JP2017507914A - テノホビルの固体形態 - Google Patents
テノホビルの固体形態 Download PDFInfo
- Publication number
- JP2017507914A JP2017507914A JP2016545902A JP2016545902A JP2017507914A JP 2017507914 A JP2017507914 A JP 2017507914A JP 2016545902 A JP2016545902 A JP 2016545902A JP 2016545902 A JP2016545902 A JP 2016545902A JP 2017507914 A JP2017507914 A JP 2017507914A
- Authority
- JP
- Japan
- Prior art keywords
- crystalline form
- xrpd
- composition
- degrees
- another embodiment
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- ZBLCZUWQCGNTJF-ZZQXMTQJSA-N CC(C)OC([C@H](C)N[P@](COCC[n]1c2ncnc(N)c2nc1)(Oc1ccccc1)=O)=O Chemical compound CC(C)OC([C@H](C)N[P@](COCC[n]1c2ncnc(N)c2nc1)(Oc1ccccc1)=O)=O ZBLCZUWQCGNTJF-ZZQXMTQJSA-N 0.000 description 1
- SGOIRFVFHAKUTI-ZCFIWIBFSA-N C[C@H](C[n]1c(ncnc2N)c2nc1)OCP(O)(O)=O Chemical compound C[C@H](C[n]1c(ncnc2N)c2nc1)OCP(O)(O)=O SGOIRFVFHAKUTI-ZCFIWIBFSA-N 0.000 description 1
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
- C07F9/65616—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings containing the ring system having three or more than three double bonds between ring members or between ring members and non-ring members, e.g. purine or analogs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Abstract
Description
本発明は、テノホビルの固体形態、ならびにかかる形態の調製、使用および単離のための方法に関する。
(R)−9−[2−(ホスホノメトキシ)プロピル]アデニン(「PMPA」)または「テノホビル」は、ホスホノメトキシヌクレオチドを作製するために使用され得る。ホスホノメトキシヌクレオチドのより効率的な合成において使用できる、テノホビルの新規固体形態を同定することが望ましい。
本発明は、テノホビルの結晶性形態、ならびにそれらに関連する使用およびプロセスを提供する。
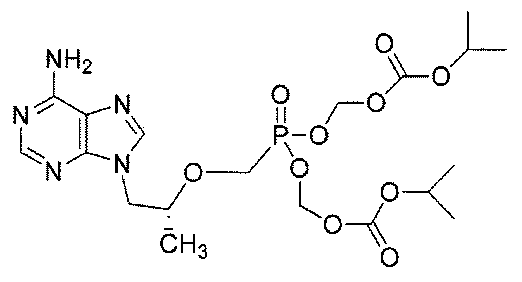
テノホビルまたは(R)−9−[2−(ホスホノメトキシ)プロピル]アデニン(PMPA)は、下記の構造:
本出願において使用される「無水」とは、相当量の水は含まない形態を指す。含水量は、当該技術分野において公知の方法、例えばカールフィッシャー滴定により決定することができる。無水形態の一例は、最大約1重量%の水、最大約0.5重量%の水、または最大約0.1重量%の水を含む。
形態Iの調製および特徴付け
形態Iは、下記のプロセスを通じた結晶化から得られる一水和物である。(PMPA、水、DMFおよびHBr)の反応混合物のpHは、約15〜30℃のNaOH水溶液を使用して1.3に調整した。PMPA溶液を約15〜30℃に維持した。この時点で、形成を促進するために混合物に対し、結晶を用いてシーディングしてもよいが、必須ではなかった。約2〜4時間かけて約15〜30℃において水性NaOHを添加し、約1.7のpHを得た。次いで、スラリーを約15〜30℃において約1時間静置した。約15〜30℃におけるpHが約2.8となるまで、水性NaOHを約15〜30℃において約4時間かけて添加した。次いで、スラリーを約0〜6℃に冷却し、約3時間静置した。次いで、混合物を濾過し、表面洗浄(top wash)用に約0〜6℃の水を4.5倍量;約15〜30℃のアセトンを2倍量加え、濾過前に約30分間攪拌し、次いで表面洗浄のために約15〜30℃のアセトンを2倍量加えた。結晶を21℃〜70℃において約24時間乾燥させた。
形態VIの調製と特徴付け
以下のプロセスから形態VIを得た。PMPA反応混合物のpHは、約19〜25℃で、NaOH水溶液またはHBrを使用して、約0.65〜0.85に調整した。PMPA水溶液を約0〜6℃に冷却し、自己結晶化のために約1〜2時間エージングした。得られたスラリーを約2時間放置し、種結晶床を形成した。約3〜4時間かけて約0〜6℃において水性NaOHを添加して、約1.1のpHとし、スラリーを約0〜6℃において約2時間静置した。約4時間かけて約0〜6℃において水性NaOHを添加し、約2.8のpHにした。次いで、スラリーを約0〜6℃において約3時間静置し、次いで、濾過して固体を分離した。
形態IVの調製と特徴付け
形態VIを周囲温度にて3週間にわたり水中に保持することにより、形態IVを得た。XRPD分析は、周囲温度にて密閉バイアル内に保持しても試料が変化しなかったことを示している。
形態IXの調製および特徴付け
水1mL中における51mgのPMPAの溶液を2〜8℃において2週間にわたり混合し、次いで冷却中に減圧濾過した。湿潤固体をXRPDにより分析し、形態IXとした。
観測されたその他の固有のXRPDパターン/物質
試験中にその他のPMPAのXRPDパターン(図5)を観測した。それらのパターン/物質を得るために使用した方法を表5に挙げる。
結晶性形態によるプロセス改善
結晶形態の使用は、上記のように、加工および製造、特に濾過速度の改善を含む多くの利点を提供する。濾過速度の改善は、製造規模で実証された。結晶形態改善の結果として、濾過時間は、以前の結晶形態の濾過時間のおよそ1/3となった。濾過改善により、プロセスのサイクル時間が短縮され、操業コストに対して良い影響がある。
Claims (24)
- CuKα1X線(波長=1.5406Å)を使用して得られた、7.5、15.0、22.5および24.8の2θ角度の度(±0.2度)で表されるピークを含むX線粉末回折(XRPD)パターンにより特徴付けられた、化合物:
- 実質的に図1に示されるXRPDを有する、請求項1に記載の結晶形態。
- 実質的に表1に示されるXRPDを有する、請求項1に記載の結晶形態。
- 約98℃にピーク最大値を有する吸熱事象を含む示差走査熱量計(DCS)温度記録図により特徴付けられた、請求項1に記載の結晶形態。
- CuKα1X線(波長=1.5406Å)を使用して得られた、6.2、12.4、18.6、24.9および37.7の2θ角度の度(±0.2度)で表されるピークを含むX線粉末回折(XRPD)パターンにより特徴付けられた、化合物:
- 実質的に図2に示されるXRPDを有する、請求項5に記載の結晶形態。
- 実質的に表3に示されるXRPDを有する、請求項5に記載の結晶形態。
- CuKα1X線(波長=1.5406Å)を使用して得られた、6.0、12.0、24.0、24.5および37.6の2θ角度の度(±0.2度)で表されるピークを含むX線粉末回折(XRPD)パターンにより特徴付けられた、化合物:
- 実質的に図3に示されるXRPDを有する、請求項8に記載の結晶形態。
- 実質的に表2に示されるXRPDを有する、請求項8に記載の結晶形態。
- CuKα1X線(波長=1.5406Å)を使用して得られた、5.9、11.3、16.5、21.5、26.1および26.8の2θ角度の度(±0.2度)で表されるピークを含むX線粉末回折(XRPD)により特徴付けられた、化合物:
- 実質的に図4に示されるXRPDを有する、請求項11に記載の結晶形態。
- 実質的に表4に示されるXRPDを有する、請求項11に記載の結晶形態。
- 請求項1に記載の結晶形態、形態Iを含み、形態IVをさらに含む組成物。
- 請求項1に記載の結晶形態、形態Iを含み、形態VIをさらに含む組成物。
- 請求項1に記載の結晶形態、形態Iを含み、形態IXをさらに含む組成物。
- 請求項1から13のいずれか一項に記載の結晶形態または請求項14から16のいずれか一項に記載の組成物をテノホビルジソプロキシル(TD)に変換する工程を含む、テノホビルジソプロキシル(TD)の製造方法。
- TDをフマル酸に接触させてテノホビルジソプロキシルフマレート(TDF)を形成する工程をさらに含む、請求項17に記載の方法。
- 請求項1から13のいずれか一項に記載の結晶形態または請求項14から16のいずれか一項に記載の組成物をテノホビルアラフェナミド(TAF)に変換する工程を含む、テノホビルアラフェナミド(TAF)の製造方法。
- 請求項1から13のいずれか一項に記載の結晶形態を含む医薬組成物。
- 薬学的に許容される担体または賦形剤をさらに含む、請求項20に記載の組成物。
- ウイルス感染を処置または予防する方法において使用するための、請求項1から13のいずれか一項に記載の結晶形態または請求項14から16のいずれか一項に記載の組成物。
- 前記ウイルス感染がヒト免疫不全ウイルス(HIV)である、請求項22に記載の方法において使用するための結晶形態または組成物。
- 前記ウイルス感染がB型肝炎ウイルス(HBV)である、請求項22に記載の方法において使用するための結晶形態または組成物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461927617P | 2014-01-15 | 2014-01-15 | |
| US61/927,617 | 2014-01-15 | ||
| PCT/US2015/010831 WO2015108780A1 (en) | 2014-01-15 | 2015-01-09 | Solid forms of tenofovir |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017228998A Division JP2018030884A (ja) | 2014-01-15 | 2017-11-29 | テノホビルの固体形態 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2017507914A true JP2017507914A (ja) | 2017-03-23 |
Family
ID=52434978
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016545902A Withdrawn JP2017507914A (ja) | 2014-01-15 | 2015-01-09 | テノホビルの固体形態 |
| JP2017228998A Pending JP2018030884A (ja) | 2014-01-15 | 2017-11-29 | テノホビルの固体形態 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017228998A Pending JP2018030884A (ja) | 2014-01-15 | 2017-11-29 | テノホビルの固体形態 |
Country Status (20)
| Country | Link |
|---|---|
| US (2) | US9487546B2 (ja) |
| EP (1) | EP3094637B1 (ja) |
| JP (2) | JP2017507914A (ja) |
| KR (1) | KR20160099655A (ja) |
| CN (1) | CN105960409A (ja) |
| AR (1) | AR099082A1 (ja) |
| AU (1) | AU2015206758B2 (ja) |
| CA (1) | CA2935965A1 (ja) |
| EA (1) | EA031174B1 (ja) |
| ES (1) | ES2762783T3 (ja) |
| HK (1) | HK1223624A1 (ja) |
| IL (1) | IL246569A0 (ja) |
| MX (1) | MX2016008884A (ja) |
| NZ (1) | NZ721832A (ja) |
| PL (1) | PL3094637T3 (ja) |
| PT (1) | PT3094637T (ja) |
| SG (1) | SG11201605415RA (ja) |
| SI (1) | SI3094637T1 (ja) |
| TW (1) | TWI660965B (ja) |
| WO (1) | WO2015108780A1 (ja) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK3236972T3 (en) | 2014-12-26 | 2021-10-04 | Univ Emory | Antivirale N4-hydroxycytidin-derivativer |
| CN106380484A (zh) * | 2016-08-29 | 2017-02-08 | 杭州百诚医药科技股份有限公司 | 一种替诺福韦艾拉酚胺的新晶型及其制备方法 |
| SG11202004403QA (en) | 2017-12-07 | 2020-06-29 | Univ Emory | N4-hydroxycytidine and derivatives and anti-viral uses related thereto |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000515866A (ja) * | 1996-07-26 | 2000-11-28 | ギリヤド サイエンシーズ,インコーポレイテッド | ヌクレオチドアナログ |
| JP2004504402A (ja) * | 2000-07-21 | 2004-02-12 | ギリアード サイエンシーズ, インコーポレイテッド | ホスホネートヌクレオチドアナログのプロドラッグならびにこれを選択および作製するための方法。 |
| WO2008007392A2 (en) * | 2006-07-12 | 2008-01-17 | Matrix Laboratories Limited | Process for the preparation of tenofovir |
| JP2012512133A (ja) * | 2008-12-17 | 2012-05-31 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | ドネペジルの多形結晶及びその製造方法 |
| WO2013025788A1 (en) * | 2011-08-16 | 2013-02-21 | Gilead Sciences, Inc. | Tenofovir alafenamide hemifumarate |
| WO2013072745A1 (en) * | 2011-11-16 | 2013-05-23 | Laurus Labs Private Limited | Process for the preparation of tenofovir |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6057305A (en) | 1992-08-05 | 2000-05-02 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Antiretroviral enantiomeric nucleotide analogs |
| US5922695A (en) | 1996-07-26 | 1999-07-13 | Gilead Sciences, Inc. | Antiviral phosphonomethyoxy nucleotide analogs having increased oral bioavarilability |
| US5935946A (en) | 1997-07-25 | 1999-08-10 | Gilead Sciences, Inc. | Nucleotide analog composition and synthesis method |
| EP1923063A3 (en) | 2003-01-14 | 2009-04-08 | Gilead Sciences, Inc. | Compositions and methods for combination antiviral therapy |
| ZA201103820B (en) * | 2010-12-13 | 2012-01-25 | Laurus Labs Private Ltd | Process for the preparation of tenofovir |
| KR102033802B1 (ko) | 2011-10-07 | 2019-10-17 | 길리애드 사이언시즈, 인코포레이티드 | 항-바이러스 뉴클레오티드 유사체의 제조 방법 |
| CN103374038B (zh) * | 2012-04-11 | 2016-04-13 | 广州白云山医药集团股份有限公司白云山制药总厂 | 一种抗病毒药物的制备方法 |
| CN102899367B (zh) * | 2012-09-24 | 2014-04-30 | 常州大学 | 一种生物法和化学法结合合成泰诺福韦的方法 |
| CN103641758B (zh) * | 2013-11-19 | 2016-03-30 | 重庆紫光化工股份有限公司 | 廉价的高纯度的d,l-蛋氨酸的制备方法 |
-
2014
- 2014-12-12 TW TW103143442A patent/TWI660965B/zh active
-
2015
- 2015-01-09 SG SG11201605415RA patent/SG11201605415RA/en unknown
- 2015-01-09 KR KR1020167019027A patent/KR20160099655A/ko active Search and Examination
- 2015-01-09 AU AU2015206758A patent/AU2015206758B2/en active Active
- 2015-01-09 CN CN201580004462.3A patent/CN105960409A/zh active Pending
- 2015-01-09 EP EP15701877.1A patent/EP3094637B1/en active Active
- 2015-01-09 PT PT157018771T patent/PT3094637T/pt unknown
- 2015-01-09 SI SI201530958T patent/SI3094637T1/sl unknown
- 2015-01-09 US US14/593,556 patent/US9487546B2/en active Active
- 2015-01-09 NZ NZ721832A patent/NZ721832A/en unknown
- 2015-01-09 PL PL15701877T patent/PL3094637T3/pl unknown
- 2015-01-09 WO PCT/US2015/010831 patent/WO2015108780A1/en active Application Filing
- 2015-01-09 JP JP2016545902A patent/JP2017507914A/ja not_active Withdrawn
- 2015-01-09 EA EA201691203A patent/EA031174B1/ru not_active IP Right Cessation
- 2015-01-09 ES ES15701877T patent/ES2762783T3/es active Active
- 2015-01-09 MX MX2016008884A patent/MX2016008884A/es unknown
- 2015-01-09 CA CA2935965A patent/CA2935965A1/en not_active Abandoned
- 2015-01-13 AR ARP150100078A patent/AR099082A1/es unknown
-
2016
- 2016-07-03 IL IL246569A patent/IL246569A0/en unknown
- 2016-10-05 US US15/285,924 patent/US20170056424A1/en not_active Abandoned
- 2016-10-17 HK HK16111943.7A patent/HK1223624A1/zh unknown
-
2017
- 2017-11-29 JP JP2017228998A patent/JP2018030884A/ja active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000515866A (ja) * | 1996-07-26 | 2000-11-28 | ギリヤド サイエンシーズ,インコーポレイテッド | ヌクレオチドアナログ |
| JP2004504402A (ja) * | 2000-07-21 | 2004-02-12 | ギリアード サイエンシーズ, インコーポレイテッド | ホスホネートヌクレオチドアナログのプロドラッグならびにこれを選択および作製するための方法。 |
| WO2008007392A2 (en) * | 2006-07-12 | 2008-01-17 | Matrix Laboratories Limited | Process for the preparation of tenofovir |
| JP2012512133A (ja) * | 2008-12-17 | 2012-05-31 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | ドネペジルの多形結晶及びその製造方法 |
| WO2013025788A1 (en) * | 2011-08-16 | 2013-02-21 | Gilead Sciences, Inc. | Tenofovir alafenamide hemifumarate |
| WO2013072745A1 (en) * | 2011-11-16 | 2013-05-23 | Laurus Labs Private Limited | Process for the preparation of tenofovir |
Non-Patent Citations (3)
| Title |
|---|
| 久保田 徳昭: "医薬品原薬・中間体製造における貧溶媒晶析の検討事例", 分かり易い貧溶媒晶析(分離技術シリーズ;28), JPN6017019627, 15 April 2013 (2013-04-15), pages 43 - 52, ISSN: 0003568102 * |
| 山野 光久: "医薬品のプロセス研究における結晶多形現象への取り組み", 有機合成化学協会誌, vol. 65, no. 9, JPN7017001749, 2007, pages 907 - 913, ISSN: 0003568103 * |
| 芦澤 一英、他, 医薬品の多形現象と晶析の科学, JPN7016001492, 20 September 2002 (2002-09-20), pages 272 - 317, ISSN: 0003568101 * |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2015206758A1 (en) | 2016-07-21 |
| AU2015206758B2 (en) | 2017-11-30 |
| US20150197535A1 (en) | 2015-07-16 |
| HK1223624A1 (zh) | 2017-08-04 |
| JP2018030884A (ja) | 2018-03-01 |
| AR099082A1 (es) | 2016-06-29 |
| CN105960409A (zh) | 2016-09-21 |
| EP3094637B1 (en) | 2019-10-16 |
| EP3094637A1 (en) | 2016-11-23 |
| PT3094637T (pt) | 2019-12-12 |
| EA201691203A1 (ru) | 2016-12-30 |
| SI3094637T1 (sl) | 2019-12-31 |
| NZ721832A (en) | 2017-09-29 |
| IL246569A0 (en) | 2016-08-31 |
| US9487546B2 (en) | 2016-11-08 |
| KR20160099655A (ko) | 2016-08-22 |
| EA031174B1 (ru) | 2018-11-30 |
| ES2762783T3 (es) | 2020-05-25 |
| SG11201605415RA (en) | 2016-07-28 |
| PL3094637T3 (pl) | 2020-04-30 |
| US20170056424A1 (en) | 2017-03-02 |
| MX2016008884A (es) | 2016-09-16 |
| TWI660965B (zh) | 2019-06-01 |
| WO2015108780A1 (en) | 2015-07-23 |
| CA2935965A1 (en) | 2015-07-23 |
| TW201609780A (zh) | 2016-03-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6966590B2 (ja) | オメカムチブメカルビルの塩及び塩を調製するプロセス | |
| JP6189299B2 (ja) | プリドピジン塩酸塩の新規な多形形態 | |
| KR102625774B1 (ko) | 오메캄티브 메카빌의 합성 | |
| JP2018030884A (ja) | テノホビルの固体形態 | |
| JP7168447B2 (ja) | ビラスチンの結晶形態及びそれらの調製方法 | |
| AU2018205995B2 (en) | Solid forms of [(1S)-1 -[(2S,4R,5R)-5-(5-amino-2-oxo-thiazolo[4,5-d]pyrimidin-3-yl)-4-hydroxy-te trahydrofuran-2-yl]propyl] acetate | |
| JP6779972B2 (ja) | 呼吸器合胞体ウイルス(rsv)感染症の処置のためのn−[(3−アミノ−3−オキセタニル)メチル]−2−(2,3−ジヒドロ−1,1−ジオキシド−1,4−ベンゾチアゼピン−4(5h)−イル)−6−メチル−4−キナゾリンアミンの結晶形 | |
| AU2023200724A1 (en) | New crystalline forms of a succinate salt of 7-cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-7h-pyrrolo[2,3-d]pyrimidine -6-carboxylic acid dimethylamide | |
| JP6761564B2 (ja) | ナトリウム・グルコース共輸送体2阻害薬のl−プロリン化合物、およびl−プロリン化合物の一水和物および結晶 | |
| WO2019011316A1 (zh) | 一种btk激酶抑制剂的结晶形式及制备方法 | |
| WO2017079678A1 (en) | Solid state forms of a pde10 inhibitor | |
| JP2019500371A (ja) | 置換アミノピラン誘導体の結晶形 | |
| WO2016058081A1 (en) | Solid forms of nilotinib hydrochloride | |
| KR102271944B1 (ko) | 시스테인 프로테아제 억제제로서 유용한 n-[1-6-(에티닐-3-옥소-헥사하이드로-푸로[3,2-b]피롤-4-카르보닐)-3-메틸-부틸]-4-[5-플루오로-2-(4-메틸-피페라지닐)티아졸-4-일]-벤즈아미드의 염의 2형 결정질 다형체 | |
| TW202033508A (zh) | 貝前列素-314d晶體及其製備方法 | |
| TW201319042A (zh) | 硫酸卡維地洛的結晶、其製備方法及其在醫藥上的應用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170531 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20170531 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20170831 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20171030 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171129 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20171228 |
|
| A761 | Written withdrawal of application |
Free format text: JAPANESE INTERMEDIATE CODE: A761 Effective date: 20180427 |