JP2011122183A - 金属の電解採取システム、および該システムを用いた電解採取方法 - Google Patents
金属の電解採取システム、および該システムを用いた電解採取方法 Download PDFInfo
- Publication number
- JP2011122183A JP2011122183A JP2009278607A JP2009278607A JP2011122183A JP 2011122183 A JP2011122183 A JP 2011122183A JP 2009278607 A JP2009278607 A JP 2009278607A JP 2009278607 A JP2009278607 A JP 2009278607A JP 2011122183 A JP2011122183 A JP 2011122183A
- Authority
- JP
- Japan
- Prior art keywords
- anode
- electrowinning
- catalyst layer
- oxide
- electrolytic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 238000005363 electrowinning Methods 0.000 title claims abstract description 93
- 229910052751 metal Inorganic materials 0.000 title claims abstract description 33
- 239000002184 metal Substances 0.000 title claims abstract description 33
- 238000000034 method Methods 0.000 title claims abstract description 13
- 239000003054 catalyst Substances 0.000 claims abstract description 68
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims abstract description 42
- HTXDPTMKBJXEOW-UHFFFAOYSA-N dioxoiridium Chemical compound O=[Ir]=O HTXDPTMKBJXEOW-UHFFFAOYSA-N 0.000 claims abstract description 38
- 229910000457 iridium oxide Inorganic materials 0.000 claims abstract description 38
- 239000008151 electrolyte solution Substances 0.000 claims abstract description 36
- 239000000758 substrate Substances 0.000 claims abstract description 27
- 229910001925 ruthenium oxide Inorganic materials 0.000 claims abstract description 25
- WOCIAKWEIIZHES-UHFFFAOYSA-N ruthenium(iv) oxide Chemical compound O=[Ru]=O WOCIAKWEIIZHES-UHFFFAOYSA-N 0.000 claims abstract description 25
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims abstract description 20
- 239000007864 aqueous solution Substances 0.000 claims abstract description 7
- 229910021645 metal ion Inorganic materials 0.000 claims abstract description 6
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 claims description 38
- 229910017052 cobalt Inorganic materials 0.000 claims description 30
- 239000010941 cobalt Substances 0.000 claims description 30
- GUTLYIVDDKVIGB-UHFFFAOYSA-N cobalt atom Chemical compound [Co] GUTLYIVDDKVIGB-UHFFFAOYSA-N 0.000 claims description 30
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 claims description 21
- 229910052725 zinc Inorganic materials 0.000 claims description 21
- 239000011701 zinc Substances 0.000 claims description 21
- 229910052759 nickel Inorganic materials 0.000 claims description 19
- BPUBBGLMJRNUCC-UHFFFAOYSA-N oxygen(2-);tantalum(5+) Chemical compound [O-2].[O-2].[O-2].[O-2].[O-2].[Ta+5].[Ta+5] BPUBBGLMJRNUCC-UHFFFAOYSA-N 0.000 claims description 10
- 229910001936 tantalum oxide Inorganic materials 0.000 claims description 10
- 229910052715 tantalum Inorganic materials 0.000 claims description 8
- GUVRBAGPIYLISA-UHFFFAOYSA-N tantalum atom Chemical compound [Ta] GUVRBAGPIYLISA-UHFFFAOYSA-N 0.000 claims description 8
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 claims description 7
- 239000002131 composite material Substances 0.000 claims description 7
- OGIDPMRJRNCKJF-UHFFFAOYSA-N titanium oxide Inorganic materials [Ti]=O OGIDPMRJRNCKJF-UHFFFAOYSA-N 0.000 claims description 7
- 229910001362 Ta alloys Inorganic materials 0.000 claims description 3
- 238000005868 electrolysis reaction Methods 0.000 abstract description 64
- 238000007086 side reaction Methods 0.000 abstract description 21
- 238000009825 accumulation Methods 0.000 abstract description 14
- 239000007795 chemical reaction product Substances 0.000 abstract description 14
- 239000010410 layer Substances 0.000 description 71
- 239000003792 electrolyte Substances 0.000 description 44
- 230000000052 comparative effect Effects 0.000 description 37
- 239000001301 oxygen Substances 0.000 description 32
- 229910052760 oxygen Inorganic materials 0.000 description 32
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 31
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 18
- 239000000460 chlorine Substances 0.000 description 18
- 229910052801 chlorine Inorganic materials 0.000 description 18
- 239000010936 titanium Substances 0.000 description 17
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 description 16
- 238000006243 chemical reaction Methods 0.000 description 16
- 229910052719 titanium Inorganic materials 0.000 description 16
- 150000002697 manganese compounds Chemical class 0.000 description 14
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 14
- 229910001429 cobalt ion Inorganic materials 0.000 description 11
- -1 cobalt oxyhydroxide Chemical class 0.000 description 11
- XLJKHNWPARRRJB-UHFFFAOYSA-N cobalt(2+) Chemical compound [Co+2] XLJKHNWPARRRJB-UHFFFAOYSA-N 0.000 description 11
- 238000000605 extraction Methods 0.000 description 11
- 230000003197 catalytic effect Effects 0.000 description 10
- RTBHLGSMKCPLCQ-UHFFFAOYSA-N [Mn].OOO Chemical compound [Mn].OOO RTBHLGSMKCPLCQ-UHFFFAOYSA-N 0.000 description 9
- 230000001965 increasing effect Effects 0.000 description 9
- 239000011248 coating agent Substances 0.000 description 8
- 238000000576 coating method Methods 0.000 description 8
- 229910001437 manganese ion Inorganic materials 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- 238000002441 X-ray diffraction Methods 0.000 description 7
- 238000004519 manufacturing process Methods 0.000 description 7
- 239000000243 solution Substances 0.000 description 7
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 6
- 229910000978 Pb alloy Inorganic materials 0.000 description 6
- 229910001245 Sb alloy Inorganic materials 0.000 description 6
- 239000011241 protective layer Substances 0.000 description 6
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 6
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- 150000002739 metals Chemical class 0.000 description 4
- 230000001105 regulatory effect Effects 0.000 description 4
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 3
- PTFCDOFLOPIGGS-UHFFFAOYSA-N Zinc dication Chemical compound [Zn+2] PTFCDOFLOPIGGS-UHFFFAOYSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 229910045601 alloy Inorganic materials 0.000 description 3
- 239000000956 alloy Substances 0.000 description 3
- 229910003460 diamond Inorganic materials 0.000 description 3
- 239000010432 diamond Substances 0.000 description 3
- 239000012153 distilled water Substances 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 229910000510 noble metal Inorganic materials 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 229910052707 ruthenium Inorganic materials 0.000 description 3
- 238000005070 sampling Methods 0.000 description 3
- 238000005979 thermal decomposition reaction Methods 0.000 description 3
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- 229910010413 TiO 2 Inorganic materials 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 150000001875 compounds Chemical class 0.000 description 2
- 238000000151 deposition Methods 0.000 description 2
- 230000008021 deposition Effects 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 238000009713 electroplating Methods 0.000 description 2
- 230000002708 enhancing effect Effects 0.000 description 2
- 238000007733 ion plating Methods 0.000 description 2
- 229910052741 iridium Inorganic materials 0.000 description 2
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 2
- 235000006408 oxalic acid Nutrition 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 2
- 239000004810 polytetrafluoroethylene Substances 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 239000006104 solid solution Substances 0.000 description 2
- 238000004544 sputter deposition Methods 0.000 description 2
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 229910018916 CoOOH Inorganic materials 0.000 description 1
- 229910021503 Cobalt(II) hydroxide Inorganic materials 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229910003174 MnOOH Inorganic materials 0.000 description 1
- VEQPNABPJHWNSG-UHFFFAOYSA-N Nickel(2+) Chemical compound [Ni+2] VEQPNABPJHWNSG-UHFFFAOYSA-N 0.000 description 1
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical compound [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 description 1
- JODOMBGKVAIYRQ-UHFFFAOYSA-N [Nb].[Ta].[Ti] Chemical compound [Nb].[Ta].[Ti] JODOMBGKVAIYRQ-UHFFFAOYSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 238000005280 amorphization Methods 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- FPCJKVGGYOAWIZ-UHFFFAOYSA-N butan-1-ol;titanium Chemical compound [Ti].CCCCO.CCCCO.CCCCO.CCCCO FPCJKVGGYOAWIZ-UHFFFAOYSA-N 0.000 description 1
- 239000000919 ceramic Substances 0.000 description 1
- 238000005229 chemical vapour deposition Methods 0.000 description 1
- ASKVAEGIVYSGNY-UHFFFAOYSA-L cobalt(ii) hydroxide Chemical compound [OH-].[OH-].[Co+2] ASKVAEGIVYSGNY-UHFFFAOYSA-L 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 238000005336 cracking Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 230000005611 electricity Effects 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 238000005530 etching Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 150000004687 hexahydrates Chemical class 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- LZPOAQXQOPXSQT-UHFFFAOYSA-F iridium(3+) tantalum(5+) octachloride Chemical compound [Ir+3].[Cl-].[Ta+5].[Cl-].[Cl-].[Cl-].[Cl-].[Cl-].[Cl-].[Cl-] LZPOAQXQOPXSQT-UHFFFAOYSA-F 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 150000004706 metal oxides Chemical class 0.000 description 1
- 239000002923 metal particle Substances 0.000 description 1
- 238000001465 metallisation Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 229910001453 nickel ion Inorganic materials 0.000 description 1
- 229910052758 niobium Inorganic materials 0.000 description 1
- 239000010955 niobium Substances 0.000 description 1
- GUCVJGMIXFAOAE-UHFFFAOYSA-N niobium atom Chemical compound [Nb] GUCVJGMIXFAOAE-UHFFFAOYSA-N 0.000 description 1
- RJSRQTFBFAJJIL-UHFFFAOYSA-N niobium titanium Chemical compound [Ti].[Nb] RJSRQTFBFAJJIL-UHFFFAOYSA-N 0.000 description 1
- 150000002926 oxygen Chemical class 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 238000000197 pyrolysis Methods 0.000 description 1
- 239000010802 sludge Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 230000001629 suppression Effects 0.000 description 1
- VSSLEOGOUUKTNN-UHFFFAOYSA-N tantalum titanium Chemical compound [Ti].[Ta] VSSLEOGOUUKTNN-UHFFFAOYSA-N 0.000 description 1
- ZTWIEIFKPFJRLV-UHFFFAOYSA-K trichlororuthenium;trihydrate Chemical compound O.O.O.Cl[Ru](Cl)Cl ZTWIEIFKPFJRLV-UHFFFAOYSA-K 0.000 description 1
- 229910052726 zirconium Inorganic materials 0.000 description 1
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25C—PROCESSES FOR THE ELECTROLYTIC PRODUCTION, RECOVERY OR REFINING OF METALS; APPARATUS THEREFOR
- C25C1/00—Electrolytic production, recovery or refining of metals by electrolysis of solutions
- C25C1/16—Electrolytic production, recovery or refining of metals by electrolysis of solutions of zinc, cadmium or mercury
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25C—PROCESSES FOR THE ELECTROLYTIC PRODUCTION, RECOVERY OR REFINING OF METALS; APPARATUS THEREFOR
- C25C1/00—Electrolytic production, recovery or refining of metals by electrolysis of solutions
- C25C1/06—Electrolytic production, recovery or refining of metals by electrolysis of solutions or iron group metals, refractory metals or manganese
- C25C1/08—Electrolytic production, recovery or refining of metals by electrolysis of solutions or iron group metals, refractory metals or manganese of nickel or cobalt
-
- C—CHEMISTRY; METALLURGY
- C25—ELECTROLYTIC OR ELECTROPHORETIC PROCESSES; APPARATUS THEREFOR
- C25C—PROCESSES FOR THE ELECTROLYTIC PRODUCTION, RECOVERY OR REFINING OF METALS; APPARATUS THEREFOR
- C25C7/00—Constructional parts, or assemblies thereof, of cells; Servicing or operating of cells
- C25C7/02—Electrodes; Connections thereof
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P10/00—Technologies related to metal processing
- Y02P10/20—Recycling
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- Materials Engineering (AREA)
- Metallurgy (AREA)
- Organic Chemistry (AREA)
- Electrolytic Production Of Metals (AREA)
Abstract
【解決手段】本発明に係る電解採取システムは、電解液中に配置された陽極と陰極との間に所定の電解電流を流して、該陰極上に所望の金属を析出させるものであって、電解液は、上記所望の金属のイオンを含む硫酸系または塩化物系の水溶液であり、陽極は、非晶質の酸化イリジウムまたは非晶質の酸化ルテニウムを含む触媒層を導電性基体上に形成したものである。
【選択図】なし
Description
(1)電解初期には電解電圧を下げることができるが、マンガン化合物が蓄積するにつれて電解電圧が高くなり、結局、電解時に消費される電力が大きくなってしまう、
という問題があった。
(2)陽極上での電流分布が不均一となり、陰極上での金属の析出が不均一となる、
(3)陰極上でデンドライト成長した金属が陽極に到達し、陽極と陰極とがショートする、
(4)上記ショートの問題を回避するために陽極と陰極との極間距離を長くする必要があるので、電解液のオーム損により電解電圧が高くなる、
といった問題があった。
また、上記の問題を回避するべく、陽極からマンガン化合物を除去する作業を行うと、
(5)その間は電解を休止しなければならず、連続的な電解を行うことができない、
(6)マンガン化合物を除去する際に触媒層が損傷し、不溶性電極の耐久性が低下する、
といった問題があった。
さらに、従来のコバルトの電解採取では、
(7)本来陰極で還元されるべき+2価のコバルトイオンが、オキシ水酸化コバルトを生成するために陽極付近で消費されてしまう、
(8)陽極に設置されたアノードバッグの外側までオキシ水酸化コバルトが成長し、アノードバックの外側で塩素が発生して、電解採取を行う環境中に人体に有害な塩素が放出される、
といった問題も発生していた。
(実施例1)
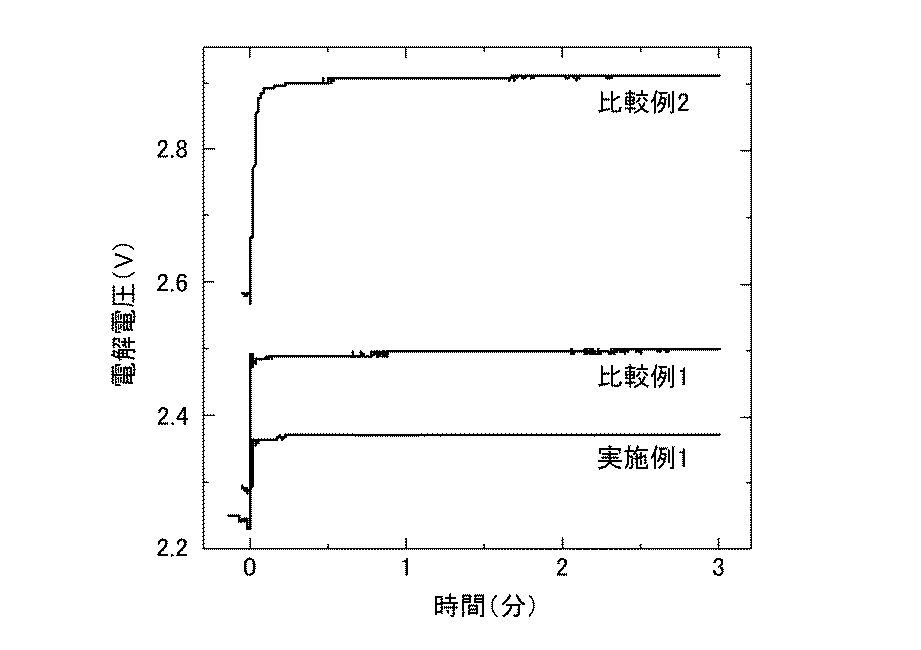
実施例1では、亜鉛イオンを含む硫酸系電解液と、導電性基体上に非晶質の酸化イリジウムを含む触媒層を形成した陽極と、陽極とともに電解液中に配置された陰極とからなる、本発明に係る第1の電解採取システムを用いて亜鉛の電解採取を行った。
比較例1では、触媒層を形成する際の熱分解温度を360℃から470℃に変えた以外は実施例1と同じ構成の電解採取システムを用いて、実施例1と同じ条件で亜鉛の電解採取を行った。
比較例2では、市販のPb−Sb(Sb 5%)合金電極を陽極とした以外は実施例1と同じ構成の電解採取システムを用いて、実施例1と同じ条件で亜鉛の電解採取を行った。
(実施例2)
実施例2では、コバルトイオンを含む硫酸系電解液と、導電性基体上に非晶質の酸化イリジウムを含む触媒層を形成した陽極と、陽極とともに電解液中に配置された陰極とからなる、本発明に係る第1の電解採取システムを用いてコバルトの電解採取を行った。
比較例3では、比較例1と同じ陽極、すなわちチタン板上に結晶質の酸化イリジウムを含む触媒層を形成し、電解に作用する電極面積を1cm2に規制した陽極を使用した。それ以外は実施例2と同じ構成の電解採取システムを用いてコバルトの電解採取を行った。
比較例4では、市販のPb−Sb合金電極(Sb 5%)を陽極とした以外は実施例2と同じ構成の電解採取システムを用いてコバルトの電解採取を行った。
(実施例3)
実施例3では、ニッケルイオンを含む塩化物系電解液と、導電性基体上に非晶質の酸化イリジウムを含む触媒層を形成した陽極と、陽極とともに電解液中に配置された陰極とからなる、本発明に係る第1の電解採取システムを用いてニッケルの電解採取を行った。
比較例5では、比較例1と同じ陽極、すなわちチタン板上に結晶質の酸化イリジウムを含む触媒層を形成し、電解に作用する電極面積を1cm2に規制した陽極を使用した。それ以外は実施例3と同じ構成の電解採取システムを用いてニッケルの電解採取を行った。
比較例6では、市販のPb−Sb合金電極(Sb 5%)を陽極とした以外は実施例3と同じ構成の電解採取システムを用いてニッケルの電解採取を行った。
(実施例4)
実施例4では、コバルトイオンを含む塩化物系電解液と、導電性基体上に非晶質の酸化ルテニウムを含む触媒層を形成した陽極と、陽極とともに電解液中に配置された陰極とからなる、本発明に係る第2の電解採取システムを用いてコバルトの電解採取を行った。
比較例7では、触媒層を形成する際の熱分解温度を340℃から450℃に変えた以外は実施例4と同じ構成の電解採取システムを用いてコバルトの電解採取を行った。
以上、本発明に係る電解採取システムの好ましい実施形態について説明してきたが、本発明はこれらの構成に限定されるものではなく、種々の変形例が考えられる。
同様に、実施例4に係る電解採取システムの触媒層は、少なくとも非晶質の酸化ルテニウムを含んでいればよく、酸化チタンを省略することができる。
同様に、実施例4に係る電解システムの触媒層は、耐久性を向上させるべく導電性基体上に結晶質の酸化ルテニウムを含む保護層を形成した上に形成してもよい。このような保護層には、結晶質の酸化ルテニウムと酸化チタンからなる複合酸化物層が特に好適である。
Claims (7)
- 電解液中に配置された陽極と陰極との間に所定の電解電流を流して、該陰極上に所望の金属を析出させる電解採取システムであって、
前記電解液は、前記金属のイオンを含む硫酸系または塩化物系の水溶液であり、
前記陽極は、非晶質の酸化イリジウムを含む触媒層を導電性基体上に形成したものである、
ことを特徴とする電解採取システム。 - 前記触媒層は、さらに非晶質の酸化タンタルを含んでいることを特徴とする請求項1に記載の電解採取システム。
- 電解液中に配置された陽極と陰極との間に所定の電解電流を流して、該陰極上に所望の金属を析出させる電解採取システムであって、
前記電解液は、前記金属のイオンを含む塩化物系の水溶液であり、
前記陽極は、非晶質の酸化ルテニウムを含む触媒層を導電性基体上に形成したものである、
ことを特徴とする電解採取システム。 - 前記触媒層の前記非晶質の酸化ルテニウムが、非晶質の酸化ルテニウムと酸化チタンの複合酸化物であることを特徴とする請求項3に記載の電解採取システム。
- 前記陽極は、前記導電性基体と前記触媒層との間に形成された、タンタルまたはタンタルの合金からなる中間層を有することを特徴とする請求項1〜4のいずれかに記載の電解採取システム。
- 前記金属は、亜鉛、ニッケル、コバルトのいずれか1つであることを特徴とする請求項1〜5のいずれかに記載の電解採取システム。
- 請求項1〜6のいずれかに記載の電解採取システムを用いて所望の金属を採取することを特徴とする電解採取方法。
Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009278607A JP5013438B2 (ja) | 2009-12-08 | 2009-12-08 | 金属の電解採取用陽極および電解採取方法 |
| EP10835830.0A EP2535443B1 (en) | 2009-12-08 | 2010-11-22 | Nickel electrowinning method |
| NO16164168A NO3088569T3 (ja) | 2009-12-08 | 2010-11-22 | |
| CN201510702213.3A CN105386086A (zh) | 2009-12-08 | 2010-11-22 | 金属的电解提取用阳极的用途 |
| US13/514,858 US20120247971A1 (en) | 2009-12-08 | 2010-11-22 | Metal electrowinning anode and electrowinning method |
| EP16164168.3A EP3088569B1 (en) | 2009-12-08 | 2010-11-22 | Nickel electrowinning method |
| CA2783302A CA2783302C (en) | 2009-12-08 | 2010-11-22 | Metal electrowinning anode and electrowinning method |
| PCT/JP2010/070809 WO2011070908A1 (ja) | 2009-12-08 | 2010-11-22 | 金属の電解採取システム、および該システムを用いた電解採取方法 |
| CN201080054561.XA CN102686783B (zh) | 2009-12-08 | 2010-11-22 | 阳极作为镍的电解提取用阳极的用途 |
| AU2010329192A AU2010329192B2 (en) | 2009-12-08 | 2010-11-22 | Metal electrowinning anode and electrowinning method |
| US15/484,536 US20170218531A1 (en) | 2009-12-08 | 2017-04-11 | Metal electrowinning anode and electrowinning method |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009278607A JP5013438B2 (ja) | 2009-12-08 | 2009-12-08 | 金属の電解採取用陽極および電解採取方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2011122183A true JP2011122183A (ja) | 2011-06-23 |
| JP2011122183A5 JP2011122183A5 (ja) | 2012-06-07 |
| JP5013438B2 JP5013438B2 (ja) | 2012-08-29 |
Family
ID=44145455
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009278607A Expired - Fee Related JP5013438B2 (ja) | 2009-12-08 | 2009-12-08 | 金属の電解採取用陽極および電解採取方法 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20120247971A1 (ja) |
| EP (2) | EP2535443B1 (ja) |
| JP (1) | JP5013438B2 (ja) |
| CN (2) | CN102686783B (ja) |
| AU (1) | AU2010329192B2 (ja) |
| CA (1) | CA2783302C (ja) |
| NO (1) | NO3088569T3 (ja) |
| WO (1) | WO2011070908A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5008043B1 (ja) * | 2011-09-13 | 2012-08-22 | 学校法人同志社 | 塩素発生用陽極 |
| WO2013038928A1 (ja) * | 2011-09-13 | 2013-03-21 | 学校法人同志社 | 電解めっき用陽極および該陽極を用いる電解めっき法 |
| US20140054180A1 (en) * | 2011-03-25 | 2014-02-27 | The Doshisha | Anode for electrowinning and method for electrowinning using same |
| JP2016003346A (ja) * | 2014-06-16 | 2016-01-12 | 住友金属鉱山株式会社 | 不溶性電極の付着物除去方法 |
| JP2019081919A (ja) * | 2017-10-30 | 2019-05-30 | 学校法人同志社 | 電解法 |
| JP2021070843A (ja) * | 2019-10-30 | 2021-05-06 | 住友金属鉱山株式会社 | 廃リチウムイオン電池からの銅、ニッケル、コバルトの回収方法 |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013029186A1 (en) | 2011-09-01 | 2013-03-07 | Trudel Simon | Electrocatalytic materials and methods for manufacturing same |
| DE102013202143A1 (de) * | 2013-02-08 | 2014-08-14 | Bayer Materialscience Ag | Katalysatorbeschichtung und Verfahren zu ihrer Herstellung |
| CN105132947B (zh) * | 2015-09-24 | 2018-02-02 | 苏州铂瑞电极工业有限公司 | 一种铜回收电极组 |
| KR102126183B1 (ko) * | 2017-11-29 | 2020-06-24 | 한국과학기술연구원 | 고분자 전해질 막 물 전기분해장치의 확산층 및 산소 전극 복합층 및 그 제조 방법, 이를 이용한 고분자 전해질 막 물 전기 분해 장치 |
| CN116555845A (zh) * | 2022-01-27 | 2023-08-08 | 马赫内托特殊阳极有限公司 | 一种电极及其用途和制备方法 |
| WO2023208026A1 (zh) * | 2022-04-28 | 2023-11-02 | 中国石油化工股份有限公司 | 一种过渡金属掺杂的铱基复合催化剂及其制备和应用 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003293180A (ja) * | 2002-04-02 | 2003-10-15 | Takayuki Shimamune | 電解槽及び電解方法 |
| JP2004238697A (ja) * | 2003-02-07 | 2004-08-26 | Daiso Co Ltd | 酸素発生用電極 |
| JP2008507626A (ja) * | 2004-07-22 | 2008-03-13 | フェルプス ドッジ コーポレイション | 貫流式電解採取用電解槽中で電解採取することにより銅粉末を生成するためのシステムおよび方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3562008A (en) * | 1968-10-14 | 1971-02-09 | Ppg Industries Inc | Method for producing a ruthenium coated titanium electrode |
| US4155821A (en) * | 1974-11-25 | 1979-05-22 | Falconbridge Nickel Mines Limited | Electrowinning metal from chloride solution |
| USRE34191E (en) * | 1989-05-31 | 1993-03-09 | Eco-Tec Limited | Process for electroplating metals |
| US6139705A (en) * | 1998-05-06 | 2000-10-31 | Eltech Systems Corporation | Lead electrode |
| JP4524248B2 (ja) | 2005-12-12 | 2010-08-11 | ペルメレック電極株式会社 | 銅採取方法 |
| US8124556B2 (en) * | 2008-05-24 | 2012-02-28 | Freeport-Mcmoran Corporation | Electrochemically active composition, methods of making, and uses thereof |
| EP2287364B1 (en) * | 2008-06-09 | 2013-07-10 | The Doshisha | Method for electrolytic winning of zinc |
-
2009
- 2009-12-08 JP JP2009278607A patent/JP5013438B2/ja not_active Expired - Fee Related
-
2010
- 2010-11-22 EP EP10835830.0A patent/EP2535443B1/en not_active Not-in-force
- 2010-11-22 CN CN201080054561.XA patent/CN102686783B/zh not_active Expired - Fee Related
- 2010-11-22 US US13/514,858 patent/US20120247971A1/en not_active Abandoned
- 2010-11-22 CN CN201510702213.3A patent/CN105386086A/zh active Pending
- 2010-11-22 AU AU2010329192A patent/AU2010329192B2/en not_active Ceased
- 2010-11-22 WO PCT/JP2010/070809 patent/WO2011070908A1/ja active Application Filing
- 2010-11-22 EP EP16164168.3A patent/EP3088569B1/en not_active Not-in-force
- 2010-11-22 NO NO16164168A patent/NO3088569T3/no unknown
- 2010-11-22 CA CA2783302A patent/CA2783302C/en not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003293180A (ja) * | 2002-04-02 | 2003-10-15 | Takayuki Shimamune | 電解槽及び電解方法 |
| JP2004238697A (ja) * | 2003-02-07 | 2004-08-26 | Daiso Co Ltd | 酸素発生用電極 |
| JP2008507626A (ja) * | 2004-07-22 | 2008-03-13 | フェルプス ドッジ コーポレイション | 貫流式電解採取用電解槽中で電解採取することにより銅粉末を生成するためのシステムおよび方法 |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140054180A1 (en) * | 2011-03-25 | 2014-02-27 | The Doshisha | Anode for electrowinning and method for electrowinning using same |
| RU2561565C1 (ru) * | 2011-09-13 | 2015-08-27 | Дзе Досиса | Анод для выделения хлора |
| WO2013038927A1 (ja) * | 2011-09-13 | 2013-03-21 | 学校法人同志社 | 塩素発生用陽極 |
| WO2013038928A1 (ja) * | 2011-09-13 | 2013-03-21 | 学校法人同志社 | 電解めっき用陽極および該陽極を用いる電解めっき法 |
| CN103797160A (zh) * | 2011-09-13 | 2014-05-14 | 学校法人同志社 | 析氯用阳极 |
| CN103827360A (zh) * | 2011-09-13 | 2014-05-28 | 学校法人同志社 | 电解镀敷用阳极及使用该阳极的电解镀敷法 |
| JP5008043B1 (ja) * | 2011-09-13 | 2012-08-22 | 学校法人同志社 | 塩素発生用陽極 |
| KR101577668B1 (ko) * | 2011-09-13 | 2015-12-15 | 학교법인 도시샤 | 염소 발생용 양극 |
| CN103827360B (zh) * | 2011-09-13 | 2016-04-27 | 学校法人同志社 | 电镀用阳极及使用该阳极的电镀法 |
| US9556534B2 (en) | 2011-09-13 | 2017-01-31 | The Doshisha | Anode for electroplating and method for electroplating using anode |
| JP2016003346A (ja) * | 2014-06-16 | 2016-01-12 | 住友金属鉱山株式会社 | 不溶性電極の付着物除去方法 |
| JP2019081919A (ja) * | 2017-10-30 | 2019-05-30 | 学校法人同志社 | 電解法 |
| JP2021070843A (ja) * | 2019-10-30 | 2021-05-06 | 住友金属鉱山株式会社 | 廃リチウムイオン電池からの銅、ニッケル、コバルトの回収方法 |
| JP7341395B2 (ja) | 2019-10-30 | 2023-09-11 | 住友金属鉱山株式会社 | 廃リチウムイオン電池からの銅、ニッケル、コバルトの回収方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5013438B2 (ja) | 2012-08-29 |
| CN105386086A (zh) | 2016-03-09 |
| CA2783302A1 (en) | 2011-06-16 |
| EP2535443A4 (en) | 2015-05-20 |
| EP3088569A1 (en) | 2016-11-02 |
| AU2010329192A1 (en) | 2012-07-12 |
| CA2783302C (en) | 2014-07-08 |
| EP2535443A1 (en) | 2012-12-19 |
| WO2011070908A1 (ja) | 2011-06-16 |
| AU2010329192B2 (en) | 2013-06-13 |
| NO3088569T3 (ja) | 2018-06-02 |
| CN102686783A (zh) | 2012-09-19 |
| CN102686783B (zh) | 2015-11-25 |
| US20120247971A1 (en) | 2012-10-04 |
| EP3088569B1 (en) | 2018-01-03 |
| EP2535443B1 (en) | 2016-07-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5013438B2 (ja) | 金属の電解採取用陽極および電解採取方法 | |
| JP2011122183A5 (ja) | ||
| JP4916040B1 (ja) | 電解採取用陽極および該陽極を用いた電解採取法 | |
| JP5008043B1 (ja) | 塩素発生用陽極 | |
| JP4516618B2 (ja) | コバルトの電解採取用陽極および電解採取法 | |
| WO2009151044A1 (ja) | 亜鉛およびコバルトの電解採取用陽極、並びに電解採取方法 | |
| JP4516617B2 (ja) | 亜鉛の電解採取用陽極および電解採取法 | |
| US20170218531A1 (en) | Metal electrowinning anode and electrowinning method | |
| JP2019081919A (ja) | 電解法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120413 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20120413 |
|
| A871 | Explanation of circumstances concerning accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A871 Effective date: 20120413 |
|
| TRDD | Decision of grant or rejection written | ||
| A975 | Report on accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A971005 Effective date: 20120502 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120509 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120530 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150615 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5013438 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |