JP3770414B2 - 新規なクロマトグラフイー分離条件を用いるα−1プロテイナーゼ阻害剤の精製 - Google Patents
新規なクロマトグラフイー分離条件を用いるα−1プロテイナーゼ阻害剤の精製 Download PDFInfo
- Publication number
- JP3770414B2 JP3770414B2 JP23322895A JP23322895A JP3770414B2 JP 3770414 B2 JP3770414 B2 JP 3770414B2 JP 23322895 A JP23322895 A JP 23322895A JP 23322895 A JP23322895 A JP 23322895A JP 3770414 B2 JP3770414 B2 JP 3770414B2
- Authority
- JP
- Japan
- Prior art keywords
- protein
- alpha
- chromatography
- proteins
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/81—Protease inhibitors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/81—Protease inhibitors
- C07K14/8107—Endopeptidase (E.C. 3.4.21-99) inhibitors
- C07K14/811—Serine protease (E.C. 3.4.21) inhibitors
- C07K14/8121—Serpins
- C07K14/8125—Alpha-1-antitrypsin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10S—TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10S530/00—Chemistry: natural resins or derivatives; peptides or proteins; lignins or reaction products thereof
- Y10S530/827—Proteins from mammals or birds
- Y10S530/829—Blood
- Y10S530/83—Plasma; serum
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Gastroenterology & Hepatology (AREA)
- Pulmonology (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Analytical Chemistry (AREA)
- Peptides Or Proteins (AREA)
- Treatment Of Liquids With Adsorbents In General (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Medicines Containing Plant Substances (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description
【発明の属する技術分野】
本発明は一般にタンパク質の精製に関する。本発明は、詳細には、活性α−1PIがカラムに結合しないが、汚染タンパク質は結合するような条件下で陽イオン交換によって血漿画分からα−1プロテイナーゼ阻害剤を精製する方法に関する。
【0002】
【従来の技術】
α−1プロテイナーゼ阻害剤(α−1 PI)は約55,000ダルトンの分子量を有する糖タンパク質である。α−1 PIは、例えばトリプシン、キモトリプシン、膵臓エラスターゼ、皮膚コラゲナーゼ、レニン、ウロキナーゼのようなプロテイナーゼ類の阻害剤であり、そして多形核リンパ球のプロテアーゼ類の阻害剤である。現在、α−1 PIは肺中のリンパ球エラスターゼの阻害剤としての治療上の用途を有する。このプロテアーゼの機能は外来タンパク質を破壊することである。エラスターゼ活性を調節するのに十分な量のα−1 PIが存在しない場合には、エラスターゼは肺組織を破壊する。やがて、このアンバランスは慢性の肺組織損傷及び気腫を生じさせる。α−1 PIの補給はこのタイプの気腫の治療に成功裏に用いられてきている。
【0003】
現在、α−1 PIの需要は入手可能な供給を超えている。α−1 PI遺伝子は微生物、細胞系及びヒツジ中に移されて発現されてきた。しかしながら、満足できる組換え産物はいまだ生産されていない。ヒト血漿が治療用α−1 PIのいまだ唯一の承認された給源である。α−1 PIは代償療法に用いられ、そして長期間にわたって規則的に患者に与えられる。痕跡量の不純物が患者の免疫応答を刺激し得るので、治療を成功させるには高純度の産物が不可欠である。α−1 PIの給源である血漿は限られているので、α−1 PIを高純度に精製する方法が必要になる。高収率でかつ高純度でα−1 PIを提供する実用的な方法は今日まで得られていない。
【0004】
ヒト血漿からα−1 PIを精製する種々の方法が記載されている。Bollenら、米国特許第4,629,567号明細書(1986)は、酵母、大腸菌及びヒト血漿からα−1 PIを精製するために5つの異なるクロマトグラフィー工程を用いている。5つの工程は、DEAEイオン交換、チオール−ジスルフィド交換、ヘパリンアフィニティー、亜鉛−キレートクロマトグラフィー及びアミノヘキシル・イオン交換を含む。純度と収率のデータは示されていない。
【0005】
Novikaら、Gematol.Transfuziol.34:46−50(1989)は、血液製品の製造の際の副生成物からの単離方法を報告している。彼らはアフィニティー、DEAEセルロース、及びゲル瀘過クロマトグラフィーを用いている。純度と収率のデータは得られていない。
【0006】
Podiareneら、Vopr.Med.Khim.35:96−99(1989)は、モノクローナル抗体を用いるアフィニティークロマトグラフィーを使用してヒト血漿からα−1 PIを単離する、単一工程手法を報告している。20%の収率でα−1 PI活性は61.1倍増大した。
【0007】
Burnoufら、Vox.Sang.52,291−297(1987)は、血漿上清A(コーン(Cohn)の画分II + IIIに等しい)から出発し、DEAEクロマトグラフィー及びサイズ排除クロマトグラフィーを用いて、80〜90%の純度の(SDS−PAGEによる)α−1 PIを製造し、純度を36倍増大させた。上清Aからの回収率は65〜70%であった。
【0008】
Heinfら、Eur.Respir.J.9:16S−20S(1990)は、出発物質としてコーンの画分IV−1を用い、そしてポリエチレングリコールによる分別沈殿及び引き続いてDEAEセファロース(商標)上の陰イオンクロマトグラフィーを利用する方法を述べている。最終生成物は45%の収率で得られ、約60%の純度を有する。
【0009】
Dubinら、Prep.Biochem.20:63−70(1990)は2工程クロマトグラフィー精製を示している。最初にα−1 PI、CI阻害剤、α−1 抗キモトリプシン及びインター−α−トリプシン阻害剤をブルー・セファロース(Blue Sepharose)(商標)から溶離し、次にα−1
PIをゲル瀘過で精製した。純度と収率のデータは得られていない。
【0010】
Ballieuxらは、4−フェニルブチルアミン・アフィニティークロマトグラフィー、陽イオン交換そして最後に免疫アフィニティー工程を用いて、化膿啖からα−1 PIとプロテイナーゼ−3の複合体を精製した(Ballieux,B.E.ら,J.Immunol.Methods 159:63−70(1993))。陽イオン交換工程で用いた緩衝液のpHは7.0であった。用いた条件下では、ほとんどの啖タンパク質は樹脂に結合したが、α−1 PI及びプロテイナーゼ−3は結合することなく通過した。
【0011】
Jordanら、米国特許第4,749,783号明細書(1988)は、ウイルス不活化工程の後に、製剤中の生物学的に不活性なタンパク質をアフィニティークロマトグラフィーで除去する方法を記述している。タンパク質の天然型と変性型間の分離は、アフィニティー樹脂に対する天然タンパク質の生物学的活性に基づくのであって、天然タンパク質と変性タンパク質間の物理的相違に基づくものではなかった。
【0012】
【発明が解決しようとする課題】
これらの方法のいずれも、精製工程として、低pH、低塩濃度及び中程度のタンパク質濃度での強陽イオン樹脂による通液型(flow through)クロマトグラフィーを用いていない。意外なことに、このような条件下で、活性α−1 PIのみがカラムを流れる。クロマトグラフィーカラムにより90%の収率を達成しそして陽イオンカラムに2回かけた後に約95%の純度が達成するように、方法の手はずを決めることができる。本発明は、大規模で、高純度でかつ高収率でヒト血漿からα−1 PIを精製する改良された方法を提供する。
【0013】
用語の定義
「活性α−1 PI」又は「天然α−1 PI」は、試験管内でのエラスターゼアッセイでエラスターゼ活性の阻害を示すα−1 PIを意味する。
【0014】
「不活性α−1 PI」又は「変性α−1 PI」は、試験管内でのエラスターゼアッセイでエラスターゼ活性に対して効果を生じないα−1 PIを意味する。
【0015】
「高度に精製された」は20%未満の汚染タンパク質を含むことを意味する。
「不活性α−1 PIを実質的に含まない」は10%未満の不活性α−1 PIを含むことを意味する。
【0016】
「不活性ウイルス類を実質的に含まない」は、認められているウイルス不活化工程(例えば、低温殺菌又は化学処理)に供されたために、低減された活性ウイルス含量を有することを意味する。一般に、このことは少なくとも対数約4のモデルウイルス力価の低減を意味する。
【0017】
すべての伝導率測定は25℃でなされた。
【0018】
【課題を解決するための手段】
本発明は、タンパク質含有水性溶液からα−1 PIを精製する方法であって、不活性(又は変性)α−1 PIを含む他のタンパク質類が媒体(又はイオン交換樹脂)に結合するのに対して、活性α−1 PIが該媒体(又は該イオン交換樹脂)に結合しないことを確保するのに十分なpH、イオン強度及びタンパク質濃度の条件下で、陽イオン樹脂クロマトグラフィー媒体を用いる通液型クロマトグラフィーによる方法である。好適な実施態様において、方法は以下の工程を含む:
(1) タンパク質溶液を約≦10 mmho/cmのイオン強度に透析又は透析瀘過し;
(2) 溶液のpHを約≦6.0に調整し;
(3) タンパク質溶液を約≦10 mgタンパク質/mLに調整し;
(4) 溶液を、陽イオン交換クロマトグラフィー樹脂を通過させ;そして
(5) クロマトグラフィーの通過液画分を精製α−1 PIとして回収する。
【0019】
本発明はヒト血漿からα−1 PIを精製する改良された方法である。この方法は、低イオン強度緩衝液によるpH≦6.0での陽イオン交換クロマトグラフィーを含む。精製α−1 PIを含む通過液画分を回収する。陽イオン交換クロマトグラフィーはα−1 PIに特異的であり、そしてこれを、コーン(Cohn)の画分IV−1懸濁液からの直接的な2工程精製方法(陽イオン交換カラム及び引き続く第2の陽イオン交換カラム)として、実際に用いることができる。
【0020】
この手法は十分に多方面に適用できるので、陽イオンクロマトグラフィーは、コーンの画分流出液 II + III、コーンの画分IV−1ペースト(現在、好適な出発物質)及び精製α−1 PIの範囲に及ぶ、どのような数の出発物質にも働くであろうし、そしてなお実質的に精製された生成物を生じさせる。
【0021】
製薬学的製剤の大規模製造用のオプションとして、ウイルス不活化を含む多種の追加的工程を加えて、収率を最適化し、ウイルス安全性を改良しそして規制コンプライアンスを確保することができる。これらの工程を含むことができるが、これらに限定されるものではない:
(1) 弱イオン交換樹脂(DEAE)による最初のクロマトグラフィー;
(2) 強陰イオン交換樹脂(QAE)による最初のクロマトグラフィー;
(3) 風乾又は溶液中での低温殺菌のいずれかを利用するウイルス不活化;
(4) あり得るウイルス汚染物を除去するためのウイルス除去瀘過;
(5) ウイルス不活化のための、例えば溶剤洗浄剤処理のような化学処理;及び
(6) 陽イオンクロマトグラフィーに先立って、出発物質を部分的に精製するための沈殿工程。
【0022】
タンパク質の荷電基を変えることにより、pHはイオン交換クロマトグラフィーにおいて重要な役割を果たす。それはクロマトグラフィー樹脂に対するタンパク質の結合挙動を、非結合から結合に、或いはその逆に変える。本発明に用いられる強陽イオン交換樹脂リガンドは、樹脂ビーズに直接結合しているか或いは短い炭素を主とする鎖を介して結合しているスルホネート基(−SO3 -)である。リガンドは1〜14のpH領域を通じて負電荷を有する。所定のpHでタンパク質の純有効表面電荷が負である場合には、タンパク質は遅滞なくカラムを流れる。一般に、溶液のpHがそのpIより高い場合にはタンパク質は負電荷を有する。
【0023】
良好に特性決定されたヒト血漿中の2種類のタンパク質はα−1 PI及びアルブミンであり、これらはそれぞれ4.8及び5.3の平均pIを有する。両タンパク質はpH5.45で負に荷電されてなければならず、かつ、陽イオン交換カラムを流れなければならない。意外なことに、低塩濃度及びpH5.45で、アルブミン及び変性(又は不活性)α−1 PIは樹脂に結合し、そして天然(又は活性)α−1 PIのみがカラムを流れることを、これらの実験は示している。α−1 PIについてのこの観察は、タンパク質のpIのみならずその3次構造がイオン交換において重要であることを示唆している。α−1 PIの天然型が明らかに負に荷電した表面を与えるのに対して、変性型の表面はより正に荷電され得る。したがって、変性タンパク質は幸運にも陽イオン樹脂に結合する。α−1 PIの天然型は、より高いpHでより相当安定である。安定性と陽イオン交換クロマトグラフィーによるα−1 PIの精製の両者を考慮した場合、pH5.45が実際上最低のpHとなる。
【0024】
イオン交換樹脂とタンパク質溶液の間の平衡は、溶液のイオン強度、タンパク質濃度、pH及び樹脂上の特異的なリガンドにより影響される。Yamamotoら、Biotechnol.Bioeng.25:1373−1391(1983)は、低タンパク質濃度で、溶液のイオン強度の関数としての分配係数に関連する半実験式を述べている。式は次のとおりである:
K = A(I)B + Kcrt
式中、Kは分配係数を表し、A及びBは実験的定数を表し、Iは溶液のイオン強度を表し、そしてKcrtは静電相互作用が無視できる高イオン強度におけるタンパク質の分配係数を表す。異なるタンパク質の分配係数は、所定のイオン強度で異なる。したがって、種々のタンパク質は、同一条件下で異なる速度で移動する。これを、このクロマトグラフィーにおけるタンパク質分離に適用する。低塩濃度では、添加工程中にほとんどのタンパク質の移動速度は樹脂に結合することにより遅れる。そのユニークな表面電荷特性により、天然α−1 PIはカラムを通過する。通過液緩衝液のイオン強度が増大する場合には、他のタンパク質と樹脂との相互作用が変わり、そしてより高いパーセンテージのタンパク質がカラムを流れるであろう。したがって、溶液のイオン強度を増大させると、カラムを流れるα−1 PIの純度が低減するであろう。
【0025】
また、塩濃度は、例えばNa+のような陽イオンを樹脂上の水素イオンで逆に交換することにより、溶離液のpHを変える。塩濃度を上げると、樹脂から溶離液により多くのH+が移動し、その結果溶離液のpHが低下する。したがって、最初のタンパク質溶液を平衡緩衝液で限外瀘過する場合には、添加溶液のイオン強度は平衡緩衝液と等しいか或いはわずかに高くなるはずである。添加中に、pHは同一のままに残るか或いはほんのわずか低下するはずである。添加後に、平衡緩衝液を洗浄液として使用すると、イオン強度が低下してpHが高くなり得る。したがって、pHを維持するために、この洗浄液に、わずかに高いイオン強度の緩衝液を用いることができる。
【0026】
上述したように、また、高タンパク質濃度は樹脂に結合したタンパク質の平衡に影響を及ぼす。タンパク質濃度が増大する場合には、吸着等温式は通常飽和曲線を示す(Yamamotoら、”Ion Exchange Chromatography of Proteins”,1988,Chromatographic Science Series)。したがって、結合の飽和レベルに近づくときに、結合した不純物の総量は最大値に達する。最大値に達した後には、試料のタンパク質濃度が増大するにつれて、カラムを通過する不純物の相対的パーセンテージは増大する。したがって、タンパク質濃度は、不純物の吸着曲線の直線領域に入るほど十分に低い場合に最適である。
【0027】
タンパク質は溶液のpHを緩衝化する傾向があるので、タンパク質濃度はまた溶離液のpHに影響を及ぼす。クロマトグラフィー媒体に結合することによりタンパク質は溶液から選択的に除去されるので、溶液の緩衝能自体(すなわち、pH)が変わる。pH変動はカラムに吸着されているタンパク質の相対パーセンテージに依存するので、クロマトグラフィーに対する影響は複雑である。吸着は、カラムの流形、溶液の緩衝能、タンパク質混合物の変性した性質及び種々のタンパク質の競争的結合により、影響を受ける。
【0028】
より高タンパク質濃度を添加すると、陽イオン交換カラムは、陽イオン交換カラムの充填が進行するにつれて溶離液中のpHの連続的な上昇をもたらすことが、我々の実験で示されている。増大したpHはα−1 PIの純度を低下させる。一般にはより希釈されたタンパク質の溶液がα−1 PI精製クロマトグラフィーのためのより良い出発物質であるが、大規模に精製するために陽イオン交換カラムの充填を増大させるためには、不純物曲線及びpH効果の許容し得る範囲内でより高い濃度を用いることができる。
【0029】
【実施例】
実施例1
好適な実施の形態
我々の好適な実施態様では、強陽イオン交換カラムにかけるのに先立って、コーンの画分IV−1(Fr.IV−1)懸濁液からα−1 PIを部分的に精製するために、陰イオン交換クロマトグラフィーを用いる。比活性によるとコーンの画分IV−1懸濁液は約10%のα−1 PIである。他の血漿画分と同様に、IV−1懸濁液は、種々のタンパク質、例えばリポタンパク質、免疫グロブリン、グロブリン、メタプロテイン等を含む。リポタンパク質は特別の問題を生じさせる。それらがクロマトグラフィー樹脂に結合する場合には、それらを除去するのが困難であり、そして樹脂の孔を塞ぎ、樹脂ベッドを加圧することができる。また、タンパク質残渣が樹脂上に蓄積するので結合能が低減する。Fr.IV−1懸濁液からリポタンパク質及び他の不純物を除去するために、強イオンクロマトグラフィーの代わりに、第1の工程としてDEAEイオン交換クロマトグラフィーを用いる。DEAE充填条件はリポタンパク質の主要部分がカラムに結合することなく、通過するような条件である。DEAEイオン交換クロマトグラフィーのα−1 PI溶離条件はα−1 PIについて95〜100%の回収率を得るように選択される。
【0030】
IV−1懸濁液を≧5 mmho/cmの伝導率でかつpH8.0に調整した。次にタンパク質溶液をDEAE樹脂上にかける。α−1 PIを、20mM 第二リン酸ナトリウム及び95mM 塩化ナトリウム、pH8.0で溶離した。DEAE溶離液を、20 mM第一リン酸ナトリウム及び5mM 塩化ナトリウム、pH6.5で透析瀘過する。透析瀘過した溶離液をpH5.45に調整し、次に強陽イオン樹脂上にかける。α−1 PIはカラムを流れる。通過液をpH7.0に調整し、そして0.15M 塩化ナトリウムを添加する。
【0031】
この時点で所望であれば、α−1 PIを凍結することができる。ウイルス不活化のために、必要であればα−1 PI溶液を解凍し、60mM ヒスチジン及び5mM 塩化カルシウムでpH6.5に調整し、次に凍結乾燥する。次に凍結乾燥物を80℃で72時間加熱して、ウイルスを不活化する。次に凍結乾燥物を精製水に溶解し、そして37%(w/v)スクロース及び0.38M シトレートを安定剤として添加する。エンベロープをもつウイルスに対する溶剤洗浄剤処理として、リン酸0.3%トリ−n−ブチル(TNBP)及び0.2%コール酸ナトリウムを添加する。30℃で3時間インキュベーションした後、TNBP及びコール酸塩を透析瀘過で除去する。
【0032】
ウイルス不活化した溶液を20mM 第一リン酸ナトリウム及び5mM 塩化ナトリウム、pH6.5で透析瀘過する。透析瀘過した溶液のpHを5.45に調整し、そして第2の強陽イオン樹脂カラム上にかけて、残存するいずれの汚染物も除去しそしてウイルス不活化工程で変性したα−1 PIも除去する。α−1 PIの天然型はカラムを流れる。回収した通過液のpHを7.0に調整し、0.15M 塩化ナトリウムを添加し、さらなるウイルス不活化工程として15μMフィルターを通過させる。他のタンパク質を実質的に含まない、高度に精製されたα−1 PI(〉95%)を製造する。DEAEクロマトグラフィーにより、リポタンパク質が除去され、そしてα−1 PI純度が20%に増大する。第1の陽イオンカラム処理は約90%の回収率で約60〜70%の純度のα−1
PIを生じさせた。第2の陽イオンカラム処理は最終生成物としての95%の純度を達成する。陰イオンカラムを1M NaCl及び1M NaOHで洗浄して、結合タンパク質を除去する。2つの陽イオンカラムに結合したタンパク質を、1M 塩化ナトリウム及び引き続いて1M 水酸化ナトリウムで除去する。
【0033】
実施例2
本実施例では、コーンの画分流出液 II + IIIを出発物質とした。それを、5℃で20mM 第1リン酸ナトリウム及び5mM 第1塩化ナトリウム、pH6.5で透析瀘過し、アルコールを除去してイオン強度を低下させた。次に溶液のpHを5.45に調整し、そして既に平衡化した強陽イオン交換カラムにかけた。α−1 PIに有意に富む画分として通過液を回収した。出発物質中の汚染タンパク質は陽イオン交換カラム上に保持された。
【0034】
陽イオン交換カラムを1回通過させた後、α−1 PIを、コーンの画分流出液 II + III中の他のタンパク質から実質的に精製した。SDS−PGAEによると、通過液の純度は82%α−1 PIであった。比活性は、全タンパク質1mg当たり0.03mgのエラスターゼ阻害活性から全タンパク質1mg当たり0.59mgのエラスターゼ阻害活性へと、20倍増大した。
【0035】
実施例3
本実施例では、出発物質として市販の部分的に精製したα−1 PI(プロラスチン(Prolastin)(商標)、Miles,Inc.社)を用いた。プロラスチン(商標)は0.1M NaCl及び0.02M リン酸ナトリウムでpH7.0に緩衝化されている。α−1 PIの濃度は約30mg/mLであり、そしてタンパク質濃度は約60mg/mLである。アルブミンは典型的には全タンパク質の12%であり、そしてIgAは典型的には約1mg/mL(全タンパク質の2.5%)で存在する。プロラスチン(商標)を5mM NaCl及び20mM 第1リン酸ナトリウムで透析瀘過して、イオン強度を低下させた。溶液のpHを5.45に下げ、タンパク質濃度を5.3mg/mLに低減し、そして溶液を強陽イオン交換カラムにかけた。通過液を精製したα−1 PIとして回収し、そしてこれは、SDS−PGAEによるとα−1 PIの単量体及び二量体のみ(それぞれ95.4%及び4.6%のタンパク質)を示した。次に溶液を0.15M NaClでpH7.0に安定化し、そして瀘過し又は濃縮した後、凍結乾燥して安定な最終生成物を得た。
【0036】
結合タンパク質画分を溶離し、そしてSDS−PGAEで電気泳動にかけた。クーマシーブルーで可視化された44%のタンパク質はα−1 PI分子量領域内であった。この画分をエラスターゼ阻害活性アッセイしたところ、α−1 PI活性は示されなかった。このことは、このバンドのタンパク質が不活化α−1
PIであることを示唆する。
【0037】
上記実施例を次の表に要約する。
【0038】
【表1】

上記の説明により、タンパク質精製の当業者は変法に想到するであろうと考えられる。したがって、上記の実施例は具体例としてのみ解釈されるべきであり、本発明の範囲は上記の特許請求の範囲によってのみ制限されるものと意図される。
【0040】
本発明の主な特徴及び態様は以下の通りである。
【0041】
1. α−1プロテイナーゼ阻害剤及び他のタンパク質類を含む水性溶液中の、該α−1プロテイナーゼ阻害剤を精製する方法であって、
(A) 該活性α−1プロテイナーゼ阻害剤はイオン交換樹脂に結合しないが該溶液中の他のタンパク質類は結合するように、該水性溶液中のpH、イオ ン強度及びタンパク質濃度を調整する工程;及び
(B) 該溶液を、該イオン交換樹脂を通過させ、そして通過液中のα−1プロテイナーゼ阻害剤を回収する工程、
を含んでなる該方法。
【0042】
2.工程(A)の水性溶液のpHが約6.0未満であるか或いは等しい上記1記載の方法。
【0043】
3.水性溶液のイオン強度が約10mmho/cm未満であるか或いは等しい上記1記載の方法。
【0044】
4.水性溶液のタンパク質濃度が約10mg/mL未満であるか或いは等しい上記1記載の方法。
【0045】
5.工程が一回より多く実施される上記1記載の方法。
【0046】
6.精製工程間に溶液に対してウイルス不活化工程が実施される上記5記載の方法。
【0047】
7.該ウイルス不活化が約60℃より高いか或いはそれと等しい温度で、約10時間より多いか或いは等しい時間で、α−1プロテイナーゼ阻害剤を加熱することを含んでなる上記6記載の方法。
【0048】
8.該ウイルス不活化が化学剤を添加することを含んでなる上記6記載の方法。
【0049】
9.該ウイルス不活化が溶液にリン酸トリ−n−ブチル及び洗浄剤を添加することを含んでなる上記8記載の方法。
【0050】
10. 活性ウイルス類を実質的に含有せず、不活性α−1プロテイナーゼ阻害剤を実質的に含有せず、そして全タンパク質1mg当たり少なくとも約0.9mgのエラスターゼ阻害活性を有する活性α−1プロテイナーゼ阻害剤を含んでなる、高度に精製したα−1プロテイナーゼ阻害剤調製物。
【0051】
11.α−1プロテイナーゼ阻害剤の純度が90%より高い上記10記載の調製物。
【0052】
12.製薬学的に許容できるキャリアー中の上記10記載の産物。
【図面の簡単な説明】
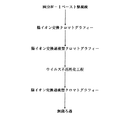
【図1】本発明に従いα−1 PIを精製するための、一般的なCohn(コーン)の分別法、画分から誘導されたタンパク質、並びに2つの出発物質[コーンの画分IV−1並びにコーンの流出液 II & III]を説明するフローチャートである。
【図2】我々のα−1 PI精製方法の好適な工程を示すフローチャートである。
【図3】従来技術と比べた場合の本発明の方法による純度及び比活性の改良を説明する棒グラフである。
Claims (6)
- α−1プロテイナーゼ阻害剤及び他のタンパク質類を含む水性溶液からα−1プロテイナーゼ阻害剤を精製する方法であって、
(A) 該水性溶液中の活性α−1プロテイナーゼ阻害剤は強陽イオン交換樹脂に結合しないが、他のタンパク質類は結合するように、該水性溶液中のpHを5.45〜6.0に、イオン強度を10mmho/cm以下に、そしてタンパク質濃度を10mg/mL以下に調整する工程;及び
(B) 該水性溶液を、該強陽イオン交換樹脂を通過させ、そしてα−1プロテイナーゼ阻害剤を含有する通過液を回収する工程、
を含んでなる該方法。 - 工程(A)及び(B)が一回より多く実施される請求項 1 記載の方法。
- 精製工程間に水性溶液にウイルス不活化工程が実施される請求項 2 記載の方法。
- ウイルス不活化工程が60℃より高い温度に10時間より長くα−1プロテイナーゼ阻害剤を加熱することを含んでなる請求項3記載の方法。
- ウイルス不活化工程が化学剤を添加することを含んでなる請求項4記載の方法。
- ウイルス不活化工程が水性溶液にリン酸トリ−n−ブチル及び洗浄剤を添加することを含んでなる請求項5記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US295119 | 1994-08-24 | ||
| US08/295,119 US5610285A (en) | 1994-08-24 | 1994-08-24 | Purification of α-1 proteinase inhibitor using novel chromatographic separation conditions |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH08176198A JPH08176198A (ja) | 1996-07-09 |
| JP3770414B2 true JP3770414B2 (ja) | 2006-04-26 |
Family
ID=23136291
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP23322895A Expired - Lifetime JP3770414B2 (ja) | 1994-08-24 | 1995-08-21 | 新規なクロマトグラフイー分離条件を用いるα−1プロテイナーゼ阻害剤の精製 |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US5610285A (ja) |
| EP (1) | EP0698615B1 (ja) |
| JP (1) | JP3770414B2 (ja) |
| KR (1) | KR100451266B1 (ja) |
| CN (2) | CN1161371C (ja) |
| AT (1) | ATE187462T1 (ja) |
| CA (2) | CA2692337A1 (ja) |
| DE (1) | DE69513751T2 (ja) |
| DK (1) | DK0698615T3 (ja) |
| ES (1) | ES2139794T3 (ja) |
| GR (1) | GR3032728T3 (ja) |
| HK (1) | HK1072372A1 (ja) |
| PT (1) | PT698615E (ja) |
Families Citing this family (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0871671A1 (en) * | 1995-09-07 | 1998-10-21 | PPL Therapeutics (Scotland) Limited | Purification of alpha-1 proteinase inhibitor |
| SE9600796D0 (sv) * | 1996-02-29 | 1996-02-29 | Pharmacia Biotech Ab | A method of preparing a liquid mixture |
| US5616693A (en) * | 1996-07-01 | 1997-04-01 | Alpha Therapeutic Corporation | Process for seperating alpha-1-proteinase inhibitor from COHN IV1 +1V4 paste |
| GB2318732A (en) | 1996-11-01 | 1998-05-06 | Johnson & Johnson Medical | Wound healing compositions containing alpha-1-antitrypsin |
| AT407114B (de) * | 1997-06-10 | 2000-12-27 | Immuno Ag | Alpha 1-antitrypsin-präparation sowie verfahren zu deren herstellung |
| AUPP521298A0 (en) * | 1998-08-12 | 1998-09-03 | Life Therapeutics Limited | Purification of fibrinogen |
| US6093804A (en) * | 1998-09-24 | 2000-07-25 | American National Red Cross | Method for purification of alpha-1 proteinase inhibitor |
| AUPP790898A0 (en) | 1998-12-23 | 1999-01-28 | Life Therapeutics Limited | Renal dialysis |
| US20050224355A1 (en) * | 1999-12-23 | 2005-10-13 | Brendon Conlan | Removal of biological contaminants |
| AUPP790698A0 (en) | 1998-12-23 | 1999-01-28 | Life Therapeutics Limited | Separation of microorganisms |
| WO2000051624A2 (en) * | 1999-03-05 | 2000-09-08 | The Trustees Of University Technology Corporation | Methods and compositions useful in inhibiting apoptosis |
| US6849605B1 (en) * | 1999-03-05 | 2005-02-01 | The Trustees Of University Technology Corporation | Inhibitors of serine protease activity, methods and compositions for treatment of viral infections |
| AU3511500A (en) | 1999-03-05 | 2000-09-21 | Trustees Of University Technology Corporation, The | Inhibitors of serine protease activity, methods and compositions for treatment of nitric oxide-induced clinical conditions |
| WO2000051625A1 (en) * | 1999-03-05 | 2000-09-08 | The Trustees Of University Technology Corporation | Inhibitors of serine protease activity, methods and compositions for treatment of herpes viruses |
| AUPP971399A0 (en) | 1999-04-12 | 1999-05-06 | Life Therapeutics Limited | Separation of plasma components |
| US6462180B1 (en) * | 1999-11-24 | 2002-10-08 | Bayer Corporation | Method of preparing α-1 proteinase inhibitor |
| US7077942B1 (en) * | 1999-12-23 | 2006-07-18 | Gradipore Limited | Removal of biological contaminants |
| AUPQ691400A0 (en) | 2000-04-14 | 2000-05-11 | Life Therapeutics Limited | Separation of micromolecules |
| AUPQ697300A0 (en) | 2000-04-18 | 2000-05-11 | Life Therapeutics Limited | Separation apparatus |
| WO2001078877A1 (en) | 2000-04-18 | 2001-10-25 | Gradipore Limited | Electrophoresis separation and treatment of samples |
| US6923896B2 (en) * | 2000-09-22 | 2005-08-02 | The Texas A&M University System | Electrophoresis apparatus and method |
| CN1235667C (zh) * | 2000-10-06 | 2006-01-11 | 格拉迪普有限公司 | 多口分离装置及方法 |
| CA2432641A1 (en) | 2000-12-14 | 2002-06-20 | Bayer Corporation | Method of preparing alpha-1 proteinase inhibitor |
| AUPR222300A0 (en) * | 2000-12-21 | 2001-01-25 | Life Therapeutics Limited | Electrophoresis device and method |
| US7777006B2 (en) | 2002-12-31 | 2010-08-17 | Csl Behring L.L.C. | Method for purification of alpha-1-antitrypsin |
| US20040220242A1 (en) * | 2003-05-02 | 2004-11-04 | Leland Shapiro | Inhibitors of serine protease activity, methods and compositions for treatment of nitric oxide induced clinical conditions |
| US7850970B2 (en) * | 2003-08-26 | 2010-12-14 | The Regents Of The University Of Colorado | Inhibitors of serine protease activity and their use in methods and compositions for treatment of bacterial infections |
| ES2305845T5 (es) * | 2003-09-22 | 2012-04-12 | Kamada Ltd. | Preparación a gran escala de un inhibidor de proteinasa alfa-1 y su utilización |
| CN1314447C (zh) * | 2004-04-12 | 2007-05-09 | 爱华生物科技(香港)有限公司 | 一种具有抑制蛋白酶作用的药物的制备方法 |
| EP1909810B2 (en) | 2005-06-07 | 2017-08-23 | The Regents of the University of Colorado | Inhibitors of serine protease activity and their use in methods and compositions for treatment of graft rejection and promotion of graft survival |
| US7807435B2 (en) * | 2005-08-11 | 2010-10-05 | Baxter International Inc. | Method for the purification of alpha-1 proteinase inhibitor (a1PI) |
| US20080124303A1 (en) * | 2005-12-12 | 2008-05-29 | Cavit Sciences, Inc | Methods and compositions for treatment of viral infections |
| EP2066174B1 (en) | 2006-09-12 | 2017-11-08 | Beth Israel Deaconess Medical Center, Inc. | Compositions containing alpha-1-antitrypsin and methods for use |
| MX2009012786A (es) * | 2007-06-01 | 2009-12-10 | Hoffmann La Roche | Purificacion de inmunoglobulina. |
| WO2009025754A2 (en) | 2007-08-17 | 2009-02-26 | Csl Behring Gmbh | Methods for purification of alpha-1-antitrypsin and apolipoprotein a-i |
| EP2321336A1 (en) * | 2008-07-18 | 2011-05-18 | Talecris Biotherapeutics, Inc. | Method of preparing alpha-1 proteinase inhibitor |
| KR101104637B1 (ko) * | 2010-02-26 | 2012-01-12 | 한국철도기술연구원 | 무개화차의 컨테이너 고정장치 |
| US8772462B2 (en) | 2010-05-26 | 2014-07-08 | Baxter International Inc. | Removal of serine proteases by treatment with finely divided silicon dioxide |
| AU2010202125B1 (en) | 2010-05-26 | 2010-09-02 | Takeda Pharmaceutical Company Limited | A method to produce an immunoglobulin preparation with improved yield |
| EP2723370A4 (en) | 2011-06-24 | 2015-06-03 | Univ Colorado Regents | COMPOSITIONS, PROCESS AND USE OF ALPHA-1-ANTITRYPHIN FUSION MOLECULES |
| WO2013098672A2 (en) | 2011-12-30 | 2013-07-04 | Grifols, S.A. | Alpha1-proteinase inhibitor for delaying the onset or progression of pulmonary exacerbations |
| KR20140137347A (ko) | 2012-01-10 | 2014-12-02 | 더 리젠츠 오브 더 유니버시티 오브 콜로라도, 어 바디 코포레이트 | 알파-1 안티트립신 융합 분자용 조성물, 방법 및 용도 |
| WO2014137903A2 (en) | 2013-03-08 | 2014-09-12 | Genzyme Corporation | Integrated continuous manufacturing of therapeutic protein drug substances |
| CA2943938A1 (en) | 2013-03-29 | 2014-10-02 | The Regents Of The University Of Colorado, A Body Corporate | Compositions and methods for preparing a subject for organ or non-organ implantation |
| TWI709569B (zh) | 2014-01-17 | 2020-11-11 | 美商健臻公司 | 無菌層析樹脂及其用於製造方法的用途 |
| TWI671312B (zh) | 2014-01-17 | 2019-09-11 | 美商健臻公司 | 無菌層析法及製法 |
| ES2749383T3 (es) | 2014-11-06 | 2020-03-20 | Hoffmann La Roche | Variantes de la región Fc con unión al FcRn modificada y procedimientos de uso |
| US11369703B2 (en) | 2018-08-31 | 2022-06-28 | Genzyme Corporation | Sterile chromatography resin and use thereof in manufacturing processes |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1189674B (de) * | 1963-06-12 | 1965-03-25 | Behringwerke Ag | Verfahren zur Isolierung von alpha-Antitrypsin |
| US4379087A (en) * | 1982-06-17 | 1983-04-05 | Cutter Laboratories, Inc. | Method of preparing alpha-1-proteinase inhibitor |
| US4752576A (en) * | 1984-06-14 | 1988-06-21 | Chiron Corporation | Expression of α-1 antitrypsin in yeast |
| US4656254A (en) * | 1985-12-02 | 1987-04-07 | Miles Laboratories, Inc. | Method of preparing alpha-1-proteinase inhibitor and antithrombin III |
| US4629567A (en) * | 1986-03-07 | 1986-12-16 | Smithkline-Rit | Alpha-1-antiprotease purification |
| US4749783A (en) | 1986-07-11 | 1988-06-07 | Miles Laboratories, Inc. | Viral inactivation and purification of active proteins |
| FR2610633B1 (fr) * | 1987-02-05 | 1992-09-18 | Lille Transfusion Sanguine | Procede d'obtention d'un concentre d'a 1-antitrypsine a partir de plasma humain et son utilisation a titre de medicament |
| US5319072A (en) * | 1992-01-10 | 1994-06-07 | Alpha Therapeutic Corporation | Human antithrombin-III preparation |
-
1994
- 1994-08-24 US US08/295,119 patent/US5610285A/en not_active Expired - Lifetime
-
1995
- 1995-08-11 AT AT95112630T patent/ATE187462T1/de active
- 1995-08-11 PT PT95112630T patent/PT698615E/pt unknown
- 1995-08-11 DK DK95112630T patent/DK0698615T3/da active
- 1995-08-11 ES ES95112630T patent/ES2139794T3/es not_active Expired - Lifetime
- 1995-08-11 DE DE69513751T patent/DE69513751T2/de not_active Expired - Lifetime
- 1995-08-11 EP EP95112630A patent/EP0698615B1/en not_active Expired - Lifetime
- 1995-08-14 CA CA002692337A patent/CA2692337A1/en not_active Abandoned
- 1995-08-14 CA CA002156007A patent/CA2156007C/en not_active Expired - Lifetime
- 1995-08-21 JP JP23322895A patent/JP3770414B2/ja not_active Expired - Lifetime
- 1995-08-23 KR KR1019950026050A patent/KR100451266B1/ko not_active IP Right Cessation
- 1995-08-23 CN CNB951087789A patent/CN1161371C/zh not_active Expired - Lifetime
- 1995-08-23 CN CNB2004100694447A patent/CN100548372C/zh not_active Expired - Fee Related
-
2000
- 2000-02-23 GR GR20000400427T patent/GR3032728T3/el unknown
-
2005
- 2005-06-17 HK HK05105079.8A patent/HK1072372A1/xx not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| CA2692337A1 (en) | 1996-02-25 |
| DE69513751D1 (de) | 2000-01-13 |
| EP0698615B1 (en) | 1999-12-08 |
| ES2139794T3 (es) | 2000-02-16 |
| HK1072372A1 (en) | 2005-08-26 |
| CA2156007A1 (en) | 1996-02-25 |
| EP0698615A1 (en) | 1996-02-28 |
| AU3011195A (en) | 1996-03-07 |
| CN1125231A (zh) | 1996-06-26 |
| AU700638B2 (en) | 1999-01-07 |
| CN1161371C (zh) | 2004-08-11 |
| KR960007617A (ko) | 1996-03-22 |
| ATE187462T1 (de) | 1999-12-15 |
| CA2156007C (en) | 2010-03-09 |
| PT698615E (pt) | 2000-05-31 |
| CN100548372C (zh) | 2009-10-14 |
| DK0698615T3 (da) | 2000-05-15 |
| CN1572321A (zh) | 2005-02-02 |
| GR3032728T3 (en) | 2000-06-30 |
| DE69513751T2 (de) | 2000-04-27 |
| KR100451266B1 (ko) | 2004-12-23 |
| US5610285A (en) | 1997-03-11 |
| JPH08176198A (ja) | 1996-07-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3770414B2 (ja) | 新規なクロマトグラフイー分離条件を用いるα−1プロテイナーゼ阻害剤の精製 | |
| US11224643B2 (en) | Methods of treatment using alpha-1-antitrypsin compositions | |
| US20110237781A1 (en) | Method of preparing alpha-1 proteinase inhibitor | |
| ZA200601206B (en) | Process for preparing an alpha-1-antitrypsin solution | |
| US6093804A (en) | Method for purification of alpha-1 proteinase inhibitor | |
| US6462180B1 (en) | Method of preparing α-1 proteinase inhibitor | |
| EP1343809B1 (en) | Method of preparing alpha-1 proteinase inhibitor | |
| AU2015200572A1 (en) | Method for purification of alpha-1-antitrypsin |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20050607 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20050906 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20050909 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20051207 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20060124 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20060202 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100217 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100217 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110217 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120217 Year of fee payment: 6 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130217 Year of fee payment: 7 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130217 Year of fee payment: 7 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140217 Year of fee payment: 8 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |