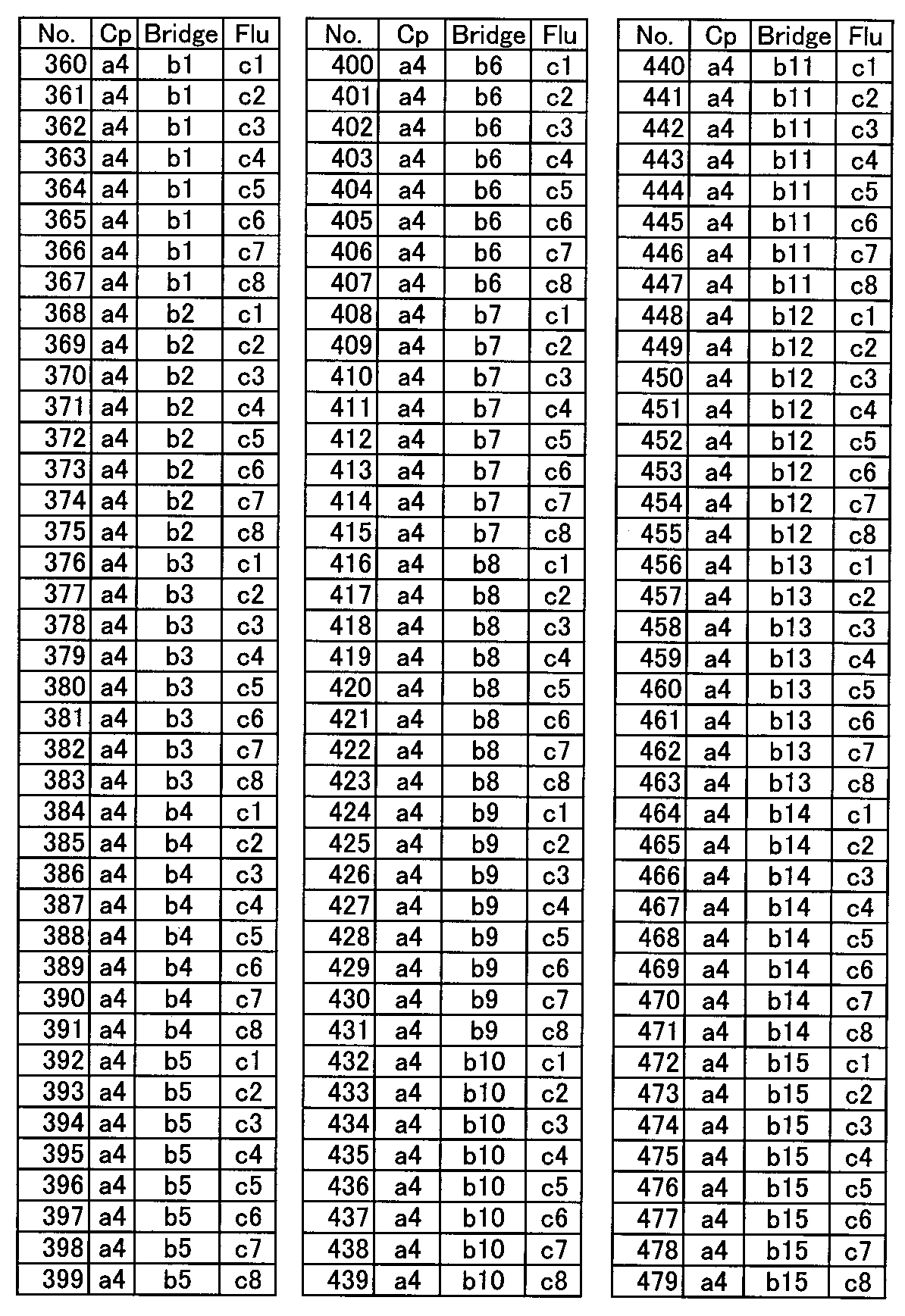
以下に本発明における上記メタロセン化合物の具体例を示すが、特にこれによって本発明の範囲が限定されるものではない。まずメタロセン化合物のMQj(金属部分)を除いたリガンド構造を、表記上、Cp(シクロペンタジエニル環部分)、Bridge(架橋部分)、Flu(フルオレニル環部分)の3つに分け、それぞれの部分構造の具体例を、及びそれらの組み合わせによるリガンド構造の具体例を以下に示す。
尚、Cp、BridgeおよびFluの具体例において、黒丸(●)で示した点は、それぞれBridgeとCpおよびFluとの結合点を表す。
〔Cpの具体例〕
【0014】
【化3】
〔Bridgeの具体例〕
【0016】
【化4】
〔Fluの具体例〕
【0018】
【化5】
【0019】
【化6】
【化7】
【化8】
【化9】
【化10】
【化11】
【化12】
【化13】
【化14】
【化15】
【0029】
【化16】
架橋メタロセン化合物の製造方法
本発明の架橋メタロセン化合物は公知の方法によって製造可能であり、特に製造法が限定されるわけではない。公知の製造方法として例えば、J. Organomet. Chem., 361, 37(1998)が挙げられる。例えば、一般式[1]の化合物は次のステップによって製造可能である。
まず一般式[1]の前駆体化合物[7]は、製法[A]または[B]のような方法で製造することができる。
【0031】
【化17】
【化18】
【0033】
さらに一般式[1]のシクロペニタジエニル配位子側の前駆体である、例えばR2とR4が共に水素原子である[11]は、例えば製法[C]のような方法で選択的に製造することができる。
【0034】
【化19】
【0035】
また、[11]の別法による製造法として、製法[D]や製法[E]のような方法もあるが、これらの方法ではR1とR3が隣り合った異性体[12]を副生することがあるため、R1とR3の組合せや反応条件等により、[12]を副生しない場合に限り製法[D]や製法[E]のような方法を採用することができる。
【0036】
【化20】
【化21】
【0038】
さらにR3がCR17R18R19で表される置換基の場合には、一般式[F]のような方法によっても[11]を製造することができる。
【0039】
【化22】
この方法においてもR1とR3が隣り合った異性体[12]を副生することがあるため、R1とR3の組合せや反応条件等により、[12]を副生しない場合に限り[F]のような方法を採用することができる。
【0040】
上記一般式[A]〜[F]の反応に用いられるアルカリ金属としては、リチウム、ナトリウムまたはカリウムが挙げられ、アルカリ土類金属としてはマグネシウム、カルシウムが挙げられる。また、ハロゲンとしては、フッ素、塩素、臭素、ヨウ素が挙げられる。アニオン配位子の具体例としては、メトキシ、tert-ブトキシ、フェノキシ等のアルコキシ基、アセテート、ベンゾエート等のカルボキシレート基、メシレート、トシレート等のスルホネート基等が挙げられる。
次に、一般式[7]の前駆体化合物からメタロセン化合物を製造する例を以下に示すが、これは発明の範囲を制限するものではなく、公知のいかなる方法で製造されてもよい。
【0041】
一般式[A]または[B]の反応で得られた一般式[7]の前駆体化合物は、有機溶媒中でアルカリ金属、水素化アルカリ金属または有機アルカリ金属と、反応温度が−80℃〜200℃の範囲で接触させることで、ジアルカリ金属塩とする。上記反応で用いられる有機溶媒としては、ペンタン、ヘキサン、ヘプタン、シクロヘキサン、デカリン等の脂肪族炭化水素、またはベンゼン、トルエン、キシレン等の芳香族炭化水素、またはTHF、ジエチルエーテル、ジオキサン、1、2−ジメトキシエタン等のエーテル、またはジクロロメタン、クロロホルム等のハロゲン化炭化水素等が挙げられる。
また上記反応で用いられるアルカリ金属としては、リチウム、ナトリウム、カリウム等が挙げられ、水素化アルカリ金属としては、水素化ナトリウム、水素化カリウム等が挙げられ、有機アルカリ金属としては、メチルリチウム、ブチルリチウム、フェニルリチウム等が挙げられる。
次に上記の該ジアルカリ金属塩を、一般式[20]
MZk …[20]
(式中、Mは周期表第4族から選ばれた金属であり、Zはハロゲン、アニオン配位子または孤立電子対で配位可能な中性配位子から同一または異なる組合せで選んでもよく、kは3〜6の整数である。)
で表される化合物と、有機溶媒中で反応させることで、一般式[1]で表されるメタロセン化合物を合成することができる。
【0042】
一般式[20]で表される化合物の好ましい具体的として、三価または四価のチタニウムフッ化物、塩化物、臭化物及びヨウ化物、四価のジルコニウムフッ化物、塩化物、臭化物及びヨウ化物、四価のハフニウムフッ化物、塩化物、臭化物及びヨウ化物、またはこれらのTHF、ジエチルエーテル、ジオキサンまたは1,2−ジメトキシエタン等のエーテル類との錯体を挙げることができる。
また、用いられる有機溶媒としては前記と同様のものを挙げることができる。該ジアルカリ金属塩と一般式[20]で表される化合物との反応は、好ましくは等モル反応で行い、前記の有機溶媒中で、反応温度が−80℃〜200℃の範囲で行うことができる。
【0043】
反応で得られたメタロセン化合物は、抽出、再結晶、昇華等の方法により、単離・精製を行うことができる。また、このような方法で得られる本発明の架橋メタロセン化合物は、プロトン核磁気共鳴スペクトル、13C核磁気共鳴スペクトル、質量分析、および元素分析などの分析手法を用いることによって同定される。
【0044】
架橋メタロセン化合物をオレフィン重合用触媒に供する際の好ましい態様
次に本発明の架橋メタロセン化合物を、オレフィン重合触媒として用いる場合の好ましい態様について説明する。
本発明の架橋メタロセン化合物をオレフィン重合触媒として用いる場合、触媒成分は、
(A)前記の架橋メタロセン化合物
(B)(B-1) 有機金属化合物、(B-2) 有機オキシアルミニウム化合物、および(B-3)架橋メタロセン化合物(A)と反応してイオン対を形成する化合物、から選ばれる少なくても1種の化合物、さらに必要に応じて、
(C)粒子状担体
から構成される。
以下、各成分について具体的に説明する。
【0045】
(B-1)有機金属化合物
本発明で用いられる(B-1) 有機金属化合物として、具体的には下記のような周期律表第1、2族および第12、13族の有機金属化合物が用いられる。
(B-1a) 一般式 Ra mAl(ORb)n Hp Xq
(式中、Ra およびRb は、互いに同一でも異なっていてもよく、炭素原子数が1〜15、好ましくは1〜4の炭化水素基を示し、Xはハロゲン原子を示し、mは0<m≦3、nは0≦n<3、pは0≦p<3、qは0≦q<3の数であり、かつm+n+p+q=3である。)で表される有機アルミニウム化合物。このような化合物の具体例として、トリメチルアルミニウム、トリエチルアルミニウム、トリイソブチルアルミニウム、ジイソブチルアルミニウムハイドライドを例示することができる。
【0046】
(B-1b) 一般式 M2 AlRa 4
(式中、M2 はLi、NaまたはKを示し、Ra は炭素原子数が1〜15、好ましくは1〜4の炭化水素基を示す。)で表される周期律表第1族金属とアルミニウムとの錯アルキル化物。このような化合物としては、LiAl(C2H5)4、LiAl(C7H15)4 などを例示することができる。
(B-1c) 一般式 Ra Rb M3
(式中、Ra およびRb は、互いに同一でも異なっていてもよく、炭素原子数が1〜15、好ましくは1〜4の炭化水素基を示し、M3 はMg、ZnまたはCdである。)で表される周期律表第2族または第12族金属のジアルキル化合物。
上記の有機金属化合物(B-1)のなかでは、有機アルミニウム化合物が好ましい。また、このような有機金属化合物(B-1)は、1種単独で用いてもよいし2種以上組み合わせて用いてもよい。
【0047】
(B-2)有機アルミニウムオキシ化合物
本発明で用いられる(B-2) 有機アルミニウムオキシ化合物は、従来公知のアルミノキサンであってもよく、また特開平2−78687号公報に例示されているようなベンゼン不溶性の有機アルミニウムオキシ化合物であってもよい。
【0048】
従来公知のアルミノキサンは、たとえば下記のような方法によって製造することができ、通常、炭化水素溶媒の溶液として得られる。(1)吸着水を含有する化合物または結晶水を含有する塩類、たとえば塩化マグネシウム水和物、硫酸銅水和物、硫酸アルミニウム水和物、硫酸ニッケル水和物、塩化第1セリウム水和物などの炭化水素媒体懸濁液に、トリアルキルアルミニウムなどの有機アルミニウム化合物を添加して、吸着水または結晶水と有機アルミニウム化合物とを反応させる方法。
(2)ベンゼン、トルエン、エチルエーテル、テトラヒドロフランなどの媒体中で、トリアルキルアルミニウムなどの有機アルミニウム化合物に直接水、氷または水蒸気を作用させる方法。
(3)デカン、ベンゼン、トルエンなどの媒体中でトリアルキルアルミニウムなどの有機アルミニウム化合物に、ジメチルスズオキシド、ジブチルスズオキシドなどの有機スズ酸化物を反応させる方法。
【0049】
なお該アルミノキサンは、少量の有機金属成分を含有してもよい。また回収された上記のアルミノキサンの溶液から溶媒または未反応有機アルミニウム化合物を蒸留して除去した後、溶媒に再溶解またはアルミノキサンの貧溶媒に懸濁させてもよい。
アルミノキサンを調製する際に用いられる有機アルミニウム化合物として具体的には、前記(B-1a)に属する有機アルミニウム化合物として例示したものと同様の有機アルミニウム化合物を挙げることができる。
これらのうち、トリアルキルアルミニウム、トリシクロアルキルアルミニウムが好ましく、トリメチルアルミニウムが特に好ましい。
上記のような有機アルミニウム化合物は、1種単独でまたは2種以上組み合せて用いられる。
【0050】
また本発明で用いられるベンゼン不溶性の有機アルミニウムオキシ化合物は、60℃のベンゼンに溶解するAl成分がAl原子換算で通常10%以下、好ましくは5%以下、特に好ましくは2%以下であるもの、すなわち、ベンゼンに対して不溶性または難溶性であるものが好ましい。これらの有機アルミニウムオキシ化合物(B-2)は、1種単独でまたは2種以上組み合せて用いられる。
【0051】
(B-3) 遷移金属化合物と反応してイオン対を形成する化合物
本発明の架橋メタロセン化合物(A)と反応してイオン対を形成する化合物(B-3) (以下、「イオン化イオン性化合物」という。)としては、特開平1−501950号公報、特開平1−502036号公報、特開平3−179005号公報、特開平3−179006号公報、特開平3−207703号公報、特開平3−207704号公報、USP−5321106号などに記載されたルイス酸、イオン性化合物、ボラン化合物およびカルボラン化合物などを挙げることができる。さらに、ヘテロポリ化合物およびイソポリ化合物も挙げることができる。このようなイオン化イオン性化合物(B-3)は、1種単独でまたは2種以上組み合せて用いられる。
本発明の架橋メタロセン化合物をオレフィン重合触媒として使用する場合、助触媒成分としてのメチルアルミノキサンなどの有機アルミニウムオキシ化合物(B-2)とを併用すると、オレフィン化合物に対して特に高い重合活性を示す。
【0052】
また、本発明に係るオレフィン重合用触媒は、上記遷移金属化合物(A)、(B-1) 有機金属化合物、(B-2) 有機アルミニウムオキシ化合物、および(B-3) イオン化イオン性化合物から選ばれる少なくとも1種の化合物(B)とともに、必要に応じて担体(C)を用いることもできる。
【0053】
(C)担体
本発明で用いられる(C)担体は、無機または有機の化合物であって、顆粒状ないしは微粒子状の固体である。このうち無機化合物としては、多孔質酸化物、無機塩化物、粘土、粘土鉱物またはイオン交換性層状化合物が好ましい。
【0054】
多孔質酸化物として、具体的にはSiO2、Al2O3、MgO、ZrO、TiO2、B2O3、CaO、ZnO、BaO、ThO2など、またはこれらを含む複合物または混合物を使用、例えば天然または合成ゼオライト、SiO2-MgO、SiO2-Al2O3、SiO2-TiO2 、SiO2-V2O5 、SiO2-Cr2O3、SiO2-TiO2-MgOなどを使用することができる。これらのうち、SiO2および/またはAl2O3を主成分とするものが好ましい。このような多孔質酸化物は、種類および製法によりその性状は異なるが、本発明に好ましく用いられる担体は、粒径が10〜300μm、好ましくは20〜200μmであって、比表面積が50〜1000m2/g、好ましくは100〜700m2/gの範囲にあり、細孔容積が0.3〜3.0cm3/gの範囲にあることが望ましい。このような担体は、必要に応じて100〜1000℃、好ましくは150〜700℃で焼成して使用される。
【0055】
無機塩化物としては、MgCl2、MgBr2、MnCl2、MnBr2等が用いられる。無機塩化物は、そのまま用いてもよいし、ボールミル、振動ミルにより粉砕した後に用いてもよい。また、アルコールなどの溶媒に無機塩化物を溶解させた後、析出剤によって微粒子状に析出させたものを用いることもできる。
【0056】
本発明で用いられる粘土は、通常粘土鉱物を主成分として構成される。また、本発明で用いられるイオン交換性層状化合物は、イオン結合などによって構成される面が互いに弱い結合力で平行に積み重なった結晶構造を有する化合物であり、含有するイオンが交換可能なものである。大部分の粘土鉱物はイオン交換性層状化合物である。また、これらの粘土、粘土鉱物、イオン交換性層状化合物としては、天然産のものに限らず、人工合成物を使用することもできる。また、粘土、粘土鉱物またはイオン交換性層状化合物として、粘土、粘土鉱物、また、六方細密パッキング型、アンチモン型、CdCl2型、CdI2型などの層状の結晶構造を有するイオン結晶性化合物などを例示することができる。このような粘土、粘土鉱物としては、カオリン、ベントナイト、木節粘土、ガイロメ粘土、アロフェン、ヒシンゲル石、パイロフィライト、ウンモ群、モンモリロナイト群、バーミキュライト、リョクデイ石群、パリゴルスカイト、カオリナイト、ナクライト、ディッカイト、ハロイサイトなどが挙げられ、イオン交換性層状化合物としては、α-Zr(HAsO4)2・H2O、α-Zr(HPO4)2、α-Zr(KPO4)2・3H2O、α-Ti(HPO4)2、α-Ti(HAsO4)2・H2O、α-Sn(HPO4)2・H2O、γ-Zr(HPO4)2、γ-Ti(HPO4)2、γ-Ti(NH4PO4)2・H2Oなどの多価金属の結晶性酸性塩などが挙げられる。本発明で用いられる粘土、粘土鉱物には、化学処理を施すことも好ましい。化学処理としては、表面に付着している不純物を除去する表面処理、粘土の結晶構造に影響を与える処理など、何れも使用できる。化学処理として具体的には、酸処理、アルカリ処理、塩類処理、有機物処理などが挙げられる。
【0057】
本発明で用いられるイオン交換性層状化合物は、イオン交換性を利用し、層間の交換性イオンを別の大きな嵩高いイオンと交換することにより、層間が拡大した状態の層状化合物であってもよい。このような嵩高いイオンは、層状構造を支える支柱的な役割を担っており、通常、ピラーと呼ばれる。また、このように層状化合物の層間に別の物質を導入することをインターカレーションという。インターカレーションするゲスト化合物としては、TiCl4、ZrCl4などの陽イオン性無機化合物、Ti(OR)4、Zr(OR)4、PO(OR)3、B(OR)3などの金属アルコキシド(Rは炭化水素基など)、[Al13O4(OH)24]7+、[Zr4(OH)14]2+、[Fe3O(OCOCH3)6]+などの金属水酸化物イオンなどが挙げられる。これらの化合物は単独でまたは2種以上組み合わせて用いられる。また、これらの化合物をインターカレーションする際に、Si(OR)4、Al(OR)3、Ge(OR)4などの金属アルコキシド(Rは炭化水素基など)などを加水分解して得た重合物、SiO2などのコロイド状無機化合物などを共存させることもできる。また、ピラーとしては、上記金属水酸化物イオンを層間にインターカレーションした後に加熱脱水することにより生成する酸化物などが挙げられる。これらのうち、好ましいものは粘土または粘土鉱物であり、特に好ましいものはモンモリロナイト、バーミキュライト、ペクトライト、テニオライトおよび合成雲母である。
【0058】
有機化合物としては、粒径が10〜300μmの範囲にある顆粒状ないしは微粒子状固体を挙げることができる。具体的には、エチレン、プロピレン、1-ブテン、4-メチル-1-ペンテンなどの炭素原子数が2〜14のα−オレフィンを主成分として生成される(共)重合体またはビニルシクロヘキサン、スチレンを主成分として生成される(共)重合体、およびそれらの変成体を例示することができる。
【0059】
本発明に係るオレフィン重合用触媒は、本発明の架橋メタロセン化合物(A)、(B-1) 有機金属化合物、(B-2) 有機アルミニウムオキシ化合物、および(B-3) イオン化イオン性化合物から選ばれる少なくとも1種の化合物(B)、必要に応じて担体(C)と共に、必要に応じて後述するような特定の有機化合物成分(D)を含むこともできる。
【0060】
(D)有機化合物成分
本発明において、(D)有機化合物成分は、必要に応じて、重合性能および生成ポリマーの物性を向上させる目的で使用される。このような有機化合物としては、アルコール類、フェノール性化合物、カルボン酸、リン化合物およびスルホン酸塩等が挙げられるが、この限りではない。
【0061】
重合の際には、各成分の使用法、添加順序は任意に選ばれるが、以下のような方法が例示される。
(1) 成分(A)を単独で重合器に添加する方法。
(2) 成分(A)をおよび成分(B)を任意の順序で重合器に添加する方法。
(3) 成分(A)を担体(C)に担持した触媒成分、成分(B)を任意の順序で重合器に添加する方法。
(4) 成分(B)を担体(C)に担持した触媒成分、成分(A)を任意の順序で重合器に添加する方法。
(5) 成分(A)と成分(B)とを担体(C)に担持した触媒成分を重合器に添加する方法。
【0062】
上記(2) 〜(5) の各方法においては、各触媒成分の少なくとも2つ以上は予め接触されていてもよい。
成分(B)が担持されている上記(4)、(5)の各方法においては、必要に応じて担持されていない成分(B)を、任意の順序で添加してもよい。この場合成分(B)は、同一でも異なっていてもよい。
また、上記の成分(C)に成分(A)が担持された固体触媒成分、成分(C)に成分(A)および成分(B)が担持された固体触媒成分は、オレフィンが予備重合されていてもよく、予備重合された固体触媒成分上に、さらに、触媒成分が担持されていてもよい。
【0063】
本発明に係るオレフィンの重合方法では、上記のようなオレフィン重合用触媒の存在下に、オレフィンを重合または共重合することによりオレフィン重合体を得る。
本発明では、重合は溶液重合、懸濁重合などの液相重合法または気相重合法のいずれにおいても実施できる。液相重合法において用いられる不活性炭化水素媒体として具体的には、プロパン、ブタン、ペンタン、ヘキサン、ヘプタン、オクタン、デカン、ドデカン、灯油などの脂肪族炭化水素;シクロペンタン、シクロヘキサン、メチルシクロペンタンなどの脂環族炭化水素;ベンゼン、トルエン、キシレンなどの芳香族炭化水素;エチレンクロリド、クロルベンゼン、ジクロロメタンなどのハロゲン化炭化水素またはこれらの混合物などを挙げることができ、オレフィン自身を溶媒として用いることもできる。
【0064】
上記のようなオレフィン重合用触媒を用いて、オレフィンの重合を行うに際して、成分(A)は、反応容積1リットル当り、通常10-9〜10-1モル、好ましくは10-8〜10-2モルになるような量で用いられる。
成分(B-1)は、成分(B-1)と、成分(A)中の全遷移金属原子(M)とのモル比〔(B-1)/M〕が通常0.01〜5000、好ましくは0.05〜2000となるような量で用いられる。成分(B-2)は、成分(B-2)中のアルミニウム原子と、成分(A)中の全遷移金属(M)とのモル比〔(B-2)/M〕が、通常10〜5000、好ましくは20〜2000となるような量で用いられる。成分(B-3)は、成分(B-3)と、成分(A)中の遷移金属原子(M)とのモル比〔(B-3)/M〕が、通常1〜10、好ましくは1〜5となるような量で用いられる。
【0065】
成分(D)は、成分(B)が成分(B-1)の場合には、モル比〔(D)/(B-1)〕が通常0.01〜10、好ましくは0.1〜5となるような量で、成分(B)が成分(B-2)の場合には、モル比〔(D)/(B-2)〕
が通常0.01〜2、好ましくは0.005〜1となるような量で、成分(B)が成分(B-3)の場合は、モル比(D)/(B-3)〕が通常0.01〜10、好ましくは0.1〜5となるような量で用いられる。
【0066】
また、このようなオレフィン重合触媒を用いたオレフィンの重合温度は、通常−50〜+200℃、好ましくは0〜170℃の範囲である。重合圧力は、通常常圧〜10Mpaゲージ圧、好ましくは常圧〜5Mpaゲージ圧の条件下であり、重合反応は、回分式、半連続式、連続式のいずれの方法においても行うことができる。さらに重合を反応条件の異なる2段以上に分けて行うことも可能である。得られるオレフィン重合体の分子量は、重合系に水素を存在させるか、または重合温度を変化させることによっても調節することができる。さらに、使用する成分(B)の量により調節することもできる。水素を添加する場合、その量はオレフィン1kgあたり0.001〜100NL程度が適当である。
【0067】
モノマー本発明において、重合反応に供給されるオレフィンは、エチレンおよびα-オレフィンから選ばれる1種以上のモノマーであり、モノマーの少なくとも1種がエチレンまたはプロピレンである。α-オレフィンとしては、炭素原子数が3〜20、好ましくは3〜10の直鎖状または分岐状のα−オレフィン、たとえばプロピレン、1-ブテン、2-ブテン、1-ペンテン、3-メチル-1-ブテン、1-ヘキセン、4-メチル-1-ペンテン、3-メチル-1-ペンテン、1-オクテン、1-デセン、1-ドデセン、1-テトラデセン、1-ヘキサデセン、1-オクタデセン、1-エイコセなどである。また本発明の重合方法においては、炭素原子数が3〜30、好ましくは3〜20の環状オレフィン、たとえばシクロペンテン、シクロヘプテン、ノルボルネン、5-メチル-2-ノルボルネン、テトラシクロドデセン、2-メチル1,4,5,8-ジメタノ-1,2,3,4,4a,5,8,8a-オクタヒドロナフタレン;極性モノマー、たとえば、アクリル酸、メタクリル酸、フマル酸、無水マレイン酸、イタコン酸、無水イタコン酸、ビシクロ(2,2,1)-5-ヘプテン-2,3-ジカルボン酸無水物などのα,β−不飽和カルボン酸、およびこれらのナトリウム塩、カリウム塩、リチウム塩、亜鉛塩、マグネシウム塩、カルシウム塩などの金属塩;アクリル酸メチル、アクリル酸エチル、アクリル酸n-プロピル、アクリル酸イソプロピル、アクリル酸n-ブチル、アクリル酸イソブチル、アクリル酸 tert-ブチル、アクリル酸2-エチルヘキシル、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸n-プロピル、メタクリル酸イソプロピル、メタクリル酸n-ブチル、メタクリル酸イソブチルなどのα,β−不飽和カルボン酸エステル;酢酸ビニル、プロピオン酸ビニル、カプロン酸ビニル、カプリン酸ビニル、ラウリン酸ビニル、ステアリン酸ビニル、トリフルオロ酢酸ビニルなどのビニルエステル類;アクリル酸グリシジル、メタクリル酸グリシジル、イタコン酸モノグリシジルエステルなどの不飽和グリシジルなどを挙げることができる。また、ビニルシクロヘキサン、ジエンまたはポリエン;芳香族ビニル化合物、例えばスチレン、o-メチルスチレン、m-メチルスチレン、p-メチルスチレン、o,p-ジメチルスチレン、o-エチルスチレン、m-エチルスチレン、p-エチルスチレンなどのモノもしくはポリアルキルスチレン;メトキシスチレン、エトキシスチレン、ビニル安息香酸、ビニル安息香酸メチル、ビニルベンジルアセテート、ヒドロキシスチレン、o-クロロスチレン、p-クロロスチレン、ジビニルベンゼンなどの官能基含有スチレン誘導体;および3- フェニルプロピレン、4-フェニルプロピレン、α- メチルスチレンなどを反応系に共存させて重合を進めることもできる。
【0068】
本発明の重合方法においては、モノマーの少なくとも1種がエチレンまたはプロピレンである。モノマーが二種以上である場合には、エチレン、プロピレンまたは(エチレン+プロピレン)が全体モノマー量の50モル%以上であることが好ましい。
【0069】
次に、本発明の遷移金属化合物を含む触媒の存在下、オレフィンの重合によって得られる重合体の物性・性状を測定する方法について述べる。
【0070】
〔重量平均分子量(Mw)、数平均分子量(Mn)〕
ウォーターズ社製GPC−150Cを用い、以下のようにして測定した。分離カラムは、TSKgel GMH6−HT及びTSKgel GMH6−HTLであり、カラムサイズはそれぞれ内径7.5mm、長さ600mmであり、カラム温度は140℃とし、移動相にはo-ジクロロベンゼン(和光純薬工業)および酸化防止剤としてBHT(武田薬品)0.025重量%を用い、1.0ml/分で移動させ、試料濃度は0.1重量%とし、試料注入量は500マイクロリットルとし、検出器として示差屈折計を用いた。標準ポリスチレンは、分子量がMw<1000およびMw>4×106 については東ソー社製を用い、1000≦Mw≦4×106 についてはプレッシャーケミカル社製を用いた。
〔極限粘度([η])〕
デカリン溶媒を用いて、135℃で測定した値である。すなわち造粒ペレット約20mgをデカリン15mlに溶解し、135℃のオイルバス中で比粘度ηspを測定する。このデカリン溶液にデカリン溶媒を5ml追加して希釈後、同様にして比粘度ηspを測定する。この希釈操作をさらに2回繰り返し、濃度(C)を0に外挿した時のηsp/Cの値を極限粘度として求める。
[η]=lim(ηsp/C) (C→0)
〔メルトフローレート(MFR10)〕
ASTM D−1238の標準法により、190℃、10kg荷重下で測定された数値である。
〔密度〕
190℃、2.16kg荷重におけるMFR測定後のストランドを、120℃で1時間熱処理し、1時間かけて室温まで徐冷したのち、密度勾配管法により測定した。
〔融点(Tm)〕
示差走査熱量測定(DSC)によって、240℃で10分間保持した重合体サンプルを、30℃まで冷却して5分間保持した後に、10℃/分で昇温させたときの結晶溶融ピークから算出した。
【0071】
【実施例】
以下、実施例に基づいて本発明をさらに具体的に説明するが、本発明はこれら実施例に限定されるものではない。
合成例で得られた化合物の構造は、270MHz 1H-NMR(日本電子GSH−270)、FD−質量分析(日本電子SX−102A)等を用いて決定した。
【0072】
〔合成例1〕
[エチレン(シクロペンタジエニル)(2,7−tertブチルフルオレニル)ジルコニウムジクロライドの合成]
(1)2、7− tert ブチル−9−(2−ヒドロキシエチル)フルオレン
氷冷下で2,7−tertブチルフルオレン(5.00g、17.96mmol)のTHF溶液にn−ブチルリチウム/ヘキサン溶液(12.27ml、19.39mmol)を窒素雰囲気下で滴下し、さらに室温で14時間攪拌した。この溶液を−78℃に冷却し2−ブロモエチルトリメチルシラノール(5.31g、26.94mmol)をゆっくりと加えた。そのまま攪拌を続け、ゆっくりと一晩かけて室温にした。10%塩酸水溶液(60ml)をゆっくりと加えて反応を停止し、飽和炭酸水素ナトリウム水溶液を加えて中和した。この反応混合物からエーテル抽出を行い、無水硫酸マグネシウムで乾燥、溶媒を除きOilを得た。これを酢酸エチル/ヘキサン=1/5を展開液としたシリカゲルカラムを通じて精製し、該当フラクションの溶媒を留去し白色固体を得た(5.00g、15.50mmol、86.33%)。分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.62(d、2H)、7.50(d、2H)、7.40(dd、2H)、4.06(t、1H)、3.64(t、2H)、2.28(q、2H)、1.37(s、18H)
【0073】
(2)2、7− tert ブチル−9−(2−ブロモエチル)フルオレン
氷冷下で2、7−tertブチル−9−(2−ヒドロキシエチル)フルオレン(4.00g、12.40mmol)の塩化メチレン溶液にトリエチルアミン(2.62ml,18.54mmol)を加え、メシルクロライド(1.27ml、18.55mmol)を窒素雰囲気下でゆっくり加え、0℃で1時間攪拌し、更に室温で122時間攪拌した。生成したスラリーに水を加えて反応を停止し、塩化メチレンで抽出した(20ml×4回)。無水硫酸マグネシウムで乾燥、溶媒を除きOilを得た。これをアセトン(50ml)に溶解し、臭化リチウム(5.39g、62.06mmol)のアセトン溶液(100ml)に室温で加えた。38時間後溶媒を留去した後にエーテルを加えて懸濁液とし更に水を加えた。エーテル層を分離し、無水硫酸マグネシウムで乾燥、溶媒を除き固体を得た。これをヘキサンを展開液としたシリカゲルカラムを通じて精製し、該当フラクションの溶媒を留去し白色固体を得た(4.52g、11.73mmol、94.6%)。
分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.63(d、2H)、7.51(d、2H)、7.40(dd、2H)、4.09(t、1H)、3.36(t、2H)、2.48(q、2H)、1.38(s、18H)
【0074】
(3)1−(シクロペンタジエニル)−2−(2,7− tert ブチル−9−フルオレニル)エタン
9−(2−ブロモエチル)フルオレン(1.98g、5.14mmol)のTHF(40ml)溶液にヘキサメチルホスホルアミド(3.00ml、17.34mmol)を加えて−78℃に冷却し、2Mナトリウムシクロペンタジエン/THF溶液(2.8ml、5.6mmol)のTHF溶液を加えた後にゆっくりと室温にしそのまま21時間攪拌した。水を加えて反応を停止してエーテルで抽出(50ml×3)し、このエーテル溶液を水で洗浄した(50ml×2)。これを無水硫酸マグネシウムで乾燥、溶媒を除き粘性固体を得た。ヘキサンを展開液としたシリカゲルカラムを通じて精製し、該当フラクションの溶媒を留去し白色固体を得た(0.99g、2.67mmol、52.0%)。分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.63(d、2H)、7.52(d、2H)、7.38(dd、2H)、6.41−6.00(m、3H)、3.97(t、1H)、2.84(dd、2H)、2.27(bm、4H)、1.38(s、18H)
【0075】
(4)[エチレン(シクロペンタジエニル)(2,7− tert ブチルフルオレニル)ジルコニウムジクロライド
1−(シクロペンタジエニル)−2−(2,7−tertブチル−9−フルオレニル)エタン(0.50 g、1.35 mmol)のジエチルエーテル(40ml)溶液を0℃に冷却し、1.57M n−ブチルリチウム/ヘキサン溶液(1.77ml、2.77mmol)を窒素雰囲気下で滴下した。滴下後室温に戻し終夜攪拌し、−78℃まで冷却し、四塩化ジルコニウム・テトラヒドロフラン錯体(1:2)(284mg、1.22 mmol)を添加し、一晩かけてゆっくり室温にした。溶媒を乾固し、グローブボックス内でヘキサンを加えろ過し、ヘキサン可溶成分を取り除いた。次に塩化メチレン可溶成分をろ過にて分取し、溶媒を留去し、塩化メチレンで再結晶をすると橙色固体を得た(0.40g、0.75mmol、62.0%)。分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.89―7.86(m、2H)、7.65−61(m、2H)、7.54(m、2H)、6.20(t、2H)、5.88(t、2H)、3.85(t、2H)、3.61(t、2H)、1.39(s、18H)
FD−MASS m/z=530(M+)
【0076】
〔合成例2〕
[エチレン(シクロペンタジエニル)(3,6−tertブチルフルオレニル)ジルコニウムジクロライドの合成]
(1)3、6− tert ブチル−9−(2−ヒドロキシエチル)フルオレン
氷冷下で3,6−tertブチルフルオレン(2.38g、8.5mmol)のTHF溶液にn−ブチルリチウム/ヘキサン溶液(6.2ml、10.3mmol)を窒素雰囲気下で滴下し、さらに室温で4時間攪拌した。この溶液を−78℃に冷却し2−ブロモエチルトリメチルシラノール(2.5g)をゆっくりと加えた。そのまま攪拌を続け、ゆっくりと一晩かけて室温にした。10日後10%塩酸水溶液(30ml)をゆっくりと加えて反応を停止し、飽和炭酸水素ナトリウム水溶液を加えて中和した。この反応混合物からエーテル抽出を行い、無水硫酸マグネシウムで乾燥、溶媒を除き固体を得た。これをヘキサンを展開液としたシリカゲルカラムを通じて精製し、該当フラクションの溶媒を留去し白色固体を得た(1.96g、6.42mmol、75.5%)。分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.78(d、2H)、7.43(dd、2H)、7.34(dd、2H)、4.03(t、1H)、3.64(t、2H)、2.26(q、2H)、1.41(s、18H)
【0077】
(2)3、6− tert ブチル−9−(2−ブロモエチル)フルオレン
氷冷下で3、6−tertブチル−9−(2−ヒドロキシエチル)フルオレン(1.88g、6.15mmol)の塩化メチレン溶液にトリエチルアミン(1.3ml,9.2mmol)を加え、メシルクロライド(0.63ml、9.2mmol)を窒素雰囲気下でゆっくり加え、0℃で1時間攪拌し、更に室温で24時間攪拌した。これに水を加えて反応を停止し、塩化メチレンで抽出した(20ml×4回)。無水硫酸マグネシウムで乾燥、溶媒を除き固体を得た。この固体をアセトン(25ml)に溶解し、臭化リチウム(2.67g、30.8mmol)のアセトン溶液(40ml)に室温で加えた。40時間後溶媒を留去した後にエーテルを加えて、更に水を加えた。エーテル層を分離し、無水硫酸マグネシウムで乾燥、溶媒を除き固体を得た。これを少量のペンタンで洗浄し白色固体を得た(1.96g、5.1mmol、82.8%)。
分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.78(d、2H)、7.43(d、2H)、7.34(dd、2H)、4.06(t、1H)、3.34(t、2H)、2.46(q、2H)、1.41(s、18H)
FD−MASS m/z=384(M+)、386
【0078】
(3)1−(シクロペンタジエニル)−2−(3,6− tert ブチル−9−フルオレニル)エタン
−78℃下でシクロペンタジエン(0.21g、3.18mmol)のTHF溶液にn−ブチルリチウム/ヘキサン溶液(2.13ml、3.34mmol)を窒素雰囲気下で滴下し、さらに室温で1時間攪拌した。次に3、6−tertブチル−9−(2−ブロモエチル)フルオレン(1.10g、2.85mmol)のTHF(30ml)溶液にヘキサメチルホスホルアミド(1.47ml、8.5mmol)を加えて−78℃に冷却し、前述の溶液を加えた後にゆっくりと室温にしそのまま14時間攪拌した。水を加えて反応を停止してエーテルで抽出(50ml×3)し、このエーテル溶液を水で洗浄した(50ml×2)。これを無水硫酸マグネシウムで乾燥、溶媒を除き粘性固体を得た。これを少量のメタノールで洗浄した後、ヘキサンを展開液としたシリカゲルカラムを通じて精製し、該当フラクションの溶媒を留去し白色固体を得た(0.75g、2.02mmol、70.9%)。分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.78(d、2H)、7.44(dd、2H)、7.32(dt、2H)、6.41−6.00(m、3H)、3.94(t、1H)、2.90(dd、2H)、2.32−2.17(m、4H)、1.42(s、18H)
【0079】
(4)[エチレン(シクロペンタジエニル)(3,6− tert ブチルフルオレニル)ジルコニウムジクロライド
1−(シクロペンタジエニル)−2−(3,6−tertブチル−9−フルオレニル)エタン(0.50 g、1.35 mmol)のジエチルエーテル(40ml)溶液を0℃に冷却し、1.57M n−ブチルリチウム/ヘキサン溶液(1.77ml、2.77mmol)を窒素雰囲気下で滴下した。滴下後室温に戻し終夜攪拌し、−78℃まで冷却し、四塩化ジルコニウム・テトラヒドロフラン錯体(1:2)(258 mg、1.11 mmol)を添加し、一晩かけてゆっくり室温に戻した。溶媒を乾固し、グローブボックス内でヘキサンを加え、ろ過し、ヘキサン可溶成分を取り除いた。次に塩化メチレン可溶成分をろ過にて分取し、溶媒を留去し、塩化メチレンで再結晶をすると黄橙色固体を得た(0.05g、0.09mmol、8.5%)。分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.94(m、2H)、7.62−7.42(m、4H)、6.20(t、2H)、5.95(t、2H)、3.82(t、2H)、3.58(t、2H)、1.45(s、18H)
FD−MASS m/z=530(M+)
【0080】
〔合成例3〕
[1,1,2,2−テトラメチルジシレン−1−(シクロペンタジエニル)−2−(フルオレニル)ジルコニウムジクロリドの合成]
(1)1−クロロ−2−フルオレニル - テトラメチルジシラン
磁気攪拌子を備えた100mlギルダールフラスコを充分に窒素置換した後、フルオレン(1.70g、10.2mmol)を入れ、ジエチルエーテル30mlを加えて溶解させた。氷浴で冷やしながら1.59M n-ブチルリチウム/ヘキサン溶液(6.5ml、10mmol)を添加した後、窒素雰囲気下室温で18時間攪拌し、溶液を得た。一方、50ml滴下漏斗、三方コックおよび磁気攪拌子を備えた200ml四口フラスコを充分に窒素置換した後、ヘキサン60mlおよび1,2−ジクロロテトラメチルジシラン(2.0ml、10mmol)を入れた。これをメタノール/ドライアイス浴で冷却しながら、先程の溶液を滴下漏斗を用いて2時間かけて徐々に添加した。その後室温まで徐々に昇温しながら窒素雰囲気下で18時間攪拌し、スラリーを得た。固体を濾過して除いた後、溶媒を減圧下で留去し、黄色固体として1−クロロ−2−フルオレニル-テトラメチルジシランを得た(3.17g、10mmol、98.0%)。1−クロロ−2−フルオレニル-テトラメチルジシランの分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ−0.38(s, 6H), 0.32(s, 6H), 3.92(s, 1H), 7.1−7.3(m, 4H),7.4−7.5(m,2H), 7.7−7.8(m, 2H)
【0081】
(2)1−シクロペンタジエニル−2−フルオレニル - テトラメチルジシラン
三方コックおよび磁気攪拌子を備えた200ml二口フラスコを充分に窒素置換した後、1−クロロ−2−フルオレニル-テトラメチルジシラン(3.17g、10mmol)を入れ、ジエチルエーテル40mlを加えた。ヘキサメチルリン酸トリアミド4.0mlを加えた後、氷水浴で冷却しながら2.0M シクロペンタジエニルナトリウム/テトラヒドロフラン溶液(10.2ml、20mmol)を徐々に添加した。室温まで徐々に昇温した後、窒素雰囲気下室温で3日間攪拌しスラリーを得た。これに飽和塩化アンモニウム水溶液100mlを加え、300ml分液漏斗を用いて水層を除き、有機層を得た。さらに、除いた水層をジエチルエーテル30mlで抽出し、先程の有機層と合わせた。これを水100mlで2回、飽和食塩水100mlで1回洗い、無水硫酸マグネシウムで1時間乾燥した後、濾過して硫酸マグネシウムを除き、濾液の溶媒を留去して油状物を得た。カラムクロマトグラフ(溶媒:ヘキサン)による分離精製を行い、固体を得た。この後、この固体のヘキサン溶液を低温(約−10℃)にして再結晶し、白色固体として1−シクロペンタジエニル−2−フルオレニル-テトラメチルジシランを得た(1.256g、3.622mmol、36.2%)。1−シクロペンタジエニル−2−フルオレニル-テトラメチルジシランの分析値を下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ−0.73(s, 6H), 0.37(s, 6H), 3.95(s, 1H), 5.7−6.7(bs, 4H),7.2−7.4(m, 4H), 7.5−7.6(m, 2H), 7.8−7.9(m,2H)
FD−MASS m/z=346(M+)
【0082】
(3)1,1,2,2−テトラメチルジシレン−1− ( シクロペンタジエニル ) −2− ( フルオレニル ) ジルコニウムジクロリド
磁気攪拌子を備えた100mlギルダールフラスコを充分に窒素置換した後、1−シクロペンタジエニル−2−フルオレニル-テトラメチルジシラン(484mg、1.40mmol)を入れ、ジエチルエーテル20mlを加えた。氷水浴で冷やしながら、1.58M n-ブチルリチウム/ヘキサン溶液(1.8ml、2.8mmol)を加えた。窒素雰囲気下室温で23時間攪拌した後、溶媒を留去し固体を得た。この固体に、メタノール/ドライアイス浴で約−78℃に冷却した。塩化メチレン20mlを加え、この溶液に、予め四塩化ジルコニウム・テトラヒドロフラン錯体(1:2)(478mg、1.27mmol)を塩化メチレン20mlに溶かした後メタノール/ドライアイス浴で約−78℃に冷却した溶液を約−78℃に保ちながらゆっくり加えた。溶液を加えた後、室温まで徐々に昇温しながら窒素雰囲気下で42時間攪拌したところスラリーが得られた。溶媒を留去して得られた黄色固体を塩化メチレンで抽出し、さらに溶媒を留去して1,1,2,2−テトラメチルジシレン−1−(シクロペンタジエニル)−2−(フルオレニル)ジルコニウムジクロリド(355mg、0.701mmol、55.2%)を得た。1,1,2,2−テトラメチルジシレン−1−(シクロペンタジエニル)−2−(フルオレニル)ジルコニウムジクロリドの分析値を下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ0.60(s, 6H), 0.71(s, 6H), 6.07(dd , 2H), 6.22(dd, 2H), 7.3−7.5(m, 4H), 7.7−7.9(m, 2H), 8.1−8.3(m, 2H)FD−MASS m/z=506(M+)
【0083】
〔合成例4〕
[ 1,1,2,2−テトラメチルジシレン−1−(シクロペンタジエニル)−2−(2,7−ジ-t-ブチルフルオレニル)ジルコニウムジクロリドの合成]
(1)1−クロロ−2− ( 2,7−ジ -t- ブチルフルオレニル )- テトラメチ ルジシラン
三方コックおよび磁気攪拌子を備えた300ml二口フラスコを充分に窒素置換した後、2,7−ジ-t-ブチルフルオレン(1.939g、6.965mmol)を入れ、ジエチルエーテル100mlを加えて溶解させた。氷浴で冷やしながら1.56M n-ブチルリチウム/ヘキサン溶液(4.5ml、7.0mmol)を添加した後、窒素雰囲気下室温で17時間攪拌し溶液を得た。一方、200ml滴下漏斗、三方コックおよび磁気攪拌子を備えた500ml三口フラスコを充分に窒素置換した後、ヘキサン200mlおよび1,2−ジクロロテトラメチルジシラン(1.3ml、7.0mmol)を入れた。これをメタノール/ドライアイス浴で冷却しながら、先程の溶液を滴下漏斗を用いて2時間かけて徐々に添加した。その後室温まで徐々に昇温しながら窒素雰囲気下で24時間攪拌しスラリーを得た。この固体を濾過して除いた後、溶媒を減圧下で留去し、薄黄色固体として1−クロロ−2−(2,7−ジ-t-ブチルフルオレニル)-テトラメチルジシランを得た(3.0g、7.0mmol、100%)。1−クロロ−2−(2,7−ジ-t-ブチルフルオレニル)-テトラメチルジシランの分析値を下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ0.44(s,6H),0.45(s, 6H),1.36(s,18H), 3.92(s, 1H),7.3−7.4(m,2H), 7.5−7.6(m, 2H), 7.70(d, 2H)
【0084】
(2)1−シクロペンタジエニル−2− ( 2,7−ジ -t- ブチルフルオレニル )- テトラメチルジシラン
三方コックおよび磁気攪拌子を備えた300ml二口フラスコを充分に窒素置換した後、1−クロロ−2−フルオレニル-テトラメチルジシラン(3.0g、7.0mmol)を入れ、ジエチルエーテル100mlを加えて溶液とした。ヘキサメチルリン酸トリアミド5.0mlを加えた後、氷水浴で冷却しながら2.0M シクロペンタジエニルナトリウム/テトラヒドロフラン溶液(7.0ml、14mmol)を徐々に添加した。室温まで徐々に昇温した後、窒素雰囲気下室温で5日間攪拌し、スラリーを得た。これに1N塩酸100mlを加え、300ml分液漏斗を用いて水層を除き、有機層を得た。この有機層を水100mlで2回、飽和食塩水100mlで1回洗い、無水硫酸マグネシウムで1.5時間乾燥した後、濾過して硫酸マグネシウムを除き、濾液の溶媒を留去して油状物を得た。カラムクロマトグラフ(溶媒:ヘキサン)による分離精製を行い、黄白色固体として1−シクロペンタジエニル−2−(2,7−ジ-t-ブチルフルオレニル)-テトラメチルジシランを得た(1.33g、3.90mmol、56%)。1−シクロペンタジエニル−2−(2,7−ジ-t-ブチルフルオレニル)-テトラメチルジシランの分析値を下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ−0.83(s, 6H),0.41(s, 6H), 1.36(s, 18H), 3.88(s, 1H),5.9−6.6(bs, 4H), 7.34(dd, 2H), 7.54(d, 2H), 7.70(d,2H)
FD−MASS m/z=458(M+)
【0085】
(3)1,1,2,2−テトラメチルジシレン−1− ( シクロペンタジエニル ) −2− ( 2,7−ジ -t- ブチルフルオレニル ) ジルコニウムジクロリド
磁気攪拌子を備えた100mlギルダールフラスコを充分に窒素置換した後、1−シクロペンタジエニル−2−(2,7−ジ-t-ブチルフルオレニル)-テトラメチルジシラン(512mg、1.12mmol)を入れ、ジエチルエーテル30mlを加えて溶液とした。氷水浴で冷やしながら、1.56M n-ブチルリチウム/ヘキサン溶液(1.50ml、2.34mmol)を加えた。窒素雰囲気下室温で19時間攪拌した後、溶媒を留去し、固体を得た。この固体に、メタノール/ドライアイス浴で約−78℃に冷却した塩化メチレン20mlを加えた。その後この溶液に、予め四塩化ジルコニウム・テトラヒドロフラン錯体(1:2)(380mg、1.01mmol)を塩化メチレン20mlに溶かした後メタノール/ドライアイス浴で約−78℃に冷却した溶液を−78℃に保ちながら加えた。溶液を加えた後、室温まで徐々に昇温しながら窒素雰囲気下で3日間攪拌したところスラリーが得られた。溶媒を留去して得られた固体をヘキサンで抽出し、さらに溶媒を留去して1,1,2,2−テトラメチルジシレン−1− (シクロペンタジエニル)−2−(2,7−ジ-t-ブチルフルオレニル)ジルコニウムジクロリド(408mg、0,659mmol、 65.3%)を橙色固体として得た。1,1,2,2−テトラメチルジシレン−1−(シクロペンタジエニル)−2−(2,7−ジ-t-ブチルフルオレニル)ジルコニウムジクロリドの分析値を下に示す。1H−NMR(270MHz、CDCl3中、TMS基準)δ0.60(s, 6H), 0.73(s, 6H), 1.40(s, 18H), 6.00(dd, 2H),6.23(dd, 2H), 7.47(dd, 2H), 7.67(dd, 2H), 8.05(dd, 2H)
FD−MASS m/z=618(M+)
【0086】
〔合成例5〕
[エチレン(シクロペンタジエニル)(オクタメチルオクタヒドロジベンゾフルオレニル)ジルコニウムジクロリドの合成]
(1)9−(2−ヒドロキシエチル)オクタメチルオクタヒドロジベンゾフルオレン
氷冷下でオクタメチルオクタヒドロジベンゾフルオレン(5.00g、12.93mmol)のTHF溶液にn−ブチルリチウム/ヘキサン溶液(8.84ml、13.97mmol)を窒素雰囲気下で滴下し、さらに室温で14時間攪拌した。この溶液を−78℃に冷却し、2−ブロモエチルトリメチルシラノール(3.82g、フルオレンに対して1.5当量)をゆっくりと加えた。そのまま攪拌を続け、一晩かけて室温にした。19時間後、10%塩酸水溶液(60ml)をゆっくりと加えて反応を停止し、飽和炭酸水素ナトリウム水溶液を加えて中和した。この反応混合物からエーテル抽出を行い、無水硫酸マグネシウムで乾燥、溶媒を除き固体を得た。これを酢酸エチル/ヘキサン=1/5を展開液としたシリカゲルカラムを通じて精製し、該当フラクションの溶媒を留去し白色固体を得た(4.14g、9.61mmol、74.4%)。分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.7−7.38(s+s、4H)、3.95(t、1H)、3.69(t、2H)、2.23(q、2H)、1.72(s、8H)、1.45−1.2(d+d、12H+12H)
【0087】
(2)9−(2−ブロモエチル)オクタメチルオクタヒドロジベンゾフルオレン氷冷下で9−(2−ヒドロキシエチル)オクタメチルオクタヒドロジベンゾフルオレン(3.50g、8.13mmol)の塩化メチレン溶液にトリエチルアミン(1.22ml,9.38mmol)を加え、メシルクロライド(0.83ml、12.12mmol)を窒素雰囲気下でゆっくり加え、0℃で1時間攪拌し、更に室温で24時間攪拌した。生成したスラリーに水を加えて反応を停止し、塩化メチレンで抽出した(20ml×4回)。無水硫酸マグネシウムで乾燥、溶媒を除き白色固体を得た。この固体をアセトン50mlに溶解し、臭化リチウム(3.53g、40.7mmol)のアセトン溶液100mlに室温で加えた。40時間後溶媒を留去した後にエーテルを加え、更に水を加えた。エーテル層を分離し、無水硫酸マグネシウムで乾燥、溶媒を除き固体を得た。これをヘキサンを展開液としたシリカゲルカラムを通じて精製し、該当フラクションの溶媒を留去し白色固体を得た(3.56g、7.21mmol、88.8%)。分析値を以下に示す。分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.61(s、2H)、7.38(s、2H)、4.00(t、1H)、3.43(t、2H)、2.40(q、2H)、1.72(s、8H)、1.38−1.32(d+d、12H+12H)
FD−MASS m/z=384(M+)、386
【0088】
(3)1−シクロペンタジエニル−2− ( オクタメチルオクタヒドロジベンゾフルオレニル ) エタン
−78℃下で、9−(2−ブロモエチル)オクタメチルオクタヒドロジベンゾフルオレン(2.50g、5.07mmol)のTHF(40ml)溶液にヘキサメチルホスホルアミド(2.66ml、15.4mmol)を加えて−78℃に冷却し、2.0M シクロペンタジエニルナトリウム/テトラヒドロフラン溶液(2.80ml、5.60mmol)を加えた後にゆっくりと室温にしそのまま12時間攪拌した。水を加えて反応を停止してエーテルで抽出(50ml×3)し、このエーテル溶液を水で洗浄した(50ml×2)。これを無水硫酸マグネシウムで乾燥、溶媒を除き粘性固体を得た。これをヘキサンを展開液としたシリカゲルカラムを通じて精製し、該当フラクションの溶媒を留去し薄黄色固体を得た(0.51g、1.07mmol、21.0%)。分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.61(s、2H)、7.40(d、2H)、6.41−6.00(m、3H)、3.87(t、1H)、2.90(dd、2H)、2.41−2.14(m、4H)、1.72(s、8H)、1.43−1.26(d+s、12H+12H)
【0089】
(4)エチレン ( シクロペンタジエニル )( オクタメチルオクタヒドロジベンゾフルオレニル ) ジルコニウムジクロリド
磁気攪拌子を備えた100mlギルダールフラスコを充分に窒素置換した後、1−シクロペンタジエニル−2−(オクタメチルオクタヒドロジベンゾフルオレニル)エタン(0.50g、1.04mmol)を入れ、ジエチルエーテル50mlを加えて溶液とした。氷水浴で冷やしながら、1.56M n-ブチルリチウム/ヘキサン溶液(1.40ml、2.18mmol)を徐々に加えた後、窒素雰囲気下室温で15時間攪拌してスラリーを得た。メタノール/ドライアイス浴で冷やしながら四塩化ジルコニウム・テトラヒドロフラン錯体(1:2)(0.388g、1.03mmol)を加えた後、室温まで徐々に昇温し、窒素雰囲気下室温で24時間攪拌してスラリーを得た。減圧下で溶媒を留去して得られた固体をヘキサンで洗浄し、続いて塩化メチレンで抽出し、この溶液の溶媒を減圧下で留去し、橙色固体としてエチレン(シクロペンタジエニル)(オクタメチルオクタヒドロジベンゾフルオレニル)ジルコニウムジクロリド(0.440g、0.689mmol、66.2%)を得た。エチレン(シクロペンタジエニル)(オクタメチルオクタヒドロジベンゾフルオレニル)ジルコニウムジクロリドの分析値を下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ1.31(s, 6H), 1.39(s, 6H), 1.42(s,6H), 1.51(s, 6H), 1.6−1.9(m, 8H), 3.56(t, 2H), 3.79(t, 2H), 5.83(t, 2H), 6.14(t,2H), 7.49(s, 2H), 7.90(s, 2H)
FD−MASS m/z=638(M+)
【0090】
〔合成例6〕
[エチレン(4−tertブチル−2−メチルシクロペンタジエニル)(3,6−tertブチルフルオレニル)ジルコニウムジクロライドの合成]
(1)3、6− tert ブチル−9−(2−ヒドロキシエチル)フルオレン
氷冷下で3,6−tertブチルフルオレン(2.38g、8.5mmol)のTHF溶液にn−ブチルリチウム/ヘキサン溶液(6.2ml、10.3mmol)を窒素雰囲気下で滴下し、さらに室温で4時間攪拌した。この溶液を−78℃に冷却し2−ブロモエチルトリメチルシラノール(2.5g)をゆっくりと加えた。そのまま攪拌を続け、ゆっくりと一晩かけて室温にした。10日後10%塩酸水溶液(30ml)をゆっくりと加えて反応を停止し、飽和炭酸水素ナトリウム水溶液を加えて中和した。この反応混合物からエーテル抽出を行い、無水硫酸マグネシウムで乾燥、溶媒を除き白色固体を得た。これをヘキサンを展開液としたシリカゲルカラムを通じて精製し、該当フラクションの溶媒を留去し白色固体を得た(1.96g、6.42mmol、75.5%)。分析値を以下に示す。1H−NMR(270MHz、CDCl3中、TMS基準)δ7.78(d、2H)、7.43(dd、2H)、7.34(dd、2H)、4.03(t、1H)、3.64(t、2H)、2.26(q、2H)、1.41(s、18H)
【0091】
(2)3、6− tert ブチル−9−(2−ブロモエチル)フルオレン
氷冷下で3、6−tertブチル−9−(2−ヒドロキシエチル)フルオレン(1.88g、6.15mmol)の塩化メチレン溶液にトリエチルアミン(1.3ml,9.2mmol)を加え、メシルクロライド(0.63ml、9.2mmol)を窒素雰囲気下でゆっくり加え、0℃で1時間攪拌し、更に室温で24時間攪拌した。生成したスラリーに水を加えて反応を停止し、塩化メチレンで抽出した(20ml×4回)。無水硫酸マグネシウムで乾燥、溶媒を除き固体を得た。この固体をアセトン25mlに溶解し、臭化リチウム(2.67g、30.8mmol)のアセトン溶液40mlに室温で加えた。40時間後溶媒を留去した後にエーテルを加え、更に水を加えた。エーテル層を分離し、無水硫酸マグネシウムで乾燥し、溶媒を除き固体を得た。これを少量のペンタンで洗浄し白色固体を得た(1.96g、5.1mmol、82.8%)。
分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.78(d、2H)、7.43(d、2H)、7.34(dd、2H)、4.06(t、1H)、3.34(t、2H)、2.46(q、2H)、1.41(s、18H)
FD−MASS m/z=384(M+)、386
【0092】
(3)(4− tert ブチル−2−メチルシクロペンタジエニル)(3,6− tert ブチル−9−フルオレニル)エタン
氷冷下で1−tertブチル−3−メチルシクロペンタジエン(0.50g、3.7mmol)のTHF溶液にn−ブチルリチウム/ヘキサン溶液(1.9ml、3.1mmol)を窒素雰囲気下で滴下し、さらに室温で21時間攪拌した。次に3、6−tertブチル−9−(2−ブロモエチル)フルオレン(1.1g、2.8mmol)のTHF(50ml)溶液にヘキサメチルホスホルアミド(1.47ml、8.5mmol)を加えて−78℃に冷却し、前述の溶液を加えた後にゆっくりと室温にしそのまま2日間攪拌した。この溶液から少量サンプリングし、ガスクロマトグラフィーで原料が残っていないことを確認した後、水を加えて反応を停止してエーテルで抽出(50ml×3)し、このエーテル溶液を水で洗浄した(50ml×2)。これを無水硫酸マグネシウムで乾燥、溶媒を除き粘性固体を得た。これを少量のメタノールで洗浄した後、酢酸エチル/ヘキサン(1/9)を展開液としたシリカゲルカラムを通じて精製し、該当フラクションの溶媒を留去し淡黄色粘性固体を得た(1.16g、2.68mmol、95.7%)。分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.78(d、2H)、7.43(d、2H)、7.32(dt、2H)、5.89(d、1H)、3.89(t、1H)、2.80(d、2H)、2.04−2.76(m、4H)、1.74(d、3H)、1.42(s、18H)
FD−MASS m/z=440(M+)、441
【0093】
(4)エチレン(4− tert ブチル−2−メチルシクロペンタジエニル)(3,6− tert ブチルフルオレニル)ジルコニウムジクロライド
氷冷下で1−(4−tertブチル−2−メチルシクロペンタジエニル)−2−(3,6−tertブチル−9−フルオレニル)エタン(0.50g、1.16mmol)のTHF(40ml)溶液にn−ブチルリチウム/ヘキサン溶液(1.51ml、2.44mmol)を窒素雰囲気下で滴下し、さらに室温で15時間攪拌した。反応混合物から溶媒を減圧下で除去し、固体を得た。この固体に−78℃で塩化メチレン100mlを加えて攪拌溶解し、次いでこの溶液を−78℃に冷却した四塩化ジルコニウム・テトラヒドロフラン錯体(1:2)(0.39g、1.04mmol)の塩化メチレン(3ml)懸濁液に加え、−78℃で12時間攪拌し、徐々に昇温して室温で一昼夜撹拌した。この反応懸濁液を濾過し、溶媒を除去し、固体を得た。さらに、この固体をヘキサンで抽出、濾液を減圧下で濃縮した後、−25℃に保ち固体を得た。この固体を少量のジエチルエーテルで洗浄し、橙色固体を得た(4.6mg、7.7μmol、6.6%)。分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.92(dd、2H)、7.56(d、2H)、7.42(td、2H)、6.00(d、1H)、5.82(d、1H)、3.97−3.60(m、4H)、2.00(s、3H)、1.44(s、18H)、1.03(s、9H)
FD−MASS m/z=600(M+)、598、602
【0094】
〔合成例7〕
[エチレン(4−tertブチルシクロペンタジエニル)(3,6−tertブチルフルオレニル)ジルコニウムジクロライドの合成]
(1)1−(4− tert ブチルシクロペンタジエニル)−2−(3,6− tert ブチル−9−フルオレニル)エタン
氷冷下でtertブチルシクロペンタジエン(0.66g、5.40mmol)のTHF溶液にn−ブチルリチウム/ヘキサン溶液(1.9ml、3.04mmol)を窒素雰囲気下で滴下し、さらに室温で20時間攪拌した。次に3、6−tertブチル−9−(2−ブロモエチル)フルオレン(0.98g、2.54mmol)のTHF(60ml)溶液にヘキサメチルホスホルアミド(1.32ml、7.62mmol)を加えて−78℃に冷却し、前述の溶液を加えた後にゆっくりと室温にしそのまま2日間攪拌した。この溶液から少量サンプリングし、ガスクロマトグラフィーで原料が残っていないことを確認した後、水を加えて反応を停止してエーテルで抽出(50ml×3)し、このエーテル溶液を水で洗浄した(50ml×2)。これを無水硫酸マグネシウムで乾燥、溶媒を除き粘性固体を得た。これを少量のメタノールで洗浄した後、酢酸エチル/ヘキサン(1/9)を展開液としたシリカゲルカラムを通じて精製し、該当フラクションの溶媒を留去し黄色粘性固体を得た(1.05g、2.46mmol、97.0%)。分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.77(d、2H)、7.43(d、2H)、7.33(dt、2H)、6.20、5.89、5.81、5.72、2.87、2.78(4H)、3.93(t、1H)、2.04−2.76(m、4H)、1.41(s、18H)、1.09(d、3H)
【0095】
(2)エチレン(4− tert ブチルシクロペンタジエニル)(3,6− tert ブチルフルオレニル)ジルコニウムジクロライド
氷冷下で1−(4−tertブチルシクロペンタジエニル)−2−(3,6−tertブチル−9−フルオレニル)エタン(1.05g、2.46mmol)のTHF(40ml)溶液にn−ブチルリチウム/ヘキサン溶液(3.2ml、5.2mmol)を窒素雰囲気下で滴下し、さらに室温で20時間攪拌した。反応混合物から溶媒を減圧下で除去し固体を得た。この固体に−78℃で塩化メチレン100mlを加えて攪拌溶解し、次いでこの溶液を−78℃に冷却した塩化ジルコニウム・テトラヒドロフラン錯体(1:2)(0.79g、2.10mmol)の塩化メチレン(3ml)懸濁液に加え、−78℃で12時間攪拌し、徐々に昇温して室温で一昼夜撹拌した。この反応懸濁液を濾過し、溶媒を除去し粘性液体を得た。少量のヘキサンを加えて−25℃に保ち、析出してきた固体を濾別した後、濾液を濃縮して−25℃に保った。この操作を2回繰り返した後に析出してきた固体を、極少量のジエチルエーテルで洗浄した後に減圧乾燥し橙色固体を得た(88mg、0.15mmol、7.1%)。分析値を以下に示す。
1H−NMR(270MHz、CDCl3中、TMS基準)δ7.93(d、2H)、7.57(dt、2H)、7.46(td、2H)、6.15(t、1H)、6.10(t、1H)、5.85(t、1H)、3.98−3.40(m、4H)、1.46(s、18H)、1.12(s、9H)
FD−MASS m/z=586(M+)、584、588
【0096】
〔実施例1〕−エチレン重合−
充分に窒素置換した内容量500mlのガラス製オートクレーブにトルエン400mlを装入し、エチレンを100リットル/時間の量で流通させ、75℃で10分間保持させておいた。これに、メチルアルミノキサンのトルエン溶液(Al=1.53mol/l)を1.30mmol添加し、次いで上記合成例1で合成したエチレン(シクロペンタジエニル)(2,7−tertブチルフルオレニル)ジルコニウムジクロライドのトルエン溶液2.0μmolを加え重合を開始した。エチレンガスを100リットル/時間の量で連続的に供給し、常圧下、75℃で1.5分間重合を行った後、少量のメタノールを添加し重合を停止した。ポリマー溶液を大過剰のメタノールに加え、ポリマーを析出させ、80℃で12時間、減圧乾燥を行った結果、ポリマー3.31gが得られた。重合活性は66.2kg−PE/mmol−Zr・hrであり、得られたポリマーの[η]は3.5dl/gであった。
【0097】
〔実施例2〕−エチレン重合−
充分に窒素置換した内容量500mlのガラス製オートクレーブにトルエン400mlを装入し、エチレンを100l/時間の量で流通させ、75℃で10分間保持させておいた。これに、メチルアルミノキサンのトルエン溶液(Al=1.53mol/l)を1.30mmol添加し、次いで上記合成例2で合成したエチレン(シクロペンタジエニル)(3,6−tertブチルフルオレニル)ジルコニウムジクロライドのトルエン溶液2.0μmolを加え重合を開始した。エチレンガスを100l/時間の量で連続的に供給し、常圧下、75℃で2分間重合を行った後、少量のメタノールを添加し重合を停止した。ポリマー溶液を大過剰のメタノールに加え、ポリマーを析出させ、80℃で12時間、減圧乾燥を行った結果、ポリマー4.19gが得られた。重合活性は62.9kg−PE/mmol−Zr・hrであり、得られたポリマーの[η]は2.99dl/gであった。
【0098】
〔実施例3〕−エチレン重合−
充分に窒素置換した内容量500mlのガラス製オートクレーブにトルエン400mlを装入し、エチレンを120l/時間の量で流通させ、75℃で10分間保持させておいた。これに、メチルアルミノキサンのトルエン溶液(Al=1.53mol/l)を0.52mmol添加し、次いで上記合成例5で合成したエチレン(シクロペンタジエニル)(オクタメチルオクタヒドロジベンゾフルオレニル)ジルコニウムジクロリドのトルエン溶液0.8μmolを加え重合を開始した。エチレンガスを120l/時間の量で連続的に供給し、常圧下、75℃で2分間重合を行った後、少量のメタノールを添加し重合を停止した。ポリマー溶液を大過剰のメタノールに加え、ポリマーを析出させ、80℃で12時間、減圧乾燥を行った結果、ポリマー4.85gが得られた。重合活性は181.9kg−PE/mmol−Zr・hrであり、得られたポリマーの[η]は4.6dl/gであった。
【0099】
〔実施例4〕−エチレン重合−
充分に窒素置換した内容量500mlのガラス製オートクレーブにトルエン400mlを装入し、エチレンを100l/時間の量で流通させ、75℃で10分間保持させておいた。これに、メチルアルミノキサンのトルエン溶液(Al=1.53mol/l)を1.30mmol添加し、次いで上記合成例7で合成したエチレン(4−tertブチルシクロペンタジエニル)(3,6−tertブチルフルオレニル)ジルコニウムジクロライドのトルエン溶液2.0μmolを加え重合を開始した。エチレンガスを100l/時間の量で連続的に供給し、常圧下、75℃で2分間重合を行った後、少量のメタノールを添加し重合を停止した。ポリマー溶液を大過剰のメタノールに加え、ポリマーを析出させ、80℃で12時間、減圧乾燥を行った結果、ポリマー2.64gが得られた。重合活性は39.6kg−PE/mmol−Zr・hrであった。
【0100】
〔実施例5〕−エチレン/ヘキセン共重合−
[固体触媒成分の調製]
200℃で3時間乾燥したシリカ8.5kgを33lのトルエンで懸濁状にした後、メチルアルミノキサン溶液(Al=1.42mol/l)82.7lを30分で滴下した。次いで1.5時間かけて115℃まで昇温し 、その温度で4時間反応させた。その後60℃まで降温し、上澄み液をデカンテーション法によって除去した。得られた固体触媒成分をトルエンで3回洗浄した後、トルエンで再懸濁化して固体触媒成分(a)を得た(全容積150l)。
【0101】
[担持触媒の調製]
充分に窒素置換した100mlの二つ口フラスコ中に、トルエン5mlに懸濁させた上記で調整した固体触媒成分(a)をアルミニウム換算で94.3μmol入れ、その懸濁液を攪拌しながら、室温下(23℃)、上記合成例1で合成したエチレン(シクロペンタジエニル)(2,7−ジ-tert-ブチルフルオレニル)ジルコニウムジクロリドのトルエン溶液0.378μmolを加えた後、室温で60分攪拌した。攪拌を停止後、上澄み液をデカンテーションで取り除き、n−ヘプタン10mlを用いて洗浄を4回行い、固体触媒成分(b)を得た。
【0102】
[エチレン/ヘキセン共重合]
充分に窒素置換した1000mlのオートクレーブにn−ヘプタン500mlを入れ、1mol/lトリイソブチルアルミニウム0.25ml(0.25mmol)、1−ヘキセン3.0ml、上記で得た固体触媒成分(b)を投入し、エチレンガスで8.0kg/cm2Gに加圧し、80℃で重合を開始した。重合中は8.0kg/cm2Gに保つようにエチレンガスを添加し、30分間重合した。重合後、脱圧し、メタノールを加えて触媒を失活させた後、ポリマーを濾過、洗浄し、真空下80℃で12時間乾燥した。得られたポリマーは21.7gであった。重合活性は57.5kg−Polymer/mmol−Zr・hrであった。
このポリマーについて、MFR10、密度、Mw/Mnを測定した。
その結果、MI100.01g/10分↓、密度0.926g/cm3、Mw=309,640、Mw/Mn=2.98であった。
【0103】
〔比較例1〕−エチレン重合−
充分に窒素置換した内容量500mlのガラス製オートクレーブにトルエン400mlを装入し、エチレンを100l/時間の量で流通させ、75℃で10分間保持させておいた。これに、メチルアルミノキサンのトルエン溶液(Al=1.53mol/l)を0.52mmol添加し、次いでジメチルメチレン(シクロペンタジエニル)(フルオレニル)ジルコニウムジクロリドのトルエン溶液0.8μmolを加え重合を開始した。エチレンガスを100l/時間の量で連続的に供給し、常圧下、75℃で6分間重合を行った後、少量のメタノールを添加し重合を停止した。ポリマー溶液を大過剰のメタノールに加え、ポリマーを析出させ、80℃で12時間、減圧乾燥を行った結果、ポリマー1.64gが得られた。重合活性は20.5kg−PE/mmol−Zr・hrであり、得られたポリマーの[η]は3.08dl/gであった。
【0104】
〔実施例6〕−プロピレン重合−
充分に窒素置換した内容量500mlのガラス製オートクレーブにトルエン250mlを装入し、プロピレンを150リットル/時間の量で流通させ、25℃で20分間保持させておいた。一方、充分に窒素置換した内容量30mlの枝付きフラスコにマグネチックスターラーを入れ、これにメチルアルミノキサンのトルエン溶液(Al=1.53mol/l)を5.00mmol、次いで上記合成例1で合成したエチレン(シクロペンタジエニル)(2,7−tertブチルフルオレニル)ジルコニウムジクロライドのトルエン溶液5.0μmolを加え、15分間攪拌した。この溶液を、プロピレンを流通させておいたガラス製オートクレーブのトルエンに加え、重合を開始した。プロピレンガスを150リットル/時間の量で連続的に供給し、常圧下、25℃で30分間重合を行った後、少量のメタノールを添加し重合を停止した。ポリマー溶液を大過剰のメタノールに加え、ポリマーを析出させ、80℃で12時間、減圧乾燥を行った結果、ポリマー11.23gが得られた。重合活性は4.49kg−PP/mmol−Zr・hrであり、得られたポリマーの[η]は2.04dl/g、Tm=111.6℃であった。
【0105】
〔実施例7〕−プロピレン重合−
充分に窒素置換した内容量500mlのガラス製オートクレーブにトルエン250mlを装入し、プロピレンを150リットル/時間の量で流通させ、50℃で20分間保持させておいた。一方、充分に窒素置換した内容量30mlの枝付きフラスコにマグネチックスターラーを入れ、これにメチルアルミノキサンのトルエン溶液(Al=1.53mol/l)を5.00mmol、次いで上記合成例1で合成したエチレン(シクロペンタジエニル)(2,7−tertブチルフルオレニル)ジルコニウムジクロライドのトルエン溶液5.0μmolを加え、15分間攪拌した。この溶液を、プロピレンを流通させておいたガラス製オートクレーブのトルエンに加え、重合を開始した。プロピレンガスを150リットル/時間の量で連続的に供給し、常圧下、50℃で40分間重合を行った後、少量のメタノールを添加し重合を停止した。ポリマー溶液を大過剰のメタノールに加え、ポリマーを析出させ、80℃で12時間、減圧乾燥を行った結果、ポリマー9.80gが得られた。重合活性は2.94kg−PP/mmol−Zr・hrであり、得られたポリマーの[η]は0.72dl/gであった。
【0106】
〔実施例8〕−プロピレン重合−
充分に窒素置換した内容量500mlのガラス製オートクレーブにトルエン250mlを装入し、プロピレンを150リットル/時間の量で流通させ、25℃で20分間保持させておいた。一方、充分に窒素置換した内容量30mlの枝付きフラスコにマグネチックスターラーを入れ、これにメチルアルミノキサンのトルエン溶液(Al=1.53mol/l)を5.00mmol、次いで上記合成例5で合成したエチレン(シクロペンタジエニル)(オクタメチルオクタヒドロジベンゾフルオレニル)ジルコニウムジクロリドのトルエン溶液5.0μmolを加え、20分間攪拌した。この溶液を、プロピレンを流通させておいたガラス製オートクレーブのトルエンに加え、重合を開始した。プロピレンガスを150リットル/時間の量で連続的に供給し、常圧下、25℃で60分間重合を行った後、少量のメタノールを添加し重合を停止した。ポリマー溶液を大過剰のメタノールに加え、ポリマーを析出させ、80℃で12時間、減圧乾燥を行った結果、ポリマー1.94gが得られた。重合活性は0.39kg−PP/mmol−Zr・hrであり、得られたポリマーの[η]は3.19dl/g、Tm=152.3℃であった。
【0107】
〔実施例9〕−プロピレン重合−
充分に窒素置換した内容量500mlのガラス製オートクレーブにトルエン250mlを装入し、プロピレンを150リットル/時間の量で流通させ、50℃で20分間保持させておいた。一方、充分に窒素置換した内容量30mlの枝付きフラスコにマグネチックスターラーを入れ、これにメチルアルミノキサンのトルエン溶液(Al=1.53mol/l)を5.00mmol、次いで上記合成例5で合成したエチレン(シクロペンタジエニル)(オクタメチルオクタヒドロジベンゾフルオレニル)ジルコニウムジクロリドのトルエン溶液5.0μmolを加え、20分間攪拌した。この溶液を、プロピレンを流通させておいたガラス製オートクレーブのトルエンに加え、重合を開始した。プロピレンガスを150リットル/時間の量で連続的に供給し、常圧下、50℃で60分間重合を行った後、少量のメタノールを添加し重合を停止した。ポリマー溶液を大過剰のメタノールに加え、ポリマーを析出させ、80℃で12時間、減圧乾燥を行った結果、ポリマー10.7gが得られた。重合活性は2.14kg−PP/mmol−Zr・hrであり、得られたポリマーの[η]は1.35dl/gであった。
【0108】
〔比較例2〕−プロピレン重合−
充分に窒素置換した内容量500mlのガラス製オートクレーブにトルエン250mlを装入し、プロピレンを150リットル/時間の量で流通させ、25℃で20分間保持させておいた。一方、充分に窒素置換した内容量30mlの枝付きフラスコにマグネチックスターラーを入れ、これにメチルアルミノキサンのトルエン溶液(Al=1.53mol/l)を5.00mmol、次いでジメチルメチレン(シクロペンタジエニル)(フルオレニル)ジルコニウムジクロリドのトルエン溶液5.0μmolを加え、20分間攪拌した。この溶液を、プロピレンを流通させておいたガラス製オートクレーブのトルエンに加え、重合を開始した。プロピレンガスを150リットル/時間の量で連続的に供給し、常圧下、25℃で60分間重合を行った後、少量のメタノールを添加し重合を停止した。ポリマー溶液を大過剰のメタノールに加え、ポリマーを析出させ、80℃で12時間、減圧乾燥を行った結果、ポリマー6.8gが得られた。重合活性は1.36kg−PP/mmol−Zr・hrであり、得られたポリマーのTm=140.0℃であった。
【0109】
〔比較例3〕−プロピレン重合−
充分に窒素置換した内容量500mlのガラス製オートクレーブにトルエン250mlを装入し、プロピレンを150リットル/時間の量で流通させ、50℃で20分間保持させておいた。一方、充分に窒素置換した内容量30mlの枝付きフラスコにマグネチックスターラーを入れ、これにメチルアルミノキサンのトルエン溶液(Al=1.53mol/l)を5.00mmol、次いでジメチルメチレン(シクロペンタジエニル)(フルオレニル)ジルコニウムジクロリドのトルエン溶液5.0μmolを加え、20分間攪拌した。この溶液を、プロピレンを流通させておいたガラス製オートクレーブのトルエンに加え、重合を開始した。プロピレンガスを150リットル/時間の量で連続的に供給し、常圧下、50℃で60分間重合を行った後、少量のメタノールを添加し重合を停止した。ポリマー溶液を大過剰のメタノールに加え、ポリマーを析出させ、80℃で12時間、減圧乾燥を行った結果、ポリマー1.93gが得られた。重合活性は0.39kg−PP/mmol−Zr・hrであり、得られたポリマーの[η]は0.8dl/g、Tm=115.0℃であった。
【0110】
以上の結果について、エチレン重合に関する実施例1、2、3、4比較例1の結果を表1に、プロピレン重合に関する実施例6,7,8、9比較例2、3の結果を表2にまとまた。
【0111】
【表1】
【表2】
【発明の効果】
本発明の架橋メタロセン化合物を含む触媒存在下ではオレフィン単独重合または共重合させることによって、高い重合活性でもって、高融点かつ高分子量オレフィン単独重合体または共重合体を与える。
Claims (7)
- 下記一般式[1]で表されることを特徴とする架橋メタロセン化合物。
- 前記一般式[1]において、n=1または2であり、Y1とY2の両方が、炭素原子又はケイ素原子であることを特徴とする請求項1記載の架橋メタロセン化合物。
- 前記一般式[1]において、R6とR7が互いに結合して脂肪族環を形成し、R10とR11が互いに結合して脂肪族環を形成していることを特徴とする請求項1または2に記載の架橋メタロセン化合物。
- 請求項1〜3のいずれか1項に記載の架橋メタロセン化合物を含むオレフィン重合用触媒。
- (A)請求項1〜3のいずれか1項に記載の架橋メタロセン化合物と、
(B) (B−1) 有機金属化合物、
(B−2) 有機アルミニウムオキシ化合物、
(B−3) メタロセン化合物(A)と反応してイオン対を形成する化合物、
とから選ばれる少なくとも1種の化合物とからなるオレフィン重合用触媒。 - 請求項4または5に記載のオレフィン重合触媒の存在下で、エチレンおよびα−オレフィンから選ばれる1種以上のモノマーを重合する方法であって、モノマーの少なくとも1種がエチレンまたはプロピレンであることを特徴とするオレフィンの重合方法。
- 前記一般式[1]で表される架橋メタロセン化合物が、担持された形態で用いられることを特徴とする請求項6に記載のオレフィンの重合方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002342815A JP4367687B2 (ja) | 2002-11-26 | 2002-11-26 | オレフィン重合用の架橋メタロセン化合物およびそれを用いたオレフィンの重合方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002342815A JP4367687B2 (ja) | 2002-11-26 | 2002-11-26 | オレフィン重合用の架橋メタロセン化合物およびそれを用いたオレフィンの重合方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2004175707A JP2004175707A (ja) | 2004-06-24 |
| JP4367687B2 true JP4367687B2 (ja) | 2009-11-18 |
Family
ID=32704763
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002342815A Expired - Lifetime JP4367687B2 (ja) | 2002-11-26 | 2002-11-26 | オレフィン重合用の架橋メタロセン化合物およびそれを用いたオレフィンの重合方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP4367687B2 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018131543A1 (ja) | 2017-01-16 | 2018-07-19 | 三井化学株式会社 | 自動車ギア用潤滑油組成物 |
| US10040884B2 (en) | 2014-03-28 | 2018-08-07 | Mitsui Chemicals, Inc. | Ethylene/α-olefin copolymers and lubricating oils |
| WO2021070811A1 (ja) | 2019-10-07 | 2021-04-15 | 三井化学株式会社 | エチレン・α-オレフィン共重合体組成物およびその用途 |
| WO2021153532A1 (ja) | 2020-01-30 | 2021-08-05 | 三井化学株式会社 | ポリアミド組成物 |
| WO2022154126A1 (ja) | 2021-01-18 | 2022-07-21 | 三井化学株式会社 | 水分散体組成物、当該水分散体組成物の製造方法およびエチレン・α-オレフィン共重合体酸変性物 |
| WO2023002947A1 (ja) | 2021-07-20 | 2023-01-26 | 三井化学株式会社 | 潤滑油用粘度調整剤および作動油用潤滑油組成物 |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7605208B2 (en) | 2005-10-31 | 2009-10-20 | Mitsui Chemicals, Inc. | Process for producing thermoplastic resin composition |
| EP1995037B1 (en) | 2006-03-10 | 2011-07-06 | Mitsui Chemicals, Inc. | Process for production of moldings by the inflation method |
| US20090166911A1 (en) | 2006-03-27 | 2009-07-02 | Mitsui Chemicals, Inc. | Process for Producing Molded Article by T-Die Molding |
| JP2007261201A (ja) | 2006-03-29 | 2007-10-11 | Mitsui Chemicals Inc | ブロー成形による成形体の製造方法 |
| EP2002955A4 (en) | 2006-03-30 | 2009-09-16 | Mitsui Chemicals Inc | METHOD FOR PRODUCING A MOLDED OBJECT BY INJECTION MOLDING |
| WO2009081792A1 (ja) * | 2007-12-21 | 2009-07-02 | Mitsui Chemicals, Inc. | エチレン/α-オレフィン/非共役ポリエン共重合体の製造方法 |
| JP5357635B2 (ja) * | 2009-06-19 | 2013-12-04 | 三井化学株式会社 | ゴム組成物およびその用途 |
| US8609793B2 (en) | 2010-10-07 | 2013-12-17 | Chevron Phillips Chemical Company Lp | Catalyst systems containing a bridged metallocene |
| US8629292B2 (en) | 2010-10-07 | 2014-01-14 | Chevron Phillips Chemical Company Lp | Stereoselective synthesis of bridged metallocene complexes |
| US8637616B2 (en) | 2010-10-07 | 2014-01-28 | Chevron Philips Chemical Company Lp | Bridged metallocene catalyst systems with switchable hydrogen and comonomer effects |
| CN105367611B (zh) * | 2015-10-13 | 2019-04-05 | 华东理工大学 | 一种烯烃高温聚合茂金属催化剂及其制备方法和在烯烃聚合中的应用 |
| KR102580620B1 (ko) | 2019-03-29 | 2023-09-19 | 미쓰이 가가쿠 가부시키가이샤 | 수지 조성물 |
-
2002
- 2002-11-26 JP JP2002342815A patent/JP4367687B2/ja not_active Expired - Lifetime
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10040884B2 (en) | 2014-03-28 | 2018-08-07 | Mitsui Chemicals, Inc. | Ethylene/α-olefin copolymers and lubricating oils |
| US10329366B2 (en) | 2014-03-28 | 2019-06-25 | Mitsui Chemicals, Inc. | Ethylene/α-olefin copolymers and lubricating oils |
| WO2018131543A1 (ja) | 2017-01-16 | 2018-07-19 | 三井化学株式会社 | 自動車ギア用潤滑油組成物 |
| WO2021070811A1 (ja) | 2019-10-07 | 2021-04-15 | 三井化学株式会社 | エチレン・α-オレフィン共重合体組成物およびその用途 |
| WO2021153532A1 (ja) | 2020-01-30 | 2021-08-05 | 三井化学株式会社 | ポリアミド組成物 |
| WO2022154126A1 (ja) | 2021-01-18 | 2022-07-21 | 三井化学株式会社 | 水分散体組成物、当該水分散体組成物の製造方法およびエチレン・α-オレフィン共重合体酸変性物 |
| WO2023002947A1 (ja) | 2021-07-20 | 2023-01-26 | 三井化学株式会社 | 潤滑油用粘度調整剤および作動油用潤滑油組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2004175707A (ja) | 2004-06-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4367686B2 (ja) | オレフィン重合用の架橋メタロセン化合物およびそれを用いたオレフィンの重合方法 | |
| JP4367687B2 (ja) | オレフィン重合用の架橋メタロセン化合物およびそれを用いたオレフィンの重合方法 | |
| EP2511305B1 (en) | Bridged metallocene compound for olefin polymerization and method of polymerizing olefin using the same | |
| EP1988104B1 (en) | Propylene copolymer, polypropylene composition, use thereof, transition metal compounds, and catalysts for olefin polymerization | |
| JPWO2006126610A1 (ja) | 遷移金属化合物、オレフィン重合用触媒およびオレフィン系重合体の製造方法 | |
| JP4367688B2 (ja) | オレフィン重合用の架橋メタロセン化合物およびそれを用いたオレフィンの重合方法 | |
| JP4367689B2 (ja) | オレフィン重合用の架橋メタロセン化合物およびそれを用いたオレフィンの重合方法 | |
| JP4610859B2 (ja) | オレフィン重合用の架橋メタロセン化合物およびそれを用いたオレフィンの重合方法 | |
| JP5153326B2 (ja) | フルオレン誘導体、遷移金属化合物、オレフィン重合用触媒およびオレフィン系重合体の製造方法 | |
| JP4111814B2 (ja) | オレフィンの重合方法 | |
| JP4389071B2 (ja) | プロピレン系重合体 | |
| JP2004189667A (ja) | オレフィン重合用の架橋メタロセン化合物およびそれを用いたオレフィンの重合方法 | |
| JP4350636B2 (ja) | プロピレン系重合体 | |
| JP2006124452A (ja) | プロピレン系重合体 | |
| JP2006124453A (ja) | プロピレン系重合体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050715 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20070703 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20081202 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090130 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090811 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20090817 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090819 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4367687 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120904 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120904 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130904 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130904 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |