JP3769293B2 - 変性された固体酸化物触媒の製造方法 - Google Patents
変性された固体酸化物触媒の製造方法 Download PDFInfo
- Publication number
- JP3769293B2 JP3769293B2 JP50520695A JP50520695A JP3769293B2 JP 3769293 B2 JP3769293 B2 JP 3769293B2 JP 50520695 A JP50520695 A JP 50520695A JP 50520695 A JP50520695 A JP 50520695A JP 3769293 B2 JP3769293 B2 JP 3769293B2
- Authority
- JP
- Japan
- Prior art keywords
- catalyst
- hours
- metal
- parts
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C5/00—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms
- C07C5/22—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms by isomerisation
- C07C5/27—Rearrangement of carbon atoms in the hydrocarbon skeleton
- C07C5/2702—Catalytic processes not covered by C07C5/2732 - C07C5/31; Catalytic processes covered by both C07C5/2732 and C07C5/277 simultaneously
- C07C5/2724—Catalytic processes not covered by C07C5/2732 - C07C5/31; Catalytic processes covered by both C07C5/2732 and C07C5/277 simultaneously with metals
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/06—Silicon, titanium, zirconium or hafnium; Oxides or hydroxides thereof
- B01J21/066—Zirconium or hafnium; Oxides or hydroxides thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/28—Molybdenum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/30—Tungsten
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/64—Platinum group metals with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/652—Chromium, molybdenum or tungsten
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/64—Platinum group metals with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/652—Chromium, molybdenum or tungsten
- B01J23/6527—Tungsten
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/0009—Use of binding agents; Moulding; Pressing; Powdering; Granulating; Addition of materials ameliorating the mechanical properties of the product catalyst
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G25/00—Compounds of zirconium
- C01G25/02—Oxides
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01G—COMPOUNDS CONTAINING METALS NOT COVERED BY SUBCLASSES C01D OR C01F
- C01G41/00—Compounds of tungsten
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C5/00—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms
- C07C5/22—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms by isomerisation
- C07C5/27—Rearrangement of carbon atoms in the hydrocarbon skeleton
- C07C5/2767—Changing the number of side-chains
- C07C5/277—Catalytic processes
- C07C5/2791—Catalytic processes with metals
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01P—INDEXING SCHEME RELATING TO STRUCTURAL AND PHYSICAL ASPECTS OF SOLID INORGANIC COMPOUNDS
- C01P2002/00—Crystal-structural characteristics
- C01P2002/50—Solid solutions
- C01P2002/52—Solid solutions containing elements as dopants
- C01P2002/54—Solid solutions containing elements as dopants one element only
-
- C—CHEMISTRY; METALLURGY
- C10—PETROLEUM, GAS OR COKE INDUSTRIES; TECHNICAL GASES CONTAINING CARBON MONOXIDE; FUELS; LUBRICANTS; PEAT
- C10G—CRACKING HYDROCARBON OILS; PRODUCTION OF LIQUID HYDROCARBON MIXTURES, e.g. BY DESTRUCTIVE HYDROGENATION, OLIGOMERISATION, POLYMERISATION; RECOVERY OF HYDROCARBON OILS FROM OIL-SHALE, OIL-SAND, OR GASES; REFINING MIXTURES MAINLY CONSISTING OF HYDROCARBONS; REFORMING OF NAPHTHA; MINERAL WAXES
- C10G2400/00—Products obtained by processes covered by groups C10G9/00 - C10G69/14
- C10G2400/10—Lubricating oil
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Inorganic Chemistry (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Description
パラフィン、特に軽質パラフィンの異性化は確立された精製プロセスであって、アルキル化装置への追加のフィード原料を供給するため、或いは比較的低いオクタン価の直鎖パラフィンをガソリンプール中に混合することのできるオクタン価のより高い分枝異性体へ転化するために伝統的に用いられている。直鎖パラフィン、例えばn−ブタン、n−ペンタンおよびn−ヘキサンは対応するイソパラフィンへ種々の異性化方法によって転化されており、その方法には種々の触媒を使用することができる。例えば、米国特許第3,766,286号;同第3,852,184号;同第3,855,346号;同第3,839,489号;同第4,144,282号および同第4,814,544号に開示されているように、非再生性(non-regenerable)のルイス酸およびブレンステッド酸触媒を使用することもできる。この種の工業的方法は、フィリップス・ペトロリアム・カンパニー(Phillips Petroleum Company)(接触異性化(Catalytic Isomerization))およびシェル・デベロプメント・カンパニー(Shell Development Company)(液相異性化(Liquid Phase Isomerization))を含む種々の企業によって開発されている。
種々の工業的異性化方法において用いられるその他の種類の触媒には、多孔質担体上の金属水素化/脱水素化成分、通常は白金を含む。この方法の1つの例は、水素および白金触媒の存在下で異性化が行われるペネックス法(Penex Process)(UOP)である。イソ−ケル法(Iso-Kel Process)(M. W. Kellogg)も水素の循環と共に高価な金属触媒を使用し、ペンタファイニング(Pentafining)(アルコ/エンゲルハルト(Arco/Englehardt))およびブタマー(Butamer)(UOP)法も外部水素循環と共に担体に担持された白金を使用する。この種の方法は、例えば、米国特許第4,834,866号(シュミット(Schmidt))および同第4,783,575号(シュミット)に開示されている。
モレキュラーシーブを含んでなる担体に担持される金属成分を利用する異性化方法は、米国特許第3,842,114号(シー(Sie));同第3,836,597号(シー);同第4,778,944号(ザーキー(Zarchy))および同第4,374,296号(ハーグ(Haag))に開示されている。
米国特許第4,783,575号(シュミット)および同第4,834,866号(シュミット)に開示されているように、パラフィン異性化触媒は、改質器フィード原料から環状の芳香族前駆物質を除くための開環触媒としても使用され得る。例えば、ベンゼンの生成を最小にするために、ベンゼンの前駆物質であるシクロヘキサンを異性化して、一部だけが改質器において芳香族化された分枝パラフィンの混合物とすることができる。米国特許第3,631,117号には、開環およびパラフィン異性化のための触媒として、ゼオライトに担持される周期表第VIII族金属を使用する環状炭化水素の水素化異性化(hydroisomerization)の方法が記載されている。芳香族前駆物質、特にシクロヘキサンを開環させるためにパラフィン異性化を利用することは、自動車ガソリンの芳香族化合物含有率、特にベンゼン含有率が環境的規制のために制限されるので、将来的により重要になるであろう。
本発明の1つの要旨によれば、
(i)第VIB族金属のオキシアニオン(oxyanion)によって第IVB族金属酸化物を変性させることにより生成する酸性固体生成物、および
(ii)固体生成物に付着する水素化/脱水素化成分
を含んでなる触媒が提供される。
本発明のもう1つの要旨によれば、上記の本発明の1つの要旨において提供される触媒を製造する方法であって、
(a)第IVB族金属の水酸化物もしくは水和された酸化物を、第VIB族金属のオキシアニオンのソースを含んでなる水溶液に接触させて、酸素、第IVB族金属および第VI族金属を含んでなる酸性固体生成物を生成させる工程、ならびに
(b)工程(a)の固体生成物に水素化/脱水素化成分を付着させる工程
を含んでなる方法が提供される。
本発明の更にもう1つの要旨によれば、C4〜C8パラフィンフィードを異性化する方法であって、フィードを上記の本発明の1つの要旨において提供される触媒に接触させることを含んでなる方法が提供される。
ここで述べる触媒は、第IVB族金属の酸化物、好ましくはジルコニアまたはチタニアを含んでなる。この第IVB族金属酸化物は、第VIB族金属のオキシアニオン、例えばタングステンのオキシアニオン、例えばタングステート(tungstate、タングステン酸基)により変性される。第VIB族金属のオキシアニオンによる第IVB族金属酸化物の変性は、この物質に酸官能性を付与する。第VIB族金属オキシアニオン、特にタングステートによる、第IVB族金属酸化物、特にジルコニアの変性は、米国特許第5,113,034号;日本国特開平1−288339号およびプロシーディングズ・ナインス・インターナショナル・コングレス・オン・キャタリシス(Proceedings 9th International Congress on Catalysis)、第4巻、第1727〜1735頁(1988年)中のケイ・アラタ(K. Arata)およびエム・ヒノ(M. Hino)による論文などに開示されている。
更に、水素化/脱水素化成分が、第VIB族金属のオキシアニオンによる第IVB族金属酸化物の変性によって生成する酸性固体生成物と組み合わされる。この水素化/脱水素化成分は、有機化合物、例えば、場合により1またはそれ以上のヘテロ原子、例えば酸素、窒素、金属もしくは硫黄により置換されていることもある炭化水素への水素の付加またはそのような有機化合物からの水素の除去を触媒する物質の能力を付与するものであり、その場合、十分な水素化または脱水素化条件下で変性された金属酸化物に有機化合物を接触させる。
水素化/脱水素化成分の例には、第VIII族金属(例えば、Pt、Pd、Ir、Rh、Os、Ru、Ni、CoおよびFe)、第IVA族金属(例えば、SnおよびPb)、第VB族金属(例えば、SbおよびBi)ならびに第VIIB族金属(例えば、Mn、TcおよびRe)の酸化物、水酸化物または遊離金属(即ち、原子価0の状態)の形態が含まれる。触媒は好ましくは、1またはそれ以上の貴金属(例えばPt、Pd、Ir、Rh、0sもしくはRu)の1またはそれ以上の触媒形態を含むことが好ましい。そのような貴金属または非貴金属の触媒形態の組み合わせ、例えばPtとSnの組み合わせなどを用いることもできる。水素化/脱水素化成分の金属の原子価状態は、例えば、この成分が酸化物または水酸化物の形態である場合には、還元された原子価状態であることが好ましい。この金属の還元された原子価状態は、還元剤、例えば水素が反応へのフィード中に含まれる場合に反応の途中でその場にて達成することができる。
本発明の説明のためには、第VIB族金属のオキシアニオンにより変性された第IVB族金属酸化物という表現は、元素分析によって、第IVB族金属、第VIB族金属および酸素を含んでなり、別々に形成された第VIB族金属酸化物またはオキシアニオンと混合された、別々に形成された第IVB族金属酸化物の単なる混合物よりも更に酸性である(多くの酸性度(acidity)を有する)物質を意味することを意図している。本発明の、第VIB族金属、例えばダングステンのオキシアニオンにより変性された第IVB族金属、例えばジルコニウムの酸化物は、第IVB族金属酸化物のソースと、第VIB族金属酸化物またはオキシアニオンのソースとの間の現実の化学的相互作用によって生じるものと考えられている。
この化学的相互作用は、Proceedings 9th International Congress on Catalysis、第4巻、第1727〜1735頁(1988年)中のケイ・アラタおよびエム・ヒノによる前述の論文において説明されている。この論文では、スルフェートがある特定の金属、例えばZrの水酸化物または酸化物と反応する場合に固体超酸が生成することが示唆されている。これらの超酸は、金属、例えばZrに配位する二座(bidentate)スルフェートイオンの構造を有すると言われている。この論文において、タングステートがZrの水酸化物または酸化物と反応する場合に超酸も生成することが更に示唆されている。得られるタングステート変性ジルコニア物質は、スルフェートおよびジルコニウムを含んでなる前述の超酸と類似する構造を有すると理論付けられており、その場合に二座構造においてタングステン原子が硫黄原子を置換している。
本発明の触媒がアラタおよびヒノによる前述の論文において示唆されている二座構造を有し得ると考えられているが、第VIB族金属のオキシアニオンにより変性された第IVB族金属酸化物に存在する触媒的活性部材(catalytically activesite)の特定の構造は、まだ確認されておらず、本発明の触媒成分がいずれかの特定の構造に限定されるべきであることを意図しているのではない。
他の元素、例えばアルカリ(第IA族)またはアルカリ土類(第IIA族)化合物は、触媒の触媒的特性を変えるために場合によって本発明の触媒に加えることができる。そのようなアルカリまたはアルカリ土類化合物を本発明の触媒に加えることにより、触媒の成分それ自体、例えばPtまたはWの触媒特性を、水素化/脱水素化成分または酸成分として機能するそれらの能力の点で向上させることができる。
本発明の触媒の第IVB族金属(例えば、Ti、ZrもしくはHf)および第VIB族金属(例えば、Cr、MoもしくはW)の化学種は、これらの化学種のいずれかの特定の原子価状態に限定されるものではない。これらの化学種は、触媒中においてその化学種が採り得るいずれのプラスの酸化価(oxidation value)で存在してもよい。触媒が、例えばタングステンを含んでなる場合、触媒を還元条件に付することにより、特定の反応、例えばn−ヘキサンの異性化などを触媒する触媒の全体としての触媒的性能を向上させることができる。
本発明の触媒を調製するために用いる第IVB族金属酸化物の適当なソースには、そのような酸化物、例えばオキシクロリド、クロリド、ニトレートなど、特にジルコニウムまたはチタニウムのそのようなものを生成することができる化合物が含まれる。そのような金属のアルコキシドを第IVB族金属酸化物の前駆物質またはソースとして使用することもできる。そのようなアルコキシドの例には、ジルコニウムn−プロポキシドおよびチタニウムi−プロポキシドが含まれる。第IVB族金属酸化物の好ましいソースは水酸化ジルコニウム、即ち、Zr(OH)4および水和ジルコニアである。水和ジルコニアという表現は、橋架け酸素原子を介して他のジルコニウム原子に共有結合的に結合されるジルコニウム原子、即ち、Zr−O−Zrを有してなり、更に利用することのできる表面ヒドロキシル基を有してなる物質を意味することを意図している。これらの利用することのできる表面ヒドロキシル基は、第IVB族金属、例えばタングステンのアニオンのソースと反応して本発明の酸性触媒成分を生成すると考えられている。Proceedings 9th International Congress on Catalysis、第4巻、第1727〜1735頁(1988年)中のケイ・アラタおよびエム・ヒノによる前述の論文において示唆されているように、100℃〜400℃の温度におけるZr(OH)4の予備焼成によって、タングステートとより好ましく相互作用する化学種が生成する。この予備焼成によって、ZrOH基が縮合し、表面ヒドロキシル基を有する高分子状のジルコニア化学種が生成すると考えられている。この高分子状の化学種を本明細書では水和ジルコニアの形態と称する。
好ましい態様において、水和された第IVB族金属酸化物、例えば水和ジルコニアは、第VIB族金属オキシアニオン、例えばタングステートのソースと接触する前に水熱処理に付される。特に、水和された第IVB族金属酸化物は、7またはそれ以上のpH値を有する水溶液において還流することが好ましい。何らかの理論により拘束されることを望むわけではないが、水熱処理は金属酸化物の表面積を増大させるので有利であるということが理論付けられている。おそらく、後に用いられるタングステートのソースとのより望ましい相互作用が促進されるように、水熱処理が水和ジルコニアの表面ヒドロキシル基を変化させるということも理論的に可能である。
水熱条件は、少なくとも50℃、例えば少なくとも80℃、例えば少なくとも100℃の温度を含むことがある。水熱処理は、大気圧より高い圧力にて封止された容器内で行なうことができる。しかしながら処理の好ましいモードには、還流条件下において開放された容器を用いることが含まれる。液体媒体中における水和第IVB族金属酸化物をかき混ぜること、例えば還流する液体の作用および/または撹拌により、液体媒体と水和酸化物との効果的な相互作用が促進される。水和された酸化物と液体媒体との接触の時間は、少なくとも1時間、例えば少なくとも8時間であってよい。この処理のための液体媒体は、約7またはそれ以上のpH値、例えば9またはそれ以上のpH値を有していてもよい。適当な液体媒体には、水、(NH4 +、Na+、K+、Mg2+およびCa2+の水酸化物を含む)水酸化物溶液、(NH4 +、Na+、K+、Mg2+およびCa2+の炭酸塩および重炭酸塩を含む)炭酸塩および重炭酸塩溶液、ピリジンおよびその誘導体ならびにアルキス/ヒドロキシルアミンが含まれる。
第VIB族金属、好ましくはモリブデンまたはタングステンのオキシアニオンの適当なソースには、メタタングステン酸アンモニウムまたはメタモリブデン酸アンモニウム、塩化タングステンまたは塩化モリブデン、タングステンカルボニルまたはモリブデンカルボニル、タングステン酸またはモリブデン酸およびタングステン酸ナトリウムまたはモリブデン酸ナトリウムが含まれるが、これらに限定されるものではない。
本発明の触媒の水素化/脱水素化成分は、第VIII族金属、例えば白金、イリジウム、オスミウム、パラジウム、ロジウム、ルテニウム、ニッケル、コバルト、鉄およびこれらの二種またはそれ以上の混合物から誘導することができる。これらの成分は、場合によって、第IVA族金属、好ましくはSnから誘導される成分および/または第VIIB族金属、好ましくはレニウムもしくはマンガンから誘導される成分と混合することができる。これらの成分を触媒に添加するのは、従来技術において既知の方法、例えばイオン交換、含浸または物理的混合などにより行うことができる。例えば、これらの金属の塩の溶液を残りの触媒成分に、それぞれの成分を組み合わせるのに十分な条件下において接触させることができる。金属含有塩は水溶性であることが好ましい。そのような塩の例には、クロロ白金酸、テトラアミン白金錯体、塩化白金、硫酸スズおよび塩化スズが含まれる。
本発明の触媒の調製は、例えば、第VIB族金属のアニオン、好ましくはタングステートもしくはモリブデートを含む水溶液を用いて、第IVB族金属の水酸化物または酸化物、特に水和された酸化物を含浸させ、続いて乾燥させることにより行うことができる。得られる固体酸性生成物(solid acidic product)は続いて、好ましくは酸化的雰囲気において、約500℃〜900℃、好ましくは700℃〜850℃、更に好ましくは750〜825℃の温度で焼成する。焼成時間は48時間まで、好ましくは0.5〜24時間、更に好ましくは1.0〜10時間であってよい。最も好ましい態様において、焼成は約800℃にて1〜3時間行なう。触媒の水素化/脱水素化成分(第VIII族金属、第VIIB族金属など)は、焼成工程の後で、従来技術において既知の技術、例えば含浸、共含浸(coimpregnation)、共沈澱(coprecipitation)および物理的混合などによって固体酸性生成物に組み合わ(または結合)される。水素化/脱水素化成分と残りの触媒成分との混合は、以下において説明するように、これらの残りの成分をバインダーまたはマトリックス物質と組み合わせる前または後において行うこともできる。
ジルコニウムの水酸化物または水和された酸化物のソースを使用する場合、全体としての物質の所望の酸性度を付与する理論化された化学反応を導くために、この材料とタングステンのオキシアニオンのソースとの組み合わせを、例えば500℃を越える温度にて焼成することが必要である。しかしながら、ジルコニアのより反応性のソースを用いる場合には、そのように高い焼成温度は必ずしも必要としないことも有り得る。
本発明の触媒において、第IVB族酸化物ではジルコニウム酸化物が好ましく、第VIB族アニオンではタングステートが好ましく、そして水素化/脱水素化成分では白金および/または白金−スズが好ましい。
定量的に言えば、本発明の触媒の元素分析により、第IVB族金属、第VIB族金属および酸素の存在が示されるであろう。そのような分析において測定される酸素の量は、幾つかの要因、例えば第IVB族および第VIB族金属の原子価状態、水素化/脱水素化成分の形態、含水率などに依存することになる。従って、本発明の触媒の組成を特徴付ける場合、存在する酸素の量はあまり有益な情報ではない。機能的な用語を用いると、本発明の触媒中の第VIB族金属のオキシアニオンの量は、第IVB族酸化物の酸性度を増大する量として表現することができる。この量を本明細書では酸性度増大量と称する。本発明の触媒の元素分析を用いて、触媒中における第IVB族金属および第VIB族金属の相対的な量を測定することができる。これらの量から、XO2/YO3の形式でモル比を計算することができ、ここでXは第IVB族金属を示しており、XO2の形態であると推定され、Yは第VIB族金属を示しており、YO3の形態であると仮定されている。しかしながら、これらの形態の酸化物、即ち、XO2およびYO3は必ずしも実際に存在しているのではなく、本明細書では本発明の触媒中のXおよびYの相対的な量を単に計算するために用いているということが理解されるであろう。本発明の触媒は、1000まで、例えば300まで、例えば2〜100、例えば4〜30のXO2/YO3の形式で表される計算モル比を有することができ、ここでXは少なくとも1つの第IVB族金属(例えばTi、ZrまたはHf)であり、Yは少なくとも1つの第VIB族金属(例えば、Cr、MoまたはW)である。
触媒中に存在する水素化/脱水素化成分の量は、特にこの成分が貴金属である場合、触媒の0.001〜5重量%、好ましくは0.1〜2重量%を含んでなることが都合がよい。
本明細書において説明する触媒は、C4〜C8パラフィンの異性化および好ましくはn−ヘキサンの2,2−ジメチルブタンへの異性化のための触媒として用いることができる。適当なフィードは、ノルマルおよび/または1つの分枝を有する低オクタン価のC4〜C8炭化水素の実質量を含有する。フィードは、開環反応を受け得るC6およびC7環状パラフィンもかなりの量で含有してよい。
本発明の異性化プロセスは、500℃以下、好ましくは90〜425℃、更に好ましくは150〜370℃の温度、および100〜20000kPa(1〜200気圧)、好ましくは100〜10000kPa(1〜100気圧)、更に好ましくは500〜5000kPa(5〜50気圧)の圧力で、炭化水素フィードを液相または気相のいずれかで、固体触媒に接触させることにより行なうことができる。異性化プロセスは、水素の存在下または不存在下のいずれかで行うことができ、更に好ましくは水素の存在下において行なわれる。水素:炭化水素のモル比は、典型的には、0.01:1〜10:1、好ましくは0.5:1〜2:1の範囲である。
本発明の触媒とその性能を向上させる別の物質とを混合することが好ましいことがある。そのような物質には、活性および非活性物質ならびに合成および/または天然産のゼオライト、ならびに無機物質、例えばクレイ、シリカおよび/または金属酸化物などが含まれる。特に、触媒は、マトリックス材料と複合化されて触媒の最終形態を形成してもよく、そのための常套のマトリックス材料としては、例えばアルミナ、シリカ−アルミナおよびシリカが適しており、非酸性バインダーとしてシリカが好ましいとされる。他のバインダー物質、例えばチタニア、ジルコニアおよび他の金属酸化物またはクレイなどを用いることもできる。活性触媒とマトリックスとの複合化は、活性触媒:マトリックスが重量比で80:20〜20:80、例えば80:20〜50:50の範囲であってよい。複合化は、材料を一緒に混練し、続いて押し出して所望の最終触媒粒状物にペレット化することを含む常套の手段によって行うことができる。
触媒は、常套の予備硫化処理、例えば硫化水素の存在下で加熱することにより、金属成分の酸化物形態を対応する硫化物形態に転化する処理によって処理することができる。
本発明の異性化プロセスにおいて、n−パラフィンおよびモノメチル分枝パラフィン成分は、一般により良好なオクタン価向上剤(octane booster)である分枝度のより高いパラフィンに異性化される。説明のため、ピー・エイチ・エメット(P. H. Emmett)編、Catalysis、第VI巻(1958年)からの純粋な炭化水素のオクタン価の以下の表を見ると、これらの反応の重要性がわかる。
本発明の触媒は、C4〜C8パラフィン系炭化水素を、純粋な化合物としてまたは混合物としてのいずれかで異性化するために用いることができる。精製操作において、パラフィンは通常、混合物で存在しており、C4〜C8物質に加えて、この沸点範囲外で沸騰する炭化水素を含有することがあり、シクロパラフィンおよび芳香族化合物が存在することもある。従って、フィードは、C4〜C8パラフィン、例えばブタン、ペンタン、ヘキサンを含んでなることになり、これらは製油所ストリーム、例えば、溶媒抽出装置からのラフィネーカット(raffinate cuts)、改質器フィード原料またはエチレン分解器からの熱分解ガソリンなどの中に存在し得る。フィードは、環状炭化水素を例えばC6+ナフサの形態で含むことがあり、金属成分が組み合わせられた触媒の存在下においてそのようなフィード中の環状物質は開環反応を受けてパラフィンを生成し、そのパラフィンが続いて異性化されて、分留(または分画)により環状化合物から分離され得るイソパラフィンとなり、環状物質はなくなるまでリサイクルされる。純粋なパラフィンフィード(C4〜C8)に加えて、かなりのレベルのオレフィンを含有するパラフィン−オレフィン混合フィードを用いることもできる。
特に触媒が多孔質バインダー物質を含んでなる場合、触媒中にルイスまたはブレンステッド酸活性を有する追加の物質を含ませることによって、より高い異性化活性が提供され得る。この目的のために、液体および固体の酸物質の両者を用いることができる。適当な追加の酸性物質の例には、三塩化アルミニウム、三フッ化ホウ素および三フッ化ホウ素の錯体、例えば水、低級アルコールまたはエステルなどとの錯体が含まれる。添加し得る最大量は、担体物質、特にバインダー物質の、添加された成分を収着する能力によって定まり、実験によって容易に決定することができる。
本発明の触媒は、1段または多段触媒床において単独の異性化触媒として用いることもできるし、他の異性化触媒と組み合わせて用いることもできる。例えば、米国特許第4,783,575号および同第4,834,866号に記載されているように、フィードを、本発明の触媒を含んでなる触媒床に最初に接触させ、続いて別の触媒、例えばモルデナイトに担持させたPt、ゼオライトベータに担持させたPtまたは塩素化した白金−アルミナ触媒を含んでなるもう1つの触媒床に接触させることもできる。第1触媒床の温度は、第2触媒床の温度より高くてもよい。本発明の触媒を広範な開環反応を起こさせるために使用する必要がある場合、特に最初の触媒床においては、比較的高い温度、例えば500℃もの温度および/または比較的高い圧力、例えば20000kPa(200気圧)もの圧力が用いてよい。
本発明の触媒の使用を異性化反応において用いることは上記の説明で強調してきたが、この触媒が種々の有機化合物、例えば炭化水素化合物の転化プロセス、特に2機能触媒((1)酸性機能および(2)水素化/脱水素化機能)を使用することが必要なプロセスに有用であることが理解されるであろう。そのような転化プロセスには、100℃〜700℃の温度、10〜3000kPa(0.1〜30気圧)の圧力、0.1〜20重量時間空間速度および0〜20の水素/炭化水素モル比を含む反応条件での炭化水素の水素化分解;300℃〜700℃の温度、10〜1000kPa(0.1〜10気圧)の圧力および0.1〜20の重量時間空間速度を含む反応条件での炭化水素化合物の脱水素化;100℃〜700℃の温度、10〜6000kPa(0.1〜60気圧)の圧力、0.5〜400の重量時間空間速度および0〜20の水素/炭化水素モル比を含む反応条件での芳香族化合物へのパラフィンの転化;100℃〜700℃の温度、10〜6000kPa(0.1〜60気圧)の圧力、0.5〜400重量時間空間速度および0〜20の水素/炭化水素モル比を含む反応条件での芳香族化合物、例えばベンゼン、トルエン、およびキシレンへのオレフィンの転化;200℃〜500℃の温度、100〜20000kPa(1〜200気圧)の圧力、10〜1000重量時間空間速度および0.3/1〜20/1の芳香族炭化水素/ポリアルキル芳香族炭化水素モル比および0〜20の水素/炭化水素モル比を含む反応条件を用いる、ポリアルキル芳香族炭化水素の存在下における芳香族炭化水素のアルキル交換反応;ならびに−25℃〜400℃、例えば75℃〜200℃の温度、100kPa以下から34500kPa(大気圧以下から5000psig)、例えば100〜7000kPa(1〜1000psig)の圧力、1:2〜500:1、例えば5:1〜100:1の全パラフィン:全オレフィンのモル比およびオレフィン基準で0.01〜100、例えば0.05〜5の重量時間空間速度を含む反応条件での、パラフィンからオレフィンへの水素移動(transferring)が含まれるが、これらの例に限定されない。
本発明の触媒は、種々の水素処理反応、例えばフィード原料から例えば金属、窒素および/または硫黄などの元素を、特にヘテロ原子の形態でそれらの元素を含む残渣油などからの金属、窒素および/または硫黄の除去などに用いることもできる。これらの水素処理反応は、金属、窒素および/または硫黄を除去するのに十分な条件下で、十分な量の水素と共にフィード原料を本発明の触媒に接触させることを含んでなる。
実施例(参考例)1
この実施例は、タングステート変性ジルコニア触媒の調製を説明する。ジルコニルクロリド(ZrOCl2・8H2O)1重量部を10M NH4OH溶液3重量部に加えた。得られるスラリー(Zr(OH)4)を濾過し、蒸留した脱イオン水5部で洗浄した後、140℃で8時間風乾した。得られるZr(OH)4約7.5重量部を、メタタングステン酸アンモニウム((NH4)6H6W12O40)1部を含む水溶液2.2部を初期湿式法(incipient wetness、ジルコニア触媒にタングステートを含浸させるのに丁度十分な量のメタタングステン酸アンモニウム溶液を用いて湿潤させる標準的な含浸法)により含浸させた。得られる物質を120℃にて2時間乾燥した後、流通空気中で800℃にて2時間焼成した。試料を触媒試験に付する前に、流通窒素中で500℃にて1時間焼成した。この試料は、ZrO2/WO3の計算モル比が11.6であった。
実施例(参考例)2
H2PtCl6および(NH4)6H6W12O40をZr(OH)4に初期湿式で同時含浸させること(incipient wetness co-impregnation)により、白金およびタングステート変性ジルコニア触媒を調製した。特に、Zr(OH)4181.8重量部に対し、(NH4)6H6W12O4024.4部およびH2PtCl61部を含む水溶液54.5部を初期湿式含浸により添加した。得られる物質を続いて120℃にて2時間乾燥し、その後800℃にて2時間空気焼成した。この白金含有触媒は、触媒試験に付する前に、流通窒素中で500℃にて1時間焼成した後、300℃にて約2時間、流通水素により還元した。この触媒はZrO2/WO3の計算モル比が11.6であり、触媒の全重量基準で100ppmのPtを含有していた。
実施例(参考例)3
同時(共)含浸段階において、より多くのH2PtCl6を使用したこと以外は、実施例2と同様の方法で触媒を調製した。この触媒はZrO2/WO3の計算モル比が11.6であり、触媒の全重量基準で0.2重量%のPtを含有していた。
実施例(参考例)4
共含浸段階において、より多くのH2PtCl6を使用したこと以外は、実施例2と同様の方法で触媒を調製した。この触媒はZrO2/WO3の計算モル比が11.6であり、触媒の全重量基準で2重量%のPtを含有していた。
実施例(参考例)5〜8
n−ヘキサンの異性化において、実施例1〜4の触媒を試験した。n−ヘキサン異性化反応は、固定床下降流反応器(fixed-bed down-flow reactor)において行った。高圧ポンプを用いて、液体のn−ヘキサンを反応器に供給した。マス・フロー・コントローラ(mass flow controller)を通して水素を装入した。ガスクロマトグラフィーにより生成物を分析した。実験は、260℃、触媒1cc当り1時間当たり1.8cc n−C6の重量時間空間速度(LHSV)、3200kPa(450psig)およびH2/n−C6モル比1.4にて行った。
表1に示す実験結果は、触媒に少量の白金を添加することによって、所望する高オクタン価のジメチルブタンを生成するn−ヘキサン異性化活性が大きく向上することを示している。
以下の表中において、次に示す略語は以下の意味を有する:n−C6(n−ヘキサン);3−MP(3−メチルペンタン);2−MP(2−メチルペンタン);2,3−DMB(2,3−ジメチルブタン);2,2−DMB(2,2−ジメチルブタン);i−C5(イソペンタン);n−C5(n−ペンタン);C4−(4またはそれ以下の炭素原子を有する炭化水素);C7+(7またはそれ以上の炭素原子を有する炭化水素);CH(シクロヘキサン);MCP(メチルシクロペンタン);BZ(ベンゼン);C3−(3またはそれ以下の炭素原子を有する炭化水素);i−C4(イソブタン);n−C4(n−ブタン);およびC5+(5またはそれ以上の炭素原子を有する炭化水素)。
実施例3の触媒(Pt 0.2重量%およびZrO2/WO3の計算モル比11.6)を、より低い温度、220℃およびより低いLHSVにて試験した。結果を表2に示すが、異性化物(isomerate)の収率が高いことを示している。
この実施例では、800℃空気焼成工程の後で、タングステート変性ジルコニア物質に白金を加えた。実施例1に記載した調製法で、Zr(OH)472.5重量部を、(NH4)6H6W12O4012.2部を含む水溶液21.7部を用いて含浸した。得られる物質を120℃にて2時間乾燥した後、空気中で800℃にて2時間焼成した。室温まで冷却した後、もう1つの(第2回目)初期湿式含浸を行った:この場合には、蒸留水21.7部に溶解したH2PtCl61部を添加した。触媒を120℃にて2時間乾燥し、流通空気中で350℃にて3時間焼成し、続いて300℃にて約2時間、水素により還元した。この試料は、触媒の全重量基準で0.5重量%のPtを含有していた。
実施例(参考例)12
実施例11の白金およびタングステート変性ジルコニア触媒を、260℃、3200kPa(450psig)、LHSV=0.6hr-1およびH2/n−C6モル比1.4にて、n−ヘキサン異性化について試験した。結果を表3に示す。
10M NH4OH溶液中におけるZr(O)Cl2の迅速な加水分解によって、ジルコニウムヒドロキシド(Zr(OH)4)を合成した。続いて、スラリーを30分間微粉砕して濾過し、脱イオン水により洗浄し、4時間真空乾燥して、140℃にて8時間乾燥した。
メタタングステン酸アンモニウム(NH4)6H6W12O40を用いてZr(OH)4を含浸することによって、タングステート変性ジルコニアを調製した。得られた試料を120℃にて2時間乾燥した後、800℃にて焼成した。物質を周囲温度に冷却した後、H2PtCl6を用いる初期湿式法によりPtを添加した。白金含有触媒を流通空気中で400℃にて2時間焼成し、その後、流通水素中で300℃にて約2時間還元した。触媒は、ZrO2/WO3の計算モル比が11.6であり、触媒の全重量基準で0.5重量%のPtを含有していた。
実施例(参考例)14および15
これらの実施例により、C6環状炭化水素の開環およびn−ヘキサン異性化を実施例13の触媒上で同時に行って得られる結果を説明する。これらの実験において、表4に示す組成を有する合成フィード原料を用いた。生成物の組成および操作条件を表5に示す。結果は、本発明の触媒が開環について高い活性で示す一方で、高いC5+収量およびパラフィンのより高分枝度パラフィンへの高い異性化選択性が維持されることを示している。
この実施例では、水和ZrO2担体の調製を説明する。ジルコニルクロライドZrOCl2・8H2O)1重量部を、H2O 10部に溶解し、濃NH4OH(aq)を溶液のpHが〜9(9以下で9に近い値)になるまで加えた。得られるスラリー(Zr(OH)4)を濾過し、蒸留した脱イオン水10部で洗浄した。固体を130℃にて16時間乾燥した。
実施例(参考例)17
この実施例では、実施例16で説明したジルコニア担体からのWOx/ZrO2触媒の調製について説明する。実施例16から得られる乾燥生成物約5.6重量部に、メタタングステン酸アンモニウム(NH4)6H6W12O401部を含む水溶液4.2部を初期湿式法により含浸させた。得られる物質を空気中で乾燥した後、空気中にて825℃で3時間焼成した。
実施例(参考例)18
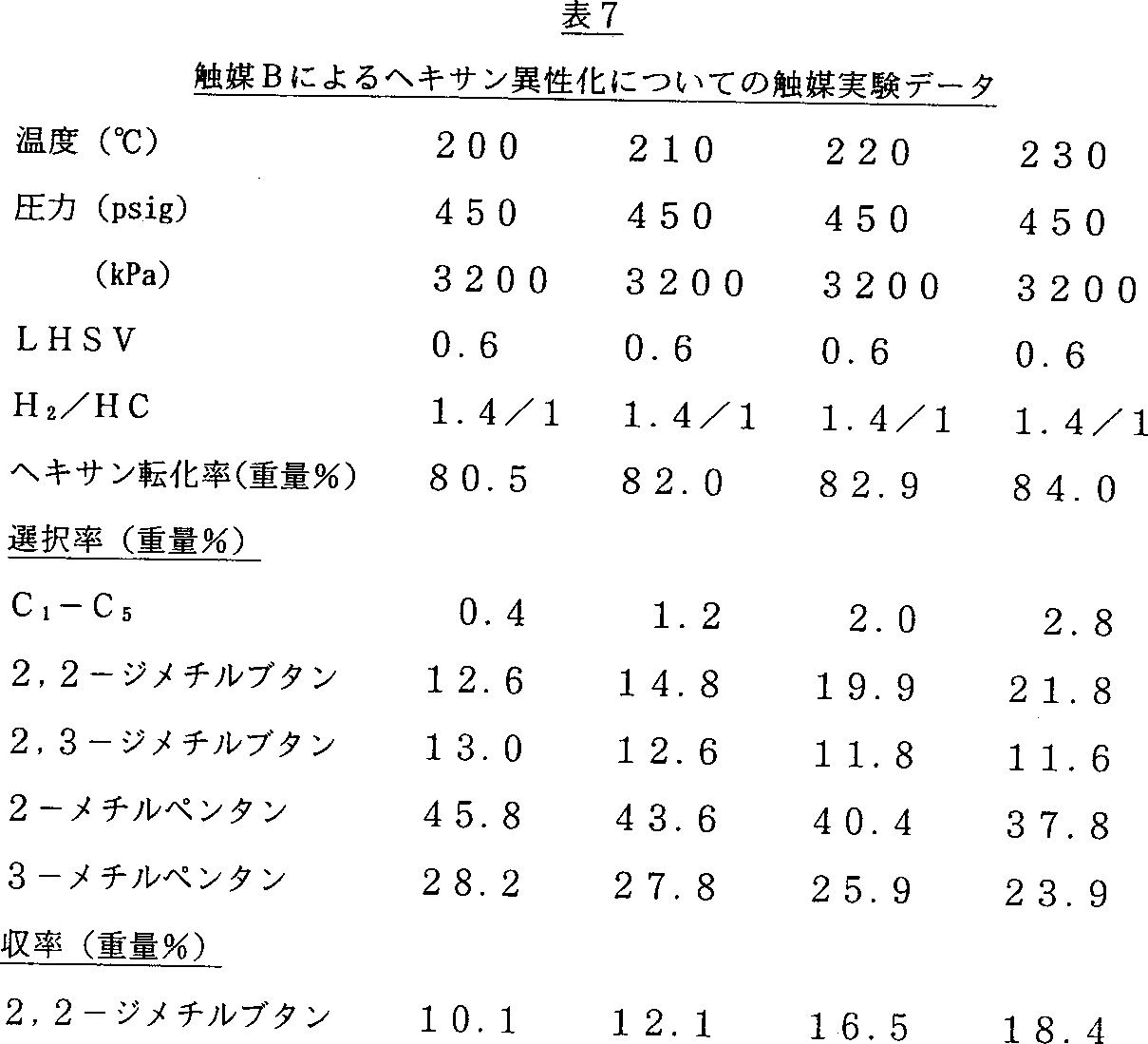
この実施例では、実施例17において説明した、得られた生成物からのPt/WOx/ZrO2触媒の調製および使用について説明する。H2PtCl6 8%溶液1部に、H2O 2.5部を加えた。この混合物を続いて、実施例17からの(130℃にて)乾燥した生成物7部の初期湿式法による含浸に使用した。続いて、触媒を空気中で300℃にて2時間焼成した。この触媒を触媒Aと称する。触媒の実験において、300℃および大気圧にてH2(100cc/分)による触媒Aの還元を4時間行った。続いて装置を所望の条件にし、ヘキサンフィードを導入した。触媒実験のデータおよび結果を表6に示す。
実施例(参考例)19
この実施例では、実施例16において述べたジルコニア担体を用いるもう1つのWOx/ZrO2触媒の調製を説明する。実施例16からの乾燥生成物約2.4重量部に、メタタングステン酸アンモニウム1部を含有する水溶液2.6部を初期湿式法により含浸させた。得られる物質を空気中で乾燥した後、空気中で825℃にて3時間焼成した。
実施例(参考例)20
この実施例では、実施例19において説明した、得られた生成物からのPt/WOx/ZrO2触媒の調製および使用について説明する。H2PtCl6 8%溶液1部に、H2O 2.5部を添加した。続いてこの混合物を実施例19からの(130℃にて)乾燥した生成物の7部の初期湿式法による含浸に使用した。続いて、触媒を空気中で300℃にて2時間焼成した。この触媒を触媒Bと称する。触媒の実験において、300℃および大気圧にてH2(100cc/分)による触媒Bの還元を18時間行った。続いて装置を所望の条件にし、ヘキサンフィードを導入した。触媒実験のデータおよび結果を表7に示す。
実施例(参考例)21
この実施例では、水熱処理されたジルコニア担体の調製を説明する。実施例16からの濾過された湿潤ケーキ1重量部を、蒸留した脱イオン水10部と混合し、濃NH4OH(aq)によって混合物のpHを〜9(9以下で9に近い値)に調節した。この混合物を16時間還流させ、冷却し、濾過し、水10部で洗浄した。この固体を130℃で16時間風乾した。
実施例(参考例)22
この実施例では、実施例21において説明したジルコニア担体からのWOx/ZrO2触媒の調製を説明する。実施例21からの乾燥生成物約5.6重量部に、メタタングステン酸アンモニウム1部を含む水溶液4.2部を初期湿式法により含浸させた。得られる物質を空気中で乾燥し、空気中で825℃にて3時間焼成した。
実施例(参考例)23
この実施例では、実施例22において得られた生成物からのPt/WOx/ZrO2触媒の調製および使用を説明する。H2PtCl6 8%溶液1部にH2O 2.5部を添加した。続いて、この混合物を実施例22からの(130℃での)乾燥生成物7部の初期湿式法による含浸に用いた。続いてこの触媒を空気中で300℃にて2時間焼成した。この触媒を触媒Cと称する。触媒の実験において、300℃および大気圧にてH2(100cc/分)による触媒Cの還元を4時間行った。続いて装置を所望の条件にして、ヘキサンフィードを導入した。触媒実験のデータおよび結果を表8に示す。
実施例(参考例)24
この実施例では、実施例21において説明したジルコニア担体を用いる、もう1つのWOx/ZrO2触媒の調製を説明する。実施例21からの乾燥生成物約3.4重量部に、メタタングステン酸アンモニウム1部を含む水溶液2.6部を初期湿式法により含浸させた。得られる物質を空気中で乾燥した後、空気中で825℃にて3時間焼成した。
実施例(参考例)25
この実施例では、実施例24において説明した得られた生成物からのPt/WOx/ZrO2触媒の調製および使用を説明する。H2PtCl6 8%溶液1部に、H2O 2.5部を加えた。続いてこの混合物を実施例24からの(130℃における)乾燥生成物7部の初期湿式法による含浸に使用した。続いて、触媒を空気中で300℃にて2時間焼成した。この触媒を触媒Dと称する。触媒の実験において、300℃および大気圧にてH2(100cc/分)による触媒Dの還元を18時間行った。続いて装置を所望の条件にして、ヘキサンフィードを導入した。触媒実験のデータおよび結果を表9に示す。
同等のH2還元時間において、塩基溶液と共に加熱される処理がなされた触媒(触媒CおよびD)は、処理されていない触媒(触媒AおよびB)よりも種々の温度において2,2−ジメチルブタンの異性化生成物について向上した収率を示した。
この実施例では、pHが〜7(7以下で7に近い値)にて、更に水熱処理されたジルコニア担体の調製を説明する。実施例16からの濾過された湿潤ケーキ1重量部を、蒸留された脱イオン水10部と混合し、この混合物を撹拌しながら16時間還流させた。混合物を冷却し、濾過し、水10部で洗浄した。固体を130℃にて16時間風乾した。
実施例27
この実施例では、実施例26において説明したジルコニア担体からのWOx/ZrO2触媒の調製を説明する。実施例26からの乾燥生成物約5.6重量部に、メタタングステン酸アンモニウム1部を含む水溶液4.2部を初期湿式法により含浸させた。得られる物質を空気中で乾燥して、空気中で825℃にて3時間焼成した。
実施例28
この実施例では、実施例27において得られた生成物からのPt/WOx/ZrO2触媒の調製および使用を説明する。H2PtCl6 8%溶液1部にH2O 2.5部を加えた。続いて、この混合物を、実施例27からの(130℃における)乾燥生成物7部の初期湿式法による含浸に用いた。続いて、触媒を空気中で300℃にて2時間焼成した。この触媒を触媒Eと称する。触媒の実験において、300℃および大気圧にてH2(100cc/分)による触媒Eの還元を18時間行った。続いて装置を所望の条件にして、ヘキサンフィードを導入した。触媒のデータおよび結果を表10に示す。対応するH2還元時間において、pH7〜9で水溶液と共に加熱される処理がなされた触媒(触媒C、DおよびE)は、処理されていない触媒(触媒AおよびB)よりも、種々の温度において2,2−ジメチルブタンの異性化生成物について向上した収率を示した。
この実施例では、含水ジルコニア担体の調製を説明する。ジルコニルクロリド(ZrOCl2・8H2O)1重量部をH2O 10部に溶解し、溶液のpHが〜9(9以下で9に近い値)になるまで濃NH4OH(aq)を加えた。得られるスラリー(Zr(OH)4)を濾過し、蒸留した脱イオン水10部で洗浄した。固体を蒸留した脱イオン水10部と混合し、NH4OH(aq)によって混合物のpHを〜9(9以下で9に近い値)に調節した。この混合物を16時間還流させ、冷却し、濾過し、水10部で洗浄した。固体を130℃で16時間風乾した。
実施例30
この実施例では、実施例29において説明したジルコニア担体からのWOx/ZrO2触媒の調製を説明する。実施例29からの乾燥生成物約3.3重量部に、メタタングステン酸アンモニウム1部を含む水溶液2.6部を初期湿式法により含浸させた。得られる物質を空気中で乾燥した後、空気中で825℃にて3時間焼成した。得られる生成物を触媒Fと称する。
実施例31
メタタングステン酸アンモニウム1.17部を使用すること以外は、触媒Fと同様にして触媒Gを調製した。
実施例32
メタタングステン酸アンモニウム1.67部を使用すること以外は、触媒Fと同様にして触媒Hを調製した。
実施例33−35
焼成後、H2O 2.5部および8%H2PtCl6 1部の溶液を用いて、初期湿式法により触媒F、GおよびHにPtを含浸させた。触媒を風乾した後、空気中で300℃にて2時間乾燥した。
実施例36および37
実施例33からの触媒Fのヘキサン異性化についての試験を行った。2つの独立したランにおいて、フィードヘキサンに接触させる前に、新しい触媒のH2(100cc/分)による処理を300℃にて、4時間および18時間行った。実験の条件および接触の結果を表11に示す。
実施例38および39
実施例34からの触媒Gのヘキサン異性化についての試験を、実施例36および37と同様にして行った。実験条件および接触の結果を表12に示す。
実施例40および41
実施例35からの触媒Hのヘキサン異性化についての試験を行った。2つの独立したランにおいて、フィードヘキサンに接触させる前に、新しい触媒のH2(100cc/分)による処理を300℃にて、4時間および72時間行った。実験の条件および接触の結果を表13に示す。
触媒F、GおよびHについては、4時間に代えて18時間水素を用いて処理した同じ触媒を用いた場合、一定温度において、異性化生成物の向上した収率が観察された。触媒Hについては、72時間のH2予備処理(pretreatment)を含む追加の実験を行った。72時間の予備処理の後でもヘキサン異性化活性が依然として存在していたが、2,2−ジメチルブタンの収率は、一定温度において、4時間のH2予備処理後に得られた収率よりも、著しく低かった。
Claims (3)
- (a)水熱処理に付された第IVB族金属の水和酸化物を、第VIB族金属のオキシアニオンのソースを含んでなる水溶液に接触させて、酸素、第IVB族金属および第VI族金属を含んでなる酸性固体生成物を生成させる工程;
(b)前記酸性固体生成物を500〜900℃の温度で焼成する工程;および
(c)前記焼成した固体生成物に水素化/脱水素化成分を付着させる工程
を含んでなる炭化水素化合物の転化プロセス用の触媒を製造する方法。 - 水熱処理が、水和された酸化物と水または水酸化物の水溶液との接触を、少なくとも7のpHおよび少なくとも80℃の温度にて行うことを含む請求項1記載の方法。
- 第IVB族金属の水酸化物または水和された酸化物が、工程(a)の前に、100〜400℃の温度で焼成される請求項1記載の方法。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US9588493A | 1993-07-22 | 1993-07-22 | |
| US08/095,884 | 1993-07-22 | ||
| US13683893A | 1993-10-18 | 1993-10-18 | |
| US08/136,838 | 1993-10-18 | ||
| PCT/US1994/007904 WO1995003121A1 (en) | 1993-07-22 | 1994-07-13 | Modified solid oxide catalyst and process for producing same |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPH09501867A JPH09501867A (ja) | 1997-02-25 |
| JP3769293B2 true JP3769293B2 (ja) | 2006-04-19 |
Family
ID=26790720
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP50520695A Expired - Fee Related JP3769293B2 (ja) | 1993-07-22 | 1994-07-13 | 変性された固体酸化物触媒の製造方法 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US5780382A (ja) |
| EP (1) | EP0710152B1 (ja) |
| JP (1) | JP3769293B2 (ja) |
| KR (1) | KR100327099B1 (ja) |
| AU (1) | AU688351B2 (ja) |
| CA (1) | CA2166288C (ja) |
| DE (1) | DE69420849T2 (ja) |
| ES (1) | ES2136201T3 (ja) |
| TW (1) | TW309449B (ja) |
| WO (1) | WO1995003121A1 (ja) |
Families Citing this family (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5510309A (en) * | 1994-05-02 | 1996-04-23 | Mobil Oil Corporation | Method for preparing a modified solid oxide |
| US6124232A (en) * | 1996-10-16 | 2000-09-26 | Mobil Oil Corporation | Method for preparing an acidic solid oxide |
| US6184430B1 (en) * | 1996-12-05 | 2001-02-06 | The United States Of America As Represented By The United States Department Of Energy | Hydrocracking and hydroisomerization of long-chain alkanes and polyolefins over metal-promoted anion-modified transition metal oxides |
| JPH11181448A (ja) * | 1997-12-25 | 1999-07-06 | Cosmo Sogo Kenkyusho Kk | 軽質炭化水素油の異性化方法 |
| DE19854635A1 (de) * | 1998-11-26 | 2000-05-31 | Basf Ag | Trägerkatalysator und dessen Einsatz in der Hydrierung |
| EP1142636A4 (en) * | 1998-12-17 | 2002-06-05 | Petroleum Energy Center Found | CATALYST FOR HYDRODESULFORMING / ISOMERIZATION OF LIGHT HYDROCARBON OILS, METHOD FOR THE PRODUCTION THEREOF AND FOR HYDRODESULFORATION / ISOMERIZATION OF LIGHT CARBON HYDROGEN WITH THE AID OF THE CATALYST |
| US6703356B1 (en) * | 2000-03-23 | 2004-03-09 | Exxonmobil Research And Engineering Company | Synthetic hydrocarbon fluids |
| US7396518B2 (en) * | 2002-03-28 | 2008-07-08 | Hitachi Zosen Corporation | High-temperature denitration catalysts and processes for preparing same |
| EP1357167A1 (en) * | 2002-04-18 | 2003-10-29 | Haldor Topsoe A/S | Process for production of high quality gasoline with low aromatic content |
| US6818589B1 (en) * | 2002-06-18 | 2004-11-16 | Uop Llc | Isomerization catalyst and processes |
| US7414007B2 (en) * | 2002-09-13 | 2008-08-19 | Uop Llc | Isomerization catalyst and process |
| WO2004024319A1 (en) * | 2002-09-13 | 2004-03-25 | Uop Llc | Isomerization catalyst and process |
| ES2341950T3 (es) * | 2002-09-25 | 2010-06-30 | Haldor Topsoe A/S | Proceso de isomerizacion de parafinas c7+ y catalizador para el mismo. |
| US7344631B2 (en) | 2002-10-08 | 2008-03-18 | Exxonmobil Research And Engineering Company | Oxygenate treatment of dewaxing catalyst for greater yield of dewaxed product |
| US7132042B2 (en) * | 2002-10-08 | 2006-11-07 | Exxonmobil Research And Engineering Company | Production of fuels and lube oils from fischer-tropsch wax |
| US20040065584A1 (en) * | 2002-10-08 | 2004-04-08 | Bishop Adeana Richelle | Heavy lube oil from fischer- tropsch wax |
| US6846778B2 (en) * | 2002-10-08 | 2005-01-25 | Exxonmobil Research And Engineering Company | Synthetic isoparaffinic premium heavy lubricant base stock |
| US7201838B2 (en) * | 2002-10-08 | 2007-04-10 | Exxonmobil Research And Engineering Company | Oxygenate treatment of dewaxing catalyst for greater yield of dewaxed product |
| US7494953B2 (en) * | 2003-09-17 | 2009-02-24 | Haldor Topsoe A/S | Process for the preparation of an isomerisation catalyst |
| EP1732686B1 (en) | 2004-03-29 | 2011-09-14 | Showa Denko K.K. | Process for producing an oxygen-containing compound using a palladium, tungsten and zirconium-based catalyst |
| DE102004040522A1 (de) * | 2004-08-20 | 2006-02-23 | Süd-Chemie AG | Saurer wolframhaltiger Katalysator |
| KR100991481B1 (ko) * | 2004-10-26 | 2010-11-04 | 유오피 엘엘씨 | 이성화 촉매 및 방법 |
| US7833933B2 (en) * | 2005-11-28 | 2010-11-16 | Haldor Topsøe A/S | Process for the preparation of a paraffin isomerization catalyst |
| US20070119748A1 (en) * | 2005-11-28 | 2007-05-31 | Konrad Herbst | Process for the preparation of a paraffin isomerization catalyst |
| US20080016768A1 (en) | 2006-07-18 | 2008-01-24 | Togna Keith A | Chemically-modified mixed fuels, methods of production and used thereof |
| US20080032886A1 (en) * | 2006-08-03 | 2008-02-07 | Abb Lummus Global, Inc. | Doped solid acid catalyst composition, process of conversion using same and conversion products thereof |
| JP4325745B2 (ja) * | 2007-03-27 | 2009-09-02 | Dic株式会社 | ポリエステル製造用固体酸触媒、及びそれを用いるポリエステルの製造方法 |
| US8153548B2 (en) | 2010-04-19 | 2012-04-10 | King Fahd University Of Petroleum & Minerals | Isomerization catalyst |
| US8366907B2 (en) * | 2010-08-02 | 2013-02-05 | Battelle Memorial Institute | Deoxygenation of fatty acids for preparation of hydrocarbons |
| DE102010050312A1 (de) | 2010-11-03 | 2012-05-03 | Süd-Chemie AG | Ammoniak-Oxidationskatalysator mit geringer N2O Nebenproduktbildung |
| US8785709B2 (en) | 2011-03-30 | 2014-07-22 | University Of Louisville Research Foundation, Inc. | Catalytic isomerisation of linear olefinic hydrocarbons |
| US9249078B2 (en) | 2011-10-07 | 2016-02-02 | Exxonmobil Chemical Patents Inc. | Mixed metal oxide catalysts and use thereof |
| WO2020245621A1 (en) * | 2019-06-04 | 2020-12-10 | Toyota Motor Europe | Supported oxide nh3-scr catalysts with dual site surface species and synthesis processes |
| RU2736047C1 (ru) * | 2020-05-25 | 2020-11-11 | Общество с ограниченной ответственностью "Алвега" (ООО "Алвега") | Способ приготовления катализатора |
| CN111569861B (zh) * | 2020-05-29 | 2023-04-07 | 河北工业大学 | 一种将异构烷烃转化为正构烷烃的反应用催化剂及其制备方法 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2566814B2 (ja) * | 1988-05-13 | 1996-12-25 | 株式会社ジャパンエナジー | 炭化水素転化用固体酸触媒及びその製造方法 |
| US4956519A (en) * | 1988-09-21 | 1990-09-11 | Sun Refining And Marketing Company | Catalyst for hydrocarbon conversion and conversion process utilizing the same |
| US4918041A (en) * | 1988-09-21 | 1990-04-17 | Sun Refining And Marketing Company | Catalyst for hydrocarbon conversion and conversion process utilizing the same |
| US5176897A (en) * | 1989-05-01 | 1993-01-05 | Allied-Signal Inc. | Catalytic destruction of organohalogen compounds |
| DE69200983T2 (de) * | 1991-03-20 | 1995-06-08 | Mitsubishi Gas Chemical Co | Verfahren zur Herstellung von Wasserstoffsuperoxyd. |
| US5113034A (en) * | 1991-08-05 | 1992-05-12 | Exxon Research And Engineering Company | Dimerization catalyst and process therefor |
| US5283041A (en) * | 1992-08-13 | 1994-02-01 | Engelhard Corporation | Catalytic incineration of organic compounds |
| CA2103876A1 (en) * | 1992-08-27 | 1994-02-28 | Stuart Leon Soled | Group viii metal containing tungsten oxide silica modified zirconia as acid catalyst |
| US5396011A (en) * | 1992-12-28 | 1995-03-07 | Mallinckrodt Chemical, Inc. | Catalytic alkylation of aromatic compounds with alkenes |
-
1994
- 1994-07-13 DE DE69420849T patent/DE69420849T2/de not_active Expired - Lifetime
- 1994-07-13 JP JP50520695A patent/JP3769293B2/ja not_active Expired - Fee Related
- 1994-07-13 WO PCT/US1994/007904 patent/WO1995003121A1/en not_active Ceased
- 1994-07-13 ES ES94923479T patent/ES2136201T3/es not_active Expired - Lifetime
- 1994-07-13 CA CA002166288A patent/CA2166288C/en not_active Expired - Fee Related
- 1994-07-13 AU AU73331/94A patent/AU688351B2/en not_active Ceased
- 1994-07-13 KR KR1019960700225A patent/KR100327099B1/ko not_active Expired - Fee Related
- 1994-07-13 EP EP94923479A patent/EP0710152B1/en not_active Expired - Lifetime
- 1994-08-05 TW TW083107205A patent/TW309449B/zh not_active IP Right Cessation
-
1995
- 1995-07-31 US US08/509,717 patent/US5780382A/en not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| TW309449B (ja) | 1997-07-01 |
| AU688351B2 (en) | 1998-03-12 |
| CA2166288A1 (en) | 1995-02-02 |
| US5780382A (en) | 1998-07-14 |
| ES2136201T3 (es) | 1999-11-16 |
| DE69420849T2 (de) | 2000-04-20 |
| EP0710152B1 (en) | 1999-09-22 |
| EP0710152A4 (en) | 1996-07-31 |
| JPH09501867A (ja) | 1997-02-25 |
| AU7333194A (en) | 1995-02-20 |
| KR960703671A (ko) | 1996-08-31 |
| KR100327099B1 (ko) | 2002-08-17 |
| DE69420849D1 (de) | 1999-10-28 |
| WO1995003121A1 (en) | 1995-02-02 |
| CA2166288C (en) | 2005-05-17 |
| EP0710152A1 (en) | 1996-05-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3769293B2 (ja) | 変性された固体酸化物触媒の製造方法 | |
| US5719097A (en) | Catalyst comprising a modified solid oxide | |
| US6080904A (en) | Isomerization process | |
| US5854170A (en) | Method for preparing a modified solid oxide | |
| AU716802B2 (en) | Method for preparing a modified solid oxide | |
| US5345026A (en) | Ring opening process | |
| Sinfelt | Catalytic reforming of hydrocarbons | |
| US4918041A (en) | Catalyst for hydrocarbon conversion and conversion process utilizing the same | |
| US5516964A (en) | Hydrocarbon isomerization using solid superacid catalysts comprising platinum metal | |
| US5382731A (en) | Combined paraffin isomerization/ring opening process | |
| Yori et al. | n-Butane isomerization on Pt/WO3–ZrO2: effect of the Pt incorporation | |
| CA2456324A1 (en) | Non-zeolitic nanocomposite materials for solid acid catalysis | |
| CA2570504C (en) | High-activity isomerization catalyst and process | |
| US5629257A (en) | Solid superacid catalysts comprising platinum metal | |
| AU683913B2 (en) | Combined paraffin isomerization/ring opening process | |
| US4263132A (en) | Catalytic reforming and hydrocracking of organic compounds employing promoted zinc titanate as the catalytic agent | |
| TW309515B (ja) | ||
| Di Gregorio et al. | Activation and isomerization of hydrocarbons over WO3/ZrO2 catalysts: II. Influence of tungsten loading on catalytic activity: Mechanistic studies and correlation with surface reducibility and tungsten surface species | |
| Al-Tabbakh et al. | Synthesis and characterization of sulfated zirconia catalyst for light naphtha isomerization process | |
| Lukinskas et al. | Chromium promotion of tungstated zirconia catalysts for the isomerization of n-alkanes | |
| CA2169637C (en) | Combined paraffin isomerization/ring opening process | |
| Yunusov et al. | CHEMICAL SCIENCES | |
| US5494569A (en) | Hydrocracking using solid superacid catalysts comprising platinum metal |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20040727 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20041026 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20041213 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050125 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20050419 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20050719 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20050915 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20051018 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20051206 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20060110 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20060206 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20090210 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20100210 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110210 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20110210 Year of fee payment: 5 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120210 Year of fee payment: 6 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120210 Year of fee payment: 6 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130210 Year of fee payment: 7 |
|
| LAPS | Cancellation because of no payment of annual fees |