JP4351908B2 - 減圧においてアルカリ性のカプロラクタム生成物を蒸留する方法 - Google Patents
減圧においてアルカリ性のカプロラクタム生成物を蒸留する方法 Download PDFInfo
- Publication number
- JP4351908B2 JP4351908B2 JP2003523225A JP2003523225A JP4351908B2 JP 4351908 B2 JP4351908 B2 JP 4351908B2 JP 2003523225 A JP2003523225 A JP 2003523225A JP 2003523225 A JP2003523225 A JP 2003523225A JP 4351908 B2 JP4351908 B2 JP 4351908B2
- Authority
- JP
- Japan
- Prior art keywords
- caprolactam
- product
- alkaline
- caprolactam product
- bases
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D201/00—Preparation, separation, purification or stabilisation of unsubstituted lactams
- C07D201/16—Separation or purification
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D223/00—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom
- C07D223/02—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom not condensed with other rings
- C07D223/06—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom not condensed with other rings with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D223/08—Oxygen atoms
- C07D223/10—Oxygen atoms attached in position 2
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Vaporization, Distillation, Condensation, Sublimation, And Cold Traps (AREA)
- Separation, Recovery Or Treatment Of Waste Materials Containing Plastics (AREA)
Description
上記式中、
v=加えられたHCl溶液のミリリットル、
t= HCl溶液のモル濃度(=0.01)
a=試料の重量(グラム)
である。
上記式中、
v=加えられたNaOH溶液のミリリットル、
t= NaOH溶液のモル濃度(=0.01)
a=試料の重量(グラム)
である。
PAN:ISO DIS 8660‐プラスチック‐カプロラクタムの過マンガン酸塩指数の測定‐Spectometric法、第1版ISO 8660の改訂;1988年
E290:ISO 7059‐工業使用のためのカプロラクタム‐290nmの波長における吸収の測定
揮発性塩基(VB) ISO 8661‐工業使用のためのカプロラクタム‐揮発性塩基含有量の測定‐蒸留後滴定法
アルカリ度:0.01M塩酸の水溶液による滴定
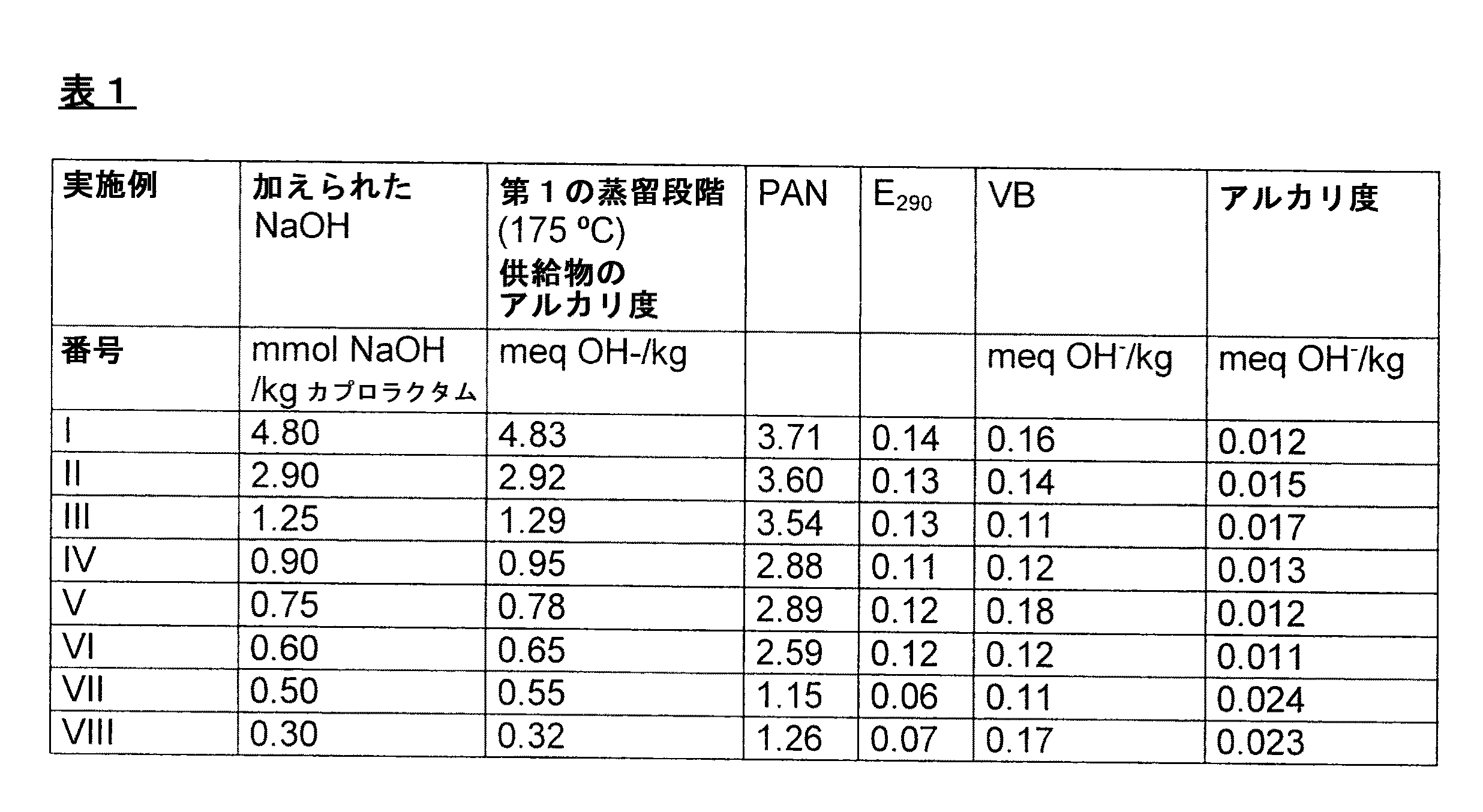
純粋なカプロラクタムの製造のための連続法において、カプロラクタム生成物の流れは、発煙硫酸の存在下にシクロヘキサノンオキシムをベックマン転位し、該ベックマン転位混合物をアンモニアで中和し、抽出技術により該中和されたベッマン転位混合物からカプロラクタムを分離することにより連続的に製造された。該流れは、イオン交換体による精製、水素化及び第一の脱水を含む一連の精製段階に付された。得られたカプロラクタム生成物の流れは、約85重量%のカプロラクタム、約15重量%の水、及び不純物を含んでおり、かつ次の規格(PAN=2.6、E290=0.32、VB=0.44meq/kg、アルカリ度=0.02 meq/kg)を有していた。この流れに、カプロラクタムの1キログラム当り4.80ミリモルのNaOHが(15%の水性溶液として)連続的に加えられた。得られたアルカリ性のカプロラクタム生成物の流れは、直列のエバポレーターにおいて脱水された。ここで、該エバポレーターの温度は80〜125℃で変化した。該エバポレーター内及び間の合計滞留時間は3時間であった。結果として、約0.5重量%の水を含むアルカリ性のカプロラクタム生成物が得られた。該アルカリ性のカプロラクタム生成物において、少なくとも90%の加えられた塩基が、ナトリウムアミノカプロエートを形成するために反応されたと思われた。該直列のエバポレーターを出るアルカリ性のカプロラクタム生成物(アルカリ度4.83meq/kg)は、減圧下における二段階で蒸留された。第一の段階において、低沸点不純物及び水が、175℃の(塔底)温度、及び5.2kPaの圧力で蒸留カラムにおいて分離された。ここで、滞留時間は数分間であった。第二の段階において、高沸点不純物が、133℃の(塔底)温度、1.2kPaの圧力で蒸留カラムにおいて分離された。ここで、滞留時間は1時間であった。得られた精製されたカプロラクタムの規格は、表1に示されている。
異なる量のNaOHが加えられて、第一の蒸留段階(175℃における蒸留)の供給物のアルカリ度のために異なる値をもたらすと言う相違をもって、実施例Iが繰返された。蒸留後に得られたカプロラクタムの規格が表1に示されている。
Claims (20)
- (i)カプロラクタム、(ii)有機不純物、並びに(iii)アルカリ水酸化物及びアルカリアミノカプロエートより成る群から選ばれる一つ以上の塩基を含むアルカリ性のカプロラクタム生成物を減圧で蒸留することを含む、精製されたカプロラクタムを得る方法において、アルカリ性のカプロラクタム生成物のアルカリ度が、カプロラクタムの1kg当り2ミリ当量より小さいことを特徴とする方法。
- アルカリ性のカプロラクタム生成物のアルカリ度が、カプロラクタムの1kg当り0.05ミリ当量より大きいところの請求項1記載の方法。
- アルカリ性のカプロラクタム生成物のアルカリ度が、カプロラクタムの1kg当り0.15ミリ当量より大きいところの請求項2記載の方法。
- 該一つ以上の塩基が、水酸化ナトリウム、ナトリウムアミノカプロエート、水酸化カリウム、カリウムアミノカプロエートより成る群から選らばれるところの請求項1〜3のいずれか一つに記載の方法。
- 該一つ以上の塩基の少なくとも75モル%が、アルカリアミノカプロエートであるところの請求項1〜3のいずれか一つに記載の方法。
- 該アルカリ性のカプロラクタム生成物が、少なくとも95重量%のカプロラクタムを含むところの請求項1〜5のいずれか一つに記載の方法。
- カプロラクタム生成物が、(i)カプロラクタム及び(ii)不純物を含み、かつアルカリ水酸化物及びアルカリアミノカプロエートより成る群から選ばれる一つ以上の塩基を上記アルカリ性のカプロラクタム生成物をもたらすような量で、該カプロラクタム生成物に加えることを含むところの、カプロラクタム生成物の精製のための請求項1〜6のいずれか一つに記載の方法。
- カプロラクタム生成物が、カプロラクタムの1kg当り0〜5ミリ当量の酸度を有するか、中性であるか、又はカプロラクタムの1kg当り0〜5ミリ当量のアルカリ度を有するところの請求項7記載の方法。
- 該一つ以上の塩基をカプロラクタムの1kg当り10ミリモルより少ない量で該カプロラクタム生成物に加えることを含むところの請求項7又は8記載の方法。
- カプロラクタムの1kg当り0.05〜10ミリモルの該一つ以上の塩基を該カプロラクタム生成物に加えることを含むところの請求項9記載の方法。
- カプロラクタムの1kg当り0.15〜3ミリモルの該一つ以上の塩基を該カプロラクタム生成物に加えることを含むところの請求項10記載の方法。
- カプロラクタム生成物が、少なくとも15重量%のカプロラクタムを含むところの請求項7〜11のいずれか一つに記載の方法。
- カプロラクタム生成物が水を含み、かつカプロラクタム生成物中の水及びカプロラクタムの合計量が少なくとも95重量%であるところの請求項12記載の方法。
- 該カプロラクタム生成物にアルカリ水酸化物を加えて、アルカリ性の生成物をもたらし、そしてアルカリ性の生成物中の該アルカリ水酸化物の少なくとも一部を転化して、該蒸留に先立ってアルカリアミノカプロエートを形成することを含むところの請求項7〜13のいずれか一つに記載の方法。
- カプロラクタムが、ベックマン転位により得られるところの請求項1〜14のいずれか一つに記載の方法。
- 100〜200℃の温度でアルカリ性のカプロラクタム生成物を蒸留することを含むところの請求項1〜15のいずれか一つに記載の方法。
- 10kPaより低い圧力でアルカリ性のカプロラクタム生成物を蒸留することを含むところの請求項1〜16のいずれか一つに記載の方法。
- 該蒸留が、アルカリ性のカプロラクタム生成物から低沸点不純物を分離すること、及び/又はアルカリ性のカプロラクタム生成物から高沸点不純物を分離することを含むところの請求項1〜17のいずれか一つに記載の方法。
- 該蒸留が、第一段階において、アリカリ性のカプロラクタム生成物から低沸点不純物を塔頂生成物として分離する一方、高沸点不純物を含むアリカリ性のカプロラクタム生成物を塔底生成物とし、そして第二段階において、該塔底生成物から高沸点不純物を分離し、そして塔頂生成物としてカプロクタムを回収することを含むところの請求項18記載の方法。
- 連続法であるところの請求項1〜19のいずれか一つに記載の方法。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP01203214 | 2001-08-27 | ||
| EP01203217 | 2001-08-27 | ||
| EP01203215 | 2001-08-27 | ||
| PCT/NL2002/000558 WO2003018562A1 (en) | 2001-08-27 | 2002-08-23 | Process for distilling alkaline caprolactam product at reduced pressure |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2005502668A JP2005502668A (ja) | 2005-01-27 |
| JP2005502668A5 JP2005502668A5 (ja) | 2006-01-05 |
| JP4351908B2 true JP4351908B2 (ja) | 2009-10-28 |
Family
ID=27224307
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003523225A Expired - Fee Related JP4351908B2 (ja) | 2001-08-27 | 2002-08-23 | 減圧においてアルカリ性のカプロラクタム生成物を蒸留する方法 |
| JP2003523214A Expired - Fee Related JP4368197B2 (ja) | 2001-08-27 | 2002-08-23 | インサイツ製造されたアルカリアミノカプロエートを使用して水性のカプロラクタム生成物からカプロラクタムを回収する方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003523214A Expired - Fee Related JP4368197B2 (ja) | 2001-08-27 | 2002-08-23 | インサイツ製造されたアルカリアミノカプロエートを使用して水性のカプロラクタム生成物からカプロラクタムを回収する方法 |
Country Status (18)
| Country | Link |
|---|---|
| US (2) | US20050029086A1 (ja) |
| EP (2) | EP1423369B1 (ja) |
| JP (2) | JP4351908B2 (ja) |
| KR (2) | KR20040031007A (ja) |
| CN (2) | CN1252050C (ja) |
| AT (1) | ATE498611T1 (ja) |
| AU (1) | AU2002313611A1 (ja) |
| BR (2) | BR0212118A (ja) |
| CO (2) | CO5560549A2 (ja) |
| CZ (1) | CZ2004296A3 (ja) |
| DE (1) | DE60239205D1 (ja) |
| EA (2) | EA005831B1 (ja) |
| ES (1) | ES2429363T3 (ja) |
| MX (1) | MX298589B (ja) |
| MY (2) | MY144316A (ja) |
| PL (2) | PL367897A1 (ja) |
| TW (2) | TW568793B (ja) |
| WO (2) | WO2003018562A1 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1423369B1 (en) * | 2001-08-27 | 2013-07-17 | DSM IP Assets B.V. | Process for distilling alkaline caprolactam product at reduced pressure |
| US7022844B2 (en) | 2002-09-21 | 2006-04-04 | Honeywell International Inc. | Amide-based compounds, production, recovery, purification and uses thereof |
| US8841445B2 (en) | 2012-12-19 | 2014-09-23 | Basf Se | Process for preparing purified caprolactam from the Beckmann rearrangement of cyclohexane oxime |
| JP6320413B2 (ja) * | 2012-12-19 | 2018-05-09 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | シクロヘキサノンオキシムのベックマン転位からの精製カプロラクタムの製造方法 |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE202870C (ja) | ||||
| CH549020A (de) * | 1970-12-22 | 1974-05-15 | Inventa Ag | Verfahren zur reinigung von lactamen. |
| NL7308100A (ja) * | 1973-06-12 | 1974-12-16 | ||
| DE3030735A1 (de) * | 1980-08-14 | 1982-03-25 | Basf Ag, 6700 Ludwigshafen | Verfahren zur gewinnung von caprolactam durch spaltung von oligomeren des caprolactams |
| NL8102280A (nl) † | 1981-05-09 | 1982-12-01 | Stamicarbon | Werkwijze voor het zuiveren van epsilon-caprolactam. |
| US4720328A (en) * | 1981-06-26 | 1988-01-19 | Akzona Incorporated | Method for removing impurities from caprolactam |
| DD202870A1 (de) * | 1981-09-29 | 1983-10-05 | Leuna Werke Veb | Destillative reinigung von epsilon-caprolactam |
| IT1222419B (it) * | 1987-07-31 | 1990-09-05 | Friuli Chim Spa | Procedimento di purificazione del caprolattame |
| DE3729853A1 (de) * | 1987-09-05 | 1989-03-23 | Basf Ag | Verfahren zur aufarbeitung von destillationsrueckstaenden, die bei der reinigung von caprolactam anfallen |
| US5496641A (en) * | 1991-06-13 | 1996-03-05 | Nippon Sheet Glass Co., Ltd. | Plastic lens |
| DE4407222A1 (de) * | 1994-03-04 | 1995-09-07 | Basf Ag | Verfahren zur Rückgewinnung von Caprolactam aus Oliogo- und/oder Polymeren des Caprolactams |
| DE19500041A1 (de) † | 1995-01-03 | 1996-07-04 | Basf Ag | Verfahren zur kontinuierlichen Reinigung von aus 6-Aminocapronitril hergestelltem Roh-Caprolactam |
| FR2809395B1 (fr) † | 2000-05-26 | 2002-07-19 | Rhodia Polyamide Intermediates | Procede de purification de lactames |
| JP2002145863A (ja) † | 2000-11-02 | 2002-05-22 | Mitsubishi Chemicals Corp | ε−カプロラクタムの精製方法 |
| ATE498610T1 (de) † | 2001-03-01 | 2011-03-15 | Dsm Ip Assets Bv | Verfahren zur rückgewinnung und aufreinigung von caprolactam aus einem organischem lösungsmittel |
| EP1423369B1 (en) * | 2001-08-27 | 2013-07-17 | DSM IP Assets B.V. | Process for distilling alkaline caprolactam product at reduced pressure |
-
2002
- 2002-08-23 EP EP02753297.7A patent/EP1423369B1/en not_active Expired - Lifetime
- 2002-08-23 WO PCT/NL2002/000558 patent/WO2003018562A1/en not_active Ceased
- 2002-08-23 US US10/487,673 patent/US20050029086A1/en not_active Abandoned
- 2002-08-23 EA EA200400360A patent/EA005831B1/ru not_active IP Right Cessation
- 2002-08-23 WO PCT/NL2002/000559 patent/WO2003018550A1/en not_active Ceased
- 2002-08-23 KR KR20047002830A patent/KR20040031007A/ko not_active Ceased
- 2002-08-23 DE DE60239205T patent/DE60239205D1/de not_active Expired - Lifetime
- 2002-08-23 KR KR1020047002816A patent/KR100907146B1/ko not_active Expired - Lifetime
- 2002-08-23 BR BR0212118-2A patent/BR0212118A/pt not_active IP Right Cessation
- 2002-08-23 AU AU2002313611A patent/AU2002313611A1/en not_active Abandoned
- 2002-08-23 PL PL02367897A patent/PL367897A1/xx unknown
- 2002-08-23 JP JP2003523225A patent/JP4351908B2/ja not_active Expired - Fee Related
- 2002-08-23 AT AT02753298T patent/ATE498611T1/de not_active IP Right Cessation
- 2002-08-23 ES ES02753297T patent/ES2429363T3/es not_active Expired - Lifetime
- 2002-08-23 EA EA200400359A patent/EA006481B1/ru not_active IP Right Cessation
- 2002-08-23 BR BR0212067-4A patent/BR0212067A/pt not_active IP Right Cessation
- 2002-08-23 JP JP2003523214A patent/JP4368197B2/ja not_active Expired - Fee Related
- 2002-08-23 EP EP02753298.5A patent/EP1423361B8/en not_active Expired - Lifetime
- 2002-08-23 CN CNB028166736A patent/CN1252050C/zh not_active Expired - Lifetime
- 2002-08-23 CN CNB028208226A patent/CN1284774C/zh not_active Expired - Lifetime
- 2002-08-23 MX MXPA04001793 patent/MX298589B/es active IP Right Grant
- 2002-08-23 CZ CZ2004296A patent/CZ2004296A3/cs unknown
- 2002-08-23 US US10/486,727 patent/US7615137B2/en not_active Expired - Lifetime
- 2002-08-23 PL PL02370035A patent/PL370035A1/xx not_active IP Right Cessation
- 2002-08-26 TW TW091119258A patent/TW568793B/zh not_active IP Right Cessation
- 2002-08-26 TW TW091119262A patent/TWI311989B/zh not_active IP Right Cessation
- 2002-08-27 MY MYPI20023187A patent/MY144316A/en unknown
- 2002-08-27 MY MYPI20023186A patent/MY144901A/en unknown
-
2004
- 2004-02-18 CO CO04013833A patent/CO5560549A2/es not_active Application Discontinuation
- 2004-03-09 CO CO04021423A patent/CO5560607A2/es not_active Application Discontinuation
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8231765B2 (en) | Process for the purification of lactams | |
| JP2005510562A (ja) | カプロラクタムの回収方法 | |
| JP4351908B2 (ja) | 減圧においてアルカリ性のカプロラクタム生成物を蒸留する方法 | |
| ES2360508T5 (es) | Procedimiento para recuperar caprolactama a partir de un producto de caprolactama acuoso utilizando aminocaproato alcalino preparado in situ | |
| WO1997003956A1 (en) | PROCESS FOR THE PREPARATION OF ε-CAPROLACTAM | |
| KR101155354B1 (ko) | ε-카프롤락탐의 제조방법 | |
| JP3789504B2 (ja) | ε−カプロラクタムの製造方法 | |
| MXPA04001794A (es) | Procedimiento para recuperar caprolactama a partir de un producto acuoso de caprolactama utilizando aminocaproato alcalino preparado in situ. | |
| JP2005504056A5 (ja) | ||
| CZ2004297A3 (cs) | Způsob regenerace kaprolaktamu z vodného kaprolaktamového produktu za použití in sítu připraveného alkalického aminokapronátu | |
| JP4239288B2 (ja) | ε−カプロラクタムの製造方法 | |
| JP2001114758A (ja) | カプロラクタムの製造方法 | |
| JPH08176102A (ja) | ε−カプロラクタムの製造方法 | |
| JPH08193061A (ja) | ε−カプロラクタムの製造方法 | |
| JPH0931052A (ja) | ε−カプロラクタムの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050822 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050822 |
|
| RD03 | Notification of appointment of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7423 Effective date: 20060721 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20060901 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20081126 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20081216 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090316 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090324 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090416 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090721 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090727 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120731 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120731 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130731 Year of fee payment: 4 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |