JP2008525005A - 新規の物質、および、それらの使用 - Google Patents
新規の物質、および、それらの使用 Download PDFInfo
- Publication number
- JP2008525005A JP2008525005A JP2007547622A JP2007547622A JP2008525005A JP 2008525005 A JP2008525005 A JP 2008525005A JP 2007547622 A JP2007547622 A JP 2007547622A JP 2007547622 A JP2007547622 A JP 2007547622A JP 2008525005 A JP2008525005 A JP 2008525005A
- Authority
- JP
- Japan
- Prior art keywords
- substance
- hcap18
- cells
- cell
- cancer cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Abandoned
Links
Images










Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4723—Cationic antimicrobial peptides, e.g. defensins
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
- C07K16/3076—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells against structure-related tumour-associated moieties
- C07K16/3092—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells against structure-related tumour-associated moieties against tumour-associated mucins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/31—Immunoglobulins specific features characterized by aspects of specificity or valency multispecific
Abstract
本発明は、癌細胞の増殖を阻害する物質を提供し、ここにおいて、該物質は、hCAP18/LL−37の生物活性を阻害する。好ましい実施態様において、該物質は、hCAP18/LL−37の転写、翻訳および/または結合特性を変化させる。好ましくは、該物質は、短鎖干渉RNA(siRNA)分子、アンチセンスオリゴヌクレオチド、および、hCAP18/LL−37への結合親和性を有する化合物からなる群より選択される。本発明はさらに、患者における癌細胞の増殖を阻害する方法、加えて癌を診断するための方法およびキットを提供する。
【選択図】図1
【選択図】図1
Description
本発明は、癌の治療と診断に使用するための物質に関する。特に本発明は、癌細胞の増殖を阻害することができる物質を提供する。
抗菌性タンパク質は、先天性免疫システムにおける主要なエフェクターである。ヒトカテリシジン(cathelicidin)抗菌性タンパク質であるhCAP18は、ヒトでしか知られていないカテリシジンであるが、これは、保存されたカテリン(cathelin)ドメインと、LL−37と呼ばれる可変性のC末端とからなる(Gudmundsson等,1996,Eur J Biochem 1238:325〜32;Zanetti等,1995,FEBS Lett 374:1〜5)。ホロタンパク質の細胞外のタンパク質分解プロセシングによりLL−37ペプチドが放出されるが、これは、広範な抗菌活性(Gudmundsson等,1995,Proc Natl Acad Sci USA 92:7085〜9;Agerberth等,1995,Proc Natl Acad Sci USA 92:195〜99)を有し、加えて、宿主細胞に対して作用も有しそのうちのいくつかは、Gタンパク質結合受容体、ホルミルペプチド受容体様1(FPRL1)が介在する(Yang等,2000,J Exp Med 192:1069〜74;Koczulla等,2003,J Clin Invest 111:1665〜72)。hCAP18は、白血球に存在し(Cowland等,1995,FEBS Lett 368:173〜76)、皮膚およびその他の上皮で発現され、そこで炎症に付随してアップレギュレートされる(Cowland等,1995,FEBS Lett 368:173〜76;Frohm等,1997,J Biol Chem 272:15258〜63)、および、傷害(Dorschner等,2001,J Invest Dermatol 117:91〜97;Heilborn等,2003,J Invest Dermatol 120:379〜89)は、先天性のバリア保護における役割と一致する。近年、カテリシジンなどの抗菌性タンパク質は、腫瘍に対する非特異的な宿主側防御においても役割を果たすことが提唱されている(Winder等,1998,Biochem Biophys Res Commun 242:608〜12;Ohtake等,1999,Br J Cancer 181:393〜403)。
本発明の第一の形態は、癌細胞の増殖を阻害するための物質を提供し、ここにおいて、該物質は、hCAP18/LL−37の生物活性を阻害する(すなわち、インビボで阻害することができる)。
ここでは、「物質」には、あらゆる化学物質が含まれ、例えばオリゴヌクレオチド、ポリヌクレオチド、ポリペプチド、ペプチド模倣剤、および、小さい化合物である。
従って、本発明は、hCAP18/LL−37の生物活性を、直接的に(例えば、タンパク質の生物活性を減少させることによって)、または、間接的に(例えば、hCAP18/LL−37の発現を減少させることによって)阻害することができる物質を提供する。
好ましい実施態様において、該物質は、hCAP18/LL−37の転写、翻訳および/または結合特性を変化させることによってhCAP18/LL−37の生物活性を阻害する。
このような物質は、当業界周知の方法を用いて同定することができ、例えば:
(a)hCAP18/LL−37のmRNAの発現レベルに対する試験物質の作用を決定すること(例えばサザンブロッティング、または、関連するハイブリダイゼーション技術で);
(b)hCAP18/LL−37のタンパク質レベルに対する試験物質の作用を決定すること(例えば抗hCAP18/LL−37抗体を用いたイムノアッセイで);および、
(c)hCAP18/LL−37の活性の機能的なマーカー(例えばErbB2のリン酸化)に対する試験物質の作用を決定すること、
によって同定することができる。
(a)hCAP18/LL−37のmRNAの発現レベルに対する試験物質の作用を決定すること(例えばサザンブロッティング、または、関連するハイブリダイゼーション技術で);
(b)hCAP18/LL−37のタンパク質レベルに対する試験物質の作用を決定すること(例えば抗hCAP18/LL−37抗体を用いたイムノアッセイで);および、
(c)hCAP18/LL−37の活性の機能的なマーカー(例えばErbB2のリン酸化)に対する試験物質の作用を決定すること、
によって同定することができる。
本発明の好ましい実施態様において、物質は、hCAP18/LL−37の転写阻害剤である。
本発明のその代わりの実施態様において、該物質は、hCAP18/LL−37の翻訳阻害剤である。
本発明のさらなる実施態様において、該物質は、hCAP18/LL−37の結合特性の阻害剤である。例えば、該物質は、hCAP18/LL−37がその受容体に結合できなくなるように、hCAP18/LL−37のコンフォメーションを変化させるものでもよい。
当業者であれば当然であるが、該物質はまた、hCAP18/LL−37受容体の機能を直接ブロックすることによって、すなわちhCAP18/LL−37受容体アンタゴニストとして作用することによって、hCAP18/LL−37の生物活性を阻害するものでもよい。好ましくは、hCAP18/LL−37受容体は、FPRL1である。
さらに本発明のその他の実施態様において、該物質は、hCAP18/LL−37またはそのmRNAの安定性を調節すること(例えば減少させること)によって、hCAP18/LL−37の生物活性を阻害する。
有利には、該物質は、hCAP18/LL−37の生物活性を選択的に阻害することができる。
「選択的に」とは、ここでは、該物質は、癌細胞中の他のタンパク質の活性を調節するよりも大きい程度にhCAP18/LL−37の生物活性を阻害することを意味する。好ましくは、該物質は、hCAP18/LL−37の生物活性だけを阻害するが、当然のことながら、癌細胞内のその他のタンパク質の発現や活性は、hCAP18/LL−37の選択的な阻害の下流の結果として変化している可能性がある。従って、我々は、遺伝子発現および/または癌細胞の増殖に非特異的な作用を有する物質を排除する。
当業者であれば当然であるが、本発明の物質によるhCAP18/LL−37の生物活性の阻害は、全体的であってもよいし、または、部分的であってもよい。例えば、該物質は、hCAP18/LL−37の生物活性を、該物質に晒されていない癌細胞におけるhCAP18/LL−37の生物活性と比較して、少なくとも10%、好ましくは少なくとも20%、30%、40%、50%、60%、70%、80%、または、90%、最も好ましくは100%阻害する可能性がある。好ましい実施態様において、該物質は、hCAP18/LL−37の生物活性を、該物質に晒されていない癌細胞におけるhCAP18/LL−37の生物活性と比較して50%またはそれ以上阻害することができる。
好ましくは、該物質は、以下のタイプの物質から選択される:
(a)短鎖干渉RNA(siRNA)分子;
(b)アンチセンスオリゴヌクレオチド;および、
(c)hCAP18/LL−37への結合親和性を有する化合物、例えばポリペプチド。
(a)短鎖干渉RNA(siRNA)分子;
(b)アンチセンスオリゴヌクレオチド;および、
(c)hCAP18/LL−37への結合親和性を有する化合物、例えばポリペプチド。
あるいは、該物質は低分子量の阻害化合物であってもよく、例えば、ビタミンDに対するアンタゴニスト、例えばZK159222(シエーリング社(Schering AG))、および、TEI−9647(帝人医学研究所(Tejin Institute for Medical Research),東京)、ならびに、ビタミンAに対するアンタゴニスト、例えばAGN193109(アレルゲン・ファーマシューティカルズ(Allergen Pharmaceuticals))。
本発明の第一の形態の好ましい実施態様において、該物質は、短鎖干渉RNA(siRNA)分子である。
RNA干渉は、2工程からなるプロセスである。第一の工程(開始工程と名付ける)は、入ってきたdsRNAを21〜23個のヌクレオチド(nt)の短鎖干渉(siRNA)に消化するが、これは恐らく、ATP依存性の様式で、dsRNA(直接導入された、または、導入遺伝子またはウイルスを介して導入された)をプロセシングする(切断する)dsRNAに特異的なリボヌクレアーゼのRNアーゼIIIファミリーの一種であるダイサーの作用によってなされる。連続的な切断現象によって、RNAを、それぞれ2個のヌクレオチドからなる3’オーバーハングを有する19〜21bpの二重鎖(siRNA)にまで分解する(HutvagnerおよびZamore,2002,Curr.Opin.Genetics and Development 12:225〜232;Bernstein,2001,Nature 409:363〜366)。
エフェクター工程において、siRNA二重鎖はヌクレアーゼ複合体に結合して、RNA誘導型サイレンシング複合体(RISC)を形成する。siRNA二重鎖のATP依存性の巻き戻しが、RISCの活性化に必要である。次に、活性RISCは、塩基対形成による相互作用で相同的に転写することを標的として、mRNAを、siRNAの3’末端から12個のヌクレオチドからなるフラグメントに切断する(HutvagnerおよびZamore,2002,上記;Hammond等,2001,Nat.Rev.Gen.2:110〜119(2001);Sharp,2001,Genes.Dev.15:485〜90)。切断メカニズムをさらに解明すべきであるが、調査によれば、RISCはそれぞれ、単一のsiRNA、および、RNアーゼを含むことが示されている(HutvagnerおよびZamore,2002,上記)。
RNAiの驚くべき効力を考慮して、RNAi経路内の増幅工程が示唆されている。増幅は、入ってきたdsRNAをコピーしてより多くのsiRNAを生成することによって起こる場合もあるし、または、形成されたsiRNAの複製によって起こる場合もある。あるいは、または、それに加えて、増幅は、RISCの転換を複数行うことによって実行されてもよい(Hammond等,2001,上記;HutvagnerおよびZamore,2002,上記)。RNAiに関するさらなる情報は、以下の総論で見出すことができる:Tuschl,2001,Chem.Biochem.2:239〜245,Cullen,2002,Nat.Immunol.3:597〜599およびBrantl,2002,Biochem.Biophys Act.1575:15〜25。
本発明との使用に適したRNAi分子の合成は、以下のように実行することができる。まず、hCAP18/LL−37のmRNA配列を、AAジヌクレオチド配列に関してAUG開始コドンの下流でスキャンする。各AAの出現と、3’に隣接する19個のヌクレオチドの出現を、可能性があるsiRNA標的部位として記録する。好ましくは、調節タンパク質結合部位において非翻訳領域(UTR)がより豊富であるため、siRNA標的部位はオープンリーディングフレームから選択される。UTR結合タンパク質および/または翻訳開始複合体は、siRNAエンドヌクレアーゼ複合体の結合に干渉する可能性がある(Tuschl,ChemBiochem.2:239〜245)。しかし当然ながら、非翻訳領域に向けられたsiRNAも、有効である可能性がある。
第二に、配列アライメントソフトウェア、例えばBLAST(www.ncbi.nlm.nih.gov/BLAST/)を用いて、可能性がある標的部位を適切なゲノムデータベース(例えばヒト、マウス、ラットなど)と比較する。他のコード配列に対して有意な相同性を示す推定上の標的部位を、フィルターをかけて除外する。
条件にあった標的配列を、siRNA合成のためのテンプレートとして選択する。好ましい配列は、低いG/C含量を含む配列であり、なぜならこれらは、遺伝子サイレンシングの媒介において、55%より高いG/C含量を有するものと比較してより有効であることが証明されているためである。好ましくは、評価するために、標的遺伝子の長さに従って数種の標的部位が選択される。選択されたsiRNAをよりよく評価するために、好ましくは、ネガティブコントロールが一緒に用いられる。好ましくは、ネガティブコントロールのsiRNAは、選択されたsiRNAと同じヌクレオチド組成を含むが、ゲノムに対する有意な相同性はない。従って、好ましくは、siRNAのスクランブル化したヌクレオチド配列が用いられる(ただしこれらは、その他のあらゆる遺伝子に対する有意な相同性をまったく示さない)。
好ましくは、siRNA分子は、配列番号1のヌクレオチド配列のフラグメント、または、このようなフラグメントの変異体を含む。
hCAP18/LL−37のmRNA(登録番号MN004345)

あるいは、siRNA分子は、ENSG00000164047(ゲノム配列)から誘導されたヌクレオチド配列のフラグメントを含む。
「フラグメント」は、ここでは、少なくとも10個のヌクレオチド、例えば少なくとも15、16、17、18、19、20、21、22、23、24または25個のヌクレオチドを意味する。
「変異体」は、ここでは、配列番号1のフラグメントと少なくとも90%の配列同一性を共有するヌクレオチド配列、例えば少なくとも95%、96%、97%、98%、または、99%の配列同一性を共有するヌクレオチド配列を意味する。
2種のポリヌクレオチド間の配列同一性(%)は、適切なコンピュータープログラム、例えばウィスコンシン大学のジェネティック・コンピューティング・グループ(University of Wisconsin Genetic Computing Group)のギャップ・プログラム(GAP program)を用いて決定してもよく、当然のことながら、同一性(%)は、配列が最適に並べられたポリヌクレオチドに関して計算される。
このようなアライメントは、別の方法で行って、クラスタルWプログラム(Clustal W program,Thompson等,1994,Nuc.Acid Res.22:4673〜4680で説明されている通り)を用いてもよい。
用いられるパラメーターは、以下のようなものが可能である:
Fastのペアワイズアライメントパラメーター:K−tuple(ワード)サイズ;1、ウィンドウサイズ:5、ギャップペナルティー;3、トップの対角線の数;5。スコア付けの方法:xパーセント。
マルチアライメントのパラメーター:ギャップオープンペナルティー;10、ギャップ伸長ペナルティー;0.05。
スコアリングマトリックス:BLOSUM。
Fastのペアワイズアライメントパラメーター:K−tuple(ワード)サイズ;1、ウィンドウサイズ:5、ギャップペナルティー;3、トップの対角線の数;5。スコア付けの方法:xパーセント。
マルチアライメントのパラメーター:ギャップオープンペナルティー;10、ギャップ伸長ペナルティー;0.05。
スコアリングマトリックス:BLOSUM。
あるいは、局所的な配列アライメントを決定するためには、BESTFITプログラムを用いてもよい。
siRNA分子が、長さが19〜23個のヌクレオチドであることが有利である。
本発明の第一の形態の好ましいその代わりの実施態様において、該物質はアンチセンスオリゴヌクレオチドである。
hCAP18/LL−37のレベル/活性を効率的に減少させるのに用いることができるアンチセンス分子の設計には、アンチセンス法に重要な2つの観点の考察が必要である。第一の観点は、癌細胞の細胞質へのオリゴヌクレオチドの送達であり、一方で、第二の観点は、細胞内で指示されたmRNAの翻訳を阻害するようにして、それらに特異的に結合するオリゴヌクレオチドの設計である。
従来技術では、多種多様の細胞型にオリゴヌクレオチドを効率的に送達するのに用いることができる多数の送達方策を教示している(例えば、Luft,1998,J Mol Med 76:75〜6;Kronenwett等,1998,Blood 91:852〜62;Rajur等,1997,Bioconjug Chem 8:935〜40;Lavigne等,1997,Biochem Biophys Res Commun 237:566〜71;Aoki等,1997,Biochem Biophys Res Commun 231:540〜5を参照)。
加えて、標的mRNAとオリゴヌクレオチドとの両方における構造的な変化のエネルギー論を明らかにする熱力学的なサイクルに基づき、それらの標的mRNAに対して最高の予想された結合親和性を有する配列を同定するためのアルゴリズムが利用可能である(例えば、Walton等,1999,Biotechnol Bioeng 65:1〜9を参照)。
また、インビトロでのシステムを用いて特異的なオリゴヌクレオチドの効率を設計および予想するためのアプローチもいくつか知られている(例えば、Matveeva等,1998,Nature biotechnology 16:1374〜1375を参照)。
いくつかの臨床的な追跡によって、アンチセンスオリゴヌクレオチドの安全性、実行可能性および活性が実証されている。例えば、癌の治療に適したアンチセンスオリゴヌクレオチドの使用が成功している(Holmlund等,1999,Curr Opin Mol Ther 1:372〜85;Gerwitz,1999,Curr Opin Mol Ther 1:297〜306)。さらに近年、ヒトヘパラナーゼ遺伝子発現のアンチセンスが介在する抑制は、マウスモデルにおいて、ヒト癌細胞の胸膜の内転移を阻害することが報告されている(Uno等,2001,Cancer Res 61:7855〜60)。
従って、当業者であれば、hCAP18/LL−37の発現をダウンレギュレートするのに適したアンチセンス法を容易に設計および実施することができる。
好ましくは、このようなアンチセンスオリゴヌクレオチドは、配列番号1に記載のヌクレオチドのフラグメント、または、このようなフラグメントの変異体を含む。
有利には、該アンチセンスオリゴヌクレオチドは、長さが15〜35個の塩基である。例えば、20−merのオリゴヌクレオチドは、上皮増殖因子受容体のmRNAの発現を阻害することが示されており(Witters等,Breast Cancer Res Treat 53:41〜50(1999))、さらに、25−merのオリゴヌクレオチドは、副腎皮質刺激ホルモンの発現を90%を超えて減少させることが示されている(Frankel等,J Neurosurg 91:261〜7(1999))。しかし当然ながら、この範囲外の長さを有するオリゴヌクレオチド、例えば10、11、12、13もしくは14個の塩基、または、36,37,38,39もしくは40個の塩基からなるオリゴヌクレオチドを使用することが望ましい場合がある。
さらに当業者であれば当然ながら、オリゴヌクレオチドは、細胞の内因性ヌクレアーゼによって分解されるか、または、不活性化される。この問題を克服するために、例えば、天然に存在するホスホジエステル結合がその他の結合で置き換えられた改変インターヌクレオチド結合を有する、改変オリゴヌクレオチドを使用することが可能である。例えば、Agrawal等(1988)Proc.Natl.Acad.Sci.USA 85,7079〜7083では、HIV−1の組織培養において、オリゴヌクレオチドホスホルアミデートおよびホスホロチオエートを用いたところ阻害が増加したことが示されている。Sarin等(1988)Proc.Natl.Acad.Sci.USA 85,7448〜7451では、オリゴヌクレオチドメチルホスホナートを用いたところ、HIV−1の阻害が増加したことが実証されている。Agrawal等(1989)Proc.Natl.Acad.Sci.USA 86,7790〜7794では、ヌクレオチド配列に特異的なオリゴヌクレオチドホスホロチオエートを用いたところ、初期に感染させた細胞培養と、慢性的に感染させた細胞培養の両方においてHIV−1複製が阻害されることが示されている。Leither等(1990)Proc.Natl.Acad.Sci.USA 87,3430〜3434は、組織培養において、オリゴヌクレオチドホスホロチオエートによりインフルエンザウイルス複製が阻害されることを報告している。
人工的な結合を有するオリゴヌクレオチドは、インビボでの分解に耐性であることが示されている。例えば、Shaw等(1991)は、Nucleic Acids Res.19,747〜750で、その他の改変されていないオリゴヌクレオチドが、所定のキャッピング構造によって3’末端でブロックされると、それらはヌクレアーゼに対してインビボでより耐性になること、さらに、キャッピングされていないオリゴヌクレオチドのホスホロチオエートが、インビボで分解されないことを報告している。
オリゴヌクレオチドのホスホロチオエートを合成することによるH−ホスホナート法の詳細な説明は、AgrawalおよびTang(1990)Tetrahedron Letters 31,7541〜7544で示されており、そこでの教示は参照により本明細書に加入させる。オリゴヌクレオチドのメチルホスホナート、ホスホロジチオエート、ホスホルアミデート、リン酸エステル、架橋ホスホルアミデート、および、架橋ホスホロチオエートの合成は、当業界既知である。例えば、AgrawalおよびGoodchild(1987)Tetrahedron Letters 28,3539;Nielsen等(1988)Tetrahedron Letters 29,2911;Jager等(1988)Biochemistry 27,7237;Uznanski等(1987)Tetrahedron Letters 28,3401;Bannwarth(1988)Helv.Chim.Acta.71,1517;CrosstickおよびVyle(1989)Tetrahedron Letters 30,4693;Agrawal等(1990)Proc.Natl.Acad.Sci.USA 87,1401〜1405を参照(これらの教示は、この参照により本明細書に含まれる)。また、その他の合成または生産方法も可能である。好ましい実施態様において、上記オリゴヌクレオチドはデオキシリボ核酸(DNA)であるが、リボ核酸(RNA)配列を合成して適用してもよい。
好ましくは、本発明において有用なオリゴヌクレオチドは、内因性の核酸分解酵素による分解に耐性を有するように設計される。インビボでのオリゴヌクレオチドの分解により、長さが減少したオリゴヌクレオチドの分解産物が生産される。このような分解産物は、非特異的なハイブリダイゼーションに関わる可能性がより高く、さらに、それに相当する全長の対応物に比べて有効である可能性はより低い。従って、体内での分解に対して耐性であり、さらに、標的化した細胞に到達することができるオリゴヌクレオチドを使用することが望ましい。本発明のオリゴヌクレオチドは、天然型ホスホジエステル結合を1個またはそれ以上の内部の人工的なインターヌクレオチド結合で置換することによって、例えば、結合内のリン酸を硫黄で置換することによって、インビボでの分解に対してより耐性にすることができる。使用可能な結合の例としては、ホスホロチオエート、メチルホスホナート、スルホン、硫酸塩、ケチル、ホスホロジチオエート、様々なホスホルアミデート、リン酸エステル、架橋ホスホロチオエート、および、架橋ホスホルアミデートが挙げられる。その他のインターヌクレオチド結合は当業界周知であるため、このような例は制限するものではなく、例証である。これらのホスホジエステルインターヌクレオチド結合が置換された結合を1個またはそれ以上有するオリゴヌクレオチドの合成は当業界周知であり、例えば、混合型のインターヌクレオチド結合を有するオリゴヌクレオチドを製造する合成経路が挙げられる。
オリゴヌクレオチドは、「キャッピング」によって、または、5’または3’末端のヌクレオチドに類似の基を取り込むことによって、内因性の酵素による伸長に対して耐性にすることができる。キャッピングのための試薬は、アプライド・バイオシステムズ社(Applied Biosystems Inc,フォスターシティー,カリフォルニア州)製のアミノ−リンクIITM(Amino−Link IITM)として市販されている。キャッピングに関する方法は、例えば、Shaw等(1991)Nucleic Acids Res.19,747〜750およびAgrawal等(1991)Proc.Natl.Acad.Sci.USA 88(17),7595〜7599によって説明されている。
オリゴヌクレオチドをヌクレアーゼの攻撃に対して耐性にするさらなる方法は、それらを「自己安定化させること」であり、例えば、Tang等(1993)Nucl.Acids Res.21,2729〜2735で説明されている通りである。自己安定化したオリゴヌクレオチドは、それらの3’末端でヘアピンループ構造を有し、ヘビ毒ホスホジエステラーゼ、DNAポリメラーゼI、および、ウシ胎児血清による分解に対して高い耐性を示す。このようなオリゴヌクレオチドの自己安定化した領域は、ハイブリダイゼーションの際に相補的な核酸に干渉せず、さらに、マウスにおける薬物動態学的研究および安定性の研究によれば、それらの直鎖状の対応物と比べて、自己安定化したオリゴヌクレオチドはインビボで持久性が増加することが示されている。
本発明の物質のさらなる好ましい実施態様において、該物質は、hCAP18/LL−37への結合親和性を有する化合物であり、例えばタンパク質、および、炭水化物である。
「結合親和性を有する化合物」とは、ここでは、インビボで、すなわち癌細胞の内部にhCAP18/LL−37が存在するような生理学的条件下で、hCAP18/LL−37に結合することができる化合物を意味する。
例えば、本化合物は、hCAP18/LL−37の活性部位に実質的に可逆的に結合してもよいし、または、実質的に不可逆的に結合してもよい。さらなる例において、本化合物は、hCAP18/LL−37のリガンドまたは受容体への結合に干渉するように、hCAP18/LL−37の活性部位ではない部分に結合してもよい。さらにその他の実施例において、本化合物は、アロステリック作用によってタンパク質活性を減少させるように、hCAP18/LL−37の一部に結合してもよい。このアロステリック作用は、hCAP18/LL−37の活性の天然の調節であってもよく、例えば「上流の活性化因子」によるhCAP18/LL−37の活性化に関与するアロステリック作用である。
試験化合物とhCAP18/LL−37との相互作用を検出する方法は当業界周知である。例えば、イオンスプレーマススペクトロスコピー/HPLC法を用いた限外ろ過、またはその他の物理的方法や分析方法を用いてもよい。加えて、蛍光エネルギー共鳴移動(FRET)法を用いてもよく、ここにおいて、2つの蛍光標識された物体の結合は、互いに近接した際の蛍光標識相互の作用を測定することによって測定することもできる。
高分子(例えばDNA、RNA、タンパク質およびリン脂質)へのポリペプチドの結合を検出する代替法としては、表面プラズモン共鳴分析が挙げられ、これは例えば、Plant等,1995,Analyt Biochem 226(2),342〜348で説明されている通りである。例えば放射標識または蛍光標識で標識されたポリペプチドを利用する方法も可能である。
このようなポリペプチドに結合することができる化合物を同定するさらなる方法は、ポリペプチドを化合物に晒し、化合物の前記ポリペプチドへのあらゆる結合を検出および/または測定する方法である。化合物のポリペプチドへの結合に関する結合定数を決定してもよい。化合物のポリペプチドへの結合を検出および/または測定する(定量する)のに適切な方法は当業者周知であり、例えば、ハイスループット操作が可能な方法(例えばチップベースの方法)を用いて行ってもよい。VLSIPSTMと呼ばれる新しい技術により、何十万またはそれを超える種類の様々な分子プローブを含む極めて小さいチップの生産が可能になっている。これらの生物学的なチップまたはアレイは、アレイ中に配置されたプローブを有し、それぞれのプローブには特定の位置が割り当てられている。それぞれの位置に例えば10ミクロンの目盛りを有する生物学的なチップが生産されている。このようなチップは、チップ上で標的分子がプローブのいずれかと相互作用するかどうかを決定するのに用いることができる。選択した試験条件下でアレイを標的分子に晒した後、スキャン装置によってアレイ中の各位置を試験し、その位置において標的分子はプローブとの相互作用を有するかどうかを決定することができる。
hCAP18/LL−37への結合親和性を有する化合物を同定するその他の方法は、酵母2ハイブリッドシステムであり、ここにおいて、本発明のポリペプチドを、hCAP18/LL−37に結合するタンパク質を「捕獲する」ために用いることができる。酵母2ハイブリッドシステムは、FieldsおよびSong,Nature 340:245〜246(1989)で説明されている。
この本発明の形態の好ましい実施態様において、該物質は、hCAP18/LL−37へのリガンド結合能力を有する化合物である。
例えば、該物質は、hCAP18/LL−37受容体(例えばFPRL1)の可溶性フラグメントが可能である。あるいは、該物質は、抗体を模擬する高い親和性を有する分子(いわゆる「アフィボディ(affibody)」)が可能である(例えば、US5,831,012、および、www.affibody.seを参照)。これらのリガンドは、小さく単純なタンパク質であり、プロテインA(細菌スタフィロコッカス・アウレウス(Staphylococcus aureus)由来の表面タンパク質)のIgG結合ドメインのうち1つの足場をベースとした3個のヘリックスバンドルで構成される。この足場は、親和性リガンドとして優れた特徴を有し、どのような所定の標的タンパク質にも高い親和性で結合するように設計することができる。
しかしながら、有利には、hCAP18/LL−37への結合親和性を有する化合物は、ポリペプチドであるか、または、ポリペプチドを含む。
例えば、本化合物は抗体であってもよく、例えばモノクローナル、または、ポリクローナル抗体、または、それらの抗原結合フラグメントである。
「抗体」には、ここでは、実質的に無傷の抗体分子、加えて、キメラ抗体、ヒト化抗体、ヒト抗体(ここにおいて、少なくとも1個のアミノ酸が、天然に存在するヒト抗体と比べて突然変異している)、単鎖抗体、二重特異性抗体、抗体重鎖、抗体軽鎖、抗体重鎖および/または軽鎖のホモ二量体およびヘテロ二量体、および、抗原結合フラグメント、ならびに、それらの誘導体が含まれる。
「抗原結合フラグメント」は、ここでは、hCAP18/LL−37に結合することができる抗体の機能的なフラグメントを意味する。
好ましくは、抗原結合フラグメントは、Fvフラグメント(例えば、単鎖Fv、および、ジスルフィド結合Fv)、Fab様フラグメント(例えば、Fabフラグメント、Fab’フラグメント、および、F(ab)2フラグメント)、単一の可変性ドメイン(例えば、VhおよびVlドメイン)、ならびに、ドメイン抗体(dAbs、単鎖様式と二重鎖様式[すなわちdAb−リンカー−dAb]を含む)からなる群より選択される。
抗体全体よりも抗体フラグメントを用いる利点は数倍にもなる。フラグメントの大きさがより小さいと、より優れた固形組織への貫入のような薬理学的特性が改善される可能性がある。さらに、抗原結合フラグメント、例えばFab、Fv、ScFvおよびdAb抗体フラグメントは、E.Coliで発現させ、分泌させてもよく、従って、大量の前記フラグメントを容易に生産することができる。
また、抗体およびそれらの抗原結合フラグメントの改変型も本発明の範囲内でであり、例えば、ポリエチレングリコールまたはその他の適切なポリマーが共有結合したことによって改変された改変型である。
抗体および抗体フラグメントの製造方法は当業界周知である。例えば、抗体は、インビボでの抗体分子生産の誘導、免疫グロブリンライブラリーのスクリーニング(Orlandi等,1989.Proc.Natl.Acad.Sci.U.S.A.86:3833〜3837;Winter等,1991,Nature 349:293〜299)、または、培養中の細胞系によるモノクローナル抗体分子の生成を用いた数種の方法のいずれか一つによって製造することができる。これらとしては、これらに限定されないが、ハイブリドーマ技術、ヒトB細胞ハイブリドーマ技術、および、エプスタイン−バーウイルス(EBV)−ハイブリドーマ技術が挙げられる(Kohler等,1975.Nature 256:4950497;Kozbor等,1985.J.Immunol.Methods 81:31〜42;Cote等,1983.Proc.Natl.Acad.Sci.USA 80:2026〜2030;Cole等,1984.Mol.Cell.Biol.62:109〜120)。
選択された抗原に適したモノクローナル抗体は、既知の技術によって製造してもよく、例えば、“Monoclonal Antibodies:A manual of techniques”,H Zola(CRC Press,1988)、および、“Monoclonal Hybridoma Antibodies:Techniques and Applications”,J G R Hurrell(CRC Press,1982)で開示された技術によって製造してもよい。
抗体フラグメントは、当業界周知の方法を用いて得ることができる(例えば、HarlowおよびLane,1988,“Antibodies:A Laboratory Manual”,Cold Spring Harbor Laboratory,New Yorkを参照)。例えば、本発明に係る抗体フラグメントは、抗体を加水分解によるタンパク質分解により、または、このフラグメントをコードするDNAをE.coliまたは哺乳動物細胞(例えばチャイニーズハムスター卵巣細胞の培養物、またはその他のタンパク質発現系)中で発現させることによって製造できる。あるいは、抗体フラグメントは、従来の方法による抗体全体のペプシンまたはパパイン消化により得ることができる。
当業者であれば当然であるが、ヒトの治療または診断法には、好ましくはヒト化された抗体が用いられる。非ヒト(例えばマウス)抗体のヒト化された形態は、好ましくは、非ヒト抗体から誘導された部分が最も少ない遺伝操作されたキメラ抗体または抗体フラグメントである。ヒト化抗体としては、ヒト抗体(レシピエント抗体)の相補性決定領域が、望ましい官能性を有するマウス、ラットまたはウサギのようなヒト以外の種(ドナー抗体)の相補性決定領域由来の残基で置き換えられた抗体が挙げられる。場合によっては、ヒト抗体のFvフレームワークの残基は、それに対応する非ヒトの残基で置き換えられる。ヒト化抗体はまた、レシピエント抗体でも、導入された相補決定領域またはフレームワーク配列でも見出されていない残基を含んでいてもよい。一般的に、ヒト化抗体は、少なくとも1個、典型的には2個の可変ドメインの実質的に全てを含むと予想され、ここにおいて、全ての、または、実質的に全ての相補決定領域は、非ヒト抗体の相補決定領域に相当し、全ての、または、実質的に全てのフレームワーク領域は、関連するヒトコンセンサス配列のフレームワーク領域に相当する。また、ヒト化された抗体は、最適には、抗体の定常領域(例えばFc領域)の少なくとも一部も含み、典型的にはヒト抗体から誘導された上記領域の少なくとも一部である(例えば、Jones等,1986.Nature 321:522〜525;Riechmann等,1988,Nature 332:323〜329;Presta,1992,Curr.Op.Struct.Biol.2:593〜596を参照)。
非ヒト抗体をヒト化する方法は当業界周知である。一般的に、ヒト化抗体は、1またはそれ以上のアミノ酸残基がヒト以外の源からそこに導入されている。これらの非ヒトアミノ酸残基(導入された残基と称されることが多い)は、典型的には、導入された可変ドメインから取り入れられる。ヒト化は、実質的に、説明されている通りにして、ヒト相補決定領域をそれに対応するげっ歯類の相補決定領域で置換することによって行うことができる(例えば、Jones等,1986,Nature 321:522〜525;Reichmann等,1988.Nature 332:323〜327;Verhoeyen等,1988,Science 239:1534〜1536l;US4,816,567を参照)。従って、このようなヒト化抗体はキメラ抗体であり、ここにおいて、実質的に無傷のヒト可変ドメイン以外は、非ヒトの種由来のそれに対応する配列で置換されている。実際には、ヒト化抗体は、典型的にはヒト抗体が可能であり、ここにおいて、いくつかの相補決定領域残基、場合によってはいくつかのフレームワーク残基は、げっ歯類の抗体における類似した部位由来の残基で置換されている。
また、ヒト抗体は、当業界既知の様々な技術を用いて同定することができ、このような技術としては、ファージディスプレイライブラリーが挙げられる(例えば、HoogenboomおよびWinter,1991,J.Mol.Biol.227:381;Marks等,1991,J.Mol.Biol.222:581;Cole等,1985,In:Monoclonal antibodies and Cancer Therapy,Alan R.Liss,pp.77;Boerner等,1991.J.Immunol.147:86〜95を参照)。
適切な抗体が得られたら、それらを活性に関して試験してもよく、例えばELISAで試験される。
本発明の第一の形態の具体的に好ましい実施態様において、該物質は、癌細胞に選択的に送達することができる、または、癌細胞によって選択的に活性化することができる。
「選択的に」は、ここでは、該物質のhCAP18/LL−37の生物活性に対する阻害作用が、癌細胞で、または、癌細胞内で選択的に作用することを意味する(ただし、該物質を癌細胞部位に局所投与することに起因する場合を除く)。
物質を癌細胞のような特定の細胞型に標的化する方法は当業界周知である(例えば、VasirおよびLabhasetwar,2005,Technol Cancer Res Treat.4(4):363〜74;Brannon−PeppasおよびBlanchette,2004,Adv Drug Deliv Rev.56(11):1649〜59およびZhaoおよびLee,2004,Adv Drug Deliv Rev.56(8):1193〜204を参照)。
例えば、該物質は、標的細胞に特異的な部分を含んでいてもよい。
「標的細胞に特異的な」部分とは、ここでは、標的癌細胞上の物体を認識して結合する1またはそれ以上の結合部位を含む物質の部分を意味する。標的細胞と接触すると、標的細胞に特異的な部分は、阻害剤部分と共に内在化され得る。
「標的細胞に特異的な」部分とは、ここでは、標的癌細胞上の物体を認識して結合する1またはそれ以上の結合部位を含む物質の部分を意味する。標的細胞と接触すると、標的細胞に特異的な部分は、阻害剤部分と共に内在化され得る。
標的細胞特異的な部分によって認識された物体は、標的癌細胞上で優勢に、好ましくは独占的に発現される。標的細胞に特異的な部分は、同じ標的細胞型で発現された異なる物体に対する1またはそれ以上の結合部位、または、2またはそれ以上の異なる標的細胞型で発現された異なる物体に対する1またはそれ以上の結合部位を含む可能性がある。
好ましくは、標的細胞特異的な部分は、高い結合活性を有する標的細胞を認識する。
「高い結合活性」は、ここでは、標的細胞特異的な部分が、少なくともKd=10-6M、好ましくは少なくともKd=10-9M適切にはKd=10-10M、より適切にはKd=10-11M、さらにより適切にはKd=10-12M、より好ましくはKd=10-15Mの結合定数を有する標的細胞を認識することを意味し、または、さらにKd=10-18Mの結合定数を有する標的細胞を認識することも意味する。
「高い結合活性」は、ここでは、標的細胞特異的な部分が、少なくともKd=10-6M、好ましくは少なくともKd=10-9M適切にはKd=10-10M、より適切にはKd=10-11M、さらにより適切にはKd=10-12M、より好ましくはKd=10-15Mの結合定数を有する標的細胞を認識することを意味し、または、さらにKd=10-18Mの結合定数を有する標的細胞を認識することも意味する。
認識される物体は、腫瘍細胞によって発現される適切なあらゆる物体が可能である。このような認識される物体は、抗原であることが多い。
抗原の例としては、表1で列挙したものが挙げられる。
その他の抗原としては、アルファフェトプロテイン、Ca−125、前立腺に特異的な抗原、および、上皮増殖因子受容体ファミリーの種類、すなわちEGFR、erb B3およびerb B4が挙げられる。
有利には、標的細胞に特異的な部分は、抗体(例えばモノクローナル抗体)、または、それらの抗原結合フラグメントである。好ましくは、このような抗体はヒト化抗体である。
都合のよい形態としては、標的細胞特異的な部分は、標的細胞に関する結合部位を2またはそれ以上を含み、ここにおいて、標的細胞に特異的な部分は、抗体またはそれらの2価フラグメントである。前記標的細胞に特異的な部分は、それぞれ互いに同じ物体を認識する「アーム」を有していてもよいし、または、異なる物体を認識する「アーム」を有していてもよい。
本発明の物質の一実施態様において、標的細胞に特異的な部分は、同じ標的細胞上で異なる分子を認識する2つの「アーム」を有し、ここにおいて、この同じ標的細胞上の分子は、その細胞型に限定されないが、2〜3種のその他の細胞型に存在する可能性がある。例えば、標的細胞特異的な部分の1つの「アーム」は、細胞型I、IIおよびIIIで分子を認識する可能性があるが、それに対して、他方の「アーム」は、細胞型I、IVおよびVで分子を認識する可能性がある。従って、このような標的細胞特異的な部分を含む本発明の物質は、細胞型II、IIIおよびIVと比較して、細胞型Iへの大きい特異性を有すると予想される。この本発明の形態は、完全に標的細胞特異的な分子はほとんど発見されていないため特に有益であり、それに対して、いくつかの細胞型に存在し本発明のこの形態において有用な分子がよく知られている。このような分子は、一般的に、交叉反応性抗体が既知の細胞表面抗原である。
表1に列挙した抗原の多くに結合すると予想されるモノクローナル抗体はすでに既知であるが、どのような場合においても、モノクローナル抗体テクノロジーに関する現在の技術を用いれば、ほとんどの抗原(上記参照)に対する抗体を製造することができる。
認識される物体は抗原性であってもよいし、または、抗原性でなくてもよく、ただし、これらは、その他の何らかの方法で、認識し、選択的に結合してもよい。例えば上記物体は、特徴的な細胞表面受容体が可能であり、例えば、黒色腫細胞で大量に発現されるメラニン細胞刺激ホルモン(MSH)に関する受容体である。あるいは、上記物体は、標的細胞中で誘導される物体であってもよい。続いて、細胞特異的部分は、非免疫的な意味で、例えば細胞表面酵素に関する基質またはそれらの類似体として、または、メッセンジャーとして、上記物体に特異的に結合する化合物またはそれらの一部であり得る。
好ましくは、高い結合活性の標的細胞に特異的な部分は、2またはそれ以上の異なる標的細胞に関する結合部位を含む。
標的細胞に関する異なる結合部位は、標的細胞上で発現された異なる物体に向けられた2種またはそれ以上の異なる抗体、または、それらのフラグメントであってもよいし、または、そうでなくてもよい。あるいは、標的細胞に関する異なる結合部位は、その他のいくつかの非免疫的な意味で、その標的細胞を認識し、選択的に結合することであってもよい。
当然のことながら、標的化部分は、本発明の阻害剤と、2つの部分の機能的な活性が保持されるようなあらゆる適切な手段で結合させてもよい。例えば、標的化部分と阻害剤部分がいずれもポリペプチドである場合、それらが互いに融合して融合ポリペプチドを形成してもよい。このような融合の例は当業者周知である。
本発明における選択的な阻害物質のその代わりの実施態様において、該物質は、癌細胞によって選択的に活性化されたプロドラッグである。
用語「プロドラッグ」は、本願で用いられる場合、癌細胞への細胞毒性が、親となる薬物と比較してより少なく、さらに、酵素による活性化またはより活性な親の形態への変換が可能な、医薬活性物質の前駆体または誘導体の形態を意味する(例えば、D.E.V.Wilman“Prodrugs in Cancer Chemotherapy” Biochemical Society Transactions 14,375〜382(615th Meeting,Belfast 1986)and V.J.Stella等“Prodrugs:A Chemical Approach to Targeted Drug Delivery” Directed Drug Delivery R.Borchardt等(編集)247〜267頁(Humana Press 1985)を参照)。
このようなプロドラッグ物質を生産する適切な方法は当業界周知である(例えば、Denny,2004,Cancer Invest.22(4):604〜19;Rooseboom等,2004,Pharmacol Rev.2004 56(1):53〜102;WO03/106491を参照)。
プロドラッグ活性化のための酵素の選択には、数種の要素を考慮する必要がある。このような要素としては、酵素の分子量および物理特性、生理学的条件下でのそれらの活性および安定性、および、酵素が生成する薬物の性質が挙げられる。
これらの方策の適応性は、多数の機構的に別個の抗癌剤を放出し得る多種多様な酵素の使用に応じる。特に価値があるのは、単一のMab−酵素複合体が、相乗的な活性を有する治療上有効な用量の機構的に別個の抗癌剤を生成できることである。これは、免疫原性の理由で重要であることがわかる。このような観点において、多くのβ−ラクタマーゼは、それらの好ましい速度論と広範な基質の特異性、加えて、セファロスポリン基質の3’位に結合した置換基を除去するそれらの能力のために、多大な可能性を有している(Svensson等(1992)“Mab−β−lactamase conjugates for the activation of a cephalosporin mustard prodrug”Bioconjugate Chem.3,176〜181を参照)。
多様なプロドラッグの活性化には、哺乳動物由来と非哺乳動物由来の酵素の両方が用いられている(Senter等,1993.Generation of cytotoxic agents by targeted enzymes.Bioconjugate 4.3〜9;Senter等,1991.Activation of prodrugs by antibody−enzyme conjugates.In Immunobiology of Proteins and Peptides VI,ed.M.Z.Atassi.Plenum Press,New York,97〜105頁)。哺乳動物由来の酵素は、減少した免疫原性のために有利である可能性があるが、それらが作用を与えるプロドラッグは、対応する内因性酵素の基質である可能性もある。
当業者であれば当然であるが、本発明の物質は、異なるタイプの癌細胞の増殖を阻害するのに用いてもよい。しかしながら、好ましい実施態様において、癌細胞(上皮性の癌細胞)は、上皮細胞または扁平細胞である。
有利には、癌細胞は、乳房、胆管、脳、結腸、胃、生殖器、肺および気道、皮膚、胆嚢、肝臓、上咽頭、神経細胞、腎臓、前立腺、リンパ腺、ならびに、消化管の癌細胞からなる群より選択される。
好ましくは、癌細胞は乳癌細胞である。より好ましくは、乳癌細胞はエルストン(Elston)のグレードがIIIの細胞である。最も好ましくは、乳癌細胞は転移性である。
本発明の第二の形態は、本発明の第一の形態に係る物質、および、製薬的に「許容できる賦形剤、希釈剤またはキャリアーを含む医薬組成物を提供する。従って、本発明はさらに、癌細胞の増殖を阻害するための医薬品を提供する。
本明細書で用いられるように、「医薬製剤」は、本発明に係る治療上有効な製剤を意味する。
本明細書で用いられる「治療有効量」または「有効量」または「治療上有効な」は、所定の条件および投与の処方計画で治療効果を提供する量を意味する。これは、必要な添加剤および希釈剤、すなわちキャリアーまたは投与用の基剤に関して望ましい治療効果が生じるように計算された活性物質の既定量である。さらに、これは、宿主の活性、機能および応答における臨床的に顕著な欠陥を減少させる、最も好ましくは予防するのに十分な量を意味することを目的とする。あるいは、治療有効量は、宿主の臨床的に顕著な状態に改善をもたらすのに十分な量である。当業者であれば当然であるが、化合物の量は、それらの特定の活性に応じて様々であってよい。適切な用量は、必要な希釈剤に関して望ましい治療効果が生じるように計算された既定量の活性組成物を含んでいてもよい。本発明の組成物の製造方法と使用において、治療有効量の活性成分が提供される。治療有効量は、通常の技術を有する医療または獣医学に関する作業者によって、患者の特徴、例えば年齢、体重、性別、状態、合併症、当業界周知のその他の病気などに基づき決定することができる。
従って、好ましい実施態様において、本発明は、癌細胞中のhCAP18/LL−37の活性を阻害するのに十分な量の本発明の物質、および、製薬上許容できるキャリアーを含む医薬製剤を提供する。
当業者であれば当然であるが、このような物質またはそれらの製剤の有効量は、単回大量投与(すなわち急性投与)として、または、より好ましくは長期にわたる連続した投与(すなわち長期にわたる投与)として送達してもよい。
本発明の物質は、使用される化合物の有効性/毒性、および、その使用される化合物の効能に応じて様々な濃度で製剤化することができる。好ましくは、この製剤は、本発明の物質を、0.1μM〜1mM、より好ましくは1μM〜100μM、5μM〜50μM、10μM〜50μM、20μM〜40μM、最も好ましくは約30μMの濃度で含む。インビトロでの用途の場合、製剤は、本発明の化合物を、より低い濃度で、例えば0.0025μM〜1μMで含んでいてもよい。
当業者であれば当然であるが、本発明の物質は、一般的に、目的とする投与経路および標準的な製薬上の実施を考慮して選択された適切な製薬用賦形剤、希釈剤またはキャリアーとの混合物として投与されると予想される(例えば、Remington:The Science and Practice of Pharmacy,第19版,1995,Alfonso Gennaro編,Mack Publishing Company,ペンシルベニア州,米国を参照)。
例えば、本発明の物質は、錠剤、カプセル、膣坐剤、エリキシル、溶液または懸濁液の形態で、経口、口腔または舌下投与することができ、これらはさらに、即時放出、遅延放出または制御放出の用途のための矯味矯臭薬剤または着色剤を含んでいてもよい。また、本発明の物質は、海綿体内注射によって投与してもよい。
このような錠剤は、賦形剤、例えば微結晶性セルロース、ラクトース、クエン酸ナトリウム、炭酸カルシウム、第二リン酸カルシウムおよびグリシン、崩壊剤、例えばスターチ(好ましくはトウモロコシ、ジャガイモまたはタピオカスターチ)、ナトリウムスターチグリコレート、クロスカルメロースナトリウム、および、ある種のケイ酸塩複合体、および、顆粒結合剤(granulation binder)、例えばポリビニルピロリドン、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシ−プロピルセルロース(HPC)、スクロース、ゼラチン、および、アラビアゴムを含んでいてもよい。加えて、潤滑剤、例えばステアリン酸マグネシウム、ステアリン酸、ベヘン酸グリセリルおよびタルクが含まれていてもよい。
またゼラチンカプセルにおいては、充填剤として類似のタイプの固形組成物を用いてもよい。これに関する好ましい賦形剤としては、ラクトース、スターチ、セルロース、乳糖、または、高分子量ポリエチレングリコールが挙げられる。水性懸濁液および/またはエリキシルの場合、本発明の化合物は、様々な甘味剤または矯味矯臭薬剤、着色物質または色素、乳化剤および/または懸濁化剤、および、希釈剤、例えば水、エタノール、プロピレングリコールおよびグリセリン、ならびに、それらの組み合わせと混合されていてもよい。
また、本発明の物質は、非経口投与することもでき、例えば、静脈内、関節内、動脈内、腹腔内、クモ膜下、心室内、胸骨内、頭蓋内、筋肉内または皮下に投与するか、または、輸液技術によって投与してもよい。これらは滅菌水溶液の形態で用いられることが最も良く、滅菌水溶液は、その他の物質を含んでいてもよく、例えば、溶液を血液と等張にするのに適した量の塩またはグルコースを含んでいてもよい。このような水溶液は、必要に応じて、適切に(好ましくはpH3〜9に)緩衝化されているべきである。滅菌条件下での適切な非経口製剤の製造は、当業者周知の標準的な製薬技術によって容易に達成される。
非経口投与に適した製剤としては、水性および非水性滅菌注射液が挙げられ、これは、抗酸化剤、緩衝液、静菌薬、および、製剤を対象のレシピエントの血液と等張にする溶質を含んでいてもよく;さらに、水性および非水性滅菌懸濁液が挙げられ、これは、懸濁化剤および増粘剤を含んでいてもよい。このような製剤は、単位用量または複数回投与のための容器、例えば密封したアンプルおよびバイアルの中に入れてもよく、さらに、凍結乾燥した(freeze−dried)(凍結乾燥した(lyophilised))状態でに保存してもよく、このような製剤は、使用直前に注射用の滅菌液状キャリアー(例えば水)を添加するだけでよい。即時型の注射液および懸濁液は、これまで説明した種類の滅菌粉末、顆粒および錠剤から製造することもできる。
人間の患者への経口および非経口投与のためには、本発明の物質の1日の用量レベルは、通常、成人一人当たり1〜1000mg(すなわち約0.015〜15mg/kg)と予想され、1回の用量で投与してもよいし、または、複数回の用量で投与してもよい。
また、本発明の物質は、鼻腔内投与してもよく、または、吸入法によって投与してもよく、都合のよい形態としては、乾燥粉末の吸入器の形態で送達されるか、または、加圧容器、ポンプ、スプレーまたはネブライザーからのエアロゾルスプレー噴射の形態で適切な噴射剤を使用して送達され、ここにおいて、適切な噴射剤としては、例えばジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、ヒドロフルオロアルカン、例えば1,1,1,2−テトラフルオロエタン(HFA134A3または1,1,1,2,3,3,3−ヘプタフルオロプロパン(HFA227EA3)、二酸化炭素、またはその他の適切なガスが挙げられる。加圧されたエアロゾルの場合、投与単位は、定量を送達するバルブを提供することによって決定してもよい。加圧容器、ポンプ、スプレーまたはネブライザーは、例えばエタノールと噴射剤(溶媒として)との混合物を用いた活性化合物の溶液または懸濁液を含んでいてもよく、この溶液または懸濁液は、加えて、潤滑剤(例えばトリオレイン酸ソルビタン)を含んでいてもよい。吸入器または注入器に使用するためのカプセルおよびカートリッジ(例えばゼラチンで製造された)は、本発明の化合物と、適切な粉末ベース(例えばラクトースまたはスターチ)との粉末混合物が含まれるように製剤化されてもよい。
エアロゾルまたは乾燥粉末製剤は、好ましくは、定量または「一吹き」それぞれが、患者に送達するための本発明の化合物を少なくとも1mg含むように調整される。当然のことながら、エアロゾルを用いた場合の総体的な1日用量は患者に応じて様々であると予想され、単回投与で投与してもよいし、または、より一般的には一日で複数回の用量で投与してもよい。
あるいは、本発明の物質は、坐剤または膣坐剤の形態で投与することができ、または、ローション、溶液、クリーム、軟膏または散粉剤の形態で局所に塗布してもよい。また、本発明の化合物は、例えば皮膚用パッチ剤の使用によって、経皮投与してもよい。また、これらは眼の経路で投与してもよい。
眼に使用する場合、本発明の物質は、等張のpH調節した滅菌生理食塩水中の微粉化した懸濁液として、または、好ましくは、場合により塩化ベンザルコニウムのような保存剤と組み合わせた、等張のpH調節した滅菌生理食塩水の溶液として、製剤化することができる。あるいは、本発明の物質は、ワセリンのような軟膏に製剤化しもよい。
皮膚に局所塗布するために、本発明の物質は、例えば以下:ミネラルオイル、液体ワセリン、白色ワセリン、プロピレングリコール、ポリオキシエチレンポリオキシプロピレン化合物、乳化ろう、および、水のうち1種またはそれ以上の混合物に懸濁または溶解させた活性化合物を含む適切な軟膏として製剤化することができる。あるいは、本発明の物質は、例えば以下:ミネラルオイル、ソルビタンモノステアレート、ポリエチレングリコール、流動パラフィン、ポリソルベート60、セチルエステルワックス、セテアリルアルコール、2−オクチルドデカノール、ベンジルアルコール、および、水のうち1種またはそれ以上の混合物に懸濁または溶解させた適切なローションまたはクリームとして製剤化することができる。
口への局所投与に適した製剤としては、ロゼンジ(これは、フレーバーを付けた基剤、一般的にはスクロースおよびアラビアゴム、または、トラガカント中に活性成分を含む);香錠(これは、不活性な基剤、例えばゼラチンおよびグリセリン、または、スクロースおよびアラビアゴム中に活性成分を含む);および、うがい薬(これは、適切な液状キャリアー中に活性成分を含む)が挙げられる。
該物質がポリペプチドの場合、持続放出性の薬物送達システムを使用することが好ましい場合があり、例えばマイクロスフェアである。これらは特に、注射の頻度が少なくなるように設計される。このようなシステムの例は、ニュートロピン(Nutropin)デポー剤であり、これは、生分解性のマイクロスフェアに組換えヒト成長ホルモン(rhGH)をカプセル封入したものであり、一度注射されると、ある期間持続的にrhGHを徐放する。
あるいは、本発明のポリペプチド剤は、必要な部位に薬物を直接放出する外科的に埋め込まれた装置によって投与することができる。
また、タンパク質およびポリペプチドを投与するために、エレクトロポレーション治療(EPT)システムを用いることもできる。細胞にパルス電場を送達する装置は、細胞膜の薬物に対する透過性を高め、それにより、細胞内薬物送達がかなり強化される。
また、タンパク質およびポリペプチドは、エレクトロインコーポレーション(electroincorporation;EI)によって送達することもできる。皮膚表面上の直径30ミクロン以下の小さい粒子が、エレクトロポレーションで用いられる電気パルスと同一な、または、それに類似した電気パルスを受けると、EIが起こる。EIにおいて、これらの粒子は角質層を通過して、皮膚のより深い層に侵入する。このような粒子は、薬物または遺伝子を充填していてもよいし、もしくはそれらで被覆されていてもよく、または、単に、皮膚に孔を生成する「弾丸」(そこを通過して薬物が入る)として作用してもよい。
タンパク質およびポリペプチドを送達する代替法は、注射用の感熱性のReGelである。体温より低温では、ReGelは注射可能な液体であるが、体温では、それは即座にゲルの貯蔵所を形成し、それが既知の安全な生分解性ポリマーにゆっくり浸透し溶解する。バイオポリマーが溶解するにつれて、活性な薬物が長期にわたり送達される。
また、タンパク質およびポリペプチド医薬は、経口的に送達することもできる。このようなシステムの一つは、体内の経口によるビタミンB12摂取のための天然のプロセスを用いて、タンパク質およびポリペプチドを共に送達するものである。ビタミンB12摂取システムに便乗することによって、タンパク質またはポリペプチドは腸壁を通過して移動が可能になり、ビタミンB12類似体と薬物との複合体が生産され、この複合体は、複合体のビタミンB12部分における内因子(IF)への有意な親和性と、複合体の薬物部分の有意な生物活性との両方を保持する。
また、本発明のオリゴヌクレオチドまたはポリヌクレオチド剤の投与方法は、当業界でもよく知られている(Dass,2002,J Pharm Pharmacol.54(1):3〜27;Dass,2001,Drug Deliv.8(4):191〜213;Lebedeva等,2000,Eur J Pharm Biopharm.50(1):101〜19;Pierce等,2005,Mini Rev Med Chem.5(1):41〜55;LysikおよびWu−Pong,2003,J Pharm Sci.2003 2(8):1559〜73;Dass,2004,Biotechnol Appl Biochem.40(Pt 2):113〜22;Medina,2004,Curr Pharm Des.10(24):2981〜9を参照)。
例えば、本発明のコンストラクトは、レトロウイルスに関連する方法によってコンストラクトが細胞のゲノムに挿入されるようにして、細胞に導入することもできる。例えば、Kuriyama等(1991)Cell Struc.and Func.16,503〜510では、精製したレトロウイルスが投与されている。上述のようなポリヌクレオチドを含むレトロウイルスのDNAコンストラクトは、当業界周知の方法を用いて作製することができる。このようなコンストラクトから活性なレトロウイルスを生産するために、通常、10%ウシ胎仔血清(FCS)を含むダルベッコ改変イーグル培地(DMEM)中で増殖させたエコトロピック・サイ2(ecotropic psi2)パッケージング細胞系を使用する。細胞系のトランスフェクションは、都合のよい形態として、リン酸カルシウムの共沈殿によってなされ、安定な形質転換株が、G418を最終濃度1mg/mlまで添加することによって選択される(レトロウイルスのコンストラクトが、neoR遺伝子を含むと仮定する)。独立したコロニーを単離し、拡張し、培養上清を除去し、孔サイズ0.45μmのフィルターでろ過し、−70℃で保存する。腫瘍細胞にレトロウイルスを導入するためには、10μg/mlのポリブレンが添加されたレトロウイルス上清を直接注入することが便利である。直径10mmを超える腫瘍の場合、レトロウイルス上清を0.1ml〜1ml;好ましくは0.5ml注入することが適切である。
あるいは、Culver等(1992)Science 256,1550〜1552で説明されているように、レトロウイルスを生産する細胞が注射される。このようにして導入されたレトロウイルス生産細胞は、腫瘍塊内でインサイチュでベクターの連続生産が起こるように、レトロウイルスベクター粒子を活発に生産するように加工される。従って、増殖中の細胞は、レトロウイルスベクター生産細胞と混合されると、インビボでうまく形質導入を起こすことができる。
また、標的化されたレトロウイルスも本発明での使用に利用可能である;例えば、特異的な結合親和性を付与する配列を、予め存在するウイルスenv遺伝子に組み入れてもよい(このような標的化ベクターおよびその他の遺伝子治療のための標的化ベクターの総論として、MillerおよびVile(1995)Faseb J.9,190〜199を参照)。
その他の方法は、限られた時間、または、ゲノムに組み込んだ後より長い時間、コンストラクトを細胞で発現させるための、コンストラクトの細胞への単純な送達に関する方法である。後者のアプローチの例としては、リポソームが挙げられる(Nassander等(1992)Cancer Res.52,646〜653)。
イムノリポソームの製造のために、MartinおよびPapahadjopoulos(1982)J.Biol.Chem.257,286〜288の方法に従って、MPB−PE(N−[4−(p−マレイミドフェニル)ブチリル]−ホスファチジルエタノールアミン)が合成される。MPB−PEはリポソームの二重層に取り込まれ、抗体またはそれらのフラグメントを、リポソーム表面に共有結合させることができる。都合のよい形態としては、このようなリポソームは、標的細胞への送達のために本発明の物質(例えば、DNAまたはその他の遺伝学的なコンストラクト)が充填されており、これは例えば、上記物質の溶液中で前記リポソームを形成し、続いて、孔径0.6μmおよび0.2μmを有するポリカーボネートメンブレンフィルターを介して0.8MPa以下の窒素圧下で連続的に押出すことによってなされる。押出し後、捕獲されたDNAコンストラクトは、80000×gで45分間超遠心分離することによって遊離のDNAコンストラクトから分離される。脱酸素した緩衝液中で新たに製造されたMPB−PE−リポソームを、新たに製造された抗体(またはそれらのフラグメント)と混合し、窒素雰囲気中、4℃で一定して転倒回転させて、一晩カップリング反応を行う。イムノリポソームは、80000×gで45分間超遠心分離することによって未結合の抗体から分離される。イムノリポソームは、腹腔内に注射してもよいし、または、腫瘍に直接注射してもよい。
その他の送達方法としては、抗体−ポリリジン架橋を介したアデノウイルスを有する外来DNA(Curiel Prog.Med.Virol.40,1〜18を参照)、および、キャリアーとしてのトランスフェリン−ポリカチオン結合体(Wagner等(1990)Proc.Natl.Acad.Sci.USA 87,3410〜3414)が挙げられる。これらの方法はまず、本発明のオリゴヌクレオチド剤を用いてポリカチオン−抗体複合体を形成し(この抗体は、野生型アデノウイルス、または、変異型アデノウイルスのいずれかに特異的である)、この段階で抗体に結合する新しいエピトープが導入されている。このオリゴヌクレオチド剤に、リン酸骨格との静電的相互作用を介してポリカチオン部分が結合する。アデノウイルスは、変化していないファイバーとペントンタンパク質を含むため、細胞の内側に入り、自身と共に本発明のオリゴヌクレオチド剤を細胞に運搬する。ポリカチオンがポリリジンである場合が好ましい。
また、オリゴヌクレオチド剤はアデノウイルスによって送達されてもよく、ここにおいて、オリゴヌクレオチド剤は、例えば以下で説明されるようにアデノウイルス粒子内に存在する。
代替法で、受容体介在エンドサイトーシスを利用してDNA高分子を細胞に運搬する高効率の核酸送達システムが用いられる。これは、鉄−輸送タンパク質トランスフェリンを、核酸と結合するポリカチオンに結合させることによって達成される。ヒトトランスフェリン、もしくは、ニワトリ相同体コンアルブミン、またはそれらの組み合わせは、小さいDNA結合タンパク質プロタミンに、または、様々なサイズのポリリジンにジスルフィド結合を介して共有結合する。これらの改変トランスフェリン分子は、それらの同源の受容体と結合し、効率的な細胞への鉄輸送を媒介するそれらの能力を維持している。トランスフェリン−ポリカチオン分子は、核酸サイズとは無関係に(短いオリゴヌクレオチドから、21キロ塩基対のDNAに至る)、本発明のDNAコンストラクトまたはその他の遺伝学的コンストラクトと共に電気泳動で安定な複合体を形成する。トランスフェリン−ポリカチオン複合体と、本発明のDNAコンストラクトまたはその他の遺伝学的コンストラクトとが腫瘍細胞に供給されると、細胞中でコンストラクトからの高レベルの発現が期待される。
また、Cotten等(1992)Proc.Natl.Acad.Sci.USA 89,6094〜6098の方法によって生産された欠陥のある、または化学的に不活性化されたアデノウイルス粒子のエンドソームの破壊活性を用いた、本発明のDNAコンストラクトまたはその他の遺伝学的コンストラクトの高効率の受容体介在送達を用いてもよい。このアプローチは、アデノウイルスは、リソソームを経由する通路ではなく、例えば本発明のDNAコンストラクトまたはその他の遺伝学的コンストラクトに結合しているトランスフェリンの存在下でエンドソームからそれらのDNAが放出されるように適合されるという事実に依存するようであり、このコンストラクトは、アデノウイルス粒子と同じ経路で細胞に取り込まれる。
このアプローチは、複合型のレトロウイルスコンストラクトを使用するする必要がない;レトロウイルス感染に付随して起こるようなゲノムの永続的な改変が起こらない;および、標的化された発現系は、標的化された送達システムとカップリングされるため、その他の細胞型への毒性を減少させるという利点を有する。
当然のことながら、「裸のDNA」、および、カチオン性および中性脂質と複合体化したDNAも、本発明のDNAを治療しようとする個体の細胞に導入することにおいて有用であり得る。遺伝子治療への非ウイルスアプローチは、Ledley(1995)Human Gene Therapy 6,1129〜1144で説明されている。
また、その代わりの標的化送達システムも既知であり、例えばWO94/10323で説明されている改変アデノウイルスシステムであり、ここにおいて、DNAは、典型的にはアデノウイルスまたはアデノウイルス様粒子内に保持されている。Michael等(1995)Gene Therapy 2,660〜668では、ファイバータンパク質に細胞選択的な部分を付加するためのアデノウイルスの改変を説明している。また、Bischoff等(1996)Science 274,373〜376で説明されているのような、p53欠失ヒト腫瘍細胞中で選択的に複製する突然変異アデノウイルスも、細胞に本発明の遺伝学的コンストラクトを運搬するのに有用である。従って、当然のことながら、本発明のさらなる形態は、本発明の遺伝学的コンストラクトを含むウイルスまたはウイルス様粒子を提供する。その他の適切なウイルスまたはウイルス様粒子としては、HSV、AAV、ワクシニア、および、パルボウイルスが挙げられる。
さらなる実施態様において、hCAP18/LL−37の機能を選択的に予防する物質は、標的化hCAP18/LL−37のmRNAまたはDNAを切断することができるリボザイムである。前記リボザイムを発現する遺伝子は、アンチセンス分子の場合と実質的に同様に、および、アンチセンス分子での賦形剤と実質的に同じ賦形剤を用いて投与してもよい。
本明細書で開示されたウイルスまたはウイルス様粒子のゲノムにコード可能なリボザイムは、US5,180,818、US5,168,053、US5,149,796、US5,116,742、US5,093,246、および、US4,987,071で説明されている。
当然のことながら、細胞特異的プロモーター構成要素から、アンチセンス分子またはリボザイムが発現されることが望ましい場合がある。
本発明の製剤の具体的に好ましい実施態様において、該製剤は、本発明の物質の癌細胞への標的化された送達が可能である。
さらに当業者であれば当然ながら、本発明の物質および医薬製剤は、医学分野と獣医学分野の両方において有用性を有する。従って、本発明の物質は、ヒトの治療で用いてもよいし、ヒト以外の動物(例えばウマ、イヌおよびネコ)の治療で用いてもよい。しかしながら、好ましくは患者はヒトである。
本発明の第三形態は、患者における癌細胞の増殖を阻害する方法を提供し、該方法は、患者に、本発明の第一の形態に係る物質、または、本発明の第二の形態に係る医薬製剤を投与することを含む。
好ましくは、患者はヒトである。
好ましくは、患者はヒトである。
有利には、該物質は、癌細胞に選択的に送達されるか、または、癌細胞によって選択的に活性化される。
従って、本発明はさらに、医薬に使用するための、本発明の第一の形態に係る物質を提供する。
好ましくは、該物質は、癌の治療に使用するためである。
好ましくは、該物質は、癌の治療に使用するためである。
「治療」は、ここでは、患者の治療的処置と予防的処置の両方を含める。用語「予防」は、本明細書で説明されているポリペプチドまたは製剤を、患者または被検体において癌を予防する、または、癌の可能性を減少させるために使用することを包含するものとして用いられる。
上記で考察されたように、用語「有効量」は、本明細書において、治療される病気または状態に好都合な変化を起こすために使用可能な、本発明に係る化合物の濃度または量を説明するために用いられ、ここにおいて、このような変化は、治療された病気または状態に応じて、治療された病状または状態の緩和、好都合な生理学的な結果、回復または低減、状態または病状が発生する見込みの予防または減少である。本発明の物質が組み合わせて用いられる場合、それぞれの物質は、有効量で用いてもよく、ここにおいて、有効量は相乗効果を生じる量を含んでいてもよい。
本発明のさらにその他の形態は、癌細胞の増殖を阻害するための医薬品の製造における、本発明の第一の形態に係る物質を提供する。
有利には、癌細胞(上皮性の癌細胞)は上皮細胞である。あるいは、癌細胞は扁平細胞であってもよい。
有利には、癌細胞(上皮性の癌細胞)は上皮細胞である。あるいは、癌細胞は扁平細胞であってもよい。
都合のよい形態としては、癌細胞は、乳房、胆管、脳、結腸、胃、生殖器、肺および気道、皮膚、胆嚢、肝臓、上咽頭、神経細胞、腎臓、前立腺、リンパ腺、ならびに、消化管の癌細胞からなる群より選択される。
好ましくは、癌細胞は乳癌細胞である。より好ましくは、乳癌細胞はエルストン(Elston)のグレードがIIIの細胞である。最も好ましくは、乳癌細胞は転移性である。
本発明の第六の形態は、患者における癌細胞を検出する方法を提供し、該方法は、以下の工程を含む:
(a)試験しようとする患者からの細胞サンプルを提供する工程;
(b)該細胞によって生産されたhCAP18/LL−37の量を(直接的または間接的に)測定する工程;および、
(c)工程(b)で測定されたhCAP18/LL−37の量と、健康な細胞によって生産されたhCAP18/LL−37の量とを比較する工程(ここにおいて、該患者からの細胞サンプルにおけるhCAP18/LL−37の生産のレベルが、健康な細胞におけるレベルと比較して高いことは、該細胞が癌細胞であることを示す)。
従って、本方法は、患者の癌を診断する方法を提供する。
(a)試験しようとする患者からの細胞サンプルを提供する工程;
(b)該細胞によって生産されたhCAP18/LL−37の量を(直接的または間接的に)測定する工程;および、
(c)工程(b)で測定されたhCAP18/LL−37の量と、健康な細胞によって生産されたhCAP18/LL−37の量とを比較する工程(ここにおいて、該患者からの細胞サンプルにおけるhCAP18/LL−37の生産のレベルが、健康な細胞におけるレベルと比較して高いことは、該細胞が癌細胞であることを示す)。
従って、本方法は、患者の癌を診断する方法を提供する。
好ましい実施態様において、工程(a)における細胞は、乳房、胆管、脳、結腸、胃、生殖器、肺および気道、皮膚、胆嚢、肝臓、上咽頭、神経細胞、腎臓、前立腺、リンパ腺、ならびに、消化管の細胞からなる群より選択される。
例えば、工程(a)における細胞サンプルは、腫瘍由来でもよいし、または、腫瘍の可能性がある組織由来でもよい。
例えば、工程(a)における細胞サンプルは、腫瘍由来でもよいし、または、腫瘍の可能性がある組織由来でもよい。
好ましくは、工程(b)は、該細胞サンプルと、hCAP18/LL−37に結合する物質とを接触させること、続いて、それに結合したhCAP18/LL−37の量を検出することを含む。
サンプル中のタンパク質検出に適した方法は、当業界周知であり、例えばラジオイムノアッセイおよびELISAである。
サンプル中のタンパク質検出に適した方法は、当業界周知であり、例えばラジオイムノアッセイおよびELISAである。
有利には、hCAP18/LL−37に結合する物質は、抗体、または、それらの抗原結合フラグメントである。
一実施態様において、工程(b)は、(i)該細胞と、hCAP18/LL−37に結合する物質とを接触させること、および、(ii)抗体、または、それらの抗原結合フラグメントを用いて、該物質に結合するhCAP18/LL−37の量を検出することを含む。例えば、工程(b)は、ELISAによって行ってもよい。
その代わりの実施態様において、工程(b)は、該細胞中のhCAP18/LL−37のmRNAの量を測定することを含む。適切な方法は当業界周知である(例えば、Molecular Cloning:a Laboratory Manual,3rd edition,SambrookおよびRussell,2001,Cold Spring Harbor Laboratory Pressを参照)。
都合のよい形態としては、工程(b)は、サザンブロット、または、RT−PCRによって行われる。
本発明の第七の形態は、患者における癌の進行をモニターする方法を提供し、該方法は:
(a)第一の時点で患者から回収された細胞のサンプルを提供すること、および、本発明の第六の形態に記載の方法を用いて、そこに含まれる癌細胞を検出すること;
(b)第二の時点で患者から回収された細胞のサンプルを提供すること’、および、本発明の第六の形態に記載の方法を用いて、そこに含まれる癌細胞を検出すること;および、
(c)工程(a)および(b)で測定された癌細胞の数を比較すること(ここにおいて、工程(b)で測定された癌細胞の数が、工程(a)と比較して高いことは、癌が進行していることの指標である)、
を含む。
(a)第一の時点で患者から回収された細胞のサンプルを提供すること、および、本発明の第六の形態に記載の方法を用いて、そこに含まれる癌細胞を検出すること;
(b)第二の時点で患者から回収された細胞のサンプルを提供すること’、および、本発明の第六の形態に記載の方法を用いて、そこに含まれる癌細胞を検出すること;および、
(c)工程(a)および(b)で測定された癌細胞の数を比較すること(ここにおいて、工程(b)で測定された癌細胞の数が、工程(a)と比較して高いことは、癌が進行していることの指標である)、
を含む。
本発明のさらなる形態は、hCAP18/LL−37、または、hCAP18/LL−37のmRNAに結合する物質を含む、本発明の第六の形態に記載の方法を行うための診断キットを提供する。
好ましい実施態様において、本キットは、hCAP18/LL−37に結合することができる抗体、または、それらの抗原結合フラグメントを含む(上記参照)。本キットは、例えばELISAに使用するための、このような一次抗体に結合することができる二次抗体をさらに含んでいてもよい。
その代わりの実施態様において、本キットは、hCAP18/LL−37のmRNAに選択的にハイブリダイズすることができるオリゴヌクレオチドを含む。好ましくは、このようなオリゴヌクレオチドは、配列番号1に記載のヌクレオチドの配列フラグメント、または、それらの変異体を含む、または、それらからなる。当然のことながら、プローブは、高いストリンジェンシー条件下でhCAP18/LL−37のmRNAに選択的にハイブリダイズすることができるものと予想される。有利には、本キットは、hCAP18/LL−37のmRNAのPCR増幅に適したプライマー対を含む。
標的mRNAに選択的にハイブリダイズすることができるPCRプライマーおよびその他のハイブリダイゼーションプローブの設計、および、それらの使用方法は当業界周知であり、例えば、上記のSambrookおよびRussellを参照。
好ましい実施態様において、hCAP18/LL−37に結合する物質、および/または、hCAP18/LL−37のmRNAに選択的にハイブリダイズすることができる物質は、検出可能な部分を含む。
「検出可能な」は、ここでは、検出可能な種の生産に直接的または間接的に関与する単一の原子および分子を含める。適切な検出可能な成分は医薬品化学において周知であり、これらの成分のポリペプチドおよびタンパク質への結合も当業界周知である。検出可能な成分の例としては、これらに限定されないが、以下が挙げられる:放射性同位体(例えば、3H、14C、35S、123I、125I、131I、99Tc、111In、90Y、188Re)、放射性核種(例えば、11C、18F、64Cu)、蛍光標識(例えば、FITC、ローダミン、ランタニド・リン(lanthanide phosphor)、カルボシアニン)、酵素標識(例えば、ホースラディッシュペルオキシダーゼ、β−ガラクトシダーゼ、ルシフェラーゼ、アルカリホスファターゼ)、化学発光性のビオチニル基、および、第二のレポーターによって認識された既定のポリペプチドエピトープ(例えば、ロイシンジッパー対の配列、二次抗体のための結合部位、金属結合ドメイン、エピトープタグ)。いくつかの実施態様において、標識は、立体障害が起こる可能性を減少させるために様々な長さのスペーサーアームによって取り付けられる。
好ましくは、本診断キットは、少なくとも1回の分析に十分な量の診断薬を、別々に包装された試薬として含む。また、包装された試薬の使用説明書も通常含まれる。このような説明書は通常、試薬濃度、および/または、少なくとも1つの分析法パラメーター、例えば、混合する試薬とサンプルの相対量、試薬/サンプル混合物の維持期間、温度、緩衝液条件などを説明する具体的な表現を含む。
本発明の好ましい形態を、以下の図を参照しながら、以下の非限定的な実施例で説明する:
図1−hCAP18/LL−37は、乳癌で高度に発現される
(A)グレードIIIの乳管内乳癌(患者番号7,表2)の切片であり、島状の間質(st)を取り囲んで腫瘍細胞中のhCAP18タンパク質に関する強い免疫反応性を示す(赤色の沈殿)。(B)インサイチュハイブリダイゼーションは、同じ組織からの切片中のhCAP18のmRNAに適合するシグナルを示す。強いオートラジオグラフィーシグナルは、暗視野照明下では白色の粒子のように見える。(C)癌細胞の高出力の表示は、免疫反応性を欠いている腫瘍細胞に隣接する免疫反応細胞を強く実証している。(D)血管内のhCAP18免疫反応性の乳癌細胞である。(E)カテリン組換えペプチドを用いた免疫吸着法によって、hCAP18の免疫反応性が完全になくなった(図1Aと同じ組織)。(F)免疫吸着法の際のポジティブコントロールとしての、hCAP18に関する一般的な免疫染色である(図1Aと同じ組織)。(G)正常な乳腺上皮は、hCAP18に対して弱い免疫反応性を示す。顕微鏡写真(A,C〜G)は、hCAP18抗体(1:500希釈)を用いて得られた結果を示す。スケールバー(A,B)=100μm;(C,D)=25μm;(E,F,G)=10μm。
図1−hCAP18/LL−37は、乳癌で高度に発現される
(A)グレードIIIの乳管内乳癌(患者番号7,表2)の切片であり、島状の間質(st)を取り囲んで腫瘍細胞中のhCAP18タンパク質に関する強い免疫反応性を示す(赤色の沈殿)。(B)インサイチュハイブリダイゼーションは、同じ組織からの切片中のhCAP18のmRNAに適合するシグナルを示す。強いオートラジオグラフィーシグナルは、暗視野照明下では白色の粒子のように見える。(C)癌細胞の高出力の表示は、免疫反応性を欠いている腫瘍細胞に隣接する免疫反応細胞を強く実証している。(D)血管内のhCAP18免疫反応性の乳癌細胞である。(E)カテリン組換えペプチドを用いた免疫吸着法によって、hCAP18の免疫反応性が完全になくなった(図1Aと同じ組織)。(F)免疫吸着法の際のポジティブコントロールとしての、hCAP18に関する一般的な免疫染色である(図1Aと同じ組織)。(G)正常な乳腺上皮は、hCAP18に対して弱い免疫反応性を示す。顕微鏡写真(A,C〜G)は、hCAP18抗体(1:500希釈)を用いて得られた結果を示す。スケールバー(A,B)=100μm;(C,D)=25μm;(E,F,G)=10μm。
図2−乳癌において、hCAP−18/LL−37は免疫ブロッティングで検出される
患者の臨床データは表2に示す(サンプル1〜10)。組換えカテリン(C)とLL−37ペプチド(L)を大きさの参照として用いた。正常な乳房組織は、レーン1に示される。エルストングレードIの腫瘍は、レーン2、4および5に示される。グレードIIの腫瘍は、レーン3に示され、グレードIIIの腫瘍は、レーン6〜10に示される。全ての組織において、無傷の未加工の18kDaのホロタンパク質に相当する免疫反応を示すバンドが見つかった。5種のグレードIIIの腫瘍(番号7〜10)のうち4種に、加工したLL−37ペプチド(4kD)が見られた。
患者の臨床データは表2に示す(サンプル1〜10)。組換えカテリン(C)とLL−37ペプチド(L)を大きさの参照として用いた。正常な乳房組織は、レーン1に示される。エルストングレードIの腫瘍は、レーン2、4および5に示される。グレードIIの腫瘍は、レーン3に示され、グレードIIIの腫瘍は、レーン6〜10に示される。全ての組織において、無傷の未加工の18kDaのホロタンパク質に相当する免疫反応を示すバンドが見つかった。5種のグレードIIIの腫瘍(番号7〜10)のうち4種に、加工したLL−37ペプチド(4kD)が見られた。
図3−上皮細胞におけるhCAP18のトランスジェニック発現は、細胞増殖を高める
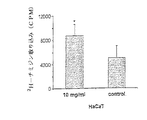
(A)上のパネル、左のレーン;HEK293抽出物における抗LL37抗血清での免疫ブロッティングである。バイシストロン性のベクターhCAP18+EGFP(hCAP18/E)でトランスフェクションされた細胞は、hCAP18タンパク質の発現を示す。上のパネル、右のレーン;EGFP(E)のみでトランスフェクションされたHEK293細胞である。下のパネル;HEK293細胞(hCAP18/E)は、コントロール細胞(E)と比較してより有意に高い増殖速度を示した(フローサイトメトリーで評価)。ポンソー染色は、ローディング用のコントロールとして示される。(B)上のパネル、左のレーン;(A)で説明されているようなトランスフェクションされたHaCaT細胞である。下のパネル;hCAP18でトランスフェクションされたHaCaT細胞は、コントロール細胞と比較してより有意に高い増殖速度を示す(3H−チミジン取り込みで評価)。
(A)上のパネル、左のレーン;HEK293抽出物における抗LL37抗血清での免疫ブロッティングである。バイシストロン性のベクターhCAP18+EGFP(hCAP18/E)でトランスフェクションされた細胞は、hCAP18タンパク質の発現を示す。上のパネル、右のレーン;EGFP(E)のみでトランスフェクションされたHEK293細胞である。下のパネル;HEK293細胞(hCAP18/E)は、コントロール細胞(E)と比較してより有意に高い増殖速度を示した(フローサイトメトリーで評価)。ポンソー染色は、ローディング用のコントロールとして示される。(B)上のパネル、左のレーン;(A)で説明されているようなトランスフェクションされたHaCaT細胞である。下のパネル;hCAP18でトランスフェクションされたHaCaT細胞は、コントロール細胞と比較してより有意に高い増殖速度を示す(3H−チミジン取り込みで評価)。
図4−合成LL−37ペプチドを用いた治療により、上皮細胞の細胞増殖が増加する
血清飢餓によって72時間同調させ、次に、10μg/mlの合成の生物学的に活性なLL−37ペプチドで36時間処理した(DMEM+5%FCS+PEST中で)HaCaT細胞は、未処理の(コントロール)HaCaT細胞と比較して有意に増加した細胞増殖を示す。増殖速度を[3H]−チミジンの取り込みを用いて評価した。
血清飢餓によって72時間同調させ、次に、10μg/mlの合成の生物学的に活性なLL−37ペプチドで36時間処理した(DMEM+5%FCS+PEST中で)HaCaT細胞は、未処理の(コントロール)HaCaT細胞と比較して有意に増加した細胞増殖を示す。増殖速度を[3H]−チミジンの取り込みを用いて評価した。
図5−LL−37受容体FPRL1は、乳癌および正常な乳腺上皮で発現される
(A)腫瘍細胞中のFPRL1受容体に対して顕著な免疫反応性を有する、エルストンのグレード2の乳管内乳癌(患者番号12、表2)の切片(赤色の沈殿)である。(B)管内領域でFPRL1に対する免疫反応性を示す正常な乳腺上皮の切片である(赤色の沈殿)。顕微鏡写真は、1:400で希釈したFPRL1抗血清を用いてで得られた結果を示す。スケールバー(A)=50μm;(B)=10μm。(C)免疫ブロッティングにより、LL−37受容体、FPRL1が、正常な(N)組織と乳癌(T)組織の両方で発現されたことが明らかになった。(D)リアルタイムPCRにより、バイシストロン性のベクターhCAP18+EGFP(hCAP18/E)でトランスフェクションされたHaCaTは、FPRL1受容体mRNAの有意に増加した発現を示す。EGFP(E)のみでトランスフェクションされたHaCaT細胞を、コントロールとして利用した。
(A)腫瘍細胞中のFPRL1受容体に対して顕著な免疫反応性を有する、エルストンのグレード2の乳管内乳癌(患者番号12、表2)の切片(赤色の沈殿)である。(B)管内領域でFPRL1に対する免疫反応性を示す正常な乳腺上皮の切片である(赤色の沈殿)。顕微鏡写真は、1:400で希釈したFPRL1抗血清を用いてで得られた結果を示す。スケールバー(A)=50μm;(B)=10μm。(C)免疫ブロッティングにより、LL−37受容体、FPRL1が、正常な(N)組織と乳癌(T)組織の両方で発現されたことが明らかになった。(D)リアルタイムPCRにより、バイシストロン性のベクターhCAP18+EGFP(hCAP18/E)でトランスフェクションされたHaCaTは、FPRL1受容体mRNAの有意に増加した発現を示す。EGFP(E)のみでトランスフェクションされたHaCaT細胞を、コントロールとして利用した。
図6−エストロゲン受容体(ER)、および、リンパ節(N)陽性の乳房腫瘍における、hCAP18/LL−37の発現の増加(対数目盛で表示)である
RNAを、140種の乳房腫瘍と4種の罹患していない乳房組織サンプルから抽出し、プライマーとしてランダムヘキサマーを用いて逆転写した。hCAP18転写物の発現を、標準的なプロトコールに従ってリアルタイムPCRでcDNA10ngを用いて決定した。このサンプルを18S−RNAの定量化によって標準化した。罹患していないサンプルの平均の発現を、任意に1に設定した。平均および偏差はAnova統計によって評価される。
RNAを、140種の乳房腫瘍と4種の罹患していない乳房組織サンプルから抽出し、プライマーとしてランダムヘキサマーを用いて逆転写した。hCAP18転写物の発現を、標準的なプロトコールに従ってリアルタイムPCRでcDNA10ngを用いて決定した。このサンプルを18S−RNAの定量化によって標準化した。罹患していないサンプルの平均の発現を、任意に1に設定した。平均および偏差はAnova統計によって評価される。
図7−HEK細胞(A)およびHaCat細胞(B)において、LL−37は、カンプトテシンにより誘導されたアポトーシスを阻害する
A.サブコンフルエントな細胞を、様々なLL−37(1/2および4μM)濃度で24時間処理した。培地単独で処理した細胞をコントロール細胞(a)として用いた。次に、この細胞を、6μMカンプトテシンの非存在下(a〜d)または存在下(e〜h)でさらに24時間培養した。回収した細胞を、2色のアポトーシス分析とフローサイトメトリーで加工し、蛍光強度に従い象限解析によって、生存可能(蛍光がない、または、ほんのわずか)、アポトーシス(FL−1YOPRO色素に関して陽性)、および、壊死(FL−1とPI蛍光の両方に関して陽性)と分類した。この図は、それぞれ3連で行われた3つの独立した実験の代表を示す。
B.アポトーシス細胞の定量分析である。
A.サブコンフルエントな細胞を、様々なLL−37(1/2および4μM)濃度で24時間処理した。培地単独で処理した細胞をコントロール細胞(a)として用いた。次に、この細胞を、6μMカンプトテシンの非存在下(a〜d)または存在下(e〜h)でさらに24時間培養した。回収した細胞を、2色のアポトーシス分析とフローサイトメトリーで加工し、蛍光強度に従い象限解析によって、生存可能(蛍光がない、または、ほんのわずか)、アポトーシス(FL−1YOPRO色素に関して陽性)、および、壊死(FL−1とPI蛍光の両方に関して陽性)と分類した。この図は、それぞれ3連で行われた3つの独立した実験の代表を示す。
B.アポトーシス細胞の定量分析である。
図8−HEKn細胞をLL−37で前処理することによって、カンプトテシンにより誘導されたアポトーシスから保護することを示すHEKn細胞のフローサイトメトリー解析である
1または2μMのLL−37で24時間処理した細胞(b,c,e,f)、および、未処理細胞(a,d)を、24時間カンプトテシンで処理してアポトーシスを受けるように誘導した(d〜f)。次に、非アポトーシス群(2N〜4N)、および、アポトーシス群(sub2N)を検出するために、細胞をフローサイトメトリーで解析した。グラフは、それぞれ三連で行われた3つの実験の代表的な結果である。
1または2μMのLL−37で24時間処理した細胞(b,c,e,f)、および、未処理細胞(a,d)を、24時間カンプトテシンで処理してアポトーシスを受けるように誘導した(d〜f)。次に、非アポトーシス群(2N〜4N)、および、アポトーシス群(sub2N)を検出するために、細胞をフローサイトメトリーで解析した。グラフは、それぞれ三連で行われた3つの実験の代表的な結果である。
図9−LL−37は、HEKn細胞においてカスパーゼ−3のカンプトテシンの活性化を減少させる
この細胞を、LL−37およびカンプトテシン存在または非存在下で培養し、次に、回収し、インビトロでVDVAD−AMCを加水分解することによってカスパーゼ−3の活性に関して解析した。カンプトテシンと24時間インキュベートすることにより誘導されたカスパーゼ活性化は、この細胞をLL−37で処理することによって減少した。値は、それぞれ三連で行われた3つの異なる実験の平均である。
この細胞を、LL−37およびカンプトテシン存在または非存在下で培養し、次に、回収し、インビトロでVDVAD−AMCを加水分解することによってカスパーゼ−3の活性に関して解析した。カンプトテシンと24時間インキュベートすることにより誘導されたカスパーゼ活性化は、この細胞をLL−37で処理することによって減少した。値は、それぞれ三連で行われた3つの異なる実験の平均である。
図10−LL−37の処理は、IAP2の発現を高める
HEKn細胞を2μMのLL−37で処理し、刺激後の異なる時点で回収した。これらの細胞からのRNAを逆転写し、IAP−2の発現をリアルタイムPCRによって決定した。各時点でのIAP−2の転写レベルは、18S−RNAに対して、未処理細胞(コントロールであり、各時点における1として設定される)に相対的に標準化して示される。
HEKn細胞を2μMのLL−37で処理し、刺激後の異なる時点で回収した。これらの細胞からのRNAを逆転写し、IAP−2の発現をリアルタイムPCRによって決定した。各時点でのIAP−2の転写レベルは、18S−RNAに対して、未処理細胞(コントロールであり、各時点における1として設定される)に相対的に標準化して示される。
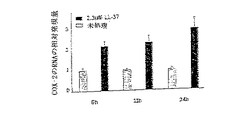
図11−LL−37は、HEK細胞におけるプロスタグランジンエンドペルオキシドシンターゼ−2(COX−2)の発現を高める
HEKn細胞を2μMのLL−37で処理し、刺激の6、12および24時間後に回収した。未処理細胞を、各時点におけるコントロールとして用いた(薄灰色のカラム)。これらの細胞からのRNAを逆転写し、コントロールに対するCOX−2の相対的な発現を、上述したようにqPCRによって決定した。
HEKn細胞を2μMのLL−37で処理し、刺激の6、12および24時間後に回収した。未処理細胞を、各時点におけるコントロールとして用いた(薄灰色のカラム)。これらの細胞からのRNAを逆転写し、コントロールに対するCOX−2の相対的な発現を、上述したようにqPCRによって決定した。
図12−COX−2阻害は、LL−37によって誘導されたIAP−2の増加に反対に作用する
HEKn細胞を、特異的なCOX−2阻害剤SC−791(25nM)を用いて、または、それを用いないで処理し、さらに6時間、LL−37(2μM)と共に、または、それを用いないでインキュベートした。IAP−2のmRNA遺伝子のレベルをRT−PCRによって解析したところ、未処理コントロールサンプルと関連することが示された。
HEKn細胞を、特異的なCOX−2阻害剤SC−791(25nM)を用いて、または、それを用いないで処理し、さらに6時間、LL−37(2μM)と共に、または、それを用いないでインキュベートした。IAP−2のmRNA遺伝子のレベルをRT−PCRによって解析したところ、未処理コントロールサンプルと関連することが示された。
実施例A−抗菌性タンパク質hCAP18/LL−37は乳癌で高度に発現され、上皮細胞の増殖因子と推測される
序論
この実施例において、一連の乳癌におけるhCAP18/LL−37の発現パターンを調査したところ、腫瘍細胞ではhCAP18のmRNAとタンパク質は著しいアップレギュレーションを示したが、隣接する間質では示さなかった。興味深いことに、最高の組織学的なグレードを有する腫瘍のなかでも、最高のレベルのhCAP18タンパク質が検出されたが、それに対していくつかの低いグレードの腫瘍では、そのhCAP18レベルは正常な乳房組織で検出されたレベルと等しかった。これらの発見は、抗菌性ペプチドに関して提唱されている仮説の抗腫瘍作用とは明らかに対照的であるが、上皮の修復および血管新生におけるhCAP18/LL−37に関する役割を示唆する近年の発見と一致する(5,10)。さらにhCAP18/LL−37が増殖促進因子であると裏付けるために、我々はここにおいて、合成の生物学的に活性なLL−37ペプチドでの処理と、hCAP18のトランスジェニック発現の両方によって上皮細胞の増殖が有意に強化されたことを示す。
序論
この実施例において、一連の乳癌におけるhCAP18/LL−37の発現パターンを調査したところ、腫瘍細胞ではhCAP18のmRNAとタンパク質は著しいアップレギュレーションを示したが、隣接する間質では示さなかった。興味深いことに、最高の組織学的なグレードを有する腫瘍のなかでも、最高のレベルのhCAP18タンパク質が検出されたが、それに対していくつかの低いグレードの腫瘍では、そのhCAP18レベルは正常な乳房組織で検出されたレベルと等しかった。これらの発見は、抗菌性ペプチドに関して提唱されている仮説の抗腫瘍作用とは明らかに対照的であるが、上皮の修復および血管新生におけるhCAP18/LL−37に関する役割を示唆する近年の発見と一致する(5,10)。さらにhCAP18/LL−37が増殖促進因子であると裏付けるために、我々はここにおいて、合成の生物学的に活性なLL−37ペプチドでの処理と、hCAP18のトランスジェニック発現の両方によって上皮細胞の増殖が有意に強化されたことを示す。
材料および方法
組織
28人の乳癌患者からの凍結させた腫瘍組織を、ダンデリード病院病理学部(Department of Pathology,Danderyd Hospital,ストックホルム,スウェーデン)から得た(表2)。これらの腫瘍を、確立されたガイドライン(13)に従ってエルストン(Elston)およびエリス(Ellis)のI〜IIIに沿ってスコア付けした。サイクリンAを、増殖マーカーとして用いた(ノボカストラ・ラボラトリーズ,ニューキャッスル・アポン・タイン,イギリス)。エストロゲン受容体の状態を、慣例的手順で加工したパラフィン切片で評価した。乳癌に罹った8人の患者、および、乳房の縮小手術を受けた2人の健康な個体からの非浸潤性の乳房組織を、コントロールとして利用した。全てのサンプルを同じ病理学者(B.S.)が試験し、正常と分類した(表2)。記入されたインフォームド・コンセントを全ての患者に提出してもらった。この研究は、倫理地方委員会(Regional Committee of Ethics)によって承認された。
組織
28人の乳癌患者からの凍結させた腫瘍組織を、ダンデリード病院病理学部(Department of Pathology,Danderyd Hospital,ストックホルム,スウェーデン)から得た(表2)。これらの腫瘍を、確立されたガイドライン(13)に従ってエルストン(Elston)およびエリス(Ellis)のI〜IIIに沿ってスコア付けした。サイクリンAを、増殖マーカーとして用いた(ノボカストラ・ラボラトリーズ,ニューキャッスル・アポン・タイン,イギリス)。エストロゲン受容体の状態を、慣例的手順で加工したパラフィン切片で評価した。乳癌に罹った8人の患者、および、乳房の縮小手術を受けた2人の健康な個体からの非浸潤性の乳房組織を、コントロールとして利用した。全てのサンプルを同じ病理学者(B.S.)が試験し、正常と分類した(表2)。記入されたインフォームド・コンセントを全ての患者に提出してもらった。この研究は、倫理地方委員会(Regional Committee of Ethics)によって承認された。
hCAP18に関するインサイチュハイブリダイゼーション
サンプル0〜17については、本質的に8で説明されている通りに、pBluescriptにサブクローニングした435bpのhCAP18全長cDNAを用いて、インビトロで[35S]で標識されたアンチセンスおよびセンスプローブを転写し、インサイチュハイブリダイゼーションを行った(表2)。
サンプル0〜17については、本質的に8で説明されている通りに、pBluescriptにサブクローニングした435bpのhCAP18全長cDNAを用いて、インビトロで[35S]で標識されたアンチセンスおよびセンスプローブを転写し、インサイチュハイブリダイゼーションを行った(表2)。
免疫組織化学
サンプル0〜17で免疫組織化学を行った(表2)。以前に10で説明されている通りに、間接ペルオキシダーゼ法に従って、ベクタステイン(Vectastain)キット(ベクター・ラボラトリーズ(Vector Laboratories),バーリンゲーム,米国)を用いて、組換えhCAP1814に対するカテリンの親和性で精製したウサギ抗血清を1:500の希釈度で用いた。その染色の特異性を確認するために、以前に10で報告されている通りにして免疫吸着法を行った。FPRL1受容体を検出するために、間接ペルオキシダーゼ法に従って、親和性によって精製したヤギポリクローナル抗体(サンタ・クルーズ・バイオテクノロジー(Santa Cruz Biotechnology),サンタクルーズ,カルフォルニア州)を1:400の希釈度で用いた。
サンプル0〜17で免疫組織化学を行った(表2)。以前に10で説明されている通りに、間接ペルオキシダーゼ法に従って、ベクタステイン(Vectastain)キット(ベクター・ラボラトリーズ(Vector Laboratories),バーリンゲーム,米国)を用いて、組換えhCAP1814に対するカテリンの親和性で精製したウサギ抗血清を1:500の希釈度で用いた。その染色の特異性を確認するために、以前に10で報告されている通りにして免疫吸着法を行った。FPRL1受容体を検出するために、間接ペルオキシダーゼ法に従って、親和性によって精製したヤギポリクローナル抗体(サンタ・クルーズ・バイオテクノロジー(Santa Cruz Biotechnology),サンタクルーズ,カルフォルニア州)を1:400の希釈度で用いた。
タンパク質抽出およびウェスタンブロット解析
凍結させた腫瘍組織16〜60mgを、電気ホモジナイザーを用いてリシン緩衝液中でホモジナイズした。標準的なプロトコール(15)に従って、SDS含有サンプル緩衝液中で腫瘍組織および細胞系由来のタンパク質を抽出した。タンパク質濃度を分光光度分析によって決定し、SDS含有サンプル緩衝液で等しいタンパク質濃度になるように調節した(16)。hCAP18/LL−37を検出するために、抽出物を16.5%トリス−トリシンの即席ゲル(Ready gel,バイオ・ラッド・ラボラトリーズ(Bio−Rad Laboratories),ハーキュリーズ,カリフォルニア州)上で分離した。組換えカテリン17と合成LL−37ペプチドを大きさの参照として用いた。ERK1/2およびFPRL1を検出するために、タンパク質を、それぞれ12%および8%のトリス−グリシンゲル上で分離した。各サンプル中でほぼ等量のタンパク質がブロットされたことを確認するために、一次抗体でインキュベートする前に、フィルターを3%ポンソーS(Ponceau S)の3%TCA溶液(シグマ・アルドリッチ,米国)で可逆的に染色した。親和性で精製した抗カテリン抗血清(17)、親和性で精製した抗LL−37抗血清(10)、抗FPRL1抗血清(sc18191,サンタ・クルーズ・バイオテクノロジー,カリフォルニア州)、および、モノクローナル抗ERK1/2抗体(セル・シグナリング・テクノロジー(Cell Signaling Technology),ビバリー,マサチューセッツ州)はいずれも、1:1000の希釈度で用いられた。ニトロセルロースフィルター(シュライヒャー&シュエル(Schleicher&Schuell),ダッセル,ドイツ)上にエレクトロブロッティングし、一次抗体と、IgGに結合したホースラディッシュペルオキシダーゼ(サンタ・クルーズ・バイオテクノロジー,サンタクルーズ,カルフォルニア州)とで連続的にインキュベートした後、強化された化学発光(ECL,アマシャム・バイオサイエンス(Amersham Biosciences)、ピスカタウェイ,ニュージャージー州)からのシグナルを、CCDカメラ(LAS1000,フジフィルム(Fujifilm),東京,日本)で捕捉した。
凍結させた腫瘍組織16〜60mgを、電気ホモジナイザーを用いてリシン緩衝液中でホモジナイズした。標準的なプロトコール(15)に従って、SDS含有サンプル緩衝液中で腫瘍組織および細胞系由来のタンパク質を抽出した。タンパク質濃度を分光光度分析によって決定し、SDS含有サンプル緩衝液で等しいタンパク質濃度になるように調節した(16)。hCAP18/LL−37を検出するために、抽出物を16.5%トリス−トリシンの即席ゲル(Ready gel,バイオ・ラッド・ラボラトリーズ(Bio−Rad Laboratories),ハーキュリーズ,カリフォルニア州)上で分離した。組換えカテリン17と合成LL−37ペプチドを大きさの参照として用いた。ERK1/2およびFPRL1を検出するために、タンパク質を、それぞれ12%および8%のトリス−グリシンゲル上で分離した。各サンプル中でほぼ等量のタンパク質がブロットされたことを確認するために、一次抗体でインキュベートする前に、フィルターを3%ポンソーS(Ponceau S)の3%TCA溶液(シグマ・アルドリッチ,米国)で可逆的に染色した。親和性で精製した抗カテリン抗血清(17)、親和性で精製した抗LL−37抗血清(10)、抗FPRL1抗血清(sc18191,サンタ・クルーズ・バイオテクノロジー,カリフォルニア州)、および、モノクローナル抗ERK1/2抗体(セル・シグナリング・テクノロジー(Cell Signaling Technology),ビバリー,マサチューセッツ州)はいずれも、1:1000の希釈度で用いられた。ニトロセルロースフィルター(シュライヒャー&シュエル(Schleicher&Schuell),ダッセル,ドイツ)上にエレクトロブロッティングし、一次抗体と、IgGに結合したホースラディッシュペルオキシダーゼ(サンタ・クルーズ・バイオテクノロジー,サンタクルーズ,カルフォルニア州)とで連続的にインキュベートした後、強化された化学発光(ECL,アマシャム・バイオサイエンス(Amersham Biosciences)、ピスカタウェイ,ニュージャージー州)からのシグナルを、CCDカメラ(LAS1000,フジフィルム(Fujifilm),東京,日本)で捕捉した。
ELISA
これまで説明されている(17)サンドイッチELISAを用いて、正常な乳腺および腫瘍組織からのタンパク質抽出物中のhCAP18を定量した。
これまで説明されている(17)サンドイッチELISAを用いて、正常な乳腺および腫瘍組織からのタンパク質抽出物中のhCAP18を定量した。
リアルタイムPCRによるhCAP18の発現解析
4種の正常なサンプルと4種の腫瘍からのRNAを、キアゲン(Qiagen)のRNイージー(RNeasy)キット(オペロン・バイオテクノロジーズ(Operon Biotechnologies),コローニュ,ドイツ)で抽出し、第一の鎖の合成キット(アマシャム・バイオサイエンス,ノーウォーク,コネチカット州)を用いて逆転写した。RNAを、リアルタイムPCRによって、標準的なプロトコールに従って、ABIプリズム7700(ABI Prism 7700,アプライド・バイオシステムズ)でcDNA(10ng)を用いて定量した。このサンプルを3連で評価した。プライマーの配列は、5’−GTCACCAGAGGATTGTGACTTCAA−3’[配列番号2]、および、5’−TTGAGGGTCACTGTCCCCATA−3’[配列番号3]であり、蛍光原性プローブの配列は、6−FAM−5’−CCGCTTCACCAGCCCGTCCTT−3’−BHQ1[配列番号4]である。このサンプルを18S−RNAの定量化によって標準化した(アッセイ・オン・デマンド(Assay on Demand),アプライド・バイオシステムズ)。正常なサンプルの平均の発現を、任意に1に設定した。
4種の正常なサンプルと4種の腫瘍からのRNAを、キアゲン(Qiagen)のRNイージー(RNeasy)キット(オペロン・バイオテクノロジーズ(Operon Biotechnologies),コローニュ,ドイツ)で抽出し、第一の鎖の合成キット(アマシャム・バイオサイエンス,ノーウォーク,コネチカット州)を用いて逆転写した。RNAを、リアルタイムPCRによって、標準的なプロトコールに従って、ABIプリズム7700(ABI Prism 7700,アプライド・バイオシステムズ)でcDNA(10ng)を用いて定量した。このサンプルを3連で評価した。プライマーの配列は、5’−GTCACCAGAGGATTGTGACTTCAA−3’[配列番号2]、および、5’−TTGAGGGTCACTGTCCCCATA−3’[配列番号3]であり、蛍光原性プローブの配列は、6−FAM−5’−CCGCTTCACCAGCCCGTCCTT−3’−BHQ1[配列番号4]である。このサンプルを18S−RNAの定量化によって標準化した(アッセイ・オン・デマンド(Assay on Demand),アプライド・バイオシステムズ)。正常なサンプルの平均の発現を、任意に1に設定した。
合成LL−37ペプチド
LL−37ペプチドを合成し、HPLCによって98%の純度まで精製した(ポリペプチド・ラボラトリーズ社(PolyPeptide Laboratories A/S、Hillerod,デンマーク)。ペプチドの生物活性を抗菌分析(18)で確認した。
LL−37ペプチドを合成し、HPLCによって98%の純度まで精製した(ポリペプチド・ラボラトリーズ社(PolyPeptide Laboratories A/S、Hillerod,デンマーク)。ペプチドの生物活性を抗菌分析(18)で確認した。
上皮細胞のLL−37ペプチドの治療
自然に不死化したヒトケラチノサイト細胞系(HaCaT)(19)を、10%FCS(ウシ胎仔血清、ハイ−クローン(Hy−Clone),ブール・ノルディック社(Boule Nordic AB),フッディンゲ,スウェーデン)、および、抗生物質(PEST=ペニシリン50U/l、および、ストレプトマイシン50mg/ml,ギブコ・BRL)が補足されたDMEM(ダルベッコ改変イーグル培地,ギブコ・BRL(Gibco BRL),ライフ・テクノロジーズ(Life Technologies),スコットランド)中で培養した。細胞を70%の密集度で回収し、培地(DMEM+10%FCS、および、PEST)100μl中で2000個の細胞を96ウェルプレートにシーディングした。12時間後に、培地を血清非含有の培地(DMEM+PEST)に交換し、細胞を血清飢餓によってG0/G1に72時間同調させ、次に、合成の生物学的に活性なLL−37ペプチドを10μg/ml含む培地(DMEM+5%FCS+PEST)100μlで処理した。DMEM+5%FCSおよびPESTのみで処理した細胞を、コントロールとして利用した。この実験を四連で行った。細胞増殖を、[3H]−チミジン取り込みによって評価した。細胞を、12時間の間に、1μCi/ウェルの[3H]−チミジン(20.00Ci/mmol,パーキン・エルマー・ライフサイエンス社(Perkin Elmer Life Sciences Inc.),ボストン,マサチューセッツ州)で処理し、グラスファイバーフィルター(ワラック(Wallac)Oy Turku,フィンランド)上に回収した(ハーベスター96(Harvester 96),トムテック(Tomtec),Orage,コネチカット州)。液体シンチレーションカウンター(マイクロベータ・プラス(Microbeta Pluss),Wallac Sveriges AB)を用いて、[3H]−チミジンの取り込みを決定した。この実験を6連で2回繰り返した。
自然に不死化したヒトケラチノサイト細胞系(HaCaT)(19)を、10%FCS(ウシ胎仔血清、ハイ−クローン(Hy−Clone),ブール・ノルディック社(Boule Nordic AB),フッディンゲ,スウェーデン)、および、抗生物質(PEST=ペニシリン50U/l、および、ストレプトマイシン50mg/ml,ギブコ・BRL)が補足されたDMEM(ダルベッコ改変イーグル培地,ギブコ・BRL(Gibco BRL),ライフ・テクノロジーズ(Life Technologies),スコットランド)中で培養した。細胞を70%の密集度で回収し、培地(DMEM+10%FCS、および、PEST)100μl中で2000個の細胞を96ウェルプレートにシーディングした。12時間後に、培地を血清非含有の培地(DMEM+PEST)に交換し、細胞を血清飢餓によってG0/G1に72時間同調させ、次に、合成の生物学的に活性なLL−37ペプチドを10μg/ml含む培地(DMEM+5%FCS+PEST)100μlで処理した。DMEM+5%FCSおよびPESTのみで処理した細胞を、コントロールとして利用した。この実験を四連で行った。細胞増殖を、[3H]−チミジン取り込みによって評価した。細胞を、12時間の間に、1μCi/ウェルの[3H]−チミジン(20.00Ci/mmol,パーキン・エルマー・ライフサイエンス社(Perkin Elmer Life Sciences Inc.),ボストン,マサチューセッツ州)で処理し、グラスファイバーフィルター(ワラック(Wallac)Oy Turku,フィンランド)上に回収した(ハーベスター96(Harvester 96),トムテック(Tomtec),Orage,コネチカット州)。液体シンチレーションカウンター(マイクロベータ・プラス(Microbeta Pluss),Wallac Sveriges AB)を用いて、[3H]−チミジンの取り込みを決定した。この実験を6連で2回繰り返した。
HEK293、および、HaCaT細胞中のhCAP18のトランスジェニック発現
16bpの5’非翻訳領域を含む全コード配列を含むイメージクローン(Image clone)3057931由来のBfalフラグメント(20)を、バイシストロン性ベクターpIRES2−EGFP(BDバイオサイエンス(BD Biosciences),ベッドフォード,マサチューセッツ州)のSma1部位にサブクローニングした。
標準条件下でフージーン(Fugene,ロシュ・ダイアグノスティックス(Roche Diagnostics),インディアナポリス,インディアナ州)を用いて、HEK293とHaCaT細胞をトランスフェクションし、2週間で、400ng/mlのG418(インビトロジェン(Invitrogen),ペーズリー,イギリス)を用いて選択した。MoFlo(R) 高速セルソーティングフローサイトメーター(ダコ・サイトメーション(DakoCytomation),フォートコリンズ,コロラド州)で、データ解析のためのSummitTMソフトウェアを用いて、細胞をEGFP発現に関してソートし、それらのCAP18発現を免疫ブロッティングによって定量した。同様にして、コントロール細胞系を、EGFPのみを発現するベクターでのトランスフェクションによって確立した。この細胞系は、数ヶ月の連続培養中ずっと、選択をまったくしなくてもCAP18の安定な発現を維持した。この実験を30連で2回繰り返した。
16bpの5’非翻訳領域を含む全コード配列を含むイメージクローン(Image clone)3057931由来のBfalフラグメント(20)を、バイシストロン性ベクターpIRES2−EGFP(BDバイオサイエンス(BD Biosciences),ベッドフォード,マサチューセッツ州)のSma1部位にサブクローニングした。
標準条件下でフージーン(Fugene,ロシュ・ダイアグノスティックス(Roche Diagnostics),インディアナポリス,インディアナ州)を用いて、HEK293とHaCaT細胞をトランスフェクションし、2週間で、400ng/mlのG418(インビトロジェン(Invitrogen),ペーズリー,イギリス)を用いて選択した。MoFlo(R) 高速セルソーティングフローサイトメーター(ダコ・サイトメーション(DakoCytomation),フォートコリンズ,コロラド州)で、データ解析のためのSummitTMソフトウェアを用いて、細胞をEGFP発現に関してソートし、それらのCAP18発現を免疫ブロッティングによって定量した。同様にして、コントロール細胞系を、EGFPのみを発現するベクターでのトランスフェクションによって確立した。この細胞系は、数ヶ月の連続培養中ずっと、選択をまったくしなくてもCAP18の安定な発現を維持した。この実験を30連で2回繰り返した。
hCAP18で安定してトランスフェクションされたHEK293およびHaCaT細胞の増殖分析
hCAP18でトランスフェクションされたHEK293細胞を70%の密集度で回収し、24ウェルプレートにシーディングした。24時間後に、培地を交換し、細胞を、5%FCSおよびPESTが補足された培地(Optimem,ギブコ・BRL,ライフ・テクノロジーズ,スコットランド)2ml中で培養した。6日目に細胞を回収し、フローサイトメトリー(ベクトン・ディッキンソン(Becton Dickinson),ベッドフォード,マサチューセッツ州)で計数した。トリパンブルーを用いて細胞生存率を測定した;いずれの条件下でも、トリパンブルー陽性の細胞は5%未満であった。全ての条件は3連で行われた。EGFPのみを発現するベクターでトランスフェクションされたHEK293細胞をコントロールとして利用した。
hCAP18でトランスフェクションされたHEK293細胞を70%の密集度で回収し、24ウェルプレートにシーディングした。24時間後に、培地を交換し、細胞を、5%FCSおよびPESTが補足された培地(Optimem,ギブコ・BRL,ライフ・テクノロジーズ,スコットランド)2ml中で培養した。6日目に細胞を回収し、フローサイトメトリー(ベクトン・ディッキンソン(Becton Dickinson),ベッドフォード,マサチューセッツ州)で計数した。トリパンブルーを用いて細胞生存率を測定した;いずれの条件下でも、トリパンブルー陽性の細胞は5%未満であった。全ての条件は3連で行われた。EGFPのみを発現するベクターでトランスフェクションされたHEK293細胞をコントロールとして利用した。
hCAP18でトランスフェクションされたHaCaT細胞を70%の密集度で回収し、10%FCS+PESTを含むDMEM中で、ウェルあたり2000個の細胞で96ウェルプレートにシーディングした。12時間後に、培地を、5%FCS+PESTが補足されたDMEMに交換した。24時間培養した後に、この細胞を1μCi/ウェルの[3H]−チミジンで12時間処理し、回収し、上述のようにして解析した。EGFPのみを発現するベクターでトランスフェクションされたHaCaT細胞をコントロールとして利用した。
FPRL1の発現解析
HaCaT細胞からのRNAをRNイージーキット(キアゲン)で抽出し、第一の鎖の合成キット(アマシャム(Amersham)−ファルマシア(Pharmacia))を用いて逆転写した。FPRL1のRNAを、リアルタイムPCRによって定量し、上述のように18S−RNAに対して標準化した。プライマーの配列は、5’−TCTGCTGGCTACACTGTTCTGC−3’[配列番号2]、および、5’−GACCCCGAGGACAAAGGTG−3’[配列番号3]であり、蛍光原性プローブの配列は、6−FAM−5’−CCCAAGCACCACCAATGGGAGGA−3’−BHQ1[配列番号4]である。
HaCaT細胞からのRNAをRNイージーキット(キアゲン)で抽出し、第一の鎖の合成キット(アマシャム(Amersham)−ファルマシア(Pharmacia))を用いて逆転写した。FPRL1のRNAを、リアルタイムPCRによって定量し、上述のように18S−RNAに対して標準化した。プライマーの配列は、5’−TCTGCTGGCTACACTGTTCTGC−3’[配列番号2]、および、5’−GACCCCGAGGACAAAGGTG−3’[配列番号3]であり、蛍光原性プローブの配列は、6−FAM−5’−CCCAAGCACCACCAATGGGAGGA−3’−BHQ1[配列番号4]である。
百日咳毒素の分析
hCAP18/LL−37により誘導された上皮細胞増殖の刺激を媒介することにFPRL1が関与するかを評価するために、HaCaT細胞を、Gタンパク質結合受容体阻害剤である百日咳毒素で処理した。LL−37処理の24時間前に、細胞を百日咳毒素(シグマ−アルドリッチ(Sigma−Aldrich)、スイス)でプレインキュベートした(最終的な毒素濃度は20ng/ml)。細胞をシーディングして48時間後に培地を交換し、HaCaT細胞を、合成の生物学的に活性なLL−37ペプチドをそれぞれ5または10μg/ml含む培地(DMEM+5%FCS、および、PEST)100μlで処理した。DMEM+5%FCS、および、PESTのみで処理した細胞を、コントロールとして利用した。
hCAP18/LL−37により誘導された上皮細胞増殖の刺激を媒介することにFPRL1が関与するかを評価するために、HaCaT細胞を、Gタンパク質結合受容体阻害剤である百日咳毒素で処理した。LL−37処理の24時間前に、細胞を百日咳毒素(シグマ−アルドリッチ(Sigma−Aldrich)、スイス)でプレインキュベートした(最終的な毒素濃度は20ng/ml)。細胞をシーディングして48時間後に培地を交換し、HaCaT細胞を、合成の生物学的に活性なLL−37ペプチドをそれぞれ5または10μg/ml含む培地(DMEM+5%FCS、および、PEST)100μlで処理した。DMEM+5%FCS、および、PESTのみで処理した細胞を、コントロールとして利用した。
LL−37で処理したHaCaT細胞におけるリン酸化ERK1/2の分析
HaCaT細胞を10%の密集度でシーディングし、0.2%FCSを含むDMEM中で36時間維持した。さらに48時間、細胞を、それぞれ1%または5%FCSを含むDMEM中で、LL−37の存在下(10μg/ml)または非存在下で、培地を毎日交換して培養した。10ng/mlのEGFを、ポジティブコントロールとして利用した。リン酸化ERK1/2の発現を、マウスモノクローナル抗体(セル・シグナリング・テクノロジー(Cell Signaling Technology),ビバリー,マサチューセッツ州)を用いたウェスタンブロット解析によって評価した。
HaCaT細胞を10%の密集度でシーディングし、0.2%FCSを含むDMEM中で36時間維持した。さらに48時間、細胞を、それぞれ1%または5%FCSを含むDMEM中で、LL−37の存在下(10μg/ml)または非存在下で、培地を毎日交換して培養した。10ng/mlのEGFを、ポジティブコントロールとして利用した。リン酸化ERK1/2の発現を、マウスモノクローナル抗体(セル・シグナリング・テクノロジー(Cell Signaling Technology),ビバリー,マサチューセッツ州)を用いたウェスタンブロット解析によって評価した。
統計的分析
値は、細胞の平均数、または、カウント毎分(CPM)±SDとして示される。群と群との比較は、両側t検定で解析した。結果は、P値が0.05未満の場合に統計学的に有意とみなした。腫瘍における発現を解析するために、有意性のレベルを0.05未満として、hCAP18タンパク質レベルで片側t検定を行った。
値は、細胞の平均数、または、カウント毎分(CPM)±SDとして示される。群と群との比較は、両側t検定で解析した。結果は、P値が0.05未満の場合に統計学的に有意とみなした。腫瘍における発現を解析するために、有意性のレベルを0.05未満として、hCAP18タンパク質レベルで片側t検定を行った。
結果
hCAP18/LL−37は乳癌で発現される
患者の詳細は表2に示す。
hCAP18/LL−37は乳癌で発現される
患者の詳細は表2に示す。
インサイチュハイブリダイゼーションによれば、健康なコントロール(図1G)からの乳房組織、および、非浸潤性の乳癌(示さず)において、hCAP18のmRNAに関する低いシグナル(示さず)、および、hCAP18タンパク質に関する弱い免疫反応性がみられた。全ての乳癌組織において、腫瘍細胞ではhCAP18に対する免疫反応性を示し、間質では示さなかった(図1A,C,D)。腫瘍細胞個体群はhCAP18の免疫反応性に関して均一ではなく、強い陽性を示す細胞は、検出可能なhCAP18を欠いている細胞に隣接していることがわかった(図1C)。カテリン組換えタンパク質を用いた免疫吸着法では、hCAP18の免疫反応性は無効になった(図1E,F)。インサイチュハイブリダイゼーションによって、hCAP18のmRNAに関する陽性シグナルが、免疫組織化学で得られた発現パターンとぴったりと適合する同じ領域で検出された(図IB)。シグナル強度は様々であり、高いグレードの腫瘍のなかでも最も顕著であった。コントロール切片は、hCAP18のmRNAに特異的なシグナルを欠失したセンスhCAP18cRNAプローブとハイブリダイズした(示さず)。
ELISAによる乳癌組織抽出物中のhCAP18タンパク質の定量化によって、エルストンIグレードの腫瘍とIIグレードの腫瘍とで差がないことが解明されたが、最高の悪性腫瘍グレードの腫瘍におけるhCAP18レベルは明らかにそれらより高かった(表2)。エルストンIIIグレードと、それ以外の腫瘍との差は統計学的に有意であった(p<0.01)。13のグレードIIIの腫瘍のうち10におけるhCAP18の濃度が、5ng/mg総タンパク質に達するか、または、それを超過していた。それ以外の18の腫瘍サンプルのうちこのレベルに達したのは、わずか2つだけだった。また我々は、エルストンIまたはIIの腫瘍と類似したレベルであることが解明された健康な乳房組織の4つの試料も分析した。ELISAにより得られた発現パターンを確かめるために、我々は、4つの正常なサンプルと、4つの腫瘍サンプルにリアルタイムPCRを行った。転写を定量した結果は、タンパク質発現に関するデータと一致していた(表2)。
免疫ブロッティングによって、調査した全ての腫瘍と正常な乳房組織は、無傷の未加工の18kDaのホロタンパク質に相当する免疫反応を示すバンドを示した(図2)。5つの調査したグレードIIIの腫瘍のうち4つにおいて(表2,サンプル6〜10)、我々はまた、LL−37、加工したhCAP18タンパク質に相当するバンドも検出した(図2)。用いられた抗血清はhCAP18ホロタンパク質に対して上昇し、LL−37はカテリンペプチドに対する親和性で精製されたにもかかわらず、LL−37が高濃度で検出された(10)。
hCAP18/LL−37は、上皮細胞の増殖を高める
hCAP18(hCAP18/E)発現ベクターでトランスフェクションされたHEK293およびHaCaT細胞は、EGFPのみを発現するベクターでトランスフェクションされたコントロール細胞(E)よりも有意に高い増殖速度を示した(図3AおよびB)。トランスフェクションされたHEK293、および、HaCaT細胞からのタンパク質抽出物を免疫ブロッティングすることによって、我々は、これらのhCAP18ベクターを含む細胞は、ホロタンパク質を生産することを確認した(図3AおよびB)、および、この細胞培地中で、LL−37に相当する4kDの免疫反応を示すバンドを検出した(データ示さず)。加えて、5%ウシ胎仔血清で培養し、10μg/mlの合成の生物学的に活性なLL−37ペプチドで処理したHaCaT細胞から、細胞増殖が有意に増加することが示された(図4)。
hCAP18(hCAP18/E)発現ベクターでトランスフェクションされたHEK293およびHaCaT細胞は、EGFPのみを発現するベクターでトランスフェクションされたコントロール細胞(E)よりも有意に高い増殖速度を示した(図3AおよびB)。トランスフェクションされたHEK293、および、HaCaT細胞からのタンパク質抽出物を免疫ブロッティングすることによって、我々は、これらのhCAP18ベクターを含む細胞は、ホロタンパク質を生産することを確認した(図3AおよびB)、および、この細胞培地中で、LL−37に相当する4kDの免疫反応を示すバンドを検出した(データ示さず)。加えて、5%ウシ胎仔血清で培養し、10μg/mlの合成の生物学的に活性なLL−37ペプチドで処理したHaCaT細胞から、細胞増殖が有意に増加することが示された(図4)。
LL−37受容体FPRL1は乳癌で発現される
Gタンパク質結合受容体、FPRL1は、真核生物細胞においてLL−37によって誘導された作用に介在することが示されており(4,5)、本発明の設定において、それらの考えられる役割を評価するために、我々は、乳房組織におけるFPRL1タンパク質の発現調査したところ、乳癌細胞と正常な腺上皮の両方でFPRL1に関する強い免疫反応性が見出された(図5A,B)。免疫ブロッティングにより、FPRL1は両方の組織で発現されたことを確認した(図5C)。加えて、HaCaT細胞において、hCAP18のトランスジェニック発現は、FPRL1のmRNAの発現を有意に増加させ(図5D)、これは、hCAP18/LL−37シグナル伝達のFPRL1の関与をさらに裏付ける可能性がある。しかしながら、百日咳毒素でのHaCat細胞の前処理は、これらの細胞の増殖を止めなかったが、約50%抑制し(示さず)、これは、FPRL1は、これらの細胞において、hCAP18/LL−37の増殖刺激作用の媒介に特異的に関与していない可能性を示す。上皮細胞増殖の活性化におけるERK1/2の関与の可能性を試験するために、我々は、HaCaT細胞を合成の生物学的に活性なLL−37で処理したが、ERK1/2の有意な活性化はみられず、これは、EGFRは、HaCaT細胞増殖においてLL−37の刺激作用の媒介に関与しないことを示す。
Gタンパク質結合受容体、FPRL1は、真核生物細胞においてLL−37によって誘導された作用に介在することが示されており(4,5)、本発明の設定において、それらの考えられる役割を評価するために、我々は、乳房組織におけるFPRL1タンパク質の発現調査したところ、乳癌細胞と正常な腺上皮の両方でFPRL1に関する強い免疫反応性が見出された(図5A,B)。免疫ブロッティングにより、FPRL1は両方の組織で発現されたことを確認した(図5C)。加えて、HaCaT細胞において、hCAP18のトランスジェニック発現は、FPRL1のmRNAの発現を有意に増加させ(図5D)、これは、hCAP18/LL−37シグナル伝達のFPRL1の関与をさらに裏付ける可能性がある。しかしながら、百日咳毒素でのHaCat細胞の前処理は、これらの細胞の増殖を止めなかったが、約50%抑制し(示さず)、これは、FPRL1は、これらの細胞において、hCAP18/LL−37の増殖刺激作用の媒介に特異的に関与していない可能性を示す。上皮細胞増殖の活性化におけるERK1/2の関与の可能性を試験するために、我々は、HaCaT細胞を合成の生物学的に活性なLL−37で処理したが、ERK1/2の有意な活性化はみられず、これは、EGFRは、HaCaT細胞増殖においてLL−37の刺激作用の媒介に関与しないことを示す。
考察
本発明の研究において、我々は、hCAP18/LL−37は、正常な乳腺上皮で構成的に生産されることを実証している。これは、ヒトの抗菌バリア保護におけるLL−37の役割と一致しており、さらに、正常な静止状態の上皮におけるLL−37の構成的発現は、傷害や炎症に付随して観察される顕著な発現に対して低いことが見出されたという初期の報告とも一致する(7〜10)。これまでに、抗菌性ペプチドの構成的発現は、様々な外分泌腺で検出されており、例えば、ヒトカテリシジンLL−37が汗腺で、カテリシジンCRAMPがマウス唾液腺で、ベータ−デフェンシンがヒト唾液腺で検出されている(21〜23)。Bals等(1998)によって、乳腺におけるヒトベータ−デフェンシン−2(hBD−2)のmRNAの発現が報告されており、近年他のグループが、非授乳婦の乳腺組織において構成的なhBD−1発現を見出し、加えて授乳中の乳房組織や母乳においても見出した(26)。
本発明の研究において、我々は、hCAP18/LL−37は、正常な乳腺上皮で構成的に生産されることを実証している。これは、ヒトの抗菌バリア保護におけるLL−37の役割と一致しており、さらに、正常な静止状態の上皮におけるLL−37の構成的発現は、傷害や炎症に付随して観察される顕著な発現に対して低いことが見出されたという初期の報告とも一致する(7〜10)。これまでに、抗菌性ペプチドの構成的発現は、様々な外分泌腺で検出されており、例えば、ヒトカテリシジンLL−37が汗腺で、カテリシジンCRAMPがマウス唾液腺で、ベータ−デフェンシンがヒト唾液腺で検出されている(21〜23)。Bals等(1998)によって、乳腺におけるヒトベータ−デフェンシン−2(hBD−2)のmRNAの発現が報告されており、近年他のグループが、非授乳婦の乳腺組織において構成的なhBD−1発現を見出し、加えて授乳中の乳房組織や母乳においても見出した(26)。
hCAP18/LL−37は、乳癌細胞で発現されるという我々の発見は新規である。興味深いことに、hCAP18の生産は、正常な乳房の上皮、または、低いグレードの腫瘍と比較して、高いグレードの腫瘍の乳房上皮において最も顕著に増加した。しかしながら、hCAP18の発現は普遍的でもなければ一様でもなく、すなわち、全ての癌細胞がhCAP18に関して陽性というわけではなく、明らかに、陽性細胞は検出可能なhCAP18のmRNAとタンパク質を欠いている細胞に隣接して見出され(図1C)、発現の程度は、あらゆる腫瘍タイプの細胞のなかでも相当ばらつきがあった。これは、腎細胞癌においてヒトアルファ−デフェンシンに関して示唆されているように、hCAP18の複雑だが厳密に制御された調節を反映している可能性がある。
我々の研究において、最高の組織学的なグレードを有する腫瘍のなかでも、最高のhCAP18/LL−37のレベルが検出された。一方では高いグレードの腫瘍と、低いグレードの乳房組織や正常な乳房組織とのhCAP18発現の差は統計学的に有意であるにもかかわらず、厳密な相関はない。全ての群で、hCAP18を発現する腫瘍が健康なサンプルのレベルで観察され、グレードIの腫瘍のうち2つは、別の方法でのみ観察されたグレードIIIの腫瘍のなかでも比較的高い発現を示した。しかしながら、このサンプル番号に限定すると、我々の観察は、悪性腫瘍の程度と、hCAP18/LL−37の発現との相関がある可能性を示唆する。乳癌におけるhCAP18の過剰発現は、hCAP18の調節に影響を及ぼす細胞内経路の欠陥が原因で起こる可能性があること、さらに、hCAP18の発現は、腫瘍に増殖の利益を提供するよりむしろ、これらの変化を反映する、と主張することもできる。しかしながら、本明細書で示したインビトロでの研究を組み合わせて、我々は、これらのデータは、腫瘍増殖の促進においてLL−37が役割を有する可能性を強調していると考える。
癌腫における抗菌性ペプチドの生物学的な役割は不明瞭である。様々な口腔癌で、ヒトアルファ−およびベータ−デフェンシンの両方における高いhBD−2タンパク質濃度と著しい免疫反応性が見出されており、これらの抗菌性ペプチドの高いレベルは、感染および/またはサイトカイン刺激の結果の可能性があると示唆されている(28〜30)。その他の研究では、昆虫から単離された抗菌性ペプチド、例えばメリチンおよびセクロピン関連ペプチドは、哺乳動物の腫瘍細胞に抗腫瘍作用を発揮することが提唱されている(31〜34)。その上、ヒト膀胱癌細胞系におけるセクロピンおよびメリチンに関するコード配列のベクターが介在する送達と発現は、ヌードマウスで腫瘍原性を抑制した(11)。同様に、ブタカテリシジンPR−39のトランスジェニック発現は、ヒト肝細胞癌の浸潤能を減少させた。
癌における抗菌性ペプチドの機能を解明するためにさらなる研究が必要であるが、これらのペプチドの多機能の役割はますます明らかになりつつある。直接的な膜作用による病原体不活性化に加えて、LL−37は、インビトロで走化性効果を発揮し、ヒト好中球、単球、T細胞の部分集合および肥満細胞の移動を誘導する(4,35,36)。この走化性活性は、LL−37の、FPRL1、百日咳毒素感受性の膜結合型Gタンパク質結合受容体への結合に依存する(4)。hCAP18/LL−37に関してさらに示唆された機能としては、皮膚創傷の再上皮化および新血管形成を促進することによる、上皮修復および血管新生における役割が挙げられる(5,10)。
従って、本明細書で示された乳癌細胞における著しいhCAP18/LL−37発現は、これらの腫瘍細胞における増殖の利益を反映している可能性がある。この仮説を検証するために、我々は、ヒト上皮細胞系HEK293とHaCaTを、hCAP18発現ベクターでトランスフェクションしたところ、トランスフェクションされた細胞の増殖が有意に増加することを見出した。加えて、合成の生物学的に活性なLL−37ペプチドは、HaCaT細胞の増殖を有意に増加させた。これらの発見は、明らかに、抗菌性ペプチドに関して提唱されている示唆された抗腫瘍作用とは対照的であるが、Mueller等による、ヒトアルファ−デフェンシンは、腎細胞癌(RCC)の進行を調節する可能性があるという近年の発見と一致する。これらのデフェンシンは、RCCの腫瘍細胞、加えて腎臓の正常な尿細管上皮細胞中に見出されており、さらに、生理学的な濃度で腫瘍細胞増殖を刺激した。
我々のインビトロでの研究によれば、LL−37は、部分的にFPRL1を介して上皮細胞の増殖を刺激することが示唆され、これは、受容体を百日咳毒素でブロックすることによって、外因性のLL−37の増殖作用を約50%減少させるためであり、場合によっては、その他の受容体の関与も示されている。近年の研究において、LL−37は、マイトジェン活性化タンパク質キナーゼ/細胞外シグナル調節性キナーゼ(MAPK/ERKキナーゼ=MEK)を、上皮増殖因子受容体(EGFR)の転写促進を介して活性化することによって気道上皮細胞を活性化することが示唆されている(37)。しかしながら、我々の実験において、我々は、ERK1/2の有意な活性化をまったく検出しなかった。
すなわち、本明細書で示された結果から、LL−37は、腫瘍の増殖を促進することが示される。
参考文献
1. Gudmundsson GH, Agerberth B, Odeberg J, Bergman T, Olsson B, Salcedo R. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur J Biochem 1996;238:325-32.
2. Zanetti M, Gennaro R, Romeo D. Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett 1995;374:1-5.
3. Agerberth B, Gunne H, Odeberg J, Kogner P, Boman HG, Gudmundsson GH. FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc Natl Acad Sci U S A 1995;92:195-99.
4. Yang D, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, Oppenheim JJ, Chertov O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med 2000;192:1069-74.
5. Koczulla R, von Degenfeld G, Kupatt C, Krotz F, Zahler S, Gloe T, Issbrucker K, Unterberger P, Zaiou M, Lebherz C, Karl A, Raake P, et al. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J Clin Invest 2003;111:1665-72.
6. Cowland JB, Johnsen AH, Borregaard N. hCAP-18, a cathelin/pro-bactenecin-like protein of human neutrophil specific granules. FEBS Lett 1995;368:173-76.
7. Frohm M, Agerberth B, Ahangari G, Stahle-Backdahl M, Liden S, Wigzell H, Gudmundsson GH. The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. J Biol Chem 1997;272:15258-63.
8. Frohm Nilsson M, Sandstedt B, Sorensen O, Weber G, Borregaard N, Stahle-Backdahl M. The human cationic antimicrobial protein (hCAP18), a peptide antibiotic, is widely expressed in human squamous epithelia and colocalizes with interleukin-6. Infect Immun 1999;67:2561-66.
9. Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, Agerberth B, Gudmundsson GH, Gallo RL. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J Invest Dermatol 2001;117:91-97.
10. Heilborn JD, Nilsson MF, Kratz G, Weber G, Sorensen O, Borregaard N, Stahle-Backdahl M. The cathelicidin anti-microbial peptide LL-37 is involved in re-epithelialization of human skin wounds and is lacking in chronic ulcer epithelium. J Invest Dermatol 2003;120:379-89.
11. Winder D, Gunzburg WH, Erfle V, Salmons B. Expression of antimicrobial peptides has an antitumour effect in human cells. Biochem Biophys Res Commun 1998;242:608-12.
12. Ohtake T, Fujimoto Y, Ikuta K, Saito H, Ohhira M, Ono M, Kohgo Y. Proline-rich antimicrobial peptide, PR-39 gene transduction altered invasive activity and actin structure in human hepatocellular carcinoma cells. Br J Cancer 1999;81:393-403.
13. Pathology NCGfBS. Pathology reporting in breast cancer screening. second edition,NHSBSP Publication 1995.
14. Sorensen O, Bratt T, Johnsen AH, Madsen MT, Borregaard N. The human antibacterial cathelicidin, hCAP-18, is bound to lipoproteins in plasma. J Biol Chem 1999;274:22445-51.
15. Ausubel FM, Brent R, R.E. K, Moore DM, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology: Wiley & Sons, Hoboken, NJ., 2003.
16. Schaffner W, Weissmann C. A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal Biochem 1973;56:502-14.
17. Sorensen O, Cowland JB, Askaa J, Borregaard N. An ELISA for hCAP-18, the cathelicidin present in human neutrophils and plasma. J Immunol Methods 1997;206:53-59.
18. Frohm M, Gunne H, Bergman AC, Agerberth B, Bergman T, Boman A, Liden S, Jornvall H, Boman HG. Biochemical and antibacterial analysis of human wound and blister fluid. Eur J Biochem 1996;237:86-92.
19. Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 1988;106:761-71.
20. Lennon G, Auffray C, Polymeropoulos M, Soares MB. The I.M.A.G.E. Consortium: an integrated molecular analysis of genomes and their expression. Genomics 1996;33:151-2.
21. Murakami M, Ohtake T, Dorschner RA, Gallo RL. Cathelicidin antimicrobial pe
ptides are expressed in salivary glands and saliva. J Dent Res 2002;81:845-50.
22. Murakami M, Ohtake T, Dorschner RA, Schittek B, Garbe C, Gallo RL. Cathelicidin anti-microbial peptide expression in sweat, an innate defense system for the skin. J Invest Dermatol 2002;119:1090-5.
23. Sahasrabudhe KS, Kimball JR, Morton TH, Weinberg A, Dale BA. Expression of the antimicrobial peptide, human beta-defensin 1, in duct cells of minor salivary glands and detection in saliva. J Dent Res 2000;79:1669-74.
24. Bals R, Wang X, Wu Z, Freeman T, Bafna V, Zasloff M, Wilson JM. Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J Clin Invest 1998;102:874-80.
25. Tunzi CR, Harper PA, Bar-Oz B, Valore EV, Semple JL, Watson-MacDonell J, Ganz T, Ito S. Beta-defensin expression in human mammary gland epithelia. Pediatr Res 2000;48:30-5.
26. Jia HP, Starner T, Ackermann M, Kirby P, Tack BF, McCray PB, Jr. Abundant human beta-defensin-1 expression in milk and mammary gland epithelium. J Pediatr 2001;138:109-12.
27. Mueller CA, Markovic-Lipkovski J, Klatt T, Gamper J, Schwarz G, Beck H, Deeg M, Kalbacher H, Widmann S, Wessels JT, Becker V, Muller GA, et al. Human alpha-defensins HNPs-1, -2, and -3 in renal cell carcinoma: influences on tumour cell proliferation. Am J Pathol 2002;160:1311-24.
28. Mizukawa N, Sawaki K, Yamachika E, Fukunaga J, Ueno T, Takagi S, Sugahara T. Presence of human beta-defensin-2 in oral squamous cell carcinoma. Anticancer Res 2000;20:2005-7.
29. Mizukawa N, Sawaki K, Nagatsuka H, Kamio M, Yamachika E, Fukunaga J, Ueno T, Takagi S, Sugahara T. Human alpha-and beta-defensin immunoreactivity in oral mucoepidermoid carcinomas. Anticancer Res 2001;21:2171-4.
30. Sawaki K, Mizukawa N, Yamaai T, Yoshimoto T, Nakano M, Sugahara T. High concentration of beta-defensin-2 in oral squamous cell carcinoma. Anticancer Res 2002;22:2103-7.
31. Jaynes JM, Julian GR, Jeffers GW, White KL, Enright FM. In vitro cytocidal effect of lytic peptides on several transformed mammalian cell lines. Pept Res 1989;2:157-60.
32. Sharma SV. Melittin-induced hyperactivation of phospholipase A2 activity and calcium influx in ras-transformed cells. Oncogene 1993;8:939-47.
33. Moore AJ, Devine DA, Bibby MC. Preliminary experimental anticancer activity
of cecropins. Pept Res 1994;7:265-9.
34. Hui L, Leung K, Chen HM. The combined effects of antibacterial peptide cecropin A and anti-cancer agents on leukemia cells. Anticancer Res 2002;22:2811-6.
35. Agerberth B, Charo J, Werr J, Olsson B, Idali F, Lindbom L, Kiessling R, Jornvall H, Wigzell H, Gudmundsson GH. The human antimicrobial and chemotactic peptides LL-37 and alpha- defensins are expressed by specific lymphocyte and monocyte populations. Blood 2000;96:3086-93.
36. Niyonsaba F, Iwabuchi K, Someya A, Hirata M, Matsuda H, Ogawa H, Nagaoka I.
A cathelicidin family of human antibacterial peptide LL-37 induces mast cell chemotaxis. Immunology 2002;106:20-6.
37. Tjabringa GS, Aarbiou J, Ninaber DK, Drijfhout JW, Sorensen OE, Borregaard N, Rabe KF, Hiemstra PS. The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J Immunol 2003;171:6690-6.
1. Gudmundsson GH, Agerberth B, Odeberg J, Bergman T, Olsson B, Salcedo R. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur J Biochem 1996;238:325-32.
2. Zanetti M, Gennaro R, Romeo D. Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett 1995;374:1-5.
3. Agerberth B, Gunne H, Odeberg J, Kogner P, Boman HG, Gudmundsson GH. FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc Natl Acad Sci U S A 1995;92:195-99.
4. Yang D, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, Oppenheim JJ, Chertov O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med 2000;192:1069-74.
5. Koczulla R, von Degenfeld G, Kupatt C, Krotz F, Zahler S, Gloe T, Issbrucker K, Unterberger P, Zaiou M, Lebherz C, Karl A, Raake P, et al. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. J Clin Invest 2003;111:1665-72.
6. Cowland JB, Johnsen AH, Borregaard N. hCAP-18, a cathelin/pro-bactenecin-like protein of human neutrophil specific granules. FEBS Lett 1995;368:173-76.
7. Frohm M, Agerberth B, Ahangari G, Stahle-Backdahl M, Liden S, Wigzell H, Gudmundsson GH. The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. J Biol Chem 1997;272:15258-63.
8. Frohm Nilsson M, Sandstedt B, Sorensen O, Weber G, Borregaard N, Stahle-Backdahl M. The human cationic antimicrobial protein (hCAP18), a peptide antibiotic, is widely expressed in human squamous epithelia and colocalizes with interleukin-6. Infect Immun 1999;67:2561-66.
9. Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, Agerberth B, Gudmundsson GH, Gallo RL. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J Invest Dermatol 2001;117:91-97.
10. Heilborn JD, Nilsson MF, Kratz G, Weber G, Sorensen O, Borregaard N, Stahle-Backdahl M. The cathelicidin anti-microbial peptide LL-37 is involved in re-epithelialization of human skin wounds and is lacking in chronic ulcer epithelium. J Invest Dermatol 2003;120:379-89.
11. Winder D, Gunzburg WH, Erfle V, Salmons B. Expression of antimicrobial peptides has an antitumour effect in human cells. Biochem Biophys Res Commun 1998;242:608-12.
12. Ohtake T, Fujimoto Y, Ikuta K, Saito H, Ohhira M, Ono M, Kohgo Y. Proline-rich antimicrobial peptide, PR-39 gene transduction altered invasive activity and actin structure in human hepatocellular carcinoma cells. Br J Cancer 1999;81:393-403.
13. Pathology NCGfBS. Pathology reporting in breast cancer screening. second edition,NHSBSP Publication 1995.
14. Sorensen O, Bratt T, Johnsen AH, Madsen MT, Borregaard N. The human antibacterial cathelicidin, hCAP-18, is bound to lipoproteins in plasma. J Biol Chem 1999;274:22445-51.
15. Ausubel FM, Brent R, R.E. K, Moore DM, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology: Wiley & Sons, Hoboken, NJ., 2003.
16. Schaffner W, Weissmann C. A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal Biochem 1973;56:502-14.
17. Sorensen O, Cowland JB, Askaa J, Borregaard N. An ELISA for hCAP-18, the cathelicidin present in human neutrophils and plasma. J Immunol Methods 1997;206:53-59.
18. Frohm M, Gunne H, Bergman AC, Agerberth B, Bergman T, Boman A, Liden S, Jornvall H, Boman HG. Biochemical and antibacterial analysis of human wound and blister fluid. Eur J Biochem 1996;237:86-92.
19. Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 1988;106:761-71.
20. Lennon G, Auffray C, Polymeropoulos M, Soares MB. The I.M.A.G.E. Consortium: an integrated molecular analysis of genomes and their expression. Genomics 1996;33:151-2.
21. Murakami M, Ohtake T, Dorschner RA, Gallo RL. Cathelicidin antimicrobial pe
ptides are expressed in salivary glands and saliva. J Dent Res 2002;81:845-50.
22. Murakami M, Ohtake T, Dorschner RA, Schittek B, Garbe C, Gallo RL. Cathelicidin anti-microbial peptide expression in sweat, an innate defense system for the skin. J Invest Dermatol 2002;119:1090-5.
23. Sahasrabudhe KS, Kimball JR, Morton TH, Weinberg A, Dale BA. Expression of the antimicrobial peptide, human beta-defensin 1, in duct cells of minor salivary glands and detection in saliva. J Dent Res 2000;79:1669-74.
24. Bals R, Wang X, Wu Z, Freeman T, Bafna V, Zasloff M, Wilson JM. Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J Clin Invest 1998;102:874-80.
25. Tunzi CR, Harper PA, Bar-Oz B, Valore EV, Semple JL, Watson-MacDonell J, Ganz T, Ito S. Beta-defensin expression in human mammary gland epithelia. Pediatr Res 2000;48:30-5.
26. Jia HP, Starner T, Ackermann M, Kirby P, Tack BF, McCray PB, Jr. Abundant human beta-defensin-1 expression in milk and mammary gland epithelium. J Pediatr 2001;138:109-12.
27. Mueller CA, Markovic-Lipkovski J, Klatt T, Gamper J, Schwarz G, Beck H, Deeg M, Kalbacher H, Widmann S, Wessels JT, Becker V, Muller GA, et al. Human alpha-defensins HNPs-1, -2, and -3 in renal cell carcinoma: influences on tumour cell proliferation. Am J Pathol 2002;160:1311-24.
28. Mizukawa N, Sawaki K, Yamachika E, Fukunaga J, Ueno T, Takagi S, Sugahara T. Presence of human beta-defensin-2 in oral squamous cell carcinoma. Anticancer Res 2000;20:2005-7.
29. Mizukawa N, Sawaki K, Nagatsuka H, Kamio M, Yamachika E, Fukunaga J, Ueno T, Takagi S, Sugahara T. Human alpha-and beta-defensin immunoreactivity in oral mucoepidermoid carcinomas. Anticancer Res 2001;21:2171-4.
30. Sawaki K, Mizukawa N, Yamaai T, Yoshimoto T, Nakano M, Sugahara T. High concentration of beta-defensin-2 in oral squamous cell carcinoma. Anticancer Res 2002;22:2103-7.
31. Jaynes JM, Julian GR, Jeffers GW, White KL, Enright FM. In vitro cytocidal effect of lytic peptides on several transformed mammalian cell lines. Pept Res 1989;2:157-60.
32. Sharma SV. Melittin-induced hyperactivation of phospholipase A2 activity and calcium influx in ras-transformed cells. Oncogene 1993;8:939-47.
33. Moore AJ, Devine DA, Bibby MC. Preliminary experimental anticancer activity
of cecropins. Pept Res 1994;7:265-9.
34. Hui L, Leung K, Chen HM. The combined effects of antibacterial peptide cecropin A and anti-cancer agents on leukemia cells. Anticancer Res 2002;22:2811-6.
35. Agerberth B, Charo J, Werr J, Olsson B, Idali F, Lindbom L, Kiessling R, Jornvall H, Wigzell H, Gudmundsson GH. The human antimicrobial and chemotactic peptides LL-37 and alpha- defensins are expressed by specific lymphocyte and monocyte populations. Blood 2000;96:3086-93.
36. Niyonsaba F, Iwabuchi K, Someya A, Hirata M, Matsuda H, Ogawa H, Nagaoka I.
A cathelicidin family of human antibacterial peptide LL-37 induces mast cell chemotaxis. Immunology 2002;106:20-6.
37. Tjabringa GS, Aarbiou J, Ninaber DK, Drijfhout JW, Sorensen OE, Borregaard N, Rabe KF, Hiemstra PS. The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J Immunol 2003;171:6690-6.
実施例B−エストロゲン受容体(ER)およびリンパ節(N)陽性乳房腫瘍におけるhCAP18/LL−37発現の増加
材料および方法
140種の乳房腫瘍、および、4種の罹患していない乳房組織サンプルからRNAを抽出し、プライマーとしてランダムヘキサマーを用いて逆転写した。hCAP18転写物の発現を、標準的なプロトコールに従って(上述した通り)cDNA10ngを用いてリアルタイムPCRによって決定した。
材料および方法
140種の乳房腫瘍、および、4種の罹患していない乳房組織サンプルからRNAを抽出し、プライマーとしてランダムヘキサマーを用いて逆転写した。hCAP18転写物の発現を、標準的なプロトコールに従って(上述した通り)cDNA10ngを用いてリアルタイムPCRによって決定した。
結果および考察
結果を図6に示す。罹患していないサンプルの平均の発現を、任意に1に設定した。平均および偏差をAnova統計によって評価した。
hCAP18の発現は、ER陽性の腫瘍で、リンパ節が発達している場合、リンパ節が無い場合よりも有意に高い(約5倍)。
結果を図6に示す。罹患していないサンプルの平均の発現を、任意に1に設定した。平均および偏差をAnova統計によって評価した。
hCAP18の発現は、ER陽性の腫瘍で、リンパ節が発達している場合、リンパ節が無い場合よりも有意に高い(約5倍)。
実施例C−hCAP18/LL−37活性を阻害する物質の同定
多数の明確なインビトロおよびインビボでの分析を用いて、hCAP18/LL−37の腫瘍発達への寄与の様々な観点を実証することができ、すなわち、マウスにおける、周知のシグナル伝達経路の誘導、細胞増殖の刺激、および、アポトーシス抑制、コロニー増殖の刺激、および、増殖から独立した定着、基底膜を越える浸潤の刺激、および、最終的に、腫瘍増殖と転移形成の促進を実証することができる。
多数の明確なインビトロおよびインビボでの分析を用いて、hCAP18/LL−37の腫瘍発達への寄与の様々な観点を実証することができ、すなわち、マウスにおける、周知のシグナル伝達経路の誘導、細胞増殖の刺激、および、アポトーシス抑制、コロニー増殖の刺激、および、増殖から独立した定着、基底膜を越える浸潤の刺激、および、最終的に、腫瘍増殖と転移形成の促進を実証することができる。
上述の特徴は、これまで一次ケラチノサイトおよび上皮細胞系においてはhCAP18/LL−37に原因があると考えられているが、これらも乳癌細胞系で実証することができた。適切な標的細胞系はMCF−7であり、これは、hCAP18を発現しない悪性が低いエストロゲン受容体陽性細胞系である。上記の観点を調査するために、組換えプラスミドからhCAP18を発現するMCF−7誘導体を確立した。hCAP18タンパク質とパラ分泌型LL−37の自己分泌活性は異なると予想されるため、インビトロでの分析はまた、この細胞において、実験の性質上LL−37を外的に添加することが内因性の生産の代わりに許容される場合、LL−37を外的に添加することによって行われた。
上記の実験は、hCAP18/LL−37の全体の腫瘍形成性の可能性を評価するために行われるが、これらの作用の抗hCAP18/LL−37活性を有する物質による阻害を実証するためにさらなる実験を行うことができる。例えば、合成された活性タンパク質LL−37は、抗体による阻害の標的としてみなされる可能性がある。その上、このタンパク質の生産は、プロモーターのブロックを介したhCAP18遺伝子の転写を阻害することによって阻害することができ、加えて、RNA干渉による転写の転写後のダウンレギュレーションによっても阻害することができる。
従って、インビトロで作用のメカニズムと標的を実証するための実験と、インビボで、マウスにおける腫瘍において体内での作用を実証するための実験を両方行ってもよい。このようなインビトロでの実験は上述のMCF−7誘導体で行ってもよい。hCAP18遺伝子における調節の実験のために、hCAP18を低いが明らかに検出可能なレベルで発現する乳癌細胞系ZR7S−1が用いられた。
以下で説明する実験については、特に他の指定がない限り以下の条件が用いられた:細胞は、10%ウシ胎仔血清を含むダルベッコ改変イーグル培地で増殖させた。トランスジェニック系のための培地は、導入遺伝子を維持するために150μg/mlのハイグロマイシン、および、400μg/mlのゲネチシンを含む。実験を3日間未満継続させる場合、この細胞の使用の24時間前に抗生物質を除去し、ウシ胎仔血清濃度を、それぞれ特定の実験に応じて必要な濃度に調節した。LL−37を2μMの濃度で添加した。
細胞増殖の刺激(Heilborn 2005を参照)
トランスジェニック細胞系および細胞系は70%の密集度で回収し、10%FCSを含むDMEM中で、ウェルあたり2000個で96ウェルプレートにシーディングした。24時間後、培地を、LL−37が添加された、または、添加されていない5%FCSを含む培地に交換した。24時間培養した後、この細胞を1μCi/ウェルの[3H]−チミジン20Ci/mmolで12時間処理し、グラスファイバーフィルター(ワラック,Turku,フィンランド)上に回収した(ハーベスター96;トムテック,Orage,コネチカット州)。[3H]−チミジンの取り込みは、液体シンチレーションカウンター(マイクロベータ・プラス,ワラック)を用いて決定した。実験の統計的な有意性を確認するために、各条件/細胞系あたり24ウェルを用いた。
トランスジェニック細胞系および細胞系は70%の密集度で回収し、10%FCSを含むDMEM中で、ウェルあたり2000個で96ウェルプレートにシーディングした。24時間後、培地を、LL−37が添加された、または、添加されていない5%FCSを含む培地に交換した。24時間培養した後、この細胞を1μCi/ウェルの[3H]−チミジン20Ci/mmolで12時間処理し、グラスファイバーフィルター(ワラック,Turku,フィンランド)上に回収した(ハーベスター96;トムテック,Orage,コネチカット州)。[3H]−チミジンの取り込みは、液体シンチレーションカウンター(マイクロベータ・プラス,ワラック)を用いて決定した。実験の統計的な有意性を確認するために、各条件/細胞系あたり24ウェルを用いた。
細胞増殖分析(Lu 2005,Ludes−Meyers 2002を参照)
この分析は、hCAP18/LL−37により細胞増殖が刺激されるかどうかを決定するために、[3H]−チミジン取り込み分析の補足として行われた。MCF−7誘導体を、各時点において三連で、50細胞/mm2の密度で平板培養した(6ウェルプレート中、約50000細胞/ウェル)。総細胞数を血球計数器で2日毎に定量した。細胞生存率は、トリパンブルーを用いることによって評価した。
この分析は、hCAP18/LL−37により細胞増殖が刺激されるかどうかを決定するために、[3H]−チミジン取り込み分析の補足として行われた。MCF−7誘導体を、各時点において三連で、50細胞/mm2の密度で平板培養した(6ウェルプレート中、約50000細胞/ウェル)。総細胞数を血球計数器で2日毎に定量した。細胞生存率は、トリパンブルーを用いることによって評価した。
hCAP18/LL−37によるアポトーシスの阻害
アポトーシス誘導のために、5%FCSを含む6ウェルプレートの培地にこの細胞を25000細胞/ウェルでシーディングし、トポイソメラーゼI阻害剤カンプトテシン(CAM)(シグマ・ケミカル社(Sigma Chemical Co.),セントルイス,ミズーリ州)で、6μMで24時間処理した。
hCAP18/LL−37によるアポトーシス抑制を確認するために、2つのアポトーシスのパラメーター、DNA含量およびカスパーゼ−3活性を評価した。統計的な有意性を確認するために、全ての実験を6連で行った。
アポトーシス誘導のために、5%FCSを含む6ウェルプレートの培地にこの細胞を25000細胞/ウェルでシーディングし、トポイソメラーゼI阻害剤カンプトテシン(CAM)(シグマ・ケミカル社(Sigma Chemical Co.),セントルイス,ミズーリ州)で、6μMで24時間処理した。
hCAP18/LL−37によるアポトーシス抑制を確認するために、2つのアポトーシスのパラメーター、DNA含量およびカスパーゼ−3活性を評価した。統計的な有意性を確認するために、全ての実験を6連で行った。
フローサイトメトリーによるDNA含量の解析(Warburton 2005を参照)
この解析のために、ヨウ化プロピジウム(PI)/RNアーゼ染色緩衝液(ベクトン・ディッキンソン,サンノゼ,カリフォルニア州)を用いた。冷70%(V/V)エタノール中でトリプシン処理した細胞を固定し、それを使用するまで4℃で保存した。PIのDNAへの取り込みによってDNA含量を測定した。蛍光解析は、FACScanフローサイトメーター(ベクトン・アンド・ディッキンソン,マウンテンビュー,カリフォルニア州,米国)を用いて行われた。細胞のサイズと粒度を決定するために、粒子のFSC(前方光散乱)とSSC(側方光散乱)を同時に測定した。PIで染色された核の赤色の蛍光は、FL−4ウィンドウ(600nmのバンドパスフィルター、および、35nmの帯域幅)で検出した。蛍光強度は、細胞のDNA含量に比例する。各ヒストグラムは、2部に分割される:(1)非同調の非アポトーシス性の生きた細胞を示すM1領域(2N〜4Nに等しいDNA含量を有するG1、SおよびG2相における細胞);および、(2)生きた細胞に比べて少量のDNAを含むアポトーシス細胞を示すM2領域。
この解析のために、ヨウ化プロピジウム(PI)/RNアーゼ染色緩衝液(ベクトン・ディッキンソン,サンノゼ,カリフォルニア州)を用いた。冷70%(V/V)エタノール中でトリプシン処理した細胞を固定し、それを使用するまで4℃で保存した。PIのDNAへの取り込みによってDNA含量を測定した。蛍光解析は、FACScanフローサイトメーター(ベクトン・アンド・ディッキンソン,マウンテンビュー,カリフォルニア州,米国)を用いて行われた。細胞のサイズと粒度を決定するために、粒子のFSC(前方光散乱)とSSC(側方光散乱)を同時に測定した。PIで染色された核の赤色の蛍光は、FL−4ウィンドウ(600nmのバンドパスフィルター、および、35nmの帯域幅)で検出した。蛍光強度は、細胞のDNA含量に比例する。各ヒストグラムは、2部に分割される:(1)非同調の非アポトーシス性の生きた細胞を示すM1領域(2N〜4Nに等しいDNA含量を有するG1、SおよびG2相における細胞);および、(2)生きた細胞に比べて少量のDNAを含むアポトーシス細胞を示すM2領域。
カスパーゼ-3活性分析
蛍光基質VDVAD−AMC(100μM)、および、DEVD−AMC(50μM)を用いて、カスパーゼ−3酵素活性を測定した。細胞溶解産物は、反応緩衝液(100mMのHEPES、10%スクロース、5mMジチオスレイトール(DTT)、10〜6%のNP−40および0.1%CHAPS(pH7.25))と合わせ、マイクロタイタープレートに添加した。蛍光発生基質の切断は、フルオロスキャンII(Fluoroscan II)プレートリーダー(ラボシステムズ(Labsystems),ストックホルム,スウェーデン)でAMCの解離によってモニターした。蛍光単位は、遊離のAMCから作製した標準曲線を用いてAMCのpmolに変換した。データは、線形回帰によって解析した。
蛍光基質VDVAD−AMC(100μM)、および、DEVD−AMC(50μM)を用いて、カスパーゼ−3酵素活性を測定した。細胞溶解産物は、反応緩衝液(100mMのHEPES、10%スクロース、5mMジチオスレイトール(DTT)、10〜6%のNP−40および0.1%CHAPS(pH7.25))と合わせ、マイクロタイタープレートに添加した。蛍光発生基質の切断は、フルオロスキャンII(Fluoroscan II)プレートリーダー(ラボシステムズ(Labsystems),ストックホルム,スウェーデン)でAMCの解離によってモニターした。蛍光単位は、遊離のAMCから作製した標準曲線を用いてAMCのpmolに変換した。データは、線形回帰によって解析した。
コロニー形成分析(Ludes−Meyers 2002,Wang 2005を参照)
この分析は、近接する支持細胞がない場合の持続的に増殖する細胞の能力を決定するために用いた。この特性は、この細胞の腫瘍を形成する能力を反映するとみなされる。
この分析は、近接する支持細胞がない場合の持続的に増殖する細胞の能力を決定するために用いた。この特性は、この細胞の腫瘍を形成する能力を反映するとみなされる。
トランスジェニック細胞を、サブコンフルエントな状態で維持し、最大70%の密集度でトリプシン処理し、続いて5、10および25細胞/ml増殖培地の濃度に希釈した。細胞を3〜7日間(基本的に3〜4倍になるのに十分な日数で)増殖させ、コロニー一つあたりの細胞数を決定した。組換えプラスミドが緑色蛍光タンパク質を発現すると、蛍光顕微鏡で細胞およびコロニーを計数することができる。コロニー形成の結果は、コロニー一つあたりの蛍光性の細胞の分布として表示され、培地中にLL−37が存在する場合と存在しない場合における、様々なトランスジェニック系の間の分布を比較するために、ウィルコクソンの順位和検定が用いた。MCF−7細胞は、β−エストロゲンの存在下でのみ腫瘍を形成することがわかっているため、トランスジェニック細胞の増殖に対する1〜5nMエストロゲンの作用を分析することができる。
増殖から独立した定着の分析(Fiucci 2002を参照)
この分析は、この細胞の転移を形成する能力を反映する。細胞は、300μ/mlトリプシン、20μ/mlエラスターゼ、および、1mMのEDTAの混合物を用いた処理によって製造した。エラスターゼの存在は、単一の細胞懸濁液への細胞の解離を容易にする。細胞を増殖培地に懸濁し、最終濃度500細胞/mlの溶融した0.7%シープラーク(Seaplaque)低融点アガロース、および、0.35%アガロースを含む増殖培地を1:1で混合した。この混合液1mlを、6ウェルプレートで、凝固した培地/0.6%アガロースの2mlの層上で平板培養した。この細胞を、3〜4日毎に、増殖培地100μlを添加して供給した。2週間後、培養の上部層を、0.2%p−ヨードニトロテトラゾリウムバイオレット(シグマ)で染色し、そして、直径が100μmより大きいコロニーを計数した。この分析は3連で行われ、2回繰り返した。
この分析は、この細胞の転移を形成する能力を反映する。細胞は、300μ/mlトリプシン、20μ/mlエラスターゼ、および、1mMのEDTAの混合物を用いた処理によって製造した。エラスターゼの存在は、単一の細胞懸濁液への細胞の解離を容易にする。細胞を増殖培地に懸濁し、最終濃度500細胞/mlの溶融した0.7%シープラーク(Seaplaque)低融点アガロース、および、0.35%アガロースを含む増殖培地を1:1で混合した。この混合液1mlを、6ウェルプレートで、凝固した培地/0.6%アガロースの2mlの層上で平板培養した。この細胞を、3〜4日毎に、増殖培地100μlを添加して供給した。2週間後、培養の上部層を、0.2%p−ヨードニトロテトラゾリウムバイオレット(シグマ)で染色し、そして、直径が100μmより大きいコロニーを計数した。この分析は3連で行われ、2回繰り返した。
浸潤の誘導(Fiucci 2002を参照)
マトリゲル(Matrigel)は基底膜とみなされ、これはEHS肉腫から生成された。マトリゲルは、基底膜成分(コラーゲン、ラミニンおよびプロテオグリカン)だけでなく、マトリックス分解酵素/それらの阻害剤および増殖因子もを含む。腫瘍細胞のマトリゲルへの浸潤は、腫瘍進行において役割を果たす細胞外マトリックス受容体とマトリックス分解酵素の関与を特徴付けるために用いられた。
マトリゲル(Matrigel)は基底膜とみなされ、これはEHS肉腫から生成された。マトリゲルは、基底膜成分(コラーゲン、ラミニンおよびプロテオグリカン)だけでなく、マトリックス分解酵素/それらの阻害剤および増殖因子もを含む。腫瘍細胞のマトリゲルへの浸潤は、腫瘍進行において役割を果たす細胞外マトリックス受容体とマトリックス分解酵素の関与を特徴付けるために用いられた。
マトリゲルを血清非含有の冷増殖培地で2mg/mlに希釈し、100μlを24−ウェルのトランスウェルの上部チャンバーに置き、ゲル化のために37℃で一晩インキュベートした。細胞を回収し、1%FCSを含む培地に106/mlの濃度で再懸濁した。マトリゲルを蠕虫血清非含有の培地で洗浄した後、この細胞懸濁液100μlを上に置いた。下部のチャンバーを化学誘引物質として調整培地を含む増殖培地600μlで充填した。このチャンバーを、細胞インキュベーターで6時間〜24時間インキュベートした。トランスウェルを染色し(ディフ−クイック染色溶液,フィッシャー・サイエンティフィック(Fisher Scientific))、非浸潤性の細胞を綿棒で削り取った後、光学顕微鏡で浸潤された細胞を計数した。
SCIDマウスにおける腫瘍増殖(Wang 2005 Warburton 2005を参照)
サブコンフルエントに増殖させた細胞を回収し、2.5×107細胞/mlで冷PBSに懸濁した。そのうち200μl分を、4〜6週齢の雌SCIDマウスの脂肪体または尾部の静脈に皮下注射した。通常、MCF−7細胞の腫瘍形成はエストロゲンに依存するため、60日にわたりβ−エストラジオールを1日あたり1.7mg放出されるようにペレットを皮下に埋め込んだ。これらのマウスを1週間あたり2回触診し、触診でわかる最初の腫瘍が見出された日付を記録した。腫瘍の増殖を手動でモニターし、最後に腫瘍の直径が1cmになったらマウスを殺した。腫瘍の数、サイズおよび分布を記録した。マウスを切片化することによって小さい転移が検出される、組換えプラスミドから発現されたGFPから蛍光を誘導するためにスキャンした。加えて、脾臓および肝臓をコラーゲン化して、蛍光性の細胞の数を決定するためにFACSソーターで単一の細胞懸濁液を評価した。
サブコンフルエントに増殖させた細胞を回収し、2.5×107細胞/mlで冷PBSに懸濁した。そのうち200μl分を、4〜6週齢の雌SCIDマウスの脂肪体または尾部の静脈に皮下注射した。通常、MCF−7細胞の腫瘍形成はエストロゲンに依存するため、60日にわたりβ−エストラジオールを1日あたり1.7mg放出されるようにペレットを皮下に埋め込んだ。これらのマウスを1週間あたり2回触診し、触診でわかる最初の腫瘍が見出された日付を記録した。腫瘍の増殖を手動でモニターし、最後に腫瘍の直径が1cmになったらマウスを殺した。腫瘍の数、サイズおよび分布を記録した。マウスを切片化することによって小さい転移が検出される、組換えプラスミドから発現されたGFPから蛍光を誘導するためにスキャンした。加えて、脾臓および肝臓をコラーゲン化して、蛍光性の細胞の数を決定するためにFACSソーターで単一の細胞懸濁液を評価した。
hCAP18転写に対するアンタゴニストを用いた阻害の研究(Agarwal 1998,Andela 2004,Toell 2001,Ishizuka 2005,Weber 2005を参照)
現在既知のhCAP18転写の最も強い誘導物質はビタミンDである。ビタミンD受容体は、hCAP18プロモーターにおける反応要素に結合することによってhCAP18転写を直接活性化する。このようにして低分子物質はhCAP18の発現を制御できるため、阻害化合物を系統的にスクリーニングすることは有意義である。初期の研究によれば、エストロゲンとその代謝産物のいくつかは作用を有さないことが示された。ビタミンAは、皮膚ケラチノサイトにおける転写を阻害するが、乳癌細胞系ZR−75−1における転写を刺激する。これらの発見に基づいて、ビタミンDに対するアンタゴニスト、例えばZK159222(シエーリング社(Schering AG))、および、TEI−9647(帝人医学研究所,東京)、および、ビタミンAに対するアンタゴニスト、例えばAGN193109(アレルゲン・ファーマシューティカルズ)は、本発明の方法に使用するための可能性がある物質である。
現在既知のhCAP18転写の最も強い誘導物質はビタミンDである。ビタミンD受容体は、hCAP18プロモーターにおける反応要素に結合することによってhCAP18転写を直接活性化する。このようにして低分子物質はhCAP18の発現を制御できるため、阻害化合物を系統的にスクリーニングすることは有意義である。初期の研究によれば、エストロゲンとその代謝産物のいくつかは作用を有さないことが示された。ビタミンAは、皮膚ケラチノサイトにおける転写を阻害するが、乳癌細胞系ZR−75−1における転写を刺激する。これらの発見に基づいて、ビタミンDに対するアンタゴニスト、例えばZK159222(シエーリング社(Schering AG))、および、TEI−9647(帝人医学研究所,東京)、および、ビタミンAに対するアンタゴニスト、例えばAGN193109(アレルゲン・ファーマシューティカルズ)は、本発明の方法に使用するための可能性がある物質である。
hCAP18転写阻害剤のスクリーニング(Weber 2005を参照)
ZR75−1細胞を25%の密集度で平板培養し、イソプロパノールまたはDMSOに100μMで溶解させた可能性がある阻害剤(最終濃度100nM)で処理した。24時間後、RNAを、キアゲンRNイージーキット(オペロン・バイオテクノロジーズ,コローニュ,ドイツ)で抽出し、第一の鎖の合成キット(アマシャム・バイオサイエンス,ノーウォーク,コネチカット州)を用いて逆転写した。RNAは、リアルタイムPCRによって、標準的なプロトコールに従って、ABIプリズム7700(ABI Prism 7700)(アプライド・バイオシステムズ)でcDNA(5ng)を用いて定量した。このサンプルは3連で評価した。プライマーの配列は、5’−GTCACCAGAGGATTGTGACTTCAA−3’[配列番号2]、および、5’−TTGAGGGTCACTGTCCCCATA−3’[配列番号3]であり、蛍光原性プローブの配列は、6−FAM−5’−CCGCTTCACCAGCCCGTCCTT−3’−BHQ1[配列番号4]である。このサンプルを、18S−RNAの定量化によって標準化する(アッセイ・オン・デマンド,アプライド・バイオシステムズ)。
ZR75−1細胞を25%の密集度で平板培養し、イソプロパノールまたはDMSOに100μMで溶解させた可能性がある阻害剤(最終濃度100nM)で処理した。24時間後、RNAを、キアゲンRNイージーキット(オペロン・バイオテクノロジーズ,コローニュ,ドイツ)で抽出し、第一の鎖の合成キット(アマシャム・バイオサイエンス,ノーウォーク,コネチカット州)を用いて逆転写した。RNAは、リアルタイムPCRによって、標準的なプロトコールに従って、ABIプリズム7700(ABI Prism 7700)(アプライド・バイオシステムズ)でcDNA(5ng)を用いて定量した。このサンプルは3連で評価した。プライマーの配列は、5’−GTCACCAGAGGATTGTGACTTCAA−3’[配列番号2]、および、5’−TTGAGGGTCACTGTCCCCATA−3’[配列番号3]であり、蛍光原性プローブの配列は、6−FAM−5’−CCGCTTCACCAGCCCGTCCTT−3’−BHQ1[配列番号4]である。このサンプルを、18S−RNAの定量化によって標準化する(アッセイ・オン・デマンド,アプライド・バイオシステムズ)。
阻害の調節メカニズムを調査するために、プロモーターの活性は、hCAP18プロモーターがルシフェラーゼレポーター遺伝子を制御する組換えプラスミドの使用によって決定した。ZR75−1細胞を25%の密集度で6ウェルプレート中で平板培養し、jetPEI(qBiogene)6μlと複合体化したレポータープラスミドをウェルあたり3μgで用いてトランスフェクションした。トランスフェクションの14時間後、上述したように阻害剤を添加した。24時間後、この細胞を溶解させ、分析システム(プロメガ(Promega))でルシフェラーゼ活性を測定した。コトランスフェクションされたプラスミド(pEF1/lacZ,インビトロジェン)200ngから発現されたβ−ガラクトシダーゼに対して、活性を標準化した。各実験は、分析ごとに少なくとも2回、3連で行った。
転写後の阻害研究(Wang 2005,Zhang 2005,Roh 2000を参照)
現在最も有効な転写後の阻害は、RNA干渉であり、これは基本的には、標的mRNAの配列特異的で触媒性の分解を誘導するものである。短鎖干渉RNAがトランスフェクションされる細胞系において、転写における最も有効な標的部位が、最も容易にスクリーニングすることができる。RNA干渉の効率は、定量PCRとウェスタンブロット解析によってモニターした。次に、うまくいく標的部位は、腫瘍部位で標的分子を発現するように設計された組換えベクターで発現することができる。
現在最も有効な転写後の阻害は、RNA干渉であり、これは基本的には、標的mRNAの配列特異的で触媒性の分解を誘導するものである。短鎖干渉RNAがトランスフェクションされる細胞系において、転写における最も有効な標的部位が、最も容易にスクリーニングすることができる。RNA干渉の効率は、定量PCRとウェスタンブロット解析によってモニターした。次に、うまくいく標的部位は、腫瘍部位で標的分子を発現するように設計された組換えベクターで発現することができる。
短鎖干渉RNAは、hCAP18のコード配列に基づき設計、合成した。このRNAは、二本鎖の19−merと、いずれかの末端に2塩基のdTdT−オーバーハングとからなる。設計のために、商業的なsiRNAデータベースが、最も簡単に利用することができ、ここにおいて、標的部位は予め選択されている(hCAP18の場合、例えば、アンビオン(Ambion)のsiRNA番号14586、14402、146365)。ZR75−1細胞またはトランスジェニックMCF−7細胞は、インターフェロン関連の経路のような非特異的な応答を回避するために最終濃度は最大10nMでsiRNAでトランスフェクションした。コントロールsiRNAは、標的配列に2〜3個のミスマッチを含む。トランスフェクションの24〜72時間後に細胞を回収し、hCAP18の発現を、上述のようなリアルタイムPCRと定量的なウェスタンブロット解析によって決定した。ウェスタンブロット解析のために、SDS含有サンプル緩衝液中でタンパク質を抽出し、15%トリス−グリシンゲルで分離し、上述のようなニトロセルロースフィルターにエレクトロブロッティングした。フィルターを3%ポンソーSで可逆的に染色し、その後、親和性で精製した抗LL−37抗血清と1/1000の希釈度でインキュベートした。強化された化学発光を用いた、HRPに結合した二次IgGからのシグナルを捕捉し、上述のようにして評価した。
インビボでの実験は、hCAP18発現の最も有効な転写阻害剤にあわせて組み立てた。12匹のマウスに、トランスジェニックhCAP18を発現するMCF−7細胞を注射した。そのマウスの半分に、毎日、オリーブ油(シグマ)50μlに溶解させた阻害剤30mgを皮下注射した。腫瘍形成の発生、サイズおよび分布は、上述したように分析した。インビボでのアプローチに有効なsiRNAは、hCAP18発現を少なくとも90%ダウンレギュレートすると予想される。
マウスにおいて腫瘍形成を妨害するために、hCAP18発現のダウンレギュレーションを数週間維持する必要がある。大量のsiRNAを尾部の静脈に毎日注射することが可能であると証明されたが、治療アプローチとしての現実性はほとんどないようである。その代わりとしては、生物/腫瘍細胞におけるsiRNAの安定な発現が挙げられる。RNAをヘアピンとして発現し、次にこれをsiRNAに細胞内で変換するプラスミドが設計された(例えばpSuper、オリジーン社(OriGene Inc))。我々の最初のアプローチとして、上記実験によって決定された標的siRNA配列をpSuperにクローニングし、このコンストラクトで腫瘍細胞をトランスフェクションし、その後、マウスに埋め込んだ。現在、治療アプローチ、すなわち腫瘍形成後の発現ベクターの送達はまだ開発されていないが、リポソームを用いたプラスミドの送達、および、レトロウイルスの構築、および、この目的のためのアデノウイルス発現ベクターは世界的に研究されている。
RNAiの代替法は、「古典的な」アンチセンス法である。標的転写物に相補的な、長さが20〜3bpの一本鎖オリゴヌクレオチドは、この細胞培地に直接添加されるか、または、マウスに注射した。このオリゴヌクレオチドの主鎖は、一般的には、キャリアー基質をまったく用いなくても、その分解が阻害され、さらにその摂取が容易になるように改変される。しかしながら、阻害メカニズムは非触媒性であり、主鎖の改変は、細胞毒性や非特異的な副作用を高めるため、本方法では、高い用量を必要とする。マウス実験において、典型的な投与範囲は、100〜500μg/動物/日である。
抗体を用いたLL−37活性の阻害(Warburton 2005を参照)
ニワトリ抗体を生産し、計画した実験に十分なラージスケールで親和性で精製した。LL−37に対する抗体は、LL−37活性を阻害できることがこれまでに示されている。それゆえにインビトロでの実験は必要ではない。
ニワトリ抗体を生産し、計画した実験に十分なラージスケールで親和性で精製した。LL−37に対する抗体は、LL−37活性を阻害できることがこれまでに示されている。それゆえにインビトロでの実験は必要ではない。
インビボでの実験のために、上述したようにマウスで腫瘍を誘導した。腫瘍が触診可能である場合、用いて抗体を、最初は1〜100mg/kg抗体で週2回注射した。
次に、腫瘍の発達における抗体の作用を評価した。
次に、腫瘍の発達における抗体の作用を評価した。
参考文献
1. Agarwal C et al.: AGN193109 is a highly effective antagonist of retinoid action in human ectocervical epithelial cells. J Biol Chem 271: 12209-12212 (1996).
2. Andela VB, Rosier RN: The proteosome inhibitor MGI32 attenuates retinoic acid receptor trans-activation and enhances trans-repression of nuclear factor B. Potential relevance to chemo-preventive interventions with retinoids. Molecular Cancer 3: 8-19 (2004).
3. Fiucci G, Ravid D, Reich R, Liscovitch M: Caveolin-1 inhibits anchorage-independent growth, anoikis and invasiveness in MCF-7 human breast cancer cells. Oncogene 21: 2365-2375 (2002).
4. Heilborn J et al.:Antimicrobial protein hCAP18/LL-37 is highly expressed in breast cancer and is a putative growth factor for epithelial cells. Int J Cancer
114:713-9 (2005).
5. Ishizuka S et al.: (23S)-25-Dehydro-1{alpha}-hydroxyvitamin D3-26,23-lactone, a vitamin D receptor antagonist that inhibits osteoclast formation and bone resorption in bone marrow cultures from patients with Paget's disease. Endocrinology. 146:2023-2030 (2005).
6. Lu C, Shen Q et al.: cFos is critical for MCF-7 breast cancer cell growth. Oncogene 24: 6516-6524 (2005).
7. Ludes-Meyers JH et al.: AP-1 blockade inhibits the growth of normal and malignant breast cells. Oncogene 20: 2771-2780 (2001).
8. Roh H et al.:HER2/neu antisense targeting of human breast carcinoma. Oncogene. 2000 Dec 11;19(53):6138-43.
9. Toell A et al.: Different molecular mechanisms of vitamin D3 receptor antagonists. Mol Pharmacol 59: 1478-1485 (2001).
10. Wang Y-h et al.: Knockdown of c-Myc expression by RNAi inhibits MCF-7 breast tumour cells growth in vitro and in vivo. Breast Cancer Res 7: R220-R228 (2005).
11. Warburton C et al.: Treatment of HER-2/neu overexpressing breast cancer xenograft models with trastuzumab (Herceptin) and gefitinib (ZD1839): drug combination effects on tumour growth, HER-2/neu and epidermal growth factor receptor expression, and viable hypoxic cell fraction. Clin Cancer Res. 10:2512-24 (2004).
12. Weber G et al.: Vitamin D induces the antimicrobial protein hCAP18 in human
skin. J Invest Dermatol. 124:1080-2 (2005).
13. Zhang M et al.: Silencing the epidermal growth factor receptor gene with RNAi may be developed as a potential therapy for non small cell lung cancer. Genet Vaccines Ther. 3:5-16 (2005).
1. Agarwal C et al.: AGN193109 is a highly effective antagonist of retinoid action in human ectocervical epithelial cells. J Biol Chem 271: 12209-12212 (1996).
2. Andela VB, Rosier RN: The proteosome inhibitor MGI32 attenuates retinoic acid receptor trans-activation and enhances trans-repression of nuclear factor B. Potential relevance to chemo-preventive interventions with retinoids. Molecular Cancer 3: 8-19 (2004).
3. Fiucci G, Ravid D, Reich R, Liscovitch M: Caveolin-1 inhibits anchorage-independent growth, anoikis and invasiveness in MCF-7 human breast cancer cells. Oncogene 21: 2365-2375 (2002).
4. Heilborn J et al.:Antimicrobial protein hCAP18/LL-37 is highly expressed in breast cancer and is a putative growth factor for epithelial cells. Int J Cancer
114:713-9 (2005).
5. Ishizuka S et al.: (23S)-25-Dehydro-1{alpha}-hydroxyvitamin D3-26,23-lactone, a vitamin D receptor antagonist that inhibits osteoclast formation and bone resorption in bone marrow cultures from patients with Paget's disease. Endocrinology. 146:2023-2030 (2005).
6. Lu C, Shen Q et al.: cFos is critical for MCF-7 breast cancer cell growth. Oncogene 24: 6516-6524 (2005).
7. Ludes-Meyers JH et al.: AP-1 blockade inhibits the growth of normal and malignant breast cells. Oncogene 20: 2771-2780 (2001).
8. Roh H et al.:HER2/neu antisense targeting of human breast carcinoma. Oncogene. 2000 Dec 11;19(53):6138-43.
9. Toell A et al.: Different molecular mechanisms of vitamin D3 receptor antagonists. Mol Pharmacol 59: 1478-1485 (2001).
10. Wang Y-h et al.: Knockdown of c-Myc expression by RNAi inhibits MCF-7 breast tumour cells growth in vitro and in vivo. Breast Cancer Res 7: R220-R228 (2005).
11. Warburton C et al.: Treatment of HER-2/neu overexpressing breast cancer xenograft models with trastuzumab (Herceptin) and gefitinib (ZD1839): drug combination effects on tumour growth, HER-2/neu and epidermal growth factor receptor expression, and viable hypoxic cell fraction. Clin Cancer Res. 10:2512-24 (2004).
12. Weber G et al.: Vitamin D induces the antimicrobial protein hCAP18 in human
skin. J Invest Dermatol. 124:1080-2 (2005).
13. Zhang M et al.: Silencing the epidermal growth factor receptor gene with RNAi may be developed as a potential therapy for non small cell lung cancer. Genet Vaccines Ther. 3:5-16 (2005).
実施例D−培養ヒトケラチノサイトにおいて、ヒト抗菌性ペプチドLL−37はアポトーシスを阻害し、アポトーシスタンパク質の阻害剤(IAP−2)の発現をアップレギュレートする
序論
カテリシジンは、多くの哺乳動物種に見出される抗菌性ペプチドファミリーである。これらは、高度に保存されたアミノ末端ドメイン、カテリン、および、タンパク質分解によって放出されて抗菌活性を付与する可変性カルボキシ末端ドメインからなる(1,2)。このファミリーのうち、18kDaのヒト陽イオン性抗菌タンパク質であるヒトカテリシジン(hCAP18)だけが、主として、好中球、数種の上皮および粘膜細胞(皮膚、気管支、頬粘膜、食道、子宮頚、膣、精巣上体および唾液腺)によって生産される(3〜8)。C末端の37個のアミノ酸ドメイン、LL−37は、膜安定性を破壊することによって(11,12)広範囲の微生物に対する抗菌活性(9、10)を示す。
序論
カテリシジンは、多くの哺乳動物種に見出される抗菌性ペプチドファミリーである。これらは、高度に保存されたアミノ末端ドメイン、カテリン、および、タンパク質分解によって放出されて抗菌活性を付与する可変性カルボキシ末端ドメインからなる(1,2)。このファミリーのうち、18kDaのヒト陽イオン性抗菌タンパク質であるヒトカテリシジン(hCAP18)だけが、主として、好中球、数種の上皮および粘膜細胞(皮膚、気管支、頬粘膜、食道、子宮頚、膣、精巣上体および唾液腺)によって生産される(3〜8)。C末端の37個のアミノ酸ドメイン、LL−37は、膜安定性を破壊することによって(11,12)広範囲の微生物に対する抗菌活性(9、10)を示す。
抗菌性の機能のほかに、このペプチドは、その他の生物活性にも関与しており、例えば、走化性、および、血液および上皮細胞におけるサイトカイン放出(8,13)、上皮細胞増殖の誘導(14)、および、血管新生(15)に関与している。近年の研究によれば、LL−37は創傷治癒の際に高度に発現され、インビトロでのヒトケラチノサイトの増殖に影響を与え、創傷の再上皮化に関与することが示されている(16,17)。
材料および方法
細胞培養
包皮の表皮ケラチノサイトを、カスケード・バイオロジクス(Cascade Biologics)(カスケード・バイオロジクス,ユージーン,オレゴン州)から得て、0,06mMのカルシウム、0.2%v/vBPE、5μg/mlのウシインスリン、0.18μg/mlのヒドロコルチゾン、5μg/mlのウシトランスフェリン、0.2ng/mlのヒト上皮増殖因子、100U/mlのペニシリンG、100μg/ml硫酸ストレプトマイシン、および、0.25μg/mlのアンフォテリシンB(全てカスケード・バイオロジクス)が補足されたエピライフ(EpiLife)基礎培地(カスケード・バイオロジクス)中で、37℃、および、5%CO2で培養した。細胞を、0.025%(w/v)トリプシン、および、0.01%(w/v)EDTA(カスケード・バイオロジクス)を用いて週1回継代させた。
細胞培養
包皮の表皮ケラチノサイトを、カスケード・バイオロジクス(Cascade Biologics)(カスケード・バイオロジクス,ユージーン,オレゴン州)から得て、0,06mMのカルシウム、0.2%v/vBPE、5μg/mlのウシインスリン、0.18μg/mlのヒドロコルチゾン、5μg/mlのウシトランスフェリン、0.2ng/mlのヒト上皮増殖因子、100U/mlのペニシリンG、100μg/ml硫酸ストレプトマイシン、および、0.25μg/mlのアンフォテリシンB(全てカスケード・バイオロジクス)が補足されたエピライフ(EpiLife)基礎培地(カスケード・バイオロジクス)中で、37℃、および、5%CO2で培養した。細胞を、0.025%(w/v)トリプシン、および、0.01%(w/v)EDTA(カスケード・バイオロジクス)を用いて週1回継代させた。
HaCaTケラチノサイトを、10%ウシ胎仔血清(ハイクローン,ブール・ノルディック社,フッディンゲ,スウェーデン)、2mMグルタミン、ペニシリン(50U/L,ギブコ−BRL)、および、ストレプトマイシン(50mg/ml,ギブコ−BRL)が補足されたDMEM(ダルベッコ改変イーグル培地;ギブコ−BRL(Gibco−BRL)テクノロジー,ペーズリー,イギリス)中で培養した。
細胞の処理
25,000細胞/ウェルを6ウェルプレート培養皿で平板培養した。平板培養して48時間後に、細胞を、0、0.23、0.46、0.69、0.9、1.15、2.3、4.6および11.5μMのLL−37で処理した。各処理は三連で行った。これらのペプチドを、培地(HaCaT細胞には5%FCSを含むDMEM、および、HEKn細胞には0.1%FCSが補足されたエピライフ基礎培地)で希釈した。この細胞を合計で24時間にわたり処理した。LL−37の刺激の後、この細胞に、トポイソメラーゼI阻害剤であるカンプトテンシン(CAM)(シグマ・ケミカル社,セントルイス,ミズーリ州)のジメチルスルホキシド溶液を最終濃度6μMまで添加した。解析の前に、細胞をさらに24時間インキュベートした。
25,000細胞/ウェルを6ウェルプレート培養皿で平板培養した。平板培養して48時間後に、細胞を、0、0.23、0.46、0.69、0.9、1.15、2.3、4.6および11.5μMのLL−37で処理した。各処理は三連で行った。これらのペプチドを、培地(HaCaT細胞には5%FCSを含むDMEM、および、HEKn細胞には0.1%FCSが補足されたエピライフ基礎培地)で希釈した。この細胞を合計で24時間にわたり処理した。LL−37の刺激の後、この細胞に、トポイソメラーゼI阻害剤であるカンプトテンシン(CAM)(シグマ・ケミカル社,セントルイス,ミズーリ州)のジメチルスルホキシド溶液を最終濃度6μMまで添加した。解析の前に、細胞をさらに24時間インキュベートした。
フローサイトメトリーによるアポトーシスの評価
2種のアポトーシスのパラメーター、すなわち膜の完全性の喪失とDNAの減少を、フローサイトメトリーによって評価した。
2種のアポトーシスのパラメーター、すなわち膜の完全性の喪失とDNAの減少を、フローサイトメトリーによって評価した。
膜の完全性の評価:LL−37での刺激、および、CAMを用いた処理の後に、細胞をトリプシン処理によって回収した。次に、この細胞を冷ダルベッコリン酸緩衝食塩水(PBS)で洗浄し、血球計数器(ホイザー・サイエンティフィック(Hausser Scientific),ホーシャム,ペンシルベニア州)で計数した。細胞濃度をPBS中で1×106細胞/mlに調節した。ヨウ化プロピジウムとYO−PRO−1(モレキュラープローブス(Molecular Probes),ユージーン,オレゴン州)色素で染色することによってアポトーシスを検出した。染色された細胞をFACScan(ベクトン−ディッキンソン,サンノゼ,カリフォルニア州)で解析した。前方光散乱(FSC)および側方光散乱(SSC)特性に基づき解析するために、細胞をゲーティングした。YO−PROおよびPI陽性細胞のパーセンテージを示すセルクエスト(Cell Quest)ソフトウェア(ベクトン・ディッキンソン)を用いて、YO−PROとヨウ化プロピジウム(PI)を示す細胞の解析を行った。YO−PROで緑色に染色された細胞をアポトーシスとみなした。ヨウ化プロピジウムで赤色に染色された細胞を壊死とした。生きている生存可能な細胞は、色素をわずかしか吸収しないか、または、まったく吸収しなかった。
フローサイトメトリーによるDNA含量の解析:この解析のために、PI/RNアーゼ染色緩衝液(ベクトン・ディッキンソン)を用いた。冷70%(v/v)エタノール中でトリプシン処理した細胞を固定し、それらを使用するまで4℃で保存した。ヨウ化プロピジウムのDNAへの取り込みによってDNA含量を測定した。蛍光解析を、FACScanフローサイトメーター(ベクトン・ディッキンソン、マウンテンビュー,カリフォルニア州,米国)を用いて行った。細胞のサイズと粒度を決定するために、粒子のFSC(前方光散乱)およびSSC(側方光散乱)を同時に測定した。PIで染色された核の赤色の蛍光を、FL−4ウィンドウ(600nmバンドパスフィルター、および、35nmの帯域幅)で検出した。蛍光強度は、細胞のDNA含量に比例していた。各ヒストグラムを2部に分割した:(1)非同調の非アポトーシス性の生きた細胞を示すM1領域(2N〜4Nに等しいDNA含量を有するG1、SおよびG2相における細胞);および、(2)生きた細胞に比べて少量のDNAを含むアポトーシス細胞を示すM2領域。
カスパーゼ−3活性分析
上述したようにして蛍光基質VDVAD−AMC(100μM)、および、DEVD−AMC(50μM)を用いてカスパーゼ−3酵素活性を概算した。簡単に言えば、細胞溶解産物を反応緩衝液(100mMのHEPES、10%スクロース、5mMジチオスレイトール(DTT)、10〜6%のNP−40および0.1%CHAPS(pH7.25))と合わせ、マイクロタイタープレートに添加した。蛍光発生基質の切断を、フルオロスキャンIIプレートリーダー(ラボシステムズ,ストックホルム,スウェーデン)でAMCの解離によってモニターした。蛍光単位を、遊離のAMCから作製した標準曲線を用いてAMCのpmolに変換した。データを線形回帰によって解析し、1分あたり放出されたAMCのpmolとして表示した。
上述したようにして蛍光基質VDVAD−AMC(100μM)、および、DEVD−AMC(50μM)を用いてカスパーゼ−3酵素活性を概算した。簡単に言えば、細胞溶解産物を反応緩衝液(100mMのHEPES、10%スクロース、5mMジチオスレイトール(DTT)、10〜6%のNP−40および0.1%CHAPS(pH7.25))と合わせ、マイクロタイタープレートに添加した。蛍光発生基質の切断を、フルオロスキャンIIプレートリーダー(ラボシステムズ,ストックホルム,スウェーデン)でAMCの解離によってモニターした。蛍光単位を、遊離のAMCから作製した標準曲線を用いてAMCのpmolに変換した。データを線形回帰によって解析し、1分あたり放出されたAMCのpmolとして表示した。
RT−PCR
様々な処理の後、キアゲンのRNアーゼイ(RNasy)キット(オペロン・バイオテクノロジーズ,コローニュ,ドイツ)を製造元の指示に従って用いることによって細胞からトータルRNAを単離した。第一鎖合成キット(アマシャム・バイオサイエンス)を用いて逆転写を行った。
様々な処理の後、キアゲンのRNアーゼイ(RNasy)キット(オペロン・バイオテクノロジーズ,コローニュ,ドイツ)を製造元の指示に従って用いることによって細胞からトータルRNAを単離した。第一鎖合成キット(アマシャム・バイオサイエンス)を用いて逆転写を行った。
結果
CAMで誘導されたアポトーシスのLL−37による阻害
我々は、アポトーシス誘導物質として広く用いられている薬物であるCAMにより誘導された細胞死およびアポトーシスに対するLL−37の作用を試験した。処理後、膜の完全性とDNA断片化を2つの方法を用いてフローサイトメトリーで解析した。第一の方法は、細胞膜の透過性の増加を区別することが可能な2種の染色システムであり;未処理細胞を、染色強度に関して解析し(図7)、3タイプ:生存可能な染色されていない細胞、アポトーシスのYO−PROで染色された細胞、および、YO−PROとPLの両方で染色された壊死の細胞に分類した。第二の方法では、エタノールで透過させた細胞におけるDNA含量をPIで解析した。アポトーシス細胞は、断片化のために、より少ないDNA量を示した。
CAMで誘導されたアポトーシスのLL−37による阻害
我々は、アポトーシス誘導物質として広く用いられている薬物であるCAMにより誘導された細胞死およびアポトーシスに対するLL−37の作用を試験した。処理後、膜の完全性とDNA断片化を2つの方法を用いてフローサイトメトリーで解析した。第一の方法は、細胞膜の透過性の増加を区別することが可能な2種の染色システムであり;未処理細胞を、染色強度に関して解析し(図7)、3タイプ:生存可能な染色されていない細胞、アポトーシスのYO−PROで染色された細胞、および、YO−PROとPLの両方で染色された壊死の細胞に分類した。第二の方法では、エタノールで透過させた細胞におけるDNA含量をPIで解析した。アポトーシス細胞は、断片化のために、より少ないDNA量を示した。
HaCaTおよびHEKn細胞においてアポトーシスが誘導された用量および時間を選択するために、前もって濃度および時点の実験を行った(データ示さず)。6μMのCAMに24時間晒した場合、HaCaTとケラチノサイトの両方が、アポトーシスに特徴的な形態学的な変化、例えば膜の完全性とDNAのプロファイルにおける変化を示した(図7)。この細胞を、LL−37単独で、または、LL−37およびCAMで同時に24時間処理した場合、アポトーシス細胞の量の変化は観察されなかった(データ示さず)。しかしながら、高濃度のLL−37(4.6および11.5μM)は細胞毒性り、壊死の(ただしアポトーシス細胞ではない)分画が増加した。同時に処理する代わりに、CAMによるアポトーシス誘導前にこの細胞をLL−37に24時間晒した場合は、この結果は異なる。LL−37は、HEKnおよびHaCaT細胞において、0.23μMから2,3μMまでの濃度で、DNA断片化と膜透過性の喪失の両方を阻害した(図7および8)。これらの条件下でも、LL−37は、より高い濃度で(4.6μM、および、11.5μM)細胞死を引き起こした。毒性濃度でさえも、アポトーシス細胞分画は低いままであり、これは、LL−37による細胞死は、非アポトーシスのメカニズムによって生じることを示す。
LL−37は、HaCaTおよびHEKn細胞におけるCAMによるカスパーゼ−3の誘導を阻害する
カスパーゼ−3はアポトーシス経路における主要なタンパク質の1つであり、アポトーシスの初期の指標であることから、我々は、LL−37で治療した、または処理していないケラチノサイトにおけるカスパーゼ−3活性を測定することにした。ケラチノサイトにおいて、最初の12時間のCAM処理の間カスパーゼ活性は有意に増加し、24時間でピークに達した。2.3μMのLL−37での前処理によって、カンプトテンシンで誘導されたカスパーゼの活性化が減少した(図9a、および、b)。これらのデータによれば、LL−37は、カスパーゼ活性を減少させることによってヒトケラチノサイトにおけるアポトーシスを阻害することが示唆される。
カスパーゼ−3はアポトーシス経路における主要なタンパク質の1つであり、アポトーシスの初期の指標であることから、我々は、LL−37で治療した、または処理していないケラチノサイトにおけるカスパーゼ−3活性を測定することにした。ケラチノサイトにおいて、最初の12時間のCAM処理の間カスパーゼ活性は有意に増加し、24時間でピークに達した。2.3μMのLL−37での前処理によって、カンプトテンシンで誘導されたカスパーゼの活性化が減少した(図9a、および、b)。これらのデータによれば、LL−37は、カスパーゼ活性を減少させることによってヒトケラチノサイトにおけるアポトーシスを阻害することが示唆される。
LL−37は、ヒトケラチノサイトにおいてIAP−2発現を誘導する
アポトーシスタンパク質の阻害剤(IAP)の種類は、主要なアポトーシスの調節因子と説明されている。このファミリーは、カスパーゼ−3活性を阻害する能力を有する。ブタカテリシジン種のPR39の抗アポトーシス機能は、IAP−2発現の誘導と関連を示している。それゆえに、我々は、RT−PCRによって、異なるLL−37濃度、異なる時点で処理したケラチノサイトにおけるIAP−2のHEKn細胞における発現を解析した。2.3μMのLL−37での刺激により、刺激後6時間から12時間にかけてIAP−2のmRNAが増加した(図10)。2.3μMより低い濃度のLL−37は、c−IAP−2レベルに影響を与えなかった(データ示さず)
アポトーシスタンパク質の阻害剤(IAP)の種類は、主要なアポトーシスの調節因子と説明されている。このファミリーは、カスパーゼ−3活性を阻害する能力を有する。ブタカテリシジン種のPR39の抗アポトーシス機能は、IAP−2発現の誘導と関連を示している。それゆえに、我々は、RT−PCRによって、異なるLL−37濃度、異なる時点で処理したケラチノサイトにおけるIAP−2のHEKn細胞における発現を解析した。2.3μMのLL−37での刺激により、刺激後6時間から12時間にかけてIAP−2のmRNAが増加した(図10)。2.3μMより低い濃度のLL−37は、c−IAP−2レベルに影響を与えなかった(データ示さず)
LL−37で刺激されたケラチノサイトは、COX−2をアップレギュレートする
以前の研究では、上皮細胞におけるCOX−2の阻害によりIAP−2の発現が減少するため、COX−2はIAP−2発現のレギュレーターと同定されている。それゆえに、我々は、2.3μMのLL−37で処理したHEKnにおけるCOX−2の発現を解析した。mRNAレベルの時間依存性の増加が見出され(図11)、12時間後に明らかに最高のレベルに達した。LL−37刺激の後に、COX−2がIAP−2発現の増加を媒介するかどうかを分析するために、HEKn細胞を、25nMの特異的なCOX−2阻害剤SC−791で12時間前処理し、次に、2μMのLL−37で刺激した。これらの条件下で、LL−37処理は、これまで観察されたようにIAP−2のmRNAレベルを増加させなかった(図12)。これらのデータによれば、LL−37は、COX−2酵素の発現の増加によってIAP−2レベルを増加させることが示唆される。
以前の研究では、上皮細胞におけるCOX−2の阻害によりIAP−2の発現が減少するため、COX−2はIAP−2発現のレギュレーターと同定されている。それゆえに、我々は、2.3μMのLL−37で処理したHEKnにおけるCOX−2の発現を解析した。mRNAレベルの時間依存性の増加が見出され(図11)、12時間後に明らかに最高のレベルに達した。LL−37刺激の後に、COX−2がIAP−2発現の増加を媒介するかどうかを分析するために、HEKn細胞を、25nMの特異的なCOX−2阻害剤SC−791で12時間前処理し、次に、2μMのLL−37で刺激した。これらの条件下で、LL−37処理は、これまで観察されたようにIAP−2のmRNAレベルを増加させなかった(図12)。これらのデータによれば、LL−37は、COX−2酵素の発現の増加によってIAP−2レベルを増加させることが示唆される。
考察
皮膚の傷害後に抗菌性ペプチドLL−37発現が増加することが報告されている。このペプチドの発現は、感染の予防だけでなく、走化性、移動、血管新生および再上皮化のような様々な創傷治癒関連プロセスへの活発な関与によって、創傷治癒の促進に関連している。
皮膚の傷害後に抗菌性ペプチドLL−37発現が増加することが報告されている。このペプチドの発現は、感染の予防だけでなく、走化性、移動、血管新生および再上皮化のような様々な創傷治癒関連プロセスへの活発な関与によって、創傷治癒の促進に関連している。
本発明のデータによれば、LL−37も、アポトーシスへの阻害作用によってケラチノサイトにおける細胞生存の促進に関与することが示される。
略語
hCAP18/LL−37,ヒトカテリシジン抗菌性ペプチド18kDa/LL−37;IAP−2,アポトーシスタンパク質−2阻害剤;COX−2,シクロオキシゲナーゼ−2;VDVAD−AMC,カスパーゼ3基質;HEKn,新生児由来ヒト表皮ケラチノサイト;DMEN,ダルベッコ改変イーグル培地;PBS,リン酸緩衝生理食塩水;DEVDアーゼ(DEVDase),Asp−Glu−Val−Aspプロテアーゼ活性CHAPS;3−[(3−コラミドプロピル)ジメチル−アンモニオ]プロパン−1−スルホン酸,CAM:カンプトテシン。
hCAP18/LL−37,ヒトカテリシジン抗菌性ペプチド18kDa/LL−37;IAP−2,アポトーシスタンパク質−2阻害剤;COX−2,シクロオキシゲナーゼ−2;VDVAD−AMC,カスパーゼ3基質;HEKn,新生児由来ヒト表皮ケラチノサイト;DMEN,ダルベッコ改変イーグル培地;PBS,リン酸緩衝生理食塩水;DEVDアーゼ(DEVDase),Asp−Glu−Val−Aspプロテアーゼ活性CHAPS;3−[(3−コラミドプロピル)ジメチル−アンモニオ]プロパン−1−スルホン酸,CAM:カンプトテシン。
参考文献
1. Zanetti, M., Gennaro, R., and Romeo, D. (1995) FEBS Lett 374(1), 1-5
2. Sorensen, O. E., Follin, P., Johnsen, A. H., Calafat, J., Tjabringa, G. S., Hiemstra, P. S., and Borregaard, N. (2001) Blood 97(12), 3951-3959
3. Sorensen, O., Arnljots, K., Cowland, J. B., Bainton, D. F., and Borregaard, N. (1997) Blood 90(7), 2796-2803
4. Bals, R., Wang, X., Zasloff, M., and Wilson, J. M. (1998) Proc Natl Acad Sci
U S A 95(16), 9541-9546
5. Frohm, M., Agerberth, B., Ahangari, G., Stahle-Backdahl, M., Liden, S., Wigzell, H., and Gudmundsson, G. H. (1997) J Biol Chem 272(24), 15258-15263
6. Agerberth, B., Gunne, H., Odeberg, J., Kogner, P., Boman, H. G., and Gudmundsson, G. H. (1995) Proc Natl Acad Sci U S A 92(1), 195-199
7. Frohm Nilsson, M., Sandstedt, B., Sorensen, O., Weber, G., Borregaard, N., and Stahle-Backdahl, M. (1999) Infect Immun 67(5), 2561-2566
8. De, Y., Chen, Q., Schmidt, A. P., Anderson, G. M., Wang, J. M., Wooters, J.,
Oppenheim, J. J., and Chertov, O. (2000) J Exp Med 192(7), 1069-1074
9. Zasloff, M. (2002) Nature 415(6870), 389-395
10. Dorschner, R. A., Pestonjamasp, V. K., Tamakuwala, S., Ohtake, T., Rudisill
, J., Nizet, V., Agerberth, B., Gudmundsson, G. H., and Gallo, R. L. (2001) J Invest Dermatol 117(1), 91-97
11. Oren, Z., Lerman, J. C., Gudmundsson, G. H., Agerberth, B., and Shai, Y. (1999) Biochem J 341 ( Pt 3), 501-513
12. Ramanathan, B., Davis, E. G., Ross, C. R., and Blecha, F. (2002) Microbes Infect 4(3), 361-372
13. Braff, M. H., Hawkins, M. A., Di Nardo, A., Lopez-Garcia, B., Howell, M. D., Wong, C., Lin, K., Streib, J. E., Dorschner, R., Leung, D. Y., and Gallo, R. L. (2005) J Immunol 174(7), 4271-4278
14. Shaykhiev, R., Beisswenger, C., Kandler, K., Senske, J., Puchner, A., Damm,
T., Behr, J., and Bals, R. (2005) Am J Physiol Lung Cell Mol Physiol 289(5), L842-L848
15. Koczulla, R., von Degenfeld, G., Kupatt, C., Krotz, F., Zahler, S., Gloe, T., Issbrucker, K., Unterberger, P., Zaiou, M., Lebherz, C., Karl, A., Raake, P., Pfosser, A., Boekstegers, P., Welsch, U., Hiemstra, P. S., Vogelmeier, C., Gallo, R. L., Clauss, M., and Bals, R. (2003) J Clin Invest 111(11), 1665-1672
16. Heilborn, J. D., Nilsson, M. F., Jimenez, C. I., Sandstedt, B., Borregaard,
N., Tham, E., Sorensen, O. E., Weber, G., and Stahle, M. (2005) Int J Cancer 114(5), 713-719
17. Heilborn, J. D., Nilsson, M. F., Kratz, G., Weber, G., Sorensen, O., Borregaard, N., and Stahle-Backdahl, M. (2003) J Invest Dermatol 120(3), 379-389
1. Zanetti, M., Gennaro, R., and Romeo, D. (1995) FEBS Lett 374(1), 1-5
2. Sorensen, O. E., Follin, P., Johnsen, A. H., Calafat, J., Tjabringa, G. S., Hiemstra, P. S., and Borregaard, N. (2001) Blood 97(12), 3951-3959
3. Sorensen, O., Arnljots, K., Cowland, J. B., Bainton, D. F., and Borregaard, N. (1997) Blood 90(7), 2796-2803
4. Bals, R., Wang, X., Zasloff, M., and Wilson, J. M. (1998) Proc Natl Acad Sci
U S A 95(16), 9541-9546
5. Frohm, M., Agerberth, B., Ahangari, G., Stahle-Backdahl, M., Liden, S., Wigzell, H., and Gudmundsson, G. H. (1997) J Biol Chem 272(24), 15258-15263
6. Agerberth, B., Gunne, H., Odeberg, J., Kogner, P., Boman, H. G., and Gudmundsson, G. H. (1995) Proc Natl Acad Sci U S A 92(1), 195-199
7. Frohm Nilsson, M., Sandstedt, B., Sorensen, O., Weber, G., Borregaard, N., and Stahle-Backdahl, M. (1999) Infect Immun 67(5), 2561-2566
8. De, Y., Chen, Q., Schmidt, A. P., Anderson, G. M., Wang, J. M., Wooters, J.,
Oppenheim, J. J., and Chertov, O. (2000) J Exp Med 192(7), 1069-1074
9. Zasloff, M. (2002) Nature 415(6870), 389-395
10. Dorschner, R. A., Pestonjamasp, V. K., Tamakuwala, S., Ohtake, T., Rudisill
, J., Nizet, V., Agerberth, B., Gudmundsson, G. H., and Gallo, R. L. (2001) J Invest Dermatol 117(1), 91-97
11. Oren, Z., Lerman, J. C., Gudmundsson, G. H., Agerberth, B., and Shai, Y. (1999) Biochem J 341 ( Pt 3), 501-513
12. Ramanathan, B., Davis, E. G., Ross, C. R., and Blecha, F. (2002) Microbes Infect 4(3), 361-372
13. Braff, M. H., Hawkins, M. A., Di Nardo, A., Lopez-Garcia, B., Howell, M. D., Wong, C., Lin, K., Streib, J. E., Dorschner, R., Leung, D. Y., and Gallo, R. L. (2005) J Immunol 174(7), 4271-4278
14. Shaykhiev, R., Beisswenger, C., Kandler, K., Senske, J., Puchner, A., Damm,
T., Behr, J., and Bals, R. (2005) Am J Physiol Lung Cell Mol Physiol 289(5), L842-L848
15. Koczulla, R., von Degenfeld, G., Kupatt, C., Krotz, F., Zahler, S., Gloe, T., Issbrucker, K., Unterberger, P., Zaiou, M., Lebherz, C., Karl, A., Raake, P., Pfosser, A., Boekstegers, P., Welsch, U., Hiemstra, P. S., Vogelmeier, C., Gallo, R. L., Clauss, M., and Bals, R. (2003) J Clin Invest 111(11), 1665-1672
16. Heilborn, J. D., Nilsson, M. F., Jimenez, C. I., Sandstedt, B., Borregaard,
N., Tham, E., Sorensen, O. E., Weber, G., and Stahle, M. (2005) Int J Cancer 114(5), 713-719
17. Heilborn, J. D., Nilsson, M. F., Kratz, G., Weber, G., Sorensen, O., Borregaard, N., and Stahle-Backdahl, M. (2003) J Invest Dermatol 120(3), 379-389
実施例E−本発明の標的化した物質の生産およびインビボでの使用
抗hCAP18/LL−37抗体/抗HMFGl抗体融合体の発現
阻害剤部分(抗hCAP18/LL−37抗体)、および、標的化部分(抗多形性の上皮のムチン抗体、HMFG1)を含む融合タンパク質を、NSO骨髄腫細胞で発現させた。
抗hCAP18/LL−37抗体/抗HMFGl抗体融合体の発現
阻害剤部分(抗hCAP18/LL−37抗体)、および、標的化部分(抗多形性の上皮のムチン抗体、HMFG1)を含む融合タンパク質を、NSO骨髄腫細胞で発現させた。
簡単に言えば、NSO骨髄腫細胞は、エレクトロポレーションによって、融合タンパク質の軽鎖および重鎖成分をコードする発現ベクターでコトランスフェクションした。次に、トランスフェクトマス(transfectomas)を選択し、ELISA分析で抗体産生に関してスクリーニングした。
患者への投与
このような融合タンパク質を滅菌注射用水溶液に製剤化した。
次に、このような製剤を、乳癌に罹った患者に静脈内(IV)輸液によって90分間かけて投与した。
用量は、医師によって決定される各患者それぞれの必要条件に従って選択した。しかしながら、典型的には、4mg/kgの初期用量が用いられ、続いて、週1回、2mg/kgの維持量が用いられる。
このような融合タンパク質を滅菌注射用水溶液に製剤化した。
次に、このような製剤を、乳癌に罹った患者に静脈内(IV)輸液によって90分間かけて投与した。
用量は、医師によって決定される各患者それぞれの必要条件に従って選択した。しかしながら、典型的には、4mg/kgの初期用量が用いられ、続いて、週1回、2mg/kgの維持量が用いられる。
病気の進行のモニター
次に、融合タンパク質を用いた処理の乳癌の進行に与える影響を、従来のマンモグラフィーによってモニターした。
次に、融合タンパク質を用いた処理の乳癌の進行に与える影響を、従来のマンモグラフィーによってモニターした。
Claims (68)
- 癌細胞の増殖を阻害するための物質であって、ここにおいて、該物質は、hCAP18/LL−37の生物活性を阻害する、上記物質。
- 物質は、hCAP18/LL−37の転写、翻訳および/または結合特性を変化させることによってhCAP18/LL−37の生物活性を阻害する、請求項1に記載の物質。
- 物質は、hCAP18/LL−37の転写阻害剤である、請求項1または2に記載の物質。
- 物質は、hCAP18/LL−37の翻訳阻害剤である、請求項1または2に記載の物質。
- 物質は、hCAP18/LL−37の結合特性の阻害剤である、請求項1または2に記載の物質。
- 物質は、hCAP18/LL−37受容体アンタゴニストである、請求項1に記載の物質。
- hCAP18/LL−37受容体は、FPRL1である、請求項6に記載の物質。
- 物質は、癌細胞中のhCAP18/LL−37の生物活性を選択的に阻害することができる、請求項1〜7のいずれか一項に記載の物質。
- 物質は、該物質に晒されていない癌細胞におけるhCAP18/LL−37の生物活性と比較して、hCAP18/LL−37の生物活性を50%またはそれ以上阻害することができる、請求項1〜8のいずれか一項に記載の物質。
- 物質は、短鎖干渉RNA(siRNA)分子、アンチセンスオリゴヌクレオチド、hCAP18/LL−37への結合親和性を有する化合物、および、低分子量の阻害化合物からなる群より選択される、請求項1〜9のいずれか一項に記載の物質。
- 物質は、短鎖干渉RNA(siRNA)分子である、請求項1〜10のいずれか一項に記載の物質。
- siRNA分子は、配列番号1に記載のヌクレオチドの配列のフラグメント、または、それらの変異体を含む、請求項11に記載の物質。
- siRNA分子は、長さが19〜23個のヌクレオチドである、請求項11または12に記載の物質。
- 物質は、アンチセンスオリゴヌクレオチドである、請求項1〜10のいずれか一項に記載の物質。
- アンチセンスオリゴヌクレオチドは、配列番号1に記載のヌクレオチドのフラグメント、または、それらの変異体を含む、請求項14に記載の物質。
- アンチセンスオリゴヌクレオチドは、長さが15〜35個のヌクレオチドである、請求項14または15に記載の物質。
- 物質は、hCAP18/LL−37への結合親和性を有する化合物である、請求項1〜10のいずれか一項に記載の物質。
- 物質は、低分子量の阻害化合物である、請求項1〜10のいずれか一項に記載の物質。
- 化合物はポリペプチドである、請求項17に記載の物質。
- ポリペプチドは、抗体、または、それらの抗原結合フラグメントである、請求項19に記載の物質。
- 抗体、または、それらの抗原結合フラグメントは、Fvフラグメント、Fab様フラグメント、単一の可変性ドメイン、および、ドメイン抗体からなる群より選択される、請求項20に記載の物質。
- 抗体、または、それらの抗原結合フラグメントは、ヒト化されている、請求項20または21に記載の物質。
- 化合物は、hCAP18/LL−37へのリガンド結合能力を有する、請求項17に記載の物質。
- 物質は、癌細胞に選択的に送達することができる、または、癌細胞によって選択的に活性化することができる、請求項1〜23のいずれか一項に記載の物質。
- 物質は、標的細胞に特異的な部分を含む、請求項24に記載の物質。
- 標的細胞に特異的な部分は、抗体、または、それらの抗原結合フラグメントである、請求項25に記載の物質。
- 抗体、または、それらの抗原結合フラグメントは、ヒト化されている、請求項26に記載の物質。
- 抗体、または、それらの抗原結合フラグメントは、癌細胞の表面で発現された抗原への特異性を有する、請求項26または27に記載の物質。
- 癌細胞の表面で発現された抗原は、C46、85A12、H17E2、NR−LU−10、HMFG1、SM−3(IgG1)、W14、L6(IgG2a)、1F5(IgG2a)、アルファフェトプロテイン、Ca−125、前立腺に特異的な抗原、および、上皮増殖因子受容体ファミリーの種類からなる群より選択される、請求項28に記載の物質。
- 物質は、癌細胞によって選択的に活性化されたプロドラッグである、請求項24に記載の物質。
- 癌細胞は上皮細胞である、請求項1〜30のいずれか一項に記載の物質。
- 癌細胞は扁平細胞である、請求項1〜31のいずれか一項に記載の物質。
- 癌細胞は、乳房、胆管、脳、結腸、胃、生殖器、肺および気道、皮膚、胆嚢、肝臓、上咽頭、神経細胞、腎臓、前立腺、リンパ腺、ならびに、消化管の癌細胞からなる群より選択される、請求項1〜32のいずれか一項に記載の物質。
- 癌細胞は乳癌細胞である、請求項33に記載の物質。
- 乳癌細胞はエルストン(Elston)グレードIIIの細胞である、請求項34に記載の物質。
- 乳癌細胞は転移性である、請求項1〜35のいずれか一項に記載の物質。
- 請求項1〜36のいずれか一項に記載の物質、および、製薬上許容できる賦形剤、希釈剤またはキャリアーを含む医薬組成物。
- 非経口投与に適した、請求項37に記載の医薬組成物。
- 組成物は、癌細胞を標的とした物質の送達が可能である、請求項37または38に記載の医薬組成物。
- 患者における癌細胞の増殖を阻害する方法であって、該方法は、該患者に、請求項1〜36のいずれか一項に記載の物質、または、請求項37〜39のいずれか一項に記載の医薬組成物を投与することを含む、上記方法。
- 患者はヒトである、請求項40に記載の方法。
- 物質は、癌細胞に選択的に送達されるか、または、癌細胞によって選択的に活性化される、請求項40または41に記載の方法。
- 医薬用途のための、請求項1〜36のいずれか一項に記載の物質。
- 癌の治療に使用するための、請求項43に記載の物質。
- 癌細胞の増殖を阻害するための医薬品の製造における、請求項1〜36のいずれか一項に記載の物質の使用。
- 癌細胞は上皮細胞である、請求項40〜42に記載の方法、または、請求項45に記載の使用。
- 癌細胞は扁平細胞である、請求項40〜42に記載の方法、または、請求項45に記載の使用。
- 癌細胞は、乳房、胆管、脳、結腸、胃、生殖器、肺および気道、皮膚、胆嚢、肝臓、上咽頭、神経細胞、腎臓、前立腺、リンパ腺、ならびに、消化管の癌細胞からなる群より選択される、請求項40〜42に記載の方法、または、請求項45に記載の使用。
- 癌細胞は乳癌細胞である、請求項48に記載の方法または使用。
- 乳癌細胞はエルストングレードIIIの細胞である、請求項49に記載の方法または使用。
- 乳癌細胞は転移性である、請求項49または50に記載の方法または使用。
- 患者における癌細胞を検出する方法であって、該方法は、
(a)試験しようとする患者からの細胞サンプルを提供する工程;
(b)該細胞によって生産されたhCAP18/LL−37の量を測定する工程;および、
(c)工程(b)で測定されたhCAP18/LL−37の量と、健康な細胞によって生産されたhCAP18/LL−37の量とを比較する工程
を含み、ここにおいて、患者からの細胞サンプルにおけるhCAP18/LL−37の生産のレベルが、健康な細胞におけるレベルと比較して高いことは、該細胞が癌細胞であることを示す、上記方法。 - 工程(a)における細胞は、乳房、胆管、脳、結腸、胃、生殖器、肺および気道、皮膚、胆嚢、肝臓、上咽頭、神経細胞、腎臓、前立腺、リンパ腺、ならびに、消化管の細胞からなる群より選択される、請求項52に記載の方法。
- 工程(a)における細胞サンプルは、腫瘍由来であるか、または、腫瘍の可能性がある組織由来である、請求項52または53に記載の方法。
- 工程(b)は、細胞サンプルと、hCAP18/LL−37に結合する物質とを接触させること、続いて、それに結合したhCAP18/LL−37の量を検出することを含む、請求項52〜54のいずれか一項に記載の方法。
- hCAP18/LL−37に結合する物質は、抗体、または、それらの抗原結合フラグメントである、請求項55に記載の方法。
- 工程(b)は、(i)細胞と、hCAP18/LL−37に結合する物質とを接触させること、および、(ii)抗体、または、それらの抗原結合フラグメントを用いて、該物質に結合するhCAP18/LL−37の量を検出することを含む、請求項55または56に記載の方法。
- 工程(b)は、ELISAによって行われる、請求項55〜57のいずれか一項に記載の方法。
- 工程(b)は、細胞中のhCAP18/LL−37のmRNAの量を測定することを含む、請求項52〜54のいずれか一項に記載の方法。
- 工程(b)は、サザンブロット、または、RT−PCRによって行われる、請求項59に記載の方法。
- 患者における癌の進行をモニターする方法であって、該方法は、
(a)第一の時点で該患者から回収された細胞のサンプルを提供すること、および、請求項52〜60のいずれか一項に記載の方法を用いて、そこに含まれる癌細胞を検出すること;
(b)第二の時点で該患者から回収された細胞のサンプルを提供すること、および、請求項52〜60のいずれか一項に記載の方法を用いて、そこに含まれる癌細胞を検出すること;および、
(c)工程(a)および(b)で測定された癌細胞の数を比較すること
を含み、ここにおいて、工程(b)で測定された癌細胞の数が、工程(a)と比較して高いことは、癌が進行していることの指標である、上記方法。 - hCAP18/LL−37、または、それらをコードするmRNAに結合する物質を含む、請求項52〜61のいずれか一項に記載の方法を行うための診断キット。
- hCAP18/LL−37に結合することができる抗体、または、それらの抗原結合フラグメントを含む、請求項62に記載の診断キット。
- hCAP18/LL−37に結合することができる抗体、または、それらの抗原結合フラグメントに結合することができる二次抗体をさらに含む、請求項63に記載の診断キット。
- 物質は、hCAP18/LL−37のmRNAに選択的にハイブリダイズすることができるオリゴヌクレオチドである、請求項62に記載の診断キット。
- オリゴヌクレオチドは、配列番号1に記載のヌクレオチドの配列のフラグメント、もしくは、それらの変異体を含む、または、それらからなる、請求項65に記載の診断キット。
- オリゴヌクレオチドは、検出可能な部分を含む、請求項66に記載の診断キット。
- 検出可能な部分は、放射性同位体、放射性核種、蛍光標識、酵素標識、化学発光法による、ビオチニル基、および、第二のレポーターによって認識された既定のポリペプチドエピトープからなる群より選択される、請求項67に記載の診断キット。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US63775904P | 2004-12-22 | 2004-12-22 | |
| PCT/GB2005/004900 WO2006067402A2 (en) | 2004-12-22 | 2005-12-16 | Agents inibiting the cathelin-like protein cap18/ll-37 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2008525005A true JP2008525005A (ja) | 2008-07-17 |
| JP2008525005A5 JP2008525005A5 (ja) | 2009-02-12 |
Family
ID=36283015
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007547622A Abandoned JP2008525005A (ja) | 2004-12-22 | 2005-12-16 | 新規の物質、および、それらの使用 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7741275B2 (ja) |
| EP (1) | EP1846444A2 (ja) |
| JP (1) | JP2008525005A (ja) |
| CN (1) | CN101142233A (ja) |
| CA (1) | CA2593956A1 (ja) |
| WO (1) | WO2006067402A2 (ja) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0608797D0 (en) * | 2006-05-04 | 2006-06-14 | Lipopeptide Ab | Novel agents and the use thereof |
| CA2655022A1 (en) * | 2006-06-20 | 2007-12-27 | Lipopeptide Ab | Use cathelicidin antimicrobial protein (hcap18 ) as anticancer agent |
| US20120315290A1 (en) * | 2006-12-15 | 2012-12-13 | Board Of Regents, The University Of Texas System | Inhibitors of ll-37 mediated immune reactivity to self nucleic acids |
| CN101571549B (zh) * | 2008-04-30 | 2013-08-14 | 上海泽润生物科技有限公司 | Vero细胞HCP检测试剂盒及其应用 |
| CA2780741C (en) | 2009-10-12 | 2023-04-04 | Smith Holdings, Llc | Methods and compositions for modulating gene expression using oligonucleotide based drugs administered in vivo or in vitro |
| CN111973805B (zh) * | 2019-08-16 | 2022-03-01 | 苏州吉美瑞生医学科技有限公司 | 抗菌肽hCAP18/LL-37在抗感染生物工程肺中的应用 |
| CA3190519A1 (en) * | 2020-08-26 | 2022-03-03 | Cila Therapeutics Inc. | Inhalable therapeutic agents |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4816567A (en) * | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US5116742A (en) * | 1986-12-03 | 1992-05-26 | University Patents, Inc. | RNA ribozyme restriction endoribonucleases and methods |
| US4987071A (en) * | 1986-12-03 | 1991-01-22 | University Patents, Inc. | RNA ribozyme polymerases, dephosphorylases, restriction endoribonucleases and methods |
| US5168053A (en) * | 1989-03-24 | 1992-12-01 | Yale University | Cleavage of targeted RNA by RNAase P |
| US5149796A (en) * | 1989-08-31 | 1992-09-22 | City Of Hope | Chimeric DNA-RNA catalytic sequences |
| US5180818A (en) * | 1990-03-21 | 1993-01-19 | The University Of Colorado Foundation, Inc. | Site specific cleavage of single-stranded dna |
| GB9223084D0 (en) | 1992-11-04 | 1992-12-16 | Imp Cancer Res Tech | Compounds to target cells |
| SE9400088D0 (sv) * | 1994-01-14 | 1994-01-14 | Kabi Pharmacia Ab | Bacterial receptor structures |
| PL178438B1 (pl) | 1994-02-04 | 2000-04-28 | Scotia Lipidteknik Ab | Preparat dwuwarstwy lipidowej, środek farmaceutyczny zawierający preparat dwuwarstwy lipidowej i sposób wytwarzania środka farmaceutycznego |
| AU691250B2 (en) | 1994-02-04 | 1998-05-14 | Lipocore Holding Ab | Lipophilic carrier preparations |
| BR9506681A (pt) | 1994-02-04 | 1997-11-18 | Scotia Lipidteknik Ab | Emulsão oleo em água e composição farmacêutica |
| EP1358888A1 (en) | 2002-02-28 | 2003-11-05 | Robert Bals | The human peptide antibiotic LL-37/hCAP-18 is an inducer of angiogenesis |
| SE0201863D0 (en) | 2002-06-18 | 2002-06-18 | Cepep Ab | Cell penetrating peptides |
| US20060057134A1 (en) * | 2002-07-22 | 2006-03-16 | Teruo Kirikae | Antibody against antibacterial peptide and utiliazation thereof |
| US20060115480A1 (en) | 2002-12-19 | 2006-06-01 | Yitzchak Hillman | Disease treatment via antimicrobial peptide inhibitors |
| SE0300207D0 (sv) * | 2003-01-29 | 2003-01-29 | Karolinska Innovations Ab | New use and composition |
| WO2004098536A2 (en) * | 2003-03-06 | 2004-11-18 | The Regents Of The University Of California | Anti-viral activity of cathelicidin peptides |
-
2005
- 2005-12-16 CA CA002593956A patent/CA2593956A1/en not_active Abandoned
- 2005-12-16 JP JP2007547622A patent/JP2008525005A/ja not_active Abandoned
- 2005-12-16 EP EP05847503A patent/EP1846444A2/en not_active Withdrawn
- 2005-12-16 US US11/793,708 patent/US7741275B2/en not_active Expired - Fee Related
- 2005-12-16 WO PCT/GB2005/004900 patent/WO2006067402A2/en active Application Filing
- 2005-12-16 CN CNA2005800482770A patent/CN101142233A/zh active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| EP1846444A2 (en) | 2007-10-24 |
| WO2006067402A3 (en) | 2006-09-08 |
| CA2593956A1 (en) | 2006-06-29 |
| WO2006067402A2 (en) | 2006-06-29 |
| CN101142233A (zh) | 2008-03-12 |
| US20080187530A1 (en) | 2008-08-07 |
| US7741275B2 (en) | 2010-06-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Massfelder et al. | Parathyroid hormone-related protein is an essential growth factor for human clear cell renal carcinoma and a target for the von Hippel-Lindau tumor suppressor gene | |
| US7741275B2 (en) | Agents and use thereof | |
| JP2008525479A (ja) | トレフォイル因子およびそれを用いた増殖性疾患の処置方法 | |
| JP2009541287A (ja) | 抗癌剤としてのカテリシジン抗菌タンパク質(hCAP18)の使用 | |
| EP1814595A1 (en) | Treatment of cancer with a combination of an agent that perturbs the egf signaling pathway and an oligonucleotide that reduces clusterin levels | |
| EP2684568B1 (en) | EEF2-derived peptides for the treatment or prevention of cancer | |
| US20070298445A1 (en) | Cancer Therapeutic | |
| US9518098B2 (en) | Uses of NANOG inhibitors and related methods | |
| JP2009535391A (ja) | 乳癌の治療に使用するためのhCAP18/LL−37の阻害剤 | |
| WO2006016574A1 (ja) | RNAiを利用した抗腫瘍剤 | |
| US7838495B2 (en) | Compositions and methods of use of EPB1, and ErbB3 binding protein | |
| WO2020081528A1 (en) | Method and system for treating cancer utilizing tinagl 1 | |
| JP2009535392A (ja) | 併用療法製品およびその使用 | |
| US20110217259A1 (en) | IL-16 as a target for diagnosis and therapy of hematological malignancies and solid tumors | |
| WO2023088464A1 (zh) | Cd300ld抑制剂及其在制备肿瘤免疫治疗产品中的用途 | |
| KR20190112977A (ko) | Nanog 매개 질환의 치료 또는 진단을 위한 hsp90aa1의 용도 | |
| DK2257299T3 (en) | Modulation of SRPX2-mediated angiogenesis | |
| KR102134954B1 (ko) | CEACAM6 발현 억제를 위한 siRNA 및 이를 포함하는 선암 치료제 | |
| WO2023212566A1 (en) | Compositions and methods for preventing t cell exhaustion | |
| CN115786270A (zh) | 工程化的巨噬细胞及其在治疗纤维化疾病中的应用 | |
| WO2018222432A1 (en) | A predictive biomarker for let-7 microrna-based therapeutics for the treatment of human cancer | |
| KR20140045345A (ko) | 질환을 치료하고, 진단하고, 모니터링하기 위한 조성물과 방법 | |
| CN117534729A (zh) | Tmem176b的竞争性多肽、药物组合物及用途 | |
| KR20190112978A (ko) | Nanog 매개 질환의 치료 또는 진단을 위한 cry1의 용도 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20081212 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20081212 |
|
| A762 | Written abandonment of application |
Free format text: JAPANESE INTERMEDIATE CODE: A762 Effective date: 20090903 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20090903 |