JP2009523156A - 癌の処置のためのγ線照射と組み合わせた化合物の使用 - Google Patents
癌の処置のためのγ線照射と組み合わせた化合物の使用 Download PDFInfo
- Publication number
- JP2009523156A JP2009523156A JP2008549886A JP2008549886A JP2009523156A JP 2009523156 A JP2009523156 A JP 2009523156A JP 2008549886 A JP2008549886 A JP 2008549886A JP 2008549886 A JP2008549886 A JP 2008549886A JP 2009523156 A JP2009523156 A JP 2009523156A
- Authority
- JP
- Japan
- Prior art keywords
- compound
- alkyl
- use according
- tppii
- unbranched
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images











































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/06—Tripeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61N—ELECTROTHERAPY; MAGNETOTHERAPY; RADIATION THERAPY; ULTRASOUND THERAPY
- A61N5/00—Radiation therapy
- A61N5/10—X-ray therapy; Gamma-ray therapy; Particle-irradiation therapy
- A61N2005/1092—Details
- A61N2005/1098—Enhancing the effect of the particle by an injected agent or implanted device
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61N—ELECTROTHERAPY; MAGNETOTHERAPY; RADIATION THERAPY; ULTRASOUND THERAPY
- A61N5/00—Radiation therapy
- A61N5/10—X-ray therapy; Gamma-ray therapy; Particle-irradiation therapy
Abstract
【課題】
【解決手段】TPPII(トリペプチジルペプチダーゼII)阻害剤は、γ線照射癌療法の効力を強化するか、腫瘍細胞のin vivoγ線照射感受性を増加させるために有用である。適当な化合物には、一般式RN1RN2N−A1−A2−A3−CO−RC1のトリペプチド化合物が含まれ、RN1、RN2、A1、A2、A3およびRC1は本明細書で定義された通りであり、その例としては、例えばトリペプチド配列GLAおよびGPGが含まれる。γ線照射と併用した処理の間、TPPII阻害剤を注射したマウスで、完全なin vivo腫瘍退縮が観察される。
【選択図】
【解決手段】TPPII(トリペプチジルペプチダーゼII)阻害剤は、γ線照射癌療法の効力を強化するか、腫瘍細胞のin vivoγ線照射感受性を増加させるために有用である。適当な化合物には、一般式RN1RN2N−A1−A2−A3−CO−RC1のトリペプチド化合物が含まれ、RN1、RN2、A1、A2、A3およびRC1は本明細書で定義された通りであり、その例としては、例えばトリペプチド配列GLAおよびGPGが含まれる。γ線照射と併用した処理の間、TPPII阻害剤を注射したマウスで、完全なin vivo腫瘍退縮が観察される。
【選択図】
Description
本発明は、癌の処置のためのγ線照射と組み合わせた化合物の使用に関する。
癌療法の分野において、アポトーシス耐性は、通常、照射療法耐性、即ち、癌細胞がγ線照射に遭遇しても死に至らないことの原因となる現象である。癌患者の腫瘍は当初はしばしば処置に応答し、療法への耐性を獲得するのは後になってからのことである。腫瘍細胞の療法耐性は、療法の失敗および患者の死亡の非常に一般的な原因である。
今回、本発明者らは、特定の化合物を用いてγ線照射癌療法を強化できることを発見した。本発明は、in vitroでのDNA損傷応答およびin vivoでの癌療法耐性におけるTPPII(トリペプチジル−ペプチダーゼII)の役割に関する本発明者らの研究に由来する。TPPIIは、ショウジョウバエからヒトまでの多細胞生物で発現される、独特の138kDaサブユニットで構築される。ショウジョウバエからのデータは、TPPII複合体が、約6MDaの本来の構造物と2つのねじれた鎖を形成する、反復サブユニットからなることを示唆する。TPPIIは、唯一の既知のサイトゾルサブチリシン様セリンペプチダーゼである。細菌のサブチリシンは完全に研究された酵素であり、結晶構造および酵素機能に関する多数の報告がある(Gupta,R.、Beg,Q.K.およびLorenz,P.、2002、「Bacterial alkaline proteases:molecular approaches and industrial applications」、Appl Microbiol Biotechnol.59:15〜32)。
したがって、第1の態様から、本発明は、γ線照射癌療法の効力を強化するか、腫瘍細胞のin vivoγ線照射感受性を増加させることにおける使用のための化合物を提供し、前記化合物はTPPII阻害剤である。
本明細書で用いるように、用語「癌療法」は、癌性の病状の処置、ならびに前癌状態の予防療法および治療を含む。
本明細書で用いるように、用語「腫瘍細胞」は、癌性または前癌性の細胞を含む。そのような細胞は、癌性または前癌性の欠陥を有することがある。したがって、細胞は、悪性の進行に特徴的な1つまたはいくつかの変化を獲得していることがある。
本発明は、γ線照射耐性の腫瘍の処置を可能にするだけでなく、γ線照射で処置可能な腫瘍においてさえ、より低い用量のγ線照射の使用を可能にする点で有利である。
さらなる態様から、本発明は、γ線照射癌療法の効力を強化するか、腫瘍細胞のin vivoγ線照射感受性を増加させるために用いる化合物を提供し、前記化合物は下記式(i)
(i)RN1RN2N−A1−A2−A3−CO−RC1
から選択されるか、その薬学的に許容される塩であり、上式で、A1、A2およびA3は、標準の1文字略語または名称に従う以下の定義を有するアミノ酸残基であり:
A1は、G、A、V、L、I、P、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシンであり、
A2は、G、A、V、L、I、P、F、W、C、S、K、R、2−アミノ酪酸、ノルバリン、ノルロイシン、tert−ブチルアラニン、α−メチルロイシン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、α−メチルバリン、tert−ブチルグリシン、2−アリルグリシン、オルニチンまたはα,γ−ジアミノ酪酸であり、
A3は、G、A、V、L、I、P、F、W、D、E、Y、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシンであり、
RN1およびRN2は、それぞれペプチドのN末端に結合し、同じであるかまたは異なり、それぞれ独立して
RN3、
(リンカー1)−RN3、
CO−(リンカー1)−RN3、
CO−O−(リンカー1)−RN3、
CO−N−((リンカー1)−RN3)RN4、または
SO2−(リンカー1)−RN3
であり、
(リンカー1)は存在しなくてもよく、即ち単結合でもよく、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであることができ、
RN3およびRN4は、同じであるかまたは異なり、水素または以下の必要に応じて置換された基:
飽和または不飽和で分枝または非分枝C1〜6アルキル、
飽和または不飽和で分枝または非分枝C3〜12シクロアルキル、
ベンジル、
フェニル、
ナフチル、
単環式または二環式C1〜10ヘテロアリール、または
非芳香族C1〜10ヘテロシクリル
のいずれかであり、RN3および/またはRN4には、0、1または2個(同じかまたは異なる)の必要に応じた置換基があり得、該置換基は、
ヒドロキシ−、
チオ−、
アミノ−、
カルボン酸、
飽和または不飽和で分枝または非分枝C1〜6アルキルオキシ、
飽和または不飽和で分枝または非分枝C3〜12シクロアルキル、
N−、O−またはS−アセチル、
カルボン酸の飽和または不飽和で分枝または非分枝C1〜6アルキルエステル、
カルボン酸の飽和または不飽和で分枝または非分枝C3〜12シクロアルキルエステル
フェニル、
単環式または二環式C1〜10ヘテロアリール、
非芳香族C1〜10ヘテロシクリル、または
ハロゲン
でよく、RC1はトリペプチドのC末端に結合され、
O−RC2、
O−(リンカー2)−RC2、
N((リンカー2)RC2)RC3、または
N(リンカー2)RC2−NRC3RC4
であり、(リンカー2)は存在しない、即ち単結合、またはC1〜6アルキルもしくはC2〜4アルケニル、好ましくは単結合、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであってもよく、
RC2、RC3およびRC4は、同じであるかまたは異なり、水素または以下の必要に応じて置換された基:
飽和または不飽和で分枝または非分枝C1〜6アルキル、
飽和または不飽和で分枝または非分枝C3〜12シクロアルキル、
ベンジル、
フェニル、
ナフチル、
単環式または二環式C1〜10ヘテロアリール、または
非芳香族C1〜10ヘテロシクリル
のいずれかであり、RC2および/またはRC3および/またはRC4には、0、1または2個(同じかまたは異なる)の必要に応じた置換基があり得、該置換基は、
ヒドロキシ−、
チオ−、
アミノ−、
カルボン酸、
飽和または不飽和で分枝または非分枝C1〜6アルキルオキシ、
飽和または不飽和で分枝または非分枝C3〜12シクロアルキル、
N−、O−またはS−アセチル、
カルボン酸の飽和または不飽和で分枝または非分枝C1〜6アルキルエステル、
カルボン酸の飽和または不飽和で分枝または非分枝C3〜12シクロアルキルエステル
フェニル、
ハロゲン、
単環式または二環式C1〜10ヘテロアリール、または
非芳香族C1〜10ヘテロシクリル
の1つまたは複数でよい。
(i)RN1RN2N−A1−A2−A3−CO−RC1
から選択されるか、その薬学的に許容される塩であり、上式で、A1、A2およびA3は、標準の1文字略語または名称に従う以下の定義を有するアミノ酸残基であり:
A1は、G、A、V、L、I、P、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシンであり、
A2は、G、A、V、L、I、P、F、W、C、S、K、R、2−アミノ酪酸、ノルバリン、ノルロイシン、tert−ブチルアラニン、α−メチルロイシン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、α−メチルバリン、tert−ブチルグリシン、2−アリルグリシン、オルニチンまたはα,γ−ジアミノ酪酸であり、
A3は、G、A、V、L、I、P、F、W、D、E、Y、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシンであり、
RN1およびRN2は、それぞれペプチドのN末端に結合し、同じであるかまたは異なり、それぞれ独立して
RN3、
(リンカー1)−RN3、
CO−(リンカー1)−RN3、
CO−O−(リンカー1)−RN3、
CO−N−((リンカー1)−RN3)RN4、または
SO2−(リンカー1)−RN3
であり、
(リンカー1)は存在しなくてもよく、即ち単結合でもよく、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであることができ、
RN3およびRN4は、同じであるかまたは異なり、水素または以下の必要に応じて置換された基:
飽和または不飽和で分枝または非分枝C1〜6アルキル、
飽和または不飽和で分枝または非分枝C3〜12シクロアルキル、
ベンジル、
フェニル、
ナフチル、
単環式または二環式C1〜10ヘテロアリール、または
非芳香族C1〜10ヘテロシクリル
のいずれかであり、RN3および/またはRN4には、0、1または2個(同じかまたは異なる)の必要に応じた置換基があり得、該置換基は、
ヒドロキシ−、
チオ−、
アミノ−、
カルボン酸、
飽和または不飽和で分枝または非分枝C1〜6アルキルオキシ、
飽和または不飽和で分枝または非分枝C3〜12シクロアルキル、
N−、O−またはS−アセチル、
カルボン酸の飽和または不飽和で分枝または非分枝C1〜6アルキルエステル、
カルボン酸の飽和または不飽和で分枝または非分枝C3〜12シクロアルキルエステル
フェニル、
単環式または二環式C1〜10ヘテロアリール、
非芳香族C1〜10ヘテロシクリル、または
ハロゲン
でよく、RC1はトリペプチドのC末端に結合され、
O−RC2、
O−(リンカー2)−RC2、
N((リンカー2)RC2)RC3、または
N(リンカー2)RC2−NRC3RC4
であり、(リンカー2)は存在しない、即ち単結合、またはC1〜6アルキルもしくはC2〜4アルケニル、好ましくは単結合、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであってもよく、
RC2、RC3およびRC4は、同じであるかまたは異なり、水素または以下の必要に応じて置換された基:
飽和または不飽和で分枝または非分枝C1〜6アルキル、
飽和または不飽和で分枝または非分枝C3〜12シクロアルキル、
ベンジル、
フェニル、
ナフチル、
単環式または二環式C1〜10ヘテロアリール、または
非芳香族C1〜10ヘテロシクリル
のいずれかであり、RC2および/またはRC3および/またはRC4には、0、1または2個(同じかまたは異なる)の必要に応じた置換基があり得、該置換基は、
ヒドロキシ−、
チオ−、
アミノ−、
カルボン酸、
飽和または不飽和で分枝または非分枝C1〜6アルキルオキシ、
飽和または不飽和で分枝または非分枝C3〜12シクロアルキル、
N−、O−またはS−アセチル、
カルボン酸の飽和または不飽和で分枝または非分枝C1〜6アルキルエステル、
カルボン酸の飽和または不飽和で分枝または非分枝C3〜12シクロアルキルエステル
フェニル、
ハロゲン、
単環式または二環式C1〜10ヘテロアリール、または
非芳香族C1〜10ヘテロシクリル
の1つまたは複数でよい。
式(i)の一般式で示されるNおよびCOは、それぞれアミノ酸残基A1の窒素原子およびアミノ酸残基A3のカルボニル基である。
さらなる態様から、本発明は、γ線照射癌療法の効力を強化するか、腫瘍細胞のin vivoγ線照射感受性を増加させる方法であって、それを必要とする患者にTPPII阻害剤または式(i)から選択される化合物もしくはその薬学的に許容される塩の治療有効量を投与することを含む方法を提供する。前記γ線照射癌療法に対する耐性を減少させるために、化合物をγ線照射癌療法と組み合わせて投与することができる。
好ましくは、化合物と組み合わせたγ線照射の投与は、腫瘍が治療されるまで、好ましくは腫瘍が消えるまで繰り返す。
同様に、さらなる態様から、本発明は、γ線照射癌療法の効力を強化するか、腫瘍細胞のin vivoγ線照射感受性を増加させるための医薬品の製造における、TPPII阻害剤または式(i)から選択される化合物もしくはその薬学的に許容される塩の使用を提供する。
理論によって束縛されることを望むものではないが、本発明は、癌の治療においてTPPII阻害剤がγ線照射との併用で有用であることを認識すると考えることができる。
さらなる態様から、本発明は、式(i)の化合物またはその薬学的に許容される塩、および薬学的に許容される希釈剤または担体を含む医薬組成物を提供する。
さらなる態様から、本発明は、医薬品として使用するための式(i)の化合物またはその薬学的に許容される塩を提供する。
さらなる態様から、本発明は、γ線照射癌療法の効力を強化するか、腫瘍細胞のin vivoγ線照射感受性を増加させるのに適当な化合物を同定する方法であって、TPPIIをスクリーニングする化合物と接触させ、その化合物がTPPIIの活性を阻害するか否かを同定することを含む方法を提供する。
本発明は、γ線照射に対する細胞の応答における、TPPIIの必須の役割を認識する。比較的低い用量のγ線照射による処置の間でも、TPPII阻害剤を注入したマウスで完全なin vivo腫瘍退縮が観察された。
本出願は、発明者Rickard GlasおよびHong Xuによる「Use of peptides and peptidomimetic compounds」という名称の2006年1月13日に出願の米国特許仮出願60/759088の優先権を主張し、その内容は、その出願が癌の処置のためのγ線照射との組合せに関する限り、本明細書で完全に組み込まれる。米国特許仮出願60/759099の出願および本出願の間で、本発明者らは、本発明の基礎をなす生物学的機構の理解を深めたさらなる実験を実施した。しかし、γ線照射と組み合わせたTPPII阻害剤が癌の処置のために有用であるとの認識の点で、およびこの場合に好ましい特定の化学構造を特定する点で、本出願は前の優先権出願と合致している。
理論によって束縛されることを望むものではないが、下記の本発明者らのデータは、その機構のいくつかの点は未解明であるが、TPPIIがPIKKによるシグナル伝達を制御することを示す。TPPIIは、DNA修復病巣への制御因子の補充および/または結合で、直接的または間接的な役割をすることができ、これらの因子がPIKKと相互作用してそれによって活性化されることを可能にする。例えば、TPPIIはγ線照射後のATMおよびp53の間の相互作用を制御すると考えられる。ATM、ATRおよびDNA−PKcsはp53の安定化においてある程度の冗長性を有し、複数のN末端部位がp53リン酸化を、また複数のPIKKが同じ部位を標的にする(Bode,AM、Dong,Z.Post−translational modification of p53 in tumorigenesis.Nat Rev Cancer.2004;4:793〜805)。
TPPIIは、比較的広い範囲の基質を受け入れる。式(i)に含まれるすべての化合物は、ペプチドまたはペプチド類似体である。式(i)の化合物は、当技術分野で知られている方法によって、容易に合成することができ(例えばGanellinら、J.Med.Chem.2000,43,664〜674を参照)、または市販品を容易に入手することができる(例えばBachem AGから)。好ましい態様では、化合物は式(i)から選択することができる。そのようなトリペプチドおよび誘導体は、特に有効な治療剤である。
本発明に従い、γ線照射癌療法の効力を強化するか、腫瘍細胞のin vivoγ線照射感受性を増加させるために用いる化合物は、in vivoでTPPII阻害剤であることが知られている化合物でよい。
例えば、該化合物は、Winterら、Journal of Molecular Graphics and Modelling 2005、23、409〜418においてTPPII阻害剤として特定された化合物から選択することができる。
それらの化合物は特にTPPIIファルマコフォアに適しているので、化合物は以下の式(ii):

から選択することができ、上式で、R’はH、CH3、CH2CH3、CH2CH2CH3またはCH(CH3)2であり、
R”は、H、CH2CH3、CH2CH2CH3、CH(CH3)2、CH2CH2CH2CH3、CH2CH(CH3)2、CH(CH3)CH2CH3またはC(CH3)3であり、
R”’は、H、CH3、OCH3、F、ClまたはBrである。
R”は、H、CH2CH3、CH2CH2CH3、CH(CH3)2、CH2CH2CH2CH3、CH2CH(CH3)2、CH(CH3)CH2CH3またはC(CH3)3であり、
R”’は、H、CH3、OCH3、F、ClまたはBrである。
式(ii)の化合物は、公知の方法によって合成可能である(例えば、Winterら、Journal of Molecular Graphics and Modelling 2005、23、409〜418およびBreslinら、Bioorg.Med.Chem.Lett.2003、13、4467〜4471を参照)。
また、例として、化合物は、Schwartzらの米国特許第6335360号でTPPII阻害剤として特定された化合物から選択することができる。そのような化合物は、以下の式(iii)のものを含む。
上式で、
各R1は同じであるかまたは異なることができ、ハロゲン、OH;ハロゲンおよびOHからなる群から選択される1つまたは複数の基で必要に応じて置換されたC1〜C6アルキル;ハロゲンおよびOHからなる群から選択される1つまたは複数の基で必要に応じて置換された(C1〜C6)アルケニル;ハロゲンおよびOH、X(C1〜C6)アルキルからなる群から選択される1つまたは複数の基で必要に応じて置換された(C1〜C6)アルキニル(XがS、OまたはOCOであり、アルキルがハロゲンおよびOHからなる群から選択される1つまたは複数の基で必要に応じて置換される);少なくとも1つのハロゲンで必要に応じて置換されたSO2(C1〜C6)アルキル、YSO3H、YSO2(C1〜C6)アルキルであって、YがOまたはNHであり、アルキルが少なくとも1つのハロゲン、ジラジカル−X1−(C1〜C2)アルキレン−X1−で必要に応じて置換されており、X1がOまたはSであり;インドリン環と縮合したベンゼン環;
からなる群から選択され、nは、0〜4であり、
R2は、R4がハロゲンおよびOHからなる群から選択される1つまたは複数の基によって置換されたC1〜C6アルキルであるCH2R4;ZがOもしくはSであり、pが0〜5であり、qが0〜5であり、ただしp+qが0〜5である(CH2)pZ(CH2)qCH3;(C2〜C6)不飽和アルキル;または(C3〜C6)シクロアルキルであり、
あるいは、R2は(C1〜C6)アルキルまたはO(C1〜C6)アルキルであり、それぞれは少なくとも1つのハロゲンで必要に応じて置換されており、
R3はH;少なくとも1つのハロゲンで必要に応じて置換された(C1〜C6)アルキル;(CH2)pZR5(式中、pが1〜3であり、ZがOまたはSであり、R5がHまたは(C1〜C3)アルキルである);ベンジルである。
各R1は同じであるかまたは異なることができ、ハロゲン、OH;ハロゲンおよびOHからなる群から選択される1つまたは複数の基で必要に応じて置換されたC1〜C6アルキル;ハロゲンおよびOHからなる群から選択される1つまたは複数の基で必要に応じて置換された(C1〜C6)アルケニル;ハロゲンおよびOH、X(C1〜C6)アルキルからなる群から選択される1つまたは複数の基で必要に応じて置換された(C1〜C6)アルキニル(XがS、OまたはOCOであり、アルキルがハロゲンおよびOHからなる群から選択される1つまたは複数の基で必要に応じて置換される);少なくとも1つのハロゲンで必要に応じて置換されたSO2(C1〜C6)アルキル、YSO3H、YSO2(C1〜C6)アルキルであって、YがOまたはNHであり、アルキルが少なくとも1つのハロゲン、ジラジカル−X1−(C1〜C2)アルキレン−X1−で必要に応じて置換されており、X1がOまたはSであり;インドリン環と縮合したベンゼン環;
からなる群から選択され、nは、0〜4であり、
R2は、R4がハロゲンおよびOHからなる群から選択される1つまたは複数の基によって置換されたC1〜C6アルキルであるCH2R4;ZがOもしくはSであり、pが0〜5であり、qが0〜5であり、ただしp+qが0〜5である(CH2)pZ(CH2)qCH3;(C2〜C6)不飽和アルキル;または(C3〜C6)シクロアルキルであり、
あるいは、R2は(C1〜C6)アルキルまたはO(C1〜C6)アルキルであり、それぞれは少なくとも1つのハロゲンで必要に応じて置換されており、
R3はH;少なくとも1つのハロゲンで必要に応じて置換された(C1〜C6)アルキル;(CH2)pZR5(式中、pが1〜3であり、ZがOまたはSであり、R5がHまたは(C1〜C3)アルキルである);ベンジルである。
式(iii)の化合物は、公知の方法によって容易に合成が可能である(例えば、Schwartzらの米国特許第6335360号を参照)。
しかし、化合物は、式(i)および(ii)、より好ましくは式(i)から選択することが好ましい。
化合物が、RN1、RN2およびRC1が上でまたは下の好ましい実施形態のいずれかで定義された通りであり、
A1は、G、A、V、L、I、P、S、T、C、N、Q、2−アミノ酪酸、ノルバリン、ノルロイシン、tert−ブチルアラニン、α−メチルロイシン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、α−メチルバリン、tert−ブチルグリシンまたは2−アリルグリシンであり、
A2は、G、A、V、L、I、P、S、T、C、N、Q、F、Y、W、K、R、ヒスチジン、2−アミノ酪酸、ノルバリン、ノルロイシン、tert−ブチルアラニン、α−メチルロイシン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、α−メチルバリン、tert−ブチルグリシン、2−アリルグリシン、オルニチン、α,γ−ジアミノ酪酸または4,5−デヒドロ−リジンであり、
A3は、G、A、V、L、I、P、S、T、C、N、Q、D、E、F、Y、W、2−アミノ酪酸、ノルバリン、ノルロイシン、tert−ブチルアラニン、α−メチルロイシン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、α−メチルバリン、tert−ブチルグリシンまたは2−アリルグリシンである、
式(i)の化合物であることも可能である。
A1は、G、A、V、L、I、P、S、T、C、N、Q、2−アミノ酪酸、ノルバリン、ノルロイシン、tert−ブチルアラニン、α−メチルロイシン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、α−メチルバリン、tert−ブチルグリシンまたは2−アリルグリシンであり、
A2は、G、A、V、L、I、P、S、T、C、N、Q、F、Y、W、K、R、ヒスチジン、2−アミノ酪酸、ノルバリン、ノルロイシン、tert−ブチルアラニン、α−メチルロイシン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、α−メチルバリン、tert−ブチルグリシン、2−アリルグリシン、オルニチン、α,γ−ジアミノ酪酸または4,5−デヒドロ−リジンであり、
A3は、G、A、V、L、I、P、S、T、C、N、Q、D、E、F、Y、W、2−アミノ酪酸、ノルバリン、ノルロイシン、tert−ブチルアラニン、α−メチルロイシン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、α−メチルバリン、tert−ブチルグリシンまたは2−アリルグリシンである、
式(i)の化合物であることも可能である。
式(i)の好ましい化合物
式(i)の化合物の様々な基および特定の例が好ましい。
式(i)の化合物の様々な基および特定の例が好ましい。
一般に、特にA2位置が天然(L)立体配置のアミノ酸が好ましい。
一般に、RN1が水素であり、
RN2が、
RN3、
(リンカー1)−RN3、
CO−(リンカー1)−RN3、または
CO−O−(リンカー1)−RN3
であり、上式で、
(リンカー1)は存在しなくてもよく、即ち単結合でもよく、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであり得、
RN3は、水素または以下の置換されていない基:
飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキル、
ベンジル、
フェニル、または
単環式ヘテロアリール
のいずれかであることが好ましい。
RN2が、
RN3、
(リンカー1)−RN3、
CO−(リンカー1)−RN3、または
CO−O−(リンカー1)−RN3
であり、上式で、
(リンカー1)は存在しなくてもよく、即ち単結合でもよく、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであり得、
RN3は、水素または以下の置換されていない基:
飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキル、
ベンジル、
フェニル、または
単環式ヘテロアリール
のいずれかであることが好ましい。
一般に、RC1は、
O−RC2、
O−(リンカー2)−RC2、または
NH−(リンカー2)RC2
であり、上式で、
(リンカー2)は存在しない、即ち単結合、C1〜6アルキルもしくはC2〜4アルケニル、好ましくは単結合、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであってもよく、
RC2は水素または以下の置換されていない基:
飽和または不飽和で分枝または非分枝C1〜5アルキル、
ベンジル、
フェニル、または
単環式C1〜10ヘテロアリール
のいずれかであることが好ましい。
O−RC2、
O−(リンカー2)−RC2、または
NH−(リンカー2)RC2
であり、上式で、
(リンカー2)は存在しない、即ち単結合、C1〜6アルキルもしくはC2〜4アルケニル、好ましくは単結合、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであってもよく、
RC2は水素または以下の置換されていない基:
飽和または不飽和で分枝または非分枝C1〜5アルキル、
ベンジル、
フェニル、または
単環式C1〜10ヘテロアリール
のいずれかであることが好ましい。
一般には、N末端の置換基に関して、
RN1は水素であり、
RN2は、水素、C(=O)−O−(リンカー1)−RN3またはC(=O)−(リンカー1)−RN3であり、
(リンカー1)は、CH2またはCH=CHであり、
RN3は、フェニルまたは2−フリルであることがさらに好ましい。
RN1は水素であり、
RN2は、水素、C(=O)−O−(リンカー1)−RN3またはC(=O)−(リンカー1)−RN3であり、
(リンカー1)は、CH2またはCH=CHであり、
RN3は、フェニルまたは2−フリルであることがさらに好ましい。
さらに、
RN1は水素であり、
RN2は、水素、C(=O)−OCH2PhまたはC(=O)−CH=CH−(2−フリル)であることが好ましい。
RN1は水素であり、
RN2は、水素、C(=O)−OCH2PhまたはC(=O)−CH=CH−(2−フリル)であることが好ましい。
N末端置換基のための他の好ましいグループは、
RN1が水素であり、
RN2が、ベンジルオキシカルボニル、ベンジル、ベンゾイル、tert−ブチルオキシカルボニル、9−フルオレニルメトキシカルボニルまたはFAであり、より好ましくはベンジルオキシカルボニルまたはFAであるものである。
RN1が水素であり、
RN2が、ベンジルオキシカルボニル、ベンジル、ベンゾイル、tert−ブチルオキシカルボニル、9−フルオレニルメトキシカルボニルまたはFAであり、より好ましくはベンジルオキシカルボニルまたはFAであるものである。
一般には、C末端の置換基に関して、
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキルフェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキル−フェニル、より好ましくはOHであることが好ましい。
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキルフェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキル−フェニル、より好ましくはOHであることが好ましい。
いくつかの好ましいグループは、以下の通りである。
グループ(i)(a):
A1は、G、A、V、L、I、P、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシンであり、
A2は、G、A、V、L、I、P、F、W、C、S、K、R、2−アミノ酪酸、ノルバリン、ノルロイシン、tert−ブチルアラニン、α−メチルロイシン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、α−メチルバリン、tert−ブチルグリシン、2−アリルグリシン、オルニチンまたはα,γ−ジアミノ酪酸であり、
A3は、G、A、V、L、I、P、F、W、D、E、Y、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシンであり、
RN1はHであり、
RN2は、水素、C(=O)−O−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、またはC(=O)−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキルフェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキルフェニルである。
A1は、G、A、V、L、I、P、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシンであり、
A2は、G、A、V、L、I、P、F、W、C、S、K、R、2−アミノ酪酸、ノルバリン、ノルロイシン、tert−ブチルアラニン、α−メチルロイシン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、α−メチルバリン、tert−ブチルグリシン、2−アリルグリシン、オルニチンまたはα,γ−ジアミノ酪酸であり、
A3は、G、A、V、L、I、P、F、W、D、E、Y、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシンであり、
RN1はHであり、
RN2は、水素、C(=O)−O−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、またはC(=O)−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキルフェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキルフェニルである。
グループ(i)(b):
A1は、G、Aまたは2−アミノ酪酸であり、
A2は、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、2−アリルグリシン、P、2−アミノ酪酸、α−メチルロイシン、α−メチルバリンまたはtert−ブチルグリシンであり、
A3は、G、A、V、P、2−アミノ酪酸またはノルバリンであり、
RN1はHであり、
RN2は、水素、C(=O)−O−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、またはC(=O)−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキル−フェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキル−フェニルである。
A1は、G、Aまたは2−アミノ酪酸であり、
A2は、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、2−アリルグリシン、P、2−アミノ酪酸、α−メチルロイシン、α−メチルバリンまたはtert−ブチルグリシンであり、
A3は、G、A、V、P、2−アミノ酪酸またはノルバリンであり、
RN1はHであり、
RN2は、水素、C(=O)−O−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、またはC(=O)−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキル−フェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキル−フェニルである。
グループ(i)(c):
A1は、G、Aまたは2−アミノ酪酸であり、
A2は、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシンまたは2−アリルグリシンであり、
A3は、G、A、V、P、2−アミノ酪酸またはノルバリンであり、
RN1はHであり、
RN2は、水素、C(=O)−O−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、またはC(=O)−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキルフェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキル−フェニルである。
A1は、G、Aまたは2−アミノ酪酸であり、
A2は、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシンまたは2−アリルグリシンであり、
A3は、G、A、V、P、2−アミノ酪酸またはノルバリンであり、
RN1はHであり、
RN2は、水素、C(=O)−O−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、またはC(=O)−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキルフェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキル−フェニルである。
グループ(i)(d):
A1は、GまたはAであり、
A2は、L、Iまたはノルロイシンであり、
A3は、GまたはAであり、
RN1はHであり、
RN2は、水素、C(=O)−O−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、またはC(=O)−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキル−フェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキル−フェニルである。
A1は、GまたはAであり、
A2は、L、Iまたはノルロイシンであり、
A3は、GまたはAであり、
RN1はHであり、
RN2は、水素、C(=O)−O−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、またはC(=O)−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキル−フェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキル−フェニルである。
特定の好ましい化合物の第1セットは、
A1がGであり、
A2がLであり、
A3が、G、A、V、L、I、P、F、W、D、E、Y、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシン、より好ましくはG、A、V、P、2−アミノ酪酸またはノルバリン、より好ましくはGまたはAであり、
RN1が水素であり、
RN2がベンジルオキシカルボニルであり、
RC1がOHである化合物である。
A1がGであり、
A2がLであり、
A3が、G、A、V、L、I、P、F、W、D、E、Y、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシン、より好ましくはG、A、V、P、2−アミノ酪酸またはノルバリン、より好ましくはGまたはAであり、
RN1が水素であり、
RN2がベンジルオキシカルボニルであり、
RC1がOHである化合物である。
特定の好ましい化合物の第2セットは、
A1がGであり、
A2がG、A、V、L、I、P、F、W、C、S、2−アミノ酪酸、ノルバリン、ノルロイシン、tert−ブチルアラニン、α−メチルロイシン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、α−メチルバリン、tert−ブチルグリシンまたは2−アリルグリシン、より好ましくは、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、2−アリルグリシン、P、2−アミノ酪酸、α−メチルロイシン、α−メチルバリンまたはtert−ブチルグリシン、より好ましくは、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシンまたは2−アリルグリシン、より好ましくは、L、Iまたはノルロイシンであり、
A3がAであり、
RN1が水素であり、
RN2がベンジルオキシカルボニルであり、
RC1がOHである化合物である。
A1がGであり、
A2がG、A、V、L、I、P、F、W、C、S、2−アミノ酪酸、ノルバリン、ノルロイシン、tert−ブチルアラニン、α−メチルロイシン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、α−メチルバリン、tert−ブチルグリシンまたは2−アリルグリシン、より好ましくは、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、2−アリルグリシン、P、2−アミノ酪酸、α−メチルロイシン、α−メチルバリンまたはtert−ブチルグリシン、より好ましくは、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシンまたは2−アリルグリシン、より好ましくは、L、Iまたはノルロイシンであり、
A3がAであり、
RN1が水素であり、
RN2がベンジルオキシカルボニルであり、
RC1がOHである化合物である。
特定の好ましい化合物の第3セットは、
A1が、G、A、V、L、I、P、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシン、より好ましくはG、Aまたは2−アミノ酪酸、より好ましくはGまたはAであり、
A2がLであり、
A3がAであり、
RN1が水素であり、
RN2がベンジルオキシカルボニルであり、
RC1がOHである化合物である。
A1が、G、A、V、L、I、P、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシン、より好ましくはG、Aまたは2−アミノ酪酸、より好ましくはGまたはAであり、
A2がLであり、
A3がAであり、
RN1が水素であり、
RN2がベンジルオキシカルボニルであり、
RC1がOHである化合物である。
好ましくは、配列A1−A2−A3は、GLA、GLF、GVA、GIA、GPAまたはALA、最も好ましくはGLAであり、
RN1は水素であり、
RN2はベンジルオキシカルボニルであり、
RC1はOHである。
RN1は水素であり、
RN2はベンジルオキシカルボニルであり、
RC1はOHである。
アルキル基が飽和または不飽和と記載される場合、これはアルキル、アルケニルおよびアルキニル炭化水素部分を包含する。
C1〜6アルキルは、好ましくはC1〜4アルキル、より好ましくはメチル、エチル、n−プロピル、イソプロピルまたはブチル(分枝または非分枝)、最も好ましくはメチルである。
C3〜12シクロアルキルは、好ましくはC5〜10シクロアルキル、より好ましくはC5〜7シクロアルキルである。
「アリール」は、芳香族基、好ましくはフェニルまたはナフチルである。
語の一部としての「ヘテロ」は、1つまたは複数の、好ましくはN、OおよびSから選択されるヘテロ原子を含むことを意味する。
「ヘテロアリール」は、好ましくはピリジル、ピロリル、キノリニル、フラニル、チエニル、オキサジアゾリル、チアジアゾリル、チアゾリル、オキサゾリル、ピラゾリル、トリアゾリル、テトラゾリル、イソオキサゾリル、イソチアゾリル、イミダゾリル、ピリミジニル、インドリル、ピラジニル、インダゾリル、ピリミジニル、チオフェネチル、ピラニル、カルバゾリル、アクリジニル、キノリニル、ベンズイミダゾリル、ベンズチアゾリル、プリニル、シノリニルまたはプテリジニルである。
「非芳香族のヘテロシクリル」は、好ましくはピロリジニル、ピペリジル、ピペラジニル、モルホリニル、テトラヒドロフラニルまたは単糖である。
「ハロゲン」は、好ましくはClまたはF、より好ましくはClである。
さらなる好ましい式(i)の化合物
一般に、A1は、好ましくは、G、Aまたは2−アミノ酪酸、より好ましくはGまたはAから選択することができる。
一般に、A1は、好ましくは、G、Aまたは2−アミノ酪酸、より好ましくはGまたはAから選択することができる。
一般に、A2は、好ましくは、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、2−アリルグリシン、P、K、2−アミノ酪酸、α−メチルロイシン、α−メチルバリンまたはtert−ブチルグリシン、より好ましくは、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、2−アリルグリシン、PまたはK、より好ましくは、L、I、ノルロイシン、PまたはK、より好ましくはLまたはPから選択することができる。
一般に、A3は、好ましくは、G、A、V、P、2−アミノ酪酸またはノルバリン、より好ましくはGまたはAから選択することができる。
一般に、RN1は水素であることが好ましい。
一般に、RN2は、好ましくは、
RN3、
(リンカー1)−RN3、
CO−(リンカー1)−RN3、または
CO−O−(リンカー1)−RN3
であり、上式で、
(リンカー1)は存在しない、即ち単結合、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであってもよく、
RN3は、水素または以下の置換されていない基:
飽和または不飽和で分枝または非分枝C1〜4アルキル、
ベンジル、
フェニル、もしくは
単環式ヘテロアリール
のいずれかである。
RN3、
(リンカー1)−RN3、
CO−(リンカー1)−RN3、または
CO−O−(リンカー1)−RN3
であり、上式で、
(リンカー1)は存在しない、即ち単結合、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであってもよく、
RN3は、水素または以下の置換されていない基:
飽和または不飽和で分枝または非分枝C1〜4アルキル、
ベンジル、
フェニル、もしくは
単環式ヘテロアリール
のいずれかである。
一般に、RN2は、より好ましくは、水素、ベンジルオキシカルボニル、ベンジル、ベンゾイル、tert−ブチルオキシカルボニル、9−フルオレニルメトキシカルボニルまたはFAであり、より好ましくは、水素、ベンジルオキシカルボニルまたはFAである。
一般に、RC1が
O−RC2、
O−(リンカー2)−RC2、または
NH−(リンカー2)RC2
であり、上式で、
(リンカー2)は存在しない、即ち単結合、C1〜6アルキルもしくはC2〜4アルケニル、好ましくは単結合、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであってもよく、
RC2は水素または以下の置換されていない基:
飽和または不飽和で分枝または非分枝C1〜5アルキル、
ベンジル、
フェニル、もしくは
単環式C1〜10ヘテロアリール
のいずれかであることが好ましい。
O−RC2、
O−(リンカー2)−RC2、または
NH−(リンカー2)RC2
であり、上式で、
(リンカー2)は存在しない、即ち単結合、C1〜6アルキルもしくはC2〜4アルケニル、好ましくは単結合、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであってもよく、
RC2は水素または以下の置換されていない基:
飽和または不飽和で分枝または非分枝C1〜5アルキル、
ベンジル、
フェニル、もしくは
単環式C1〜10ヘテロアリール
のいずれかであることが好ましい。
一般に、RC1は、より好ましくは、OH、O−C1〜6アルキル、O−C1〜6アルキル−フェニル、NH2、NH−C1〜6アルキルまたはNH−C1〜6アルキル−フェニル、より好ましくは、OH、O−C1〜6アルキル、NH2またはNH−C1〜6アルキル、より好ましくは、OHまたはNH2である。
特に興味がある化合物には、A2がPであるものが含まれる。
特に興味がある化合物には、RC1がNH2であるものが含まれる。
一般に、以下のアミノ酸、F、W、D、EおよびYは、A3により不適当である。同様に、一般に、A3は、Pおよび/またはEでないものが選択されることがあるが、それは、これらを含む化合物はより低い活性を示すからである。
好ましい式(ii)の化合物
式(ii)の化合物は、好ましくは、
R’が、CH2CH3またはCH2CH2CH3であり、
R”が、CH2CH2CH3またはCH(CH3)2であり、
R”’が、HまたはClである化合物である。
式(ii)の化合物は、好ましくは、
R’が、CH2CH3またはCH2CH2CH3であり、
R”が、CH2CH2CH3またはCH(CH3)2であり、
R”’が、HまたはClである化合物である。
好ましい式(iii)の化合物
式(iii)の化合物の様々な好ましいグループおよび特定の例は、別にとられたSchwartzらのUS6,335,360B1の請求項のいずれかに規定される通りである。
式(iii)の化合物の様々な好ましいグループおよび特定の例は、別にとられたSchwartzらのUS6,335,360B1の請求項のいずれかに規定される通りである。
式(i)の治療化合物の一例は、Z−GLA−OH、即ち、N末端がZ基で誘導体化され、C末端が誘導体化されないトリペプチドGLAである。Zは、ベンジルオキシカルボニルを表す。これは、RN1がH、RN2がZ、A1がG、A2がL、A3がA、RC1がOHである、式(i)の化合物である。この化合物はBachem AGから市販され、真核生物TPPIIの細菌同族体、サブチリシンを抑制することが発見された。Z−GLA−OHは低コストであり、γ線照射による療法に耐性である腫瘍の排除を誘導するために、in vivoでよく効く。療法耐性の癌の新規治療法は、公衆衛生上かなり関心がある。
好ましい化合物には、GLAを含むもの、例えば、Z−GLA−OH、Bn−GLA−OH、FA−GLA−OHおよびH−GLA−OH、例えばZ−GLA−OHが含まれるが、本発明に従い、本明細書での任意の化合物または化合物群のいかなる開示も、配列A1A2A3はGLAではないとのただし書き、または化合物はZ−GLA−OH、Bn−GLA−OH、FA−GLA−OHまたはH−GLA−OHからなる群から選択されないとのただし書き、または化合物はZ−GLA−OHではないとのただし書きに必要に応じて従うことができる。
標準のγ線照射処置に応答しない腫瘍の処置では、悪性疾患患者でそのような処置を改善するために、例えば、固形腫瘍でそのような処置に対するin vivo応答を増加させるために、本明細書で記載されるZ−GLA−OHまたは他の化合物を投与することができる。
他の好ましい化合物には、A1A2A3がGPGであるもの、例えばGPG−NH2またはZ−GPG−NH2が含まれる。
当業者は、本明細書で記載される化合物は、任意の適当な方法で投与することができることに気付くであろう。例えば、投与は、非経口的、例えば静脈内もしくは皮下、経口、経皮、鼻腔内、吸入、または経直腸であることができる。好ましい一実施形態では、化合物は注射によって投与される。
本発明の医薬組成物に用いる薬学的に許容される付加塩の例には、塩酸、臭化水素酸、リン酸、メタリン酸、硝酸および硫酸などの無機酸、ならびに、酒石酸、酢酸、クエン酸、リンゴ酸、乳酸、フマル酸、安息香酸、グリコール酸、グルコン酸、コハク酸およびアリールスルホン酸などの有機酸に由来するものが含まれる。本明細書で記載される薬学的に許容される賦形剤、例えば、媒体、アジュバント、担体または希釈剤は、当技術分野の技術者に周知であり、容易に入手できる。薬学的に許容される担体は、活性化合物に対して化学的に不活性であり、使用条件下で有害な副作用および毒性を及ぼさないものでよい。医薬製剤は、例えば、Remington:The Science and Practice of Pharmacy、19th ed.、Mack Printing Company、Easton、Pennsylvania(1995)で見られる。
組成物は任意の投与経路、例えば経口、静脈内、経皮または皮下、経鼻、筋肉内または腹腔内投与のために調製することができる。担体または他の材料の正確な性質は、投与経路によって決まる。非経口投与のためには、非経口的に許容される水溶液を使用し、それは発熱物質を含まず、必要なpH、等張性および安定性を有する。当業者は適当な溶液を調製する能力が十分あり、多数の方法が文献で記載されている。薬剤送達方法の簡単な総説は、例えば、Langer、Science 249:1527〜1533(1990)でも見られる。
本発明との関連で、哺乳動物、特にヒトに投与する用量は、相応な時間枠の間哺乳動物で治療反応を実現するのに十分であるべきである。当業者は、投薬量が、患者の年齢、病状および体重、ならびに、疾患の段階/重症度を含めて、様々な因子によって決まることを理解するであろう。用量は、投与の経路(投与形態)、タイミングおよび頻度によっても決まる。経口投与の場合、投薬量は、例えば約0.01mg〜約10g、好ましくは約0.01mg〜約1000mg、より好ましくは約10mg〜約1000mg/日の化合物、またはその薬学的に許容される塩の対応量でよい。
化合物は、γ線照射の前、その間、またはその後に投与することができる。
TPPII活性の抑制についてどのように化合物をスクリーニングするべきかは、当業者に明白である。TPPIIタンパク質は第1段階で精製することができ、TPPIIに好ましい蛍光原基質を第2の段階で用いることができる。これは、TPPII活性を測定する効果的な方法になる。
特に高レベルの精製を達成する必要はなく、スクリーニング法で用いるのに十分な品質のTPPIIを得るために、従来の単純な技術を用いることができる。TPPIIの精製の一つの非限定的な例では、、100×106個の細胞(例えばEL−4細胞)を沈殿させ、ガラスビーズおよびホモジナイゼーション緩衝液(50mMトリス塩基pH7.5、250mMショ糖、5mM MgCl2、1mM DTT)中でボルテックスすることによって溶解した。細胞溶解物を分画遠心分離にかけた。最初に、細胞のホモジネートを14,000rpmで15分間遠心分離し、次に、上清を超遠心管へ移した。次に、試料を100,000×gで1時間超遠心分離し、上清(ほとんどの生化学文献ではサイトゾルと表される)を100,000×gで5時間遠心分離すると、高分子量サイトゾルタンパク質/タンパク質複合体が沈殿した。生じたペレットを50mMトリス塩基pH7.5、30%グリセロール、5mM MgCl2および1mM DTTに溶解し、1μgの高分子量タンパク質をペプチダーゼアッセイの酵素として用いた。
TPPIIの活性は、例えば基質AAF−AMC(Sigma、St.Louis、MO)を用いて試験することが可能である。これは、例えば、50mMトリ塩基pH7.5、5mM MgCl2および1mM DTTで構成される100μlの試験緩衝液中で、100μMの濃度で用いることができる。900μlの1%SDS溶液による希釈を用いて、反応を停止することが可能である。切断活性は、例えば、LS50B Luminescence Spectrometer(Perkin Elmer、Boston、MA)における、460nmでの発光で測定することができる。
本発明で使用する化合物は、(i)マウスへ腫瘍細胞を接種する段階と、(ii)前記マウスへγ線照射し、前記マウスへ化合物を投与する段階と、(iii)定期間隔で腫瘍サイズを測定する段階とを含むin vivoモデルで用いたときの、対照実験と比較して部分的または好ましくは完全な腫瘍退縮をもたらすものと定義することができる。γ線照射段階は、対照実験では省略される。腫瘍成長実験のさらなる詳細および実施例は、以下に記す。γ線照射処置の直後に化合物を注入することが好都合であることがわかったが、本発明は、この投与順序に限定されるものと理解すべきではない。
本発明で使用する化合物は、γ線照射と一緒に、例えば本明細書で記載される方法で使用されると、in vivoで部分的または好ましくは完全な腫瘍退縮をもたらす。
本発明で用いる化合物は、十分に血清安定性であり、即ちin vivoで、それらは所望の治療効果を発揮するのに十分長い期間、それらの同一性を保持する。
理論によって束縛されることを望まないが、本発明は、これから要約する添付図面に関して、下記のそれには限定されない実施例でさらに詳細に記載される。
[実施例]
[実施例]
用いた材料および方法は、以下の通りであった。
細胞および培養条件。EL−4は、C57BI/6マウス系統に由来する、ベンツピレンによって誘導されたリンパ腫細胞系統である。EL−4.wtおよびEL−4.TPPIIiは、pSUPERベクターでトランスフェクトされたEL−4細胞であり(Brummelkamp,TR、Bernards,R、Agami,R.A system for stable expression of short interfering RNAs in mammalian cells.Science 2002;296:550〜3)、空に対してTPPIIに対するsiRNAを含有する。HeLa細胞は、ヒト子宮頸癌細胞である。YAC−1は、A/Snマウス系統に由来する、モロニー白血病ウイルスによって誘導されたリンパ腫細胞系である。ALCは、C57BI/6マウス系統に由来する、放射線白血病ウイルスD−RadLVによって誘導されたT細胞リンパ腫である。DNA合成の測定のために、細胞を96穴プレートに播種し、3H−チミジンを16時間または36時間後に加え、6時間インキュベートした後に洗浄した。ストレスの誘導のために、細胞を500〜1000ラドでγ線照射したか、50%〜75%リン酸緩衝食塩水(PBS)中での増殖によって飢えさせ、37℃および5.3%CO2でインキュベートした。
酵素阻害剤、NLVSはキモトリプシンのペプチダーゼ活性を優先的に標的とするプロテアソーム阻害剤であって、生細胞でのプロテアソーム分解を効率的に抑制する。ブタビンダイドは文献で記載される(Rose,C、Vargas,F、Facchinetti,P、Bourgeat,P、Bambal,RB、Bishop,PBら.Characterization and inhibition of a cholecystokinin−inactivating serine peptidase.Nature 1996;380:403〜9)。Z−Gly−Leu−Ala−OH(Z−GLA−OH)は、TPPIIのそれと相同である活性部位を有する細菌酵素、サブチリシン(Bachem、Weil am Rhein、Germany)の阻害剤である。ワートマニン(Wortmannin)は、PIKK(PI3−キナーゼ関連)−ファミリーキナーゼ(Sigma、St.Louis、MO)の阻害剤である。すべての阻害剤はDMSOで溶解し、使用時まで−20℃で保存した。
タンパク質精製、ペプチダーゼアッセイおよびDNA断片の分析。100×106個の細胞を沈殿させ、ガラスビーズおよびホモジナイゼーション緩衝液(50mMトリス塩基pH7.5、250mMショ糖、5mM MgCl2、1mM DTT)中でボルテックスすることによって溶解した。細胞溶解物を分画遠心分離にかけ、そこでは、100,000×gで1時間の遠心分離からの上清(サイトゾル)を100,000×gの遠心分離に3〜5時間かけ、高分子量サイトゾルタンパク質/タンパク質複合体を沈殿させた。生じたペレットを50mMトリス塩基pH7.5、30%グリセロール、5mM MgCl2および1mM DTTに溶解し、1μgの高分子量タンパク質を、TPPII発現のためのペプチダーゼアッセイまたはウェスタンブロットの酵素として用いた。TPPIIの活性を試験するために、50mMトリ塩基pH7.5、5mM MgCl2および1mM DTTで構成される100μlの試験緩衝液中で、基質AAF−AMC(Sigma、St.Louis、MO)を100μMの濃度で用いた。切断活性は、LS50B Luminescence Spectrometer(Perkin Elmer、Boston、MA)における、460nmでの発光により測定した。DNA断片の分析のために、細胞を12穴プレートに106細胞数/mlで播種し、アポトーシス誘導剤として通常用いられるDNAトポイソメラーゼII阻害剤である、25μMのエトポシドに、飢餓(50%PBS)まで曝露させた。細胞を12穴プレートに106細胞/mlで播種し、示した時間、通常18〜24時間インキュベートした。EL−4対照および適応細胞からのDNAを標準のクロロホルム抽出によって精製し、アポトーシス細胞からDNAを検出するために、2.5μgのDNAを1.8%アガロースゲルに加えた。
プラスミドおよび遺伝子トランスフェクション。TPPII siRNAを発現するpSUPER(Brummelkamp,TR、Bernards,R、Agami,R.A system for stable expression of short interfering RNAs in mammalian cells.Science 2002;296:550〜3)プラスミドは、以下の通りに構築した。非リン酸化DNAオリゴマー(Thermo Hybaid、Ulm、Germany)を、3μg/μlの濃度に再懸濁した。各オリゴ対の1μlを48μlのアニーリング緩衝液(100mM KAc;30mM HEPES−KOH pH7.4;2mM MgAc)と混合し、95℃で4分間、70℃で10分間加熱し、次に、室温に徐々に冷却した。アニールしたオリゴマーの2μlを100ngのpSUPERプラスミド(BgIIIおよびHindIIIで消化した)と混合し、連結し、形質転換し、既述の通り、Amp/LPプレートに平板培養した(Brummelkamp,TR、Bernards,R、Agami,R.A system for stable expression of short interfering RNAs in mammalian cells.Science 2002;296:550〜3)。コロニーは、EcoRI−HindIII消化およびDNAシークエンシングにより、挿入断片の存在についてスクリーニングした。アニールさせたオリゴマー対は以下の通りであり、pSUPER−TPPIIiについては、フォワードプライマーは
5'GATCCCCGATGTATGGGAGAGGCCTTTCAAGAGAAGGCCTCTCCCATACATCTTTTTGGAAA-3'であり、リバースプライマーは
5'AGCTTTTCCAAAAAGATGTATGGGAGAGGCCTTCTCTTGAAAGGCCTCTCCCATACATCGGG-3'であった。
5'GATCCCCGATGTATGGGAGAGGCCTTTCAAGAGAAGGCCTCTCCCATACATCTTTTTGGAAA-3'であり、リバースプライマーは
5'AGCTTTTCCAAAAAGATGTATGGGAGAGGCCTTCTCTTGAAAGGCCTCTCCCATACATCGGG-3'であった。
安定したトランスフェクタントの生成のために、5×106個の細胞をPBSで洗浄し、次に、Bio−Rad遺伝子パルサー内の500μlのPBSに再懸濁し、10μgのDNAと960μFで250Vでパルスし、G418に対する耐性によって選択した。
抗体および抗血清。以下の分子を、指定した抗体によって検出した。AktをウサギAkt抗血清により(Cell Signaling Technology、Beverly、MA);ホスホ−Akt(Ser473)を193H2ウサギホスホ−Akt抗血清により(Cell Signaling Technology、Beverly、MA);γ−H2AXをウサギ抗γ−H2AXにより(Cell Signaling Technology、Beverly、MA);Mre11をポリクローナルウサギ抗ヒトMre11により(Cell Signaling Technology、Beverly、MA);p21をSX118により(R&D Systems、Minneapolis、MN);p53(R&D Systems、Minneapolis、MN);Rae−1をモノクローナルラット抗マウスRae−1、199215により(R&D Systems、Minneapolis、MN);XIAPをモノクローナルマウス抗ヒトXIAP、117320により(R&D Systems、Minneapolis、MN)。TPPIIの検出のために、ニワトリTPPII抗血清を用いた(Immunsystem、Uppsala、Sweden)。ウェスタンブロット法は、標準の技術によって実施した。タンパク質濃度は、BCAタンパク質検定用試薬(Pierce Chemical Co.)で測定した。特記されていない場合は、SDS/PAGEによる分離のために、レーンにつき5μgのタンパク質をロードした。
免疫組織化学。細胞を、サイトスピン(cytospin)を通してガラスカバースリップに付着させ、アセトン:メタノール(1:1)で1時間固定させ、次に、スライドをBSS緩衝液で1時間再水和した。第1の抗体を加え、BSSで短時間洗浄するまで1時間そのままにし、その後、二次コンジュゲート体(抗ウサギFITC)を加えて1時間インキュベートした。次に、スライドを洗浄し、Hoescht333258で30分間染色した。最後に、スライドにDABCOマウント緩衝液をマウントし、分析まで4℃に保った。
フローサイトメトリー。細胞表面Rae−1抗原の染色のため、0.5〜1.0×106個の細胞を、20μg/mlのRae−1モノクローナル抗体199215(R&D Systems、Minneapolis、MN)の50μlと一緒にインキュベートし、氷上で30分間インキュベートした。PBSで洗浄の後、ビオチン化ポリクローナルラビット抗ラットIg(Dako Cytomation、Glostrup、Denmark)およびストレプトアビジン−FITC(Pharmingen、San Diego、CA)と逐次的にインキュベートし、各段階の後にはPBSで洗浄した。蛍光は、FACScaliburによって定量化した。生細胞のフローサイトメトリー細胞選別は、細胞と2μg/mlのヨウ化プロピジウム(PI)との5分間のインキュベーション、ならびに、以降のFACSvantageによるPI+およびPI−集団への選別によって実施した。
腫瘍増殖実験。腫瘍細胞をPBSで洗浄し、接種につき200μlの量で再懸濁した。次に、細胞をマウス当たり106個右側脇腹に接種し、腫瘍の増殖は週2回の測定によって監視した。マウスの抗腫瘍治療の開始は、腫瘍増殖が各マウスで開始した時期に従い、ある程度個体別に調整した。抗腫瘍免疫応答を抑制するために、腫瘍接種前にマウスを4Gyで照射した。腫瘍体積は、(a1×a2×a3)/2(数字aiは腫瘍の直径、幅および深さを表す)に従って、腫瘍増殖を有するマウスの平均体積として計算した。第1の触診の時間は、マウスによって異なったが、一般増殖パターンはマウスのすべてで実質的に類似していた。ほとんどの図の中で、小さな腫瘍に対する治療効果、即ち完全な排除の存在をよりよく視覚化するために、対数尺度を用いる。in vivoでのTPPIIの抑制のために、体重1kgにつき、200μlのPBSに希釈した13.8mg(14μlの50mM溶液/マウス)のズブチリシン阻害剤Z−Gly−Leu−Ala−OH(Z−GLA−OH、Bachem、Weil am Rhein、Germany)で週2回腹腔内注射を行った。すべてのγ線照射は、全身照射であった。
レトロウイルス形質導入および移植。実施例7に関して、c−Mycの配列は、以下のプライマー5'ACGTGAATTCCACCATGCCCCTCAACGTTAGCTTCおよび3'TACGTCTCGAGCTTACGCACAAGAGTTCCGTAGを用いてPCRによってヒトcDNA(脳)から増幅し、レトロウイルス発現ベクターpMSCV−IRES−EYFPのEcoRI部位に挿入した。hBcI−xLはpLXIN−hBcI−xLから切り取り(Djerbi,M.、Darreh−Shori,T.、Zhivotovsky,B.およびGrandien,A.Characterization of the human FLICE−inhibitory protein locus and comparison of the anti−apoptotic activity of four different flip isoforms.Scand J Immunol.54、180〜9、2001)、pMSCV−IRES−EGFPのEcoRI部位に挿入した。レトロウイルス粒子の生成、造血幹細胞の濃縮および形質導入、ならびに移植は、既報の通りに実施した(Nyakeriga,A.M.、Djerbi,M.、Malinowski,M.M.およびGrandien,A.Simultaneous expression and detection of multiple retroviral constructs in haematopoietic cells after bone marrow transplantation.Scand J Immunol.61、545〜50、2005)。簡潔には、レトロウイルスベクターを、LipofectAMINE2000試薬(Invitrogen、Life Technologies Inc.、Paisley、UK)を用いてPhoenix−Ecoパッケジング細胞に一時的にトランスフェクトし、ウイルス粒子を含むウイルス上清を収集し、5−フルオロウラシル処理マウスの骨髄から得た系統陰性細胞に形質導入するのに用いた。その後これらの細胞を、致死的照射を受けたレシピエントマウスに注入した。移植後の7〜14日の間に、マウスは急性骨髄性白血病様疾患を発症した。そのようなマウスの脾臓からの細胞は、グルタミンおよびウシ胎仔血清を加えた通常のRPMI培地で、in vitroで増殖させることができた。
GFPおよびYFP発現の検出は、Cyan(商標)ADPサイトメーター(Dako、Glostrup、Denmark)を用いて実施したが、そこでは、488nmでの励起の後に、最初にシグナルを分離するために525nmロングパス二色性ミラーを使用し、次にEGFPの検出のために510/21nmバンドパスフィルタを、EYFPのために550/30nmバンドパスフィルタを用いた。データは、FlowJoソフトウェア(Tree Star,Inc.、San Carlos、CA)を用いて分析した。
略語リスト。ATM、毛細血管拡張性運動失調症変異;BRCT、BRCAのC末端リピート;NLVS、4−ヒドロキシ−5−ヨード−3−ニトロフェニルアセチル−Leu−Leu−Leu−ビニルスルホン;PI、ヨウ化プロピジウム;PIKK、ホスホイノシチド−3−OH−キナーゼ関連キナーゼ;TPPII、トリペプチジル−ペプチダーゼII;FA、3−(2−フリル)アクリロイル;YFP、黄色蛍光タンパク質、GFP、緑色蛍光タンパク質。
本明細書では、化学物質およびアミノ酸のために標準の略語を用いる。
略語 代替略語
A アラニン Ala
R アルギニン Arg
N アスパラギン Asn
D アスパラギン酸 Asp
C システイン Cys
E グルタミン酸 Glu
Q グルタミン Gln
G グリシン Gly
H ヒスチジン His
I イソロイシン Ile
L ロイシン Leu
K リジン Lys
M メチオニン Met
F フェニルアラニン Phe
P プロリン Pro
S セリン Ser
T スレオニン Thr
W トリプトファン Trp
Y チロシン Tyr
V バリン Val
本発明は、いくつかの非天然αアミノ酸も利用する。
A アラニン Ala
R アルギニン Arg
N アスパラギン Asn
D アスパラギン酸 Asp
C システイン Cys
E グルタミン酸 Glu
Q グルタミン Gln
G グリシン Gly
H ヒスチジン His
I イソロイシン Ile
L ロイシン Leu
K リジン Lys
M メチオニン Met
F フェニルアラニン Phe
P プロリン Pro
S セリン Ser
T スレオニン Thr
W トリプトファン Trp
Y チロシン Tyr
V バリン Val
本発明は、いくつかの非天然αアミノ酸も利用する。
略語 側鎖
Abu 2−アミノ酪酸 CH2CH3
Nva ノルバリン CH2CH2CH3
Nle ノルロイシン CH2CH2CH2CH3
tert−ブチルアラニン CH2C(CH3)3
α−メチルロイシン (CH3)(CH2C(CH3)CH3)
4,5−デヒドロ−ロイシン CH2C(=CH2)CH3
アロ−イソロイシン CH(CH3)CH2CH3
α−メチルバリン (CH3)CH(CH3)(CH3)
tert−ブチルグリシン C(CH3)3
2−アリルグリシン CH2CH=CH2
Orn オルニチン CH2CH2CH2NH2
Dab α,γ−ジアミノ酪酸 CH2CH2NH2
4,5−デヒドロ−リジン CH2CH=CHCH2NH2
Abu 2−アミノ酪酸 CH2CH3
Nva ノルバリン CH2CH2CH3
Nle ノルロイシン CH2CH2CH2CH3
tert−ブチルアラニン CH2C(CH3)3
α−メチルロイシン (CH3)(CH2C(CH3)CH3)
4,5−デヒドロ−ロイシン CH2C(=CH2)CH3
アロ−イソロイシン CH(CH3)CH2CH3
α−メチルバリン (CH3)CH(CH3)(CH3)
tert−ブチルグリシン C(CH3)3
2−アリルグリシン CH2CH=CH2
Orn オルニチン CH2CH2CH2NH2
Dab α,γ−ジアミノ酪酸 CH2CH2NH2
4,5−デヒドロ−リジン CH2CH=CHCH2NH2
γ線照射によって誘導される細胞周期停止は、TPPII発現に依存する。
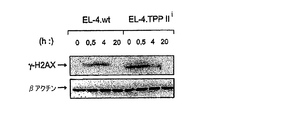
TPPII発現は数種類のストレスによって増加するので、これがPIKKによって制御されるのか否かを試験した。TPPII抗血清によるT細胞リンパ腫系EL−4のウェスタンブロット分析により、TPPII発現はγ線照射によって増加することがわかった。さらに、この増加は、PIKK阻害剤である1μMワートマニンで処理したγ線照射EL−4細胞では見られず、それはむしろTPPII発現を減少させた(図1A)。プロテアソーム阻害剤であるNLVSによる処理は、ワートマニン処理γ線照射EL−4細胞でTPPIIの下方制御を抑制し、TPPIIが、PIKKシグナル伝達の非存在下で、プロテアソームによって分解されることを示唆した(図1A)。TPPIIがPIKKによって媒介される細胞応答でなんらかの役割を果たすか否かをさらに研究するために、pSUPERベクターによってコードされる、TPPIIに対してsiRNAを発現する安定したEL−4トランスフェクタントを作製した(EL−4.TPPIIiと表される、[Brummelkamp,TR、Bernards,R、Agami,R.A system for stable expression of short interfering RNAs in mammalian cells.Science 2002;296:550〜3])。EL−4.TPPIIi細胞は、EL−4.wt細胞(空のpSUPERベクターでトランスフェクトされる、図1B)と比較して、TPPIIの発現および活性をともに抑制していた。PIKKファミリーメンバーのメンバーがシグナル伝達を制御する細胞ストレス応答を誘発するために、γ線照射(5Gy)を用いた。TPPIIは、以前、溶解性のサイトゾルペプチダーゼと報告された(Reits,E、Neijssen,J、Herberts,C、Benckhuijsen,W、Janssen,L、Drijfhout,JWら.A major role for TPPII in trimming proteasomal degradation products for MHC class I antigen presentation.Immunity 2004;20:495〜506)が、今回、γ線照射EL−4細胞(図1C)の核内へのTPPIIの急速な移行が発見された。これは、TPPIIの免疫組織化学的分析によって検出された通り、EL−4細胞のγ線照射曝露から1時間後にすでに明白であった。類似した応答が、ALCおよびYAC−1リンパ腫ならびにルイスの肺癌(LLC)細胞で観察された。
DNA損傷に応じてDNA合成を停止させるために、PIKKの活性化が必要とされる(Bakkenist,CJ、Kastan MB.Initiating cellular stress responses.Cell 2004;118:9〜17)(McKinnon,PJ.ATM and ataxia telangiectasia.EMBO Rep.2004;5:772〜6)。DNA合成が、γ線照射EL−4.wt対照で抑制されることが観察されたが、曝露後36時間まで、EL−4.TPPIIi細胞で高レベルのγ線照射耐性DNA合成が見られた(3H−チミジン取り込みで測定、図1D)。これらのデータは、TPPIIが、γ線照射に応じてEL−4細胞のDNA合成を停止させるために重要なことを示唆した。EL−4.TPPIIi細胞はγ線照射曝露の後、G2/Mでほとんど一様に停止したが、EL−4.wt対照細胞はG1およびG2/Mの停止を示し、EL−4.TPPIIi細胞でのG1/Sチェックポイントの非存在が示唆された(図1E)。しかし、γ−H2AX(Ser139−リン酸化H2AX、図1F)のウェスタンブロット法で測定されたように、γ線照射EL−4.TPPIIi細胞でDNA損傷の初期検出はまだ見られた。H2AXはATM活性化に応じてリン酸化され、それは、DNA修復巣の形成を誘発する(Bakkenist,CJ、Kastan MB.Initiating cellular stress responses.Cell 2004;118:9〜17)。ゆえに、TPPIIはγ線照射曝露後急速に核内に移行し、EL−4細胞でDNA合成を効率的に停止させるために必要であるが、H2AXのリン酸化のためには必要ではない。
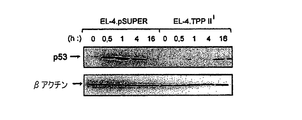
TPPII発現が抑制された細胞でのp53の安定化の失敗。
転写因子p53は多種のストレスに応じて細胞周期停止を開始し、その発現はPIKKによる直接リン酸化によって制御される。γ線照射EL−4.wt細胞の細胞溶解物のウェスタンブロット分析によって、p53レベルの上昇を認めたが、EL−4.TPPIIi細胞のそれらは低レベルを示した(図2A)。しかし、NLVSによる処理はγ線照射EL−4.TPPIIi細胞のp53発現を増加させ、p53はまだ合成されていたものの、EL−4.TPPIIi細胞のプロテアソームによって分解されたことが示唆される。p53の転写標的であるp21は、γ線照射曝露の後、EL−4.wt対照細胞と比較して、EL−4.TPPIIi細胞で弱く発現された(図2B)。さらに、TPPIIを安定して過剰発現するEL−4.pcDNA−TPPII細胞は、EL−4.pcDNA3細胞と比較して、γ線照射曝露後にp53レベルの増加を示した(Wang,EW、Kessler,BM、Borodovsky,A、Cravatt,BF、Bogyo,M、Ploegh,HLら.Integration of the ubiquitin−proteasome pathway with a cytosolic oligopeptidase activity.Proc Natl Acad Sci USA.2000;97:9990〜5)(図2C)。p53およびTPPIIが物理的に連結するか否かを試験するために、次に、p53のN末端に対する抗血清を用いて免疫共沈降実験を実施し、その後、TPPIIのウェスタンブロット分析を実施した。EL−4−pSUPER細胞の溶解物からのp53免疫沈降物において、γ線照射によって増加したTPPIIiレベルを検出した(図2D、上)。これは、EL−4.TPPIIi細胞からの溶解物および1μMワートマニンで処理したEL−4.wt細胞の溶解物では観察されなかった(図2D)。これらのデータは、TPPIIとp53との間の、γ線照射によって誘導された物理的リンクを裏付けるものであった。p53発現は、γ線照射YAC−1およびALCリンパ腫細胞でTPPII依存性であることも発見され、そこでは、pSUPER−TPPIIiの安定的発現の後、p53は実質的に検出不可能であった(図2E)。ルイスの肺癌(LLC)細胞でp53の発現を発見することはできなかった(図2E)。γ線照射への曝露より前にも本発明者らの対照腫瘍細胞系のいくつかでかなりのレベルのp53に気づいたが、これは、形質転換細胞でのしばしば上方制御されるDNA損傷応答と同調する現象である(Bartkova,J、Horejsi,Z、Koed,K、Kramer,A、Tort,F、Zieger,Kら.Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions.Nature 2005;434:907〜13)(Bartkova,J、Horejsi,Z、Koed,K、Kramer,A、Tort,F、Zieger,Kら.DNA damage response as a candidate anti−cancer barrier in early human tumorigenesis.Nature 2005;434:864〜70)。p53の効率的な安定化のために、TPPIIの発現が必要であると結論した。
TPPIIは、PIKKシグナル伝達に依存するいくつかの経路の活性化を制御する。
TPPII発現はp53の安定化のための要件であったので、本発明者らはEL−4.wt対EL−4.TPPIIi細胞でのそれらの状態を比較することによって、PIKKシグナル伝達に依存する他のストレス誘発性経路も試験した(Gasser,S、Orsulic,S、Brown,EJ、Raulet,DH.The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor.Nature 2005;436:1186〜90)(Viniegra,JG、Martinez,N、Modirassari,P、Losa,JH、Parada Cobo,C、Lobo,VJら.Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM.J Biol Chem.2005;280:4029〜36)(Feng,J、Park,J、Cron,P、Hess,D、Hemmings,BA.Identification of a PKB/Akt hydrophobic motif Ser−473 kinase as DNA−dependent protein kinase.J Biol Chem 2004;279:41189〜96)(Sarbassov,DD、Guertin,DA、Ali,SM、Sabatini,DM.Phosphorylation and regulation of Akt/PKB by the rictor−mTOR complex.Science 2005;307:1098〜101)。AktキナーゼのSer473リン酸化は、ATM、DNA−PKまたはmTORによるPIKKシグナル伝達を必要とするが、そのメカニズムの詳細は論争されている(Viniegra,JG、Martinez,N、Modirassari,P、Losa,JH、Parada Cobo,C、Lobo,VJら.Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM.J Biol Chem.2005;280:4029〜36)(Feng,J、Park,J、Cron,P、Hess,D、Hemmings,BA.Identification of a PKB/Akt hydrophobic motif Ser−473 kinase as DNA−dependent protein kinase.J Biol Chem 2004;279:41189〜96)(Sarbassov,DD、Guertin,DA、Ali,SM、Sabatini,DM.Phosphorylation and regulation of Akt/PKB by the rictor−mTOR complex.Science 2005;307:1098〜101)。EL−4.wt細胞の溶解物で、かなりのレベルのホスホ−Ser473−Aktを検出した。しかし、EL−4.TPPIIi細胞は非常に低いレベルのホスホ−Ser473−Aktを示し、一方、Aktの総発現は類似していた(図3A)。さらに、本発明者らはEL−4.pcDNA3対照細胞と比較して、EL−4.pcDNA3−TPPIIでAktのSer473リン酸化の増加が見られ、TPPII発現がAkt−Ser−473リン酸化を制御することをさらに裏付ける(図3B)。Aktキナーゼは細胞生存シグナルの形質導入にとって重要であり、多くの腫瘍で過度に活性化される。正常な培地(5%血清)では、EL−4.wtと比較して、EL−4.TPPIIi細胞は増殖速度の増加を示したが、死細胞の蓄積増加も示した(図3C)。さらに、血清濃度を1%まで低下させることによって、EL−4.wt細胞と比較してこの蓄積は促進され、細胞生存機構がTPPIIの非存在下で損なわれたことが示唆される(図3C)。さらに、EL−4.pcDNA3−TPPII細胞は、0.5%血清で限定的な増殖を達成することができたが、EL−4.pcDNA3細胞はそうでなかった(図3D)。これらの表現型は、TPPII発現が、in vitro培養中のAkt Ser473リン酸化および細胞生存にとって重要なことを示す。Aktキナーゼの直接の基質であるXIAP(Dan,HC、Sun,M、Kaneko,S、Feldman,RI、Nicosia,SV、Wang,HGら.Akt phosphorylation and stabilization of X−linked inhibitor of apoptosis protein(XIAP).J Biol Chem.2004;279:5405〜12)は、IAPファミリーの分子、即ち、腫瘍細胞で通常過剰発現する内因性のカスパーゼ阻害剤のメンバーである。TPPIIの上方制御は、EL−4.pcDNA3−TPPII細胞で、c−IAP−1およびXIAP分子の発現の増加を引き起こす。エトポシドによる処理により、XIAPの発現は、EL−4.TPPIIi細胞と比較して、EL−4.wt細胞でかなり高く、分解速度が遅いことがわかった(図3E)。さらに、ATMおよびATRキナーゼの活性化はNKG2Dリガンドの発現を媒介し、それによって、免疫系に対して進行中のDNA損傷応答を有する細胞を検出させる(Gasser,S、Orsulic,S、Brown,EJ、Raulet,DH.The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor.Nature 2005;436:1186〜90).フローサイトメトリー計測により、本発明者らはEL−4.wt細胞上でRae−1の発現を検出したが、EL−4.TPPIIi細胞上ではわずかな量のRae−1発現が検出された(図3F)。ALCリンパ腫細胞上でRae−1リガンドの発現を検出することはできなかったが、安定的pSUPER−TPPIIiトランスフェクタント(YAC−1.TPPIIi)が細胞表面でわずかなレベルのRae−1リガンドを発現するので、マウスYAC−1リンパ腫の分析もRae−1発現がTPPII発現依存性であることを示した(図3G)。これらのデータは、PIKKによって活性化されるいくつかのストレス誘発経路は、TPPII発現を必要とすることを示す。
TPPIIのBRCT様モチーフは、γ線照射に応じてp53の安定化を必要とした。
BRCAのC末端のリピート(BRCT)−ドメインは、DNA損傷シグナル伝達経路を制御するタンパク質内にしばしば含まれ、そこでそれらは、ATM基質との相互作用を制御する(Bork,P、Hofmann,K、Bucher,P、Neuwald,AF、Altschul,SF、Koonin,EV.A superfamily of conserved domains in DNA damage−responsive cell cycle checkpoint proteins.FASEB J.1997;11:68〜76)(Manke,IA、Lowery,DM、Nguyen,A、Yaffe,MB.BRCT repeats as phosphopeptide−binding modules involved in protein targeting.Science 2003;302:636〜9)(Yu,X、Chini,CC、He,M、Mer,G、Chen,J.The BRCT domain is a phospho−protein binding domain.Science 2003;302:639〜42)。すべてではないが最もBRCTモチーフの要件に合致する、位置725のGG−二重項を中心とするTPPIIの1領域を本発明者らは発見した(図4A)。本発明者らは多くのBRCT配列に存在する特徴的なGly−Gly二重項(*で表示、図4A)の部位特異的突然変異誘発を実施し、それを、本発明者らのpcDNA3−TPPIIベクターにおいてGly−Gluに変異させた。EL−4.TPPIIi細胞でこのプラスミドを発現させるために、位置725の突然変異(TPPIIwt/G725Eと表した)に加えて、3つのサイレント突然変異を、pSUPER−TPPIIiでコードされたsiRNAと相互作用するヌクレオチドの間のTPPIIの3’領域に挿入した(このプラスミドはTPPIIwtと表した)。TPPIIwtおよびTPPIIwt/G725E突然変異分子が、pSUPER−TPPIIiでコトランスフェクトしたEL−4細胞で安定して発現されることがわかった(図4B)。さらに、p53の発現を、γ線照射に曝露させたEL−4.TPPIIwtおよびEL−4.TPPIIwt/G725Eトランスフェクタント細胞で分析した。EL−4.TPPIIwt/G725E細胞が、EL−4.TPPIIwt対照細胞と比較して、p53のかなり低い発現を示すことがわかった(図4C)。さらに、γ線照射の存在下および非存在下の両方で、EL−4.TPPIIwt/G725E細胞の溶解物からのp53免疫沈降物でTPPIIを検出することができなかったが、TPPIIは、EL−4.TPPIIwt対照細胞を用いると検出された(図4D)。本発明者らはTPPIIは、DNA損傷シグナル伝達にとって重要なBRCT様ドメインを有すると結論した。
制御因子はDNA損傷部位に共存して、下流側応答を活性化させる(Al Rashid,ST、Dellaire,G、Cuddihy,A、Jalali,F、Vaid,M、Coackley,Cら.Evidence for the direct binding of phosphorylated p53 to sites of DNA breaks in vivo.Cancer Res.2005;65:10810〜21)(Lisby,M、Barlow,JH、Burgess,RC、Rothstein,R.Choreography of the DNA damage response:spatiotemporal relationships among checkpoint and repair proteins.Cell.2004;118:699〜713)。TPPII発現が抑制された細胞でのp53安定化の失敗の背後にある可能な理由は、p53をそのような部位に補充することができないということである。EL−4.wt対EL−4.TPPIIi細胞からのp53免疫沈降物での、DNA修復巣成分の存在を調べた。ウェスタンブロット法で測定されたように、本発明者らはEL−4.wtからのp53免疫沈降物でATMを検出したが、EL−4.TPPIIi細胞からは検出しなかった(図4E)。本発明者らはまたEL−4.wtにおいて、γ線照射後にp53結合タンパク質でDNA修復巣タンパク質53BP1およびMre11も検出されたが、EL−4.TPPIIi細胞では検出されなかった(図4F、G)。さらに、NLVS処理EL−4.TPPIIi細胞も、p53免疫沈降物でATM、53BP1およびMre11を示すことができなかった(図4E〜G)。p53およびATMがDNA修復巣成分と近接して見られる事実は、あるp53アイソフォームがこれらの巣に集積することと一致し、そこでは、それらはATMキナーゼと相互作用することができる(Al Rashid,ST、Dellaire,G、Cuddihy,A、Jalali,F、Vaid,M、Coackley,Cら.Evidence for the direct binding of phosphorylated p53 to sites of DNA breaks in vivo.Cancer Res.2005;65:10810〜21)。本発明者らのデータは、p53とATM、ならびにDNA修復巣成分53BP1およびMre11との間の物理的リンクが、TPPIIを必要とすることを裏付ける。
TPPII発現は、EL−4腫瘍のγ線照射耐性をin vivoで制御する。
PIKKは、新規癌療法の開発のための可能な標的分子である(Choudhury,A、Cuddihy,A、Bristow,RG.Radiation and new molecular agents part I:targeting ATM−ATR checkpoints,DNA repair,and the proteasome.Semin Radiat Oncol 2006;16:51〜8)。TPPII媒介性の成長調節がin vivo腫瘍増殖にとって重要であるか否かを調べるために、同系のC57BI/6マウスに、106個のEL−4.wt対照またはEL−4.TPPIIi細胞を接種した。本発明者らはEL−4.wtおよびEL−4.TPPIIi細胞の両方がほぼ同じ速度で腫瘍を確立し、成長することを見出し、単独で考慮すると、TPPIIがin vivoでのEL−4腫瘍の増殖にとって重要ではないことが示唆される(図5A、B、対照と表示されたパネル)。しかし、加えて、本発明者らはEL−4.wtまたはEL−4.TPPIIi細胞の腫瘍を有するマウスを、4Gy(400ラド)のγ線照射で2〜4回処理した。これは、γ線照射にもかかわらず成長し続けた106個のEL−4.wt細胞を接種した後の腫瘍サイズに、わずかな影響を及ぼすことを本発明者は見出した(図5A、γ線照射は矢印で示した)。対照的に、EL−4.TPPIIi細胞の腫瘍を有するマウスはγ線照射処理に応答して、定着腫瘍の完全な退縮がもたらされた(図5B)。これらのデータは、EL−4.ATMiまたはEL−4.TPPIIwt/G725E細胞の腫瘍で得られたものに類似していたが、その訳は、これらも、in vivoでγ線照射に耐えることができなかったからである(図5C、D)。該データは、腫瘍細胞のin vivoでのγ線照射感受性を増加させる標的としての、TPPIIを支持する。
トリ−ペプチド−ベースのTPPII阻害剤は、in vivoで腫瘍を放射線増感させる。
TPPIIは、細菌のズブチリシンと相同性の触媒ドメインを有する、ズブチリシン型のセリンペプチダーゼである(Tomkinson,B、Wernstedt,C、Hellman,U、Zetterqvist,O.Active site of tripeptidyl peptidase II from human erythrocytes is of the subtilisin type.Proc Natl Acad Sci USA.1987;84:7508〜12)。基質AAF−AMCのTPPII切断の抑制で観察されるように、約10nMのKi50を有するトリ−ペプチドズブチリシン阻害剤Z−Gly−Leu−Ala−OH(Z−GLA−OH)は、効率的にTPPIIを抑制し、その効率はブタビンダイド(7nMのKi50を有する)で観察されたよりもわずかに低いことがわかった(図6A)。さらに、Z−GLA−OHは、血清中で比較的安定していた。
in vivoでの腫瘍のγ線照射の間の触媒性TPPII抑制の影響を試験するために、確立されたEL−4腫瘍を有するC57BI/6マウスを、4Gyのγ線照射線量(1線量/週)および毎週2回のZ−GLA−OHによる注射(13.8mg/kg体重)に曝露させた。毎週の4Gyのγ線照射線量は、C57BI/6対照マウスの確立されたEL−4腫瘍の増殖に、わずかな影響を及ぼした。これに対して、Z−GLA−OHを注射後、3〜4回の400ラドの線量のγ線照射の後に、すべての試験マウスで完全な腫瘍退縮が観察された(図6B)。これらの腫瘍がもはや明瞭でなくなったとき、処理を中止したが、腫瘍の再成長は全観察期間(3カ月間)観察されなかった。Z−GLA−OH注射の存在下で、γ線照射線量の滴定(titration)も、3Gyの線量に曝露させたマウスでEL−4腫瘍の完全な退縮を示したが、より低い線量のγ線照射も腫瘍増殖を減少させ、一部の完全な排除を認めた(2Gyでは5匹中2匹のマウス、1Gyでは4匹中1匹、図6C)。Z−GLA−OH化合物の滴定は、6.9mg/kgのZ−GLA−OHを接種した後、ほとんどのマウス(4匹中3匹)でγ線照射に応じた完全な腫瘍排除を示したが、3.5mg/kg以下の用量は、腫瘍退縮という点で部分的効果(4匹中2匹、3Gyのγ線照射線量を用いた、図6C、右パネル)を示した。
γ線照射に対するin vivo耐性を含む、腫瘍治療抵抗性の背後にある一般的な理由の1つは、p53突然変異である(El−Deiry,WS.The role of p53 in chemosensitivity and radiosensitivity.Oncogene 2003;22:7486〜95)。p53突然変異腫瘍もTPPII阻害剤の存在下でγ線照射に応答するか否かを試験するために、本発明者らは同系のC57BI/6マウスに106個のルイスの肺癌(LLC)細胞を同様に接種した。LLC腫瘍は、4Gyの反復γ線照射線量に実質的に非感受性であることがわかり、Z−GLA−OH単独は(γ線照射がない場合)いかなる影響も及ぼさなかった(図6D)。対照的に、Z−GLA−OHを注射したマウスで、γ線照射に対する確立されたLLC腫瘍の完全な退縮が観察された(図6D)。保護された−ジペプチドZ−GL−OHは、LLC腫瘍のTPPII抑制および放射線増感に関して無効であることがわかったが、N末端の保護Z基は、in vivoでの抗腫瘍効果のために必ずしも厳密に必要ではなかった。TPPIIは、ヒトおよびマウスの間でアミノ酸レベルで96%の同一性を有する進化上保存された酵素であり、γ線照射に応じて、Z−GLA−OH処理SCIDマウスで、ヒトHeLa子宮頸癌細胞の強い腫瘍退縮も観察された(図6E)。低い線量のγ線照射(1.5Gy/線量)を用いたが、その訳は、SCIDマウスはかなり低い放射線抵抗性を有するからである。
毒性研究は、予備研究で、Z−GLA−OHが100mg/kgまでの用量で、単剤としてわずかな影響をin vivoで及ぼしたことを示す。さらに、本発明者らのマウスは、研究の後、長期間生存した。ここで用いたγ線照射プロトコルはもっぱら全身照射であったので、Z−GLA−OHが分布したすべての組織は、γ線照射およびZ−GLA−OHの組合せに曝露させられた。これは、併用処理の対処可能な毒性を示唆する。
新たに形質転換された白血病細胞のin vivoでの放射線増感。
原発腫瘍により類似する腫瘍細胞を確立するために、本発明者らはc−MycおよびBcI−xLをコードする2つの別々のベクター(pMSCV−BcI−xL−IRES−EGFPおよびpMSCV−c−Myc−IRES−EYFP)を有するレトロウイルス発現系を用いた。DBA/2骨髄細胞を、レトロウイルスによりこれらのBcI−xLおよびc−Mycを発現するベクターに感染させ、γ線照射同系マウスに移植した。ベクターによってコードされた緑色蛍光タンパク質(GFP)対黄色蛍光タンパク質(YFP)は、レトロウイルス遺伝子発現の監視を可能にした(Nyakeriga,A.M.、Djerbi,M.、Malinowski,M.M.およびGrandien,A.Simultaneous expression and detection of multiple retroviral constructs in haematopoietic cells after bone marrow transplantation.Scand J Immunol.61、545〜50、2005)。移植から7〜14日後、本発明者らは脾臓および骨髄にYFP+/GFP+骨髄(CD11b+Gr1+)芽細胞の大量の蓄積を観察した(脾臓について示す、図7A)。これらのDBA/2−c−Myc/BcI−xL細胞を同系のDBA/2マウスの皮下に接種し、約3週後に、さらなる2〜3週内に1000mm3を超えるサイズに成長した明瞭な腫瘍を観察した(図7B)。DBA/2−c−Myc/BcI−xL細胞を接種したすべてのマウスにおいて、固定臓器の組織学的分析によって観察されたように、肝臓への腫瘍拡散を認めた(図7H)。これらの悪性細胞は、原発腫瘍からの細胞を対照として用いて、脾臓、肺および肝臓のYFP+/GFP+細胞を示すフローサイトメトリーによっても検出された(図7C〜G)。γ線照射(4Gy/線量、1線量/週)による処理により、わずかに減少した増殖が観察されたが、DBA/2−c−Myc/BcI−xL腫瘍はまだ1週未満の遅れで1000mm3を超えるサイズに到達し、肝転移も存在した(図7B)。対照的に、Z−GLA−OH(13.8mg/kg体重)を投与された確立したDBA/2−c−Myc/BcI−xL腫瘍を有するマウスは、4Gy線量のγ線照射に応じて完全な腫瘍退縮を示した(図7B)。さらに、これらのZ−GLA−OH処理マウスの肺、脾臓および肝臓で、腫瘍細胞を発見することはできなかった(図7F、G、J)。Z−GLA−OHだけを投与されたマウスでは腫瘍サイズの減少が観察されなかったので、この治療応答のためにγ線照射が必要とされた(図7B)。これらのデータは、Z−GLA−OHから観察される放射線増感効果が特定の腫瘍の欠陥に依存する可能性は低いことを裏付けるが、増殖およびアポトーシスを脱制御する単純な2ヒット戦略によって新たに形質転換された細胞で観察することができる。
ジ−ペプチド、トリ−ペプチドおよび誘導体のin vitro試験。
表1は、任意であるが相対的である蛍光定量的ユニットにおける、AAF−AMC(H−Ala−Ala−7−アミド−4−メチルクマリン)のいくつかの濃度の化合物による切断の抑制に関するin vitroデータを含む。試験した大部分の化合物で、多少の有益効果が見られる。
TPPIIタンパク質を濃縮し、次に、TPPIIに好ましい蛍光原基質AAF−AMCを用いた。100×106個の細胞を沈殿させ、ガラスビーズおよびホモジナイゼーション緩衝液(50mMトリス塩基pH7.5、250mMショ糖、5mM MgCl2、1mM DTT)中でボルテックスすることによって溶解した。細胞溶解物を、分画遠心分離にかけた。最初に、細胞のホモジネートを14,000rpmで15分間遠心分離し、次に、上清を超遠心管へ移した。次に、試料を100,000×gで1時間超遠心分離し、上清(ほとんどの生化学文献ではサイトゾルと表される)を100,000×gで5時間遠心分離すると、高分子量サイトゾルタンパク質/タンパク質複合体が沈殿した。生じたペレットを50mMトリス塩基pH7.5、30%グリセロール、5mM MgCl2および1mM DTTに溶解し、1μgの高分子量タンパク質をペプチダーゼアッセイの酵素として用いた。
TPPIIの活性を試験するために、50mMトリベースpH7.5、5mM MgCl2および1mM DTTで構成される100μlの試験緩衝液中で、基質およびAAF−AMC(Sigma、St.Louis、MO)を100μMの濃度で用いた。反応を停止するために、900μlの1%SDS溶液による希釈を用いた。切断活性は、LS50B Luminescence Spectrometer(Perkin Elmer、Boston、MA)における、460nmでの発光により測定した。
FA=3−(2−フリル)アクリロイル;PBS=リン酸緩衝食塩水。各化合物名の最初のテキスト(Z、FA、H、その他)は、N末端の置換基である;Hは、N末端が遊離のNH2であることを示す。各化合物名の終わりのテキスト(OH、NBu、その他)は、C末端の置換基である;OHは、C末端が遊離のCO2Hであることを示す。
ジ−ペプチド、トリ−ペプチドおよび誘導体のin vivo試験。
表2は、LLC(ルイスの肺癌)を有する4匹のマウスの群における、mm3で表した腫瘍体積を示すin vivoデータを含む。腫瘍体積が1000mm3を超えたとき、マウスを屠殺した。一部のマウスには、化合物を単独で投与し、他には、さらに放射線を投与した。7、10、14、18および21日後に、マウスに化合物を投与し、一部にはγ線照射(400ラド)も投与した。照射と併用すると、一部の化合物は優れた結果を示した。ジペプチド誘導体Z−GL−OHがin vivoと同様にin vitroで性能が劣る事実は、in vitroの結果をin vivoでの結果に外挿することができるという理論を支持する。
Z−GLA−OHのさらなるin vivo試験
表3は、上記のEL−4腫瘍モデルに従う、7〜8匹のマウスの群におけるmm3で表した腫瘍体積を示す、さらなるin vivoデータを含む。0日目に、1,000,000個のEL−4リンパ腫細胞を皮下に接種した。22日後まで、明瞭な腫瘍は観察されなかった。各処理時(週2回)、明瞭な腫瘍を有するマウスに、400ラド照射を単独で、または14μlのZ−GLA−OHの50mM溶液と併用して投与した。明瞭な腫瘍をもたないマウスは処理せず、即ち、腫瘍が排除されたマウスにおいては処理を中止し、マウスを経過観察した。表3は、優れた結果を、即ち、単なる腫瘍増殖の停止、容積の減少および腫瘍増殖の遅れではなく、確立腫瘍の完全な排除を示す。
表3は、上記のEL−4腫瘍モデルに従う、7〜8匹のマウスの群におけるmm3で表した腫瘍体積を示す、さらなるin vivoデータを含む。0日目に、1,000,000個のEL−4リンパ腫細胞を皮下に接種した。22日後まで、明瞭な腫瘍は観察されなかった。各処理時(週2回)、明瞭な腫瘍を有するマウスに、400ラド照射を単独で、または14μlのZ−GLA−OHの50mM溶液と併用して投与した。明瞭な腫瘍をもたないマウスは処理せず、即ち、腫瘍が排除されたマウスにおいては処理を中止し、マウスを経過観察した。表3は、優れた結果を、即ち、単なる腫瘍増殖の停止、容積の減少および腫瘍増殖の遅れではなく、確立腫瘍の完全な排除を示す。
すべてのマウスは、0日目に400ラド(1Gy=100ラド)でγ線照射したが、これは、腫瘍受容を向上させる標準手順である。化合物は腹腔内に接種したが、腫瘍は常に皮下に接種した。
Z−GLA−OHと同じ方法で、GPG−NH2およびZ−GPG−NH2を試験した。これらを、腫瘍担持マウスに13.8mg/kgで週2回注射し、in vivoでγ線照射増感を媒介するそれらの能力について、Z−GLA−OHと比較した。GPG−NH2およびZ−GPG−NH2は、γ線照射後、確立EL−4腫瘍の完全な退縮を媒介することがわかった。
TPPIIは、Mre11巣形成のために必要とされる
図1Cで示し、実施例1で述べるように、TPPIIはγ線照射細胞の核に迅速に移行する。さらなる免疫細胞化学的実験の結果を、図9に示す。TPPIIは巣を形成しないようであり、代わりに、点状の外観を示したようである(図9、抑制されたTPPII発現、LLC、ALCおよびYAC−1を有する細胞について示す)。TPPII発現が抑制された細胞が、γ線照射曝露後にMre11巣を集めることができなかったこの失敗は、本発明でのTPPII阻害剤の使用を、さらに裏付ける。
図1Cで示し、実施例1で述べるように、TPPIIはγ線照射細胞の核に迅速に移行する。さらなる免疫細胞化学的実験の結果を、図9に示す。TPPIIは巣を形成しないようであり、代わりに、点状の外観を示したようである(図9、抑制されたTPPII発現、LLC、ALCおよびYAC−1を有する細胞について示す)。TPPII発現が抑制された細胞が、γ線照射曝露後にMre11巣を集めることができなかったこの失敗は、本発明でのTPPII阻害剤の使用を、さらに裏付ける。
Claims (43)
- γ線照射癌療法の効力を強化するか、腫瘍細胞のin vivoγ線照射感受性を増加させるための使用のための化合物であって、TPPII阻害剤である化合物。
- 前記化合物が、式(i)から選択されるか、その薬学的に許容される塩である、請求項1に記載の使用のための化合物
(i)RN1RN2N−A1−A2−A3−CO−RC1
[式中、A1、A2およびA3は、標準の1文字略語または名称に従う以下の定義を有するアミノ酸残基であり:
A1は、G、A、V、L、I、P、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシンであり、
A2は、G、A、V、L、I、P、F、W、C、S、K、R、2−アミノ酪酸、ノルバリン、ノルロイシン、tert−ブチルアラニン、α−メチルロイシン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、α−メチルバリン、tert−ブチルグリシン、2−アリルグリシン、オルニチンまたはα,γ−ジアミノ酪酸であり、
A3は、G、A、V、L、I、P、F、W、D、E、Y、2−アミノ酪酸、ノルバリンまたはtert−ブチルグリシンであり、
RN1およびRN2は、それぞれペプチドのN末端に結合し、同じであるかまたは異なり、それぞれ独立して
RN3、
(リンカー1)−RN3、
CO−(リンカー1)−RN3、
CO−O−(リンカー1)−RN3、
CO−N−((リンカー1)−RN3)RN4、または
SO2−(リンカー1)−RN3
であり、(リンカー1)は存在しなくてもよく、即ち単結合でもよく、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであることができ、
RN3およびRN4は、同じであるかまたは異なり、水素または以下の必要に応じて置換された基:
飽和または不飽和で分枝または非分枝C1〜6アルキル、
飽和または不飽和で分枝または非分枝C3〜12シクロアルキル、
ベンジル、
フェニル、
ナフチル、
単環式または二環式C1〜10ヘテロアリール、または
非芳香族C1〜10ヘテロシクリル
のいずれかであり、RN3および/またはRN4には、0、1または2個(同じかまたは異なる)の必要に応じた置換基があり得、該置換基は、
ヒドロキシ−、
チオ−、
アミノ−、
カルボン酸、
飽和または不飽和で分枝または非分枝C1〜6アルキルオキシ、
飽和または不飽和で分枝または非分枝C3〜12シクロアルキル、
N−、O−またはS−アセチル、
カルボン酸の飽和または不飽和で分枝または非分枝C1〜6アルキルエステル、
カルボン酸の飽和または不飽和で分枝または非分枝C3〜12シクロアルキルエステル
フェニル、
単環式または二環式C1〜10ヘテロアリール、
非芳香族C1〜10ヘテロシクリル、または
ハロゲン
であってもよく、RC1は、トリペプチドのC末端に結合し、
O−RC2、
O−(リンカー2)−RC2、
N((リンカー2)RC2)RC3、または
N(リンカー2)RC2−NRC3RC4、
であり、(リンカー2)は存在しない、即ち単結合、またはC1〜6アルキルもしくはC2〜4アルケニル、好ましくは単結合、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであってもよく、
RC2、RC3およびRC4は、同じであるかまたは異なり、水素または以下の必要に応じて置換された基:
飽和または不飽和で分枝または非分枝C1〜6アルキル、
飽和または不飽和で分枝または非分枝C3〜12シクロアルキル、
ベンジル、
フェニル、
ナフチル、
単環式または二環式C1〜10ヘテロアリール、または
非芳香族C1〜10ヘテロシクリル
のいずれかであり、RC2および/またはRC3および/またはRC4のそれぞれには、0、1または2個(同じかまたは異なる)の必要に応じた置換基があり得、該置換基は、
ヒドロキシ−、
チオ−、
アミノ−、
カルボン酸、
飽和または不飽和で分枝または非分枝C1〜6アルキルオキシ、
飽和または不飽和で分枝または非分枝C3〜12シクロアルキル、
N−、O−またはS−アセチル、
カルボン酸の飽和または不飽和で分枝または非分枝C1〜6アルキルエステル、
カルボン酸の飽和または不飽和で分枝または非分枝C3〜12シクロアルキルエステル
フェニル、
ハロゲン、
単環式または二環式C1〜10ヘテロアリール、または
非芳香族C1〜10ヘテロシクリル
の1つまたは複数でよい]。 - 式(i)の前記化合物が、
RN1は水素であり、
RN2は、水素、C(=O)−O−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換され、またはC(=O)−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換され、
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキル−フェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキルフェニルである、請求項2に記載の使用のための化合物。 - 式(i)の前記化合物が、
A1は、G、Aまたは2−アミノ酪酸であり、
A2は、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、2−アリルグリシン、P、2−アミノ酪酸、α−メチルロイシン、α−メチルバリンまたはtert−ブチルグリシンであり、
A3は、G、A、V、P、2−アミノ酪酸またはノルバリンであり、
RN1はHであり、
RN2は、水素、C(=O)−O−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換され、またはC(=O)−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換され、
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキルフェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキルフェニルである、請求項3に記載の使用のための化合物。 - 式(i)の前記化合物が、
A1は、G、Aまたは2−アミノ酪酸であり、
A2は、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシンまたは2−アリルグリシンであり、
A3は、G、A、V、P、2−アミノ酪酸またはノルバリンであり、
RN1はHであり、
RN2は、水素、C(=O)−O−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換され、またはC(=O)−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換され、
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキル−フェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキル−フェニルである、請求項4に記載の使用のための化合物。 - 式(i)の前記化合物が、
A1は、GまたはAであり、
A2は、L、Iまたはノルロイシンであり、
A3は、GまたはAであり、
RN1は水素であり、
RN2は、水素、C(=O)−O−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、またはC(=O)−飽和もしくは不飽和で分枝もしくは非分枝C1〜4アルキルであり、フェニルもしくは2−フリルで必要に応じて置換されており、
RC1は、OH、O−C1〜6アルキル、O−C1〜6アルキル−フェニル、NH−C1〜6アルキルまたはNH−C1〜6アルキル−フェニルである、請求項5に記載の使用のための化合物。 - RN1は水素であり、
RN2は、水素、C(=O)−OCH2PhまたはC(=O)−CH=CH−(2−フリル)であり、
RC1は、OH、O−C1〜6アルキルまたはNH−C1〜6アルキルである、
請求項2から6のいずれかに記載の使用のための化合物。 - 式(i)の前記化合物が、Z−GLA−OH、Bn−GLA−OH、FA−GLA−OHまたはH−GLA−OHである、請求項7に記載の使用のための化合物。
- 式(i)の前記化合物がZ−GLA−OHである、請求項8に記載の使用のための化合物。
- A1が、G、Aまたは2−アミノ酪酸である、請求項2に記載の使用のための化合物。
- A1がGまたはAである、請求項10に記載の使用のための化合物。
- A2が、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、2−アリルグリシン、P、K、2−アミノ酪酸、α−メチルロイシン、α−メチルバリンまたはtert−ブチルグリシンである、請求項2、10または11のいずれかに記載の使用のための化合物。
- A2が、L、I、ノルロイシン、V、ノルバリン、tert−ブチルアラニン、4,5−デヒドロ−ロイシン、アロ−イソロイシン、2−アリルグリシン、PまたはKである、請求項12に記載の使用のための化合物。
- A2がL、I、ノルロイシン、PまたはKである、請求項13に記載の使用のための化合物。
- A2がLまたはPである、請求項14に記載の使用のための化合物。
- A2がPである、請求項15に記載の使用のための化合物。
- A3が、G、A、V、P、2−アミノ酪酸またはノルバリンである、請求項2または10から16のいずれかに記載の使用のための化合物。
- A3がGまたはAである、請求項17に記載の使用のための化合物。
- RN1が水素である、請求項2または10から18のいずれかに記載の使用のための化合物。
- RN2が、
RN3、
(リンカー1)−RN3、
CO−(リンカー1)−RN3、または
CO−O−(リンカー1)−RN3
であり、上式で、
(リンカー1)は存在しなくてもよく、即ち単結合でもよく、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであることができ、
RN3は、水素または以下の置換されていない基
飽和または不飽和で分枝または非分枝C1〜4アルキル、
ベンジル、
フェニル、または
単環式ヘテロアリール
のいずれかである、請求項2または10から19のいずれかに記載の使用のための化合物。 - RN2が、水素、ベンジルオキシカルボニル、ベンジル、ベンゾイル、tert−ブチルオキシカルボニル、9−フルオレニルメトキシカルボニルまたはFAである、請求項20に記載の使用のための化合物。
- RN2が、水素、ベンジルオキシカルボニルまたはFAである、請求項21に記載の使用のための化合物。
- RC1が、
O−RC2、
O−(リンカー2)−RC2、または
NH−(リンカー2)RC2
であり、上式で、
(リンカー2)は存在しない、即ち単結合、C1〜6アルキルもしくはC2〜4アルケニル、好ましくは単結合、またはCH2、CH2CH2、CH2CH2CH2、CH2CH2CH2CH2もしくはCH=CHであってもよく、
RC2は、水素または以下の置換されていない基:
飽和または不飽和で分枝または非分枝C1〜5アルキル、
ベンジル、
フェニル、または
単環式C1〜10ヘテロアリール
のいずれかである、請求項2または10から22のいずれかに記載の使用のための化合物。 - RC1が、OH、O−C1〜6アルキル、O−C1〜6アルキル−フェニル、NH2、NH−C1〜6アルキルまたはNH−C1〜6アルキル−フェニルである、請求項23に記載の使用のための化合物。
- RC1が、OH、O−C1〜6アルキル、NH2またはNH−C1〜6アルキルである、請求項24に記載の使用のための化合物。
- RC1がOHまたはNH2である、請求項25に記載の使用のための化合物。
- RC1がNH2である、請求項26に記載の使用のための化合物。
- 前記化合物が、GPG−NH2、Z−GPG−NH2、Bn−GPG−NH2、FA−GPG−NH2、GPG−OH、Z−GPG−OH、Bn−GPG−OHまたはFA−GPG−OHである、請求項2に記載の使用のための化合物。
- 前記化合物がGPG−NH2である、請求項28に記載の使用のための化合物。
- 前記化合物が、ALG−NH2、Z−ALG−NH2、Bn−ALG−NH2、FA−ALG−NH2、ALG−OH、Z−ALG−OH、Bn−ALG−OHまたはFA−ALG−OHである、請求項2に記載の使用のための化合物。
- 前記化合物がALG−NH2である、請求項30に記載の使用のための化合物。
- A3が、F、W、D、EまたはYではない、請求項2から31のいずれかに記載の使用のための化合物。
- A3がPではない、請求項2から32のいずれかに記載の使用のための化合物。
- A3がEではない、請求項2から33のいずれかに記載の使用のための化合物。
- γ線照射癌療法の効力を強化するか、腫瘍細胞のin vivoγ線照射感受性を増加させる方法であって、それを必要とする患者に請求項1から34のいずれかに記載の化合物の治療有効量を投与することを含む、方法。
- γ線照射癌療法の効力を強化するか、腫瘍細胞のin vivoγ線照射感受性を増加させる医薬の製造での化合物の使用であって、前記化合物が請求項1から34のいずれかに記載のものである使用。
- γ線照射癌療法の効力を強化するか、腫瘍細胞のin vivoγ線照射感受性を増加させるのに適当な化合物を同定する方法であって、TPPIIをスクリーニングする化合物と接触させ、その化合物がTPPIIの活性を阻害するか否かを同定することを含む、方法。
- 請求項2から34のいずれかに記載の式(i)の構造を有する化合物、および薬学的に許容される希釈剤または担体を含む医薬組成物。
- 請求項2から34のいずれかに記載の構造を有する化合物、および薬学的に許容される希釈剤または担体を含む医薬組成物であって、前記化合物が、シンナモイル−IFP−エチルアミド、GPE−OH、GGF−OH、GVF−OH、AAA−OHまたはIPI−OHではない医薬組成物。
- A3がプロリンではない、請求項38または39に記載の医薬組成物。
- 化合物がGPE−OHではない、請求項38から40のいずれかに記載の医薬組成物。
- RC1がNH2ではない、請求項38から41のいずれかに記載の医薬組成物。
- 医薬として使用するための、請求項38から42のいずれかに記載の化合物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US75908806P | 2006-01-13 | 2006-01-13 | |
| US60/759,088 | 2006-01-13 | ||
| PCT/EP2007/050363 WO2007080194A2 (en) | 2006-01-13 | 2007-01-15 | Use of tpp ii inhibitors in combination with gamma-irradiation for the treatment of cancer |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009523156A true JP2009523156A (ja) | 2009-06-18 |
| JP2009523156A5 JP2009523156A5 (ja) | 2010-02-25 |
Family
ID=36609604
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008549887A Pending JP2009523157A (ja) | 2006-01-13 | 2007-01-15 | 虚血および神経変性の処置のための化合物 |
| JP2008549886A Pending JP2009523156A (ja) | 2006-01-13 | 2007-01-15 | 癌の処置のためのγ線照射と組み合わせた化合物の使用 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008549887A Pending JP2009523157A (ja) | 2006-01-13 | 2007-01-15 | 虚血および神経変性の処置のための化合物 |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US20100168038A1 (ja) |
| EP (2) | EP1971357A2 (ja) |
| JP (2) | JP2009523157A (ja) |
| KR (1) | KR20080085035A (ja) |
| CN (1) | CN101370509A (ja) |
| AU (1) | AU2007204314A1 (ja) |
| CA (1) | CA2636533A1 (ja) |
| WO (2) | WO2007088099A2 (ja) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2007355462A1 (en) * | 2007-06-25 | 2008-12-31 | Oncoreg Ab | TPP II inhibitors for use in the treatment of autoimmune and inflammatory diseases and transplant rejection |
| WO2009000297A1 (en) * | 2007-06-25 | 2008-12-31 | Oncoreg Ab | Tpp ii inhibitors for use in combination with chemotherapy for the treatment of cancer |
| ES2552764T3 (es) * | 2007-10-15 | 2015-12-02 | The Salk Institute For Biological Studies | Métodos para el tratamiento de varias enfermedades y afecciones, y compuestos útiles para los mismos |
| MY159958A (en) | 2009-12-18 | 2017-02-15 | Idenix Pharmaceuticals Inc | 5,5-fused arylene or heteroarylene hepatitis c virus inhibitors |
| CN103189067A (zh) * | 2010-06-16 | 2013-07-03 | 密执安大学评议会 | Wdr5与其结合配偶体的相互作用的抑制及治疗方法 |
| ES2703499T3 (es) * | 2010-12-22 | 2019-03-11 | Salk Inst Biological Studies | Péptidos antagonistas de CRF cíclicos |
| SG11201602161XA (en) | 2013-09-23 | 2016-04-28 | Wolff August Gmbh & Co Kg Arzneimittel Dr | Anti-inflammatory tripeptides |
| EP3171941B1 (en) * | 2014-07-24 | 2021-03-24 | Naurex Inc. | N-methyl-d-aspartate receptor modulators and methods of making and using same |
| CN111603560A (zh) * | 2020-06-22 | 2020-09-01 | 泉州台商投资区秋鑫茶业有限公司 | 茶叶γ-氨基丁酸在肿瘤放射治疗上的应用 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06504035A (ja) * | 1990-08-22 | 1994-05-12 | トリペプ アクチボラゲット | ヒト免疫不全ウィルス感染を阻止するペプチドとその使用法 |
| JP2003506410A (ja) * | 1999-08-09 | 2003-02-18 | トリペップ アクチ ボラゲット | ウィルス感染性を阻止するペプチドおよびその使用方法 |
| JP2004531480A (ja) * | 2001-02-05 | 2004-10-14 | ノイロテル・アーゲー | 神経変性疾患の処置のためのトリペプチド誘導体 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5627035A (en) * | 1990-08-22 | 1997-05-06 | Syntello Vaccine Development Ab | Peptides that block human immunodeficiency virus and methods of use thereof |
| EP1042288B1 (en) * | 1997-12-23 | 2007-11-07 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | Tripeptidyl peptidase inhibitors |
| EP1436317A1 (en) * | 2001-09-19 | 2004-07-14 | Tripep Ab | Molecules that block viral infectivity and methods of use thereof |
| US20040097422A1 (en) * | 2002-06-14 | 2004-05-20 | Karl Munger | Methods of use for tripeptidyl peptidase II inhibitors as anticancer agents |
| WO2005073397A1 (en) * | 2004-01-31 | 2005-08-11 | Bayer Healthcare Ag | Diagnostics and therapeutics for diseases associated with tripeptidyl-peptidase 2(tpp2) |
-
2007
- 2007-01-15 CN CNA200780002382XA patent/CN101370509A/zh active Pending
- 2007-01-15 WO PCT/EP2007/050364 patent/WO2007088099A2/en active Application Filing
- 2007-01-15 AU AU2007204314A patent/AU2007204314A1/en not_active Abandoned
- 2007-01-15 EP EP07703879A patent/EP1971357A2/en not_active Withdrawn
- 2007-01-15 EP EP07726197A patent/EP2160196A2/en not_active Withdrawn
- 2007-01-15 KR KR1020087017003A patent/KR20080085035A/ko not_active Application Discontinuation
- 2007-01-15 WO PCT/EP2007/050363 patent/WO2007080194A2/en active Application Filing
- 2007-01-15 JP JP2008549887A patent/JP2009523157A/ja active Pending
- 2007-01-15 CA CA002636533A patent/CA2636533A1/en not_active Abandoned
- 2007-01-15 JP JP2008549886A patent/JP2009523156A/ja active Pending
- 2007-01-15 US US12/160,787 patent/US20100168038A1/en not_active Abandoned
- 2007-01-15 US US12/160,786 patent/US20090227521A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH06504035A (ja) * | 1990-08-22 | 1994-05-12 | トリペプ アクチボラゲット | ヒト免疫不全ウィルス感染を阻止するペプチドとその使用法 |
| JP2003506410A (ja) * | 1999-08-09 | 2003-02-18 | トリペップ アクチ ボラゲット | ウィルス感染性を阻止するペプチドおよびその使用方法 |
| JP2004531480A (ja) * | 2001-02-05 | 2004-10-14 | ノイロテル・アーゲー | 神経変性疾患の処置のためのトリペプチド誘導体 |
Non-Patent Citations (3)
| Title |
|---|
| JPN6012029641; J. Med. Chem. Vol.43, No.4, 2000, 664-674 * |
| JPN6012029642; Tetrahedron Vol.50, No.21, 1994, p.6333-46 * |
| JPN6012029643; Bulletin of Experimental Biology and Medicine Vol.136, No.4, 2003, p.319-322 * |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2007204314A1 (en) | 2007-07-19 |
| WO2007080194A3 (en) | 2008-02-14 |
| US20100168038A1 (en) | 2010-07-01 |
| KR20080085035A (ko) | 2008-09-22 |
| US20090227521A1 (en) | 2009-09-10 |
| WO2007080194A2 (en) | 2007-07-19 |
| JP2009523157A (ja) | 2009-06-18 |
| CA2636533A1 (en) | 2007-07-19 |
| CN101370509A (zh) | 2009-02-18 |
| EP1971357A2 (en) | 2008-09-24 |
| WO2007088099A3 (en) | 2007-11-08 |
| WO2007088099A2 (en) | 2007-08-09 |
| EP2160196A2 (en) | 2010-03-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2009523156A (ja) | 癌の処置のためのγ線照射と組み合わせた化合物の使用 | |
| JP6030622B2 (ja) | 癌の阻害剤としてのmuc−1細胞質ドメインペプチド | |
| CN105377288A (zh) | Xbp1、cd138和cs1肽、包括所述肽的药物组合物及使用所述肽和组合物的方法 | |
| US20140186381A1 (en) | Epitope peptides derived from vascular endothelial growth factor receptor 1 and vaccines containing these peptides | |
| US20120121595A1 (en) | FRAGMENT OF SECRETED HEAT SHOCK PROTEIN-90ALPHA (Hsp90ALPHA) AS VACCINES OR EPITOPE FOR MONOCLONAL ANTIBODY DRUGS OR TARGET FOR SMALL MOLECULE DRUGS AGAINST A RANGE OF SOLID HUMAN TUMORS | |
| AU2016230125A1 (en) | Peptide derived from GPC3, pharmaceutical composition for treatment or prevention of cancer using same, immunity inducer, and method for producing antigen-presenting cells | |
| KR20110139256A (ko) | Sparc 혈관형성 영역과 사용방법 | |
| US20080045463A1 (en) | Methods For Lowering Hif-1 Mediated Gene Expression | |
| US20110060120A1 (en) | Immunogenic treatment of cancer by peptides inducing the plasma membrane exposure of erp57 | |
| WO2012174591A1 (en) | Prevention and treatment of acute inflammatory conditions | |
| US20100240591A1 (en) | Tpp ii inhibitors for use in the treatment of autoimmune and inflammatory diseases and transplant rejection | |
| US8940701B2 (en) | Compounds for, and methods of, treating cancer and inhibiting invasion and metastases | |
| WO2009000297A1 (en) | Tpp ii inhibitors for use in combination with chemotherapy for the treatment of cancer | |
| EP3503924B1 (en) | Bcl-w polypeptides and mimetics for treating or preventing chemotherapy-induced peripheral neuropathy and hearing loss | |
| WO2013009165A1 (en) | A peptide capable of binding with a human leukocyte antigen (hla) molecule, a cancer vaccine derived from said peptide and use of said cancer vaccine | |
| Shevtsov et al. | Therapeutic implications of heat shock proteins in cancer | |
| JP7291327B2 (ja) | ペプチド誘導体及びそれを含む医薬組成物 | |
| JP2019510015A (ja) | Bcl−2 l10/ip3受容体の相互作用の阻害剤 | |
| US20140155331A1 (en) | Novel high affinity bivalent helically constrained peptide against cancer | |
| JP2017505755A (ja) | 転移がんの治療および予防のための医薬組成物および方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100106 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20100106 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120612 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20121106 |